User login
Chemo-free induction regimen shines in MCL
SAN DIEGO – A chemotherapy-free induction regimen of ibrutinib and rituximab was well tolerated and achieved an overall response rate of 100% among patients with newly diagnosed mantle cell lymphoma (MCL), according to results of a small single-center phase II trial.
A total of 72% of patients had complete responses to induction, while 28% had partial responses, and 100% had complete responses to consolidation, reported Michael Wang, MD, at the annual meeting of the American Society of Hematology. The findings highlight a “window of opportunity” to effectively treat de novo MCL in young, fit patients while potentially sparing them from repeated cycles of intensive chemoimmunotherapy, the investigators noted.
Established treatments for MCL include eight cycles of rituximab–hyper-CVAD (hyper-fractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone) alternating with rituximab–methotrexate–Ara-C (cytarabine). Median overall survival with this regimen exceeds 10 years, but during that decade, more than 6% of patients will develop myeloid neoplasms related to treatment, noted Dr. Wang of the University of Texas MD Anderson Cancer Center in Houston.
For the study, patients up to 65 years old with newly diagnosed, untreated MCL underwent induction with continuous daily ibrutinib (560 mg), plus rituximab (375 mg/m2) administered weekly for 4 weeks during cycle 1 and on day 1 of cycles 3-12. Consolidation consisted of rituximab plus hyper-CVAD, alternating every 28 days with rituximab plus high-dose methotrexate–Ara-C. Complete responders to induction received four cycles of chemoimmunotherapy, while progressors and partial responders received chemoimmunotherapy for two cycles beyond the point of complete remission.
Among 36 evaluable patients, 28% had a partial response and the rest had complete responses to induction. Moreover, all 19 patients who completed consolidation had a complete response. During induction, the most common toxicities were fatigue, diarrhea, rash, and myalgia, nearly all of which were mild or moderate in severity. During consolidation, two patients developed severe neutropenia, one patient developed severe febrile neutropenia, and one developed a severe increase in liver enzymes. There were no treatment-related deaths.
“The toxicity after intensive immune-chemotherapy in shortened cycles [is] much improved compared to historical controls, but longer follow-up is needed,” the researchers wrote. “This unprecedented efficacy and safety may provide a window of opportunity for less chemoimmunotherapy needed for consolidation.”
Dr. Wang disclosed ties to Janssen, Onyx, BeiGene, Kite Pharma, Asana Biosciences, Juno Therapeutics, Acerta Pharma, and Celgene.
SAN DIEGO – A chemotherapy-free induction regimen of ibrutinib and rituximab was well tolerated and achieved an overall response rate of 100% among patients with newly diagnosed mantle cell lymphoma (MCL), according to results of a small single-center phase II trial.
A total of 72% of patients had complete responses to induction, while 28% had partial responses, and 100% had complete responses to consolidation, reported Michael Wang, MD, at the annual meeting of the American Society of Hematology. The findings highlight a “window of opportunity” to effectively treat de novo MCL in young, fit patients while potentially sparing them from repeated cycles of intensive chemoimmunotherapy, the investigators noted.
Established treatments for MCL include eight cycles of rituximab–hyper-CVAD (hyper-fractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone) alternating with rituximab–methotrexate–Ara-C (cytarabine). Median overall survival with this regimen exceeds 10 years, but during that decade, more than 6% of patients will develop myeloid neoplasms related to treatment, noted Dr. Wang of the University of Texas MD Anderson Cancer Center in Houston.
For the study, patients up to 65 years old with newly diagnosed, untreated MCL underwent induction with continuous daily ibrutinib (560 mg), plus rituximab (375 mg/m2) administered weekly for 4 weeks during cycle 1 and on day 1 of cycles 3-12. Consolidation consisted of rituximab plus hyper-CVAD, alternating every 28 days with rituximab plus high-dose methotrexate–Ara-C. Complete responders to induction received four cycles of chemoimmunotherapy, while progressors and partial responders received chemoimmunotherapy for two cycles beyond the point of complete remission.
Among 36 evaluable patients, 28% had a partial response and the rest had complete responses to induction. Moreover, all 19 patients who completed consolidation had a complete response. During induction, the most common toxicities were fatigue, diarrhea, rash, and myalgia, nearly all of which were mild or moderate in severity. During consolidation, two patients developed severe neutropenia, one patient developed severe febrile neutropenia, and one developed a severe increase in liver enzymes. There were no treatment-related deaths.
“The toxicity after intensive immune-chemotherapy in shortened cycles [is] much improved compared to historical controls, but longer follow-up is needed,” the researchers wrote. “This unprecedented efficacy and safety may provide a window of opportunity for less chemoimmunotherapy needed for consolidation.”
Dr. Wang disclosed ties to Janssen, Onyx, BeiGene, Kite Pharma, Asana Biosciences, Juno Therapeutics, Acerta Pharma, and Celgene.
SAN DIEGO – A chemotherapy-free induction regimen of ibrutinib and rituximab was well tolerated and achieved an overall response rate of 100% among patients with newly diagnosed mantle cell lymphoma (MCL), according to results of a small single-center phase II trial.
A total of 72% of patients had complete responses to induction, while 28% had partial responses, and 100% had complete responses to consolidation, reported Michael Wang, MD, at the annual meeting of the American Society of Hematology. The findings highlight a “window of opportunity” to effectively treat de novo MCL in young, fit patients while potentially sparing them from repeated cycles of intensive chemoimmunotherapy, the investigators noted.
