User login
Breakdown
The patient's tachycardia and leukocytosis suggest sepsis. Potential sources include soft tissue infection or osteomyelitis from his sacral ulcers, Clostridium difficile, or a urinary tract infection. Impaired visceral sensation from his spinal cord injury may dampen his response to an intra‐abdominal process, such as mesenteric ischemia or toxic megacolon. Records from other hospitals should be reviewed to assess the acuity of change in his WBC count, hemoglobin, and creatinine. His anemia may be from chronic inflammation (eg, osteomyelitis), renal insufficiency, hemolysis, or occult blood loss, including retroperitoneal and gastrointestinal sources. His kidney injury may be from tubular necrosis in the setting of sepsis or obstructive uropathy related to a neurogenic bladder.
Potential contributors to his PEA and cardiovascular collapse are drug use (cocaine), alcohol withdrawal, infection, hypovolemia, myocardial ischemia, or heart failure. Severe hemorrhage, hyperkalemia, or acidosis from acute kidney injury and sepsis could also account for his cardiac arrest. His paraplegia and hospitalization raise the risk of venous thromboembolism, which can lead to PEA from pulmonary embolus and prolonged hypoxia.
His profound anemia is the likely cause of his PEA arrest and severe lactic acidosis. Massive hemolysis is most likely given no overt evidence of bleeding to account for the precipitous fall in hematocrit. Hemolysis can result from disorders intrinsic or extrinsic to the red blood cell (RBC). Intrinsic defects are usually congenital and involve the membrane, hemoglobin, or metabolic enzymes within the RBC. Extrinsic hemolysis arises from processes that injure the RBC from the outside: antibodies, infections, and mechanical shearing.
A rapidly declining platelet count is seen in microangiopathic hemolytic conditions such as disseminated intravascular coagulation (DIC) or thrombotic thrombocytopenic purpura (TTP), where platelets are consumed along with RBCs; sepsis makes DIC more likely. Autoimmune hemolytic anemia (AIHA) is sometimes accompanied by immune thrombocytopenia. AIHA arises from antibodies that are idiopathic or produced in response to infection, autoimmune conditions (eg, systemic lupus erythematosus), lymphoproliferative disease, or drugs (eg, ‐lactam antibiotics). The antiphospholipid syndrome can lead to thrombocytopenia, hemolysis, and kidney injury. Devitalized tissue in his sacral ulcers may predispose the patient to infection with Clostridium perfringens, which can elaborate enzymes that trigger massive hemolysis.
Because automated hemoglobin measurement is performed by spectrophotometry (light absorption and scatter), high concentrations of poorly soluble autoantibodies can increase the turbidity of the sample and preclude the measurement of hemoglobin concentration. This could lead to the report of interfering substances.
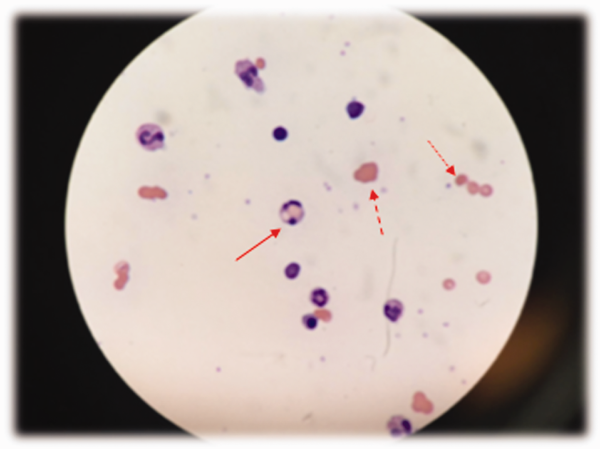
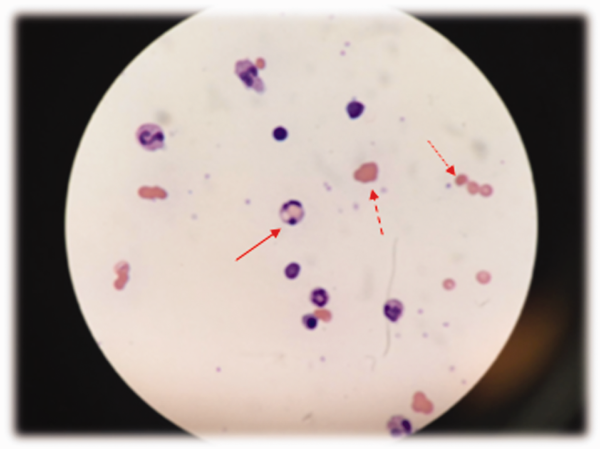
Low haptoglobin, elevated LDH, and hyperbilirubinemia confirm hemolysis. A more robust reticulocytosis is expected in the face of profound anemia, but the patient may also suffer from a concomitant hypoproliferative state (eg, nutritional deficiency). More likely, the rapidity of his decline outpaced the marrow's response, which can be delayed by days.
