User login
Asymptomatic Papules on the Neck
THE DIAGNOSIS: White Fibrous Papulosis
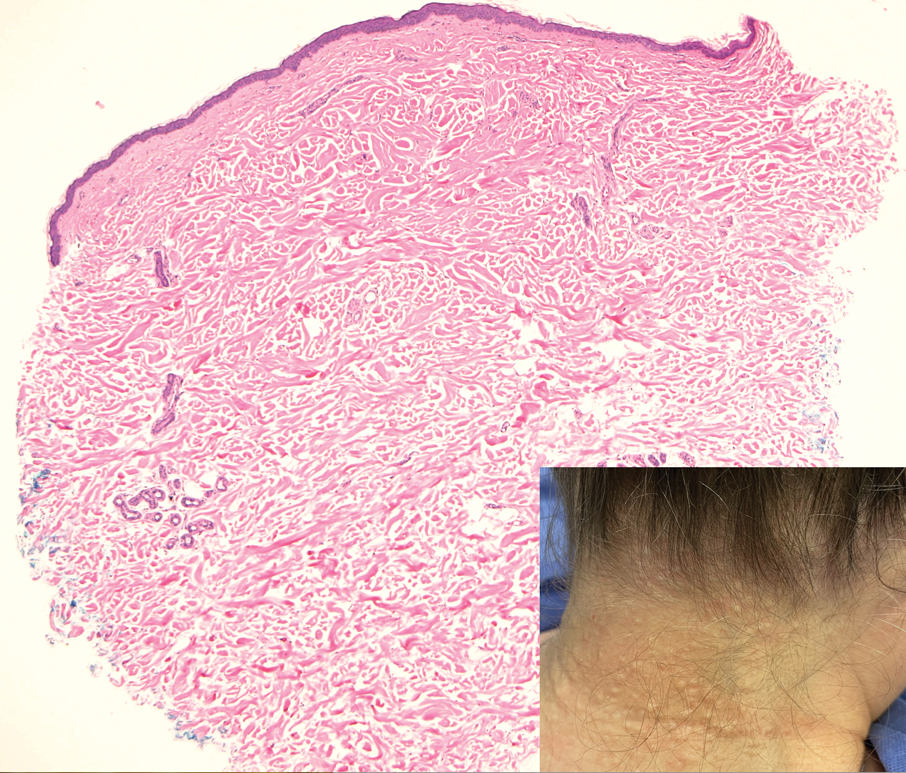
Given the histopathology findings, location on a sun-exposed site, lack of any additional systemic signs or symptoms, and no family history of similar lesions to suggest an underlying genetic condition, a diagnosis of white fibrous papulosis (WFP) was made. White fibrous papulosis is a relatively rare cutaneous disorder that was first reported by Shimizu et al1 in 1985. It is characterized by numerous grouped, 2- to 3-mm, white to flesh-colored papules that in most cases are confined to the neck in middle-aged to elderly individuals; however, cases involving the upper trunk and axillae also have been reported.1-3 The etiology of this condition is unclear but is thought to be related to aging and chronic exposure to UV light. Although treatment is not required, various modalities including tretinoin, excision, and laser therapy have been trialed with varying success.2,4 Our patient elected not to proceed with treatment.
Histologically, WFP may manifest similarly to connective tissue nevi; the overall architecture is nonspecific with focally thickened collagen and often elastic fibers that may be normal to reduced and/or fragmented, as well as an overall decrease in superficial dermal elastic tissue.3,5 Therefore, the differential diagnosis may include connective tissue nevi and require clinical correlation to make a correct diagnosis.
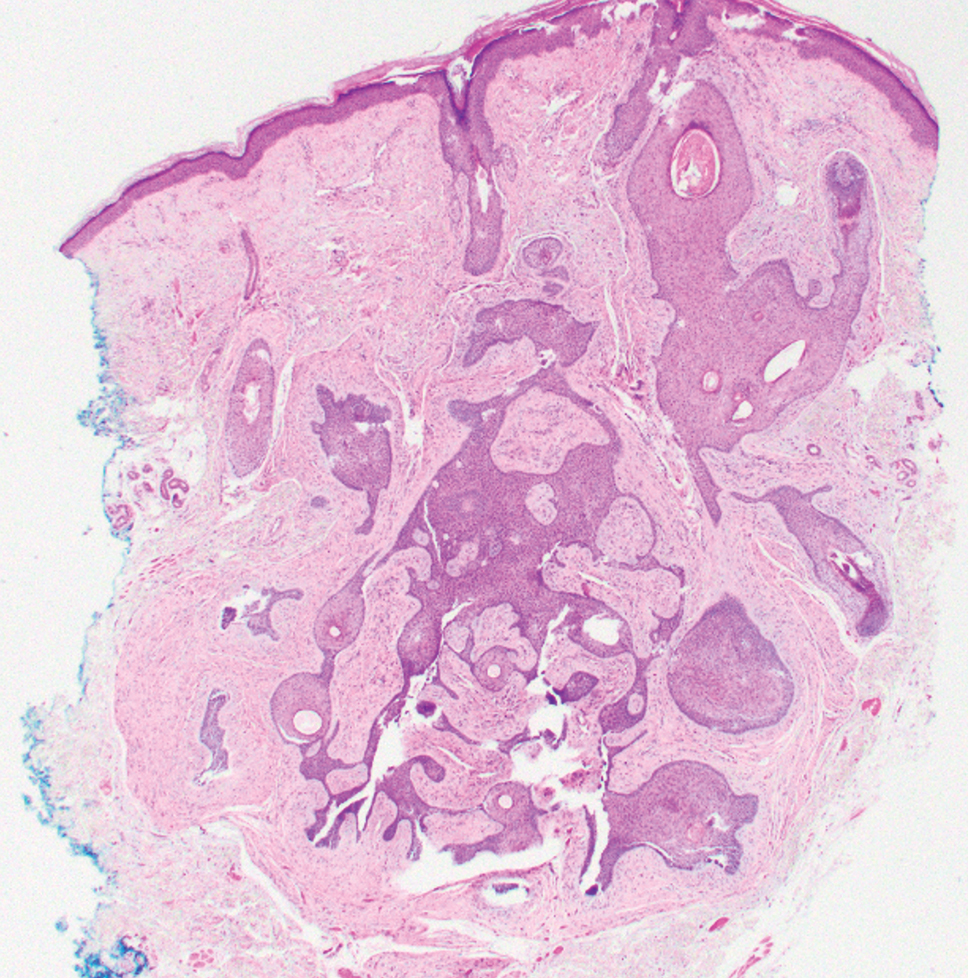
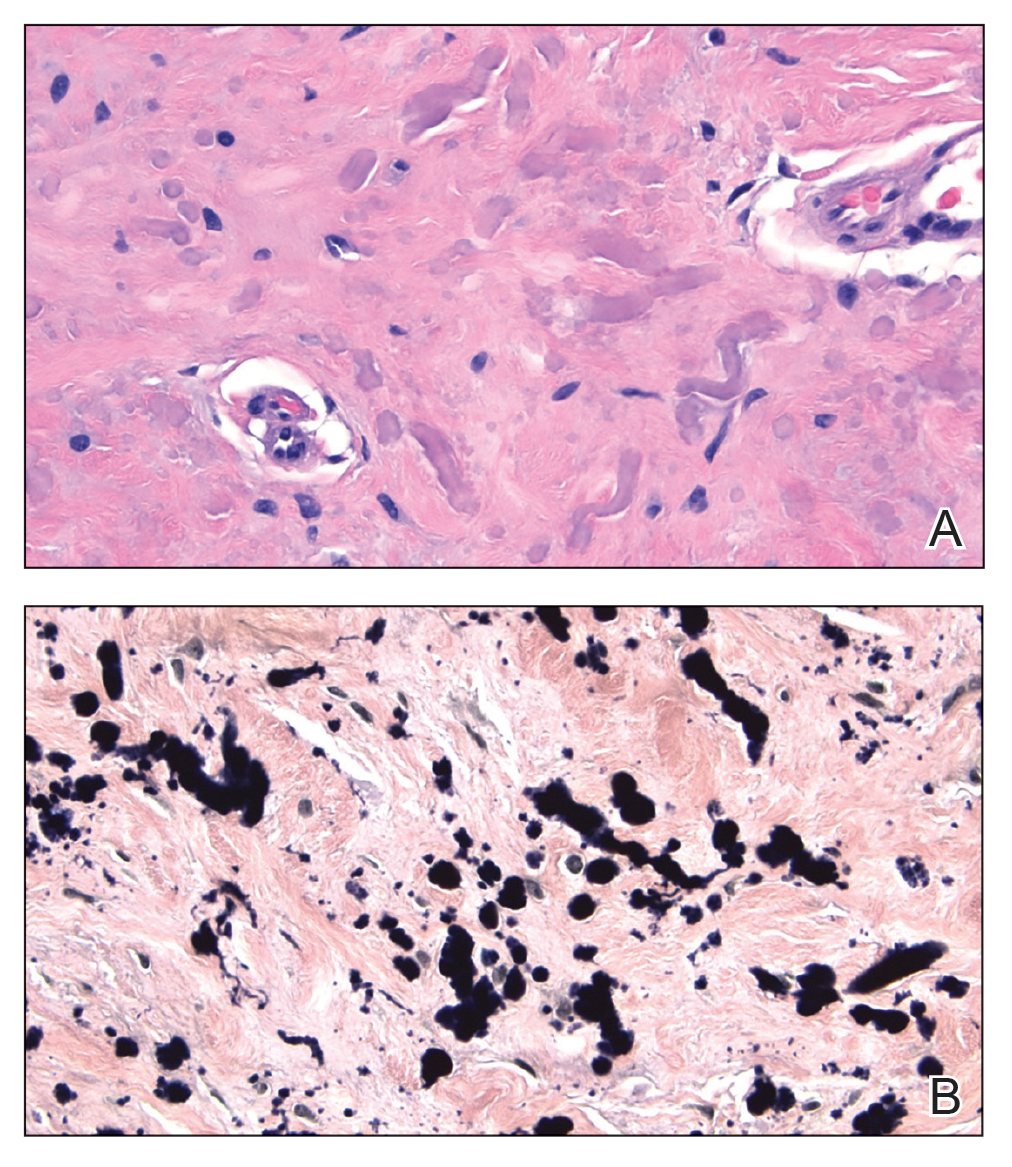
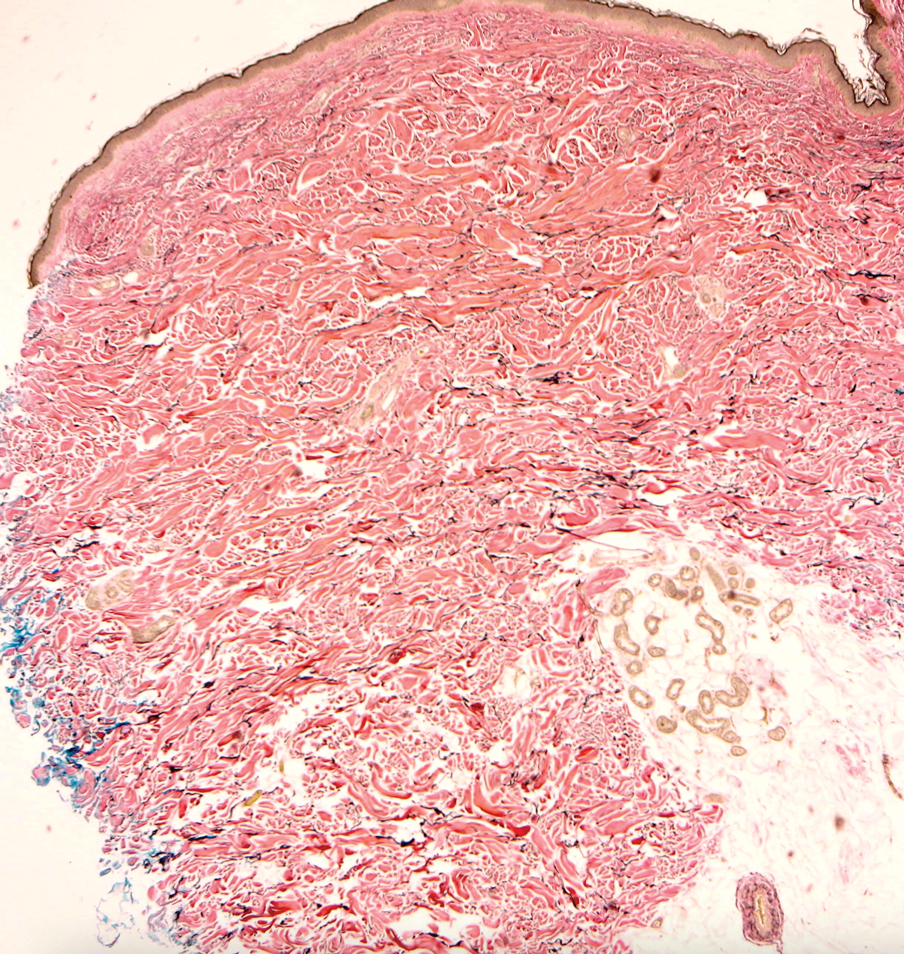
Pseudoxanthoma elasticum (PXE) is an autosomalrecessive disorder most commonly related to mutations in the ATP binding cassette subfamily C member 6 (ABCC6) gene that tends to manifest clinically on the neck and flexural extremities.6 This disease affects elastic fibers, which may become calcified over time. Pseudoxanthoma elasticum is associated with ocular complications relating to the Bruch membrane of the retina and angioid streaks; choroidal neovascularization involving the damaged Bruch membrane and episodes of acute retinopathy may result in vision loss in later stages of the disease.7 Involvement of the elastic laminae of arteries can be associated with cardiovascular and cerebrovascular complications such as stroke, coronary artery disease, claudication, and aneurysms. Involvement of the gastrointestinal or genitourinary tracts also may occur and most commonly manifests with bleeding. Pathologic alterations in the elastic fibers of the lungs also have been reported in patients with PXE.8 Histologically, PXE exhibits increased abnormally clumped and fragmented elastic fibers in the superficial dermis, often with calcification (Figure 1). Pseudo-PXE related to D-penicillamine use often lacks calcification and has a bramble bush appearance.9

Fibrofolliculomas may manifest alone or in association with an underlying condition such as Birt-Hogg-Dubé syndrome, in which lesions are most frequently seen scattered on the scalp, face, ears, neck, or upper trunk.10 This condition is related to a folliculin (FLCN) gene germline mutation. Birt-Hogg-Dubé syndrome also may be associated with acrochordons, trichodiscomas, renal cancer, and lung cysts with or without spontaneous pneumothorax. Less frequently noted findings include oral papules, epidermal cysts, angiofibromas, lipomas/angiolipomas, parotid gland tumors, and thyroid neoplasms. Connective tissue nevi/collagenomas can appear clinically similar to fibrofolliculomas; true connective tissue nevi are reported less commonly in Birt-Hogg-Dubé syndrome.11 Histologically, a fibrofolliculoma manifests with epidermal strands originating from a hair follicle associated with prominent surrounding connective tissue (Figure 2).
