User login
A 41-year-old man presented with pain in the left upper quadrant for 4 days. The pain was constant, was worse on inspiration, and did not radiate. He denied fevers, night sweats, nausea, vomiting, diarrhea, and urinary symptoms. He had been diagnosed with multiple sclerosis a few years earlier, and he had undergone aortofemoral bypass surgery on the left side 2 years ago. He denied smoking or using illicit drugs and described himself as a social drinker.
In the emergency room, he appeared comfortable. He was afebrile, blood pressure 136/69 mm Hg, pulse rate 98 per minute, and respiratory rate 16. All pulses were palpable and equal, the jugular venous pressure was not elevated, and no cardiac murmurs were heard. The abdomen was tender in the left upper quadrant, with no guarding or rigidity. Examination of the nervous, musculoskeletal, and respiratory systems was unremarkable. Skin examination revealed only scars from previous surgery.
LABORATORY AND IMAGING RESULTS
- White blood cell count 7.2 × 109/L (reference range 4.0–10.0) with a normal differential
- Hemoglobin 134 g/dL (140–180)
- Platelet count 167 × 109/L (150–400)
- Renal and liver panels were normal
- Erythrocyte sedimentation rate 30 mm/hour
- C-reactive protein level 14.3 mg/L
- D-dimer level 2,670 ng/mL (< 500)
- International normalized ratio (INR) 1.0 (0.9–1.3)
- Activated partial thromboplastin time (aPTT) 44 seconds (25–38)
- Fibrinogen level 3.0 g/L (1.8–3.5)
- Urinalysis negative for leukocytes and casts.
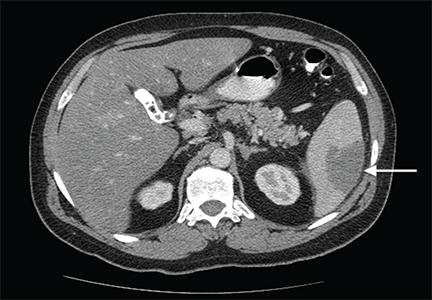
Computed tomography of the abdomen showed a wedge-shaped area of hypodensity along the inferolateral aspect of the spleen measuring 6 × 3.8 cm, consistent with a recent infarct (Figure 1). There was also evidence of a previous infarct in the posterolateral aspect of the spleen. Splenic, celiac, superior mesenteric, and inferior mesenteric arteries were patent.
1. Given these findings, which of the following diagnoses should be considered?
- Subacute infective endocarditis
- Inherited thrombophilia
- Antiphospholipid syndrome
All three diagnoses should be considered in this case.
Endocarditis
Embolism from a source in the heart caused by subacute bacterial endocarditis is more common than the other two conditions listed here and must be excluded.
Our patient lacks key features of this condition: he has no predisposing factors (artificial valve, cyanotic congenital heart disease, previous endocarditis, intravenous drug abuse); no constitutional symptoms of fever, night sweats, and weight loss; no findings on examination of skin and cardiovascular systems; and a normal white blood cell count. Nevertheless, even though the absence of these features makes bacterial endocarditis unlikely, it does not exclude it. Blood cultures and transesophageal echocardiography are indicated to rule out bacterial endocarditis.
We obtained serial blood cultures, which were negative, and transesophageal echocardiography showed normal valves and no evidence of thrombus or vegetation, thus excluding a cardiac source of emboli.
Thrombophilia
Our patient has a history of recurrent thromboembolic episodes, and this warrants testing to rule out an inherited thrombophilia. A family history of thromboembolic disease should also be sought.1
In our patient, tests for prothrombotic activity including protein C chromogen, activated protein C ratio, free protein S, functional protein S, antithrombin factor V Leiden, and the prothrombin 20210G>A mutation were either negative or within the reference range. A negative family history of thromboembolic disease and the negative laboratory tests make inherited thrombophilia unlikely in our patient.
Sickle cell disease, polycythemia vera, and essential thrombocythemia may also cause splenic infarction but can be ruled out in this patient on the basis of history and initial blood tests.
Antiphospholipid syndrome
A history of vascular disease (aortofemoral bypass surgery), a recent splenic infarct, and an elevated aPTT makes antiphospholipid syndrome the likeliest diagnosis in this patient.
Appropriate tests are for lupus anticoagulant, immunoglobulin G (IgG) or IgM cardiolipin antibody, and beta-2 glycoprotein 1 (beta-2 GP1) antibody, as well as the dilute Russell viper venom time (dRVVT) and the dRVVT ratio. The IgG and IgM cardiolipin antibody and beta-2 GP1 antibody tests have the same diagnostic value, and only medium to high titers should be considered positive.
Our patient’s IgG cardiolipin antibody level was in the normal range at 15 IgG phospholipid units (reference range 0–22); his IgM cardiolipin antibody level was high at 41 IgM phospholipid units (0–10). The dRVVT was 57 seconds (24–42), and the dRVVT ratio was 2.0 (0.0–1.3).
