User login
Development and Validation of an Administrative Algorithm to Identify Veterans With Epilepsy
Development and Validation of an Administrative Algorithm to Identify Veterans With Epilepsy
Epilepsy affects about 4.5 million people in the United States and 150,000 new individuals are diagnosed each year.1,2 In 2019, epilepsy-attributable health care spending for noninstitutionalized people was around $5.4 billion and total epilepsy-attributable and epilepsy or seizure health care-related costs totaled $54 billion.3
Accurate surveillance of epilepsy in large health care systems can potentially improve health care delivery and resource allocation. A 2012 Institute of Medicine (IOM) report identified 13 recommendations to guide public health action on epilepsy, including validation of standard definitions for case ascertainment, identification of epilepsy through screening programs or protocols, and expansion of surveillance to better understand disease burden.4
A systematic review of validation studies concluded that it is reasonable to use administrative data to identify people with epilepsy in epidemiologic research. Combining The International Classification of Diseases (ICD) codes for epilepsy (ICD-10, G40-41; ICD-9, 345) with antiseizure medications (ASMs) could provide high positive predictive values (PPVs) and combining symptoms codes for convulsions (ICD-10, R56; ICD-9, 780.3, 780.39) with ASMs could lead to high sensitivity.5 However, identifying individuals with epilepsy from administrative data in large managed health care organizations is challenging.6 The IOM report noted that large managed health care organizations presented varying incidence and prevalence estimates due to differing methodology, geographic area, demographics, and definitions of epilepsy.
The Veterans Health Administration (VHA) is the largest integrated US health care system, providing care to > 9.1 million veterans.7 To improve the health and well-being of veterans with epilepsy (VWEs), a network of sites was established in 2008 called the US Department of Veterans Affairs (VA) Epilepsy Centers of Excellence (ECoE). Subsequent to the creation of the ECoE, efforts were made to identify VWEs within VHA databases.8,9 Prior to fiscal year (FY) 2016, the ECoE adopted a modified version of a well-established epilepsy diagnostic algorithm developed by Holden et al for large managed care organizations.10 The original algorithm identified patients by cross-matching ASMs with ICD-9 codes for an index year. But it failed to capture a considerable number of stable patients with epilepsy in the VHA due to incomplete documentation, and had false positives due to inclusion of patients identified from diagnostic clinics. The modified algorithm the ECoE used prior to FY 2016 considered additional prior years and excluded encounters from diagnostic clinics. The result was an improvement in the sensitivity and specificity of the algorithm. Researchers evaluating 500 patients with epilepsy estimated that the modified algorithm had a PPV of 82.0% (95% CI, 78.6%-85.4%).11
After implementation of ICD-10 codes in the VHA in FY 2016, the task of reliably and efficiently identifying VWE led to a 3-tier algorithm. This article presents a validation of the different tiers of this algorithm after the implementation of ICD-10 diagnosis codes and summarizes the surveillance data collected over the years within the VHA showing the trends of epilepsy.
Methods
The VHA National Neurology office commissioned a Neurology Cube dashboard in FY 2021 in collaboration with VHA Support Service Center (VSSC) for reporting and surveillance of VWEs as a quality improvement initiative. The Neurology Cube uses a 3-tier system for identifying VWE in the VHA databases. VSSC programmers extract data from the VHA Corporate Data Warehouse (CDW) and utilize Microsoft SQL Server and Microsoft Power BI for Neurology Cube reports. The 3-tier system identifies VWE and divides them into distinct groups. The first tier identifies VWE with the highest degree of confidence; Tiers 2 and 3 represent identification with successively lesser degrees of confidence (Figure 1).
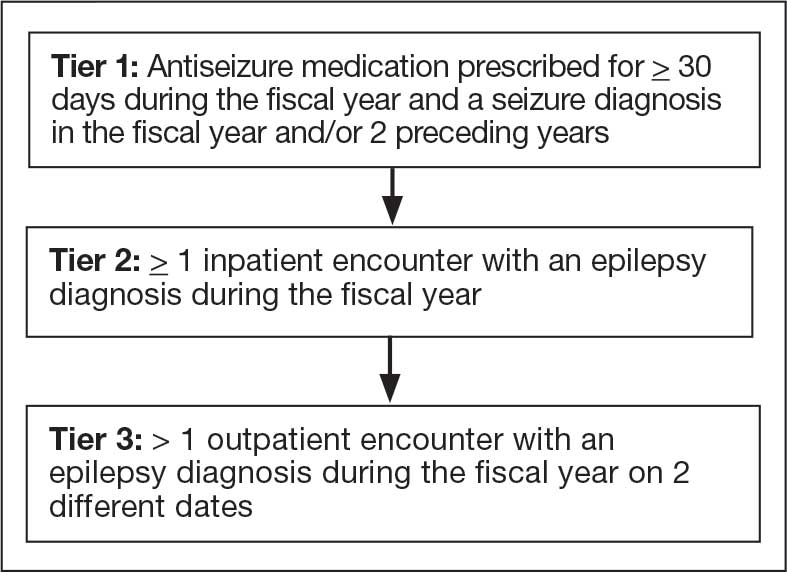
Tier 1
Definition. For a given index year and the preceding 2 years, any of following diagnosis codes on ≥ 1 clinical encounter are considered: 345.xx (epilepsy in ICD-9), 780.3x (other convulsions in ICD-9), G40.xxx (epilepsy in ICD-10), R40.4 (transient alteration of awareness), R56.1 (posttraumatic seizures), or R56.9 (unspecified convulsions). To reduce false positive rates, EEG clinic visits, which may include long-term monitoring, are excluded. Patients identified with ICD codes are then evaluated for an ASM prescription for ≥ 30 days during the index year. ASMs are listed in Appendix 1.
Validation. The development and validation of ICD-9 diagnosis codes crossmatched with an ASM prescription in the VHA has been published elsewhere.11 In FY 2017, after implementation of ICD-10 diagnostic codes, Tier 1 development and validation was performed in 2 phases. Even though Tier 1 study phases were conducted and completed during FY 2017, the patients for Tier 1 were identified from evaluation of FY 2016 data (October 1, 2015, to September 30, 2016). After the pilot analysis, the Tier 1 definition was implemented, and a chart review of 625 randomized patients was conducted at 5 sites for validation. Adequate preliminary data was not available to perform a sample size estimation for this study. Therefore, a practical target of 125 patients was set for Tier 1 from each site to obtain a final sample size of 625 patients. This second phase validated that the crossmatch of ICD-10 diagnosis codes with ASMs had a high PPV for identifying VWE.
Tiers 2 and 3
Definitions. For an index year, Tier 2 includes patients with ≥ 1 inpatient encounter documentation of either ICD-9 345.xx or ICD-10 G40.xxx, excluding EEG clinics. Tier 3 Includes patients who have had ≥ 2 outpatient encounters with diagnosis codes 345.xx or G40.xxx on 2 separate days, excluding EEG clinics. Tiers 2 and 3 do not require ASM prescriptions; this helps to identify VWEs who may be getting their medications outside of VHA or those who have received a new diagnosis.
Validations. Tiers 2 and 3 were included in the epilepsy identification algorithm in FY 2021 after validation was performed on a sample of 8 patients in each tier. Five patients were subsequently identified as having epilepsy in Tier 2 and 6 patients were identified in Tier 3. A more comprehensive validation of Tiers 2 and 3 was performed during FY 2022 that included patients at 5 sites seen during FY 2019 to FY 2022. Since yearly trends showed only about 8% of total patients were identified as having epilepsy through Tiers 2 and 3 we sought ≥ 20 patients per tier for the 5 sites for a total of 200 patients to ensure representation across the VHA. The final count was 126 patients for Tier 2 and 174 patients for Tier 3 (n = 300).
Gold Standard Criteria for Epilepsy Diagnosis
We used the International League Against Epilepsy (ILAE) definition of epilepsy for the validation of the 3 algorithm tiers. ILAE defines epilepsy as ≥ 2 unprovoked (or reflex) seizures occurring > 24 hours apart or 1 unprovoked (or reflex) seizure and a probability of further seizures similar to the general recurrence risk (≥ 60%) after 2 unprovoked seizures, occurring over the next 10 years.12
A standard protocol was provided to evaluators to identify patients using the VHA Computerized Patient Record System (Appendix 1). After review, evaluators categorized each patient in 1 of 4 ways: (1) Yes, definite: The patient’s health care practitioner (HCP) believes the patient has epilepsy and is treating with medication; (2) Yes, uncertain: The HCP has enough suspicion of epilepsy that a medication is prescribed, but uncertainty is expressed of the diagnosis; (3) No, definite: The HCP does not believe the patient has epilepsy and is therefore not treating with medication for seizure; (4) No, uncertain: The HCP is not treating with medication for epilepsy, because the diagnostic suspicion is not high enough, but there is suspicion for epilepsy.
As a quality improvement operational project, the Epilepsy National Program Office approved this validation project and determined that institutional review board approval was not required.
Statistical Analysis
Counts and percentages were computed for categories of epilepsy status. PPV of each tier was estimated with asymptotic 95% CIs.
Results
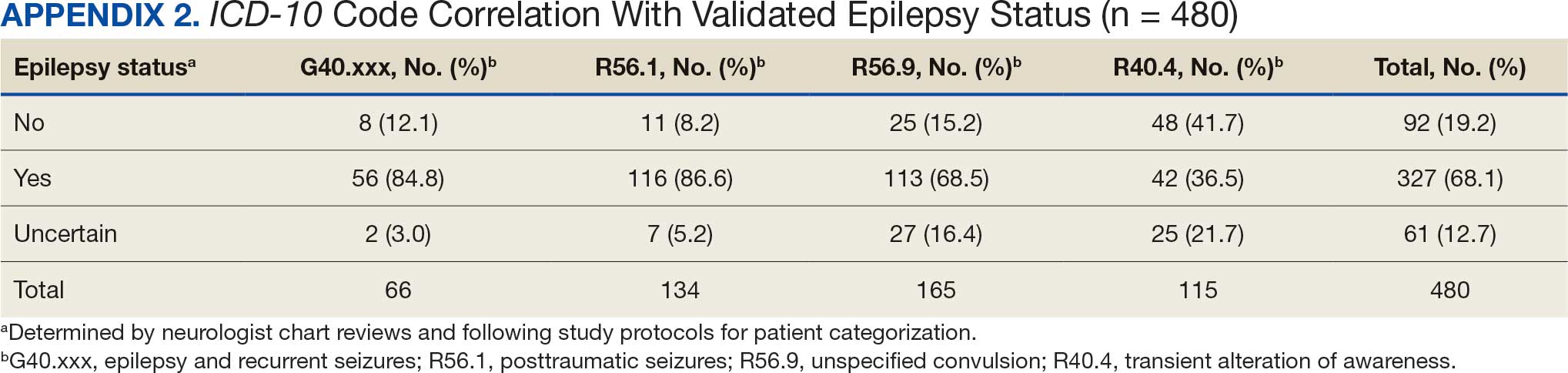
ICD-10 codes for 480 patients were evaluated in Tier 1 phase 1; 13.8% were documented with G40.xxx, 27.9% with R56.1, 34.4% with R56.9, and 24.0% with R40.4 (Appendix 2). In total, 68.1% fulfilled the criteria of epilepsy, 19.2% did not, and 12.7% were uncertain). From the validation of Tier 1 phase 2 (n = 625), the PPV of the algorithm for patients presumed to have epilepsy (definite and uncertain) was 85.1% (95% CI, 82.1%-87.8%) (Table).
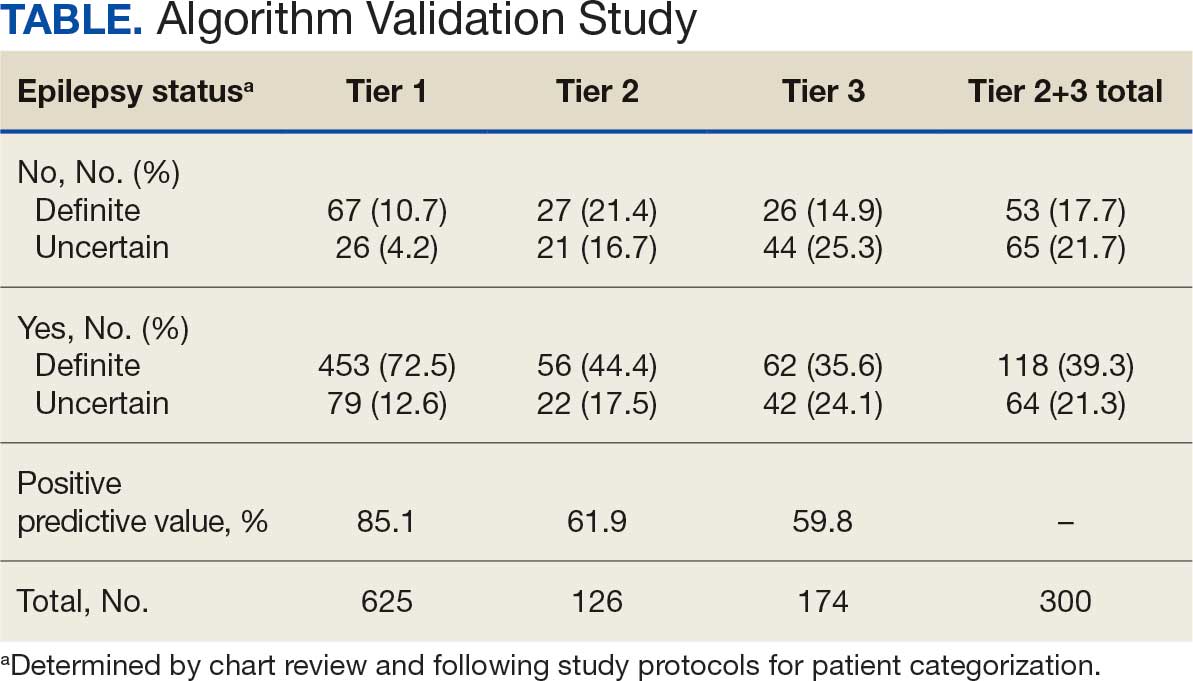
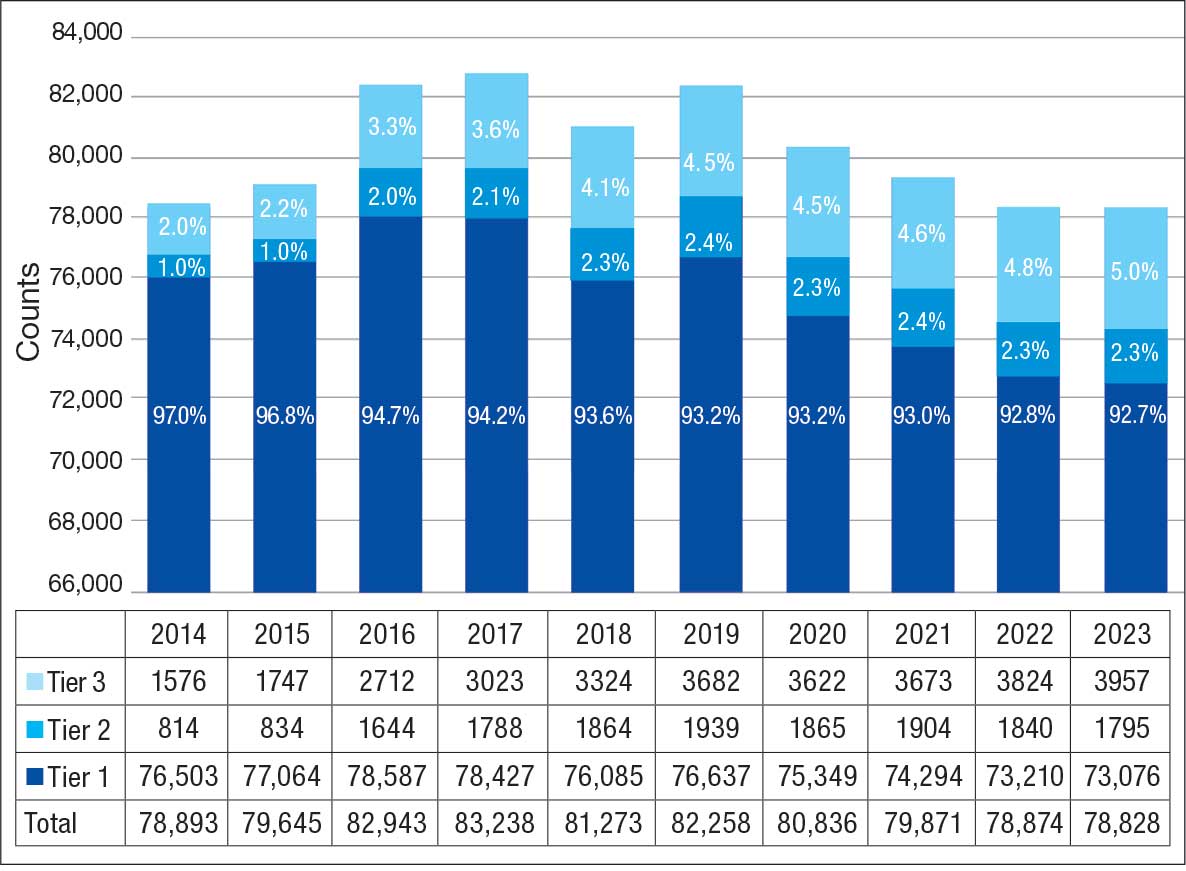
Of 300 patients evaluated, 126 (42.0%) were evaluated for Tier 2 with a PPV of 61.9% (95% CI, 53.4%-70.4%), and 174 (58.0%) patients were evaluated for Tier 3 with a PPV of 59.8% (95% CI, 52.5%-67.1%. The PPV of the algorithm for patients presumed to have epilepsy (definite and uncertain) were combined to calculate the PPV. Estimates of VHA VWE counts were computed for each tier from FY 2014 to FY 2023 using the VSSC Neurology Cube (Figure 2). For all years, > 92% patients were classified using the Tier 1 definition.
Discussion
The development and validation of the 3-tier diagnostic algorithm represents an important advancement in the surveillance and management of epilepsy among veterans within the VHA. The validation of this algorithm also demonstrates its practical utility in a large, integrated health care system.
Specific challenges were encountered when attempting to use pre-existing algorithms; these challenges included differences in the usage patterns of diagnostic codes and the patterns of ASM use within the VHA. These challenges prompted the need for a tailored approach, which led to the development of this algorithm. The inclusion of additional ICD-10 codes led to further revisions and subsequent validation. While many of the basic concepts of the algorithm, including ICD codes and ASMs, could work in other institutions, it would be wise for health care organizations to develop their own algorithms because of certain variables, including organizational size, patient demographics, common comorbidities, and the specific configurations of electronic health records and administrative data systems.
Studies have shown that ICD-10 codes for epilepsy (G40.* and/or R56.9) perform well in identifying epilepsy whether they are assigned by neurologists (sensitivity, 97.7%; specificity, 44.1%; PPV, 96.2%; negative predictive value, 57.7%), or in emergency department or hospital discharges (PPV, 75.5%).13,14 The pilot study of the algorithm’s Tier 1 development (phase 1) evaluated whether the selected ICD-10 diagnostic codes accurately included the VWE population within the VHA and revealed that while most codes (eg, epilepsy [G40.xxx]; posttraumatic seizures [R56.1]; and unspecified convulsions [R56.9]), had a low false positive rate (< 16%), the R40.4 code (transient alteration of awareness) had a higher false positivity of 42%. While this is not surprising given the broad spectrum of conditions that can manifest as transient alteration of awareness, it underscores the inherent challenges in diagnosing epilepsy using diagnosis codes.
In phase 2, the Tier 1 algorithm was validated as effective for identifying VWE in the VHA system, as its PPV was determined to be high (85%). In comparison, Tiers 2 and 3, whose criteria did not require data on VHA prescribed ASM use, had lower tiers of epilepsy predictability (PPV about 60% for both). This was thought to be acceptable because Tiers 2 and 3 represent a smaller population of the identified VWEs (about 8%). These VWEs may otherwise have been missed, partly because veterans are not required to get ASMs from the VHA.
Upon VHA implementation in FY 2021, this diagnostic algorithm exhibited significant clinical utility when integrated within the VSSC Neurology Cube. It facilitated an efficient approach to identifying VWEs using readily available databases. This led to better tracking of real-time epilepsy cases, which facilitated improving current resource allocation and targeted intervention strategies such as identification of drug-resistant epilepsy patients, optimizing strategies for telehealth and patient outreach for awareness of epilepsy care resources within VHA. Meanwhile, data acquired by the algorithm over the decade since its development (FY 2014 to FY 2023) contributed to more accurate epidemiologic information and identification of historic trends. Development of the algorithm represents one of the ways ECoEs have led to improved care for VWEs. ECoEs have been shown to improve health care for veterans in several metrics.15
A strength of this study is the rigorous multitiered validation process to confirm the diagnostic accuracy of ICD-10 codes against the gold standard ILAE definition of epilepsy to identify “definite” epilepsy cases within the VHA. The use of specific ICD codes further enhances the precision of epilepsy diagnoses. The inclusion of ASMs, which are sometimes prescribed for conditions other than epilepsy, could potentially inflate false positive rates.16
This study focused exclusively on the identification and validation of definite epilepsy cases within the VHA VSSC database, employing more stringent diagnostic criteria to ensure the highest level of certainty in ascertaining epilepsy. It is important to note there is a separate category of probable epilepsy, which involves a broader set of diagnostic criteria. While not covered in this study, probable epilepsy would be subject to future research and validation, which could provide insights into a wider spectrum of epilepsy diagnoses. Such future research could help refine the algorithm’s applicability and accuracy and potentially lead to more comprehensive surveillance and management strategies in clinical practice.
This study highlights the inherent challenges in leveraging administrative data for disease identification, particularly for conditions such as epilepsy, where diagnostic clarity can be complex. However, other conditions such as multiple sclerosis have noted similar success with the use of VHA administrative data for categorizing disease.17
Limitations
The algorithm discussed in this article is, in and of itself, generalizable. However, the validation process was unique to the VHA patient population, limiting the generalizability of the findings. Documentation practices and HCP attitudes within the VHA may differ from those in other health care settings. Identifying people with epilepsy can be challenging because of changing definitions of epilepsy over time. In addition to clinical evaluation, EEG and magnetic resonance imaging results, response to ASM treatment, and video-EEG monitoring of habitual events all can help establish the diagnosis. Therefore, studies may vary in how inclusive or exclusive the criteria are. ASMs such as gabapentin, pregabalin, carbamazepine, lamotrigine, topiramate, and valproate are used to treat other conditions, including headaches, generalized pain, and mood disorders. Consequently, including these ASMs in the Tier 1 definition may have increased the false positive rate. Additional research is needed to evaluate whether excluding these ASMs from the algorithm based on specific criteria (eg, dose of ASM used) can further refine the algorithm to identify patients with epilepsy.
Further refinement of this algorithm may also occur as technology changes. Future electronic health records may allow better tracking of different epilepsy factors, the integration of additional diagnostic criteria, and the use of natural language processing or other forms of artificial intelligence.
Conclusions
This study presents a significant step forward in epilepsy surveillance within the VHA. The algorithm offers a robust tool for identifying VWEs with good PPVs, facilitating better resource allocation and targeted care. Despite its limitations, this research lays a foundation for future advancements in the management and understanding of epilepsy within large health care systems. Since this VHA algorithm is based on ASMs and ICD diagnosis codes from patient records, other large managed health care systems also may be able to adapt this algorithm to their data specifications.

- Kobau R, Luncheon C, Greenlund K. Active epilepsy prevalence among U.S. adults is 1.1% and differs by educational level-National Health Interview Survey, United States, 2021. Epilepsy Behav. 2023;142:109180. doi:10.1016/j.yebeh.2023.109180
- GBD 2017 US Neurological Disorders Collaborators, Feigin VL, Vos T, et al. Burden of neurological disorders across the US from 1990-2017: a global burden of disease study. JAMA Neurol. 2021;78:165-176. doi:10.1001/jamaneurol.2020.4152
- Moura LMVR, Karakis I, Zack MM, et al. Drivers of US health care spending for persons with seizures and/or epilepsies, 2010-2018. Epilepsia. 2022;63:2144-2154. doi:10.1111/epi.17305
- Institute of Medicine. Epilepsy Across the Spectrum: Promoting Health and Understanding. The National Academies Press; 2012. Accessed November 11, 2025. www.nap.edu/catalog/13379
- Mbizvo GK, Bennett KH, Schnier C, Simpson CR, Duncan SE, Chin RFM. The accuracy of using administrative healthcare data to identify epilepsy cases: A systematic review of validation studies. Epilepsia. 2020;61:1319-1335. doi:10.1111/epi.16547
- Montouris GD. How will primary care physicians, specialists, and managed care treat epilepsy in the new millennium? Neurology. 2000;55:S42-S44.
- US Department of Veterans Affairs. Veterans Health Administration: About VHA. Accessed November 11, 2025. https://www.va.gov/health/aboutvha.asp
- Veterans’ Mental Health and Other Care Improvements Act of 2008, S 2162, 110th Cong (2008). Accessed November 11, 2025. https://www.congress.gov/bill/110th-congress/senate-bill/2162
- Rehman R, Kelly PR, Husain AM, Tran TT. Characteristics of Veterans diagnosed with seizures within Veterans Health Administration. J Rehabil Res Dev. 2015;52(7):751-762. doi:10.1682/JRRD.2014.10.0241
- Holden EW, Grossman E, Nguyen HT, et al. Developing a computer algorithm to identify epilepsy cases in managed care organizations. Dis Manag. 2005;8:1-14. doi:10.1089/dis.2005.8.1
- Rehman R, Everhart A, Frontera AT, et al. Implementation of an established algorithm and modifications for the identification of epilepsy patients in the Veterans Health Administration. Epilepsy Res. 2016;127:284-290. doi:10.1016/j.eplepsyres.2016.09.012
- Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55:475-482. doi:10.1111/epi.12550
- Smith JR, Jones FJS, Fureman BE, et al. Accuracy of ICD-10-CM claims-based definitions for epilepsy and seizure type. Epilepsy Res. 2020;166:106414. doi:10.1016/j.eplepsyres.2020.106414
- Jetté N, Reid AY, Quan H, et al. How accurate is ICD coding for epilepsy? Epilepsia. 2010;51:62-69. doi:10.1111/j.1528-1167.2009.02201.x
- Kelly P, Chinta R, Privitera G. Do centers of excellence reduce health care costs? Evidence from the US Veterans Health Administration Centers for Epilepsy. Glob Bus Organ Excell. 2015;34:18-29.
- Haneef Z, Rehman R, Husain AM. Association between standardized mortality ratio and utilization of care in US veterans with drug-resistant epilepsy compared with all US veterans and the US general population. JAMA Neurol. 2022;79:879-887. doi:10.1001/jamaneurol.2022.2290
- Culpepper WJ, Marrie RA, Langer-Gould A, et al. Validation of an algorithm for identifying MS cases in administrative health claims datasets. Neurology. 2019;92:e1016-e1028 doi:10.1212/WNL.0000000000007043
Epilepsy affects about 4.5 million people in the United States and 150,000 new individuals are diagnosed each year.1,2 In 2019, epilepsy-attributable health care spending for noninstitutionalized people was around $5.4 billion and total epilepsy-attributable and epilepsy or seizure health care-related costs totaled $54 billion.3
Accurate surveillance of epilepsy in large health care systems can potentially improve health care delivery and resource allocation. A 2012 Institute of Medicine (IOM) report identified 13 recommendations to guide public health action on epilepsy, including validation of standard definitions for case ascertainment, identification of epilepsy through screening programs or protocols, and expansion of surveillance to better understand disease burden.4
A systematic review of validation studies concluded that it is reasonable to use administrative data to identify people with epilepsy in epidemiologic research. Combining The International Classification of Diseases (ICD) codes for epilepsy (ICD-10, G40-41; ICD-9, 345) with antiseizure medications (ASMs) could provide high positive predictive values (PPVs) and combining symptoms codes for convulsions (ICD-10, R56; ICD-9, 780.3, 780.39) with ASMs could lead to high sensitivity.5 However, identifying individuals with epilepsy from administrative data in large managed health care organizations is challenging.6 The IOM report noted that large managed health care organizations presented varying incidence and prevalence estimates due to differing methodology, geographic area, demographics, and definitions of epilepsy.
The Veterans Health Administration (VHA) is the largest integrated US health care system, providing care to > 9.1 million veterans.7 To improve the health and well-being of veterans with epilepsy (VWEs), a network of sites was established in 2008 called the US Department of Veterans Affairs (VA) Epilepsy Centers of Excellence (ECoE). Subsequent to the creation of the ECoE, efforts were made to identify VWEs within VHA databases.8,9 Prior to fiscal year (FY) 2016, the ECoE adopted a modified version of a well-established epilepsy diagnostic algorithm developed by Holden et al for large managed care organizations.10 The original algorithm identified patients by cross-matching ASMs with ICD-9 codes for an index year. But it failed to capture a considerable number of stable patients with epilepsy in the VHA due to incomplete documentation, and had false positives due to inclusion of patients identified from diagnostic clinics. The modified algorithm the ECoE used prior to FY 2016 considered additional prior years and excluded encounters from diagnostic clinics. The result was an improvement in the sensitivity and specificity of the algorithm. Researchers evaluating 500 patients with epilepsy estimated that the modified algorithm had a PPV of 82.0% (95% CI, 78.6%-85.4%).11
After implementation of ICD-10 codes in the VHA in FY 2016, the task of reliably and efficiently identifying VWE led to a 3-tier algorithm. This article presents a validation of the different tiers of this algorithm after the implementation of ICD-10 diagnosis codes and summarizes the surveillance data collected over the years within the VHA showing the trends of epilepsy.
