User login
A 16-Year-Old Hispanic Male with a History of Hyperlipidemia Reports a Pruritic Rash on His Neck and Chest
Discussion
Given the patient’s recent dietary changes, particularly his switch to a ketogenic diet, he was diagnosed with prurigo pigmentosa and treated with doxycycline, which cleared the rash. Prurigo pigmentosa is a rare inflammatory dermatosis characterized by net-like or reticulated pink, and later hyperpigmented, papules and plaques. Although the condition predominantly affects young women of East Asian descent, cases have been reported worldwide, highlighting the importance of considering this diagnosis in diverse populations, including children. Here, we describe a case of prurigo pigmentosa in a young male who had recently adopted a ketogenic diet for weight loss.
The association between prurigo pigmentosa and dietary changes, particularly ketosis, is becoming increasingly recognized. This condition is strongly linked to ketosis, a metabolic state marked by the production of ketone bodies (e.g., beta-hydroxybutyrate and acetoacetate) during carbohydrate restriction, fasting, or ketogenic diets, as seen in our patient. These ketone bodies may act as irritants or trigger oxidative stress and inflammatory cascades in the skin.
Ketoacidosis, particularly in prolonged or intense ketosis, is thought to alter the local skin microenvironment, promoting activation of inflammatory cytokines and immune cells. The ketogenic state is believed to generate oxidative stress through increased free fatty acid oxidation, leading to the production of reactive oxygen species (ROS). ROS can induce apoptosis of keratinocytes and inflammation in the epidermis, which is predominantly mediated by neutrophilic infiltration, as seen in histopathological findings. Elevated levels of pro-inflammatory cytokines, such as interleukin-8 (IL-8) and tumor necrosis factor-alpha (TNF-α), have been implicated in neutrophil recruitment and activation. IL-8 is particularly important for guiding neutrophils to areas of injury.
Secondary hyperpigmentation, a hallmark of this condition, is thought to result from melanin-laden macrophages and persistent melanocyte activation in response to inflammation at the dermo-epidermal junction.
The condition progresses in three stages. In the early stage, lesions appear as pruritic, urticarial plaques. These evolve into crusted erythematous papules and papulovesicles in the middle stage, as observed in our patient. Finally, in the late stage, the lesions mature into smooth, hyperpigmented plaques. Each stage of prurigo pigmentosa has distinct histopathological features.
Differential Diagnosis
The differential diagnosis for prurigo pigmentosa includes several conditions that may present similarly. Allergic contact dermatitis (ACD) can initially mimic the erythematous papules of prurigo pigmentosa, but the absence of a clear allergen exposure and failure to improve with avoidance measures makes ACD less likely. Psoriasis is another possibility, as its erythematous plaques may overlap with prurigo pigmentosa. However, the lack of silvery scales and chronicity makes psoriasis less likely in this case. Eczema, or atopic dermatitis, typically presents with pruritic, ill-defined plaques, often in flexural areas, which were not observed in this patient. Flagellate dermatitis, often caused by exposure to bleomycin or consumption of shiitake mushrooms, can present with linear erythematous lesions resembling prurigo pigmentosa. However, the absence of relevant exposures and a flagellate pattern in this patient rules out this diagnosis.
This case highlights the growing recognition of prurigo pigmentosa in the context of dietary trends, especially ketogenic diets, which have become popular for weight loss and other health benefits. Pediatric populations, in particular, may adopt such diets for various reasons and require careful monitoring, as their physiological responses may differ from those in adults. Prurigo pigmentosa has also been reported in a teenager girl with a history of anorexia nervosa, who was in a ketotic state.
Treatment options for prurigo pigmentosa include antibiotics such as minocycline or doxycycline, or macrolides for 4–10 weeks. Other treatment modalities include dapsone, Q-switch Nd:YAG laser, narrow-band ultraviolet B (UVB) phototherapy, and topical treatments like crisaborole and tacrolimus.
Early recognition of this condition is crucial to avoid unnecessary interventions and to allow for resolution through dietary modification. Dermatologists and pediatricians should maintain a high index of suspicion for prurigo pigmentosa in patients presenting with characteristic eruptions and a history of dietary ketosis.
Catalina Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
Suggested Reading
1. Mufti A et al. Clinical Manifestations and Treatment Outcomes in Prurigo Pigmentosa (Nagashima Disease): A Systematic Review of the Literature. JAAD Int. 2021 Apr 10:3:79-87. doi: 10.1016/j.jdin.2021.03.003.
2. Yang J et al. Use of Minocycline for the Treatment of Prurigo Pigmentosa with Intraepidermal Vesiculation: A Case Report. J Int Med Res. 2021 May;49(5):3000605211015593. doi: 10.1177/03000605211015593.
3. Capucilli P et al. Prurigo Pigmentosa: An Itchy, Urticarial Eruption Confused for Food Allergy. J Allergy Clin Immunol Pract. 2018 Jul-Aug;6(4):1381-1382. doi: 10.1016/j.jaip.2018.02.033.
Discussion
Given the patient’s recent dietary changes, particularly his switch to a ketogenic diet, he was diagnosed with prurigo pigmentosa and treated with doxycycline, which cleared the rash. Prurigo pigmentosa is a rare inflammatory dermatosis characterized by net-like or reticulated pink, and later hyperpigmented, papules and plaques. Although the condition predominantly affects young women of East Asian descent, cases have been reported worldwide, highlighting the importance of considering this diagnosis in diverse populations, including children. Here, we describe a case of prurigo pigmentosa in a young male who had recently adopted a ketogenic diet for weight loss.
The association between prurigo pigmentosa and dietary changes, particularly ketosis, is becoming increasingly recognized. This condition is strongly linked to ketosis, a metabolic state marked by the production of ketone bodies (e.g., beta-hydroxybutyrate and acetoacetate) during carbohydrate restriction, fasting, or ketogenic diets, as seen in our patient. These ketone bodies may act as irritants or trigger oxidative stress and inflammatory cascades in the skin.
Ketoacidosis, particularly in prolonged or intense ketosis, is thought to alter the local skin microenvironment, promoting activation of inflammatory cytokines and immune cells. The ketogenic state is believed to generate oxidative stress through increased free fatty acid oxidation, leading to the production of reactive oxygen species (ROS). ROS can induce apoptosis of keratinocytes and inflammation in the epidermis, which is predominantly mediated by neutrophilic infiltration, as seen in histopathological findings. Elevated levels of pro-inflammatory cytokines, such as interleukin-8 (IL-8) and tumor necrosis factor-alpha (TNF-α), have been implicated in neutrophil recruitment and activation. IL-8 is particularly important for guiding neutrophils to areas of injury.
Secondary hyperpigmentation, a hallmark of this condition, is thought to result from melanin-laden macrophages and persistent melanocyte activation in response to inflammation at the dermo-epidermal junction.
The condition progresses in three stages. In the early stage, lesions appear as pruritic, urticarial plaques. These evolve into crusted erythematous papules and papulovesicles in the middle stage, as observed in our patient. Finally, in the late stage, the lesions mature into smooth, hyperpigmented plaques. Each stage of prurigo pigmentosa has distinct histopathological features.
Differential Diagnosis
The differential diagnosis for prurigo pigmentosa includes several conditions that may present similarly. Allergic contact dermatitis (ACD) can initially mimic the erythematous papules of prurigo pigmentosa, but the absence of a clear allergen exposure and failure to improve with avoidance measures makes ACD less likely. Psoriasis is another possibility, as its erythematous plaques may overlap with prurigo pigmentosa. However, the lack of silvery scales and chronicity makes psoriasis less likely in this case. Eczema, or atopic dermatitis, typically presents with pruritic, ill-defined plaques, often in flexural areas, which were not observed in this patient. Flagellate dermatitis, often caused by exposure to bleomycin or consumption of shiitake mushrooms, can present with linear erythematous lesions resembling prurigo pigmentosa. However, the absence of relevant exposures and a flagellate pattern in this patient rules out this diagnosis.
This case highlights the growing recognition of prurigo pigmentosa in the context of dietary trends, especially ketogenic diets, which have become popular for weight loss and other health benefits. Pediatric populations, in particular, may adopt such diets for various reasons and require careful monitoring, as their physiological responses may differ from those in adults. Prurigo pigmentosa has also been reported in a teenager girl with a history of anorexia nervosa, who was in a ketotic state.
Treatment options for prurigo pigmentosa include antibiotics such as minocycline or doxycycline, or macrolides for 4–10 weeks. Other treatment modalities include dapsone, Q-switch Nd:YAG laser, narrow-band ultraviolet B (UVB) phototherapy, and topical treatments like crisaborole and tacrolimus.
Early recognition of this condition is crucial to avoid unnecessary interventions and to allow for resolution through dietary modification. Dermatologists and pediatricians should maintain a high index of suspicion for prurigo pigmentosa in patients presenting with characteristic eruptions and a history of dietary ketosis.
Catalina Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
Suggested Reading
1. Mufti A et al. Clinical Manifestations and Treatment Outcomes in Prurigo Pigmentosa (Nagashima Disease): A Systematic Review of the Literature. JAAD Int. 2021 Apr 10:3:79-87. doi: 10.1016/j.jdin.2021.03.003.
2. Yang J et al. Use of Minocycline for the Treatment of Prurigo Pigmentosa with Intraepidermal Vesiculation: A Case Report. J Int Med Res. 2021 May;49(5):3000605211015593. doi: 10.1177/03000605211015593.
3. Capucilli P et al. Prurigo Pigmentosa: An Itchy, Urticarial Eruption Confused for Food Allergy. J Allergy Clin Immunol Pract. 2018 Jul-Aug;6(4):1381-1382. doi: 10.1016/j.jaip.2018.02.033.
Discussion
Given the patient’s recent dietary changes, particularly his switch to a ketogenic diet, he was diagnosed with prurigo pigmentosa and treated with doxycycline, which cleared the rash. Prurigo pigmentosa is a rare inflammatory dermatosis characterized by net-like or reticulated pink, and later hyperpigmented, papules and plaques. Although the condition predominantly affects young women of East Asian descent, cases have been reported worldwide, highlighting the importance of considering this diagnosis in diverse populations, including children. Here, we describe a case of prurigo pigmentosa in a young male who had recently adopted a ketogenic diet for weight loss.
The association between prurigo pigmentosa and dietary changes, particularly ketosis, is becoming increasingly recognized. This condition is strongly linked to ketosis, a metabolic state marked by the production of ketone bodies (e.g., beta-hydroxybutyrate and acetoacetate) during carbohydrate restriction, fasting, or ketogenic diets, as seen in our patient. These ketone bodies may act as irritants or trigger oxidative stress and inflammatory cascades in the skin.
Ketoacidosis, particularly in prolonged or intense ketosis, is thought to alter the local skin microenvironment, promoting activation of inflammatory cytokines and immune cells. The ketogenic state is believed to generate oxidative stress through increased free fatty acid oxidation, leading to the production of reactive oxygen species (ROS). ROS can induce apoptosis of keratinocytes and inflammation in the epidermis, which is predominantly mediated by neutrophilic infiltration, as seen in histopathological findings. Elevated levels of pro-inflammatory cytokines, such as interleukin-8 (IL-8) and tumor necrosis factor-alpha (TNF-α), have been implicated in neutrophil recruitment and activation. IL-8 is particularly important for guiding neutrophils to areas of injury.
Secondary hyperpigmentation, a hallmark of this condition, is thought to result from melanin-laden macrophages and persistent melanocyte activation in response to inflammation at the dermo-epidermal junction.
The condition progresses in three stages. In the early stage, lesions appear as pruritic, urticarial plaques. These evolve into crusted erythematous papules and papulovesicles in the middle stage, as observed in our patient. Finally, in the late stage, the lesions mature into smooth, hyperpigmented plaques. Each stage of prurigo pigmentosa has distinct histopathological features.
Differential Diagnosis
The differential diagnosis for prurigo pigmentosa includes several conditions that may present similarly. Allergic contact dermatitis (ACD) can initially mimic the erythematous papules of prurigo pigmentosa, but the absence of a clear allergen exposure and failure to improve with avoidance measures makes ACD less likely. Psoriasis is another possibility, as its erythematous plaques may overlap with prurigo pigmentosa. However, the lack of silvery scales and chronicity makes psoriasis less likely in this case. Eczema, or atopic dermatitis, typically presents with pruritic, ill-defined plaques, often in flexural areas, which were not observed in this patient. Flagellate dermatitis, often caused by exposure to bleomycin or consumption of shiitake mushrooms, can present with linear erythematous lesions resembling prurigo pigmentosa. However, the absence of relevant exposures and a flagellate pattern in this patient rules out this diagnosis.
This case highlights the growing recognition of prurigo pigmentosa in the context of dietary trends, especially ketogenic diets, which have become popular for weight loss and other health benefits. Pediatric populations, in particular, may adopt such diets for various reasons and require careful monitoring, as their physiological responses may differ from those in adults. Prurigo pigmentosa has also been reported in a teenager girl with a history of anorexia nervosa, who was in a ketotic state.
Treatment options for prurigo pigmentosa include antibiotics such as minocycline or doxycycline, or macrolides for 4–10 weeks. Other treatment modalities include dapsone, Q-switch Nd:YAG laser, narrow-band ultraviolet B (UVB) phototherapy, and topical treatments like crisaborole and tacrolimus.
Early recognition of this condition is crucial to avoid unnecessary interventions and to allow for resolution through dietary modification. Dermatologists and pediatricians should maintain a high index of suspicion for prurigo pigmentosa in patients presenting with characteristic eruptions and a history of dietary ketosis.
Catalina Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
Suggested Reading
1. Mufti A et al. Clinical Manifestations and Treatment Outcomes in Prurigo Pigmentosa (Nagashima Disease): A Systematic Review of the Literature. JAAD Int. 2021 Apr 10:3:79-87. doi: 10.1016/j.jdin.2021.03.003.
2. Yang J et al. Use of Minocycline for the Treatment of Prurigo Pigmentosa with Intraepidermal Vesiculation: A Case Report. J Int Med Res. 2021 May;49(5):3000605211015593. doi: 10.1177/03000605211015593.
3. Capucilli P et al. Prurigo Pigmentosa: An Itchy, Urticarial Eruption Confused for Food Allergy. J Allergy Clin Immunol Pract. 2018 Jul-Aug;6(4):1381-1382. doi: 10.1016/j.jaip.2018.02.033.
Case Report
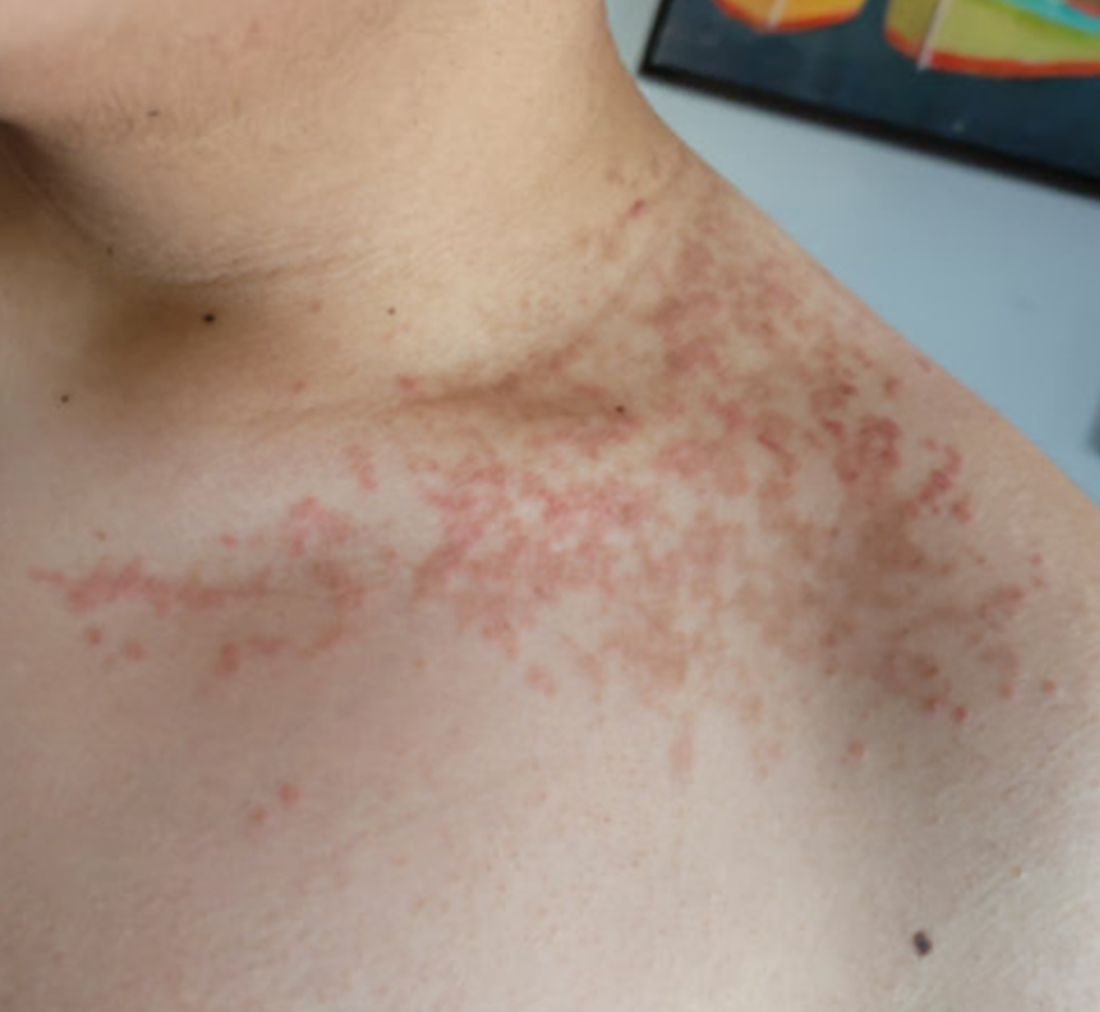
A 16-year-old Hispanic male with a history of hyperlipidemia presents to his pediatrician's office for a routine well-child check-up. He reports a pruritic rash on his neck and chest that has been present for the past 1.5 weeks. The rash is itchy, and although a cream from Mexico initially helped, it has not been effective recently. The patient mentions that he has increased his gym workouts and has been training for basketball. He has a history of obesity but has lost almost 100 pounds in the last 6 months. Most recently, he has stopped consuming carbohydrates and has been fasting in the mornings.
There is no history of eczema or psoriasis, either in the patient or his family.
Physical Examination
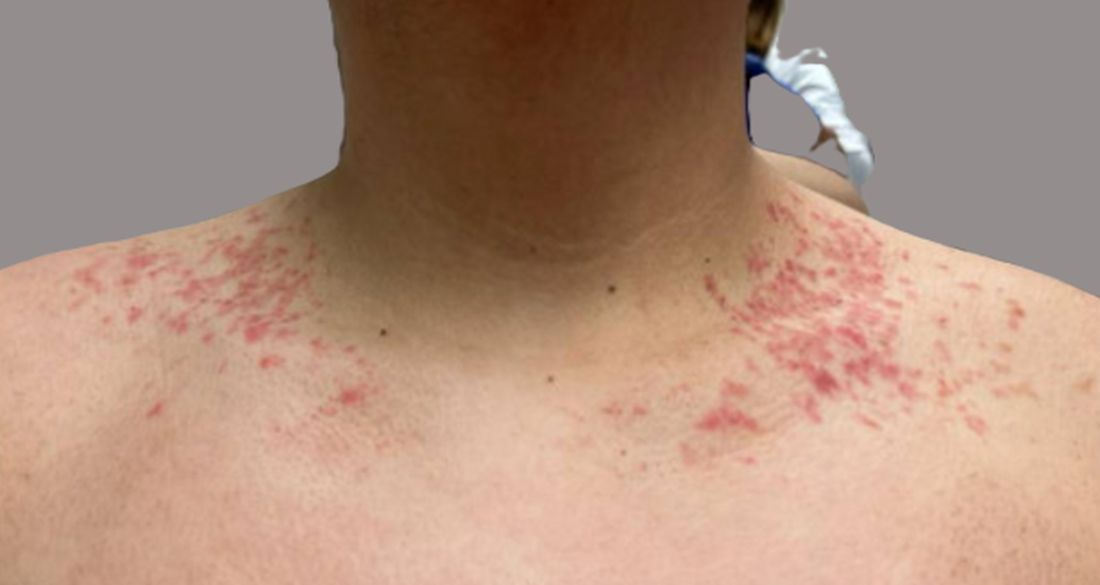
The patient weighs 147 pounds, a significant decrease from his previous weight of 270 pounds 6 months ago. Other vital signs are within normal limits.
On physical examination, the patient presents with net-like, pink, scaly plaques on his neck, with no other rashes on the body (see Pictures 1 and 2).
A 7-Year-Old Boy Presents With Dark Spots on His Scalp and Areas of Poor Hair Growth
Given the trichoscopic findings, scrapings from the scaly areas were taken and revealed hyphae, confirming the diagnosis of tinea capitis. A fungal culture identified Trichophyton tonsurans as the causative organism.
Tinea capitis is the most common dermatophyte infection in children. Risk factors include participation in close-contact sports like wrestling or jiu-jitsu, attendance at daycare for younger children, African American hair care practices, pet ownership (particularly cats and rodents), and living in overcrowded conditions.
Diagnosis of tinea capitis requires a thorough clinical history to identify potential risk factors. On physical examination, patchy hair loss with associated scaling should raise suspicion for tinea capitis. Inflammatory signs, such as pustules and swelling, may suggest the presence of a kerion, further supporting the diagnosis. Although some practitioners use Wood’s lamp to help with diagnosis, its utility is limited. It detects fluorescence in Microsporum species (exothrix infections) but not in Trichophyton species (endothrix infections).
Trichoscopy can be a valuable tool when inflammation is minimal, and only hair loss and scaling are observed. Trichoscopic findings suggestive of tinea capitis include comma hairs, corkscrew hairs (as seen in this patient), Morse code-like hairs, zigzag hairs, bent hairs, block hairs, and i-hairs. Other common, though not characteristic, findings include broken hairs, black dots, perifollicular scaling, and diffuse scaling.
KOH (potassium hydroxide) analysis is another useful method for detecting fungal elements, though it does not identify the specific fungus and may not be available in all clinical settings. Mycologic culture remains the gold standard for diagnosing tinea capitis, though results can take 3-4 weeks. Newer diagnostic techniques, such as PCR analysis and MALDI-TOF/MS, offer more rapid identification of the causative organism.

The differential diagnosis includes:
- Seborrheic dermatitis, which presents with greasy, yellowish scales and itching, with trichoscopy showing twisted, coiled hairs and yellowish scaling.
- Psoriasis, which can mimic tinea capitis but presents with well-demarcated red plaques and silvery-white scales. Trichoscopy shows red dots and uniform scaling.
- Alopecia areata, which causes patchy hair loss without inflammation or scaling, with trichoscopic findings of exclamation mark hairs, black dots, and yellow dots.
- Trichotillomania, a hair-pulling disorder, which results in irregular patches of hair loss. Trichoscopy shows broken hairs of varying lengths, V-sign hairs, and flame-shaped residues at follicular openings.
