User login
Diagnosis: Systemic sclerosis
Systemic sclerosis, or scleroderma, is a rare connective tissue disorder in which excessive collagen is deposited in the skin and internal organs. This disease predominantly affects women (3-6:1) between the ages of 20 and 60 years with no apparent racial predominance. Effective treatment is critical, as scleroderma carries a poor prognosis, with a mortality rate of up to 50% at 5 years in severe cases. The pathogenesis of systemic sclerosis is unknown, but three pathways are implicated, including immune deregulation, vascular abnormalities, and abnormal fibroblast activation.
Clinical presentation is variable because of the involvement of multiple organ systems. Common features include cutaneous pruritus, skin thickening, Raynaud's phenomenon, difficulty swallowing, shortness of breath, palpitations, nonproductive cough, and joint pain and swelling, as well as muscle pain and weakness. Laboratory findings may include elevated erythrocyte sedimentation rate, thrombocytopenia, hypergammaglobulinemia, increased urea and creatinine levels, and elevated C-reactive protein. Antinuclear antibodies are usually elevated, especially Scl-70, antimitochondrial, and anticentromere antibodies. Cardiac and pulmonary function should be assessed upon diagnosis. A Doppler echocardiogram may detect cardiac abnormalities, and chest x-ray or high-resolution CT is used to assess for pulmonary fibrosis.
Despite the severity of the disease, there are no Food and Drug Administration-approved disease-modifying agents for the treatment of scleroderma, and management often focuses on symptom relief. For example, patients with kidney involvement should be placed on an ACE inhibitor or angiotensin II inhibitor therapy, and patients with gastrointestinal tract involvement should use proton pump inhibitors and H2 blockers to control reflux. Bosentan and pentoxifylline, which target vascular abnormalities, also may help improve skin fibrosis. Steroids show benefits in the early stages of the disease, but carry a risk of scleroderma renal crisis with doses greater than 15 mg of prednisone daily. Mycophenolate mofetil and sirolimus have immunomodulatory and antifibrotic properties, which may be of benefit in this disease.
Cyclophosphamide is reserved for more severe cases. Other treatment modalities include rituximab, intravenous immunoglobulin, and autologous stem cell transplantation.
Diagnosis: Systemic sclerosis
Systemic sclerosis, or scleroderma, is a rare connective tissue disorder in which excessive collagen is deposited in the skin and internal organs. This disease predominantly affects women (3-6:1) between the ages of 20 and 60 years with no apparent racial predominance. Effective treatment is critical, as scleroderma carries a poor prognosis, with a mortality rate of up to 50% at 5 years in severe cases. The pathogenesis of systemic sclerosis is unknown, but three pathways are implicated, including immune deregulation, vascular abnormalities, and abnormal fibroblast activation.
Clinical presentation is variable because of the involvement of multiple organ systems. Common features include cutaneous pruritus, skin thickening, Raynaud's phenomenon, difficulty swallowing, shortness of breath, palpitations, nonproductive cough, and joint pain and swelling, as well as muscle pain and weakness. Laboratory findings may include elevated erythrocyte sedimentation rate, thrombocytopenia, hypergammaglobulinemia, increased urea and creatinine levels, and elevated C-reactive protein. Antinuclear antibodies are usually elevated, especially Scl-70, antimitochondrial, and anticentromere antibodies. Cardiac and pulmonary function should be assessed upon diagnosis. A Doppler echocardiogram may detect cardiac abnormalities, and chest x-ray or high-resolution CT is used to assess for pulmonary fibrosis.
Despite the severity of the disease, there are no Food and Drug Administration-approved disease-modifying agents for the treatment of scleroderma, and management often focuses on symptom relief. For example, patients with kidney involvement should be placed on an ACE inhibitor or angiotensin II inhibitor therapy, and patients with gastrointestinal tract involvement should use proton pump inhibitors and H2 blockers to control reflux. Bosentan and pentoxifylline, which target vascular abnormalities, also may help improve skin fibrosis. Steroids show benefits in the early stages of the disease, but carry a risk of scleroderma renal crisis with doses greater than 15 mg of prednisone daily. Mycophenolate mofetil and sirolimus have immunomodulatory and antifibrotic properties, which may be of benefit in this disease.