Methods
The VHA National Neurology office commissioned a Neurology Cube dashboard in FY 2021 in collaboration with VHA Support Service Center (VSSC) for reporting and surveillance of VWEs as a quality improvement initiative. The Neurology Cube uses a 3-tier system for identifying VWE in the VHA databases. VSSC programmers extract data from the VHA Corporate Data Warehouse (CDW) and utilize Microsoft SQL Server and Microsoft Power BI for Neurology Cube reports. The 3-tier system identifies VWE and divides them into distinct groups. The first tier identifies VWE with the highest degree of confidence; Tiers 2 and 3 represent identification with successively lesser degrees of confidence (Figure 1).
Tier 1
Definition. For a given index year and the preceding 2 years, any of following diagnosis codes on ≥ 1 clinical encounter are considered: 345.xx (epilepsy in ICD-9), 780.3x (other convulsions in ICD-9), G40.xxx (epilepsy in ICD-10), R40.4 (transient alteration of awareness), R56.1 (posttraumatic seizures), or R56.9 (unspecified convulsions). To reduce false positive rates, EEG clinic visits, which may include long-term monitoring, are excluded. Patients identified with ICD codes are then evaluated for an ASM prescription for ≥ 30 days during the index year. ASMs are listed in Appendix 1.
Validation. The development and validation of ICD-9 diagnosis codes crossmatched with an ASM prescription in the VHA has been published elsewhere.11 In FY 2017, after implementation of ICD-10 diagnostic codes, Tier 1 development and validation was performed in 2 phases. Even though Tier 1 study phases were conducted and completed during FY 2017, the patients for Tier 1 were identified from evaluation of FY 2016 data (October 1, 2015, to September 30, 2016). After the pilot analysis, the Tier 1 definition was implemented, and a chart review of 625 randomized patients was conducted at 5 sites for validation. Adequate preliminary data was not available to perform a sample size estimation for this study. Therefore, a practical target of 125 patients was set for Tier 1 from each site to obtain a final sample size of 625 patients. This second phase validated that the crossmatch of ICD-10 diagnosis codes with ASMs had a high PPV for identifying VWE.
Tiers 2 and 3
Definitions. For an index year, Tier 2 includes patients with ≥ 1 inpatient encounter documentation of either ICD-9 345.xx or ICD-10 G40.xxx, excluding EEG clinics. Tier 3 Includes patients who have had ≥ 2 outpatient encounters with diagnosis codes 345.xx or G40.xxx on 2 separate days, excluding EEG clinics. Tiers 2 and 3 do not require ASM prescriptions; this helps to identify VWEs who may be getting their medications outside of VHA or those who have received a new diagnosis.
Validations. Tiers 2 and 3 were included in the epilepsy identification algorithm in FY 2021 after validation was performed on a sample of 8 patients in each tier. Five patients were subsequently identified as having epilepsy in Tier 2 and 6 patients were identified in Tier 3. A more comprehensive validation of Tiers 2 and 3 was performed during FY 2022 that included patients at 5 sites seen during FY 2019 to FY 2022. Since yearly trends showed only about 8% of total patients were identified as having epilepsy through Tiers 2 and 3 we sought ≥ 20 patients per tier for the 5 sites for a total of 200 patients to ensure representation across the VHA. The final count was 126 patients for Tier 2 and 174 patients for Tier 3 (n = 300).
Gold Standard Criteria for Epilepsy Diagnosis
We used the International League Against Epilepsy (ILAE) definition of epilepsy for the validation of the 3 algorithm tiers. ILAE defines epilepsy as ≥ 2 unprovoked (or reflex) seizures occurring > 24 hours apart or 1 unprovoked (or reflex) seizure and a probability of further seizures similar to the general recurrence risk (≥ 60%) after 2 unprovoked seizures, occurring over the next 10 years.12
A standard protocol was provided to evaluators to identify patients using the VHA Computerized Patient Record System (Appendix 1). After review, evaluators categorized each patient in 1 of 4 ways: (1) Yes, definite: The patient’s health care practitioner (HCP) believes the patient has epilepsy and is treating with medication; (2) Yes, uncertain: The HCP has enough suspicion of epilepsy that a medication is prescribed, but uncertainty is expressed of the diagnosis; (3) No, definite: The HCP does not believe the patient has epilepsy and is therefore not treating with medication for seizure; (4) No, uncertain: The HCP is not treating with medication for epilepsy, because the diagnostic suspicion is not high enough, but there is suspicion for epilepsy.
As a quality improvement operational project, the Epilepsy National Program Office approved this validation project and determined that institutional review board approval was not required.
Statistical Analysis
Counts and percentages were computed for categories of epilepsy status. PPV of each tier was estimated with asymptotic 95% CIs.
Results
ICD-10 codes for 480 patients were evaluated in Tier 1 phase 1; 13.8% were documented with G40.xxx, 27.9% with R56.1, 34.4% with R56.9, and 24.0% with R40.4 (Appendix 2). In total, 68.1% fulfilled the criteria of epilepsy, 19.2% did not, and 12.7% were uncertain). From the validation of Tier 1 phase 2 (n = 625), the PPV of the algorithm for patients presumed to have epilepsy (definite and uncertain) was 85.1% (95% CI, 82.1%-87.8%) (Table).
Of 300 patients evaluated, 126 (42.0%) were evaluated for Tier 2 with a PPV of 61.9% (95% CI, 53.4%-70.4%), and 174 (58.0%) patients were evaluated for Tier 3 with a PPV of 59.8% (95% CI, 52.5%-67.1%. The PPV of the algorithm for patients presumed to have epilepsy (definite and uncertain) were combined to calculate the PPV. Estimates of VHA VWE counts were computed for each tier from FY 2014 to FY 2023 using the VSSC Neurology Cube (Figure 2). For all years, > 92% patients were classified using the Tier 1 definition.
Discussion
The development and validation of the 3-tier diagnostic algorithm represents an important advancement in the surveillance and management of epilepsy among veterans within the VHA. The validation of this algorithm also demonstrates its practical utility in a large, integrated health care system.
Specific challenges were encountered when attempting to use pre-existing algorithms; these challenges included differences in the usage patterns of diagnostic codes and the patterns of ASM use within the VHA. These challenges prompted the need for a tailored approach, which led to the development of this algorithm. The inclusion of additional ICD-10 codes led to further revisions and subsequent validation. While many of the basic concepts of the algorithm, including ICD codes and ASMs, could work in other institutions, it would be wise for health care organizations to develop their own algorithms because of certain variables, including organizational size, patient demographics, common comorbidities, and the specific configurations of electronic health records and administrative data systems.
Studies have shown that ICD-10 codes for epilepsy (G40.* and/or R56.9) perform well in identifying epilepsy whether they are assigned by neurologists (sensitivity, 97.7%; specificity, 44.1%; PPV, 96.2%; negative predictive value, 57.7%), or in emergency department or hospital discharges (PPV, 75.5%).13,14 The pilot study of the algorithm’s Tier 1 development (phase 1) evaluated whether the selected ICD-10 diagnostic codes accurately included the VWE population within the VHA and revealed that while most codes (eg, epilepsy [G40.xxx]; posttraumatic seizures [R56.1]; and unspecified convulsions [R56.9]), had a low false positive rate (< 16%), the R40.4 code (transient alteration of awareness) had a higher false positivity of 42%. While this is not surprising given the broad spectrum of conditions that can manifest as transient alteration of awareness, it underscores the inherent challenges in diagnosing epilepsy using diagnosis codes.
In phase 2, the Tier 1 algorithm was validated as effective for identifying VWE in the VHA system, as its PPV was determined to be high (85%). In comparison, Tiers 2 and 3, whose criteria did not require data on VHA prescribed ASM use, had lower tiers of epilepsy predictability (PPV about 60% for both). This was thought to be acceptable because Tiers 2 and 3 represent a smaller population of the identified VWEs (about 8%). These VWEs may otherwise have been missed, partly because veterans are not required to get ASMs from the VHA.
Upon VHA implementation in FY 2021, this diagnostic algorithm exhibited significant clinical utility when integrated within the VSSC Neurology Cube. It facilitated an efficient approach to identifying VWEs using readily available databases. This led to better tracking of real-time epilepsy cases, which facilitated improving current resource allocation and targeted intervention strategies such as identification of drug-resistant epilepsy patients, optimizing strategies for telehealth and patient outreach for awareness of epilepsy care resources within VHA. Meanwhile, data acquired by the algorithm over the decade since its development (FY 2014 to FY 2023) contributed to more accurate epidemiologic information and identification of historic trends. Development of the algorithm represents one of the ways ECoEs have led to improved care for VWEs. ECoEs have been shown to improve health care for veterans in several metrics.15
A strength of this study is the rigorous multitiered validation process to confirm the diagnostic accuracy of ICD-10 codes against the gold standard ILAE definition of epilepsy to identify “definite” epilepsy cases within the VHA. The use of specific ICD codes further enhances the precision of epilepsy diagnoses. The inclusion of ASMs, which are sometimes prescribed for conditions other than epilepsy, could potentially inflate false positive rates.16
This study focused exclusively on the identification and validation of definite epilepsy cases within the VHA VSSC database, employing more stringent diagnostic criteria to ensure the highest level of certainty in ascertaining epilepsy. It is important to note there is a separate category of probable epilepsy, which involves a broader set of diagnostic criteria. While not covered in this study, probable epilepsy would be subject to future research and validation, which could provide insights into a wider spectrum of epilepsy diagnoses. Such future research could help refine the algorithm’s applicability and accuracy and potentially lead to more comprehensive surveillance and management strategies in clinical practice.
This study highlights the inherent challenges in leveraging administrative data for disease identification, particularly for conditions such as epilepsy, where diagnostic clarity can be complex. However, other conditions such as multiple sclerosis have noted similar success with the use of VHA administrative data for categorizing disease.17
Limitations
The algorithm discussed in this article is, in and of itself, generalizable. However, the validation process was unique to the VHA patient population, limiting the generalizability of the findings. Documentation practices and HCP attitudes within the VHA may differ from those in other health care settings. Identifying people with epilepsy can be challenging because of changing definitions of epilepsy over time. In addition to clinical evaluation, EEG and magnetic resonance imaging results, response to ASM treatment, and video-EEG monitoring of habitual events all can help establish the diagnosis. Therefore, studies may vary in how inclusive or exclusive the criteria are. ASMs such as gabapentin, pregabalin, carbamazepine, lamotrigine, topiramate, and valproate are used to treat other conditions, including headaches, generalized pain, and mood disorders. Consequently, including these ASMs in the Tier 1 definition may have increased the false positive rate. Additional research is needed to evaluate whether excluding these ASMs from the algorithm based on specific criteria (eg, dose of ASM used) can further refine the algorithm to identify patients with epilepsy.
Further refinement of this algorithm may also occur as technology changes. Future electronic health records may allow better tracking of different epilepsy factors, the integration of additional diagnostic criteria, and the use of natural language processing or other forms of artificial intelligence.
Conclusions
This study presents a significant step forward in epilepsy surveillance within the VHA. The algorithm offers a robust tool for identifying VWEs with good PPVs, facilitating better resource allocation and targeted care. Despite its limitations, this research lays a foundation for future advancements in the management and understanding of epilepsy within large health care systems. Since this VHA algorithm is based on ASMs and ICD diagnosis codes from patient records, other large managed health care systems also may be able to adapt this algorithm to their data specifications.
Epilepsy affects about 4.5 million people in the United States and 150,000 new individuals are diagnosed each year.1,2 In 2019, epilepsy-attributable health care spending for noninstitutionalized people was around $5.4 billion and total epilepsy-attributable and epilepsy or seizure health care-related costs totaled $54 billion.3
Accurate surveillance of epilepsy in large health care systems can potentially improve health care delivery and resource allocation. A 2012 Institute of Medicine (IOM) report identified 13 recommendations to guide public health action on epilepsy, including validation of standard definitions for case ascertainment, identification of epilepsy through screening programs or protocols, and expansion of surveillance to better understand disease burden.4
A systematic review of validation studies concluded that it is reasonable to use administrative data to identify people with epilepsy in epidemiologic research. Combining The International Classification of Diseases (ICD) codes for epilepsy (ICD-10, G40-41; ICD-9, 345) with antiseizure medications (ASMs) could provide high positive predictive values (PPVs) and combining symptoms codes for convulsions (ICD-10, R56; ICD-9, 780.3, 780.39) with ASMs could lead to high sensitivity.5 However, identifying individuals with epilepsy from administrative data in large managed health care organizations is challenging.6 The IOM report noted that large managed health care organizations presented varying incidence and prevalence estimates due to differing methodology, geographic area, demographics, and definitions of epilepsy.
The Veterans Health Administration (VHA) is the largest integrated US health care system, providing care to > 9.1 million veterans.7 To improve the health and well-being of veterans with epilepsy (VWEs), a network of sites was established in 2008 called the US Department of Veterans Affairs (VA) Epilepsy Centers of Excellence (ECoE). Subsequent to the creation of the ECoE, efforts were made to identify VWEs within VHA databases.8,9 Prior to fiscal year (FY) 2016, the ECoE adopted a modified version of a well-established epilepsy diagnostic algorithm developed by Holden et al for large managed care organizations.10 The original algorithm identified patients by cross-matching ASMs with ICD-9 codes for an index year. But it failed to capture a considerable number of stable patients with epilepsy in the VHA due to incomplete documentation, and had false positives due to inclusion of patients identified from diagnostic clinics. The modified algorithm the ECoE used prior to FY 2016 considered additional prior years and excluded encounters from diagnostic clinics. The result was an improvement in the sensitivity and specificity of the algorithm. Researchers evaluating 500 patients with epilepsy estimated that the modified algorithm had a PPV of 82.0% (95% CI, 78.6%-85.4%).11
After implementation of ICD-10 codes in the VHA in FY 2016, the task of reliably and efficiently identifying VWE led to a 3-tier algorithm. This article presents a validation of the different tiers of this algorithm after the implementation of ICD-10 diagnosis codes and summarizes the surveillance data collected over the years within the VHA showing the trends of epilepsy.
Methods
The VHA National Neurology office commissioned a Neurology Cube dashboard in FY 2021 in collaboration with VHA Support Service Center (VSSC) for reporting and surveillance of VWEs as a quality improvement initiative. The Neurology Cube uses a 3-tier system for identifying VWE in the VHA databases. VSSC programmers extract data from the VHA Corporate Data Warehouse (CDW) and utilize Microsoft SQL Server and Microsoft Power BI for Neurology Cube reports. The 3-tier system identifies VWE and divides them into distinct groups. The first tier identifies VWE with the highest degree of confidence; Tiers 2 and 3 represent identification with successively lesser degrees of confidence (Figure 1).
Tier 1
Definition. For a given index year and the preceding 2 years, any of following diagnosis codes on ≥ 1 clinical encounter are considered: 345.xx (epilepsy in ICD-9), 780.3x (other convulsions in ICD-9), G40.xxx (epilepsy in ICD-10), R40.4 (transient alteration of awareness), R56.1 (posttraumatic seizures), or R56.9 (unspecified convulsions). To reduce false positive rates, EEG clinic visits, which may include long-term monitoring, are excluded. Patients identified with ICD codes are then evaluated for an ASM prescription for ≥ 30 days during the index year. ASMs are listed in Appendix 1.
Validation. The development and validation of ICD-9 diagnosis codes crossmatched with an ASM prescription in the VHA has been published elsewhere.11 In FY 2017, after implementation of ICD-10 diagnostic codes, Tier 1 development and validation was performed in 2 phases. Even though Tier 1 study phases were conducted and completed during FY 2017, the patients for Tier 1 were identified from evaluation of FY 2016 data (October 1, 2015, to September 30, 2016). After the pilot analysis, the Tier 1 definition was implemented, and a chart review of 625 randomized patients was conducted at 5 sites for validation. Adequate preliminary data was not available to perform a sample size estimation for this study. Therefore, a practical target of 125 patients was set for Tier 1 from each site to obtain a final sample size of 625 patients. This second phase validated that the crossmatch of ICD-10 diagnosis codes with ASMs had a high PPV for identifying VWE.
Tiers 2 and 3
Definitions. For an index year, Tier 2 includes patients with ≥ 1 inpatient encounter documentation of either ICD-9 345.xx or ICD-10 G40.xxx, excluding EEG clinics. Tier 3 Includes patients who have had ≥ 2 outpatient encounters with diagnosis codes 345.xx or G40.xxx on 2 separate days, excluding EEG clinics. Tiers 2 and 3 do not require ASM prescriptions; this helps to identify VWEs who may be getting their medications outside of VHA or those who have received a new diagnosis.
Validations. Tiers 2 and 3 were included in the epilepsy identification algorithm in FY 2021 after validation was performed on a sample of 8 patients in each tier. Five patients were subsequently identified as having epilepsy in Tier 2 and 6 patients were identified in Tier 3. A more comprehensive validation of Tiers 2 and 3 was performed during FY 2022 that included patients at 5 sites seen during FY 2019 to FY 2022. Since yearly trends showed only about 8% of total patients were identified as having epilepsy through Tiers 2 and 3 we sought ≥ 20 patients per tier for the 5 sites for a total of 200 patients to ensure representation across the VHA. The final count was 126 patients for Tier 2 and 174 patients for Tier 3 (n = 300).
Gold Standard Criteria for Epilepsy Diagnosis
We used the International League Against Epilepsy (ILAE) definition of epilepsy for the validation of the 3 algorithm tiers. ILAE defines epilepsy as ≥ 2 unprovoked (or reflex) seizures occurring > 24 hours apart or 1 unprovoked (or reflex) seizure and a probability of further seizures similar to the general recurrence risk (≥ 60%) after 2 unprovoked seizures, occurring over the next 10 years.12
A standard protocol was provided to evaluators to identify patients using the VHA Computerized Patient Record System (Appendix 1). After review, evaluators categorized each patient in 1 of 4 ways: (1) Yes, definite: The patient’s health care practitioner (HCP) believes the patient has epilepsy and is treating with medication; (2) Yes, uncertain: The HCP has enough suspicion of epilepsy that a medication is prescribed, but uncertainty is expressed of the diagnosis; (3) No, definite: The HCP does not believe the patient has epilepsy and is therefore not treating with medication for seizure; (4) No, uncertain: The HCP is not treating with medication for epilepsy, because the diagnostic suspicion is not high enough, but there is suspicion for epilepsy.
As a quality improvement operational project, the Epilepsy National Program Office approved this validation project and determined that institutional review board approval was not required.
Statistical Analysis
Counts and percentages were computed for categories of epilepsy status. PPV of each tier was estimated with asymptotic 95% CIs.
Results
ICD-10 codes for 480 patients were evaluated in Tier 1 phase 1; 13.8% were documented with G40.xxx, 27.9% with R56.1, 34.4% with R56.9, and 24.0% with R40.4 (Appendix 2). In total, 68.1% fulfilled the criteria of epilepsy, 19.2% did not, and 12.7% were uncertain). From the validation of Tier 1 phase 2 (n = 625), the PPV of the algorithm for patients presumed to have epilepsy (definite and uncertain) was 85.1% (95% CI, 82.1%-87.8%) (Table).
Of 300 patients evaluated, 126 (42.0%) were evaluated for Tier 2 with a PPV of 61.9% (95% CI, 53.4%-70.4%), and 174 (58.0%) patients were evaluated for Tier 3 with a PPV of 59.8% (95% CI, 52.5%-67.1%. The PPV of the algorithm for patients presumed to have epilepsy (definite and uncertain) were combined to calculate the PPV. Estimates of VHA VWE counts were computed for each tier from FY 2014 to FY 2023 using the VSSC Neurology Cube (Figure 2). For all years, > 92% patients were classified using the Tier 1 definition.
Discussion
The development and validation of the 3-tier diagnostic algorithm represents an important advancement in the surveillance and management of epilepsy among veterans within the VHA. The validation of this algorithm also demonstrates its practical utility in a large, integrated health care system.
Specific challenges were encountered when attempting to use pre-existing algorithms; these challenges included differences in the usage patterns of diagnostic codes and the patterns of ASM use within the VHA. These challenges prompted the need for a tailored approach, which led to the development of this algorithm. The inclusion of additional ICD-10 codes led to further revisions and subsequent validation. While many of the basic concepts of the algorithm, including ICD codes and ASMs, could work in other institutions, it would be wise for health care organizations to develop their own algorithms because of certain variables, including organizational size, patient demographics, common comorbidities, and the specific configurations of electronic health records and administrative data systems.
Studies have shown that ICD-10 codes for epilepsy (G40.* and/or R56.9) perform well in identifying epilepsy whether they are assigned by neurologists (sensitivity, 97.7%; specificity, 44.1%; PPV, 96.2%; negative predictive value, 57.7%), or in emergency department or hospital discharges (PPV, 75.5%).13,14 The pilot study of the algorithm’s Tier 1 development (phase 1) evaluated whether the selected ICD-10 diagnostic codes accurately included the VWE population within the VHA and revealed that while most codes (eg, epilepsy [G40.xxx]; posttraumatic seizures [R56.1]; and unspecified convulsions [R56.9]), had a low false positive rate (< 16%), the R40.4 code (transient alteration of awareness) had a higher false positivity of 42%. While this is not surprising given the broad spectrum of conditions that can manifest as transient alteration of awareness, it underscores the inherent challenges in diagnosing epilepsy using diagnosis codes.
In phase 2, the Tier 1 algorithm was validated as effective for identifying VWE in the VHA system, as its PPV was determined to be high (85%). In comparison, Tiers 2 and 3, whose criteria did not require data on VHA prescribed ASM use, had lower tiers of epilepsy predictability (PPV about 60% for both). This was thought to be acceptable because Tiers 2 and 3 represent a smaller population of the identified VWEs (about 8%). These VWEs may otherwise have been missed, partly because veterans are not required to get ASMs from the VHA.
Upon VHA implementation in FY 2021, this diagnostic algorithm exhibited significant clinical utility when integrated within the VSSC Neurology Cube. It facilitated an efficient approach to identifying VWEs using readily available databases. This led to better tracking of real-time epilepsy cases, which facilitated improving current resource allocation and targeted intervention strategies such as identification of drug-resistant epilepsy patients, optimizing strategies for telehealth and patient outreach for awareness of epilepsy care resources within VHA. Meanwhile, data acquired by the algorithm over the decade since its development (FY 2014 to FY 2023) contributed to more accurate epidemiologic information and identification of historic trends. Development of the algorithm represents one of the ways ECoEs have led to improved care for VWEs. ECoEs have been shown to improve health care for veterans in several metrics.15
A strength of this study is the rigorous multitiered validation process to confirm the diagnostic accuracy of ICD-10 codes against the gold standard ILAE definition of epilepsy to identify “definite” epilepsy cases within the VHA. The use of specific ICD codes further enhances the precision of epilepsy diagnoses. The inclusion of ASMs, which are sometimes prescribed for conditions other than epilepsy, could potentially inflate false positive rates.16
This study focused exclusively on the identification and validation of definite epilepsy cases within the VHA VSSC database, employing more stringent diagnostic criteria to ensure the highest level of certainty in ascertaining epilepsy. It is important to note there is a separate category of probable epilepsy, which involves a broader set of diagnostic criteria. While not covered in this study, probable epilepsy would be subject to future research and validation, which could provide insights into a wider spectrum of epilepsy diagnoses. Such future research could help refine the algorithm’s applicability and accuracy and potentially lead to more comprehensive surveillance and management strategies in clinical practice.
This study highlights the inherent challenges in leveraging administrative data for disease identification, particularly for conditions such as epilepsy, where diagnostic clarity can be complex. However, other conditions such as multiple sclerosis have noted similar success with the use of VHA administrative data for categorizing disease.17
Limitations
The algorithm discussed in this article is, in and of itself, generalizable. However, the validation process was unique to the VHA patient population, limiting the generalizability of the findings. Documentation practices and HCP attitudes within the VHA may differ from those in other health care settings. Identifying people with epilepsy can be challenging because of changing definitions of epilepsy over time. In addition to clinical evaluation, EEG and magnetic resonance imaging results, response to ASM treatment, and video-EEG monitoring of habitual events all can help establish the diagnosis. Therefore, studies may vary in how inclusive or exclusive the criteria are. ASMs such as gabapentin, pregabalin, carbamazepine, lamotrigine, topiramate, and valproate are used to treat other conditions, including headaches, generalized pain, and mood disorders. Consequently, including these ASMs in the Tier 1 definition may have increased the false positive rate. Additional research is needed to evaluate whether excluding these ASMs from the algorithm based on specific criteria (eg, dose of ASM used) can further refine the algorithm to identify patients with epilepsy.
Further refinement of this algorithm may also occur as technology changes. Future electronic health records may allow better tracking of different epilepsy factors, the integration of additional diagnostic criteria, and the use of natural language processing or other forms of artificial intelligence.
Conclusions
This study presents a significant step forward in epilepsy surveillance within the VHA. The algorithm offers a robust tool for identifying VWEs with good PPVs, facilitating better resource allocation and targeted care. Despite its limitations, this research lays a foundation for future advancements in the management and understanding of epilepsy within large health care systems. Since this VHA algorithm is based on ASMs and ICD diagnosis codes from patient records, other large managed health care systems also may be able to adapt this algorithm to their data specifications.
- Kobau R, Luncheon C, Greenlund K. Active epilepsy prevalence among U.S. adults is 1.1% and differs by educational level-National Health Interview Survey, United States, 2021. Epilepsy Behav. 2023;142:109180. doi:10.1016/j.yebeh.2023.109180
- GBD 2017 US Neurological Disorders Collaborators, Feigin VL, Vos T, et al. Burden of neurological disorders across the US from 1990-2017: a global burden of disease study. JAMA Neurol. 2021;78:165-176. doi:10.1001/jamaneurol.2020.4152
- Moura LMVR, Karakis I, Zack MM, et al. Drivers of US health care spending for persons with seizures and/or epilepsies, 2010-2018. Epilepsia. 2022;63:2144-2154. doi:10.1111/epi.17305
- Institute of Medicine. Epilepsy Across the Spectrum: Promoting Health and Understanding. The National Academies Press; 2012. Accessed November 11, 2025. www.nap.edu/catalog/13379
- Mbizvo GK, Bennett KH, Schnier C, Simpson CR, Duncan SE, Chin RFM. The accuracy of using administrative healthcare data to identify epilepsy cases: A systematic review of validation studies. Epilepsia. 2020;61:1319-1335. doi:10.1111/epi.16547
- Montouris GD. How will primary care physicians, specialists, and managed care treat epilepsy in the new millennium? Neurology. 2000;55:S42-S44.
- US Department of Veterans Affairs. Veterans Health Administration: About VHA. Accessed November 11, 2025. https://www.va.gov/health/aboutvha.asp
- Veterans’ Mental Health and Other Care Improvements Act of 2008, S 2162, 110th Cong (2008). Accessed November 11, 2025. https://www.congress.gov/bill/110th-congress/senate-bill/2162
- Rehman R, Kelly PR, Husain AM, Tran TT. Characteristics of Veterans diagnosed with seizures within Veterans Health Administration. J Rehabil Res Dev. 2015;52(7):751-762. doi:10.1682/JRRD.2014.10.0241
- Holden EW, Grossman E, Nguyen HT, et al. Developing a computer algorithm to identify epilepsy cases in managed care organizations. Dis Manag. 2005;8:1-14. doi:10.1089/dis.2005.8.1
- Rehman R, Everhart A, Frontera AT, et al. Implementation of an established algorithm and modifications for the identification of epilepsy patients in the Veterans Health Administration. Epilepsy Res. 2016;127:284-290. doi:10.1016/j.eplepsyres.2016.09.012
- Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55:475-482. doi:10.1111/epi.12550
- Smith JR, Jones FJS, Fureman BE, et al. Accuracy of ICD-10-CM claims-based definitions for epilepsy and seizure type. Epilepsy Res. 2020;166:106414. doi:10.1016/j.eplepsyres.2020.106414
- Jetté N, Reid AY, Quan H, et al. How accurate is ICD coding for epilepsy? Epilepsia. 2010;51:62-69. doi:10.1111/j.1528-1167.2009.02201.x
- Kelly P, Chinta R, Privitera G. Do centers of excellence reduce health care costs? Evidence from the US Veterans Health Administration Centers for Epilepsy. Glob Bus Organ Excell. 2015;34:18-29.
- Haneef Z, Rehman R, Husain AM. Association between standardized mortality ratio and utilization of care in US veterans with drug-resistant epilepsy compared with all US veterans and the US general population. JAMA Neurol. 2022;79:879-887. doi:10.1001/jamaneurol.2022.2290
- Culpepper WJ, Marrie RA, Langer-Gould A, et al. Validation of an algorithm for identifying MS cases in administrative health claims datasets. Neurology. 2019;92:e1016-e1028 doi:10.1212/WNL.0000000000007043
- Kobau R, Luncheon C, Greenlund K. Active epilepsy prevalence among U.S. adults is 1.1% and differs by educational level-National Health Interview Survey, United States, 2021. Epilepsy Behav. 2023;142:109180. doi:10.1016/j.yebeh.2023.109180
- GBD 2017 US Neurological Disorders Collaborators, Feigin VL, Vos T, et al. Burden of neurological disorders across the US from 1990-2017: a global burden of disease study. JAMA Neurol. 2021;78:165-176. doi:10.1001/jamaneurol.2020.4152
- Moura LMVR, Karakis I, Zack MM, et al. Drivers of US health care spending for persons with seizures and/or epilepsies, 2010-2018. Epilepsia. 2022;63:2144-2154. doi:10.1111/epi.17305
- Institute of Medicine. Epilepsy Across the Spectrum: Promoting Health and Understanding. The National Academies Press; 2012. Accessed November 11, 2025. www.nap.edu/catalog/13379
- Mbizvo GK, Bennett KH, Schnier C, Simpson CR, Duncan SE, Chin RFM. The accuracy of using administrative healthcare data to identify epilepsy cases: A systematic review of validation studies. Epilepsia. 2020;61:1319-1335. doi:10.1111/epi.16547
- Montouris GD. How will primary care physicians, specialists, and managed care treat epilepsy in the new millennium? Neurology. 2000;55:S42-S44.