Elastofibroma dorsi is a benign tumor of connective tissue that most commonly manifests clinically as a solitary subcutaneous mass on the back near the inferior angle of the scapula; it typically develops below the rhomboid major and latissimus dorsi muscles.12 The pathogenesis is uncertain, but some patients have reported a family history of the condition or a history of repetitive shoulder movement/trauma prior to onset; the mass may be asymptomatic or associated with pain and/or swelling. Those affected tend to be older than 50 years.13 Histologically, thickened and rounded to beaded elastic fibers are seen admixed with collagen (Figure 3).
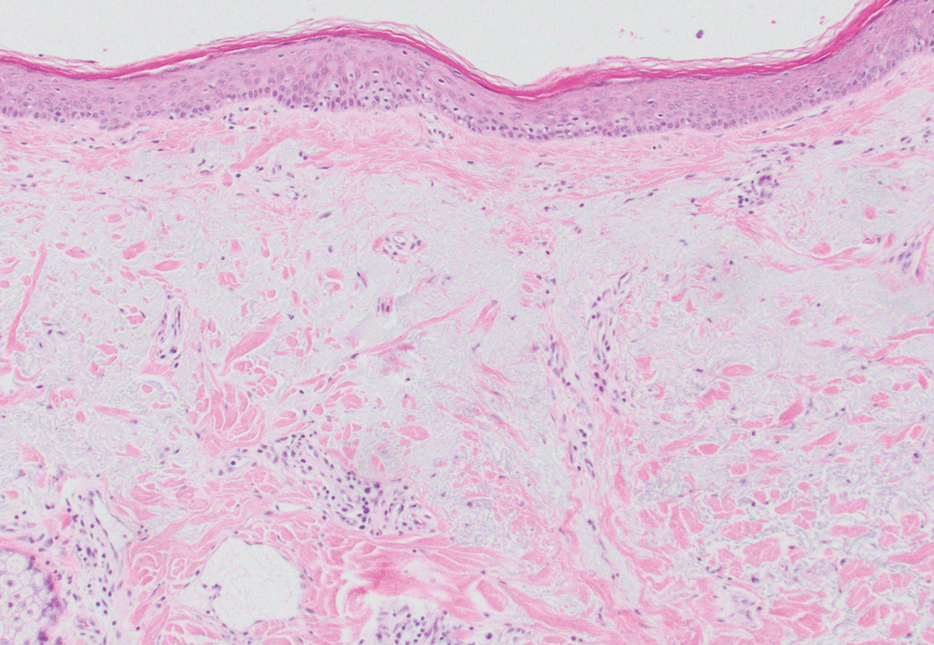
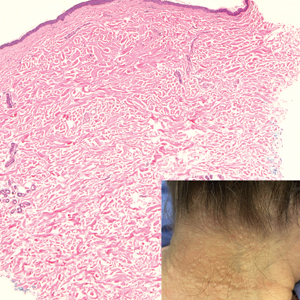
Actinic (solar) elastosis frequently is encountered in many skin biopsies and is caused by chronic photodamage. More hypertrophic variants, such as papular or nodular solar elastosis, may clinically manifest similarly to WFP.14 Histologically, actinic elastosis manifests as a considerable increase in elastic tissue in the papillary and superficial reticular dermis (Figure 4).
- Shimizu H, Nishikawa T, Kimura S. White fibrous papulosis of the neck: review of our 16 cases. Nihon Hifuka Gakkai Zasshi. 1985;95:1077-1084.
- Teo W, Pang S. White fibrous papulosis of the chest and back. J Am Acad Dermatol. 2012;66:AB33.
- Dokic Y, Tschen J. White fibrous papulosis of the axillae and neck. Cureus. 2020;12:E7635.
- Lueangarun S, Panchaprateep R. White fibrous papulosis of the neck treated with fractionated 1550-nm erbium glass laser: a case report. J Lasers Med Sci. 2016;7:256-258.
- Rios-Gomez M, Ramos-Garibay JA, Perez-Santana ME, et al. White fibrous papulosis of the neck: a case report. Cureus. 2022;14:E25661.
- Váradi A, Szabó Z, Pomozi V, et al. ABCC6 as a target in pseudoxanthoma elasticum. Curr Drug Targets. 2011;12:671-682.
- Gliem M, Birtel J, Müller PL, et al. Acute retinopathy in pseudoxanthoma elasticum. JAMA Ophthalmol. 2019;137:1165-1173.
- Germain DP. Pseudoxanthoma elasticum. Orphanet J Rare Dis. 2017;12:85. doi:10.1186/s13023-017-0639-8
- Chisti MA, Binamer Y, Alfadley A, et al. D-penicillamine-induced pseudo-pseudoxanthoma elasticum and extensive elastosis perforans serpiginosa with excellent response to acitretin. Ann Saudi Med. 2019;39:56-60.
- Criscito MC, Mu EW, Meehan SA, et al. Dermoscopic features of a solitary fibrofolliculoma on the left cheek. J Am Acad Dermatol. 2017;76(2 suppl 1):S8-S9.
- Sattler EC, Steinlein OK. Birt-Hogg-Dubé syndrome. In: Adam MP, Everman DB, Mirzaa GM, et al, eds. GeneReviews® [Internet]. Updated January 30, 2020. Accessed February 23, 2023. https://www.ncbi.nlm.nih.gov/books/NBK1522
- Patnayak R, Jena A, Settipalli S, et al. Elastofibroma: an uncommon tumor revisited. J Cutan Aesthet Surg. 2016;9:34-37. doi:10.4103/0974- 2077.178543
- Chandrasekar CR, Grimer RJ, Carter SR, et al. Elastofibroma dorsi: an uncommon benign pseudotumour. Sarcoma. 2008;2008:756565. doi:10.1155/2008/756565
- Kwittken J. Papular elastosis. Cutis. 2000;66:81-83.
THE DIAGNOSIS: White Fibrous Papulosis
Given the histopathology findings, location on a sun-exposed site, lack of any additional systemic signs or symptoms, and no family history of similar lesions to suggest an underlying genetic condition, a diagnosis of white fibrous papulosis (WFP) was made. White fibrous papulosis is a relatively rare cutaneous disorder that was first reported by Shimizu et al1 in 1985. It is characterized by numerous grouped, 2- to 3-mm, white to flesh-colored papules that in most cases are confined to the neck in middle-aged to elderly individuals; however, cases involving the upper trunk and axillae also have been reported.1-3 The etiology of this condition is unclear but is thought to be related to aging and chronic exposure to UV light. Although treatment is not required, various modalities including tretinoin, excision, and laser therapy have been trialed with varying success.2,4 Our patient elected not to proceed with treatment.
Histologically, WFP may manifest similarly to connective tissue nevi; the overall architecture is nonspecific with focally thickened collagen and often elastic fibers that may be normal to reduced and/or fragmented, as well as an overall decrease in superficial dermal elastic tissue.3,5 Therefore, the differential diagnosis may include connective tissue nevi and require clinical correlation to make a correct diagnosis.
Pseudoxanthoma elasticum (PXE) is an autosomalrecessive disorder most commonly related to mutations in the ATP binding cassette subfamily C member 6 (ABCC6) gene that tends to manifest clinically on the neck and flexural extremities.6 This disease affects elastic fibers, which may become calcified over time. Pseudoxanthoma elasticum is associated with ocular complications relating to the Bruch membrane of the retina and angioid streaks; choroidal neovascularization involving the damaged Bruch membrane and episodes of acute retinopathy may result in vision loss in later stages of the disease.7 Involvement of the elastic laminae of arteries can be associated with cardiovascular and cerebrovascular complications such as stroke, coronary artery disease, claudication, and aneurysms. Involvement of the gastrointestinal or genitourinary tracts also may occur and most commonly manifests with bleeding. Pathologic alterations in the elastic fibers of the lungs also have been reported in patients with PXE.8 Histologically, PXE exhibits increased abnormally clumped and fragmented elastic fibers in the superficial dermis, often with calcification (Figure 1). Pseudo-PXE related to D-penicillamine use often lacks calcification and has a bramble bush appearance.9
Fibrofolliculomas may manifest alone or in association with an underlying condition such as Birt-Hogg-Dubé syndrome, in which lesions are most frequently seen scattered on the scalp, face, ears, neck, or upper trunk.10 This condition is related to a folliculin (FLCN) gene germline mutation. Birt-Hogg-Dubé syndrome also may be associated with acrochordons, trichodiscomas, renal cancer, and lung cysts with or without spontaneous pneumothorax. Less frequently noted findings include oral papules, epidermal cysts, angiofibromas, lipomas/angiolipomas, parotid gland tumors, and thyroid neoplasms. Connective tissue nevi/collagenomas can appear clinically similar to fibrofolliculomas; true connective tissue nevi are reported less commonly in Birt-Hogg-Dubé syndrome.11 Histologically, a fibrofolliculoma manifests with epidermal strands originating from a hair follicle associated with prominent surrounding connective tissue (Figure 2).
Elastofibroma dorsi is a benign tumor of connective tissue that most commonly manifests clinically as a solitary subcutaneous mass on the back near the inferior angle of the scapula; it typically develops below the rhomboid major and latissimus dorsi muscles.12 The pathogenesis is uncertain, but some patients have reported a family history of the condition or a history of repetitive shoulder movement/trauma prior to onset; the mass may be asymptomatic or associated with pain and/or swelling. Those affected tend to be older than 50 years.13 Histologically, thickened and rounded to beaded elastic fibers are seen admixed with collagen (Figure 3).
Actinic (solar) elastosis frequently is encountered in many skin biopsies and is caused by chronic photodamage. More hypertrophic variants, such as papular or nodular solar elastosis, may clinically manifest similarly to WFP.14 Histologically, actinic elastosis manifests as a considerable increase in elastic tissue in the papillary and superficial reticular dermis (Figure 4).
THE DIAGNOSIS: White Fibrous Papulosis
Given the histopathology findings, location on a sun-exposed site, lack of any additional systemic signs or symptoms, and no family history of similar lesions to suggest an underlying genetic condition, a diagnosis of white fibrous papulosis (WFP) was made. White fibrous papulosis is a relatively rare cutaneous disorder that was first reported by Shimizu et al1 in 1985. It is characterized by numerous grouped, 2- to 3-mm, white to flesh-colored papules that in most cases are confined to the neck in middle-aged to elderly individuals; however, cases involving the upper trunk and axillae also have been reported.1-3 The etiology of this condition is unclear but is thought to be related to aging and chronic exposure to UV light. Although treatment is not required, various modalities including tretinoin, excision, and laser therapy have been trialed with varying success.2,4 Our patient elected not to proceed with treatment.
Histologically, WFP may manifest similarly to connective tissue nevi; the overall architecture is nonspecific with focally thickened collagen and often elastic fibers that may be normal to reduced and/or fragmented, as well as an overall decrease in superficial dermal elastic tissue.3,5 Therefore, the differential diagnosis may include connective tissue nevi and require clinical correlation to make a correct diagnosis.
Pseudoxanthoma elasticum (PXE) is an autosomalrecessive disorder most commonly related to mutations in the ATP binding cassette subfamily C member 6 (ABCC6) gene that tends to manifest clinically on the neck and flexural extremities.6 This disease affects elastic fibers, which may become calcified over time. Pseudoxanthoma elasticum is associated with ocular complications relating to the Bruch membrane of the retina and angioid streaks; choroidal neovascularization involving the damaged Bruch membrane and episodes of acute retinopathy may result in vision loss in later stages of the disease.7 Involvement of the elastic laminae of arteries can be associated with cardiovascular and cerebrovascular complications such as stroke, coronary artery disease, claudication, and aneurysms. Involvement of the gastrointestinal or genitourinary tracts also may occur and most commonly manifests with bleeding. Pathologic alterations in the elastic fibers of the lungs also have been reported in patients with PXE.8 Histologically, PXE exhibits increased abnormally clumped and fragmented elastic fibers in the superficial dermis, often with calcification (Figure 1). Pseudo-PXE related to D-penicillamine use often lacks calcification and has a bramble bush appearance.9
Fibrofolliculomas may manifest alone or in association with an underlying condition such as Birt-Hogg-Dubé syndrome, in which lesions are most frequently seen scattered on the scalp, face, ears, neck, or upper trunk.10 This condition is related to a folliculin (FLCN) gene germline mutation. Birt-Hogg-Dubé syndrome also may be associated with acrochordons, trichodiscomas, renal cancer, and lung cysts with or without spontaneous pneumothorax. Less frequently noted findings include oral papules, epidermal cysts, angiofibromas, lipomas/angiolipomas, parotid gland tumors, and thyroid neoplasms. Connective tissue nevi/collagenomas can appear clinically similar to fibrofolliculomas; true connective tissue nevi are reported less commonly in Birt-Hogg-Dubé syndrome.11 Histologically, a fibrofolliculoma manifests with epidermal strands originating from a hair follicle associated with prominent surrounding connective tissue (Figure 2).
