User login
FDA approves Sympazan for Lennox-Gastaut syndrome
, according to a release from its developer. Final approval came after the orphan drug designation period for the previously marketed formulation, Onfi, came to an end in October.

LGS is a severe form of epilepsy; it can present with multiple types of seizures, as well as intellectual disabilities. Patients with LGS can have difficulty swallowing tablets or large volumes of oral suspension – which was previously the only way clobazam was delivered – because of physical limitations or behavioral or compliance issues. According to the press release from Aquestive Therapeutics, the Sympazan oral film might be able to get around those difficulties and reduce care burdens, especially with patients who are resistant to or even combative about treatment.
The approval is based on multiple pharmacokinetic studies that altogether showed that the oral film is bioequivalent to clobazam tablets and has a similar safety profile.
In a phase 3 study of 238 patients with LGS, clobazam tablets were shown to reduce drop seizures (those that involved falls) by 41% at low doses and by 68% at high doses versus a reduction of 12% seen with placebo (P less than .05 for all doses vs. placebo).
There is a risk of profound sedation when clobazam is used alongside benzodiazepines; there is also a risk of sedation and somnolence if it is used concomitantly with alcohol or other CNS depressants. Other risks associated with clobazam include suicidal ideation and behavior, serious dermatologic reactions, and physical and psychological dependence. The most common adverse reactions included constipation, pyrexia, lethargy, and drooling.
Full prescribing information can be found on the FDA website.
, according to a release from its developer. Final approval came after the orphan drug designation period for the previously marketed formulation, Onfi, came to an end in October.
LGS is a severe form of epilepsy; it can present with multiple types of seizures, as well as intellectual disabilities. Patients with LGS can have difficulty swallowing tablets or large volumes of oral suspension – which was previously the only way clobazam was delivered – because of physical limitations or behavioral or compliance issues. According to the press release from Aquestive Therapeutics, the Sympazan oral film might be able to get around those difficulties and reduce care burdens, especially with patients who are resistant to or even combative about treatment.
The approval is based on multiple pharmacokinetic studies that altogether showed that the oral film is bioequivalent to clobazam tablets and has a similar safety profile.
In a phase 3 study of 238 patients with LGS, clobazam tablets were shown to reduce drop seizures (those that involved falls) by 41% at low doses and by 68% at high doses versus a reduction of 12% seen with placebo (P less than .05 for all doses vs. placebo).
There is a risk of profound sedation when clobazam is used alongside benzodiazepines; there is also a risk of sedation and somnolence if it is used concomitantly with alcohol or other CNS depressants. Other risks associated with clobazam include suicidal ideation and behavior, serious dermatologic reactions, and physical and psychological dependence. The most common adverse reactions included constipation, pyrexia, lethargy, and drooling.
Full prescribing information can be found on the FDA website.
, according to a release from its developer. Final approval came after the orphan drug designation period for the previously marketed formulation, Onfi, came to an end in October.
LGS is a severe form of epilepsy; it can present with multiple types of seizures, as well as intellectual disabilities. Patients with LGS can have difficulty swallowing tablets or large volumes of oral suspension – which was previously the only way clobazam was delivered – because of physical limitations or behavioral or compliance issues. According to the press release from Aquestive Therapeutics, the Sympazan oral film might be able to get around those difficulties and reduce care burdens, especially with patients who are resistant to or even combative about treatment.
The approval is based on multiple pharmacokinetic studies that altogether showed that the oral film is bioequivalent to clobazam tablets and has a similar safety profile.
In a phase 3 study of 238 patients with LGS, clobazam tablets were shown to reduce drop seizures (those that involved falls) by 41% at low doses and by 68% at high doses versus a reduction of 12% seen with placebo (P less than .05 for all doses vs. placebo).
There is a risk of profound sedation when clobazam is used alongside benzodiazepines; there is also a risk of sedation and somnolence if it is used concomitantly with alcohol or other CNS depressants. Other risks associated with clobazam include suicidal ideation and behavior, serious dermatologic reactions, and physical and psychological dependence. The most common adverse reactions included constipation, pyrexia, lethargy, and drooling.
Full prescribing information can be found on the FDA website.
FDA approval of powerful opioid tinged with irony
The timing of the Food and Drug Administration’s Nov. 2 approval of the medication Dsuvia, a sublingual formulation of the synthetic opioid sufentanil, is interesting – to say the least. Dsuvia is a powerful pain medication, said to be 10 times more potent than fentanyl and 1,000 times more potent than morphine. The medication, developed by AcelRx Pharmaceuticals for use in medically supervised settings, has an indication for moderate to severe pain, and is packaged in single-dose applicators.

The chairperson of the FDA’s Anesthetic and Analgesics Drug Product Advisory Committee, Raeford E. Brown Jr., MD, a professor of pediatric anesthesia at the University of Kentucky, Lexington, could not be present Oct. 12 at the committee vote recommending approval. With the consumer advocacy group Public Citizen, Dr. Brown wrote a letter to FDA leaders detailing concerns about the new formulation of sufentanil.
“It is my observation,” Dr. Brown wrote, “that once the FDA approves an opioid compound, there are no safeguards as to the population that will be exposed, the postmarketing analysis of prescribing behavior, or the ongoing analysis of the risks of the drug to the general population relative to its benefit to the public health. Briefly stated, for all of the opioids that have been marketed in the last 10 years, there has not been sufficient demonstration of safety, nor has there been postmarketing assessment of who is taking the drug, how often prescribing is inappropriate, and whether there was ever a reason to risk the health of the general population by having one more opioid on the market.”
Dr. Brown went on to detail his concerns about sufentanil. In the intravenous formulation, the medication has been in use for more than two decades.
“It is so potent that abusers of this intravenous formulation often die when they inject the first dose; I have witnessed this in resuscitating physicians, medical students, technicians, and other health care providers, some successfully, as a part of my duties as a clinician in a major academic medical center. Because it is so potent, the dosing volume, whether in the IV formulation or the sublingual form, can be quite small. It is thus an extremely divertible drug, and I predict that we will encounter diversion, abuse, and death within the early months of its availability on the market.”
The letter finishes by criticizing the fact that the full Drug Safety and Risk Management Advisory Committee was not invited to the Oct. 12 meeting, and finally, about the ease of diversion among health care professionals – and anesthesiologists in particular.
Meanwhile, Scott Gottlieb, MD, commissioner of the FDA, posted a lengthy explanation on the organization’s website on Nov. 2, after the vote. In his statement on the agency’s approval of Dsuvia and the FDA’s future consideration of new opioids, Dr. Gottlieb explains: “To address concerns about the potential risks associated with Dsuvia, this product will have strong limitations on its use. It can’t be dispensed to patients for home use and should not be used for more than 72 hours. And it should only be administered by a health care provider using a single-dose applicator. That means it won’t be available at retail pharmacies for patients to take home. These measures to restrict the use of this product only within a supervised health care setting, and not for home use, are important steps to help prevent misuse and abuse of Dsuvia, as well reduce the potential for diversion. Because of the risks of addiction, abuse, and misuse with opioids, Dsuvia also is to be reserved for use in patients for whom alternative pain treatment options have not been tolerated, or are not expected to be tolerated, where existing treatment options have not provided adequate analgesia, or where these alternatives are not expected to provide adequate analgesia.”
In addition to the statement posted on the FDA’s website, Dr. Gottlieb made the approval of Dsuvia the topic of his weekly #SundayTweetorial on Nov. 4. In this venue, Dr. Gottlieb posts tweets on a single topic. On both Twitter and the FDA website, he noted that a major factor in the approval of Dsuvia was advantages it might convey for pain control to soldiers on the battlefield, where oral medications might take time to work and intravenous access might not be possible.
One tweet read: “Whether there’s a need for another powerful opioid in the throes of a massive crisis of addiction is a critical question. As a public health agency, we have an obligation to address this question for patients with pain, for the addiction crisis, for innovators, for all Americans.”
Another tweet stated, “While Dsuvia brings another highly potent opioid to market it fulfills a limited, unmet medical need in treating our nation’s soldiers on the battlefield. That’s why the Pentagon worked closely with the sponsor on developing Dsuvia. FDA committed to prioritize needs of our troops.”
in possible deaths from misdirected use of a very potent agent. And while the new opioid may have been geared toward unmet military needs, Dsuvia will be available for use in civilian medical facilities as well.
There is some irony to the idea that a pharmaceutical company would continue to develop opioids when there is so much need for nonaddictive agents for pain control and so much pressure on physicians to limit access of opiates to pain patients. We are left to stand by and watch as yet another potent opioid preparation is introduced.
Dr. Miller is coauthor of “Committed: The Battle Over Involuntary Psychiatric Care” (Baltimore: Johns Hopkins University Press, 2016), and assistant professor of psychiatry and behavioral sciences at Johns Hopkins University, Baltimore.
The timing of the Food and Drug Administration’s Nov. 2 approval of the medication Dsuvia, a sublingual formulation of the synthetic opioid sufentanil, is interesting – to say the least. Dsuvia is a powerful pain medication, said to be 10 times more potent than fentanyl and 1,000 times more potent than morphine. The medication, developed by AcelRx Pharmaceuticals for use in medically supervised settings, has an indication for moderate to severe pain, and is packaged in single-dose applicators.
The chairperson of the FDA’s Anesthetic and Analgesics Drug Product Advisory Committee, Raeford E. Brown Jr., MD, a professor of pediatric anesthesia at the University of Kentucky, Lexington, could not be present Oct. 12 at the committee vote recommending approval. With the consumer advocacy group Public Citizen, Dr. Brown wrote a letter to FDA leaders detailing concerns about the new formulation of sufentanil.
“It is my observation,” Dr. Brown wrote, “that once the FDA approves an opioid compound, there are no safeguards as to the population that will be exposed, the postmarketing analysis of prescribing behavior, or the ongoing analysis of the risks of the drug to the general population relative to its benefit to the public health. Briefly stated, for all of the opioids that have been marketed in the last 10 years, there has not been sufficient demonstration of safety, nor has there been postmarketing assessment of who is taking the drug, how often prescribing is inappropriate, and whether there was ever a reason to risk the health of the general population by having one more opioid on the market.”
Dr. Brown went on to detail his concerns about sufentanil. In the intravenous formulation, the medication has been in use for more than two decades.
“It is so potent that abusers of this intravenous formulation often die when they inject the first dose; I have witnessed this in resuscitating physicians, medical students, technicians, and other health care providers, some successfully, as a part of my duties as a clinician in a major academic medical center. Because it is so potent, the dosing volume, whether in the IV formulation or the sublingual form, can be quite small. It is thus an extremely divertible drug, and I predict that we will encounter diversion, abuse, and death within the early months of its availability on the market.”
The letter finishes by criticizing the fact that the full Drug Safety and Risk Management Advisory Committee was not invited to the Oct. 12 meeting, and finally, about the ease of diversion among health care professionals – and anesthesiologists in particular.
Meanwhile, Scott Gottlieb, MD, commissioner of the FDA, posted a lengthy explanation on the organization’s website on Nov. 2, after the vote. In his statement on the agency’s approval of Dsuvia and the FDA’s future consideration of new opioids, Dr. Gottlieb explains: “To address concerns about the potential risks associated with Dsuvia, this product will have strong limitations on its use. It can’t be dispensed to patients for home use and should not be used for more than 72 hours. And it should only be administered by a health care provider using a single-dose applicator. That means it won’t be available at retail pharmacies for patients to take home. These measures to restrict the use of this product only within a supervised health care setting, and not for home use, are important steps to help prevent misuse and abuse of Dsuvia, as well reduce the potential for diversion. Because of the risks of addiction, abuse, and misuse with opioids, Dsuvia also is to be reserved for use in patients for whom alternative pain treatment options have not been tolerated, or are not expected to be tolerated, where existing treatment options have not provided adequate analgesia, or where these alternatives are not expected to provide adequate analgesia.”
In addition to the statement posted on the FDA’s website, Dr. Gottlieb made the approval of Dsuvia the topic of his weekly #SundayTweetorial on Nov. 4. In this venue, Dr. Gottlieb posts tweets on a single topic. On both Twitter and the FDA website, he noted that a major factor in the approval of Dsuvia was advantages it might convey for pain control to soldiers on the battlefield, where oral medications might take time to work and intravenous access might not be possible.
One tweet read: “Whether there’s a need for another powerful opioid in the throes of a massive crisis of addiction is a critical question. As a public health agency, we have an obligation to address this question for patients with pain, for the addiction crisis, for innovators, for all Americans.”
Another tweet stated, “While Dsuvia brings another highly potent opioid to market it fulfills a limited, unmet medical need in treating our nation’s soldiers on the battlefield. That’s why the Pentagon worked closely with the sponsor on developing Dsuvia. FDA committed to prioritize needs of our troops.”
in possible deaths from misdirected use of a very potent agent. And while the new opioid may have been geared toward unmet military needs, Dsuvia will be available for use in civilian medical facilities as well.
There is some irony to the idea that a pharmaceutical company would continue to develop opioids when there is so much need for nonaddictive agents for pain control and so much pressure on physicians to limit access of opiates to pain patients. We are left to stand by and watch as yet another potent opioid preparation is introduced.
Dr. Miller is coauthor of “Committed: The Battle Over Involuntary Psychiatric Care” (Baltimore: Johns Hopkins University Press, 2016), and assistant professor of psychiatry and behavioral sciences at Johns Hopkins University, Baltimore.
The timing of the Food and Drug Administration’s Nov. 2 approval of the medication Dsuvia, a sublingual formulation of the synthetic opioid sufentanil, is interesting – to say the least. Dsuvia is a powerful pain medication, said to be 10 times more potent than fentanyl and 1,000 times more potent than morphine. The medication, developed by AcelRx Pharmaceuticals for use in medically supervised settings, has an indication for moderate to severe pain, and is packaged in single-dose applicators.
The chairperson of the FDA’s Anesthetic and Analgesics Drug Product Advisory Committee, Raeford E. Brown Jr., MD, a professor of pediatric anesthesia at the University of Kentucky, Lexington, could not be present Oct. 12 at the committee vote recommending approval. With the consumer advocacy group Public Citizen, Dr. Brown wrote a letter to FDA leaders detailing concerns about the new formulation of sufentanil.