Treatment of tinea capitis requires systemic antifungals and topical agents to prevent fungal spore spread. Several treatment guidelines are available from different institutions. Griseofulvin (FDA-approved for patients > 2 years of age) has been widely used, particularly for Microsporum canis infections. However, due to limited availability in many countries, terbinafine (FDA-approved for patients > 4 years of age) is now commonly used as first-line therapy, especially for Trichophyton species. Treatment typically lasts 4-6 weeks, and post-treatment cultures may be recommended to confirm mycologic cure.
Concerns about drug resistance have emerged, particularly for terbinafine-resistant dermatophytes linked to mutations in the squalene epoxidase enzyme. Resistance may be driven by limited antifungal availability and poor adherence to prolonged treatment regimens. While fluconazole and itraconazole are used off-label, growing evidence supports their effectiveness, although one large trial showed suboptimal cure rates with fluconazole.
Though systemic antifungals are generally safe, hepatotoxicity remains a concern, especially in patients with hepatic conditions or other comorbidities. Lab monitoring is advised for patients on prolonged or multiple therapies, or for those with coexisting conditions. The decision to conduct lab monitoring should be discussed with parents, balancing the very low risk of hepatotoxicity in healthy children against their comfort level.
An alternative to systemic therapy is photodynamic therapy (PDT), which has been reported as successful in treating tinea capitis infections, particularly in cases of T. mentagrophytes and M. canis. However, large-scale trials are needed to confirm PDT’s efficacy and safety.
In conclusion, children presenting with hair loss, scaling, and associated dark spots on the scalp should be evaluated for fungal infection. While trichoscopy can aid in diagnosis, fungal culture remains the gold standard for confirmation.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Rudnicka L et al. Hair shafts in trichoscopy: clues for diagnosis of hair and scalp diseases. Dermatol Clin. 2013 Oct;31(4):695-708, x. doi: 10.1016/j.det.2013.06.007.
Gupta AK et al. An update on tinea capitis in children. Pediatr Dermatol. 2024 Aug 7. doi: 10.1111/pde.15708.
Anna Waskiel-Burnat et al. Trichoscopy of tinea capitis: A systematic review. Dermatol Ther (Heidelb). 2020 Feb;10(1):43-52. doi: 10.1007/s13555-019-00350-1.
Given the trichoscopic findings, scrapings from the scaly areas were taken and revealed hyphae, confirming the diagnosis of tinea capitis. A fungal culture identified Trichophyton tonsurans as the causative organism.
Tinea capitis is the most common dermatophyte infection in children. Risk factors include participation in close-contact sports like wrestling or jiu-jitsu, attendance at daycare for younger children, African American hair care practices, pet ownership (particularly cats and rodents), and living in overcrowded conditions.
Diagnosis of tinea capitis requires a thorough clinical history to identify potential risk factors. On physical examination, patchy hair loss with associated scaling should raise suspicion for tinea capitis. Inflammatory signs, such as pustules and swelling, may suggest the presence of a kerion, further supporting the diagnosis. Although some practitioners use Wood’s lamp to help with diagnosis, its utility is limited. It detects fluorescence in Microsporum species (exothrix infections) but not in Trichophyton species (endothrix infections).
Trichoscopy can be a valuable tool when inflammation is minimal, and only hair loss and scaling are observed. Trichoscopic findings suggestive of tinea capitis include comma hairs, corkscrew hairs (as seen in this patient), Morse code-like hairs, zigzag hairs, bent hairs, block hairs, and i-hairs. Other common, though not characteristic, findings include broken hairs, black dots, perifollicular scaling, and diffuse scaling.
KOH (potassium hydroxide) analysis is another useful method for detecting fungal elements, though it does not identify the specific fungus and may not be available in all clinical settings. Mycologic culture remains the gold standard for diagnosing tinea capitis, though results can take 3-4 weeks. Newer diagnostic techniques, such as PCR analysis and MALDI-TOF/MS, offer more rapid identification of the causative organism.
The differential diagnosis includes:
- Seborrheic dermatitis, which presents with greasy, yellowish scales and itching, with trichoscopy showing twisted, coiled hairs and yellowish scaling.
- Psoriasis, which can mimic tinea capitis but presents with well-demarcated red plaques and silvery-white scales. Trichoscopy shows red dots and uniform scaling.
- Alopecia areata, which causes patchy hair loss without inflammation or scaling, with trichoscopic findings of exclamation mark hairs, black dots, and yellow dots.
- Trichotillomania, a hair-pulling disorder, which results in irregular patches of hair loss. Trichoscopy shows broken hairs of varying lengths, V-sign hairs, and flame-shaped residues at follicular openings.
Treatment of tinea capitis requires systemic antifungals and topical agents to prevent fungal spore spread. Several treatment guidelines are available from different institutions. Griseofulvin (FDA-approved for patients > 2 years of age) has been widely used, particularly for Microsporum canis infections. However, due to limited availability in many countries, terbinafine (FDA-approved for patients > 4 years of age) is now commonly used as first-line therapy, especially for Trichophyton species. Treatment typically lasts 4-6 weeks, and post-treatment cultures may be recommended to confirm mycologic cure.
Concerns about drug resistance have emerged, particularly for terbinafine-resistant dermatophytes linked to mutations in the squalene epoxidase enzyme. Resistance may be driven by limited antifungal availability and poor adherence to prolonged treatment regimens. While fluconazole and itraconazole are used off-label, growing evidence supports their effectiveness, although one large trial showed suboptimal cure rates with fluconazole.
Though systemic antifungals are generally safe, hepatotoxicity remains a concern, especially in patients with hepatic conditions or other comorbidities. Lab monitoring is advised for patients on prolonged or multiple therapies, or for those with coexisting conditions. The decision to conduct lab monitoring should be discussed with parents, balancing the very low risk of hepatotoxicity in healthy children against their comfort level.
An alternative to systemic therapy is photodynamic therapy (PDT), which has been reported as successful in treating tinea capitis infections, particularly in cases of T. mentagrophytes and M. canis. However, large-scale trials are needed to confirm PDT’s efficacy and safety.
In conclusion, children presenting with hair loss, scaling, and associated dark spots on the scalp should be evaluated for fungal infection. While trichoscopy can aid in diagnosis, fungal culture remains the gold standard for confirmation.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Rudnicka L et al. Hair shafts in trichoscopy: clues for diagnosis of hair and scalp diseases. Dermatol Clin. 2013 Oct;31(4):695-708, x. doi: 10.1016/j.det.2013.06.007.
Gupta AK et al. An update on tinea capitis in children. Pediatr Dermatol. 2024 Aug 7. doi: 10.1111/pde.15708.
Anna Waskiel-Burnat et al. Trichoscopy of tinea capitis: A systematic review. Dermatol Ther (Heidelb). 2020 Feb;10(1):43-52. doi: 10.1007/s13555-019-00350-1.
Given the trichoscopic findings, scrapings from the scaly areas were taken and revealed hyphae, confirming the diagnosis of tinea capitis. A fungal culture identified Trichophyton tonsurans as the causative organism.
Tinea capitis is the most common dermatophyte infection in children. Risk factors include participation in close-contact sports like wrestling or jiu-jitsu, attendance at daycare for younger children, African American hair care practices, pet ownership (particularly cats and rodents), and living in overcrowded conditions.
Diagnosis of tinea capitis requires a thorough clinical history to identify potential risk factors. On physical examination, patchy hair loss with associated scaling should raise suspicion for tinea capitis. Inflammatory signs, such as pustules and swelling, may suggest the presence of a kerion, further supporting the diagnosis. Although some practitioners use Wood’s lamp to help with diagnosis, its utility is limited. It detects fluorescence in Microsporum species (exothrix infections) but not in Trichophyton species (endothrix infections).
Trichoscopy can be a valuable tool when inflammation is minimal, and only hair loss and scaling are observed. Trichoscopic findings suggestive of tinea capitis include comma hairs, corkscrew hairs (as seen in this patient), Morse code-like hairs, zigzag hairs, bent hairs, block hairs, and i-hairs. Other common, though not characteristic, findings include broken hairs, black dots, perifollicular scaling, and diffuse scaling.
KOH (potassium hydroxide) analysis is another useful method for detecting fungal elements, though it does not identify the specific fungus and may not be available in all clinical settings. Mycologic culture remains the gold standard for diagnosing tinea capitis, though results can take 3-4 weeks. Newer diagnostic techniques, such as PCR analysis and MALDI-TOF/MS, offer more rapid identification of the causative organism.
The differential diagnosis includes:
- Seborrheic dermatitis, which presents with greasy, yellowish scales and itching, with trichoscopy showing twisted, coiled hairs and yellowish scaling.
- Psoriasis, which can mimic tinea capitis but presents with well-demarcated red plaques and silvery-white scales. Trichoscopy shows red dots and uniform scaling.
- Alopecia areata, which causes patchy hair loss without inflammation or scaling, with trichoscopic findings of exclamation mark hairs, black dots, and yellow dots.
- Trichotillomania, a hair-pulling disorder, which results in irregular patches of hair loss. Trichoscopy shows broken hairs of varying lengths, V-sign hairs, and flame-shaped residues at follicular openings.
Treatment of tinea capitis requires systemic antifungals and topical agents to prevent fungal spore spread. Several treatment guidelines are available from different institutions. Griseofulvin (FDA-approved for patients > 2 years of age) has been widely used, particularly for Microsporum canis infections. However, due to limited availability in many countries, terbinafine (FDA-approved for patients > 4 years of age) is now commonly used as first-line therapy, especially for Trichophyton species. Treatment typically lasts 4-6 weeks, and post-treatment cultures may be recommended to confirm mycologic cure.
Concerns about drug resistance have emerged, particularly for terbinafine-resistant dermatophytes linked to mutations in the squalene epoxidase enzyme. Resistance may be driven by limited antifungal availability and poor adherence to prolonged treatment regimens. While fluconazole and itraconazole are used off-label, growing evidence supports their effectiveness, although one large trial showed suboptimal cure rates with fluconazole.
Though systemic antifungals are generally safe, hepatotoxicity remains a concern, especially in patients with hepatic conditions or other comorbidities. Lab monitoring is advised for patients on prolonged or multiple therapies, or for those with coexisting conditions. The decision to conduct lab monitoring should be discussed with parents, balancing the very low risk of hepatotoxicity in healthy children against their comfort level.
An alternative to systemic therapy is photodynamic therapy (PDT), which has been reported as successful in treating tinea capitis infections, particularly in cases of T. mentagrophytes and M. canis. However, large-scale trials are needed to confirm PDT’s efficacy and safety.
In conclusion, children presenting with hair loss, scaling, and associated dark spots on the scalp should be evaluated for fungal infection. While trichoscopy can aid in diagnosis, fungal culture remains the gold standard for confirmation.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Rudnicka L et al. Hair shafts in trichoscopy: clues for diagnosis of hair and scalp diseases. Dermatol Clin. 2013 Oct;31(4):695-708, x. doi: 10.1016/j.det.2013.06.007.
Gupta AK et al. An update on tinea capitis in children. Pediatr Dermatol. 2024 Aug 7. doi: 10.1111/pde.15708.
Anna Waskiel-Burnat et al. Trichoscopy of tinea capitis: A systematic review. Dermatol Ther (Heidelb). 2020 Feb;10(1):43-52. doi: 10.1007/s13555-019-00350-1.
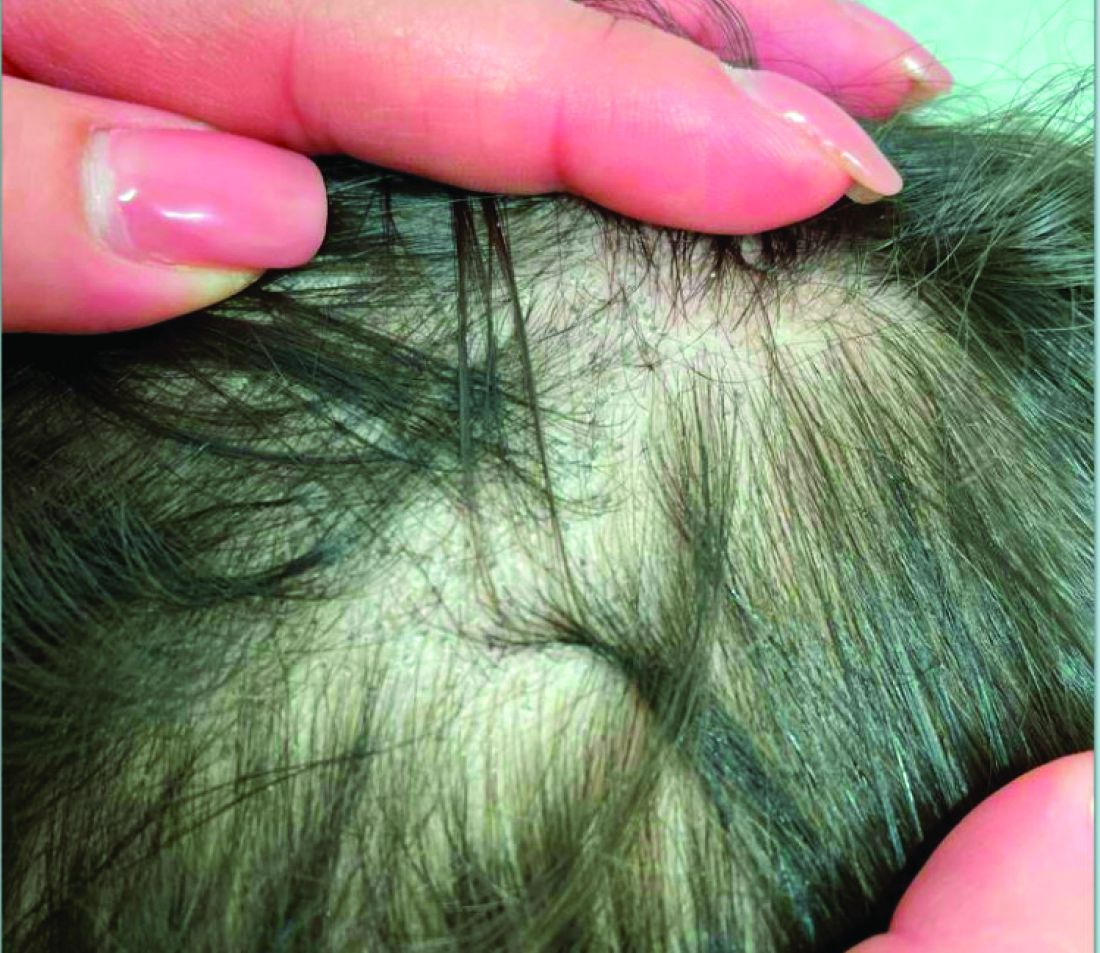
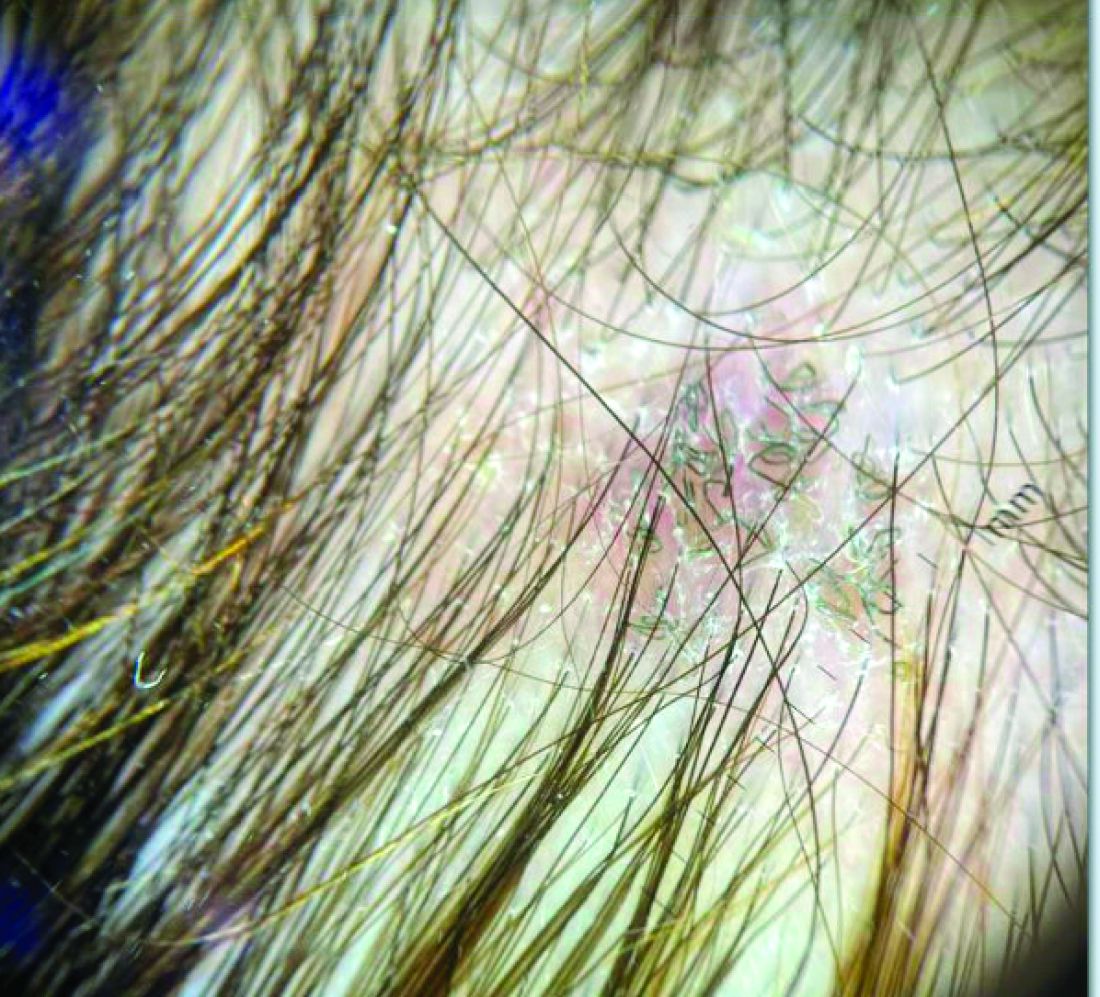
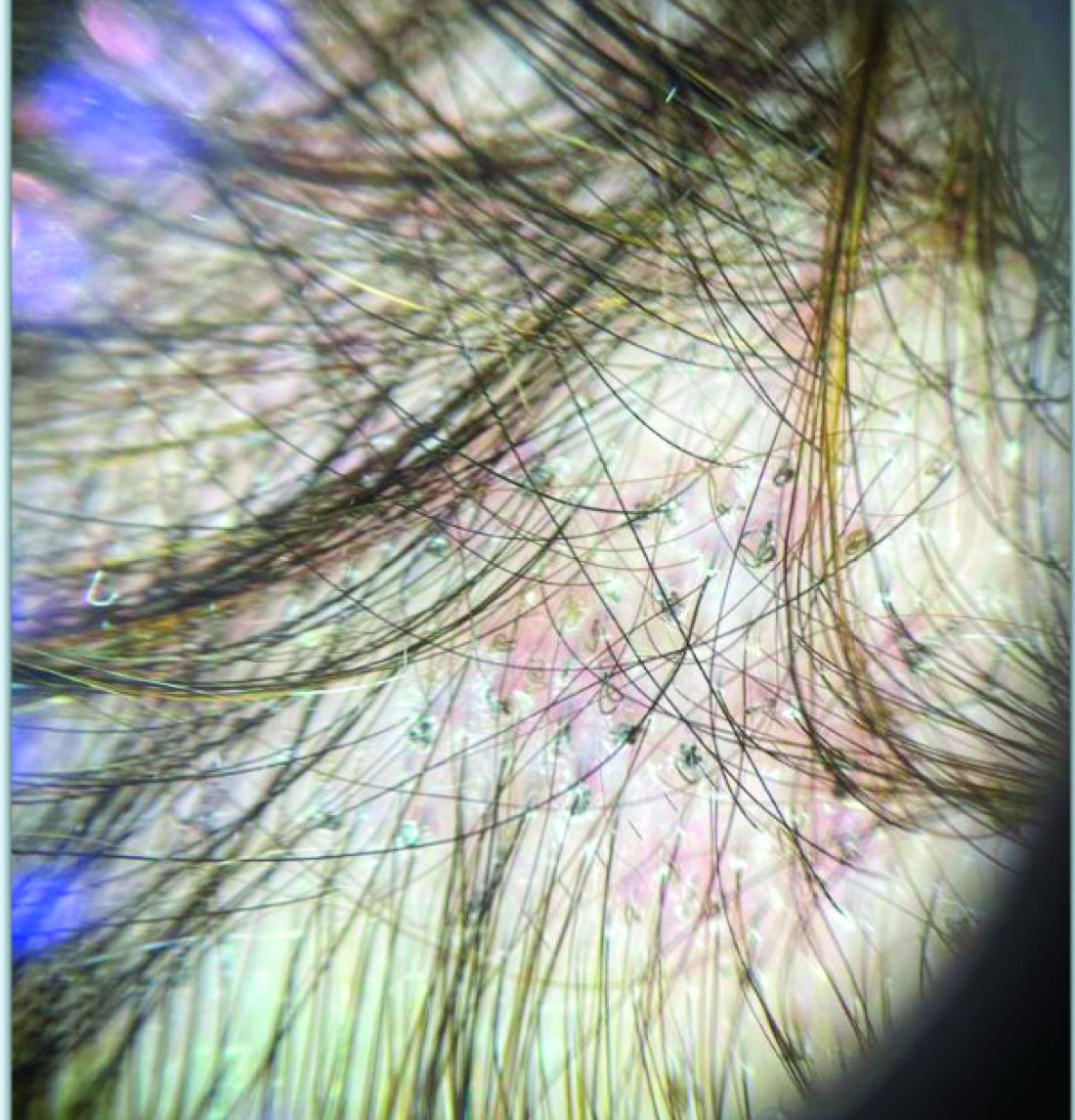
A 7-Month-Old Female Presented With Nail Changes
Given the clinical presentation and the absence of other systemic or dermatological findings, the diagnosis of chevron nails was made.
Discussion
The condition is characterized by transverse ridges on the nails that converge towards the center, forming a V or chevron shape. This condition was first described by Perry et al. and later by Shuster et al., who explained that the condition might result from axial growth of the nail with synchronous growth occurring from a chevron-shaped growing edge of the nail root. Alternatively, Shuster suggested that sequential growth, with localized variation in the nail production rate, could propagate a wave from the center of the nail to the edge.
The etiology of chevron nails is not well understood, but it is believed to result from temporary disruptions in the nail matrix, possibly related to minor illness or physiological stress during infancy.
In the case of our 7-month-old patient, the history of mild upper respiratory infections might have contributed to the development of chevron nails. However, the lack of other significant illness, skin involvement, or systemic findings supports the benign and self-limiting nature of this condition. Parents were reassured that chevron nails typically resolve on their own as the child grows and that no specific treatment is necessary.
Differential Diagnosis
The differential diagnosis of transverse nail changes in children includes other conditions such as trachyonychia, lichen planus, Darier disease, and pachyonychia congenita.
