User login
Painful Purple Toes
The Diagnosis: Blue Toe Syndrome
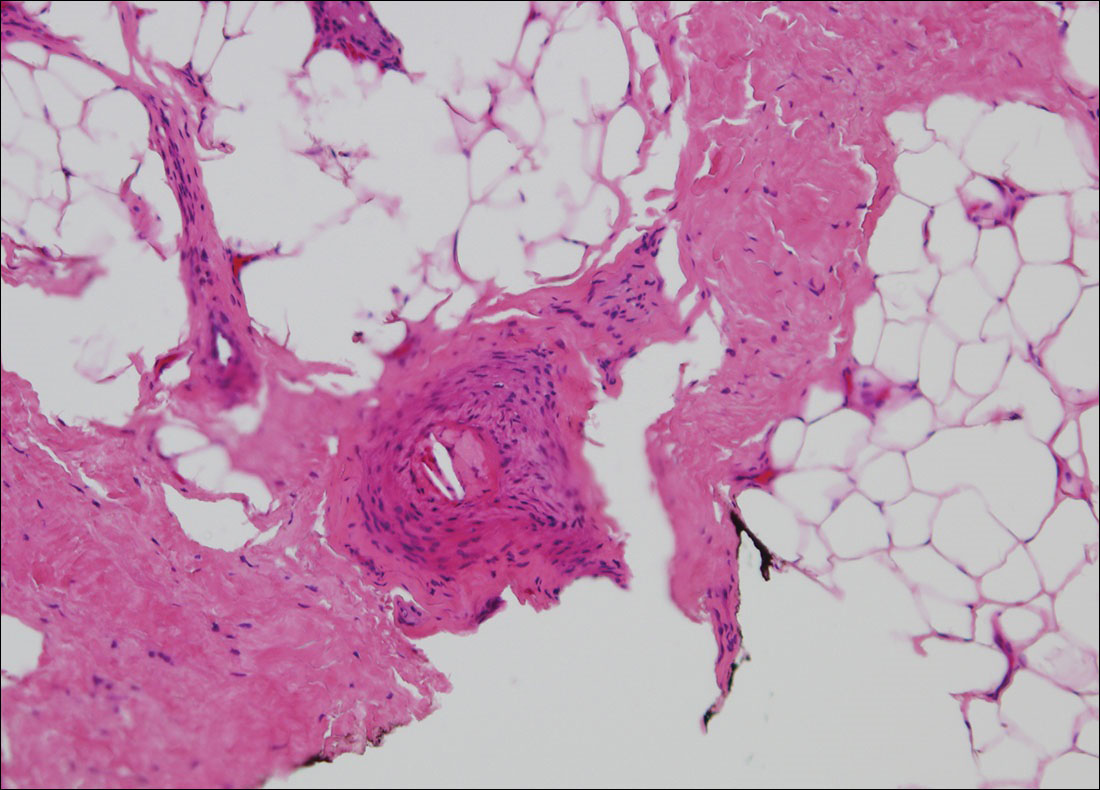
The clinical manifestation suggested blue toe syndrome. A variety of causes for blue toe syndrome are known such as embolism, thrombosis, vasoconstrictive disorders, infectious and noninfectious inflammation, extensive venous thrombosis, and abnormal circulating blood.1 Among them, only emboli from atherosclerotic plaques give rise to typical cholesterol clefts on skin biopsy (Figure 1). Such atheroemboli often are an iatrogenic complication, especially those caused by invasive percutaneous procedures or damage to the arterial walls from vascular surgery. However, spontaneous plaque hemorrhage or shearing forces of the circulating blood can disrupt atheromatous plaques and cause embolization of the cholesterol crystals, which was likely to be the case in our patient because no preceding trigger events were noted.
Other clinical features also are seen in atheroembolism. Approximately half of patients with atheroembolism develop clinical kidney disease.2 Almost all iatrogenic cases have acute or subacute reduction in glomerular filtration rate of at least to 50% level, whereas the spontaneous cases present as stable chronic renal failure.3 Approximately 20% of patients with atheroembolism also have involvement of digestive organs.4,5 Abdominal pain, diarrhea, and gastrointestinal blood loss are common features; bowel infarction and perforation occasionally occur.5 Pancreatitis is another common complication, and serum amylase levels are raised in approximately 50% of patients.6 Atheroemboli may reach the eyes and brain. They occasionally can cause loss of vision,7 as well as transient ischemic attacks, strokes, and gradual deterioration in cerebral function.3 Blood eosinophilia, which occurs in approximately 60% of patients, is an important finding.3,8
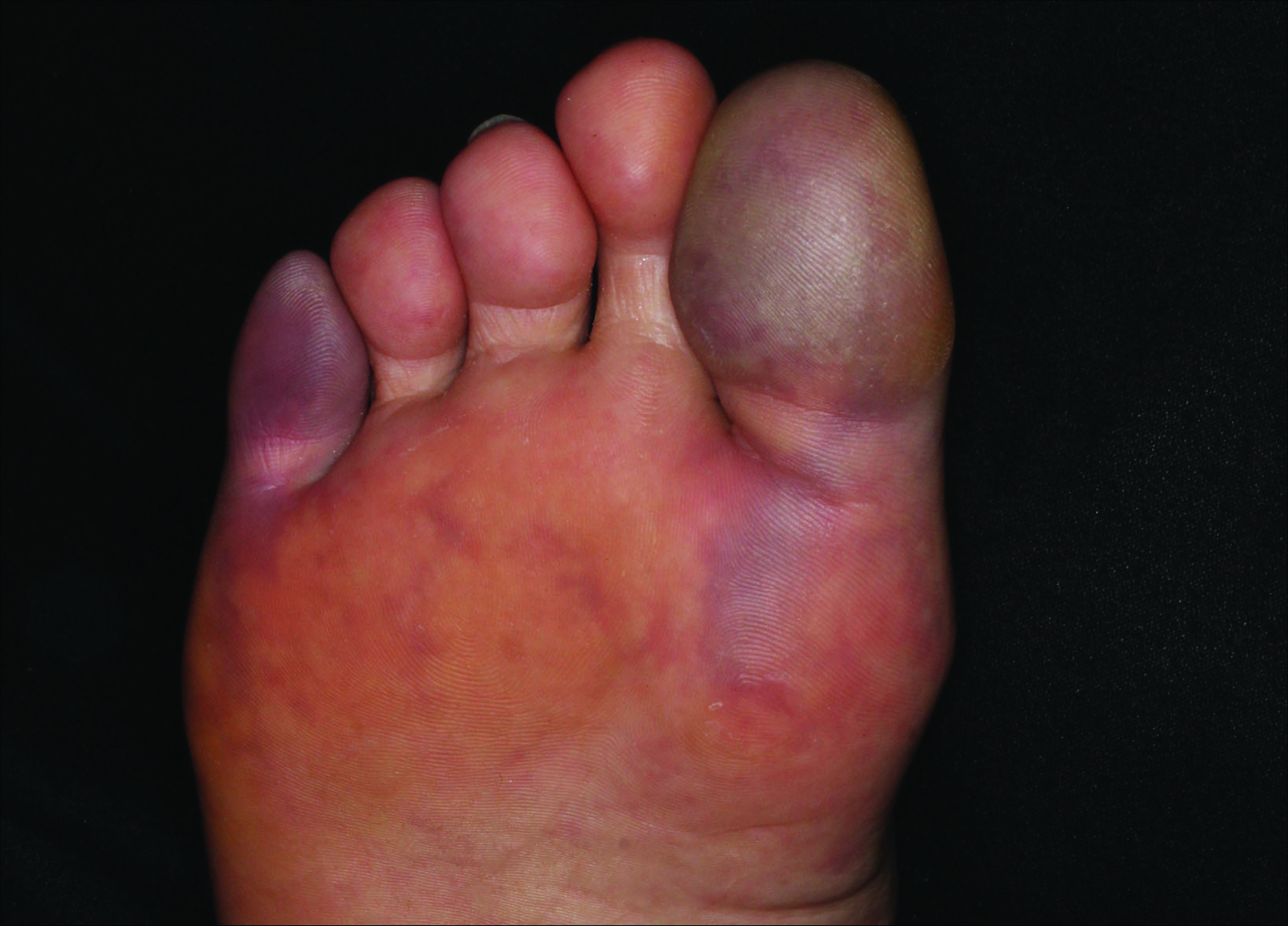
Although there is no specific therapy for atheroembolism, the use of antiplatelet agents is considered reasonable because they are beneficial in preventing myocardial infarction in patients with atherosclerosis.9 In our case, the livedo reticularis cleared, as did the coldness on the affected toes after 2 weeks of sarpogrelate hydrochloride administration; however, development of necrotic change was noted (Figure 2). Necrotic change on the hallux disappeared after 2 weeks.