“It is my observation,” Dr. Brown wrote, “that once the FDA approves an opioid compound, there are no safeguards as to the population that will be exposed, the postmarketing analysis of prescribing behavior, or the ongoing analysis of the risks of the drug to the general population relative to its benefit to the public health. Briefly stated, for all of the opioids that have been marketed in the last 10 years, there has not been sufficient demonstration of safety, nor has there been postmarketing assessment of who is taking the drug, how often prescribing is inappropriate, and whether there was ever a reason to risk the health of the general population by having one more opioid on the market.”
Dr. Brown went on to detail his concerns about sufentanil. In the intravenous formulation, the medication has been in use for more than two decades.
“It is so potent that abusers of this intravenous formulation often die when they inject the first dose; I have witnessed this in resuscitating physicians, medical students, technicians, and other health care providers, some successfully, as a part of my duties as a clinician in a major academic medical center. Because it is so potent, the dosing volume, whether in the IV formulation or the sublingual form, can be quite small. It is thus an extremely divertible drug, and I predict that we will encounter diversion, abuse, and death within the early months of its availability on the market.”
The letter finishes by criticizing the fact that the full Drug Safety and Risk Management Advisory Committee was not invited to the Oct. 12 meeting, and finally, about the ease of diversion among health care professionals – and anesthesiologists in particular.
Meanwhile, Scott Gottlieb, MD, commissioner of the FDA, posted a lengthy explanation on the organization’s website on Nov. 2, after the vote. In his statement on the agency’s approval of Dsuvia and the FDA’s future consideration of new opioids, Dr. Gottlieb explains: “To address concerns about the potential risks associated with Dsuvia, this product will have strong limitations on its use. It can’t be dispensed to patients for home use and should not be used for more than 72 hours. And it should only be administered by a health care provider using a single-dose applicator. That means it won’t be available at retail pharmacies for patients to take home. These measures to restrict the use of this product only within a supervised health care setting, and not for home use, are important steps to help prevent misuse and abuse of Dsuvia, as well reduce the potential for diversion. Because of the risks of addiction, abuse, and misuse with opioids, Dsuvia also is to be reserved for use in patients for whom alternative pain treatment options have not been tolerated, or are not expected to be tolerated, where existing treatment options have not provided adequate analgesia, or where these alternatives are not expected to provide adequate analgesia.”
In addition to the statement posted on the FDA’s website, Dr. Gottlieb made the approval of Dsuvia the topic of his weekly #SundayTweetorial on Nov. 4. In this venue, Dr. Gottlieb posts tweets on a single topic. On both Twitter and the FDA website, he noted that a major factor in the approval of Dsuvia was advantages it might convey for pain control to soldiers on the battlefield, where oral medications might take time to work and intravenous access might not be possible.
One tweet read: “Whether there’s a need for another powerful opioid in the throes of a massive crisis of addiction is a critical question. As a public health agency, we have an obligation to address this question for patients with pain, for the addiction crisis, for innovators, for all Americans.”
Another tweet stated, “While Dsuvia brings another highly potent opioid to market it fulfills a limited, unmet medical need in treating our nation’s soldiers on the battlefield. That’s why the Pentagon worked closely with the sponsor on developing Dsuvia. FDA committed to prioritize needs of our troops.”
in possible deaths from misdirected use of a very potent agent. And while the new opioid may have been geared toward unmet military needs, Dsuvia will be available for use in civilian medical facilities as well.
There is some irony to the idea that a pharmaceutical company would continue to develop opioids when there is so much need for nonaddictive agents for pain control and so much pressure on physicians to limit access of opiates to pain patients. We are left to stand by and watch as yet another potent opioid preparation is introduced.
Dr. Miller is coauthor of “Committed: The Battle Over Involuntary Psychiatric Care” (Baltimore: Johns Hopkins University Press, 2016), and assistant professor of psychiatry and behavioral sciences at Johns Hopkins University, Baltimore.
FDA approves sufentanil for adults with acute pain
The Food and Drug Administration on Nov. 2 approved sufentanil (Dsuvia) for managing acute pain in adult patients in certified, medically supervised health care settings.
Sufentanil, an opioid analgesic manufactured by AcelRx Pharmaceuticals, was approved as a 30-mcg sublingual tablet. The efficacy of Dsuvia was shown in a randomized, clinical trial where patients who received the drug demonstrated significantly greater pain relief after both 15 minutes and 12 hours, compared with placebo.
“As a single-dose, noninvasive medication with a rapid reduction in pain intensity, Dsuvia represents an important alternative for health care providers to offer patients for acute pain management,” David Leiman, MD, of the department of surgery at the University of Texas, Houston, said in the AcelRx press statement.
FDA Commissioner Scott Gottlieb, MD, commented on the approval amid concerns expressed by some, such as the advocacy group Public Citizen, that the drug is “more than 1,000 times more potent than morphine,” and that approval could lead to diversion and abuse – particularly in light of the U.S. opioid epidemic.
In his statement, Dr. Gottlieb identified one broad, significant issue. “Why do we need an oral formulation of sufentanil – a more potent form of fentanyl that’s been approved for intravenous and epidural use in the U.S. since 1984 – on the market?”
In particular, he focused on the needs of the military. The Department of Defense has taken interest in sufentanil as it fulfills a small but specific battlefield need, namely as a means of pain relief in battlefield situations where soldiers cannot swallow oral medication and access to intravenous medication is limited.

Dr. Gottlieb made clear that sufentanil was meant only to be taken in controlled settings and will have strong limitations on its use. It cannot be prescribed for home use, and treatment should be limited to 72 hours. It can only be delivered by health care professionals using a single-dose applicator and will not be available in pharmacies. It is only to be used in patients who have not tolerated or are expected not to tolerate alternative methods of pain management.
“The FDA has implemented a REMS [Risk Evaluation and Mitigation Strategy] that reflects the potential risks associated with this product and mandates that Dsuvia will only be made available for use in a certified medically supervised heath care setting, including its use on the battlefield,” Dr. Gottlieb said.
However, he recognized that the debate runs deeper than how the FDA should mitigate risk over a new drug, and “as a public health agency, we have an obligation to address this question openly and directly. As a physician and regulator, I won’t bypass legitimate questions and concerns related to our role in addressing the opioid crisis,” he said.
Find Dr. Gottlieb’s full statement on the FDA website.
The Food and Drug Administration on Nov. 2 approved sufentanil (Dsuvia) for managing acute pain in adult patients in certified, medically supervised health care settings.
Sufentanil, an opioid analgesic manufactured by AcelRx Pharmaceuticals, was approved as a 30-mcg sublingual tablet. The efficacy of Dsuvia was shown in a randomized, clinical trial where patients who received the drug demonstrated significantly greater pain relief after both 15 minutes and 12 hours, compared with placebo.
“As a single-dose, noninvasive medication with a rapid reduction in pain intensity, Dsuvia represents an important alternative for health care providers to offer patients for acute pain management,” David Leiman, MD, of the department of surgery at the University of Texas, Houston, said in the AcelRx press statement.
FDA Commissioner Scott Gottlieb, MD, commented on the approval amid concerns expressed by some, such as the advocacy group Public Citizen, that the drug is “more than 1,000 times more potent than morphine,” and that approval could lead to diversion and abuse – particularly in light of the U.S. opioid epidemic.
In his statement, Dr. Gottlieb identified one broad, significant issue. “Why do we need an oral formulation of sufentanil – a more potent form of fentanyl that’s been approved for intravenous and epidural use in the U.S. since 1984 – on the market?”
In particular, he focused on the needs of the military. The Department of Defense has taken interest in sufentanil as it fulfills a small but specific battlefield need, namely as a means of pain relief in battlefield situations where soldiers cannot swallow oral medication and access to intravenous medication is limited.
Dr. Gottlieb made clear that sufentanil was meant only to be taken in controlled settings and will have strong limitations on its use. It cannot be prescribed for home use, and treatment should be limited to 72 hours. It can only be delivered by health care professionals using a single-dose applicator and will not be available in pharmacies. It is only to be used in patients who have not tolerated or are expected not to tolerate alternative methods of pain management.
“The FDA has implemented a REMS [Risk Evaluation and Mitigation Strategy] that reflects the potential risks associated with this product and mandates that Dsuvia will only be made available for use in a certified medically supervised heath care setting, including its use on the battlefield,” Dr. Gottlieb said.
However, he recognized that the debate runs deeper than how the FDA should mitigate risk over a new drug, and “as a public health agency, we have an obligation to address this question openly and directly. As a physician and regulator, I won’t bypass legitimate questions and concerns related to our role in addressing the opioid crisis,” he said.
Find Dr. Gottlieb’s full statement on the FDA website.
The Food and Drug Administration on Nov. 2 approved sufentanil (Dsuvia) for managing acute pain in adult patients in certified, medically supervised health care settings.
Sufentanil, an opioid analgesic manufactured by AcelRx Pharmaceuticals, was approved as a 30-mcg sublingual tablet. The efficacy of Dsuvia was shown in a randomized, clinical trial where patients who received the drug demonstrated significantly greater pain relief after both 15 minutes and 12 hours, compared with placebo.
“As a single-dose, noninvasive medication with a rapid reduction in pain intensity, Dsuvia represents an important alternative for health care providers to offer patients for acute pain management,” David Leiman, MD, of the department of surgery at the University of Texas, Houston, said in the AcelRx press statement.
FDA Commissioner Scott Gottlieb, MD, commented on the approval amid concerns expressed by some, such as the advocacy group Public Citizen, that the drug is “more than 1,000 times more potent than morphine,” and that approval could lead to diversion and abuse – particularly in light of the U.S. opioid epidemic.
In his statement, Dr. Gottlieb identified one broad, significant issue. “Why do we need an oral formulation of sufentanil – a more potent form of fentanyl that’s been approved for intravenous and epidural use in the U.S. since 1984 – on the market?”
In particular, he focused on the needs of the military. The Department of Defense has taken interest in sufentanil as it fulfills a small but specific battlefield need, namely as a means of pain relief in battlefield situations where soldiers cannot swallow oral medication and access to intravenous medication is limited.
Dr. Gottlieb made clear that sufentanil was meant only to be taken in controlled settings and will have strong limitations on its use. It cannot be prescribed for home use, and treatment should be limited to 72 hours. It can only be delivered by health care professionals using a single-dose applicator and will not be available in pharmacies. It is only to be used in patients who have not tolerated or are expected not to tolerate alternative methods of pain management.
“The FDA has implemented a REMS [Risk Evaluation and Mitigation Strategy] that reflects the potential risks associated with this product and mandates that Dsuvia will only be made available for use in a certified medically supervised heath care setting, including its use on the battlefield,” Dr. Gottlieb said.
However, he recognized that the debate runs deeper than how the FDA should mitigate risk over a new drug, and “as a public health agency, we have an obligation to address this question openly and directly. As a physician and regulator, I won’t bypass legitimate questions and concerns related to our role in addressing the opioid crisis,” he said.
Find Dr. Gottlieb’s full statement on the FDA website.
Appendix linked to Parkinson’s disease in series of unexpected findings
Appendectomy has been associated with a reduced risk of Parkinson’s disease (PD), which supports the potential for a reservoir of aggregated alpha-synuclein in the appendix to affect risk of the condition, according to new epidemiologic and translational evidence from two data sets that promotes a new and emerging theory for PD etiology.
When placed into the context of other recent studies, these epidemiologic data “point to the appendix as a site of origin for Parkinson’s and provide a path forward for devising new treatment strategies,” reported senior author Viviane Labrie, PhD, of the Van Andel Research Institute (VARI) in Grand Rapids, Mich.
The epidemiologic data was the most recent step in a series of findings summarized in a newly published paper in Science Translational Medicine. As the researchers explained, it is relevant to a separate body of evidence that alpha-synuclein, a protein that serves as the hallmark of PD when it appears in Lewy bodies, can be isolated in the nerve fibers and nerve cells of the appendix.
“We have shown that alpha-synuclein proteins, including the truncated forms observed in Lewy bodies, are abundant in the appendix,” reported first author Bryan A. Killinger, PhD, also at VARI, in a press teleconference. He said this finding is likely to explain the reduced risk of PD from appendectomy.
In the largest of the epidemiologic studies, the effect of appendectomy on subsequent risk of PD was evaluated through the health records from more than 1.6 million individuals in Sweden. The incidence of PD was found to be 19.3% lower among 551,647 patients who had an appendectomy, compared with controls.
In addition, the data showed that when PD did occur after appendectomy, it was delayed on average by 3.6 years. It is notable that appendectomy was not associated with protection from PD in patients with a familial link to PD, a group they said comprises less than 10% of cases.
In patients with PD, nonmotor symptoms often include GI tract dysfunction, which can, in some cases, be part of a prodromal presentation that precedes the onset of classical PD symptoms by several years, the authors reported. However, the new research upends previous conceptions of disease. The demonstration of abundant alpha-synuclein in the appendix coupled with the protective effect of appendectomy, suggests that PD may originate in the GI tract and then spread to the central nervous system (CNS) rather than the other way around.
“The vermiform appendix was once considered to be an unnecessary organ. Although there is now good evidence that the appendix plays a major role in the regulation of the immune system, including the regulation of gut bacteria, our work suggests it is also mediates risk of Parkinson’s,” Dr. Labrie said in the teleconference.
In the paper, numerous pieces of the puzzle are brought together to suggest that alpha-synuclein in the appendix is linked to alpha-synuclein in the CNS. Many of the findings along this investigative pathway were described as surprising. For example, immunohistochemistry studies revealed high amounts of alpha-synuclein in nearly every sample of appendiceal tissue examined, including normal and inflamed tissue, tissue from individuals with PD and those without, and tissues from young and old individuals.
