User login
Pharmacoresistant epilepsy: From pathogenesis to current and emerging therapies
Although more than 10 new antiepileptic drugs have been developed in the past decade, epilepsy remains resistant to drug therapy in about one-third of patients. Approximately 20% of patients with primary generalized epilepsy and up to 60% of patients who have focal epilepsy develop drug resistance during the course of their condition, which for many is lifelong.1
Those who get no response or only a partial response to drugs continue to have incapacitating seizures that lead to significant neuropsychiatric and social impairment, lower quality of life, greater morbidity, and a higher risk of death.
Managing these patients is a challenge and requires a structured multidisciplinary approach in specialized clinics. Newer research, particularly in pharmacogenomics, holds promise of therapy that more closely suits an individual’s profile and type of epilepsy.
This article reviews recent developments in the pathogenesis and treatment of pharmacoresistant epilepsy, placing these topics in clinical context to facilitate and enhance the physician’s ability to manage it.
THE COSTS OF RESISTANT EPILEPSY
A US study in the early 1990s estimated that the annual cost of refractory epilepsy in adults exceeds $11,745 per person2; the cost would be considerably higher today. Another study found that costs correlate with severity of illness and that patients who have intractable seizures incur a cost eight times greater than in those whose epilepsy is controlled.3
Higher risk of death
In any given interval of time, people with pharmacoresistant epilepsy are about two to 10 times more likely to die compared with the general population.4 The risk is inversely linked to seizure control.
“Sudden unexpected death in epilepsy”is the most frequent type of death in patients with pharmacoresistant epilepsy. This category excludes deaths from trauma or drowning. The death can be witnessed or unwitnessed and with or without evidence of a seizure (but not documented status epilepticus). Postmortem examination does not reveal a toxic or anatomic cause of death, and the underlying mechanisms remain unknown. However, the risk is closely associated with drug resistance (which manifests with uncontrolled convulsive seizures and need for polytherapy with antiepileptic drugs).5
Case-control studies have shown that the risk of sudden unexpected death is closely and inversely associated with seizure control; the rate is significantly higher in patients who have a higher frequency of convulsive seizures.6 In addition, freedom from seizures, achieved after successful epilepsy surgery, reduces the risk of death from all causes.7
Other causes of death in patients with epilepsy may be directly related to seizures (accidental trauma, drowning, burns) or to the underlying condition causing the seizures. Furthermore, people with epilepsy are at higher risk of suicide than the general population.
CONCEPT OF PHARMACORESISTANCE AS IT PERTAINS TO EPILEPSY
There is no uniformly accepted definition of pharmacoresistant epilepsy. Most studies defined it according to the number of antiepileptic drugs the patient had tried without success, the frequency of seizures, the duration of illness, and the period of remission. Its true definition awaits a better understanding of underlying mechanisms.
Nevertheless, a useful operational definition at present is failure to control seizures despite a trial of two or three drugs that are suitable for the type of epilepsy and have been appropriately prescribed at maximum tolerated doses. This is because the chances of controlling epilepsy decline sharply after failure of the second or third antiepileptic drug trial. In fact, some clinicians would argue against trying another antiepileptic drug in these patients, who may be candidates for surgical procedures that have high rates of success.8
Common causes of treatment failure, such as poor compliance or inappropriate selection of first-line antiepileptic drugs, should be addressed early on by the treating physician. Nonadherence to the prescribed regimen is a very common cause of uncontrolled seizures, so it is critical to maintain a good rapport with the patient and to inquire about reasons for noncompliance.
Factors that have been associated with treatment-resistant epilepsy include:
- Early onset of seizures
- Long history of poor seizure control
- Having more than one type of seizure
- Remote symptomatic etiology (eg, patients with a history of brain infection or head trauma)
- Certain structural abnormalities (eg, cortical dysplasia)
- Certain abnormalities on electroencephalography (EEG)
- Cognitive disability
- History of status epilepticus.
When to consider referral
A topic of debate is how long a patient must have active epilepsy before he or she is considered to have pharmacoresistance and should be referred to a specialist center.
Both the rate of remission and the time needed to achieve remission depend on multiple factors such as the type and etiology of the epilepsy and the definition of sustained intractability.
Importantly, the prognosis for most patients with newly diagnosed epilepsy, whether good or bad, becomes apparent within a few years of starting treatment. Although pharmacoresistant epilepsy will become refractory within 8 years in some patients, in others a second drug may not fail for more than 1 or 2 decades after diagnosis. Nonetheless, a history of a lack of a sustained seizure-free period for 12 consecutive months, in spite of two or three suitable and tolerated antiepileptic drugs, is a definite red flag for clinicians and should prompt referral to a specialist center.9,10 The National Association of Epilepsy Centers recommends referral to a specialized epilepsy center if seizure control is not achieved by a general neurologist within a period of 9 months.11

AN APPROACH TO PHARMACORESISTANT EPILEPSY FOR THE NONSPECIALIST
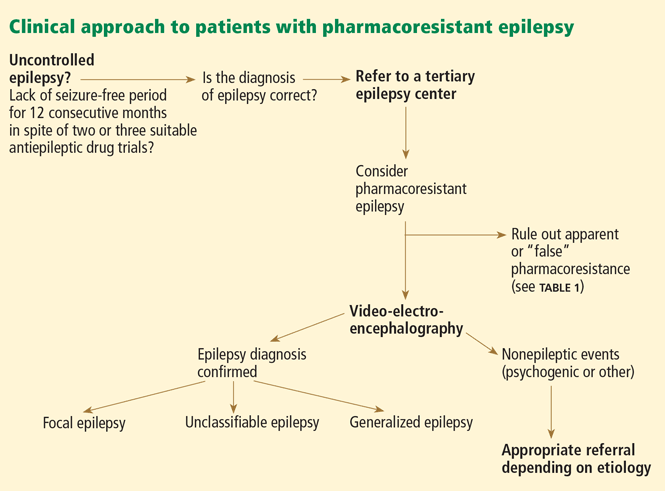
Review and confirm the diagnosis of epilepsy with the help of a careful history, video-EEG, and imaging.
When seizures cannot be controlled with drugs, it is important to verify that the events in question are indeed epileptic. Continuous video-EEG monitoring may be necessary to capture and characterize the clinical manifestations and corresponding EEG changes.14 When a typical spell is not associated with an EEG change, one can often make the diagnosis of nonepileptic events, which commonly are psychogenic nonepileptic seizures. Of note, however: scalp EEG may fail to pick up ictal EEG changes in focal seizures arising from a small or deeply situated focus: for example, as in focal sensorimotor seizures from restricted perirolandic cortex.15,16
Identify the cause, type of seizure or seizures, and syndromic classification, if any.
Review past and present medications, doses, efficacy, and side effects. Consider the possibility of drug interactions.
Different drugs may have different pharmacokinetic and dose-efficacy curves. With most of the newer-generation antiepileptic compounds such as lamotrigine (Lamictal), levetiracetam (Keppra), pregabalin (Lyrica), and topiramate (Topamax), efficacy may continue to increase in some patients as the dose is increased without reaching toxicity. An important exception is phenytoin (Dilantin): due to its nonlinear and saturable pharmacokinetics, even minor dose increases may lead to a large increase in phenytoin concentration (the drug accumulates as elimination becomes saturated). A high degree of individual variability exists, which is determined by factors that include the patient’s age, genetic and enzymatic profile, comorbidities, and concurrent medications.17 Understanding these relationships in individual patients facilitates dose selection and titration and enhances compliance. Testing serum drug levels may help when compliance is questionable.
Choose antiepileptic drugs primarily on the basis of the type of seizures and the individual clinical scenario: Which drug is likely to be most efficacious with the fewest side effects, and which one is appropriate for the patient’s comorbidities and concomitant medications?
When changing the dosage, withdrawing a drug, or adding a second antiepileptic drug, always do it in a systematic way, one step at a time. Reassess the response to the change before moving to the next step.
Discuss issues such as seizure precautions, lifestyle modifications, psychosocial dysfunction, and sudden unexpected death. Offer access to epilepsy-specialist nurses and epilepsy support groups such as those offered by the Epilepsy Foundation of America, the agency dedicated to the welfare of individuals with epilepsy in the United States (http://epilepsyfoundation.ning.com/).
PATTERNS OF DRUG RESISTANCE
Epidemiologic studies suggest three different patterns of drug resistance in epilepsy: de novo, progressive, and waxing-and-waning.
De novo drug resistance
In some patients, resistance is present from the time of onset of the very first seizure, before any antiepileptic drug is even started. One landmark study showed that patients with newly diagnosed epilepsy for whom the first drug was ineffective had only an 11% probability of future success, compared with 41% to 55% in patients who had had to stop taking the drug because of intolerable side effects or idiosyncratic reactions.10 Most patients for whom the first drug fails will be resistant to most and often all antiepileptic drugs.18 These results suggest that seizures in newly diagnosed patients are either easy to control or difficult to control right from the start.
Progressive drug resistance
In some patients, epilepsy is initially controlled but then gradually becomes refractory. This pattern may be seen, for example, in childhood epilepsies or in patients with hippocampal sclerosis.19,20
Waxing and waning resistance
In some patients, epilepsy has a waxing-and-waning pattern: ie, it alternates between a remitting (pharmacoresponsive) and relapsing (pharmacoresistant) course. Patients thought to have drug-resistant epilepsy may become seizure-free when other drugs are tried. Changes in drug bioavailability, local concentration of the drug in the brain, receptor changes, the development of tolerance, and interactions with new medications may be implicated, though the exact mechanism is not understood.21
BIOLOGIC BASIS OF PHARMACORESISTANT EPILEPSY
Pharmacoresistance is not unique to epilepsy: it is now recognized in diverse brain disorders, including depression and schizophrenia,22 and in other diseases affecting the brain, such as human immunodeficiency virus infection and many forms of cancer.23
Multiple drug resistance is characterized by insensitivity to a broad spectrum of drugs that presumably act on different receptors and by different mechanisms.24
Conceptually, the variable response to antiepileptic drugs can be attributed to factors related to the disease, the patient, and the drugs, or to other unknown factors. These factors are not mutually exclusive and may be either constitutive or acquired during the course of the disease.
Factors related to the disease (independent of the host)
These factors include etiology, epilepsy progression resulting in persistent changes of the epileptogenic network, and alterations of drug targets (ie, the “target” or pharmacodynamic hypothesis that reduced sensitivity to antiepileptic drugs is due to seizure-related alterations of specific drug targets25,26) or drug uptake into the brain27 (the “transporter” or pharmacokinetic hypothesis that the drugs are ineffective due to intrinsic or acquired overexpression of multidrug transporter proteins that hamper local drug delivery to target sites).28
Factors related to the drugs
Several drug-related factors have been implicated, such as the development of tolerance, lack of antiepileptogenic (disease-modifying) actions to interrupt the ongoing process of epileptogenesis rather than only suppressing seizures, and paucity of drugs with specific mechanisms of action tailored to difficult-tocontrol epilepsies.29
Patient characteristics
Variability in response (efficacy and adverse effects) to each antiepileptic drug can be due to interindividual differences in any of four interrelated fundamental factors: DNA, RNA, proteins, or metabolites. The field of study that aims to assess the effect of DNA variations (genotype) on a patient’s clinical response to a drug (phenotype) is known as pharmacogenetics. Age-related changes in pharmacokinetic and pharmacodynamic variables may contribute to age-dependent pharmacoresistance. Least studied are environmental factors that may play a role in the development or expression of pharmacoresistance.
NONPHARMACOLOGIC TREATMENTS
Ketogenic diet
The ketogenic diet, an important nonpharmacologic alternative, is usually reserved for young patients with difficult-to-control seizures. Originally developed almost a century ago, the diet mimics the biochemical changes associated with starvation. It is a strict regimen, high in fat and low in carbohydrate and protein (typically in a ratio of 4:1 or 3:1 in adolescents and very young children).