The most common cause of a combined elevation of the INR/PT and aPTT in a critically ill patient is DIC. Although no schistocytes were detected on the peripheral smear, they can be absent in up to 50% of DIC cases. TTP is associated with hemolytic anemia, kidney injury, and thrombocytopenia, but it generally does not cause coagulopathy.
The combination of red cell agglutination and hemophagocytosis suggests that the RBCs are coated with autoantibodies that cross‐link the cells and make them targets for phagocytosis by neutrophils in the circulation. This is distinct from the hemophagocytic syndrome, a rare immune activation syndrome characterized by macrophage phagocytosis of RBCs in the reticuloendothelial system. The blood smear also shows microspherocytes, which are seen in AIHA and hereditary spherocytosis.
Acute tubular necrosis could result from sepsis, ischemic injury from DIC, hypotension during cardiac arrest, or heme pigment toxicity. Urine sediment should be reviewed for dysmorphic RBCs or RBC casts that would indicate glomerulonephritis (eg, from an underlying autoimmune process associated with AIHA).
Urine hemoglobin that is disproportionate to the degree of hematuria suggests hemoglobinuria, which in turn defines the hemolysis as intravascular. Processes that directly lyse RBCs in circulation via mechanical shearing, activation of complement, infection of the RBC, or enzymatic or oxidative destruction of the membrane cause intravascular hemolysis. Leading considerations include microangiopathy (eg, DIC, TTP), clostridial sepsis, and AIHA.
AIHA can be broadly classified as warm or cold. Warm AIHA is caused by immunoglobulin IgG antibodies that bind most avidly at body temperature. Because warm AIHA does not activate complement, patients present with evidence of extravascular hemolysis that is typically chronic and mild to moderate in severity. It does not typically cause the acute, fulminant, intravascular hemolytic condition seen here.
Cold AIHA is characterized by autoantibodies that bind at lower temperatures and comes in 2 forms: cold agglutinin disease and (rarely) paroxysmal cold hemoglobinuria (PCH). Cold agglutinins are most often IgM antibodies produced in response to infection (Mycoplasma pneumoniae, infectious mononucleosis), drugs, or a hematologic malignancy. These IgM antibodies bind RBCs, causing them to agglutinate, and fix complement (including C3) to the surface of RBCs when blood circulates to cooler parts of the body. This results in complement activation, formation of the membrane attack complex, and intravascular hemolysis when bound and activated complement is present in large numbers. Acute infection can increase the complement available for binding to the surface of RBCs. Through a slightly different mechanism, PCH causes intravascular hemolysis through direct IgG activation of complement fixed to the surface of RBCs. During a hemolytic episode the direct antibody test (DAT) is positive using anti‐C3 and negative for IgG.
Based on the patient's clinical evidence of intravascular hemolysis and a suspected autoimmune etiology, the leading diagnosis at this time is cold AIHA.
The DAT detects IgG or complement adherent to RBCs. This patient has tested positive for both IgG and C3, though much more strongly for IgG, suggesting an unusual ability of the patient's IgG to activate complement. The phenomenon of mixed AIHA, in which the patient has both warm‐ and cold‐reacting antibodies, is rare.
Regarding infections associated with AIHA, there is no cough or rash to suggest M pneumoniae, and there is no sore throat, fever, lymphadenopathy, splenomegaly, or atypical lymphocytosis to suggest infectious mononucleosis. He should be tested for human immunodeficiency virus, which is also associated with AIHA. His leukocytosis may raise suspicion for an underlying hematologic malignancy, but he does not have blasts, dysplastic leukocytes, or lymphocytosis on his peripheral blood smear. Systemic lupus erythematosus can be associated with AIHA, thrombocytopenia, and renal failure, but he lacks the more common clinical manifestations of rash, arthralgias, and fever.
