User login
© 2017 Society of Hospital Medicine
The “Things We Do for No Reason” series reviews practices which have become common parts of hospital care but which may provide little value to our patients. Practices reviewed in the TWDFNR series do not represent “black and white” conclusions or clinical practice standards, but are meant as a starting place for research and active discussions among hospitalists and patients. We invite you to be part of that discussion. https://www.choosingwisely.org/

Ammonia is predominantly generated in the gut by intestinal bacteria and enzymes and detoxified primarily in the liver. Since the 1930s, ammonia has been identified as the principal culprit in hepatic encephalopathy (HE). Many physicians utilize serum ammonia to diagnose, assess severity, and determine the resolution of HE in patients with chronic liver disease (CLD) despite research showing that ammonia levels are unhelpful in all of these clinical circumstances. HE in patients with CLD is a clinical diagnosis of exclusion that should not be based on ammonia levels.
CASE PRESENTATION
A 62-year-old man diagnosed with cirrhosis due to Hepatitis C and alcoholism was brought to the emergency department for alteration in mentation. He had scant melenic stools 5 days preceding his admission and did not exhibit overt signs or symptoms of infection. His systemic examination was normal except for somnolence, disorientation to space and time, asterixis, and ascites. His lab parameters were within normal limits except for an elevated blood urea nitrogen and thrombocytopenia. His blood cultures did not grow any organisms, and paracentesis ruled out spontaneous bacterial peritonitis. During his hospital stay, he underwent esophageal variceal banding and was effectively managed with lactulose and rifaximin. The patient was alert, fully oriented, and without asterixis at the time of discharge 6 days later. Would an elevated venous ammonia level at admission alter management? If the ammonia level was elevated, would serial ammonia measurements affect management?
BACKGROUND

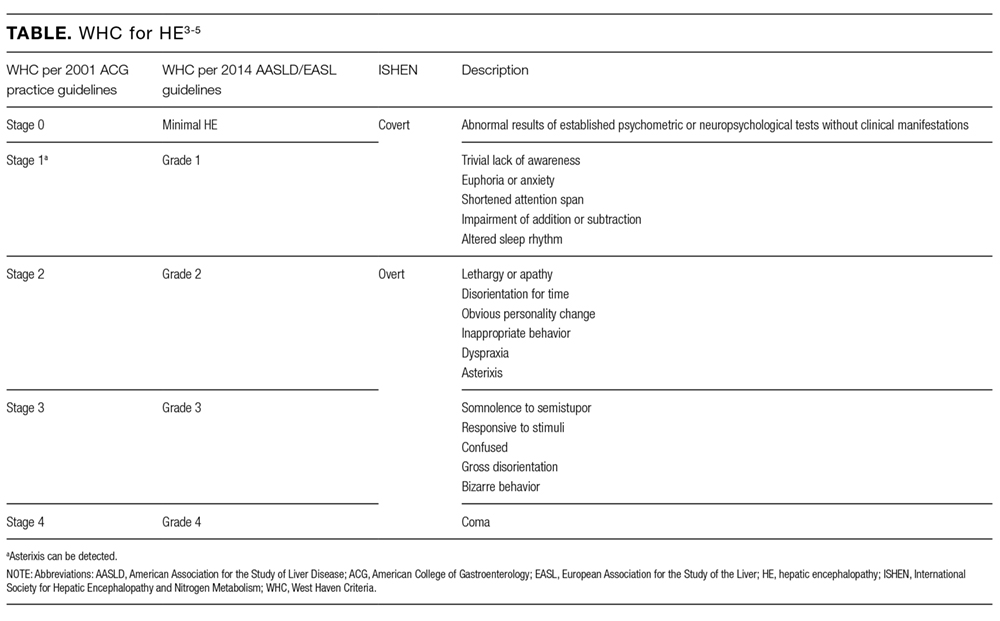
The colonic microbiome produces ammonia from dietary nitrogen. In health, approximately 85% of it is detoxified by the liver and excreted as urea in urine, while muscle and brain tissue metabolize the remaining 15%. The process of transamination and the urea cycle prevents this metabolic product from accumulating in the body. The elevated levels of nitrogenous toxins, including ammonia, in the systemic circulation of patients with CLD occur due to hepatocellular dysfunction and/or portosystemic shunting. This hyperammonemia is compounded by reduced peripheral metabolism of ammonia by muscle as a consequence of cachexia and muscle atrophy. Astrocytes synthesize glutamine excessively in the setting of hyperammonemia, resulting in astrocyte swelling and the generation of reactive oxygen species. Astrocyte swelling, free radical generation, and increased inhibitory function of gamma-Aminobutyric Acid result in cerebral dysfunction.1,2 HE manifests as a broad spectrum of neurological or psychiatric abnormalities ranging from subclinical alterations to coma and was commonly graded on the West Haven Criteria (WHC) of 0 to 4 (Table).3 The Grade 0 from the previous WHC, referenced in many trials included in this article, has been replaced with minimal HE in the newly updated WHC by the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver.4,5
WHY YOU MIGHT THINK AMMONIA LEVELS HELP TO GUIDE TREATMENT OF HE IN PATIENTS WITH CLD
The ammonia hypothesis posits that ammonia is key in the pathogenesis of HE.6-10 Some of the common precipitants of HE—gastrointestinal bleeding, infection, and renal failure—promote hyperammonemia.11 HE is treated with nonabsorbable disaccharides (lactulose and lactitol) and rifaximin, which reduce the serum concentration of ammonia. Given these associations between HE and ammonia, physicians have for decades tested serum ammonia levels to diagnose HE and chart its resolution. In a study conducted by the Bavarian Society of Gastroenterology,12 60% of the respondents to an anonymous questionnaire regularly performed ammonia analysis in all their patients with liver cirrhosis, believing that it efficiently diagnosed HE.