- Hirschmann JV, Raugi GJ. Blue (or purple) toe syndrome. J Am Acad Dermatol. 2009;60:1-20; quiz 21-22.
- Scolari F, Ravani P, Gaggi R, et al. The challenge of diagnosing atheroembolic renal disease: clinical features and prognostic factors. Circulation. 2007;116:298-304.
- Scolari F, Tardanico R, Zani R, et al. Cholesterol crystal embolism: a recognizable cause of renal disease. Am J Kidney Dis. 2000;36:1089-1109.
- Moolenaar W, Lamers CB. Cholesterol crystal embolization in the Netherlands. Arch Intern Med. 1996;156:653-657.
- Ben-Horin S, Bardan E, Barshack I, et al. Cholesterol crystal embolization to the digestive system: characterization of a common, yet overlooked presentation of atheroembolism. Am J Gastroenterol. 2003;98:1471-1479.
- Mayo RR, Swartz RD. Redefining the incidence of clinically detectable atheroembolism. Am J Med. 1996;100:524-529.
- Gittinger JW Jr, Kershaw GR. Retinal cholesterol emboli in the diagnosis of renal atheroembolism. Arch Intern Med. 1998;158:1265-1267.
- Kasinath BS, Corwin HL, Bidani AK, et al. Eosinophilia in the diagnosis of atheroembolic renal disease. Am J Nephrol. 1987;7:173-177.
- Quinones A, Saric M. The cholesterol emboli syndrome in atherosclerosis. Curr Atheroscler Rep. 2013;15:315.
The Diagnosis: Blue Toe Syndrome
The clinical manifestation suggested blue toe syndrome. A variety of causes for blue toe syndrome are known such as embolism, thrombosis, vasoconstrictive disorders, infectious and noninfectious inflammation, extensive venous thrombosis, and abnormal circulating blood.1 Among them, only emboli from atherosclerotic plaques give rise to typical cholesterol clefts on skin biopsy (Figure 1). Such atheroemboli often are an iatrogenic complication, especially those caused by invasive percutaneous procedures or damage to the arterial walls from vascular surgery. However, spontaneous plaque hemorrhage or shearing forces of the circulating blood can disrupt atheromatous plaques and cause embolization of the cholesterol crystals, which was likely to be the case in our patient because no preceding trigger events were noted.
Other clinical features also are seen in atheroembolism. Approximately half of patients with atheroembolism develop clinical kidney disease.2 Almost all iatrogenic cases have acute or subacute reduction in glomerular filtration rate of at least to 50% level, whereas the spontaneous cases present as stable chronic renal failure.3 Approximately 20% of patients with atheroembolism also have involvement of digestive organs.4,5 Abdominal pain, diarrhea, and gastrointestinal blood loss are common features; bowel infarction and perforation occasionally occur.5 Pancreatitis is another common complication, and serum amylase levels are raised in approximately 50% of patients.6 Atheroemboli may reach the eyes and brain. They occasionally can cause loss of vision,7 as well as transient ischemic attacks, strokes, and gradual deterioration in cerebral function.3 Blood eosinophilia, which occurs in approximately 60% of patients, is an important finding.3,8
Although there is no specific therapy for atheroembolism, the use of antiplatelet agents is considered reasonable because they are beneficial in preventing myocardial infarction in patients with atherosclerosis.9 In our case, the livedo reticularis cleared, as did the coldness on the affected toes after 2 weeks of sarpogrelate hydrochloride administration; however, development of necrotic change was noted (Figure 2). Necrotic change on the hallux disappeared after 2 weeks.
The Diagnosis: Blue Toe Syndrome
The clinical manifestation suggested blue toe syndrome. A variety of causes for blue toe syndrome are known such as embolism, thrombosis, vasoconstrictive disorders, infectious and noninfectious inflammation, extensive venous thrombosis, and abnormal circulating blood.1 Among them, only emboli from atherosclerotic plaques give rise to typical cholesterol clefts on skin biopsy (Figure 1). Such atheroemboli often are an iatrogenic complication, especially those caused by invasive percutaneous procedures or damage to the arterial walls from vascular surgery. However, spontaneous plaque hemorrhage or shearing forces of the circulating blood can disrupt atheromatous plaques and cause embolization of the cholesterol crystals, which was likely to be the case in our patient because no preceding trigger events were noted.
Other clinical features also are seen in atheroembolism. Approximately half of patients with atheroembolism develop clinical kidney disease.2 Almost all iatrogenic cases have acute or subacute reduction in glomerular filtration rate of at least to 50% level, whereas the spontaneous cases present as stable chronic renal failure.3 Approximately 20% of patients with atheroembolism also have involvement of digestive organs.4,5 Abdominal pain, diarrhea, and gastrointestinal blood loss are common features; bowel infarction and perforation occasionally occur.5 Pancreatitis is another common complication, and serum amylase levels are raised in approximately 50% of patients.6 Atheroemboli may reach the eyes and brain. They occasionally can cause loss of vision,7 as well as transient ischemic attacks, strokes, and gradual deterioration in cerebral function.3 Blood eosinophilia, which occurs in approximately 60% of patients, is an important finding.3,8
Although there is no specific therapy for atheroembolism, the use of antiplatelet agents is considered reasonable because they are beneficial in preventing myocardial infarction in patients with atherosclerosis.9 In our case, the livedo reticularis cleared, as did the coldness on the affected toes after 2 weeks of sarpogrelate hydrochloride administration; however, development of necrotic change was noted (Figure 2). Necrotic change on the hallux disappeared after 2 weeks.
- Hirschmann JV, Raugi GJ. Blue (or purple) toe syndrome. J Am Acad Dermatol. 2009;60:1-20; quiz 21-22.
- Scolari F, Ravani P, Gaggi R, et al. The challenge of diagnosing atheroembolic renal disease: clinical features and prognostic factors. Circulation. 2007;116:298-304.
- Scolari F, Tardanico R, Zani R, et al. Cholesterol crystal embolism: a recognizable cause of renal disease. Am J Kidney Dis. 2000;36:1089-1109.
- Moolenaar W, Lamers CB. Cholesterol crystal embolization in the Netherlands. Arch Intern Med. 1996;156:653-657.
- Ben-Horin S, Bardan E, Barshack I, et al. Cholesterol crystal embolization to the digestive system: characterization of a common, yet overlooked presentation of atheroembolism. Am J Gastroenterol. 2003;98:1471-1479.
- Mayo RR, Swartz RD. Redefining the incidence of clinically detectable atheroembolism. Am J Med. 1996;100:524-529.
- Gittinger JW Jr, Kershaw GR. Retinal cholesterol emboli in the diagnosis of renal atheroembolism. Arch Intern Med. 1998;158:1265-1267.
- Kasinath BS, Corwin HL, Bidani AK, et al. Eosinophilia in the diagnosis of atheroembolic renal disease. Am J Nephrol. 1987;7:173-177.
- Quinones A, Saric M. The cholesterol emboli syndrome in atherosclerosis. Curr Atheroscler Rep. 2013;15:315.
- Hirschmann JV, Raugi GJ. Blue (or purple) toe syndrome. J Am Acad Dermatol. 2009;60:1-20; quiz 21-22.
- Scolari F, Ravani P, Gaggi R, et al. The challenge of diagnosing atheroembolic renal disease: clinical features and prognostic factors. Circulation. 2007;116:298-304.
- Scolari F, Tardanico R, Zani R, et al. Cholesterol crystal embolism: a recognizable cause of renal disease. Am J Kidney Dis. 2000;36:1089-1109.
- Moolenaar W, Lamers CB. Cholesterol crystal embolization in the Netherlands. Arch Intern Med. 1996;156:653-657.
- Ben-Horin S, Bardan E, Barshack I, et al. Cholesterol crystal embolization to the digestive system: characterization of a common, yet overlooked presentation of atheroembolism. Am J Gastroenterol. 2003;98:1471-1479.
- Mayo RR, Swartz RD. Redefining the incidence of clinically detectable atheroembolism. Am J Med. 1996;100:524-529.
