User login
Systemic sclerosis (SSc), also called scleroderma, is a rare but serious autoimmune connective tissue disease that has multiple fluctuating pathologic manifestations throughout its temporal course. Estimates have shown that the incidence is 10 to 20 cases per 1 million, and the prevalence is 4 to 253 cases per 1 million.1,2 Given the rarity of this incurable condition, it is vital that primary care providers (PCPs) are able to recognize its unique features early to limit and prevent acute and chronic complications. This case report discusses a patient’s journey with late-diagnosed scleroderma in order to convey these broad manifestations and what providers can do to manage it with their patients.
Case Presentation
Mr. P is a 60-year-old African American male with a history of hypertension, recurrent digital ulcers, pulmonary hypertension (PH), interstitial lung disease (ILD), kidney involvement, congestive heart failure (CHF), and gastroesophageal reflux disease (GERD). Mr. P’s workup began in his late 40s with resistant hypertension, resistant GERD, and multiple hospitalizations for hypertensive urgency. It was not until he was 54 years old that he was diagnosed with mixed connective tissue disorder with sclerodermatous predominance.
Review of systems throughout his medical examinations in his 50s were notable for skin tightening over his hands and shoulders, skin hypopigmentation over his scalp and face, and hair loss. Mr. P was found to have Raynaud phenomenon beginning with his original presentation and digital ulceration without complications of gangrene or autoamputation. Aggregate physical examinations were notable for digital ulceration, skin tightening/sclerodactyly, and telangiectasia. Serologic markers were notable for the following:
- Positive ANA (antinuclear antibody) with titer of 1:1,280/homogenous pattern;
- Positive anti-RNP (antiribonucleoprotein) with titer of 171.2;
- Positive anti-Scl-70 (antitopoisomerase I) with titer of 108.1;
- Positive anti-SM (anti-Smith antibody) with titer of 30.2;
- Positive anti-Ro (SSA) with titer of 107.6;
- Negative anti-La (SSB) with titer of 1.3;
- Negative anti-dsDNA (anti-double stranded DNA) with titer of 9; and
- Negative ACA (anticentromere antibody).
Early transthoracic echocardiograms revealed an ejection fraction (EF) initially at 55% with evidence of left ventricular hypertrophy. Following treatment with phosphodiesterase-5 inhibitors (PDE-5 inhibitors) and endothelin-1 antagonists for pulmonary hypertension, serial transthoracic echocardiograms showed improvement in his EF. 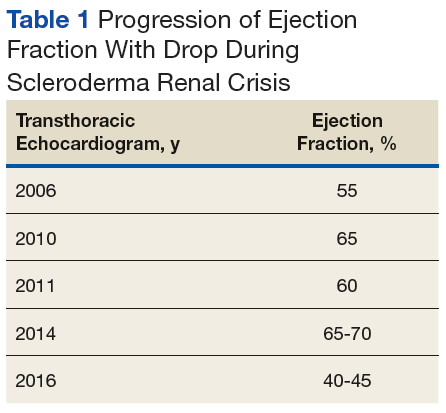
A chest X-ray did not show signs of ILD, but a subsequent high-resolution computed tomography (HRCT) scan was consistent with chronic ILD with a main pulmonary artery diameter of 3 cm (Figure 1). 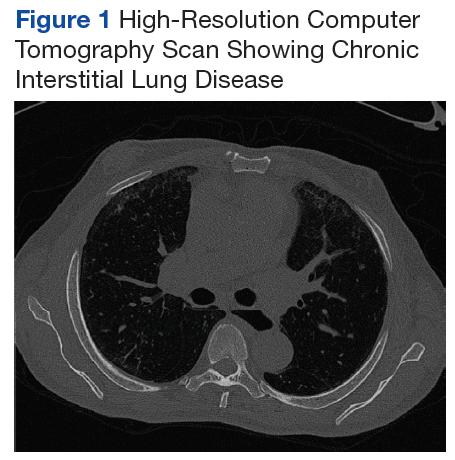
In subsequent years, Mr. P was hospitalized several times, secondary to digit pain, ulcerations, and osteomyelitis. His first episode was 1 year after his scleroderma diagnosis, when he was hospitalized for 6 days for complications of SSc and finger pain. The following year, he had a 3-day hospitalization for hypertensive urgency and right third-digit osteomyelitis, treated initially with IV fluids, levofloxacin, and vancomycin, and then ceftaroline for 1 month. Throughout the next 6 years, Mr. P presented multiple times with fingertip ulcerations and was followed in the Infectious Disease clinic for recurrent osteomyelitis. He found some relief with systemic antibiotics, including augmentin, minocycline, moxifloxacin, and doxycycline.
At age 59, he was hospitalized for scleroderma renal crisis (SRC). Early in his disease, his kidney function was normal, but the SRC was discovered after an abrupt rise in his blood pressure (BP) and an increase in serum creatinine (SCr) from 1.2 mg/dL at baseline to 3.06 mg/dL. The presence of brown granular cast in his urine prompted a renal biopsy that showed thrombotic microangiopathy with schistocytes. Mr. P was started on captopril and remained stable with outpatient follow-up for this renal complication.
Discussion
Initial presentation of SSc can occur along a spectrum of its pathophysiology. A more severe presentation, like the one seen in Mr. P, seems to occur more frequently in African American patients relative to white patients.3 Differentiating between the 2 types—diffuse vs limited SSc—is vital to managing patients and disease progression. Limited-type SSc is more common (60%) and less severe with slower progression than is diffuse SSc. 
Diffuse-type SSc (35%) includes features such as skin thickening and tightening, ILD, SRC, tendon friction rubs (palpable crepitus over tendons), and skin pigment changes.4 The specific involvement of the renal and cardiopulmonary systems accounts for the higher mortality rates in the diffuse-type.5
Many patients with SSc require periodic hospitalizations throughout their life for the acute complications of the disease. Hospitalized patients often range from age 45 to 64 years and are more often female. However, of hospitalized patients with SSc, in-hospital death rates are higher among men.3,6 Although these rates have decreased as the pathogenesis of SSc has become better understood, it is important to note that in-hospital mortality in 1995 for all patients with SSc was 7.1% and mean length of stay was 7.5 days, and in 2002 to 2003, 6.3% and 6.6 days, respectively.3 Though the burden of this disease has decreased, mortality and hospitalizations continue to persist at high rates. Understanding the pathogenesis, progression, and treatments of SSc are essential to aiding patients with this diagnosis.
Skin Involvement
A common finding and presentation for patients with SSc is related to skin involvement. Common patient complaints and exam findings include calcinosis along extensor tendons and digits, Raynaud phenomenon (seen in more than 95% of patients), sclerodactyly, telangiectasias, hyper/hypopigmentation, and pruritus.4 These findings are useful in diagnosing and monitoring patients for disease progression.