2. What further investigations are indicated before starting treatment?
- No further investigations required
- Repeat testing for phospholipid antibodies in 12 weeks
- Test for antinuclear antibodies
Antiphospholipid antibodies may appear transiently in certain infections, such as syphilis, Lyme disease, Epstein-Barr virus, cytomegalovirus, hepatitis C, and human immunodeficiency virus. Therefore, the presence of antiphospholipid antibodies must be confirmed over time, with two positive results at least 12 weeks apart.2
When repeated 12 weeks later, our patient’s IgG anticardiolipin antibody level was 14 GPL units, and the IgM anticardiolipin antibody level was 30 MPL units; the dRVVT was 55 seconds, and the dRVVT ratio was 1.8. These results, along with a history of recurrent arterial thrombosis, confirmed antiphospholipid syndrome.
The 2009 update of the International Society of Thrombosis and Haemostasis guidelines recommend two tests, the dRVVT and the aPTT, since no single test is 100% sensitive for lupus anticoagulant.3 The dRVVT has a high specificity for lupus anticoagulant in patients at high risk of thrombosis.
A SYNDROME WITH A WIDE RANGE OF EFFECTS AND COMPLICATIONS
Antiphospholipid syndrome is a systemic autoimmune disease that manifests as arterial and venous thrombosis and as obstetric complications. Thrombosis tends to be recurrent and may involve any site. For example, it can cause blurred vision in one or both eyes; amaurosis fugax; visual field defects; central or branch retinal artery or vein occlusion; deep vein thrombosis; pulmonary embolism; myocardial infarction; transient ischemic attack and stroke; cerebral vein thrombosis; and portal, renal, and mesenteric infarction involving veins or arteries.4 Pulmonary capillaritis may cause diffuse alveolar hemorrhage. Livedo reticularis, digital gangrene, cutaneous necrosis, splinter hemorrhages, chorea, and transverse myelopathy may also occur.
Obstetric complications of antiphospholipid syndrome include recurrent miscarriage and pregnancy loss at or after 10 weeks of gestation, eclampsia, preeclampsia, and placental insufficiency.5 The syndrome also has a potentially lethal variant characterized by multiorgan thrombosis affecting mainly small vessels.
The diagnosis of antiphospholipid syndrome requires relevant clinical features and symptoms and the presence of at least one of the antiphospholipid antibodies. Because the rate of false-positive tests for antiphospholipid antibodies ranges from 3% to 20% in the general population, asymptomatic patients should not be tested.6
Antiphospholipid syndrome may occur in the setting of other autoimmune diseases, most commonly systemic lupus erythematosus, when it is termed “secondary” antiphospholipid syndrome. Although only 40% of patients with lupus have antiphospholipid antibodies and less than 40% will have a thrombotic event, thrombotic antiphospholipid syndrome is a major adverse prognostic factor in these patients.7,8 Therefore, it is prudent to consider systemic lupus erythematosus and to do appropriate tests if the patient has other features suggestive of lupus, such as renal, skin, or musculoskeletal lesions.
In our patient, antinuclear antibody testing was positive, with a titer of 1:320, and showed a finely speckled staining pattern. Tests for antibodies to Sjögren syndrome A and B antigens were negative. The complement C3 level was 1.28 g/L (reference range 0.74–1.85) and the C4 level was 0.24 g/L (0.16–0.44). Although the speckled staining pattern can be seen in lupus, it is more common in Sjögren syndrome, mixed connective tissue disease, scleroderma, and CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia).9 Moreover, normal levels of complement C3 and C4, in the absence of clinical features, make lupus unlikely. Similarly, our patient had no clinical features of other connective tissue disorders. Therefore, he had primary antiphospholipid syndrome.
3. How should this patient be managed?
- Antiplatelet therapy
- Warfarin to maintain an INR between 2.0 and 3.0
- Warfarin to maintain an INR above 3.0
The risk of recurrent thrombosis is high in patients who test positive for lupus anticoagulant, and the risk is highest in patients who are also positive for anticardiolipin and anti-beta-2 GP1 antibodies: the incidence of thrombosis is 12.2% at 1 year, 26.1% at 5 years, 44.2% at 10 years.10
Since our patient is positive for lupus anticoagulant (prolonged aPTT and elevated dRVVT, both indicating lupus anticoagulant positivity) and for anticardiolipin antibodies (anti-beta-2 GP1 not tested), his risk of recurrent thrombosis is high, and he requires lifelong anticoagulation therapy.
The intensity of anticoagulation in different subgroups of patients is controversial. Based on retrospective trials, indefinite anticoagulation at an INR of 2.0 to 3.0 has been suggested for patients with antiphospholipid syndrome presenting with venous thrombosis, and more intense anticoagulation with an INR above 3.0 in patients with recurrent or arterial thrombosis.11 The combination of warfarin with an INR between 2.0 and 3.0 and aspirin 100 mg daily has also been proposed for patients with arterial thrombosis.12
Modifiable risk factors such as smoking, obesity, and use of estrogens should be addressed in all patients with antiphospholipid syndrome.