WHY SERUM AMMONIA LEVELS DO NOT HELP IN THE DIAGNOSIS OR MANAGEMENT OF HE IN CLD PATIENTS
Accuracy of Serum Ammonia
Multiple factors affect the accuracy of ammonia levels. First, fist clenching or the use of a tourniquet during the process of phlebotomy can falsely increase ammonia levels.13 Second, some authors have argued that the source of the ammonia sample matters. Kramer et al.14 reported that partial pressure of ammonia correlated closely with the degree of clinical and electrophysiological abnormalities of HE. However, Nicolao et al.15 and Ong et al.16 showed that the blood ammonia levels, whether measured by total venous, total arterial, or partial pressure methods, were equivalent. Third, ammonia levels are dependent on the time to processing of the specimen. Inaccurate results may occur if the blood sample is not immediately placed on ice after collection or if it is not centrifuged within 15 minutes of collection.17,18
Ammonia Levels and Diagnosis of HE
Even with proper collection and processing, ammonia levels in patients with CLD do not reliably diagnose HE. Gundling et al.19 determined the sensitivity and specificity of venous ammonia levels ≥ 55 µmol/L to diagnose HE to be 47.2% and 78.3%, respectively, by using a gold standard of the WHC and the critical flicker frequency test (a psychophysiologic test). The positive predictive and negative predictive values of ammonia were 77.3% and 48.6%, with an overall diagnostic accuracy of 59.3%. Approximately 60% of the patients with Grade 3 WHC HE had a normal ammonia level in this study. Ong et al16 found that only 31% of patients with CLD and no evidence of HE had a normal ammonia level.In other words, CLD patients with normal ammonia levels can have HE, and patients with elevated ammonia levels may have normal cognitive functioning.
Furthermore, ammonia levels are not a valid tool to diagnose HE even with an oral glutamine challenge.20 Most importantly, HE is a clinical diagnosis reached following the exclusion of other likely causes of cerebral dysfunction, independent of the ammonia level.
Ammonia Levels and Staging HE
The grading of HE was introduced to assess the response to an intervention in patients with HE enrolled in clinical trials.21 Tools like the WHC (Table) categorize the severity of HE. Nicolao et al.15 noted significant overlap in the levels of ammonia between patients with HE Grades 1 and 2 when compared with patients with Grades 3 and 4. This considerable overlap in levels of ammonia was more evident among patients with Grades 0 to 2 per Ong’s study.16 Most importantly, hospitalists do not need ammonia levels to determine that a patient has HE Grade 3 or HE Grade 4 symptoms, as the stage is graded on clinical grounds only. Once other causes for cerebral dysfunction have been ruled out, the ammonia level does not add to the clinical picture.
Serial Ammonia Levels and Resolution of HE
If the ammonia hypothesis is the sole explanation for the pathogenesis of HE, then the resolution of HE symptoms should be associated with normalization of ammonia levels. Physicians have commonly followed ammonia levels serially throughout a hospital stay. Nicolao et al.15 evaluated the association of ammonia with HE. They noted that some of the CLD patients had unchanged or increasing levels of ammonia despite overt neurological improvement from their HE.15 Some have argued that the normalization of ammonia levels lag behind the clinical improvement by 48 hours after resolution of symptoms. In the Nicolao et al.15 study, ammonia levels for almost all of the patients did not normalize 48 hours after resolution of neurologic symptoms. Moreover, 29% of the patients were noted to have higher venous ammonia levels 48 hours after the resolution of neurologic symptoms.15 These data underscore why serial measurements of ammonia in patients with CLD are not useful. For patients with overt symptoms, clinicians can determine improvement based on serial exams.
RECOMMENDATIONS
- HE is a diagnosis of exclusion and is made on clinical grounds.
- Do not check serum ammonia levels in patients with CLD to diagnose HE, to assess the severity of HE, or to determine whether HE is resolving.
- Use your clinical evaluation to determine the severity and course of HE.
- Treatment should be tailored according to clinical findings, not ammonia levels.
CONCLUSION
The attraction of the ammonia theory to explain HE continues to lead physicians to check and follow blood ammonia levels in patients with CLD and suspected HE. However, ammonia measurement, as in the clinical vignette, should be replaced by a thorough clinical evaluation to rule out other causes for altered mental status. Serial exams of the patient should guide management, not ammonia levels.