Established treatments for MCL include eight cycles of rituximab–hyper-CVAD (hyper-fractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone) alternating with rituximab–methotrexate–Ara-C (cytarabine). Median overall survival with this regimen exceeds 10 years, but during that decade, more than 6% of patients will develop myeloid neoplasms related to treatment, noted Dr. Wang of the University of Texas MD Anderson Cancer Center in Houston.
For the study, patients up to 65 years old with newly diagnosed, untreated MCL underwent induction with continuous daily ibrutinib (560 mg), plus rituximab (375 mg/m2) administered weekly for 4 weeks during cycle 1 and on day 1 of cycles 3-12. Consolidation consisted of rituximab plus hyper-CVAD, alternating every 28 days with rituximab plus high-dose methotrexate–Ara-C. Complete responders to induction received four cycles of chemoimmunotherapy, while progressors and partial responders received chemoimmunotherapy for two cycles beyond the point of complete remission.
Among 36 evaluable patients, 28% had a partial response and the rest had complete responses to induction. Moreover, all 19 patients who completed consolidation had a complete response. During induction, the most common toxicities were fatigue, diarrhea, rash, and myalgia, nearly all of which were mild or moderate in severity. During consolidation, two patients developed severe neutropenia, one patient developed severe febrile neutropenia, and one developed a severe increase in liver enzymes. There were no treatment-related deaths.
“The toxicity after intensive immune-chemotherapy in shortened cycles [is] much improved compared to historical controls, but longer follow-up is needed,” the researchers wrote. “This unprecedented efficacy and safety may provide a window of opportunity for less chemoimmunotherapy needed for consolidation.”
Dr. Wang disclosed ties to Janssen, Onyx, BeiGene, Kite Pharma, Asana Biosciences, Juno Therapeutics, Acerta Pharma, and Celgene.
AT ASH 2016
Key clinical point: Induction with ibrutinib and rituximab achieved a 100% response rate in newly diagnosed mantle cell lymphoma, enabling patients to receive less intensive consolidation.
Major finding: Rates of complete response were 72% for induction and 100% for induction plus consolidation.
Data source: A single-center phase II trial of 36 patients with newly diagnosed, untreated mantle cell lymphoma.
Disclosures: Dr. Wang disclosed ties to Janssen, Onyx, BeiGene, Kite Pharma, Asana Biosciences, Juno Therapeutics, Acerta Pharma, and Celgene.
Rituximab vanquished MRD in mantle cell lymphoma
SAN DIEGO – Rituximab can at least temporarily vanquish minimal residual disease (MRD) in mantle cell lymphoma (MCL) patients who relapse after induction therapy and autologous stem cell transplantation (ASCT), researchers reported at the annual meeting of the American Society of Hematology.
Of 58 patients whose MCL relapsed after induction therapy and ASCT, 82% converted back to an MRD-negative state after receiving 4 weekly doses of rituximab (375 mg/m2), Arne Kolstad, MD, PhD, and his associates. The data “strongly suggest that preemptive rituximab treatment delayed clinical relapse in MCL,” they wrote in their abstract. They recommended molecular and clinical monitoring after ASCT, not only “as an alternative to maintenance therapy for all MCL patients” but to identify MRD-positive candidates for clinical trials.
The study was an analysis of the Nordic Lymphoma Group phase II MCL2 and MCL3 trials (NTC 00514475), in which patients received six alternating cycles of R-CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone plus rituximab) and R-Ara-C (rituximab-cytarabine). followed by high-dose ASCT. In MCL3, responders who fell short of complete remission also received intensification with yttium-90 ibritumomab tiuxetan (0.4 mCi/kg) 1 week before treatment with BEAM/C (carmustine, etoposide, cytarabine, and melphalan or cyclophosphamide). Patients were evaluated 2-3 months after completing ASCT, and then every 6 months for 5 years or until relapse. Survivors were followed for a median of 8.5 years, noted Dr. Kolstad, who is with Oslo University Hospital in Norway.
Among 183 patients who underwent polymerase chain reaction–based testing for markers of MRD, median time to molecular relapse was 55 months. However, the relapse-free survival curve did not plateau – patients in all risk groups continued to relapse 5-10 years after undergoing ASCT, the researchers said. “Hence, it is fair to consider MCL as a chronic incurable lymphoma entity, and novel approaches will be necessary to change the natural course of this disease,” they wrote.
After controlling for potential confounders, significant predictors of molecular relapse included high MCL international prognostic index at diagnosis (hazard ratio, 1.9; 95% confidence interval 1.4-2.7; P = .0001) and detection of MRD before patients underwent ASCT (HR, 2.5; 95% CI, 1.5-4.1; P = .0005). Minimal residual disease predicted clinical relapse and shorter survival (P less than .001 for both associations). In contrast, the 86 patients who remained in continuous molecular remission had a 76% chance of having at least a 10-year clinical remission, the investigators said.
Minimal residual disease was assessed by testing bone marrow and blood samples with combined standard nested and quantitative real-time polymerase chain reaction (PCR) for Bcl-1 or IgH rearrangement. They defined molecular relapse as conversion from a negative to a positive result on standard nested PCR, or, for patients who were MRD positive after ASCT, as a more than fivefold rise in real-time quantitative PCR levels in two consecutive bone marrow samples.
Oslo University sponsored the trials. Dr. Kolstad reported ties to Nordic Nanovector, Bayer Schering Pharma, Merck, and Roche.