In pregnant women with complications such as preeclampsia, low-dose aspirin can be used, and in women with a history of miscarriage, the combination of low-dose aspirin and heparin is recommended throughout the prenatal period.4
In patients who have recurrent thrombosis despite adequate anticoagulation, an expert committee12 has proposed that alternative regimens could include long-term low-molecular-weight heparin instead of warfarin, the combination of warfarin and aspirin, or warfarin and hydroxychloroquine. Adding a statin can also be considered.
Treatment of catastrophic antiphospholipid syndrome is based on expert opinion. A combination of anticoagulation, corticosteroids, plasma exchange, intravenous immunoglobulins, and rituximab has been tried, but the mortality rate remains high.13
OUR PATIENT'S COURSE
Our patient was started on warfarin, with a target INR above 3.0, and was doing well at 6 months of follow-up.
- De Stefano V, Rossi E. Testing for inherited thrombophilia and consequences for antithrombotic prophylaxis in patients with venous thromboembolism and their relatives. A review of the Guidelines from Scientific Societies and Working Groups. Thromb Haemost 2013; 110:697–705.
- Galli M. Interpretation and recommended testing for antiphospholipid antibodies. Semin Thromb Hemost 2012; 38:348–352.
- Pengo V, Tripodi A, Reber G, et al; Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost 2009; 7:1737–1740.
- Keeling D, Mackie I, Moore GW, Greer IA, Greaves M; British Committee for Standards in Haematology. Guidelines on the investigation and management of antiphospholipid syndrome. Br J Haematol 2012; 157:47–58.
- Misita CP, Moll S. Antiphospholipid antibodies. Circulation 2005; 112:e39–e44.
- Rand JH, Wolgast LR. Do’s and don’t’s in diagnosing antiphospholipid syndrome. Hematology Am Soc Hematol Educ Program 2012; 2012:455–459.
- Mok CC, Tang SS, To CH, Petri M. Incidence and risk factors of thromboembolism in systemic lupus erythematosus: a comparison of three ethnic groups. Arthritis Rheum 2005; 52:2774–2782.
- Ruiz-Irastorza G, Egurbide MV, Ugalde J, Aguirre C. High impact of antiphospholipid syndrome on irreversible organ damage and survival of patients with systemic lupus erythematosus. Arch Intern Med 2004; 164:77–82.
- Locht H, Pelck R, Manthorpe R. Clinical manifestations correlated to the prevalence of autoantibodies in a large (n = 321) cohort of patients with primary Sjögren’s syndrome: a comparison of patients initially diagnosed according to the Copenhagen classification criteria with the American-European consensus criteria. Autoimmun Rev 2005; 4:276–281.
- Pengo V, Ruffatti A, Legnani C, et al. Clinical course of high-risk patients diagnosed with antiphospholipid syndrome. J Thromb Haemost 2010; 8:237–242.
- Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet 2010; 376:1498–1509.
- Ruiz-Irastorza G, Cuadrado MJ, Ruiz-Arruza I, et al. Evidence-based recommendations for the prevention and long-term management of thrombosis in antiphospholipid antibody-positive patients: report of a task force at the 13th International Congress on antiphospholipid antibodies. Lupus 2011; 20:206–218.
- Cervera R. Update on the diagnosis, treatment, and prognosis of the catastrophic antiphospholipid syndrome. Curr Rheumatol Rep 2010; 12:70–76.
A 41-year-old man presented with pain in the left upper quadrant for 4 days. The pain was constant, was worse on inspiration, and did not radiate. He denied fevers, night sweats, nausea, vomiting, diarrhea, and urinary symptoms. He had been diagnosed with multiple sclerosis a few years earlier, and he had undergone aortofemoral bypass surgery on the left side 2 years ago. He denied smoking or using illicit drugs and described himself as a social drinker.
In the emergency room, he appeared comfortable. He was afebrile, blood pressure 136/69 mm Hg, pulse rate 98 per minute, and respiratory rate 16. All pulses were palpable and equal, the jugular venous pressure was not elevated, and no cardiac murmurs were heard. The abdomen was tender in the left upper quadrant, with no guarding or rigidity. Examination of the nervous, musculoskeletal, and respiratory systems was unremarkable. Skin examination revealed only scars from previous surgery.
LABORATORY AND IMAGING RESULTS
- White blood cell count 7.2 × 109/L (reference range 4.0–10.0) with a normal differential
- Hemoglobin 134 g/dL (140–180)
- Platelet count 167 × 109/L (150–400)
- Renal and liver panels were normal
- Erythrocyte sedimentation rate 30 mm/hour
- C-reactive protein level 14.3 mg/L
- D-dimer level 2,670 ng/mL (< 500)
- International normalized ratio (INR) 1.0 (0.9–1.3)
- Activated partial thromboplastin time (aPTT) 44 seconds (25–38)
- Fibrinogen level 3.0 g/L (1.8–3.5)
- Urinalysis negative for leukocytes and casts.
