User login
Imiquimod-Induced Hypopigmentation Following Treatment of Periungual Verruca Vulgaris
Imiquimod is derived from the imidazoquinoline family and works by activating both innate and adaptive immune pathways. Imiquimod binds to toll-like receptor 7 located on monocytes, macrophages, and dendritic cells,1 which allows nuclear factor κβ light chain enhancer of activated B cells to induce production of proinflammatory cytokines, including IFN-α and tumor necrosis factor α, as well as IL-1, IL-6, IL-8, IL-10, and IL-12.2 These proinflammatory cytokines play a role in the innate immunity, triggering upregulation of the adaptive immune pathway and activating type 1 helper T cells, cytotoxic T cells, and natural killer cells. These cells have antiviral and antitumoral effects that lend to their significance in coordinating innate and adaptive immune mechanisms.3 More specifically, imiquimod enhances dendritic cell migration to regional lymph nodes and induces apoptosis via activation of proapoptotic B-cell lymphoma 2 proteins.1,2 Imiquimod has been approved by the US Food and Drug Administration (FDA) to treat external genitalia and perianal condyloma acuminata, actinic keratoses (AKs), and superficial basal cell carcinoma (BCC). It often is used off label for antiviral or antitumoral therapy in Bowen disease, squamous cell carcinoma, lentigo maligna, vulvar intraepithelial neoplasia, molluscum contagiosum, common warts, and leishmaniasis.1,2 Imiquimod is generally well tolerated; erythema and irritation at the application site are the most common side effects, with pigmentary change being less common.
Case Report
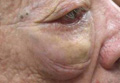
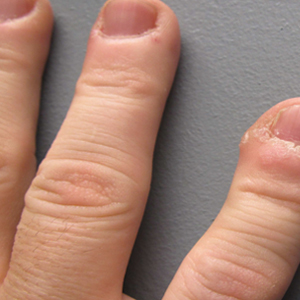
A 51-year-old man with a medical history of vitamin D deficiency, vitamin B12 deficiency, tinea pedis, and BCC presented with periungual verruca vulgaris on the right fifth digit and left thumb (Figure 1). The patient was prescribed imiquimod cream 5% to be applied 3 times weekly for 3 months. At 5-month follow-up the patient reported new-onset vitiligolike patches of depigmentation on the hands and feet that abruptly began 3 months after initiating treatment with imiquimod. On examination he had several depigmented patches with well-defined irregular borders on the bilateral dorsal hands and right foot as well as the right elbow (Figure 2). There was no personal or family history of vitiligo, thyroid disease, or autoimmune disease. Thyroid function studies and autoimmune panel were unremarkable. The patient also denied applying imiquimod to areas other than the periungual region of the right fifth digit and left thumb. He declined a biopsy of the lesions and was given a prescription for tacrolimus ointment 0.1% for twice-daily application. At 3-month follow-up the depigmented patches had spread. The patient is currently on 5-fluorouracil cream 5%. Despite loss of pigmentation, the periungual verruca vulgaris has persisted as well as depigmentation.

 and the right elbow (B).")
Comment
Imiquimod therapy is commonly used to treat conditions for which an antiviral or antitumor immune response is necessary for treatment and full resolution of skin conditions. It can yield positive results in conditions that are difficult to treat, such as periungual verruca vulgaris.4 The most common adverse effects of imiquimod include localized inflammation and application-site reactions. Pigment changes, though less common, also have been reported. From 1997 to 2003, 1257 cases of imiquimod adverse effects were reported to the FDA. There were 68 reported cases of pigmentary change, of which 51 documented vitiligo, hypopigmentation, or depigmentation. The others reported hyperpigmentation following imiquimod use.4 The imiquimod package insert lists application-site hypopigmentation as a possible adverse effect.5 Imiquimod-induced hypopigmentation and depigmentation have been reported in the peer-reviewed literature.4,6-14 Pigment loss has been reported in imiquimod treatment of condyloma acuminata, superficial BCC, nodular BCC, and extramammary Paget disease.6-8 Duration of therapy to onset of pigment loss ranged from 7 to 28 weeks.9 Imiquimod dosing varied among reported cases, ranging from 3 times weekly to daily application. Interestingly, hypopigmentation or depigmentation are not commonly associated with imiquimod use for the treatment of AKs, which Burnett and Kouba9 proposed may be due to the twice weekly imiquimod dosing regimen recommended by the FDA for the treatment of AK (below the minimum threshold for pigment loss). Our patient applied imiquimod cream 5% to periungual verruca vulgaris 3 times weekly for 3 months and may have developed vitiligolike depigmentation because he met this theoretical dosage threshold. Further research is necessary to confirm a dosage-related threshold for the development of depigmentation. Imiquimod-induced pigment loss has mainly been limited to the site of application.
Depigmentation was limited to the application site the majority of the time; however, depigmentation at adjacent sites has been reported.10 This finding was consistent with the proposed notion that cytokines induced by imiquimod have localized paracrine activity.11 Our patient was unique in that his depigmentation was present at the site of application, adjacent to the site of application, and at distant sites. He applied imiquimod only to the periungual area of the right fifth digit and left thumb but experienced depigmentation at several other sites. Although it is possible that our patient unintentionally spread imiquimod on the distant sites, it is less likely that the application would have been sufficient to cause depigmentation. Although systemic absorption of topical medications varies depending on multiple factors, the systemic absorption of imiquimod is minimal with mild systemic side effects reported, including headache, myalgia, and influenzalike symptoms.5 Thus, it is possible that our patient developed distant vitiligolike depigmentation as a systemic side effect of imiquimod therapy. Although our patient declined to have a biopsy performed, Gowda et al15 reported biopsy-proven vitiligo, demonstrating the absence of melanin and melanocytes following the use of imiquimod.
Several mechanisms have been proposed for imiquimod-induced depigmentation. For example, imiquimod may induce melanocyte apoptosis by increasing the levels of several proinflammatory and proapoptotic cytokines.16 Imiquimod-induced melanocyte apoptosis appears to involve elevated caspase-3, decreased B-cell lymphoma 2, altered mitogen-activated protein kinase expression, and ubiquitin-mediated proteolysis.13,17 Additionally, increased levels of IL-6 appear to increase melanocyte-binding molecules and increase melanocyte-leukocyte interactions. Another proposed theory targets toll-like receptor 7 on melanocytes that are acted on directly by imiquimod.11,17 In contrast, development of vitiligo following trauma (Koebner phenomenon) is not uncommon, and the immune effects induced by imiquimod may mimic those seen with trauma.14 Further research is needed to elucidate the mechanism by which imiquimod causes vitiligolike depigmentation.
Unfortunately, the depigmentation seen with imiquimod generally is permanent. Stefanaki et al10 showed repigmentation on cessation of imiquimod use. Our patient’s depigmentation remains unchanged despite treatment with tacrolimus ointment. Although it is possible for vitiligo to occur de novo without obvious inciting event or laboratory abnormality, the timeline and number of other cases in the literature make ours highly suspect for imiquimod-induced depigmentation.
Conclusion
Imiquimod is a commonly used immune-enhancing medication with an increasing list of off-label uses. Prior to prescribing imiquimod for a benign skin condition, clinicians should be cognizant of the potential for localized or possibly even distant depigmentation. We report a case of distant depigmentation following the use of imiquimod for periungual verruca vulgaris.
- Ganjian S, Ourian AJ, Shamtoub G, et al. Off-label indications for imiquimod. Dermatol Online J. 2009;15:4.
- Skinner RB Jr. Imiquimod. Dermatol Clin. 2003;21:291-300.
- Murphy K, Travers P, Walport M. Innate immunity. In: Murphy K, Travers P, Walport M, eds. Janeway’s Immunobiology. 7th ed. New York, NY: Garland Science. 2008:39-108.
- Brown T, Zirvi M, Cotsarelis G, et al. Vitiligo-like hypopigmentation associated with imiquimod treatment of genital warts. J Am Acad Dermatol. 2005;52:715-716.