Trachyonychia, also known as “sandpaper nails,” trachyonychia is characterized by the roughening of the nail surface, giving it a dull and ridged appearance. The condition may affect all 20 nails and is often associated with underlying dermatological conditions such as lichen planus or alopecia areata. Unlike chevron nails, trachyonychia presents with more diffuse nail changes and does not typically feature the distinct V-shaped ridging seen in this patient.
Lichen planus is an inflammatory condition that can affect the skin, mucous membranes, and nails. Nail involvement in lichen planus can lead to longitudinal ridging, thinning, and sometimes even complete nail loss. The absence of other characteristic features of lichen planus, such as violaceous papules on the skin or white lacy patterns on mucous membranes (Wickham striae), makes this diagnosis less likely in our patient.
Darier disease, also known as keratosis follicularis, is a genetic disorder characterized by greasy, warty papules primarily on seborrheic areas of the skin, nail abnormalities, and sometimes mucosal involvement. Nail changes in Darier disease include longitudinal red and white streaks, V-shaped notching at the free edge of the nails, and subungual hyperkeratosis. These nail changes are more severe and distinct than the simple transverse ridging seen in chevron nails. The absence of other clinical signs of Darier disease, such as skin papules or characteristic nail notching, makes this diagnosis unlikely in our patient.
Pachyonychia congenita is a rare genetic disorder characterized by thickened nails (pachyonychia), painful plantar keratoderma, and sometimes oral leukokeratosis. The condition typically presents with significant nail thickening and other systemic findings, which were absent in our patient. The distinct pattern of V-shaped ridging observed in chevron nails does not align with the typical presentation of pachyonychia congenita.
Next Steps
No specific treatment is required for chevron nails. The condition is typically self-resolving, and the nails usually return to a normal appearance as the child continues to grow. Parents were advised to monitor the nails for any changes or new symptoms and were reassured about the benign nature of the findings. Follow-up was scheduled to ensure the resolution of the condition as the child develops.

Conclusion
Chevron nails are an important consideration in the differential diagnosis of transverse nail ridging in infants and young children. While the condition is benign and self-limiting, it is crucial to differentiate it from other nail dystrophies, such as trachyonychia, lichen planus, Darier disease, and pachyonychia congenita, which may require further investigation or intervention. Awareness of chevron nails can help prevent unnecessary worry and provide reassurance to parents and caregivers.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
Suggested Reading
Delano S, Belazarian L. Chevron nails: A normal variant in the pediatric population. Pediatr Dermatol. 2014 Jan-Feb;31(1):e24-5. doi: 10.1111/pde.12193.
John JM et al. Chevron nail — An under-recognised normal variant of nail development. Arch Dis Child. 2024 Jul 18;109(8):648. doi: 10.1136/archdischild-2024-326975.
Shuster S. The significance of chevron nails. Br J Dermatol. 1996;135:151–152. doi: 10.1046/j.1365-2133.1996.d01-961.x.
Starace M et al. Nail disorders in children. Skin Appendage Disord. 2018 Oct;4(4):217-229. doi: 10.1159/000486020.
Given the clinical presentation and the absence of other systemic or dermatological findings, the diagnosis of chevron nails was made.
Discussion
The condition is characterized by transverse ridges on the nails that converge towards the center, forming a V or chevron shape. This condition was first described by Perry et al. and later by Shuster et al., who explained that the condition might result from axial growth of the nail with synchronous growth occurring from a chevron-shaped growing edge of the nail root. Alternatively, Shuster suggested that sequential growth, with localized variation in the nail production rate, could propagate a wave from the center of the nail to the edge.
The etiology of chevron nails is not well understood, but it is believed to result from temporary disruptions in the nail matrix, possibly related to minor illness or physiological stress during infancy.
In the case of our 7-month-old patient, the history of mild upper respiratory infections might have contributed to the development of chevron nails. However, the lack of other significant illness, skin involvement, or systemic findings supports the benign and self-limiting nature of this condition. Parents were reassured that chevron nails typically resolve on their own as the child grows and that no specific treatment is necessary.
Differential Diagnosis
The differential diagnosis of transverse nail changes in children includes other conditions such as trachyonychia, lichen planus, Darier disease, and pachyonychia congenita.
Trachyonychia, also known as “sandpaper nails,” trachyonychia is characterized by the roughening of the nail surface, giving it a dull and ridged appearance. The condition may affect all 20 nails and is often associated with underlying dermatological conditions such as lichen planus or alopecia areata. Unlike chevron nails, trachyonychia presents with more diffuse nail changes and does not typically feature the distinct V-shaped ridging seen in this patient.
Lichen planus is an inflammatory condition that can affect the skin, mucous membranes, and nails. Nail involvement in lichen planus can lead to longitudinal ridging, thinning, and sometimes even complete nail loss. The absence of other characteristic features of lichen planus, such as violaceous papules on the skin or white lacy patterns on mucous membranes (Wickham striae), makes this diagnosis less likely in our patient.
Darier disease, also known as keratosis follicularis, is a genetic disorder characterized by greasy, warty papules primarily on seborrheic areas of the skin, nail abnormalities, and sometimes mucosal involvement. Nail changes in Darier disease include longitudinal red and white streaks, V-shaped notching at the free edge of the nails, and subungual hyperkeratosis. These nail changes are more severe and distinct than the simple transverse ridging seen in chevron nails. The absence of other clinical signs of Darier disease, such as skin papules or characteristic nail notching, makes this diagnosis unlikely in our patient.
Pachyonychia congenita is a rare genetic disorder characterized by thickened nails (pachyonychia), painful plantar keratoderma, and sometimes oral leukokeratosis. The condition typically presents with significant nail thickening and other systemic findings, which were absent in our patient. The distinct pattern of V-shaped ridging observed in chevron nails does not align with the typical presentation of pachyonychia congenita.
Next Steps
No specific treatment is required for chevron nails. The condition is typically self-resolving, and the nails usually return to a normal appearance as the child continues to grow. Parents were advised to monitor the nails for any changes or new symptoms and were reassured about the benign nature of the findings. Follow-up was scheduled to ensure the resolution of the condition as the child develops.
Conclusion
Chevron nails are an important consideration in the differential diagnosis of transverse nail ridging in infants and young children. While the condition is benign and self-limiting, it is crucial to differentiate it from other nail dystrophies, such as trachyonychia, lichen planus, Darier disease, and pachyonychia congenita, which may require further investigation or intervention. Awareness of chevron nails can help prevent unnecessary worry and provide reassurance to parents and caregivers.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
Suggested Reading
Delano S, Belazarian L. Chevron nails: A normal variant in the pediatric population. Pediatr Dermatol. 2014 Jan-Feb;31(1):e24-5. doi: 10.1111/pde.12193.
John JM et al. Chevron nail — An under-recognised normal variant of nail development. Arch Dis Child. 2024 Jul 18;109(8):648. doi: 10.1136/archdischild-2024-326975.
Shuster S. The significance of chevron nails. Br J Dermatol. 1996;135:151–152. doi: 10.1046/j.1365-2133.1996.d01-961.x.
Starace M et al. Nail disorders in children. Skin Appendage Disord. 2018 Oct;4(4):217-229. doi: 10.1159/000486020.
Given the clinical presentation and the absence of other systemic or dermatological findings, the diagnosis of chevron nails was made.
Discussion
The condition is characterized by transverse ridges on the nails that converge towards the center, forming a V or chevron shape. This condition was first described by Perry et al. and later by Shuster et al., who explained that the condition might result from axial growth of the nail with synchronous growth occurring from a chevron-shaped growing edge of the nail root. Alternatively, Shuster suggested that sequential growth, with localized variation in the nail production rate, could propagate a wave from the center of the nail to the edge.
The etiology of chevron nails is not well understood, but it is believed to result from temporary disruptions in the nail matrix, possibly related to minor illness or physiological stress during infancy.
In the case of our 7-month-old patient, the history of mild upper respiratory infections might have contributed to the development of chevron nails. However, the lack of other significant illness, skin involvement, or systemic findings supports the benign and self-limiting nature of this condition. Parents were reassured that chevron nails typically resolve on their own as the child grows and that no specific treatment is necessary.
Differential Diagnosis
The differential diagnosis of transverse nail changes in children includes other conditions such as trachyonychia, lichen planus, Darier disease, and pachyonychia congenita.
Trachyonychia, also known as “sandpaper nails,” trachyonychia is characterized by the roughening of the nail surface, giving it a dull and ridged appearance. The condition may affect all 20 nails and is often associated with underlying dermatological conditions such as lichen planus or alopecia areata. Unlike chevron nails, trachyonychia presents with more diffuse nail changes and does not typically feature the distinct V-shaped ridging seen in this patient.
Lichen planus is an inflammatory condition that can affect the skin, mucous membranes, and nails. Nail involvement in lichen planus can lead to longitudinal ridging, thinning, and sometimes even complete nail loss. The absence of other characteristic features of lichen planus, such as violaceous papules on the skin or white lacy patterns on mucous membranes (Wickham striae), makes this diagnosis less likely in our patient.
Darier disease, also known as keratosis follicularis, is a genetic disorder characterized by greasy, warty papules primarily on seborrheic areas of the skin, nail abnormalities, and sometimes mucosal involvement. Nail changes in Darier disease include longitudinal red and white streaks, V-shaped notching at the free edge of the nails, and subungual hyperkeratosis. These nail changes are more severe and distinct than the simple transverse ridging seen in chevron nails. The absence of other clinical signs of Darier disease, such as skin papules or characteristic nail notching, makes this diagnosis unlikely in our patient.
Pachyonychia congenita is a rare genetic disorder characterized by thickened nails (pachyonychia), painful plantar keratoderma, and sometimes oral leukokeratosis. The condition typically presents with significant nail thickening and other systemic findings, which were absent in our patient. The distinct pattern of V-shaped ridging observed in chevron nails does not align with the typical presentation of pachyonychia congenita.
Next Steps
No specific treatment is required for chevron nails. The condition is typically self-resolving, and the nails usually return to a normal appearance as the child continues to grow. Parents were advised to monitor the nails for any changes or new symptoms and were reassured about the benign nature of the findings. Follow-up was scheduled to ensure the resolution of the condition as the child develops.
Conclusion
Chevron nails are an important consideration in the differential diagnosis of transverse nail ridging in infants and young children. While the condition is benign and self-limiting, it is crucial to differentiate it from other nail dystrophies, such as trachyonychia, lichen planus, Darier disease, and pachyonychia congenita, which may require further investigation or intervention. Awareness of chevron nails can help prevent unnecessary worry and provide reassurance to parents and caregivers.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
Suggested Reading
Delano S, Belazarian L. Chevron nails: A normal variant in the pediatric population. Pediatr Dermatol. 2014 Jan-Feb;31(1):e24-5. doi: 10.1111/pde.12193.
John JM et al. Chevron nail — An under-recognised normal variant of nail development. Arch Dis Child. 2024 Jul 18;109(8):648. doi: 10.1136/archdischild-2024-326975.
Shuster S. The significance of chevron nails. Br J Dermatol. 1996;135:151–152. doi: 10.1046/j.1365-2133.1996.d01-961.x.
Starace M et al. Nail disorders in children. Skin Appendage Disord. 2018 Oct;4(4):217-229. doi: 10.1159/000486020.
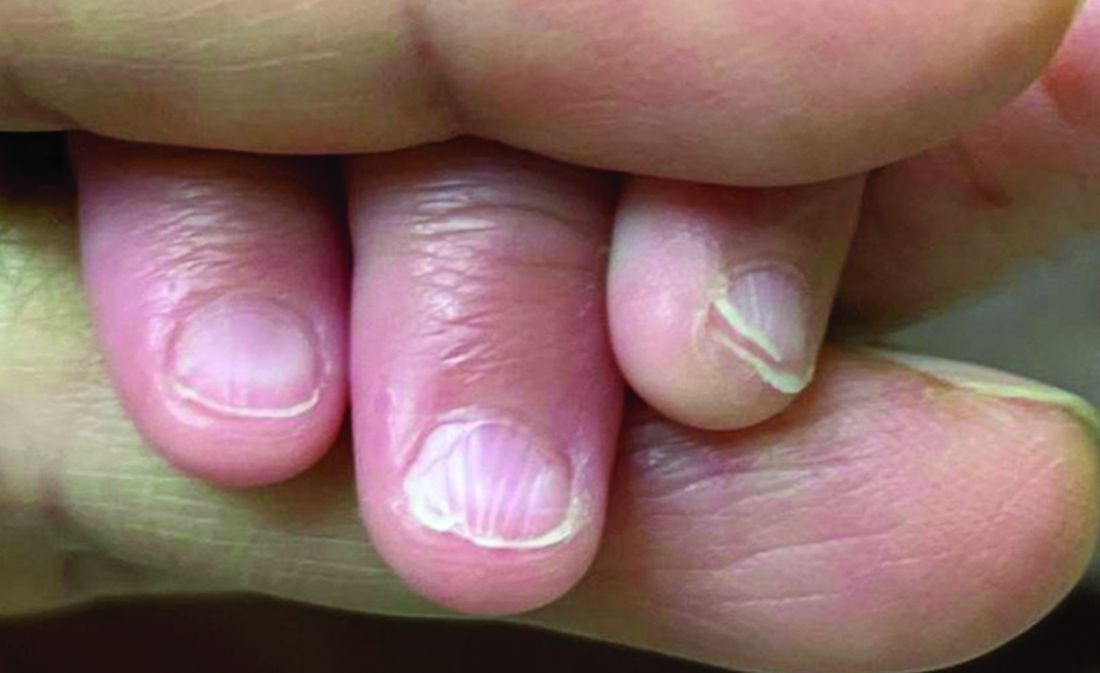
There was no family history of similar nail findings and no relatives had a history of chronic skin conditions or congenital nail disorders.
On physical examination, several of the child’s fingernails exhibited distinct longitudinal ridges, with a characteristic pattern where the ridges converged at the center of the nail, forming a V-shape. There were no other concerning dermatologic findings, such as rashes, plaques, or erosions, and the skin and hair appeared otherwise normal. The rest of the physical exam was unremarkable.
A 7-year-old female presents with persistent pimples on the nose and cheeks for approximately 1 year
Diagnosis
During the visit, skin scrapings were performed, revealing several Demodex mites, confirming the diagnosis of demodicosis.
Demodex folliculorum and Demodex brevis are the two common species implicated. The life cycle of Demodex spp. occurs in the sebaceous glands, leading to mechanical and chemical irritation of the skin.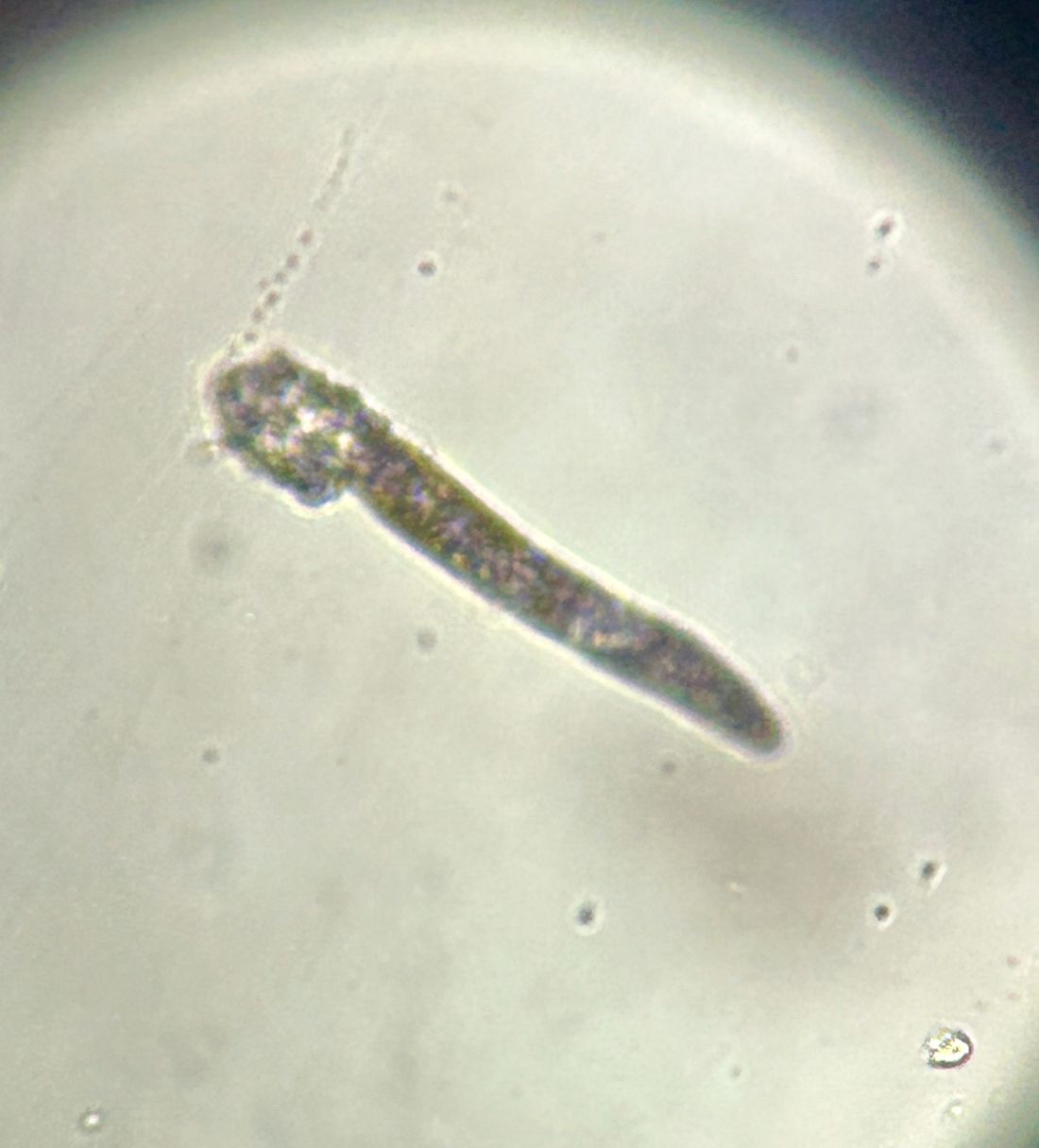
Various immune responses are also triggered, such as a keratinocyte response via Toll-like receptor 2. Patients usually present with non-specific symptoms such as skin erythema, irritation, peeling, and dryness on the cheeks, eyelids, and paranasal areas. Patients may develop a maculopapular or rosacea-like rash.
Diagnosis is often made through microscopic examination of a skin sample in KOH solution. In rare occasions, a skin surface standardization biopsy method may be used, which determines the density of mites per 1 cm2. Dermoscopy may identify spiky white structures. Molecular methods such as PCR can be used but are not standard.
The differential diagnosis may include acne, rosacea, folliculitis, and Candida infection. Demodicosis can be differentiated by history and further studies including dermoscopy.
Acne vulgaris is an inflammatory disease of the skin’s pilosebaceous unit, primarily involving the face and trunk. It can present with comedones, papules, pustules, and nodules. Secondary signs suggestive of acne vulgaris include scars, erythema, and hyperpigmentation. All forms of acne share a common pathogenesis resulting in the formation of microcomedones, precursors for all clinical acne lesions. In this patient, the absence of microcomedones and the presence of primary inflammatory papules localized to the nose and cheeks suggested an alternative diagnosis.

Rosacea was also considered in the differential diagnosis. Rosacea is an inflammatory dermatosis characterized by erythema, telangiectasia, recurrent flushing, and inflammatory lesions including papulopustules and swelling, primarily affecting the face. The pathogenesis of rosacea is not fully understood but is suggested to involve immune-mediated responses. Vascular dysregulation and reactive oxygen species damage keratinocytes, fibroblasts, and endothelial cells. A higher incidence of rosacea in those with a family history and UV exposure is a known trigger. Demodex folliculorum and Helicobacter pylori are also implicated. Occasionally, Demodex infestation and rosacea may co-occur, and treatment with topical metronidazole can be helpful.
Folliculitis is an infection and inflammation of the hair follicles, forming pustules or erythematous papules over hair-covered skin. It is commonly caused by bacterial infection but can also be due to fungi, viruses, and noninfectious causes such as eosinophilic folliculitis. Diagnosis is clinical, based on physical exam and history, such as recent increased sweating or scratching. KOH prep can be used for Malassezia folliculitis and skin biopsy for eosinophilic folliculitis. Treatment targets the underlying cause. Most bacterial folliculitis cases resolve without treatment, but topical antibiotics may be used. Fungal folliculitis requires oral antifungals, and herpes simplex folliculitis can be treated with antiviral medications.
Cutaneous candidiasis is an infection of the skin by various Candida species, commonly C. albicans. Superficial infections of the skin and mucous membranes, such as intertrigo, are common types. Risk factors include immunosuppression, endocrine disorders, or compromised blood flow. Increased humidity, occlusion, broken skin barriers, and altered skin microbial flora contribute to Candida infection. Diagnosis is clinical but can be confirmed by KOH prep, microscopy, and culture. Treatment involves anti-inflammatory, antibacterial, and antifungal medications. Topical clotrimazole, nystatin, and miconazole are commonly used. Recurrence is prevented by keeping the affected area dry with barrier creams.
Therapeutic goals include arresting mite reproduction, elimination, and preventing recurrent infestations. Treatment may last several months, and the choice of drug depends on patient factors. There have been no standardized treatment studies or long-term effectiveness analyses. Antibiotics such as tetracycline, metronidazole, doxycycline, and ivermectin may be used to prevent proliferation. Permethrin, benzyl benzoate, crotamiton, lindane, and sulfur have also been used. Metronidazole is a common treatment for demodicosis, as was used in our patient for several weeks until the lesions cleared. Systemic metronidazole therapy may be indicated for reducing Demodex spp. density. Severe cases, particularly in immunocompromised individuals, may require oral ivermectin. Appropriate hygiene is important for prevention, such as washing the face with non-oily cleansers and laundering linens regularly.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Mr. Lee is a medical student at the University of California San Diego.
Suggested Reading
Chudzicka-Strugała I et al. Demodicosis in different age groups and alternative treatment options—A review. J Clin Med. 2023 Feb 19;12(4):1649. doi: 10.3390/jcm12041649.
Eichenfield DZ et al. Management of acne vulgaris: A review. JAMA. 2021 Nov 23;326(20):2055-2067. doi: 10.1001/jama.2021.17633.
Sharma A et al. Rosacea management: A comprehensive review. J Cosmet Dermatol. 2022 May;21(5):1895-1904. doi: 10.1111/jocd.14816.
Taudorf EH et al. Cutaneous candidiasis — an evidence-based review of topical and systemic treatments to inform clinical practice. J Eur Acad Dermatol Venereol. 2019 Oct;33(10):1863-1873. doi: 10.1111/jdv.15782.
Winters RD, Mitchell M. Folliculitis. [Updated 2023 Aug 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK547754/
Diagnosis
During the visit, skin scrapings were performed, revealing several Demodex mites, confirming the diagnosis of demodicosis.
Demodex folliculorum and Demodex brevis are the two common species implicated. The life cycle of Demodex spp. occurs in the sebaceous glands, leading to mechanical and chemical irritation of the skin.