Disclosure
The authors report no conflicts of interest.
Do you think this is a low-value practice? Is this truly a “Thing We Do for No Reason”? Let us know what you do in your practice and propose ideas for other “Things We Do for No Reason” topics. Please join in the conversation online at Twitter (#TWDFNR)/Facebook, and don’t forget to “Like It” on Facebook or retweet it on Twitter.
1. Tapper EB, Jiang ZG, Patwardhan VR. Refining the ammonia hypothesis: A physiology-driven approach to the treatment of hepatic encephalopathy. Mayo Clin Proc. 2015;90:646-658. PubMed
2. Parekh PJ, Balart LA. Ammonia and Its Role in the Pathogenesis of Hepatic Encephalopathy. Clin Liver Dis. 2015;19:529-537. PubMed
3. Blei AT, Córdoba J. Hepatic Encephalopathy. Am J Gastroenterol. 2001;96:1968-1976. PubMed
4. Vilstrup H, Amodio P, Bajaj J, et al. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study Of Liver Diseases and the European Association for the Study of the Liver. Hepatology. 2014;60:715-735. PubMed
5. Bajaj JS, Cordoba J, Mullen KD, et al. Review Article: the design of clinical trials in Hepatic Encephalopathy - an International Society for Hepatic Encephalopathy and Nitrogen Metabolism (ISHEN) consensus statement. Aliment Pharmacol Ther. 2011;33:739-747. PubMed
6. Ahboucha S, Butterworth RF. Pathophysiology of hepatic encephalopathy: A new look at GABA from the molecular standpoint. Metab Brain Dis. 2004;19:331-343. PubMed
7. Butterworth RF. Pathophysiology of Hepatic Encephalopathy: A New Look at Ammonia. 2003;17:1-7. PubMed
8. Schafer DF, Fowler JM, Munson PJ, Thakur AK, Waggoner JG, Jones EA. Gamma-aminobutyric acid and benzodiazepine receptors in an animal model of fulminant hepatic failure. J Lab Clin Med. 1983;102:870-880. PubMed
9. Michalak A, Rose C, Butterworth J, Butterworth RF. Neuroactive amino acids and glutamate (NMDA) receptors in frontal cortex of rats with experimental acute liver failure. Hepatology. 1996;24:908-13. PubMed
10. Bassett ML, Mullen KD, Scholz B, Fenstermacher JD, Jones EA. Increased brain uptake of gamma-aminobutyric acid in a rabbit model of hepatic encephalopathy. Gastroenterology. 1990;98:747-757. PubMed
11. Clay AS, Hainline BE. Hyperammonemia in the ICU. Chest. 2007;132:1368-1378. PubMed
12. Gundling F, Seidl H, Schmidt T, Schepp W. Blood ammonia level in liver cirrhosis: a conditio sine qua non to confirm hepatic encephalopathy? Eur J Gastroenterol Hepatol. 2008;20:246-247. PubMed
13. Stahl J. Studies of the Blood Ammonia in Liver Disease: Its Diagnostic, Prognostic and Therapeutic Significance. Ann Intern Med. 1963;58:1–24. PubMed
14. Kramer L, Tribl B, Gendo A, et al. Partial pressure of ammonia versus ammonia in hepatic encephalopathy. Hepatology. 2000;31:30-34. PubMed
15. Nicolao F, Masini A, Manuela M, Attili AF, Riggio O. Role of determination of partial pressure of ammonia in cirrhotic patients with or without hepatic encephalopathy. J Hepatol. 2003;38:441-446. PubMed
16. Ong JP, Aggarwal A, Krieger D, et al. Correlation between ammonia levels and the severity of hepatic encephalopathy. Am J Med. 2003;114:188-193. PubMed
17. Da Fonseca-Wollheim F. Preanalytical increase of ammonia in blood specimens from healthy subjects. Clin Chem. 1990;36:1483-1487. PubMed
18. Howanitz JH, Howanitz PJ, Skrodzki CA, Iwanski JA. Influences of specimen processing and storage conditions on results for plasma ammonia. Clin Chem. 1984;30:906-908. PubMed
19. Gundling F, Zelihic E, Seidl H, et al. How to diagnose hepatic encephalopathy in the emergency department. Ann Hepatol. 2013;12:108-114. PubMed
20. Ditisheim S, Giostra E, Burkhard PR, et al. A capillary blood ammonia bedside test following glutamine load to improve the diagnosis of hepatic encephalopathy in cirrhosis. BMC Gastroenterol. 2011;11:134. PubMed
21. Conn HO, Leevy CM, Vlahcevic ZR, et al. Comparison of lactulose and neomycin in the treatment of chronic portal-systemic encephalopathy. A double blind controlled trial. Gastroenterology. 1977;72:573-583. PubMed
© 2017 Society of Hospital Medicine
The “Things We Do for No Reason” series reviews practices which have become common parts of hospital care but which may provide little value to our patients. Practices reviewed in the TWDFNR series do not represent “black and white” conclusions or clinical practice standards, but are meant as a starting place for research and active discussions among hospitalists and patients. We invite you to be part of that discussion. https://www.choosingwisely.org/
Ammonia is predominantly generated in the gut by intestinal bacteria and enzymes and detoxified primarily in the liver. Since the 1930s, ammonia has been identified as the principal culprit in hepatic encephalopathy (HE). Many physicians utilize serum ammonia to diagnose, assess severity, and determine the resolution of HE in patients with chronic liver disease (CLD) despite research showing that ammonia levels are unhelpful in all of these clinical circumstances. HE in patients with CLD is a clinical diagnosis of exclusion that should not be based on ammonia levels.