- US Department of Veterans Affairs. Veterans Health Administration: About VHA. Accessed November 11, 2025. https://www.va.gov/health/aboutvha.asp
- Veterans’ Mental Health and Other Care Improvements Act of 2008, S 2162, 110th Cong (2008). Accessed November 11, 2025. https://www.congress.gov/bill/110th-congress/senate-bill/2162
- Rehman R, Kelly PR, Husain AM, Tran TT. Characteristics of Veterans diagnosed with seizures within Veterans Health Administration. J Rehabil Res Dev. 2015;52(7):751-762. doi:10.1682/JRRD.2014.10.0241
- Holden EW, Grossman E, Nguyen HT, et al. Developing a computer algorithm to identify epilepsy cases in managed care organizations. Dis Manag. 2005;8:1-14. doi:10.1089/dis.2005.8.1
- Rehman R, Everhart A, Frontera AT, et al. Implementation of an established algorithm and modifications for the identification of epilepsy patients in the Veterans Health Administration. Epilepsy Res. 2016;127:284-290. doi:10.1016/j.eplepsyres.2016.09.012
- Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55:475-482. doi:10.1111/epi.12550
- Smith JR, Jones FJS, Fureman BE, et al. Accuracy of ICD-10-CM claims-based definitions for epilepsy and seizure type. Epilepsy Res. 2020;166:106414. doi:10.1016/j.eplepsyres.2020.106414
- Jetté N, Reid AY, Quan H, et al. How accurate is ICD coding for epilepsy? Epilepsia. 2010;51:62-69. doi:10.1111/j.1528-1167.2009.02201.x
- Kelly P, Chinta R, Privitera G. Do centers of excellence reduce health care costs? Evidence from the US Veterans Health Administration Centers for Epilepsy. Glob Bus Organ Excell. 2015;34:18-29.
- Haneef Z, Rehman R, Husain AM. Association between standardized mortality ratio and utilization of care in US veterans with drug-resistant epilepsy compared with all US veterans and the US general population. JAMA Neurol. 2022;79:879-887. doi:10.1001/jamaneurol.2022.2290
- Culpepper WJ, Marrie RA, Langer-Gould A, et al. Validation of an algorithm for identifying MS cases in administrative health claims datasets. Neurology. 2019;92:e1016-e1028 doi:10.1212/WNL.0000000000007043
Development and Validation of an Administrative Algorithm to Identify Veterans With Epilepsy
Development and Validation of an Administrative Algorithm to Identify Veterans With Epilepsy

Clinical Presentation of Subacute Combined Degeneration in a Patient With Chronic B12 Deficiency
Subacute combined degeneration (SCD) is an acquired neurologic complication of vitamin B12 (cobalamin) or, rarely, vitamin B9 (folate) deficiency. SCD is characterized by progressive demyelination of the dorsal and lateral spinal cord, resulting in peripheral neuropathy; gait ataxia; impaired proprioception, vibration, and fine touch; optic neuropathy; and cognitive impairment.1 In addition to SCD, other neurologic manifestations of B12 deficiency include dementia, depression, visual symptoms due to optic atrophy, and behavioral changes.2 The prevalence of SCD in the US has not been well documented, but B12 deficiency is reported at 6% in those aged < 60 years and 20% in those > 60 years.3
Causes of B12 and B9 deficiency include advanced age, low nutritional intake (eg, vegan diet), impaired absorption (eg, inflammatory bowel disease, autoimmune pernicious anemia, gastrectomy, pancreatic disease), alcohol use, tapeworm infection, medications, and high metabolic states.2,4 Impaired B12 absorption is common in patients taking medications, such as metformin and proton pump inhibitors (PPI), due to suppression of ileal membrane transport and intrinsic factor activity.5-7 B-vitamin deficiency can be exacerbated by states of increased cellular turnover, such as polycythemia vera, due to elevated DNA synthesis.
Patients may experience permanent neurologic damage when the diagnosis and treatment of SCD are missed or delayed. Early diagnosis of SCD can be challenging due to lack of specific hematologic markers. In addition, many other conditions such as diabetic neuropathy, malnutrition, toxic neuropathy, sarcoidosis, HIV, multiple sclerosis, polycythemia vera, and iron deficiency anemia have similar presentations and clinical findings.8 Anemia and/or macrocytosis are not specific to B12 deficiency.4 In addition, patients with B12 deficiency may have a normal complete blood count (CBC); those with concomitant iron deficiency may have minimal or no mean corpuscular volume (MCV) elevation.4 In patients suspected to have B12 deficiency based on clinical presentation or laboratory findings of macrocytosis, serum methylmalonic acid (MMA) can serve as a direct measure of B12 activity, with levels > 0.75 μmol/L almost always indicating cobalamin deficiency. 9 On the other hand, plasma total homocysteine (tHcy) is a sensitive marker for B12 deficiency. The active form of B12, holotranscobalamin, has also emerged as a specific measure of B12 deficiency.9 However, in patients with SCD, measurement of these markers may be unnecessary due to the severity of their clinical symptoms.
The diagnosis of SCD is further complicated because not all individuals who develop B12 or B9 deficiency will develop SCD. It is difficult to determine which patients will develop SCD because the minimum level of serum B12 required for normal function is unknown, and recent studies indicate that SCD may occur even at low-normal B12 and B9 levels.2,4,10 Commonly, a serum B12 level of < 200 pg/mL is considered deficient, while a level between 200 and 300 pg/mL is considered borderline.4 The goal level of serum B12 is > 300 pg/mL, which is considered normal.4 While serologic findings of B-vitamin deficiency are only moderately specific, radiographic findings are highly sensitive and specific for SCD. According to Briani and colleagues, the most consistent finding in SCD on magnetic resonance imaging (MRI) is a “symmetrical, abnormally increased T2 signal intensity, commonly confined to posterior or posterior and lateral columns in the cervical and thoracic spinal cord.”2
We present a case of SCD in a patient with low-normal vitamin B12 levels who presented with progressive sensorimotor deficits and vision loss. The patient was subsequently diagnosed with SCD by radiologic workup. His course was complicated by worsening neurologic deficits despite B12 replacement. The progression of his clinical symptoms demonstrates the need for prompt, aggressive B12 replacement in patients diagnosed with SCD.
Case Presentation
A 63-year-old man presented for neurologic evaluation of progressive gait disturbance, paresthesia, blurred vision, and increasing falls despite use of a walker. Pertinent medical history included polycythemia vera requiring phlebotomy for approximately 9 years, alcohol use disorder (18 servings weekly), type 2 diabetes mellitus, and a remote episode of transient ischemic attack (TIA). The patient reported a 5-year history of burning pain in all extremities. A prior physician diagnosis attributed the symptoms to polyneuropathy secondary to iron deficiency anemia in the setting of chronic phlebotomy for polycythemia vera and high erythrogenesis. He was prescribed gabapentin 600 mg 3 times daily for pain control. B12 deficiency was considered an unlikely etiology due to a low-normal serum level of 305 pg/mL (reference range, 190-950 pg/mL) and normocytosis, with MCV of 88 fL (reference range, 80-100 fL). The patient also reported a 3-year history of blurred vision, which was initially attributed to be secondary to diabetic retinopathy. One week prior to presenting to our clinic, he was evaluated by ophthalmology for new-onset, bilateral central visual field defects, and he was diagnosed with nutritional optic neuropathy.
Ophthalmology suspected B12 deficiency. Notable findings included reduced deep tendon reflexes (DTRs) in the upper extremities and absent DTRs in the lower extremities, reduced sensation to light touch in all extremities, absent sensation to pinprick, vibration, and temperature in the lower extremities, positive Romberg sign, and a wide-based antalgic gait with the ankles externally rotated bilaterally (Table 1)
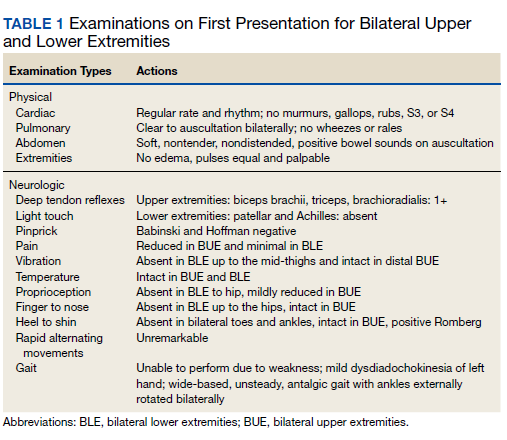
Previous cardiac evaluation failed to provide a diagnosis for syncopal episodes. MRI of the brain revealed nonspecific white matter changes consistent with chronic microvascular ischemic disease. Electromyography was limited due to pain but showed severe peripheral neuropathy. Laboratory results showed megalocytosis, low-normal serum B12 levels, and low serum folate levels (Table 2). The patient was diagnosed with polyneuropathy and was given intramuscular (IM) vitamin B12 1000 mcg once and a daily multivitamin (containing 25 mcg of B12). He was counseled on alcohol abstinence and medication adherence and was scheduled for follow-up in 3 months. He continued outpatient phlebotomy every 6 weeks for polycythemia.
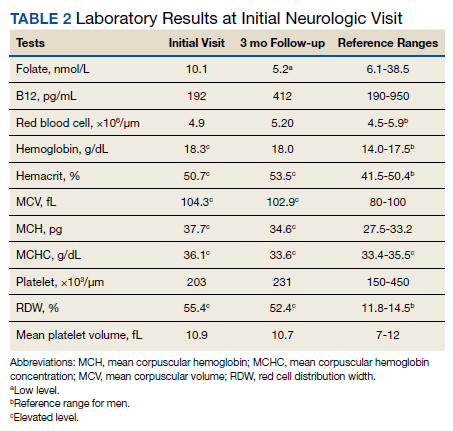
At 3-month follow-up, the patient reported medication adherence, continued alcohol use, and worsening of symptoms. Falls, which now occurred 2 to 3 times weekly despite proper use of a walker, were described as sudden loss of bilateral lower extremity strength without loss of consciousness, palpitations, or other prodrome. Laboratory results showed minimal changes. Physical examination of the patient demonstrated similar deficits as on initial presentation. The patient received one additional B12 1000 mcg IM. Gabapentin was replaced with pregabalin 75 mg twice daily due to persistent uncontrolled pain and paresthesia. The patient was scheduled for a 3-month followup (6 months from initial visit) and repeat serology.
At 6-month follow-up, the patient showed continued progression of disease with significant difficulty using the walker, worsening falls, and wheelchair use required. Physical examination showed decreased sensation bilaterally up to the knees, absent bilateral patellar and Achilles reflexes, and unsteady gait. Laboratory results showed persistent subclinical B12 deficiency. MRI of the brain and spine showed high T2 signaling in a pattern highly specific for SCD. A formal diagnosis of SCD was made. The patient received an additional B12 1000 mcg IM once. Follow-up phone call with the patient 1 month later revealed no progression or improvement of symptoms.
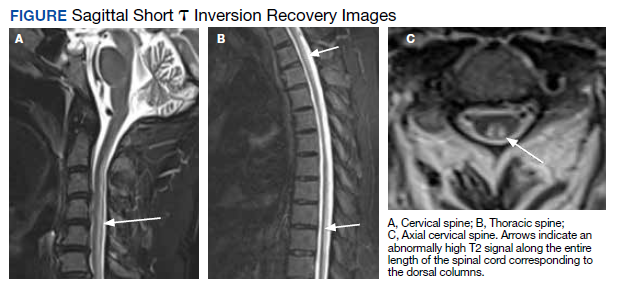
Radiographic Findings
MRI of the cervical and thoracic spine demonstrated abnormal high T2 signal starting from C2 and extending along the course of the cervical and thoracic spinal cord (Figure). MRI in SCD classically shows symmetric, bilateral high T2 signal within the dorsal columns; on axial images, there is typically an inverted “V” sign.2,4 There can also be abnormal cerebral white matter change; however, MRI of the brain in this patient did not show any abnormalities.2 The imaging differential for this appearance includes other metabolic deficiencies/toxicities: copper deficiency; vitamin E deficiency; methotrexateinduced myelopathy, and infectious causes: HIV vacuolar myelopathy; and neurosyphilis (tabes dorsalis).4
Discussion
This case demonstrates the clinical and radiographic findings of SCD and underscores the need for high-intensity dosing of B12 replacement in patients with SCD to prevent progression of the disease and development of morbidities.
Symptoms of SCD may manifest even when the vitamin levels are in low-normal levels. Its presentation is often nonspecific, thus radiologic workup is beneficial to elucidate the clinical picture. We support the use of spinal MRI in patients with clinical suspicion of SCD to help rule out other causes of myelopathy. However, an MRI is not indicated in all patients with B12 deficiency, especially those without myelopathic symptoms. Additionally, follow-up spinal MRIs are useful in monitoring the progression or improvement of SCD after B12 replacement.2 It is important to note that the MRI findings in SCD are not specific to B12 deficiency; other causes may present with similar radiographic findings.4 Therefore, radiologic findings must be correlated with a patient’s clinical presentation.
B12 replacement improves and may resolve clinical symptoms and abnormal radiographic findings of SCD. The treatment duration of B12 deficiency depends on the underlying etiology. Reversible causes, such as metformin use > 4 months, PPI use > 12 months, and dietary deficiency, require treatment until appropriate levels are reached and symptoms are resolved.4,11 The need for chronic metformin and PPI use should also be reassessed regularly. In patients who require long-term metformin use, IM administration of B12 1000 mcg annually should be considered, which will ensure adequate storage for more than 1 year.12,13 In patients who require long-term PPI use, the risk and benefits of continued use should be measured, and if needed, the lowest possible effective PPI dose is recommended.14 Irreversible causes of B12 deficiency, such as advanced age, prior gastrectomy, chronic pancreatitis, or autoimmune pernicious anemia, require lifelong supplementation of B12.4,11
In general, oral vitamin B12 replacement at 1000 to 2000 mcg daily may be as effective as parenteral replacement in patients with mild to moderate deficiency or neurologic symptoms.11 On the other hand, patients with SCD often require parenteral replacement of B12 due to the severity of their deficiency or neurologic symptoms, need for more rapid improvement in symptoms, and prevention of irreversible neurological deficits. 4,11 Appropriate B12 replacement in SCD requires intensive initial therapy which may involve IM B12 1000 mcg every other day for 2 weeks and additional IM supplementation every 2 to 3 months afterward until resolution of deficiency.4,14 IM replacement may also be considered in patients who are nonadherent to oral replacement or have an underlying gastrointestinal condition that impairs enteral absorption.4,11
B12 deficiency is frequently undertreated and can lead to progression of disease with significant morbidity. The need for highintensity dosing of B12 replacement is crucial in patients with SCD. Failure to respond to treatment, as shown from the lack of improvement of serum markers or symptoms, likely suggests undertreatment, treatment nonadherence, iron deficiency anemia, an unidentified malabsorption syndrome, or other diagnoses. In our case, significant undertreatment, compounded by his suspected iron deficiency anemia secondary to his polycythemia vera and chronic phlebotomies, are the most likel etiologies for his lack of clinical improvement.
Multiple factors may affect the prognosis of SCD. Males aged < 50 years with absence of anemia, spinal cord atrophy, Romberg sign, Babinski sign, or sensory deficits on examination have increased likelihood of eventual recovery of signs and symptoms of SCD; those with less spinal cord involvement (< 7 cord segments), contrast enhancement, and spinal cord edema also have improved outcomes.4,15
Conclusion
SCD is a rare but serious complication of chronic vitamin B12 deficiency that presents with a variety of neurological findings and may be easily confused with other illnesses. The condition is easily overlooked or misdiagnosed; thus, it is crucial to differentiate B12 deficiency from other common causes of neurologic symptoms. Specific findings on MRI are useful to support the clinical diagnosis of SCD and guide clinical decisions. Given the prevalence of B12 deficiency in the older adult population, clinicians should remain alert to the possibility of these conditions in patients who present with progressive neuropathy. Once a patient is diagnosed with SCD secondary to a B12 deficiency, appropriate B12 replacement is critical. Appropriate B12 replacement is aggressive and involves IM B12 1000 mcg every other day for 2 to 3 weeks, followed by additional IM administration every 2 months before transitioning to oral therapy. As seen in this case, failure to adequately replenish B12 can lead to progression or lack of resolution of SCD symptoms.
1. Gürsoy AE, Kolukısa M, Babacan-Yıldız G, Celebi A. Subacute Combined Degeneration of the Spinal Cord due to Different Etiologies and Improvement of MRI Findings. Case Rep Neurol Med. 2013;2013:159649. doi:10.1155/2013/159649
2. Briani C, Dalla Torre C, Citton V, et al. Cobalamin deficiency: clinical picture and radiological findings. Nutrients. 2013;5(11):4521-4539. Published 2013 Nov 15. doi:10.3390/nu5114521
3. Hunt A, Harrington D, Robinson S. Vitamin B12 deficiency. BMJ. 2014;349:g5226. Published 2014 Sep 4. doi:10.1136/bmj.g5226
4. Qudsiya Z, De Jesus O. Subacute combined degeneration of the spinal cord. [Updated 2021 Feb 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Updated August 30, 2021. Accessed January 5, 2022. https://www.ncbi.nlm.nih.gov/books /NBK559316/
5. de Jager J, Kooy A, Lehert P, et al. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: randomised placebo controlled trial. BMJ. 2010;340:c2181. Published 2010 May 20. doi:10.1136/bmj.c2181
6. Aroda VR, Edelstein SL, Goldberg RB, et al. Longterm Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study. J Clin Endocrinol Metab. 2016;101(4):1754-1761. doi:10.1210/jc.2015-3754
7. Lam JR, Schneider JL, Zhao W, Corley DA. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin B12 deficiency. JAMA. 2013;310(22):2435-2442. doi:10.1001/jama.2013.280490
8. Mihalj M, Titlic´ M, Bonacin D, Dogaš Z. Sensomotor axonal peripheral neuropathy as a first complication of polycythemia rubra vera: A report of 3 cases. Am J Case Rep. 2013;14:385-387. Published 2013 Sep 25. doi:10.12659/AJCR.884016
9. Devalia V, Hamilton MS, Molloy AM; British Committee for Standards in Haematology. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br J Haematol. 2014;166(4):496-513. doi:10.1111/bjh.12959
10. Cao J, Xu S, Liu C. Is serum vitamin B12 decrease a necessity for the diagnosis of subacute combined degeneration?: A meta-analysis. Medicine (Baltimore). 2020;99(14):e19700.doi:10.1097/MD.0000000000019700
11. Langan RC, Goodbred AJ. Vitamin B12 Deficiency: Recognition and Management. Am Fam Physician. 2017;96(6):384-389.
12. Mazokopakis EE, Starakis IK. Recommendations for diagnosis and management of metformin-induced vitamin B12 (Cbl) deficiency. Diabetes Res Clin Pract. 2012;97(3):359-367. doi:10.1016/j.diabres.2012.06.001
13. Mahajan R, Gupta K. Revisiting Metformin: Annual Vitamin B12 Supplementation may become Mandatory with Long-Term Metformin Use. J Young Pharm. 2010;2(4):428-429. doi:10.4103/0975-1483.71621
14. Parks NE. Metabolic and Toxic Myelopathies. Continuum (Minneap Minn). 2021;27(1):143-162. doi:10.1212/CON.0000000000000963
15. Vasconcelos OM, Poehm EH, McCarter RJ, Campbell WW, Quezado ZM. Potential outcome factors in subacute combined degeneration: review of observational studies. J Gen Intern Med. 2006;21(10):1063-1068. doi:10.1111/j.1525-1497.2006.00525.x
Subacute combined degeneration (SCD) is an acquired neurologic complication of vitamin B12 (cobalamin) or, rarely, vitamin B9 (folate) deficiency. SCD is characterized by progressive demyelination of the dorsal and lateral spinal cord, resulting in peripheral neuropathy; gait ataxia; impaired proprioception, vibration, and fine touch; optic neuropathy; and cognitive impairment.1 In addition to SCD, other neurologic manifestations of B12 deficiency include dementia, depression, visual symptoms due to optic atrophy, and behavioral changes.2 The prevalence of SCD in the US has not been well documented, but B12 deficiency is reported at 6% in those aged < 60 years and 20% in those > 60 years.3
Causes of B12 and B9 deficiency include advanced age, low nutritional intake (eg, vegan diet), impaired absorption (eg, inflammatory bowel disease, autoimmune pernicious anemia, gastrectomy, pancreatic disease), alcohol use, tapeworm infection, medications, and high metabolic states.2,4 Impaired B12 absorption is common in patients taking medications, such as metformin and proton pump inhibitors (PPI), due to suppression of ileal membrane transport and intrinsic factor activity.5-7 B-vitamin deficiency can be exacerbated by states of increased cellular turnover, such as polycythemia vera, due to elevated DNA synthesis.
Patients may experience permanent neurologic damage when the diagnosis and treatment of SCD are missed or delayed. Early diagnosis of SCD can be challenging due to lack of specific hematologic markers. In addition, many other conditions such as diabetic neuropathy, malnutrition, toxic neuropathy, sarcoidosis, HIV, multiple sclerosis, polycythemia vera, and iron deficiency anemia have similar presentations and clinical findings.8 Anemia and/or macrocytosis are not specific to B12 deficiency.4 In addition, patients with B12 deficiency may have a normal complete blood count (CBC); those with concomitant iron deficiency may have minimal or no mean corpuscular volume (MCV) elevation.4 In patients suspected to have B12 deficiency based on clinical presentation or laboratory findings of macrocytosis, serum methylmalonic acid (MMA) can serve as a direct measure of B12 activity, with levels > 0.75 μmol/L almost always indicating cobalamin deficiency. 9 On the other hand, plasma total homocysteine (tHcy) is a sensitive marker for B12 deficiency. The active form of B12, holotranscobalamin, has also emerged as a specific measure of B12 deficiency.9 However, in patients with SCD, measurement of these markers may be unnecessary due to the severity of their clinical symptoms.
The diagnosis of SCD is further complicated because not all individuals who develop B12 or B9 deficiency will develop SCD. It is difficult to determine which patients will develop SCD because the minimum level of serum B12 required for normal function is unknown, and recent studies indicate that SCD may occur even at low-normal B12 and B9 levels.2,4,10 Commonly, a serum B12 level of < 200 pg/mL is considered deficient, while a level between 200 and 300 pg/mL is considered borderline.4 The goal level of serum B12 is > 300 pg/mL, which is considered normal.4 While serologic findings of B-vitamin deficiency are only moderately specific, radiographic findings are highly sensitive and specific for SCD. According to Briani and colleagues, the most consistent finding in SCD on magnetic resonance imaging (MRI) is a “symmetrical, abnormally increased T2 signal intensity, commonly confined to posterior or posterior and lateral columns in the cervical and thoracic spinal cord.”2
We present a case of SCD in a patient with low-normal vitamin B12 levels who presented with progressive sensorimotor deficits and vision loss. The patient was subsequently diagnosed with SCD by radiologic workup. His course was complicated by worsening neurologic deficits despite B12 replacement. The progression of his clinical symptoms demonstrates the need for prompt, aggressive B12 replacement in patients diagnosed with SCD.
Case Presentation
A 63-year-old man presented for neurologic evaluation of progressive gait disturbance, paresthesia, blurred vision, and increasing falls despite use of a walker. Pertinent medical history included polycythemia vera requiring phlebotomy for approximately 9 years, alcohol use disorder (18 servings weekly), type 2 diabetes mellitus, and a remote episode of transient ischemic attack (TIA). The patient reported a 5-year history of burning pain in all extremities. A prior physician diagnosis attributed the symptoms to polyneuropathy secondary to iron deficiency anemia in the setting of chronic phlebotomy for polycythemia vera and high erythrogenesis. He was prescribed gabapentin 600 mg 3 times daily for pain control. B12 deficiency was considered an unlikely etiology due to a low-normal serum level of 305 pg/mL (reference range, 190-950 pg/mL) and normocytosis, with MCV of 88 fL (reference range, 80-100 fL). The patient also reported a 3-year history of blurred vision, which was initially attributed to be secondary to diabetic retinopathy. One week prior to presenting to our clinic, he was evaluated by ophthalmology for new-onset, bilateral central visual field defects, and he was diagnosed with nutritional optic neuropathy.
Ophthalmology suspected B12 deficiency. Notable findings included reduced deep tendon reflexes (DTRs) in the upper extremities and absent DTRs in the lower extremities, reduced sensation to light touch in all extremities, absent sensation to pinprick, vibration, and temperature in the lower extremities, positive Romberg sign, and a wide-based antalgic gait with the ankles externally rotated bilaterally (Table 1)
Previous cardiac evaluation failed to provide a diagnosis for syncopal episodes. MRI of the brain revealed nonspecific white matter changes consistent with chronic microvascular ischemic disease. Electromyography was limited due to pain but showed severe peripheral neuropathy. Laboratory results showed megalocytosis, low-normal serum B12 levels, and low serum folate levels (Table 2). The patient was diagnosed with polyneuropathy and was given intramuscular (IM) vitamin B12 1000 mcg once and a daily multivitamin (containing 25 mcg of B12). He was counseled on alcohol abstinence and medication adherence and was scheduled for follow-up in 3 months. He continued outpatient phlebotomy every 6 weeks for polycythemia.
At 3-month follow-up, the patient reported medication adherence, continued alcohol use, and worsening of symptoms. Falls, which now occurred 2 to 3 times weekly despite proper use of a walker, were described as sudden loss of bilateral lower extremity strength without loss of consciousness, palpitations, or other prodrome. Laboratory results showed minimal changes. Physical examination of the patient demonstrated similar deficits as on initial presentation. The patient received one additional B12 1000 mcg IM. Gabapentin was replaced with pregabalin 75 mg twice daily due to persistent uncontrolled pain and paresthesia. The patient was scheduled for a 3-month followup (6 months from initial visit) and repeat serology.
At 6-month follow-up, the patient showed continued progression of disease with significant difficulty using the walker, worsening falls, and wheelchair use required. Physical examination showed decreased sensation bilaterally up to the knees, absent bilateral patellar and Achilles reflexes, and unsteady gait. Laboratory results showed persistent subclinical B12 deficiency. MRI of the brain and spine showed high T2 signaling in a pattern highly specific for SCD. A formal diagnosis of SCD was made. The patient received an additional B12 1000 mcg IM once. Follow-up phone call with the patient 1 month later revealed no progression or improvement of symptoms.
Radiographic Findings
MRI of the cervical and thoracic spine demonstrated abnormal high T2 signal starting from C2 and extending along the course of the cervical and thoracic spinal cord (Figure). MRI in SCD classically shows symmetric, bilateral high T2 signal within the dorsal columns; on axial images, there is typically an inverted “V” sign.2,4 There can also be abnormal cerebral white matter change; however, MRI of the brain in this patient did not show any abnormalities.2 The imaging differential for this appearance includes other metabolic deficiencies/toxicities: copper deficiency; vitamin E deficiency; methotrexateinduced myelopathy, and infectious causes: HIV vacuolar myelopathy; and neurosyphilis (tabes dorsalis).4
Discussion
This case demonstrates the clinical and radiographic findings of SCD and underscores the need for high-intensity dosing of B12 replacement in patients with SCD to prevent progression of the disease and development of morbidities.
Symptoms of SCD may manifest even when the vitamin levels are in low-normal levels. Its presentation is often nonspecific, thus radiologic workup is beneficial to elucidate the clinical picture. We support the use of spinal MRI in patients with clinical suspicion of SCD to help rule out other causes of myelopathy. However, an MRI is not indicated in all patients with B12 deficiency, especially those without myelopathic symptoms. Additionally, follow-up spinal MRIs are useful in monitoring the progression or improvement of SCD after B12 replacement.2 It is important to note that the MRI findings in SCD are not specific to B12 deficiency; other causes may present with similar radiographic findings.4 Therefore, radiologic findings must be correlated with a patient’s clinical presentation.
B12 replacement improves and may resolve clinical symptoms and abnormal radiographic findings of SCD. The treatment duration of B12 deficiency depends on the underlying etiology. Reversible causes, such as metformin use > 4 months, PPI use > 12 months, and dietary deficiency, require treatment until appropriate levels are reached and symptoms are resolved.4,11 The need for chronic metformin and PPI use should also be reassessed regularly. In patients who require long-term metformin use, IM administration of B12 1000 mcg annually should be considered, which will ensure adequate storage for more than 1 year.12,13 In patients who require long-term PPI use, the risk and benefits of continued use should be measured, and if needed, the lowest possible effective PPI dose is recommended.14 Irreversible causes of B12 deficiency, such as advanced age, prior gastrectomy, chronic pancreatitis, or autoimmune pernicious anemia, require lifelong supplementation of B12.4,11
In general, oral vitamin B12 replacement at 1000 to 2000 mcg daily may be as effective as parenteral replacement in patients with mild to moderate deficiency or neurologic symptoms.11 On the other hand, patients with SCD often require parenteral replacement of B12 due to the severity of their deficiency or neurologic symptoms, need for more rapid improvement in symptoms, and prevention of irreversible neurological deficits. 4,11 Appropriate B12 replacement in SCD requires intensive initial therapy which may involve IM B12 1000 mcg every other day for 2 weeks and additional IM supplementation every 2 to 3 months afterward until resolution of deficiency.4,14 IM replacement may also be considered in patients who are nonadherent to oral replacement or have an underlying gastrointestinal condition that impairs enteral absorption.4,11
B12 deficiency is frequently undertreated and can lead to progression of disease with significant morbidity. The need for highintensity dosing of B12 replacement is crucial in patients with SCD. Failure to respond to treatment, as shown from the lack of improvement of serum markers or symptoms, likely suggests undertreatment, treatment nonadherence, iron deficiency anemia, an unidentified malabsorption syndrome, or other diagnoses. In our case, significant undertreatment, compounded by his suspected iron deficiency anemia secondary to his polycythemia vera and chronic phlebotomies, are the most likel etiologies for his lack of clinical improvement.