Various immune responses are also triggered, such as a keratinocyte response via Toll-like receptor 2. Patients usually present with non-specific symptoms such as skin erythema, irritation, peeling, and dryness on the cheeks, eyelids, and paranasal areas. Patients may develop a maculopapular or rosacea-like rash.
Diagnosis is often made through microscopic examination of a skin sample in KOH solution. In rare occasions, a skin surface standardization biopsy method may be used, which determines the density of mites per 1 cm2. Dermoscopy may identify spiky white structures. Molecular methods such as PCR can be used but are not standard.
The differential diagnosis may include acne, rosacea, folliculitis, and Candida infection. Demodicosis can be differentiated by history and further studies including dermoscopy.
Acne vulgaris is an inflammatory disease of the skin’s pilosebaceous unit, primarily involving the face and trunk. It can present with comedones, papules, pustules, and nodules. Secondary signs suggestive of acne vulgaris include scars, erythema, and hyperpigmentation. All forms of acne share a common pathogenesis resulting in the formation of microcomedones, precursors for all clinical acne lesions. In this patient, the absence of microcomedones and the presence of primary inflammatory papules localized to the nose and cheeks suggested an alternative diagnosis.
Rosacea was also considered in the differential diagnosis. Rosacea is an inflammatory dermatosis characterized by erythema, telangiectasia, recurrent flushing, and inflammatory lesions including papulopustules and swelling, primarily affecting the face. The pathogenesis of rosacea is not fully understood but is suggested to involve immune-mediated responses. Vascular dysregulation and reactive oxygen species damage keratinocytes, fibroblasts, and endothelial cells. A higher incidence of rosacea in those with a family history and UV exposure is a known trigger. Demodex folliculorum and Helicobacter pylori are also implicated. Occasionally, Demodex infestation and rosacea may co-occur, and treatment with topical metronidazole can be helpful.
Folliculitis is an infection and inflammation of the hair follicles, forming pustules or erythematous papules over hair-covered skin. It is commonly caused by bacterial infection but can also be due to fungi, viruses, and noninfectious causes such as eosinophilic folliculitis. Diagnosis is clinical, based on physical exam and history, such as recent increased sweating or scratching. KOH prep can be used for Malassezia folliculitis and skin biopsy for eosinophilic folliculitis. Treatment targets the underlying cause. Most bacterial folliculitis cases resolve without treatment, but topical antibiotics may be used. Fungal folliculitis requires oral antifungals, and herpes simplex folliculitis can be treated with antiviral medications.
Cutaneous candidiasis is an infection of the skin by various Candida species, commonly C. albicans. Superficial infections of the skin and mucous membranes, such as intertrigo, are common types. Risk factors include immunosuppression, endocrine disorders, or compromised blood flow. Increased humidity, occlusion, broken skin barriers, and altered skin microbial flora contribute to Candida infection. Diagnosis is clinical but can be confirmed by KOH prep, microscopy, and culture. Treatment involves anti-inflammatory, antibacterial, and antifungal medications. Topical clotrimazole, nystatin, and miconazole are commonly used. Recurrence is prevented by keeping the affected area dry with barrier creams.
Therapeutic goals include arresting mite reproduction, elimination, and preventing recurrent infestations. Treatment may last several months, and the choice of drug depends on patient factors. There have been no standardized treatment studies or long-term effectiveness analyses. Antibiotics such as tetracycline, metronidazole, doxycycline, and ivermectin may be used to prevent proliferation. Permethrin, benzyl benzoate, crotamiton, lindane, and sulfur have also been used. Metronidazole is a common treatment for demodicosis, as was used in our patient for several weeks until the lesions cleared. Systemic metronidazole therapy may be indicated for reducing Demodex spp. density. Severe cases, particularly in immunocompromised individuals, may require oral ivermectin. Appropriate hygiene is important for prevention, such as washing the face with non-oily cleansers and laundering linens regularly.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Mr. Lee is a medical student at the University of California San Diego.
Suggested Reading
Chudzicka-Strugała I et al. Demodicosis in different age groups and alternative treatment options—A review. J Clin Med. 2023 Feb 19;12(4):1649. doi: 10.3390/jcm12041649.
Eichenfield DZ et al. Management of acne vulgaris: A review. JAMA. 2021 Nov 23;326(20):2055-2067. doi: 10.1001/jama.2021.17633.
Sharma A et al. Rosacea management: A comprehensive review. J Cosmet Dermatol. 2022 May;21(5):1895-1904. doi: 10.1111/jocd.14816.
Taudorf EH et al. Cutaneous candidiasis — an evidence-based review of topical and systemic treatments to inform clinical practice. J Eur Acad Dermatol Venereol. 2019 Oct;33(10):1863-1873. doi: 10.1111/jdv.15782.
Winters RD, Mitchell M. Folliculitis. [Updated 2023 Aug 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK547754/
Diagnosis
During the visit, skin scrapings were performed, revealing several Demodex mites, confirming the diagnosis of demodicosis.
Demodex folliculorum and Demodex brevis are the two common species implicated. The life cycle of Demodex spp. occurs in the sebaceous glands, leading to mechanical and chemical irritation of the skin.
Various immune responses are also triggered, such as a keratinocyte response via Toll-like receptor 2. Patients usually present with non-specific symptoms such as skin erythema, irritation, peeling, and dryness on the cheeks, eyelids, and paranasal areas. Patients may develop a maculopapular or rosacea-like rash.
Diagnosis is often made through microscopic examination of a skin sample in KOH solution. In rare occasions, a skin surface standardization biopsy method may be used, which determines the density of mites per 1 cm2. Dermoscopy may identify spiky white structures. Molecular methods such as PCR can be used but are not standard.
The differential diagnosis may include acne, rosacea, folliculitis, and Candida infection. Demodicosis can be differentiated by history and further studies including dermoscopy.
Acne vulgaris is an inflammatory disease of the skin’s pilosebaceous unit, primarily involving the face and trunk. It can present with comedones, papules, pustules, and nodules. Secondary signs suggestive of acne vulgaris include scars, erythema, and hyperpigmentation. All forms of acne share a common pathogenesis resulting in the formation of microcomedones, precursors for all clinical acne lesions. In this patient, the absence of microcomedones and the presence of primary inflammatory papules localized to the nose and cheeks suggested an alternative diagnosis.
Rosacea was also considered in the differential diagnosis. Rosacea is an inflammatory dermatosis characterized by erythema, telangiectasia, recurrent flushing, and inflammatory lesions including papulopustules and swelling, primarily affecting the face. The pathogenesis of rosacea is not fully understood but is suggested to involve immune-mediated responses. Vascular dysregulation and reactive oxygen species damage keratinocytes, fibroblasts, and endothelial cells. A higher incidence of rosacea in those with a family history and UV exposure is a known trigger. Demodex folliculorum and Helicobacter pylori are also implicated. Occasionally, Demodex infestation and rosacea may co-occur, and treatment with topical metronidazole can be helpful.
Folliculitis is an infection and inflammation of the hair follicles, forming pustules or erythematous papules over hair-covered skin. It is commonly caused by bacterial infection but can also be due to fungi, viruses, and noninfectious causes such as eosinophilic folliculitis. Diagnosis is clinical, based on physical exam and history, such as recent increased sweating or scratching. KOH prep can be used for Malassezia folliculitis and skin biopsy for eosinophilic folliculitis. Treatment targets the underlying cause. Most bacterial folliculitis cases resolve without treatment, but topical antibiotics may be used. Fungal folliculitis requires oral antifungals, and herpes simplex folliculitis can be treated with antiviral medications.
Cutaneous candidiasis is an infection of the skin by various Candida species, commonly C. albicans. Superficial infections of the skin and mucous membranes, such as intertrigo, are common types. Risk factors include immunosuppression, endocrine disorders, or compromised blood flow. Increased humidity, occlusion, broken skin barriers, and altered skin microbial flora contribute to Candida infection. Diagnosis is clinical but can be confirmed by KOH prep, microscopy, and culture. Treatment involves anti-inflammatory, antibacterial, and antifungal medications. Topical clotrimazole, nystatin, and miconazole are commonly used. Recurrence is prevented by keeping the affected area dry with barrier creams.
Therapeutic goals include arresting mite reproduction, elimination, and preventing recurrent infestations. Treatment may last several months, and the choice of drug depends on patient factors. There have been no standardized treatment studies or long-term effectiveness analyses. Antibiotics such as tetracycline, metronidazole, doxycycline, and ivermectin may be used to prevent proliferation. Permethrin, benzyl benzoate, crotamiton, lindane, and sulfur have also been used. Metronidazole is a common treatment for demodicosis, as was used in our patient for several weeks until the lesions cleared. Systemic metronidazole therapy may be indicated for reducing Demodex spp. density. Severe cases, particularly in immunocompromised individuals, may require oral ivermectin. Appropriate hygiene is important for prevention, such as washing the face with non-oily cleansers and laundering linens regularly.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Mr. Lee is a medical student at the University of California San Diego.
Suggested Reading
Chudzicka-Strugała I et al. Demodicosis in different age groups and alternative treatment options—A review. J Clin Med. 2023 Feb 19;12(4):1649. doi: 10.3390/jcm12041649.
Eichenfield DZ et al. Management of acne vulgaris: A review. JAMA. 2021 Nov 23;326(20):2055-2067. doi: 10.1001/jama.2021.17633.
Sharma A et al. Rosacea management: A comprehensive review. J Cosmet Dermatol. 2022 May;21(5):1895-1904. doi: 10.1111/jocd.14816.
Taudorf EH et al. Cutaneous candidiasis — an evidence-based review of topical and systemic treatments to inform clinical practice. J Eur Acad Dermatol Venereol. 2019 Oct;33(10):1863-1873. doi: 10.1111/jdv.15782.
Winters RD, Mitchell M. Folliculitis. [Updated 2023 Aug 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK547754/
A 7-year-old female presents with persistent pimples on the nose and cheeks for approximately 1 year. She had been treated with several topical antibiotics and acne washes without resolution of the lesions. There were no signs of early puberty, and the child had no history of medical conditions. Her mother has a history of rosacea. Physical examination revealed erythematous papules on the nose and cheeks bilaterally.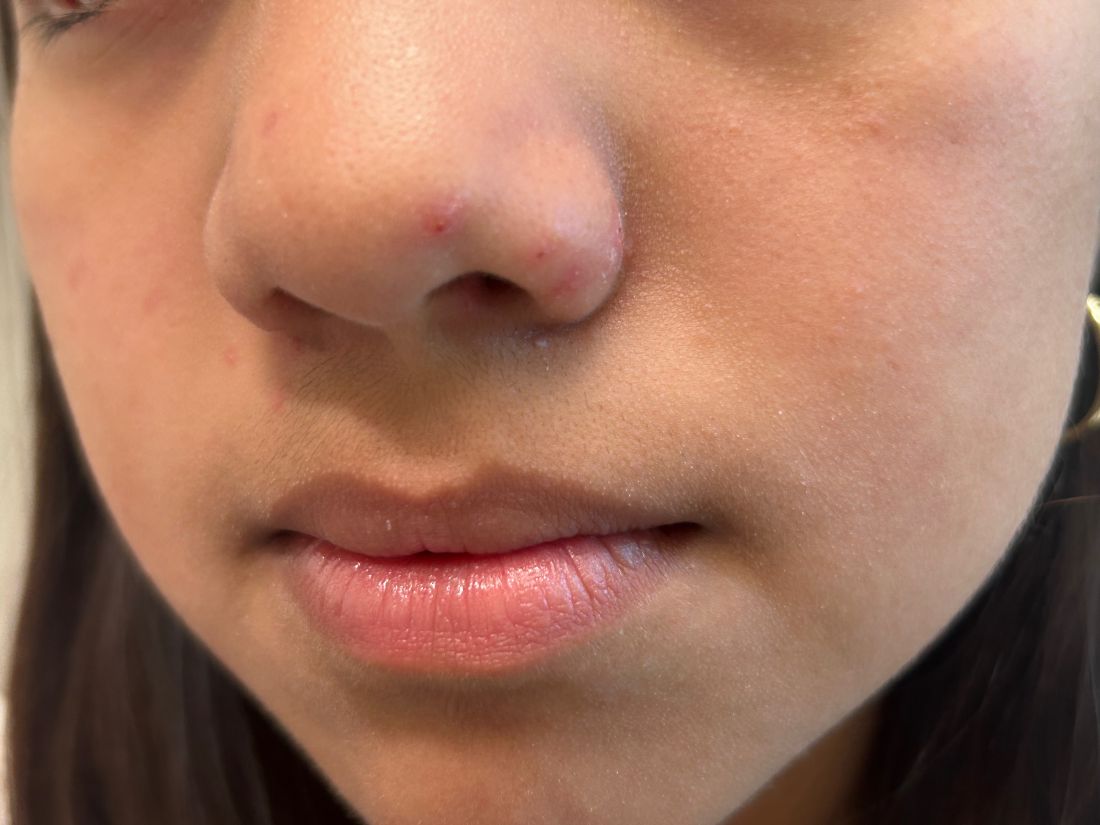
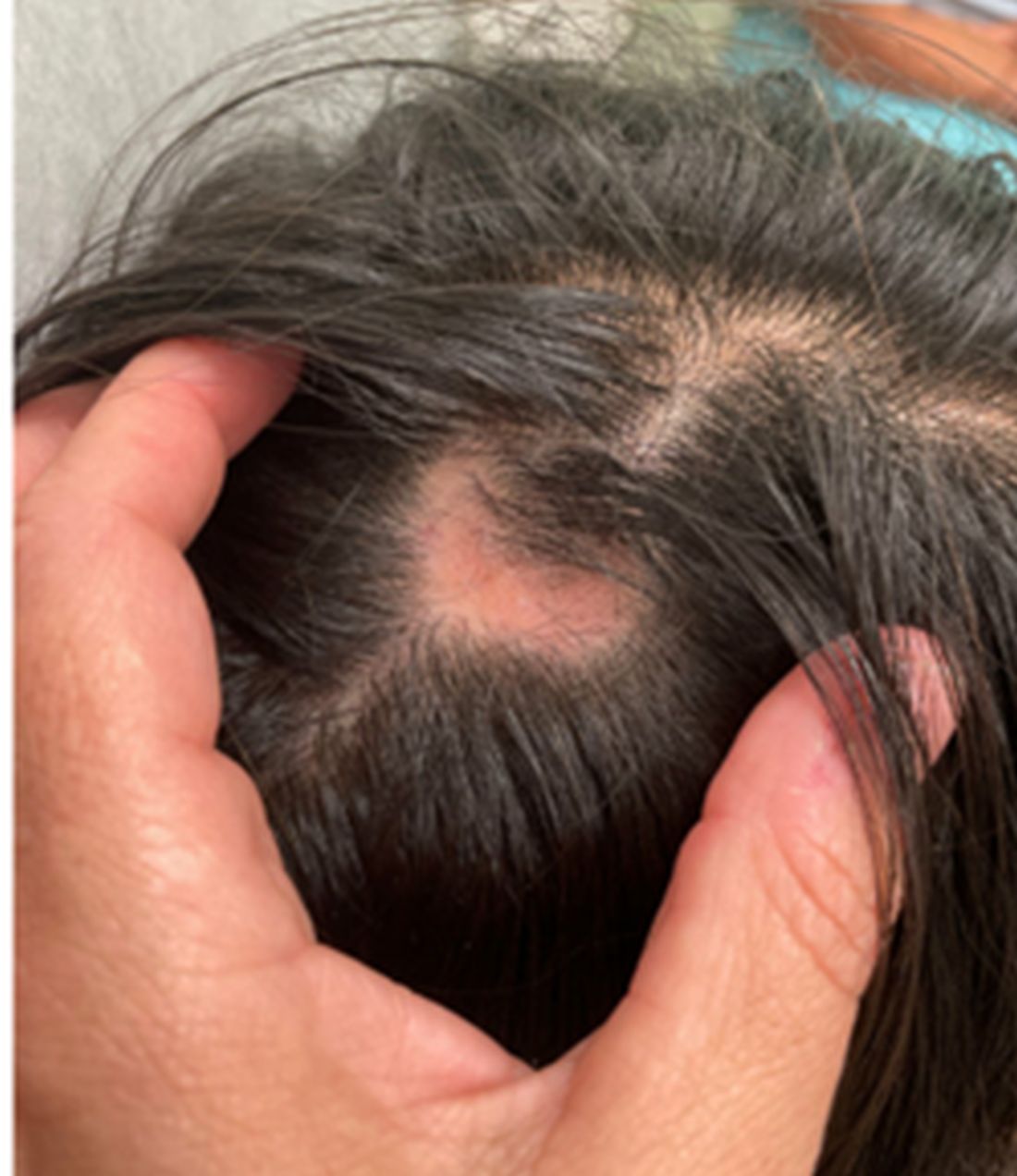
A 16-Year-Old Female Presents With Multiple Areas of Hair Loss on the Scalp
KOH analysis of the scales from the scalp areas revealed no fungal elements. Given the observed erythema and scaling, a punch biopsy was conducted. Histopathological examination of the biopsy sample displayed interface inflammation affecting both the infundibular and lower portions of hair follicles. The presence of folliculosebaceous units transitioning from intermediate to terminal size follicles was noted. A perifollicular, peri eccrine, superficial, and deep perivascular lymphoplasmacytic infiltrate was identified, alongside increased dermal mucin, findings consistent with a diagnosis of discoid lupus erythematosus.
Subsequent laboratory investigations were largely unremarkable, except for an elevated ANA titer (1:320, with a speckled pattern). The patient was initiated on a treatment regimen comprising intralesional triamcinolone and oral hydroxychloroquine (Plaquenil).
Discussion
It predominantly affects adults, yet pediatric cases account for 5%-7% of DLE diagnoses, with a significant predominance in females. Pediatric scalp DLE is particularly concerning due to its potential for causing scarring and permanent hair loss, which can significantly impact the psychological wellbeing of affected children.
The pathogenesis of DLE is multifactorial, involving genetic predispositions, environmental factors like UV light exposure, and immunological mechanisms leading to skin damage.
In children, DLE typically presents as well-demarcated, erythematous plaques with scale and follicular plugging, primarily affecting the scalp. Lesions may also exhibit changes in pigmentation, atrophy, and telangiectasia. The scalp involvement often leads to scarring alopecia, which can be distressing for pediatric patients. Unlike systemic lupus erythematosus (SLE), DLE is usually limited to the skin without systemic involvement. The progression of DLE to systemic lupus erythematosus in children has been previously described to be 22.2%. In a recent report of 201 pediatric cases of DLE, 12% of the cases progressed to systemic lupus erythematosus (SLE) and 14.5% had concurrent SLE. The onset of symptoms before the age of 10 years was the only statistically significant predictor for progression to SLE. Pruritus is a common symptom and may be correlated with disease activity.
The differential diagnosis for this patient encompassed a variety of conditions, including tinea capitis, alopecia areata, trichotillomania, and lichen planopilaris, each considered based on clinical presentation but ultimately excluded through clinical, microscopic, and biopsy findings.
Management strategies for pediatric scalp DLE aim at preventing disease progression, minimizing scarring, and addressing aesthetic concerns. These include the use of topical and intralesional corticosteroids, calcineurin inhibitors, and antimalarial agents like hydroxychloroquine, alongside stringent photoprotection to mitigate UV-triggered exacerbations.
Conclusion
The prognosis for pediatric scalp DLE can be favorable with timely and appropriate management, underscoring the importance of early diagnosis and intervention to prevent scarring and hair loss. However, ongoing surveillance is crucial for monitoring potential progression to systemic lupus erythematosus, albeit a low-risk transformation.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. George PM and Tunnessen WW. Childhood discoid lupus erythematosus. Arch Dermatol. 1993;129(5):613-617.
2. Hawat T et al. Pediatric discoid lupus erythematosus: Short report. Dermatol Ther. 2022 Jan;35(1):e15170. doi: 10.1111/dth.15170.
3. Arkin LM et al. Practice-based differences in paediatric discoid lupus erythematosus. Br J Dermatol. 2019 Oct;181(4):805-810. doi: 10.1111/bjd.17780.
KOH analysis of the scales from the scalp areas revealed no fungal elements. Given the observed erythema and scaling, a punch biopsy was conducted. Histopathological examination of the biopsy sample displayed interface inflammation affecting both the infundibular and lower portions of hair follicles. The presence of folliculosebaceous units transitioning from intermediate to terminal size follicles was noted. A perifollicular, peri eccrine, superficial, and deep perivascular lymphoplasmacytic infiltrate was identified, alongside increased dermal mucin, findings consistent with a diagnosis of discoid lupus erythematosus.
Subsequent laboratory investigations were largely unremarkable, except for an elevated ANA titer (1:320, with a speckled pattern). The patient was initiated on a treatment regimen comprising intralesional triamcinolone and oral hydroxychloroquine (Plaquenil).
Discussion
It predominantly affects adults, yet pediatric cases account for 5%-7% of DLE diagnoses, with a significant predominance in females. Pediatric scalp DLE is particularly concerning due to its potential for causing scarring and permanent hair loss, which can significantly impact the psychological wellbeing of affected children.
The pathogenesis of DLE is multifactorial, involving genetic predispositions, environmental factors like UV light exposure, and immunological mechanisms leading to skin damage.
In children, DLE typically presents as well-demarcated, erythematous plaques with scale and follicular plugging, primarily affecting the scalp. Lesions may also exhibit changes in pigmentation, atrophy, and telangiectasia. The scalp involvement often leads to scarring alopecia, which can be distressing for pediatric patients. Unlike systemic lupus erythematosus (SLE), DLE is usually limited to the skin without systemic involvement. The progression of DLE to systemic lupus erythematosus in children has been previously described to be 22.2%. In a recent report of 201 pediatric cases of DLE, 12% of the cases progressed to systemic lupus erythematosus (SLE) and 14.5% had concurrent SLE. The onset of symptoms before the age of 10 years was the only statistically significant predictor for progression to SLE. Pruritus is a common symptom and may be correlated with disease activity.
The differential diagnosis for this patient encompassed a variety of conditions, including tinea capitis, alopecia areata, trichotillomania, and lichen planopilaris, each considered based on clinical presentation but ultimately excluded through clinical, microscopic, and biopsy findings.
Management strategies for pediatric scalp DLE aim at preventing disease progression, minimizing scarring, and addressing aesthetic concerns. These include the use of topical and intralesional corticosteroids, calcineurin inhibitors, and antimalarial agents like hydroxychloroquine, alongside stringent photoprotection to mitigate UV-triggered exacerbations.
Conclusion
The prognosis for pediatric scalp DLE can be favorable with timely and appropriate management, underscoring the importance of early diagnosis and intervention to prevent scarring and hair loss. However, ongoing surveillance is crucial for monitoring potential progression to systemic lupus erythematosus, albeit a low-risk transformation.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. George PM and Tunnessen WW. Childhood discoid lupus erythematosus. Arch Dermatol. 1993;129(5):613-617.
2. Hawat T et al. Pediatric discoid lupus erythematosus: Short report. Dermatol Ther. 2022 Jan;35(1):e15170. doi: 10.1111/dth.15170.