SAN DIEGO – Rituximab can at least temporarily vanquish minimal residual disease (MRD) in mantle cell lymphoma (MCL) patients who relapse after induction therapy and autologous stem cell transplantation (ASCT), researchers reported at the annual meeting of the American Society of Hematology.
Of 58 patients whose MCL relapsed after induction therapy and ASCT, 82% converted back to an MRD-negative state after receiving 4 weekly doses of rituximab (375 mg/m2), Arne Kolstad, MD, PhD, and his associates. The data “strongly suggest that preemptive rituximab treatment delayed clinical relapse in MCL,” they wrote in their abstract. They recommended molecular and clinical monitoring after ASCT, not only “as an alternative to maintenance therapy for all MCL patients” but to identify MRD-positive candidates for clinical trials.
The study was an analysis of the Nordic Lymphoma Group phase II MCL2 and MCL3 trials (NTC 00514475), in which patients received six alternating cycles of R-CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone plus rituximab) and R-Ara-C (rituximab-cytarabine). followed by high-dose ASCT. In MCL3, responders who fell short of complete remission also received intensification with yttium-90 ibritumomab tiuxetan (0.4 mCi/kg) 1 week before treatment with BEAM/C (carmustine, etoposide, cytarabine, and melphalan or cyclophosphamide). Patients were evaluated 2-3 months after completing ASCT, and then every 6 months for 5 years or until relapse. Survivors were followed for a median of 8.5 years, noted Dr. Kolstad, who is with Oslo University Hospital in Norway.
Among 183 patients who underwent polymerase chain reaction–based testing for markers of MRD, median time to molecular relapse was 55 months. However, the relapse-free survival curve did not plateau – patients in all risk groups continued to relapse 5-10 years after undergoing ASCT, the researchers said. “Hence, it is fair to consider MCL as a chronic incurable lymphoma entity, and novel approaches will be necessary to change the natural course of this disease,” they wrote.
After controlling for potential confounders, significant predictors of molecular relapse included high MCL international prognostic index at diagnosis (hazard ratio, 1.9; 95% confidence interval 1.4-2.7; P = .0001) and detection of MRD before patients underwent ASCT (HR, 2.5; 95% CI, 1.5-4.1; P = .0005). Minimal residual disease predicted clinical relapse and shorter survival (P less than .001 for both associations). In contrast, the 86 patients who remained in continuous molecular remission had a 76% chance of having at least a 10-year clinical remission, the investigators said.
Minimal residual disease was assessed by testing bone marrow and blood samples with combined standard nested and quantitative real-time polymerase chain reaction (PCR) for Bcl-1 or IgH rearrangement. They defined molecular relapse as conversion from a negative to a positive result on standard nested PCR, or, for patients who were MRD positive after ASCT, as a more than fivefold rise in real-time quantitative PCR levels in two consecutive bone marrow samples.
Oslo University sponsored the trials. Dr. Kolstad reported ties to Nordic Nanovector, Bayer Schering Pharma, Merck, and Roche.
SAN DIEGO – Rituximab can at least temporarily vanquish minimal residual disease (MRD) in mantle cell lymphoma (MCL) patients who relapse after induction therapy and autologous stem cell transplantation (ASCT), researchers reported at the annual meeting of the American Society of Hematology.
Of 58 patients whose MCL relapsed after induction therapy and ASCT, 82% converted back to an MRD-negative state after receiving 4 weekly doses of rituximab (375 mg/m2), Arne Kolstad, MD, PhD, and his associates. The data “strongly suggest that preemptive rituximab treatment delayed clinical relapse in MCL,” they wrote in their abstract. They recommended molecular and clinical monitoring after ASCT, not only “as an alternative to maintenance therapy for all MCL patients” but to identify MRD-positive candidates for clinical trials.
The study was an analysis of the Nordic Lymphoma Group phase II MCL2 and MCL3 trials (NTC 00514475), in which patients received six alternating cycles of R-CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone plus rituximab) and R-Ara-C (rituximab-cytarabine). followed by high-dose ASCT. In MCL3, responders who fell short of complete remission also received intensification with yttium-90 ibritumomab tiuxetan (0.4 mCi/kg) 1 week before treatment with BEAM/C (carmustine, etoposide, cytarabine, and melphalan or cyclophosphamide). Patients were evaluated 2-3 months after completing ASCT, and then every 6 months for 5 years or until relapse. Survivors were followed for a median of 8.5 years, noted Dr. Kolstad, who is with Oslo University Hospital in Norway.
Among 183 patients who underwent polymerase chain reaction–based testing for markers of MRD, median time to molecular relapse was 55 months. However, the relapse-free survival curve did not plateau – patients in all risk groups continued to relapse 5-10 years after undergoing ASCT, the researchers said. “Hence, it is fair to consider MCL as a chronic incurable lymphoma entity, and novel approaches will be necessary to change the natural course of this disease,” they wrote.
After controlling for potential confounders, significant predictors of molecular relapse included high MCL international prognostic index at diagnosis (hazard ratio, 1.9; 95% confidence interval 1.4-2.7; P = .0001) and detection of MRD before patients underwent ASCT (HR, 2.5; 95% CI, 1.5-4.1; P = .0005). Minimal residual disease predicted clinical relapse and shorter survival (P less than .001 for both associations). In contrast, the 86 patients who remained in continuous molecular remission had a 76% chance of having at least a 10-year clinical remission, the investigators said.