CASE PRESENTATION
A 62-year-old man diagnosed with cirrhosis due to Hepatitis C and alcoholism was brought to the emergency department for alteration in mentation. He had scant melenic stools 5 days preceding his admission and did not exhibit overt signs or symptoms of infection. His systemic examination was normal except for somnolence, disorientation to space and time, asterixis, and ascites. His lab parameters were within normal limits except for an elevated blood urea nitrogen and thrombocytopenia. His blood cultures did not grow any organisms, and paracentesis ruled out spontaneous bacterial peritonitis. During his hospital stay, he underwent esophageal variceal banding and was effectively managed with lactulose and rifaximin. The patient was alert, fully oriented, and without asterixis at the time of discharge 6 days later. Would an elevated venous ammonia level at admission alter management? If the ammonia level was elevated, would serial ammonia measurements affect management?
BACKGROUND
The colonic microbiome produces ammonia from dietary nitrogen. In health, approximately 85% of it is detoxified by the liver and excreted as urea in urine, while muscle and brain tissue metabolize the remaining 15%. The process of transamination and the urea cycle prevents this metabolic product from accumulating in the body. The elevated levels of nitrogenous toxins, including ammonia, in the systemic circulation of patients with CLD occur due to hepatocellular dysfunction and/or portosystemic shunting. This hyperammonemia is compounded by reduced peripheral metabolism of ammonia by muscle as a consequence of cachexia and muscle atrophy. Astrocytes synthesize glutamine excessively in the setting of hyperammonemia, resulting in astrocyte swelling and the generation of reactive oxygen species. Astrocyte swelling, free radical generation, and increased inhibitory function of gamma-Aminobutyric Acid result in cerebral dysfunction.1,2 HE manifests as a broad spectrum of neurological or psychiatric abnormalities ranging from subclinical alterations to coma and was commonly graded on the West Haven Criteria (WHC) of 0 to 4 (Table).3 The Grade 0 from the previous WHC, referenced in many trials included in this article, has been replaced with minimal HE in the newly updated WHC by the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver.4,5
WHY YOU MIGHT THINK AMMONIA LEVELS HELP TO GUIDE TREATMENT OF HE IN PATIENTS WITH CLD
The ammonia hypothesis posits that ammonia is key in the pathogenesis of HE.6-10 Some of the common precipitants of HE—gastrointestinal bleeding, infection, and renal failure—promote hyperammonemia.11 HE is treated with nonabsorbable disaccharides (lactulose and lactitol) and rifaximin, which reduce the serum concentration of ammonia. Given these associations between HE and ammonia, physicians have for decades tested serum ammonia levels to diagnose HE and chart its resolution. In a study conducted by the Bavarian Society of Gastroenterology,12 60% of the respondents to an anonymous questionnaire regularly performed ammonia analysis in all their patients with liver cirrhosis, believing that it efficiently diagnosed HE.
WHY SERUM AMMONIA LEVELS DO NOT HELP IN THE DIAGNOSIS OR MANAGEMENT OF HE IN CLD PATIENTS
Accuracy of Serum Ammonia
Multiple factors affect the accuracy of ammonia levels. First, fist clenching or the use of a tourniquet during the process of phlebotomy can falsely increase ammonia levels.13 Second, some authors have argued that the source of the ammonia sample matters. Kramer et al.14 reported that partial pressure of ammonia correlated closely with the degree of clinical and electrophysiological abnormalities of HE. However, Nicolao et al.15 and Ong et al.16 showed that the blood ammonia levels, whether measured by total venous, total arterial, or partial pressure methods, were equivalent. Third, ammonia levels are dependent on the time to processing of the specimen. Inaccurate results may occur if the blood sample is not immediately placed on ice after collection or if it is not centrifuged within 15 minutes of collection.17,18
Ammonia Levels and Diagnosis of HE
Even with proper collection and processing, ammonia levels in patients with CLD do not reliably diagnose HE. Gundling et al.19 determined the sensitivity and specificity of venous ammonia levels ≥ 55 µmol/L to diagnose HE to be 47.2% and 78.3%, respectively, by using a gold standard of the WHC and the critical flicker frequency test (a psychophysiologic test). The positive predictive and negative predictive values of ammonia were 77.3% and 48.6%, with an overall diagnostic accuracy of 59.3%. Approximately 60% of the patients with Grade 3 WHC HE had a normal ammonia level in this study. Ong et al16 found that only 31% of patients with CLD and no evidence of HE had a normal ammonia level.In other words, CLD patients with normal ammonia levels can have HE, and patients with elevated ammonia levels may have normal cognitive functioning.