Many of the listed skin manifestations affect patients’ quality of life (QOL) but are not directly associated with mortality. However, a common and feared complication includes skin ulcers and osteomyelitis, seen in 48% and 7.7% of patients, respectively.7 Digit ulcers, areas with loss of dermis and epidermis distal to the proximal interphalangeal joints (Figures 2A and 2B), are significant because they parallel a more rapid progression of internal organ involvement.8 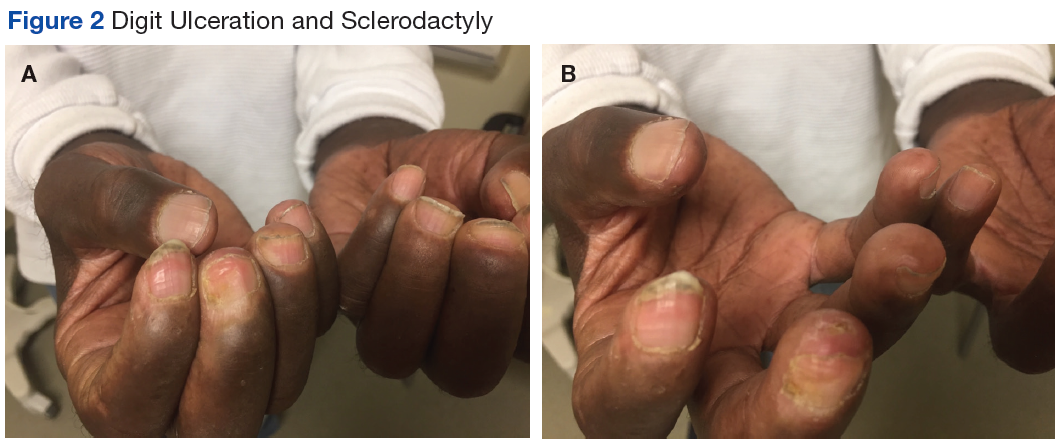
Mr. P required multiple hospitalizations and antibiotic regimens for painful digit ulcers complicated by osteomyelitis (Figure 3). 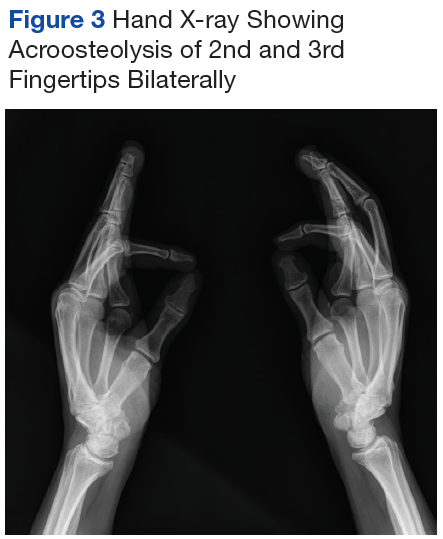
Treatment usually is aimed at infections that complicate these skin ulcers and are based on site-specific cultures. Preventive measures are aimed at the risk factors associated with digit ulcers, including decreased whole-body warmth, direct trauma to digits, smoking, and vasoconstrictors (eg, cocaine, sympathomimetics).8 Some patients may prevent ulcers by using D-penicillamine, mycophenolate mofetil, and cyclophosphamide, although definitive treatment has not been found.4 Calcium channel blockers, PDE-5 inhibitors, endothelin receptor antagonists, and prostacyclin analogues also have been used to reduce the severity of Raynaud phenomenon attacks and to decrease the number of digital ulcers (in addition to their beneficial effects on pulmonary involvement).8 Additional pharmacologic agents that have been linked to an improvement in Raynaud phenomenon and digital ulcers include statins, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, intravenous N-acetylcysteine, vitamin E gel, and surgical options (eg, revascularization and sympathectomy).8
Pulmonary Involvement
Systemic sclerosis can lead to 2 complications in the lungs, both present in Mr. P: PAH (mean pulmonary arterial pressure > 25 mm Hg) and ILD, which together make lung involvement the leading cause of death for these patients. Limited-type SSc usually is restricted to PAH, whereas diffuse-type SSc usually leads to ILD.4
On diagnosis of SSc, an HRCT is indicated to assess the degree of lung involvement. Mr. P’s HRCT showed a 3-cm main pulmonary artery, suggestive of PAH, in addition to evidence of ILD seen on the chest X-ray (Figure 4). 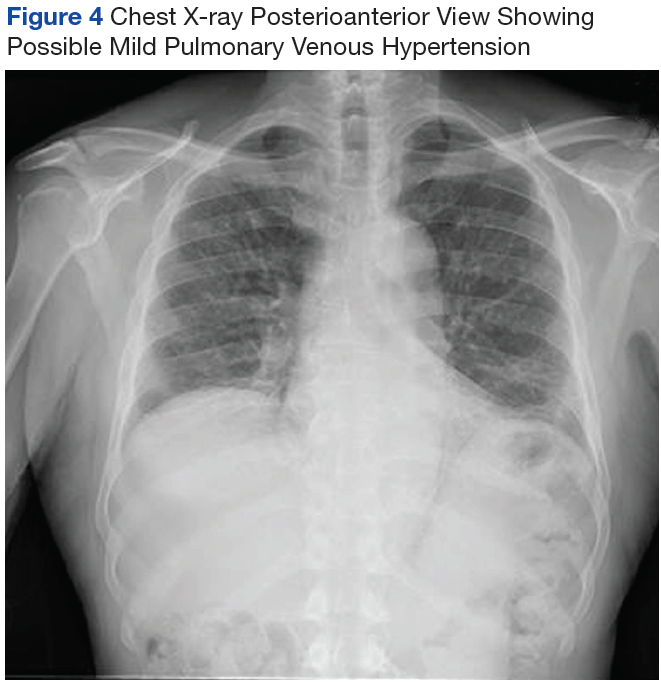
Pulmonary arterial hypertension is a common finding in patients with SSc and carries a severe prognosis. Risk factors for the development of PAH, when not present on initial diagnosis, include limited-type SSc; late age of onset; Raynaud phenomenon; decreased DLCO, FVC/DLCO < 1.6; increased N-terminal pro-B-type natriuretic peptide (NT-proBNP) serum levels; and the presence of antibodies.9 Patients with both SSc and PAH have a 50% to 87% 1-year survival, whereas patients with idiopathic PAH without connective tissue disease have an 88% 1-year survival.9
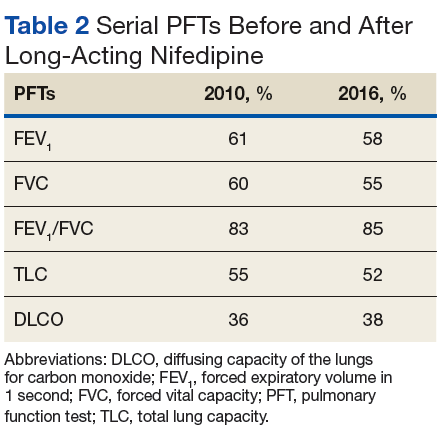
Many patients with lung involvement are asymptomatic, but some have findings of crackles and interstitial thickening on chest X-ray that can progress to cyanosis and right heart failure (cor pulmonale).10 On discovery of lung involvement (Figure 5), it is important to follow up with a PFT, primarily because the outcomes and prognosis for patients with SSc are correlated with the presenting severity of ILD and the subsequent progression of their DLCO.4,10 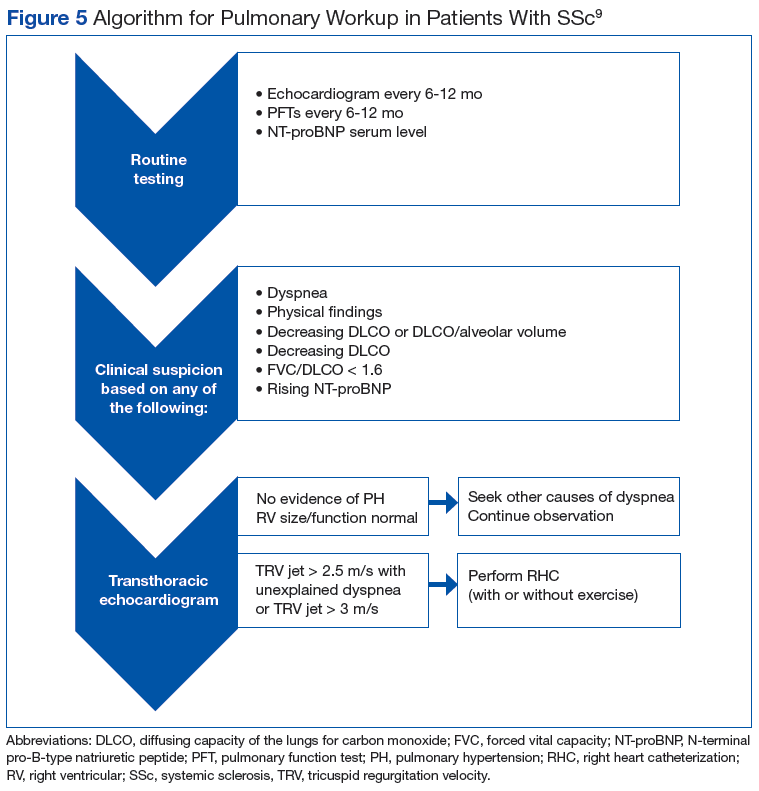
Treatment is directed at PAH and ILD separately. For PAH, a PDE-5 inhibitor, such as tadalafil or sildenafil, and an endothelin-1 receptor antagonist, such as ambrisentan or bosentan, are indicated. Other PAH treatments include diuretics and prostacyclin analogues (eg, epoprostenol, treprostinil, or iloprost) in addition to warfarin if patients have a history of thrombotic events.4 Mr. P’s pulmonologists deferred treatment with prostacyclin analogues given the potential for adverse effects with endothelin-1 receptor inhibitors, PDE-5 inhibitors, and calcium-channel blockers, although combination studies with bosentan and inhaled iloprost have shown promise.11
Like the skin manifestations of SSc, ILD lacks definitive treatment to prevent disease progression. However, some patients benefit from cyclophosphamide, followed by mycophenolate mofetil—which is usually better tolerated—azathioprine, haematopoietic stem cell transplant as rescue therapy, and lung transplant for life-saving treatment.10
Renal Involvement
Renal involvement in patients with SSc can have profound effects on QOL. Even without clinical renal involvement, glomerular filtration rate (GFR) usually decreases with the progression of vascular damage correlated with age and disease duration.12 Patients with a history of digital ulcers, like Mr. P, usually have a lower GFR than that of patients without digital ulcers.12 Monitoring renal function is vital in caring for these patients, because SRC occurs in 2% to 15% of all patients with SSc.13,14
Scleroderma renal crisis is generally defined as an abruptly elevated BP (> 140/90 mm Hgor > 30 mm Hg rise from baseline) with acute renal failure (elevated SCr) and decreased urine output.14 Given the rarity of SSc in the population, diagnosis of SRC requires high clinical suspicion. In the case of Mr. P, a workup involving serum analysis, urinalysis, and renal biopsy allowed for a definitive diagnosis. A renal biopsy can show microangiopathic hemolytic anemia and confirm SRC, although it may not be necessary in patients with known SSc presenting with new hypertension, rising creatinine, and unremarkable urine sediment on microscopy.14,15
Although an acute SRC can be difficult to predict, monitoring renal function and attention to key factors can assist in discovering this SSc complication. Scleroderma renal crisis usually occurs within the first 4 years of SSc diagnosis, often paralleling rapid progression of skin thickening and tightening with higher rates in both African Americans and males.13,14 Additional predictive factors include diffuse skin involvement, rapid progression of skin involvement, positive anti-RNA polymerase III antibodies, new anemia, new cardiac events (eg, pericardial effusion, pericarditis, left ventricular insufficiency), CHF, tendon friction rubs, arthritis, and recent (within 3 months) high-dose glucocorticoid use.4,13-15