Minimal residual disease was assessed by testing bone marrow and blood samples with combined standard nested and quantitative real-time polymerase chain reaction (PCR) for Bcl-1 or IgH rearrangement. They defined molecular relapse as conversion from a negative to a positive result on standard nested PCR, or, for patients who were MRD positive after ASCT, as a more than fivefold rise in real-time quantitative PCR levels in two consecutive bone marrow samples.
Oslo University sponsored the trials. Dr. Kolstad reported ties to Nordic Nanovector, Bayer Schering Pharma, Merck, and Roche.
AT ASH 2016
Key clinical point:
Major finding: Among 58 patients who relapsed after induction therapy and autologous stem cell transplantation, 82% converted back to an MRD-negative state with 4 weekly doses of rituximab (375 mg/m2).
Data source: A study of 183 patients with mantle cell lymphoma from the Nordic MCL2 and MCL3 trials.
Disclosures: Oslo University sponsored the trials. Dr. Kolstad reported ties to Nordic Nanovector, Bayer Schering Pharma, Merck, and Roche.
FDA issues CRL for IV formulation of antiemetic agent

chemotherapy
Photo by Rhoda Baer
The US Food and Drug Administration (FDA) has issued a complete response letter (CRL) regarding the new drug application (NDA) for an intravenous (IV) formulation of rolapitant.
An oral formulation of rolapitant, marketed as VARUBI®, is FDA-approved for use in combination with other antiemetic agents to prevent delayed nausea and vomiting associated with initial and repeat courses of emetogenic cancer chemotherapy in adults.
The NDA for rolapitant IV is for the same indication.
The FDA requested additional information regarding the in vitro method utilized to demonstrate comparability of drug product produced at the 2 proposed commercial manufacturers for rolapitant IV that were included in the NDA.
TESARO Inc., the company developing rolapitant IV, said it is working to provide the requested information.
The CRL did not identify concerns related to the safety or efficacy of rolapitant IV or request additional clinical studies. No concerns were raised regarding the active pharmaceutical ingredient, which is also used for VARUBI®.
TESARO identified potential deficiencies at the original contract manufacturer for rolapitant IV, secured a second drug product supplier, and included data from this manufacturer in the NDA.
During the NDA review, the FDA requested and TESARO provided in vitro data to demonstrate comparability of drug product made at the 2 manufacturing sites.
“TESARO is committed to bringing this new intravenous formulation of rolapitant to physicians and patients to enable additional flexibility and choice of antiemetic regimens, and we plan to address FDA’s questions expeditiously and complete this application, which we expect to enable approval in the first half of 2017,” said Mary Lynne Hedley, PhD, president and chief operating officer of TESARO. ![]()
chemotherapy
Photo by Rhoda Baer
The US Food and Drug Administration (FDA) has issued a complete response letter (CRL) regarding the new drug application (NDA) for an intravenous (IV) formulation of rolapitant.
An oral formulation of rolapitant, marketed as VARUBI®, is FDA-approved for use in combination with other antiemetic agents to prevent delayed nausea and vomiting associated with initial and repeat courses of emetogenic cancer chemotherapy in adults.
The NDA for rolapitant IV is for the same indication.
The FDA requested additional information regarding the in vitro method utilized to demonstrate comparability of drug product produced at the 2 proposed commercial manufacturers for rolapitant IV that were included in the NDA.
TESARO Inc., the company developing rolapitant IV, said it is working to provide the requested information.
The CRL did not identify concerns related to the safety or efficacy of rolapitant IV or request additional clinical studies. No concerns were raised regarding the active pharmaceutical ingredient, which is also used for VARUBI®.
TESARO identified potential deficiencies at the original contract manufacturer for rolapitant IV, secured a second drug product supplier, and included data from this manufacturer in the NDA.
During the NDA review, the FDA requested and TESARO provided in vitro data to demonstrate comparability of drug product made at the 2 manufacturing sites.
“TESARO is committed to bringing this new intravenous formulation of rolapitant to physicians and patients to enable additional flexibility and choice of antiemetic regimens, and we plan to address FDA’s questions expeditiously and complete this application, which we expect to enable approval in the first half of 2017,” said Mary Lynne Hedley, PhD, president and chief operating officer of TESARO. ![]()
chemotherapy
Photo by Rhoda Baer
The US Food and Drug Administration (FDA) has issued a complete response letter (CRL) regarding the new drug application (NDA) for an intravenous (IV) formulation of rolapitant.
An oral formulation of rolapitant, marketed as VARUBI®, is FDA-approved for use in combination with other antiemetic agents to prevent delayed nausea and vomiting associated with initial and repeat courses of emetogenic cancer chemotherapy in adults.
The NDA for rolapitant IV is for the same indication.
The FDA requested additional information regarding the in vitro method utilized to demonstrate comparability of drug product produced at the 2 proposed commercial manufacturers for rolapitant IV that were included in the NDA.
TESARO Inc., the company developing rolapitant IV, said it is working to provide the requested information.
The CRL did not identify concerns related to the safety or efficacy of rolapitant IV or request additional clinical studies. No concerns were raised regarding the active pharmaceutical ingredient, which is also used for VARUBI®.
TESARO identified potential deficiencies at the original contract manufacturer for rolapitant IV, secured a second drug product supplier, and included data from this manufacturer in the NDA.
During the NDA review, the FDA requested and TESARO provided in vitro data to demonstrate comparability of drug product made at the 2 manufacturing sites.