Furthermore, ammonia levels are not a valid tool to diagnose HE even with an oral glutamine challenge.20 Most importantly, HE is a clinical diagnosis reached following the exclusion of other likely causes of cerebral dysfunction, independent of the ammonia level.
Ammonia Levels and Staging HE
The grading of HE was introduced to assess the response to an intervention in patients with HE enrolled in clinical trials.21 Tools like the WHC (Table) categorize the severity of HE. Nicolao et al.15 noted significant overlap in the levels of ammonia between patients with HE Grades 1 and 2 when compared with patients with Grades 3 and 4. This considerable overlap in levels of ammonia was more evident among patients with Grades 0 to 2 per Ong’s study.16 Most importantly, hospitalists do not need ammonia levels to determine that a patient has HE Grade 3 or HE Grade 4 symptoms, as the stage is graded on clinical grounds only. Once other causes for cerebral dysfunction have been ruled out, the ammonia level does not add to the clinical picture.
Serial Ammonia Levels and Resolution of HE
If the ammonia hypothesis is the sole explanation for the pathogenesis of HE, then the resolution of HE symptoms should be associated with normalization of ammonia levels. Physicians have commonly followed ammonia levels serially throughout a hospital stay. Nicolao et al.15 evaluated the association of ammonia with HE. They noted that some of the CLD patients had unchanged or increasing levels of ammonia despite overt neurological improvement from their HE.15 Some have argued that the normalization of ammonia levels lag behind the clinical improvement by 48 hours after resolution of symptoms. In the Nicolao et al.15 study, ammonia levels for almost all of the patients did not normalize 48 hours after resolution of neurologic symptoms. Moreover, 29% of the patients were noted to have higher venous ammonia levels 48 hours after the resolution of neurologic symptoms.15 These data underscore why serial measurements of ammonia in patients with CLD are not useful. For patients with overt symptoms, clinicians can determine improvement based on serial exams.
RECOMMENDATIONS
- HE is a diagnosis of exclusion and is made on clinical grounds.
- Do not check serum ammonia levels in patients with CLD to diagnose HE, to assess the severity of HE, or to determine whether HE is resolving.
- Use your clinical evaluation to determine the severity and course of HE.
- Treatment should be tailored according to clinical findings, not ammonia levels.
CONCLUSION
The attraction of the ammonia theory to explain HE continues to lead physicians to check and follow blood ammonia levels in patients with CLD and suspected HE. However, ammonia measurement, as in the clinical vignette, should be replaced by a thorough clinical evaluation to rule out other causes for altered mental status. Serial exams of the patient should guide management, not ammonia levels.
Disclosure
The authors report no conflicts of interest.
Do you think this is a low-value practice? Is this truly a “Thing We Do for No Reason”? Let us know what you do in your practice and propose ideas for other “Things We Do for No Reason” topics. Please join in the conversation online at Twitter (#TWDFNR)/Facebook, and don’t forget to “Like It” on Facebook or retweet it on Twitter.
© 2017 Society of Hospital Medicine
The “Things We Do for No Reason” series reviews practices which have become common parts of hospital care but which may provide little value to our patients. Practices reviewed in the TWDFNR series do not represent “black and white” conclusions or clinical practice standards, but are meant as a starting place for research and active discussions among hospitalists and patients. We invite you to be part of that discussion. https://www.choosingwisely.org/
Ammonia is predominantly generated in the gut by intestinal bacteria and enzymes and detoxified primarily in the liver. Since the 1930s, ammonia has been identified as the principal culprit in hepatic encephalopathy (HE). Many physicians utilize serum ammonia to diagnose, assess severity, and determine the resolution of HE in patients with chronic liver disease (CLD) despite research showing that ammonia levels are unhelpful in all of these clinical circumstances. HE in patients with CLD is a clinical diagnosis of exclusion that should not be based on ammonia levels.
CASE PRESENTATION
A 62-year-old man diagnosed with cirrhosis due to Hepatitis C and alcoholism was brought to the emergency department for alteration in mentation. He had scant melenic stools 5 days preceding his admission and did not exhibit overt signs or symptoms of infection. His systemic examination was normal except for somnolence, disorientation to space and time, asterixis, and ascites. His lab parameters were within normal limits except for an elevated blood urea nitrogen and thrombocytopenia. His blood cultures did not grow any organisms, and paracentesis ruled out spontaneous bacterial peritonitis. During his hospital stay, he underwent esophageal variceal banding and was effectively managed with lactulose and rifaximin. The patient was alert, fully oriented, and without asterixis at the time of discharge 6 days later. Would an elevated venous ammonia level at admission alter management? If the ammonia level was elevated, would serial ammonia measurements affect management?