Multiple factors may affect the prognosis of SCD. Males aged < 50 years with absence of anemia, spinal cord atrophy, Romberg sign, Babinski sign, or sensory deficits on examination have increased likelihood of eventual recovery of signs and symptoms of SCD; those with less spinal cord involvement (< 7 cord segments), contrast enhancement, and spinal cord edema also have improved outcomes.4,15
Conclusion
SCD is a rare but serious complication of chronic vitamin B12 deficiency that presents with a variety of neurological findings and may be easily confused with other illnesses. The condition is easily overlooked or misdiagnosed; thus, it is crucial to differentiate B12 deficiency from other common causes of neurologic symptoms. Specific findings on MRI are useful to support the clinical diagnosis of SCD and guide clinical decisions. Given the prevalence of B12 deficiency in the older adult population, clinicians should remain alert to the possibility of these conditions in patients who present with progressive neuropathy. Once a patient is diagnosed with SCD secondary to a B12 deficiency, appropriate B12 replacement is critical. Appropriate B12 replacement is aggressive and involves IM B12 1000 mcg every other day for 2 to 3 weeks, followed by additional IM administration every 2 months before transitioning to oral therapy. As seen in this case, failure to adequately replenish B12 can lead to progression or lack of resolution of SCD symptoms.
Subacute combined degeneration (SCD) is an acquired neurologic complication of vitamin B12 (cobalamin) or, rarely, vitamin B9 (folate) deficiency. SCD is characterized by progressive demyelination of the dorsal and lateral spinal cord, resulting in peripheral neuropathy; gait ataxia; impaired proprioception, vibration, and fine touch; optic neuropathy; and cognitive impairment.1 In addition to SCD, other neurologic manifestations of B12 deficiency include dementia, depression, visual symptoms due to optic atrophy, and behavioral changes.2 The prevalence of SCD in the US has not been well documented, but B12 deficiency is reported at 6% in those aged < 60 years and 20% in those > 60 years.3
Causes of B12 and B9 deficiency include advanced age, low nutritional intake (eg, vegan diet), impaired absorption (eg, inflammatory bowel disease, autoimmune pernicious anemia, gastrectomy, pancreatic disease), alcohol use, tapeworm infection, medications, and high metabolic states.2,4 Impaired B12 absorption is common in patients taking medications, such as metformin and proton pump inhibitors (PPI), due to suppression of ileal membrane transport and intrinsic factor activity.5-7 B-vitamin deficiency can be exacerbated by states of increased cellular turnover, such as polycythemia vera, due to elevated DNA synthesis.
Patients may experience permanent neurologic damage when the diagnosis and treatment of SCD are missed or delayed. Early diagnosis of SCD can be challenging due to lack of specific hematologic markers. In addition, many other conditions such as diabetic neuropathy, malnutrition, toxic neuropathy, sarcoidosis, HIV, multiple sclerosis, polycythemia vera, and iron deficiency anemia have similar presentations and clinical findings.8 Anemia and/or macrocytosis are not specific to B12 deficiency.4 In addition, patients with B12 deficiency may have a normal complete blood count (CBC); those with concomitant iron deficiency may have minimal or no mean corpuscular volume (MCV) elevation.4 In patients suspected to have B12 deficiency based on clinical presentation or laboratory findings of macrocytosis, serum methylmalonic acid (MMA) can serve as a direct measure of B12 activity, with levels > 0.75 μmol/L almost always indicating cobalamin deficiency. 9 On the other hand, plasma total homocysteine (tHcy) is a sensitive marker for B12 deficiency. The active form of B12, holotranscobalamin, has also emerged as a specific measure of B12 deficiency.9 However, in patients with SCD, measurement of these markers may be unnecessary due to the severity of their clinical symptoms.
The diagnosis of SCD is further complicated because not all individuals who develop B12 or B9 deficiency will develop SCD. It is difficult to determine which patients will develop SCD because the minimum level of serum B12 required for normal function is unknown, and recent studies indicate that SCD may occur even at low-normal B12 and B9 levels.2,4,10 Commonly, a serum B12 level of < 200 pg/mL is considered deficient, while a level between 200 and 300 pg/mL is considered borderline.4 The goal level of serum B12 is > 300 pg/mL, which is considered normal.4 While serologic findings of B-vitamin deficiency are only moderately specific, radiographic findings are highly sensitive and specific for SCD. According to Briani and colleagues, the most consistent finding in SCD on magnetic resonance imaging (MRI) is a “symmetrical, abnormally increased T2 signal intensity, commonly confined to posterior or posterior and lateral columns in the cervical and thoracic spinal cord.”2
We present a case of SCD in a patient with low-normal vitamin B12 levels who presented with progressive sensorimotor deficits and vision loss. The patient was subsequently diagnosed with SCD by radiologic workup. His course was complicated by worsening neurologic deficits despite B12 replacement. The progression of his clinical symptoms demonstrates the need for prompt, aggressive B12 replacement in patients diagnosed with SCD.
Case Presentation
A 63-year-old man presented for neurologic evaluation of progressive gait disturbance, paresthesia, blurred vision, and increasing falls despite use of a walker. Pertinent medical history included polycythemia vera requiring phlebotomy for approximately 9 years, alcohol use disorder (18 servings weekly), type 2 diabetes mellitus, and a remote episode of transient ischemic attack (TIA). The patient reported a 5-year history of burning pain in all extremities. A prior physician diagnosis attributed the symptoms to polyneuropathy secondary to iron deficiency anemia in the setting of chronic phlebotomy for polycythemia vera and high erythrogenesis. He was prescribed gabapentin 600 mg 3 times daily for pain control. B12 deficiency was considered an unlikely etiology due to a low-normal serum level of 305 pg/mL (reference range, 190-950 pg/mL) and normocytosis, with MCV of 88 fL (reference range, 80-100 fL). The patient also reported a 3-year history of blurred vision, which was initially attributed to be secondary to diabetic retinopathy. One week prior to presenting to our clinic, he was evaluated by ophthalmology for new-onset, bilateral central visual field defects, and he was diagnosed with nutritional optic neuropathy.
Ophthalmology suspected B12 deficiency. Notable findings included reduced deep tendon reflexes (DTRs) in the upper extremities and absent DTRs in the lower extremities, reduced sensation to light touch in all extremities, absent sensation to pinprick, vibration, and temperature in the lower extremities, positive Romberg sign, and a wide-based antalgic gait with the ankles externally rotated bilaterally (Table 1)
Previous cardiac evaluation failed to provide a diagnosis for syncopal episodes. MRI of the brain revealed nonspecific white matter changes consistent with chronic microvascular ischemic disease. Electromyography was limited due to pain but showed severe peripheral neuropathy. Laboratory results showed megalocytosis, low-normal serum B12 levels, and low serum folate levels (Table 2). The patient was diagnosed with polyneuropathy and was given intramuscular (IM) vitamin B12 1000 mcg once and a daily multivitamin (containing 25 mcg of B12). He was counseled on alcohol abstinence and medication adherence and was scheduled for follow-up in 3 months. He continued outpatient phlebotomy every 6 weeks for polycythemia.
At 3-month follow-up, the patient reported medication adherence, continued alcohol use, and worsening of symptoms. Falls, which now occurred 2 to 3 times weekly despite proper use of a walker, were described as sudden loss of bilateral lower extremity strength without loss of consciousness, palpitations, or other prodrome. Laboratory results showed minimal changes. Physical examination of the patient demonstrated similar deficits as on initial presentation. The patient received one additional B12 1000 mcg IM. Gabapentin was replaced with pregabalin 75 mg twice daily due to persistent uncontrolled pain and paresthesia. The patient was scheduled for a 3-month followup (6 months from initial visit) and repeat serology.
At 6-month follow-up, the patient showed continued progression of disease with significant difficulty using the walker, worsening falls, and wheelchair use required. Physical examination showed decreased sensation bilaterally up to the knees, absent bilateral patellar and Achilles reflexes, and unsteady gait. Laboratory results showed persistent subclinical B12 deficiency. MRI of the brain and spine showed high T2 signaling in a pattern highly specific for SCD. A formal diagnosis of SCD was made. The patient received an additional B12 1000 mcg IM once. Follow-up phone call with the patient 1 month later revealed no progression or improvement of symptoms.
Radiographic Findings
MRI of the cervical and thoracic spine demonstrated abnormal high T2 signal starting from C2 and extending along the course of the cervical and thoracic spinal cord (Figure). MRI in SCD classically shows symmetric, bilateral high T2 signal within the dorsal columns; on axial images, there is typically an inverted “V” sign.2,4 There can also be abnormal cerebral white matter change; however, MRI of the brain in this patient did not show any abnormalities.2 The imaging differential for this appearance includes other metabolic deficiencies/toxicities: copper deficiency; vitamin E deficiency; methotrexateinduced myelopathy, and infectious causes: HIV vacuolar myelopathy; and neurosyphilis (tabes dorsalis).4
Discussion
This case demonstrates the clinical and radiographic findings of SCD and underscores the need for high-intensity dosing of B12 replacement in patients with SCD to prevent progression of the disease and development of morbidities.
Symptoms of SCD may manifest even when the vitamin levels are in low-normal levels. Its presentation is often nonspecific, thus radiologic workup is beneficial to elucidate the clinical picture. We support the use of spinal MRI in patients with clinical suspicion of SCD to help rule out other causes of myelopathy. However, an MRI is not indicated in all patients with B12 deficiency, especially those without myelopathic symptoms. Additionally, follow-up spinal MRIs are useful in monitoring the progression or improvement of SCD after B12 replacement.2 It is important to note that the MRI findings in SCD are not specific to B12 deficiency; other causes may present with similar radiographic findings.4 Therefore, radiologic findings must be correlated with a patient’s clinical presentation.
B12 replacement improves and may resolve clinical symptoms and abnormal radiographic findings of SCD. The treatment duration of B12 deficiency depends on the underlying etiology. Reversible causes, such as metformin use > 4 months, PPI use > 12 months, and dietary deficiency, require treatment until appropriate levels are reached and symptoms are resolved.4,11 The need for chronic metformin and PPI use should also be reassessed regularly. In patients who require long-term metformin use, IM administration of B12 1000 mcg annually should be considered, which will ensure adequate storage for more than 1 year.12,13 In patients who require long-term PPI use, the risk and benefits of continued use should be measured, and if needed, the lowest possible effective PPI dose is recommended.14 Irreversible causes of B12 deficiency, such as advanced age, prior gastrectomy, chronic pancreatitis, or autoimmune pernicious anemia, require lifelong supplementation of B12.4,11
In general, oral vitamin B12 replacement at 1000 to 2000 mcg daily may be as effective as parenteral replacement in patients with mild to moderate deficiency or neurologic symptoms.11 On the other hand, patients with SCD often require parenteral replacement of B12 due to the severity of their deficiency or neurologic symptoms, need for more rapid improvement in symptoms, and prevention of irreversible neurological deficits. 4,11 Appropriate B12 replacement in SCD requires intensive initial therapy which may involve IM B12 1000 mcg every other day for 2 weeks and additional IM supplementation every 2 to 3 months afterward until resolution of deficiency.4,14 IM replacement may also be considered in patients who are nonadherent to oral replacement or have an underlying gastrointestinal condition that impairs enteral absorption.4,11
B12 deficiency is frequently undertreated and can lead to progression of disease with significant morbidity. The need for highintensity dosing of B12 replacement is crucial in patients with SCD. Failure to respond to treatment, as shown from the lack of improvement of serum markers or symptoms, likely suggests undertreatment, treatment nonadherence, iron deficiency anemia, an unidentified malabsorption syndrome, or other diagnoses. In our case, significant undertreatment, compounded by his suspected iron deficiency anemia secondary to his polycythemia vera and chronic phlebotomies, are the most likel etiologies for his lack of clinical improvement.
Multiple factors may affect the prognosis of SCD. Males aged < 50 years with absence of anemia, spinal cord atrophy, Romberg sign, Babinski sign, or sensory deficits on examination have increased likelihood of eventual recovery of signs and symptoms of SCD; those with less spinal cord involvement (< 7 cord segments), contrast enhancement, and spinal cord edema also have improved outcomes.4,15
Conclusion
SCD is a rare but serious complication of chronic vitamin B12 deficiency that presents with a variety of neurological findings and may be easily confused with other illnesses. The condition is easily overlooked or misdiagnosed; thus, it is crucial to differentiate B12 deficiency from other common causes of neurologic symptoms. Specific findings on MRI are useful to support the clinical diagnosis of SCD and guide clinical decisions. Given the prevalence of B12 deficiency in the older adult population, clinicians should remain alert to the possibility of these conditions in patients who present with progressive neuropathy. Once a patient is diagnosed with SCD secondary to a B12 deficiency, appropriate B12 replacement is critical. Appropriate B12 replacement is aggressive and involves IM B12 1000 mcg every other day for 2 to 3 weeks, followed by additional IM administration every 2 months before transitioning to oral therapy. As seen in this case, failure to adequately replenish B12 can lead to progression or lack of resolution of SCD symptoms.
1. Gürsoy AE, Kolukısa M, Babacan-Yıldız G, Celebi A. Subacute Combined Degeneration of the Spinal Cord due to Different Etiologies and Improvement of MRI Findings. Case Rep Neurol Med. 2013;2013:159649. doi:10.1155/2013/159649
2. Briani C, Dalla Torre C, Citton V, et al. Cobalamin deficiency: clinical picture and radiological findings. Nutrients. 2013;5(11):4521-4539. Published 2013 Nov 15. doi:10.3390/nu5114521
3. Hunt A, Harrington D, Robinson S. Vitamin B12 deficiency. BMJ. 2014;349:g5226. Published 2014 Sep 4. doi:10.1136/bmj.g5226
4. Qudsiya Z, De Jesus O. Subacute combined degeneration of the spinal cord. [Updated 2021 Feb 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Updated August 30, 2021. Accessed January 5, 2022. https://www.ncbi.nlm.nih.gov/books /NBK559316/
5. de Jager J, Kooy A, Lehert P, et al. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: randomised placebo controlled trial. BMJ. 2010;340:c2181. Published 2010 May 20. doi:10.1136/bmj.c2181
6. Aroda VR, Edelstein SL, Goldberg RB, et al. Longterm Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study. J Clin Endocrinol Metab. 2016;101(4):1754-1761. doi:10.1210/jc.2015-3754
7. Lam JR, Schneider JL, Zhao W, Corley DA. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin B12 deficiency. JAMA. 2013;310(22):2435-2442. doi:10.1001/jama.2013.280490
8. Mihalj M, Titlic´ M, Bonacin D, Dogaš Z. Sensomotor axonal peripheral neuropathy as a first complication of polycythemia rubra vera: A report of 3 cases. Am J Case Rep. 2013;14:385-387. Published 2013 Sep 25. doi:10.12659/AJCR.884016
9. Devalia V, Hamilton MS, Molloy AM; British Committee for Standards in Haematology. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br J Haematol. 2014;166(4):496-513. doi:10.1111/bjh.12959
10. Cao J, Xu S, Liu C. Is serum vitamin B12 decrease a necessity for the diagnosis of subacute combined degeneration?: A meta-analysis. Medicine (Baltimore). 2020;99(14):e19700.doi:10.1097/MD.0000000000019700
11. Langan RC, Goodbred AJ. Vitamin B12 Deficiency: Recognition and Management. Am Fam Physician. 2017;96(6):384-389.
12. Mazokopakis EE, Starakis IK. Recommendations for diagnosis and management of metformin-induced vitamin B12 (Cbl) deficiency. Diabetes Res Clin Pract. 2012;97(3):359-367. doi:10.1016/j.diabres.2012.06.001
13. Mahajan R, Gupta K. Revisiting Metformin: Annual Vitamin B12 Supplementation may become Mandatory with Long-Term Metformin Use. J Young Pharm. 2010;2(4):428-429. doi:10.4103/0975-1483.71621
14. Parks NE. Metabolic and Toxic Myelopathies. Continuum (Minneap Minn). 2021;27(1):143-162. doi:10.1212/CON.0000000000000963
15. Vasconcelos OM, Poehm EH, McCarter RJ, Campbell WW, Quezado ZM. Potential outcome factors in subacute combined degeneration: review of observational studies. J Gen Intern Med. 2006;21(10):1063-1068. doi:10.1111/j.1525-1497.2006.00525.x
1. Gürsoy AE, Kolukısa M, Babacan-Yıldız G, Celebi A. Subacute Combined Degeneration of the Spinal Cord due to Different Etiologies and Improvement of MRI Findings. Case Rep Neurol Med. 2013;2013:159649. doi:10.1155/2013/159649
2. Briani C, Dalla Torre C, Citton V, et al. Cobalamin deficiency: clinical picture and radiological findings. Nutrients. 2013;5(11):4521-4539. Published 2013 Nov 15. doi:10.3390/nu5114521
3. Hunt A, Harrington D, Robinson S. Vitamin B12 deficiency. BMJ. 2014;349:g5226. Published 2014 Sep 4. doi:10.1136/bmj.g5226
4. Qudsiya Z, De Jesus O. Subacute combined degeneration of the spinal cord. [Updated 2021 Feb 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Updated August 30, 2021. Accessed January 5, 2022. https://www.ncbi.nlm.nih.gov/books /NBK559316/
5. de Jager J, Kooy A, Lehert P, et al. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: randomised placebo controlled trial. BMJ. 2010;340:c2181. Published 2010 May 20. doi:10.1136/bmj.c2181
6. Aroda VR, Edelstein SL, Goldberg RB, et al. Longterm Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study. J Clin Endocrinol Metab. 2016;101(4):1754-1761. doi:10.1210/jc.2015-3754
7. Lam JR, Schneider JL, Zhao W, Corley DA. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin B12 deficiency. JAMA. 2013;310(22):2435-2442. doi:10.1001/jama.2013.280490
8. Mihalj M, Titlic´ M, Bonacin D, Dogaš Z. Sensomotor axonal peripheral neuropathy as a first complication of polycythemia rubra vera: A report of 3 cases. Am J Case Rep. 2013;14:385-387. Published 2013 Sep 25. doi:10.12659/AJCR.884016
9. Devalia V, Hamilton MS, Molloy AM; British Committee for Standards in Haematology. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br J Haematol. 2014;166(4):496-513. doi:10.1111/bjh.12959
10. Cao J, Xu S, Liu C. Is serum vitamin B12 decrease a necessity for the diagnosis of subacute combined degeneration?: A meta-analysis. Medicine (Baltimore). 2020;99(14):e19700.doi:10.1097/MD.0000000000019700
11. Langan RC, Goodbred AJ. Vitamin B12 Deficiency: Recognition and Management. Am Fam Physician. 2017;96(6):384-389.
12. Mazokopakis EE, Starakis IK. Recommendations for diagnosis and management of metformin-induced vitamin B12 (Cbl) deficiency. Diabetes Res Clin Pract. 2012;97(3):359-367. doi:10.1016/j.diabres.2012.06.001
13. Mahajan R, Gupta K. Revisiting Metformin: Annual Vitamin B12 Supplementation may become Mandatory with Long-Term Metformin Use. J Young Pharm. 2010;2(4):428-429. doi:10.4103/0975-1483.71621
14. Parks NE. Metabolic and Toxic Myelopathies. Continuum (Minneap Minn). 2021;27(1):143-162. doi:10.1212/CON.0000000000000963
15. Vasconcelos OM, Poehm EH, McCarter RJ, Campbell WW, Quezado ZM. Potential outcome factors in subacute combined degeneration: review of observational studies. J Gen Intern Med. 2006;21(10):1063-1068. doi:10.1111/j.1525-1497.2006.00525.x
Hemiballismus in Patients With Poorly Controlled Type 2 Diabetes Mellitus
Hemiballismus is an acquired hyperkinetic movement disorder characterized by unilateral, involuntary, often large-amplitude limb movements. Ballistic movements are now considered to be on the choreiform spectrum.1 Movements usually involve both the arm and leg, and in half of cases, facial movements such as tongue clucking and grimacing are seen.2,3 Presentations of hemiballismus vary in severity from intermittent to nearly continuous movements, which, in some cases, may lead to exhaustion, injury, or disability. Some patients are unable to ambulate or feed themselves with the affected limb.
Background
The 2 most common causes of hemichorea-hemiballismus are stroke and hyperglycemia, with an incidence of 4% and unknown incidence, respectively.1,3,4 Other causes include HIV, traumatic brain injury, encephalitis, vasculitis, mass effect, multiple sclerosis, and adverse drug reactions. 4-7 Acute or subacute hemiballismus is classically attributed to a lesion in subthalamic nucleus (STN), but this is true only in a minority of cases. Hemiballismus can be caused by any abnormality in various subnuclei of the basal ganglia, including the classic location in the STN, striatum, and globus pallidus.4 Evidence shows the lesions typically involve a functional network connected to the posterolateral putamen.8
Although not commonly recognized, hyperglycemia in patients with type 2 diabetes mellitus (T2DM) is the second most common cause of hemichoreahemiballismus. 3 Over the past 90 years, numerous case reports have described patients with DM with acute and subacute onset of hemiballistic and hemichoreiform movements while in a hyperglycemic state or after its resolution. Reported cases have been limited to small numbers of patients with only a few larger-scale reviews of more than 20 patients.7,9 Most reported cases involve geriatric patients and more commonly, females of Eastern Asian descent with an average age of onset of 71 years.4,10 Patients typically present with glucose levels from 500 to 1,000 mg/dL and hemoglobin A1c (HbA1c) levels almost double the normal values. Interestingly, neuroimaging findings in these patients have consistently shown hyperintense signal in the contralateral basal ganglia on T1-weighted magnetic resonance images (MRIs). Noncontrast computed tomography (CT) shows well-defined unilateral increased density in the contralateral basal ganglia without mass effect.1,9,11
This report aims to illustrate and enhance the understanding of hemiballismus associated with hyperglycemia. One patient presented to the US Department of Veterans Affairs (VA) Bay Pines VA Healthcare System (BPVAHCS) in Florida, which motivated us to search for other similar cases. We reviewed the charts of 2 other patients who presented to BPVAHCS over the past 10 years. The first case presented with severe hyperglycemia and abnormal movements that were not clearly diagnosed as hemiballismus. MRI findings were characteristic and assisted in making the diagnosis. The second case was misdiagnosed as hemiballismus secondary to ischemic stroke. The third case was initially diagnosed as conversion disorder until movements worsened and the correct diagnosis of hyperglycemia-induced hemichorea hemiballismus was confirmed by the pathognomonic neuroimaging findings.
Case Presentations
Case 1
A 65-year-old male with a history of uncontrolled T2DM presented with repetitive twitching and kicking movements that involved his left upper and lower extremities for 3 weeks. The patient reported that he did not take his medications or follow the recommended diabetes diet. His HbA1c on admission was 12.2% with a serum glucose of 254 mg/dL. The MRI showed a hyperintense T1 signal within the right basal ganglia including the right caudate with sparing of the internal capsule (Figure 1). There was no associated mass effect or restricted diffusion. It was compatible with a diagnosis of hyperglycemia- induced hemichorea-hemiballismus. The patient was advised to resume taking glipizide 10 mg daily, metformin 1,000 mg by mouth twice daily, and to begin 10 units of 70/30 insulin aspart 15 minutes before meals twice daily, and to follow a low carbohydrate diet, with reduce dietary intake of sugar. At his 1-month follow-up visit, the patient reported an improvement in his involuntary movements. At the 5-month follow-up, the patient’s HbA1c level was 10.4% and his hyperkinetic movements had completely resolved.
Case 2
of T2DM, hypertension, and hyperlipidemia was admitted due to increased jerky movements in the left upper extremity. On admission, his vital signs were within normal limits and his physical examination demonstrated choreoathetoid movements with ballistic components of his left upper extremity. His laboratory results showed a glucose level of 528 mg/dL with a HbA1c of 16.3%. An initial CT obtained in the emergency department (ED) demonstrated a well-defined hyperdensity in the striatal (caudate and lentiform nucleus) region (Figure 2). There was no associated edema/mass effect that would be typical for an intracranial hemorrhage.
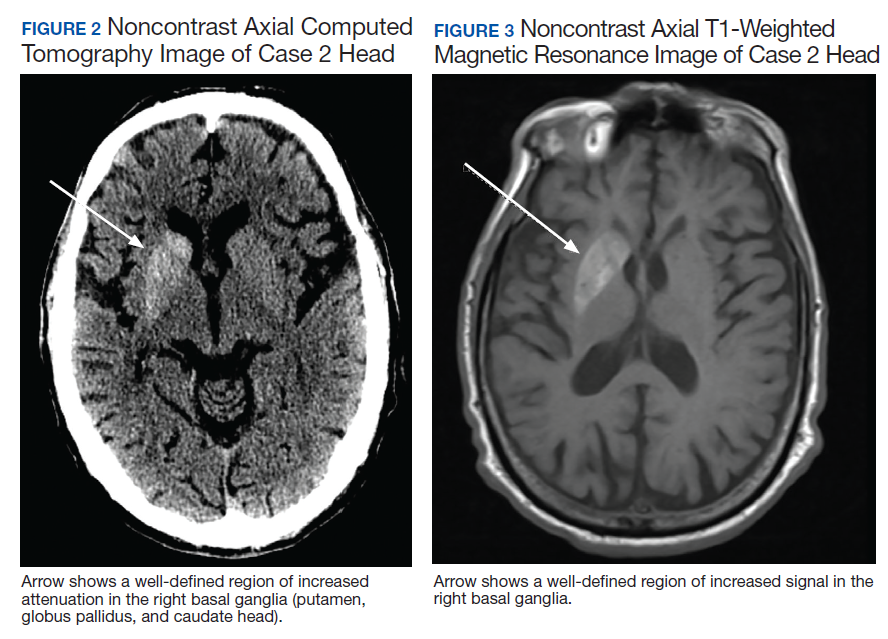
An MRI obtained 1 week later showed hyperintense TI signal corresponding to the basal ganglia (Figure 3). In addition, there was a questionable lacunar infarct in the right internal capsule. Due to lack of awareness regarding hyperglycemic associated basal ganglia changes, the patient’s movement disorder was presumed to be ischemic in etiology. The patient was prescribed oral amantadine 100 mg 3 times daily for the hemiballismus in conjunction with treatment of his T2DM. The only follow-up occurred 5 weeks later, which showed no improvement of uncontrollable movements. Imaging at that time (not available) indicated the persistence of the abnormal signal in the right basal ganglia. This patient died later that year without further follow-up.
Case 3
A 78-year-old white male with a history of syncope, transient ischemic attacks (TIAs), and poorly controlled T2DM presented with a 1-month history of progressively worsening involuntary, left-sided movements that began in his left shoulder and advanced to involve his arm, hand, and leg, and the left side of his face with grimacing and clucking of his tongue. Three weeks earlier, the patient had been discharged from the ED with a diagnosis of conversion disorder particularly because he experienced decreased movements when given a dose of Vitamin D. It was overlooked that administration of haloperidol had occurred a few hours before, and because the sounds made by his tongue were not felt to be consistent with a known movement disorder. A MRI of the brain was read as normal.
The patient returned 3 weeks later (the original presentation) due to his inability to perform activities of daily living because of his worsening involuntary movements. On admission, his HbA1c was 11.1% and his glucose was 167 mg/dL. On chart review, it was revealed that the patient’s HbA1c had been > 9% for the past 3 years with an increase from 10.1% to 11.1% in the 3 months preceding the onset of his symptoms.
On admission a MRI showed a unilateral right-sided T1 hyperintensity in the basal ganglia, no acute ischemia (Figure 4). In retrospect, subtle increased T1 signal can be seen on the earlier MRI (Figure 5). In view of the patient’s left-sided symptoms, DM, and MRI findings, a diagnosis of hyperglycemia-induced hemichorea- hemiballismus was made as the etiology of the patient’s symptoms.
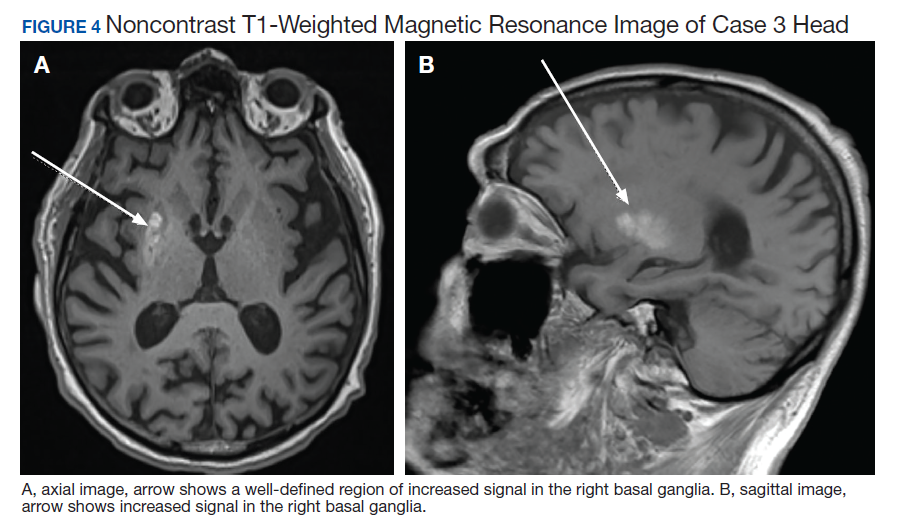
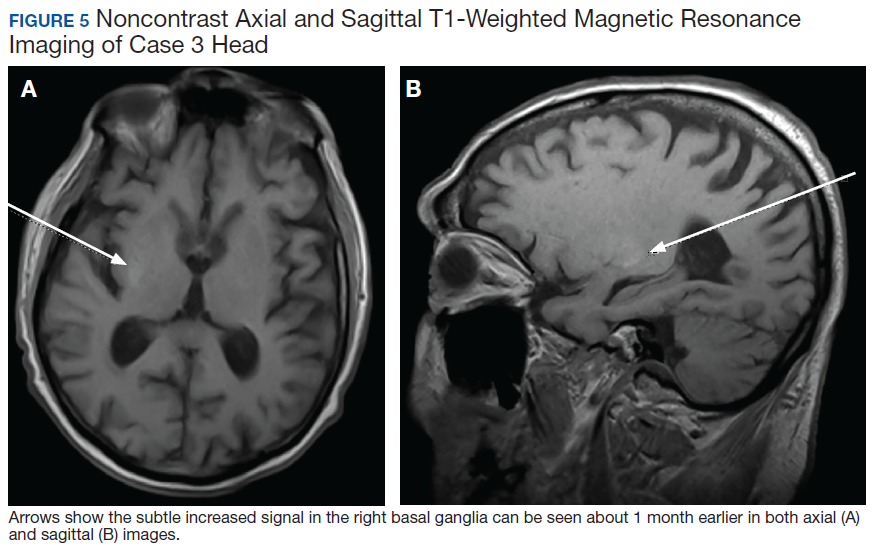
The patient was prescribed numerous medications to control his hyperkinesia including (and in combination): benztropine, gabapentin, baclofen, diphenhydramine, benzodiazepines, risperidone, olanzapine, and valproic acid, which did not control his movements. Ultimately, his hyperglycemic hemiballismus improved with tight glycemic control and oral tetrabenazine 12.5 mg twice daily. This patient underwent a protracted course of treatment with 17 days of inpatient medical admission, 3 weeks inpatient rehabilitation, and subsequent transfer to an assisted living facility.