The presentation of SRC can be nonspecific, often resembling findings related to acute kidney injury. Patients may report malaise, fatigue, fever, headache, seizure, blurred vision, or dyspnea.13 Clinical parametersto examine include systolic BP > 140 mm Hgand/or diastolic BP > 90 mm Hg (or an abrupt rise of > 30 or > 20, respectively), SCr increase by 50%+ or > 120% of the upper normal limit, proteinuria at 2+, high protein:creatinine ratio, hematuria > 2+ or > 10 red blood cells (RBCs), platelets < 100,000/mm3, and hypertensive encephalopathy.13 Mr. P presented with fatigue, dyspnea, an abrupt rise in BP (156/96 mm Hg), foamy urine, bilateral lower extremity edema (3.9 g/dL albumin), 2+ RBCs, and a SCr of 2.46 mg/dL on admission (a 205% increase from his last baseline of 1.2 mg/dL).
Treatments have had a large impact on the mortality rates of SRC. Following the introduction of ACE inhibitors, mortality from SRC has decreased from 76% to < 10% over recent decades.13 In addition to improving survival, these medications also have improved hospitalization outcomes for these patients.3 Captopril is often the medication of choice given its rapid onset and short duration of action relative to other medications in the same class, such as benazepril, enalapril, fosinopril, lisinopril, quinapril, and ramipril.4,13 Current clinical trials are assessing the specific renal benefits of endothelin-1 receptor antagonists, which often are used for pulmonary and skin involvement.14 For patients presenting with acute SRC and uremia, dialysis may be necessary (typically predicted by elevated NT-proBNP serum levels), while the decision for transplantation may be indicated at least 2 years after SRC onset and resolution.4,13-15
Once the acute crisis resolves, it is important to discuss prognosis with patients. The 1-, 2-, 3-, 5-, and 10-year survival rates are 82%, 74%, 71%, 59%, and 47%, respectively; however, when looking at only male patients at 10 years, the survival rate is 17%.13 Prevention of SRC can be addressed with daily BP checks and advising patients to seek medical care if they notice a consecutive 2-day abrupt rise.13
Cardiac Involvement
Systemic sclerosis has significant effects on patients’ heart function. The introduction of ACE inhibitors has shifted mortality in SSc patients from predominantly SRC to cardiac causes.6 Cardiac involvement can occur through a range of processes, including abnormal cardiac conduction, CHF, diastolic dysfunction, mitral valve nodular thickening, and pericardial effusion.3 Even though patients often present with skin findings, an initial cardiac workup is crucial to understanding disease progression and patient prognosis. The severity of the cutaneous manifestations often predicts the degree of diastolic dysfunction.16
Clinical evidence of cardiac involvement is seen in 20% to 25% of patients with SSc and is associated with a 70% mortality at 5 years when symptoms are evident.16 Additionally, right ventricular dysfunction at presentation is the strongest marker for all-mortality prognosis, representing the degree of pulmonary involvement, and includes findings such as progressive shortness of breath and systemic edema.9
Given the increasing survival of patients with SSc, cardiac involvement is becoming more evident and prominent. Direct treatments for cardiac manifestations are based on the causative feature, namely, focusing on pulmonary and renal involvement, which can be assessed with periodic echocardiograms evaluating left ventricular EF.
Gastrointestinal Involvement
Along with skin manifestations, the gastrointestinal (GI) involvement of SSc can have a significant impact on patients’ QOL without direct contribution to mortality. One of Mr. P’s earliest symptoms that led to a diagnosis of SSc was GERD, which caused a chronic cough, dental erosions, esophageal erosions, duodenal ulcers, dysphagia, abdominal pain, halitosis, pharyngitis, and weight loss. Esophageal involvement occurs in up to 96% of patients with SSc and can include motility abnormalities (eg, strictures and/or muscle dysfunction), lower esophageal sphincter abnormalities, and Barrett esophagus.17
Additional symptoms of the GI system linked with SSc can occur anywhere along the GI tract and include gastric antral vascular ectasia, causing GI bleeds and pernicious anemia, gastroparesis, bacterial overgrowth, intestinal malabsorption, pseudo-obstruction due to hypomotility, fecal incontinence due to anorectal involvement, and rarely, primary biliary cirrhosis.4,17 Decreased mobility of the oral aperture secondary to skin thickening and tightening also can contribute to malnutrition by decreasing oral intake.
Treatments are supportive and target symptom relief. Chronic treatment of GERD is often necessary and includes antacids, histamine-2 receptor blockers, and proton pump inhibitors.4 Other medications that can help with symptom relief include motility agents (such as metoclopramide, domperidone, prucalopride, tegaserod, and macrolides), osmotic laxatives, and ursodeoxycholic acid for primary biliary cirrhosis.17 Surgical intervention should be considered depending on the severity and progression of involvement within the GI tract. Behavioral changes that can improve patient symptoms include facial grimacing and other mouth-stretching exercises, frequent smaller meals followed by maintaining a vertical posture, and high fiber diets.17
Conclusion
Systemic sclerosis is an autoimmune and connective tissue disease with a pathophysiology that can manifest throughout the body. The organ systems that impact patient outcomes include skin, pulmonary, renal, cardiac, and GI. Primary care providers caring for patients diagnosed with SSc should monitor acute management and disease progression in all these systems. Important acute events that can impact morbidity, mortality, and/or QOL include Raynaud phenomenon, SRC, and pericardial effusion. Chronic manifestations that may be present on diagnosis of SSc or may develop while a patient is under a provider’s care include sclerodactyly, tendon calcinosis, PAH, ILD, chronic kidney injury, chronic cardiac damage, GERD, and esophageal dysmotility. While this discussion serves as a pertinent overview of patients with SSc, it is summative, and providers are encouraged to seek a stronger understanding of both the common and rarer manifestations within each of their patients.
1. Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41(5):778-799.
2. Silman AG. Scleroderma. In: Epidemiology of the Rheumatic Diseases. 2nd ed. Silman AJ, Hochberg MC, eds. Oxford, UK: Oxford University Press. 1993:chap 8.
3. Chung L, Krishnan E, Chakravarty EF. Hospitalizations and mortality in systemic sclerosis: results from the Nationwide Inpatient Sample. Rheumatology (Oxford). 2007;46(12):1808-1813.