Elastofibroma dorsi is a benign tumor of connective tissue that most commonly manifests clinically as a solitary subcutaneous mass on the back near the inferior angle of the scapula; it typically develops below the rhomboid major and latissimus dorsi muscles.12 The pathogenesis is uncertain, but some patients have reported a family history of the condition or a history of repetitive shoulder movement/trauma prior to onset; the mass may be asymptomatic or associated with pain and/or swelling. Those affected tend to be older than 50 years.13 Histologically, thickened and rounded to beaded elastic fibers are seen admixed with collagen (Figure 3).
Actinic (solar) elastosis frequently is encountered in many skin biopsies and is caused by chronic photodamage. More hypertrophic variants, such as papular or nodular solar elastosis, may clinically manifest similarly to WFP.14 Histologically, actinic elastosis manifests as a considerable increase in elastic tissue in the papillary and superficial reticular dermis (Figure 4).
- Shimizu H, Nishikawa T, Kimura S. White fibrous papulosis of the neck: review of our 16 cases. Nihon Hifuka Gakkai Zasshi. 1985;95:1077-1084.
- Teo W, Pang S. White fibrous papulosis of the chest and back. J Am Acad Dermatol. 2012;66:AB33.
- Dokic Y, Tschen J. White fibrous papulosis of the axillae and neck. Cureus. 2020;12:E7635.
- Lueangarun S, Panchaprateep R. White fibrous papulosis of the neck treated with fractionated 1550-nm erbium glass laser: a case report. J Lasers Med Sci. 2016;7:256-258.
- Rios-Gomez M, Ramos-Garibay JA, Perez-Santana ME, et al. White fibrous papulosis of the neck: a case report. Cureus. 2022;14:E25661.
- Váradi A, Szabó Z, Pomozi V, et al. ABCC6 as a target in pseudoxanthoma elasticum. Curr Drug Targets. 2011;12:671-682.
- Gliem M, Birtel J, Müller PL, et al. Acute retinopathy in pseudoxanthoma elasticum. JAMA Ophthalmol. 2019;137:1165-1173.
- Germain DP. Pseudoxanthoma elasticum. Orphanet J Rare Dis. 2017;12:85. doi:10.1186/s13023-017-0639-8
- Chisti MA, Binamer Y, Alfadley A, et al. D-penicillamine-induced pseudo-pseudoxanthoma elasticum and extensive elastosis perforans serpiginosa with excellent response to acitretin. Ann Saudi Med. 2019;39:56-60.
- Criscito MC, Mu EW, Meehan SA, et al. Dermoscopic features of a solitary fibrofolliculoma on the left cheek. J Am Acad Dermatol. 2017;76(2 suppl 1):S8-S9.
- Sattler EC, Steinlein OK. Birt-Hogg-Dubé syndrome. In: Adam MP, Everman DB, Mirzaa GM, et al, eds. GeneReviews® [Internet]. Updated January 30, 2020. Accessed February 23, 2023. https://www.ncbi.nlm.nih.gov/books/NBK1522
- Patnayak R, Jena A, Settipalli S, et al. Elastofibroma: an uncommon tumor revisited. J Cutan Aesthet Surg. 2016;9:34-37. doi:10.4103/0974- 2077.178543
- Chandrasekar CR, Grimer RJ, Carter SR, et al. Elastofibroma dorsi: an uncommon benign pseudotumour. Sarcoma. 2008;2008:756565. doi:10.1155/2008/756565
- Kwittken J. Papular elastosis. Cutis. 2000;66:81-83.
- Shimizu H, Nishikawa T, Kimura S. White fibrous papulosis of the neck: review of our 16 cases. Nihon Hifuka Gakkai Zasshi. 1985;95:1077-1084.
- Teo W, Pang S. White fibrous papulosis of the chest and back. J Am Acad Dermatol. 2012;66:AB33.
- Dokic Y, Tschen J. White fibrous papulosis of the axillae and neck. Cureus. 2020;12:E7635.
- Lueangarun S, Panchaprateep R. White fibrous papulosis of the neck treated with fractionated 1550-nm erbium glass laser: a case report. J Lasers Med Sci. 2016;7:256-258.
- Rios-Gomez M, Ramos-Garibay JA, Perez-Santana ME, et al. White fibrous papulosis of the neck: a case report. Cureus. 2022;14:E25661.
- Váradi A, Szabó Z, Pomozi V, et al. ABCC6 as a target in pseudoxanthoma elasticum. Curr Drug Targets. 2011;12:671-682.
- Gliem M, Birtel J, Müller PL, et al. Acute retinopathy in pseudoxanthoma elasticum. JAMA Ophthalmol. 2019;137:1165-1173.
- Germain DP. Pseudoxanthoma elasticum. Orphanet J Rare Dis. 2017;12:85. doi:10.1186/s13023-017-0639-8
- Chisti MA, Binamer Y, Alfadley A, et al. D-penicillamine-induced pseudo-pseudoxanthoma elasticum and extensive elastosis perforans serpiginosa with excellent response to acitretin. Ann Saudi Med. 2019;39:56-60.
- Criscito MC, Mu EW, Meehan SA, et al. Dermoscopic features of a solitary fibrofolliculoma on the left cheek. J Am Acad Dermatol. 2017;76(2 suppl 1):S8-S9.
- Sattler EC, Steinlein OK. Birt-Hogg-Dubé syndrome. In: Adam MP, Everman DB, Mirzaa GM, et al, eds. GeneReviews® [Internet]. Updated January 30, 2020. Accessed February 23, 2023. https://www.ncbi.nlm.nih.gov/books/NBK1522
- Patnayak R, Jena A, Settipalli S, et al. Elastofibroma: an uncommon tumor revisited. J Cutan Aesthet Surg. 2016;9:34-37. doi:10.4103/0974- 2077.178543
- Chandrasekar CR, Grimer RJ, Carter SR, et al. Elastofibroma dorsi: an uncommon benign pseudotumour. Sarcoma. 2008;2008:756565. doi:10.1155/2008/756565
- Kwittken J. Papular elastosis. Cutis. 2000;66:81-83.
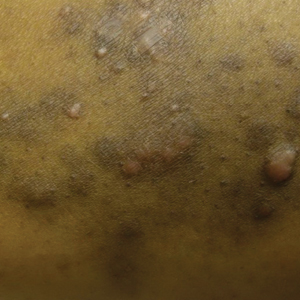
A 70-year-old woman with a history of osteoporosis and breast cancer presented for evaluation of asymptomatic, 2- to 3-mm, white to flesh-colored papules concentrated on the inferior occipital scalp and posterior neck (inset) for at least several months. She had no additional systemic signs or symptoms, and there was no family history of similar skin findings. A punch biopsy was performed.
Antineutrophil Cytoplasmic Antibody Vasculitis Induced by Hydralazine
To the Editor:
Hydralazine-induced antineutrophil cytoplasmic antibody vasculitis (HIAV) is a rare side effect that may develop in patients treated with hydralazine. Without early recognition and hydralazine cessation, patients often develop acute renal failure and pulmonary hemorrhage that may result in death. We present a case of HIAV.
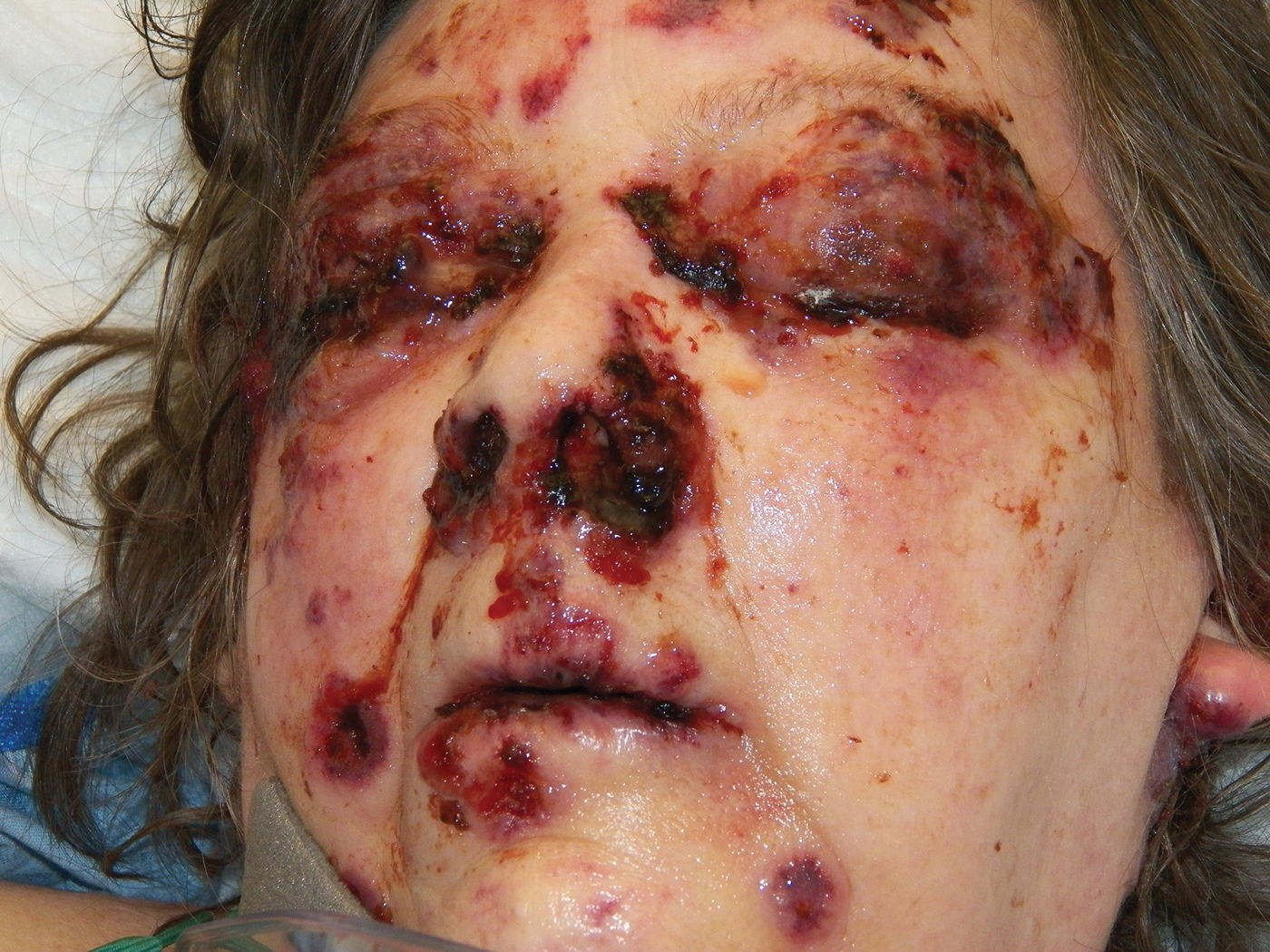
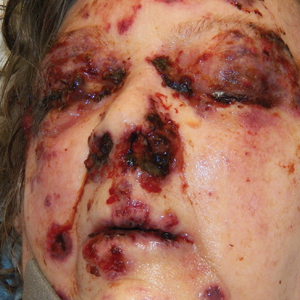
A 67-year-old woman presented with progressive, tense, hemorrhagic, and necrotic bullae on both sides of the face and neck as well as the extremities of 2 weeks’ duration. She had a history of hypertension and a thyroid nodule after unilateral thyroid lobectomy. A review of symptoms was positive for worsening dyspnea and progressive generalized weakness. Noteworthy medications included amlodipine, metoprolol, levothyroxine, and oral hydralazine 75 mg 3 times daily for 13 months.