3. Arkin LM et al. Practice-based differences in paediatric discoid lupus erythematosus. Br J Dermatol. 2019 Oct;181(4):805-810. doi: 10.1111/bjd.17780.
KOH analysis of the scales from the scalp areas revealed no fungal elements. Given the observed erythema and scaling, a punch biopsy was conducted. Histopathological examination of the biopsy sample displayed interface inflammation affecting both the infundibular and lower portions of hair follicles. The presence of folliculosebaceous units transitioning from intermediate to terminal size follicles was noted. A perifollicular, peri eccrine, superficial, and deep perivascular lymphoplasmacytic infiltrate was identified, alongside increased dermal mucin, findings consistent with a diagnosis of discoid lupus erythematosus.
Subsequent laboratory investigations were largely unremarkable, except for an elevated ANA titer (1:320, with a speckled pattern). The patient was initiated on a treatment regimen comprising intralesional triamcinolone and oral hydroxychloroquine (Plaquenil).
Discussion
It predominantly affects adults, yet pediatric cases account for 5%-7% of DLE diagnoses, with a significant predominance in females. Pediatric scalp DLE is particularly concerning due to its potential for causing scarring and permanent hair loss, which can significantly impact the psychological wellbeing of affected children.
The pathogenesis of DLE is multifactorial, involving genetic predispositions, environmental factors like UV light exposure, and immunological mechanisms leading to skin damage.
In children, DLE typically presents as well-demarcated, erythematous plaques with scale and follicular plugging, primarily affecting the scalp. Lesions may also exhibit changes in pigmentation, atrophy, and telangiectasia. The scalp involvement often leads to scarring alopecia, which can be distressing for pediatric patients. Unlike systemic lupus erythematosus (SLE), DLE is usually limited to the skin without systemic involvement. The progression of DLE to systemic lupus erythematosus in children has been previously described to be 22.2%. In a recent report of 201 pediatric cases of DLE, 12% of the cases progressed to systemic lupus erythematosus (SLE) and 14.5% had concurrent SLE. The onset of symptoms before the age of 10 years was the only statistically significant predictor for progression to SLE. Pruritus is a common symptom and may be correlated with disease activity.
The differential diagnosis for this patient encompassed a variety of conditions, including tinea capitis, alopecia areata, trichotillomania, and lichen planopilaris, each considered based on clinical presentation but ultimately excluded through clinical, microscopic, and biopsy findings.
Management strategies for pediatric scalp DLE aim at preventing disease progression, minimizing scarring, and addressing aesthetic concerns. These include the use of topical and intralesional corticosteroids, calcineurin inhibitors, and antimalarial agents like hydroxychloroquine, alongside stringent photoprotection to mitigate UV-triggered exacerbations.
Conclusion
The prognosis for pediatric scalp DLE can be favorable with timely and appropriate management, underscoring the importance of early diagnosis and intervention to prevent scarring and hair loss. However, ongoing surveillance is crucial for monitoring potential progression to systemic lupus erythematosus, albeit a low-risk transformation.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. George PM and Tunnessen WW. Childhood discoid lupus erythematosus. Arch Dermatol. 1993;129(5):613-617.
2. Hawat T et al. Pediatric discoid lupus erythematosus: Short report. Dermatol Ther. 2022 Jan;35(1):e15170. doi: 10.1111/dth.15170.
3. Arkin LM et al. Practice-based differences in paediatric discoid lupus erythematosus. Br J Dermatol. 2019 Oct;181(4):805-810. doi: 10.1111/bjd.17780.
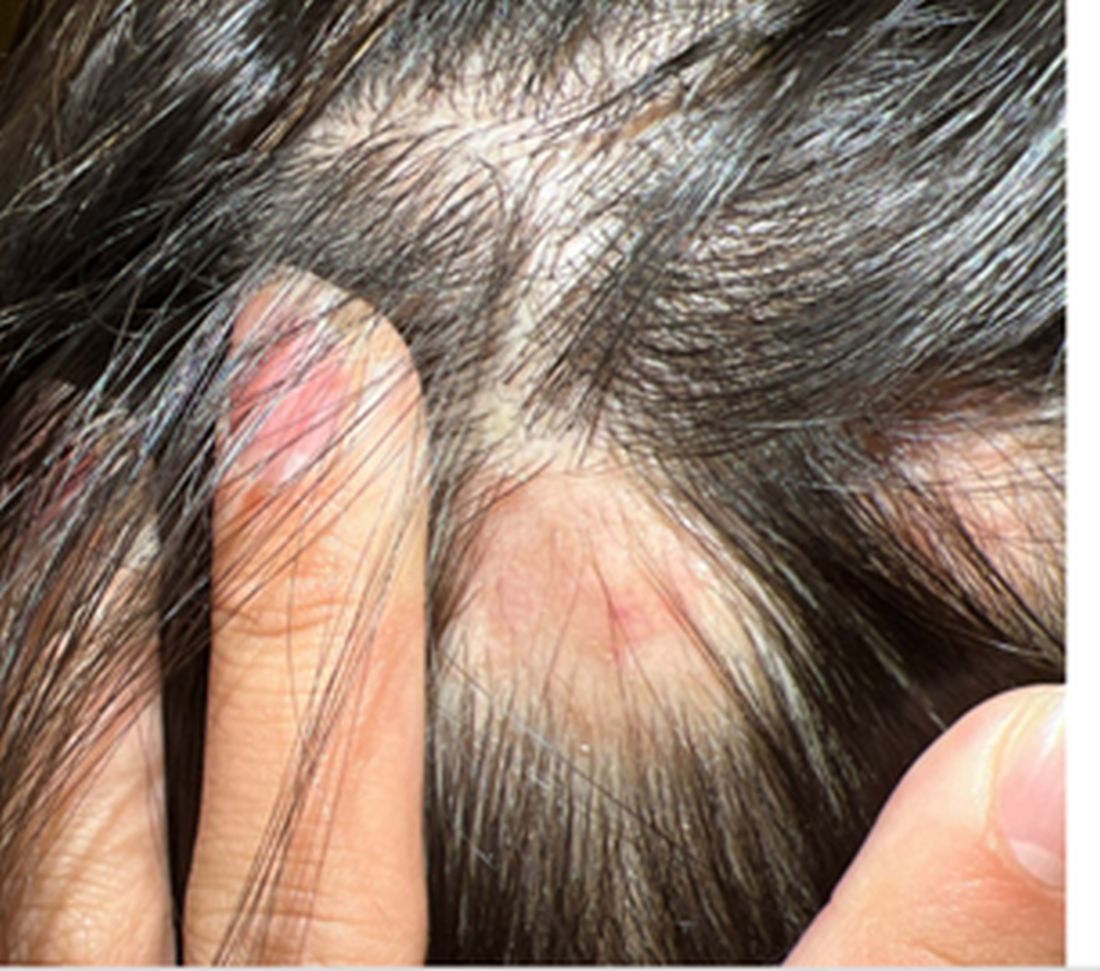
Upon physical examination, the patient exhibited several patches of alopecia with accompanying perifollicular scaling, crusting, and erythema on the affected areas. Examination of her face revealed comedones and papules.
An 8-Year-Old Male With Asymptomatic Brown Rough Plaques on the Dorsum of the Right Hand and Fingers, Accompanied by Widening of the Knuckles
During examination, the patient was observed repetitively cracking his knuckles, making a fist with the right hand, placing the left hand on top, and rubbing the hand, a behavior he routinely did multiple times daily. The observed pattern of finger involvement on the dorsum of the right hand corresponded to areas subjected to significant pressure during the described activity. Consequently, a diagnosis of lichen simplex chronicus (LSC) secondary to mechanical rubbing, along with associated pachydermodactyly on the fingers of the right hand, was established.
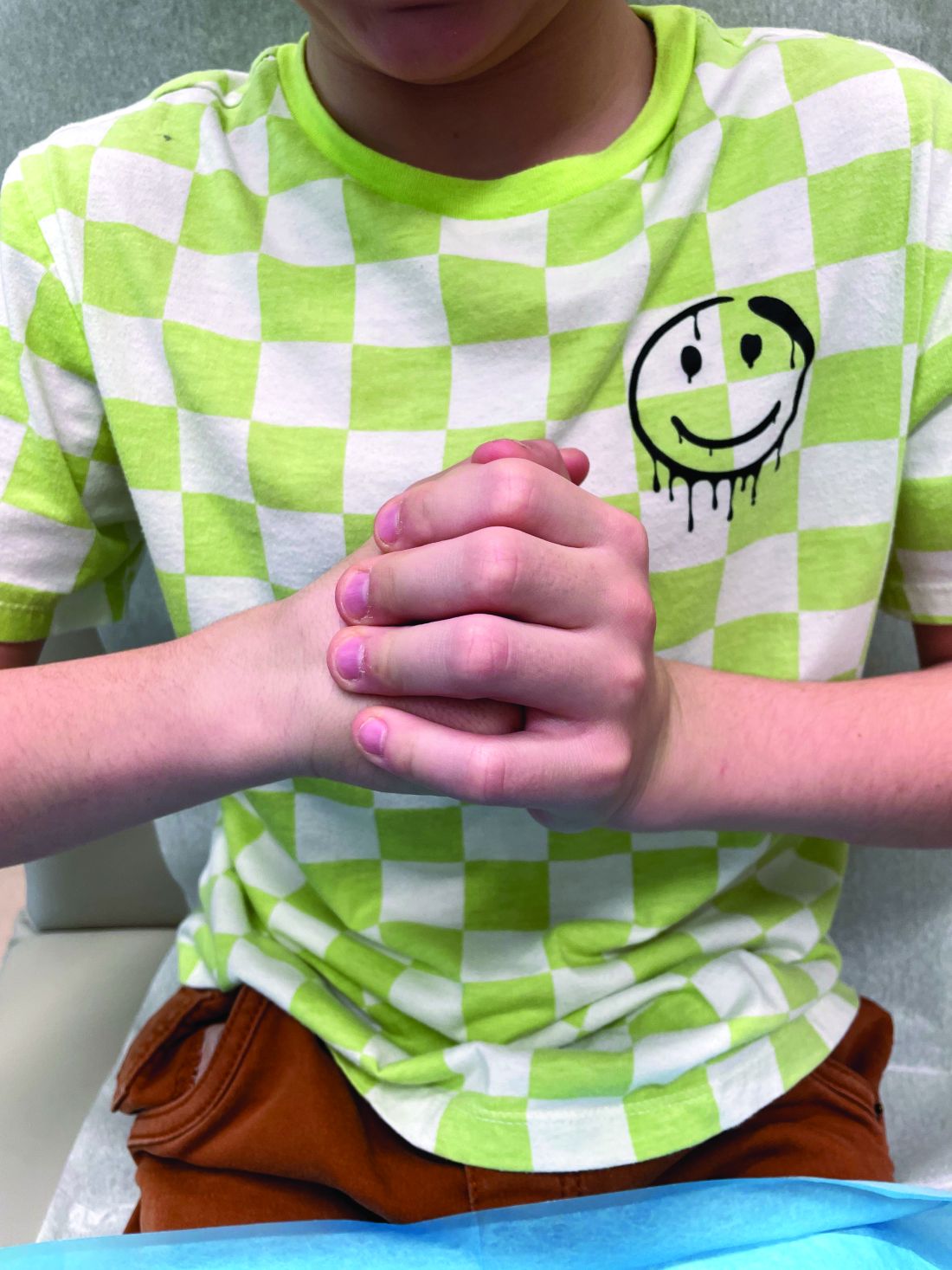
Lichen simplex chronicus and pachydermodactyly are both attributed to microtrauma inflicted upon the skin. Lichen simplex chronicus often constitutes a diagnosis of exclusion and is characterized by repetitive trauma-induced keratinocyte proliferation and melanocyte activation, resulting in hyperpigmentation and skin thickening. Although typically observed in women between the fourth and fifth decades of life, LSC is rarely reported in children. In adults, LSC-related rubbing or scratching frequently arises from chronic pruritic dermatitis such as eczema or psoriasis, neurodermatitis from dysesthesia, or habitual movements, as exhibited by this young patient. While generally benign, LSC may become infected. In rare instances, malignant transformation may occur.
The association with pachydermodactyly implicates microtrauma, necessitating careful observation and questioning to elucidate the cause, as demonstrated in this case. Lesions are typically hyperpigmented, though cases of associated hypopigmentation or depigmentation have been documented. Affected areas typically fall within the patient’s hand and finger reach, with lesion improvement over several months achievable through trigger avoidance.
Pachydermodactyly, a rare but benign fibromatosis around the proximal interphalangeal joints, is often misdiagnosed as juvenile idiopathic arthritis, potentially leading to unnecessary treatments and patient anxiety. Microtrauma history due to digit manipulation is prevalent among affected individuals, with most also exhibiting neuropsychiatric disorders. Histological examination of pachydermodactyly reveals hypergranulosis and dermal thickening, accompanied by increased fibroblasts and collagen types I, III, and V, differing from the epidermal changes seen in LSC.
The differential diagnosis also included phytophotodermatitis, a phototoxic dermatologic reaction following exposure to ultraviolet light subsequent to contact with furocoumarin-containing plant chemicals. However, the persistence of the patient’s lesions for over a year precluded this diagnosis. Secondary hyperpigmentation was also contemplated but excluded due to the absence of preceding inflammatory dermatitis.
Treatment of LSC primarily involves identifying and addressing any underlying conditions, repairing the skin barrier, reducing inflammation, and modifying behaviors contributing to chronic microtrauma, as observed in this patient. Topical corticosteroids may aid in decreasing epidermal thickening and discoloration, though lesion resolution necessitates behavior cessation.
It’s important to identify these types of skin changes in children to avoid unnecessary medical treatments for these benign conditions.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
Suggested Reading
Seier JA, Dissemond J. Lichen Simplex Chronicus Due to Mechanical Irritation. Dtsch Arztebl Int. 2022 Nov 18;119(46):802. doi: 10.3238/arztebl.m2022.0213.
Small S et al. A 12-Year-Old Boy Presenting With Unilateral Proximal Interphalangeal Joint Swelling. BMJ Case Rep. 2011 Apr 13:2011:bcr0120113719. doi: 10.1136/bcr.01.2011.3719.
Voicu C et al Lichen Simplex Chronicus as an Essential Part of the Dermatologic Masquerade. Open Access Maced J Med Sci. 2017 Jul 24;5(4):556-557. doi: 10.3889/oamjms.2017.133.
During examination, the patient was observed repetitively cracking his knuckles, making a fist with the right hand, placing the left hand on top, and rubbing the hand, a behavior he routinely did multiple times daily. The observed pattern of finger involvement on the dorsum of the right hand corresponded to areas subjected to significant pressure during the described activity. Consequently, a diagnosis of lichen simplex chronicus (LSC) secondary to mechanical rubbing, along with associated pachydermodactyly on the fingers of the right hand, was established.
Lichen simplex chronicus and pachydermodactyly are both attributed to microtrauma inflicted upon the skin. Lichen simplex chronicus often constitutes a diagnosis of exclusion and is characterized by repetitive trauma-induced keratinocyte proliferation and melanocyte activation, resulting in hyperpigmentation and skin thickening. Although typically observed in women between the fourth and fifth decades of life, LSC is rarely reported in children. In adults, LSC-related rubbing or scratching frequently arises from chronic pruritic dermatitis such as eczema or psoriasis, neurodermatitis from dysesthesia, or habitual movements, as exhibited by this young patient. While generally benign, LSC may become infected. In rare instances, malignant transformation may occur.
The association with pachydermodactyly implicates microtrauma, necessitating careful observation and questioning to elucidate the cause, as demonstrated in this case. Lesions are typically hyperpigmented, though cases of associated hypopigmentation or depigmentation have been documented. Affected areas typically fall within the patient’s hand and finger reach, with lesion improvement over several months achievable through trigger avoidance.
Pachydermodactyly, a rare but benign fibromatosis around the proximal interphalangeal joints, is often misdiagnosed as juvenile idiopathic arthritis, potentially leading to unnecessary treatments and patient anxiety. Microtrauma history due to digit manipulation is prevalent among affected individuals, with most also exhibiting neuropsychiatric disorders. Histological examination of pachydermodactyly reveals hypergranulosis and dermal thickening, accompanied by increased fibroblasts and collagen types I, III, and V, differing from the epidermal changes seen in LSC.
The differential diagnosis also included phytophotodermatitis, a phototoxic dermatologic reaction following exposure to ultraviolet light subsequent to contact with furocoumarin-containing plant chemicals. However, the persistence of the patient’s lesions for over a year precluded this diagnosis. Secondary hyperpigmentation was also contemplated but excluded due to the absence of preceding inflammatory dermatitis.
Treatment of LSC primarily involves identifying and addressing any underlying conditions, repairing the skin barrier, reducing inflammation, and modifying behaviors contributing to chronic microtrauma, as observed in this patient. Topical corticosteroids may aid in decreasing epidermal thickening and discoloration, though lesion resolution necessitates behavior cessation.
It’s important to identify these types of skin changes in children to avoid unnecessary medical treatments for these benign conditions.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
Suggested Reading
Seier JA, Dissemond J. Lichen Simplex Chronicus Due to Mechanical Irritation. Dtsch Arztebl Int. 2022 Nov 18;119(46):802. doi: 10.3238/arztebl.m2022.0213.
Small S et al. A 12-Year-Old Boy Presenting With Unilateral Proximal Interphalangeal Joint Swelling. BMJ Case Rep. 2011 Apr 13:2011:bcr0120113719. doi: 10.1136/bcr.01.2011.3719.
Voicu C et al Lichen Simplex Chronicus as an Essential Part of the Dermatologic Masquerade. Open Access Maced J Med Sci. 2017 Jul 24;5(4):556-557. doi: 10.3889/oamjms.2017.133.
During examination, the patient was observed repetitively cracking his knuckles, making a fist with the right hand, placing the left hand on top, and rubbing the hand, a behavior he routinely did multiple times daily. The observed pattern of finger involvement on the dorsum of the right hand corresponded to areas subjected to significant pressure during the described activity. Consequently, a diagnosis of lichen simplex chronicus (LSC) secondary to mechanical rubbing, along with associated pachydermodactyly on the fingers of the right hand, was established.
Lichen simplex chronicus and pachydermodactyly are both attributed to microtrauma inflicted upon the skin. Lichen simplex chronicus often constitutes a diagnosis of exclusion and is characterized by repetitive trauma-induced keratinocyte proliferation and melanocyte activation, resulting in hyperpigmentation and skin thickening. Although typically observed in women between the fourth and fifth decades of life, LSC is rarely reported in children. In adults, LSC-related rubbing or scratching frequently arises from chronic pruritic dermatitis such as eczema or psoriasis, neurodermatitis from dysesthesia, or habitual movements, as exhibited by this young patient. While generally benign, LSC may become infected. In rare instances, malignant transformation may occur.
The association with pachydermodactyly implicates microtrauma, necessitating careful observation and questioning to elucidate the cause, as demonstrated in this case. Lesions are typically hyperpigmented, though cases of associated hypopigmentation or depigmentation have been documented. Affected areas typically fall within the patient’s hand and finger reach, with lesion improvement over several months achievable through trigger avoidance.
Pachydermodactyly, a rare but benign fibromatosis around the proximal interphalangeal joints, is often misdiagnosed as juvenile idiopathic arthritis, potentially leading to unnecessary treatments and patient anxiety. Microtrauma history due to digit manipulation is prevalent among affected individuals, with most also exhibiting neuropsychiatric disorders. Histological examination of pachydermodactyly reveals hypergranulosis and dermal thickening, accompanied by increased fibroblasts and collagen types I, III, and V, differing from the epidermal changes seen in LSC.
The differential diagnosis also included phytophotodermatitis, a phototoxic dermatologic reaction following exposure to ultraviolet light subsequent to contact with furocoumarin-containing plant chemicals. However, the persistence of the patient’s lesions for over a year precluded this diagnosis. Secondary hyperpigmentation was also contemplated but excluded due to the absence of preceding inflammatory dermatitis.
Treatment of LSC primarily involves identifying and addressing any underlying conditions, repairing the skin barrier, reducing inflammation, and modifying behaviors contributing to chronic microtrauma, as observed in this patient. Topical corticosteroids may aid in decreasing epidermal thickening and discoloration, though lesion resolution necessitates behavior cessation.
It’s important to identify these types of skin changes in children to avoid unnecessary medical treatments for these benign conditions.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
Suggested Reading
Seier JA, Dissemond J. Lichen Simplex Chronicus Due to Mechanical Irritation. Dtsch Arztebl Int. 2022 Nov 18;119(46):802. doi: 10.3238/arztebl.m2022.0213.
Small S et al. A 12-Year-Old Boy Presenting With Unilateral Proximal Interphalangeal Joint Swelling. BMJ Case Rep. 2011 Apr 13:2011:bcr0120113719. doi: 10.1136/bcr.01.2011.3719.
Voicu C et al Lichen Simplex Chronicus as an Essential Part of the Dermatologic Masquerade. Open Access Maced J Med Sci. 2017 Jul 24;5(4):556-557. doi: 10.3889/oamjms.2017.133.
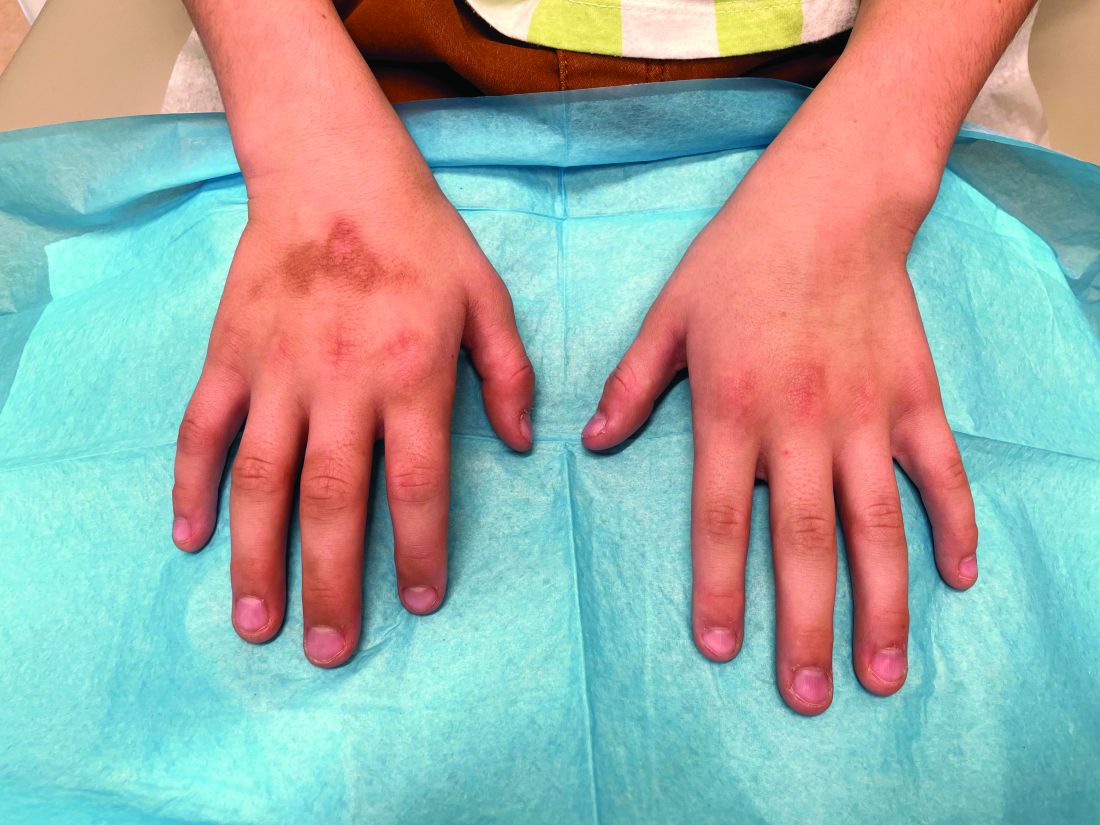
The patient was otherwise healthy, with no current medication intake, and he engaged in baseball and soccer activities. Upon physical examination, a hyperpigmented lichenified irregular plaque was observed on the dorsum of the right hand, along with irregular hyperpigmented macules and plaques on the fingers. Fusiform widening of the interphalangeal joints on the second, third, and fourth fingers of the right hand was noted, without associated pain, edema, or erythema.