4. Hinchcliff M, Varga J. Systemic sclerosis/scleroderma: a treatable multisystem disease. Am Fam Physician. 2008;78(8):961-968.
5. Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010;69(10):1809-1815.
6. Piga M, Casula L, Sanna S, et al. Population-based analysis of hospitalizations for patients with systemic sclerosis in a West-European region over the period 2001-2012. Rheumatol Int. 2016;36(1):73-81.
7. Giuggioli D, Manfredi A, Colaci M, Lumetti F, Ferri C. Osteomyelitis complicating scleroderma digital ulcers. Clin Rheumatol. 2013;32(5):623-627.
8. Schiopu E, Impens AJ, Phillips K. Digital ischemia in scleroderma spectrum of diseases. Int J Rheumatol. 2010;2010:923743.
9. Hassoun PM. Therapies for scleroderma-related pulmonary arterial hypertension. Expert Rev Respir Med. 2009;3(2):187-196.
10. Giacomelli R, Liakouli V, Berardicurti O, et al. Interstitial lung disease in systemic sclerosis: current and future treatment. Rheumatol Int. 2017;37(6):853-863.
11. McLaughlin V, Humbert M, Coghlan G, Nash P, Steen V. Pulmonary arterial hypertension: the most devastating vascular complication of systemic sclerosis. Rheumatology (Oxford). 2009; (suppl 3):iii25-iii31.
12. Gigante A, Barbano B, Granata G, et al. Evaluation of estimated glomerular filtration rate and clinical variables in systemic sclerosis patients. Clin Nephrol. 2016;85(6):326-331.
13. Bose N, Chiesa-Vottero A, Chatterjee S. Scleroderma renal crisis. Semin Arthritis Rheum. 2015;44(6):687-694.
14. Woodworth TG, Suliman YA, Furst DE, Clements P. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat Rev Nephrol. 2016;12(11):678-691.
15. Mouthon L, Bérezné A, Bussone G, Noël LH, Villiger PM, Guillevin L. Scleroderma renal crisis: a rare but severe complication of systemic sclerosis. Clin Rev Allergy Immunol. 2011;40(2):84-91.
16. Champion HC. The heart in scleroderma. Rheum Dis Clin North Am. 2008;34(1):181-90.
17. Tian XP, Zhang X. Gastrointestinal complications of systemic sclerosis. World J Gastroenterol. 2013;19(41):7062-7068.
Systemic sclerosis (SSc), also called scleroderma, is a rare but serious autoimmune connective tissue disease that has multiple fluctuating pathologic manifestations throughout its temporal course. Estimates have shown that the incidence is 10 to 20 cases per 1 million, and the prevalence is 4 to 253 cases per 1 million.1,2 Given the rarity of this incurable condition, it is vital that primary care providers (PCPs) are able to recognize its unique features early to limit and prevent acute and chronic complications. This case report discusses a patient’s journey with late-diagnosed scleroderma in order to convey these broad manifestations and what providers can do to manage it with their patients.
Case Presentation
Mr. P is a 60-year-old African American male with a history of hypertension, recurrent digital ulcers, pulmonary hypertension (PH), interstitial lung disease (ILD), kidney involvement, congestive heart failure (CHF), and gastroesophageal reflux disease (GERD). Mr. P’s workup began in his late 40s with resistant hypertension, resistant GERD, and multiple hospitalizations for hypertensive urgency. It was not until he was 54 years old that he was diagnosed with mixed connective tissue disorder with sclerodermatous predominance.
Review of systems throughout his medical examinations in his 50s were notable for skin tightening over his hands and shoulders, skin hypopigmentation over his scalp and face, and hair loss. Mr. P was found to have Raynaud phenomenon beginning with his original presentation and digital ulceration without complications of gangrene or autoamputation. Aggregate physical examinations were notable for digital ulceration, skin tightening/sclerodactyly, and telangiectasia. Serologic markers were notable for the following:
- Positive ANA (antinuclear antibody) with titer of 1:1,280/homogenous pattern;
- Positive anti-RNP (antiribonucleoprotein) with titer of 171.2;
- Positive anti-Scl-70 (antitopoisomerase I) with titer of 108.1;
- Positive anti-SM (anti-Smith antibody) with titer of 30.2;
- Positive anti-Ro (SSA) with titer of 107.6;
- Negative anti-La (SSB) with titer of 1.3;
- Negative anti-dsDNA (anti-double stranded DNA) with titer of 9; and
- Negative ACA (anticentromere antibody).
Early transthoracic echocardiograms revealed an ejection fraction (EF) initially at 55% with evidence of left ventricular hypertrophy. Following treatment with phosphodiesterase-5 inhibitors (PDE-5 inhibitors) and endothelin-1 antagonists for pulmonary hypertension, serial transthoracic echocardiograms showed improvement in his EF.
A chest X-ray did not show signs of ILD, but a subsequent high-resolution computed tomography (HRCT) scan was consistent with chronic ILD with a main pulmonary artery diameter of 3 cm (Figure 1).
In subsequent years, Mr. P was hospitalized several times, secondary to digit pain, ulcerations, and osteomyelitis. His first episode was 1 year after his scleroderma diagnosis, when he was hospitalized for 6 days for complications of SSc and finger pain. The following year, he had a 3-day hospitalization for hypertensive urgency and right third-digit osteomyelitis, treated initially with IV fluids, levofloxacin, and vancomycin, and then ceftaroline for 1 month. Throughout the next 6 years, Mr. P presented multiple times with fingertip ulcerations and was followed in the Infectious Disease clinic for recurrent osteomyelitis. He found some relief with systemic antibiotics, including augmentin, minocycline, moxifloxacin, and doxycycline.
At age 59, he was hospitalized for scleroderma renal crisis (SRC). Early in his disease, his kidney function was normal, but the SRC was discovered after an abrupt rise in his blood pressure (BP) and an increase in serum creatinine (SCr) from 1.2 mg/dL at baseline to 3.06 mg/dL. The presence of brown granular cast in his urine prompted a renal biopsy that showed thrombotic microangiopathy with schistocytes. Mr. P was started on captopril and remained stable with outpatient follow-up for this renal complication.
Discussion
Initial presentation of SSc can occur along a spectrum of its pathophysiology. A more severe presentation, like the one seen in Mr. P, seems to occur more frequently in African American patients relative to white patients.3 Differentiating between the 2 types—diffuse vs limited SSc—is vital to managing patients and disease progression. Limited-type SSc is more common (60%) and less severe with slower progression than is diffuse SSc.
Diffuse-type SSc (35%) includes features such as skin thickening and tightening, ILD, SRC, tendon friction rubs (palpable crepitus over tendons), and skin pigment changes.4 The specific involvement of the renal and cardiopulmonary systems accounts for the higher mortality rates in the diffuse-type.5
Many patients with SSc require periodic hospitalizations throughout their life for the acute complications of the disease. Hospitalized patients often range from age 45 to 64 years and are more often female. However, of hospitalized patients with SSc, in-hospital death rates are higher among men.3,6 Although these rates have decreased as the pathogenesis of SSc has become better understood, it is important to note that in-hospital mortality in 1995 for all patients with SSc was 7.1% and mean length of stay was 7.5 days, and in 2002 to 2003, 6.3% and 6.6 days, respectively.3 Though the burden of this disease has decreased, mortality and hospitalizations continue to persist at high rates. Understanding the pathogenesis, progression, and treatments of SSc are essential to aiding patients with this diagnosis.
Skin Involvement
A common finding and presentation for patients with SSc is related to skin involvement. Common patient complaints and exam findings include calcinosis along extensor tendons and digits, Raynaud phenomenon (seen in more than 95% of patients), sclerodactyly, telangiectasias, hyper/hypopigmentation, and pruritus.4 These findings are useful in diagnosing and monitoring patients for disease progression.
Many of the listed skin manifestations affect patients’ quality of life (QOL) but are not directly associated with mortality. However, a common and feared complication includes skin ulcers and osteomyelitis, seen in 48% and 7.7% of patients, respectively.7 Digit ulcers, areas with loss of dermis and epidermis distal to the proximal interphalangeal joints (Figures 2A and 2B), are significant because they parallel a more rapid progression of internal organ involvement.8
Mr. P required multiple hospitalizations and antibiotic regimens for painful digit ulcers complicated by osteomyelitis (Figure 3).