- Gittinger JW Jr, Kershaw GR. Retinal cholesterol emboli in the diagnosis of renal atheroembolism. Arch Intern Med. 1998;158:1265-1267.
- Kasinath BS, Corwin HL, Bidani AK, et al. Eosinophilia in the diagnosis of atheroembolic renal disease. Am J Nephrol. 1987;7:173-177.
- Quinones A, Saric M. The cholesterol emboli syndrome in atherosclerosis. Curr Atheroscler Rep. 2013;15:315.
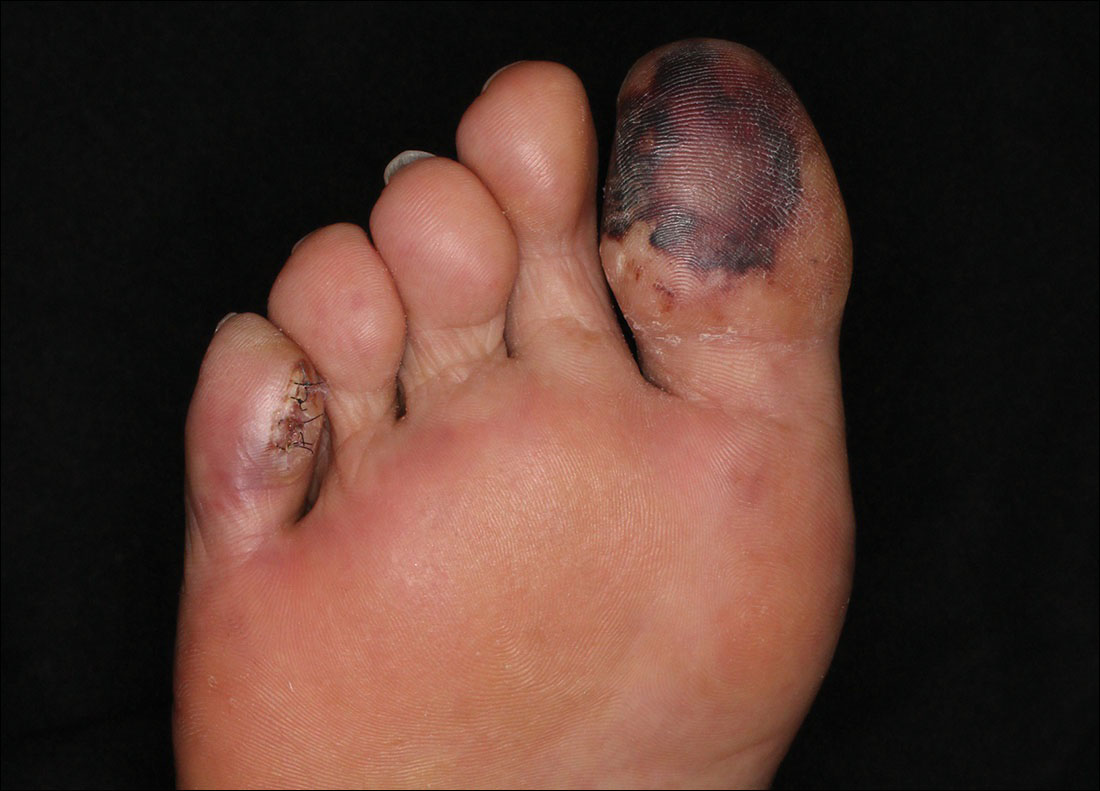
A 63-year-old man presented with sudden onset of severe pain in the right hallux and fifth toe of 3 days' duration. The patient had hypertension and hyperlipidemia with a 45-year history of smoking and had not undergone any vascular procedures. Physical examination revealed relatively well-defined cyanotic change with remarkable coldness on the affected toes as well as livedo reticularis on the underside of the toes. All peripheral pulses were present. Laboratory investigation revealed no remarkable changes with eosinophil counts within reference range and normal renal function. A biopsy taken from the fifth toe revealed thrombotic arterioles with cholesterol clefts.
Brown Macule on the Waist
The best diagnosis is:
a. granular cell tumor
b. intradermal nevus
c. Langerhans cell disease
d. mastocytosis
e. multicentric reticulohistiocytosis
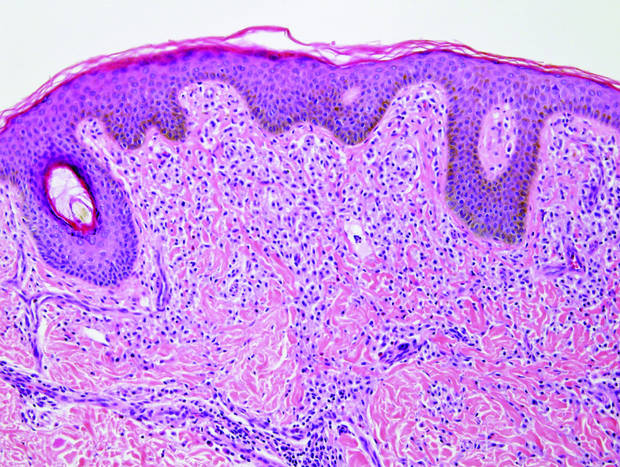
|
| Monomorphic cell infiltrate in the upper dermis (H&E, original magnification ×100). |
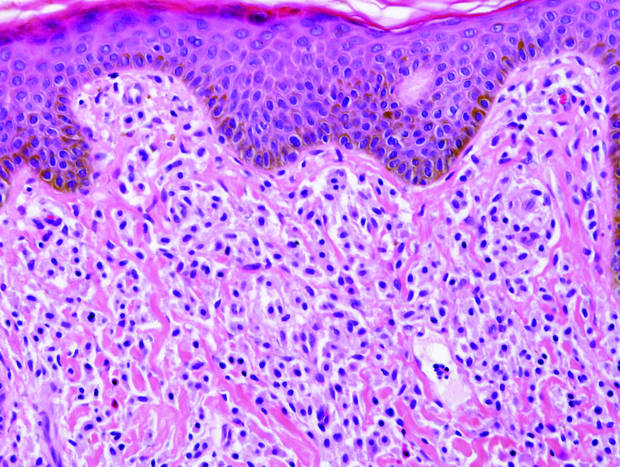
|
| A closer view reveals cuboidal or spindle cells with basal hyperpigmentation (H&E, original magnification ×200). |
Continue to the next page for the diagnosis >>
Mastocytosis
Mastocytosis is a clonal proliferation of mast cells in the skin and various systems of the body including the bone marrow, liver, lymph nodes, and gastrointestinal tract.1,2 Mast cell proliferation is closely associated with germline and acquired activating KIT mutations.3-5 Adult-onset mastocytosis is likely to involve several organs, whereas pediatric mastocytosis usually affects only the skin and is self-limiting. Patients with profound mast cell infiltration in the skin or other organs are likely to have attacks of flushing, palpitation, or diarrhea resulting from the degranulation of mast cells and release of histamine.6,7 In a majority of patients with advanced systemic mastocytosis, mast cells are positive for the Ki-1 antigen (CD30), whereas in most patients with indolent systemic mastocytosis, only a few mast cells are positive for CD30.8 Recently, CD30 was reported as a new drug target in patients with CD30+ advanced systemic mastocytosis.9 Because the skin frequently is involved and easily accessible in comparison with other organs, skin biopsy often is performed to establish a diagnosis of mastocytosis. Cutaneous mastocytosis comprises urticaria pigmentosa, solitary mastocytoma, diffuse cutaneous mastocytosis, and telangiectasia macularis eruptiva perstans; approximately 80% of all cases have urticaria pigmentosa.10-12 In cutaneous mastocytosis, skin biopsy typically shows monomorphous mast cell infiltrate mostly in the upper third of the dermis. The density of mast cells varies according to the clinical variant. For example, a lesion of telangiectasia macularis eruptiva perstans has only a perivascular mast cell infiltrate, whereas a solitary mastocytoma has sheets of mast cells in the dermis, sometimes extending into the subcutis. A skin biopsy of the brown macule on the waist showed a number of cuboidal or spindle mast cells in the upper dermis with occasional eosinophils. These mast cells are monomorphous, and no mitotic figures, necrotic cells, or atypical cells are seen. Mast cells have metachromatic granules in the cytoplasm, which can be seen with toluidine blue or Giemsa stain. CD117 (c-kit) also is positive. Mast cells in urticaria pigmentosa easily may be mistaken for nevus cells. Hyperpigmentation of the basal layer, a characteristic feature seen in urticaria pigmentosa, also may erroneously suggest a diagnosis of a melanocytic nevus.