“TESARO is committed to bringing this new intravenous formulation of rolapitant to physicians and patients to enable additional flexibility and choice of antiemetic regimens, and we plan to address FDA’s questions expeditiously and complete this application, which we expect to enable approval in the first half of 2017,” said Mary Lynne Hedley, PhD, president and chief operating officer of TESARO. ![]()
Venetoclax approved to treat CLL in Australia

venetoclax (US version)
Photo courtesy of Abbvie
The Australian Therapeutic Goods Administration (TGA) has approved the BCL-2 inhibitor venetoclax (Venclexta™, formerly ABT-199) for use in certain patients with chronic lymphocytic leukemia (CLL).
The drug is now approved to treat Australian patients with relapsed or refractory CLL who have 17p deletion or no other treatment options.
Venetoclax is being developed by AbbVie and Genentech, a member of the Roche Group. The drug is jointly commercialized by the companies in the US and by AbbVie outside of the US.
Now that venetoclax has been approved by the TGA, it can be registered on the Australian Register of Therapeutic Goods and legally marketed and sold in Australia.
To make the drug affordable to the Australian public, the manufacturer can apply to the Pharmaceutical Benefits Advisory Committee to have the cost of the drug subsidized by the Australian government on the Pharmaceutical Benefits Scheme (PBS).
Venetoclax is not listed on the PBS. Historically, the delay between TGA approval and PBS listing ranges from 14 months to 31 months for cancer drugs.
Phase 2 trials
Venetoclax has produced high objective response rates (ORR) in two phase 2 trials of CLL patients.
In one of these trials, researchers tested venetoclax in 107 patients with previously treated CLL and 17p deletion. The results were published in The Lancet Oncology in June 2016.
The ORR in this trial was 79%. At the time of analysis, the median duration of response had not been reached. The same was true for progression-free survival and overall survival.
The progression-free survival estimate for 12 months was 72%, and the overall survival estimate was 87%.
The incidence of treatment-emergent adverse events was 96%, and the incidence of serious adverse events was 55%.
Grade 3 laboratory tumor lysis syndrome (TLS) was reported in 5 patients. Three of these patients continued on venetoclax, but 2 patients required a dose interruption of 1 day each.
In the second trial, researchers tested venetoclax in 64 patients with CLL who had failed treatment with ibrutinib and/or idelalisib. Results from this trial were presented at the 2016 ASH Annual Meeting.
The ORR was 67%. At 11.8 months of follow-up, the median duration of response, progression-free survival, and overall survival had not been reached. The estimated 12-month progression-free survival was 80%.
The incidence of adverse events was 100%, and the incidence of serious adverse events was 53%. No clinical TLS was observed, but 1 patient met Howard criteria for laboratory TLS.
In the past, TLS has caused deaths in patients receiving venetoclax. In response, AbbVie stopped dose-escalation in patients receiving the drug and suspended enrollment in phase 1 trials.
However, researchers subsequently found that a modified dosing schedule, prophylaxis, and patient monitoring can reduce the risk of TLS. ![]()
venetoclax (US version)
Photo courtesy of Abbvie
The Australian Therapeutic Goods Administration (TGA) has approved the BCL-2 inhibitor venetoclax (Venclexta™, formerly ABT-199) for use in certain patients with chronic lymphocytic leukemia (CLL).
The drug is now approved to treat Australian patients with relapsed or refractory CLL who have 17p deletion or no other treatment options.
Venetoclax is being developed by AbbVie and Genentech, a member of the Roche Group. The drug is jointly commercialized by the companies in the US and by AbbVie outside of the US.
Now that venetoclax has been approved by the TGA, it can be registered on the Australian Register of Therapeutic Goods and legally marketed and sold in Australia.
To make the drug affordable to the Australian public, the manufacturer can apply to the Pharmaceutical Benefits Advisory Committee to have the cost of the drug subsidized by the Australian government on the Pharmaceutical Benefits Scheme (PBS).
Venetoclax is not listed on the PBS. Historically, the delay between TGA approval and PBS listing ranges from 14 months to 31 months for cancer drugs.
Phase 2 trials
Venetoclax has produced high objective response rates (ORR) in two phase 2 trials of CLL patients.
In one of these trials, researchers tested venetoclax in 107 patients with previously treated CLL and 17p deletion. The results were published in The Lancet Oncology in June 2016.
The ORR in this trial was 79%. At the time of analysis, the median duration of response had not been reached. The same was true for progression-free survival and overall survival.
The progression-free survival estimate for 12 months was 72%, and the overall survival estimate was 87%.
The incidence of treatment-emergent adverse events was 96%, and the incidence of serious adverse events was 55%.
Grade 3 laboratory tumor lysis syndrome (TLS) was reported in 5 patients. Three of these patients continued on venetoclax, but 2 patients required a dose interruption of 1 day each.
In the second trial, researchers tested venetoclax in 64 patients with CLL who had failed treatment with ibrutinib and/or idelalisib. Results from this trial were presented at the 2016 ASH Annual Meeting.
The ORR was 67%. At 11.8 months of follow-up, the median duration of response, progression-free survival, and overall survival had not been reached. The estimated 12-month progression-free survival was 80%.
The incidence of adverse events was 100%, and the incidence of serious adverse events was 53%. No clinical TLS was observed, but 1 patient met Howard criteria for laboratory TLS.
In the past, TLS has caused deaths in patients receiving venetoclax. In response, AbbVie stopped dose-escalation in patients receiving the drug and suspended enrollment in phase 1 trials.