Treatment usually is aimed at infections that complicate these skin ulcers and are based on site-specific cultures. Preventive measures are aimed at the risk factors associated with digit ulcers, including decreased whole-body warmth, direct trauma to digits, smoking, and vasoconstrictors (eg, cocaine, sympathomimetics).8 Some patients may prevent ulcers by using D-penicillamine, mycophenolate mofetil, and cyclophosphamide, although definitive treatment has not been found.4 Calcium channel blockers, PDE-5 inhibitors, endothelin receptor antagonists, and prostacyclin analogues also have been used to reduce the severity of Raynaud phenomenon attacks and to decrease the number of digital ulcers (in addition to their beneficial effects on pulmonary involvement).8 Additional pharmacologic agents that have been linked to an improvement in Raynaud phenomenon and digital ulcers include statins, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, intravenous N-acetylcysteine, vitamin E gel, and surgical options (eg, revascularization and sympathectomy).8
Pulmonary Involvement
Systemic sclerosis can lead to 2 complications in the lungs, both present in Mr. P: PAH (mean pulmonary arterial pressure > 25 mm Hg) and ILD, which together make lung involvement the leading cause of death for these patients. Limited-type SSc usually is restricted to PAH, whereas diffuse-type SSc usually leads to ILD.4
On diagnosis of SSc, an HRCT is indicated to assess the degree of lung involvement. Mr. P’s HRCT showed a 3-cm main pulmonary artery, suggestive of PAH, in addition to evidence of ILD seen on the chest X-ray (Figure 4).
Pulmonary arterial hypertension is a common finding in patients with SSc and carries a severe prognosis. Risk factors for the development of PAH, when not present on initial diagnosis, include limited-type SSc; late age of onset; Raynaud phenomenon; decreased DLCO, FVC/DLCO < 1.6; increased N-terminal pro-B-type natriuretic peptide (NT-proBNP) serum levels; and the presence of antibodies.9 Patients with both SSc and PAH have a 50% to 87% 1-year survival, whereas patients with idiopathic PAH without connective tissue disease have an 88% 1-year survival.9
Many patients with lung involvement are asymptomatic, but some have findings of crackles and interstitial thickening on chest X-ray that can progress to cyanosis and right heart failure (cor pulmonale).10 On discovery of lung involvement (Figure 5), it is important to follow up with a PFT, primarily because the outcomes and prognosis for patients with SSc are correlated with the presenting severity of ILD and the subsequent progression of their DLCO.4,10
Treatment is directed at PAH and ILD separately. For PAH, a PDE-5 inhibitor, such as tadalafil or sildenafil, and an endothelin-1 receptor antagonist, such as ambrisentan or bosentan, are indicated. Other PAH treatments include diuretics and prostacyclin analogues (eg, epoprostenol, treprostinil, or iloprost) in addition to warfarin if patients have a history of thrombotic events.4 Mr. P’s pulmonologists deferred treatment with prostacyclin analogues given the potential for adverse effects with endothelin-1 receptor inhibitors, PDE-5 inhibitors, and calcium-channel blockers, although combination studies with bosentan and inhaled iloprost have shown promise.11
Like the skin manifestations of SSc, ILD lacks definitive treatment to prevent disease progression. However, some patients benefit from cyclophosphamide, followed by mycophenolate mofetil—which is usually better tolerated—azathioprine, haematopoietic stem cell transplant as rescue therapy, and lung transplant for life-saving treatment.10
Renal Involvement
Renal involvement in patients with SSc can have profound effects on QOL. Even without clinical renal involvement, glomerular filtration rate (GFR) usually decreases with the progression of vascular damage correlated with age and disease duration.12 Patients with a history of digital ulcers, like Mr. P, usually have a lower GFR than that of patients without digital ulcers.12 Monitoring renal function is vital in caring for these patients, because SRC occurs in 2% to 15% of all patients with SSc.13,14
Scleroderma renal crisis is generally defined as an abruptly elevated BP (> 140/90 mm Hgor > 30 mm Hg rise from baseline) with acute renal failure (elevated SCr) and decreased urine output.14 Given the rarity of SSc in the population, diagnosis of SRC requires high clinical suspicion. In the case of Mr. P, a workup involving serum analysis, urinalysis, and renal biopsy allowed for a definitive diagnosis. A renal biopsy can show microangiopathic hemolytic anemia and confirm SRC, although it may not be necessary in patients with known SSc presenting with new hypertension, rising creatinine, and unremarkable urine sediment on microscopy.14,15
Although an acute SRC can be difficult to predict, monitoring renal function and attention to key factors can assist in discovering this SSc complication. Scleroderma renal crisis usually occurs within the first 4 years of SSc diagnosis, often paralleling rapid progression of skin thickening and tightening with higher rates in both African Americans and males.13,14 Additional predictive factors include diffuse skin involvement, rapid progression of skin involvement, positive anti-RNA polymerase III antibodies, new anemia, new cardiac events (eg, pericardial effusion, pericarditis, left ventricular insufficiency), CHF, tendon friction rubs, arthritis, and recent (within 3 months) high-dose glucocorticoid use.4,13-15
The presentation of SRC can be nonspecific, often resembling findings related to acute kidney injury. Patients may report malaise, fatigue, fever, headache, seizure, blurred vision, or dyspnea.13 Clinical parametersto examine include systolic BP > 140 mm Hgand/or diastolic BP > 90 mm Hg (or an abrupt rise of > 30 or > 20, respectively), SCr increase by 50%+ or > 120% of the upper normal limit, proteinuria at 2+, high protein:creatinine ratio, hematuria > 2+ or > 10 red blood cells (RBCs), platelets < 100,000/mm3, and hypertensive encephalopathy.13 Mr. P presented with fatigue, dyspnea, an abrupt rise in BP (156/96 mm Hg), foamy urine, bilateral lower extremity edema (3.9 g/dL albumin), 2+ RBCs, and a SCr of 2.46 mg/dL on admission (a 205% increase from his last baseline of 1.2 mg/dL).
Treatments have had a large impact on the mortality rates of SRC. Following the introduction of ACE inhibitors, mortality from SRC has decreased from 76% to < 10% over recent decades.13 In addition to improving survival, these medications also have improved hospitalization outcomes for these patients.3 Captopril is often the medication of choice given its rapid onset and short duration of action relative to other medications in the same class, such as benazepril, enalapril, fosinopril, lisinopril, quinapril, and ramipril.4,13 Current clinical trials are assessing the specific renal benefits of endothelin-1 receptor antagonists, which often are used for pulmonary and skin involvement.14 For patients presenting with acute SRC and uremia, dialysis may be necessary (typically predicted by elevated NT-proBNP serum levels), while the decision for transplantation may be indicated at least 2 years after SRC onset and resolution.4,13-15
Once the acute crisis resolves, it is important to discuss prognosis with patients. The 1-, 2-, 3-, 5-, and 10-year survival rates are 82%, 74%, 71%, 59%, and 47%, respectively; however, when looking at only male patients at 10 years, the survival rate is 17%.13 Prevention of SRC can be addressed with daily BP checks and advising patients to seek medical care if they notice a consecutive 2-day abrupt rise.13
Cardiac Involvement
Systemic sclerosis has significant effects on patients’ heart function. The introduction of ACE inhibitors has shifted mortality in SSc patients from predominantly SRC to cardiac causes.6 Cardiac involvement can occur through a range of processes, including abnormal cardiac conduction, CHF, diastolic dysfunction, mitral valve nodular thickening, and pericardial effusion.3 Even though patients often present with skin findings, an initial cardiac workup is crucial to understanding disease progression and patient prognosis. The severity of the cutaneous manifestations often predicts the degree of diastolic dysfunction.16
Clinical evidence of cardiac involvement is seen in 20% to 25% of patients with SSc and is associated with a 70% mortality at 5 years when symptoms are evident.16 Additionally, right ventricular dysfunction at presentation is the strongest marker for all-mortality prognosis, representing the degree of pulmonary involvement, and includes findings such as progressive shortness of breath and systemic edema.9
Given the increasing survival of patients with SSc, cardiac involvement is becoming more evident and prominent. Direct treatments for cardiac manifestations are based on the causative feature, namely, focusing on pulmonary and renal involvement, which can be assessed with periodic echocardiograms evaluating left ventricular EF.