- Aldara [package insert]. Bristol, TN: Graceway Pharmaceuticals, LLC; 2007.
- Kwon HH, Cho KH. Induction of vitiligo-like hypopigmentation after imiquimod treatment of extramammary Paget’s disease. Ann Dermatol. 2012;24:482-484.
- Mendonca CO, Yates VM. Permanent facial hypopigmentation following treatment with imiquimod. Clin Exp Dermatol. 2006;31:721-722.
- Zhang R, Zhu W. Genital vitiligo following use of imiquimod 5% cream. Indian J Dermatol. 2011;56:335-336.
- Burnett CT, Kouba DJ. Imiquimod-induced depigmentation: report of two cases and review of the literature. Dermatol Surg. 2012;38:1872-1875.
- Stefanaki C, Nicolaidou E, Hadjivassiliou M. Imiquimod-induced vitiligo in a patient with genital warts. J Eur Acad Dermatol Venereol. 2006;20:755-756.
- Al-Dujaili Z, Hsu S. Imiquimod-induced vitiligo. Dermatol Online J. 2007;13:10.
- Mashiah J, Brenner S. Possible mechanisms in the induction of vitiligo-like hypopigmentation by topical imiquimod. Clin Exp Dermatol. 2007;33:74-76.
- Grahovac M, Ehmann LM, Flaig M, et al. Giant basal cell carcinoma. Improvement and vitiligo-like hypopigmentation after intermittent treatment with 5% imiquimod. Acta Dermatovenerol Croat. 2012;20:275-278.
- Serrão VV, Páris FR, Feio AB. Genital vitiligo-like depigmentation following use of imiquimod 5% cream. Eur J Dermatol. 2008;18:342-343.
- Gowda S, Tillman DK, Fitzpatrick JE, et al. Imiquimod-induced vitiligo after treatment of nodular basal cell carcinoma. J Cutan Pathol. 2009;36:878-881.
- Kim CH, Ahn JH, Kang SU, et al. Imiquimod induces apoptosis of human melanocytes. Arch Dermatol Res. 2010;302:301-306.
- Eapen BR. Vitiligo, psoriasis, and imiquimod: fitting all into the same pathway. Indian J Dermatol Venereol Leprol. 2008;74:169.
Imiquimod is derived from the imidazoquinoline family and works by activating both innate and adaptive immune pathways. Imiquimod binds to toll-like receptor 7 located on monocytes, macrophages, and dendritic cells,1 which allows nuclear factor κβ light chain enhancer of activated B cells to induce production of proinflammatory cytokines, including IFN-α and tumor necrosis factor α, as well as IL-1, IL-6, IL-8, IL-10, and IL-12.2 These proinflammatory cytokines play a role in the innate immunity, triggering upregulation of the adaptive immune pathway and activating type 1 helper T cells, cytotoxic T cells, and natural killer cells. These cells have antiviral and antitumoral effects that lend to their significance in coordinating innate and adaptive immune mechanisms.3 More specifically, imiquimod enhances dendritic cell migration to regional lymph nodes and induces apoptosis via activation of proapoptotic B-cell lymphoma 2 proteins.1,2 Imiquimod has been approved by the US Food and Drug Administration (FDA) to treat external genitalia and perianal condyloma acuminata, actinic keratoses (AKs), and superficial basal cell carcinoma (BCC). It often is used off label for antiviral or antitumoral therapy in Bowen disease, squamous cell carcinoma, lentigo maligna, vulvar intraepithelial neoplasia, molluscum contagiosum, common warts, and leishmaniasis.1,2 Imiquimod is generally well tolerated; erythema and irritation at the application site are the most common side effects, with pigmentary change being less common.
Case Report
A 51-year-old man with a medical history of vitamin D deficiency, vitamin B12 deficiency, tinea pedis, and BCC presented with periungual verruca vulgaris on the right fifth digit and left thumb (Figure 1). The patient was prescribed imiquimod cream 5% to be applied 3 times weekly for 3 months. At 5-month follow-up the patient reported new-onset vitiligolike patches of depigmentation on the hands and feet that abruptly began 3 months after initiating treatment with imiquimod. On examination he had several depigmented patches with well-defined irregular borders on the bilateral dorsal hands and right foot as well as the right elbow (Figure 2). There was no personal or family history of vitiligo, thyroid disease, or autoimmune disease. Thyroid function studies and autoimmune panel were unremarkable. The patient also denied applying imiquimod to areas other than the periungual region of the right fifth digit and left thumb. He declined a biopsy of the lesions and was given a prescription for tacrolimus ointment 0.1% for twice-daily application. At 3-month follow-up the depigmented patches had spread. The patient is currently on 5-fluorouracil cream 5%. Despite loss of pigmentation, the periungual verruca vulgaris has persisted as well as depigmentation.
Comment
Imiquimod therapy is commonly used to treat conditions for which an antiviral or antitumor immune response is necessary for treatment and full resolution of skin conditions. It can yield positive results in conditions that are difficult to treat, such as periungual verruca vulgaris.4 The most common adverse effects of imiquimod include localized inflammation and application-site reactions. Pigment changes, though less common, also have been reported. From 1997 to 2003, 1257 cases of imiquimod adverse effects were reported to the FDA. There were 68 reported cases of pigmentary change, of which 51 documented vitiligo, hypopigmentation, or depigmentation. The others reported hyperpigmentation following imiquimod use.4 The imiquimod package insert lists application-site hypopigmentation as a possible adverse effect.5 Imiquimod-induced hypopigmentation and depigmentation have been reported in the peer-reviewed literature.4,6-14 Pigment loss has been reported in imiquimod treatment of condyloma acuminata, superficial BCC, nodular BCC, and extramammary Paget disease.6-8 Duration of therapy to onset of pigment loss ranged from 7 to 28 weeks.9 Imiquimod dosing varied among reported cases, ranging from 3 times weekly to daily application. Interestingly, hypopigmentation or depigmentation are not commonly associated with imiquimod use for the treatment of AKs, which Burnett and Kouba9 proposed may be due to the twice weekly imiquimod dosing regimen recommended by the FDA for the treatment of AK (below the minimum threshold for pigment loss). Our patient applied imiquimod cream 5% to periungual verruca vulgaris 3 times weekly for 3 months and may have developed vitiligolike depigmentation because he met this theoretical dosage threshold. Further research is necessary to confirm a dosage-related threshold for the development of depigmentation. Imiquimod-induced pigment loss has mainly been limited to the site of application.
Depigmentation was limited to the application site the majority of the time; however, depigmentation at adjacent sites has been reported.10 This finding was consistent with the proposed notion that cytokines induced by imiquimod have localized paracrine activity.11 Our patient was unique in that his depigmentation was present at the site of application, adjacent to the site of application, and at distant sites. He applied imiquimod only to the periungual area of the right fifth digit and left thumb but experienced depigmentation at several other sites. Although it is possible that our patient unintentionally spread imiquimod on the distant sites, it is less likely that the application would have been sufficient to cause depigmentation. Although systemic absorption of topical medications varies depending on multiple factors, the systemic absorption of imiquimod is minimal with mild systemic side effects reported, including headache, myalgia, and influenzalike symptoms.5 Thus, it is possible that our patient developed distant vitiligolike depigmentation as a systemic side effect of imiquimod therapy. Although our patient declined to have a biopsy performed, Gowda et al15 reported biopsy-proven vitiligo, demonstrating the absence of melanin and melanocytes following the use of imiquimod.
Several mechanisms have been proposed for imiquimod-induced depigmentation. For example, imiquimod may induce melanocyte apoptosis by increasing the levels of several proinflammatory and proapoptotic cytokines.16 Imiquimod-induced melanocyte apoptosis appears to involve elevated caspase-3, decreased B-cell lymphoma 2, altered mitogen-activated protein kinase expression, and ubiquitin-mediated proteolysis.13,17 Additionally, increased levels of IL-6 appear to increase melanocyte-binding molecules and increase melanocyte-leukocyte interactions. Another proposed theory targets toll-like receptor 7 on melanocytes that are acted on directly by imiquimod.11,17 In contrast, development of vitiligo following trauma (Koebner phenomenon) is not uncommon, and the immune effects induced by imiquimod may mimic those seen with trauma.14 Further research is needed to elucidate the mechanism by which imiquimod causes vitiligolike depigmentation.