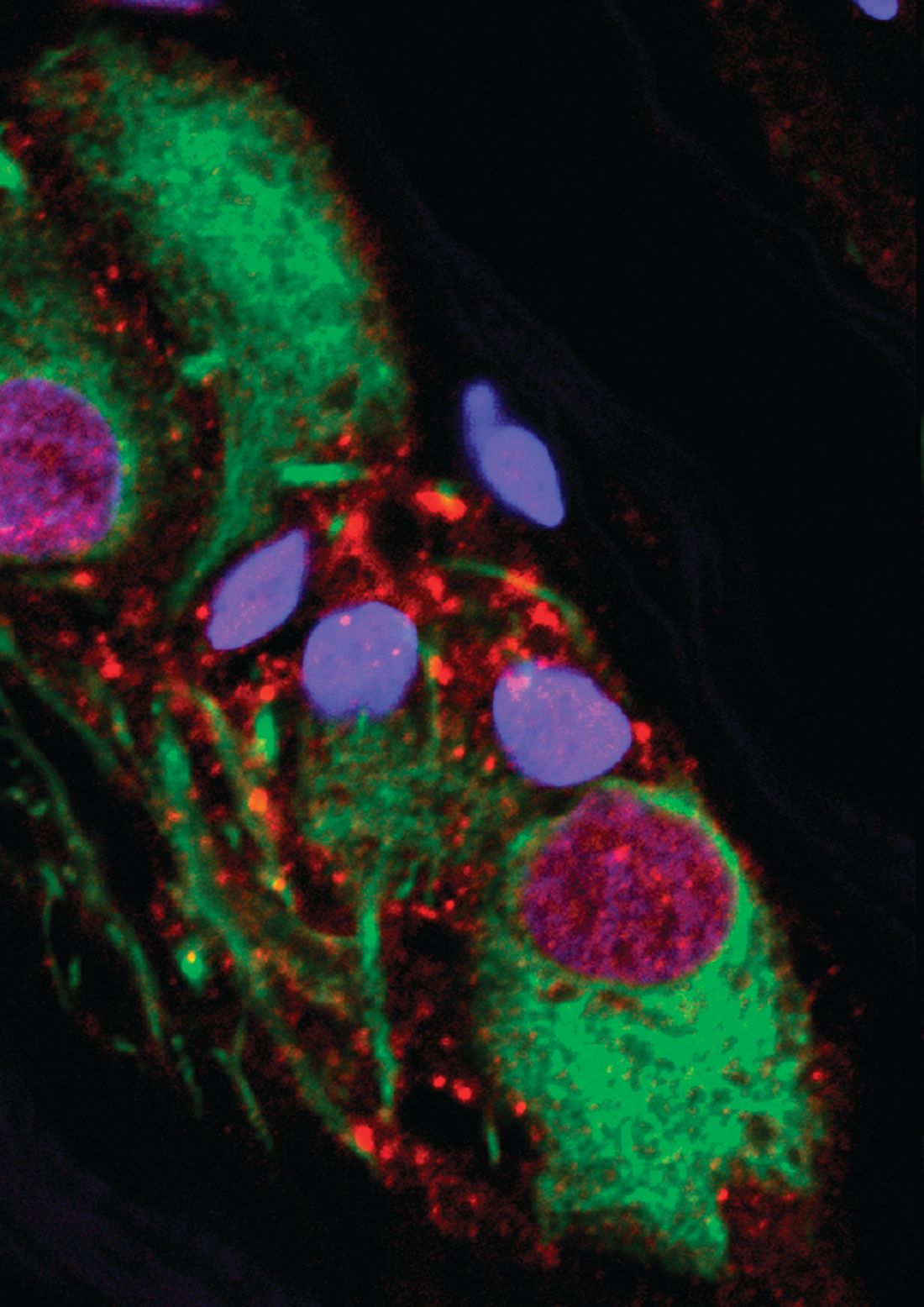
“The normal tissue, as well as appendiceal tissue from PD patients, contained high levels of alpha-synuclein in the truncated forms analogous to those seen in Lewy body pathology,” Dr. Killinger said. Based on these and other findings, he believes that alpha-synuclein in the appendix forms a reservoir for seeding the aggregates involved in the pathology of PD, although he acknowledged that it is not yet clear how the proteins in the appendix find their way to the brain.
From these data, it appears that most individuals with an intact appendix have alpha-synuclein in the nerve fibers, but Dr. Labrie pointed out that the only about 1% of the population develops PD. She speculated that there is “some confluence of events,” such as an environmental trigger altering the GI microbiome, that mediates ultimate risk of PD, but she noted that these events may take place decades before signs and symptoms of PD develop. The data appear to be a substantial reorientation in understanding PD.
“We have shown that the appendix is a hub for the accumulation of clumped forms of alpha-synuclein proteins, which are implicated in Parkinson’s,” Dr. Killinger said. “This knowledge will be invaluable as we explore new prevention and treatment strategies.”
The research was funded by a variety of governmental and private grants to individual authors. Dr. Killinger and Dr. Labrie report no financial relationships relevant to this study.
SOURCE: Killinger BA et al. Sci Transl Med. 2018;10:eaar5380.
Appendectomy has been associated with a reduced risk of Parkinson’s disease (PD), which supports the potential for a reservoir of aggregated alpha-synuclein in the appendix to affect risk of the condition, according to new epidemiologic and translational evidence from two data sets that promotes a new and emerging theory for PD etiology.
When placed into the context of other recent studies, these epidemiologic data “point to the appendix as a site of origin for Parkinson’s and provide a path forward for devising new treatment strategies,” reported senior author Viviane Labrie, PhD, of the Van Andel Research Institute (VARI) in Grand Rapids, Mich.
The epidemiologic data was the most recent step in a series of findings summarized in a newly published paper in Science Translational Medicine. As the researchers explained, it is relevant to a separate body of evidence that alpha-synuclein, a protein that serves as the hallmark of PD when it appears in Lewy bodies, can be isolated in the nerve fibers and nerve cells of the appendix.
“We have shown that alpha-synuclein proteins, including the truncated forms observed in Lewy bodies, are abundant in the appendix,” reported first author Bryan A. Killinger, PhD, also at VARI, in a press teleconference. He said this finding is likely to explain the reduced risk of PD from appendectomy.
In the largest of the epidemiologic studies, the effect of appendectomy on subsequent risk of PD was evaluated through the health records from more than 1.6 million individuals in Sweden. The incidence of PD was found to be 19.3% lower among 551,647 patients who had an appendectomy, compared with controls.
In addition, the data showed that when PD did occur after appendectomy, it was delayed on average by 3.6 years. It is notable that appendectomy was not associated with protection from PD in patients with a familial link to PD, a group they said comprises less than 10% of cases.
In patients with PD, nonmotor symptoms often include GI tract dysfunction, which can, in some cases, be part of a prodromal presentation that precedes the onset of classical PD symptoms by several years, the authors reported. However, the new research upends previous conceptions of disease. The demonstration of abundant alpha-synuclein in the appendix coupled with the protective effect of appendectomy, suggests that PD may originate in the GI tract and then spread to the central nervous system (CNS) rather than the other way around.
“The vermiform appendix was once considered to be an unnecessary organ. Although there is now good evidence that the appendix plays a major role in the regulation of the immune system, including the regulation of gut bacteria, our work suggests it is also mediates risk of Parkinson’s,” Dr. Labrie said in the teleconference.
In the paper, numerous pieces of the puzzle are brought together to suggest that alpha-synuclein in the appendix is linked to alpha-synuclein in the CNS. Many of the findings along this investigative pathway were described as surprising. For example, immunohistochemistry studies revealed high amounts of alpha-synuclein in nearly every sample of appendiceal tissue examined, including normal and inflamed tissue, tissue from individuals with PD and those without, and tissues from young and old individuals.
“The normal tissue, as well as appendiceal tissue from PD patients, contained high levels of alpha-synuclein in the truncated forms analogous to those seen in Lewy body pathology,” Dr. Killinger said. Based on these and other findings, he believes that alpha-synuclein in the appendix forms a reservoir for seeding the aggregates involved in the pathology of PD, although he acknowledged that it is not yet clear how the proteins in the appendix find their way to the brain.
From these data, it appears that most individuals with an intact appendix have alpha-synuclein in the nerve fibers, but Dr. Labrie pointed out that the only about 1% of the population develops PD. She speculated that there is “some confluence of events,” such as an environmental trigger altering the GI microbiome, that mediates ultimate risk of PD, but she noted that these events may take place decades before signs and symptoms of PD develop. The data appear to be a substantial reorientation in understanding PD.
“We have shown that the appendix is a hub for the accumulation of clumped forms of alpha-synuclein proteins, which are implicated in Parkinson’s,” Dr. Killinger said. “This knowledge will be invaluable as we explore new prevention and treatment strategies.”
The research was funded by a variety of governmental and private grants to individual authors. Dr. Killinger and Dr. Labrie report no financial relationships relevant to this study.
SOURCE: Killinger BA et al. Sci Transl Med. 2018;10:eaar5380.
Appendectomy has been associated with a reduced risk of Parkinson’s disease (PD), which supports the potential for a reservoir of aggregated alpha-synuclein in the appendix to affect risk of the condition, according to new epidemiologic and translational evidence from two data sets that promotes a new and emerging theory for PD etiology.
When placed into the context of other recent studies, these epidemiologic data “point to the appendix as a site of origin for Parkinson’s and provide a path forward for devising new treatment strategies,” reported senior author Viviane Labrie, PhD, of the Van Andel Research Institute (VARI) in Grand Rapids, Mich.
The epidemiologic data was the most recent step in a series of findings summarized in a newly published paper in Science Translational Medicine. As the researchers explained, it is relevant to a separate body of evidence that alpha-synuclein, a protein that serves as the hallmark of PD when it appears in Lewy bodies, can be isolated in the nerve fibers and nerve cells of the appendix.
“We have shown that alpha-synuclein proteins, including the truncated forms observed in Lewy bodies, are abundant in the appendix,” reported first author Bryan A. Killinger, PhD, also at VARI, in a press teleconference. He said this finding is likely to explain the reduced risk of PD from appendectomy.
In the largest of the epidemiologic studies, the effect of appendectomy on subsequent risk of PD was evaluated through the health records from more than 1.6 million individuals in Sweden. The incidence of PD was found to be 19.3% lower among 551,647 patients who had an appendectomy, compared with controls.
In addition, the data showed that when PD did occur after appendectomy, it was delayed on average by 3.6 years. It is notable that appendectomy was not associated with protection from PD in patients with a familial link to PD, a group they said comprises less than 10% of cases.
In patients with PD, nonmotor symptoms often include GI tract dysfunction, which can, in some cases, be part of a prodromal presentation that precedes the onset of classical PD symptoms by several years, the authors reported. However, the new research upends previous conceptions of disease. The demonstration of abundant alpha-synuclein in the appendix coupled with the protective effect of appendectomy, suggests that PD may originate in the GI tract and then spread to the central nervous system (CNS) rather than the other way around.
“The vermiform appendix was once considered to be an unnecessary organ. Although there is now good evidence that the appendix plays a major role in the regulation of the immune system, including the regulation of gut bacteria, our work suggests it is also mediates risk of Parkinson’s,” Dr. Labrie said in the teleconference.
In the paper, numerous pieces of the puzzle are brought together to suggest that alpha-synuclein in the appendix is linked to alpha-synuclein in the CNS. Many of the findings along this investigative pathway were described as surprising. For example, immunohistochemistry studies revealed high amounts of alpha-synuclein in nearly every sample of appendiceal tissue examined, including normal and inflamed tissue, tissue from individuals with PD and those without, and tissues from young and old individuals.
“The normal tissue, as well as appendiceal tissue from PD patients, contained high levels of alpha-synuclein in the truncated forms analogous to those seen in Lewy body pathology,” Dr. Killinger said. Based on these and other findings, he believes that alpha-synuclein in the appendix forms a reservoir for seeding the aggregates involved in the pathology of PD, although he acknowledged that it is not yet clear how the proteins in the appendix find their way to the brain.
From these data, it appears that most individuals with an intact appendix have alpha-synuclein in the nerve fibers, but Dr. Labrie pointed out that the only about 1% of the population develops PD. She speculated that there is “some confluence of events,” such as an environmental trigger altering the GI microbiome, that mediates ultimate risk of PD, but she noted that these events may take place decades before signs and symptoms of PD develop. The data appear to be a substantial reorientation in understanding PD.
“We have shown that the appendix is a hub for the accumulation of clumped forms of alpha-synuclein proteins, which are implicated in Parkinson’s,” Dr. Killinger said. “This knowledge will be invaluable as we explore new prevention and treatment strategies.”
The research was funded by a variety of governmental and private grants to individual authors. Dr. Killinger and Dr. Labrie report no financial relationships relevant to this study.
SOURCE: Killinger BA et al. Sci Transl Med. 2018;10:eaar5380.
FROM SCIENCE TRANSLATIONAL MEDICINE
Key clinical point:
Major finding: A 19.3% reduction in risk of PD from appendectomy may relate to alpha-synuclein in the appendix.
Study details: Series of related epidemiologic and translational studies.
Disclosures: The research was funded by a variety of governmental and private grants to individual authors. Dr. Killinger and Dr. Labrie report no financial relationships relevant to this study.
Source: Killinger BA et al. Sci Transl Med. 2018;10:eaar5380.
Funding for NIH BRAIN Initiative reaches new heights
The National Institutes of Health’s Brain Research through Advancing Innovative Neurotechnologies (BRAIN) Initiative will finish 2018 with its largest round of grant funding ever, giving $220 million to more than 200 research awards, and bringing this year’s total to more than $400 million, according to an announcement from the agency.
The BRAIN Initiative began in 2013 with the objective of revolutionizing our understanding of the human brain by accelerating the development and application of innovative technologies that will allow researchers to show how individual cells and complex neural circuits interact in both time and space and thereby seek new ways to treat, cure, and prevent brain disorders.
In the current round of funding that was authorized by Congress through the regular appropriations process and the 21st Century Cures Act, new projects include the creation of a wireless optical tomography cap for scanning human brain activity; the development of a noninvasive brain-computer interface system for improving the lives of paralysis patients; and the testing of noninvasive brain stimulation devices for treating schizophrenia, attention deficit disorders, and other brain diseases; the development of self-growing biological electrodes for recording brain activity; and the creation of an indestructible hydrogel system to help map neural circuits, according to the announcement.
Not all of the research involves technological advancement. In fact, one line of funding involves neuroethics. For instance, for epilepsy syndromes in the latest round of funding for 2018, researchers aim to explore ethical issues confronting families and clinicians when considering new treatment options for drug-resistant epilepsy in children.
The NIH is also leveraging some of the BRAIN Initiative funding toward finding new, nonaddictive pain treatments as part of the its HEAL (Helping to End Addiction Long-term) Initiative, such as support for research on the fundamental neurobiology of endogenous opioid systems.
The National Institutes of Health’s Brain Research through Advancing Innovative Neurotechnologies (BRAIN) Initiative will finish 2018 with its largest round of grant funding ever, giving $220 million to more than 200 research awards, and bringing this year’s total to more than $400 million, according to an announcement from the agency.