Computed tomography of the abdomen showed a wedge-shaped area of hypodensity along the inferolateral aspect of the spleen measuring 6 × 3.8 cm, consistent with a recent infarct (Figure 1). There was also evidence of a previous infarct in the posterolateral aspect of the spleen. Splenic, celiac, superior mesenteric, and inferior mesenteric arteries were patent.
1. Given these findings, which of the following diagnoses should be considered?
- Subacute infective endocarditis
- Inherited thrombophilia
- Antiphospholipid syndrome
All three diagnoses should be considered in this case.
Endocarditis
Embolism from a source in the heart caused by subacute bacterial endocarditis is more common than the other two conditions listed here and must be excluded.
Our patient lacks key features of this condition: he has no predisposing factors (artificial valve, cyanotic congenital heart disease, previous endocarditis, intravenous drug abuse); no constitutional symptoms of fever, night sweats, and weight loss; no findings on examination of skin and cardiovascular systems; and a normal white blood cell count. Nevertheless, even though the absence of these features makes bacterial endocarditis unlikely, it does not exclude it. Blood cultures and transesophageal echocardiography are indicated to rule out bacterial endocarditis.
We obtained serial blood cultures, which were negative, and transesophageal echocardiography showed normal valves and no evidence of thrombus or vegetation, thus excluding a cardiac source of emboli.
Thrombophilia
Our patient has a history of recurrent thromboembolic episodes, and this warrants testing to rule out an inherited thrombophilia. A family history of thromboembolic disease should also be sought.1
In our patient, tests for prothrombotic activity including protein C chromogen, activated protein C ratio, free protein S, functional protein S, antithrombin factor V Leiden, and the prothrombin 20210G>A mutation were either negative or within the reference range. A negative family history of thromboembolic disease and the negative laboratory tests make inherited thrombophilia unlikely in our patient.
Sickle cell disease, polycythemia vera, and essential thrombocythemia may also cause splenic infarction but can be ruled out in this patient on the basis of history and initial blood tests.
Antiphospholipid syndrome
A history of vascular disease (aortofemoral bypass surgery), a recent splenic infarct, and an elevated aPTT makes antiphospholipid syndrome the likeliest diagnosis in this patient.
Appropriate tests are for lupus anticoagulant, immunoglobulin G (IgG) or IgM cardiolipin antibody, and beta-2 glycoprotein 1 (beta-2 GP1) antibody, as well as the dilute Russell viper venom time (dRVVT) and the dRVVT ratio. The IgG and IgM cardiolipin antibody and beta-2 GP1 antibody tests have the same diagnostic value, and only medium to high titers should be considered positive.
Our patient’s IgG cardiolipin antibody level was in the normal range at 15 IgG phospholipid units (reference range 0–22); his IgM cardiolipin antibody level was high at 41 IgM phospholipid units (0–10). The dRVVT was 57 seconds (24–42), and the dRVVT ratio was 2.0 (0.0–1.3).
2. What further investigations are indicated before starting treatment?
- No further investigations required
- Repeat testing for phospholipid antibodies in 12 weeks
- Test for antinuclear antibodies
Antiphospholipid antibodies may appear transiently in certain infections, such as syphilis, Lyme disease, Epstein-Barr virus, cytomegalovirus, hepatitis C, and human immunodeficiency virus. Therefore, the presence of antiphospholipid antibodies must be confirmed over time, with two positive results at least 12 weeks apart.2
When repeated 12 weeks later, our patient’s IgG anticardiolipin antibody level was 14 GPL units, and the IgM anticardiolipin antibody level was 30 MPL units; the dRVVT was 55 seconds, and the dRVVT ratio was 1.8. These results, along with a history of recurrent arterial thrombosis, confirmed antiphospholipid syndrome.
The 2009 update of the International Society of Thrombosis and Haemostasis guidelines recommend two tests, the dRVVT and the aPTT, since no single test is 100% sensitive for lupus anticoagulant.3 The dRVVT has a high specificity for lupus anticoagulant in patients at high risk of thrombosis.
A SYNDROME WITH A WIDE RANGE OF EFFECTS AND COMPLICATIONS
Antiphospholipid syndrome is a systemic autoimmune disease that manifests as arterial and venous thrombosis and as obstetric complications. Thrombosis tends to be recurrent and may involve any site. For example, it can cause blurred vision in one or both eyes; amaurosis fugax; visual field defects; central or branch retinal artery or vein occlusion; deep vein thrombosis; pulmonary embolism; myocardial infarction; transient ischemic attack and stroke; cerebral vein thrombosis; and portal, renal, and mesenteric infarction involving veins or arteries.4 Pulmonary capillaritis may cause diffuse alveolar hemorrhage. Livedo reticularis, digital gangrene, cutaneous necrosis, splinter hemorrhages, chorea, and transverse myelopathy may also occur.