Granular cell tumors predominantly affect the oral cavity, but the skin also can be involved. It comprises a fascicular infiltrate of large and polygonal cells with characteristic eosinophilic granular cytoplasm in the dermis (Figure 1).13 Cell membranes are not always distinct. Although the nuclei usually are small and centrally located, irregular and plump nuclei with distinct nucleoli also may be seen. The overlying epidermis tends to be hyperplastic. Granular cell tumor is considered a group of lesions of varying histogenesis. Cases in which tumors originated from a neural crest–derived peripheral nerve–related cell as well as a Schwann cell have been reported.14,15 The origin of granular cell tumors is controversial.
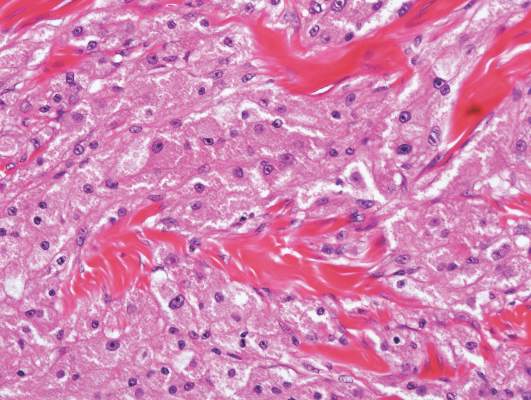
|
| Figure 1. Granular cell tumor showing fascicles of large and polygonal cells with characteristic eosinophilic granular cytoplasm in the dermis (H&E, original magnification ×200). |

|
| Figure 2. Intradermal nevus showing nests with melanin in the uppermost area of the lesion and neurotized nevus cells in the lower part (H&E, original magnification ×100). Pseudovascular spaces are seen on the right side. |
Intradermal nevus usually has nests and cords of nevus cells in the upper dermis. The uppermost melanocytes often contain a moderate amount of melanin, whereas nevus cells in the mid and lower dermis usually do not contain melanin (Figure 2). Shrinkage during tissue processing maycause clefts between nevus cells, resulting in pseudovascular spaces.16 The deeper dermis may have a neuroid appearance with spindle-shaped cells and Meissner corpuscle–like structures.17
Although Langerhans cell disease was formerly known as Langerhans cell histiocytosis and subdivided into several clinical subtypes, including Letterer-Siwe disease, Hand-Schüller-Christian disease, and eosinophilic granuloma, these clinical subtypes commonly overlapped. Langerhans cell disease is now used as a terminology that encompasses all subtypes.18,19 Langerhans cell disease is characterized by a proliferation of Langerhans cells with a variable mixture of other inflammatory cells. The constituent cells are large and ovoid with a distinct folded or lobulated, often kidney-shaped nucleus.20 Langerhans cells usually infiltrate the upper dermis and occasionally the epidermis (Figure 3). CD1a, HLA-DR, S-100 protein, and langerin are positive in Langerhans cells.21

|
| Figure 3. Langerhans cell disease showing an infiltrate of large and ovoid Langerhans cells with a distinct folded or lobulated, often kidney-shaped nucleus in the upper dermis and epidermis (H&E, original magnification ×200). |

|
| Figure 4. Multicentric reticulohistiocytosis showing a mixture of mononuclear and multinucleate histiocytes with abundant eosinophilic and finely granular cytoplasm (H&E, original magnification ×200). |
Multicentric reticulohistiocytosis is characterized by a combination of papulonodular cutaneous lesions and severe arthropathy.22 An irregular mixture of mononuclear and multinucleate histiocytes showing abundant eosinophilic and finely granular cytoplasm, often with a ground-glass appearance, is seen along with lymphocytic infiltration (Figure 4).23 A few giant cells may be seen in early lesions; older lesions more commonly have giant cells and fibrosis.
1. Arock M, Valent P. Pathogenesis, classification and treatment of mastocytosis: state of the art in 2010 and future perspectives. Expert Rev Hematol. 2010;3:497-516.
2. Pardanani A. Systemic mastocytosis in adults: 2013 update on diagnosis, risk stratification, and management. Am J Hematol. 2013;88:612-624.
3. Orfao A, Garcia-Montero AC, Sanchez L, et al. Recent advances in the understanding of mastocytosis: the role of KIT mutations. Br J Haematol. 2007;138:12-30.
4. Yanagihori H, Oyama N, Nakamura K, et al. c-KIT mutations in patients with childhood-onset mastocytosis and genotype-phenotype correlation. J Mol Diagn. 2005;7:252-257.
5. Bodemer C, Hermine O, Palmérini F, et al. Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J Invest Dermatol. 2010;130:804-815.
6. Kettelhut BV, Metcalfe DD. Pediatric mastocytosis. Ann Allergy. 1994;73:197-202; quiz 202-207.
7. Longley J, Duffy TP, Kohn S. The mast cell and mast cell disease. J Am Acad Dermatol. 1995;32:545-561; quiz 562-564.
8. Sotlar K, Cerny-Reiterer S, Petat-Dutter K, et al. Aberrant expression of CD30 in neoplastic mast cells in high-grade mastocytosis. Mod Pathol. 2011;24:585-595.
9. Blatt K, Cerny-Reiterer S, Schwaab J, et al. Identification of the Ki-1 antigen (CD30) as a novel therapeutic target in systemic mastocytosis [published online October 20, 2015]. Blood. 2015;126:2832-2841.
10. Kiszewski AE, Duran-Mckinster C, Orozco-Covarrubias L, et al. Cutaneous mastocytosis in children: a clinical analysis of 71 cases. J Eur Acad Dermatol Venereol. 2004;18:285-290.
11. Akoglu G, Erkin G, Cakir B, et al. Cutaneous mastocytosis: demographic aspects and clinical features of 55 patients. J Eur Acad Dermatol Venereol. 2006;20:969-973.
12. Sarkany RP, Monk BE, Handfield-Jones SE. Telangiectasia macularis eruptiva perstans: a case report and review of the literature. Clin Exp Dermatol. 1998;23:38-39.
13. Lack EE, Worsham GF, Callihan MD, et al. Granular cell tumor: a clinicopathologic study of 110 patients. J Surg Oncol. 1980;13:301-316.
14. Buley ID, Gatter KC, Kelly PM, et al. Granular cell tumours revisited. an immunohistological and ultrastructural study. Histopathology. 1988;12:263-274.
15. Penneys NS, Adachi K, Ziegels-Weissman J, et al. Granular cell tumors of the skin contain myelin basic protein. Arch Pathol Lab Med. 1983;107:302-303.
16. Modlin RL, Gottlieb B, Taylor C, et al. Identification of cells lining pseudovascular spaces of benign pigmented nevi. Am J Dermatopathol. 1984;(suppl 6):25-29.
17. Fullen DR, Reed JA, Finnerty B, et al. S100A6 preferentially labels type C nevus cells and nevic corpuscles: additional support for Schwannian differentiation of intradermal nevi. J Cutan Pathol. 2001;28:393-399.
18. Newman B, Hu W, Nigro K, et al. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol. 2007;56:302-316.
19. Weedon D. Cutaneous infiltrates—non-lymphoid. In: Weedon D, ed. Weedon’s Skin Pathology. 3rd ed. Amsterdam, Netherlands: Elsevier; 2010:937-970.
20. Harrist TJ, Bhan AK, Murphy GF, et al. Histiocytosis-X: in situ characterization of cutaneous infiltrates with monoclonal antibodies. Am J Clin Pathol. 1983;79:294-300.
21. Lau SK, Chu PG, Weiss LM. Immunohistochemical expression of langerin in Langerhans cell histiocytosis and non-Langerhans cell histiocytic disorders. Am J Surg Pathol. 2008;32:615-619.
22. Lesher JL Jr, Allen BS. Multicentric reticulohistiocytosis. J Am Acad Dermatol. 1984;11:713-723.
23. Heathcote JG, Guenther LC, Wallace AC. Multicentric reticulohistiocytosis: a report of a case and a review of the pathology. Pathology. 1985;17:601-608.