The BRAIN Initiative began in 2013 with the objective of revolutionizing our understanding of the human brain by accelerating the development and application of innovative technologies that will allow researchers to show how individual cells and complex neural circuits interact in both time and space and thereby seek new ways to treat, cure, and prevent brain disorders.
In the current round of funding that was authorized by Congress through the regular appropriations process and the 21st Century Cures Act, new projects include the creation of a wireless optical tomography cap for scanning human brain activity; the development of a noninvasive brain-computer interface system for improving the lives of paralysis patients; and the testing of noninvasive brain stimulation devices for treating schizophrenia, attention deficit disorders, and other brain diseases; the development of self-growing biological electrodes for recording brain activity; and the creation of an indestructible hydrogel system to help map neural circuits, according to the announcement.
Not all of the research involves technological advancement. In fact, one line of funding involves neuroethics. For instance, for epilepsy syndromes in the latest round of funding for 2018, researchers aim to explore ethical issues confronting families and clinicians when considering new treatment options for drug-resistant epilepsy in children.
The NIH is also leveraging some of the BRAIN Initiative funding toward finding new, nonaddictive pain treatments as part of the its HEAL (Helping to End Addiction Long-term) Initiative, such as support for research on the fundamental neurobiology of endogenous opioid systems.
The National Institutes of Health’s Brain Research through Advancing Innovative Neurotechnologies (BRAIN) Initiative will finish 2018 with its largest round of grant funding ever, giving $220 million to more than 200 research awards, and bringing this year’s total to more than $400 million, according to an announcement from the agency.
The BRAIN Initiative began in 2013 with the objective of revolutionizing our understanding of the human brain by accelerating the development and application of innovative technologies that will allow researchers to show how individual cells and complex neural circuits interact in both time and space and thereby seek new ways to treat, cure, and prevent brain disorders.
In the current round of funding that was authorized by Congress through the regular appropriations process and the 21st Century Cures Act, new projects include the creation of a wireless optical tomography cap for scanning human brain activity; the development of a noninvasive brain-computer interface system for improving the lives of paralysis patients; and the testing of noninvasive brain stimulation devices for treating schizophrenia, attention deficit disorders, and other brain diseases; the development of self-growing biological electrodes for recording brain activity; and the creation of an indestructible hydrogel system to help map neural circuits, according to the announcement.
Not all of the research involves technological advancement. In fact, one line of funding involves neuroethics. For instance, for epilepsy syndromes in the latest round of funding for 2018, researchers aim to explore ethical issues confronting families and clinicians when considering new treatment options for drug-resistant epilepsy in children.
The NIH is also leveraging some of the BRAIN Initiative funding toward finding new, nonaddictive pain treatments as part of the its HEAL (Helping to End Addiction Long-term) Initiative, such as support for research on the fundamental neurobiology of endogenous opioid systems.
CT opens extended window for stroke thrombolysis
MONTREAL – An extended time window for thrombolytic treatment of acute ischemic stroke patients using tissue plasminogen activator out to 9 hours from stroke onset was safe and effective using CT perfusion imaging and automated imaging processing software to select suitable patients in the EXTEND trial. This result matches the groundbreaking finding reported earlier in 2018 that used MRI to select patients for extended thrombolysis.

“To reproduce our results you need to set up CT perfusion” as well as the RAPID software for automated image processing to identify patients with a small infarct core and a large area of salvageable brain, said Henry Ma, MD, a stroke neurologist at Monash University, Melbourne, as he reported results from the trial at the World Stroke Congress. “EXTEND is the first positive thrombolysis trial in an extended time window using automated penumbral imaging.”
The new finding, from a trial with 225 randomized stroke patients, was especially notable because, by showing the validity of CT imaging for patient selection, it makes applying the extended time window for thrombolytic therapy more feasible for U.S. and Canadian stroke centers where CT imaging is much more common than MRI. A report from European investigators published in August 2018 from the WAKE-UP trial showed that thrombolysis with tissue plasminogen activator (tPA) was safe and effective when administered to patients who woke up with an acute ischemic stroke that had occurred more than 4.5 hours before treatment, but this study exclusively used MRI for patient selection (N Engl J Med. 2018 Aug 16;379[7]:611-22).
“In North America, our systems are more equipped for using CT,” commented Ashfaq Shuaib, MD, a professor of medicine and neurologist at the University of Alberta, Edmonton. Based on the WAKE-UP results, “MR would be preferred, but what we’ve been doing [since the WAKE-UP report] is if we see a CT scan that’s good we go ahead” with thrombolysis.

“Biologically, it doesn’t matter whether you use MR or CT; they both index the same underlying pathology. We’ve been hesitant to go beyond the MR finding from WAKE-UP, where there were data, but the findings from EXTEND were right in line with the WAKE-UP results, and that’s all we need to be reassured” that CT perfusion imaging also works for patient selection, commented Jeffrey L. Saver, MD, professor of medicine and director of the Comprehensive Stroke Center at the University of California, Los Angeles.
CT perfusion imaging and automated image processing “worked to select stroke patients” for an extended time window for treatment with mechanical thrombectomy in the DAWN (N Engl J Med. 2018 Jan 4;378[1]:11-21) and DEFUSE 3 (N Engl J Med. 2018 Feb 22;378[8]:308-18) trials, a history that makes the new finding of successfully using CT imaging to select patients who qualify for extended use of thrombolysis “a convincing result,” Dr. Saver said in an interview. The new EXTEND findings “will have a major impact” on using an extended time window for thrombolysis in U.S. practice, he predicted.
The EXTEND trial (Int J Stroke. 2012 Jan 1;7[1]:74-80) ran at 22 sites in Australia, 11 sites in Taiwan, and 1 center in New Zealand. Recruitment of patients into the study stopped early, after enrolling 225 patients, in June 2018, when results from WAKE-UP came out.
The EXTEND investigators enrolled patients who were either 4.5-9 hours out from the onset of their stroke or patients with a wake-up stroke with an uncertain onset. Participating centers could use either CT perfusion or MRI to identify candidates for treatment, and all used the RAPID software for image processing to identify patients with a perfusion lesion of at least 10 mL and an ischemic core volume no greater than 70 mL. Dr. Ma did not report what percentage of patients underwent imaging with each of these methods, but hinted that clinicians had used CT for a majority of the cases. The study randomized patients to receive either 0.9 mg/kg tPA or placebo, and by the trial protocol none of the enrolled patients received treatment with mechanical thrombectomy.
The trial’s primary endpoint was the percentage of patients with a modified Rankin Scale score of 0 or 1 at 90 days after their stroke, which was achieved by 44% more patients in the tPA group relative to the placebo arm after adjustment for age and baseline stroke severity, a statistically significant difference. The results were also positive for several secondary endpoints, such as recanalization 24 hours after treatment, which occurred in 67% of patients treated with tPA and 37% of the control patients, a statistically significant 68% relative improvement with thrombolysis.
Mortality at 90 days was similar in the two arms – 9% among the placebo patients and 12% among those who received tPA. The rate of symptomatic intracranial hemorrhage 36 hours after treatment was significantly higher among patients treated with tPA at 6%, compared with 1% in the placebo group, but the magnitude of this adverse effect was consistent with rates of intracranial hemorrhages previously reported in other studies of thrombolytic treatment for acute ischemic stroke, Dr. Ma said. The small number of increased intracranial hemorrhages “was not associated with increased mortality, and did not negate the positive result of an improved rate of excellent functional outcomes.”
These findings will likely spur further adoption of imaging processing software of the type used in EXTEND by U.S. stroke centers, Dr. Saver predicted.
“More and more centers have been getting this [software], and now they have two reasons to have it: to identify patients for an extended window for mechanical thrombectomy and to identify patients for an extended window for thrombolysis. It is a compelling case to have the imaging software as widely disseminated as possible. Centers that want to do the best for patients should have this imaging-processing software,” Dr. Saver said.
Dr. Ma and Dr. Shuaib reported no disclosures. Dr. Saver has received research funding and personal fees from Medtronic-Abbott and Neuravia.
SOURCE: Ma H et al. Int J. Stroke. 2018 Oct;13(2S):235, Abstract 1014.
Ever since results from the WAKE-UP trial came out earlier in 2018, we at the University of Cincinnati have been imaging acute ischemic stroke patients who presented outside the standard 4.5-hour time limit for thrombolysis with MRI to see if they qualify for an extended window for thrombolysis. But this has been a cumbersome and redundant process because our default imaging method is CT, so we have been imaging potential candidates for an extended thrombolytic window twice, first with CT and then later with MRI.

The EXTEND findings also provide a further reason for U.S. stroke centers to purchase and use some type of imaging processing software if they don’t already have it, either the RAPID software that was used in EXTEND or one of the several similar software packages that are now available. Several primary stroke centers in my area still do not currently use this software, although its use has been quickly spreading and it will now be increasingly hard for these centers to hold off acquiring it. Fortunately the increased competition among software vendors who sell this type of software has meant that the price has been dropping.
Pooja Khatri, MD , is a professor of neurology and director of acute stroke at the University of Cincinnati. She has been a consultant to Biogen, Greenwich, and PTC Therapeutics. She made these comments in an interview.
Ever since results from the WAKE-UP trial came out earlier in 2018, we at the University of Cincinnati have been imaging acute ischemic stroke patients who presented outside the standard 4.5-hour time limit for thrombolysis with MRI to see if they qualify for an extended window for thrombolysis. But this has been a cumbersome and redundant process because our default imaging method is CT, so we have been imaging potential candidates for an extended thrombolytic window twice, first with CT and then later with MRI.
The EXTEND findings also provide a further reason for U.S. stroke centers to purchase and use some type of imaging processing software if they don’t already have it, either the RAPID software that was used in EXTEND or one of the several similar software packages that are now available. Several primary stroke centers in my area still do not currently use this software, although its use has been quickly spreading and it will now be increasingly hard for these centers to hold off acquiring it. Fortunately the increased competition among software vendors who sell this type of software has meant that the price has been dropping.
Pooja Khatri, MD , is a professor of neurology and director of acute stroke at the University of Cincinnati. She has been a consultant to Biogen, Greenwich, and PTC Therapeutics. She made these comments in an interview.
Ever since results from the WAKE-UP trial came out earlier in 2018, we at the University of Cincinnati have been imaging acute ischemic stroke patients who presented outside the standard 4.5-hour time limit for thrombolysis with MRI to see if they qualify for an extended window for thrombolysis. But this has been a cumbersome and redundant process because our default imaging method is CT, so we have been imaging potential candidates for an extended thrombolytic window twice, first with CT and then later with MRI.
The EXTEND findings also provide a further reason for U.S. stroke centers to purchase and use some type of imaging processing software if they don’t already have it, either the RAPID software that was used in EXTEND or one of the several similar software packages that are now available. Several primary stroke centers in my area still do not currently use this software, although its use has been quickly spreading and it will now be increasingly hard for these centers to hold off acquiring it. Fortunately the increased competition among software vendors who sell this type of software has meant that the price has been dropping.
Pooja Khatri, MD , is a professor of neurology and director of acute stroke at the University of Cincinnati. She has been a consultant to Biogen, Greenwich, and PTC Therapeutics. She made these comments in an interview.
MONTREAL – An extended time window for thrombolytic treatment of acute ischemic stroke patients using tissue plasminogen activator out to 9 hours from stroke onset was safe and effective using CT perfusion imaging and automated imaging processing software to select suitable patients in the EXTEND trial. This result matches the groundbreaking finding reported earlier in 2018 that used MRI to select patients for extended thrombolysis.
“To reproduce our results you need to set up CT perfusion” as well as the RAPID software for automated image processing to identify patients with a small infarct core and a large area of salvageable brain, said Henry Ma, MD, a stroke neurologist at Monash University, Melbourne, as he reported results from the trial at the World Stroke Congress. “EXTEND is the first positive thrombolysis trial in an extended time window using automated penumbral imaging.”
The new finding, from a trial with 225 randomized stroke patients, was especially notable because, by showing the validity of CT imaging for patient selection, it makes applying the extended time window for thrombolytic therapy more feasible for U.S. and Canadian stroke centers where CT imaging is much more common than MRI. A report from European investigators published in August 2018 from the WAKE-UP trial showed that thrombolysis with tissue plasminogen activator (tPA) was safe and effective when administered to patients who woke up with an acute ischemic stroke that had occurred more than 4.5 hours before treatment, but this study exclusively used MRI for patient selection (N Engl J Med. 2018 Aug 16;379[7]:611-22).
“In North America, our systems are more equipped for using CT,” commented Ashfaq Shuaib, MD, a professor of medicine and neurologist at the University of Alberta, Edmonton. Based on the WAKE-UP results, “MR would be preferred, but what we’ve been doing [since the WAKE-UP report] is if we see a CT scan that’s good we go ahead” with thrombolysis.
“Biologically, it doesn’t matter whether you use MR or CT; they both index the same underlying pathology. We’ve been hesitant to go beyond the MR finding from WAKE-UP, where there were data, but the findings from EXTEND were right in line with the WAKE-UP results, and that’s all we need to be reassured” that CT perfusion imaging also works for patient selection, commented Jeffrey L. Saver, MD, professor of medicine and director of the Comprehensive Stroke Center at the University of California, Los Angeles.
CT perfusion imaging and automated image processing “worked to select stroke patients” for an extended time window for treatment with mechanical thrombectomy in the DAWN (N Engl J Med. 2018 Jan 4;378[1]:11-21) and DEFUSE 3 (N Engl J Med. 2018 Feb 22;378[8]:308-18) trials, a history that makes the new finding of successfully using CT imaging to select patients who qualify for extended use of thrombolysis “a convincing result,” Dr. Saver said in an interview. The new EXTEND findings “will have a major impact” on using an extended time window for thrombolysis in U.S. practice, he predicted.
The EXTEND trial (Int J Stroke. 2012 Jan 1;7[1]:74-80) ran at 22 sites in Australia, 11 sites in Taiwan, and 1 center in New Zealand. Recruitment of patients into the study stopped early, after enrolling 225 patients, in June 2018, when results from WAKE-UP came out.
The EXTEND investigators enrolled patients who were either 4.5-9 hours out from the onset of their stroke or patients with a wake-up stroke with an uncertain onset. Participating centers could use either CT perfusion or MRI to identify candidates for treatment, and all used the RAPID software for image processing to identify patients with a perfusion lesion of at least 10 mL and an ischemic core volume no greater than 70 mL. Dr. Ma did not report what percentage of patients underwent imaging with each of these methods, but hinted that clinicians had used CT for a majority of the cases. The study randomized patients to receive either 0.9 mg/kg tPA or placebo, and by the trial protocol none of the enrolled patients received treatment with mechanical thrombectomy.