Obstetric complications of antiphospholipid syndrome include recurrent miscarriage and pregnancy loss at or after 10 weeks of gestation, eclampsia, preeclampsia, and placental insufficiency.5 The syndrome also has a potentially lethal variant characterized by multiorgan thrombosis affecting mainly small vessels.
The diagnosis of antiphospholipid syndrome requires relevant clinical features and symptoms and the presence of at least one of the antiphospholipid antibodies. Because the rate of false-positive tests for antiphospholipid antibodies ranges from 3% to 20% in the general population, asymptomatic patients should not be tested.6
Antiphospholipid syndrome may occur in the setting of other autoimmune diseases, most commonly systemic lupus erythematosus, when it is termed “secondary” antiphospholipid syndrome. Although only 40% of patients with lupus have antiphospholipid antibodies and less than 40% will have a thrombotic event, thrombotic antiphospholipid syndrome is a major adverse prognostic factor in these patients.7,8 Therefore, it is prudent to consider systemic lupus erythematosus and to do appropriate tests if the patient has other features suggestive of lupus, such as renal, skin, or musculoskeletal lesions.
In our patient, antinuclear antibody testing was positive, with a titer of 1:320, and showed a finely speckled staining pattern. Tests for antibodies to Sjögren syndrome A and B antigens were negative. The complement C3 level was 1.28 g/L (reference range 0.74–1.85) and the C4 level was 0.24 g/L (0.16–0.44). Although the speckled staining pattern can be seen in lupus, it is more common in Sjögren syndrome, mixed connective tissue disease, scleroderma, and CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia).9 Moreover, normal levels of complement C3 and C4, in the absence of clinical features, make lupus unlikely. Similarly, our patient had no clinical features of other connective tissue disorders. Therefore, he had primary antiphospholipid syndrome.
3. How should this patient be managed?
- Antiplatelet therapy
- Warfarin to maintain an INR between 2.0 and 3.0
- Warfarin to maintain an INR above 3.0
The risk of recurrent thrombosis is high in patients who test positive for lupus anticoagulant, and the risk is highest in patients who are also positive for anticardiolipin and anti-beta-2 GP1 antibodies: the incidence of thrombosis is 12.2% at 1 year, 26.1% at 5 years, 44.2% at 10 years.10
Since our patient is positive for lupus anticoagulant (prolonged aPTT and elevated dRVVT, both indicating lupus anticoagulant positivity) and for anticardiolipin antibodies (anti-beta-2 GP1 not tested), his risk of recurrent thrombosis is high, and he requires lifelong anticoagulation therapy.
The intensity of anticoagulation in different subgroups of patients is controversial. Based on retrospective trials, indefinite anticoagulation at an INR of 2.0 to 3.0 has been suggested for patients with antiphospholipid syndrome presenting with venous thrombosis, and more intense anticoagulation with an INR above 3.0 in patients with recurrent or arterial thrombosis.11 The combination of warfarin with an INR between 2.0 and 3.0 and aspirin 100 mg daily has also been proposed for patients with arterial thrombosis.12
Modifiable risk factors such as smoking, obesity, and use of estrogens should be addressed in all patients with antiphospholipid syndrome.
In pregnant women with complications such as preeclampsia, low-dose aspirin can be used, and in women with a history of miscarriage, the combination of low-dose aspirin and heparin is recommended throughout the prenatal period.4
In patients who have recurrent thrombosis despite adequate anticoagulation, an expert committee12 has proposed that alternative regimens could include long-term low-molecular-weight heparin instead of warfarin, the combination of warfarin and aspirin, or warfarin and hydroxychloroquine. Adding a statin can also be considered.
Treatment of catastrophic antiphospholipid syndrome is based on expert opinion. A combination of anticoagulation, corticosteroids, plasma exchange, intravenous immunoglobulins, and rituximab has been tried, but the mortality rate remains high.13
OUR PATIENT'S COURSE
Our patient was started on warfarin, with a target INR above 3.0, and was doing well at 6 months of follow-up.
A 41-year-old man presented with pain in the left upper quadrant for 4 days. The pain was constant, was worse on inspiration, and did not radiate. He denied fevers, night sweats, nausea, vomiting, diarrhea, and urinary symptoms. He had been diagnosed with multiple sclerosis a few years earlier, and he had undergone aortofemoral bypass surgery on the left side 2 years ago. He denied smoking or using illicit drugs and described himself as a social drinker.
In the emergency room, he appeared comfortable. He was afebrile, blood pressure 136/69 mm Hg, pulse rate 98 per minute, and respiratory rate 16. All pulses were palpable and equal, the jugular venous pressure was not elevated, and no cardiac murmurs were heard. The abdomen was tender in the left upper quadrant, with no guarding or rigidity. Examination of the nervous, musculoskeletal, and respiratory systems was unremarkable. Skin examination revealed only scars from previous surgery.