Drug‐induced immune hemolytic anemia (DIIHA) can cause both the clinical and serologic profile of an AIHA, as seen here. DIIHA can be distinguished from mixed AIHA if hemolysis abates with discontinuation of an offending drug. His deterioration is temporally associated with drug administration at the time of admission. Cephalosporins and ‐lactams (e.g., piperacillin) are the most common causes of DIIHA, and ‐lactamases such as tazobactam have also been implicated. By exclusion of other causes, DIIHA secondary to piperacillin is most likely responsible for his massive intravascular hemolysis.
COMMENTARY
This case illustrates a dramatic presentation of fulminant intravascular hemolysis secondary to piperacillin. The incidence of DIIHA is estimated to be 1 in 1 million.[1] Historically, methyldopa and high‐dose penicillin have been responsible for the majority of cases,[2] but in recent years complex penicillins, including piperacillin, and second‐ and third‐generation cephalosporins have been implicated.[3, 4] Cases of DIIHA are often underdiagnosed or misdiagnosed, as smoldering or less severe cases may not be recognized or are attributed to other causes.
A positive DAT, suggesting immunoglobulin and/or complement binding to RBCs, is the most reliable laboratory finding in DIIHA.[5] However, a positive DAT does not identify the source of the antigen and may result in misattribution of the immune hemolysis to autoimmunity rather than to a drug. Repeated or continued administration of the offending drug (as in this case) may perpetuate or worsen the hemolysis. Drug‐specific antibody tests may help to confirm the diagnosis, but these tests are complex and take significant time for specialized laboratories to run.
Severe hemolysis should be considered when a patient has a sudden and dramatic drop in his hemoglobin level in the absence of bleeding. Because DIIHA can be rapidly progressive, discontinuing a suspected culprit drug is the most important diagnostic and therapeutic measure. Typically, when an offending drug is stopped, the hemolysis stops as well. The time course over which this occurs depends on the rapidity of drug clearance.[4] Hemodialysis or plasmapheresis may be required in cases where the medication is renally excreted, particularly in cases of concomitant kidney injury. Evidence supporting corticosteroid use in DIIHA is limited, as the offending agent is usually discontinued by the time corticosteroids are initiated.[4]
This patient's DAT confirmed both IgG and complement activation, consistent with DIIHA caused by an immune complexlike reaction. This mechanism involves the antibody binding to a mixed epitope of the drug and a RBC membrane glycoprotein.[6] The offending drug was stopped only when review of his medical records established a clear temporal association between antibiotic administration and prior hemolysis.
The 2009 Health Information Technology for Economic and Clinical Health Act created an electronic health record (EHR) incentive program (meaningful use criteria).[7] By 2012, only 6% of hospitals met all of the stage 2 criteria, which include EHR interoperability across health systems.[8] The patient's preceding hemolytic event was described in records faxed by the outside hospitals, but without EHR interoperability, the treating clinicians did not have timely access to this information. Instead, the familiar manual process of obtaining outside records involving signed forms, phone calls, fax machines, and reams of paper progressed at its usual pace. Real‐time access to health records might have guided providers to select an alternative antibiotic regimen. Instead, a communication breakdown contributed to a catastrophic drug reaction and to this tragic patient outcome.
KEY TEACHING POINTS
- In a patient presenting with acute hemolysis and a positive DAT, consider DIIHA.
- Both piperacillin and tazobactam can cause a severe, complement‐mediated immune hemolytic anemia (DIIHA).
- Drug‐induced antibodies are detected by direct antiglobulin testing, but a complete medication history is the key to diagnosis.
- Management of drug‐induced hemolytic anemia involves immediate discontinuation of the culprit medication, supportive care, and potentially corticosteroids, plasmapheresis, and/or hemodialysis to expedite removal of the offending agent.
- EHR interoperability may provide timely access to important health information across different hospitals, expedite health information exchange, and reduce adverse patient outcomes that stem from communication delays.
This case was submitted anonymously to AHRQ WebM&M on July 18, 2014, and was accepted on August 7, 2014. The case and WebM&M commentary were published online on October 26, 2015.[9] This separate commentary on the same case was later submitted to the Journal of Hospital Medicine on September 2, 2015, accepted on November 24, 2015, and published on January 22, 2016. The 2 publications are written by different authors, and although they reference the same case, they make different but valuable points.
Disclosure
Nothing to report.