The trial’s primary endpoint was the percentage of patients with a modified Rankin Scale score of 0 or 1 at 90 days after their stroke, which was achieved by 44% more patients in the tPA group relative to the placebo arm after adjustment for age and baseline stroke severity, a statistically significant difference. The results were also positive for several secondary endpoints, such as recanalization 24 hours after treatment, which occurred in 67% of patients treated with tPA and 37% of the control patients, a statistically significant 68% relative improvement with thrombolysis.
Mortality at 90 days was similar in the two arms – 9% among the placebo patients and 12% among those who received tPA. The rate of symptomatic intracranial hemorrhage 36 hours after treatment was significantly higher among patients treated with tPA at 6%, compared with 1% in the placebo group, but the magnitude of this adverse effect was consistent with rates of intracranial hemorrhages previously reported in other studies of thrombolytic treatment for acute ischemic stroke, Dr. Ma said. The small number of increased intracranial hemorrhages “was not associated with increased mortality, and did not negate the positive result of an improved rate of excellent functional outcomes.”
These findings will likely spur further adoption of imaging processing software of the type used in EXTEND by U.S. stroke centers, Dr. Saver predicted.
“More and more centers have been getting this [software], and now they have two reasons to have it: to identify patients for an extended window for mechanical thrombectomy and to identify patients for an extended window for thrombolysis. It is a compelling case to have the imaging software as widely disseminated as possible. Centers that want to do the best for patients should have this imaging-processing software,” Dr. Saver said.
Dr. Ma and Dr. Shuaib reported no disclosures. Dr. Saver has received research funding and personal fees from Medtronic-Abbott and Neuravia.
SOURCE: Ma H et al. Int J. Stroke. 2018 Oct;13(2S):235, Abstract 1014.
MONTREAL – An extended time window for thrombolytic treatment of acute ischemic stroke patients using tissue plasminogen activator out to 9 hours from stroke onset was safe and effective using CT perfusion imaging and automated imaging processing software to select suitable patients in the EXTEND trial. This result matches the groundbreaking finding reported earlier in 2018 that used MRI to select patients for extended thrombolysis.
“To reproduce our results you need to set up CT perfusion” as well as the RAPID software for automated image processing to identify patients with a small infarct core and a large area of salvageable brain, said Henry Ma, MD, a stroke neurologist at Monash University, Melbourne, as he reported results from the trial at the World Stroke Congress. “EXTEND is the first positive thrombolysis trial in an extended time window using automated penumbral imaging.”
The new finding, from a trial with 225 randomized stroke patients, was especially notable because, by showing the validity of CT imaging for patient selection, it makes applying the extended time window for thrombolytic therapy more feasible for U.S. and Canadian stroke centers where CT imaging is much more common than MRI. A report from European investigators published in August 2018 from the WAKE-UP trial showed that thrombolysis with tissue plasminogen activator (tPA) was safe and effective when administered to patients who woke up with an acute ischemic stroke that had occurred more than 4.5 hours before treatment, but this study exclusively used MRI for patient selection (N Engl J Med. 2018 Aug 16;379[7]:611-22).
“In North America, our systems are more equipped for using CT,” commented Ashfaq Shuaib, MD, a professor of medicine and neurologist at the University of Alberta, Edmonton. Based on the WAKE-UP results, “MR would be preferred, but what we’ve been doing [since the WAKE-UP report] is if we see a CT scan that’s good we go ahead” with thrombolysis.
“Biologically, it doesn’t matter whether you use MR or CT; they both index the same underlying pathology. We’ve been hesitant to go beyond the MR finding from WAKE-UP, where there were data, but the findings from EXTEND were right in line with the WAKE-UP results, and that’s all we need to be reassured” that CT perfusion imaging also works for patient selection, commented Jeffrey L. Saver, MD, professor of medicine and director of the Comprehensive Stroke Center at the University of California, Los Angeles.
CT perfusion imaging and automated image processing “worked to select stroke patients” for an extended time window for treatment with mechanical thrombectomy in the DAWN (N Engl J Med. 2018 Jan 4;378[1]:11-21) and DEFUSE 3 (N Engl J Med. 2018 Feb 22;378[8]:308-18) trials, a history that makes the new finding of successfully using CT imaging to select patients who qualify for extended use of thrombolysis “a convincing result,” Dr. Saver said in an interview. The new EXTEND findings “will have a major impact” on using an extended time window for thrombolysis in U.S. practice, he predicted.
The EXTEND trial (Int J Stroke. 2012 Jan 1;7[1]:74-80) ran at 22 sites in Australia, 11 sites in Taiwan, and 1 center in New Zealand. Recruitment of patients into the study stopped early, after enrolling 225 patients, in June 2018, when results from WAKE-UP came out.
The EXTEND investigators enrolled patients who were either 4.5-9 hours out from the onset of their stroke or patients with a wake-up stroke with an uncertain onset. Participating centers could use either CT perfusion or MRI to identify candidates for treatment, and all used the RAPID software for image processing to identify patients with a perfusion lesion of at least 10 mL and an ischemic core volume no greater than 70 mL. Dr. Ma did not report what percentage of patients underwent imaging with each of these methods, but hinted that clinicians had used CT for a majority of the cases. The study randomized patients to receive either 0.9 mg/kg tPA or placebo, and by the trial protocol none of the enrolled patients received treatment with mechanical thrombectomy.
The trial’s primary endpoint was the percentage of patients with a modified Rankin Scale score of 0 or 1 at 90 days after their stroke, which was achieved by 44% more patients in the tPA group relative to the placebo arm after adjustment for age and baseline stroke severity, a statistically significant difference. The results were also positive for several secondary endpoints, such as recanalization 24 hours after treatment, which occurred in 67% of patients treated with tPA and 37% of the control patients, a statistically significant 68% relative improvement with thrombolysis.
Mortality at 90 days was similar in the two arms – 9% among the placebo patients and 12% among those who received tPA. The rate of symptomatic intracranial hemorrhage 36 hours after treatment was significantly higher among patients treated with tPA at 6%, compared with 1% in the placebo group, but the magnitude of this adverse effect was consistent with rates of intracranial hemorrhages previously reported in other studies of thrombolytic treatment for acute ischemic stroke, Dr. Ma said. The small number of increased intracranial hemorrhages “was not associated with increased mortality, and did not negate the positive result of an improved rate of excellent functional outcomes.”
These findings will likely spur further adoption of imaging processing software of the type used in EXTEND by U.S. stroke centers, Dr. Saver predicted.
“More and more centers have been getting this [software], and now they have two reasons to have it: to identify patients for an extended window for mechanical thrombectomy and to identify patients for an extended window for thrombolysis. It is a compelling case to have the imaging software as widely disseminated as possible. Centers that want to do the best for patients should have this imaging-processing software,” Dr. Saver said.
Dr. Ma and Dr. Shuaib reported no disclosures. Dr. Saver has received research funding and personal fees from Medtronic-Abbott and Neuravia.
SOURCE: Ma H et al. Int J. Stroke. 2018 Oct;13(2S):235, Abstract 1014.
REPORTING FROM THE WORLD STROKE CONGRESS
Key clinical point:
Major finding: Patients who received thrombolysis 4.5-9 hours after stroke onset had a 44% increased rate of good outcomes, compared with controls.
Study details: EXTEND, a multicenter, controlled trial with 225 patients.
Disclosures: Dr. Ma and Dr. Shuaib had no disclosures. Dr. Saver has received research funding and personal fees from Medtronic-Abbott and Neuravia.
Source: Ma H et al. Int J. Stroke. 2018 Oct;13(2S):235, Abstract 1014.
Management of Lewy body dementia remains complex
ATLANTA – In the not-so-distant past, neurologists viewed dementia with Lewy bodies as a disorder primarily of the brain, but it turned out to be far more complex than that.

At the annual meeting of the American Neurological Association, Bradley F. Boeve, MD, described dementia with Lewy bodies (DLB) as a systemic neurologic disorder affecting the brain, including brain stem, spinal cord, and peripheral nervous system, especially the autonomic nervous system. “This leads to the complex array of clinical manifestations, which are quite different from patient to patient cross-sectionally and longitudinally,” said Dr. Boeve, the Little Family Foundation Professor of Lewy Body Dementia in the department of neurology at the Mayo Clinic, Rochester, Minn.
, he said. The four core clinical features are Parkinsonism unrelated to medications; recurrent, fully-formed visual hallucinations; fluctuations in cognition and/or arousal; and rapid eye movement (REM) sleep behavior disorder. “This is the most predictive of all four features,” Dr. Boeve said. He described REM sleep behavior disorder as a parasomnia manifested by the tendency to repeatedly “act out one’s dreams.” The dreams tend to contain a chasing/attacking theme, and behaviors mirror dream content. Injuries to the patient and bed partner can occur.
Typically, patients will present with REM sleep behavior disorder in their 50s and 60s, and sometimes in their 30s and 40s, “decades before cognitive changes begin,” he said. “This is usually followed by Parkinsonism and visual hallucinations. That’s the prototypical DLB [case], but there are many examples where this is not followed. Prominent neuropsychiatric features can also begin before any cognitive changes.”
Neuropsychological features of DLB often include impairment of executive functions and visuospatial functions. “Early in the course of Alzheimer’s disease, typically performance on memory measures – especially delayed recall – are down and the other measures are borderline or mildly impaired,” Dr. Boeve noted. “By contrast, in DLB, attention, executive function, and visuospatial measures are down, but memory is often pretty good. What’s remarkable is that in the office setting, when you take a history the person often says, ‘I’m very forgetful,’ yet in the testing environment people tend to rise to the occasion pretty well.”
Imaging isn’t always helpful in establishing a diagnosis of DLB. MRI scans, for example, “can look pretty normal, including the hippocampi,” he said. “This is really the norm in DLB and it seems to be a disconnect. The person can have significant symptoms yet their MRI scan can be pretty normal.”
In Alzheimer’s disease, 18F-fluorodeoxyglucose-PET (FDG-PET) shows temporal, parietal, and frontal hypometabolism, sparing of the sensory-motor strip and sparing of the primary occipital cortex, while in DLB, FDG-PET shows marked deficits in the occipital regions with relative sparing of the frontal and temporal lobes. Another key neuroimaging sign of DLB is the posterior cingulate island sign, which is characterized by sparing of the posterior cingulate cortex relative to the precuneus plus cuneus on FDG-PET.
In 2017, the Dementia with Lewy Bodies Consortium published updated recommendations on the diagnosis and management of the disease (Neurology. 2017;89[1]:88-100). In its consensus report, the consortium defines probable DLB as dementia plus two or more clinical features or one core clinical feature plus one or more indicative biomarker. These biomarkers include reduced dopamine transport uptake in basal ganglia by SPECT or PET; abnormal (low uptake) meta-iodobenzylguanidine (MIBG) myocardial scintigraphy, and/or polysomnographic confirmation of REM sleep without atonia.
“Neuropathologically, limbic with or without neocortical Lewy bodies and Lewy neurites are the defining characteristics of pathologically-proven DLB,” added Dr. Boeve, a member of the DLB consortium. “The classic DLB phenotype can occur in limbic-predominant DLB. Lewy bodies in the neocortex are not necessary to cause a dementia syndrome.”
He characterized management of DLB as “very complicated. Consider symptoms as they relate to cognitive impairment, neuropsychiatric features, motor features, sleep disorders, and autonomic dysfunction.” He often asks the patient/family to prioritize the three most troublesome issues they seek to change, and develops a plan based on their input.
There is no Food and Drug Administration–approved medication for DLB, but the standard of care is an acetylcholinesterase inhibitor such as donepezil. “There is evidence that memantine can provide a modest benefit,” Dr. Boeve said. “Hypersomnia is quite prominent in DLB and worthy of assessing and treating.” Clinicians must weigh the pros and cons of pharmacotherapy with each patient. “For example, in the atypical neuroleptic class [of drugs], there may be a benefit to the hallucinations and delusions in DLB but hypersomnia can worsen,” he said. “Selecting agents is challenging but worth the effort.”
Survival is lower and more rapid with DLB, compared with Alzheimer’s. Most people pass away from primary DLB-related features or failure to thrive. The second most common is pneumonia or aspiration. Median survival was 4 years after diagnosis in one study, and end-of life discussions occurred in less than half of all patients. “This is a frustrating reminder that we as clinicians are not very good at discussing important topics such as end-of-life care with patients and their families,” Dr. Boeve said. Resources that he recommends for education and support include the Lewy Body Dementia Association and The Lewy Body Society.
At the 2016 Alzheimer’s Disease-Related Dementias Summit, clinicians formed a list of DLB research priorities (Neurology 2017;89[23]:2381-91). Among them were recommendations to “initiate clinical trials in diverse populations using therapies that address symptoms that have the greatest effect on patient function and caregiver burden” and “identify novel common and rare genetic variants, epigenetic changes, and environmental influences that affect the risk for and clinical features of” the disease.
Meanwhile, several research protocols are under way, including the development of a DLB module by the U.S. Alzheimer’s Research Disease Centers and a number of DLB-focused projects from the National Institute of Neurological Disorders and Stroke (NINDS) Parkinson’s Disease Biomarkers Program. In addition, the Lewy Body Dementia Association Research Centers of Excellence program is focused on optimizing clinical care and setting up the infrastructure for clinical trials, while the North American Prodromal Synucleinopathy Consortium is conducting longitudinal studies in those with REM sleep behavior disorder.
Dr. Boeve disclosed that he has been an investigator for clinical trials sponsored by GE Healthcare, Axovant, and Biogen. He is a member of the scientific advisory board for the Tau Consortium and has received research support from the National Institute on Aging, the NINDS, the Mangurian Foundation, and the Little Family Foundation.
ATLANTA – In the not-so-distant past, neurologists viewed dementia with Lewy bodies as a disorder primarily of the brain, but it turned out to be far more complex than that.