The best diagnosis is:
a. granular cell tumor
b. intradermal nevus
c. Langerhans cell disease
d. mastocytosis
e. multicentric reticulohistiocytosis
|
|
| Monomorphic cell infiltrate in the upper dermis (H&E, original magnification ×100). |
|
|
| A closer view reveals cuboidal or spindle cells with basal hyperpigmentation (H&E, original magnification ×200). |
Continue to the next page for the diagnosis >>
Mastocytosis
Mastocytosis is a clonal proliferation of mast cells in the skin and various systems of the body including the bone marrow, liver, lymph nodes, and gastrointestinal tract.1,2 Mast cell proliferation is closely associated with germline and acquired activating KIT mutations.3-5 Adult-onset mastocytosis is likely to involve several organs, whereas pediatric mastocytosis usually affects only the skin and is self-limiting. Patients with profound mast cell infiltration in the skin or other organs are likely to have attacks of flushing, palpitation, or diarrhea resulting from the degranulation of mast cells and release of histamine.6,7 In a majority of patients with advanced systemic mastocytosis, mast cells are positive for the Ki-1 antigen (CD30), whereas in most patients with indolent systemic mastocytosis, only a few mast cells are positive for CD30.8 Recently, CD30 was reported as a new drug target in patients with CD30+ advanced systemic mastocytosis.9 Because the skin frequently is involved and easily accessible in comparison with other organs, skin biopsy often is performed to establish a diagnosis of mastocytosis. Cutaneous mastocytosis comprises urticaria pigmentosa, solitary mastocytoma, diffuse cutaneous mastocytosis, and telangiectasia macularis eruptiva perstans; approximately 80% of all cases have urticaria pigmentosa.10-12 In cutaneous mastocytosis, skin biopsy typically shows monomorphous mast cell infiltrate mostly in the upper third of the dermis. The density of mast cells varies according to the clinical variant. For example, a lesion of telangiectasia macularis eruptiva perstans has only a perivascular mast cell infiltrate, whereas a solitary mastocytoma has sheets of mast cells in the dermis, sometimes extending into the subcutis. A skin biopsy of the brown macule on the waist showed a number of cuboidal or spindle mast cells in the upper dermis with occasional eosinophils. These mast cells are monomorphous, and no mitotic figures, necrotic cells, or atypical cells are seen. Mast cells have metachromatic granules in the cytoplasm, which can be seen with toluidine blue or Giemsa stain. CD117 (c-kit) also is positive. Mast cells in urticaria pigmentosa easily may be mistaken for nevus cells. Hyperpigmentation of the basal layer, a characteristic feature seen in urticaria pigmentosa, also may erroneously suggest a diagnosis of a melanocytic nevus.
Granular cell tumors predominantly affect the oral cavity, but the skin also can be involved. It comprises a fascicular infiltrate of large and polygonal cells with characteristic eosinophilic granular cytoplasm in the dermis (Figure 1).13 Cell membranes are not always distinct. Although the nuclei usually are small and centrally located, irregular and plump nuclei with distinct nucleoli also may be seen. The overlying epidermis tends to be hyperplastic. Granular cell tumor is considered a group of lesions of varying histogenesis. Cases in which tumors originated from a neural crest–derived peripheral nerve–related cell as well as a Schwann cell have been reported.14,15 The origin of granular cell tumors is controversial.
|
|
| Figure 1. Granular cell tumor showing fascicles of large and polygonal cells with characteristic eosinophilic granular cytoplasm in the dermis (H&E, original magnification ×200). |
|
|
| Figure 2. Intradermal nevus showing nests with melanin in the uppermost area of the lesion and neurotized nevus cells in the lower part (H&E, original magnification ×100). Pseudovascular spaces are seen on the right side. |
Intradermal nevus usually has nests and cords of nevus cells in the upper dermis. The uppermost melanocytes often contain a moderate amount of melanin, whereas nevus cells in the mid and lower dermis usually do not contain melanin (Figure 2). Shrinkage during tissue processing maycause clefts between nevus cells, resulting in pseudovascular spaces.16 The deeper dermis may have a neuroid appearance with spindle-shaped cells and Meissner corpuscle–like structures.17
Although Langerhans cell disease was formerly known as Langerhans cell histiocytosis and subdivided into several clinical subtypes, including Letterer-Siwe disease, Hand-Schüller-Christian disease, and eosinophilic granuloma, these clinical subtypes commonly overlapped. Langerhans cell disease is now used as a terminology that encompasses all subtypes.18,19 Langerhans cell disease is characterized by a proliferation of Langerhans cells with a variable mixture of other inflammatory cells. The constituent cells are large and ovoid with a distinct folded or lobulated, often kidney-shaped nucleus.20 Langerhans cells usually infiltrate the upper dermis and occasionally the epidermis (Figure 3). CD1a, HLA-DR, S-100 protein, and langerin are positive in Langerhans cells.21
|
|
| Figure 3. Langerhans cell disease showing an infiltrate of large and ovoid Langerhans cells with a distinct folded or lobulated, often kidney-shaped nucleus in the upper dermis and epidermis (H&E, original magnification ×200). |
|
|
| Figure 4. Multicentric reticulohistiocytosis showing a mixture of mononuclear and multinucleate histiocytes with abundant eosinophilic and finely granular cytoplasm (H&E, original magnification ×200). |
Multicentric reticulohistiocytosis is characterized by a combination of papulonodular cutaneous lesions and severe arthropathy.22 An irregular mixture of mononuclear and multinucleate histiocytes showing abundant eosinophilic and finely granular cytoplasm, often with a ground-glass appearance, is seen along with lymphocytic infiltration (Figure 4).23 A few giant cells may be seen in early lesions; older lesions more commonly have giant cells and fibrosis.
The best diagnosis is:
a. granular cell tumor
b. intradermal nevus
c. Langerhans cell disease
d. mastocytosis
e. multicentric reticulohistiocytosis
|
|
| Monomorphic cell infiltrate in the upper dermis (H&E, original magnification ×100). |
|
|
| A closer view reveals cuboidal or spindle cells with basal hyperpigmentation (H&E, original magnification ×200). |
Continue to the next page for the diagnosis >>
Mastocytosis
Mastocytosis is a clonal proliferation of mast cells in the skin and various systems of the body including the bone marrow, liver, lymph nodes, and gastrointestinal tract.1,2 Mast cell proliferation is closely associated with germline and acquired activating KIT mutations.3-5 Adult-onset mastocytosis is likely to involve several organs, whereas pediatric mastocytosis usually affects only the skin and is self-limiting. Patients with profound mast cell infiltration in the skin or other organs are likely to have attacks of flushing, palpitation, or diarrhea resulting from the degranulation of mast cells and release of histamine.6,7 In a majority of patients with advanced systemic mastocytosis, mast cells are positive for the Ki-1 antigen (CD30), whereas in most patients with indolent systemic mastocytosis, only a few mast cells are positive for CD30.8 Recently, CD30 was reported as a new drug target in patients with CD30+ advanced systemic mastocytosis.9 Because the skin frequently is involved and easily accessible in comparison with other organs, skin biopsy often is performed to establish a diagnosis of mastocytosis. Cutaneous mastocytosis comprises urticaria pigmentosa, solitary mastocytoma, diffuse cutaneous mastocytosis, and telangiectasia macularis eruptiva perstans; approximately 80% of all cases have urticaria pigmentosa.10-12 In cutaneous mastocytosis, skin biopsy typically shows monomorphous mast cell infiltrate mostly in the upper third of the dermis. The density of mast cells varies according to the clinical variant. For example, a lesion of telangiectasia macularis eruptiva perstans has only a perivascular mast cell infiltrate, whereas a solitary mastocytoma has sheets of mast cells in the dermis, sometimes extending into the subcutis. A skin biopsy of the brown macule on the waist showed a number of cuboidal or spindle mast cells in the upper dermis with occasional eosinophils. These mast cells are monomorphous, and no mitotic figures, necrotic cells, or atypical cells are seen. Mast cells have metachromatic granules in the cytoplasm, which can be seen with toluidine blue or Giemsa stain. CD117 (c-kit) also is positive. Mast cells in urticaria pigmentosa easily may be mistaken for nevus cells. Hyperpigmentation of the basal layer, a characteristic feature seen in urticaria pigmentosa, also may erroneously suggest a diagnosis of a melanocytic nevus.