The patient's tachycardia and leukocytosis suggest sepsis. Potential sources include soft tissue infection or osteomyelitis from his sacral ulcers, Clostridium difficile, or a urinary tract infection. Impaired visceral sensation from his spinal cord injury may dampen his response to an intra‐abdominal process, such as mesenteric ischemia or toxic megacolon. Records from other hospitals should be reviewed to assess the acuity of change in his WBC count, hemoglobin, and creatinine. His anemia may be from chronic inflammation (eg, osteomyelitis), renal insufficiency, hemolysis, or occult blood loss, including retroperitoneal and gastrointestinal sources. His kidney injury may be from tubular necrosis in the setting of sepsis or obstructive uropathy related to a neurogenic bladder.
Potential contributors to his PEA and cardiovascular collapse are drug use (cocaine), alcohol withdrawal, infection, hypovolemia, myocardial ischemia, or heart failure. Severe hemorrhage, hyperkalemia, or acidosis from acute kidney injury and sepsis could also account for his cardiac arrest. His paraplegia and hospitalization raise the risk of venous thromboembolism, which can lead to PEA from pulmonary embolus and prolonged hypoxia.
His profound anemia is the likely cause of his PEA arrest and severe lactic acidosis. Massive hemolysis is most likely given no overt evidence of bleeding to account for the precipitous fall in hematocrit. Hemolysis can result from disorders intrinsic or extrinsic to the red blood cell (RBC). Intrinsic defects are usually congenital and involve the membrane, hemoglobin, or metabolic enzymes within the RBC. Extrinsic hemolysis arises from processes that injure the RBC from the outside: antibodies, infections, and mechanical shearing.
A rapidly declining platelet count is seen in microangiopathic hemolytic conditions such as disseminated intravascular coagulation (DIC) or thrombotic thrombocytopenic purpura (TTP), where platelets are consumed along with RBCs; sepsis makes DIC more likely. Autoimmune hemolytic anemia (AIHA) is sometimes accompanied by immune thrombocytopenia. AIHA arises from antibodies that are idiopathic or produced in response to infection, autoimmune conditions (eg, systemic lupus erythematosus), lymphoproliferative disease, or drugs (eg, ‐lactam antibiotics). The antiphospholipid syndrome can lead to thrombocytopenia, hemolysis, and kidney injury. Devitalized tissue in his sacral ulcers may predispose the patient to infection with Clostridium perfringens, which can elaborate enzymes that trigger massive hemolysis.
Because automated hemoglobin measurement is performed by spectrophotometry (light absorption and scatter), high concentrations of poorly soluble autoantibodies can increase the turbidity of the sample and preclude the measurement of hemoglobin concentration. This could lead to the report of interfering substances.
Low haptoglobin, elevated LDH, and hyperbilirubinemia confirm hemolysis. A more robust reticulocytosis is expected in the face of profound anemia, but the patient may also suffer from a concomitant hypoproliferative state (eg, nutritional deficiency). More likely, the rapidity of his decline outpaced the marrow's response, which can be delayed by days.
The most common cause of a combined elevation of the INR/PT and aPTT in a critically ill patient is DIC. Although no schistocytes were detected on the peripheral smear, they can be absent in up to 50% of DIC cases. TTP is associated with hemolytic anemia, kidney injury, and thrombocytopenia, but it generally does not cause coagulopathy.
The combination of red cell agglutination and hemophagocytosis suggests that the RBCs are coated with autoantibodies that cross‐link the cells and make them targets for phagocytosis by neutrophils in the circulation. This is distinct from the hemophagocytic syndrome, a rare immune activation syndrome characterized by macrophage phagocytosis of RBCs in the reticuloendothelial system. The blood smear also shows microspherocytes, which are seen in AIHA and hereditary spherocytosis.
Acute tubular necrosis could result from sepsis, ischemic injury from DIC, hypotension during cardiac arrest, or heme pigment toxicity. Urine sediment should be reviewed for dysmorphic RBCs or RBC casts that would indicate glomerulonephritis (eg, from an underlying autoimmune process associated with AIHA).
Urine hemoglobin that is disproportionate to the degree of hematuria suggests hemoglobinuria, which in turn defines the hemolysis as intravascular. Processes that directly lyse RBCs in circulation via mechanical shearing, activation of complement, infection of the RBC, or enzymatic or oxidative destruction of the membrane cause intravascular hemolysis. Leading considerations include microangiopathy (eg, DIC, TTP), clostridial sepsis, and AIHA.