Such a strict regimen is difficult to implement and maintain and requires close supervision by a dietician and physician. In addition to the practical complexities, concerns also exist about the long-term effects of the diet on the child’s growth and overall health. For these reasons, the ketogenic diet is restricted to a small group of young patients with pharmacoresistant epilepsy and is not usually used long-term. There are few data indicating when it is appropriate to terminate the diet in patients who have a favorable response, but most clinicians wean the patient after 2 to 3 years.
Reports on the use of the ketogenic diet in adults are scarce, although benefit was seen in a small series.30 No long-term follow-up data exist for adults, especially regarding the risk of atherosclerosis.
Vagus nerve stimulation
Vagus nerve stimulation is a nonpharmacologic alternative for adults and for adolescents over age 12 years who have intractable focal seizures and who are not favorable surgical candidates. 31 Its effectiveness in younger patients and in those who have intractable generalized seizures is less clear, although published uncontrolled series have reported benefit (fewer seizures and better quality of life).32–34
A device consisting of a pulse generator is implanted subcutaneously in the precordium, and a lead wire is tunneled under the skin and attached to the left vagus nerve. The generator is programmed using a telemetry wand held over the device, with settings for current intensity (typically 1–2 mA), pulse width (250–300 μsec), frequency (30 Hz), and “duty cycle” (typically 30 seconds on stimulation, followed by 3 to 5 minutes off, cycling 24 hours/day). Hence, it provides “open-loop stimulation,” ie, continuous stimulation that is not modified in response to the patient’s EEG seizure activity. Patients or caregivers can also activate the device manually (“on demand”) at the first sign or warning of an impending seizure by swiping a handheld magnet.
Common side effects such as cough, voice alteration, and hoarseness are usually stimulation-dependent and tend to diminish with time. Notably, vagus nerve stimulation has none of the cognitive side effects often encountered with increasing doses of antiepileptic drugs. As with other implantable stimulators, some safety concerns exist in patients undergoing magnetic resonance imaging (MRI).
At least one-third of patients who receive this treatment show a sustained response, defined as a 50% or greater reduction in seizures. However, few achieve freedom from seizures, and therefore this therapy is considered palliative and is reserved for patients who are not candidates for surgery or for whom surgery has failed.
Unfortunately, it has not been possible to predict which patients will benefit from vagus nerve stimulation. The American Academy of Neurology recommends that this treatment be considered only after a thorough evaluation by a subspecialist to rule out nonepileptic conditions, false pharmacoresistance, and surgically treatable types of epilepsy.31
IS THE PATIENT A CANDIDATE FOR EPILEPSY SURGERY?
- Is the epilepsy diagnosis correct?
- Is the epilepsy focal? Have the following possibilities been excluded: generalized or multifocal epilepsy, situational or provoked seizures, or an epilepsy syndrome with spontaneous remission?
- Do seizures remain poorly controlled despite adequate pharmacologic trials?
- If so, do the seizures or medication side effects significantly affect the patient’s quality of life?
- Can an epileptogenic lesion be seen on MRI, and what is the suspected etiology?
- Is there converging evidence for a single epileptogenic focus?
- Are there abnormalities elsewhere in the brain?
- What are the chances of a good outcome in terms of seizure control and improvement in quality of life?
- What are the risks of surgery, and how do these compare with the risks of not having surgery?
- What are the patient’s perceptions and attitudes toward epilepsy surgery?
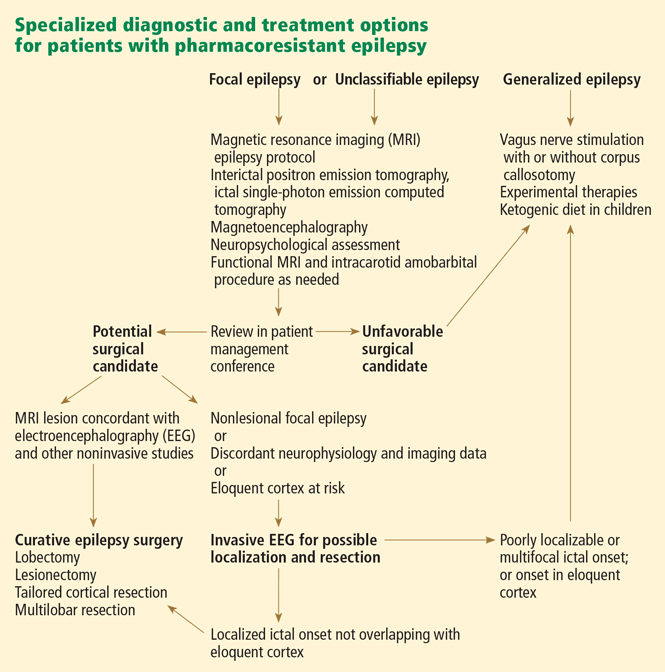
Surgical recommendations should be made after a thorough discussion of all preoperative data in a multidisciplinary patient management conference (Figure 1, Figure 2), in which epileptologists, neurosurgeons, neuroradiologists, neuropsychologists, and psychiatrists all actively participate.
Preoperative counseling is essential for the patient and his or her family, addressing the goals, risks, and benefits of the surgery. Treatment decisions should take into account the possible impact of surgery on the patient’s medical and psychosocial circumstances (risks of ongoing seizures vs surgical intervention; impact on the patient’s independence, employment status, emotional well-being, and psychiatric and other comorbidities).
EPILEPSY SURGERY: CURATIVE OR PALLIATIVE
Epilepsy surgery can be classified as curative or palliative, depending on the goal.
Curative procedures
Curative procedures include lobectomy, lesionectomy, and multilobar or hemispheric surgery (hemispherectomy).
Anterior temporal lobectomy and hippocampectomy are used for temporal lobe epilepsy and have several variations.
“Mesial temporal lobe epilepsy associated with hippocampal sclerosis,” a recognizable syndrome with complex partial seizures, stereotyped electroclinical features, and fairly typical natural history, is the most common of the focal epilepsies in adults.36 Its prognosis is poor when treated medically, but it responds well to surgery.37,38 In a landmark prospective trial, 58% of 40 patients randomized to surgical treatment were free of disabling seizures after 1 year, compared with only 8% of 40 patients treated medically.39
Lesionectomy and lobectomy are resective approaches targeting seizure foci outside the temporal lobe (most often in the frontal lobe, less commonly in the parietal or occipital lobes) or within the temporal lobe but outside the hippocampus (neocortical temporal lobe epilepsies).
The nature of the underlying substrate plays a significant role in determining the natural history,40 surgical strategy, and prognosis for freedom from seizures postoperatively. Patients with seizures due to structural lesions that are visible on MRI (“lesional epilepsies,” eg, cavernous angiomas or circumscribed lowgrade tumors) may become seizure-free after limited resections targeting the lesion itself (lesionectomy) or extending to involve part of a lobe or an entire lobe (lobectomy).
A favorable surgical outcome is much more likely if the lesion can be completely removed.41 At times, however, the lesion cannot be resected in its entirety, for example if it is located within inaccessible or essential (eloquent) cortex.
On the other hand, identifying the epileptogenic focus in patients with no visible structural abnormality on MRI (“nonlesional epilepsies”) can be challenging and usually requires intracranial investigations. In this instance, the aim of surgery is to resect regions that are electrographically abnormal. In general, the postoperative outcome is less favorable in nonlesional focal epilepsies than in lesional epilepsies.42
Multilobar resections and hemispherectomy are indicated when seizures arise from extensive, diffuse, or multiple regions of a single hemisphere.
If the neurologic function supported by the abnormal hemisphere is intact, a tailored multilobar resection aims at eliminating the epileptogenic focus without creating new deficits.
If, however, the underlying hemispheric abnormality is associated with significant contralateral hemiparesis, hemiplegia, or visual field deficits, the need to preserve function does not limit surgery, and hemispherectomy can be considered. Hemispherectomy can be the procedure of choice for selected infants and young children with catastrophic epilepsies and unilateral brain damage.43,44 The goal is to control seizures by completely disconnecting the abnormal epileptogenic hemisphere from the opposite, “good” hemisphere. A second major goal is to improve psychosocial and cognitive development by eliminating the child’s uncontrolled seizures.
Palliative procedures
Palliative procedures, in contrast to curative ones, rarely eliminate seizures entirely. It is important to determine that patients are not candidates for a more definitive, potentially curative resective procedure before considering palliative surgical options such as corpus callosotomy, multiple subpial transections, or vagus nerve stimulation.
Corpus callosotomy (transection of the corpus callosum) is performed in a small number of patients, ie, those who have disabling seizures that rapidly become generalized or injurious drop attacks and are not candidates for focal resection. By disconnecting the two hemispheres, this procedure aims to hinder the fast interhemispheric spread of seizure discharges.
Callosotomy may be complete or involve only a portion of the corpus callosum. The extent of resection has been correlated with favorable outcome.45
Some investigators report a 50% or greater reduction in seizure frequency, with drop attacks and generalized tonic-clonic seizures showing the most consistent improvement. In addition, behavior and quality of life may also improve.46
Multiple subpial transections are reserved for seizures arising from eloquent cortex (ie, from areas that cannot be removed without incurring unacceptable neurologic deficits). Therefore, the surgeon only transects the epileptogenic cortex in a vertical manner, so as to interrupt the horizontal cortical connections without resection. This approach is thought to disrupt the synchrony of seizure propagation while preserving physiologic function.
A meta-analysis of small case series suggests some decrease in seizure frequency with no or minimal neurologic compromise in up to 60% of patients.47
COMPLICATIONS OF EPILEPSY SURGERY
Resective surgery is not without risk, but often the risk is much less than that posed by uncontrolled epilepsy in the long term. Operative mortality rates vary from almost zero for temporal lobe surgery to 2.5% for hemispherectomy. The reported risk of permanent surgical morbidity varies by type of surgery from 1.1% for temporal lobe resection to about 5% for frontal lobe resection.4
NOVEL EPILEPSY THERAPIES
The failure of available antiepileptic drugs to control seizures in a substantial number of patients underscores the need to develop novel therapies such as electrical stimulation, local drug delivery, cell transplantation, and genebased therapies.48 Future targeted therapies could be coupled to seizure-forecasting systems to create “smart” implantable devices that predict, detect, and preemptively treat the seizures in a “closed-loop” fashion.
Targeted electrical stimulation
To modulate abnormal cortical hyperexcitability, electrical stimulation can be applied to the peripheral nervous system (eg, vagus nerve stimulation) or central nervous system. Central nervous system stimulation can be broadly divided into two approaches:
Direct stimulation targets presumed epileptogenic brain tissue such as the neocortex or hippocampus.
Indirect stimulation targets presumed seizure-gating networks such as in the cerebellum and various deep brain nuclei in the basal ganglia or thalamus (deep brain stimulation), which are believed to play a central role in modulating the synchronization and propagation of seizure activity.
Systematic, well-designed studies are currently under way. The budding field of electrical stimulation faces a number of challenges, which include optimizing stimulation variables and target sites, selecting favorable candidates, validating long-term safety and efficacy, evaluating long-term effects on tissue reorganization, plasticity, and epileptogenicity, and developing reliable algorithms for seizure detection and prediction.
Local drug delivery
Direct delivery of drugs into the epileptogenic brain tissue holds promise, particularly for patients whose foci cannot be surgically removed. By bypassing the systemic circulation, this approach has the potential to avoid systemic and even whole-brain side effects.
However, only a few proof-of-principle experiments have been conducted in animals, and to date no clinical study has explored the utility of intraparenchymal or intraventricular antiepileptic drug delivery in humans.
Cell and gene therapies
The emerging field of experimental cell- and gene-based neuropharmacology holds promise for location-specific therapeutic strategies. In ex vivo gene therapy, bioengineered cells capable of delivering anticonvulsant compounds might be transplanted into specific areas of the brain. On the other hand, in vivo gene therapy would involve delivering genes by viral vectors to induce the localized production of antiepileptic compounds in situ.