However, researchers subsequently found that a modified dosing schedule, prophylaxis, and patient monitoring can reduce the risk of TLS. ![]()
venetoclax (US version)
Photo courtesy of Abbvie
The Australian Therapeutic Goods Administration (TGA) has approved the BCL-2 inhibitor venetoclax (Venclexta™, formerly ABT-199) for use in certain patients with chronic lymphocytic leukemia (CLL).
The drug is now approved to treat Australian patients with relapsed or refractory CLL who have 17p deletion or no other treatment options.
Venetoclax is being developed by AbbVie and Genentech, a member of the Roche Group. The drug is jointly commercialized by the companies in the US and by AbbVie outside of the US.
Now that venetoclax has been approved by the TGA, it can be registered on the Australian Register of Therapeutic Goods and legally marketed and sold in Australia.
To make the drug affordable to the Australian public, the manufacturer can apply to the Pharmaceutical Benefits Advisory Committee to have the cost of the drug subsidized by the Australian government on the Pharmaceutical Benefits Scheme (PBS).
Venetoclax is not listed on the PBS. Historically, the delay between TGA approval and PBS listing ranges from 14 months to 31 months for cancer drugs.
Phase 2 trials
Venetoclax has produced high objective response rates (ORR) in two phase 2 trials of CLL patients.
In one of these trials, researchers tested venetoclax in 107 patients with previously treated CLL and 17p deletion. The results were published in The Lancet Oncology in June 2016.
The ORR in this trial was 79%. At the time of analysis, the median duration of response had not been reached. The same was true for progression-free survival and overall survival.
The progression-free survival estimate for 12 months was 72%, and the overall survival estimate was 87%.
The incidence of treatment-emergent adverse events was 96%, and the incidence of serious adverse events was 55%.
Grade 3 laboratory tumor lysis syndrome (TLS) was reported in 5 patients. Three of these patients continued on venetoclax, but 2 patients required a dose interruption of 1 day each.
In the second trial, researchers tested venetoclax in 64 patients with CLL who had failed treatment with ibrutinib and/or idelalisib. Results from this trial were presented at the 2016 ASH Annual Meeting.
The ORR was 67%. At 11.8 months of follow-up, the median duration of response, progression-free survival, and overall survival had not been reached. The estimated 12-month progression-free survival was 80%.
The incidence of adverse events was 100%, and the incidence of serious adverse events was 53%. No clinical TLS was observed, but 1 patient met Howard criteria for laboratory TLS.
In the past, TLS has caused deaths in patients receiving venetoclax. In response, AbbVie stopped dose-escalation in patients receiving the drug and suspended enrollment in phase 1 trials.
However, researchers subsequently found that a modified dosing schedule, prophylaxis, and patient monitoring can reduce the risk of TLS. ![]()
Combo granted orphan designation for CLL

The US Food and Drug Administration (FDA) has granted orphan drug designation for the combination of TG-1101 (ublituximab) and TGR-1202 for the treatment of patients with chronic lymphocytic leukemia (CLL).
Ublituximab is a glycoengineered anti-CD20 monoclonal antibody, and TGR-1202 is a next-generation PI3K delta inhibitor. Both drugs are being developed by TG Therapeutics, Inc.
Researchers have evaluated ublituximab and TGR-1202 in combination in a phase 1 trial of patients with relapsed or refractory CLL/small lymphocytic lymphoma (SLL) and non-Hodgkin lymphomas (NHLs).
Results were presented at the 2015 ASH Annual Meeting.
There was a 3+3 dose-escalation portion of the study and a dose-expansion phase. The patients received TGR-1202 at doses ranging from 400 mg to 1200 mg and 2 different doses of ublituximab—900 mg for patients with NHL and 600 mg or 900 mg for patients with CLL/SLL.
As of ASH, there were 58 patients evaluable for efficacy and 71 evaluable for safety.
There were 10 CLL/SLL patients exposed to higher doses of TGR-1202. Among these patients, the overall response rate was 80%. Seven patients achieved a partial response, 1 achieved a complete response, and the remaining 2 patients had stable disease.
For the entire safety population, the most common adverse events were nausea (46%), diarrhea (44%), fatigue (41%), neutropenia (30%), and infusion-related reactions (25%).
Grade 3/4 adverse events included neutropenia (25%), diarrhea (3%), fatigue (3%), dyspnea (3%), pyrexia (3%), nausea (1%), infusion-related reactions (1%), sinusitis (1%), anemia (1%), hypophosphatemia (1%), and peripheral edema (1%).
Now, the combination of ublituximab and TGR-1202 is being evaluated in the UNITY-CLL phase 3 trial for patients with previously treated or untreated CLL.
“[W]ith enrollment into our UNITY-CLL phase 3 trial currently exceeding our expectations, we expect to be able to commence a regulatory filing for the combination in 2018, and having orphan drug designation will provide certain cost-saving advantages for us during the regulatory approval process,” said Michael S. Weiss, executive chairman and chief executive officer of TG Therapeutics.
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation for the combination of TG-1101 (ublituximab) and TGR-1202 for the treatment of patients with chronic lymphocytic leukemia (CLL).
Ublituximab is a glycoengineered anti-CD20 monoclonal antibody, and TGR-1202 is a next-generation PI3K delta inhibitor. Both drugs are being developed by TG Therapeutics, Inc.