Unfortunately, the depigmentation seen with imiquimod generally is permanent. Stefanaki et al10 showed repigmentation on cessation of imiquimod use. Our patient’s depigmentation remains unchanged despite treatment with tacrolimus ointment. Although it is possible for vitiligo to occur de novo without obvious inciting event or laboratory abnormality, the timeline and number of other cases in the literature make ours highly suspect for imiquimod-induced depigmentation.
Conclusion
Imiquimod is a commonly used immune-enhancing medication with an increasing list of off-label uses. Prior to prescribing imiquimod for a benign skin condition, clinicians should be cognizant of the potential for localized or possibly even distant depigmentation. We report a case of distant depigmentation following the use of imiquimod for periungual verruca vulgaris.
Imiquimod is derived from the imidazoquinoline family and works by activating both innate and adaptive immune pathways. Imiquimod binds to toll-like receptor 7 located on monocytes, macrophages, and dendritic cells,1 which allows nuclear factor κβ light chain enhancer of activated B cells to induce production of proinflammatory cytokines, including IFN-α and tumor necrosis factor α, as well as IL-1, IL-6, IL-8, IL-10, and IL-12.2 These proinflammatory cytokines play a role in the innate immunity, triggering upregulation of the adaptive immune pathway and activating type 1 helper T cells, cytotoxic T cells, and natural killer cells. These cells have antiviral and antitumoral effects that lend to their significance in coordinating innate and adaptive immune mechanisms.3 More specifically, imiquimod enhances dendritic cell migration to regional lymph nodes and induces apoptosis via activation of proapoptotic B-cell lymphoma 2 proteins.1,2 Imiquimod has been approved by the US Food and Drug Administration (FDA) to treat external genitalia and perianal condyloma acuminata, actinic keratoses (AKs), and superficial basal cell carcinoma (BCC). It often is used off label for antiviral or antitumoral therapy in Bowen disease, squamous cell carcinoma, lentigo maligna, vulvar intraepithelial neoplasia, molluscum contagiosum, common warts, and leishmaniasis.1,2 Imiquimod is generally well tolerated; erythema and irritation at the application site are the most common side effects, with pigmentary change being less common.
Case Report
A 51-year-old man with a medical history of vitamin D deficiency, vitamin B12 deficiency, tinea pedis, and BCC presented with periungual verruca vulgaris on the right fifth digit and left thumb (Figure 1). The patient was prescribed imiquimod cream 5% to be applied 3 times weekly for 3 months. At 5-month follow-up the patient reported new-onset vitiligolike patches of depigmentation on the hands and feet that abruptly began 3 months after initiating treatment with imiquimod. On examination he had several depigmented patches with well-defined irregular borders on the bilateral dorsal hands and right foot as well as the right elbow (Figure 2). There was no personal or family history of vitiligo, thyroid disease, or autoimmune disease. Thyroid function studies and autoimmune panel were unremarkable. The patient also denied applying imiquimod to areas other than the periungual region of the right fifth digit and left thumb. He declined a biopsy of the lesions and was given a prescription for tacrolimus ointment 0.1% for twice-daily application. At 3-month follow-up the depigmented patches had spread. The patient is currently on 5-fluorouracil cream 5%. Despite loss of pigmentation, the periungual verruca vulgaris has persisted as well as depigmentation.
Comment
Imiquimod therapy is commonly used to treat conditions for which an antiviral or antitumor immune response is necessary for treatment and full resolution of skin conditions. It can yield positive results in conditions that are difficult to treat, such as periungual verruca vulgaris.4 The most common adverse effects of imiquimod include localized inflammation and application-site reactions. Pigment changes, though less common, also have been reported. From 1997 to 2003, 1257 cases of imiquimod adverse effects were reported to the FDA. There were 68 reported cases of pigmentary change, of which 51 documented vitiligo, hypopigmentation, or depigmentation. The others reported hyperpigmentation following imiquimod use.4 The imiquimod package insert lists application-site hypopigmentation as a possible adverse effect.5 Imiquimod-induced hypopigmentation and depigmentation have been reported in the peer-reviewed literature.4,6-14 Pigment loss has been reported in imiquimod treatment of condyloma acuminata, superficial BCC, nodular BCC, and extramammary Paget disease.6-8 Duration of therapy to onset of pigment loss ranged from 7 to 28 weeks.9 Imiquimod dosing varied among reported cases, ranging from 3 times weekly to daily application. Interestingly, hypopigmentation or depigmentation are not commonly associated with imiquimod use for the treatment of AKs, which Burnett and Kouba9 proposed may be due to the twice weekly imiquimod dosing regimen recommended by the FDA for the treatment of AK (below the minimum threshold for pigment loss). Our patient applied imiquimod cream 5% to periungual verruca vulgaris 3 times weekly for 3 months and may have developed vitiligolike depigmentation because he met this theoretical dosage threshold. Further research is necessary to confirm a dosage-related threshold for the development of depigmentation. Imiquimod-induced pigment loss has mainly been limited to the site of application.
Depigmentation was limited to the application site the majority of the time; however, depigmentation at adjacent sites has been reported.10 This finding was consistent with the proposed notion that cytokines induced by imiquimod have localized paracrine activity.11 Our patient was unique in that his depigmentation was present at the site of application, adjacent to the site of application, and at distant sites. He applied imiquimod only to the periungual area of the right fifth digit and left thumb but experienced depigmentation at several other sites. Although it is possible that our patient unintentionally spread imiquimod on the distant sites, it is less likely that the application would have been sufficient to cause depigmentation. Although systemic absorption of topical medications varies depending on multiple factors, the systemic absorption of imiquimod is minimal with mild systemic side effects reported, including headache, myalgia, and influenzalike symptoms.5 Thus, it is possible that our patient developed distant vitiligolike depigmentation as a systemic side effect of imiquimod therapy. Although our patient declined to have a biopsy performed, Gowda et al15 reported biopsy-proven vitiligo, demonstrating the absence of melanin and melanocytes following the use of imiquimod.
Several mechanisms have been proposed for imiquimod-induced depigmentation. For example, imiquimod may induce melanocyte apoptosis by increasing the levels of several proinflammatory and proapoptotic cytokines.16 Imiquimod-induced melanocyte apoptosis appears to involve elevated caspase-3, decreased B-cell lymphoma 2, altered mitogen-activated protein kinase expression, and ubiquitin-mediated proteolysis.13,17 Additionally, increased levels of IL-6 appear to increase melanocyte-binding molecules and increase melanocyte-leukocyte interactions. Another proposed theory targets toll-like receptor 7 on melanocytes that are acted on directly by imiquimod.11,17 In contrast, development of vitiligo following trauma (Koebner phenomenon) is not uncommon, and the immune effects induced by imiquimod may mimic those seen with trauma.14 Further research is needed to elucidate the mechanism by which imiquimod causes vitiligolike depigmentation.
Unfortunately, the depigmentation seen with imiquimod generally is permanent. Stefanaki et al10 showed repigmentation on cessation of imiquimod use. Our patient’s depigmentation remains unchanged despite treatment with tacrolimus ointment. Although it is possible for vitiligo to occur de novo without obvious inciting event or laboratory abnormality, the timeline and number of other cases in the literature make ours highly suspect for imiquimod-induced depigmentation.
Conclusion
Imiquimod is a commonly used immune-enhancing medication with an increasing list of off-label uses. Prior to prescribing imiquimod for a benign skin condition, clinicians should be cognizant of the potential for localized or possibly even distant depigmentation. We report a case of distant depigmentation following the use of imiquimod for periungual verruca vulgaris.