Discussion
The 3 cases presented in this report contribute to the evidence that severe persistent hyperglycemia can result in movement disorders that mimic those seen after basal ganglia strokes. As with Case 2, past literature describes many cases of acute hyperglycemic episodes with glucose ranging from 500 to 1,000 mg/mL presenting with hemiballismus.1,3 However, there are many cases that describe hemiballismus occurring after glycemic correction, persisting despite glycemic correction, and presenting without an acute hyperglycemic episode, but in the setting of elevated HbA1c, as in Case 3.12,13 Notably, all 3 cases in this series had marked elevation in their HbA1c levels, which suggests that a more chronic hyperglycemic state or multiple shorter periods of hyperglycemia may be necessary to produce the described hyperkinetic movements.
Case reports describe the pathognomonic T1 hyperintensity of the basal ganglia that is identified in all 3 cases presented here. While the exact etiology remains unclear, the to metabolic derangements caused by hyperviscosity of the blood in the small end arteries feeding the basal ganglia.3,11 These abnormalities in turn interrupt the signaling cascade with abnormal firing rates or firing patterns, leading to reduced inhibition of the motor thalamus and ultimately present as hemiballismus.1,3,7 While most cases presented with unilateral hyperkinesis and associated contralateral basal ganglia abnormalities, there are reports of both unilateral and bilateral movements associated with bilateral basal ganglia hyperintensities on imaging. 9 The predilection for unilateral brain lesions may be explained by the varying degree of small vessel disease in different areas of the brain leading to perfusion deficits worsened by hyper viscosity. Further research into this is required to elucidate the exact pathophysiologic mechanism.
The course of disease for patients ranges from resolution within hours of tight glycemic control to persistent movements for > 3 months with a gradual improvement in severity.12,13 Treatments center on the importance of tight glycemic control to protect against the protracted course described in Case 3. Swift recognition of this rare condition is critical because improved glycemic control decreases the severity and duration of this disease. The significant disability associated with Case 3 highlights the need for prompt recognition and early, aggressive glycemic management to prevent the progression of hemiballismus. In addition to glycemic control, various CNS medications such as typical and atypical antipsychotics and tetrabenazine are firstline therapy with chemodenervation and surgical lesioning in cases unresponsive to medication therapy.
When unrecognized, hyperglycemic hemiballismus is associated with significant morbidity and mortality. The patients presented in this report were subject to either delayed diagnosis or misdiagnosis as stroke or psychiatric disorder. The rarity of the disorder, lack of evidence delineating pathogenesis and causality, low level of awareness, and varying presentations of patients all contribute to the challenge of recognizing, diagnosing, and treating hemiballismus due to hyperglycemia. This challenge can subsequently result in deteriorating symptoms, prolonged hospital stays, and unnecessary health care costs.
Conclusion
While hemiballismus due to severe persistent hyperglycemia is rare, the goal of this report is to highlight its occurrence in patients with T2DM. Further research can help develop a standardized, effective treatment strategy for these patients. Currently, lowering and maintaining appropriate glucose and HbA1c levels is the most effective treatment approach. Potential areas of research include alternative medical and surgical treatment interventions for patients while glycemic control is being achieved or for those who fail to benefit from glycemic control alone. Some success has been demonstrated with the use of antidopaminergic medications such as atypical antipsychotics and tetrabenazine and these medications should be considered when tight, sustained glycemic control alone is not successful in treating this disorder in the acute stages. Hopefully, with increasing awareness and recognition of hemiballismus related to hyperglycemia, more large-scale clinical trials can be conducted that will result in an effective treatment strategy for this devastating disorder.
1. Hawley JS, Weiner WJ. Hemiballismus: current concepts and review. Parkinsonism Relat Disord. 2012;18(2):125‐129. doi:10.1016/j.parkreldis.2011.08.015
2. Gasca-Salas C, Lang AE. Paroxysmal Hemiballism/ Hemichorea Resulting from Transient Ischemic Attacks. Mov Disord Clin Pract. 2015;3(3):303‐305. doi:10.1002/mdc3.12268
3. Garcia-Grimshaw MA, Jimenez-Ruiz A, Ornelas-Velazquez A, Luna-Armenta A, Gutierrez-Manjarrez FA. New-onset diabetes presenting as monoballism secondary to a mixed hyperglycemic crisis. Cureus. 2018;10(6):e2882. doi:10.7759/cureus.2882
4. Postuma RB, Lang AE. Hemiballism: revisiting a classic disorder. Lancet Neurol. 2003;2(11):661‐668. doi:10.1016/s1474-4422(03)00554-4
5. Gallo BV, Shulman LM, Weiner WJ, Petito CK, Berger JR. HIV encephalitis presenting with severe generalized chorea. Neurology. 1996;46(4):1163‐1165. doi:10.1212/wnl.46.4.1163
6. Provenzale JM, Glass JP. Hemiballismus: CT and MR findings. J Comput Assist Tomogr. 1995;19(4):537‐540.
7. Hodde M, Rowe KE, Surapaneni K, Terrigno P, Brighenti A, Altschuler EL. Management of severe hemiballismus: treatment challenges in the acute inpatient rehabilitation setting: a case presentation. PMR. 2017;9(7):732‐735. doi:10.1016/j.pmrj.2016.10.023
8. Laganiere S, Boes AD, Fox MD. Network localization of hemichorea-hemiballismus. Neurology. 2016;86(23):2187‐2195. doi:10.1212/WNL.0000000000002741
9. Cosentino C, Torres L, Nuñez Y, Suarez R, Velez M, Flores M. Hemichorea/hemiballism associated with hyperglycemia: report of 20 cases. Tremor Other Hyperkinet Mov (NY). 2016;6:402. doi:10.7916/D8DN454P
10. Oh SH, Lee KY, Im JH, Lee MS. Chorea associated with non-ketotic hyperglycemia and hyperintensity basal ganglia lesion on T1-weighted brain MRI study: a metaanalysis of 53 cases including four present cases. J Neurol Sci. 2002;200(1-2):57‐62. doi:10.1016/s0022-510x(02)00133-8
11. Carrion DM, Carrion AF. Non-ketotic hyperglycaemia hemichorea-hemiballismus and acute ischaemic stroke. BMJ Case Rep. 2013;2013:bcr2012008359. doi:10.1136/bcr-2012-008359
12. Cho HS, Hong CT, Chan L. Hemichorea after hyperglycemia correction: a case report and a short review of hyperglycemia-related hemichorea at the euglycemic state. Medicine (Baltimore). 2018;97(10):e0076. doi:10.1097/MD.0000000000010076
13. Lin YC, Lin YC. Prolonged hemiballism after the remission of non-ketotic hyperosmolar syndrome. BMJ Case Rep. 2012;2012:bcr0120125627. doi:10.1136/bcr.01.2012.5627
Hemiballismus is an acquired hyperkinetic movement disorder characterized by unilateral, involuntary, often large-amplitude limb movements. Ballistic movements are now considered to be on the choreiform spectrum.1 Movements usually involve both the arm and leg, and in half of cases, facial movements such as tongue clucking and grimacing are seen.2,3 Presentations of hemiballismus vary in severity from intermittent to nearly continuous movements, which, in some cases, may lead to exhaustion, injury, or disability. Some patients are unable to ambulate or feed themselves with the affected limb.
Background
The 2 most common causes of hemichorea-hemiballismus are stroke and hyperglycemia, with an incidence of 4% and unknown incidence, respectively.1,3,4 Other causes include HIV, traumatic brain injury, encephalitis, vasculitis, mass effect, multiple sclerosis, and adverse drug reactions. 4-7 Acute or subacute hemiballismus is classically attributed to a lesion in subthalamic nucleus (STN), but this is true only in a minority of cases. Hemiballismus can be caused by any abnormality in various subnuclei of the basal ganglia, including the classic location in the STN, striatum, and globus pallidus.4 Evidence shows the lesions typically involve a functional network connected to the posterolateral putamen.8
Although not commonly recognized, hyperglycemia in patients with type 2 diabetes mellitus (T2DM) is the second most common cause of hemichoreahemiballismus. 3 Over the past 90 years, numerous case reports have described patients with DM with acute and subacute onset of hemiballistic and hemichoreiform movements while in a hyperglycemic state or after its resolution. Reported cases have been limited to small numbers of patients with only a few larger-scale reviews of more than 20 patients.7,9 Most reported cases involve geriatric patients and more commonly, females of Eastern Asian descent with an average age of onset of 71 years.4,10 Patients typically present with glucose levels from 500 to 1,000 mg/dL and hemoglobin A1c (HbA1c) levels almost double the normal values. Interestingly, neuroimaging findings in these patients have consistently shown hyperintense signal in the contralateral basal ganglia on T1-weighted magnetic resonance images (MRIs). Noncontrast computed tomography (CT) shows well-defined unilateral increased density in the contralateral basal ganglia without mass effect.1,9,11
This report aims to illustrate and enhance the understanding of hemiballismus associated with hyperglycemia. One patient presented to the US Department of Veterans Affairs (VA) Bay Pines VA Healthcare System (BPVAHCS) in Florida, which motivated us to search for other similar cases. We reviewed the charts of 2 other patients who presented to BPVAHCS over the past 10 years. The first case presented with severe hyperglycemia and abnormal movements that were not clearly diagnosed as hemiballismus. MRI findings were characteristic and assisted in making the diagnosis. The second case was misdiagnosed as hemiballismus secondary to ischemic stroke. The third case was initially diagnosed as conversion disorder until movements worsened and the correct diagnosis of hyperglycemia-induced hemichorea hemiballismus was confirmed by the pathognomonic neuroimaging findings.
Case Presentations
Case 1
A 65-year-old male with a history of uncontrolled T2DM presented with repetitive twitching and kicking movements that involved his left upper and lower extremities for 3 weeks. The patient reported that he did not take his medications or follow the recommended diabetes diet. His HbA1c on admission was 12.2% with a serum glucose of 254 mg/dL. The MRI showed a hyperintense T1 signal within the right basal ganglia including the right caudate with sparing of the internal capsule (Figure 1). There was no associated mass effect or restricted diffusion. It was compatible with a diagnosis of hyperglycemia- induced hemichorea-hemiballismus. The patient was advised to resume taking glipizide 10 mg daily, metformin 1,000 mg by mouth twice daily, and to begin 10 units of 70/30 insulin aspart 15 minutes before meals twice daily, and to follow a low carbohydrate diet, with reduce dietary intake of sugar. At his 1-month follow-up visit, the patient reported an improvement in his involuntary movements. At the 5-month follow-up, the patient’s HbA1c level was 10.4% and his hyperkinetic movements had completely resolved.
Case 2
of T2DM, hypertension, and hyperlipidemia was admitted due to increased jerky movements in the left upper extremity. On admission, his vital signs were within normal limits and his physical examination demonstrated choreoathetoid movements with ballistic components of his left upper extremity. His laboratory results showed a glucose level of 528 mg/dL with a HbA1c of 16.3%. An initial CT obtained in the emergency department (ED) demonstrated a well-defined hyperdensity in the striatal (caudate and lentiform nucleus) region (Figure 2). There was no associated edema/mass effect that would be typical for an intracranial hemorrhage.
An MRI obtained 1 week later showed hyperintense TI signal corresponding to the basal ganglia (Figure 3). In addition, there was a questionable lacunar infarct in the right internal capsule. Due to lack of awareness regarding hyperglycemic associated basal ganglia changes, the patient’s movement disorder was presumed to be ischemic in etiology. The patient was prescribed oral amantadine 100 mg 3 times daily for the hemiballismus in conjunction with treatment of his T2DM. The only follow-up occurred 5 weeks later, which showed no improvement of uncontrollable movements. Imaging at that time (not available) indicated the persistence of the abnormal signal in the right basal ganglia. This patient died later that year without further follow-up.
Case 3
A 78-year-old white male with a history of syncope, transient ischemic attacks (TIAs), and poorly controlled T2DM presented with a 1-month history of progressively worsening involuntary, left-sided movements that began in his left shoulder and advanced to involve his arm, hand, and leg, and the left side of his face with grimacing and clucking of his tongue. Three weeks earlier, the patient had been discharged from the ED with a diagnosis of conversion disorder particularly because he experienced decreased movements when given a dose of Vitamin D. It was overlooked that administration of haloperidol had occurred a few hours before, and because the sounds made by his tongue were not felt to be consistent with a known movement disorder. A MRI of the brain was read as normal.
The patient returned 3 weeks later (the original presentation) due to his inability to perform activities of daily living because of his worsening involuntary movements. On admission, his HbA1c was 11.1% and his glucose was 167 mg/dL. On chart review, it was revealed that the patient’s HbA1c had been > 9% for the past 3 years with an increase from 10.1% to 11.1% in the 3 months preceding the onset of his symptoms.
On admission a MRI showed a unilateral right-sided T1 hyperintensity in the basal ganglia, no acute ischemia (Figure 4). In retrospect, subtle increased T1 signal can be seen on the earlier MRI (Figure 5). In view of the patient’s left-sided symptoms, DM, and MRI findings, a diagnosis of hyperglycemia-induced hemichorea- hemiballismus was made as the etiology of the patient’s symptoms.
The patient was prescribed numerous medications to control his hyperkinesia including (and in combination): benztropine, gabapentin, baclofen, diphenhydramine, benzodiazepines, risperidone, olanzapine, and valproic acid, which did not control his movements. Ultimately, his hyperglycemic hemiballismus improved with tight glycemic control and oral tetrabenazine 12.5 mg twice daily. This patient underwent a protracted course of treatment with 17 days of inpatient medical admission, 3 weeks inpatient rehabilitation, and subsequent transfer to an assisted living facility.
Discussion
The 3 cases presented in this report contribute to the evidence that severe persistent hyperglycemia can result in movement disorders that mimic those seen after basal ganglia strokes. As with Case 2, past literature describes many cases of acute hyperglycemic episodes with glucose ranging from 500 to 1,000 mg/mL presenting with hemiballismus.1,3 However, there are many cases that describe hemiballismus occurring after glycemic correction, persisting despite glycemic correction, and presenting without an acute hyperglycemic episode, but in the setting of elevated HbA1c, as in Case 3.12,13 Notably, all 3 cases in this series had marked elevation in their HbA1c levels, which suggests that a more chronic hyperglycemic state or multiple shorter periods of hyperglycemia may be necessary to produce the described hyperkinetic movements.
Case reports describe the pathognomonic T1 hyperintensity of the basal ganglia that is identified in all 3 cases presented here. While the exact etiology remains unclear, the to metabolic derangements caused by hyperviscosity of the blood in the small end arteries feeding the basal ganglia.3,11 These abnormalities in turn interrupt the signaling cascade with abnormal firing rates or firing patterns, leading to reduced inhibition of the motor thalamus and ultimately present as hemiballismus.1,3,7 While most cases presented with unilateral hyperkinesis and associated contralateral basal ganglia abnormalities, there are reports of both unilateral and bilateral movements associated with bilateral basal ganglia hyperintensities on imaging. 9 The predilection for unilateral brain lesions may be explained by the varying degree of small vessel disease in different areas of the brain leading to perfusion deficits worsened by hyper viscosity. Further research into this is required to elucidate the exact pathophysiologic mechanism.
The course of disease for patients ranges from resolution within hours of tight glycemic control to persistent movements for > 3 months with a gradual improvement in severity.12,13 Treatments center on the importance of tight glycemic control to protect against the protracted course described in Case 3. Swift recognition of this rare condition is critical because improved glycemic control decreases the severity and duration of this disease. The significant disability associated with Case 3 highlights the need for prompt recognition and early, aggressive glycemic management to prevent the progression of hemiballismus. In addition to glycemic control, various CNS medications such as typical and atypical antipsychotics and tetrabenazine are firstline therapy with chemodenervation and surgical lesioning in cases unresponsive to medication therapy.
When unrecognized, hyperglycemic hemiballismus is associated with significant morbidity and mortality. The patients presented in this report were subject to either delayed diagnosis or misdiagnosis as stroke or psychiatric disorder. The rarity of the disorder, lack of evidence delineating pathogenesis and causality, low level of awareness, and varying presentations of patients all contribute to the challenge of recognizing, diagnosing, and treating hemiballismus due to hyperglycemia. This challenge can subsequently result in deteriorating symptoms, prolonged hospital stays, and unnecessary health care costs.
Conclusion
While hemiballismus due to severe persistent hyperglycemia is rare, the goal of this report is to highlight its occurrence in patients with T2DM. Further research can help develop a standardized, effective treatment strategy for these patients. Currently, lowering and maintaining appropriate glucose and HbA1c levels is the most effective treatment approach. Potential areas of research include alternative medical and surgical treatment interventions for patients while glycemic control is being achieved or for those who fail to benefit from glycemic control alone. Some success has been demonstrated with the use of antidopaminergic medications such as atypical antipsychotics and tetrabenazine and these medications should be considered when tight, sustained glycemic control alone is not successful in treating this disorder in the acute stages. Hopefully, with increasing awareness and recognition of hemiballismus related to hyperglycemia, more large-scale clinical trials can be conducted that will result in an effective treatment strategy for this devastating disorder.
Hemiballismus is an acquired hyperkinetic movement disorder characterized by unilateral, involuntary, often large-amplitude limb movements. Ballistic movements are now considered to be on the choreiform spectrum.1 Movements usually involve both the arm and leg, and in half of cases, facial movements such as tongue clucking and grimacing are seen.2,3 Presentations of hemiballismus vary in severity from intermittent to nearly continuous movements, which, in some cases, may lead to exhaustion, injury, or disability. Some patients are unable to ambulate or feed themselves with the affected limb.
Background
The 2 most common causes of hemichorea-hemiballismus are stroke and hyperglycemia, with an incidence of 4% and unknown incidence, respectively.1,3,4 Other causes include HIV, traumatic brain injury, encephalitis, vasculitis, mass effect, multiple sclerosis, and adverse drug reactions. 4-7 Acute or subacute hemiballismus is classically attributed to a lesion in subthalamic nucleus (STN), but this is true only in a minority of cases. Hemiballismus can be caused by any abnormality in various subnuclei of the basal ganglia, including the classic location in the STN, striatum, and globus pallidus.4 Evidence shows the lesions typically involve a functional network connected to the posterolateral putamen.8
Although not commonly recognized, hyperglycemia in patients with type 2 diabetes mellitus (T2DM) is the second most common cause of hemichoreahemiballismus. 3 Over the past 90 years, numerous case reports have described patients with DM with acute and subacute onset of hemiballistic and hemichoreiform movements while in a hyperglycemic state or after its resolution. Reported cases have been limited to small numbers of patients with only a few larger-scale reviews of more than 20 patients.7,9 Most reported cases involve geriatric patients and more commonly, females of Eastern Asian descent with an average age of onset of 71 years.4,10 Patients typically present with glucose levels from 500 to 1,000 mg/dL and hemoglobin A1c (HbA1c) levels almost double the normal values. Interestingly, neuroimaging findings in these patients have consistently shown hyperintense signal in the contralateral basal ganglia on T1-weighted magnetic resonance images (MRIs). Noncontrast computed tomography (CT) shows well-defined unilateral increased density in the contralateral basal ganglia without mass effect.1,9,11
This report aims to illustrate and enhance the understanding of hemiballismus associated with hyperglycemia. One patient presented to the US Department of Veterans Affairs (VA) Bay Pines VA Healthcare System (BPVAHCS) in Florida, which motivated us to search for other similar cases. We reviewed the charts of 2 other patients who presented to BPVAHCS over the past 10 years. The first case presented with severe hyperglycemia and abnormal movements that were not clearly diagnosed as hemiballismus. MRI findings were characteristic and assisted in making the diagnosis. The second case was misdiagnosed as hemiballismus secondary to ischemic stroke. The third case was initially diagnosed as conversion disorder until movements worsened and the correct diagnosis of hyperglycemia-induced hemichorea hemiballismus was confirmed by the pathognomonic neuroimaging findings.
Case Presentations
Case 1
A 65-year-old male with a history of uncontrolled T2DM presented with repetitive twitching and kicking movements that involved his left upper and lower extremities for 3 weeks. The patient reported that he did not take his medications or follow the recommended diabetes diet. His HbA1c on admission was 12.2% with a serum glucose of 254 mg/dL. The MRI showed a hyperintense T1 signal within the right basal ganglia including the right caudate with sparing of the internal capsule (Figure 1). There was no associated mass effect or restricted diffusion. It was compatible with a diagnosis of hyperglycemia- induced hemichorea-hemiballismus. The patient was advised to resume taking glipizide 10 mg daily, metformin 1,000 mg by mouth twice daily, and to begin 10 units of 70/30 insulin aspart 15 minutes before meals twice daily, and to follow a low carbohydrate diet, with reduce dietary intake of sugar. At his 1-month follow-up visit, the patient reported an improvement in his involuntary movements. At the 5-month follow-up, the patient’s HbA1c level was 10.4% and his hyperkinetic movements had completely resolved.
Case 2
of T2DM, hypertension, and hyperlipidemia was admitted due to increased jerky movements in the left upper extremity. On admission, his vital signs were within normal limits and his physical examination demonstrated choreoathetoid movements with ballistic components of his left upper extremity. His laboratory results showed a glucose level of 528 mg/dL with a HbA1c of 16.3%. An initial CT obtained in the emergency department (ED) demonstrated a well-defined hyperdensity in the striatal (caudate and lentiform nucleus) region (Figure 2). There was no associated edema/mass effect that would be typical for an intracranial hemorrhage.
An MRI obtained 1 week later showed hyperintense TI signal corresponding to the basal ganglia (Figure 3). In addition, there was a questionable lacunar infarct in the right internal capsule. Due to lack of awareness regarding hyperglycemic associated basal ganglia changes, the patient’s movement disorder was presumed to be ischemic in etiology. The patient was prescribed oral amantadine 100 mg 3 times daily for the hemiballismus in conjunction with treatment of his T2DM. The only follow-up occurred 5 weeks later, which showed no improvement of uncontrollable movements. Imaging at that time (not available) indicated the persistence of the abnormal signal in the right basal ganglia. This patient died later that year without further follow-up.
Case 3
A 78-year-old white male with a history of syncope, transient ischemic attacks (TIAs), and poorly controlled T2DM presented with a 1-month history of progressively worsening involuntary, left-sided movements that began in his left shoulder and advanced to involve his arm, hand, and leg, and the left side of his face with grimacing and clucking of his tongue. Three weeks earlier, the patient had been discharged from the ED with a diagnosis of conversion disorder particularly because he experienced decreased movements when given a dose of Vitamin D. It was overlooked that administration of haloperidol had occurred a few hours before, and because the sounds made by his tongue were not felt to be consistent with a known movement disorder. A MRI of the brain was read as normal.
The patient returned 3 weeks later (the original presentation) due to his inability to perform activities of daily living because of his worsening involuntary movements. On admission, his HbA1c was 11.1% and his glucose was 167 mg/dL. On chart review, it was revealed that the patient’s HbA1c had been > 9% for the past 3 years with an increase from 10.1% to 11.1% in the 3 months preceding the onset of his symptoms.
On admission a MRI showed a unilateral right-sided T1 hyperintensity in the basal ganglia, no acute ischemia (Figure 4). In retrospect, subtle increased T1 signal can be seen on the earlier MRI (Figure 5). In view of the patient’s left-sided symptoms, DM, and MRI findings, a diagnosis of hyperglycemia-induced hemichorea- hemiballismus was made as the etiology of the patient’s symptoms.
The patient was prescribed numerous medications to control his hyperkinesia including (and in combination): benztropine, gabapentin, baclofen, diphenhydramine, benzodiazepines, risperidone, olanzapine, and valproic acid, which did not control his movements. Ultimately, his hyperglycemic hemiballismus improved with tight glycemic control and oral tetrabenazine 12.5 mg twice daily. This patient underwent a protracted course of treatment with 17 days of inpatient medical admission, 3 weeks inpatient rehabilitation, and subsequent transfer to an assisted living facility.
Discussion
The 3 cases presented in this report contribute to the evidence that severe persistent hyperglycemia can result in movement disorders that mimic those seen after basal ganglia strokes. As with Case 2, past literature describes many cases of acute hyperglycemic episodes with glucose ranging from 500 to 1,000 mg/mL presenting with hemiballismus.1,3 However, there are many cases that describe hemiballismus occurring after glycemic correction, persisting despite glycemic correction, and presenting without an acute hyperglycemic episode, but in the setting of elevated HbA1c, as in Case 3.12,13 Notably, all 3 cases in this series had marked elevation in their HbA1c levels, which suggests that a more chronic hyperglycemic state or multiple shorter periods of hyperglycemia may be necessary to produce the described hyperkinetic movements.
Case reports describe the pathognomonic T1 hyperintensity of the basal ganglia that is identified in all 3 cases presented here. While the exact etiology remains unclear, the to metabolic derangements caused by hyperviscosity of the blood in the small end arteries feeding the basal ganglia.3,11 These abnormalities in turn interrupt the signaling cascade with abnormal firing rates or firing patterns, leading to reduced inhibition of the motor thalamus and ultimately present as hemiballismus.1,3,7 While most cases presented with unilateral hyperkinesis and associated contralateral basal ganglia abnormalities, there are reports of both unilateral and bilateral movements associated with bilateral basal ganglia hyperintensities on imaging. 9 The predilection for unilateral brain lesions may be explained by the varying degree of small vessel disease in different areas of the brain leading to perfusion deficits worsened by hyper viscosity. Further research into this is required to elucidate the exact pathophysiologic mechanism.
The course of disease for patients ranges from resolution within hours of tight glycemic control to persistent movements for > 3 months with a gradual improvement in severity.12,13 Treatments center on the importance of tight glycemic control to protect against the protracted course described in Case 3. Swift recognition of this rare condition is critical because improved glycemic control decreases the severity and duration of this disease. The significant disability associated with Case 3 highlights the need for prompt recognition and early, aggressive glycemic management to prevent the progression of hemiballismus. In addition to glycemic control, various CNS medications such as typical and atypical antipsychotics and tetrabenazine are firstline therapy with chemodenervation and surgical lesioning in cases unresponsive to medication therapy.
When unrecognized, hyperglycemic hemiballismus is associated with significant morbidity and mortality. The patients presented in this report were subject to either delayed diagnosis or misdiagnosis as stroke or psychiatric disorder. The rarity of the disorder, lack of evidence delineating pathogenesis and causality, low level of awareness, and varying presentations of patients all contribute to the challenge of recognizing, diagnosing, and treating hemiballismus due to hyperglycemia. This challenge can subsequently result in deteriorating symptoms, prolonged hospital stays, and unnecessary health care costs.
Conclusion
While hemiballismus due to severe persistent hyperglycemia is rare, the goal of this report is to highlight its occurrence in patients with T2DM. Further research can help develop a standardized, effective treatment strategy for these patients. Currently, lowering and maintaining appropriate glucose and HbA1c levels is the most effective treatment approach. Potential areas of research include alternative medical and surgical treatment interventions for patients while glycemic control is being achieved or for those who fail to benefit from glycemic control alone. Some success has been demonstrated with the use of antidopaminergic medications such as atypical antipsychotics and tetrabenazine and these medications should be considered when tight, sustained glycemic control alone is not successful in treating this disorder in the acute stages. Hopefully, with increasing awareness and recognition of hemiballismus related to hyperglycemia, more large-scale clinical trials can be conducted that will result in an effective treatment strategy for this devastating disorder.