LABORATORY AND IMAGING RESULTS
- White blood cell count 7.2 × 109/L (reference range 4.0–10.0) with a normal differential
- Hemoglobin 134 g/dL (140–180)
- Platelet count 167 × 109/L (150–400)
- Renal and liver panels were normal
- Erythrocyte sedimentation rate 30 mm/hour
- C-reactive protein level 14.3 mg/L
- D-dimer level 2,670 ng/mL (< 500)
- International normalized ratio (INR) 1.0 (0.9–1.3)
- Activated partial thromboplastin time (aPTT) 44 seconds (25–38)
- Fibrinogen level 3.0 g/L (1.8–3.5)
- Urinalysis negative for leukocytes and casts.
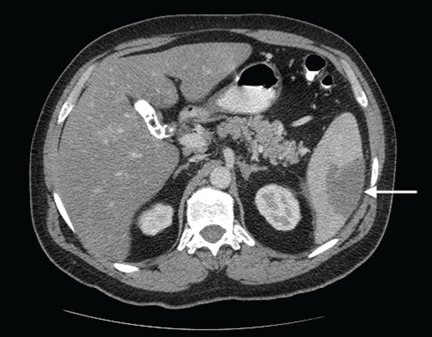
Computed tomography of the abdomen showed a wedge-shaped area of hypodensity along the inferolateral aspect of the spleen measuring 6 × 3.8 cm, consistent with a recent infarct (Figure 1). There was also evidence of a previous infarct in the posterolateral aspect of the spleen. Splenic, celiac, superior mesenteric, and inferior mesenteric arteries were patent.
1. Given these findings, which of the following diagnoses should be considered?
- Subacute infective endocarditis
- Inherited thrombophilia
- Antiphospholipid syndrome
All three diagnoses should be considered in this case.
Endocarditis
Embolism from a source in the heart caused by subacute bacterial endocarditis is more common than the other two conditions listed here and must be excluded.
Our patient lacks key features of this condition: he has no predisposing factors (artificial valve, cyanotic congenital heart disease, previous endocarditis, intravenous drug abuse); no constitutional symptoms of fever, night sweats, and weight loss; no findings on examination of skin and cardiovascular systems; and a normal white blood cell count. Nevertheless, even though the absence of these features makes bacterial endocarditis unlikely, it does not exclude it. Blood cultures and transesophageal echocardiography are indicated to rule out bacterial endocarditis.
We obtained serial blood cultures, which were negative, and transesophageal echocardiography showed normal valves and no evidence of thrombus or vegetation, thus excluding a cardiac source of emboli.
Thrombophilia
Our patient has a history of recurrent thromboembolic episodes, and this warrants testing to rule out an inherited thrombophilia. A family history of thromboembolic disease should also be sought.1
In our patient, tests for prothrombotic activity including protein C chromogen, activated protein C ratio, free protein S, functional protein S, antithrombin factor V Leiden, and the prothrombin 20210G>A mutation were either negative or within the reference range. A negative family history of thromboembolic disease and the negative laboratory tests make inherited thrombophilia unlikely in our patient.
Sickle cell disease, polycythemia vera, and essential thrombocythemia may also cause splenic infarction but can be ruled out in this patient on the basis of history and initial blood tests.
Antiphospholipid syndrome
A history of vascular disease (aortofemoral bypass surgery), a recent splenic infarct, and an elevated aPTT makes antiphospholipid syndrome the likeliest diagnosis in this patient.
Appropriate tests are for lupus anticoagulant, immunoglobulin G (IgG) or IgM cardiolipin antibody, and beta-2 glycoprotein 1 (beta-2 GP1) antibody, as well as the dilute Russell viper venom time (dRVVT) and the dRVVT ratio. The IgG and IgM cardiolipin antibody and beta-2 GP1 antibody tests have the same diagnostic value, and only medium to high titers should be considered positive.
Our patient’s IgG cardiolipin antibody level was in the normal range at 15 IgG phospholipid units (reference range 0–22); his IgM cardiolipin antibody level was high at 41 IgM phospholipid units (0–10). The dRVVT was 57 seconds (24–42), and the dRVVT ratio was 2.0 (0.0–1.3).
2. What further investigations are indicated before starting treatment?
- No further investigations required
- Repeat testing for phospholipid antibodies in 12 weeks
- Test for antinuclear antibodies
Antiphospholipid antibodies may appear transiently in certain infections, such as syphilis, Lyme disease, Epstein-Barr virus, cytomegalovirus, hepatitis C, and human immunodeficiency virus. Therefore, the presence of antiphospholipid antibodies must be confirmed over time, with two positive results at least 12 weeks apart.2
When repeated 12 weeks later, our patient’s IgG anticardiolipin antibody level was 14 GPL units, and the IgM anticardiolipin antibody level was 30 MPL units; the dRVVT was 55 seconds, and the dRVVT ratio was 1.8. These results, along with a history of recurrent arterial thrombosis, confirmed antiphospholipid syndrome.
The 2009 update of the International Society of Thrombosis and Haemostasis guidelines recommend two tests, the dRVVT and the aPTT, since no single test is 100% sensitive for lupus anticoagulant.3 The dRVVT has a high specificity for lupus anticoagulant in patients at high risk of thrombosis.