- Ganjian S, Ourian AJ, Shamtoub G, et al. Off-label indications for imiquimod. Dermatol Online J. 2009;15:4.
- Skinner RB Jr. Imiquimod. Dermatol Clin. 2003;21:291-300.
- Murphy K, Travers P, Walport M. Innate immunity. In: Murphy K, Travers P, Walport M, eds. Janeway’s Immunobiology. 7th ed. New York, NY: Garland Science. 2008:39-108.
- Brown T, Zirvi M, Cotsarelis G, et al. Vitiligo-like hypopigmentation associated with imiquimod treatment of genital warts. J Am Acad Dermatol. 2005;52:715-716.
- Aldara [package insert]. Bristol, TN: Graceway Pharmaceuticals, LLC; 2007.
- Kwon HH, Cho KH. Induction of vitiligo-like hypopigmentation after imiquimod treatment of extramammary Paget’s disease. Ann Dermatol. 2012;24:482-484.
- Mendonca CO, Yates VM. Permanent facial hypopigmentation following treatment with imiquimod. Clin Exp Dermatol. 2006;31:721-722.
- Zhang R, Zhu W. Genital vitiligo following use of imiquimod 5% cream. Indian J Dermatol. 2011;56:335-336.
- Burnett CT, Kouba DJ. Imiquimod-induced depigmentation: report of two cases and review of the literature. Dermatol Surg. 2012;38:1872-1875.
- Stefanaki C, Nicolaidou E, Hadjivassiliou M. Imiquimod-induced vitiligo in a patient with genital warts. J Eur Acad Dermatol Venereol. 2006;20:755-756.
- Al-Dujaili Z, Hsu S. Imiquimod-induced vitiligo. Dermatol Online J. 2007;13:10.
- Mashiah J, Brenner S. Possible mechanisms in the induction of vitiligo-like hypopigmentation by topical imiquimod. Clin Exp Dermatol. 2007;33:74-76.
- Grahovac M, Ehmann LM, Flaig M, et al. Giant basal cell carcinoma. Improvement and vitiligo-like hypopigmentation after intermittent treatment with 5% imiquimod. Acta Dermatovenerol Croat. 2012;20:275-278.
- Serrão VV, Páris FR, Feio AB. Genital vitiligo-like depigmentation following use of imiquimod 5% cream. Eur J Dermatol. 2008;18:342-343.
- Gowda S, Tillman DK, Fitzpatrick JE, et al. Imiquimod-induced vitiligo after treatment of nodular basal cell carcinoma. J Cutan Pathol. 2009;36:878-881.
- Kim CH, Ahn JH, Kang SU, et al. Imiquimod induces apoptosis of human melanocytes. Arch Dermatol Res. 2010;302:301-306.
- Eapen BR. Vitiligo, psoriasis, and imiquimod: fitting all into the same pathway. Indian J Dermatol Venereol Leprol. 2008;74:169.
- Ganjian S, Ourian AJ, Shamtoub G, et al. Off-label indications for imiquimod. Dermatol Online J. 2009;15:4.
- Skinner RB Jr. Imiquimod. Dermatol Clin. 2003;21:291-300.
- Murphy K, Travers P, Walport M. Innate immunity. In: Murphy K, Travers P, Walport M, eds. Janeway’s Immunobiology. 7th ed. New York, NY: Garland Science. 2008:39-108.
- Brown T, Zirvi M, Cotsarelis G, et al. Vitiligo-like hypopigmentation associated with imiquimod treatment of genital warts. J Am Acad Dermatol. 2005;52:715-716.
- Aldara [package insert]. Bristol, TN: Graceway Pharmaceuticals, LLC; 2007.
- Kwon HH, Cho KH. Induction of vitiligo-like hypopigmentation after imiquimod treatment of extramammary Paget’s disease. Ann Dermatol. 2012;24:482-484.
- Mendonca CO, Yates VM. Permanent facial hypopigmentation following treatment with imiquimod. Clin Exp Dermatol. 2006;31:721-722.
- Zhang R, Zhu W. Genital vitiligo following use of imiquimod 5% cream. Indian J Dermatol. 2011;56:335-336.
- Burnett CT, Kouba DJ. Imiquimod-induced depigmentation: report of two cases and review of the literature. Dermatol Surg. 2012;38:1872-1875.
- Stefanaki C, Nicolaidou E, Hadjivassiliou M. Imiquimod-induced vitiligo in a patient with genital warts. J Eur Acad Dermatol Venereol. 2006;20:755-756.
- Al-Dujaili Z, Hsu S. Imiquimod-induced vitiligo. Dermatol Online J. 2007;13:10.
- Mashiah J, Brenner S. Possible mechanisms in the induction of vitiligo-like hypopigmentation by topical imiquimod. Clin Exp Dermatol. 2007;33:74-76.
- Grahovac M, Ehmann LM, Flaig M, et al. Giant basal cell carcinoma. Improvement and vitiligo-like hypopigmentation after intermittent treatment with 5% imiquimod. Acta Dermatovenerol Croat. 2012;20:275-278.
- Serrão VV, Páris FR, Feio AB. Genital vitiligo-like depigmentation following use of imiquimod 5% cream. Eur J Dermatol. 2008;18:342-343.
- Gowda S, Tillman DK, Fitzpatrick JE, et al. Imiquimod-induced vitiligo after treatment of nodular basal cell carcinoma. J Cutan Pathol. 2009;36:878-881.
- Kim CH, Ahn JH, Kang SU, et al. Imiquimod induces apoptosis of human melanocytes. Arch Dermatol Res. 2010;302:301-306.
- Eapen BR. Vitiligo, psoriasis, and imiquimod: fitting all into the same pathway. Indian J Dermatol Venereol Leprol. 2008;74:169.
Practice Points
- Imiquimod commonly is used off label to treat viral and neoplastic processes.
- Clinicians should be aware of the potential for dyspigmentation or depigmentation as a side effect from treatment.
Necrobiotic Xanthogranuloma Without a Monoclonal Gammopathy
Necrobiotic xanthogranuloma (NXG) was first described in 1980 by Kossard and Winkelmann1 as a xanthomatosis associated with paraproteinemia. It is an indolent disorder characterized by indurated, yellow to violaceous red papules, plaques, or nodules often presenting with telangiectases and ulceration.2 The lesions have a predilection for the bilateral periorbital region in the majority of documented cases, consequently producing ocular findings such as periocular skin lesions, blepharoptosis, restricted ocular motility, and proptosis.3
Necrobiotic xanthogranuloma is a systemic disease that may involve extracutaneous sites such as the heart, respiratory tract, spleen, kidneys, ovaries, liver, skeletal muscle, and central nervous system.4-6 The most common sites include the respiratory tract and heart, with documented cases of pulmonary and myocardial giant cell granulomas.4 In a 2009 review, Spicknall and Mehregan7 reported an increased frequency of systemic involvement.
The distinctive histopathologic features of NXG consist of large bands of necrobiosis and a pattern of palisading histiocytic granulomas comprised of Touton giant cells, bizarre foreign body giant cells, foam cells, and cholesterol clefts.8 These histopathologic findings differentiate NXG from other clinical differential diagnoses such as necrobiosis lipoidica.
Necrobiotic xanthogranuloma is associated with paraproteinemia in 80% of documented cases, most commonly as an IgG monoclonal gammopathy.2 The etiology of this indolent disorder remains unclear despite proposed theories of its pathogenesis. Consequently, treatment proves difficult with no recommended first-line therapy and a tendency for recurrent cutaneous lesions. We report an unusual case of periorbital NXG without development of a monoclonal gammopathy.
Case Report
A 76-year-old man presented with a long-standing history (30 years) of bilateral periorbital NXG. Approximately 30 years prior to the current presentation, the patient presented to a dermatologist with dry eyes and periorbital cutaneous lesions that were originally diagnosed as xanthelasma. He later developed edema of the right periorbital region that progressed to involve the left periorbital region. He underwent surgical excision of the lesions 10 years prior to the current presentation, which showed the lesions were infiltrating into the muscle. At that time, a diagnosis of NXG was made. The department of plastic surgery at an outside institution evaluated the patient and identified no further treatment options; however, annual computed tomography scans were performed to detect disease progression.