Granular cell tumors predominantly affect the oral cavity, but the skin also can be involved. It comprises a fascicular infiltrate of large and polygonal cells with characteristic eosinophilic granular cytoplasm in the dermis (Figure 1).13 Cell membranes are not always distinct. Although the nuclei usually are small and centrally located, irregular and plump nuclei with distinct nucleoli also may be seen. The overlying epidermis tends to be hyperplastic. Granular cell tumor is considered a group of lesions of varying histogenesis. Cases in which tumors originated from a neural crest–derived peripheral nerve–related cell as well as a Schwann cell have been reported.14,15 The origin of granular cell tumors is controversial.
|
|
| Figure 1. Granular cell tumor showing fascicles of large and polygonal cells with characteristic eosinophilic granular cytoplasm in the dermis (H&E, original magnification ×200). |
|
|
| Figure 2. Intradermal nevus showing nests with melanin in the uppermost area of the lesion and neurotized nevus cells in the lower part (H&E, original magnification ×100). Pseudovascular spaces are seen on the right side. |
Intradermal nevus usually has nests and cords of nevus cells in the upper dermis. The uppermost melanocytes often contain a moderate amount of melanin, whereas nevus cells in the mid and lower dermis usually do not contain melanin (Figure 2). Shrinkage during tissue processing maycause clefts between nevus cells, resulting in pseudovascular spaces.16 The deeper dermis may have a neuroid appearance with spindle-shaped cells and Meissner corpuscle–like structures.17
Although Langerhans cell disease was formerly known as Langerhans cell histiocytosis and subdivided into several clinical subtypes, including Letterer-Siwe disease, Hand-Schüller-Christian disease, and eosinophilic granuloma, these clinical subtypes commonly overlapped. Langerhans cell disease is now used as a terminology that encompasses all subtypes.18,19 Langerhans cell disease is characterized by a proliferation of Langerhans cells with a variable mixture of other inflammatory cells. The constituent cells are large and ovoid with a distinct folded or lobulated, often kidney-shaped nucleus.20 Langerhans cells usually infiltrate the upper dermis and occasionally the epidermis (Figure 3). CD1a, HLA-DR, S-100 protein, and langerin are positive in Langerhans cells.21
|
|
| Figure 3. Langerhans cell disease showing an infiltrate of large and ovoid Langerhans cells with a distinct folded or lobulated, often kidney-shaped nucleus in the upper dermis and epidermis (H&E, original magnification ×200). |
|
|
| Figure 4. Multicentric reticulohistiocytosis showing a mixture of mononuclear and multinucleate histiocytes with abundant eosinophilic and finely granular cytoplasm (H&E, original magnification ×200). |
Multicentric reticulohistiocytosis is characterized by a combination of papulonodular cutaneous lesions and severe arthropathy.22 An irregular mixture of mononuclear and multinucleate histiocytes showing abundant eosinophilic and finely granular cytoplasm, often with a ground-glass appearance, is seen along with lymphocytic infiltration (Figure 4).23 A few giant cells may be seen in early lesions; older lesions more commonly have giant cells and fibrosis.
1. Arock M, Valent P. Pathogenesis, classification and treatment of mastocytosis: state of the art in 2010 and future perspectives. Expert Rev Hematol. 2010;3:497-516.
2. Pardanani A. Systemic mastocytosis in adults: 2013 update on diagnosis, risk stratification, and management. Am J Hematol. 2013;88:612-624.
3. Orfao A, Garcia-Montero AC, Sanchez L, et al. Recent advances in the understanding of mastocytosis: the role of KIT mutations. Br J Haematol. 2007;138:12-30.
4. Yanagihori H, Oyama N, Nakamura K, et al. c-KIT mutations in patients with childhood-onset mastocytosis and genotype-phenotype correlation. J Mol Diagn. 2005;7:252-257.
5. Bodemer C, Hermine O, Palmérini F, et al. Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J Invest Dermatol. 2010;130:804-815.
6. Kettelhut BV, Metcalfe DD. Pediatric mastocytosis. Ann Allergy. 1994;73:197-202; quiz 202-207.
7. Longley J, Duffy TP, Kohn S. The mast cell and mast cell disease. J Am Acad Dermatol. 1995;32:545-561; quiz 562-564.
8. Sotlar K, Cerny-Reiterer S, Petat-Dutter K, et al. Aberrant expression of CD30 in neoplastic mast cells in high-grade mastocytosis. Mod Pathol. 2011;24:585-595.
9. Blatt K, Cerny-Reiterer S, Schwaab J, et al. Identification of the Ki-1 antigen (CD30) as a novel therapeutic target in systemic mastocytosis [published online October 20, 2015]. Blood. 2015;126:2832-2841.
10. Kiszewski AE, Duran-Mckinster C, Orozco-Covarrubias L, et al. Cutaneous mastocytosis in children: a clinical analysis of 71 cases. J Eur Acad Dermatol Venereol. 2004;18:285-290.
11. Akoglu G, Erkin G, Cakir B, et al. Cutaneous mastocytosis: demographic aspects and clinical features of 55 patients. J Eur Acad Dermatol Venereol. 2006;20:969-973.
12. Sarkany RP, Monk BE, Handfield-Jones SE. Telangiectasia macularis eruptiva perstans: a case report and review of the literature. Clin Exp Dermatol. 1998;23:38-39.
13. Lack EE, Worsham GF, Callihan MD, et al. Granular cell tumor: a clinicopathologic study of 110 patients. J Surg Oncol. 1980;13:301-316.
14. Buley ID, Gatter KC, Kelly PM, et al. Granular cell tumours revisited. an immunohistological and ultrastructural study. Histopathology. 1988;12:263-274.
15. Penneys NS, Adachi K, Ziegels-Weissman J, et al. Granular cell tumors of the skin contain myelin basic protein. Arch Pathol Lab Med. 1983;107:302-303.
16. Modlin RL, Gottlieb B, Taylor C, et al. Identification of cells lining pseudovascular spaces of benign pigmented nevi. Am J Dermatopathol. 1984;(suppl 6):25-29.
17. Fullen DR, Reed JA, Finnerty B, et al. S100A6 preferentially labels type C nevus cells and nevic corpuscles: additional support for Schwannian differentiation of intradermal nevi. J Cutan Pathol. 2001;28:393-399.
18. Newman B, Hu W, Nigro K, et al. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol. 2007;56:302-316.
19. Weedon D. Cutaneous infiltrates—non-lymphoid. In: Weedon D, ed. Weedon’s Skin Pathology. 3rd ed. Amsterdam, Netherlands: Elsevier; 2010:937-970.
20. Harrist TJ, Bhan AK, Murphy GF, et al. Histiocytosis-X: in situ characterization of cutaneous infiltrates with monoclonal antibodies. Am J Clin Pathol. 1983;79:294-300.
21. Lau SK, Chu PG, Weiss LM. Immunohistochemical expression of langerin in Langerhans cell histiocytosis and non-Langerhans cell histiocytic disorders. Am J Surg Pathol. 2008;32:615-619.
22. Lesher JL Jr, Allen BS. Multicentric reticulohistiocytosis. J Am Acad Dermatol. 1984;11:713-723.
23. Heathcote JG, Guenther LC, Wallace AC. Multicentric reticulohistiocytosis: a report of a case and a review of the pathology. Pathology. 1985;17:601-608.
1. Arock M, Valent P. Pathogenesis, classification and treatment of mastocytosis: state of the art in 2010 and future perspectives. Expert Rev Hematol. 2010;3:497-516.
2. Pardanani A. Systemic mastocytosis in adults: 2013 update on diagnosis, risk stratification, and management. Am J Hematol. 2013;88:612-624.
3. Orfao A, Garcia-Montero AC, Sanchez L, et al. Recent advances in the understanding of mastocytosis: the role of KIT mutations. Br J Haematol. 2007;138:12-30.
4. Yanagihori H, Oyama N, Nakamura K, et al. c-KIT mutations in patients with childhood-onset mastocytosis and genotype-phenotype correlation. J Mol Diagn. 2005;7:252-257.
5. Bodemer C, Hermine O, Palmérini F, et al. Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J Invest Dermatol. 2010;130:804-815.
6. Kettelhut BV, Metcalfe DD. Pediatric mastocytosis. Ann Allergy. 1994;73:197-202; quiz 202-207.
7. Longley J, Duffy TP, Kohn S. The mast cell and mast cell disease. J Am Acad Dermatol. 1995;32:545-561; quiz 562-564.
8. Sotlar K, Cerny-Reiterer S, Petat-Dutter K, et al. Aberrant expression of CD30 in neoplastic mast cells in high-grade mastocytosis. Mod Pathol. 2011;24:585-595.