Researchers have evaluated ublituximab and TGR-1202 in combination in a phase 1 trial of patients with relapsed or refractory CLL/small lymphocytic lymphoma (SLL) and non-Hodgkin lymphomas (NHLs).
Results were presented at the 2015 ASH Annual Meeting.
There was a 3+3 dose-escalation portion of the study and a dose-expansion phase. The patients received TGR-1202 at doses ranging from 400 mg to 1200 mg and 2 different doses of ublituximab—900 mg for patients with NHL and 600 mg or 900 mg for patients with CLL/SLL.
As of ASH, there were 58 patients evaluable for efficacy and 71 evaluable for safety.
There were 10 CLL/SLL patients exposed to higher doses of TGR-1202. Among these patients, the overall response rate was 80%. Seven patients achieved a partial response, 1 achieved a complete response, and the remaining 2 patients had stable disease.
For the entire safety population, the most common adverse events were nausea (46%), diarrhea (44%), fatigue (41%), neutropenia (30%), and infusion-related reactions (25%).
Grade 3/4 adverse events included neutropenia (25%), diarrhea (3%), fatigue (3%), dyspnea (3%), pyrexia (3%), nausea (1%), infusion-related reactions (1%), sinusitis (1%), anemia (1%), hypophosphatemia (1%), and peripheral edema (1%).
Now, the combination of ublituximab and TGR-1202 is being evaluated in the UNITY-CLL phase 3 trial for patients with previously treated or untreated CLL.
“[W]ith enrollment into our UNITY-CLL phase 3 trial currently exceeding our expectations, we expect to be able to commence a regulatory filing for the combination in 2018, and having orphan drug designation will provide certain cost-saving advantages for us during the regulatory approval process,” said Michael S. Weiss, executive chairman and chief executive officer of TG Therapeutics.
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation for the combination of TG-1101 (ublituximab) and TGR-1202 for the treatment of patients with chronic lymphocytic leukemia (CLL).
Ublituximab is a glycoengineered anti-CD20 monoclonal antibody, and TGR-1202 is a next-generation PI3K delta inhibitor. Both drugs are being developed by TG Therapeutics, Inc.
Researchers have evaluated ublituximab and TGR-1202 in combination in a phase 1 trial of patients with relapsed or refractory CLL/small lymphocytic lymphoma (SLL) and non-Hodgkin lymphomas (NHLs).
Results were presented at the 2015 ASH Annual Meeting.
There was a 3+3 dose-escalation portion of the study and a dose-expansion phase. The patients received TGR-1202 at doses ranging from 400 mg to 1200 mg and 2 different doses of ublituximab—900 mg for patients with NHL and 600 mg or 900 mg for patients with CLL/SLL.
As of ASH, there were 58 patients evaluable for efficacy and 71 evaluable for safety.
There were 10 CLL/SLL patients exposed to higher doses of TGR-1202. Among these patients, the overall response rate was 80%. Seven patients achieved a partial response, 1 achieved a complete response, and the remaining 2 patients had stable disease.
For the entire safety population, the most common adverse events were nausea (46%), diarrhea (44%), fatigue (41%), neutropenia (30%), and infusion-related reactions (25%).
Grade 3/4 adverse events included neutropenia (25%), diarrhea (3%), fatigue (3%), dyspnea (3%), pyrexia (3%), nausea (1%), infusion-related reactions (1%), sinusitis (1%), anemia (1%), hypophosphatemia (1%), and peripheral edema (1%).
Now, the combination of ublituximab and TGR-1202 is being evaluated in the UNITY-CLL phase 3 trial for patients with previously treated or untreated CLL.
“[W]ith enrollment into our UNITY-CLL phase 3 trial currently exceeding our expectations, we expect to be able to commence a regulatory filing for the combination in 2018, and having orphan drug designation will provide certain cost-saving advantages for us during the regulatory approval process,” said Michael S. Weiss, executive chairman and chief executive officer of TG Therapeutics.
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
Yoga may improve QOL in kids with cancer

Photo by Bill Branson
A yoga program for children with cancer can be carried out during cancer treatment and has quality of life (QOL) benefits for the children as well as their caregivers, according to research published in Rehabilitation Oncology.
However, the program was not feasible for all patients. More than half of those initially enrolled could not complete the study due to treatment toxicity or scheduling conflicts.
Andrea Orsey, MD, of Connecticut Children’s Medical Center in Hartford, and her colleagues conducted this research to evaluate the feasibility and effectiveness of a yoga intervention for children with cancer and their families.
The team began by conducting a survey of 20 children and adolescents with cancer and their parents/guardians.
Survey respondents expressed interest in a yoga program. But they also perceived several barriers to such a program, including concerns about side effects, pain/discomfort, and physical limitations.
With these barriers in mind, Dr Orsey and her colleagues developed a yoga intervention for pediatric cancer patients, delivered by certified yoga instructors.
The program was designed to be performed in a variety of settings and tailored to the children’s physical condition or mobility issues.
A pilot evaluation included 10 children with cancer and their caregivers. Twenty-two patient/caregiver pairs were actually enrolled, but 6 pairs withdrew because of treatment toxicity, and 6 had the study window lapse due to scheduling conflicts.
Although limited by its small size, the study suggested that yoga improved health-related QOL for both caregivers and children.
The children had significant improvements in both social and emotional QOL. They had an overall improvement in fatigue, but this was not statistically significant.
Caregivers had a significant improvement in mental health but not physical health or caregiver burden.
Both caregivers and children said they were satisfied with the yoga program and would recommend it to others.