The patient presented for increasing periorbital manifestations of NXG. He denied any other remarkable medical history or any family history of NXG, malignancy, or hematologic disorders. His surgical history was exclusive to the excisional surgery of the periorbital lesions. At the time of presentation he was not taking medications and had no known drug allergies. He denied tobacco use but occasionally consumed alcohol.
On systemic inquiry the patient’s only concerns were ocular in nature and included dry, sensitive, and painful eyes. Dermatologic examination revealed substantial periorbital edema with multiple yellow indurated plaques (Figure). There were no additional findings on physical examination.
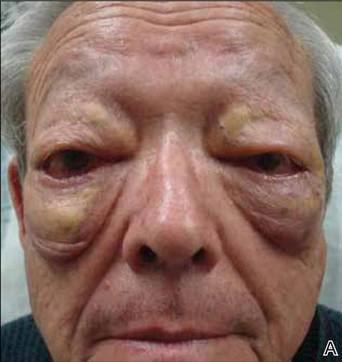
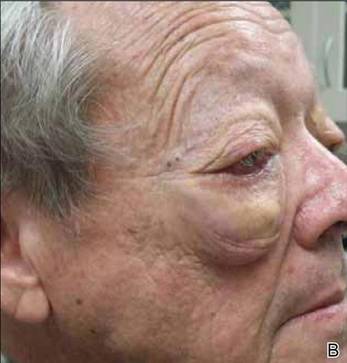
Bilateral periorbital edema with yellow indurated plaques (A). A lateral view showed substantial right periorbital edema with multiple yellow plaques (B). |
Extensive hematologic and oncologic investigations revealed the absence of a monoclonal gammopathy. Serum protein electrophoresis was negative for paraproteinemia and quantitative serum immunoglobulin testing was normal. A complete blood cell count, lipid panel, CD4 count, CD8 count, C3, C4, and computed tomography scan did not reveal any abnormalities. A complete metabolic panel identified elevated serum glucose levels (162 mg/dL [reference range, 74–118 mg/dL]), low serum albumin levels (3.3 g/dL [reference range, 3.5–4.8 g/dL]), and low serum calcium levels (8.8 mg/dL [reference range, 8.9–10.3 mg/dL]). IgG subclass (SC) proteins were mildly increasedwith an IgG SC1 of 950 mg/dL (reference range, 382–929 mg/dL), IgG SC3 of 211 mg/dL (reference range, 22–178 mg/dL), and IgG SC4 of 292 mg/dL (reference range, 4–86 mg/dL), and the plasma IgG was in the upper limit of the reference range with a value of 1591 mg/dL (reference range, 791–1643 mg/dL).
After the hematologic and oncologic workup was completed, intravenous immunoglobulin and acitretin were recommended to the patient as viable treatment options to reduce the cutaneous sequelae of NXG. A 6-month regimen of acitretin markedly improved cutaneous edema and plaque size. However, these sequelae returned to baseline just months after acitretin was discontinued.
Comment
Necrobiotic xanthogranuloma is a distinct granulomatous disorder with no predilection for sex and the average age of onset is 54 years.2 Consistent with prior reports, our patient presented with bilateral periorbital lesions and ophthalmic concerns of dryness, burning, and sensitivity. Reddy et al9 demonstrated that aggressive forms of periorbital NXG may involve ocular tissues and result in vision loss and corneal perforation. On follow-up, our patient underwent annual computed tomography scans to rule out further progression.
Eighty percent of patients diagnosed with NXG have an associated monoclonal gammopathy and 10% develop multiple myeloma.2 Our case presents an unusual variant of NXG due to the absence of a monoclonal gammopathy. Chang et al10 described a similar case of NXG without a monoclonal gammopathy and hypothesized that periorbital involvement, malignancy, and systemic involvement are the main contributing factors to the morbidity of NXG.
Our patient had mildly elevated IgG SC1, IgG SC3, and IgG SC4 levels. The most substantially elevated subclass was IgG SC4. Elevations of IgG SC4 often are associated with disorders that are allergic or autoimmune in nature, such as pemphigus vulgaris, autoimmune pancreatitis, and inflammatory pseudotumor.11 Our patient denied prior history and lacked manifestations of allergic or autoimmune disorders. A similar case was reported in a 67-year-old man with periorbital NXG and elevated IgG SC4 levels. Singh et al12 postulated that systemic elevation of IgG SC4 can be associated with NXG of the orbit.
Due to the rarity and uncertain etiology of NXG, there are no definitive first-line therapies.There have been encouraging results with intravenous immunoglobulin,13 autologous stem cell transplantation,14 lenalidomide,15 melphalan with prednisolone,16 and chlorambucil with low-dose corticosteroids.11 In 2007, Ho et al17 identified that CD20 and CD25 were both strongly expressed in tissue specimens of NXG, indicating the possible effectiveness of rituximab and denileukin diftitox, which target CD20 and CD25, respectively. It is unclear how these data pertain to patients without paraproteinemia because the treatment often is directed at the monoclonal gammopathy. These uncertainties are concerning because of the undesirable and often toxic side effects associated with these therapies. Psoralen plus UVA therapy was described as an alternative to cytotoxic drugs and immunosuppressive agents in 1 patient without paraproteinemia.18
Bullock et al19 proposed that NXG arises from a foreign body giant cell reaction resulting from circulating immune complexes precipitating in periorbital tissues. Although the relationship between the cutaneous manifestations and a monoclonal gammopathy remains poorly understood, cases of NXG without paraproteinemia challenge this theory. Our case supports the theory that there is no correlation between the histopathologic findings of NXG and the extent of the monoclonal gammopathy.
Conclusion
The etiology and pathogenesis of NXG remain elusive. We strive to attain a more sophisticated understanding of NXG to identify efficacious treatment options. Our case highlights the ambiguous association between the cutaneous lesions of NXG and a monoclonal gammopathy.
1. Kossard S, Winkelmann RK. Necrobiotic xanthogranuloma with paraproteinemia. J Am Acad Dermatol. 1980;3:257-270.
2. Mehregan DA, Winkelmann RK. Necrobiotic xanthogranuloma [published correction in Arch Dermatol. 1992;128:632]. Arch Dermatol. 1992;128:94-100.
3. Ugurlu S, Bartley GB, Gibson LE. Necrobiotic xanthogranuloma: long-term outcome of ocular and systemic involvement. Am J Ophthalmol. 2000;129:651-657.
4. Winkelmann RK, Litzow MR, Umbert IJ, et al. Giant cell granulomatous pulmonary and myocardial lesions in necrobiotic xanthogranuloma with paraproteinemia. Mayo Clin Proc. 1997;72:1028-1033.
5. Shah KC, Poonnoose SI, George R, et al. Necrobiotic xanthogranuloma with cutaneous and cerebral manifestations. case report and review of literature. J Neurosurg. 2004;100:1111-1114.
6. Umbert I, Winkelmann RK. Necrobiotic xanthogranuloma with cardiac involvement. Br J Dermatol. 1995;133:438-443.
7. Spicknall KE, Mehregan DA. Necrobiotic xanthogranuloma. Int J Dermatol. 2009;48:1-10.
8. Finan MC, Winkelmann RK. Histopathology of necrobiotic xanthogranuloma with paraproteinemia. J Cutan Pathol. 1987;14:92-99.
9. Reddy VC, Salomão DR, Garrity JA, et al. Periorbital and ocular necrobiotic xanthogranuloma leading to perforation. Arch Ophthalmol. 2010;128:1493-1494.
10. Chang SE, Lee WS, Lee MW, et al. A case of necrobiotic xanthogranuloma without paraproteinemia presenting as a solitary tumor on the thigh. Int J Dermatol. 2003;42:470-472.