Gastrointestinal Involvement
Along with skin manifestations, the gastrointestinal (GI) involvement of SSc can have a significant impact on patients’ QOL without direct contribution to mortality. One of Mr. P’s earliest symptoms that led to a diagnosis of SSc was GERD, which caused a chronic cough, dental erosions, esophageal erosions, duodenal ulcers, dysphagia, abdominal pain, halitosis, pharyngitis, and weight loss. Esophageal involvement occurs in up to 96% of patients with SSc and can include motility abnormalities (eg, strictures and/or muscle dysfunction), lower esophageal sphincter abnormalities, and Barrett esophagus.17
Additional symptoms of the GI system linked with SSc can occur anywhere along the GI tract and include gastric antral vascular ectasia, causing GI bleeds and pernicious anemia, gastroparesis, bacterial overgrowth, intestinal malabsorption, pseudo-obstruction due to hypomotility, fecal incontinence due to anorectal involvement, and rarely, primary biliary cirrhosis.4,17 Decreased mobility of the oral aperture secondary to skin thickening and tightening also can contribute to malnutrition by decreasing oral intake.
Treatments are supportive and target symptom relief. Chronic treatment of GERD is often necessary and includes antacids, histamine-2 receptor blockers, and proton pump inhibitors.4 Other medications that can help with symptom relief include motility agents (such as metoclopramide, domperidone, prucalopride, tegaserod, and macrolides), osmotic laxatives, and ursodeoxycholic acid for primary biliary cirrhosis.17 Surgical intervention should be considered depending on the severity and progression of involvement within the GI tract. Behavioral changes that can improve patient symptoms include facial grimacing and other mouth-stretching exercises, frequent smaller meals followed by maintaining a vertical posture, and high fiber diets.17
Conclusion
Systemic sclerosis is an autoimmune and connective tissue disease with a pathophysiology that can manifest throughout the body. The organ systems that impact patient outcomes include skin, pulmonary, renal, cardiac, and GI. Primary care providers caring for patients diagnosed with SSc should monitor acute management and disease progression in all these systems. Important acute events that can impact morbidity, mortality, and/or QOL include Raynaud phenomenon, SRC, and pericardial effusion. Chronic manifestations that may be present on diagnosis of SSc or may develop while a patient is under a provider’s care include sclerodactyly, tendon calcinosis, PAH, ILD, chronic kidney injury, chronic cardiac damage, GERD, and esophageal dysmotility. While this discussion serves as a pertinent overview of patients with SSc, it is summative, and providers are encouraged to seek a stronger understanding of both the common and rarer manifestations within each of their patients.
Systemic sclerosis (SSc), also called scleroderma, is a rare but serious autoimmune connective tissue disease that has multiple fluctuating pathologic manifestations throughout its temporal course. Estimates have shown that the incidence is 10 to 20 cases per 1 million, and the prevalence is 4 to 253 cases per 1 million.1,2 Given the rarity of this incurable condition, it is vital that primary care providers (PCPs) are able to recognize its unique features early to limit and prevent acute and chronic complications. This case report discusses a patient’s journey with late-diagnosed scleroderma in order to convey these broad manifestations and what providers can do to manage it with their patients.
Case Presentation
Mr. P is a 60-year-old African American male with a history of hypertension, recurrent digital ulcers, pulmonary hypertension (PH), interstitial lung disease (ILD), kidney involvement, congestive heart failure (CHF), and gastroesophageal reflux disease (GERD). Mr. P’s workup began in his late 40s with resistant hypertension, resistant GERD, and multiple hospitalizations for hypertensive urgency. It was not until he was 54 years old that he was diagnosed with mixed connective tissue disorder with sclerodermatous predominance.
Review of systems throughout his medical examinations in his 50s were notable for skin tightening over his hands and shoulders, skin hypopigmentation over his scalp and face, and hair loss. Mr. P was found to have Raynaud phenomenon beginning with his original presentation and digital ulceration without complications of gangrene or autoamputation. Aggregate physical examinations were notable for digital ulceration, skin tightening/sclerodactyly, and telangiectasia. Serologic markers were notable for the following:
- Positive ANA (antinuclear antibody) with titer of 1:1,280/homogenous pattern;
- Positive anti-RNP (antiribonucleoprotein) with titer of 171.2;
- Positive anti-Scl-70 (antitopoisomerase I) with titer of 108.1;
- Positive anti-SM (anti-Smith antibody) with titer of 30.2;
- Positive anti-Ro (SSA) with titer of 107.6;
- Negative anti-La (SSB) with titer of 1.3;
- Negative anti-dsDNA (anti-double stranded DNA) with titer of 9; and
- Negative ACA (anticentromere antibody).
Early transthoracic echocardiograms revealed an ejection fraction (EF) initially at 55% with evidence of left ventricular hypertrophy. Following treatment with phosphodiesterase-5 inhibitors (PDE-5 inhibitors) and endothelin-1 antagonists for pulmonary hypertension, serial transthoracic echocardiograms showed improvement in his EF.
A chest X-ray did not show signs of ILD, but a subsequent high-resolution computed tomography (HRCT) scan was consistent with chronic ILD with a main pulmonary artery diameter of 3 cm (Figure 1).
In subsequent years, Mr. P was hospitalized several times, secondary to digit pain, ulcerations, and osteomyelitis. His first episode was 1 year after his scleroderma diagnosis, when he was hospitalized for 6 days for complications of SSc and finger pain. The following year, he had a 3-day hospitalization for hypertensive urgency and right third-digit osteomyelitis, treated initially with IV fluids, levofloxacin, and vancomycin, and then ceftaroline for 1 month. Throughout the next 6 years, Mr. P presented multiple times with fingertip ulcerations and was followed in the Infectious Disease clinic for recurrent osteomyelitis. He found some relief with systemic antibiotics, including augmentin, minocycline, moxifloxacin, and doxycycline.
At age 59, he was hospitalized for scleroderma renal crisis (SRC). Early in his disease, his kidney function was normal, but the SRC was discovered after an abrupt rise in his blood pressure (BP) and an increase in serum creatinine (SCr) from 1.2 mg/dL at baseline to 3.06 mg/dL. The presence of brown granular cast in his urine prompted a renal biopsy that showed thrombotic microangiopathy with schistocytes. Mr. P was started on captopril and remained stable with outpatient follow-up for this renal complication.
Discussion
Initial presentation of SSc can occur along a spectrum of its pathophysiology. A more severe presentation, like the one seen in Mr. P, seems to occur more frequently in African American patients relative to white patients.3 Differentiating between the 2 types—diffuse vs limited SSc—is vital to managing patients and disease progression. Limited-type SSc is more common (60%) and less severe with slower progression than is diffuse SSc.
Diffuse-type SSc (35%) includes features such as skin thickening and tightening, ILD, SRC, tendon friction rubs (palpable crepitus over tendons), and skin pigment changes.4 The specific involvement of the renal and cardiopulmonary systems accounts for the higher mortality rates in the diffuse-type.5
Many patients with SSc require periodic hospitalizations throughout their life for the acute complications of the disease. Hospitalized patients often range from age 45 to 64 years and are more often female. However, of hospitalized patients with SSc, in-hospital death rates are higher among men.3,6 Although these rates have decreased as the pathogenesis of SSc has become better understood, it is important to note that in-hospital mortality in 1995 for all patients with SSc was 7.1% and mean length of stay was 7.5 days, and in 2002 to 2003, 6.3% and 6.6 days, respectively.3 Though the burden of this disease has decreased, mortality and hospitalizations continue to persist at high rates. Understanding the pathogenesis, progression, and treatments of SSc are essential to aiding patients with this diagnosis.
Skin Involvement
A common finding and presentation for patients with SSc is related to skin involvement. Common patient complaints and exam findings include calcinosis along extensor tendons and digits, Raynaud phenomenon (seen in more than 95% of patients), sclerodactyly, telangiectasias, hyper/hypopigmentation, and pruritus.4 These findings are useful in diagnosing and monitoring patients for disease progression.
Many of the listed skin manifestations affect patients’ quality of life (QOL) but are not directly associated with mortality. However, a common and feared complication includes skin ulcers and osteomyelitis, seen in 48% and 7.7% of patients, respectively.7 Digit ulcers, areas with loss of dermis and epidermis distal to the proximal interphalangeal joints (Figures 2A and 2B), are significant because they parallel a more rapid progression of internal organ involvement.8
Mr. P required multiple hospitalizations and antibiotic regimens for painful digit ulcers complicated by osteomyelitis (Figure 3).