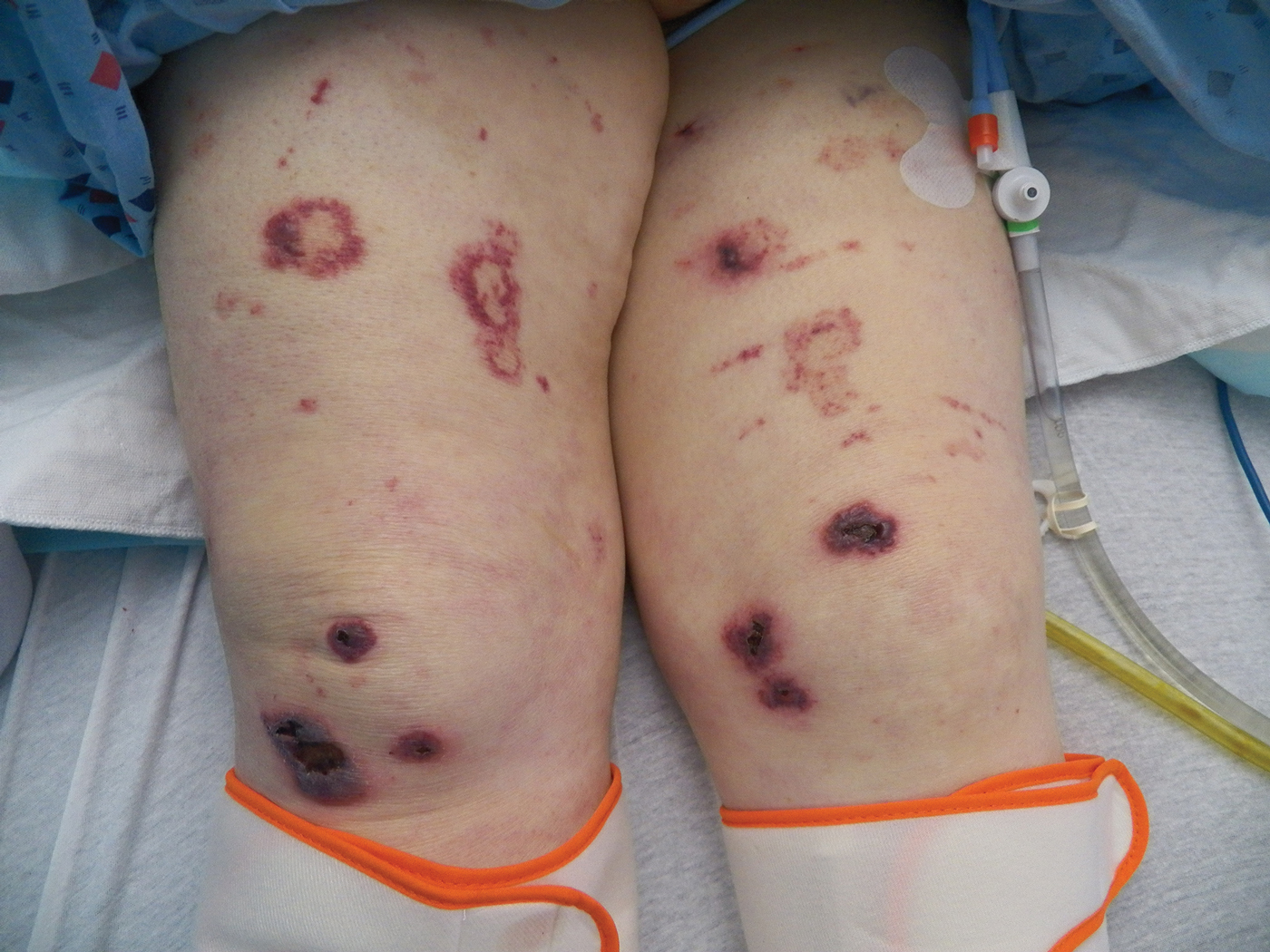
Bullae first appeared on the patient’s scalp and quickly progressed with a cephalocaudal pattern with a propensity for the eyes, nostrils, and labial mucosa (Figure 1). The tongue was covered by an eschar, and she had diffuse periorbital edema. Additionally, concentric purpuric patches were noted on the thighs and lower legs (Figure 2).
Pertinent laboratory findings included a positive antinuclear antibody titer of 1:320 and perinuclear antineutrophil cytoplasmic antibody (ANCA) titer of 1:160, along with an elevated serum creatinine level (2.31 mg/dL [reference range, 0.6–1.2 mg/dL]). Bilateral perihilar infiltrates with bilateral pleural effusions were noted on a chest radiograph.
While hospitalized, she developed pulmonary hemorrhages and a progressive decline in respiratory status. She subsequently was admitted to the medical intensive care unit. Aggressive support was administered, and several skin biopsy specimens were obtained along with an endobronchial biopsy of the right middle lobe.
Skin histopathology revealed a necrotic vasculitis (Figure 3). Direct immunofluorescence was not performed. Lung histopathology showed fragments of bronchial tissue with acute and chronic inflammation, focal necrosis, granulation tissue formation, edema, and squamous metaplasia. Together with the clinical history, these findings were consistent with HIAV.

Hydralazine was immediately discontinued, and the patient was started on 65 mg daily of intravenous methylprednisolone; methylprednisolone was later changed to oral prednisone 30 mg daily. Due to multiple organ involvement—lung and kidney—intravenous rituximab 375 mg/m2 every week for 4 weeks, per lymphoma protocol, was started. Within 2 weeks of beginning therapy, her renal function and respiratory status improved, and by week 4, the skin lesions had completely resolved. Although initially she did well on immunosuppressive therapy with resolution of all symptoms, the patient contracted Clostridium difficile–induced systemic inflammatory response syndrome after 5 weeks of therapy and died.
Hydralazine was first introduced in 1951 for adjunctive hypertension therapy due to its vasodilation effects.1-3 Since its introduction, it has been implicated in 2 important disease processes: HIAV and hydralazine-induced lupus.
Hydralazine-induced ANCA vasculitis was first documented in 1980; by 2011, multiple cases had been reported.1-7 Hydralazine-induced ANCA vasculitis has occurred in patients aged 11 to 79 years taking 50 to 300 mg daily. Symptom onset varies from 6 months to 14 years, with a mean exposure duration of 4.7 years and mean daily dose of 142 mg.1-7
Clinical manifestations range from less specific, such as fever, malaise, arthralgia, myalgia, and weight loss, to single tissue or organ involvement that may be fatal. The most frequent clinical features include kidney involvement (81%), cutaneous vasculitis (25%), arthralgia (24%), and pleuropulmonary involvement (19%). Cutaneous manifestations include but are not limited to palpable lower extremity purpura; morbilliform eruptions; and hemorrhagic blisters on the lower legs, arms, trunk, nasal septum, and uvula.1-4,8
The most commonly affected organ is the kidney, which commonly presents as hematuria, proteinuria, and elevated serum creatinine level. Histopathologically, patients most likely will have necrotizing and crescentic glomerulonephritis that is pauci-immune by immunofluorescence.7,9 The lungs are the next most commonly affected organ, with a classic presentation of cough, dyspnea, and hemoptysis in the setting of intra-alveolar hemorrhage.6,8 When both the kidneys and lungs are involved, the patient is said to have pulmonary-renal syndrome that is characterized by lung infiltrates or nodules with or without hemorrhage, hemoptysis, and pleuritis in the setting of glomerulonephritis.1,6
Clear data on incidence and prevalence of HIAV does not exist due to the rarity of the disease and the lack of prospective studies. To identify a clear incidence and prevalence, prospective longitudinal studies with larger cohorts along with better recognition and diagnosis are needed.2,8,10 A few predisposing risk factors have been identified, including older age, a cumulative dose of 100 g at the time of presentation, female sex, a history of thyroid disease, HLA-DR4 genotypes, slow hepatic acetylation, and the null gene for C4.1,3,5,9-11 Our patient was an older woman with a history of thyroid disease who had been taking oral hydralazine 75 mg 3 times daily for 13 months. During this 13-month duration, she had no dose adjustments.
Currently, the pathomechanism for HIAV is unclear and may be multifactorial. There are 4 main theories2,8-10,12,13:
1. Hydralazine and its metabolites accumulate inside neutrophils, then subsequently bind and alter the configuration of myeloperoxidase (MPO). This alteration leads to spreading of the autoimmune response to other autoantigens, making neutrophil proteins (eg, elastase, lactoferrin, nuclear antigens) immunogenic.
2. Hydralazine binds MPO in neutrophils, creating cytotoxic products that induce neutrophil apoptosis. Neutrophil apoptosis without priming then results in ANCA antigen presence on the neutrophil cell membrane and the formation of MPO-ANCA. Myeloperoxidase-ANCA then binds to these membrane-bound antigens that cause self-perpetuating, constitutive activation through cross-linking with proteinase 3 or MPO and Fcγ receptors.
3. Activated neutrophils in the presence of hydrogen peroxidase release MPO that converts hydralazine into a cytotoxic product that is immunogenic for T cells that activate ANCA-producing B cells.
4. Histone H3 trimethyl Lys27 (H3K27me3) levels are perturbed in HIAV, which leads to aberrant gene silencing of proteinase 3 and MPO.In contrast, the demethylase Jumonji domain-containing protein 3 for the H3K27me3 histone is increased in patients without HIAV. Based on this data and the data showing a role for hydralazine in reversing epigenetic silencing of tumor suppressor genes in cancer cells,13 it has been proposed that hydralazine may reverse epigenetic silencing of proteinase 3 and MPO.
Diagnosing HIAV is still difficult because physicians do not recognize the drug as the etiologic agent, there is extensive variability in duration between starting the drug and onset of symptoms, and there often is a failure to order the appropriate laboratory and invasive tests needed for evaluation and diagnosis.3,5,8,10,12 Despite these difficulties, a set of criteria and practices for diagnosis are delineated in Table 1, with the key diagnostic feature being resolution with hydralazine cessation.1,5,7,8,12
A comprehensive drug history from at least 6 months prior to presentation is essential. Biopsies also are strongly encouraged to confirm the presence of vasculitis and to determine its severity.8,12 If renal biopsies are performed, they typically show scant IgG, IgM, and C3 deposition that is characteristic of ANCA-positive pauci-immune glomerulonephritis. Compared to hydralazine-induced lupus, renal involvement in the setting of HIAV has a relative lack of immunoglobulin and complement deposition with histopathology and immunostaining.14
Laboratory test results including serum MPO-ANCA (perinuclear ANCA) with coexisting elastase and/or lactoferrin autoantibodies is characteristic of HIAV. Antinuclear antibody, antihistone, anti–double-stranded DNA, and antiphospholipid antibodies along with low complement levels also may be present.2,4,9,10,13,15 It is recommended that ANCA assays combine indirect immunofluorescence with antigen-specific enzyme-linked immunosorbent assay.8 With respect to its idiopathic counterpart, patients may only present with MPO-ANCA, while other aforementioned antibodies (eg, antihistone, anti–double-stranded DNA) are rarely found or are entirely absent.2,9 Patients with HIAV often have higher titers of MPO-ANCA.9,15 In hydralazine-induced lupus, patients rarely have MPO-ANCA.
When a diagnosis of HIAV is made, it cannot be confirmed until hydralazine is discontinued and the patient’s symptoms resolve. Therefore, it is both diagnostic and therapeutic to discontinue hydralazine when HIAV is suspected. If recognized when the patient is only presenting with nonspecific symptoms, simple hydralazine cessation may be all that is needed; however, because recognition and diagnosis of HIAV is difficult, most patients present when the disease is severe and has progressed to organ involvement.8-10
Treatment recommendations are highlighted in Table 2.8,9,12 Glucocorticoid therapy is believed to work by preventing T-cell and B-cell maturation needed to produce MPO-ANCA. Rituximab, on the other hand, is suspected to act by clearing the peripheral blood of MPO-ANCA B cells.12,16 Of note, patients with HIAV are different from their idiopathic counterparts because they usually need shorter courses of immunosuppressive therapy, long-term maintenance usually is unnecessary, and their prognosis generally is good if the offending agent is withdrawn.7-9,12 Once the appropriate therapy is instituted, vasculitic manifestations are expected to resolve 10 days to 8 months after hydralazine cessation; however, a response often is seen within 1 to 4 weeks after initiation of systemic treatment.4,8 Serum ANCA should be monitored, and there should be surveillance for the emergence of a chronic underlying vasculitis.8,12
Our patient highlights the importance of identifying individuals at risk for HIAV. We seek to increase recognition of this entity, as it is not commonly seen in a dermatologic setting and is associated with high morbidity and mortality, as seen in our patient.
- Yokogawa N, Vivino FB. Hydralazine-induced autoimmune disease: comparison to idiopathic lupus and ANCA-positive vasculitis. Mod Rheumatol. 2009;19:338-347.
- Agarwal G, Sultan G, Werner SL, et al. Hydralazine induces myeloperoxidase and proteinase 3 anti-neutrophil cytoplasmic antibody vasculitis and leads to pulmonary renal syndrome. Case Rep Nephrol. 2014;2014:868590.
- Keasberry J, Frazier J, Isbel NM, et al. Hydralazine-induced anti-neutrophilic cytoplasmic antibody-positive renal vasculitis presenting with a vasculitic syndrome, acute nephritis and a puzzling skin rash: a case report. J Med Case Rep. 2013;7:20.
- ten Holder SM, Joy MS, Falk RJ. Cutaneous and systemic manifestations of drug-induced vasculitis. Ann Pharmacother. 2002;36:130-147.
- Namas R, Rubin B, Adwar W, et al. A challenging twist in pulmonary renal syndrome. Case Rep Rheumatol. 2014;2014:516362.
- Dobre M, Wish J, Negrea L. Hydralazine-induced ANCA-positive pauci-immune glomerulonephritis. Ren Fail. 2009;31:745-748.
- Hogan JJ, Markowitz GS, Radhakrishnan J. Drug-induced glomerular disease: immune-mediated injury. Clin J Am Soc Nephrol. 2015;10:1300-1310.
- Radic M, Martinovic Kaliterna D, Radic J. Drug-induced vasculitis: a clinical and pathological review. Neth J Med. 2012;70:12-17.
- Babar F, Posner JN, Obah EA. Hydralazine-induced pauci-immune glomerulonephritis: intriguing case series misleading diagnoses. J Community Hosp Intern Med Perspect. 2016;6:30632.
- Marina VP, Malhotra D, Kaw D. Hydralazine-induced ANCA vasculitis with pulmonary renal syndrome: a rare clinical presentation. Int Urol Nephrol. 2012;44:1907-1909.
- Magro CM. Associated ANCA positive vasculitis. The Dermatologist. 2015;23(7). http://www.the-dermatologist.com/content/associated-anca-positive-vasculitis. Accessed January 30, 2020.
- Gao Y, Zhao MH. Review article: Drug-induced anti-neutrophil cytoplasmic antibody-associated vasculitis. Nephrology (Carlton). 2009;14:33-41.
- Grau RG. Drug-induced vasculitis: new insights and a changing lineup of suspects. Curr Rheumatol Rep. 2015;17:71.
- Sangala N, Lee RW, Horsfield C, et al. Combined ANCA-associated vasculitis and lupus syndrome following prolonged use of hydralazine: a timely reminder of an old foe. Int Urol Nephrol. 2010;42:503-506.
- Choi HK, Merkel PA, Walker AM, et al. Drug-associated antineutrophil cytoplasmic antibody-positive vasculitis: prevalence among patients with high titers of antimyeloperoxidase antibodies. Arthritis Rheum. 2000;43:405-413.
- Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol. 2011;335:2-13.
To the Editor:
Hydralazine-induced antineutrophil cytoplasmic antibody vasculitis (HIAV) is a rare side effect that may develop in patients treated with hydralazine. Without early recognition and hydralazine cessation, patients often develop acute renal failure and pulmonary hemorrhage that may result in death. We present a case of HIAV.
A 67-year-old woman presented with progressive, tense, hemorrhagic, and necrotic bullae on both sides of the face and neck as well as the extremities of 2 weeks’ duration. She had a history of hypertension and a thyroid nodule after unilateral thyroid lobectomy. A review of symptoms was positive for worsening dyspnea and progressive generalized weakness. Noteworthy medications included amlodipine, metoprolol, levothyroxine, and oral hydralazine 75 mg 3 times daily for 13 months.
Bullae first appeared on the patient’s scalp and quickly progressed with a cephalocaudal pattern with a propensity for the eyes, nostrils, and labial mucosa (Figure 1). The tongue was covered by an eschar, and she had diffuse periorbital edema. Additionally, concentric purpuric patches were noted on the thighs and lower legs (Figure 2).
Pertinent laboratory findings included a positive antinuclear antibody titer of 1:320 and perinuclear antineutrophil cytoplasmic antibody (ANCA) titer of 1:160, along with an elevated serum creatinine level (2.31 mg/dL [reference range, 0.6–1.2 mg/dL]). Bilateral perihilar infiltrates with bilateral pleural effusions were noted on a chest radiograph.
While hospitalized, she developed pulmonary hemorrhages and a progressive decline in respiratory status. She subsequently was admitted to the medical intensive care unit. Aggressive support was administered, and several skin biopsy specimens were obtained along with an endobronchial biopsy of the right middle lobe.
Skin histopathology revealed a necrotic vasculitis (Figure 3). Direct immunofluorescence was not performed. Lung histopathology showed fragments of bronchial tissue with acute and chronic inflammation, focal necrosis, granulation tissue formation, edema, and squamous metaplasia. Together with the clinical history, these findings were consistent with HIAV.
Hydralazine was immediately discontinued, and the patient was started on 65 mg daily of intravenous methylprednisolone; methylprednisolone was later changed to oral prednisone 30 mg daily. Due to multiple organ involvement—lung and kidney—intravenous rituximab 375 mg/m2 every week for 4 weeks, per lymphoma protocol, was started. Within 2 weeks of beginning therapy, her renal function and respiratory status improved, and by week 4, the skin lesions had completely resolved. Although initially she did well on immunosuppressive therapy with resolution of all symptoms, the patient contracted Clostridium difficile–induced systemic inflammatory response syndrome after 5 weeks of therapy and died.
Hydralazine was first introduced in 1951 for adjunctive hypertension therapy due to its vasodilation effects.1-3 Since its introduction, it has been implicated in 2 important disease processes: HIAV and hydralazine-induced lupus.
Hydralazine-induced ANCA vasculitis was first documented in 1980; by 2011, multiple cases had been reported.1-7 Hydralazine-induced ANCA vasculitis has occurred in patients aged 11 to 79 years taking 50 to 300 mg daily. Symptom onset varies from 6 months to 14 years, with a mean exposure duration of 4.7 years and mean daily dose of 142 mg.1-7
Clinical manifestations range from less specific, such as fever, malaise, arthralgia, myalgia, and weight loss, to single tissue or organ involvement that may be fatal. The most frequent clinical features include kidney involvement (81%), cutaneous vasculitis (25%), arthralgia (24%), and pleuropulmonary involvement (19%). Cutaneous manifestations include but are not limited to palpable lower extremity purpura; morbilliform eruptions; and hemorrhagic blisters on the lower legs, arms, trunk, nasal septum, and uvula.1-4,8
The most commonly affected organ is the kidney, which commonly presents as hematuria, proteinuria, and elevated serum creatinine level. Histopathologically, patients most likely will have necrotizing and crescentic glomerulonephritis that is pauci-immune by immunofluorescence.7,9 The lungs are the next most commonly affected organ, with a classic presentation of cough, dyspnea, and hemoptysis in the setting of intra-alveolar hemorrhage.6,8 When both the kidneys and lungs are involved, the patient is said to have pulmonary-renal syndrome that is characterized by lung infiltrates or nodules with or without hemorrhage, hemoptysis, and pleuritis in the setting of glomerulonephritis.1,6
Clear data on incidence and prevalence of HIAV does not exist due to the rarity of the disease and the lack of prospective studies. To identify a clear incidence and prevalence, prospective longitudinal studies with larger cohorts along with better recognition and diagnosis are needed.2,8,10 A few predisposing risk factors have been identified, including older age, a cumulative dose of 100 g at the time of presentation, female sex, a history of thyroid disease, HLA-DR4 genotypes, slow hepatic acetylation, and the null gene for C4.1,3,5,9-11 Our patient was an older woman with a history of thyroid disease who had been taking oral hydralazine 75 mg 3 times daily for 13 months. During this 13-month duration, she had no dose adjustments.
Currently, the pathomechanism for HIAV is unclear and may be multifactorial. There are 4 main theories2,8-10,12,13:
1. Hydralazine and its metabolites accumulate inside neutrophils, then subsequently bind and alter the configuration of myeloperoxidase (MPO). This alteration leads to spreading of the autoimmune response to other autoantigens, making neutrophil proteins (eg, elastase, lactoferrin, nuclear antigens) immunogenic.
2. Hydralazine binds MPO in neutrophils, creating cytotoxic products that induce neutrophil apoptosis. Neutrophil apoptosis without priming then results in ANCA antigen presence on the neutrophil cell membrane and the formation of MPO-ANCA. Myeloperoxidase-ANCA then binds to these membrane-bound antigens that cause self-perpetuating, constitutive activation through cross-linking with proteinase 3 or MPO and Fcγ receptors.
3. Activated neutrophils in the presence of hydrogen peroxidase release MPO that converts hydralazine into a cytotoxic product that is immunogenic for T cells that activate ANCA-producing B cells.
4. Histone H3 trimethyl Lys27 (H3K27me3) levels are perturbed in HIAV, which leads to aberrant gene silencing of proteinase 3 and MPO.In contrast, the demethylase Jumonji domain-containing protein 3 for the H3K27me3 histone is increased in patients without HIAV. Based on this data and the data showing a role for hydralazine in reversing epigenetic silencing of tumor suppressor genes in cancer cells,13 it has been proposed that hydralazine may reverse epigenetic silencing of proteinase 3 and MPO.
Diagnosing HIAV is still difficult because physicians do not recognize the drug as the etiologic agent, there is extensive variability in duration between starting the drug and onset of symptoms, and there often is a failure to order the appropriate laboratory and invasive tests needed for evaluation and diagnosis.3,5,8,10,12 Despite these difficulties, a set of criteria and practices for diagnosis are delineated in Table 1, with the key diagnostic feature being resolution with hydralazine cessation.1,5,7,8,12
A comprehensive drug history from at least 6 months prior to presentation is essential. Biopsies also are strongly encouraged to confirm the presence of vasculitis and to determine its severity.8,12 If renal biopsies are performed, they typically show scant IgG, IgM, and C3 deposition that is characteristic of ANCA-positive pauci-immune glomerulonephritis. Compared to hydralazine-induced lupus, renal involvement in the setting of HIAV has a relative lack of immunoglobulin and complement deposition with histopathology and immunostaining.14
Laboratory test results including serum MPO-ANCA (perinuclear ANCA) with coexisting elastase and/or lactoferrin autoantibodies is characteristic of HIAV. Antinuclear antibody, antihistone, anti–double-stranded DNA, and antiphospholipid antibodies along with low complement levels also may be present.2,4,9,10,13,15 It is recommended that ANCA assays combine indirect immunofluorescence with antigen-specific enzyme-linked immunosorbent assay.8 With respect to its idiopathic counterpart, patients may only present with MPO-ANCA, while other aforementioned antibodies (eg, antihistone, anti–double-stranded DNA) are rarely found or are entirely absent.2,9 Patients with HIAV often have higher titers of MPO-ANCA.9,15 In hydralazine-induced lupus, patients rarely have MPO-ANCA.
When a diagnosis of HIAV is made, it cannot be confirmed until hydralazine is discontinued and the patient’s symptoms resolve. Therefore, it is both diagnostic and therapeutic to discontinue hydralazine when HIAV is suspected. If recognized when the patient is only presenting with nonspecific symptoms, simple hydralazine cessation may be all that is needed; however, because recognition and diagnosis of HIAV is difficult, most patients present when the disease is severe and has progressed to organ involvement.8-10
Treatment recommendations are highlighted in Table 2.8,9,12 Glucocorticoid therapy is believed to work by preventing T-cell and B-cell maturation needed to produce MPO-ANCA. Rituximab, on the other hand, is suspected to act by clearing the peripheral blood of MPO-ANCA B cells.12,16 Of note, patients with HIAV are different from their idiopathic counterparts because they usually need shorter courses of immunosuppressive therapy, long-term maintenance usually is unnecessary, and their prognosis generally is good if the offending agent is withdrawn.7-9,12 Once the appropriate therapy is instituted, vasculitic manifestations are expected to resolve 10 days to 8 months after hydralazine cessation; however, a response often is seen within 1 to 4 weeks after initiation of systemic treatment.4,8 Serum ANCA should be monitored, and there should be surveillance for the emergence of a chronic underlying vasculitis.8,12
Our patient highlights the importance of identifying individuals at risk for HIAV. We seek to increase recognition of this entity, as it is not commonly seen in a dermatologic setting and is associated with high morbidity and mortality, as seen in our patient.
To the Editor:
Hydralazine-induced antineutrophil cytoplasmic antibody vasculitis (HIAV) is a rare side effect that may develop in patients treated with hydralazine. Without early recognition and hydralazine cessation, patients often develop acute renal failure and pulmonary hemorrhage that may result in death. We present a case of HIAV.
A 67-year-old woman presented with progressive, tense, hemorrhagic, and necrotic bullae on both sides of the face and neck as well as the extremities of 2 weeks’ duration. She had a history of hypertension and a thyroid nodule after unilateral thyroid lobectomy. A review of symptoms was positive for worsening dyspnea and progressive generalized weakness. Noteworthy medications included amlodipine, metoprolol, levothyroxine, and oral hydralazine 75 mg 3 times daily for 13 months.
Bullae first appeared on the patient’s scalp and quickly progressed with a cephalocaudal pattern with a propensity for the eyes, nostrils, and labial mucosa (Figure 1). The tongue was covered by an eschar, and she had diffuse periorbital edema. Additionally, concentric purpuric patches were noted on the thighs and lower legs (Figure 2).
Pertinent laboratory findings included a positive antinuclear antibody titer of 1:320 and perinuclear antineutrophil cytoplasmic antibody (ANCA) titer of 1:160, along with an elevated serum creatinine level (2.31 mg/dL [reference range, 0.6–1.2 mg/dL]). Bilateral perihilar infiltrates with bilateral pleural effusions were noted on a chest radiograph.
While hospitalized, she developed pulmonary hemorrhages and a progressive decline in respiratory status. She subsequently was admitted to the medical intensive care unit. Aggressive support was administered, and several skin biopsy specimens were obtained along with an endobronchial biopsy of the right middle lobe.
Skin histopathology revealed a necrotic vasculitis (Figure 3). Direct immunofluorescence was not performed. Lung histopathology showed fragments of bronchial tissue with acute and chronic inflammation, focal necrosis, granulation tissue formation, edema, and squamous metaplasia. Together with the clinical history, these findings were consistent with HIAV.
Hydralazine was immediately discontinued, and the patient was started on 65 mg daily of intravenous methylprednisolone; methylprednisolone was later changed to oral prednisone 30 mg daily. Due to multiple organ involvement—lung and kidney—intravenous rituximab 375 mg/m2 every week for 4 weeks, per lymphoma protocol, was started. Within 2 weeks of beginning therapy, her renal function and respiratory status improved, and by week 4, the skin lesions had completely resolved. Although initially she did well on immunosuppressive therapy with resolution of all symptoms, the patient contracted Clostridium difficile–induced systemic inflammatory response syndrome after 5 weeks of therapy and died.
Hydralazine was first introduced in 1951 for adjunctive hypertension therapy due to its vasodilation effects.1-3 Since its introduction, it has been implicated in 2 important disease processes: HIAV and hydralazine-induced lupus.
Hydralazine-induced ANCA vasculitis was first documented in 1980; by 2011, multiple cases had been reported.1-7 Hydralazine-induced ANCA vasculitis has occurred in patients aged 11 to 79 years taking 50 to 300 mg daily. Symptom onset varies from 6 months to 14 years, with a mean exposure duration of 4.7 years and mean daily dose of 142 mg.1-7
Clinical manifestations range from less specific, such as fever, malaise, arthralgia, myalgia, and weight loss, to single tissue or organ involvement that may be fatal. The most frequent clinical features include kidney involvement (81%), cutaneous vasculitis (25%), arthralgia (24%), and pleuropulmonary involvement (19%). Cutaneous manifestations include but are not limited to palpable lower extremity purpura; morbilliform eruptions; and hemorrhagic blisters on the lower legs, arms, trunk, nasal septum, and uvula.1-4,8
The most commonly affected organ is the kidney, which commonly presents as hematuria, proteinuria, and elevated serum creatinine level. Histopathologically, patients most likely will have necrotizing and crescentic glomerulonephritis that is pauci-immune by immunofluorescence.7,9 The lungs are the next most commonly affected organ, with a classic presentation of cough, dyspnea, and hemoptysis in the setting of intra-alveolar hemorrhage.6,8 When both the kidneys and lungs are involved, the patient is said to have pulmonary-renal syndrome that is characterized by lung infiltrates or nodules with or without hemorrhage, hemoptysis, and pleuritis in the setting of glomerulonephritis.1,6
Clear data on incidence and prevalence of HIAV does not exist due to the rarity of the disease and the lack of prospective studies. To identify a clear incidence and prevalence, prospective longitudinal studies with larger cohorts along with better recognition and diagnosis are needed.2,8,10 A few predisposing risk factors have been identified, including older age, a cumulative dose of 100 g at the time of presentation, female sex, a history of thyroid disease, HLA-DR4 genotypes, slow hepatic acetylation, and the null gene for C4.1,3,5,9-11 Our patient was an older woman with a history of thyroid disease who had been taking oral hydralazine 75 mg 3 times daily for 13 months. During this 13-month duration, she had no dose adjustments.