11. Wood AJ, Wagner MV, Abbott JJ, et al. Necrobiotic xanthogranuloma: a review of 17 cases with emphasis on clinical and pathologic correlation. Arch Dermatol. 2009;145:279-284.
12. Singh K, Rajan KD, Eberhart C. Orbital necrobiotic xanthogranuloma associated with systemic IgG4 disease. Ocul Immunol Inflamm. 2010;18:373-378.
13. Hallermann C, Tittelbach J, Norgauer J, et al. Successful treatment of necrobiotic xanthogranuloma with intravenous immunoglobulin. Arch Dermatol. 2009;146:957-960.
14. Goede JS, Misselwitz B, Taverna C, et al. Necrobiotic xanthogranuloma successfully treated with autologous stem cell transplantation. Ann Hematol. 2007;86:303-306.
15. Silapunt S, Chon SY. Generalized necrobiotic xanthogranuloma successfully treated with lenalidomide. J Drugs Dermatol. 2010;9:273-276.
16. Saeki H, Tomita M, Kai H, et al. Necrobiotic xanthogranuloma with paraproteinemia successfully treated with melphalan, prednisolone and skin graft. J Dermatol. 2007;34:795-797.
17. Ho VH, Chevez-Barrios P, Jorgensen JL, et al. Receptor expression in orbital inflammatory syndromes and implications for targeted therapy. Tissue Antigens. 2007;70:105-109.
18. Al-Niaimi FA, Dawn G, Cox NH. Necrobiotic xanthogranuloma without paraproteinemia: marked improvement with psoralen ultraviolet A treatment. Clin Exp Dermatol. 2010;35:275-277.
19. Bullock JD, Bartley GB, Campbell RJ, et al. Necrobiotic xanthogranuloma with paraproteinemia: case report and a pathogenetic theory. Trans Am Ophthalmol Soc. 1986;84:342-352.
Necrobiotic xanthogranuloma (NXG) was first described in 1980 by Kossard and Winkelmann1 as a xanthomatosis associated with paraproteinemia. It is an indolent disorder characterized by indurated, yellow to violaceous red papules, plaques, or nodules often presenting with telangiectases and ulceration.2 The lesions have a predilection for the bilateral periorbital region in the majority of documented cases, consequently producing ocular findings such as periocular skin lesions, blepharoptosis, restricted ocular motility, and proptosis.3
Necrobiotic xanthogranuloma is a systemic disease that may involve extracutaneous sites such as the heart, respiratory tract, spleen, kidneys, ovaries, liver, skeletal muscle, and central nervous system.4-6 The most common sites include the respiratory tract and heart, with documented cases of pulmonary and myocardial giant cell granulomas.4 In a 2009 review, Spicknall and Mehregan7 reported an increased frequency of systemic involvement.
The distinctive histopathologic features of NXG consist of large bands of necrobiosis and a pattern of palisading histiocytic granulomas comprised of Touton giant cells, bizarre foreign body giant cells, foam cells, and cholesterol clefts.8 These histopathologic findings differentiate NXG from other clinical differential diagnoses such as necrobiosis lipoidica.
Necrobiotic xanthogranuloma is associated with paraproteinemia in 80% of documented cases, most commonly as an IgG monoclonal gammopathy.2 The etiology of this indolent disorder remains unclear despite proposed theories of its pathogenesis. Consequently, treatment proves difficult with no recommended first-line therapy and a tendency for recurrent cutaneous lesions. We report an unusual case of periorbital NXG without development of a monoclonal gammopathy.
Case Report
A 76-year-old man presented with a long-standing history (30 years) of bilateral periorbital NXG. Approximately 30 years prior to the current presentation, the patient presented to a dermatologist with dry eyes and periorbital cutaneous lesions that were originally diagnosed as xanthelasma. He later developed edema of the right periorbital region that progressed to involve the left periorbital region. He underwent surgical excision of the lesions 10 years prior to the current presentation, which showed the lesions were infiltrating into the muscle. At that time, a diagnosis of NXG was made. The department of plastic surgery at an outside institution evaluated the patient and identified no further treatment options; however, annual computed tomography scans were performed to detect disease progression.
The patient presented for increasing periorbital manifestations of NXG. He denied any other remarkable medical history or any family history of NXG, malignancy, or hematologic disorders. His surgical history was exclusive to the excisional surgery of the periorbital lesions. At the time of presentation he was not taking medications and had no known drug allergies. He denied tobacco use but occasionally consumed alcohol.
On systemic inquiry the patient’s only concerns were ocular in nature and included dry, sensitive, and painful eyes. Dermatologic examination revealed substantial periorbital edema with multiple yellow indurated plaques (Figure). There were no additional findings on physical examination.
|
Bilateral periorbital edema with yellow indurated plaques (A). A lateral view showed substantial right periorbital edema with multiple yellow plaques (B). |
Extensive hematologic and oncologic investigations revealed the absence of a monoclonal gammopathy. Serum protein electrophoresis was negative for paraproteinemia and quantitative serum immunoglobulin testing was normal. A complete blood cell count, lipid panel, CD4 count, CD8 count, C3, C4, and computed tomography scan did not reveal any abnormalities. A complete metabolic panel identified elevated serum glucose levels (162 mg/dL [reference range, 74–118 mg/dL]), low serum albumin levels (3.3 g/dL [reference range, 3.5–4.8 g/dL]), and low serum calcium levels (8.8 mg/dL [reference range, 8.9–10.3 mg/dL]). IgG subclass (SC) proteins were mildly increasedwith an IgG SC1 of 950 mg/dL (reference range, 382–929 mg/dL), IgG SC3 of 211 mg/dL (reference range, 22–178 mg/dL), and IgG SC4 of 292 mg/dL (reference range, 4–86 mg/dL), and the plasma IgG was in the upper limit of the reference range with a value of 1591 mg/dL (reference range, 791–1643 mg/dL).
After the hematologic and oncologic workup was completed, intravenous immunoglobulin and acitretin were recommended to the patient as viable treatment options to reduce the cutaneous sequelae of NXG. A 6-month regimen of acitretin markedly improved cutaneous edema and plaque size. However, these sequelae returned to baseline just months after acitretin was discontinued.
Comment
Necrobiotic xanthogranuloma is a distinct granulomatous disorder with no predilection for sex and the average age of onset is 54 years.2 Consistent with prior reports, our patient presented with bilateral periorbital lesions and ophthalmic concerns of dryness, burning, and sensitivity. Reddy et al9 demonstrated that aggressive forms of periorbital NXG may involve ocular tissues and result in vision loss and corneal perforation. On follow-up, our patient underwent annual computed tomography scans to rule out further progression.
Eighty percent of patients diagnosed with NXG have an associated monoclonal gammopathy and 10% develop multiple myeloma.2 Our case presents an unusual variant of NXG due to the absence of a monoclonal gammopathy. Chang et al10 described a similar case of NXG without a monoclonal gammopathy and hypothesized that periorbital involvement, malignancy, and systemic involvement are the main contributing factors to the morbidity of NXG.
Our patient had mildly elevated IgG SC1, IgG SC3, and IgG SC4 levels. The most substantially elevated subclass was IgG SC4. Elevations of IgG SC4 often are associated with disorders that are allergic or autoimmune in nature, such as pemphigus vulgaris, autoimmune pancreatitis, and inflammatory pseudotumor.11 Our patient denied prior history and lacked manifestations of allergic or autoimmune disorders. A similar case was reported in a 67-year-old man with periorbital NXG and elevated IgG SC4 levels. Singh et al12 postulated that systemic elevation of IgG SC4 can be associated with NXG of the orbit.