At the annual meeting of the American Neurological Association, Bradley F. Boeve, MD, described dementia with Lewy bodies (DLB) as a systemic neurologic disorder affecting the brain, including brain stem, spinal cord, and peripheral nervous system, especially the autonomic nervous system. “This leads to the complex array of clinical manifestations, which are quite different from patient to patient cross-sectionally and longitudinally,” said Dr. Boeve, the Little Family Foundation Professor of Lewy Body Dementia in the department of neurology at the Mayo Clinic, Rochester, Minn.
, he said. The four core clinical features are Parkinsonism unrelated to medications; recurrent, fully-formed visual hallucinations; fluctuations in cognition and/or arousal; and rapid eye movement (REM) sleep behavior disorder. “This is the most predictive of all four features,” Dr. Boeve said. He described REM sleep behavior disorder as a parasomnia manifested by the tendency to repeatedly “act out one’s dreams.” The dreams tend to contain a chasing/attacking theme, and behaviors mirror dream content. Injuries to the patient and bed partner can occur.
Typically, patients will present with REM sleep behavior disorder in their 50s and 60s, and sometimes in their 30s and 40s, “decades before cognitive changes begin,” he said. “This is usually followed by Parkinsonism and visual hallucinations. That’s the prototypical DLB [case], but there are many examples where this is not followed. Prominent neuropsychiatric features can also begin before any cognitive changes.”
Neuropsychological features of DLB often include impairment of executive functions and visuospatial functions. “Early in the course of Alzheimer’s disease, typically performance on memory measures – especially delayed recall – are down and the other measures are borderline or mildly impaired,” Dr. Boeve noted. “By contrast, in DLB, attention, executive function, and visuospatial measures are down, but memory is often pretty good. What’s remarkable is that in the office setting, when you take a history the person often says, ‘I’m very forgetful,’ yet in the testing environment people tend to rise to the occasion pretty well.”
Imaging isn’t always helpful in establishing a diagnosis of DLB. MRI scans, for example, “can look pretty normal, including the hippocampi,” he said. “This is really the norm in DLB and it seems to be a disconnect. The person can have significant symptoms yet their MRI scan can be pretty normal.”
In Alzheimer’s disease, 18F-fluorodeoxyglucose-PET (FDG-PET) shows temporal, parietal, and frontal hypometabolism, sparing of the sensory-motor strip and sparing of the primary occipital cortex, while in DLB, FDG-PET shows marked deficits in the occipital regions with relative sparing of the frontal and temporal lobes. Another key neuroimaging sign of DLB is the posterior cingulate island sign, which is characterized by sparing of the posterior cingulate cortex relative to the precuneus plus cuneus on FDG-PET.
In 2017, the Dementia with Lewy Bodies Consortium published updated recommendations on the diagnosis and management of the disease (Neurology. 2017;89[1]:88-100). In its consensus report, the consortium defines probable DLB as dementia plus two or more clinical features or one core clinical feature plus one or more indicative biomarker. These biomarkers include reduced dopamine transport uptake in basal ganglia by SPECT or PET; abnormal (low uptake) meta-iodobenzylguanidine (MIBG) myocardial scintigraphy, and/or polysomnographic confirmation of REM sleep without atonia.
“Neuropathologically, limbic with or without neocortical Lewy bodies and Lewy neurites are the defining characteristics of pathologically-proven DLB,” added Dr. Boeve, a member of the DLB consortium. “The classic DLB phenotype can occur in limbic-predominant DLB. Lewy bodies in the neocortex are not necessary to cause a dementia syndrome.”
He characterized management of DLB as “very complicated. Consider symptoms as they relate to cognitive impairment, neuropsychiatric features, motor features, sleep disorders, and autonomic dysfunction.” He often asks the patient/family to prioritize the three most troublesome issues they seek to change, and develops a plan based on their input.
There is no Food and Drug Administration–approved medication for DLB, but the standard of care is an acetylcholinesterase inhibitor such as donepezil. “There is evidence that memantine can provide a modest benefit,” Dr. Boeve said. “Hypersomnia is quite prominent in DLB and worthy of assessing and treating.” Clinicians must weigh the pros and cons of pharmacotherapy with each patient. “For example, in the atypical neuroleptic class [of drugs], there may be a benefit to the hallucinations and delusions in DLB but hypersomnia can worsen,” he said. “Selecting agents is challenging but worth the effort.”
Survival is lower and more rapid with DLB, compared with Alzheimer’s. Most people pass away from primary DLB-related features or failure to thrive. The second most common is pneumonia or aspiration. Median survival was 4 years after diagnosis in one study, and end-of life discussions occurred in less than half of all patients. “This is a frustrating reminder that we as clinicians are not very good at discussing important topics such as end-of-life care with patients and their families,” Dr. Boeve said. Resources that he recommends for education and support include the Lewy Body Dementia Association and The Lewy Body Society.
At the 2016 Alzheimer’s Disease-Related Dementias Summit, clinicians formed a list of DLB research priorities (Neurology 2017;89[23]:2381-91). Among them were recommendations to “initiate clinical trials in diverse populations using therapies that address symptoms that have the greatest effect on patient function and caregiver burden” and “identify novel common and rare genetic variants, epigenetic changes, and environmental influences that affect the risk for and clinical features of” the disease.
Meanwhile, several research protocols are under way, including the development of a DLB module by the U.S. Alzheimer’s Research Disease Centers and a number of DLB-focused projects from the National Institute of Neurological Disorders and Stroke (NINDS) Parkinson’s Disease Biomarkers Program. In addition, the Lewy Body Dementia Association Research Centers of Excellence program is focused on optimizing clinical care and setting up the infrastructure for clinical trials, while the North American Prodromal Synucleinopathy Consortium is conducting longitudinal studies in those with REM sleep behavior disorder.
Dr. Boeve disclosed that he has been an investigator for clinical trials sponsored by GE Healthcare, Axovant, and Biogen. He is a member of the scientific advisory board for the Tau Consortium and has received research support from the National Institute on Aging, the NINDS, the Mangurian Foundation, and the Little Family Foundation.
ATLANTA – In the not-so-distant past, neurologists viewed dementia with Lewy bodies as a disorder primarily of the brain, but it turned out to be far more complex than that.
At the annual meeting of the American Neurological Association, Bradley F. Boeve, MD, described dementia with Lewy bodies (DLB) as a systemic neurologic disorder affecting the brain, including brain stem, spinal cord, and peripheral nervous system, especially the autonomic nervous system. “This leads to the complex array of clinical manifestations, which are quite different from patient to patient cross-sectionally and longitudinally,” said Dr. Boeve, the Little Family Foundation Professor of Lewy Body Dementia in the department of neurology at the Mayo Clinic, Rochester, Minn.
, he said. The four core clinical features are Parkinsonism unrelated to medications; recurrent, fully-formed visual hallucinations; fluctuations in cognition and/or arousal; and rapid eye movement (REM) sleep behavior disorder. “This is the most predictive of all four features,” Dr. Boeve said. He described REM sleep behavior disorder as a parasomnia manifested by the tendency to repeatedly “act out one’s dreams.” The dreams tend to contain a chasing/attacking theme, and behaviors mirror dream content. Injuries to the patient and bed partner can occur.
Typically, patients will present with REM sleep behavior disorder in their 50s and 60s, and sometimes in their 30s and 40s, “decades before cognitive changes begin,” he said. “This is usually followed by Parkinsonism and visual hallucinations. That’s the prototypical DLB [case], but there are many examples where this is not followed. Prominent neuropsychiatric features can also begin before any cognitive changes.”
Neuropsychological features of DLB often include impairment of executive functions and visuospatial functions. “Early in the course of Alzheimer’s disease, typically performance on memory measures – especially delayed recall – are down and the other measures are borderline or mildly impaired,” Dr. Boeve noted. “By contrast, in DLB, attention, executive function, and visuospatial measures are down, but memory is often pretty good. What’s remarkable is that in the office setting, when you take a history the person often says, ‘I’m very forgetful,’ yet in the testing environment people tend to rise to the occasion pretty well.”
Imaging isn’t always helpful in establishing a diagnosis of DLB. MRI scans, for example, “can look pretty normal, including the hippocampi,” he said. “This is really the norm in DLB and it seems to be a disconnect. The person can have significant symptoms yet their MRI scan can be pretty normal.”
In Alzheimer’s disease, 18F-fluorodeoxyglucose-PET (FDG-PET) shows temporal, parietal, and frontal hypometabolism, sparing of the sensory-motor strip and sparing of the primary occipital cortex, while in DLB, FDG-PET shows marked deficits in the occipital regions with relative sparing of the frontal and temporal lobes. Another key neuroimaging sign of DLB is the posterior cingulate island sign, which is characterized by sparing of the posterior cingulate cortex relative to the precuneus plus cuneus on FDG-PET.
In 2017, the Dementia with Lewy Bodies Consortium published updated recommendations on the diagnosis and management of the disease (Neurology. 2017;89[1]:88-100). In its consensus report, the consortium defines probable DLB as dementia plus two or more clinical features or one core clinical feature plus one or more indicative biomarker. These biomarkers include reduced dopamine transport uptake in basal ganglia by SPECT or PET; abnormal (low uptake) meta-iodobenzylguanidine (MIBG) myocardial scintigraphy, and/or polysomnographic confirmation of REM sleep without atonia.
“Neuropathologically, limbic with or without neocortical Lewy bodies and Lewy neurites are the defining characteristics of pathologically-proven DLB,” added Dr. Boeve, a member of the DLB consortium. “The classic DLB phenotype can occur in limbic-predominant DLB. Lewy bodies in the neocortex are not necessary to cause a dementia syndrome.”
He characterized management of DLB as “very complicated. Consider symptoms as they relate to cognitive impairment, neuropsychiatric features, motor features, sleep disorders, and autonomic dysfunction.” He often asks the patient/family to prioritize the three most troublesome issues they seek to change, and develops a plan based on their input.
There is no Food and Drug Administration–approved medication for DLB, but the standard of care is an acetylcholinesterase inhibitor such as donepezil. “There is evidence that memantine can provide a modest benefit,” Dr. Boeve said. “Hypersomnia is quite prominent in DLB and worthy of assessing and treating.” Clinicians must weigh the pros and cons of pharmacotherapy with each patient. “For example, in the atypical neuroleptic class [of drugs], there may be a benefit to the hallucinations and delusions in DLB but hypersomnia can worsen,” he said. “Selecting agents is challenging but worth the effort.”
Survival is lower and more rapid with DLB, compared with Alzheimer’s. Most people pass away from primary DLB-related features or failure to thrive. The second most common is pneumonia or aspiration. Median survival was 4 years after diagnosis in one study, and end-of life discussions occurred in less than half of all patients. “This is a frustrating reminder that we as clinicians are not very good at discussing important topics such as end-of-life care with patients and their families,” Dr. Boeve said. Resources that he recommends for education and support include the Lewy Body Dementia Association and The Lewy Body Society.
At the 2016 Alzheimer’s Disease-Related Dementias Summit, clinicians formed a list of DLB research priorities (Neurology 2017;89[23]:2381-91). Among them were recommendations to “initiate clinical trials in diverse populations using therapies that address symptoms that have the greatest effect on patient function and caregiver burden” and “identify novel common and rare genetic variants, epigenetic changes, and environmental influences that affect the risk for and clinical features of” the disease.
Meanwhile, several research protocols are under way, including the development of a DLB module by the U.S. Alzheimer’s Research Disease Centers and a number of DLB-focused projects from the National Institute of Neurological Disorders and Stroke (NINDS) Parkinson’s Disease Biomarkers Program. In addition, the Lewy Body Dementia Association Research Centers of Excellence program is focused on optimizing clinical care and setting up the infrastructure for clinical trials, while the North American Prodromal Synucleinopathy Consortium is conducting longitudinal studies in those with REM sleep behavior disorder.
Dr. Boeve disclosed that he has been an investigator for clinical trials sponsored by GE Healthcare, Axovant, and Biogen. He is a member of the scientific advisory board for the Tau Consortium and has received research support from the National Institute on Aging, the NINDS, the Mangurian Foundation, and the Little Family Foundation.
EXPERT ANALYSIS FROM ANA 2018
Click for Credit: Short-term NSAIDs; endometriosis; more
Here are 5 articles from the November issue of Clinician Reviews (individual articles are valid for one year from date of publication—expiration dates below):
1. Short-term NSAIDs appear safe for high-risk patients
To take the posttest, go to: https://bit.ly/2PgXKGx
Expires October 8, 2019
2. Chronic liver disease raises death risk in pneumonia patients
To take the posttest, go to: https://bit.ly/2NPSXXA
Expires October 8, 2019
3. Half of outpatient antibiotics prescribed with no infectious disease code
To take the posttest, go to: https://bit.ly/2pWEWxU
Expires October 6, 2019
4. Secondary fractures in older men spike soon after first, but exercise may help
To take the posttest, go to: https://bit.ly/2OCNl8A
Expires October 3, 2019
5. Consider ART for younger endometriosis patients
To take the posttest, go to: https://bit.ly/2NO1Sc4
Expires October 5, 2019
Here are 5 articles from the November issue of Clinician Reviews (individual articles are valid for one year from date of publication—expiration dates below):
1. Short-term NSAIDs appear safe for high-risk patients
To take the posttest, go to: https://bit.ly/2PgXKGx
Expires October 8, 2019
2. Chronic liver disease raises death risk in pneumonia patients
To take the posttest, go to: https://bit.ly/2NPSXXA
Expires October 8, 2019
3. Half of outpatient antibiotics prescribed with no infectious disease code
To take the posttest, go to: https://bit.ly/2pWEWxU
Expires October 6, 2019
4. Secondary fractures in older men spike soon after first, but exercise may help
To take the posttest, go to: https://bit.ly/2OCNl8A
Expires October 3, 2019
5. Consider ART for younger endometriosis patients
To take the posttest, go to: https://bit.ly/2NO1Sc4
Expires October 5, 2019
Here are 5 articles from the November issue of Clinician Reviews (individual articles are valid for one year from date of publication—expiration dates below):
1. Short-term NSAIDs appear safe for high-risk patients
To take the posttest, go to: https://bit.ly/2PgXKGx
Expires October 8, 2019
2. Chronic liver disease raises death risk in pneumonia patients
To take the posttest, go to: https://bit.ly/2NPSXXA
Expires October 8, 2019
3. Half of outpatient antibiotics prescribed with no infectious disease code
To take the posttest, go to: https://bit.ly/2pWEWxU
Expires October 6, 2019
4. Secondary fractures in older men spike soon after first, but exercise may help
To take the posttest, go to: https://bit.ly/2OCNl8A
Expires October 3, 2019
5. Consider ART for younger endometriosis patients
To take the posttest, go to: https://bit.ly/2NO1Sc4
Expires October 5, 2019
BAN2401 subanalyses attempt to ease concerns over study design and findings
BARCELONA – Alzheimer’s disease genetic risk status didn’t influence positive findings of the investigational monoclonal antibody, BAN2401, in a post hoc analysis of a phase 2 study’s secondary endpoint, Eisai said in a report released during the Clinical Trials on Alzheimer’s Disease conference.