Currently, the pathomechanism for HIAV is unclear and may be multifactorial. There are 4 main theories2,8-10,12,13:
1. Hydralazine and its metabolites accumulate inside neutrophils, then subsequently bind and alter the configuration of myeloperoxidase (MPO). This alteration leads to spreading of the autoimmune response to other autoantigens, making neutrophil proteins (eg, elastase, lactoferrin, nuclear antigens) immunogenic.
2. Hydralazine binds MPO in neutrophils, creating cytotoxic products that induce neutrophil apoptosis. Neutrophil apoptosis without priming then results in ANCA antigen presence on the neutrophil cell membrane and the formation of MPO-ANCA. Myeloperoxidase-ANCA then binds to these membrane-bound antigens that cause self-perpetuating, constitutive activation through cross-linking with proteinase 3 or MPO and Fcγ receptors.
3. Activated neutrophils in the presence of hydrogen peroxidase release MPO that converts hydralazine into a cytotoxic product that is immunogenic for T cells that activate ANCA-producing B cells.
4. Histone H3 trimethyl Lys27 (H3K27me3) levels are perturbed in HIAV, which leads to aberrant gene silencing of proteinase 3 and MPO.In contrast, the demethylase Jumonji domain-containing protein 3 for the H3K27me3 histone is increased in patients without HIAV. Based on this data and the data showing a role for hydralazine in reversing epigenetic silencing of tumor suppressor genes in cancer cells,13 it has been proposed that hydralazine may reverse epigenetic silencing of proteinase 3 and MPO.
Diagnosing HIAV is still difficult because physicians do not recognize the drug as the etiologic agent, there is extensive variability in duration between starting the drug and onset of symptoms, and there often is a failure to order the appropriate laboratory and invasive tests needed for evaluation and diagnosis.3,5,8,10,12 Despite these difficulties, a set of criteria and practices for diagnosis are delineated in Table 1, with the key diagnostic feature being resolution with hydralazine cessation.1,5,7,8,12
A comprehensive drug history from at least 6 months prior to presentation is essential. Biopsies also are strongly encouraged to confirm the presence of vasculitis and to determine its severity.8,12 If renal biopsies are performed, they typically show scant IgG, IgM, and C3 deposition that is characteristic of ANCA-positive pauci-immune glomerulonephritis. Compared to hydralazine-induced lupus, renal involvement in the setting of HIAV has a relative lack of immunoglobulin and complement deposition with histopathology and immunostaining.14
Laboratory test results including serum MPO-ANCA (perinuclear ANCA) with coexisting elastase and/or lactoferrin autoantibodies is characteristic of HIAV. Antinuclear antibody, antihistone, anti–double-stranded DNA, and antiphospholipid antibodies along with low complement levels also may be present.2,4,9,10,13,15 It is recommended that ANCA assays combine indirect immunofluorescence with antigen-specific enzyme-linked immunosorbent assay.8 With respect to its idiopathic counterpart, patients may only present with MPO-ANCA, while other aforementioned antibodies (eg, antihistone, anti–double-stranded DNA) are rarely found or are entirely absent.2,9 Patients with HIAV often have higher titers of MPO-ANCA.9,15 In hydralazine-induced lupus, patients rarely have MPO-ANCA.
When a diagnosis of HIAV is made, it cannot be confirmed until hydralazine is discontinued and the patient’s symptoms resolve. Therefore, it is both diagnostic and therapeutic to discontinue hydralazine when HIAV is suspected. If recognized when the patient is only presenting with nonspecific symptoms, simple hydralazine cessation may be all that is needed; however, because recognition and diagnosis of HIAV is difficult, most patients present when the disease is severe and has progressed to organ involvement.8-10
Treatment recommendations are highlighted in Table 2.8,9,12 Glucocorticoid therapy is believed to work by preventing T-cell and B-cell maturation needed to produce MPO-ANCA. Rituximab, on the other hand, is suspected to act by clearing the peripheral blood of MPO-ANCA B cells.12,16 Of note, patients with HIAV are different from their idiopathic counterparts because they usually need shorter courses of immunosuppressive therapy, long-term maintenance usually is unnecessary, and their prognosis generally is good if the offending agent is withdrawn.7-9,12 Once the appropriate therapy is instituted, vasculitic manifestations are expected to resolve 10 days to 8 months after hydralazine cessation; however, a response often is seen within 1 to 4 weeks after initiation of systemic treatment.4,8 Serum ANCA should be monitored, and there should be surveillance for the emergence of a chronic underlying vasculitis.8,12
Our patient highlights the importance of identifying individuals at risk for HIAV. We seek to increase recognition of this entity, as it is not commonly seen in a dermatologic setting and is associated with high morbidity and mortality, as seen in our patient.
- Yokogawa N, Vivino FB. Hydralazine-induced autoimmune disease: comparison to idiopathic lupus and ANCA-positive vasculitis. Mod Rheumatol. 2009;19:338-347.
- Agarwal G, Sultan G, Werner SL, et al. Hydralazine induces myeloperoxidase and proteinase 3 anti-neutrophil cytoplasmic antibody vasculitis and leads to pulmonary renal syndrome. Case Rep Nephrol. 2014;2014:868590.
- Keasberry J, Frazier J, Isbel NM, et al. Hydralazine-induced anti-neutrophilic cytoplasmic antibody-positive renal vasculitis presenting with a vasculitic syndrome, acute nephritis and a puzzling skin rash: a case report. J Med Case Rep. 2013;7:20.
- ten Holder SM, Joy MS, Falk RJ. Cutaneous and systemic manifestations of drug-induced vasculitis. Ann Pharmacother. 2002;36:130-147.
- Namas R, Rubin B, Adwar W, et al. A challenging twist in pulmonary renal syndrome. Case Rep Rheumatol. 2014;2014:516362.
- Dobre M, Wish J, Negrea L. Hydralazine-induced ANCA-positive pauci-immune glomerulonephritis. Ren Fail. 2009;31:745-748.
- Hogan JJ, Markowitz GS, Radhakrishnan J. Drug-induced glomerular disease: immune-mediated injury. Clin J Am Soc Nephrol. 2015;10:1300-1310.
- Radic M, Martinovic Kaliterna D, Radic J. Drug-induced vasculitis: a clinical and pathological review. Neth J Med. 2012;70:12-17.
- Babar F, Posner JN, Obah EA. Hydralazine-induced pauci-immune glomerulonephritis: intriguing case series misleading diagnoses. J Community Hosp Intern Med Perspect. 2016;6:30632.
- Marina VP, Malhotra D, Kaw D. Hydralazine-induced ANCA vasculitis with pulmonary renal syndrome: a rare clinical presentation. Int Urol Nephrol. 2012;44:1907-1909.
- Magro CM. Associated ANCA positive vasculitis. The Dermatologist. 2015;23(7). http://www.the-dermatologist.com/content/associated-anca-positive-vasculitis. Accessed January 30, 2020.
- Gao Y, Zhao MH. Review article: Drug-induced anti-neutrophil cytoplasmic antibody-associated vasculitis. Nephrology (Carlton). 2009;14:33-41.
- Grau RG. Drug-induced vasculitis: new insights and a changing lineup of suspects. Curr Rheumatol Rep. 2015;17:71.
- Sangala N, Lee RW, Horsfield C, et al. Combined ANCA-associated vasculitis and lupus syndrome following prolonged use of hydralazine: a timely reminder of an old foe. Int Urol Nephrol. 2010;42:503-506.
- Choi HK, Merkel PA, Walker AM, et al. Drug-associated antineutrophil cytoplasmic antibody-positive vasculitis: prevalence among patients with high titers of antimyeloperoxidase antibodies. Arthritis Rheum. 2000;43:405-413.
- Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol. 2011;335:2-13.
- Yokogawa N, Vivino FB. Hydralazine-induced autoimmune disease: comparison to idiopathic lupus and ANCA-positive vasculitis. Mod Rheumatol. 2009;19:338-347.
- Agarwal G, Sultan G, Werner SL, et al. Hydralazine induces myeloperoxidase and proteinase 3 anti-neutrophil cytoplasmic antibody vasculitis and leads to pulmonary renal syndrome. Case Rep Nephrol. 2014;2014:868590.
- Keasberry J, Frazier J, Isbel NM, et al. Hydralazine-induced anti-neutrophilic cytoplasmic antibody-positive renal vasculitis presenting with a vasculitic syndrome, acute nephritis and a puzzling skin rash: a case report. J Med Case Rep. 2013;7:20.
- ten Holder SM, Joy MS, Falk RJ. Cutaneous and systemic manifestations of drug-induced vasculitis. Ann Pharmacother. 2002;36:130-147.
- Namas R, Rubin B, Adwar W, et al. A challenging twist in pulmonary renal syndrome. Case Rep Rheumatol. 2014;2014:516362.
- Dobre M, Wish J, Negrea L. Hydralazine-induced ANCA-positive pauci-immune glomerulonephritis. Ren Fail. 2009;31:745-748.
- Hogan JJ, Markowitz GS, Radhakrishnan J. Drug-induced glomerular disease: immune-mediated injury. Clin J Am Soc Nephrol. 2015;10:1300-1310.
- Radic M, Martinovic Kaliterna D, Radic J. Drug-induced vasculitis: a clinical and pathological review. Neth J Med. 2012;70:12-17.
- Babar F, Posner JN, Obah EA. Hydralazine-induced pauci-immune glomerulonephritis: intriguing case series misleading diagnoses. J Community Hosp Intern Med Perspect. 2016;6:30632.
- Marina VP, Malhotra D, Kaw D. Hydralazine-induced ANCA vasculitis with pulmonary renal syndrome: a rare clinical presentation. Int Urol Nephrol. 2012;44:1907-1909.
- Magro CM. Associated ANCA positive vasculitis. The Dermatologist. 2015;23(7). http://www.the-dermatologist.com/content/associated-anca-positive-vasculitis. Accessed January 30, 2020.
- Gao Y, Zhao MH. Review article: Drug-induced anti-neutrophil cytoplasmic antibody-associated vasculitis. Nephrology (Carlton). 2009;14:33-41.
- Grau RG. Drug-induced vasculitis: new insights and a changing lineup of suspects. Curr Rheumatol Rep. 2015;17:71.
- Sangala N, Lee RW, Horsfield C, et al. Combined ANCA-associated vasculitis and lupus syndrome following prolonged use of hydralazine: a timely reminder of an old foe. Int Urol Nephrol. 2010;42:503-506.
- Choi HK, Merkel PA, Walker AM, et al. Drug-associated antineutrophil cytoplasmic antibody-positive vasculitis: prevalence among patients with high titers of antimyeloperoxidase antibodies. Arthritis Rheum. 2000;43:405-413.
- Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol. 2011;335:2-13.
Practice Points
- Hydralazine-induced antineutrophil cytoplasmic antibody vasculitis (HIAV) is a rare side effect of hydralazine treatment and can have notable morbidity and mortality.
- Incidence and prevalence of HIAV is unclear due to its rarity, but risk factors that have been identified are older age, a cumulative dose of 100 g of hydralazine at the time of presentation, female sex, thyroid disease, HLA-DR4 genotypes, slow hepatic acetylation, and the null gene for C4.
- Symptoms of HIAV can include fever, malaise, arthralgia, weight loss, or even involvement of organs such as the kidneys and lungs.
- If recognized early, cessation of hydralazine and supportive therapy generally are sufficient; however, severe cases may need management with high-dose corticosteroids, rituximab, and even plasmapheresis.
Cutaneous Rosai-Dorfman Disease
Case Report
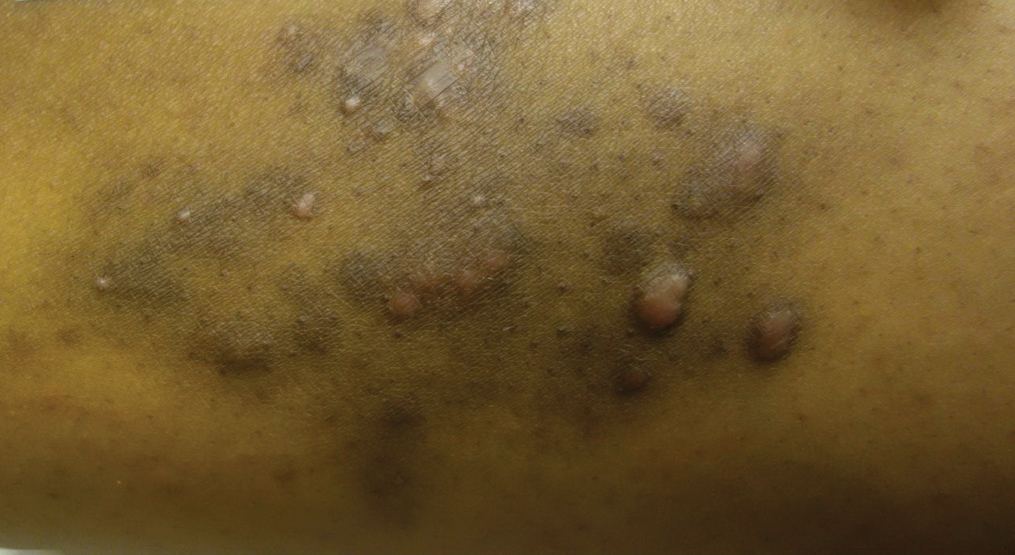
A 31-year-old black woman presented with a slow-spreading pruritic rash on the right thigh of 1 year’s duration. She had previously seen a dermatologist and was prescribed triamcinolone acetonide cream 0.1% and mupirocin ointment 2% but declined a biopsy. Review of symptoms was negative for any constitutional symptoms. Family history included hypertension and eczema with a personal history of anxiety. Clinical examination revealed grouped flesh-colored to light pink papules and plaques within a hyperpigmented patch on the right medial thigh (Figure 1).