An 18-month-old male presents with a red mark on the forehead and nose
Following the initial presentation, the lesion was initially considered an acquired port wine stain and the child was referred for laser treatment. Upon reassessment during laser treatment a few months later, the lesion had progressed to hyper- and hypopigmented plaques with associated tissue sclerosis and bone atrophy on the mid forehead, nose, and scalp. Patches of alopecia and atrophy were observed on the frontal scalp. The diagnosis was revised to linear morphea en coup de sabre and the child was referred to pediatric rheumatology and commenced treatment with methotrexate and oral corticosteroids.
Linear morphea, a rare connective tissue disorder, primarily affects girls in the first 2 decades of life. Lesions can initially present in many ways. Usually, they present as hypo- or hyperpigmented patches, but may also present as lichenoid uncolored or pink plaques resembling lichen striatus. There may also be erythematous patches mimicking a capillary malformation, as seen in our patient. A recent article reviewing the progression of the lesions from erythematous patches to sclerosis suggests it occurs between 3 and 7 months of age. Subsequent stages manifest as significant atrophy, hypo- and hyperpigmentation, and in severe cases, bone atrophy and deformity, often causing substantial cosmetic disfigurement and functional impairment.
Pathophysiologically, linear morphea involves a complex interplay of immunologic, vascular, and fibrotic processes. While the initial triggers remain elusive, dysregulated immune responses leading to endothelial injury, subsequent activation of fibroblasts and myofibroblasts, and excessive collagen deposition are implicated. Angiogenic disturbances exacerbate tissue ischemia, perpetuating the fibrotic cascade. Alterations in cytokine signaling pathways, particularly TGF-beta and interleukin-6, play pivotal roles in promoting fibrosis and modulating the inflammatory milieu.
Diagnosis of linear morphea en coup de sabre relies on clinical examination, imaging (ultrasonography, MRI, CT scan), and skin biopsy for histopathological analysis. Imaging helps evaluate tissue involvement, while histology reveals characteristic dermal sclerosis, collagen deposition, and inflammation. Early-stage histology may show telangiectatic changes, complicating its differentiation from capillary malformation.
Treatment aims to mitigate symptoms, halt disease progression, and improve cosmesis and functionality. This involves a multidisciplinary approach with systemic medications, phototherapy, physical therapy, and surgical interventions in severe cases. Early identification is crucial for systemic treatments such as methotrexate and systemic corticosteroids to arrest disease progression. Other adjunctive therapies include topical corticosteroids, calcineurin inhibitors, and phototherapy. Surgical procedures like tissue expansion or autologous fat grafting may address tissue atrophy and deformities.
Linear morphea en coup de sabre presents diagnostic and therapeutic challenges because of its rarity and variable clinical course. Collaborative efforts among dermatologists, rheumatologists, radiologists, and surgeons are essential for accurate diagnosis, evaluation, and tailored management. Continued research into pathogenesis and novel therapeutic agents is pivotal to enhance understanding and improve outcomes for those affected by this enigmatic dermatologic condition.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Gomez-Garcia LA et al. Pediatr Dermatol. 2022 Mar;39(2):275-80.
Ng SS, Tay YK. J Cosmet Laser Ther. 2015;17(5):277-80.
Nijhawan RI et al. J Am Acad Dermatol. 2011 Apr;64(4):779-82.
Following the initial presentation, the lesion was initially considered an acquired port wine stain and the child was referred for laser treatment. Upon reassessment during laser treatment a few months later, the lesion had progressed to hyper- and hypopigmented plaques with associated tissue sclerosis and bone atrophy on the mid forehead, nose, and scalp. Patches of alopecia and atrophy were observed on the frontal scalp. The diagnosis was revised to linear morphea en coup de sabre and the child was referred to pediatric rheumatology and commenced treatment with methotrexate and oral corticosteroids.
Linear morphea, a rare connective tissue disorder, primarily affects girls in the first 2 decades of life. Lesions can initially present in many ways. Usually, they present as hypo- or hyperpigmented patches, but may also present as lichenoid uncolored or pink plaques resembling lichen striatus. There may also be erythematous patches mimicking a capillary malformation, as seen in our patient. A recent article reviewing the progression of the lesions from erythematous patches to sclerosis suggests it occurs between 3 and 7 months of age. Subsequent stages manifest as significant atrophy, hypo- and hyperpigmentation, and in severe cases, bone atrophy and deformity, often causing substantial cosmetic disfigurement and functional impairment.
Pathophysiologically, linear morphea involves a complex interplay of immunologic, vascular, and fibrotic processes. While the initial triggers remain elusive, dysregulated immune responses leading to endothelial injury, subsequent activation of fibroblasts and myofibroblasts, and excessive collagen deposition are implicated. Angiogenic disturbances exacerbate tissue ischemia, perpetuating the fibrotic cascade. Alterations in cytokine signaling pathways, particularly TGF-beta and interleukin-6, play pivotal roles in promoting fibrosis and modulating the inflammatory milieu.
Diagnosis of linear morphea en coup de sabre relies on clinical examination, imaging (ultrasonography, MRI, CT scan), and skin biopsy for histopathological analysis. Imaging helps evaluate tissue involvement, while histology reveals characteristic dermal sclerosis, collagen deposition, and inflammation. Early-stage histology may show telangiectatic changes, complicating its differentiation from capillary malformation.
Treatment aims to mitigate symptoms, halt disease progression, and improve cosmesis and functionality. This involves a multidisciplinary approach with systemic medications, phototherapy, physical therapy, and surgical interventions in severe cases. Early identification is crucial for systemic treatments such as methotrexate and systemic corticosteroids to arrest disease progression. Other adjunctive therapies include topical corticosteroids, calcineurin inhibitors, and phototherapy. Surgical procedures like tissue expansion or autologous fat grafting may address tissue atrophy and deformities.
Linear morphea en coup de sabre presents diagnostic and therapeutic challenges because of its rarity and variable clinical course. Collaborative efforts among dermatologists, rheumatologists, radiologists, and surgeons are essential for accurate diagnosis, evaluation, and tailored management. Continued research into pathogenesis and novel therapeutic agents is pivotal to enhance understanding and improve outcomes for those affected by this enigmatic dermatologic condition.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Gomez-Garcia LA et al. Pediatr Dermatol. 2022 Mar;39(2):275-80.
Ng SS, Tay YK. J Cosmet Laser Ther. 2015;17(5):277-80.
Nijhawan RI et al. J Am Acad Dermatol. 2011 Apr;64(4):779-82.
Following the initial presentation, the lesion was initially considered an acquired port wine stain and the child was referred for laser treatment. Upon reassessment during laser treatment a few months later, the lesion had progressed to hyper- and hypopigmented plaques with associated tissue sclerosis and bone atrophy on the mid forehead, nose, and scalp. Patches of alopecia and atrophy were observed on the frontal scalp. The diagnosis was revised to linear morphea en coup de sabre and the child was referred to pediatric rheumatology and commenced treatment with methotrexate and oral corticosteroids.
Linear morphea, a rare connective tissue disorder, primarily affects girls in the first 2 decades of life. Lesions can initially present in many ways. Usually, they present as hypo- or hyperpigmented patches, but may also present as lichenoid uncolored or pink plaques resembling lichen striatus. There may also be erythematous patches mimicking a capillary malformation, as seen in our patient. A recent article reviewing the progression of the lesions from erythematous patches to sclerosis suggests it occurs between 3 and 7 months of age. Subsequent stages manifest as significant atrophy, hypo- and hyperpigmentation, and in severe cases, bone atrophy and deformity, often causing substantial cosmetic disfigurement and functional impairment.
Pathophysiologically, linear morphea involves a complex interplay of immunologic, vascular, and fibrotic processes. While the initial triggers remain elusive, dysregulated immune responses leading to endothelial injury, subsequent activation of fibroblasts and myofibroblasts, and excessive collagen deposition are implicated. Angiogenic disturbances exacerbate tissue ischemia, perpetuating the fibrotic cascade. Alterations in cytokine signaling pathways, particularly TGF-beta and interleukin-6, play pivotal roles in promoting fibrosis and modulating the inflammatory milieu.
Diagnosis of linear morphea en coup de sabre relies on clinical examination, imaging (ultrasonography, MRI, CT scan), and skin biopsy for histopathological analysis. Imaging helps evaluate tissue involvement, while histology reveals characteristic dermal sclerosis, collagen deposition, and inflammation. Early-stage histology may show telangiectatic changes, complicating its differentiation from capillary malformation.
Treatment aims to mitigate symptoms, halt disease progression, and improve cosmesis and functionality. This involves a multidisciplinary approach with systemic medications, phototherapy, physical therapy, and surgical interventions in severe cases. Early identification is crucial for systemic treatments such as methotrexate and systemic corticosteroids to arrest disease progression. Other adjunctive therapies include topical corticosteroids, calcineurin inhibitors, and phototherapy. Surgical procedures like tissue expansion or autologous fat grafting may address tissue atrophy and deformities.
Linear morphea en coup de sabre presents diagnostic and therapeutic challenges because of its rarity and variable clinical course. Collaborative efforts among dermatologists, rheumatologists, radiologists, and surgeons are essential for accurate diagnosis, evaluation, and tailored management. Continued research into pathogenesis and novel therapeutic agents is pivotal to enhance understanding and improve outcomes for those affected by this enigmatic dermatologic condition.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Gomez-Garcia LA et al. Pediatr Dermatol. 2022 Mar;39(2):275-80.
Ng SS, Tay YK. J Cosmet Laser Ther. 2015;17(5):277-80.
Nijhawan RI et al. J Am Acad Dermatol. 2011 Apr;64(4):779-82.
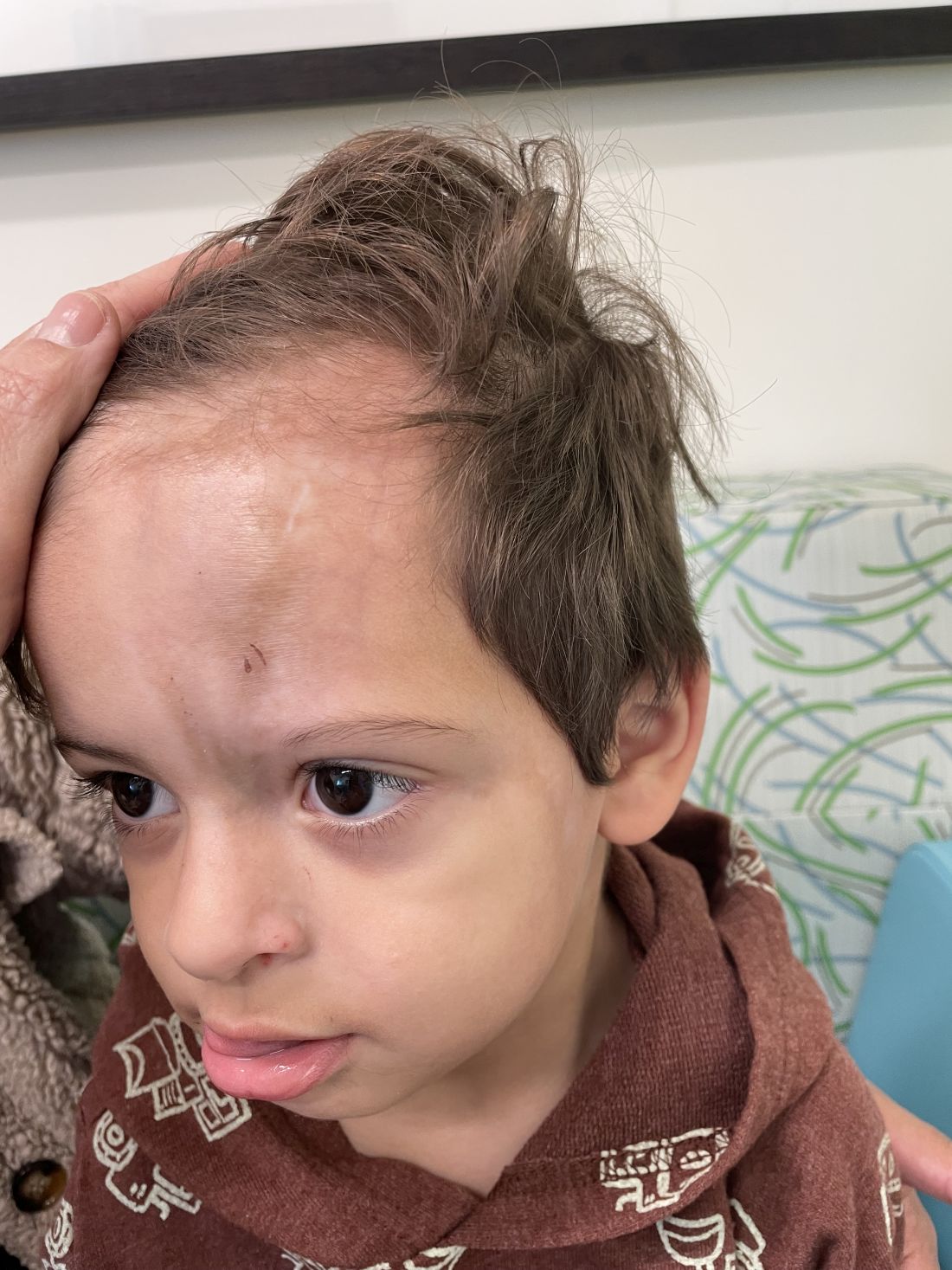
On examination, a faint pink patch was observed on the right forehead, frontal scalp, and nose. The lesion paled under pressure, with small areas of hair loss on the scalp. No atrophy was noted.
What's the diagnosis?
The lesions on the heels are consistent with piezogenic pedal papules. They seem to be more common in women and have been described in families, though a genetic link hasn’t been elucidated. PPP manifests as small, soft, compressible papules on the lateral aspects of the skin on the heels, more noticeable when the patient is standing, and can also present on the wrists and legs. While they may not be a cause for serious concern, understanding their causes, associated conditions, and management is important.
Piezogenic pedal papules are flesh-colored or slightly reddish and can range in size from a few millimeters to a centimeter or more. They are described as benign herniations of elastic tissue and subcutaneous fat through the reticular dermis. The lesions are triggered by increased pressure and compression, such as standing or the application of pressure on the heel. The exact etiology is not known. While piezogenic pedal papules are often asymptomatic, some individuals experience discomfort, itching, or mild pain, particularly when walking or applying pressure to the affected area, especially in patients with Ehlers-Danlos syndrome (EDS).
Individuals who may be at risk of developing these lesions include obese patients, individuals with pes planus, and people who have occupations that require long periods of standing. It can also be seen in athletes who participate in long-distance running or high-impact sports. Piezogenic pedal papules have been described as one of the core skin findings in patients with hypermobile Ehlers-Danlos syndrome (hEDS), which also includes skin hyperextensibility, joint hypermobility, tissue fragility with atrophic cutaneous scars, and abnormal bruising and bleeding. Our patient presented with some of these characteristics (piezogenic papules, soft elastic skin, and some joint hypermobility) but did not fulfill all the criteria for the diagnosis of hEDS or other types of EDS.
The diagnosis of hEDS is based on the 2017 diagnostic criteria checklist. To be diagnosed with hEDS, the patient may have all three parts of the diagnostic criteria. The three domains include generalized joint hypermobility (partially met by our patient), evidence of syndromic features, musculoskeletal complications, and/or family history (she had a few of these criteria, including piezogenic papules and striae), and the exclusion of alternative diagnoses (see references for the PDF checklist). As she does have some features, we diagnosed her with hypermobility spectrum disorder. There is no genetic testing available for the hypermobile spectrum disorder or the hypermobile type of EDS. Given that these patients can present with mitral valve prolapse, she was referred to a cardiac echocardiogram, which was reported as normal.
The diagnosis of PPP is made clinically, and rarely a biopsy is required. Biopsies of the lesions show hyperkeratosis, degeneration of the thin fibrous septa between fat lobules, and subsequent coalescence of fat. If the presentation is atypical, a high-frequency ultrasound can be requested to confirm the physical exam findings.
If the lesions are fixed, firm, and solitary, a diagnosis to consider is juvenile aponeurotic fibroma, which occurs more often in children and adolescents on the wrists and is less common on the ankles. If there is suspicion for this condition, a plain radiograph will show stippled calcifications.
PPP are usually asymptomatic and need no further treatment. When they are symptomatic, conservative management should be considered first, which includes behavioral modifications, weight loss, avoidance of prolonged standing, and reduced foot trauma. If these are not successful, compression socks, heel cups, and other orthotics can be recommended. Intralesional injections of betamethasone and bupivacaine have been reported as an option in patients with symptomatic PPP and a history of EDS.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
Suggested reading
Edimo CO et al. Int J Womens Dermatol. 2021 Jan. doi: 10.1016/j.ijwd.2021.01.020. Erratum in: Int J Womens Dermatol. 2021 Jul 31;7(5Part B):869-70.
Brown F, Cook C. Piezogenic Pedal Papule. 2023 Aug 16. In: StatPearls [Internet]. Treasure Island, Fla.: StatPearls Publishing, 2023. PMID: 29489228.
Levy HP. Hypermobile Ehlers-Danlos Syndrome. 2004 Oct 22 [Updated 2018 Jun 21]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 1993-2023. Available from: www.ncbi.nlm.nih.gov/books/NBK1279/.
The International Consortium on Ehlers-Danlos Syndrome & Related Disorders. Diagnostic Criteria for Hypermobile Ehlers-Danlos Syndrome (hEDS). www.ehlers-danlos.com/wp-content/uploads/2017/05/hEDS-Dx-Criteria-checklist-1.pdf.
The lesions on the heels are consistent with piezogenic pedal papules. They seem to be more common in women and have been described in families, though a genetic link hasn’t been elucidated. PPP manifests as small, soft, compressible papules on the lateral aspects of the skin on the heels, more noticeable when the patient is standing, and can also present on the wrists and legs. While they may not be a cause for serious concern, understanding their causes, associated conditions, and management is important.
Piezogenic pedal papules are flesh-colored or slightly reddish and can range in size from a few millimeters to a centimeter or more. They are described as benign herniations of elastic tissue and subcutaneous fat through the reticular dermis. The lesions are triggered by increased pressure and compression, such as standing or the application of pressure on the heel. The exact etiology is not known. While piezogenic pedal papules are often asymptomatic, some individuals experience discomfort, itching, or mild pain, particularly when walking or applying pressure to the affected area, especially in patients with Ehlers-Danlos syndrome (EDS).
Individuals who may be at risk of developing these lesions include obese patients, individuals with pes planus, and people who have occupations that require long periods of standing. It can also be seen in athletes who participate in long-distance running or high-impact sports. Piezogenic pedal papules have been described as one of the core skin findings in patients with hypermobile Ehlers-Danlos syndrome (hEDS), which also includes skin hyperextensibility, joint hypermobility, tissue fragility with atrophic cutaneous scars, and abnormal bruising and bleeding. Our patient presented with some of these characteristics (piezogenic papules, soft elastic skin, and some joint hypermobility) but did not fulfill all the criteria for the diagnosis of hEDS or other types of EDS.
The diagnosis of hEDS is based on the 2017 diagnostic criteria checklist. To be diagnosed with hEDS, the patient may have all three parts of the diagnostic criteria. The three domains include generalized joint hypermobility (partially met by our patient), evidence of syndromic features, musculoskeletal complications, and/or family history (she had a few of these criteria, including piezogenic papules and striae), and the exclusion of alternative diagnoses (see references for the PDF checklist). As she does have some features, we diagnosed her with hypermobility spectrum disorder. There is no genetic testing available for the hypermobile spectrum disorder or the hypermobile type of EDS. Given that these patients can present with mitral valve prolapse, she was referred to a cardiac echocardiogram, which was reported as normal.
The diagnosis of PPP is made clinically, and rarely a biopsy is required. Biopsies of the lesions show hyperkeratosis, degeneration of the thin fibrous septa between fat lobules, and subsequent coalescence of fat. If the presentation is atypical, a high-frequency ultrasound can be requested to confirm the physical exam findings.
If the lesions are fixed, firm, and solitary, a diagnosis to consider is juvenile aponeurotic fibroma, which occurs more often in children and adolescents on the wrists and is less common on the ankles. If there is suspicion for this condition, a plain radiograph will show stippled calcifications.
PPP are usually asymptomatic and need no further treatment. When they are symptomatic, conservative management should be considered first, which includes behavioral modifications, weight loss, avoidance of prolonged standing, and reduced foot trauma. If these are not successful, compression socks, heel cups, and other orthotics can be recommended. Intralesional injections of betamethasone and bupivacaine have been reported as an option in patients with symptomatic PPP and a history of EDS.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
Suggested reading
Edimo CO et al. Int J Womens Dermatol. 2021 Jan. doi: 10.1016/j.ijwd.2021.01.020. Erratum in: Int J Womens Dermatol. 2021 Jul 31;7(5Part B):869-70.
Brown F, Cook C. Piezogenic Pedal Papule. 2023 Aug 16. In: StatPearls [Internet]. Treasure Island, Fla.: StatPearls Publishing, 2023. PMID: 29489228.
Levy HP. Hypermobile Ehlers-Danlos Syndrome. 2004 Oct 22 [Updated 2018 Jun 21]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 1993-2023. Available from: www.ncbi.nlm.nih.gov/books/NBK1279/.
The International Consortium on Ehlers-Danlos Syndrome & Related Disorders. Diagnostic Criteria for Hypermobile Ehlers-Danlos Syndrome (hEDS). www.ehlers-danlos.com/wp-content/uploads/2017/05/hEDS-Dx-Criteria-checklist-1.pdf.
The lesions on the heels are consistent with piezogenic pedal papules. They seem to be more common in women and have been described in families, though a genetic link hasn’t been elucidated. PPP manifests as small, soft, compressible papules on the lateral aspects of the skin on the heels, more noticeable when the patient is standing, and can also present on the wrists and legs. While they may not be a cause for serious concern, understanding their causes, associated conditions, and management is important.