BACKGROUND
The colonic microbiome produces ammonia from dietary nitrogen. In health, approximately 85% of it is detoxified by the liver and excreted as urea in urine, while muscle and brain tissue metabolize the remaining 15%. The process of transamination and the urea cycle prevents this metabolic product from accumulating in the body. The elevated levels of nitrogenous toxins, including ammonia, in the systemic circulation of patients with CLD occur due to hepatocellular dysfunction and/or portosystemic shunting. This hyperammonemia is compounded by reduced peripheral metabolism of ammonia by muscle as a consequence of cachexia and muscle atrophy. Astrocytes synthesize glutamine excessively in the setting of hyperammonemia, resulting in astrocyte swelling and the generation of reactive oxygen species. Astrocyte swelling, free radical generation, and increased inhibitory function of gamma-Aminobutyric Acid result in cerebral dysfunction.1,2 HE manifests as a broad spectrum of neurological or psychiatric abnormalities ranging from subclinical alterations to coma and was commonly graded on the West Haven Criteria (WHC) of 0 to 4 (Table).3 The Grade 0 from the previous WHC, referenced in many trials included in this article, has been replaced with minimal HE in the newly updated WHC by the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver.4,5
WHY YOU MIGHT THINK AMMONIA LEVELS HELP TO GUIDE TREATMENT OF HE IN PATIENTS WITH CLD
The ammonia hypothesis posits that ammonia is key in the pathogenesis of HE.6-10 Some of the common precipitants of HE—gastrointestinal bleeding, infection, and renal failure—promote hyperammonemia.11 HE is treated with nonabsorbable disaccharides (lactulose and lactitol) and rifaximin, which reduce the serum concentration of ammonia. Given these associations between HE and ammonia, physicians have for decades tested serum ammonia levels to diagnose HE and chart its resolution. In a study conducted by the Bavarian Society of Gastroenterology,12 60% of the respondents to an anonymous questionnaire regularly performed ammonia analysis in all their patients with liver cirrhosis, believing that it efficiently diagnosed HE.
WHY SERUM AMMONIA LEVELS DO NOT HELP IN THE DIAGNOSIS OR MANAGEMENT OF HE IN CLD PATIENTS
Accuracy of Serum Ammonia
Multiple factors affect the accuracy of ammonia levels. First, fist clenching or the use of a tourniquet during the process of phlebotomy can falsely increase ammonia levels.13 Second, some authors have argued that the source of the ammonia sample matters. Kramer et al.14 reported that partial pressure of ammonia correlated closely with the degree of clinical and electrophysiological abnormalities of HE. However, Nicolao et al.15 and Ong et al.16 showed that the blood ammonia levels, whether measured by total venous, total arterial, or partial pressure methods, were equivalent. Third, ammonia levels are dependent on the time to processing of the specimen. Inaccurate results may occur if the blood sample is not immediately placed on ice after collection or if it is not centrifuged within 15 minutes of collection.17,18
Ammonia Levels and Diagnosis of HE
Even with proper collection and processing, ammonia levels in patients with CLD do not reliably diagnose HE. Gundling et al.19 determined the sensitivity and specificity of venous ammonia levels ≥ 55 µmol/L to diagnose HE to be 47.2% and 78.3%, respectively, by using a gold standard of the WHC and the critical flicker frequency test (a psychophysiologic test). The positive predictive and negative predictive values of ammonia were 77.3% and 48.6%, with an overall diagnostic accuracy of 59.3%. Approximately 60% of the patients with Grade 3 WHC HE had a normal ammonia level in this study. Ong et al16 found that only 31% of patients with CLD and no evidence of HE had a normal ammonia level.In other words, CLD patients with normal ammonia levels can have HE, and patients with elevated ammonia levels may have normal cognitive functioning.
Furthermore, ammonia levels are not a valid tool to diagnose HE even with an oral glutamine challenge.20 Most importantly, HE is a clinical diagnosis reached following the exclusion of other likely causes of cerebral dysfunction, independent of the ammonia level.
Ammonia Levels and Staging HE
The grading of HE was introduced to assess the response to an intervention in patients with HE enrolled in clinical trials.21 Tools like the WHC (Table) categorize the severity of HE. Nicolao et al.15 noted significant overlap in the levels of ammonia between patients with HE Grades 1 and 2 when compared with patients with Grades 3 and 4. This considerable overlap in levels of ammonia was more evident among patients with Grades 0 to 2 per Ong’s study.16 Most importantly, hospitalists do not need ammonia levels to determine that a patient has HE Grade 3 or HE Grade 4 symptoms, as the stage is graded on clinical grounds only. Once other causes for cerebral dysfunction have been ruled out, the ammonia level does not add to the clinical picture.
Serial Ammonia Levels and Resolution of HE
If the ammonia hypothesis is the sole explanation for the pathogenesis of HE, then the resolution of HE symptoms should be associated with normalization of ammonia levels. Physicians have commonly followed ammonia levels serially throughout a hospital stay. Nicolao et al.15 evaluated the association of ammonia with HE. They noted that some of the CLD patients had unchanged or increasing levels of ammonia despite overt neurological improvement from their HE.15 Some have argued that the normalization of ammonia levels lag behind the clinical improvement by 48 hours after resolution of symptoms. In the Nicolao et al.15 study, ammonia levels for almost all of the patients did not normalize 48 hours after resolution of neurologic symptoms. Moreover, 29% of the patients were noted to have higher venous ammonia levels 48 hours after the resolution of neurologic symptoms.15 These data underscore why serial measurements of ammonia in patients with CLD are not useful. For patients with overt symptoms, clinicians can determine improvement based on serial exams.