9. Blatt K, Cerny-Reiterer S, Schwaab J, et al. Identification of the Ki-1 antigen (CD30) as a novel therapeutic target in systemic mastocytosis [published online October 20, 2015]. Blood. 2015;126:2832-2841.
10. Kiszewski AE, Duran-Mckinster C, Orozco-Covarrubias L, et al. Cutaneous mastocytosis in children: a clinical analysis of 71 cases. J Eur Acad Dermatol Venereol. 2004;18:285-290.
11. Akoglu G, Erkin G, Cakir B, et al. Cutaneous mastocytosis: demographic aspects and clinical features of 55 patients. J Eur Acad Dermatol Venereol. 2006;20:969-973.
12. Sarkany RP, Monk BE, Handfield-Jones SE. Telangiectasia macularis eruptiva perstans: a case report and review of the literature. Clin Exp Dermatol. 1998;23:38-39.
13. Lack EE, Worsham GF, Callihan MD, et al. Granular cell tumor: a clinicopathologic study of 110 patients. J Surg Oncol. 1980;13:301-316.
14. Buley ID, Gatter KC, Kelly PM, et al. Granular cell tumours revisited. an immunohistological and ultrastructural study. Histopathology. 1988;12:263-274.
15. Penneys NS, Adachi K, Ziegels-Weissman J, et al. Granular cell tumors of the skin contain myelin basic protein. Arch Pathol Lab Med. 1983;107:302-303.
16. Modlin RL, Gottlieb B, Taylor C, et al. Identification of cells lining pseudovascular spaces of benign pigmented nevi. Am J Dermatopathol. 1984;(suppl 6):25-29.
17. Fullen DR, Reed JA, Finnerty B, et al. S100A6 preferentially labels type C nevus cells and nevic corpuscles: additional support for Schwannian differentiation of intradermal nevi. J Cutan Pathol. 2001;28:393-399.
18. Newman B, Hu W, Nigro K, et al. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol. 2007;56:302-316.
19. Weedon D. Cutaneous infiltrates—non-lymphoid. In: Weedon D, ed. Weedon’s Skin Pathology. 3rd ed. Amsterdam, Netherlands: Elsevier; 2010:937-970.
20. Harrist TJ, Bhan AK, Murphy GF, et al. Histiocytosis-X: in situ characterization of cutaneous infiltrates with monoclonal antibodies. Am J Clin Pathol. 1983;79:294-300.
21. Lau SK, Chu PG, Weiss LM. Immunohistochemical expression of langerin in Langerhans cell histiocytosis and non-Langerhans cell histiocytic disorders. Am J Surg Pathol. 2008;32:615-619.
22. Lesher JL Jr, Allen BS. Multicentric reticulohistiocytosis. J Am Acad Dermatol. 1984;11:713-723.
23. Heathcote JG, Guenther LC, Wallace AC. Multicentric reticulohistiocytosis: a report of a case and a review of the pathology. Pathology. 1985;17:601-608.
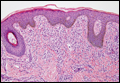
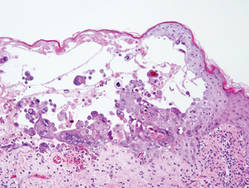
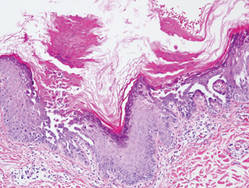
Hailey-Hailey Disease
Hailey-Hailey disease (HHD), or benign familial chronic pemphigus, typically presents as suprabasal blisters with a perivascular and interstitial lymphocytic infiltrate (Figure 1).1 Villi, or elongated dermal papillae lined with a single layer of basal cells, protrude into the bullae (Figure 2). In HHD lesions, the epidermis is thickened with scale-crust, and at least the lower half of the epidermis shows acantholysis. Despite the acantholytic changes, a few intact intercellular bridges remain, giving the appearance of a dilapidated brick wall (Figure 2). There may be dyskeratotic cells among the acantholytic cells, though they are scant in many cases. These acantholytic dyskeratotic cells have eosinophilic polygonal-shaped cytoplasm. Hailey-Hailey disease typically does not show adnexal extension of the acantholysis. Direct immunofluorescence is negative in HHD.
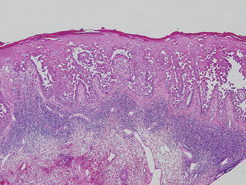
Pemphigus vulgaris is an autoimmune intraepidermal bullous disease that presents with suprabasal acantholysis (Figure 3).2 The epidermis is not thickened and acantholysis is limited to the suprabasal layer. Acantholytic cells with eosinophils and/or neutrophils are found within the bullae. Perivascular and interstitial infiltrates of lymphocytes, eosinophils, and occasionally neutrophils are seen; however, the inflammatory cell infiltrate can vary from extensive to scant. Direct immunofluorescence usually reveals IgG and/or C3 deposition on the surface of the keratinocytes throughout the epidermis.
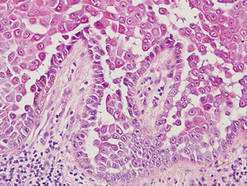
Pemphigus foliaceus is another autoimmune intraepidermal bullous disease that is characterized by acantholysis in the granular or upper spinous layers (Figure 4).3 The epidermis is not thickened. Sometimes acantholytic cells show dyskeratotic change (Figure 4). Some biopsy specimens do not contain the roof of the bullae; therefore, only erosion is seen and the diagnosis may be missed. Moreover, when only the adnexal epithelium shows acantholysis without epidermal involvement, diagnosis can be difficult.4 Acantholysis is accompanied with a superficial perivascular and interstitial inflammatory cell infiltrate consisting of lymphocytes, eosinophils, and occasionally neutrophils. The amount of inflammatory cell infiltrate may vary. Bullous impetigo and staphylococcal scalded skin syndrome reveal a similar histopathologic pattern. Direct immunofluorescence usually discloses IgG and/or C3 deposition on cell surfaces of keratinocytes in the entire or upper epidermis.
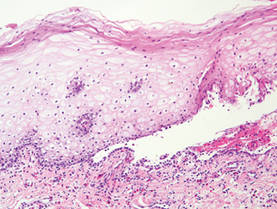
Herpesvirus infection shows ballooning (intracellular edema) of keratinocytes. Eventually acantholysis occurs and intraepidermal bullae are formed. In the bullae, virus-associated acantholytic keratinocytes, some that are multinucleated, can be easily found (Figure 5).5 These cells are larger than normal keratinocytes and have steel gray nuclei with peripheral accentuation. Some of these cells are necrotic, and the remains of necrotic multinucleated acantholytic cells are easily recognized. Adnexal epithelial cells occasionally are affected by herpesvirus infection; nuclear change is similar to the epidermis. A perivascular and interstitial infiltrate of lymphocytes and neutrophils is seen. Neutrophils accumulate within the old bullae, clinically manifesting as a pustule.
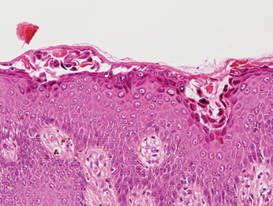
Darier disease is characterized by suprabasal clefts and acantholysis above the basal layer (Figure 6).6 Similar to HHD, villi protrude within the clefts (Figure 6). Conspicuous columns of parakeratosis above the acantholytic epidermis often are observed. Dyskeratotic cells exist among acantholytic ke-ratinocytes in the granular layer and parakeratotic column, which are known as corps ronds and crops grains, respectively. A scant to moderate lymphocytic infiltrate is found in the upper dermis.
- Hernandez-Perez E. Familial benign chronic pemphigus. Cutis. 1987;39:75-77.
- Venugopal SS, Murrell DF. Diagnosis and clinical features of pemphigus vulgaris. Dermatol Clin. 2011;29:373-380, vii.
- Dasher D, Rubenstein D, Diaz LA. Pemphigus foliaceus. Curr Dir Autoimmun. 2008;10:182-194.
- Ohata C, Akamatsu K, Imai N, et al. Localized pemphigus foliaceus exclusively involving the follicular infundibulum: a novel peau d’orange appearance. Eur J Dermatol. 2011;21:392-395.
- King DF, King LA. Giant cells in lesions of varicella and herpes zoster. Am J Dermatopathol. 1986;8:456-458.
- Burge S. Management of Darier’s disease. Clin Exp Dermatol. 1999;24:53-56.