1. Hawley JS, Weiner WJ. Hemiballismus: current concepts and review. Parkinsonism Relat Disord. 2012;18(2):125‐129. doi:10.1016/j.parkreldis.2011.08.015
2. Gasca-Salas C, Lang AE. Paroxysmal Hemiballism/ Hemichorea Resulting from Transient Ischemic Attacks. Mov Disord Clin Pract. 2015;3(3):303‐305. doi:10.1002/mdc3.12268
3. Garcia-Grimshaw MA, Jimenez-Ruiz A, Ornelas-Velazquez A, Luna-Armenta A, Gutierrez-Manjarrez FA. New-onset diabetes presenting as monoballism secondary to a mixed hyperglycemic crisis. Cureus. 2018;10(6):e2882. doi:10.7759/cureus.2882
4. Postuma RB, Lang AE. Hemiballism: revisiting a classic disorder. Lancet Neurol. 2003;2(11):661‐668. doi:10.1016/s1474-4422(03)00554-4
5. Gallo BV, Shulman LM, Weiner WJ, Petito CK, Berger JR. HIV encephalitis presenting with severe generalized chorea. Neurology. 1996;46(4):1163‐1165. doi:10.1212/wnl.46.4.1163
6. Provenzale JM, Glass JP. Hemiballismus: CT and MR findings. J Comput Assist Tomogr. 1995;19(4):537‐540.
7. Hodde M, Rowe KE, Surapaneni K, Terrigno P, Brighenti A, Altschuler EL. Management of severe hemiballismus: treatment challenges in the acute inpatient rehabilitation setting: a case presentation. PMR. 2017;9(7):732‐735. doi:10.1016/j.pmrj.2016.10.023
8. Laganiere S, Boes AD, Fox MD. Network localization of hemichorea-hemiballismus. Neurology. 2016;86(23):2187‐2195. doi:10.1212/WNL.0000000000002741
9. Cosentino C, Torres L, Nuñez Y, Suarez R, Velez M, Flores M. Hemichorea/hemiballism associated with hyperglycemia: report of 20 cases. Tremor Other Hyperkinet Mov (NY). 2016;6:402. doi:10.7916/D8DN454P
10. Oh SH, Lee KY, Im JH, Lee MS. Chorea associated with non-ketotic hyperglycemia and hyperintensity basal ganglia lesion on T1-weighted brain MRI study: a metaanalysis of 53 cases including four present cases. J Neurol Sci. 2002;200(1-2):57‐62. doi:10.1016/s0022-510x(02)00133-8
11. Carrion DM, Carrion AF. Non-ketotic hyperglycaemia hemichorea-hemiballismus and acute ischaemic stroke. BMJ Case Rep. 2013;2013:bcr2012008359. doi:10.1136/bcr-2012-008359
12. Cho HS, Hong CT, Chan L. Hemichorea after hyperglycemia correction: a case report and a short review of hyperglycemia-related hemichorea at the euglycemic state. Medicine (Baltimore). 2018;97(10):e0076. doi:10.1097/MD.0000000000010076
13. Lin YC, Lin YC. Prolonged hemiballism after the remission of non-ketotic hyperosmolar syndrome. BMJ Case Rep. 2012;2012:bcr0120125627. doi:10.1136/bcr.01.2012.5627
1. Hawley JS, Weiner WJ. Hemiballismus: current concepts and review. Parkinsonism Relat Disord. 2012;18(2):125‐129. doi:10.1016/j.parkreldis.2011.08.015
2. Gasca-Salas C, Lang AE. Paroxysmal Hemiballism/ Hemichorea Resulting from Transient Ischemic Attacks. Mov Disord Clin Pract. 2015;3(3):303‐305. doi:10.1002/mdc3.12268
3. Garcia-Grimshaw MA, Jimenez-Ruiz A, Ornelas-Velazquez A, Luna-Armenta A, Gutierrez-Manjarrez FA. New-onset diabetes presenting as monoballism secondary to a mixed hyperglycemic crisis. Cureus. 2018;10(6):e2882. doi:10.7759/cureus.2882
4. Postuma RB, Lang AE. Hemiballism: revisiting a classic disorder. Lancet Neurol. 2003;2(11):661‐668. doi:10.1016/s1474-4422(03)00554-4
5. Gallo BV, Shulman LM, Weiner WJ, Petito CK, Berger JR. HIV encephalitis presenting with severe generalized chorea. Neurology. 1996;46(4):1163‐1165. doi:10.1212/wnl.46.4.1163
6. Provenzale JM, Glass JP. Hemiballismus: CT and MR findings. J Comput Assist Tomogr. 1995;19(4):537‐540.
7. Hodde M, Rowe KE, Surapaneni K, Terrigno P, Brighenti A, Altschuler EL. Management of severe hemiballismus: treatment challenges in the acute inpatient rehabilitation setting: a case presentation. PMR. 2017;9(7):732‐735. doi:10.1016/j.pmrj.2016.10.023
8. Laganiere S, Boes AD, Fox MD. Network localization of hemichorea-hemiballismus. Neurology. 2016;86(23):2187‐2195. doi:10.1212/WNL.0000000000002741
9. Cosentino C, Torres L, Nuñez Y, Suarez R, Velez M, Flores M. Hemichorea/hemiballism associated with hyperglycemia: report of 20 cases. Tremor Other Hyperkinet Mov (NY). 2016;6:402. doi:10.7916/D8DN454P
10. Oh SH, Lee KY, Im JH, Lee MS. Chorea associated with non-ketotic hyperglycemia and hyperintensity basal ganglia lesion on T1-weighted brain MRI study: a metaanalysis of 53 cases including four present cases. J Neurol Sci. 2002;200(1-2):57‐62. doi:10.1016/s0022-510x(02)00133-8
11. Carrion DM, Carrion AF. Non-ketotic hyperglycaemia hemichorea-hemiballismus and acute ischaemic stroke. BMJ Case Rep. 2013;2013:bcr2012008359. doi:10.1136/bcr-2012-008359
12. Cho HS, Hong CT, Chan L. Hemichorea after hyperglycemia correction: a case report and a short review of hyperglycemia-related hemichorea at the euglycemic state. Medicine (Baltimore). 2018;97(10):e0076. doi:10.1097/MD.0000000000010076
13. Lin YC, Lin YC. Prolonged hemiballism after the remission of non-ketotic hyperosmolar syndrome. BMJ Case Rep. 2012;2012:bcr0120125627. doi:10.1136/bcr.01.2012.5627
Hemolytic Uremic Syndrome With Severe Neurologic Complications in an Adult (FULL)
The case of a female presenting with Shiga toxin-producing Escherichia coli and hemolytic uremic syndrome highlights a severe neurologic complication that canbe associated with these conditions.
Hemolytic uremic syndrome (HUS) is a rare illness that can be acquired through the consumption of food products contaminated with strains of Shiga toxin-producing Escherichia coli (E coli; STEC).1 Between 6% and 15% of individuals infected with STEC develop HUS, with children affected more frequently than adults.2,3 This strain of E coli releases Shiga toxin into the systemic circulation, which causes a thrombotic microangiopathy resulting in the characteristic HUS triad of symptoms: acute renal insufficiency, thrombocytopenia, and hemolytic anemia.4-6
Although neurologic features are common in HUS, they have not been extensively studied, particularly in adults. We report a case of STEC 0157:H7 subtype HUS in an adult with severe neurologic complications. This case highlights the neurological sequelae in an adult with typical STEC-HUS. The use of treatment modalities, such as plasmapheresis and eculizumab, and their use in adult typical STEC-HUS also is explored.
Case
A 53-year-old white woman with no pertinent past medical history presented to the Bay Pines Veterans Affairs Healthcare System Emergency Department with a 2-day history of abdominal pain, vomiting, nausea, diarrhea, and bright bloody stools. She returned from a cruise to the Bahamas 3 days prior, where she ate local foods, including salads. She reported no fever, shortness of breath, chest pain, headache, and cognitive difficulties. She presented with a normal mental status and neurologic exam. Apart from leukocytosis and elevated glucose level, her laboratory results at initial presentation were normal, (Table). A stool sample showed occult blood with white blood cell counts (WBCs) but was negative for Clostridium difficile. She was started on ciprofloxacin 400 mg and metronidazole 500 mg on the day of admission.
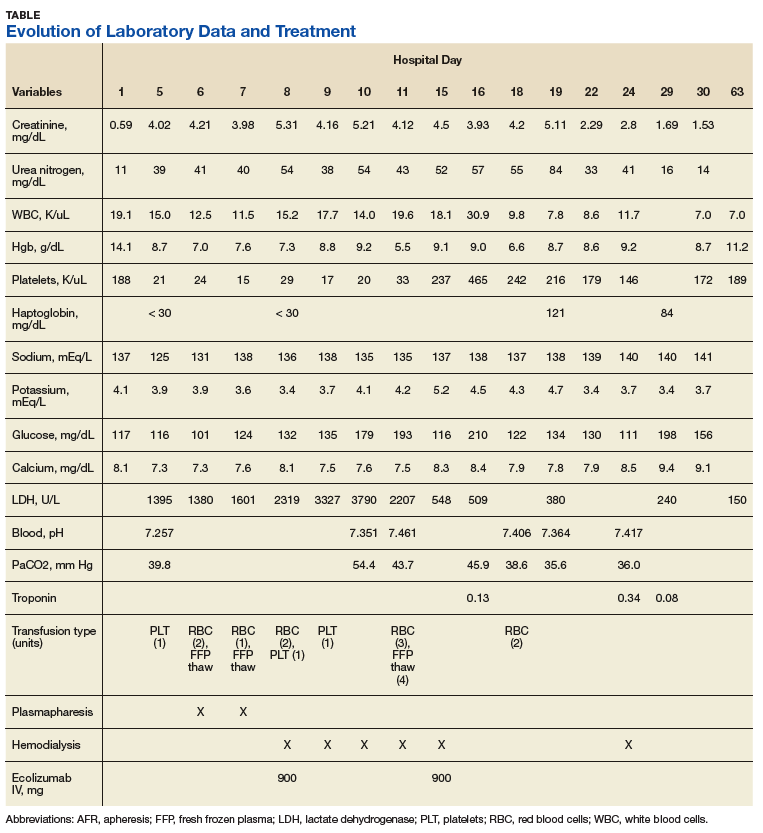
Hematuria was found on hospital day 2. On hospital day 4, the patient exhibited word finding difficulties. Blood studies revealed anemia, thrombocytopenia, leukocytosis, and increasing blood urea nitrogen (BUN) and creatinine. A computed tomography scan of the head was normal. Laboratory analysis showed schistocytes in the peripheral blood smear.
The patient’s cognitive functioning deteriorated on hospital day 5. She was not oriented to time or place. Her laboratory results showed complement level C3 at 70 mg/dL (ref: 83-193 mg/dL) complement C4 at 12 mg/dL (ref: 15-57mg/dL). The patient exhibited oliguria and hyponatremia, as well as both metabolic and respiratory acidosis; dialysis was then initiated. Results from the stool sample that was collected on hospital day 1 were received and tested positive for Shiga toxin.
At this point, the patient’s presentation of hemolytic anemia and thrombocytopenia in the setting of acute bloody diarrheal illness with known Shiga toxin, schistocytes on blood smear, and lack of pertinent medical history for other causes of this presentation made STEC-HUS the leading differential diagnosis. Plasmapheresis was ordered and performed on hospital day 6 and 7. Shiga toxin was no longer detected in the stool and antibiotics were stopped on hospital day 7.
The patient’s progressive deterioration in mental status continued on hospital day 8. She was not oriented to time or place, unable to perform simple calculations, and could not spell the word “hand” backwards. Physicians observed repetitive jerking motions of the upper extremities that were worse on the left side. An electroencephalogram (EEG) revealed right hemispheric sharp waves that were thought to be epileptiform (Figure 1). The patient began taking levetiracetam 1500 mg IV with 750 mg bid maintenance for seizure control. Plasmapheresis was discontinued due to her continued neurologic deterioration on this therapy. Consequently, eculizumab 900 mg IV was given along with the Neisseria meningitidis (N meningitidis) vaccine and a 19-day course of azithromycin 250 mg po as prophylaxis for encapsulated bacteria.
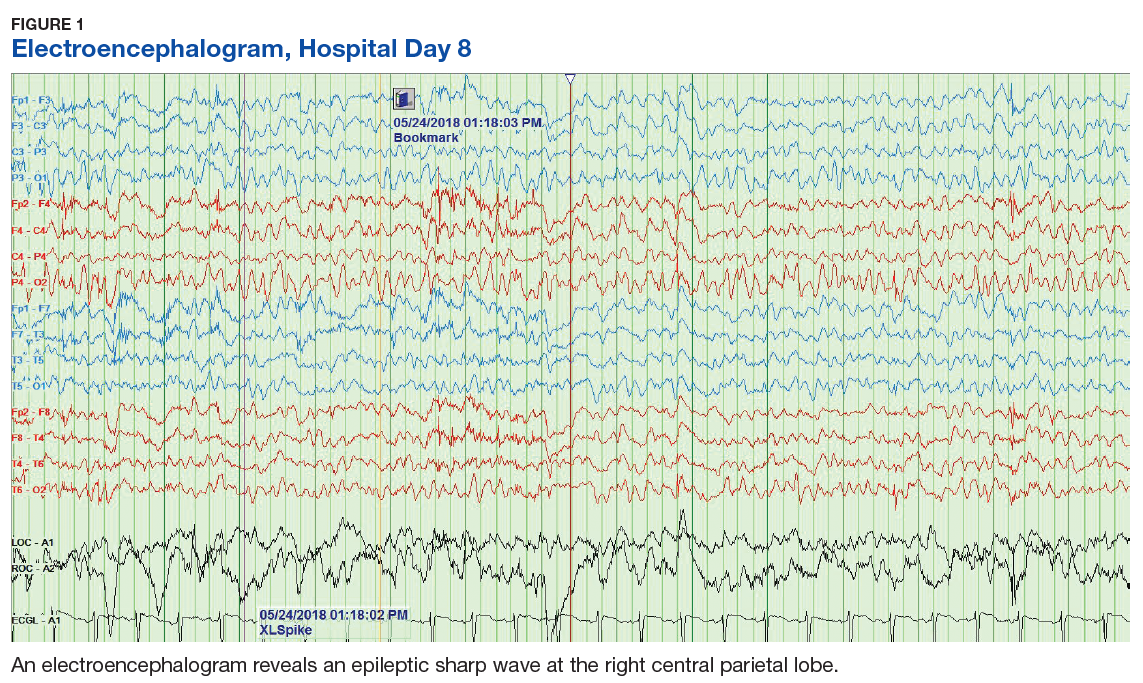
The patient continued to seize on hospital days 10 through 13. Oculocephalic maneuvers showed a tendency to keep her eyes deviated to the right. Her pupils continued to react to light. A repeat EEG showed diffuse slowing (5-6 Hz) with no epileptic activity seen (Figure 2). A second dose of eculizumab 900 mg IV was administered on hospital day 15. The patient experienced cardiac arrest on hospital day 16 and was successfully resuscitated. On hospital day 25 (10 days after receiving her second dose of eculizumab), the patient was able to speak and follow simple commands but exhibited difficulty concentrating and poor impulse control.
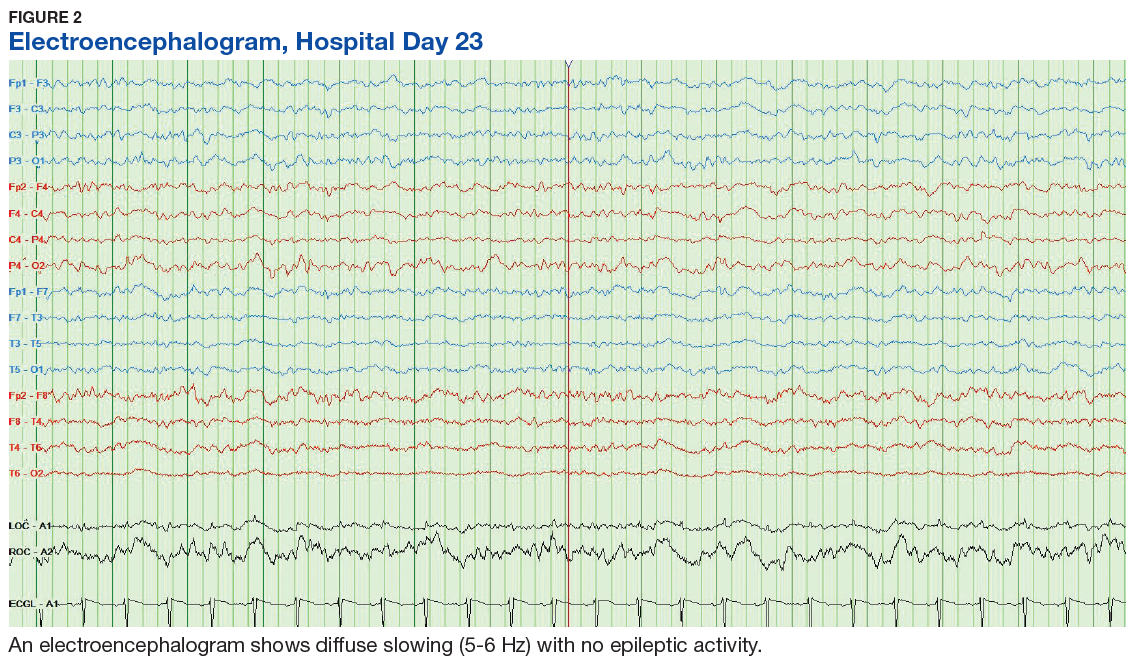
The patient was alert and oriented to person, place, time, and situation on hospital day 28 (6 days after the third and final dose of eculizumab). A neurologic exam was significant only for a slight intention tremor. She was continued on levetiracetam with a plan to be maintained on the medication for the next 6 months for seizure control. She was discharged on hospital day 30.
Twenty-eight days postdischarge (57 days postadmission), the patient showed marked recovery. She had returned to her previous employment as a business administrator on a part-time basis and exhibited no deficiencies in executive functioning or handling activities of daily living. Although she had been very active prior to this illness, she now experienced decreased physical and mental endurance; however, this gradually improved with physical therapy. She planned on returning to work on a full-time basis when she had regained her stamina. She also noticed difficulties in retaining short term memory since her discharge but believed that these symptoms were remitting. On examination her mental status and neurologic exam was significant for inability to continue serial 7s, left sided 4/5 muscle strength in quadriceps and thumb to 5th metacarpal adduction, bilateral 1+ reflexes in muscle groups tested (triceps, biceps, brachioradialis, patellar, and Achilles), loss of dull pinprick sensation bilaterally at web of hands, deficit in tandem gait while looking away, and slight intention tremor on finger to nose testing bilaterally (with left hand tremor more pronounced than right). Her complete blood count was normal. Her recovery continues to be monitored in an outpatient setting.
Discussion
HUS is characterized by 3 core clinical features: microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury.4 Schistocytes are seen on peripheral blood smear and occur due to the passage of red blood cells over the microvascular thrombi induced by the disease. HUS can be classified as typical, atypical, or occurring with a coexisting disease. Typical HUS is associated with STEC 0157:H7 subtype, a bacterium known to be acquired through contaminated food and via human-to-human transmission.6-8 In the case of typical STEC 0157:H7, the bacterium releases a verotoxin that damages the vascular endothelium, thereby leading to activation of the coagulation cascade and eventually the formation of thrombi.4 It has been hypothesized that the Shiga toxin also activates the alternative complement pathway directly, which could contribute to thrombosis.9 This would explain the findings of low complement levels in our patient. Atypical HUS is primarily attributable to mutations in the alternative complement pathway. Causes for the third type of HUS can include Streptococcus pneumoniae, HIV, drug toxicity, and alterations in the metabolism of cobalamin C.
Epidemiologically, 15.3% of children aged < 5 years develop typical HUS after exposure to STEC compared with 1.2% of adults aged 18 to 59 years. The median age of patients who developed HUS from STEC exposure was 4 years compared with 16 years for those who did not develop HUS.2
Neurologic manifestations increase mortality for HUS patients.10 These have been described in the pediatric population as alteration in consciousness (85%), seizures (71%), pyramidal syndrome (52%), and extrapyramidal syndrome with hypertonia (42%).11 Brain imaging in children has demonstrated hemorrhagic lesions involving the pons, basal ganglia, and occipital cortex.11 Blood flow to areas such as the cerebellum, brainstem, and orbitofrontal area can be compromised.10 Adult patients with HUS can present without lesions on cranial magnetic resonance imaging (MRI), but instead with transient symmetric vasogenic edema of the central brain stem.12 Unfortunately in this case, MRI was not performed because it was thought to provide limited aid in diagnosis and to avoid unnecessary testing for the acutely ill patient.
The underlying pathophysiology of neurologic manifestations in patients may be due to a metabolic disturbance, toxin-mediated damage of the vascular endothelium, or toxin-induced cytokine release resulting in death of neural cells and subsequent neuroinflammation. However, the most likely mechanism is parenchymal ischemic changes related to microangiopathy.11,13 Pediatric patients often experience seizures and altered mental status, and their EEGs display delta waves.13 This patient’s diffuse slowing on her second EEG and altered mental status suggests that the neuropathologic mechanisms for typical HUS in adults may be similar to those in children.
HUS Treatment
The treatment and management of adults with typical STEC-HUS is evolving. The patient was first suspected to have an infectious colitis and empiric antibiotics were initiated. Some studies suggest that antibiotic administration may worsen the course of HUS in children as it may lead to release and subsequent absorption of Shiga toxin in the intestine.9,14 However, there is little evidence to suggest harm or efficacy of administration in adults. It is unclear what role antibiotic administration played in the recovery time of HUS given the co-administration of other treatments such as eculizumab and plasmapheresis, but it does appear to have helped with the initial E coli infection.
Plasmapheresis was subsequently administered, due to its documented benefit in the treatment of HUS.15 However, it should be noted that even though plasmapheresis is currently used in patients with CNS involvement, it remains unproven with conflicting information on its efficacy.3,16 The mechanism of action is unclear, but it has been hypothesized that plasmapheresis prevents microangiopathy caused by microthrombi.3,16 For this reason, eculizumab is becoming the mainstay for treatment of STEC-HUS with neurologic complications given the lack of well researched alternative treatments. In this case study, the use of plasmapheresis did not result in clinical improvement, and was abandoned after 2 days of treatment.
Eculizumab is a humanized, recombinant monoclonal IgG antibody that is a terminal complement inhibitor of the alternative complement system at the final step to cleave C5.17 The Shiga toxin may directly activate the complement system via the alternative pathway, which can result in uncontrolled platelet and white blood cell activation and depletion, endothelial cell damage, and hemolysis. The galvanized complement system leads to a series of cascading events that contribute to organ damage and death.9 Eculizumab is FDA approved for use in atypical HUS.18 It also can be used off-label to treat typical-HUS in adults with neurologic complications.
Eculizumab interferes with the immune response against encapsulated bacteria because it inhibits the alternative complement pathway. Thus, vaccination against N meningitides is recommended 2 weeks prior to the administration of eculizumab. However, in situations where the risks of delaying eculizumab for 2 weeks are greater than the risk of developing an N meningitides infection, eculizumab may be given without delay.18 Given the rapid deterioration of our patient’s condition, the vaccine and eculizumab were given together with prophylactic azithromycin. Although penicillin is the standard for prophylaxis in this situation, the patient’s penicillin allergy led to the use of azithromycin 250 mg po once a day. Literature also suggests azithromycin reduces the carriage duration of E coli-induced colitis.19 As such, it is possible that some improvement in the patient’s condition could be attributed to the elimination of the pathogen and toxin.
Conclusion
Three doses of eculizumab were administered at weekly intervals, with the first dose on hospital day 8 and the final dose on hospital day 22. Prior to the first dose, the patient displayed significant decline in mental status with EEG findings of right hemisphere epileptogenic discharges. After her third dose, she was found to have a drastically improved mental status exam and a normal EEG. One week later, she was discharged home. At the time of her 1-month follow-up, she was independent in all activities of daily living and had returned to part-time work. Apart from subtle cognitive changes, the remainder of her neurologic exam was normal.
There is evidence that supports the efficacy of eculizumab in children with HUS with neurologic symptoms on dialysis.20 However, its use in adults is not well established.21 This patient required dialysis and had neurologic symptoms similar to pediatric patients described in the literature, and responded similarly to the eculizumab. The rationale for the use of eculizumab in STEC-HUS also is evidenced by in vitro demonstrations of complement activation in STEC-HUS.22-25 This case report adds to the literature supporting the use of eculizumab in adult patients with typical HUS with neurological complications. Further research is necessary to develop guidelines in the treatment of adult STEC-HUS with regards to neurologic complications.
Acknowledgments
The authors would like to thank Pete DiStaso, REEGT for his work on obtaining the electroencephalograms and Anthony Rinaldi, PsyD; Julie Cessnapalas, PsyD; and Syed Faizan Sagheer for proof-reading the article.
1. Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365(9464):1073-1086.
2. Gould LH, Demma L, Jones TF, et al. Hemolytic uremic syndrome and death in persons with Escherichia coli O157:H7 infection, foodborne diseases active surveillance network sites, 2000-2006. Clin Infect Dis. 2009;49(10):1480-1485.
3. Boyce TG, Swerdlow DL, Griffin PM. Escherichia coli O157:H7 and the hemolytic-uremic syndrome. N Engl J Med. 1995;333(6):364-368.
4. Rondeau E, Peraldi MN. Escherichia coli and the hemolytic-uremic syndrome. N Engl J Med. 1996;335(9):660-662.
5. Te Loo DM, van Hinsbergh VW, van den Heuvel LP, Monnens LA. Detection of verocytotoxin bound to circulating polymorphonuclear leukocytes of patients with hemolytic uremic syndrome. J Am Soc Nephrol. 2001;12(4):800-806.
6. Tran SL, Jenkins C, Livrelli V, Schüller S. Shiga toxin 2 translocation across intestinal epithelium is linked to virulence of Shiga toxin-producing Escherichia coli in humans. Microbiology. 2018;164(4):509-516.
7. Jokiranta TS. HUS and atypical HUS. Blood. 2017;129(21):2847-2856.
8. Ferens WA, Hovde CJ. Escherichia coli O157:H7: animal reservoir and sources of human infection. Foodborne Pathog Dis. 2011;8(4):465-487.
9. Percheron L, Gramada R, Tellier S, et al. Eculizumab treatment in severe pediatric STEC-HUS: a multicenter retrospective study. Pediatr Nephrol. 2018;33(8):1385-1394.
10. Hosaka T, Nakamagoe K, Tamaoka A. Hemolytic uremic syndrome-associated encephalopathy successfully treated with corticosteroids. Intern Med. 2017;56(21):2937-2941.
11. Nathanson S, Kwon T, Elmaleh M, et al. Acute neurological involvement in diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2010;5(7):1218-1228.
12. Wengenroth M, Hoeltje J, Repenthin J, et al. Central nervous system involvement in adults with epidemic hemolytic uremic syndrome. AJNR Am J Neuroradiol. 2013;34(5):1016-1021, S1.
13. Eriksson KJ, Boyd SG, Tasker RC. Acute neurology and neurophysiology of haemolytic-uraemic syndrome. Arch Dis Child. 2001;84(5):434-435.
14. Wong CS, Jelacic S, Habeeb RL, Watkins SL, Tarr PI. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Engl J Med. 2000;342(26):1930-1936.
15. Nguyen TC, Kiss JE, Goldman JR, Carcillo JA. The role of plasmapheresis in critical illness. Crit Care Clin. 2012;28(3):453-468, vii.
16. Loos S, Ahlenstiel T, Kranz B, et al. An outbreak of Shiga toxin-producing Escherichia coli O104:H4 hemolytic uremic syndrome in Germany: presentation and short-term outcome in children. Clin Infect Dis. 2012;55(6):753-759.
17. Hossain MA, Cheema A, Kalathil S, et al. Atypical hemolytic uremic syndrome: Laboratory characteristics, complement-amplifying conditions, renal biopsy, and genetic mutations. Saudi J Kidney Dis Transpl. 2018;29(2):276-283.
18. Soliris (eculizumab) [package insert]. Cheshire, CT: Alexion Pharmaceuticals, Inc; 2011.
19. Keenswijk W, Raes A, Vande Walle J. Is eculizumab efficacious in Shigatoxin-associated hemolytic uremic syndrome? A narrative review of current evidence. Eur J Pediatr. 2018;177(3):311-318.
20. Lapeyraque AL, Malina M, Fremeaux-Bacchi V, et al. Eculizumab in severe Shiga-toxin-associated HUS. N Engl J Med. 2011;364(26):2561-2563.
21. Pape L, Hartmann H, Bange FC, Suerbaum S, Bueltmann E, Ahlenstiel-Grunow T. Eculizumab in typical hemolytic uremic syndrome (HUS) with neurological involvement. Medicine (Baltimore). 2015;94(24):e1000.
22. Kim Y, Miller K, Michael AF. Breakdown products of C3 and factor B in hemolytic-uremic syndrome. J Lab Clin Med. 1977;89(4):845-850.
23. Monnens L, Molenaar J, Lambert PH, Proesmans W, van Munster P. The complement system in hemolytic-uremic syndrome in childhood. Clin Nephrol. 1980;13(4):168-171.
24. Thurman JM, Marians R, Emlen W, et al. Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2009;4(12):1920-1924.
25. Ståhl AL, Sartz L, Karpman D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood. 2011;117(20):5503-5513.
The case of a female presenting with Shiga toxin-producing Escherichia coli and hemolytic uremic syndrome highlights a severe neurologic complication that canbe associated with these conditions.
The case of a female presenting with Shiga toxin-producing Escherichia coli and hemolytic uremic syndrome highlights a severe neurologic complication that canbe associated with these conditions.
Hemolytic uremic syndrome (HUS) is a rare illness that can be acquired through the consumption of food products contaminated with strains of Shiga toxin-producing Escherichia coli (E coli; STEC).1 Between 6% and 15% of individuals infected with STEC develop HUS, with children affected more frequently than adults.2,3 This strain of E coli releases Shiga toxin into the systemic circulation, which causes a thrombotic microangiopathy resulting in the characteristic HUS triad of symptoms: acute renal insufficiency, thrombocytopenia, and hemolytic anemia.4-6
Although neurologic features are common in HUS, they have not been extensively studied, particularly in adults. We report a case of STEC 0157:H7 subtype HUS in an adult with severe neurologic complications. This case highlights the neurological sequelae in an adult with typical STEC-HUS. The use of treatment modalities, such as plasmapheresis and eculizumab, and their use in adult typical STEC-HUS also is explored.
Case
A 53-year-old white woman with no pertinent past medical history presented to the Bay Pines Veterans Affairs Healthcare System Emergency Department with a 2-day history of abdominal pain, vomiting, nausea, diarrhea, and bright bloody stools. She returned from a cruise to the Bahamas 3 days prior, where she ate local foods, including salads. She reported no fever, shortness of breath, chest pain, headache, and cognitive difficulties. She presented with a normal mental status and neurologic exam. Apart from leukocytosis and elevated glucose level, her laboratory results at initial presentation were normal, (Table). A stool sample showed occult blood with white blood cell counts (WBCs) but was negative for Clostridium difficile. She was started on ciprofloxacin 400 mg and metronidazole 500 mg on the day of admission.
Hematuria was found on hospital day 2. On hospital day 4, the patient exhibited word finding difficulties. Blood studies revealed anemia, thrombocytopenia, leukocytosis, and increasing blood urea nitrogen (BUN) and creatinine. A computed tomography scan of the head was normal. Laboratory analysis showed schistocytes in the peripheral blood smear.
The patient’s cognitive functioning deteriorated on hospital day 5. She was not oriented to time or place. Her laboratory results showed complement level C3 at 70 mg/dL (ref: 83-193 mg/dL) complement C4 at 12 mg/dL (ref: 15-57mg/dL). The patient exhibited oliguria and hyponatremia, as well as both metabolic and respiratory acidosis; dialysis was then initiated. Results from the stool sample that was collected on hospital day 1 were received and tested positive for Shiga toxin.