RECOMMENDATIONS
- HE is a diagnosis of exclusion and is made on clinical grounds.
- Do not check serum ammonia levels in patients with CLD to diagnose HE, to assess the severity of HE, or to determine whether HE is resolving.
- Use your clinical evaluation to determine the severity and course of HE.
- Treatment should be tailored according to clinical findings, not ammonia levels.
CONCLUSION
The attraction of the ammonia theory to explain HE continues to lead physicians to check and follow blood ammonia levels in patients with CLD and suspected HE. However, ammonia measurement, as in the clinical vignette, should be replaced by a thorough clinical evaluation to rule out other causes for altered mental status. Serial exams of the patient should guide management, not ammonia levels.
Disclosure
The authors report no conflicts of interest.
Do you think this is a low-value practice? Is this truly a “Thing We Do for No Reason”? Let us know what you do in your practice and propose ideas for other “Things We Do for No Reason” topics. Please join in the conversation online at Twitter (#TWDFNR)/Facebook, and don’t forget to “Like It” on Facebook or retweet it on Twitter.
1. Tapper EB, Jiang ZG, Patwardhan VR. Refining the ammonia hypothesis: A physiology-driven approach to the treatment of hepatic encephalopathy. Mayo Clin Proc. 2015;90:646-658. PubMed
2. Parekh PJ, Balart LA. Ammonia and Its Role in the Pathogenesis of Hepatic Encephalopathy. Clin Liver Dis. 2015;19:529-537. PubMed
3. Blei AT, Córdoba J. Hepatic Encephalopathy. Am J Gastroenterol. 2001;96:1968-1976. PubMed
4. Vilstrup H, Amodio P, Bajaj J, et al. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study Of Liver Diseases and the European Association for the Study of the Liver. Hepatology. 2014;60:715-735. PubMed
5. Bajaj JS, Cordoba J, Mullen KD, et al. Review Article: the design of clinical trials in Hepatic Encephalopathy - an International Society for Hepatic Encephalopathy and Nitrogen Metabolism (ISHEN) consensus statement. Aliment Pharmacol Ther. 2011;33:739-747. PubMed
6. Ahboucha S, Butterworth RF. Pathophysiology of hepatic encephalopathy: A new look at GABA from the molecular standpoint. Metab Brain Dis. 2004;19:331-343. PubMed
7. Butterworth RF. Pathophysiology of Hepatic Encephalopathy: A New Look at Ammonia. 2003;17:1-7. PubMed
8. Schafer DF, Fowler JM, Munson PJ, Thakur AK, Waggoner JG, Jones EA. Gamma-aminobutyric acid and benzodiazepine receptors in an animal model of fulminant hepatic failure. J Lab Clin Med. 1983;102:870-880. PubMed
9. Michalak A, Rose C, Butterworth J, Butterworth RF. Neuroactive amino acids and glutamate (NMDA) receptors in frontal cortex of rats with experimental acute liver failure. Hepatology. 1996;24:908-13. PubMed
10. Bassett ML, Mullen KD, Scholz B, Fenstermacher JD, Jones EA. Increased brain uptake of gamma-aminobutyric acid in a rabbit model of hepatic encephalopathy. Gastroenterology. 1990;98:747-757. PubMed
11. Clay AS, Hainline BE. Hyperammonemia in the ICU. Chest. 2007;132:1368-1378. PubMed
12. Gundling F, Seidl H, Schmidt T, Schepp W. Blood ammonia level in liver cirrhosis: a conditio sine qua non to confirm hepatic encephalopathy? Eur J Gastroenterol Hepatol. 2008;20:246-247. PubMed
13. Stahl J. Studies of the Blood Ammonia in Liver Disease: Its Diagnostic, Prognostic and Therapeutic Significance. Ann Intern Med. 1963;58:1–24. PubMed
14. Kramer L, Tribl B, Gendo A, et al. Partial pressure of ammonia versus ammonia in hepatic encephalopathy. Hepatology. 2000;31:30-34. PubMed
15. Nicolao F, Masini A, Manuela M, Attili AF, Riggio O. Role of determination of partial pressure of ammonia in cirrhotic patients with or without hepatic encephalopathy. J Hepatol. 2003;38:441-446. PubMed
16. Ong JP, Aggarwal A, Krieger D, et al. Correlation between ammonia levels and the severity of hepatic encephalopathy. Am J Med. 2003;114:188-193. PubMed
17. Da Fonseca-Wollheim F. Preanalytical increase of ammonia in blood specimens from healthy subjects. Clin Chem. 1990;36:1483-1487. PubMed
18. Howanitz JH, Howanitz PJ, Skrodzki CA, Iwanski JA. Influences of specimen processing and storage conditions on results for plasma ammonia. Clin Chem. 1984;30:906-908. PubMed