Due to the rarity and uncertain etiology of NXG, there are no definitive first-line therapies.There have been encouraging results with intravenous immunoglobulin,13 autologous stem cell transplantation,14 lenalidomide,15 melphalan with prednisolone,16 and chlorambucil with low-dose corticosteroids.11 In 2007, Ho et al17 identified that CD20 and CD25 were both strongly expressed in tissue specimens of NXG, indicating the possible effectiveness of rituximab and denileukin diftitox, which target CD20 and CD25, respectively. It is unclear how these data pertain to patients without paraproteinemia because the treatment often is directed at the monoclonal gammopathy. These uncertainties are concerning because of the undesirable and often toxic side effects associated with these therapies. Psoralen plus UVA therapy was described as an alternative to cytotoxic drugs and immunosuppressive agents in 1 patient without paraproteinemia.18
Bullock et al19 proposed that NXG arises from a foreign body giant cell reaction resulting from circulating immune complexes precipitating in periorbital tissues. Although the relationship between the cutaneous manifestations and a monoclonal gammopathy remains poorly understood, cases of NXG without paraproteinemia challenge this theory. Our case supports the theory that there is no correlation between the histopathologic findings of NXG and the extent of the monoclonal gammopathy.
Conclusion
The etiology and pathogenesis of NXG remain elusive. We strive to attain a more sophisticated understanding of NXG to identify efficacious treatment options. Our case highlights the ambiguous association between the cutaneous lesions of NXG and a monoclonal gammopathy.
Necrobiotic xanthogranuloma (NXG) was first described in 1980 by Kossard and Winkelmann1 as a xanthomatosis associated with paraproteinemia. It is an indolent disorder characterized by indurated, yellow to violaceous red papules, plaques, or nodules often presenting with telangiectases and ulceration.2 The lesions have a predilection for the bilateral periorbital region in the majority of documented cases, consequently producing ocular findings such as periocular skin lesions, blepharoptosis, restricted ocular motility, and proptosis.3
Necrobiotic xanthogranuloma is a systemic disease that may involve extracutaneous sites such as the heart, respiratory tract, spleen, kidneys, ovaries, liver, skeletal muscle, and central nervous system.4-6 The most common sites include the respiratory tract and heart, with documented cases of pulmonary and myocardial giant cell granulomas.4 In a 2009 review, Spicknall and Mehregan7 reported an increased frequency of systemic involvement.
The distinctive histopathologic features of NXG consist of large bands of necrobiosis and a pattern of palisading histiocytic granulomas comprised of Touton giant cells, bizarre foreign body giant cells, foam cells, and cholesterol clefts.8 These histopathologic findings differentiate NXG from other clinical differential diagnoses such as necrobiosis lipoidica.
Necrobiotic xanthogranuloma is associated with paraproteinemia in 80% of documented cases, most commonly as an IgG monoclonal gammopathy.2 The etiology of this indolent disorder remains unclear despite proposed theories of its pathogenesis. Consequently, treatment proves difficult with no recommended first-line therapy and a tendency for recurrent cutaneous lesions. We report an unusual case of periorbital NXG without development of a monoclonal gammopathy.
Case Report
A 76-year-old man presented with a long-standing history (30 years) of bilateral periorbital NXG. Approximately 30 years prior to the current presentation, the patient presented to a dermatologist with dry eyes and periorbital cutaneous lesions that were originally diagnosed as xanthelasma. He later developed edema of the right periorbital region that progressed to involve the left periorbital region. He underwent surgical excision of the lesions 10 years prior to the current presentation, which showed the lesions were infiltrating into the muscle. At that time, a diagnosis of NXG was made. The department of plastic surgery at an outside institution evaluated the patient and identified no further treatment options; however, annual computed tomography scans were performed to detect disease progression.
The patient presented for increasing periorbital manifestations of NXG. He denied any other remarkable medical history or any family history of NXG, malignancy, or hematologic disorders. His surgical history was exclusive to the excisional surgery of the periorbital lesions. At the time of presentation he was not taking medications and had no known drug allergies. He denied tobacco use but occasionally consumed alcohol.
On systemic inquiry the patient’s only concerns were ocular in nature and included dry, sensitive, and painful eyes. Dermatologic examination revealed substantial periorbital edema with multiple yellow indurated plaques (Figure). There were no additional findings on physical examination.
|
Bilateral periorbital edema with yellow indurated plaques (A). A lateral view showed substantial right periorbital edema with multiple yellow plaques (B). |
Extensive hematologic and oncologic investigations revealed the absence of a monoclonal gammopathy. Serum protein electrophoresis was negative for paraproteinemia and quantitative serum immunoglobulin testing was normal. A complete blood cell count, lipid panel, CD4 count, CD8 count, C3, C4, and computed tomography scan did not reveal any abnormalities. A complete metabolic panel identified elevated serum glucose levels (162 mg/dL [reference range, 74–118 mg/dL]), low serum albumin levels (3.3 g/dL [reference range, 3.5–4.8 g/dL]), and low serum calcium levels (8.8 mg/dL [reference range, 8.9–10.3 mg/dL]). IgG subclass (SC) proteins were mildly increasedwith an IgG SC1 of 950 mg/dL (reference range, 382–929 mg/dL), IgG SC3 of 211 mg/dL (reference range, 22–178 mg/dL), and IgG SC4 of 292 mg/dL (reference range, 4–86 mg/dL), and the plasma IgG was in the upper limit of the reference range with a value of 1591 mg/dL (reference range, 791–1643 mg/dL).
After the hematologic and oncologic workup was completed, intravenous immunoglobulin and acitretin were recommended to the patient as viable treatment options to reduce the cutaneous sequelae of NXG. A 6-month regimen of acitretin markedly improved cutaneous edema and plaque size. However, these sequelae returned to baseline just months after acitretin was discontinued.
Comment
Necrobiotic xanthogranuloma is a distinct granulomatous disorder with no predilection for sex and the average age of onset is 54 years.2 Consistent with prior reports, our patient presented with bilateral periorbital lesions and ophthalmic concerns of dryness, burning, and sensitivity. Reddy et al9 demonstrated that aggressive forms of periorbital NXG may involve ocular tissues and result in vision loss and corneal perforation. On follow-up, our patient underwent annual computed tomography scans to rule out further progression.
Eighty percent of patients diagnosed with NXG have an associated monoclonal gammopathy and 10% develop multiple myeloma.2 Our case presents an unusual variant of NXG due to the absence of a monoclonal gammopathy. Chang et al10 described a similar case of NXG without a monoclonal gammopathy and hypothesized that periorbital involvement, malignancy, and systemic involvement are the main contributing factors to the morbidity of NXG.
Our patient had mildly elevated IgG SC1, IgG SC3, and IgG SC4 levels. The most substantially elevated subclass was IgG SC4. Elevations of IgG SC4 often are associated with disorders that are allergic or autoimmune in nature, such as pemphigus vulgaris, autoimmune pancreatitis, and inflammatory pseudotumor.11 Our patient denied prior history and lacked manifestations of allergic or autoimmune disorders. A similar case was reported in a 67-year-old man with periorbital NXG and elevated IgG SC4 levels. Singh et al12 postulated that systemic elevation of IgG SC4 can be associated with NXG of the orbit.
Due to the rarity and uncertain etiology of NXG, there are no definitive first-line therapies.There have been encouraging results with intravenous immunoglobulin,13 autologous stem cell transplantation,14 lenalidomide,15 melphalan with prednisolone,16 and chlorambucil with low-dose corticosteroids.11 In 2007, Ho et al17 identified that CD20 and CD25 were both strongly expressed in tissue specimens of NXG, indicating the possible effectiveness of rituximab and denileukin diftitox, which target CD20 and CD25, respectively. It is unclear how these data pertain to patients without paraproteinemia because the treatment often is directed at the monoclonal gammopathy. These uncertainties are concerning because of the undesirable and often toxic side effects associated with these therapies. Psoralen plus UVA therapy was described as an alternative to cytotoxic drugs and immunosuppressive agents in 1 patient without paraproteinemia.18
Bullock et al19 proposed that NXG arises from a foreign body giant cell reaction resulting from circulating immune complexes precipitating in periorbital tissues. Although the relationship between the cutaneous manifestations and a monoclonal gammopathy remains poorly understood, cases of NXG without paraproteinemia challenge this theory. Our case supports the theory that there is no correlation between the histopathologic findings of NXG and the extent of the monoclonal gammopathy.