AIHA can be broadly classified as warm or cold. Warm AIHA is caused by immunoglobulin IgG antibodies that bind most avidly at body temperature. Because warm AIHA does not activate complement, patients present with evidence of extravascular hemolysis that is typically chronic and mild to moderate in severity. It does not typically cause the acute, fulminant, intravascular hemolytic condition seen here.
Cold AIHA is characterized by autoantibodies that bind at lower temperatures and comes in 2 forms: cold agglutinin disease and (rarely) paroxysmal cold hemoglobinuria (PCH). Cold agglutinins are most often IgM antibodies produced in response to infection (Mycoplasma pneumoniae, infectious mononucleosis), drugs, or a hematologic malignancy. These IgM antibodies bind RBCs, causing them to agglutinate, and fix complement (including C3) to the surface of RBCs when blood circulates to cooler parts of the body. This results in complement activation, formation of the membrane attack complex, and intravascular hemolysis when bound and activated complement is present in large numbers. Acute infection can increase the complement available for binding to the surface of RBCs. Through a slightly different mechanism, PCH causes intravascular hemolysis through direct IgG activation of complement fixed to the surface of RBCs. During a hemolytic episode the direct antibody test (DAT) is positive using anti‐C3 and negative for IgG.
Based on the patient's clinical evidence of intravascular hemolysis and a suspected autoimmune etiology, the leading diagnosis at this time is cold AIHA.
The DAT detects IgG or complement adherent to RBCs. This patient has tested positive for both IgG and C3, though much more strongly for IgG, suggesting an unusual ability of the patient's IgG to activate complement. The phenomenon of mixed AIHA, in which the patient has both warm‐ and cold‐reacting antibodies, is rare.
Regarding infections associated with AIHA, there is no cough or rash to suggest M pneumoniae, and there is no sore throat, fever, lymphadenopathy, splenomegaly, or atypical lymphocytosis to suggest infectious mononucleosis. He should be tested for human immunodeficiency virus, which is also associated with AIHA. His leukocytosis may raise suspicion for an underlying hematologic malignancy, but he does not have blasts, dysplastic leukocytes, or lymphocytosis on his peripheral blood smear. Systemic lupus erythematosus can be associated with AIHA, thrombocytopenia, and renal failure, but he lacks the more common clinical manifestations of rash, arthralgias, and fever.
Drug‐induced immune hemolytic anemia (DIIHA) can cause both the clinical and serologic profile of an AIHA, as seen here. DIIHA can be distinguished from mixed AIHA if hemolysis abates with discontinuation of an offending drug. His deterioration is temporally associated with drug administration at the time of admission. Cephalosporins and ‐lactams (e.g., piperacillin) are the most common causes of DIIHA, and ‐lactamases such as tazobactam have also been implicated. By exclusion of other causes, DIIHA secondary to piperacillin is most likely responsible for his massive intravascular hemolysis.
COMMENTARY
This case illustrates a dramatic presentation of fulminant intravascular hemolysis secondary to piperacillin. The incidence of DIIHA is estimated to be 1 in 1 million.[1] Historically, methyldopa and high‐dose penicillin have been responsible for the majority of cases,[2] but in recent years complex penicillins, including piperacillin, and second‐ and third‐generation cephalosporins have been implicated.[3, 4] Cases of DIIHA are often underdiagnosed or misdiagnosed, as smoldering or less severe cases may not be recognized or are attributed to other causes.
A positive DAT, suggesting immunoglobulin and/or complement binding to RBCs, is the most reliable laboratory finding in DIIHA.[5] However, a positive DAT does not identify the source of the antigen and may result in misattribution of the immune hemolysis to autoimmunity rather than to a drug. Repeated or continued administration of the offending drug (as in this case) may perpetuate or worsen the hemolysis. Drug‐specific antibody tests may help to confirm the diagnosis, but these tests are complex and take significant time for specialized laboratories to run.
Severe hemolysis should be considered when a patient has a sudden and dramatic drop in his hemoglobin level in the absence of bleeding. Because DIIHA can be rapidly progressive, discontinuing a suspected culprit drug is the most important diagnostic and therapeutic measure. Typically, when an offending drug is stopped, the hemolysis stops as well. The time course over which this occurs depends on the rapidity of drug clearance.[4] Hemodialysis or plasmapheresis may be required in cases where the medication is renally excreted, particularly in cases of concomitant kidney injury. Evidence supporting corticosteroid use in DIIHA is limited, as the offending agent is usually discontinued by the time corticosteroids are initiated.[4]
This patient's DAT confirmed both IgG and complement activation, consistent with DIIHA caused by an immune complexlike reaction. This mechanism involves the antibody binding to a mixed epitope of the drug and a RBC membrane glycoprotein.[6] The offending drug was stopped only when review of his medical records established a clear temporal association between antibiotic administration and prior hemolysis.