Histopathology
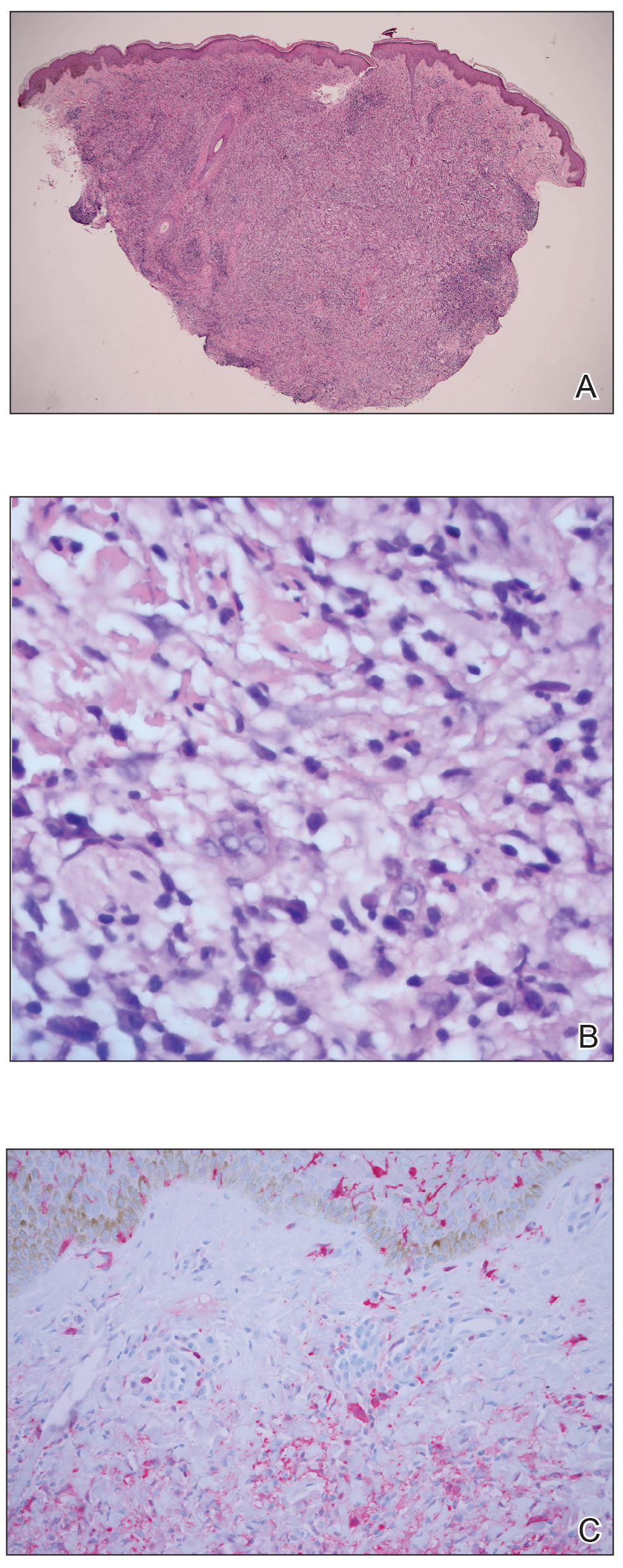
A punch biopsy was negative for fungal, bacterial, or acid-fast bacilli culture. Histopathologic evaluation demonstrated a dense dermal infiltrate of large histiocytes admixed with inflammatory cells composed predominantly of lymphocytes and plasma cells. The histiocytes within the inflammatory infiltrate had vesicular nuclei and abundant eosinophilic cytoplasm (Figure 2A). Areas of emperipolesis were noted (Figure 2B). The large histiocytes stained positive for S-100 protein (Figure 2C) and negative for CD1a.
Course and Treatment
Laboratory studies revealed leukopenia. Prior to histopathologic results, empiric treatment was started with doxycycline 100 mg twice daily for 2 weeks. Once pathology confirmed the diagnosis of Rosai-Dorfman disease (RDD), computed tomography of the chest, abdomen, and pelvis was performed and within normal limits. Due to the lack of systemic involvement, we diagnosed the rare form of purely cutaneous Rosai-Dorfman disease (CRDD). In subsequent visits, treatment with oral prednisone (40 mg daily for 1 week followed by 20 mg daily for 1 week) and intralesional triamcinolone acetonide (5 areas on the right medial thigh were injected with 1.0 mL of 10 mg/mL) was attempted with mild improvement, though the patient declined surgical excision.
Comment
Rosai-Dorfman disease (also known as sinus histiocytosis with massive lymphadenopathy) is a non–Langerhans cell histiocytosis.1 There are 2 main forms of RDD: one form that affects the lymph nodes and in certain cases the extranodal organs, and the other is purely CRDD. Cutaneous RDD is extremely rare and the etiology is unknown, though a number of viral and immune causes have been postulated. Cutaneous RDD presents as solitary or numerous papules, nodules, and/or plaques. Treatment options include steroids, methotrexate, dapsone, thalidomide, and isotretinoin, with varying efficacy reported.1
Extranodal forms occur in 43% of RDD cases, with the skin being the most common site.1 Other extranodal sites include the soft tissue, upper and lower respiratory tract, bones, genitourinary tract, oral cavity, gastrointestinal tract, orbits, testes, and rarely central nervous system involvement.2
Approximately 10% of RDD patients exhibit skin lesions, and in 3% it is contained solely in the skin.3 Pure CRDD was first documented in 1978 by Thawerani et al4 who presented the case of a 48-year-old man with a solitary nodule on the shoulder.
Cutaneous RDD and RDD may be distinct clinical entities. Cutaneous RDD has a later age of onset than RDD (median age, 43.5 years vs 20.6 years) and a female predominance (2:1 vs 1.4:1). It most commonly affects Asian and white individuals while the majority of patients with RDD are of African descent with rare reports in Asians.1
The etiology of CRDD remains unknown with hypotheses of viral and immune causes such as human herpesvirus 6, Epstein-Barr virus, and parvovirus B19. The polyclonal nature of the cell infiltrate and the clinical progression of RDD suggest a reactive process rather than a neoplastic disorder.1 Rosai-Dorfman disease has been hypothesized to be closely related to autoimmune lymphoproliferative syndrome, an inherited disorder associated with defects in Fas-mediated apoptosis.5
Histologic findings in CRDD are similar to those in RDD, with a superficial and deep perivascular infiltrate of lymphocytes and plasma cells. A diffuse and nodular dermal infiltrate of foamy histiocytes exists in a background infiltrate of lymphocytes and plasma cells. Foamy histiocytes may be seen in dermal lymphatics, and lymphoid follicles with reactive germinal centers also may be present. Emperipolesis, the presence of intact inflammatory cells within histiocytes, is common in CRDD. Less often, histiocytes may contain plasma cells, neutrophils, and red blood cells. Mitoses and nuclear atypia are rare. Cutaneous RDD histiocytes stain positive for S-100 protein, CD4, factor XIIIa, and CD68, and negative for CD1a. Birbeck granules are absent on electronic microscopy of CRDD tissue, eliminating Langerhans cell histiocytosis.1,3,5
The clinical diagnosis of CRDD is hard to confirm in the absence of lymphadenopathy. The lesions in CRDD may be solitary or numerous, usually presenting as papules, nodules, and/or plaques. More rarely, the lesions may present as pustules, acneform lesions, mimickers of vasculitis and panniculitis, macular erythema, large annular lesions resembling granuloma annulare, or even a breast mass.1,3 One case report with involvement of deep subcutaneous fat presented with flank swelling beneath papules and nodules.6
The most common site of lesions in CRDD is the face, with the eyelids and malar regions frequently involved, followed by the back, chest, thighs, flanks, and shoulders.1,5 Rarely, CRDD may be associated with other disorders, including bilateral uveitis, antinuclear antibody–positive lupus erythematosus, rheumatoid arthritis, hypothyroidism, lymphoma, and human immunodeficiency virus.1
Numerous treatments have been attempted, yet the response often is poor. Because RDD is a benign and self-limiting disease, less aggressive therapeutic approaches should be used, if possible. Surgical excision of the lesions has been helpful in certain cases.6 Cryotherapy and local radiation, topical steroids, or laser treatment also have been found to improve the condition.1,7 For refractory cases, dapsone and thalidomide have been effective. Mixed results have been observed with isotretinoin and imatinib; some patients improved whereas others did not. Utikal et al8 described a patient with complete remission of CRDD after receiving imatinib therapy; however, a different study reported a patient with CRDD who was completely resistant to this treatment.9 One case presenting on the breast did not respond to topical steroids, acitretin, and thalidomide but later responded to methotrexate.10
Conclusion
Cutaneous RDD is an unusual clinical entity with varied lesions. Generally, CRDD follows a benign clinical course, with a possibility of spontaneous remission. Further studies are required to confidently classify the etiology and variance between both RDD and CRDD.
- Fang S, Chen AJ. Facial cutaneous Rosai-Dorfman disease: a case report and literature review [published online February 5, 2015]. Exp Ther Med. 2015;9:1389-1392.
- Chen A, Fernedez A, Janik M, et al. Cutaneous Rosai-Dorfman disease. J Am Acad Dermatol. 2012;66:AB48.
- James WD, Berger T, Elston D, et al. Andrews’ Diseases of the Skin. 12th ed. Philadelphia, PA: Elsevier; 2015.
- Thawerani H, Sanchez RL, Rosai J, et al. The cutaneous manifestations of sinus histiocytosis with massive lymphadenopathy. Arch Dermatol. 1978;114:191-197.
- Bolognia JL, Jorizzo JL, Schaffer JV, et al, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Al Salamah SM, Abdullah M, Al Salamah RA, et al. Cutaneous Rosai-Dorfman disease presenting as a flank swelling. Int J Health Sci. 2014;8:434-438.
- Khan A, Musbahi E, Suchak R, et al. Cutaneous Rosai-Dorfman disease treated by surgical excision and a review of the literature. J Am Acad Dermatol. 2015;72:AB259.
- Utikal J, Ugurel S, Kurzen H, et al. Imatinib as a treatment option forsystemic non-Langerhans cell histiocytosis. Arch Dermatol. 2007;143:736-740.
- Gebhardt C, Averbeck M, Paasch V, et al. A case of cutaneous Rosai-Dorfman disease refractory to imatinib therapy. Arch Dermatol. 2009;145:571-574.
- Nadal M, Kervarrec T, Machet MC, et al. Cutaneous Rosai-Dorfman disease located on the breast: rapid effectiveness of methotrexate after failure of topical corticosteroids, acitretin and thalidomide. Acta Derm Venereol. 2015;95:758-759.
Case Report
A 31-year-old black woman presented with a slow-spreading pruritic rash on the right thigh of 1 year’s duration. She had previously seen a dermatologist and was prescribed triamcinolone acetonide cream 0.1% and mupirocin ointment 2% but declined a biopsy. Review of symptoms was negative for any constitutional symptoms. Family history included hypertension and eczema with a personal history of anxiety. Clinical examination revealed grouped flesh-colored to light pink papules and plaques within a hyperpigmented patch on the right medial thigh (Figure 1).
Histopathology
A punch biopsy was negative for fungal, bacterial, or acid-fast bacilli culture. Histopathologic evaluation demonstrated a dense dermal infiltrate of large histiocytes admixed with inflammatory cells composed predominantly of lymphocytes and plasma cells. The histiocytes within the inflammatory infiltrate had vesicular nuclei and abundant eosinophilic cytoplasm (Figure 2A). Areas of emperipolesis were noted (Figure 2B). The large histiocytes stained positive for S-100 protein (Figure 2C) and negative for CD1a.
Course and Treatment
Laboratory studies revealed leukopenia. Prior to histopathologic results, empiric treatment was started with doxycycline 100 mg twice daily for 2 weeks. Once pathology confirmed the diagnosis of Rosai-Dorfman disease (RDD), computed tomography of the chest, abdomen, and pelvis was performed and within normal limits. Due to the lack of systemic involvement, we diagnosed the rare form of purely cutaneous Rosai-Dorfman disease (CRDD). In subsequent visits, treatment with oral prednisone (40 mg daily for 1 week followed by 20 mg daily for 1 week) and intralesional triamcinolone acetonide (5 areas on the right medial thigh were injected with 1.0 mL of 10 mg/mL) was attempted with mild improvement, though the patient declined surgical excision.
Comment
Rosai-Dorfman disease (also known as sinus histiocytosis with massive lymphadenopathy) is a non–Langerhans cell histiocytosis.1 There are 2 main forms of RDD: one form that affects the lymph nodes and in certain cases the extranodal organs, and the other is purely CRDD. Cutaneous RDD is extremely rare and the etiology is unknown, though a number of viral and immune causes have been postulated. Cutaneous RDD presents as solitary or numerous papules, nodules, and/or plaques. Treatment options include steroids, methotrexate, dapsone, thalidomide, and isotretinoin, with varying efficacy reported.1
Extranodal forms occur in 43% of RDD cases, with the skin being the most common site.1 Other extranodal sites include the soft tissue, upper and lower respiratory tract, bones, genitourinary tract, oral cavity, gastrointestinal tract, orbits, testes, and rarely central nervous system involvement.2
Approximately 10% of RDD patients exhibit skin lesions, and in 3% it is contained solely in the skin.3 Pure CRDD was first documented in 1978 by Thawerani et al4 who presented the case of a 48-year-old man with a solitary nodule on the shoulder.