In fact, according to the new analysis, the positive data presented last summer probably underestimated the molecule’s benefit to homozygous carriers of the apolipoprotein E epsilon-4 (APOE4), Chad Swanson, PhD, said at the meeting.
The new data cut was based on a small fraction of the 856 patients with early Alzheimer’s disease (AD) who were enrolled in Study 201: 10 APOE4 carriers and 69 noncarriers who completed 18 months on infusions of BAN2401 at 10 mg/kg biweekly, the only beneficial dose. The subanalysis determined that carriers benefited much more than did noncarriers on the three clinical measures, said Dr. Swanson, senior director of neurology clinical research at Eisai, and the study’s director:
- The Alzheimer’s Disease Composite Score (ADCOMS), a new measure of subtle cognitive changes in early disease: Carriers, 63% less decline than placebo vs. 7% less in noncarriers.
- Alzheimer’s Disease Assessment Scale-cognitive subscale (ADAS-Cog): Carriers, 84% less decline than placebo vs. 43% less in noncarriers.
- Clinical Dementia Rating-Sum of Boxes (CDR-SB): Carriers, 60% less decline than placebo vs. a 7% worsening in noncarriers.
To strengthen the power of the analysis, Dr. Swanson pooled data from the entire 18 months of both the 10 mg/kg biweekly and the next-highest dose (10 mg/kg monthly). This increased the numbers to 273 carriers and 141 noncarriers who had at least one exposure to the antibody.
He said this corrected the APOE4 imbalance and showed a 25% slowing of ADCOMS decline in carriers, a 6% slowing in noncarriers, and a 21% slowing overall, relative to placebo.
The somewhat counterintuitive findings took even copresenter Jeffrey L. Cummings, MD, by surprise.
“I would have hypothesized a greater effect in noncarriers than carriers, but that’s the great thing about data – they challenge our assumptions,” said Dr. Cummings, director of the Center for Neurodegeneration and Translational Neuroscience and director emeritus of the Cleveland Clinic Lou Ruvo Center for Brain Health in Las Vegas and a consultant for both Eisai and Biogen, the company’s BAN2401 developmental partner. He postulated that APOE4-driven differences in plaque composition could have contributed to the observed benefit. “I think this also points to a very interesting biology that we don’t yet know. The plaque compactness is different, the distribution of diffuse plaques is different. This will bear a lot of additional analysis.”
Study 201 randomized patients with early Alzheimer’s to up to 18 months of treatment with placebo or infusions of BAN2401 at 2.5 mg/kg biweekly, 5 mg/kg monthly, 5 mg/kg biweekly, 10 mg/kg monthly, or 10 mg/kg biweekly. It had two unusual design features. First, patents were allocated to treatment arms by a Bayesian algorithm. The computer program examined results after every 50 enrollees and then allocated new subjects to what were, at that point, the most effective two doses. Additionally, the 18-month study was designed with a potential 12-month exit point, if computer modeling showed that it had at least an 80% probability of reaching at least a 25% cognitive benefit. In December, Eisai announced that the study hit just a 64% probability, missing the primary endpoint. But because Eisai felt the numbers were moving in the right direction, it continued with the additional 6 months of treatment, as allowed for in the study design, and then reanalyzed results with conventional statistics.
Thus, the successes reported at the Alzheimer’s Association International Conference in July and reanalyzed at CTAD, were secondary cognitive and functional outcomes.
Partway through the trial, a European regulatory body became alarmed at the rate of amyloid-related imaging abnormalities (ARIA) in the APOE4 carriers and excluded them from the highest-dose group. This meant that carriers comprised about 70%-80% of every unsuccessful treatment arm but just 29% of the successful one. The unbalanced randomization caused July’s skepticism. The CTAD subanalysis didn’t entirely alleviate it, and several acclaimed Alzheimer’s researchers gave it voice.
“You showed no difference in placebo rates of decline, but an increased benefit in the E4 carriers, which was really interesting,” said Reisa Sperling, MD, of Brigham and Women’s Hospital, Boston. “One difference in the E4 carriers was their rate of ARIA and how long they stayed in the study. How did you account for these issues in this analysis?”
“We didn’t specifically look at that kind of question,” Dr. Swanson said. “I think we feel that we actually see effects in both carriers and noncarriers, and we are still exploring the data and will keep looking through them to see what we can find.”
Gil Rabinovici, MD, also expressed some reservations.

“I agree that the post hoc analyses are encouraging, but I wouldn’t say they alleviate the concern when you have such a dramatic imbalance in a core feature of this disease, like APOE4 between the placebo and the high-dose group,” said Dr. Rabinovici, the Edward Fein and Pearl Landrith Endowed Professor in Memory & Aging at the University of California, San Francisco. “The people on this panel know far more than I do of the pitfalls of these kinds of analyses. We really need to see true randomization to put this issue to rest.”
Biomarkers support subanalysis finding
Dr. Swanson also presented new cerebrospinal fluid biomarker data showing changes in phosphorylated tau, neurogranin, and neurofilament light chain that support the overall 47% slowing of cognitive decline reported last summer. The 10 mg/kg biweekly and monthly groups were again combined to increase the sample size, but it remained quite small, with full data on 16 in the placebo group and 23 in the active group:
- Neurogranin, a synaptic protein that is a marker of neuronal damage, decreased by a median 58 pg/mL (11%), compared with a 13.5-pg/mL increase in the placebo group.
- Phosphorylated tau, a marker of tau pathology in the brain, decreased by a median 12 pg/mL (13%), compared with no change in the placebo group.
- Neurofilament light chain, a neuronal structural scaffold protein that is a marker of axonal degeneration, increased by a median 75 pg/mL in the active group, compared with a 156-pg/mL increase in the placebo group – a 48% difference.
The positive biomarker data bolstered the subanalysis to some extent, researchers felt. But in the end, Study 201 is just a first step for BAN2401, said Laurie Ryan, PhD, chief of the dementias of aging branch in the division of neuroscience at the National Institute on Aging.
“Today’s presentation gave us a new look at the trial data from the summer,” Dr. Ryan said in an interview. “The new analysis supports the findings previously released but is still preliminary. Nothing is definitive in a phase 2 study, so while it appears to suggest a potential positive, beneficial result, it needs further testing.”
Eisai has made its subanalysis presentation slides publicly available.
Dr. Swanson is an employee of Eisai. Dr. Cummings is a consultant for Eisai and Biogen. Dr. Sperling has consulted for numerous pharmaceutical companies. Dr. Rabinovici and Dr. Ryan have no disclosures.
SOURCE: Swanson C et al. CTAD, Symposium 3.
BARCELONA – Alzheimer’s disease genetic risk status didn’t influence positive findings of the investigational monoclonal antibody, BAN2401, in a post hoc analysis of a phase 2 study’s secondary endpoint, Eisai said in a report released during the Clinical Trials on Alzheimer’s Disease conference.
In fact, according to the new analysis, the positive data presented last summer probably underestimated the molecule’s benefit to homozygous carriers of the apolipoprotein E epsilon-4 (APOE4), Chad Swanson, PhD, said at the meeting.
The new data cut was based on a small fraction of the 856 patients with early Alzheimer’s disease (AD) who were enrolled in Study 201: 10 APOE4 carriers and 69 noncarriers who completed 18 months on infusions of BAN2401 at 10 mg/kg biweekly, the only beneficial dose. The subanalysis determined that carriers benefited much more than did noncarriers on the three clinical measures, said Dr. Swanson, senior director of neurology clinical research at Eisai, and the study’s director:
- The Alzheimer’s Disease Composite Score (ADCOMS), a new measure of subtle cognitive changes in early disease: Carriers, 63% less decline than placebo vs. 7% less in noncarriers.
- Alzheimer’s Disease Assessment Scale-cognitive subscale (ADAS-Cog): Carriers, 84% less decline than placebo vs. 43% less in noncarriers.
- Clinical Dementia Rating-Sum of Boxes (CDR-SB): Carriers, 60% less decline than placebo vs. a 7% worsening in noncarriers.
To strengthen the power of the analysis, Dr. Swanson pooled data from the entire 18 months of both the 10 mg/kg biweekly and the next-highest dose (10 mg/kg monthly). This increased the numbers to 273 carriers and 141 noncarriers who had at least one exposure to the antibody.
He said this corrected the APOE4 imbalance and showed a 25% slowing of ADCOMS decline in carriers, a 6% slowing in noncarriers, and a 21% slowing overall, relative to placebo.
The somewhat counterintuitive findings took even copresenter Jeffrey L. Cummings, MD, by surprise.
“I would have hypothesized a greater effect in noncarriers than carriers, but that’s the great thing about data – they challenge our assumptions,” said Dr. Cummings, director of the Center for Neurodegeneration and Translational Neuroscience and director emeritus of the Cleveland Clinic Lou Ruvo Center for Brain Health in Las Vegas and a consultant for both Eisai and Biogen, the company’s BAN2401 developmental partner. He postulated that APOE4-driven differences in plaque composition could have contributed to the observed benefit. “I think this also points to a very interesting biology that we don’t yet know. The plaque compactness is different, the distribution of diffuse plaques is different. This will bear a lot of additional analysis.”
Study 201 randomized patients with early Alzheimer’s to up to 18 months of treatment with placebo or infusions of BAN2401 at 2.5 mg/kg biweekly, 5 mg/kg monthly, 5 mg/kg biweekly, 10 mg/kg monthly, or 10 mg/kg biweekly. It had two unusual design features. First, patents were allocated to treatment arms by a Bayesian algorithm. The computer program examined results after every 50 enrollees and then allocated new subjects to what were, at that point, the most effective two doses. Additionally, the 18-month study was designed with a potential 12-month exit point, if computer modeling showed that it had at least an 80% probability of reaching at least a 25% cognitive benefit. In December, Eisai announced that the study hit just a 64% probability, missing the primary endpoint. But because Eisai felt the numbers were moving in the right direction, it continued with the additional 6 months of treatment, as allowed for in the study design, and then reanalyzed results with conventional statistics.
Thus, the successes reported at the Alzheimer’s Association International Conference in July and reanalyzed at CTAD, were secondary cognitive and functional outcomes.
Partway through the trial, a European regulatory body became alarmed at the rate of amyloid-related imaging abnormalities (ARIA) in the APOE4 carriers and excluded them from the highest-dose group. This meant that carriers comprised about 70%-80% of every unsuccessful treatment arm but just 29% of the successful one. The unbalanced randomization caused July’s skepticism. The CTAD subanalysis didn’t entirely alleviate it, and several acclaimed Alzheimer’s researchers gave it voice.
“You showed no difference in placebo rates of decline, but an increased benefit in the E4 carriers, which was really interesting,” said Reisa Sperling, MD, of Brigham and Women’s Hospital, Boston. “One difference in the E4 carriers was their rate of ARIA and how long they stayed in the study. How did you account for these issues in this analysis?”
“We didn’t specifically look at that kind of question,” Dr. Swanson said. “I think we feel that we actually see effects in both carriers and noncarriers, and we are still exploring the data and will keep looking through them to see what we can find.”
Gil Rabinovici, MD, also expressed some reservations.
“I agree that the post hoc analyses are encouraging, but I wouldn’t say they alleviate the concern when you have such a dramatic imbalance in a core feature of this disease, like APOE4 between the placebo and the high-dose group,” said Dr. Rabinovici, the Edward Fein and Pearl Landrith Endowed Professor in Memory & Aging at the University of California, San Francisco. “The people on this panel know far more than I do of the pitfalls of these kinds of analyses. We really need to see true randomization to put this issue to rest.”
Biomarkers support subanalysis finding
Dr. Swanson also presented new cerebrospinal fluid biomarker data showing changes in phosphorylated tau, neurogranin, and neurofilament light chain that support the overall 47% slowing of cognitive decline reported last summer. The 10 mg/kg biweekly and monthly groups were again combined to increase the sample size, but it remained quite small, with full data on 16 in the placebo group and 23 in the active group:
- Neurogranin, a synaptic protein that is a marker of neuronal damage, decreased by a median 58 pg/mL (11%), compared with a 13.5-pg/mL increase in the placebo group.
- Phosphorylated tau, a marker of tau pathology in the brain, decreased by a median 12 pg/mL (13%), compared with no change in the placebo group.
- Neurofilament light chain, a neuronal structural scaffold protein that is a marker of axonal degeneration, increased by a median 75 pg/mL in the active group, compared with a 156-pg/mL increase in the placebo group – a 48% difference.
The positive biomarker data bolstered the subanalysis to some extent, researchers felt. But in the end, Study 201 is just a first step for BAN2401, said Laurie Ryan, PhD, chief of the dementias of aging branch in the division of neuroscience at the National Institute on Aging.
“Today’s presentation gave us a new look at the trial data from the summer,” Dr. Ryan said in an interview. “The new analysis supports the findings previously released but is still preliminary. Nothing is definitive in a phase 2 study, so while it appears to suggest a potential positive, beneficial result, it needs further testing.”
Eisai has made its subanalysis presentation slides publicly available.
Dr. Swanson is an employee of Eisai. Dr. Cummings is a consultant for Eisai and Biogen. Dr. Sperling has consulted for numerous pharmaceutical companies. Dr. Rabinovici and Dr. Ryan have no disclosures.
SOURCE: Swanson C et al. CTAD, Symposium 3.
BARCELONA – Alzheimer’s disease genetic risk status didn’t influence positive findings of the investigational monoclonal antibody, BAN2401, in a post hoc analysis of a phase 2 study’s secondary endpoint, Eisai said in a report released during the Clinical Trials on Alzheimer’s Disease conference.
In fact, according to the new analysis, the positive data presented last summer probably underestimated the molecule’s benefit to homozygous carriers of the apolipoprotein E epsilon-4 (APOE4), Chad Swanson, PhD, said at the meeting.