The 2009 Health Information Technology for Economic and Clinical Health Act created an electronic health record (EHR) incentive program (meaningful use criteria).[7] By 2012, only 6% of hospitals met all of the stage 2 criteria, which include EHR interoperability across health systems.[8] The patient's preceding hemolytic event was described in records faxed by the outside hospitals, but without EHR interoperability, the treating clinicians did not have timely access to this information. Instead, the familiar manual process of obtaining outside records involving signed forms, phone calls, fax machines, and reams of paper progressed at its usual pace. Real‐time access to health records might have guided providers to select an alternative antibiotic regimen. Instead, a communication breakdown contributed to a catastrophic drug reaction and to this tragic patient outcome.
KEY TEACHING POINTS
- In a patient presenting with acute hemolysis and a positive DAT, consider DIIHA.
- Both piperacillin and tazobactam can cause a severe, complement‐mediated immune hemolytic anemia (DIIHA).
- Drug‐induced antibodies are detected by direct antiglobulin testing, but a complete medication history is the key to diagnosis.
- Management of drug‐induced hemolytic anemia involves immediate discontinuation of the culprit medication, supportive care, and potentially corticosteroids, plasmapheresis, and/or hemodialysis to expedite removal of the offending agent.
- EHR interoperability may provide timely access to important health information across different hospitals, expedite health information exchange, and reduce adverse patient outcomes that stem from communication delays.
This case was submitted anonymously to AHRQ WebM&M on July 18, 2014, and was accepted on August 7, 2014. The case and WebM&M commentary were published online on October 26, 2015.[9] This separate commentary on the same case was later submitted to the Journal of Hospital Medicine on September 2, 2015, accepted on November 24, 2015, and published on January 22, 2016. The 2 publications are written by different authors, and although they reference the same case, they make different but valuable points.
Disclosure
Nothing to report.
The patient's tachycardia and leukocytosis suggest sepsis. Potential sources include soft tissue infection or osteomyelitis from his sacral ulcers, Clostridium difficile, or a urinary tract infection. Impaired visceral sensation from his spinal cord injury may dampen his response to an intra‐abdominal process, such as mesenteric ischemia or toxic megacolon. Records from other hospitals should be reviewed to assess the acuity of change in his WBC count, hemoglobin, and creatinine. His anemia may be from chronic inflammation (eg, osteomyelitis), renal insufficiency, hemolysis, or occult blood loss, including retroperitoneal and gastrointestinal sources. His kidney injury may be from tubular necrosis in the setting of sepsis or obstructive uropathy related to a neurogenic bladder.
Potential contributors to his PEA and cardiovascular collapse are drug use (cocaine), alcohol withdrawal, infection, hypovolemia, myocardial ischemia, or heart failure. Severe hemorrhage, hyperkalemia, or acidosis from acute kidney injury and sepsis could also account for his cardiac arrest. His paraplegia and hospitalization raise the risk of venous thromboembolism, which can lead to PEA from pulmonary embolus and prolonged hypoxia.
His profound anemia is the likely cause of his PEA arrest and severe lactic acidosis. Massive hemolysis is most likely given no overt evidence of bleeding to account for the precipitous fall in hematocrit. Hemolysis can result from disorders intrinsic or extrinsic to the red blood cell (RBC). Intrinsic defects are usually congenital and involve the membrane, hemoglobin, or metabolic enzymes within the RBC. Extrinsic hemolysis arises from processes that injure the RBC from the outside: antibodies, infections, and mechanical shearing.
A rapidly declining platelet count is seen in microangiopathic hemolytic conditions such as disseminated intravascular coagulation (DIC) or thrombotic thrombocytopenic purpura (TTP), where platelets are consumed along with RBCs; sepsis makes DIC more likely. Autoimmune hemolytic anemia (AIHA) is sometimes accompanied by immune thrombocytopenia. AIHA arises from antibodies that are idiopathic or produced in response to infection, autoimmune conditions (eg, systemic lupus erythematosus), lymphoproliferative disease, or drugs (eg, ‐lactam antibiotics). The antiphospholipid syndrome can lead to thrombocytopenia, hemolysis, and kidney injury. Devitalized tissue in his sacral ulcers may predispose the patient to infection with Clostridium perfringens, which can elaborate enzymes that trigger massive hemolysis.