Endogenous anticonvulsants such as gamma-aminobutyric acid (GABA) and adenosine have been tried in various animal experiments. 49 Before they can be applied clinically, significant questions need to be addressed, including the potential for toxicity or maladaptive plasticity and long-term therapeutic safety and efficacy.
Cell transplantation is aimed at restoring the physiologic balance of neurotransmitters, and is currently being investigated for the treatment of several neurologic disorders such as Parkinson disease and Huntington disease.50 Unlike delivery of exogenous compounds, cell transplantation (heterologous fetal cell grafts or embryonic or adult stem cells) has the potential to form restorative synaptic connections and assimilate within existing cells and networks in the host tissue. An essential limitation to xenotransplantation in humans is the risk of immunologic rejection.
The future
We hope that continued progress in genomics will lead to targeted development of disease-modifying drugs that can impede or reverse the process of epileptogenesis. Advances in informatics and genetics may be harnessed to predict which patients are likely to develop pharmacoresistance, to cure certain genetic epilepsies, and to individualize antiepileptic drug selection on the basis of each person’s genetic profile.
- Siegel AM. Presurgical evaluation and surgical treatment of medically refractory epilepsy. Neurosurg Rev 2004; 27:1–18.
- Murray MI, Halpern MT, Leppik IE. Cost of refractory epilepsy in adults in the USA. Epilepsy Res 1996; 23:139–148.
- Jacoby A, Buck D, Baker G, McNamee P, Graham-Jones S, Chadwick D. Uptake and costs of care for epilepsy: findings from a U.K. regional study. Epilepsia 1998; 39:776–786.
- Chapell R, Reston J, Snyder D, Treadwell J, Treager S, Turkelson C. Management of treatment-resistant epilepsy. Evid Rep Technol Assess (Summ) 2003 Apr; 77:1–8.
- Nei M, Bagla R. Seizure-related injury and death. Curr Neurol Neurosci Rep 2007; 7:335–341.
- Langan Y, Nashef L, Sander JW. Case-control study of SUDEP. Neurology 2005; 64:1131–1133.
- Sperling MR, Feldman H, Kinman J, Liporace JD, O’Connor MJ. Seizure control and mortality in epilepsy. Ann Neurol 1999; 46:45–50.
- Berg AT. Understanding the delay before epilepsy surgery: who develops intractable focal epilepsy and when? CNS Spectr 2004; 9:136–144.
- Perucca E. Pharmacoresistance in epilepsy: how should it be defined? CNS Drugs 1998; 10:171–179.
- Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med 2000; 342:314–319.
- Gumnit RJ, Walczak TS; National Association of Epilepsy Centers. Guidelines for essential services, personnel, and facilities in specialized epilepsy centers in the United States. Epilepsia 2001; 42:804–814.
- Smith D, Defalla BA, Chadwick DW. The misdiagnosis of epilepsy and the management of refractory epilepsy in a specialist clinic. QJM 1999; 92:15–23.
- Schuele SU, Lüders HO. Intractable epilepsy: management and therapeutic alternatives. Lancet Neurol 2008; 7:514–524.
- Engel J, Burchfiel J, Ebersole J, et al. Long-term monitoring for epilepsy. Report of an IFCN committee. Electroencephalogr Clin Neurophysiol 1993; 87:437–458.
- Kanner AM, Morris HH, Lüders H, et al. Supplementary motor seizures mimicking pseudoseizures: some clinical differences. Neurology 1990; 40:1404–1407.
- Alexopoulos AV, Dinner DS. Focal motor seizures, epilepsia partialis continua, and supplementary sensorimotor seizures. In:Wyllie E, Gupta A, Lachhwani DK, editors. The Treatment of Epilepsy: Principles & Practice. Philadelphia, PA: Lippincott Williams & Wilkins, 2006:257–277.
- French JA. Refractory epilepsy: clinical overview. Epilepsia 2007; 48(suppl 1):3–7.
- Regesta G, Tanganelli P. Clinical aspects and biological bases of drug-resistant epilepsies. Epilepsy Res 1999; 34:109–122.
- Berg AT, Langfitt J, Shinnar S, et al. How long does it take for partial epilepsy to become intractable? Neurology 2003; 60:186–190.
- Berg AT, Vickrey BG, Testa FM, et al. How long does it take for epilepsy to become intractable? A prospective investigation. Ann Neurol 2006; 60:73–79.
- Löscher W, Schmidt D. Experimental and clinical evidence for loss of effect (tolerance) during prolonged treatment with antiepileptic drugs. Epilepsia 2006; 47:1253–1284.
- Löscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci 2005; 6:591–602.
- Siddiqui A, Kerb R, Weale ME, et al. Association of multidrug resistance in epilepsy with a polymorphism in the drug-transporter gene ABCB1. N Engl J Med 2003; 348:1442–1448.
- Granata T, Marchi N, Carlton E, et al. Management of the patient with medically refractory epilepsy. Expert Rev Neurother 2009; 9:1791–1802.
- Marchi N, Hallene KL, Kight KM, et al. Significance of MDR1 and multiple drug resistance in refractory human epileptic brain. BMC Med 2004; 2:37.
- Oby E, Janigro D. The blood-brain barrier and epilepsy. Epilepsia 2006; 47:1761–1774.
- Schmidt D, Löscher W. New developments in antiepileptic drug resistance: an integrative view. Epilepsy Curr 2009; 9:47–52.
- Remy S, Beck H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain 2006; 129:18–35.
- Löscher W, Schmidt D. New horizons in the development of antiepileptic drugs. Epilepsy Res 2002; 50:3–16.
- Sirven J, Whedon B, Caplan D, et al. The ketogenic diet for intractable epilepsy in adults: preliminary results. Epilepsia 1999; 40:1721–1726.
- Fisher RS, Handforth A. Reassessment: vagus nerve stimulation for epilepsy: a report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 1999; 53:666–669.
- Hallböök T, Lundgren J, Stjernqvist K, Blennow G, Strömblad LG, Rosén I. Vagus nerve stimulation in 15 children with therapy resistant epilepsy; its impact on cognition, quality of life, behaviour and mood. Seizure 2005; 14:504–513.
- Holmes MD, Silbergeld DL, Drouhard D, Wilensky AJ, Ojemann LM. Effect of vagus nerve stimulation on adults with pharmacoresistant generalized epilepsy syndromes. Seizure 2004; 13:340–345.
- Murphy JV, Torkelson R, Dowler I, Simon S, Hudson S. Vagal nerve stimulation in refractory epilepsy: the first 100 patients receiving vagal nerve stimulation at a pediatric epilepsy center. Arch Pediatr Adolesc Med 2003; 157:560–564.
- Alexopoulos AV, Najm IM. Neurosurgical management of focal epilepsies in adults. In:Panayiotopoulos CP, et al, editors. Focal Epilepsies: Seizures, Syndromes and Management. Oxford, UK: Medicinae; 2009:204–220.
- Wieser HGILAE Commission on Neurosurgery of Epilepsy. ILAE Commission Report. Mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsia 2004; 45:695–714.
- Engel J. Surgery for seizures. N Engl J Med 1996; 334:647–652.
- Engel J, Wiebe S, French J, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: temporal lobe and localized neocortical resections for epilepsy: report of the Quality Standards Subcommittee of the American Academy of Neurology, in association with the American Epilepsy Society and the American Association of Neurological Surgeons. Neurology 2003; 60:538–547.
- Wiebe S, Blume WT, Girvin JP, Eliasziw M; Effectiveness and Efficiency of Surgery for Temporal Lobe Epilepsy Study Group. A randomized, controlled trial of surgery for temporal-lobe epilepsy. N Engl J Med 2001; 345:311–318.
- Semah F, Picot MC, Adam C, et al. Is the underlying cause of epilepsy a major prognostic factor for recurrence? Neurology 1998; 51:1256–1262.
- Awad IA, Rosenfeld J, Ahl J, Hahn JF, Lüders H. Intractable epilepsy and structural lesions of the brain: mapping, resection strategies, and seizure outcome. Epilepsia 1991; 32:179–186.
- Cascino GD. Surgical treatment for extratemporal epilepsy. Curr Treat Options Neurol 2004; 6:257–262.
- González-Martínez JA, Gupta A, Kotagal P, et al. Hemispherectomy for catastrophic epilepsy in infants. Epilepsia 2005; 46:1518–1525.
- Wyllie E. Surgery for catastrophic localization-related epilepsy in infants. Epilepsia 1996; 37(suppl 1):S22–S25.
- Tanriverdi T, Olivier A, Poulin N, Andermann F, Dubeau F. Long-term seizure outcome after corpus callosotomy: a retrospective analysis of 95 patients. J Neurosurg 2009; 110:332–342.
- Asadi-Pooya AA, Sharan A, Nei M, Sperling MR. Corpus callosotomy. Epilepsy Behav 2008; 13:271–278.
- Spencer SS, Schramm J, Wyler A, et al. Multiple subpial transection for intractable partial epilepsy: an international meta-analysis. Epilepsia 2002; 43:141–145.
- Alexopoulos AV, Gonugunta V, Yang J, Boulis NM. Electrical stimulation and gene-based neuromodulation for control of medically-refractory epilepsy. Acta Neurochir Suppl 2007; 97:293–309.
- Detlev B. Cell and gene therapies for refractory epilepsy. Curr Neuropharmacol 2007; 5:115–125.
- Fisher RS, Ho J. Potential new methods for antiepileptic drug delivery. CNS Drugs 2002; 16:579–593.
Although more than 10 new antiepileptic drugs have been developed in the past decade, epilepsy remains resistant to drug therapy in about one-third of patients. Approximately 20% of patients with primary generalized epilepsy and up to 60% of patients who have focal epilepsy develop drug resistance during the course of their condition, which for many is lifelong.1
Those who get no response or only a partial response to drugs continue to have incapacitating seizures that lead to significant neuropsychiatric and social impairment, lower quality of life, greater morbidity, and a higher risk of death.
Managing these patients is a challenge and requires a structured multidisciplinary approach in specialized clinics. Newer research, particularly in pharmacogenomics, holds promise of therapy that more closely suits an individual’s profile and type of epilepsy.
This article reviews recent developments in the pathogenesis and treatment of pharmacoresistant epilepsy, placing these topics in clinical context to facilitate and enhance the physician’s ability to manage it.
THE COSTS OF RESISTANT EPILEPSY
A US study in the early 1990s estimated that the annual cost of refractory epilepsy in adults exceeds $11,745 per person2; the cost would be considerably higher today. Another study found that costs correlate with severity of illness and that patients who have intractable seizures incur a cost eight times greater than in those whose epilepsy is controlled.3
Higher risk of death
In any given interval of time, people with pharmacoresistant epilepsy are about two to 10 times more likely to die compared with the general population.4 The risk is inversely linked to seizure control.
“Sudden unexpected death in epilepsy”is the most frequent type of death in patients with pharmacoresistant epilepsy. This category excludes deaths from trauma or drowning. The death can be witnessed or unwitnessed and with or without evidence of a seizure (but not documented status epilepticus). Postmortem examination does not reveal a toxic or anatomic cause of death, and the underlying mechanisms remain unknown. However, the risk is closely associated with drug resistance (which manifests with uncontrolled convulsive seizures and need for polytherapy with antiepileptic drugs).5
Case-control studies have shown that the risk of sudden unexpected death is closely and inversely associated with seizure control; the rate is significantly higher in patients who have a higher frequency of convulsive seizures.6 In addition, freedom from seizures, achieved after successful epilepsy surgery, reduces the risk of death from all causes.7
Other causes of death in patients with epilepsy may be directly related to seizures (accidental trauma, drowning, burns) or to the underlying condition causing the seizures. Furthermore, people with epilepsy are at higher risk of suicide than the general population.