A SYNDROME WITH A WIDE RANGE OF EFFECTS AND COMPLICATIONS
Antiphospholipid syndrome is a systemic autoimmune disease that manifests as arterial and venous thrombosis and as obstetric complications. Thrombosis tends to be recurrent and may involve any site. For example, it can cause blurred vision in one or both eyes; amaurosis fugax; visual field defects; central or branch retinal artery or vein occlusion; deep vein thrombosis; pulmonary embolism; myocardial infarction; transient ischemic attack and stroke; cerebral vein thrombosis; and portal, renal, and mesenteric infarction involving veins or arteries.4 Pulmonary capillaritis may cause diffuse alveolar hemorrhage. Livedo reticularis, digital gangrene, cutaneous necrosis, splinter hemorrhages, chorea, and transverse myelopathy may also occur.
Obstetric complications of antiphospholipid syndrome include recurrent miscarriage and pregnancy loss at or after 10 weeks of gestation, eclampsia, preeclampsia, and placental insufficiency.5 The syndrome also has a potentially lethal variant characterized by multiorgan thrombosis affecting mainly small vessels.
The diagnosis of antiphospholipid syndrome requires relevant clinical features and symptoms and the presence of at least one of the antiphospholipid antibodies. Because the rate of false-positive tests for antiphospholipid antibodies ranges from 3% to 20% in the general population, asymptomatic patients should not be tested.6
Antiphospholipid syndrome may occur in the setting of other autoimmune diseases, most commonly systemic lupus erythematosus, when it is termed “secondary” antiphospholipid syndrome. Although only 40% of patients with lupus have antiphospholipid antibodies and less than 40% will have a thrombotic event, thrombotic antiphospholipid syndrome is a major adverse prognostic factor in these patients.7,8 Therefore, it is prudent to consider systemic lupus erythematosus and to do appropriate tests if the patient has other features suggestive of lupus, such as renal, skin, or musculoskeletal lesions.
In our patient, antinuclear antibody testing was positive, with a titer of 1:320, and showed a finely speckled staining pattern. Tests for antibodies to Sjögren syndrome A and B antigens were negative. The complement C3 level was 1.28 g/L (reference range 0.74–1.85) and the C4 level was 0.24 g/L (0.16–0.44). Although the speckled staining pattern can be seen in lupus, it is more common in Sjögren syndrome, mixed connective tissue disease, scleroderma, and CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia).9 Moreover, normal levels of complement C3 and C4, in the absence of clinical features, make lupus unlikely. Similarly, our patient had no clinical features of other connective tissue disorders. Therefore, he had primary antiphospholipid syndrome.
3. How should this patient be managed?
- Antiplatelet therapy
- Warfarin to maintain an INR between 2.0 and 3.0
- Warfarin to maintain an INR above 3.0
The risk of recurrent thrombosis is high in patients who test positive for lupus anticoagulant, and the risk is highest in patients who are also positive for anticardiolipin and anti-beta-2 GP1 antibodies: the incidence of thrombosis is 12.2% at 1 year, 26.1% at 5 years, 44.2% at 10 years.10
Since our patient is positive for lupus anticoagulant (prolonged aPTT and elevated dRVVT, both indicating lupus anticoagulant positivity) and for anticardiolipin antibodies (anti-beta-2 GP1 not tested), his risk of recurrent thrombosis is high, and he requires lifelong anticoagulation therapy.
The intensity of anticoagulation in different subgroups of patients is controversial. Based on retrospective trials, indefinite anticoagulation at an INR of 2.0 to 3.0 has been suggested for patients with antiphospholipid syndrome presenting with venous thrombosis, and more intense anticoagulation with an INR above 3.0 in patients with recurrent or arterial thrombosis.11 The combination of warfarin with an INR between 2.0 and 3.0 and aspirin 100 mg daily has also been proposed for patients with arterial thrombosis.12
Modifiable risk factors such as smoking, obesity, and use of estrogens should be addressed in all patients with antiphospholipid syndrome.
In pregnant women with complications such as preeclampsia, low-dose aspirin can be used, and in women with a history of miscarriage, the combination of low-dose aspirin and heparin is recommended throughout the prenatal period.4
In patients who have recurrent thrombosis despite adequate anticoagulation, an expert committee12 has proposed that alternative regimens could include long-term low-molecular-weight heparin instead of warfarin, the combination of warfarin and aspirin, or warfarin and hydroxychloroquine. Adding a statin can also be considered.
Treatment of catastrophic antiphospholipid syndrome is based on expert opinion. A combination of anticoagulation, corticosteroids, plasma exchange, intravenous immunoglobulins, and rituximab has been tried, but the mortality rate remains high.13
OUR PATIENT'S COURSE
Our patient was started on warfarin, with a target INR above 3.0, and was doing well at 6 months of follow-up.