At this point, the patient’s presentation of hemolytic anemia and thrombocytopenia in the setting of acute bloody diarrheal illness with known Shiga toxin, schistocytes on blood smear, and lack of pertinent medical history for other causes of this presentation made STEC-HUS the leading differential diagnosis. Plasmapheresis was ordered and performed on hospital day 6 and 7. Shiga toxin was no longer detected in the stool and antibiotics were stopped on hospital day 7.
The patient’s progressive deterioration in mental status continued on hospital day 8. She was not oriented to time or place, unable to perform simple calculations, and could not spell the word “hand” backwards. Physicians observed repetitive jerking motions of the upper extremities that were worse on the left side. An electroencephalogram (EEG) revealed right hemispheric sharp waves that were thought to be epileptiform (Figure 1). The patient began taking levetiracetam 1500 mg IV with 750 mg bid maintenance for seizure control. Plasmapheresis was discontinued due to her continued neurologic deterioration on this therapy. Consequently, eculizumab 900 mg IV was given along with the Neisseria meningitidis (N meningitidis) vaccine and a 19-day course of azithromycin 250 mg po as prophylaxis for encapsulated bacteria.
The patient continued to seize on hospital days 10 through 13. Oculocephalic maneuvers showed a tendency to keep her eyes deviated to the right. Her pupils continued to react to light. A repeat EEG showed diffuse slowing (5-6 Hz) with no epileptic activity seen (Figure 2). A second dose of eculizumab 900 mg IV was administered on hospital day 15. The patient experienced cardiac arrest on hospital day 16 and was successfully resuscitated. On hospital day 25 (10 days after receiving her second dose of eculizumab), the patient was able to speak and follow simple commands but exhibited difficulty concentrating and poor impulse control.
The patient was alert and oriented to person, place, time, and situation on hospital day 28 (6 days after the third and final dose of eculizumab). A neurologic exam was significant only for a slight intention tremor. She was continued on levetiracetam with a plan to be maintained on the medication for the next 6 months for seizure control. She was discharged on hospital day 30.
Twenty-eight days postdischarge (57 days postadmission), the patient showed marked recovery. She had returned to her previous employment as a business administrator on a part-time basis and exhibited no deficiencies in executive functioning or handling activities of daily living. Although she had been very active prior to this illness, she now experienced decreased physical and mental endurance; however, this gradually improved with physical therapy. She planned on returning to work on a full-time basis when she had regained her stamina. She also noticed difficulties in retaining short term memory since her discharge but believed that these symptoms were remitting. On examination her mental status and neurologic exam was significant for inability to continue serial 7s, left sided 4/5 muscle strength in quadriceps and thumb to 5th metacarpal adduction, bilateral 1+ reflexes in muscle groups tested (triceps, biceps, brachioradialis, patellar, and Achilles), loss of dull pinprick sensation bilaterally at web of hands, deficit in tandem gait while looking away, and slight intention tremor on finger to nose testing bilaterally (with left hand tremor more pronounced than right). Her complete blood count was normal. Her recovery continues to be monitored in an outpatient setting.
Discussion
HUS is characterized by 3 core clinical features: microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury.4 Schistocytes are seen on peripheral blood smear and occur due to the passage of red blood cells over the microvascular thrombi induced by the disease. HUS can be classified as typical, atypical, or occurring with a coexisting disease. Typical HUS is associated with STEC 0157:H7 subtype, a bacterium known to be acquired through contaminated food and via human-to-human transmission.6-8 In the case of typical STEC 0157:H7, the bacterium releases a verotoxin that damages the vascular endothelium, thereby leading to activation of the coagulation cascade and eventually the formation of thrombi.4 It has been hypothesized that the Shiga toxin also activates the alternative complement pathway directly, which could contribute to thrombosis.9 This would explain the findings of low complement levels in our patient. Atypical HUS is primarily attributable to mutations in the alternative complement pathway. Causes for the third type of HUS can include Streptococcus pneumoniae, HIV, drug toxicity, and alterations in the metabolism of cobalamin C.
Epidemiologically, 15.3% of children aged < 5 years develop typical HUS after exposure to STEC compared with 1.2% of adults aged 18 to 59 years. The median age of patients who developed HUS from STEC exposure was 4 years compared with 16 years for those who did not develop HUS.2
Neurologic manifestations increase mortality for HUS patients.10 These have been described in the pediatric population as alteration in consciousness (85%), seizures (71%), pyramidal syndrome (52%), and extrapyramidal syndrome with hypertonia (42%).11 Brain imaging in children has demonstrated hemorrhagic lesions involving the pons, basal ganglia, and occipital cortex.11 Blood flow to areas such as the cerebellum, brainstem, and orbitofrontal area can be compromised.10 Adult patients with HUS can present without lesions on cranial magnetic resonance imaging (MRI), but instead with transient symmetric vasogenic edema of the central brain stem.12 Unfortunately in this case, MRI was not performed because it was thought to provide limited aid in diagnosis and to avoid unnecessary testing for the acutely ill patient.
The underlying pathophysiology of neurologic manifestations in patients may be due to a metabolic disturbance, toxin-mediated damage of the vascular endothelium, or toxin-induced cytokine release resulting in death of neural cells and subsequent neuroinflammation. However, the most likely mechanism is parenchymal ischemic changes related to microangiopathy.11,13 Pediatric patients often experience seizures and altered mental status, and their EEGs display delta waves.13 This patient’s diffuse slowing on her second EEG and altered mental status suggests that the neuropathologic mechanisms for typical HUS in adults may be similar to those in children.
HUS Treatment
The treatment and management of adults with typical STEC-HUS is evolving. The patient was first suspected to have an infectious colitis and empiric antibiotics were initiated. Some studies suggest that antibiotic administration may worsen the course of HUS in children as it may lead to release and subsequent absorption of Shiga toxin in the intestine.9,14 However, there is little evidence to suggest harm or efficacy of administration in adults. It is unclear what role antibiotic administration played in the recovery time of HUS given the co-administration of other treatments such as eculizumab and plasmapheresis, but it does appear to have helped with the initial E coli infection.
Plasmapheresis was subsequently administered, due to its documented benefit in the treatment of HUS.15 However, it should be noted that even though plasmapheresis is currently used in patients with CNS involvement, it remains unproven with conflicting information on its efficacy.3,16 The mechanism of action is unclear, but it has been hypothesized that plasmapheresis prevents microangiopathy caused by microthrombi.3,16 For this reason, eculizumab is becoming the mainstay for treatment of STEC-HUS with neurologic complications given the lack of well researched alternative treatments. In this case study, the use of plasmapheresis did not result in clinical improvement, and was abandoned after 2 days of treatment.
Eculizumab is a humanized, recombinant monoclonal IgG antibody that is a terminal complement inhibitor of the alternative complement system at the final step to cleave C5.17 The Shiga toxin may directly activate the complement system via the alternative pathway, which can result in uncontrolled platelet and white blood cell activation and depletion, endothelial cell damage, and hemolysis. The galvanized complement system leads to a series of cascading events that contribute to organ damage and death.9 Eculizumab is FDA approved for use in atypical HUS.18 It also can be used off-label to treat typical-HUS in adults with neurologic complications.
Eculizumab interferes with the immune response against encapsulated bacteria because it inhibits the alternative complement pathway. Thus, vaccination against N meningitides is recommended 2 weeks prior to the administration of eculizumab. However, in situations where the risks of delaying eculizumab for 2 weeks are greater than the risk of developing an N meningitides infection, eculizumab may be given without delay.18 Given the rapid deterioration of our patient’s condition, the vaccine and eculizumab were given together with prophylactic azithromycin. Although penicillin is the standard for prophylaxis in this situation, the patient’s penicillin allergy led to the use of azithromycin 250 mg po once a day. Literature also suggests azithromycin reduces the carriage duration of E coli-induced colitis.19 As such, it is possible that some improvement in the patient’s condition could be attributed to the elimination of the pathogen and toxin.
Conclusion
Three doses of eculizumab were administered at weekly intervals, with the first dose on hospital day 8 and the final dose on hospital day 22. Prior to the first dose, the patient displayed significant decline in mental status with EEG findings of right hemisphere epileptogenic discharges. After her third dose, she was found to have a drastically improved mental status exam and a normal EEG. One week later, she was discharged home. At the time of her 1-month follow-up, she was independent in all activities of daily living and had returned to part-time work. Apart from subtle cognitive changes, the remainder of her neurologic exam was normal.
There is evidence that supports the efficacy of eculizumab in children with HUS with neurologic symptoms on dialysis.20 However, its use in adults is not well established.21 This patient required dialysis and had neurologic symptoms similar to pediatric patients described in the literature, and responded similarly to the eculizumab. The rationale for the use of eculizumab in STEC-HUS also is evidenced by in vitro demonstrations of complement activation in STEC-HUS.22-25 This case report adds to the literature supporting the use of eculizumab in adult patients with typical HUS with neurological complications. Further research is necessary to develop guidelines in the treatment of adult STEC-HUS with regards to neurologic complications.
Acknowledgments
The authors would like to thank Pete DiStaso, REEGT for his work on obtaining the electroencephalograms and Anthony Rinaldi, PsyD; Julie Cessnapalas, PsyD; and Syed Faizan Sagheer for proof-reading the article.
Hemolytic uremic syndrome (HUS) is a rare illness that can be acquired through the consumption of food products contaminated with strains of Shiga toxin-producing Escherichia coli (E coli; STEC).1 Between 6% and 15% of individuals infected with STEC develop HUS, with children affected more frequently than adults.2,3 This strain of E coli releases Shiga toxin into the systemic circulation, which causes a thrombotic microangiopathy resulting in the characteristic HUS triad of symptoms: acute renal insufficiency, thrombocytopenia, and hemolytic anemia.4-6
Although neurologic features are common in HUS, they have not been extensively studied, particularly in adults. We report a case of STEC 0157:H7 subtype HUS in an adult with severe neurologic complications. This case highlights the neurological sequelae in an adult with typical STEC-HUS. The use of treatment modalities, such as plasmapheresis and eculizumab, and their use in adult typical STEC-HUS also is explored.
Case
A 53-year-old white woman with no pertinent past medical history presented to the Bay Pines Veterans Affairs Healthcare System Emergency Department with a 2-day history of abdominal pain, vomiting, nausea, diarrhea, and bright bloody stools. She returned from a cruise to the Bahamas 3 days prior, where she ate local foods, including salads. She reported no fever, shortness of breath, chest pain, headache, and cognitive difficulties. She presented with a normal mental status and neurologic exam. Apart from leukocytosis and elevated glucose level, her laboratory results at initial presentation were normal, (Table). A stool sample showed occult blood with white blood cell counts (WBCs) but was negative for Clostridium difficile. She was started on ciprofloxacin 400 mg and metronidazole 500 mg on the day of admission.
Hematuria was found on hospital day 2. On hospital day 4, the patient exhibited word finding difficulties. Blood studies revealed anemia, thrombocytopenia, leukocytosis, and increasing blood urea nitrogen (BUN) and creatinine. A computed tomography scan of the head was normal. Laboratory analysis showed schistocytes in the peripheral blood smear.
The patient’s cognitive functioning deteriorated on hospital day 5. She was not oriented to time or place. Her laboratory results showed complement level C3 at 70 mg/dL (ref: 83-193 mg/dL) complement C4 at 12 mg/dL (ref: 15-57mg/dL). The patient exhibited oliguria and hyponatremia, as well as both metabolic and respiratory acidosis; dialysis was then initiated. Results from the stool sample that was collected on hospital day 1 were received and tested positive for Shiga toxin.
At this point, the patient’s presentation of hemolytic anemia and thrombocytopenia in the setting of acute bloody diarrheal illness with known Shiga toxin, schistocytes on blood smear, and lack of pertinent medical history for other causes of this presentation made STEC-HUS the leading differential diagnosis. Plasmapheresis was ordered and performed on hospital day 6 and 7. Shiga toxin was no longer detected in the stool and antibiotics were stopped on hospital day 7.
The patient’s progressive deterioration in mental status continued on hospital day 8. She was not oriented to time or place, unable to perform simple calculations, and could not spell the word “hand” backwards. Physicians observed repetitive jerking motions of the upper extremities that were worse on the left side. An electroencephalogram (EEG) revealed right hemispheric sharp waves that were thought to be epileptiform (Figure 1). The patient began taking levetiracetam 1500 mg IV with 750 mg bid maintenance for seizure control. Plasmapheresis was discontinued due to her continued neurologic deterioration on this therapy. Consequently, eculizumab 900 mg IV was given along with the Neisseria meningitidis (N meningitidis) vaccine and a 19-day course of azithromycin 250 mg po as prophylaxis for encapsulated bacteria.
The patient continued to seize on hospital days 10 through 13. Oculocephalic maneuvers showed a tendency to keep her eyes deviated to the right. Her pupils continued to react to light. A repeat EEG showed diffuse slowing (5-6 Hz) with no epileptic activity seen (Figure 2). A second dose of eculizumab 900 mg IV was administered on hospital day 15. The patient experienced cardiac arrest on hospital day 16 and was successfully resuscitated. On hospital day 25 (10 days after receiving her second dose of eculizumab), the patient was able to speak and follow simple commands but exhibited difficulty concentrating and poor impulse control.
The patient was alert and oriented to person, place, time, and situation on hospital day 28 (6 days after the third and final dose of eculizumab). A neurologic exam was significant only for a slight intention tremor. She was continued on levetiracetam with a plan to be maintained on the medication for the next 6 months for seizure control. She was discharged on hospital day 30.
Twenty-eight days postdischarge (57 days postadmission), the patient showed marked recovery. She had returned to her previous employment as a business administrator on a part-time basis and exhibited no deficiencies in executive functioning or handling activities of daily living. Although she had been very active prior to this illness, she now experienced decreased physical and mental endurance; however, this gradually improved with physical therapy. She planned on returning to work on a full-time basis when she had regained her stamina. She also noticed difficulties in retaining short term memory since her discharge but believed that these symptoms were remitting. On examination her mental status and neurologic exam was significant for inability to continue serial 7s, left sided 4/5 muscle strength in quadriceps and thumb to 5th metacarpal adduction, bilateral 1+ reflexes in muscle groups tested (triceps, biceps, brachioradialis, patellar, and Achilles), loss of dull pinprick sensation bilaterally at web of hands, deficit in tandem gait while looking away, and slight intention tremor on finger to nose testing bilaterally (with left hand tremor more pronounced than right). Her complete blood count was normal. Her recovery continues to be monitored in an outpatient setting.
Discussion
HUS is characterized by 3 core clinical features: microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury.4 Schistocytes are seen on peripheral blood smear and occur due to the passage of red blood cells over the microvascular thrombi induced by the disease. HUS can be classified as typical, atypical, or occurring with a coexisting disease. Typical HUS is associated with STEC 0157:H7 subtype, a bacterium known to be acquired through contaminated food and via human-to-human transmission.6-8 In the case of typical STEC 0157:H7, the bacterium releases a verotoxin that damages the vascular endothelium, thereby leading to activation of the coagulation cascade and eventually the formation of thrombi.4 It has been hypothesized that the Shiga toxin also activates the alternative complement pathway directly, which could contribute to thrombosis.9 This would explain the findings of low complement levels in our patient. Atypical HUS is primarily attributable to mutations in the alternative complement pathway. Causes for the third type of HUS can include Streptococcus pneumoniae, HIV, drug toxicity, and alterations in the metabolism of cobalamin C.
Epidemiologically, 15.3% of children aged < 5 years develop typical HUS after exposure to STEC compared with 1.2% of adults aged 18 to 59 years. The median age of patients who developed HUS from STEC exposure was 4 years compared with 16 years for those who did not develop HUS.2
Neurologic manifestations increase mortality for HUS patients.10 These have been described in the pediatric population as alteration in consciousness (85%), seizures (71%), pyramidal syndrome (52%), and extrapyramidal syndrome with hypertonia (42%).11 Brain imaging in children has demonstrated hemorrhagic lesions involving the pons, basal ganglia, and occipital cortex.11 Blood flow to areas such as the cerebellum, brainstem, and orbitofrontal area can be compromised.10 Adult patients with HUS can present without lesions on cranial magnetic resonance imaging (MRI), but instead with transient symmetric vasogenic edema of the central brain stem.12 Unfortunately in this case, MRI was not performed because it was thought to provide limited aid in diagnosis and to avoid unnecessary testing for the acutely ill patient.
The underlying pathophysiology of neurologic manifestations in patients may be due to a metabolic disturbance, toxin-mediated damage of the vascular endothelium, or toxin-induced cytokine release resulting in death of neural cells and subsequent neuroinflammation. However, the most likely mechanism is parenchymal ischemic changes related to microangiopathy.11,13 Pediatric patients often experience seizures and altered mental status, and their EEGs display delta waves.13 This patient’s diffuse slowing on her second EEG and altered mental status suggests that the neuropathologic mechanisms for typical HUS in adults may be similar to those in children.
HUS Treatment
The treatment and management of adults with typical STEC-HUS is evolving. The patient was first suspected to have an infectious colitis and empiric antibiotics were initiated. Some studies suggest that antibiotic administration may worsen the course of HUS in children as it may lead to release and subsequent absorption of Shiga toxin in the intestine.9,14 However, there is little evidence to suggest harm or efficacy of administration in adults. It is unclear what role antibiotic administration played in the recovery time of HUS given the co-administration of other treatments such as eculizumab and plasmapheresis, but it does appear to have helped with the initial E coli infection.
Plasmapheresis was subsequently administered, due to its documented benefit in the treatment of HUS.15 However, it should be noted that even though plasmapheresis is currently used in patients with CNS involvement, it remains unproven with conflicting information on its efficacy.3,16 The mechanism of action is unclear, but it has been hypothesized that plasmapheresis prevents microangiopathy caused by microthrombi.3,16 For this reason, eculizumab is becoming the mainstay for treatment of STEC-HUS with neurologic complications given the lack of well researched alternative treatments. In this case study, the use of plasmapheresis did not result in clinical improvement, and was abandoned after 2 days of treatment.
Eculizumab is a humanized, recombinant monoclonal IgG antibody that is a terminal complement inhibitor of the alternative complement system at the final step to cleave C5.17 The Shiga toxin may directly activate the complement system via the alternative pathway, which can result in uncontrolled platelet and white blood cell activation and depletion, endothelial cell damage, and hemolysis. The galvanized complement system leads to a series of cascading events that contribute to organ damage and death.9 Eculizumab is FDA approved for use in atypical HUS.18 It also can be used off-label to treat typical-HUS in adults with neurologic complications.
Eculizumab interferes with the immune response against encapsulated bacteria because it inhibits the alternative complement pathway. Thus, vaccination against N meningitides is recommended 2 weeks prior to the administration of eculizumab. However, in situations where the risks of delaying eculizumab for 2 weeks are greater than the risk of developing an N meningitides infection, eculizumab may be given without delay.18 Given the rapid deterioration of our patient’s condition, the vaccine and eculizumab were given together with prophylactic azithromycin. Although penicillin is the standard for prophylaxis in this situation, the patient’s penicillin allergy led to the use of azithromycin 250 mg po once a day. Literature also suggests azithromycin reduces the carriage duration of E coli-induced colitis.19 As such, it is possible that some improvement in the patient’s condition could be attributed to the elimination of the pathogen and toxin.
Conclusion
Three doses of eculizumab were administered at weekly intervals, with the first dose on hospital day 8 and the final dose on hospital day 22. Prior to the first dose, the patient displayed significant decline in mental status with EEG findings of right hemisphere epileptogenic discharges. After her third dose, she was found to have a drastically improved mental status exam and a normal EEG. One week later, she was discharged home. At the time of her 1-month follow-up, she was independent in all activities of daily living and had returned to part-time work. Apart from subtle cognitive changes, the remainder of her neurologic exam was normal.
There is evidence that supports the efficacy of eculizumab in children with HUS with neurologic symptoms on dialysis.20 However, its use in adults is not well established.21 This patient required dialysis and had neurologic symptoms similar to pediatric patients described in the literature, and responded similarly to the eculizumab. The rationale for the use of eculizumab in STEC-HUS also is evidenced by in vitro demonstrations of complement activation in STEC-HUS.22-25 This case report adds to the literature supporting the use of eculizumab in adult patients with typical HUS with neurological complications. Further research is necessary to develop guidelines in the treatment of adult STEC-HUS with regards to neurologic complications.
Acknowledgments
The authors would like to thank Pete DiStaso, REEGT for his work on obtaining the electroencephalograms and Anthony Rinaldi, PsyD; Julie Cessnapalas, PsyD; and Syed Faizan Sagheer for proof-reading the article.
1. Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365(9464):1073-1086.
2. Gould LH, Demma L, Jones TF, et al. Hemolytic uremic syndrome and death in persons with Escherichia coli O157:H7 infection, foodborne diseases active surveillance network sites, 2000-2006. Clin Infect Dis. 2009;49(10):1480-1485.
3. Boyce TG, Swerdlow DL, Griffin PM. Escherichia coli O157:H7 and the hemolytic-uremic syndrome. N Engl J Med. 1995;333(6):364-368.
4. Rondeau E, Peraldi MN. Escherichia coli and the hemolytic-uremic syndrome. N Engl J Med. 1996;335(9):660-662.
5. Te Loo DM, van Hinsbergh VW, van den Heuvel LP, Monnens LA. Detection of verocytotoxin bound to circulating polymorphonuclear leukocytes of patients with hemolytic uremic syndrome. J Am Soc Nephrol. 2001;12(4):800-806.
6. Tran SL, Jenkins C, Livrelli V, Schüller S. Shiga toxin 2 translocation across intestinal epithelium is linked to virulence of Shiga toxin-producing Escherichia coli in humans. Microbiology. 2018;164(4):509-516.
7. Jokiranta TS. HUS and atypical HUS. Blood. 2017;129(21):2847-2856.
8. Ferens WA, Hovde CJ. Escherichia coli O157:H7: animal reservoir and sources of human infection. Foodborne Pathog Dis. 2011;8(4):465-487.
9. Percheron L, Gramada R, Tellier S, et al. Eculizumab treatment in severe pediatric STEC-HUS: a multicenter retrospective study. Pediatr Nephrol. 2018;33(8):1385-1394.
10. Hosaka T, Nakamagoe K, Tamaoka A. Hemolytic uremic syndrome-associated encephalopathy successfully treated with corticosteroids. Intern Med. 2017;56(21):2937-2941.
11. Nathanson S, Kwon T, Elmaleh M, et al. Acute neurological involvement in diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2010;5(7):1218-1228.
12. Wengenroth M, Hoeltje J, Repenthin J, et al. Central nervous system involvement in adults with epidemic hemolytic uremic syndrome. AJNR Am J Neuroradiol. 2013;34(5):1016-1021, S1.
13. Eriksson KJ, Boyd SG, Tasker RC. Acute neurology and neurophysiology of haemolytic-uraemic syndrome. Arch Dis Child. 2001;84(5):434-435.
14. Wong CS, Jelacic S, Habeeb RL, Watkins SL, Tarr PI. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Engl J Med. 2000;342(26):1930-1936.
15. Nguyen TC, Kiss JE, Goldman JR, Carcillo JA. The role of plasmapheresis in critical illness. Crit Care Clin. 2012;28(3):453-468, vii.
16. Loos S, Ahlenstiel T, Kranz B, et al. An outbreak of Shiga toxin-producing Escherichia coli O104:H4 hemolytic uremic syndrome in Germany: presentation and short-term outcome in children. Clin Infect Dis. 2012;55(6):753-759.
17. Hossain MA, Cheema A, Kalathil S, et al. Atypical hemolytic uremic syndrome: Laboratory characteristics, complement-amplifying conditions, renal biopsy, and genetic mutations. Saudi J Kidney Dis Transpl. 2018;29(2):276-283.
18. Soliris (eculizumab) [package insert]. Cheshire, CT: Alexion Pharmaceuticals, Inc; 2011.
19. Keenswijk W, Raes A, Vande Walle J. Is eculizumab efficacious in Shigatoxin-associated hemolytic uremic syndrome? A narrative review of current evidence. Eur J Pediatr. 2018;177(3):311-318.
20. Lapeyraque AL, Malina M, Fremeaux-Bacchi V, et al. Eculizumab in severe Shiga-toxin-associated HUS. N Engl J Med. 2011;364(26):2561-2563.
21. Pape L, Hartmann H, Bange FC, Suerbaum S, Bueltmann E, Ahlenstiel-Grunow T. Eculizumab in typical hemolytic uremic syndrome (HUS) with neurological involvement. Medicine (Baltimore). 2015;94(24):e1000.
22. Kim Y, Miller K, Michael AF. Breakdown products of C3 and factor B in hemolytic-uremic syndrome. J Lab Clin Med. 1977;89(4):845-850.
23. Monnens L, Molenaar J, Lambert PH, Proesmans W, van Munster P. The complement system in hemolytic-uremic syndrome in childhood. Clin Nephrol. 1980;13(4):168-171.
24. Thurman JM, Marians R, Emlen W, et al. Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2009;4(12):1920-1924.
25. Ståhl AL, Sartz L, Karpman D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood. 2011;117(20):5503-5513.
1. Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365(9464):1073-1086.
2. Gould LH, Demma L, Jones TF, et al. Hemolytic uremic syndrome and death in persons with Escherichia coli O157:H7 infection, foodborne diseases active surveillance network sites, 2000-2006. Clin Infect Dis. 2009;49(10):1480-1485.
3. Boyce TG, Swerdlow DL, Griffin PM. Escherichia coli O157:H7 and the hemolytic-uremic syndrome. N Engl J Med. 1995;333(6):364-368.
4. Rondeau E, Peraldi MN. Escherichia coli and the hemolytic-uremic syndrome. N Engl J Med. 1996;335(9):660-662.
5. Te Loo DM, van Hinsbergh VW, van den Heuvel LP, Monnens LA. Detection of verocytotoxin bound to circulating polymorphonuclear leukocytes of patients with hemolytic uremic syndrome. J Am Soc Nephrol. 2001;12(4):800-806.
6. Tran SL, Jenkins C, Livrelli V, Schüller S. Shiga toxin 2 translocation across intestinal epithelium is linked to virulence of Shiga toxin-producing Escherichia coli in humans. Microbiology. 2018;164(4):509-516.
7. Jokiranta TS. HUS and atypical HUS. Blood. 2017;129(21):2847-2856.
8. Ferens WA, Hovde CJ. Escherichia coli O157:H7: animal reservoir and sources of human infection. Foodborne Pathog Dis. 2011;8(4):465-487.
9. Percheron L, Gramada R, Tellier S, et al. Eculizumab treatment in severe pediatric STEC-HUS: a multicenter retrospective study. Pediatr Nephrol. 2018;33(8):1385-1394.
10. Hosaka T, Nakamagoe K, Tamaoka A. Hemolytic uremic syndrome-associated encephalopathy successfully treated with corticosteroids. Intern Med. 2017;56(21):2937-2941.
11. Nathanson S, Kwon T, Elmaleh M, et al. Acute neurological involvement in diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2010;5(7):1218-1228.
12. Wengenroth M, Hoeltje J, Repenthin J, et al. Central nervous system involvement in adults with epidemic hemolytic uremic syndrome. AJNR Am J Neuroradiol. 2013;34(5):1016-1021, S1.
13. Eriksson KJ, Boyd SG, Tasker RC. Acute neurology and neurophysiology of haemolytic-uraemic syndrome. Arch Dis Child. 2001;84(5):434-435.
14. Wong CS, Jelacic S, Habeeb RL, Watkins SL, Tarr PI. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Engl J Med. 2000;342(26):1930-1936.
15. Nguyen TC, Kiss JE, Goldman JR, Carcillo JA. The role of plasmapheresis in critical illness. Crit Care Clin. 2012;28(3):453-468, vii.
16. Loos S, Ahlenstiel T, Kranz B, et al. An outbreak of Shiga toxin-producing Escherichia coli O104:H4 hemolytic uremic syndrome in Germany: presentation and short-term outcome in children. Clin Infect Dis. 2012;55(6):753-759.
17. Hossain MA, Cheema A, Kalathil S, et al. Atypical hemolytic uremic syndrome: Laboratory characteristics, complement-amplifying conditions, renal biopsy, and genetic mutations. Saudi J Kidney Dis Transpl. 2018;29(2):276-283.
18. Soliris (eculizumab) [package insert]. Cheshire, CT: Alexion Pharmaceuticals, Inc; 2011.
19. Keenswijk W, Raes A, Vande Walle J. Is eculizumab efficacious in Shigatoxin-associated hemolytic uremic syndrome? A narrative review of current evidence. Eur J Pediatr. 2018;177(3):311-318.
20. Lapeyraque AL, Malina M, Fremeaux-Bacchi V, et al. Eculizumab in severe Shiga-toxin-associated HUS. N Engl J Med. 2011;364(26):2561-2563.
21. Pape L, Hartmann H, Bange FC, Suerbaum S, Bueltmann E, Ahlenstiel-Grunow T. Eculizumab in typical hemolytic uremic syndrome (HUS) with neurological involvement. Medicine (Baltimore). 2015;94(24):e1000.
22. Kim Y, Miller K, Michael AF. Breakdown products of C3 and factor B in hemolytic-uremic syndrome. J Lab Clin Med. 1977;89(4):845-850.
23. Monnens L, Molenaar J, Lambert PH, Proesmans W, van Munster P. The complement system in hemolytic-uremic syndrome in childhood. Clin Nephrol. 1980;13(4):168-171.