19. Gundling F, Zelihic E, Seidl H, et al. How to diagnose hepatic encephalopathy in the emergency department. Ann Hepatol. 2013;12:108-114. PubMed
20. Ditisheim S, Giostra E, Burkhard PR, et al. A capillary blood ammonia bedside test following glutamine load to improve the diagnosis of hepatic encephalopathy in cirrhosis. BMC Gastroenterol. 2011;11:134. PubMed
21. Conn HO, Leevy CM, Vlahcevic ZR, et al. Comparison of lactulose and neomycin in the treatment of chronic portal-systemic encephalopathy. A double blind controlled trial. Gastroenterology. 1977;72:573-583. PubMed
1. Tapper EB, Jiang ZG, Patwardhan VR. Refining the ammonia hypothesis: A physiology-driven approach to the treatment of hepatic encephalopathy. Mayo Clin Proc. 2015;90:646-658. PubMed
2. Parekh PJ, Balart LA. Ammonia and Its Role in the Pathogenesis of Hepatic Encephalopathy. Clin Liver Dis. 2015;19:529-537. PubMed
3. Blei AT, Córdoba J. Hepatic Encephalopathy. Am J Gastroenterol. 2001;96:1968-1976. PubMed
4. Vilstrup H, Amodio P, Bajaj J, et al. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study Of Liver Diseases and the European Association for the Study of the Liver. Hepatology. 2014;60:715-735. PubMed
5. Bajaj JS, Cordoba J, Mullen KD, et al. Review Article: the design of clinical trials in Hepatic Encephalopathy - an International Society for Hepatic Encephalopathy and Nitrogen Metabolism (ISHEN) consensus statement. Aliment Pharmacol Ther. 2011;33:739-747. PubMed
6. Ahboucha S, Butterworth RF. Pathophysiology of hepatic encephalopathy: A new look at GABA from the molecular standpoint. Metab Brain Dis. 2004;19:331-343. PubMed
7. Butterworth RF. Pathophysiology of Hepatic Encephalopathy: A New Look at Ammonia. 2003;17:1-7. PubMed
8. Schafer DF, Fowler JM, Munson PJ, Thakur AK, Waggoner JG, Jones EA. Gamma-aminobutyric acid and benzodiazepine receptors in an animal model of fulminant hepatic failure. J Lab Clin Med. 1983;102:870-880. PubMed
9. Michalak A, Rose C, Butterworth J, Butterworth RF. Neuroactive amino acids and glutamate (NMDA) receptors in frontal cortex of rats with experimental acute liver failure. Hepatology. 1996;24:908-13. PubMed
10. Bassett ML, Mullen KD, Scholz B, Fenstermacher JD, Jones EA. Increased brain uptake of gamma-aminobutyric acid in a rabbit model of hepatic encephalopathy. Gastroenterology. 1990;98:747-757. PubMed
11. Clay AS, Hainline BE. Hyperammonemia in the ICU. Chest. 2007;132:1368-1378. PubMed
12. Gundling F, Seidl H, Schmidt T, Schepp W. Blood ammonia level in liver cirrhosis: a conditio sine qua non to confirm hepatic encephalopathy? Eur J Gastroenterol Hepatol. 2008;20:246-247. PubMed
13. Stahl J. Studies of the Blood Ammonia in Liver Disease: Its Diagnostic, Prognostic and Therapeutic Significance. Ann Intern Med. 1963;58:1–24. PubMed
14. Kramer L, Tribl B, Gendo A, et al. Partial pressure of ammonia versus ammonia in hepatic encephalopathy. Hepatology. 2000;31:30-34. PubMed
15. Nicolao F, Masini A, Manuela M, Attili AF, Riggio O. Role of determination of partial pressure of ammonia in cirrhotic patients with or without hepatic encephalopathy. J Hepatol. 2003;38:441-446. PubMed
16. Ong JP, Aggarwal A, Krieger D, et al. Correlation between ammonia levels and the severity of hepatic encephalopathy. Am J Med. 2003;114:188-193. PubMed
17. Da Fonseca-Wollheim F. Preanalytical increase of ammonia in blood specimens from healthy subjects. Clin Chem. 1990;36:1483-1487. PubMed
18. Howanitz JH, Howanitz PJ, Skrodzki CA, Iwanski JA. Influences of specimen processing and storage conditions on results for plasma ammonia. Clin Chem. 1984;30:906-908. PubMed
19. Gundling F, Zelihic E, Seidl H, et al. How to diagnose hepatic encephalopathy in the emergency department. Ann Hepatol. 2013;12:108-114. PubMed
20. Ditisheim S, Giostra E, Burkhard PR, et al. A capillary blood ammonia bedside test following glutamine load to improve the diagnosis of hepatic encephalopathy in cirrhosis. BMC Gastroenterol. 2011;11:134. PubMed
21. Conn HO, Leevy CM, Vlahcevic ZR, et al. Comparison of lactulose and neomycin in the treatment of chronic portal-systemic encephalopathy. A double blind controlled trial. Gastroenterology. 1977;72:573-583. PubMed