CONCEPT OF PHARMACORESISTANCE AS IT PERTAINS TO EPILEPSY
There is no uniformly accepted definition of pharmacoresistant epilepsy. Most studies defined it according to the number of antiepileptic drugs the patient had tried without success, the frequency of seizures, the duration of illness, and the period of remission. Its true definition awaits a better understanding of underlying mechanisms.
Nevertheless, a useful operational definition at present is failure to control seizures despite a trial of two or three drugs that are suitable for the type of epilepsy and have been appropriately prescribed at maximum tolerated doses. This is because the chances of controlling epilepsy decline sharply after failure of the second or third antiepileptic drug trial. In fact, some clinicians would argue against trying another antiepileptic drug in these patients, who may be candidates for surgical procedures that have high rates of success.8
Common causes of treatment failure, such as poor compliance or inappropriate selection of first-line antiepileptic drugs, should be addressed early on by the treating physician. Nonadherence to the prescribed regimen is a very common cause of uncontrolled seizures, so it is critical to maintain a good rapport with the patient and to inquire about reasons for noncompliance.
Factors that have been associated with treatment-resistant epilepsy include:
- Early onset of seizures
- Long history of poor seizure control
- Having more than one type of seizure
- Remote symptomatic etiology (eg, patients with a history of brain infection or head trauma)
- Certain structural abnormalities (eg, cortical dysplasia)
- Certain abnormalities on electroencephalography (EEG)
- Cognitive disability
- History of status epilepticus.
When to consider referral
A topic of debate is how long a patient must have active epilepsy before he or she is considered to have pharmacoresistance and should be referred to a specialist center.
Both the rate of remission and the time needed to achieve remission depend on multiple factors such as the type and etiology of the epilepsy and the definition of sustained intractability.
Importantly, the prognosis for most patients with newly diagnosed epilepsy, whether good or bad, becomes apparent within a few years of starting treatment. Although pharmacoresistant epilepsy will become refractory within 8 years in some patients, in others a second drug may not fail for more than 1 or 2 decades after diagnosis. Nonetheless, a history of a lack of a sustained seizure-free period for 12 consecutive months, in spite of two or three suitable and tolerated antiepileptic drugs, is a definite red flag for clinicians and should prompt referral to a specialist center.9,10 The National Association of Epilepsy Centers recommends referral to a specialized epilepsy center if seizure control is not achieved by a general neurologist within a period of 9 months.11
AN APPROACH TO PHARMACORESISTANT EPILEPSY FOR THE NONSPECIALIST
Review and confirm the diagnosis of epilepsy with the help of a careful history, video-EEG, and imaging.
When seizures cannot be controlled with drugs, it is important to verify that the events in question are indeed epileptic. Continuous video-EEG monitoring may be necessary to capture and characterize the clinical manifestations and corresponding EEG changes.14 When a typical spell is not associated with an EEG change, one can often make the diagnosis of nonepileptic events, which commonly are psychogenic nonepileptic seizures. Of note, however: scalp EEG may fail to pick up ictal EEG changes in focal seizures arising from a small or deeply situated focus: for example, as in focal sensorimotor seizures from restricted perirolandic cortex.15,16
Identify the cause, type of seizure or seizures, and syndromic classification, if any.
Review past and present medications, doses, efficacy, and side effects. Consider the possibility of drug interactions.
Different drugs may have different pharmacokinetic and dose-efficacy curves. With most of the newer-generation antiepileptic compounds such as lamotrigine (Lamictal), levetiracetam (Keppra), pregabalin (Lyrica), and topiramate (Topamax), efficacy may continue to increase in some patients as the dose is increased without reaching toxicity. An important exception is phenytoin (Dilantin): due to its nonlinear and saturable pharmacokinetics, even minor dose increases may lead to a large increase in phenytoin concentration (the drug accumulates as elimination becomes saturated). A high degree of individual variability exists, which is determined by factors that include the patient’s age, genetic and enzymatic profile, comorbidities, and concurrent medications.17 Understanding these relationships in individual patients facilitates dose selection and titration and enhances compliance. Testing serum drug levels may help when compliance is questionable.
Choose antiepileptic drugs primarily on the basis of the type of seizures and the individual clinical scenario: Which drug is likely to be most efficacious with the fewest side effects, and which one is appropriate for the patient’s comorbidities and concomitant medications?
When changing the dosage, withdrawing a drug, or adding a second antiepileptic drug, always do it in a systematic way, one step at a time. Reassess the response to the change before moving to the next step.
Discuss issues such as seizure precautions, lifestyle modifications, psychosocial dysfunction, and sudden unexpected death. Offer access to epilepsy-specialist nurses and epilepsy support groups such as those offered by the Epilepsy Foundation of America, the agency dedicated to the welfare of individuals with epilepsy in the United States (http://epilepsyfoundation.ning.com/).
PATTERNS OF DRUG RESISTANCE
Epidemiologic studies suggest three different patterns of drug resistance in epilepsy: de novo, progressive, and waxing-and-waning.
De novo drug resistance
In some patients, resistance is present from the time of onset of the very first seizure, before any antiepileptic drug is even started. One landmark study showed that patients with newly diagnosed epilepsy for whom the first drug was ineffective had only an 11% probability of future success, compared with 41% to 55% in patients who had had to stop taking the drug because of intolerable side effects or idiosyncratic reactions.10 Most patients for whom the first drug fails will be resistant to most and often all antiepileptic drugs.18 These results suggest that seizures in newly diagnosed patients are either easy to control or difficult to control right from the start.
Progressive drug resistance
In some patients, epilepsy is initially controlled but then gradually becomes refractory. This pattern may be seen, for example, in childhood epilepsies or in patients with hippocampal sclerosis.19,20
Waxing and waning resistance
In some patients, epilepsy has a waxing-and-waning pattern: ie, it alternates between a remitting (pharmacoresponsive) and relapsing (pharmacoresistant) course. Patients thought to have drug-resistant epilepsy may become seizure-free when other drugs are tried. Changes in drug bioavailability, local concentration of the drug in the brain, receptor changes, the development of tolerance, and interactions with new medications may be implicated, though the exact mechanism is not understood.21
BIOLOGIC BASIS OF PHARMACORESISTANT EPILEPSY
Pharmacoresistance is not unique to epilepsy: it is now recognized in diverse brain disorders, including depression and schizophrenia,22 and in other diseases affecting the brain, such as human immunodeficiency virus infection and many forms of cancer.23
Multiple drug resistance is characterized by insensitivity to a broad spectrum of drugs that presumably act on different receptors and by different mechanisms.24
Conceptually, the variable response to antiepileptic drugs can be attributed to factors related to the disease, the patient, and the drugs, or to other unknown factors. These factors are not mutually exclusive and may be either constitutive or acquired during the course of the disease.
Factors related to the disease (independent of the host)
These factors include etiology, epilepsy progression resulting in persistent changes of the epileptogenic network, and alterations of drug targets (ie, the “target” or pharmacodynamic hypothesis that reduced sensitivity to antiepileptic drugs is due to seizure-related alterations of specific drug targets25,26) or drug uptake into the brain27 (the “transporter” or pharmacokinetic hypothesis that the drugs are ineffective due to intrinsic or acquired overexpression of multidrug transporter proteins that hamper local drug delivery to target sites).28
Factors related to the drugs
Several drug-related factors have been implicated, such as the development of tolerance, lack of antiepileptogenic (disease-modifying) actions to interrupt the ongoing process of epileptogenesis rather than only suppressing seizures, and paucity of drugs with specific mechanisms of action tailored to difficult-tocontrol epilepsies.29
Patient characteristics
Variability in response (efficacy and adverse effects) to each antiepileptic drug can be due to interindividual differences in any of four interrelated fundamental factors: DNA, RNA, proteins, or metabolites. The field of study that aims to assess the effect of DNA variations (genotype) on a patient’s clinical response to a drug (phenotype) is known as pharmacogenetics. Age-related changes in pharmacokinetic and pharmacodynamic variables may contribute to age-dependent pharmacoresistance. Least studied are environmental factors that may play a role in the development or expression of pharmacoresistance.
NONPHARMACOLOGIC TREATMENTS
Ketogenic diet
The ketogenic diet, an important nonpharmacologic alternative, is usually reserved for young patients with difficult-to-control seizures. Originally developed almost a century ago, the diet mimics the biochemical changes associated with starvation. It is a strict regimen, high in fat and low in carbohydrate and protein (typically in a ratio of 4:1 or 3:1 in adolescents and very young children).
Such a strict regimen is difficult to implement and maintain and requires close supervision by a dietician and physician. In addition to the practical complexities, concerns also exist about the long-term effects of the diet on the child’s growth and overall health. For these reasons, the ketogenic diet is restricted to a small group of young patients with pharmacoresistant epilepsy and is not usually used long-term. There are few data indicating when it is appropriate to terminate the diet in patients who have a favorable response, but most clinicians wean the patient after 2 to 3 years.
Reports on the use of the ketogenic diet in adults are scarce, although benefit was seen in a small series.30 No long-term follow-up data exist for adults, especially regarding the risk of atherosclerosis.
Vagus nerve stimulation
Vagus nerve stimulation is a nonpharmacologic alternative for adults and for adolescents over age 12 years who have intractable focal seizures and who are not favorable surgical candidates. 31 Its effectiveness in younger patients and in those who have intractable generalized seizures is less clear, although published uncontrolled series have reported benefit (fewer seizures and better quality of life).32–34
A device consisting of a pulse generator is implanted subcutaneously in the precordium, and a lead wire is tunneled under the skin and attached to the left vagus nerve. The generator is programmed using a telemetry wand held over the device, with settings for current intensity (typically 1–2 mA), pulse width (250–300 μsec), frequency (30 Hz), and “duty cycle” (typically 30 seconds on stimulation, followed by 3 to 5 minutes off, cycling 24 hours/day). Hence, it provides “open-loop stimulation,” ie, continuous stimulation that is not modified in response to the patient’s EEG seizure activity. Patients or caregivers can also activate the device manually (“on demand”) at the first sign or warning of an impending seizure by swiping a handheld magnet.
Common side effects such as cough, voice alteration, and hoarseness are usually stimulation-dependent and tend to diminish with time. Notably, vagus nerve stimulation has none of the cognitive side effects often encountered with increasing doses of antiepileptic drugs. As with other implantable stimulators, some safety concerns exist in patients undergoing magnetic resonance imaging (MRI).
At least one-third of patients who receive this treatment show a sustained response, defined as a 50% or greater reduction in seizures. However, few achieve freedom from seizures, and therefore this therapy is considered palliative and is reserved for patients who are not candidates for surgery or for whom surgery has failed.
Unfortunately, it has not been possible to predict which patients will benefit from vagus nerve stimulation. The American Academy of Neurology recommends that this treatment be considered only after a thorough evaluation by a subspecialist to rule out nonepileptic conditions, false pharmacoresistance, and surgically treatable types of epilepsy.31
IS THE PATIENT A CANDIDATE FOR EPILEPSY SURGERY?
- Is the epilepsy diagnosis correct?
- Is the epilepsy focal? Have the following possibilities been excluded: generalized or multifocal epilepsy, situational or provoked seizures, or an epilepsy syndrome with spontaneous remission?
- Do seizures remain poorly controlled despite adequate pharmacologic trials?
- If so, do the seizures or medication side effects significantly affect the patient’s quality of life?
- Can an epileptogenic lesion be seen on MRI, and what is the suspected etiology?
- Is there converging evidence for a single epileptogenic focus?
- Are there abnormalities elsewhere in the brain?
- What are the chances of a good outcome in terms of seizure control and improvement in quality of life?
- What are the risks of surgery, and how do these compare with the risks of not having surgery?
- What are the patient’s perceptions and attitudes toward epilepsy surgery?
Surgical recommendations should be made after a thorough discussion of all preoperative data in a multidisciplinary patient management conference (Figure 1, Figure 2), in which epileptologists, neurosurgeons, neuroradiologists, neuropsychologists, and psychiatrists all actively participate.