Because automated hemoglobin measurement is performed by spectrophotometry (light absorption and scatter), high concentrations of poorly soluble autoantibodies can increase the turbidity of the sample and preclude the measurement of hemoglobin concentration. This could lead to the report of interfering substances.
Low haptoglobin, elevated LDH, and hyperbilirubinemia confirm hemolysis. A more robust reticulocytosis is expected in the face of profound anemia, but the patient may also suffer from a concomitant hypoproliferative state (eg, nutritional deficiency). More likely, the rapidity of his decline outpaced the marrow's response, which can be delayed by days.
The most common cause of a combined elevation of the INR/PT and aPTT in a critically ill patient is DIC. Although no schistocytes were detected on the peripheral smear, they can be absent in up to 50% of DIC cases. TTP is associated with hemolytic anemia, kidney injury, and thrombocytopenia, but it generally does not cause coagulopathy.
The combination of red cell agglutination and hemophagocytosis suggests that the RBCs are coated with autoantibodies that cross‐link the cells and make them targets for phagocytosis by neutrophils in the circulation. This is distinct from the hemophagocytic syndrome, a rare immune activation syndrome characterized by macrophage phagocytosis of RBCs in the reticuloendothelial system. The blood smear also shows microspherocytes, which are seen in AIHA and hereditary spherocytosis.
Acute tubular necrosis could result from sepsis, ischemic injury from DIC, hypotension during cardiac arrest, or heme pigment toxicity. Urine sediment should be reviewed for dysmorphic RBCs or RBC casts that would indicate glomerulonephritis (eg, from an underlying autoimmune process associated with AIHA).
Urine hemoglobin that is disproportionate to the degree of hematuria suggests hemoglobinuria, which in turn defines the hemolysis as intravascular. Processes that directly lyse RBCs in circulation via mechanical shearing, activation of complement, infection of the RBC, or enzymatic or oxidative destruction of the membrane cause intravascular hemolysis. Leading considerations include microangiopathy (eg, DIC, TTP), clostridial sepsis, and AIHA.
AIHA can be broadly classified as warm or cold. Warm AIHA is caused by immunoglobulin IgG antibodies that bind most avidly at body temperature. Because warm AIHA does not activate complement, patients present with evidence of extravascular hemolysis that is typically chronic and mild to moderate in severity. It does not typically cause the acute, fulminant, intravascular hemolytic condition seen here.
Cold AIHA is characterized by autoantibodies that bind at lower temperatures and comes in 2 forms: cold agglutinin disease and (rarely) paroxysmal cold hemoglobinuria (PCH). Cold agglutinins are most often IgM antibodies produced in response to infection (Mycoplasma pneumoniae, infectious mononucleosis), drugs, or a hematologic malignancy. These IgM antibodies bind RBCs, causing them to agglutinate, and fix complement (including C3) to the surface of RBCs when blood circulates to cooler parts of the body. This results in complement activation, formation of the membrane attack complex, and intravascular hemolysis when bound and activated complement is present in large numbers. Acute infection can increase the complement available for binding to the surface of RBCs. Through a slightly different mechanism, PCH causes intravascular hemolysis through direct IgG activation of complement fixed to the surface of RBCs. During a hemolytic episode the direct antibody test (DAT) is positive using anti‐C3 and negative for IgG.
Based on the patient's clinical evidence of intravascular hemolysis and a suspected autoimmune etiology, the leading diagnosis at this time is cold AIHA.
The DAT detects IgG or complement adherent to RBCs. This patient has tested positive for both IgG and C3, though much more strongly for IgG, suggesting an unusual ability of the patient's IgG to activate complement. The phenomenon of mixed AIHA, in which the patient has both warm‐ and cold‐reacting antibodies, is rare.
Regarding infections associated with AIHA, there is no cough or rash to suggest M pneumoniae, and there is no sore throat, fever, lymphadenopathy, splenomegaly, or atypical lymphocytosis to suggest infectious mononucleosis. He should be tested for human immunodeficiency virus, which is also associated with AIHA. His leukocytosis may raise suspicion for an underlying hematologic malignancy, but he does not have blasts, dysplastic leukocytes, or lymphocytosis on his peripheral blood smear. Systemic lupus erythematosus can be associated with AIHA, thrombocytopenia, and renal failure, but he lacks the more common clinical manifestations of rash, arthralgias, and fever.