Piezogenic pedal papules are flesh-colored or slightly reddish and can range in size from a few millimeters to a centimeter or more. They are described as benign herniations of elastic tissue and subcutaneous fat through the reticular dermis. The lesions are triggered by increased pressure and compression, such as standing or the application of pressure on the heel. The exact etiology is not known. While piezogenic pedal papules are often asymptomatic, some individuals experience discomfort, itching, or mild pain, particularly when walking or applying pressure to the affected area, especially in patients with Ehlers-Danlos syndrome (EDS).
Individuals who may be at risk of developing these lesions include obese patients, individuals with pes planus, and people who have occupations that require long periods of standing. It can also be seen in athletes who participate in long-distance running or high-impact sports. Piezogenic pedal papules have been described as one of the core skin findings in patients with hypermobile Ehlers-Danlos syndrome (hEDS), which also includes skin hyperextensibility, joint hypermobility, tissue fragility with atrophic cutaneous scars, and abnormal bruising and bleeding. Our patient presented with some of these characteristics (piezogenic papules, soft elastic skin, and some joint hypermobility) but did not fulfill all the criteria for the diagnosis of hEDS or other types of EDS.
The diagnosis of hEDS is based on the 2017 diagnostic criteria checklist. To be diagnosed with hEDS, the patient may have all three parts of the diagnostic criteria. The three domains include generalized joint hypermobility (partially met by our patient), evidence of syndromic features, musculoskeletal complications, and/or family history (she had a few of these criteria, including piezogenic papules and striae), and the exclusion of alternative diagnoses (see references for the PDF checklist). As she does have some features, we diagnosed her with hypermobility spectrum disorder. There is no genetic testing available for the hypermobile spectrum disorder or the hypermobile type of EDS. Given that these patients can present with mitral valve prolapse, she was referred to a cardiac echocardiogram, which was reported as normal.
The diagnosis of PPP is made clinically, and rarely a biopsy is required. Biopsies of the lesions show hyperkeratosis, degeneration of the thin fibrous septa between fat lobules, and subsequent coalescence of fat. If the presentation is atypical, a high-frequency ultrasound can be requested to confirm the physical exam findings.
If the lesions are fixed, firm, and solitary, a diagnosis to consider is juvenile aponeurotic fibroma, which occurs more often in children and adolescents on the wrists and is less common on the ankles. If there is suspicion for this condition, a plain radiograph will show stippled calcifications.
PPP are usually asymptomatic and need no further treatment. When they are symptomatic, conservative management should be considered first, which includes behavioral modifications, weight loss, avoidance of prolonged standing, and reduced foot trauma. If these are not successful, compression socks, heel cups, and other orthotics can be recommended. Intralesional injections of betamethasone and bupivacaine have been reported as an option in patients with symptomatic PPP and a history of EDS.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
Suggested reading
Edimo CO et al. Int J Womens Dermatol. 2021 Jan. doi: 10.1016/j.ijwd.2021.01.020. Erratum in: Int J Womens Dermatol. 2021 Jul 31;7(5Part B):869-70.
Brown F, Cook C. Piezogenic Pedal Papule. 2023 Aug 16. In: StatPearls [Internet]. Treasure Island, Fla.: StatPearls Publishing, 2023. PMID: 29489228.
Levy HP. Hypermobile Ehlers-Danlos Syndrome. 2004 Oct 22 [Updated 2018 Jun 21]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 1993-2023. Available from: www.ncbi.nlm.nih.gov/books/NBK1279/.
The International Consortium on Ehlers-Danlos Syndrome & Related Disorders. Diagnostic Criteria for Hypermobile Ehlers-Danlos Syndrome (hEDS). www.ehlers-danlos.com/wp-content/uploads/2017/05/hEDS-Dx-Criteria-checklist-1.pdf.
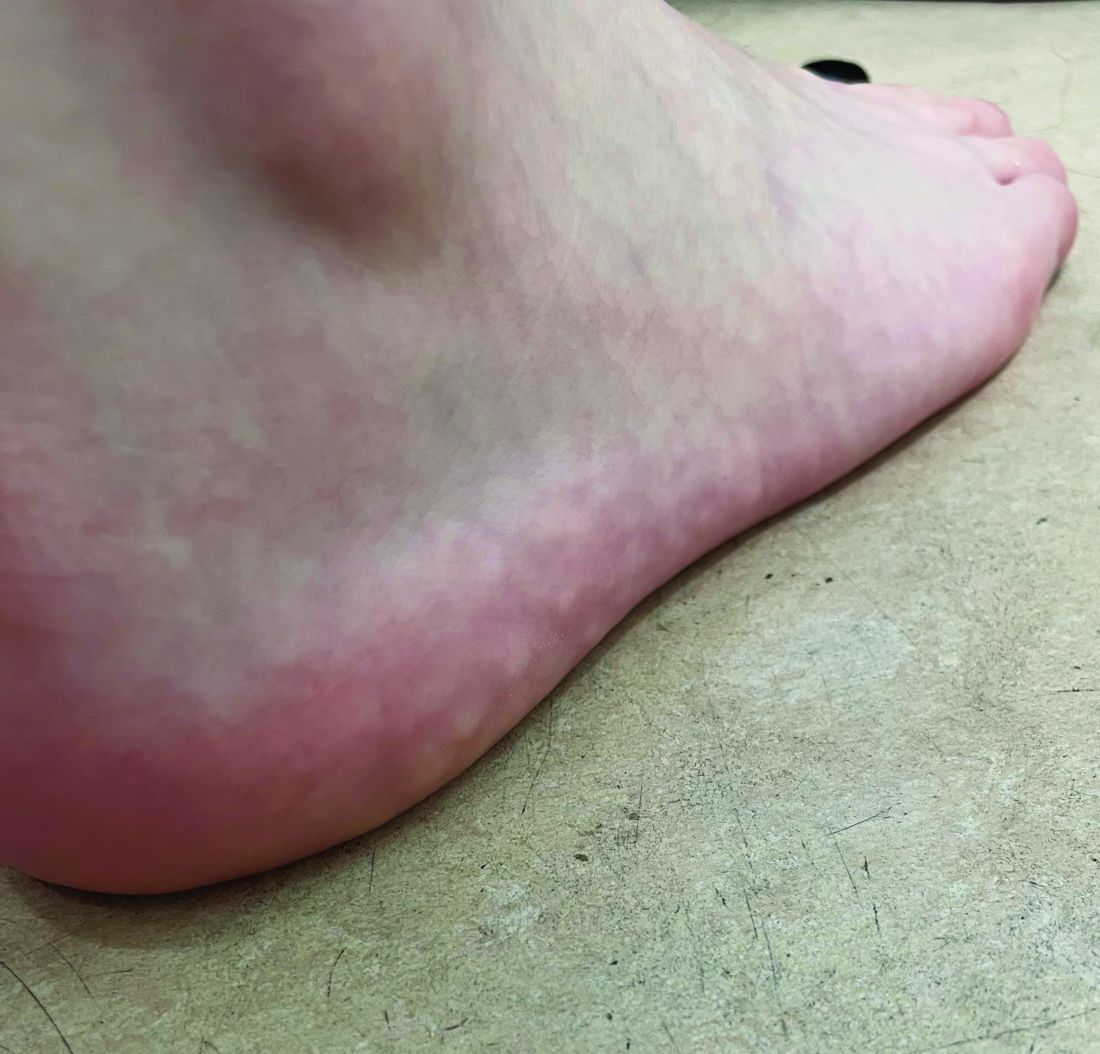
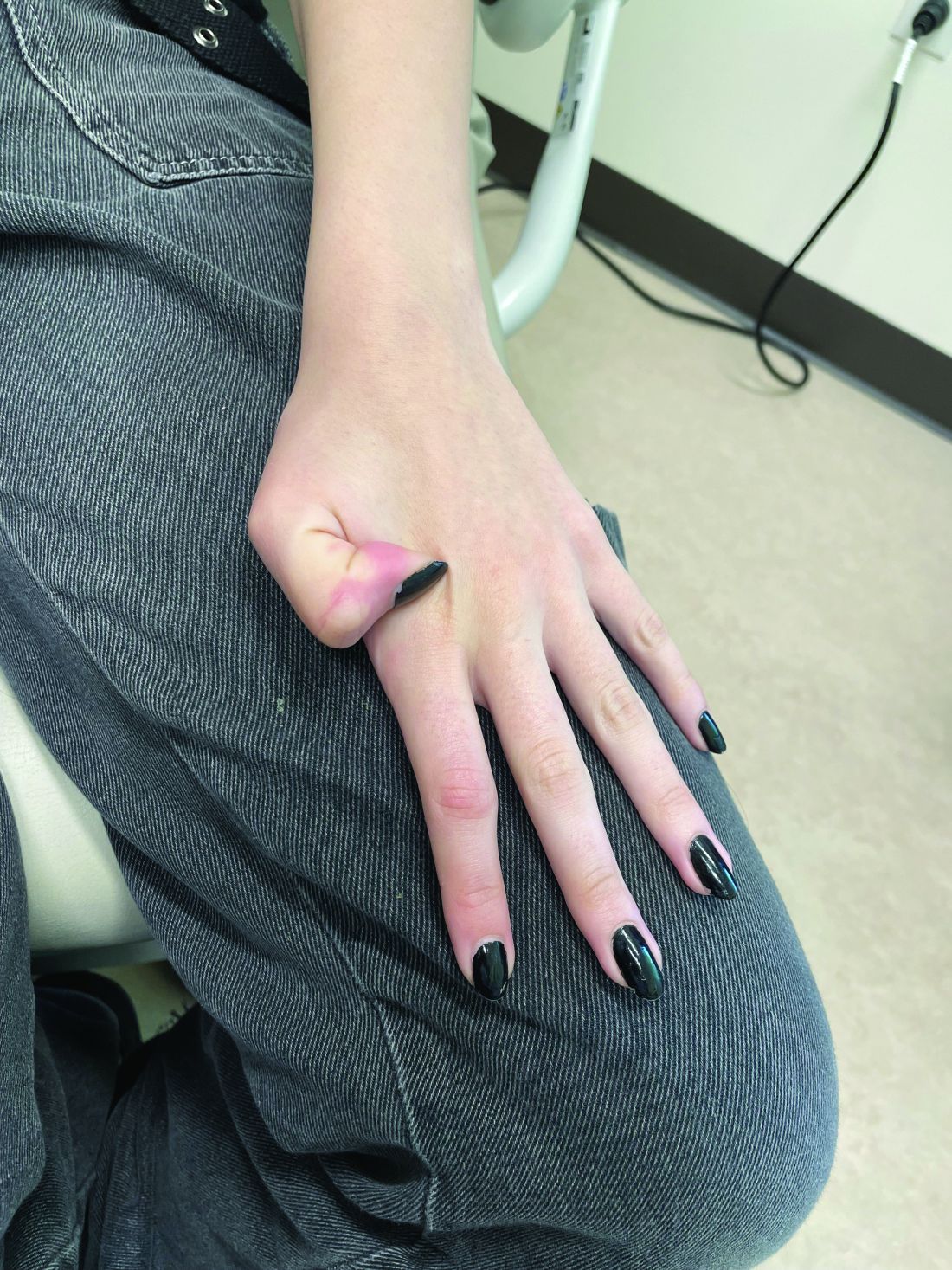
On a physical exam, she has several skin-colored soft papules on the heels when she stands up (Picture 1).
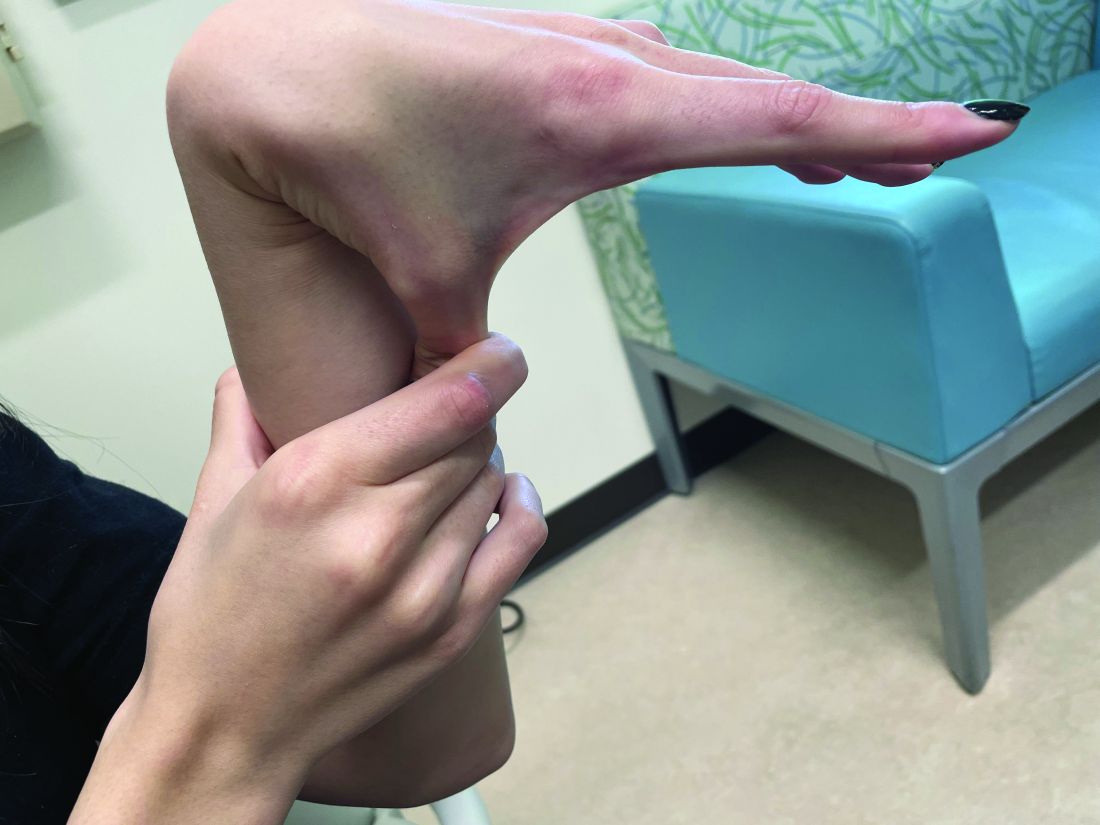
She is not able to touch the floor without bending her knees, and she has normal extension of her arms and knees. She has no bruises or abnormal scars and has some striae on her legs.
What's the diagnosis?
At the week follow-up, the lesions were unchanged and the swelling on the left lateral eyebrow was worsening. A biopsy of the yellow lesion on the back and one of the scaly papules on the abdomen was performed. A fungal and bacterial cultures were also ordered.
He was referred to ophthalmology for evaluation of the eyelid swelling and an ultrasound was requested.
The skin biopsy showed a clonal proliferation of reniform histiocytes with eosinophils within the dermis. The cells were positive for S100, CD207 (langerin), and CD1a and negative for pancytokeratin and Melan-A, supportive of the diagnosis of Langerhans cell histiocytosis (LCH).
Diagnosis
The patient was admitted to the hospital, where a skeletal survey was performed, which showed an asymmetric lucency involving the left frontal calvarium extending to the superior lateral orbital rim. The brain MRI demonstrated a destructive avidly enhancing soft-tissue process which involved the superior left orbital rim likely with some degree of intracranial extension. This lesion exerts mass effect upon surrounding structures to the left ocular globe. With the skin and skeletal findings, the patient was diagnosed with LCH. His blood count was significant for thrombocytopenia. His liver and kidney function were normal. His electrolytes were also with in normal range. He was started on chemotherapy with vinblastine and systemic corticosteroids with resolution of the rash and decrease on the size of the lesion on the orbit within a few weeks.
Infantile LCH is a rare neoplastic disorder of hematopoietic myeloid precursor cells caused by activating mutations in the mitogen-activated protein kinase (MAPK) pathway, particularly BRAF-V600E mutation. White male children are mostly affected, with a peak incidence of 1-3 years of age. Nine out of 10 children with cutaneous involvement also have multisystemic disease, such as the case of our patient. LCH is classified as single or multisystem organ disease. Two-thirds of the cases present with single system involvement. Organs most commonly affected include the bone (the skull being the most commonly affected), skin, and high-risk organs like the liver, spleen, and bone marrow, and less commonly the lungs, lymph nodes, and central nervous system. Some patients can present with fever, lethargy, and weight loss. None were noted in our patient.
Skin findings of LCH can have multiple morphologies and presentations and often described as a big mimicker. In young infants like our patient, the seborrheic dermatitis–mimicking type is often seen. In other cases, the skin lesions can appear eczematous, petechial, with scabbing, crusting, or purpura. Xanthoma-like lesions, like that one our patient had in the back, have also been described. Resistant diaper dermatitis and cradle cap should prompt the clinician to think about LCH. Lesions can be so varied that can present with hypopigmentation (vitiligo like), hyperpigmentation, varicella-like papulo-pustules, and red blue nodules within others. Oral mucosa and nail involvement can also occur.
Bone involvement can present as soft-tissue mass with swelling and pain as it occur in our patient.
Endocrinopathies have been described in patients with LCH including diabetes insipidus, growth hormone deficiency, and less likely thyroid disease.
Multidisciplinary care
The diagnosis of LCH in infants necessitates a combination of clinical, radiological, and histopathologic findings. In infants, cutaneous involvement is a frequent initial presentation, with characteristic lesions that are often misdiagnosed as other dermatologic conditions. Timely recognition of these lesions and appropriate skin biopsies for histological examination are essential steps in achieving an accurate diagnosis.
Radiological imaging, including x-rays, CT, and MRI, plays a crucial role in assessing the extent of involvement.
The management of LCH in infants requires a well-coordinated multidisciplinary approach involving pediatric oncologists, dermatologists, radiologists, orthopedic surgeons, and other relevant specialists. Treatment strategies vary depending on the extent of disease involvement and the presence of risk factors. In localized cases, observation with close monitoring may be considered, as some cases of LCH in infants may undergo spontaneous regression. However, cases with severe symptoms, extensive organ involvement, or high-risk features may require systemic therapies.
Chemotherapy agents, including vinblastine and prednisone have been utilized in the treatment of infantile LCH with varying success. The selection of treatment regimens should be tailored to each individual case, considering disease severity, potential toxicities, and long-term effects. In cases of bone lesions causing significant deformities or functional impairment, surgical intervention may be necessary. Skin only disease can be treated with topical corticosteroids.
Prognosis
Survival rates in patients with single-organ involvement without risk-organ involvement is close to 100% and with risk-organ involvement of 98% at 5 years.
Long-term follow-up is essential for infants diagnosed with LCH, as recurrence and late effects can occur even after successful treatment. Continued monitoring allows for the timely detection of relapses or the development of secondary complications.
Infants thought to have common skin conditions like eczema, seborrheic dermatitis, or diaper dermatitis not responding to treatment should be referred to pediatric dermatology for evaluation to rule out the possibility of LCH.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Krooks J et al. J Am Acad Dermatol. 2018 Jun;78(6):1035-44.
Krooks J et al. J Am Acad Dermatol. 2018 Jun;78(6):1047-56.
Leung AKC et al. World J Pediatr. 2019 Dec;15(6):536-45.
At the week follow-up, the lesions were unchanged and the swelling on the left lateral eyebrow was worsening. A biopsy of the yellow lesion on the back and one of the scaly papules on the abdomen was performed. A fungal and bacterial cultures were also ordered.
He was referred to ophthalmology for evaluation of the eyelid swelling and an ultrasound was requested.
The skin biopsy showed a clonal proliferation of reniform histiocytes with eosinophils within the dermis. The cells were positive for S100, CD207 (langerin), and CD1a and negative for pancytokeratin and Melan-A, supportive of the diagnosis of Langerhans cell histiocytosis (LCH).
Diagnosis
The patient was admitted to the hospital, where a skeletal survey was performed, which showed an asymmetric lucency involving the left frontal calvarium extending to the superior lateral orbital rim. The brain MRI demonstrated a destructive avidly enhancing soft-tissue process which involved the superior left orbital rim likely with some degree of intracranial extension. This lesion exerts mass effect upon surrounding structures to the left ocular globe. With the skin and skeletal findings, the patient was diagnosed with LCH. His blood count was significant for thrombocytopenia. His liver and kidney function were normal. His electrolytes were also with in normal range. He was started on chemotherapy with vinblastine and systemic corticosteroids with resolution of the rash and decrease on the size of the lesion on the orbit within a few weeks.
Infantile LCH is a rare neoplastic disorder of hematopoietic myeloid precursor cells caused by activating mutations in the mitogen-activated protein kinase (MAPK) pathway, particularly BRAF-V600E mutation. White male children are mostly affected, with a peak incidence of 1-3 years of age. Nine out of 10 children with cutaneous involvement also have multisystemic disease, such as the case of our patient. LCH is classified as single or multisystem organ disease. Two-thirds of the cases present with single system involvement. Organs most commonly affected include the bone (the skull being the most commonly affected), skin, and high-risk organs like the liver, spleen, and bone marrow, and less commonly the lungs, lymph nodes, and central nervous system. Some patients can present with fever, lethargy, and weight loss. None were noted in our patient.
Skin findings of LCH can have multiple morphologies and presentations and often described as a big mimicker. In young infants like our patient, the seborrheic dermatitis–mimicking type is often seen. In other cases, the skin lesions can appear eczematous, petechial, with scabbing, crusting, or purpura. Xanthoma-like lesions, like that one our patient had in the back, have also been described. Resistant diaper dermatitis and cradle cap should prompt the clinician to think about LCH. Lesions can be so varied that can present with hypopigmentation (vitiligo like), hyperpigmentation, varicella-like papulo-pustules, and red blue nodules within others. Oral mucosa and nail involvement can also occur.
Bone involvement can present as soft-tissue mass with swelling and pain as it occur in our patient.
Endocrinopathies have been described in patients with LCH including diabetes insipidus, growth hormone deficiency, and less likely thyroid disease.
Multidisciplinary care
The diagnosis of LCH in infants necessitates a combination of clinical, radiological, and histopathologic findings. In infants, cutaneous involvement is a frequent initial presentation, with characteristic lesions that are often misdiagnosed as other dermatologic conditions. Timely recognition of these lesions and appropriate skin biopsies for histological examination are essential steps in achieving an accurate diagnosis.
Radiological imaging, including x-rays, CT, and MRI, plays a crucial role in assessing the extent of involvement.
The management of LCH in infants requires a well-coordinated multidisciplinary approach involving pediatric oncologists, dermatologists, radiologists, orthopedic surgeons, and other relevant specialists. Treatment strategies vary depending on the extent of disease involvement and the presence of risk factors. In localized cases, observation with close monitoring may be considered, as some cases of LCH in infants may undergo spontaneous regression. However, cases with severe symptoms, extensive organ involvement, or high-risk features may require systemic therapies.
Chemotherapy agents, including vinblastine and prednisone have been utilized in the treatment of infantile LCH with varying success. The selection of treatment regimens should be tailored to each individual case, considering disease severity, potential toxicities, and long-term effects. In cases of bone lesions causing significant deformities or functional impairment, surgical intervention may be necessary. Skin only disease can be treated with topical corticosteroids.