Preoperative counseling is essential for the patient and his or her family, addressing the goals, risks, and benefits of the surgery. Treatment decisions should take into account the possible impact of surgery on the patient’s medical and psychosocial circumstances (risks of ongoing seizures vs surgical intervention; impact on the patient’s independence, employment status, emotional well-being, and psychiatric and other comorbidities).
EPILEPSY SURGERY: CURATIVE OR PALLIATIVE
Epilepsy surgery can be classified as curative or palliative, depending on the goal.
Curative procedures
Curative procedures include lobectomy, lesionectomy, and multilobar or hemispheric surgery (hemispherectomy).
Anterior temporal lobectomy and hippocampectomy are used for temporal lobe epilepsy and have several variations.
“Mesial temporal lobe epilepsy associated with hippocampal sclerosis,” a recognizable syndrome with complex partial seizures, stereotyped electroclinical features, and fairly typical natural history, is the most common of the focal epilepsies in adults.36 Its prognosis is poor when treated medically, but it responds well to surgery.37,38 In a landmark prospective trial, 58% of 40 patients randomized to surgical treatment were free of disabling seizures after 1 year, compared with only 8% of 40 patients treated medically.39
Lesionectomy and lobectomy are resective approaches targeting seizure foci outside the temporal lobe (most often in the frontal lobe, less commonly in the parietal or occipital lobes) or within the temporal lobe but outside the hippocampus (neocortical temporal lobe epilepsies).
The nature of the underlying substrate plays a significant role in determining the natural history,40 surgical strategy, and prognosis for freedom from seizures postoperatively. Patients with seizures due to structural lesions that are visible on MRI (“lesional epilepsies,” eg, cavernous angiomas or circumscribed lowgrade tumors) may become seizure-free after limited resections targeting the lesion itself (lesionectomy) or extending to involve part of a lobe or an entire lobe (lobectomy).
A favorable surgical outcome is much more likely if the lesion can be completely removed.41 At times, however, the lesion cannot be resected in its entirety, for example if it is located within inaccessible or essential (eloquent) cortex.
On the other hand, identifying the epileptogenic focus in patients with no visible structural abnormality on MRI (“nonlesional epilepsies”) can be challenging and usually requires intracranial investigations. In this instance, the aim of surgery is to resect regions that are electrographically abnormal. In general, the postoperative outcome is less favorable in nonlesional focal epilepsies than in lesional epilepsies.42
Multilobar resections and hemispherectomy are indicated when seizures arise from extensive, diffuse, or multiple regions of a single hemisphere.
If the neurologic function supported by the abnormal hemisphere is intact, a tailored multilobar resection aims at eliminating the epileptogenic focus without creating new deficits.
If, however, the underlying hemispheric abnormality is associated with significant contralateral hemiparesis, hemiplegia, or visual field deficits, the need to preserve function does not limit surgery, and hemispherectomy can be considered. Hemispherectomy can be the procedure of choice for selected infants and young children with catastrophic epilepsies and unilateral brain damage.43,44 The goal is to control seizures by completely disconnecting the abnormal epileptogenic hemisphere from the opposite, “good” hemisphere. A second major goal is to improve psychosocial and cognitive development by eliminating the child’s uncontrolled seizures.
Palliative procedures
Palliative procedures, in contrast to curative ones, rarely eliminate seizures entirely. It is important to determine that patients are not candidates for a more definitive, potentially curative resective procedure before considering palliative surgical options such as corpus callosotomy, multiple subpial transections, or vagus nerve stimulation.
Corpus callosotomy (transection of the corpus callosum) is performed in a small number of patients, ie, those who have disabling seizures that rapidly become generalized or injurious drop attacks and are not candidates for focal resection. By disconnecting the two hemispheres, this procedure aims to hinder the fast interhemispheric spread of seizure discharges.
Callosotomy may be complete or involve only a portion of the corpus callosum. The extent of resection has been correlated with favorable outcome.45
Some investigators report a 50% or greater reduction in seizure frequency, with drop attacks and generalized tonic-clonic seizures showing the most consistent improvement. In addition, behavior and quality of life may also improve.46
Multiple subpial transections are reserved for seizures arising from eloquent cortex (ie, from areas that cannot be removed without incurring unacceptable neurologic deficits). Therefore, the surgeon only transects the epileptogenic cortex in a vertical manner, so as to interrupt the horizontal cortical connections without resection. This approach is thought to disrupt the synchrony of seizure propagation while preserving physiologic function.
A meta-analysis of small case series suggests some decrease in seizure frequency with no or minimal neurologic compromise in up to 60% of patients.47
COMPLICATIONS OF EPILEPSY SURGERY
Resective surgery is not without risk, but often the risk is much less than that posed by uncontrolled epilepsy in the long term. Operative mortality rates vary from almost zero for temporal lobe surgery to 2.5% for hemispherectomy. The reported risk of permanent surgical morbidity varies by type of surgery from 1.1% for temporal lobe resection to about 5% for frontal lobe resection.4
NOVEL EPILEPSY THERAPIES
The failure of available antiepileptic drugs to control seizures in a substantial number of patients underscores the need to develop novel therapies such as electrical stimulation, local drug delivery, cell transplantation, and genebased therapies.48 Future targeted therapies could be coupled to seizure-forecasting systems to create “smart” implantable devices that predict, detect, and preemptively treat the seizures in a “closed-loop” fashion.
Targeted electrical stimulation
To modulate abnormal cortical hyperexcitability, electrical stimulation can be applied to the peripheral nervous system (eg, vagus nerve stimulation) or central nervous system. Central nervous system stimulation can be broadly divided into two approaches:
Direct stimulation targets presumed epileptogenic brain tissue such as the neocortex or hippocampus.
Indirect stimulation targets presumed seizure-gating networks such as in the cerebellum and various deep brain nuclei in the basal ganglia or thalamus (deep brain stimulation), which are believed to play a central role in modulating the synchronization and propagation of seizure activity.
Systematic, well-designed studies are currently under way. The budding field of electrical stimulation faces a number of challenges, which include optimizing stimulation variables and target sites, selecting favorable candidates, validating long-term safety and efficacy, evaluating long-term effects on tissue reorganization, plasticity, and epileptogenicity, and developing reliable algorithms for seizure detection and prediction.
Local drug delivery
Direct delivery of drugs into the epileptogenic brain tissue holds promise, particularly for patients whose foci cannot be surgically removed. By bypassing the systemic circulation, this approach has the potential to avoid systemic and even whole-brain side effects.
However, only a few proof-of-principle experiments have been conducted in animals, and to date no clinical study has explored the utility of intraparenchymal or intraventricular antiepileptic drug delivery in humans.
Cell and gene therapies
The emerging field of experimental cell- and gene-based neuropharmacology holds promise for location-specific therapeutic strategies. In ex vivo gene therapy, bioengineered cells capable of delivering anticonvulsant compounds might be transplanted into specific areas of the brain. On the other hand, in vivo gene therapy would involve delivering genes by viral vectors to induce the localized production of antiepileptic compounds in situ.
Endogenous anticonvulsants such as gamma-aminobutyric acid (GABA) and adenosine have been tried in various animal experiments. 49 Before they can be applied clinically, significant questions need to be addressed, including the potential for toxicity or maladaptive plasticity and long-term therapeutic safety and efficacy.
Cell transplantation is aimed at restoring the physiologic balance of neurotransmitters, and is currently being investigated for the treatment of several neurologic disorders such as Parkinson disease and Huntington disease.50 Unlike delivery of exogenous compounds, cell transplantation (heterologous fetal cell grafts or embryonic or adult stem cells) has the potential to form restorative synaptic connections and assimilate within existing cells and networks in the host tissue. An essential limitation to xenotransplantation in humans is the risk of immunologic rejection.
The future
We hope that continued progress in genomics will lead to targeted development of disease-modifying drugs that can impede or reverse the process of epileptogenesis. Advances in informatics and genetics may be harnessed to predict which patients are likely to develop pharmacoresistance, to cure certain genetic epilepsies, and to individualize antiepileptic drug selection on the basis of each person’s genetic profile.
Although more than 10 new antiepileptic drugs have been developed in the past decade, epilepsy remains resistant to drug therapy in about one-third of patients. Approximately 20% of patients with primary generalized epilepsy and up to 60% of patients who have focal epilepsy develop drug resistance during the course of their condition, which for many is lifelong.1
Those who get no response or only a partial response to drugs continue to have incapacitating seizures that lead to significant neuropsychiatric and social impairment, lower quality of life, greater morbidity, and a higher risk of death.
Managing these patients is a challenge and requires a structured multidisciplinary approach in specialized clinics. Newer research, particularly in pharmacogenomics, holds promise of therapy that more closely suits an individual’s profile and type of epilepsy.
This article reviews recent developments in the pathogenesis and treatment of pharmacoresistant epilepsy, placing these topics in clinical context to facilitate and enhance the physician’s ability to manage it.
THE COSTS OF RESISTANT EPILEPSY
A US study in the early 1990s estimated that the annual cost of refractory epilepsy in adults exceeds $11,745 per person2; the cost would be considerably higher today. Another study found that costs correlate with severity of illness and that patients who have intractable seizures incur a cost eight times greater than in those whose epilepsy is controlled.3
Higher risk of death
In any given interval of time, people with pharmacoresistant epilepsy are about two to 10 times more likely to die compared with the general population.4 The risk is inversely linked to seizure control.
“Sudden unexpected death in epilepsy”is the most frequent type of death in patients with pharmacoresistant epilepsy. This category excludes deaths from trauma or drowning. The death can be witnessed or unwitnessed and with or without evidence of a seizure (but not documented status epilepticus). Postmortem examination does not reveal a toxic or anatomic cause of death, and the underlying mechanisms remain unknown. However, the risk is closely associated with drug resistance (which manifests with uncontrolled convulsive seizures and need for polytherapy with antiepileptic drugs).5
Case-control studies have shown that the risk of sudden unexpected death is closely and inversely associated with seizure control; the rate is significantly higher in patients who have a higher frequency of convulsive seizures.6 In addition, freedom from seizures, achieved after successful epilepsy surgery, reduces the risk of death from all causes.7
Other causes of death in patients with epilepsy may be directly related to seizures (accidental trauma, drowning, burns) or to the underlying condition causing the seizures. Furthermore, people with epilepsy are at higher risk of suicide than the general population.
CONCEPT OF PHARMACORESISTANCE AS IT PERTAINS TO EPILEPSY
There is no uniformly accepted definition of pharmacoresistant epilepsy. Most studies defined it according to the number of antiepileptic drugs the patient had tried without success, the frequency of seizures, the duration of illness, and the period of remission. Its true definition awaits a better understanding of underlying mechanisms.
Nevertheless, a useful operational definition at present is failure to control seizures despite a trial of two or three drugs that are suitable for the type of epilepsy and have been appropriately prescribed at maximum tolerated doses. This is because the chances of controlling epilepsy decline sharply after failure of the second or third antiepileptic drug trial. In fact, some clinicians would argue against trying another antiepileptic drug in these patients, who may be candidates for surgical procedures that have high rates of success.8
Common causes of treatment failure, such as poor compliance or inappropriate selection of first-line antiepileptic drugs, should be addressed early on by the treating physician. Nonadherence to the prescribed regimen is a very common cause of uncontrolled seizures, so it is critical to maintain a good rapport with the patient and to inquire about reasons for noncompliance.
Factors that have been associated with treatment-resistant epilepsy include:
- Early onset of seizures
- Long history of poor seizure control
- Having more than one type of seizure
- Remote symptomatic etiology (eg, patients with a history of brain infection or head trauma)
- Certain structural abnormalities (eg, cortical dysplasia)
- Certain abnormalities on electroencephalography (EEG)
- Cognitive disability
- History of status epilepticus.
When to consider referral
A topic of debate is how long a patient must have active epilepsy before he or she is considered to have pharmacoresistance and should be referred to a specialist center.