Treatment usually is aimed at infections that complicate these skin ulcers and are based on site-specific cultures. Preventive measures are aimed at the risk factors associated with digit ulcers, including decreased whole-body warmth, direct trauma to digits, smoking, and vasoconstrictors (eg, cocaine, sympathomimetics).8 Some patients may prevent ulcers by using D-penicillamine, mycophenolate mofetil, and cyclophosphamide, although definitive treatment has not been found.4 Calcium channel blockers, PDE-5 inhibitors, endothelin receptor antagonists, and prostacyclin analogues also have been used to reduce the severity of Raynaud phenomenon attacks and to decrease the number of digital ulcers (in addition to their beneficial effects on pulmonary involvement).8 Additional pharmacologic agents that have been linked to an improvement in Raynaud phenomenon and digital ulcers include statins, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, intravenous N-acetylcysteine, vitamin E gel, and surgical options (eg, revascularization and sympathectomy).8
Pulmonary Involvement
Systemic sclerosis can lead to 2 complications in the lungs, both present in Mr. P: PAH (mean pulmonary arterial pressure > 25 mm Hg) and ILD, which together make lung involvement the leading cause of death for these patients. Limited-type SSc usually is restricted to PAH, whereas diffuse-type SSc usually leads to ILD.4
On diagnosis of SSc, an HRCT is indicated to assess the degree of lung involvement. Mr. P’s HRCT showed a 3-cm main pulmonary artery, suggestive of PAH, in addition to evidence of ILD seen on the chest X-ray (Figure 4).
Pulmonary arterial hypertension is a common finding in patients with SSc and carries a severe prognosis. Risk factors for the development of PAH, when not present on initial diagnosis, include limited-type SSc; late age of onset; Raynaud phenomenon; decreased DLCO, FVC/DLCO < 1.6; increased N-terminal pro-B-type natriuretic peptide (NT-proBNP) serum levels; and the presence of antibodies.9 Patients with both SSc and PAH have a 50% to 87% 1-year survival, whereas patients with idiopathic PAH without connective tissue disease have an 88% 1-year survival.9
Many patients with lung involvement are asymptomatic, but some have findings of crackles and interstitial thickening on chest X-ray that can progress to cyanosis and right heart failure (cor pulmonale).10 On discovery of lung involvement (Figure 5), it is important to follow up with a PFT, primarily because the outcomes and prognosis for patients with SSc are correlated with the presenting severity of ILD and the subsequent progression of their DLCO.4,10
Treatment is directed at PAH and ILD separately. For PAH, a PDE-5 inhibitor, such as tadalafil or sildenafil, and an endothelin-1 receptor antagonist, such as ambrisentan or bosentan, are indicated. Other PAH treatments include diuretics and prostacyclin analogues (eg, epoprostenol, treprostinil, or iloprost) in addition to warfarin if patients have a history of thrombotic events.4 Mr. P’s pulmonologists deferred treatment with prostacyclin analogues given the potential for adverse effects with endothelin-1 receptor inhibitors, PDE-5 inhibitors, and calcium-channel blockers, although combination studies with bosentan and inhaled iloprost have shown promise.11
Like the skin manifestations of SSc, ILD lacks definitive treatment to prevent disease progression. However, some patients benefit from cyclophosphamide, followed by mycophenolate mofetil—which is usually better tolerated—azathioprine, haematopoietic stem cell transplant as rescue therapy, and lung transplant for life-saving treatment.10
Renal Involvement
Renal involvement in patients with SSc can have profound effects on QOL. Even without clinical renal involvement, glomerular filtration rate (GFR) usually decreases with the progression of vascular damage correlated with age and disease duration.12 Patients with a history of digital ulcers, like Mr. P, usually have a lower GFR than that of patients without digital ulcers.12 Monitoring renal function is vital in caring for these patients, because SRC occurs in 2% to 15% of all patients with SSc.13,14
Scleroderma renal crisis is generally defined as an abruptly elevated BP (> 140/90 mm Hgor > 30 mm Hg rise from baseline) with acute renal failure (elevated SCr) and decreased urine output.14 Given the rarity of SSc in the population, diagnosis of SRC requires high clinical suspicion. In the case of Mr. P, a workup involving serum analysis, urinalysis, and renal biopsy allowed for a definitive diagnosis. A renal biopsy can show microangiopathic hemolytic anemia and confirm SRC, although it may not be necessary in patients with known SSc presenting with new hypertension, rising creatinine, and unremarkable urine sediment on microscopy.14,15
Although an acute SRC can be difficult to predict, monitoring renal function and attention to key factors can assist in discovering this SSc complication. Scleroderma renal crisis usually occurs within the first 4 years of SSc diagnosis, often paralleling rapid progression of skin thickening and tightening with higher rates in both African Americans and males.13,14 Additional predictive factors include diffuse skin involvement, rapid progression of skin involvement, positive anti-RNA polymerase III antibodies, new anemia, new cardiac events (eg, pericardial effusion, pericarditis, left ventricular insufficiency), CHF, tendon friction rubs, arthritis, and recent (within 3 months) high-dose glucocorticoid use.4,13-15
The presentation of SRC can be nonspecific, often resembling findings related to acute kidney injury. Patients may report malaise, fatigue, fever, headache, seizure, blurred vision, or dyspnea.13 Clinical parametersto examine include systolic BP > 140 mm Hgand/or diastolic BP > 90 mm Hg (or an abrupt rise of > 30 or > 20, respectively), SCr increase by 50%+ or > 120% of the upper normal limit, proteinuria at 2+, high protein:creatinine ratio, hematuria > 2+ or > 10 red blood cells (RBCs), platelets < 100,000/mm3, and hypertensive encephalopathy.13 Mr. P presented with fatigue, dyspnea, an abrupt rise in BP (156/96 mm Hg), foamy urine, bilateral lower extremity edema (3.9 g/dL albumin), 2+ RBCs, and a SCr of 2.46 mg/dL on admission (a 205% increase from his last baseline of 1.2 mg/dL).
Treatments have had a large impact on the mortality rates of SRC. Following the introduction of ACE inhibitors, mortality from SRC has decreased from 76% to < 10% over recent decades.13 In addition to improving survival, these medications also have improved hospitalization outcomes for these patients.3 Captopril is often the medication of choice given its rapid onset and short duration of action relative to other medications in the same class, such as benazepril, enalapril, fosinopril, lisinopril, quinapril, and ramipril.4,13 Current clinical trials are assessing the specific renal benefits of endothelin-1 receptor antagonists, which often are used for pulmonary and skin involvement.14 For patients presenting with acute SRC and uremia, dialysis may be necessary (typically predicted by elevated NT-proBNP serum levels), while the decision for transplantation may be indicated at least 2 years after SRC onset and resolution.4,13-15
Once the acute crisis resolves, it is important to discuss prognosis with patients. The 1-, 2-, 3-, 5-, and 10-year survival rates are 82%, 74%, 71%, 59%, and 47%, respectively; however, when looking at only male patients at 10 years, the survival rate is 17%.13 Prevention of SRC can be addressed with daily BP checks and advising patients to seek medical care if they notice a consecutive 2-day abrupt rise.13
Cardiac Involvement
Systemic sclerosis has significant effects on patients’ heart function. The introduction of ACE inhibitors has shifted mortality in SSc patients from predominantly SRC to cardiac causes.6 Cardiac involvement can occur through a range of processes, including abnormal cardiac conduction, CHF, diastolic dysfunction, mitral valve nodular thickening, and pericardial effusion.3 Even though patients often present with skin findings, an initial cardiac workup is crucial to understanding disease progression and patient prognosis. The severity of the cutaneous manifestations often predicts the degree of diastolic dysfunction.16
Clinical evidence of cardiac involvement is seen in 20% to 25% of patients with SSc and is associated with a 70% mortality at 5 years when symptoms are evident.16 Additionally, right ventricular dysfunction at presentation is the strongest marker for all-mortality prognosis, representing the degree of pulmonary involvement, and includes findings such as progressive shortness of breath and systemic edema.9
Given the increasing survival of patients with SSc, cardiac involvement is becoming more evident and prominent. Direct treatments for cardiac manifestations are based on the causative feature, namely, focusing on pulmonary and renal involvement, which can be assessed with periodic echocardiograms evaluating left ventricular EF.