Cutaneous RDD and RDD may be distinct clinical entities. Cutaneous RDD has a later age of onset than RDD (median age, 43.5 years vs 20.6 years) and a female predominance (2:1 vs 1.4:1). It most commonly affects Asian and white individuals while the majority of patients with RDD are of African descent with rare reports in Asians.1
The etiology of CRDD remains unknown with hypotheses of viral and immune causes such as human herpesvirus 6, Epstein-Barr virus, and parvovirus B19. The polyclonal nature of the cell infiltrate and the clinical progression of RDD suggest a reactive process rather than a neoplastic disorder.1 Rosai-Dorfman disease has been hypothesized to be closely related to autoimmune lymphoproliferative syndrome, an inherited disorder associated with defects in Fas-mediated apoptosis.5
Histologic findings in CRDD are similar to those in RDD, with a superficial and deep perivascular infiltrate of lymphocytes and plasma cells. A diffuse and nodular dermal infiltrate of foamy histiocytes exists in a background infiltrate of lymphocytes and plasma cells. Foamy histiocytes may be seen in dermal lymphatics, and lymphoid follicles with reactive germinal centers also may be present. Emperipolesis, the presence of intact inflammatory cells within histiocytes, is common in CRDD. Less often, histiocytes may contain plasma cells, neutrophils, and red blood cells. Mitoses and nuclear atypia are rare. Cutaneous RDD histiocytes stain positive for S-100 protein, CD4, factor XIIIa, and CD68, and negative for CD1a. Birbeck granules are absent on electronic microscopy of CRDD tissue, eliminating Langerhans cell histiocytosis.1,3,5
The clinical diagnosis of CRDD is hard to confirm in the absence of lymphadenopathy. The lesions in CRDD may be solitary or numerous, usually presenting as papules, nodules, and/or plaques. More rarely, the lesions may present as pustules, acneform lesions, mimickers of vasculitis and panniculitis, macular erythema, large annular lesions resembling granuloma annulare, or even a breast mass.1,3 One case report with involvement of deep subcutaneous fat presented with flank swelling beneath papules and nodules.6
The most common site of lesions in CRDD is the face, with the eyelids and malar regions frequently involved, followed by the back, chest, thighs, flanks, and shoulders.1,5 Rarely, CRDD may be associated with other disorders, including bilateral uveitis, antinuclear antibody–positive lupus erythematosus, rheumatoid arthritis, hypothyroidism, lymphoma, and human immunodeficiency virus.1
Numerous treatments have been attempted, yet the response often is poor. Because RDD is a benign and self-limiting disease, less aggressive therapeutic approaches should be used, if possible. Surgical excision of the lesions has been helpful in certain cases.6 Cryotherapy and local radiation, topical steroids, or laser treatment also have been found to improve the condition.1,7 For refractory cases, dapsone and thalidomide have been effective. Mixed results have been observed with isotretinoin and imatinib; some patients improved whereas others did not. Utikal et al8 described a patient with complete remission of CRDD after receiving imatinib therapy; however, a different study reported a patient with CRDD who was completely resistant to this treatment.9 One case presenting on the breast did not respond to topical steroids, acitretin, and thalidomide but later responded to methotrexate.10
Conclusion
Cutaneous RDD is an unusual clinical entity with varied lesions. Generally, CRDD follows a benign clinical course, with a possibility of spontaneous remission. Further studies are required to confidently classify the etiology and variance between both RDD and CRDD.
Case Report
A 31-year-old black woman presented with a slow-spreading pruritic rash on the right thigh of 1 year’s duration. She had previously seen a dermatologist and was prescribed triamcinolone acetonide cream 0.1% and mupirocin ointment 2% but declined a biopsy. Review of symptoms was negative for any constitutional symptoms. Family history included hypertension and eczema with a personal history of anxiety. Clinical examination revealed grouped flesh-colored to light pink papules and plaques within a hyperpigmented patch on the right medial thigh (Figure 1).
Histopathology
A punch biopsy was negative for fungal, bacterial, or acid-fast bacilli culture. Histopathologic evaluation demonstrated a dense dermal infiltrate of large histiocytes admixed with inflammatory cells composed predominantly of lymphocytes and plasma cells. The histiocytes within the inflammatory infiltrate had vesicular nuclei and abundant eosinophilic cytoplasm (Figure 2A). Areas of emperipolesis were noted (Figure 2B). The large histiocytes stained positive for S-100 protein (Figure 2C) and negative for CD1a.
Course and Treatment
Laboratory studies revealed leukopenia. Prior to histopathologic results, empiric treatment was started with doxycycline 100 mg twice daily for 2 weeks. Once pathology confirmed the diagnosis of Rosai-Dorfman disease (RDD), computed tomography of the chest, abdomen, and pelvis was performed and within normal limits. Due to the lack of systemic involvement, we diagnosed the rare form of purely cutaneous Rosai-Dorfman disease (CRDD). In subsequent visits, treatment with oral prednisone (40 mg daily for 1 week followed by 20 mg daily for 1 week) and intralesional triamcinolone acetonide (5 areas on the right medial thigh were injected with 1.0 mL of 10 mg/mL) was attempted with mild improvement, though the patient declined surgical excision.
Comment
Rosai-Dorfman disease (also known as sinus histiocytosis with massive lymphadenopathy) is a non–Langerhans cell histiocytosis.1 There are 2 main forms of RDD: one form that affects the lymph nodes and in certain cases the extranodal organs, and the other is purely CRDD. Cutaneous RDD is extremely rare and the etiology is unknown, though a number of viral and immune causes have been postulated. Cutaneous RDD presents as solitary or numerous papules, nodules, and/or plaques. Treatment options include steroids, methotrexate, dapsone, thalidomide, and isotretinoin, with varying efficacy reported.1
Extranodal forms occur in 43% of RDD cases, with the skin being the most common site.1 Other extranodal sites include the soft tissue, upper and lower respiratory tract, bones, genitourinary tract, oral cavity, gastrointestinal tract, orbits, testes, and rarely central nervous system involvement.2
Approximately 10% of RDD patients exhibit skin lesions, and in 3% it is contained solely in the skin.3 Pure CRDD was first documented in 1978 by Thawerani et al4 who presented the case of a 48-year-old man with a solitary nodule on the shoulder.
Cutaneous RDD and RDD may be distinct clinical entities. Cutaneous RDD has a later age of onset than RDD (median age, 43.5 years vs 20.6 years) and a female predominance (2:1 vs 1.4:1). It most commonly affects Asian and white individuals while the majority of patients with RDD are of African descent with rare reports in Asians.1
The etiology of CRDD remains unknown with hypotheses of viral and immune causes such as human herpesvirus 6, Epstein-Barr virus, and parvovirus B19. The polyclonal nature of the cell infiltrate and the clinical progression of RDD suggest a reactive process rather than a neoplastic disorder.1 Rosai-Dorfman disease has been hypothesized to be closely related to autoimmune lymphoproliferative syndrome, an inherited disorder associated with defects in Fas-mediated apoptosis.5
Histologic findings in CRDD are similar to those in RDD, with a superficial and deep perivascular infiltrate of lymphocytes and plasma cells. A diffuse and nodular dermal infiltrate of foamy histiocytes exists in a background infiltrate of lymphocytes and plasma cells. Foamy histiocytes may be seen in dermal lymphatics, and lymphoid follicles with reactive germinal centers also may be present. Emperipolesis, the presence of intact inflammatory cells within histiocytes, is common in CRDD. Less often, histiocytes may contain plasma cells, neutrophils, and red blood cells. Mitoses and nuclear atypia are rare. Cutaneous RDD histiocytes stain positive for S-100 protein, CD4, factor XIIIa, and CD68, and negative for CD1a. Birbeck granules are absent on electronic microscopy of CRDD tissue, eliminating Langerhans cell histiocytosis.1,3,5
The clinical diagnosis of CRDD is hard to confirm in the absence of lymphadenopathy. The lesions in CRDD may be solitary or numerous, usually presenting as papules, nodules, and/or plaques. More rarely, the lesions may present as pustules, acneform lesions, mimickers of vasculitis and panniculitis, macular erythema, large annular lesions resembling granuloma annulare, or even a breast mass.1,3 One case report with involvement of deep subcutaneous fat presented with flank swelling beneath papules and nodules.6
The most common site of lesions in CRDD is the face, with the eyelids and malar regions frequently involved, followed by the back, chest, thighs, flanks, and shoulders.1,5 Rarely, CRDD may be associated with other disorders, including bilateral uveitis, antinuclear antibody–positive lupus erythematosus, rheumatoid arthritis, hypothyroidism, lymphoma, and human immunodeficiency virus.1
Numerous treatments have been attempted, yet the response often is poor. Because RDD is a benign and self-limiting disease, less aggressive therapeutic approaches should be used, if possible. Surgical excision of the lesions has been helpful in certain cases.6 Cryotherapy and local radiation, topical steroids, or laser treatment also have been found to improve the condition.1,7 For refractory cases, dapsone and thalidomide have been effective. Mixed results have been observed with isotretinoin and imatinib; some patients improved whereas others did not. Utikal et al8 described a patient with complete remission of CRDD after receiving imatinib therapy; however, a different study reported a patient with CRDD who was completely resistant to this treatment.9 One case presenting on the breast did not respond to topical steroids, acitretin, and thalidomide but later responded to methotrexate.10
Conclusion
Cutaneous RDD is an unusual clinical entity with varied lesions. Generally, CRDD follows a benign clinical course, with a possibility of spontaneous remission. Further studies are required to confidently classify the etiology and variance between both RDD and CRDD.
- Fang S, Chen AJ. Facial cutaneous Rosai-Dorfman disease: a case report and literature review [published online February 5, 2015]. Exp Ther Med. 2015;9:1389-1392.
- Chen A, Fernedez A, Janik M, et al. Cutaneous Rosai-Dorfman disease. J Am Acad Dermatol. 2012;66:AB48.
- James WD, Berger T, Elston D, et al. Andrews’ Diseases of the Skin. 12th ed. Philadelphia, PA: Elsevier; 2015.
- Thawerani H, Sanchez RL, Rosai J, et al. The cutaneous manifestations of sinus histiocytosis with massive lymphadenopathy. Arch Dermatol. 1978;114:191-197.
- Bolognia JL, Jorizzo JL, Schaffer JV, et al, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Al Salamah SM, Abdullah M, Al Salamah RA, et al. Cutaneous Rosai-Dorfman disease presenting as a flank swelling. Int J Health Sci. 2014;8:434-438.
- Khan A, Musbahi E, Suchak R, et al. Cutaneous Rosai-Dorfman disease treated by surgical excision and a review of the literature. J Am Acad Dermatol. 2015;72:AB259.
- Utikal J, Ugurel S, Kurzen H, et al. Imatinib as a treatment option forsystemic non-Langerhans cell histiocytosis. Arch Dermatol. 2007;143:736-740.
- Gebhardt C, Averbeck M, Paasch V, et al. A case of cutaneous Rosai-Dorfman disease refractory to imatinib therapy. Arch Dermatol. 2009;145:571-574.
- Nadal M, Kervarrec T, Machet MC, et al. Cutaneous Rosai-Dorfman disease located on the breast: rapid effectiveness of methotrexate after failure of topical corticosteroids, acitretin and thalidomide. Acta Derm Venereol. 2015;95:758-759.
- Fang S, Chen AJ. Facial cutaneous Rosai-Dorfman disease: a case report and literature review [published online February 5, 2015]. Exp Ther Med. 2015;9:1389-1392.
- Chen A, Fernedez A, Janik M, et al. Cutaneous Rosai-Dorfman disease. J Am Acad Dermatol. 2012;66:AB48.
- James WD, Berger T, Elston D, et al. Andrews’ Diseases of the Skin. 12th ed. Philadelphia, PA: Elsevier; 2015.
- Thawerani H, Sanchez RL, Rosai J, et al. The cutaneous manifestations of sinus histiocytosis with massive lymphadenopathy. Arch Dermatol. 1978;114:191-197.
- Bolognia JL, Jorizzo JL, Schaffer JV, et al, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Al Salamah SM, Abdullah M, Al Salamah RA, et al. Cutaneous Rosai-Dorfman disease presenting as a flank swelling. Int J Health Sci. 2014;8:434-438.
- Khan A, Musbahi E, Suchak R, et al. Cutaneous Rosai-Dorfman disease treated by surgical excision and a review of the literature. J Am Acad Dermatol. 2015;72:AB259.
- Utikal J, Ugurel S, Kurzen H, et al. Imatinib as a treatment option forsystemic non-Langerhans cell histiocytosis. Arch Dermatol. 2007;143:736-740.
- Gebhardt C, Averbeck M, Paasch V, et al. A case of cutaneous Rosai-Dorfman disease refractory to imatinib therapy. Arch Dermatol. 2009;145:571-574.
- Nadal M, Kervarrec T, Machet MC, et al. Cutaneous Rosai-Dorfman disease located on the breast: rapid effectiveness of methotrexate after failure of topical corticosteroids, acitretin and thalidomide. Acta Derm Venereol. 2015;95:758-759.
Practice Points
- Rosai-Dorfman disease generally is characterized by painless cervical lymphadenopathy and systemic involvement.
- Cutaneous Rosai-Dorfman disease can clinically present with great variety, mimicking many other dermatologic conditions.
- Patients presenting with cutaneous lesions and lymphadenopathy warrant workup with systemic imaging.