Both the rate of remission and the time needed to achieve remission depend on multiple factors such as the type and etiology of the epilepsy and the definition of sustained intractability.
Importantly, the prognosis for most patients with newly diagnosed epilepsy, whether good or bad, becomes apparent within a few years of starting treatment. Although pharmacoresistant epilepsy will become refractory within 8 years in some patients, in others a second drug may not fail for more than 1 or 2 decades after diagnosis. Nonetheless, a history of a lack of a sustained seizure-free period for 12 consecutive months, in spite of two or three suitable and tolerated antiepileptic drugs, is a definite red flag for clinicians and should prompt referral to a specialist center.9,10 The National Association of Epilepsy Centers recommends referral to a specialized epilepsy center if seizure control is not achieved by a general neurologist within a period of 9 months.11
AN APPROACH TO PHARMACORESISTANT EPILEPSY FOR THE NONSPECIALIST
Review and confirm the diagnosis of epilepsy with the help of a careful history, video-EEG, and imaging.
When seizures cannot be controlled with drugs, it is important to verify that the events in question are indeed epileptic. Continuous video-EEG monitoring may be necessary to capture and characterize the clinical manifestations and corresponding EEG changes.14 When a typical spell is not associated with an EEG change, one can often make the diagnosis of nonepileptic events, which commonly are psychogenic nonepileptic seizures. Of note, however: scalp EEG may fail to pick up ictal EEG changes in focal seizures arising from a small or deeply situated focus: for example, as in focal sensorimotor seizures from restricted perirolandic cortex.15,16
Identify the cause, type of seizure or seizures, and syndromic classification, if any.
Review past and present medications, doses, efficacy, and side effects. Consider the possibility of drug interactions.
Different drugs may have different pharmacokinetic and dose-efficacy curves. With most of the newer-generation antiepileptic compounds such as lamotrigine (Lamictal), levetiracetam (Keppra), pregabalin (Lyrica), and topiramate (Topamax), efficacy may continue to increase in some patients as the dose is increased without reaching toxicity. An important exception is phenytoin (Dilantin): due to its nonlinear and saturable pharmacokinetics, even minor dose increases may lead to a large increase in phenytoin concentration (the drug accumulates as elimination becomes saturated). A high degree of individual variability exists, which is determined by factors that include the patient’s age, genetic and enzymatic profile, comorbidities, and concurrent medications.17 Understanding these relationships in individual patients facilitates dose selection and titration and enhances compliance. Testing serum drug levels may help when compliance is questionable.
Choose antiepileptic drugs primarily on the basis of the type of seizures and the individual clinical scenario: Which drug is likely to be most efficacious with the fewest side effects, and which one is appropriate for the patient’s comorbidities and concomitant medications?
When changing the dosage, withdrawing a drug, or adding a second antiepileptic drug, always do it in a systematic way, one step at a time. Reassess the response to the change before moving to the next step.
Discuss issues such as seizure precautions, lifestyle modifications, psychosocial dysfunction, and sudden unexpected death. Offer access to epilepsy-specialist nurses and epilepsy support groups such as those offered by the Epilepsy Foundation of America, the agency dedicated to the welfare of individuals with epilepsy in the United States (http://epilepsyfoundation.ning.com/).
PATTERNS OF DRUG RESISTANCE
Epidemiologic studies suggest three different patterns of drug resistance in epilepsy: de novo, progressive, and waxing-and-waning.
De novo drug resistance
In some patients, resistance is present from the time of onset of the very first seizure, before any antiepileptic drug is even started. One landmark study showed that patients with newly diagnosed epilepsy for whom the first drug was ineffective had only an 11% probability of future success, compared with 41% to 55% in patients who had had to stop taking the drug because of intolerable side effects or idiosyncratic reactions.10 Most patients for whom the first drug fails will be resistant to most and often all antiepileptic drugs.18 These results suggest that seizures in newly diagnosed patients are either easy to control or difficult to control right from the start.
Progressive drug resistance
In some patients, epilepsy is initially controlled but then gradually becomes refractory. This pattern may be seen, for example, in childhood epilepsies or in patients with hippocampal sclerosis.19,20
Waxing and waning resistance
In some patients, epilepsy has a waxing-and-waning pattern: ie, it alternates between a remitting (pharmacoresponsive) and relapsing (pharmacoresistant) course. Patients thought to have drug-resistant epilepsy may become seizure-free when other drugs are tried. Changes in drug bioavailability, local concentration of the drug in the brain, receptor changes, the development of tolerance, and interactions with new medications may be implicated, though the exact mechanism is not understood.21
BIOLOGIC BASIS OF PHARMACORESISTANT EPILEPSY
Pharmacoresistance is not unique to epilepsy: it is now recognized in diverse brain disorders, including depression and schizophrenia,22 and in other diseases affecting the brain, such as human immunodeficiency virus infection and many forms of cancer.23
Multiple drug resistance is characterized by insensitivity to a broad spectrum of drugs that presumably act on different receptors and by different mechanisms.24
Conceptually, the variable response to antiepileptic drugs can be attributed to factors related to the disease, the patient, and the drugs, or to other unknown factors. These factors are not mutually exclusive and may be either constitutive or acquired during the course of the disease.
Factors related to the disease (independent of the host)
These factors include etiology, epilepsy progression resulting in persistent changes of the epileptogenic network, and alterations of drug targets (ie, the “target” or pharmacodynamic hypothesis that reduced sensitivity to antiepileptic drugs is due to seizure-related alterations of specific drug targets25,26) or drug uptake into the brain27 (the “transporter” or pharmacokinetic hypothesis that the drugs are ineffective due to intrinsic or acquired overexpression of multidrug transporter proteins that hamper local drug delivery to target sites).28
Factors related to the drugs
Several drug-related factors have been implicated, such as the development of tolerance, lack of antiepileptogenic (disease-modifying) actions to interrupt the ongoing process of epileptogenesis rather than only suppressing seizures, and paucity of drugs with specific mechanisms of action tailored to difficult-tocontrol epilepsies.29
Patient characteristics
Variability in response (efficacy and adverse effects) to each antiepileptic drug can be due to interindividual differences in any of four interrelated fundamental factors: DNA, RNA, proteins, or metabolites. The field of study that aims to assess the effect of DNA variations (genotype) on a patient’s clinical response to a drug (phenotype) is known as pharmacogenetics. Age-related changes in pharmacokinetic and pharmacodynamic variables may contribute to age-dependent pharmacoresistance. Least studied are environmental factors that may play a role in the development or expression of pharmacoresistance.
NONPHARMACOLOGIC TREATMENTS
Ketogenic diet
The ketogenic diet, an important nonpharmacologic alternative, is usually reserved for young patients with difficult-to-control seizures. Originally developed almost a century ago, the diet mimics the biochemical changes associated with starvation. It is a strict regimen, high in fat and low in carbohydrate and protein (typically in a ratio of 4:1 or 3:1 in adolescents and very young children).
Such a strict regimen is difficult to implement and maintain and requires close supervision by a dietician and physician. In addition to the practical complexities, concerns also exist about the long-term effects of the diet on the child’s growth and overall health. For these reasons, the ketogenic diet is restricted to a small group of young patients with pharmacoresistant epilepsy and is not usually used long-term. There are few data indicating when it is appropriate to terminate the diet in patients who have a favorable response, but most clinicians wean the patient after 2 to 3 years.
Reports on the use of the ketogenic diet in adults are scarce, although benefit was seen in a small series.30 No long-term follow-up data exist for adults, especially regarding the risk of atherosclerosis.
Vagus nerve stimulation
Vagus nerve stimulation is a nonpharmacologic alternative for adults and for adolescents over age 12 years who have intractable focal seizures and who are not favorable surgical candidates. 31 Its effectiveness in younger patients and in those who have intractable generalized seizures is less clear, although published uncontrolled series have reported benefit (fewer seizures and better quality of life).32–34
A device consisting of a pulse generator is implanted subcutaneously in the precordium, and a lead wire is tunneled under the skin and attached to the left vagus nerve. The generator is programmed using a telemetry wand held over the device, with settings for current intensity (typically 1–2 mA), pulse width (250–300 μsec), frequency (30 Hz), and “duty cycle” (typically 30 seconds on stimulation, followed by 3 to 5 minutes off, cycling 24 hours/day). Hence, it provides “open-loop stimulation,” ie, continuous stimulation that is not modified in response to the patient’s EEG seizure activity. Patients or caregivers can also activate the device manually (“on demand”) at the first sign or warning of an impending seizure by swiping a handheld magnet.
Common side effects such as cough, voice alteration, and hoarseness are usually stimulation-dependent and tend to diminish with time. Notably, vagus nerve stimulation has none of the cognitive side effects often encountered with increasing doses of antiepileptic drugs. As with other implantable stimulators, some safety concerns exist in patients undergoing magnetic resonance imaging (MRI).
At least one-third of patients who receive this treatment show a sustained response, defined as a 50% or greater reduction in seizures. However, few achieve freedom from seizures, and therefore this therapy is considered palliative and is reserved for patients who are not candidates for surgery or for whom surgery has failed.
Unfortunately, it has not been possible to predict which patients will benefit from vagus nerve stimulation. The American Academy of Neurology recommends that this treatment be considered only after a thorough evaluation by a subspecialist to rule out nonepileptic conditions, false pharmacoresistance, and surgically treatable types of epilepsy.31
IS THE PATIENT A CANDIDATE FOR EPILEPSY SURGERY?
- Is the epilepsy diagnosis correct?
- Is the epilepsy focal? Have the following possibilities been excluded: generalized or multifocal epilepsy, situational or provoked seizures, or an epilepsy syndrome with spontaneous remission?
- Do seizures remain poorly controlled despite adequate pharmacologic trials?
- If so, do the seizures or medication side effects significantly affect the patient’s quality of life?
- Can an epileptogenic lesion be seen on MRI, and what is the suspected etiology?
- Is there converging evidence for a single epileptogenic focus?
- Are there abnormalities elsewhere in the brain?
- What are the chances of a good outcome in terms of seizure control and improvement in quality of life?
- What are the risks of surgery, and how do these compare with the risks of not having surgery?
- What are the patient’s perceptions and attitudes toward epilepsy surgery?
Surgical recommendations should be made after a thorough discussion of all preoperative data in a multidisciplinary patient management conference (Figure 1, Figure 2), in which epileptologists, neurosurgeons, neuroradiologists, neuropsychologists, and psychiatrists all actively participate.
Preoperative counseling is essential for the patient and his or her family, addressing the goals, risks, and benefits of the surgery. Treatment decisions should take into account the possible impact of surgery on the patient’s medical and psychosocial circumstances (risks of ongoing seizures vs surgical intervention; impact on the patient’s independence, employment status, emotional well-being, and psychiatric and other comorbidities).
EPILEPSY SURGERY: CURATIVE OR PALLIATIVE
Epilepsy surgery can be classified as curative or palliative, depending on the goal.
Curative procedures
Curative procedures include lobectomy, lesionectomy, and multilobar or hemispheric surgery (hemispherectomy).
Anterior temporal lobectomy and hippocampectomy are used for temporal lobe epilepsy and have several variations.
“Mesial temporal lobe epilepsy associated with hippocampal sclerosis,” a recognizable syndrome with complex partial seizures, stereotyped electroclinical features, and fairly typical natural history, is the most common of the focal epilepsies in adults.36 Its prognosis is poor when treated medically, but it responds well to surgery.37,38 In a landmark prospective trial, 58% of 40 patients randomized to surgical treatment were free of disabling seizures after 1 year, compared with only 8% of 40 patients treated medically.39
Lesionectomy and lobectomy are resective approaches targeting seizure foci outside the temporal lobe (most often in the frontal lobe, less commonly in the parietal or occipital lobes) or within the temporal lobe but outside the hippocampus (neocortical temporal lobe epilepsies).