Conclusion
The etiology and pathogenesis of NXG remain elusive. We strive to attain a more sophisticated understanding of NXG to identify efficacious treatment options. Our case highlights the ambiguous association between the cutaneous lesions of NXG and a monoclonal gammopathy.
1. Kossard S, Winkelmann RK. Necrobiotic xanthogranuloma with paraproteinemia. J Am Acad Dermatol. 1980;3:257-270.
2. Mehregan DA, Winkelmann RK. Necrobiotic xanthogranuloma [published correction in Arch Dermatol. 1992;128:632]. Arch Dermatol. 1992;128:94-100.
3. Ugurlu S, Bartley GB, Gibson LE. Necrobiotic xanthogranuloma: long-term outcome of ocular and systemic involvement. Am J Ophthalmol. 2000;129:651-657.
4. Winkelmann RK, Litzow MR, Umbert IJ, et al. Giant cell granulomatous pulmonary and myocardial lesions in necrobiotic xanthogranuloma with paraproteinemia. Mayo Clin Proc. 1997;72:1028-1033.
5. Shah KC, Poonnoose SI, George R, et al. Necrobiotic xanthogranuloma with cutaneous and cerebral manifestations. case report and review of literature. J Neurosurg. 2004;100:1111-1114.
6. Umbert I, Winkelmann RK. Necrobiotic xanthogranuloma with cardiac involvement. Br J Dermatol. 1995;133:438-443.
7. Spicknall KE, Mehregan DA. Necrobiotic xanthogranuloma. Int J Dermatol. 2009;48:1-10.
8. Finan MC, Winkelmann RK. Histopathology of necrobiotic xanthogranuloma with paraproteinemia. J Cutan Pathol. 1987;14:92-99.
9. Reddy VC, Salomão DR, Garrity JA, et al. Periorbital and ocular necrobiotic xanthogranuloma leading to perforation. Arch Ophthalmol. 2010;128:1493-1494.
10. Chang SE, Lee WS, Lee MW, et al. A case of necrobiotic xanthogranuloma without paraproteinemia presenting as a solitary tumor on the thigh. Int J Dermatol. 2003;42:470-472.
11. Wood AJ, Wagner MV, Abbott JJ, et al. Necrobiotic xanthogranuloma: a review of 17 cases with emphasis on clinical and pathologic correlation. Arch Dermatol. 2009;145:279-284.
12. Singh K, Rajan KD, Eberhart C. Orbital necrobiotic xanthogranuloma associated with systemic IgG4 disease. Ocul Immunol Inflamm. 2010;18:373-378.
13. Hallermann C, Tittelbach J, Norgauer J, et al. Successful treatment of necrobiotic xanthogranuloma with intravenous immunoglobulin. Arch Dermatol. 2009;146:957-960.
14. Goede JS, Misselwitz B, Taverna C, et al. Necrobiotic xanthogranuloma successfully treated with autologous stem cell transplantation. Ann Hematol. 2007;86:303-306.
15. Silapunt S, Chon SY. Generalized necrobiotic xanthogranuloma successfully treated with lenalidomide. J Drugs Dermatol. 2010;9:273-276.
16. Saeki H, Tomita M, Kai H, et al. Necrobiotic xanthogranuloma with paraproteinemia successfully treated with melphalan, prednisolone and skin graft. J Dermatol. 2007;34:795-797.
17. Ho VH, Chevez-Barrios P, Jorgensen JL, et al. Receptor expression in orbital inflammatory syndromes and implications for targeted therapy. Tissue Antigens. 2007;70:105-109.
18. Al-Niaimi FA, Dawn G, Cox NH. Necrobiotic xanthogranuloma without paraproteinemia: marked improvement with psoralen ultraviolet A treatment. Clin Exp Dermatol. 2010;35:275-277.
19. Bullock JD, Bartley GB, Campbell RJ, et al. Necrobiotic xanthogranuloma with paraproteinemia: case report and a pathogenetic theory. Trans Am Ophthalmol Soc. 1986;84:342-352.
1. Kossard S, Winkelmann RK. Necrobiotic xanthogranuloma with paraproteinemia. J Am Acad Dermatol. 1980;3:257-270.
2. Mehregan DA, Winkelmann RK. Necrobiotic xanthogranuloma [published correction in Arch Dermatol. 1992;128:632]. Arch Dermatol. 1992;128:94-100.
3. Ugurlu S, Bartley GB, Gibson LE. Necrobiotic xanthogranuloma: long-term outcome of ocular and systemic involvement. Am J Ophthalmol. 2000;129:651-657.
4. Winkelmann RK, Litzow MR, Umbert IJ, et al. Giant cell granulomatous pulmonary and myocardial lesions in necrobiotic xanthogranuloma with paraproteinemia. Mayo Clin Proc. 1997;72:1028-1033.
5. Shah KC, Poonnoose SI, George R, et al. Necrobiotic xanthogranuloma with cutaneous and cerebral manifestations. case report and review of literature. J Neurosurg. 2004;100:1111-1114.
6. Umbert I, Winkelmann RK. Necrobiotic xanthogranuloma with cardiac involvement. Br J Dermatol. 1995;133:438-443.
7. Spicknall KE, Mehregan DA. Necrobiotic xanthogranuloma. Int J Dermatol. 2009;48:1-10.
8. Finan MC, Winkelmann RK. Histopathology of necrobiotic xanthogranuloma with paraproteinemia. J Cutan Pathol. 1987;14:92-99.
9. Reddy VC, Salomão DR, Garrity JA, et al. Periorbital and ocular necrobiotic xanthogranuloma leading to perforation. Arch Ophthalmol. 2010;128:1493-1494.
10. Chang SE, Lee WS, Lee MW, et al. A case of necrobiotic xanthogranuloma without paraproteinemia presenting as a solitary tumor on the thigh. Int J Dermatol. 2003;42:470-472.
11. Wood AJ, Wagner MV, Abbott JJ, et al. Necrobiotic xanthogranuloma: a review of 17 cases with emphasis on clinical and pathologic correlation. Arch Dermatol. 2009;145:279-284.
12. Singh K, Rajan KD, Eberhart C. Orbital necrobiotic xanthogranuloma associated with systemic IgG4 disease. Ocul Immunol Inflamm. 2010;18:373-378.
13. Hallermann C, Tittelbach J, Norgauer J, et al. Successful treatment of necrobiotic xanthogranuloma with intravenous immunoglobulin. Arch Dermatol. 2009;146:957-960.
14. Goede JS, Misselwitz B, Taverna C, et al. Necrobiotic xanthogranuloma successfully treated with autologous stem cell transplantation. Ann Hematol. 2007;86:303-306.
15. Silapunt S, Chon SY. Generalized necrobiotic xanthogranuloma successfully treated with lenalidomide. J Drugs Dermatol. 2010;9:273-276.
16. Saeki H, Tomita M, Kai H, et al. Necrobiotic xanthogranuloma with paraproteinemia successfully treated with melphalan, prednisolone and skin graft. J Dermatol. 2007;34:795-797.
17. Ho VH, Chevez-Barrios P, Jorgensen JL, et al. Receptor expression in orbital inflammatory syndromes and implications for targeted therapy. Tissue Antigens. 2007;70:105-109.
18. Al-Niaimi FA, Dawn G, Cox NH. Necrobiotic xanthogranuloma without paraproteinemia: marked improvement with psoralen ultraviolet A treatment. Clin Exp Dermatol. 2010;35:275-277.
19. Bullock JD, Bartley GB, Campbell RJ, et al. Necrobiotic xanthogranuloma with paraproteinemia: case report and a pathogenetic theory. Trans Am Ophthalmol Soc. 1986;84:342-352.
Practice Points
- Necrobiotic xanthogranuloma is a rare histiocytic disease that is strongly associated with monoclonal gammopathy.
- Due to the rarity and uncertain etiology, there are no definitive first-line therapies.