Hailey-Hailey disease (HHD), or benign familial chronic pemphigus, typically presents as suprabasal blisters with a perivascular and interstitial lymphocytic infiltrate (Figure 1).1 Villi, or elongated dermal papillae lined with a single layer of basal cells, protrude into the bullae (Figure 2). In HHD lesions, the epidermis is thickened with scale-crust, and at least the lower half of the epidermis shows acantholysis. Despite the acantholytic changes, a few intact intercellular bridges remain, giving the appearance of a dilapidated brick wall (Figure 2). There may be dyskeratotic cells among the acantholytic cells, though they are scant in many cases. These acantholytic dyskeratotic cells have eosinophilic polygonal-shaped cytoplasm. Hailey-Hailey disease typically does not show adnexal extension of the acantholysis. Direct immunofluorescence is negative in HHD.
Pemphigus vulgaris is an autoimmune intraepidermal bullous disease that presents with suprabasal acantholysis (Figure 3).2 The epidermis is not thickened and acantholysis is limited to the suprabasal layer. Acantholytic cells with eosinophils and/or neutrophils are found within the bullae. Perivascular and interstitial infiltrates of lymphocytes, eosinophils, and occasionally neutrophils are seen; however, the inflammatory cell infiltrate can vary from extensive to scant. Direct immunofluorescence usually reveals IgG and/or C3 deposition on the surface of the keratinocytes throughout the epidermis.
Pemphigus foliaceus is another autoimmune intraepidermal bullous disease that is characterized by acantholysis in the granular or upper spinous layers (Figure 4).3 The epidermis is not thickened. Sometimes acantholytic cells show dyskeratotic change (Figure 4). Some biopsy specimens do not contain the roof of the bullae; therefore, only erosion is seen and the diagnosis may be missed. Moreover, when only the adnexal epithelium shows acantholysis without epidermal involvement, diagnosis can be difficult.4 Acantholysis is accompanied with a superficial perivascular and interstitial inflammatory cell infiltrate consisting of lymphocytes, eosinophils, and occasionally neutrophils. The amount of inflammatory cell infiltrate may vary. Bullous impetigo and staphylococcal scalded skin syndrome reveal a similar histopathologic pattern. Direct immunofluorescence usually discloses IgG and/or C3 deposition on cell surfaces of keratinocytes in the entire or upper epidermis.
Herpesvirus infection shows ballooning (intracellular edema) of keratinocytes. Eventually acantholysis occurs and intraepidermal bullae are formed. In the bullae, virus-associated acantholytic keratinocytes, some that are multinucleated, can be easily found (Figure 5).5 These cells are larger than normal keratinocytes and have steel gray nuclei with peripheral accentuation. Some of these cells are necrotic, and the remains of necrotic multinucleated acantholytic cells are easily recognized. Adnexal epithelial cells occasionally are affected by herpesvirus infection; nuclear change is similar to the epidermis. A perivascular and interstitial infiltrate of lymphocytes and neutrophils is seen. Neutrophils accumulate within the old bullae, clinically manifesting as a pustule.
Darier disease is characterized by suprabasal clefts and acantholysis above the basal layer (Figure 6).6 Similar to HHD, villi protrude within the clefts (Figure 6). Conspicuous columns of parakeratosis above the acantholytic epidermis often are observed. Dyskeratotic cells exist among acantholytic ke-ratinocytes in the granular layer and parakeratotic column, which are known as corps ronds and crops grains, respectively. A scant to moderate lymphocytic infiltrate is found in the upper dermis.
Hailey-Hailey disease (HHD), or benign familial chronic pemphigus, typically presents as suprabasal blisters with a perivascular and interstitial lymphocytic infiltrate (Figure 1).1 Villi, or elongated dermal papillae lined with a single layer of basal cells, protrude into the bullae (Figure 2). In HHD lesions, the epidermis is thickened with scale-crust, and at least the lower half of the epidermis shows acantholysis. Despite the acantholytic changes, a few intact intercellular bridges remain, giving the appearance of a dilapidated brick wall (Figure 2). There may be dyskeratotic cells among the acantholytic cells, though they are scant in many cases. These acantholytic dyskeratotic cells have eosinophilic polygonal-shaped cytoplasm. Hailey-Hailey disease typically does not show adnexal extension of the acantholysis. Direct immunofluorescence is negative in HHD.
Pemphigus vulgaris is an autoimmune intraepidermal bullous disease that presents with suprabasal acantholysis (Figure 3).2 The epidermis is not thickened and acantholysis is limited to the suprabasal layer. Acantholytic cells with eosinophils and/or neutrophils are found within the bullae. Perivascular and interstitial infiltrates of lymphocytes, eosinophils, and occasionally neutrophils are seen; however, the inflammatory cell infiltrate can vary from extensive to scant. Direct immunofluorescence usually reveals IgG and/or C3 deposition on the surface of the keratinocytes throughout the epidermis.
Pemphigus foliaceus is another autoimmune intraepidermal bullous disease that is characterized by acantholysis in the granular or upper spinous layers (Figure 4).3 The epidermis is not thickened. Sometimes acantholytic cells show dyskeratotic change (Figure 4). Some biopsy specimens do not contain the roof of the bullae; therefore, only erosion is seen and the diagnosis may be missed. Moreover, when only the adnexal epithelium shows acantholysis without epidermal involvement, diagnosis can be difficult.4 Acantholysis is accompanied with a superficial perivascular and interstitial inflammatory cell infiltrate consisting of lymphocytes, eosinophils, and occasionally neutrophils. The amount of inflammatory cell infiltrate may vary. Bullous impetigo and staphylococcal scalded skin syndrome reveal a similar histopathologic pattern. Direct immunofluorescence usually discloses IgG and/or C3 deposition on cell surfaces of keratinocytes in the entire or upper epidermis.
Herpesvirus infection shows ballooning (intracellular edema) of keratinocytes. Eventually acantholysis occurs and intraepidermal bullae are formed. In the bullae, virus-associated acantholytic keratinocytes, some that are multinucleated, can be easily found (Figure 5).5 These cells are larger than normal keratinocytes and have steel gray nuclei with peripheral accentuation. Some of these cells are necrotic, and the remains of necrotic multinucleated acantholytic cells are easily recognized. Adnexal epithelial cells occasionally are affected by herpesvirus infection; nuclear change is similar to the epidermis. A perivascular and interstitial infiltrate of lymphocytes and neutrophils is seen. Neutrophils accumulate within the old bullae, clinically manifesting as a pustule.
Darier disease is characterized by suprabasal clefts and acantholysis above the basal layer (Figure 6).6 Similar to HHD, villi protrude within the clefts (Figure 6). Conspicuous columns of parakeratosis above the acantholytic epidermis often are observed. Dyskeratotic cells exist among acantholytic ke-ratinocytes in the granular layer and parakeratotic column, which are known as corps ronds and crops grains, respectively. A scant to moderate lymphocytic infiltrate is found in the upper dermis.
- Hernandez-Perez E. Familial benign chronic pemphigus. Cutis. 1987;39:75-77.
- Venugopal SS, Murrell DF. Diagnosis and clinical features of pemphigus vulgaris. Dermatol Clin. 2011;29:373-380, vii.
- Dasher D, Rubenstein D, Diaz LA. Pemphigus foliaceus. Curr Dir Autoimmun. 2008;10:182-194.
- Ohata C, Akamatsu K, Imai N, et al. Localized pemphigus foliaceus exclusively involving the follicular infundibulum: a novel peau d’orange appearance. Eur J Dermatol. 2011;21:392-395.
- King DF, King LA. Giant cells in lesions of varicella and herpes zoster. Am J Dermatopathol. 1986;8:456-458.
- Burge S. Management of Darier’s disease. Clin Exp Dermatol. 1999;24:53-56.
- Hernandez-Perez E. Familial benign chronic pemphigus. Cutis. 1987;39:75-77.
- Venugopal SS, Murrell DF. Diagnosis and clinical features of pemphigus vulgaris. Dermatol Clin. 2011;29:373-380, vii.
- Dasher D, Rubenstein D, Diaz LA. Pemphigus foliaceus. Curr Dir Autoimmun. 2008;10:182-194.
- Ohata C, Akamatsu K, Imai N, et al. Localized pemphigus foliaceus exclusively involving the follicular infundibulum: a novel peau d’orange appearance. Eur J Dermatol. 2011;21:392-395.
- King DF, King LA. Giant cells in lesions of varicella and herpes zoster. Am J Dermatopathol. 1986;8:456-458.
- Burge S. Management of Darier’s disease. Clin Exp Dermatol. 1999;24:53-56.