Prognosis
Survival rates in patients with single-organ involvement without risk-organ involvement is close to 100% and with risk-organ involvement of 98% at 5 years.
Long-term follow-up is essential for infants diagnosed with LCH, as recurrence and late effects can occur even after successful treatment. Continued monitoring allows for the timely detection of relapses or the development of secondary complications.
Infants thought to have common skin conditions like eczema, seborrheic dermatitis, or diaper dermatitis not responding to treatment should be referred to pediatric dermatology for evaluation to rule out the possibility of LCH.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Krooks J et al. J Am Acad Dermatol. 2018 Jun;78(6):1035-44.
Krooks J et al. J Am Acad Dermatol. 2018 Jun;78(6):1047-56.
Leung AKC et al. World J Pediatr. 2019 Dec;15(6):536-45.
At the week follow-up, the lesions were unchanged and the swelling on the left lateral eyebrow was worsening. A biopsy of the yellow lesion on the back and one of the scaly papules on the abdomen was performed. A fungal and bacterial cultures were also ordered.
He was referred to ophthalmology for evaluation of the eyelid swelling and an ultrasound was requested.
The skin biopsy showed a clonal proliferation of reniform histiocytes with eosinophils within the dermis. The cells were positive for S100, CD207 (langerin), and CD1a and negative for pancytokeratin and Melan-A, supportive of the diagnosis of Langerhans cell histiocytosis (LCH).
Diagnosis
The patient was admitted to the hospital, where a skeletal survey was performed, which showed an asymmetric lucency involving the left frontal calvarium extending to the superior lateral orbital rim. The brain MRI demonstrated a destructive avidly enhancing soft-tissue process which involved the superior left orbital rim likely with some degree of intracranial extension. This lesion exerts mass effect upon surrounding structures to the left ocular globe. With the skin and skeletal findings, the patient was diagnosed with LCH. His blood count was significant for thrombocytopenia. His liver and kidney function were normal. His electrolytes were also with in normal range. He was started on chemotherapy with vinblastine and systemic corticosteroids with resolution of the rash and decrease on the size of the lesion on the orbit within a few weeks.
Infantile LCH is a rare neoplastic disorder of hematopoietic myeloid precursor cells caused by activating mutations in the mitogen-activated protein kinase (MAPK) pathway, particularly BRAF-V600E mutation. White male children are mostly affected, with a peak incidence of 1-3 years of age. Nine out of 10 children with cutaneous involvement also have multisystemic disease, such as the case of our patient. LCH is classified as single or multisystem organ disease. Two-thirds of the cases present with single system involvement. Organs most commonly affected include the bone (the skull being the most commonly affected), skin, and high-risk organs like the liver, spleen, and bone marrow, and less commonly the lungs, lymph nodes, and central nervous system. Some patients can present with fever, lethargy, and weight loss. None were noted in our patient.
Skin findings of LCH can have multiple morphologies and presentations and often described as a big mimicker. In young infants like our patient, the seborrheic dermatitis–mimicking type is often seen. In other cases, the skin lesions can appear eczematous, petechial, with scabbing, crusting, or purpura. Xanthoma-like lesions, like that one our patient had in the back, have also been described. Resistant diaper dermatitis and cradle cap should prompt the clinician to think about LCH. Lesions can be so varied that can present with hypopigmentation (vitiligo like), hyperpigmentation, varicella-like papulo-pustules, and red blue nodules within others. Oral mucosa and nail involvement can also occur.
Bone involvement can present as soft-tissue mass with swelling and pain as it occur in our patient.
Endocrinopathies have been described in patients with LCH including diabetes insipidus, growth hormone deficiency, and less likely thyroid disease.
Multidisciplinary care
The diagnosis of LCH in infants necessitates a combination of clinical, radiological, and histopathologic findings. In infants, cutaneous involvement is a frequent initial presentation, with characteristic lesions that are often misdiagnosed as other dermatologic conditions. Timely recognition of these lesions and appropriate skin biopsies for histological examination are essential steps in achieving an accurate diagnosis.
Radiological imaging, including x-rays, CT, and MRI, plays a crucial role in assessing the extent of involvement.
The management of LCH in infants requires a well-coordinated multidisciplinary approach involving pediatric oncologists, dermatologists, radiologists, orthopedic surgeons, and other relevant specialists. Treatment strategies vary depending on the extent of disease involvement and the presence of risk factors. In localized cases, observation with close monitoring may be considered, as some cases of LCH in infants may undergo spontaneous regression. However, cases with severe symptoms, extensive organ involvement, or high-risk features may require systemic therapies.
Chemotherapy agents, including vinblastine and prednisone have been utilized in the treatment of infantile LCH with varying success. The selection of treatment regimens should be tailored to each individual case, considering disease severity, potential toxicities, and long-term effects. In cases of bone lesions causing significant deformities or functional impairment, surgical intervention may be necessary. Skin only disease can be treated with topical corticosteroids.
Prognosis
Survival rates in patients with single-organ involvement without risk-organ involvement is close to 100% and with risk-organ involvement of 98% at 5 years.
Long-term follow-up is essential for infants diagnosed with LCH, as recurrence and late effects can occur even after successful treatment. Continued monitoring allows for the timely detection of relapses or the development of secondary complications.
Infants thought to have common skin conditions like eczema, seborrheic dermatitis, or diaper dermatitis not responding to treatment should be referred to pediatric dermatology for evaluation to rule out the possibility of LCH.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Krooks J et al. J Am Acad Dermatol. 2018 Jun;78(6):1035-44.
Krooks J et al. J Am Acad Dermatol. 2018 Jun;78(6):1047-56.
Leung AKC et al. World J Pediatr. 2019 Dec;15(6):536-45.
A 4-month male was referred to the pediatric dermatology clinic for a rash on the scalp, torso, and the diaper area since he was 2 months of age. He has been treated with nystatin, clotrimazole, and zinc oxide paste with partial improvement. After 2 months of partial improvement the rash worsened again, and he was referred to pediatric dermatology. The mother also reported asymptomatic left upper lateral eyebrow swelling noted a few weeks prior.
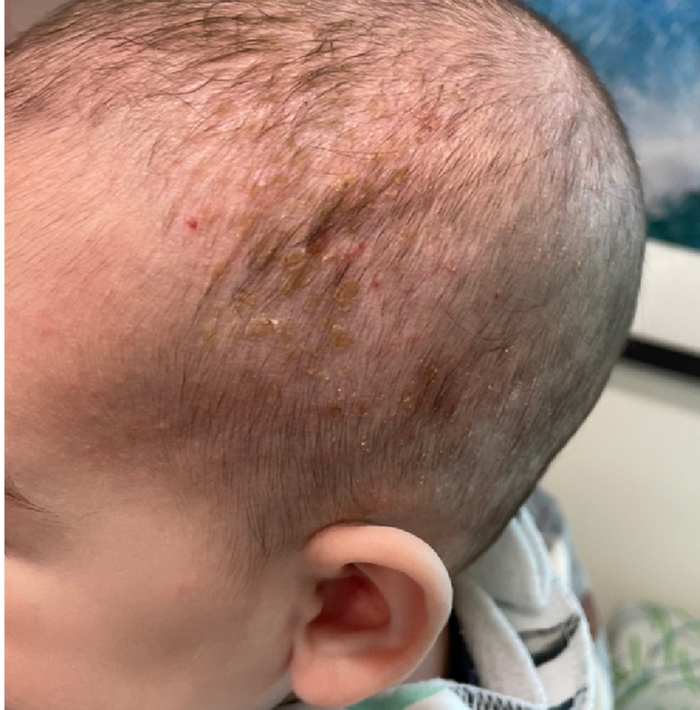
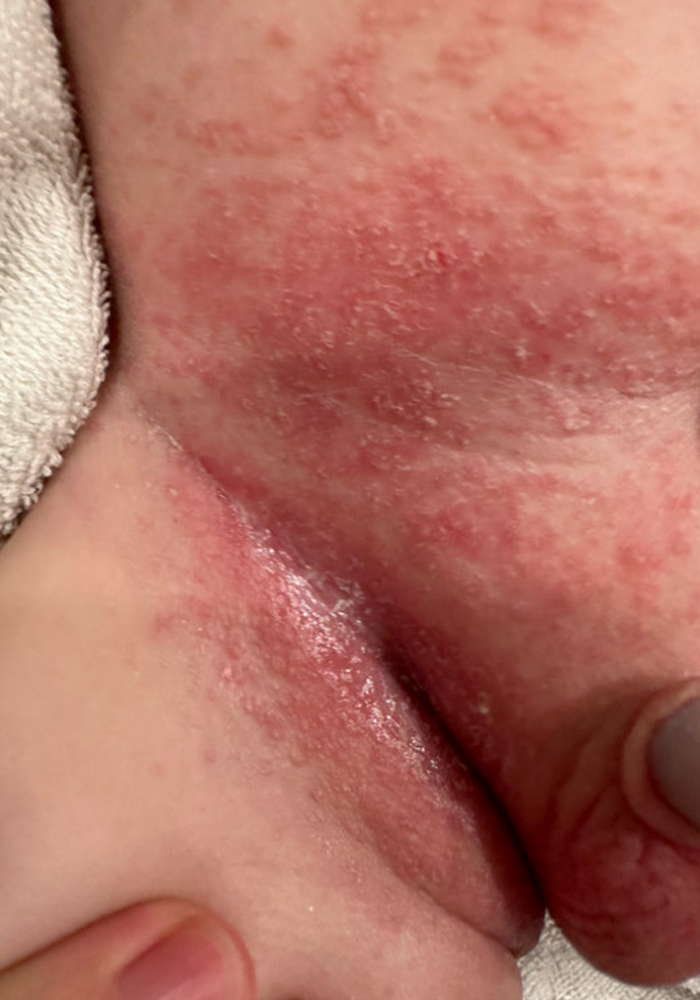
On the torso and diaper area, he had multiple scaly pink papules. On the groin he had eroded pink scaly plaques (Picture 2).
On his back he had a 3-mm yellow papule (Picture 3).
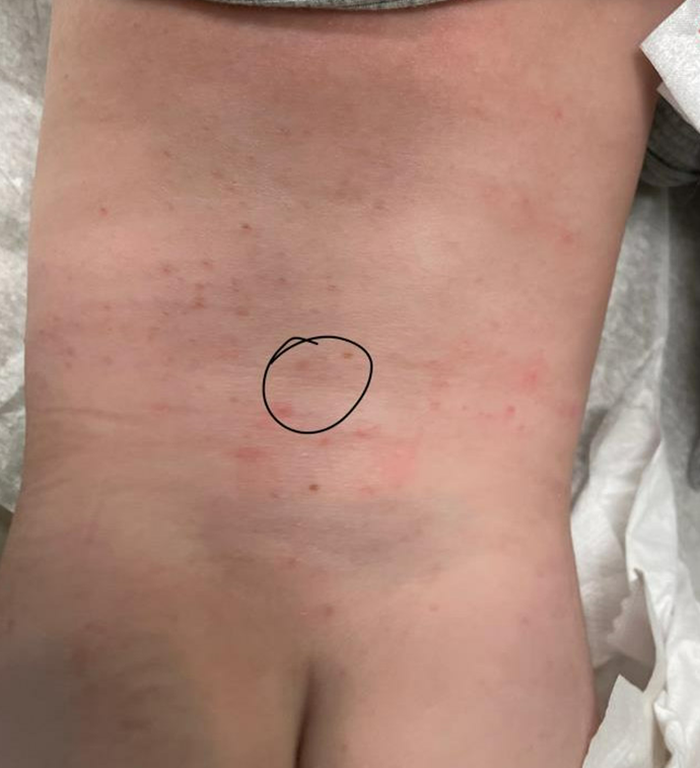
A 7-year-old male has a bumpy rash on the chin for several months
Given the presentation and the unique location of the lesions he was diagnosed with follicular keratosis of the chin (FKC).
This is a rare and poorly understood condition that can be present in older children and young teenagers. In the cases reported by Kanzaki et al.1 were two boys who presented with the condition; it was thought to be associated with rubbing of the chin with their hands when watching TV or reading. The author described improvement with habit change. This condition is usually described in boys, and some cases presented in brothers,2 suggesting a genetic predisposition. Some reports lack a history of rubbing or trauma to the area.
Histopathologic evaluation of the lesions demonstrates dilated hair follicles containing keratotic basophilic material without any signs of inflammation.
The lesions can be confused with keratosis pilaris (KP). Keratosis pilaris can be described in association with atopic dermatitis and ichthyosis, which were not present in our patient. The lesions usually present on the sides of the cheeks and lateral region of the arms and legs. Compared with follicular keratosis, KP lesions usually present with associated perifollicular erythema. Our patient did not present with lesions on the cheeks or the sides of the arms or legs. Milia can present on the chin of children, usually if there is history of rubbing or trauma, or on a scar. Milia are micro keratin cysts, usually seen in areas of the face. Lichen spinulous is described as rough small follicular papules that present in oval or circular patches that can grow up to 5 cm and spread rapidly. They usually present on the extensor surfaces of the extremities, neck, abdomen, and knees. These lesions are thought to be secondary to infections, have been associated with atopy, and have been seen in patients with atopic dermatitis. There is a probable genetic predisposition. The lesions are usually treated with gentle soaps and moisturizer containing keratolytics like urea or salicylic acid, and in some cases topical retinoids can also be tried. Follicular mucinosis can also present similarly to keratosis follicularis. The lesions present as scaly plaques or as grouped skin color papules on the face, scalp, or the neck that can also be associated with hair loss. Sometimes a biopsy needs to be done to be able to distinguish it from follicular keratosis. There is an increase of mucin around hair follicles and sebaceous glands with associated inflammation and degeneration of the follicular structures. In patients with primary follicular mucinosis the lesions can resolve spontaneously in a couple of years. Lesions can be treated with topical corticosteroids, oral antibiotics like macrolides or tetracyclines, dapsone, and phototherapy.
KFC can be treated with vitamin D analogues. It is usually unresponsive to corticosteroids, keratolytic lotions, and retinoids. Our patient was prescribed calcipotriene.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego
References
1. Kanzaki T et al. J Am Acad Dermatol. 1992;26(1):134-5.
2. Buechner AA et al. JAMA Dermatol. 2018 Jan 1;154(1):111-2.
Given the presentation and the unique location of the lesions he was diagnosed with follicular keratosis of the chin (FKC).
This is a rare and poorly understood condition that can be present in older children and young teenagers. In the cases reported by Kanzaki et al.1 were two boys who presented with the condition; it was thought to be associated with rubbing of the chin with their hands when watching TV or reading. The author described improvement with habit change. This condition is usually described in boys, and some cases presented in brothers,2 suggesting a genetic predisposition. Some reports lack a history of rubbing or trauma to the area.
Histopathologic evaluation of the lesions demonstrates dilated hair follicles containing keratotic basophilic material without any signs of inflammation.
The lesions can be confused with keratosis pilaris (KP). Keratosis pilaris can be described in association with atopic dermatitis and ichthyosis, which were not present in our patient. The lesions usually present on the sides of the cheeks and lateral region of the arms and legs. Compared with follicular keratosis, KP lesions usually present with associated perifollicular erythema. Our patient did not present with lesions on the cheeks or the sides of the arms or legs. Milia can present on the chin of children, usually if there is history of rubbing or trauma, or on a scar. Milia are micro keratin cysts, usually seen in areas of the face. Lichen spinulous is described as rough small follicular papules that present in oval or circular patches that can grow up to 5 cm and spread rapidly. They usually present on the extensor surfaces of the extremities, neck, abdomen, and knees. These lesions are thought to be secondary to infections, have been associated with atopy, and have been seen in patients with atopic dermatitis. There is a probable genetic predisposition. The lesions are usually treated with gentle soaps and moisturizer containing keratolytics like urea or salicylic acid, and in some cases topical retinoids can also be tried. Follicular mucinosis can also present similarly to keratosis follicularis. The lesions present as scaly plaques or as grouped skin color papules on the face, scalp, or the neck that can also be associated with hair loss. Sometimes a biopsy needs to be done to be able to distinguish it from follicular keratosis. There is an increase of mucin around hair follicles and sebaceous glands with associated inflammation and degeneration of the follicular structures. In patients with primary follicular mucinosis the lesions can resolve spontaneously in a couple of years. Lesions can be treated with topical corticosteroids, oral antibiotics like macrolides or tetracyclines, dapsone, and phototherapy.
KFC can be treated with vitamin D analogues. It is usually unresponsive to corticosteroids, keratolytic lotions, and retinoids. Our patient was prescribed calcipotriene.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego
References
1. Kanzaki T et al. J Am Acad Dermatol. 1992;26(1):134-5.
2. Buechner AA et al. JAMA Dermatol. 2018 Jan 1;154(1):111-2.
Given the presentation and the unique location of the lesions he was diagnosed with follicular keratosis of the chin (FKC).
This is a rare and poorly understood condition that can be present in older children and young teenagers. In the cases reported by Kanzaki et al.1 were two boys who presented with the condition; it was thought to be associated with rubbing of the chin with their hands when watching TV or reading. The author described improvement with habit change. This condition is usually described in boys, and some cases presented in brothers,2 suggesting a genetic predisposition. Some reports lack a history of rubbing or trauma to the area.
Histopathologic evaluation of the lesions demonstrates dilated hair follicles containing keratotic basophilic material without any signs of inflammation.
The lesions can be confused with keratosis pilaris (KP). Keratosis pilaris can be described in association with atopic dermatitis and ichthyosis, which were not present in our patient. The lesions usually present on the sides of the cheeks and lateral region of the arms and legs. Compared with follicular keratosis, KP lesions usually present with associated perifollicular erythema. Our patient did not present with lesions on the cheeks or the sides of the arms or legs. Milia can present on the chin of children, usually if there is history of rubbing or trauma, or on a scar. Milia are micro keratin cysts, usually seen in areas of the face. Lichen spinulous is described as rough small follicular papules that present in oval or circular patches that can grow up to 5 cm and spread rapidly. They usually present on the extensor surfaces of the extremities, neck, abdomen, and knees. These lesions are thought to be secondary to infections, have been associated with atopy, and have been seen in patients with atopic dermatitis. There is a probable genetic predisposition. The lesions are usually treated with gentle soaps and moisturizer containing keratolytics like urea or salicylic acid, and in some cases topical retinoids can also be tried. Follicular mucinosis can also present similarly to keratosis follicularis. The lesions present as scaly plaques or as grouped skin color papules on the face, scalp, or the neck that can also be associated with hair loss. Sometimes a biopsy needs to be done to be able to distinguish it from follicular keratosis. There is an increase of mucin around hair follicles and sebaceous glands with associated inflammation and degeneration of the follicular structures. In patients with primary follicular mucinosis the lesions can resolve spontaneously in a couple of years. Lesions can be treated with topical corticosteroids, oral antibiotics like macrolides or tetracyclines, dapsone, and phototherapy.
KFC can be treated with vitamin D analogues. It is usually unresponsive to corticosteroids, keratolytic lotions, and retinoids. Our patient was prescribed calcipotriene.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego
References
1. Kanzaki T et al. J Am Acad Dermatol. 1992;26(1):134-5.
2. Buechner AA et al. JAMA Dermatol. 2018 Jan 1;154(1):111-2.
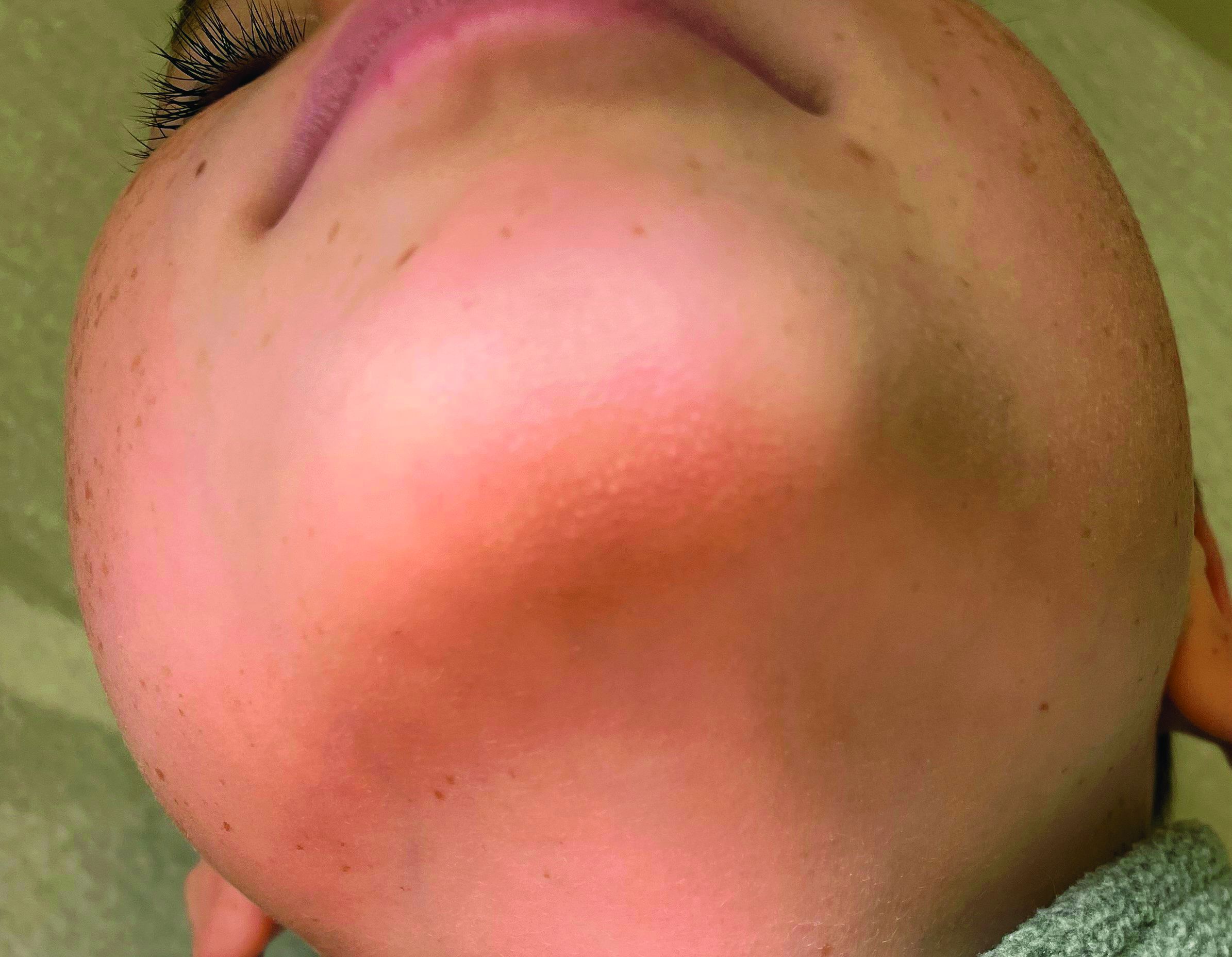
He is a healthy child with no past medical history. He is not taking any medications.
On physical exam he has follicular hyperkeratotic papules on the chin. No lesions on the axilla or thighs.