24. Thurman JM, Marians R, Emlen W, et al. Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2009;4(12):1920-1924.
25. Ståhl AL, Sartz L, Karpman D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood. 2011;117(20):5503-5513.
Adherence to Disease-Modifying Therapies in Patients With MS: A Retrospective Cohort Study
Multiple sclerosis (MS) is an autoimmune disorder in which the myelin of the brain and spinal cord is selectively targeted by immune-system cells. As a result, nerve transmission is disrupted, leading to a variety of unpredictable symptoms from weakness and a lack of balance to blindness and paralysis of the body. Clinically, MS can take 4 courses, including relapsing-remitting (RRMS), primary-progressive (PPMS), secondary-progressive, and progressive-relapsing.1 At onset, 85% of diagnosed patients have RRMS, and 10% to 15% have PPMS.2 If untreated, patients with RRMS become secondary-progressive, with progressive disability and indiscrete relapses.3 Hence, disease-modifying therapies are targeted toward decreasing the relapse rate as well as slowing the progression of the disease.4
Annually, about 16,000 veterans with MS receive health care services from the VHA.5 The C.W. Bill Young Bay Pines VA Healthcare System (BPVAHCS) is a level 1 facility that annually serves more than 105,000 veterans. The BPVAMC sees veterans with a wide variety of neurologic illnesses and has 5 full-time neurologists with subspecialty training. The BPVAHCS facility has outpatient clinics and a 200 inpatient bed facility. The Neurology Department sees 125 outpatients per week and consults on about 30 inpatients per week.
Methods
A retrospective review of BPVAHCS patients diagnosed with MS from January 2009 to July 2014 was performed with institutional review board approval. Patient data were collected from ICD-9-CM codes and kept confidential. A list of patients was collected from Neurology Clinic patient visits with “Multiple Sclerosis” on the problem list.
Patient medical records were reviewed to collect the following information: presence of rigorous diagnosis of MS, clinical course of MS in patient, presence or absence of disease-modifying therapy, and disease-modifying agents (DMAs) used.
Determining factors for DMA treatment included increasing tiredness, weakness, visual symptoms, and radiologic evidence (magnetic resonance imaging) of recurrent, active lesions. Each patient was examined on a case-by-case basis to assess whether or not the patient actually had MS and if so, whether they were being treated with DMAs. Only patients with RRMS were included. Patients were excluded from the study if they were deceased, not currently under BPVACHS care, or had symptoms of optic neuritis but were not fully indicative of MS. Patients with clinically isolated syndrome, probable diagnosis of MS, or PPMS also were excluded from the study.
Exclusion from this study was based on 2 additional premises. Patients were excluded if they discontinued an initial ABC (interferon beta-1a, interferon beta-1b, glatiramer acetate) due to DMA treatment relapse or adverse effects (AEs), such as injection site reactions, flulike symptoms, or depression. Additionally, patients who were not willing to take more DMA medications were excluded if they felt they were relatively stable (had infrequent relapses) and believed that additional medication was not worth the risk of potential AEs.
The study patients were seen and followed up by the neurologists. All the data for this study were based on interactions with the neurologists and not primary care providers (PCPs). Because MS treatment is complex, PCPs have little involvement in its management. The percentage of patients not on any DMAs was calculated from the list of BPVAHCS patients with RRMS.
The results were compared with a similar retrospective cohort study conducted using the Commercial Claims database and Medicare Supplemental and Coordination of Benefits database to identify individuals newly diagnosed with MS.6 This study was chosen because it was similar in methodology but investigated a comparable non-VA group. A 2-tailed difference between proportions test was then performed to determine whether the BPVAHCS patients with MS who were not treated with DMAs were significantly different from those from this non-VA population. Additionally, data from VA patients who were receiving DMAs were further examined and presented.
Results
At the BPVAHCS, 262 patients were diagnosed with MS and 43% were not treated with DMAs. Margolis and colleagues found that about 60% of its 11,061 newly diagnosed non-VA patients with MS remained untreated.6 Although the latter proportion is higher, a 2-tailed difference between proportions test indicates that the proportion of patients with MS being treated at the VA was significantly lower (P < .01).
Among the 148 patients who were diagnosed with MS and treated with DMA at BPVAHCS, 5 different DMAs were identified (Table). The most commonly prescribed regimen was glatiramer acetate, which was used by 56 of 148 patients (37.8%). Fifty-two patients (35.1%) used interferon beta-1a. Of the 2 interferon DMAs, beta-1a was twice as popular as beta-1b, which was prescribed to 22 (14.9%) of patients. Dimethyl fumarate (6.8%) and fingolimod (5.4%) were used sparingly, because they were new to the market (cost and availability also were factors). With time, increased efficacy and objective assessment of benefit in the reduction of the T2 lesion load may result in a greater use of these oral DMAs.7–9
Based on this evaluation, 43% of patients who were diagnosed with MS were untreated at BPVAHCS. Concern over treatment AEs, the inconvenience of injectable dosing, and patients who were not 100% service-connected and lost to follow-up because of the cost may have contributed to the poor rate of treatment.
Discussion
Injected-based DMAs, such as interferon beta-1a, interferon beta-1b, and glatiramer acetate, were first introduced in the 1990s, but these proved to be inconvenient and triggered AEs, including injection site reactions. Overall, their efficacy was about 30%, with interferon beta-1a showing a 27% reduction in relapses.10 In 2010, oral DMAs, such as fingolimod, were FDA approved. These oral DMAs were a significant improvement over injectable DMAs but still had AEs. Hence, their use was restricted to neurologists by the BPVAHCS, and rightfully so.
Still, newer and more effective oral DMAs are showing promise, such as dimethyl fumarate, teriflunomide, and alemtuzumab. These new DMAs have significantly impacted the treatment of MS as they are not only easier for patients to adhere to and for neurologists to prescribe, but most significantly, have had a 50% decrease in the rate of relapse.10 Yet, the newer oral DMAs were less commonly prescribed than the older treatments at BPVAHCS.
Since this study did not demonstrate increased use of oral DMAs at the BPVAHCS, more PCP and neurologist-focused educational programs on the use of DMAs may be beneficial. Educational programs should lead to a reevaluation of patients with MS to consider oral DMAs, which offer better efficacy and fewer AEs. The newer oral DMAs have shown a higher reduction of T2 lesions, and the significantly decreased incidence of relapses in many other medical facilities is quite promising for the BPVAHCS.7-9
The data collected at BPVAHCS were part of a quality improvement (QI) study that will be used by the Neurology Department to follow up on the patients with MS in order to implement DMA therapies. A questionnaire was developed for following up with BPVAHCS patients with MS. The primary purpose of the questionnaire is to help neurologists identify the reasons patients avoid DMA therapies and to reduce the number of BPVAHCS patients not on the most efficacious MS DMA treatment.
Conclusion
Multiple sclerosis is a disease without a cure. Current treatment strategies focus on modifying the course of the disease and managing its symptoms. However, even as promising new treatments emerge, the current literature suggests that a significant number of patients diagnosed with MS are not receiving DMAs and may not be receiving optimal treatment.11
Findings from this study indicate that although DMAs are optimal for patients with MS, they may not be prescribed as frequently at BPVAHCS as they are at a non-VA care facility. It is unclear whether this finding is explained by an educational gap, clinical differences between non-VA and VA patients, organizational factors, or a combination of these variables. Further study is warranted to examine the use of DMAs among veterans with MS and factors that facilitate or impede optimal practice. The BPVAHCS will use data from this retrospective cohort study in a QI initiative for patients with MS. Findings from the QI initiative will be reported using the Standards for Quality Improvement Reporting Excellence.12,13
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the Bay Pines VA Healthcare System.
1. National Multiple Sclerosis Society. Types of MS. National Multiple Sclerosis Society website. http://www.nationalmssociety.org/What-is-MS/Types-of-MS. Accessed April 7, 2016.
2. McKay KA, Kwan V, Duggan T, Tremlett H. Risk factors associated with the onset of relapsing-remitting and primary progressive multiple sclerosis: a systematic review. Biomed Res Int. 2015;2015:817238.
3. Gold R, Wolinsky JS, Amato MP, Comi G. Evolving expectations around early management of multiple sclerosis. Ther Adv Neurol Disord. 2010;3(6):351-367.
4. Ben-Zacharia A, Lublin FD. Talking About Initiating and Adhering to Treatment With Injectable Disease Modifying Agents. Washington, DC: National Multiple Sclerosis Society; 2009.
5. Cameron MH, Poel AJ, Haselkorn JK, Linke A, Bourdette D. Falls requiring medical attention among veterans with multiple sclerosis: a cohort study. J Rehabil Res Dev. 2011;48(1):13-20.
6. Margolis JM, Fowler R, Johnson BH, Kassed CA, Kahler K. Disease-modifying drug initiation patterns in commercially insured multiple sclerosis patients: a retrospective cohort study. BMC Neurol. 2011;11:122.
7. Johnson KP, Brooks BR, Ford CC, et al. Sustained clinical benefits of glatiramer acetate in relapsing multiple sclerosis patients observed for 6 years. Copolymer 1 Multiple Scleroisis Study Group. Mult Scler. 2000;6(4):255-266.
8. Steinberg SC, Faris RJ, Chang CF, Chan A, Tankersley MA. Impact of adherence to interferons in the treatment of multiple sclerosis: a non-experimental, retrospective, cohort study. Clin Drug Investig. 2010;30(2):89-100.
9. Agashivala N, Wu N, Abouzaid S, et al. Compliance to fingolimod and other disease modifying treatments in multiple sclerosis patients, a retrospective cohort study. BMC Neurol. 2013;13:138.
10. Williams UE, Oparah SK, Philip-Ephraim EE. Disease modifying therapy in multiple sclerosis. Int Sch Res Notices. 2014;2014:307064.
11. Lus G, Signoriello E, Maniscalco GT, Bonavita S, Signoriello S, Gallo C. Treatment withdrawal in relapsing-remitting multiple sclerosis: a retrospective cohort study. Eur J Neurol. 2016;23(3):489-493.
12. Davidoff F, Batalden P, Stevens D, Ogrinc G, Mooney S; SQUIRE development group. Publication guidelines for quality improvement studies in health care: evolution of the SQUIRE project. Qual Saf Health Care. 2008;17(suppl 1):i3–i9.
13. Ogrinc G, Mooney SE, Estrada C, et al. The SQUIRE (Standards for QUality Improvement Reporting Excellence) guidelines for quality improvement reporting: explanation and elaboration. Qual Saf Health Care. 2008;17(suppl 1):i13-i32.
Multiple sclerosis (MS) is an autoimmune disorder in which the myelin of the brain and spinal cord is selectively targeted by immune-system cells. As a result, nerve transmission is disrupted, leading to a variety of unpredictable symptoms from weakness and a lack of balance to blindness and paralysis of the body. Clinically, MS can take 4 courses, including relapsing-remitting (RRMS), primary-progressive (PPMS), secondary-progressive, and progressive-relapsing.1 At onset, 85% of diagnosed patients have RRMS, and 10% to 15% have PPMS.2 If untreated, patients with RRMS become secondary-progressive, with progressive disability and indiscrete relapses.3 Hence, disease-modifying therapies are targeted toward decreasing the relapse rate as well as slowing the progression of the disease.4
Annually, about 16,000 veterans with MS receive health care services from the VHA.5 The C.W. Bill Young Bay Pines VA Healthcare System (BPVAHCS) is a level 1 facility that annually serves more than 105,000 veterans. The BPVAMC sees veterans with a wide variety of neurologic illnesses and has 5 full-time neurologists with subspecialty training. The BPVAHCS facility has outpatient clinics and a 200 inpatient bed facility. The Neurology Department sees 125 outpatients per week and consults on about 30 inpatients per week.
Methods
A retrospective review of BPVAHCS patients diagnosed with MS from January 2009 to July 2014 was performed with institutional review board approval. Patient data were collected from ICD-9-CM codes and kept confidential. A list of patients was collected from Neurology Clinic patient visits with “Multiple Sclerosis” on the problem list.
Patient medical records were reviewed to collect the following information: presence of rigorous diagnosis of MS, clinical course of MS in patient, presence or absence of disease-modifying therapy, and disease-modifying agents (DMAs) used.
Determining factors for DMA treatment included increasing tiredness, weakness, visual symptoms, and radiologic evidence (magnetic resonance imaging) of recurrent, active lesions. Each patient was examined on a case-by-case basis to assess whether or not the patient actually had MS and if so, whether they were being treated with DMAs. Only patients with RRMS were included. Patients were excluded from the study if they were deceased, not currently under BPVACHS care, or had symptoms of optic neuritis but were not fully indicative of MS. Patients with clinically isolated syndrome, probable diagnosis of MS, or PPMS also were excluded from the study.
Exclusion from this study was based on 2 additional premises. Patients were excluded if they discontinued an initial ABC (interferon beta-1a, interferon beta-1b, glatiramer acetate) due to DMA treatment relapse or adverse effects (AEs), such as injection site reactions, flulike symptoms, or depression. Additionally, patients who were not willing to take more DMA medications were excluded if they felt they were relatively stable (had infrequent relapses) and believed that additional medication was not worth the risk of potential AEs.
The study patients were seen and followed up by the neurologists. All the data for this study were based on interactions with the neurologists and not primary care providers (PCPs). Because MS treatment is complex, PCPs have little involvement in its management. The percentage of patients not on any DMAs was calculated from the list of BPVAHCS patients with RRMS.
The results were compared with a similar retrospective cohort study conducted using the Commercial Claims database and Medicare Supplemental and Coordination of Benefits database to identify individuals newly diagnosed with MS.6 This study was chosen because it was similar in methodology but investigated a comparable non-VA group. A 2-tailed difference between proportions test was then performed to determine whether the BPVAHCS patients with MS who were not treated with DMAs were significantly different from those from this non-VA population. Additionally, data from VA patients who were receiving DMAs were further examined and presented.
Results
At the BPVAHCS, 262 patients were diagnosed with MS and 43% were not treated with DMAs. Margolis and colleagues found that about 60% of its 11,061 newly diagnosed non-VA patients with MS remained untreated.6 Although the latter proportion is higher, a 2-tailed difference between proportions test indicates that the proportion of patients with MS being treated at the VA was significantly lower (P < .01).
Among the 148 patients who were diagnosed with MS and treated with DMA at BPVAHCS, 5 different DMAs were identified (Table). The most commonly prescribed regimen was glatiramer acetate, which was used by 56 of 148 patients (37.8%). Fifty-two patients (35.1%) used interferon beta-1a. Of the 2 interferon DMAs, beta-1a was twice as popular as beta-1b, which was prescribed to 22 (14.9%) of patients. Dimethyl fumarate (6.8%) and fingolimod (5.4%) were used sparingly, because they were new to the market (cost and availability also were factors). With time, increased efficacy and objective assessment of benefit in the reduction of the T2 lesion load may result in a greater use of these oral DMAs.7–9
Based on this evaluation, 43% of patients who were diagnosed with MS were untreated at BPVAHCS. Concern over treatment AEs, the inconvenience of injectable dosing, and patients who were not 100% service-connected and lost to follow-up because of the cost may have contributed to the poor rate of treatment.
Discussion
Injected-based DMAs, such as interferon beta-1a, interferon beta-1b, and glatiramer acetate, were first introduced in the 1990s, but these proved to be inconvenient and triggered AEs, including injection site reactions. Overall, their efficacy was about 30%, with interferon beta-1a showing a 27% reduction in relapses.10 In 2010, oral DMAs, such as fingolimod, were FDA approved. These oral DMAs were a significant improvement over injectable DMAs but still had AEs. Hence, their use was restricted to neurologists by the BPVAHCS, and rightfully so.
Still, newer and more effective oral DMAs are showing promise, such as dimethyl fumarate, teriflunomide, and alemtuzumab. These new DMAs have significantly impacted the treatment of MS as they are not only easier for patients to adhere to and for neurologists to prescribe, but most significantly, have had a 50% decrease in the rate of relapse.10 Yet, the newer oral DMAs were less commonly prescribed than the older treatments at BPVAHCS.
Since this study did not demonstrate increased use of oral DMAs at the BPVAHCS, more PCP and neurologist-focused educational programs on the use of DMAs may be beneficial. Educational programs should lead to a reevaluation of patients with MS to consider oral DMAs, which offer better efficacy and fewer AEs. The newer oral DMAs have shown a higher reduction of T2 lesions, and the significantly decreased incidence of relapses in many other medical facilities is quite promising for the BPVAHCS.7-9
The data collected at BPVAHCS were part of a quality improvement (QI) study that will be used by the Neurology Department to follow up on the patients with MS in order to implement DMA therapies. A questionnaire was developed for following up with BPVAHCS patients with MS. The primary purpose of the questionnaire is to help neurologists identify the reasons patients avoid DMA therapies and to reduce the number of BPVAHCS patients not on the most efficacious MS DMA treatment.
Conclusion
Multiple sclerosis is a disease without a cure. Current treatment strategies focus on modifying the course of the disease and managing its symptoms. However, even as promising new treatments emerge, the current literature suggests that a significant number of patients diagnosed with MS are not receiving DMAs and may not be receiving optimal treatment.11
Findings from this study indicate that although DMAs are optimal for patients with MS, they may not be prescribed as frequently at BPVAHCS as they are at a non-VA care facility. It is unclear whether this finding is explained by an educational gap, clinical differences between non-VA and VA patients, organizational factors, or a combination of these variables. Further study is warranted to examine the use of DMAs among veterans with MS and factors that facilitate or impede optimal practice. The BPVAHCS will use data from this retrospective cohort study in a QI initiative for patients with MS. Findings from the QI initiative will be reported using the Standards for Quality Improvement Reporting Excellence.12,13
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the Bay Pines VA Healthcare System.
Multiple sclerosis (MS) is an autoimmune disorder in which the myelin of the brain and spinal cord is selectively targeted by immune-system cells. As a result, nerve transmission is disrupted, leading to a variety of unpredictable symptoms from weakness and a lack of balance to blindness and paralysis of the body. Clinically, MS can take 4 courses, including relapsing-remitting (RRMS), primary-progressive (PPMS), secondary-progressive, and progressive-relapsing.1 At onset, 85% of diagnosed patients have RRMS, and 10% to 15% have PPMS.2 If untreated, patients with RRMS become secondary-progressive, with progressive disability and indiscrete relapses.3 Hence, disease-modifying therapies are targeted toward decreasing the relapse rate as well as slowing the progression of the disease.4
Annually, about 16,000 veterans with MS receive health care services from the VHA.5 The C.W. Bill Young Bay Pines VA Healthcare System (BPVAHCS) is a level 1 facility that annually serves more than 105,000 veterans. The BPVAMC sees veterans with a wide variety of neurologic illnesses and has 5 full-time neurologists with subspecialty training. The BPVAHCS facility has outpatient clinics and a 200 inpatient bed facility. The Neurology Department sees 125 outpatients per week and consults on about 30 inpatients per week.
Methods
A retrospective review of BPVAHCS patients diagnosed with MS from January 2009 to July 2014 was performed with institutional review board approval. Patient data were collected from ICD-9-CM codes and kept confidential. A list of patients was collected from Neurology Clinic patient visits with “Multiple Sclerosis” on the problem list.
Patient medical records were reviewed to collect the following information: presence of rigorous diagnosis of MS, clinical course of MS in patient, presence or absence of disease-modifying therapy, and disease-modifying agents (DMAs) used.
Determining factors for DMA treatment included increasing tiredness, weakness, visual symptoms, and radiologic evidence (magnetic resonance imaging) of recurrent, active lesions. Each patient was examined on a case-by-case basis to assess whether or not the patient actually had MS and if so, whether they were being treated with DMAs. Only patients with RRMS were included. Patients were excluded from the study if they were deceased, not currently under BPVACHS care, or had symptoms of optic neuritis but were not fully indicative of MS. Patients with clinically isolated syndrome, probable diagnosis of MS, or PPMS also were excluded from the study.
Exclusion from this study was based on 2 additional premises. Patients were excluded if they discontinued an initial ABC (interferon beta-1a, interferon beta-1b, glatiramer acetate) due to DMA treatment relapse or adverse effects (AEs), such as injection site reactions, flulike symptoms, or depression. Additionally, patients who were not willing to take more DMA medications were excluded if they felt they were relatively stable (had infrequent relapses) and believed that additional medication was not worth the risk of potential AEs.
The study patients were seen and followed up by the neurologists. All the data for this study were based on interactions with the neurologists and not primary care providers (PCPs). Because MS treatment is complex, PCPs have little involvement in its management. The percentage of patients not on any DMAs was calculated from the list of BPVAHCS patients with RRMS.
The results were compared with a similar retrospective cohort study conducted using the Commercial Claims database and Medicare Supplemental and Coordination of Benefits database to identify individuals newly diagnosed with MS.6 This study was chosen because it was similar in methodology but investigated a comparable non-VA group. A 2-tailed difference between proportions test was then performed to determine whether the BPVAHCS patients with MS who were not treated with DMAs were significantly different from those from this non-VA population. Additionally, data from VA patients who were receiving DMAs were further examined and presented.
Results
At the BPVAHCS, 262 patients were diagnosed with MS and 43% were not treated with DMAs. Margolis and colleagues found that about 60% of its 11,061 newly diagnosed non-VA patients with MS remained untreated.6 Although the latter proportion is higher, a 2-tailed difference between proportions test indicates that the proportion of patients with MS being treated at the VA was significantly lower (P < .01).
Among the 148 patients who were diagnosed with MS and treated with DMA at BPVAHCS, 5 different DMAs were identified (Table). The most commonly prescribed regimen was glatiramer acetate, which was used by 56 of 148 patients (37.8%). Fifty-two patients (35.1%) used interferon beta-1a. Of the 2 interferon DMAs, beta-1a was twice as popular as beta-1b, which was prescribed to 22 (14.9%) of patients. Dimethyl fumarate (6.8%) and fingolimod (5.4%) were used sparingly, because they were new to the market (cost and availability also were factors). With time, increased efficacy and objective assessment of benefit in the reduction of the T2 lesion load may result in a greater use of these oral DMAs.7–9
Based on this evaluation, 43% of patients who were diagnosed with MS were untreated at BPVAHCS. Concern over treatment AEs, the inconvenience of injectable dosing, and patients who were not 100% service-connected and lost to follow-up because of the cost may have contributed to the poor rate of treatment.
Discussion
Injected-based DMAs, such as interferon beta-1a, interferon beta-1b, and glatiramer acetate, were first introduced in the 1990s, but these proved to be inconvenient and triggered AEs, including injection site reactions. Overall, their efficacy was about 30%, with interferon beta-1a showing a 27% reduction in relapses.10 In 2010, oral DMAs, such as fingolimod, were FDA approved. These oral DMAs were a significant improvement over injectable DMAs but still had AEs. Hence, their use was restricted to neurologists by the BPVAHCS, and rightfully so.
Still, newer and more effective oral DMAs are showing promise, such as dimethyl fumarate, teriflunomide, and alemtuzumab. These new DMAs have significantly impacted the treatment of MS as they are not only easier for patients to adhere to and for neurologists to prescribe, but most significantly, have had a 50% decrease in the rate of relapse.10 Yet, the newer oral DMAs were less commonly prescribed than the older treatments at BPVAHCS.
Since this study did not demonstrate increased use of oral DMAs at the BPVAHCS, more PCP and neurologist-focused educational programs on the use of DMAs may be beneficial. Educational programs should lead to a reevaluation of patients with MS to consider oral DMAs, which offer better efficacy and fewer AEs. The newer oral DMAs have shown a higher reduction of T2 lesions, and the significantly decreased incidence of relapses in many other medical facilities is quite promising for the BPVAHCS.7-9
The data collected at BPVAHCS were part of a quality improvement (QI) study that will be used by the Neurology Department to follow up on the patients with MS in order to implement DMA therapies. A questionnaire was developed for following up with BPVAHCS patients with MS. The primary purpose of the questionnaire is to help neurologists identify the reasons patients avoid DMA therapies and to reduce the number of BPVAHCS patients not on the most efficacious MS DMA treatment.
Conclusion
Multiple sclerosis is a disease without a cure. Current treatment strategies focus on modifying the course of the disease and managing its symptoms. However, even as promising new treatments emerge, the current literature suggests that a significant number of patients diagnosed with MS are not receiving DMAs and may not be receiving optimal treatment.11
Findings from this study indicate that although DMAs are optimal for patients with MS, they may not be prescribed as frequently at BPVAHCS as they are at a non-VA care facility. It is unclear whether this finding is explained by an educational gap, clinical differences between non-VA and VA patients, organizational factors, or a combination of these variables. Further study is warranted to examine the use of DMAs among veterans with MS and factors that facilitate or impede optimal practice. The BPVAHCS will use data from this retrospective cohort study in a QI initiative for patients with MS. Findings from the QI initiative will be reported using the Standards for Quality Improvement Reporting Excellence.12,13
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the Bay Pines VA Healthcare System.
1. National Multiple Sclerosis Society. Types of MS. National Multiple Sclerosis Society website. http://www.nationalmssociety.org/What-is-MS/Types-of-MS. Accessed April 7, 2016.
2. McKay KA, Kwan V, Duggan T, Tremlett H. Risk factors associated with the onset of relapsing-remitting and primary progressive multiple sclerosis: a systematic review. Biomed Res Int. 2015;2015:817238.
3. Gold R, Wolinsky JS, Amato MP, Comi G. Evolving expectations around early management of multiple sclerosis. Ther Adv Neurol Disord. 2010;3(6):351-367.
4. Ben-Zacharia A, Lublin FD. Talking About Initiating and Adhering to Treatment With Injectable Disease Modifying Agents. Washington, DC: National Multiple Sclerosis Society; 2009.
5. Cameron MH, Poel AJ, Haselkorn JK, Linke A, Bourdette D. Falls requiring medical attention among veterans with multiple sclerosis: a cohort study. J Rehabil Res Dev. 2011;48(1):13-20.
6. Margolis JM, Fowler R, Johnson BH, Kassed CA, Kahler K. Disease-modifying drug initiation patterns in commercially insured multiple sclerosis patients: a retrospective cohort study. BMC Neurol. 2011;11:122.
7. Johnson KP, Brooks BR, Ford CC, et al. Sustained clinical benefits of glatiramer acetate in relapsing multiple sclerosis patients observed for 6 years. Copolymer 1 Multiple Scleroisis Study Group. Mult Scler. 2000;6(4):255-266.
8. Steinberg SC, Faris RJ, Chang CF, Chan A, Tankersley MA. Impact of adherence to interferons in the treatment of multiple sclerosis: a non-experimental, retrospective, cohort study. Clin Drug Investig. 2010;30(2):89-100.
9. Agashivala N, Wu N, Abouzaid S, et al. Compliance to fingolimod and other disease modifying treatments in multiple sclerosis patients, a retrospective cohort study. BMC Neurol. 2013;13:138.
10. Williams UE, Oparah SK, Philip-Ephraim EE. Disease modifying therapy in multiple sclerosis. Int Sch Res Notices. 2014;2014:307064.
11. Lus G, Signoriello E, Maniscalco GT, Bonavita S, Signoriello S, Gallo C. Treatment withdrawal in relapsing-remitting multiple sclerosis: a retrospective cohort study. Eur J Neurol. 2016;23(3):489-493.
12. Davidoff F, Batalden P, Stevens D, Ogrinc G, Mooney S; SQUIRE development group. Publication guidelines for quality improvement studies in health care: evolution of the SQUIRE project. Qual Saf Health Care. 2008;17(suppl 1):i3–i9.
13. Ogrinc G, Mooney SE, Estrada C, et al. The SQUIRE (Standards for QUality Improvement Reporting Excellence) guidelines for quality improvement reporting: explanation and elaboration. Qual Saf Health Care. 2008;17(suppl 1):i13-i32.
1. National Multiple Sclerosis Society. Types of MS. National Multiple Sclerosis Society website. http://www.nationalmssociety.org/What-is-MS/Types-of-MS. Accessed April 7, 2016.
2. McKay KA, Kwan V, Duggan T, Tremlett H. Risk factors associated with the onset of relapsing-remitting and primary progressive multiple sclerosis: a systematic review. Biomed Res Int. 2015;2015:817238.
3. Gold R, Wolinsky JS, Amato MP, Comi G. Evolving expectations around early management of multiple sclerosis. Ther Adv Neurol Disord. 2010;3(6):351-367.
4. Ben-Zacharia A, Lublin FD. Talking About Initiating and Adhering to Treatment With Injectable Disease Modifying Agents. Washington, DC: National Multiple Sclerosis Society; 2009.
5. Cameron MH, Poel AJ, Haselkorn JK, Linke A, Bourdette D. Falls requiring medical attention among veterans with multiple sclerosis: a cohort study. J Rehabil Res Dev. 2011;48(1):13-20.
6. Margolis JM, Fowler R, Johnson BH, Kassed CA, Kahler K. Disease-modifying drug initiation patterns in commercially insured multiple sclerosis patients: a retrospective cohort study. BMC Neurol. 2011;11:122.
7. Johnson KP, Brooks BR, Ford CC, et al. Sustained clinical benefits of glatiramer acetate in relapsing multiple sclerosis patients observed for 6 years. Copolymer 1 Multiple Scleroisis Study Group. Mult Scler. 2000;6(4):255-266.
8. Steinberg SC, Faris RJ, Chang CF, Chan A, Tankersley MA. Impact of adherence to interferons in the treatment of multiple sclerosis: a non-experimental, retrospective, cohort study. Clin Drug Investig. 2010;30(2):89-100.
9. Agashivala N, Wu N, Abouzaid S, et al. Compliance to fingolimod and other disease modifying treatments in multiple sclerosis patients, a retrospective cohort study. BMC Neurol. 2013;13:138.
10. Williams UE, Oparah SK, Philip-Ephraim EE. Disease modifying therapy in multiple sclerosis. Int Sch Res Notices. 2014;2014:307064.
11. Lus G, Signoriello E, Maniscalco GT, Bonavita S, Signoriello S, Gallo C. Treatment withdrawal in relapsing-remitting multiple sclerosis: a retrospective cohort study. Eur J Neurol. 2016;23(3):489-493.
12. Davidoff F, Batalden P, Stevens D, Ogrinc G, Mooney S; SQUIRE development group. Publication guidelines for quality improvement studies in health care: evolution of the SQUIRE project. Qual Saf Health Care. 2008;17(suppl 1):i3–i9.
13. Ogrinc G, Mooney SE, Estrada C, et al. The SQUIRE (Standards for QUality Improvement Reporting Excellence) guidelines for quality improvement reporting: explanation and elaboration. Qual Saf Health Care. 2008;17(suppl 1):i13-i32.