The nature of the underlying substrate plays a significant role in determining the natural history,40 surgical strategy, and prognosis for freedom from seizures postoperatively. Patients with seizures due to structural lesions that are visible on MRI (“lesional epilepsies,” eg, cavernous angiomas or circumscribed lowgrade tumors) may become seizure-free after limited resections targeting the lesion itself (lesionectomy) or extending to involve part of a lobe or an entire lobe (lobectomy).
A favorable surgical outcome is much more likely if the lesion can be completely removed.41 At times, however, the lesion cannot be resected in its entirety, for example if it is located within inaccessible or essential (eloquent) cortex.
On the other hand, identifying the epileptogenic focus in patients with no visible structural abnormality on MRI (“nonlesional epilepsies”) can be challenging and usually requires intracranial investigations. In this instance, the aim of surgery is to resect regions that are electrographically abnormal. In general, the postoperative outcome is less favorable in nonlesional focal epilepsies than in lesional epilepsies.42
Multilobar resections and hemispherectomy are indicated when seizures arise from extensive, diffuse, or multiple regions of a single hemisphere.
If the neurologic function supported by the abnormal hemisphere is intact, a tailored multilobar resection aims at eliminating the epileptogenic focus without creating new deficits.
If, however, the underlying hemispheric abnormality is associated with significant contralateral hemiparesis, hemiplegia, or visual field deficits, the need to preserve function does not limit surgery, and hemispherectomy can be considered. Hemispherectomy can be the procedure of choice for selected infants and young children with catastrophic epilepsies and unilateral brain damage.43,44 The goal is to control seizures by completely disconnecting the abnormal epileptogenic hemisphere from the opposite, “good” hemisphere. A second major goal is to improve psychosocial and cognitive development by eliminating the child’s uncontrolled seizures.
Palliative procedures
Palliative procedures, in contrast to curative ones, rarely eliminate seizures entirely. It is important to determine that patients are not candidates for a more definitive, potentially curative resective procedure before considering palliative surgical options such as corpus callosotomy, multiple subpial transections, or vagus nerve stimulation.
Corpus callosotomy (transection of the corpus callosum) is performed in a small number of patients, ie, those who have disabling seizures that rapidly become generalized or injurious drop attacks and are not candidates for focal resection. By disconnecting the two hemispheres, this procedure aims to hinder the fast interhemispheric spread of seizure discharges.
Callosotomy may be complete or involve only a portion of the corpus callosum. The extent of resection has been correlated with favorable outcome.45
Some investigators report a 50% or greater reduction in seizure frequency, with drop attacks and generalized tonic-clonic seizures showing the most consistent improvement. In addition, behavior and quality of life may also improve.46
Multiple subpial transections are reserved for seizures arising from eloquent cortex (ie, from areas that cannot be removed without incurring unacceptable neurologic deficits). Therefore, the surgeon only transects the epileptogenic cortex in a vertical manner, so as to interrupt the horizontal cortical connections without resection. This approach is thought to disrupt the synchrony of seizure propagation while preserving physiologic function.
A meta-analysis of small case series suggests some decrease in seizure frequency with no or minimal neurologic compromise in up to 60% of patients.47
COMPLICATIONS OF EPILEPSY SURGERY
Resective surgery is not without risk, but often the risk is much less than that posed by uncontrolled epilepsy in the long term. Operative mortality rates vary from almost zero for temporal lobe surgery to 2.5% for hemispherectomy. The reported risk of permanent surgical morbidity varies by type of surgery from 1.1% for temporal lobe resection to about 5% for frontal lobe resection.4
NOVEL EPILEPSY THERAPIES
The failure of available antiepileptic drugs to control seizures in a substantial number of patients underscores the need to develop novel therapies such as electrical stimulation, local drug delivery, cell transplantation, and genebased therapies.48 Future targeted therapies could be coupled to seizure-forecasting systems to create “smart” implantable devices that predict, detect, and preemptively treat the seizures in a “closed-loop” fashion.
Targeted electrical stimulation
To modulate abnormal cortical hyperexcitability, electrical stimulation can be applied to the peripheral nervous system (eg, vagus nerve stimulation) or central nervous system. Central nervous system stimulation can be broadly divided into two approaches:
Direct stimulation targets presumed epileptogenic brain tissue such as the neocortex or hippocampus.
Indirect stimulation targets presumed seizure-gating networks such as in the cerebellum and various deep brain nuclei in the basal ganglia or thalamus (deep brain stimulation), which are believed to play a central role in modulating the synchronization and propagation of seizure activity.
Systematic, well-designed studies are currently under way. The budding field of electrical stimulation faces a number of challenges, which include optimizing stimulation variables and target sites, selecting favorable candidates, validating long-term safety and efficacy, evaluating long-term effects on tissue reorganization, plasticity, and epileptogenicity, and developing reliable algorithms for seizure detection and prediction.
Local drug delivery
Direct delivery of drugs into the epileptogenic brain tissue holds promise, particularly for patients whose foci cannot be surgically removed. By bypassing the systemic circulation, this approach has the potential to avoid systemic and even whole-brain side effects.
However, only a few proof-of-principle experiments have been conducted in animals, and to date no clinical study has explored the utility of intraparenchymal or intraventricular antiepileptic drug delivery in humans.
Cell and gene therapies
The emerging field of experimental cell- and gene-based neuropharmacology holds promise for location-specific therapeutic strategies. In ex vivo gene therapy, bioengineered cells capable of delivering anticonvulsant compounds might be transplanted into specific areas of the brain. On the other hand, in vivo gene therapy would involve delivering genes by viral vectors to induce the localized production of antiepileptic compounds in situ.
Endogenous anticonvulsants such as gamma-aminobutyric acid (GABA) and adenosine have been tried in various animal experiments. 49 Before they can be applied clinically, significant questions need to be addressed, including the potential for toxicity or maladaptive plasticity and long-term therapeutic safety and efficacy.
Cell transplantation is aimed at restoring the physiologic balance of neurotransmitters, and is currently being investigated for the treatment of several neurologic disorders such as Parkinson disease and Huntington disease.50 Unlike delivery of exogenous compounds, cell transplantation (heterologous fetal cell grafts or embryonic or adult stem cells) has the potential to form restorative synaptic connections and assimilate within existing cells and networks in the host tissue. An essential limitation to xenotransplantation in humans is the risk of immunologic rejection.
The future
We hope that continued progress in genomics will lead to targeted development of disease-modifying drugs that can impede or reverse the process of epileptogenesis. Advances in informatics and genetics may be harnessed to predict which patients are likely to develop pharmacoresistance, to cure certain genetic epilepsies, and to individualize antiepileptic drug selection on the basis of each person’s genetic profile.
- Siegel AM. Presurgical evaluation and surgical treatment of medically refractory epilepsy. Neurosurg Rev 2004; 27:1–18.
- Murray MI, Halpern MT, Leppik IE. Cost of refractory epilepsy in adults in the USA. Epilepsy Res 1996; 23:139–148.
- Jacoby A, Buck D, Baker G, McNamee P, Graham-Jones S, Chadwick D. Uptake and costs of care for epilepsy: findings from a U.K. regional study. Epilepsia 1998; 39:776–786.
- Chapell R, Reston J, Snyder D, Treadwell J, Treager S, Turkelson C. Management of treatment-resistant epilepsy. Evid Rep Technol Assess (Summ) 2003 Apr; 77:1–8.
- Nei M, Bagla R. Seizure-related injury and death. Curr Neurol Neurosci Rep 2007; 7:335–341.
- Langan Y, Nashef L, Sander JW. Case-control study of SUDEP. Neurology 2005; 64:1131–1133.
- Sperling MR, Feldman H, Kinman J, Liporace JD, O’Connor MJ. Seizure control and mortality in epilepsy. Ann Neurol 1999; 46:45–50.
- Berg AT. Understanding the delay before epilepsy surgery: who develops intractable focal epilepsy and when? CNS Spectr 2004; 9:136–144.
- Perucca E. Pharmacoresistance in epilepsy: how should it be defined? CNS Drugs 1998; 10:171–179.
- Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med 2000; 342:314–319.
- Gumnit RJ, Walczak TS; National Association of Epilepsy Centers. Guidelines for essential services, personnel, and facilities in specialized epilepsy centers in the United States. Epilepsia 2001; 42:804–814.
- Smith D, Defalla BA, Chadwick DW. The misdiagnosis of epilepsy and the management of refractory epilepsy in a specialist clinic. QJM 1999; 92:15–23.
- Schuele SU, Lüders HO. Intractable epilepsy: management and therapeutic alternatives. Lancet Neurol 2008; 7:514–524.
- Engel J, Burchfiel J, Ebersole J, et al. Long-term monitoring for epilepsy. Report of an IFCN committee. Electroencephalogr Clin Neurophysiol 1993; 87:437–458.
- Kanner AM, Morris HH, Lüders H, et al. Supplementary motor seizures mimicking pseudoseizures: some clinical differences. Neurology 1990; 40:1404–1407.
- Alexopoulos AV, Dinner DS. Focal motor seizures, epilepsia partialis continua, and supplementary sensorimotor seizures. In:Wyllie E, Gupta A, Lachhwani DK, editors. The Treatment of Epilepsy: Principles & Practice. Philadelphia, PA: Lippincott Williams & Wilkins, 2006:257–277.
- French JA. Refractory epilepsy: clinical overview. Epilepsia 2007; 48(suppl 1):3–7.
- Regesta G, Tanganelli P. Clinical aspects and biological bases of drug-resistant epilepsies. Epilepsy Res 1999; 34:109–122.
- Berg AT, Langfitt J, Shinnar S, et al. How long does it take for partial epilepsy to become intractable? Neurology 2003; 60:186–190.
- Berg AT, Vickrey BG, Testa FM, et al. How long does it take for epilepsy to become intractable? A prospective investigation. Ann Neurol 2006; 60:73–79.
- Löscher W, Schmidt D. Experimental and clinical evidence for loss of effect (tolerance) during prolonged treatment with antiepileptic drugs. Epilepsia 2006; 47:1253–1284.
- Löscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci 2005; 6:591–602.
- Siddiqui A, Kerb R, Weale ME, et al. Association of multidrug resistance in epilepsy with a polymorphism in the drug-transporter gene ABCB1. N Engl J Med 2003; 348:1442–1448.
- Granata T, Marchi N, Carlton E, et al. Management of the patient with medically refractory epilepsy. Expert Rev Neurother 2009; 9:1791–1802.
- Marchi N, Hallene KL, Kight KM, et al. Significance of MDR1 and multiple drug resistance in refractory human epileptic brain. BMC Med 2004; 2:37.
- Oby E, Janigro D. The blood-brain barrier and epilepsy. Epilepsia 2006; 47:1761–1774.
- Schmidt D, Löscher W. New developments in antiepileptic drug resistance: an integrative view. Epilepsy Curr 2009; 9:47–52.
- Remy S, Beck H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain 2006; 129:18–35.
- Löscher W, Schmidt D. New horizons in the development of antiepileptic drugs. Epilepsy Res 2002; 50:3–16.
- Sirven J, Whedon B, Caplan D, et al. The ketogenic diet for intractable epilepsy in adults: preliminary results. Epilepsia 1999; 40:1721–1726.
- Fisher RS, Handforth A. Reassessment: vagus nerve stimulation for epilepsy: a report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 1999; 53:666–669.
- Hallböök T, Lundgren J, Stjernqvist K, Blennow G, Strömblad LG, Rosén I. Vagus nerve stimulation in 15 children with therapy resistant epilepsy; its impact on cognition, quality of life, behaviour and mood. Seizure 2005; 14:504–513.
- Holmes MD, Silbergeld DL, Drouhard D, Wilensky AJ, Ojemann LM. Effect of vagus nerve stimulation on adults with pharmacoresistant generalized epilepsy syndromes. Seizure 2004; 13:340–345.
- Murphy JV, Torkelson R, Dowler I, Simon S, Hudson S. Vagal nerve stimulation in refractory epilepsy: the first 100 patients receiving vagal nerve stimulation at a pediatric epilepsy center. Arch Pediatr Adolesc Med 2003; 157:560–564.