The new data cut was based on a small fraction of the 856 patients with early Alzheimer’s disease (AD) who were enrolled in Study 201: 10 APOE4 carriers and 69 noncarriers who completed 18 months on infusions of BAN2401 at 10 mg/kg biweekly, the only beneficial dose. The subanalysis determined that carriers benefited much more than did noncarriers on the three clinical measures, said Dr. Swanson, senior director of neurology clinical research at Eisai, and the study’s director:
- The Alzheimer’s Disease Composite Score (ADCOMS), a new measure of subtle cognitive changes in early disease: Carriers, 63% less decline than placebo vs. 7% less in noncarriers.
- Alzheimer’s Disease Assessment Scale-cognitive subscale (ADAS-Cog): Carriers, 84% less decline than placebo vs. 43% less in noncarriers.
- Clinical Dementia Rating-Sum of Boxes (CDR-SB): Carriers, 60% less decline than placebo vs. a 7% worsening in noncarriers.
To strengthen the power of the analysis, Dr. Swanson pooled data from the entire 18 months of both the 10 mg/kg biweekly and the next-highest dose (10 mg/kg monthly). This increased the numbers to 273 carriers and 141 noncarriers who had at least one exposure to the antibody.
He said this corrected the APOE4 imbalance and showed a 25% slowing of ADCOMS decline in carriers, a 6% slowing in noncarriers, and a 21% slowing overall, relative to placebo.
The somewhat counterintuitive findings took even copresenter Jeffrey L. Cummings, MD, by surprise.
“I would have hypothesized a greater effect in noncarriers than carriers, but that’s the great thing about data – they challenge our assumptions,” said Dr. Cummings, director of the Center for Neurodegeneration and Translational Neuroscience and director emeritus of the Cleveland Clinic Lou Ruvo Center for Brain Health in Las Vegas and a consultant for both Eisai and Biogen, the company’s BAN2401 developmental partner. He postulated that APOE4-driven differences in plaque composition could have contributed to the observed benefit. “I think this also points to a very interesting biology that we don’t yet know. The plaque compactness is different, the distribution of diffuse plaques is different. This will bear a lot of additional analysis.”
Study 201 randomized patients with early Alzheimer’s to up to 18 months of treatment with placebo or infusions of BAN2401 at 2.5 mg/kg biweekly, 5 mg/kg monthly, 5 mg/kg biweekly, 10 mg/kg monthly, or 10 mg/kg biweekly. It had two unusual design features. First, patents were allocated to treatment arms by a Bayesian algorithm. The computer program examined results after every 50 enrollees and then allocated new subjects to what were, at that point, the most effective two doses. Additionally, the 18-month study was designed with a potential 12-month exit point, if computer modeling showed that it had at least an 80% probability of reaching at least a 25% cognitive benefit. In December, Eisai announced that the study hit just a 64% probability, missing the primary endpoint. But because Eisai felt the numbers were moving in the right direction, it continued with the additional 6 months of treatment, as allowed for in the study design, and then reanalyzed results with conventional statistics.
Thus, the successes reported at the Alzheimer’s Association International Conference in July and reanalyzed at CTAD, were secondary cognitive and functional outcomes.
Partway through the trial, a European regulatory body became alarmed at the rate of amyloid-related imaging abnormalities (ARIA) in the APOE4 carriers and excluded them from the highest-dose group. This meant that carriers comprised about 70%-80% of every unsuccessful treatment arm but just 29% of the successful one. The unbalanced randomization caused July’s skepticism. The CTAD subanalysis didn’t entirely alleviate it, and several acclaimed Alzheimer’s researchers gave it voice.
“You showed no difference in placebo rates of decline, but an increased benefit in the E4 carriers, which was really interesting,” said Reisa Sperling, MD, of Brigham and Women’s Hospital, Boston. “One difference in the E4 carriers was their rate of ARIA and how long they stayed in the study. How did you account for these issues in this analysis?”
“We didn’t specifically look at that kind of question,” Dr. Swanson said. “I think we feel that we actually see effects in both carriers and noncarriers, and we are still exploring the data and will keep looking through them to see what we can find.”
Gil Rabinovici, MD, also expressed some reservations.
“I agree that the post hoc analyses are encouraging, but I wouldn’t say they alleviate the concern when you have such a dramatic imbalance in a core feature of this disease, like APOE4 between the placebo and the high-dose group,” said Dr. Rabinovici, the Edward Fein and Pearl Landrith Endowed Professor in Memory & Aging at the University of California, San Francisco. “The people on this panel know far more than I do of the pitfalls of these kinds of analyses. We really need to see true randomization to put this issue to rest.”
Biomarkers support subanalysis finding
Dr. Swanson also presented new cerebrospinal fluid biomarker data showing changes in phosphorylated tau, neurogranin, and neurofilament light chain that support the overall 47% slowing of cognitive decline reported last summer. The 10 mg/kg biweekly and monthly groups were again combined to increase the sample size, but it remained quite small, with full data on 16 in the placebo group and 23 in the active group:
- Neurogranin, a synaptic protein that is a marker of neuronal damage, decreased by a median 58 pg/mL (11%), compared with a 13.5-pg/mL increase in the placebo group.
- Phosphorylated tau, a marker of tau pathology in the brain, decreased by a median 12 pg/mL (13%), compared with no change in the placebo group.
- Neurofilament light chain, a neuronal structural scaffold protein that is a marker of axonal degeneration, increased by a median 75 pg/mL in the active group, compared with a 156-pg/mL increase in the placebo group – a 48% difference.
The positive biomarker data bolstered the subanalysis to some extent, researchers felt. But in the end, Study 201 is just a first step for BAN2401, said Laurie Ryan, PhD, chief of the dementias of aging branch in the division of neuroscience at the National Institute on Aging.
“Today’s presentation gave us a new look at the trial data from the summer,” Dr. Ryan said in an interview. “The new analysis supports the findings previously released but is still preliminary. Nothing is definitive in a phase 2 study, so while it appears to suggest a potential positive, beneficial result, it needs further testing.”
Eisai has made its subanalysis presentation slides publicly available.
Dr. Swanson is an employee of Eisai. Dr. Cummings is a consultant for Eisai and Biogen. Dr. Sperling has consulted for numerous pharmaceutical companies. Dr. Rabinovici and Dr. Ryan have no disclosures.
SOURCE: Swanson C et al. CTAD, Symposium 3.
REPORTING FROM CTAD
Statins cut all-cause mortality in spinal cord injury
ATLANTA – Statin use among a cohort of veterans with traumatic spinal cord injury reduced all-cause mortality, results from a novel observational study showed.

“This is the first clinical study to show that administration of statins irrespective of the lipid levels reduces all-cause mortality, not just cardiovascular mortality,” lead study author Meheroz H. Rabadi, MD, said in an interview in advance of the annual meeting of the American Neurological Association. “This clinical study confirms the impression of several prior studies in animal models with spinal cord injury, which have shown the anti-inflammatory and neuro-protective effects of statins.”
To determine whether statin use in a cohort of patients with traumatic spinal cord injuries (SCI) reduced overall and cause-specific mortality, Dr. Rabadi and his colleagues retrospectively reviewed the medical charts and death records of 163 individuals with SCI who were treated at the Oklahoma City Veterans Administration Medical Center Spinal Cord Injury & Disease, Multiple Sclerosis, and ALS Program, an outpatient clinic, from 2000 to 2014. They collected data on statin use, duration of statin use, and intensity of statin therapy, as well as cause-specific mortality.
Of the 163 subjects studied, 75 (46%) had taken statins for an average of 5.7 years, and had greater cardiovascular risk burdens than those who had not taken statins. The mortality rate for patients on statins, however, was 33.8-49.9 per 1,000 person-years, compared with 47.4-66.8 deaths per 1,000 person-years among those who had not taken statins. Kaplan-Meier survival curves showed a significant difference between the two groups (P less than .0052). Within the statin group, neither duration nor average intensity of statin therapy affected mortality.
“We were surprised to note statins reduced pneumonia-related mortality in patients with SCI,” Dr. Rabadi said. “Since our publication there have been several publications, including a meta-analysis of statins reducing community-acquired pneumonia-related mortality and reducing the need for mechanical ventilation or ICU admission (see CHEST 2015;148:523-32, Clin Med (Lond) 2017;17(5):403-7, and Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2018;40(1):30-40). Another surprise was neither the intensity, duration, or types of statin affected the result.”
He acknowledged certain limitations of the analysis, including its retrospective design, its relatively small sample size, and the fact that most of the subjects were non-Hispanic white men. “Routine prescription of statins in any dose in patients with SCI – even if the lipid profile is normal – is more beneficial than detrimental over the long haul,” concluded Dr. Rabadi, who also directs the Oklahoma VAMC Stroke Program. “Nearly all our patients with SCI continue to be on varying doses of statins.”
Dr. Rabadi reported having no financial disclosures.
dbrunk@mdedge.com
SOURCE: Ann Neurol. 2018;84[S22]:S127. Abstract S302.
ATLANTA – Statin use among a cohort of veterans with traumatic spinal cord injury reduced all-cause mortality, results from a novel observational study showed.
“This is the first clinical study to show that administration of statins irrespective of the lipid levels reduces all-cause mortality, not just cardiovascular mortality,” lead study author Meheroz H. Rabadi, MD, said in an interview in advance of the annual meeting of the American Neurological Association. “This clinical study confirms the impression of several prior studies in animal models with spinal cord injury, which have shown the anti-inflammatory and neuro-protective effects of statins.”
To determine whether statin use in a cohort of patients with traumatic spinal cord injuries (SCI) reduced overall and cause-specific mortality, Dr. Rabadi and his colleagues retrospectively reviewed the medical charts and death records of 163 individuals with SCI who were treated at the Oklahoma City Veterans Administration Medical Center Spinal Cord Injury & Disease, Multiple Sclerosis, and ALS Program, an outpatient clinic, from 2000 to 2014. They collected data on statin use, duration of statin use, and intensity of statin therapy, as well as cause-specific mortality.
Of the 163 subjects studied, 75 (46%) had taken statins for an average of 5.7 years, and had greater cardiovascular risk burdens than those who had not taken statins. The mortality rate for patients on statins, however, was 33.8-49.9 per 1,000 person-years, compared with 47.4-66.8 deaths per 1,000 person-years among those who had not taken statins. Kaplan-Meier survival curves showed a significant difference between the two groups (P less than .0052). Within the statin group, neither duration nor average intensity of statin therapy affected mortality.
“We were surprised to note statins reduced pneumonia-related mortality in patients with SCI,” Dr. Rabadi said. “Since our publication there have been several publications, including a meta-analysis of statins reducing community-acquired pneumonia-related mortality and reducing the need for mechanical ventilation or ICU admission (see CHEST 2015;148:523-32, Clin Med (Lond) 2017;17(5):403-7, and Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2018;40(1):30-40). Another surprise was neither the intensity, duration, or types of statin affected the result.”
He acknowledged certain limitations of the analysis, including its retrospective design, its relatively small sample size, and the fact that most of the subjects were non-Hispanic white men. “Routine prescription of statins in any dose in patients with SCI – even if the lipid profile is normal – is more beneficial than detrimental over the long haul,” concluded Dr. Rabadi, who also directs the Oklahoma VAMC Stroke Program. “Nearly all our patients with SCI continue to be on varying doses of statins.”
Dr. Rabadi reported having no financial disclosures.
dbrunk@mdedge.com
SOURCE: Ann Neurol. 2018;84[S22]:S127. Abstract S302.
ATLANTA – Statin use among a cohort of veterans with traumatic spinal cord injury reduced all-cause mortality, results from a novel observational study showed.
“This is the first clinical study to show that administration of statins irrespective of the lipid levels reduces all-cause mortality, not just cardiovascular mortality,” lead study author Meheroz H. Rabadi, MD, said in an interview in advance of the annual meeting of the American Neurological Association. “This clinical study confirms the impression of several prior studies in animal models with spinal cord injury, which have shown the anti-inflammatory and neuro-protective effects of statins.”
To determine whether statin use in a cohort of patients with traumatic spinal cord injuries (SCI) reduced overall and cause-specific mortality, Dr. Rabadi and his colleagues retrospectively reviewed the medical charts and death records of 163 individuals with SCI who were treated at the Oklahoma City Veterans Administration Medical Center Spinal Cord Injury & Disease, Multiple Sclerosis, and ALS Program, an outpatient clinic, from 2000 to 2014. They collected data on statin use, duration of statin use, and intensity of statin therapy, as well as cause-specific mortality.
Of the 163 subjects studied, 75 (46%) had taken statins for an average of 5.7 years, and had greater cardiovascular risk burdens than those who had not taken statins. The mortality rate for patients on statins, however, was 33.8-49.9 per 1,000 person-years, compared with 47.4-66.8 deaths per 1,000 person-years among those who had not taken statins. Kaplan-Meier survival curves showed a significant difference between the two groups (P less than .0052). Within the statin group, neither duration nor average intensity of statin therapy affected mortality.
“We were surprised to note statins reduced pneumonia-related mortality in patients with SCI,” Dr. Rabadi said. “Since our publication there have been several publications, including a meta-analysis of statins reducing community-acquired pneumonia-related mortality and reducing the need for mechanical ventilation or ICU admission (see CHEST 2015;148:523-32, Clin Med (Lond) 2017;17(5):403-7, and Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2018;40(1):30-40). Another surprise was neither the intensity, duration, or types of statin affected the result.”
He acknowledged certain limitations of the analysis, including its retrospective design, its relatively small sample size, and the fact that most of the subjects were non-Hispanic white men. “Routine prescription of statins in any dose in patients with SCI – even if the lipid profile is normal – is more beneficial than detrimental over the long haul,” concluded Dr. Rabadi, who also directs the Oklahoma VAMC Stroke Program. “Nearly all our patients with SCI continue to be on varying doses of statins.”
Dr. Rabadi reported having no financial disclosures.
dbrunk@mdedge.com
SOURCE: Ann Neurol. 2018;84[S22]:S127. Abstract S302.
AT ANA 2018
Key clinical point:
Major finding: The mortality rate for patients on statins was 33.8-49.9 per 1,000 person-years, compared with 47.4-66.8 deaths per 1,000 person-years among those who had not taken statins (P less than .0052).
Study details: A retrospective review of 163 individuals with traumatic spinal cord injuries.
Disclosures: Dr. Rabadi reported having no financial disclosures.
Source: Ann Neurol. 2018;84[S22]:S127. Abstract S302.