Cyclophosphamide is reserved for more severe cases. Other treatment modalities include rituximab, intravenous immunoglobulin, and autologous stem cell transplantation.
Diagnosis: Systemic sclerosis
Systemic sclerosis, or scleroderma, is a rare connective tissue disorder in which excessive collagen is deposited in the skin and internal organs. This disease predominantly affects women (3-6:1) between the ages of 20 and 60 years with no apparent racial predominance. Effective treatment is critical, as scleroderma carries a poor prognosis, with a mortality rate of up to 50% at 5 years in severe cases. The pathogenesis of systemic sclerosis is unknown, but three pathways are implicated, including immune deregulation, vascular abnormalities, and abnormal fibroblast activation.
Clinical presentation is variable because of the involvement of multiple organ systems. Common features include cutaneous pruritus, skin thickening, Raynaud's phenomenon, difficulty swallowing, shortness of breath, palpitations, nonproductive cough, and joint pain and swelling, as well as muscle pain and weakness. Laboratory findings may include elevated erythrocyte sedimentation rate, thrombocytopenia, hypergammaglobulinemia, increased urea and creatinine levels, and elevated C-reactive protein. Antinuclear antibodies are usually elevated, especially Scl-70, antimitochondrial, and anticentromere antibodies. Cardiac and pulmonary function should be assessed upon diagnosis. A Doppler echocardiogram may detect cardiac abnormalities, and chest x-ray or high-resolution CT is used to assess for pulmonary fibrosis.
Despite the severity of the disease, there are no Food and Drug Administration-approved disease-modifying agents for the treatment of scleroderma, and management often focuses on symptom relief. For example, patients with kidney involvement should be placed on an ACE inhibitor or angiotensin II inhibitor therapy, and patients with gastrointestinal tract involvement should use proton pump inhibitors and H2 blockers to control reflux. Bosentan and pentoxifylline, which target vascular abnormalities, also may help improve skin fibrosis. Steroids show benefits in the early stages of the disease, but carry a risk of scleroderma renal crisis with doses greater than 15 mg of prednisone daily. Mycophenolate mofetil and sirolimus have immunomodulatory and antifibrotic properties, which may be of benefit in this disease.
Cyclophosphamide is reserved for more severe cases. Other treatment modalities include rituximab, intravenous immunoglobulin, and autologous stem cell transplantation.
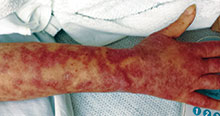
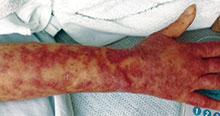
This case and photo were submitted by Dr. Andrew R. Styperek, Houston Methodist Hospital and Dr. Leonard H. Goldberg of DermSurgery Associates, both in Houston. Dr. Bilu Martin is in private practice at Premier Dermatology, MD in Aventura, Fla. To submit your case for possible publication, send an e-mail to dermnews@frontlinemedcom.com. A 27-year-old white female was admitted to the hospital with fever and shortness of breath. Dermatology was consulted for evaluation of a long history of chronic skin itchiness and eczema, which had never resolved despite topical therapy. The patient denied any skeletal, tooth, or lung abnormalities. Her past medical history was significant for chronic cytomegalovirus infection of the right eye, resulting in extirpation of the orbit. She also had a history of eczema herpeticum. On physical exam, she had a patch of gauze over her right orbit, significant soft tissue loss of the nose, and numerous diffuse pink/red eczematous plaques over her arms, trunk, legs, and face. Of note, she also had multiple umbilicated and verrucous papules scattered over her body.