Drug‐induced immune hemolytic anemia (DIIHA) can cause both the clinical and serologic profile of an AIHA, as seen here. DIIHA can be distinguished from mixed AIHA if hemolysis abates with discontinuation of an offending drug. His deterioration is temporally associated with drug administration at the time of admission. Cephalosporins and ‐lactams (e.g., piperacillin) are the most common causes of DIIHA, and ‐lactamases such as tazobactam have also been implicated. By exclusion of other causes, DIIHA secondary to piperacillin is most likely responsible for his massive intravascular hemolysis.
COMMENTARY
This case illustrates a dramatic presentation of fulminant intravascular hemolysis secondary to piperacillin. The incidence of DIIHA is estimated to be 1 in 1 million.[1] Historically, methyldopa and high‐dose penicillin have been responsible for the majority of cases,[2] but in recent years complex penicillins, including piperacillin, and second‐ and third‐generation cephalosporins have been implicated.[3, 4] Cases of DIIHA are often underdiagnosed or misdiagnosed, as smoldering or less severe cases may not be recognized or are attributed to other causes.
A positive DAT, suggesting immunoglobulin and/or complement binding to RBCs, is the most reliable laboratory finding in DIIHA.[5] However, a positive DAT does not identify the source of the antigen and may result in misattribution of the immune hemolysis to autoimmunity rather than to a drug. Repeated or continued administration of the offending drug (as in this case) may perpetuate or worsen the hemolysis. Drug‐specific antibody tests may help to confirm the diagnosis, but these tests are complex and take significant time for specialized laboratories to run.
Severe hemolysis should be considered when a patient has a sudden and dramatic drop in his hemoglobin level in the absence of bleeding. Because DIIHA can be rapidly progressive, discontinuing a suspected culprit drug is the most important diagnostic and therapeutic measure. Typically, when an offending drug is stopped, the hemolysis stops as well. The time course over which this occurs depends on the rapidity of drug clearance.[4] Hemodialysis or plasmapheresis may be required in cases where the medication is renally excreted, particularly in cases of concomitant kidney injury. Evidence supporting corticosteroid use in DIIHA is limited, as the offending agent is usually discontinued by the time corticosteroids are initiated.[4]
This patient's DAT confirmed both IgG and complement activation, consistent with DIIHA caused by an immune complexlike reaction. This mechanism involves the antibody binding to a mixed epitope of the drug and a RBC membrane glycoprotein.[6] The offending drug was stopped only when review of his medical records established a clear temporal association between antibiotic administration and prior hemolysis.
The 2009 Health Information Technology for Economic and Clinical Health Act created an electronic health record (EHR) incentive program (meaningful use criteria).[7] By 2012, only 6% of hospitals met all of the stage 2 criteria, which include EHR interoperability across health systems.[8] The patient's preceding hemolytic event was described in records faxed by the outside hospitals, but without EHR interoperability, the treating clinicians did not have timely access to this information. Instead, the familiar manual process of obtaining outside records involving signed forms, phone calls, fax machines, and reams of paper progressed at its usual pace. Real‐time access to health records might have guided providers to select an alternative antibiotic regimen. Instead, a communication breakdown contributed to a catastrophic drug reaction and to this tragic patient outcome.
KEY TEACHING POINTS
- In a patient presenting with acute hemolysis and a positive DAT, consider DIIHA.
- Both piperacillin and tazobactam can cause a severe, complement‐mediated immune hemolytic anemia (DIIHA).
- Drug‐induced antibodies are detected by direct antiglobulin testing, but a complete medication history is the key to diagnosis.
- Management of drug‐induced hemolytic anemia involves immediate discontinuation of the culprit medication, supportive care, and potentially corticosteroids, plasmapheresis, and/or hemodialysis to expedite removal of the offending agent.
- EHR interoperability may provide timely access to important health information across different hospitals, expedite health information exchange, and reduce adverse patient outcomes that stem from communication delays.
This case was submitted anonymously to AHRQ WebM&M on July 18, 2014, and was accepted on August 7, 2014. The case and WebM&M commentary were published online on October 26, 2015.[9] This separate commentary on the same case was later submitted to the Journal of Hospital Medicine on September 2, 2015, accepted on November 24, 2015, and published on January 22, 2016. The 2 publications are written by different authors, and although they reference the same case, they make different but valuable points.
Disclosure
Nothing to report.