- De Stefano V, Rossi E. Testing for inherited thrombophilia and consequences for antithrombotic prophylaxis in patients with venous thromboembolism and their relatives. A review of the Guidelines from Scientific Societies and Working Groups. Thromb Haemost 2013; 110:697–705.
- Galli M. Interpretation and recommended testing for antiphospholipid antibodies. Semin Thromb Hemost 2012; 38:348–352.
- Pengo V, Tripodi A, Reber G, et al; Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost 2009; 7:1737–1740.
- Keeling D, Mackie I, Moore GW, Greer IA, Greaves M; British Committee for Standards in Haematology. Guidelines on the investigation and management of antiphospholipid syndrome. Br J Haematol 2012; 157:47–58.
- Misita CP, Moll S. Antiphospholipid antibodies. Circulation 2005; 112:e39–e44.
- Rand JH, Wolgast LR. Do’s and don’t’s in diagnosing antiphospholipid syndrome. Hematology Am Soc Hematol Educ Program 2012; 2012:455–459.
- Mok CC, Tang SS, To CH, Petri M. Incidence and risk factors of thromboembolism in systemic lupus erythematosus: a comparison of three ethnic groups. Arthritis Rheum 2005; 52:2774–2782.
- Ruiz-Irastorza G, Egurbide MV, Ugalde J, Aguirre C. High impact of antiphospholipid syndrome on irreversible organ damage and survival of patients with systemic lupus erythematosus. Arch Intern Med 2004; 164:77–82.
- Locht H, Pelck R, Manthorpe R. Clinical manifestations correlated to the prevalence of autoantibodies in a large (n = 321) cohort of patients with primary Sjögren’s syndrome: a comparison of patients initially diagnosed according to the Copenhagen classification criteria with the American-European consensus criteria. Autoimmun Rev 2005; 4:276–281.
- Pengo V, Ruffatti A, Legnani C, et al. Clinical course of high-risk patients diagnosed with antiphospholipid syndrome. J Thromb Haemost 2010; 8:237–242.
- Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet 2010; 376:1498–1509.
- Ruiz-Irastorza G, Cuadrado MJ, Ruiz-Arruza I, et al. Evidence-based recommendations for the prevention and long-term management of thrombosis in antiphospholipid antibody-positive patients: report of a task force at the 13th International Congress on antiphospholipid antibodies. Lupus 2011; 20:206–218.
- Cervera R. Update on the diagnosis, treatment, and prognosis of the catastrophic antiphospholipid syndrome. Curr Rheumatol Rep 2010; 12:70–76.
- De Stefano V, Rossi E. Testing for inherited thrombophilia and consequences for antithrombotic prophylaxis in patients with venous thromboembolism and their relatives. A review of the Guidelines from Scientific Societies and Working Groups. Thromb Haemost 2013; 110:697–705.
- Galli M. Interpretation and recommended testing for antiphospholipid antibodies. Semin Thromb Hemost 2012; 38:348–352.
- Pengo V, Tripodi A, Reber G, et al; Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost 2009; 7:1737–1740.
- Keeling D, Mackie I, Moore GW, Greer IA, Greaves M; British Committee for Standards in Haematology. Guidelines on the investigation and management of antiphospholipid syndrome. Br J Haematol 2012; 157:47–58.
- Misita CP, Moll S. Antiphospholipid antibodies. Circulation 2005; 112:e39–e44.
- Rand JH, Wolgast LR. Do’s and don’t’s in diagnosing antiphospholipid syndrome. Hematology Am Soc Hematol Educ Program 2012; 2012:455–459.
- Mok CC, Tang SS, To CH, Petri M. Incidence and risk factors of thromboembolism in systemic lupus erythematosus: a comparison of three ethnic groups. Arthritis Rheum 2005; 52:2774–2782.
- Ruiz-Irastorza G, Egurbide MV, Ugalde J, Aguirre C. High impact of antiphospholipid syndrome on irreversible organ damage and survival of patients with systemic lupus erythematosus. Arch Intern Med 2004; 164:77–82.
- Locht H, Pelck R, Manthorpe R. Clinical manifestations correlated to the prevalence of autoantibodies in a large (n = 321) cohort of patients with primary Sjögren’s syndrome: a comparison of patients initially diagnosed according to the Copenhagen classification criteria with the American-European consensus criteria. Autoimmun Rev 2005; 4:276–281.
- Pengo V, Ruffatti A, Legnani C, et al. Clinical course of high-risk patients diagnosed with antiphospholipid syndrome. J Thromb Haemost 2010; 8:237–242.
- Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet 2010; 376:1498–1509.
- Ruiz-Irastorza G, Cuadrado MJ, Ruiz-Arruza I, et al. Evidence-based recommendations for the prevention and long-term management of thrombosis in antiphospholipid antibody-positive patients: report of a task force at the 13th International Congress on antiphospholipid antibodies. Lupus 2011; 20:206–218.
- Cervera R. Update on the diagnosis, treatment, and prognosis of the catastrophic antiphospholipid syndrome. Curr Rheumatol Rep 2010; 12:70–76.