- Alexopoulos AV, Najm IM. Neurosurgical management of focal epilepsies in adults. In:Panayiotopoulos CP, et al, editors. Focal Epilepsies: Seizures, Syndromes and Management. Oxford, UK: Medicinae; 2009:204–220.
- Wieser HGILAE Commission on Neurosurgery of Epilepsy. ILAE Commission Report. Mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsia 2004; 45:695–714.
- Engel J. Surgery for seizures. N Engl J Med 1996; 334:647–652.
- Engel J, Wiebe S, French J, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: temporal lobe and localized neocortical resections for epilepsy: report of the Quality Standards Subcommittee of the American Academy of Neurology, in association with the American Epilepsy Society and the American Association of Neurological Surgeons. Neurology 2003; 60:538–547.
- Wiebe S, Blume WT, Girvin JP, Eliasziw M; Effectiveness and Efficiency of Surgery for Temporal Lobe Epilepsy Study Group. A randomized, controlled trial of surgery for temporal-lobe epilepsy. N Engl J Med 2001; 345:311–318.
- Semah F, Picot MC, Adam C, et al. Is the underlying cause of epilepsy a major prognostic factor for recurrence? Neurology 1998; 51:1256–1262.
- Awad IA, Rosenfeld J, Ahl J, Hahn JF, Lüders H. Intractable epilepsy and structural lesions of the brain: mapping, resection strategies, and seizure outcome. Epilepsia 1991; 32:179–186.
- Cascino GD. Surgical treatment for extratemporal epilepsy. Curr Treat Options Neurol 2004; 6:257–262.
- González-Martínez JA, Gupta A, Kotagal P, et al. Hemispherectomy for catastrophic epilepsy in infants. Epilepsia 2005; 46:1518–1525.
- Wyllie E. Surgery for catastrophic localization-related epilepsy in infants. Epilepsia 1996; 37(suppl 1):S22–S25.
- Tanriverdi T, Olivier A, Poulin N, Andermann F, Dubeau F. Long-term seizure outcome after corpus callosotomy: a retrospective analysis of 95 patients. J Neurosurg 2009; 110:332–342.
- Asadi-Pooya AA, Sharan A, Nei M, Sperling MR. Corpus callosotomy. Epilepsy Behav 2008; 13:271–278.
- Spencer SS, Schramm J, Wyler A, et al. Multiple subpial transection for intractable partial epilepsy: an international meta-analysis. Epilepsia 2002; 43:141–145.
- Alexopoulos AV, Gonugunta V, Yang J, Boulis NM. Electrical stimulation and gene-based neuromodulation for control of medically-refractory epilepsy. Acta Neurochir Suppl 2007; 97:293–309.
- Detlev B. Cell and gene therapies for refractory epilepsy. Curr Neuropharmacol 2007; 5:115–125.
- Fisher RS, Ho J. Potential new methods for antiepileptic drug delivery. CNS Drugs 2002; 16:579–593.
- Siegel AM. Presurgical evaluation and surgical treatment of medically refractory epilepsy. Neurosurg Rev 2004; 27:1–18.
- Murray MI, Halpern MT, Leppik IE. Cost of refractory epilepsy in adults in the USA. Epilepsy Res 1996; 23:139–148.
- Jacoby A, Buck D, Baker G, McNamee P, Graham-Jones S, Chadwick D. Uptake and costs of care for epilepsy: findings from a U.K. regional study. Epilepsia 1998; 39:776–786.
- Chapell R, Reston J, Snyder D, Treadwell J, Treager S, Turkelson C. Management of treatment-resistant epilepsy. Evid Rep Technol Assess (Summ) 2003 Apr; 77:1–8.
- Nei M, Bagla R. Seizure-related injury and death. Curr Neurol Neurosci Rep 2007; 7:335–341.
- Langan Y, Nashef L, Sander JW. Case-control study of SUDEP. Neurology 2005; 64:1131–1133.
- Sperling MR, Feldman H, Kinman J, Liporace JD, O’Connor MJ. Seizure control and mortality in epilepsy. Ann Neurol 1999; 46:45–50.
- Berg AT. Understanding the delay before epilepsy surgery: who develops intractable focal epilepsy and when? CNS Spectr 2004; 9:136–144.
- Perucca E. Pharmacoresistance in epilepsy: how should it be defined? CNS Drugs 1998; 10:171–179.
- Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med 2000; 342:314–319.
- Gumnit RJ, Walczak TS; National Association of Epilepsy Centers. Guidelines for essential services, personnel, and facilities in specialized epilepsy centers in the United States. Epilepsia 2001; 42:804–814.
- Smith D, Defalla BA, Chadwick DW. The misdiagnosis of epilepsy and the management of refractory epilepsy in a specialist clinic. QJM 1999; 92:15–23.
- Schuele SU, Lüders HO. Intractable epilepsy: management and therapeutic alternatives. Lancet Neurol 2008; 7:514–524.
- Engel J, Burchfiel J, Ebersole J, et al. Long-term monitoring for epilepsy. Report of an IFCN committee. Electroencephalogr Clin Neurophysiol 1993; 87:437–458.
- Kanner AM, Morris HH, Lüders H, et al. Supplementary motor seizures mimicking pseudoseizures: some clinical differences. Neurology 1990; 40:1404–1407.
- Alexopoulos AV, Dinner DS. Focal motor seizures, epilepsia partialis continua, and supplementary sensorimotor seizures. In:Wyllie E, Gupta A, Lachhwani DK, editors. The Treatment of Epilepsy: Principles & Practice. Philadelphia, PA: Lippincott Williams & Wilkins, 2006:257–277.
- French JA. Refractory epilepsy: clinical overview. Epilepsia 2007; 48(suppl 1):3–7.
- Regesta G, Tanganelli P. Clinical aspects and biological bases of drug-resistant epilepsies. Epilepsy Res 1999; 34:109–122.
- Berg AT, Langfitt J, Shinnar S, et al. How long does it take for partial epilepsy to become intractable? Neurology 2003; 60:186–190.
- Berg AT, Vickrey BG, Testa FM, et al. How long does it take for epilepsy to become intractable? A prospective investigation. Ann Neurol 2006; 60:73–79.
- Löscher W, Schmidt D. Experimental and clinical evidence for loss of effect (tolerance) during prolonged treatment with antiepileptic drugs. Epilepsia 2006; 47:1253–1284.
- Löscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci 2005; 6:591–602.
- Siddiqui A, Kerb R, Weale ME, et al. Association of multidrug resistance in epilepsy with a polymorphism in the drug-transporter gene ABCB1. N Engl J Med 2003; 348:1442–1448.
- Granata T, Marchi N, Carlton E, et al. Management of the patient with medically refractory epilepsy. Expert Rev Neurother 2009; 9:1791–1802.
- Marchi N, Hallene KL, Kight KM, et al. Significance of MDR1 and multiple drug resistance in refractory human epileptic brain. BMC Med 2004; 2:37.
- Oby E, Janigro D. The blood-brain barrier and epilepsy. Epilepsia 2006; 47:1761–1774.
- Schmidt D, Löscher W. New developments in antiepileptic drug resistance: an integrative view. Epilepsy Curr 2009; 9:47–52.
- Remy S, Beck H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain 2006; 129:18–35.
- Löscher W, Schmidt D. New horizons in the development of antiepileptic drugs. Epilepsy Res 2002; 50:3–16.
- Sirven J, Whedon B, Caplan D, et al. The ketogenic diet for intractable epilepsy in adults: preliminary results. Epilepsia 1999; 40:1721–1726.
- Fisher RS, Handforth A. Reassessment: vagus nerve stimulation for epilepsy: a report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 1999; 53:666–669.
- Hallböök T, Lundgren J, Stjernqvist K, Blennow G, Strömblad LG, Rosén I. Vagus nerve stimulation in 15 children with therapy resistant epilepsy; its impact on cognition, quality of life, behaviour and mood. Seizure 2005; 14:504–513.
- Holmes MD, Silbergeld DL, Drouhard D, Wilensky AJ, Ojemann LM. Effect of vagus nerve stimulation on adults with pharmacoresistant generalized epilepsy syndromes. Seizure 2004; 13:340–345.
- Murphy JV, Torkelson R, Dowler I, Simon S, Hudson S. Vagal nerve stimulation in refractory epilepsy: the first 100 patients receiving vagal nerve stimulation at a pediatric epilepsy center. Arch Pediatr Adolesc Med 2003; 157:560–564.
- Alexopoulos AV, Najm IM. Neurosurgical management of focal epilepsies in adults. In:Panayiotopoulos CP, et al, editors. Focal Epilepsies: Seizures, Syndromes and Management. Oxford, UK: Medicinae; 2009:204–220.
- Wieser HGILAE Commission on Neurosurgery of Epilepsy. ILAE Commission Report. Mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsia 2004; 45:695–714.
- Engel J. Surgery for seizures. N Engl J Med 1996; 334:647–652.
- Engel J, Wiebe S, French J, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: temporal lobe and localized neocortical resections for epilepsy: report of the Quality Standards Subcommittee of the American Academy of Neurology, in association with the American Epilepsy Society and the American Association of Neurological Surgeons. Neurology 2003; 60:538–547.
- Wiebe S, Blume WT, Girvin JP, Eliasziw M; Effectiveness and Efficiency of Surgery for Temporal Lobe Epilepsy Study Group. A randomized, controlled trial of surgery for temporal-lobe epilepsy. N Engl J Med 2001; 345:311–318.
- Semah F, Picot MC, Adam C, et al. Is the underlying cause of epilepsy a major prognostic factor for recurrence? Neurology 1998; 51:1256–1262.
- Awad IA, Rosenfeld J, Ahl J, Hahn JF, Lüders H. Intractable epilepsy and structural lesions of the brain: mapping, resection strategies, and seizure outcome. Epilepsia 1991; 32:179–186.
- Cascino GD. Surgical treatment for extratemporal epilepsy. Curr Treat Options Neurol 2004; 6:257–262.
- González-Martínez JA, Gupta A, Kotagal P, et al. Hemispherectomy for catastrophic epilepsy in infants. Epilepsia 2005; 46:1518–1525.
- Wyllie E. Surgery for catastrophic localization-related epilepsy in infants. Epilepsia 1996; 37(suppl 1):S22–S25.
- Tanriverdi T, Olivier A, Poulin N, Andermann F, Dubeau F. Long-term seizure outcome after corpus callosotomy: a retrospective analysis of 95 patients. J Neurosurg 2009; 110:332–342.
- Asadi-Pooya AA, Sharan A, Nei M, Sperling MR. Corpus callosotomy. Epilepsy Behav 2008; 13:271–278.
- Spencer SS, Schramm J, Wyler A, et al. Multiple subpial transection for intractable partial epilepsy: an international meta-analysis. Epilepsia 2002; 43:141–145.
- Alexopoulos AV, Gonugunta V, Yang J, Boulis NM. Electrical stimulation and gene-based neuromodulation for control of medically-refractory epilepsy. Acta Neurochir Suppl 2007; 97:293–309.
- Detlev B. Cell and gene therapies for refractory epilepsy. Curr Neuropharmacol 2007; 5:115–125.
- Fisher RS, Ho J. Potential new methods for antiepileptic drug delivery. CNS Drugs 2002; 16:579–593.
KEY POINTS
- When seizures have failed to respond to two or three appropriate antiepileptic drugs, the chance of significant benefit from other drugs is 10% or less.
- The biologic basis of pharmacoresistance is multifactorial and varies from one patient to another.
- Social and lifestyle factors, including alcohol misuse and nonadherence to prescribed antiepileptic drugs, can contribute to or masquerade as pharmacoresistance.
- Current options for patients with pharmacoresistant epilepsy are surgery (the best option when feasible), vagus nerve stimulation, investigational drugs or devices, and aggressive combination treatment with available antiepileptic drugs.