Gastrointestinal Involvement
Along with skin manifestations, the gastrointestinal (GI) involvement of SSc can have a significant impact on patients’ QOL without direct contribution to mortality. One of Mr. P’s earliest symptoms that led to a diagnosis of SSc was GERD, which caused a chronic cough, dental erosions, esophageal erosions, duodenal ulcers, dysphagia, abdominal pain, halitosis, pharyngitis, and weight loss. Esophageal involvement occurs in up to 96% of patients with SSc and can include motility abnormalities (eg, strictures and/or muscle dysfunction), lower esophageal sphincter abnormalities, and Barrett esophagus.17
Additional symptoms of the GI system linked with SSc can occur anywhere along the GI tract and include gastric antral vascular ectasia, causing GI bleeds and pernicious anemia, gastroparesis, bacterial overgrowth, intestinal malabsorption, pseudo-obstruction due to hypomotility, fecal incontinence due to anorectal involvement, and rarely, primary biliary cirrhosis.4,17 Decreased mobility of the oral aperture secondary to skin thickening and tightening also can contribute to malnutrition by decreasing oral intake.
Treatments are supportive and target symptom relief. Chronic treatment of GERD is often necessary and includes antacids, histamine-2 receptor blockers, and proton pump inhibitors.4 Other medications that can help with symptom relief include motility agents (such as metoclopramide, domperidone, prucalopride, tegaserod, and macrolides), osmotic laxatives, and ursodeoxycholic acid for primary biliary cirrhosis.17 Surgical intervention should be considered depending on the severity and progression of involvement within the GI tract. Behavioral changes that can improve patient symptoms include facial grimacing and other mouth-stretching exercises, frequent smaller meals followed by maintaining a vertical posture, and high fiber diets.17
Conclusion
Systemic sclerosis is an autoimmune and connective tissue disease with a pathophysiology that can manifest throughout the body. The organ systems that impact patient outcomes include skin, pulmonary, renal, cardiac, and GI. Primary care providers caring for patients diagnosed with SSc should monitor acute management and disease progression in all these systems. Important acute events that can impact morbidity, mortality, and/or QOL include Raynaud phenomenon, SRC, and pericardial effusion. Chronic manifestations that may be present on diagnosis of SSc or may develop while a patient is under a provider’s care include sclerodactyly, tendon calcinosis, PAH, ILD, chronic kidney injury, chronic cardiac damage, GERD, and esophageal dysmotility. While this discussion serves as a pertinent overview of patients with SSc, it is summative, and providers are encouraged to seek a stronger understanding of both the common and rarer manifestations within each of their patients.
1. Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41(5):778-799.
2. Silman AG. Scleroderma. In: Epidemiology of the Rheumatic Diseases. 2nd ed. Silman AJ, Hochberg MC, eds. Oxford, UK: Oxford University Press. 1993:chap 8.
3. Chung L, Krishnan E, Chakravarty EF. Hospitalizations and mortality in systemic sclerosis: results from the Nationwide Inpatient Sample. Rheumatology (Oxford). 2007;46(12):1808-1813.
4. Hinchcliff M, Varga J. Systemic sclerosis/scleroderma: a treatable multisystem disease. Am Fam Physician. 2008;78(8):961-968.
5. Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010;69(10):1809-1815.
6. Piga M, Casula L, Sanna S, et al. Population-based analysis of hospitalizations for patients with systemic sclerosis in a West-European region over the period 2001-2012. Rheumatol Int. 2016;36(1):73-81.
7. Giuggioli D, Manfredi A, Colaci M, Lumetti F, Ferri C. Osteomyelitis complicating scleroderma digital ulcers. Clin Rheumatol. 2013;32(5):623-627.
8. Schiopu E, Impens AJ, Phillips K. Digital ischemia in scleroderma spectrum of diseases. Int J Rheumatol. 2010;2010:923743.
9. Hassoun PM. Therapies for scleroderma-related pulmonary arterial hypertension. Expert Rev Respir Med. 2009;3(2):187-196.
10. Giacomelli R, Liakouli V, Berardicurti O, et al. Interstitial lung disease in systemic sclerosis: current and future treatment. Rheumatol Int. 2017;37(6):853-863.
11. McLaughlin V, Humbert M, Coghlan G, Nash P, Steen V. Pulmonary arterial hypertension: the most devastating vascular complication of systemic sclerosis. Rheumatology (Oxford). 2009; (suppl 3):iii25-iii31.
12. Gigante A, Barbano B, Granata G, et al. Evaluation of estimated glomerular filtration rate and clinical variables in systemic sclerosis patients. Clin Nephrol. 2016;85(6):326-331.
13. Bose N, Chiesa-Vottero A, Chatterjee S. Scleroderma renal crisis. Semin Arthritis Rheum. 2015;44(6):687-694.
14. Woodworth TG, Suliman YA, Furst DE, Clements P. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat Rev Nephrol. 2016;12(11):678-691.
15. Mouthon L, Bérezné A, Bussone G, Noël LH, Villiger PM, Guillevin L. Scleroderma renal crisis: a rare but severe complication of systemic sclerosis. Clin Rev Allergy Immunol. 2011;40(2):84-91.
16. Champion HC. The heart in scleroderma. Rheum Dis Clin North Am. 2008;34(1):181-90.
17. Tian XP, Zhang X. Gastrointestinal complications of systemic sclerosis. World J Gastroenterol. 2013;19(41):7062-7068.
1. Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41(5):778-799.
2. Silman AG. Scleroderma. In: Epidemiology of the Rheumatic Diseases. 2nd ed. Silman AJ, Hochberg MC, eds. Oxford, UK: Oxford University Press. 1993:chap 8.
3. Chung L, Krishnan E, Chakravarty EF. Hospitalizations and mortality in systemic sclerosis: results from the Nationwide Inpatient Sample. Rheumatology (Oxford). 2007;46(12):1808-1813.
4. Hinchcliff M, Varga J. Systemic sclerosis/scleroderma: a treatable multisystem disease. Am Fam Physician. 2008;78(8):961-968.
5. Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010;69(10):1809-1815.
6. Piga M, Casula L, Sanna S, et al. Population-based analysis of hospitalizations for patients with systemic sclerosis in a West-European region over the period 2001-2012. Rheumatol Int. 2016;36(1):73-81.
7. Giuggioli D, Manfredi A, Colaci M, Lumetti F, Ferri C. Osteomyelitis complicating scleroderma digital ulcers. Clin Rheumatol. 2013;32(5):623-627.
8. Schiopu E, Impens AJ, Phillips K. Digital ischemia in scleroderma spectrum of diseases. Int J Rheumatol. 2010;2010:923743.
9. Hassoun PM. Therapies for scleroderma-related pulmonary arterial hypertension. Expert Rev Respir Med. 2009;3(2):187-196.
10. Giacomelli R, Liakouli V, Berardicurti O, et al. Interstitial lung disease in systemic sclerosis: current and future treatment. Rheumatol Int. 2017;37(6):853-863.
11. McLaughlin V, Humbert M, Coghlan G, Nash P, Steen V. Pulmonary arterial hypertension: the most devastating vascular complication of systemic sclerosis. Rheumatology (Oxford). 2009; (suppl 3):iii25-iii31.
12. Gigante A, Barbano B, Granata G, et al. Evaluation of estimated glomerular filtration rate and clinical variables in systemic sclerosis patients. Clin Nephrol. 2016;85(6):326-331.
13. Bose N, Chiesa-Vottero A, Chatterjee S. Scleroderma renal crisis. Semin Arthritis Rheum. 2015;44(6):687-694.
14. Woodworth TG, Suliman YA, Furst DE, Clements P. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat Rev Nephrol. 2016;12(11):678-691.
15. Mouthon L, Bérezné A, Bussone G, Noël LH, Villiger PM, Guillevin L. Scleroderma renal crisis: a rare but severe complication of systemic sclerosis. Clin Rev Allergy Immunol. 2011;40(2):84-91.
16. Champion HC. The heart in scleroderma. Rheum Dis Clin North Am. 2008;34(1):181-90.
17. Tian XP, Zhang X. Gastrointestinal complications of systemic sclerosis. World J Gastroenterol. 2013;19(41):7062-7068.