Dr Orsey and her colleagues hope this pilot study will help guide future efforts to provide yoga to children with cancer and their families.
The researchers noted that a key issue will be coordinating yoga sessions with the medical demands of chemotherapy. ![]()
Photo by Bill Branson
A yoga program for children with cancer can be carried out during cancer treatment and has quality of life (QOL) benefits for the children as well as their caregivers, according to research published in Rehabilitation Oncology.
However, the program was not feasible for all patients. More than half of those initially enrolled could not complete the study due to treatment toxicity or scheduling conflicts.
Andrea Orsey, MD, of Connecticut Children’s Medical Center in Hartford, and her colleagues conducted this research to evaluate the feasibility and effectiveness of a yoga intervention for children with cancer and their families.
The team began by conducting a survey of 20 children and adolescents with cancer and their parents/guardians.
Survey respondents expressed interest in a yoga program. But they also perceived several barriers to such a program, including concerns about side effects, pain/discomfort, and physical limitations.
With these barriers in mind, Dr Orsey and her colleagues developed a yoga intervention for pediatric cancer patients, delivered by certified yoga instructors.
The program was designed to be performed in a variety of settings and tailored to the children’s physical condition or mobility issues.
A pilot evaluation included 10 children with cancer and their caregivers. Twenty-two patient/caregiver pairs were actually enrolled, but 6 pairs withdrew because of treatment toxicity, and 6 had the study window lapse due to scheduling conflicts.
Although limited by its small size, the study suggested that yoga improved health-related QOL for both caregivers and children.
The children had significant improvements in both social and emotional QOL. They had an overall improvement in fatigue, but this was not statistically significant.
Caregivers had a significant improvement in mental health but not physical health or caregiver burden.
Both caregivers and children said they were satisfied with the yoga program and would recommend it to others.
Dr Orsey and her colleagues hope this pilot study will help guide future efforts to provide yoga to children with cancer and their families.
The researchers noted that a key issue will be coordinating yoga sessions with the medical demands of chemotherapy. ![]()
Photo by Bill Branson
A yoga program for children with cancer can be carried out during cancer treatment and has quality of life (QOL) benefits for the children as well as their caregivers, according to research published in Rehabilitation Oncology.
However, the program was not feasible for all patients. More than half of those initially enrolled could not complete the study due to treatment toxicity or scheduling conflicts.
Andrea Orsey, MD, of Connecticut Children’s Medical Center in Hartford, and her colleagues conducted this research to evaluate the feasibility and effectiveness of a yoga intervention for children with cancer and their families.
The team began by conducting a survey of 20 children and adolescents with cancer and their parents/guardians.
Survey respondents expressed interest in a yoga program. But they also perceived several barriers to such a program, including concerns about side effects, pain/discomfort, and physical limitations.
With these barriers in mind, Dr Orsey and her colleagues developed a yoga intervention for pediatric cancer patients, delivered by certified yoga instructors.
The program was designed to be performed in a variety of settings and tailored to the children’s physical condition or mobility issues.
A pilot evaluation included 10 children with cancer and their caregivers. Twenty-two patient/caregiver pairs were actually enrolled, but 6 pairs withdrew because of treatment toxicity, and 6 had the study window lapse due to scheduling conflicts.
Although limited by its small size, the study suggested that yoga improved health-related QOL for both caregivers and children.
The children had significant improvements in both social and emotional QOL. They had an overall improvement in fatigue, but this was not statistically significant.
Caregivers had a significant improvement in mental health but not physical health or caregiver burden.
Both caregivers and children said they were satisfied with the yoga program and would recommend it to others.
Dr Orsey and her colleagues hope this pilot study will help guide future efforts to provide yoga to children with cancer and their families.
The researchers noted that a key issue will be coordinating yoga sessions with the medical demands of chemotherapy. ![]()
US cancer cases may near 1.7 million in 2017

patient and her father
Photo by Rhoda Baer
The US may see nearly 1.7 million new cancer cases in 2017 and more than 600,000 cancer-related deaths, according to a report from the American Cancer Society (ACS).
In addition to estimates for 2017, the report, “Cancer Statistics 2017,” includes the most recent data on cancer incidence, mortality, and survival in the US.
The report was published in CA: A Cancer Journal for Clinicians.
The report projects there will be 1,688,780 new cancer cases and 600,920 cancer deaths in the US this year.
This includes:
- 80,500 new cases of lymphoma and 21,210 lymphoma deaths
- 62,130 new cases of leukemia and 24,500 leukemia deaths
- 30,280 new cases of myeloma and 12,590 myeloma deaths.
The report also shows that, from 2004 to 2013, the overall cancer incidence rate was stable in women and declined by about 2% per year in men. From 2005 to 2014, the cancer death rate declined by about 1.5% annually in both men and women.
Overall, the cancer death rate dropped 25% from its peak of 215.1 (per 100,000 population) in 1991 to 161.2 (per 100,000 population) in 2014, the latest year for which data was available. This translates to about 2,143,200 fewer cancer deaths.
“The continuing drops in the cancer death rate are a powerful sign of the potential we have to reduce cancer’s deadly toll,” said Otis W. Brawley, MD, chief medical officer of the ACS.
He said the decrease in cancer death rates is the result of steady reductions in smoking and advances in early detection and treatment. The decrease is driven by decreasing death rates for the 4 major cancer sites—lung, breast, colorectal, and prostate.
The report also shows that racial disparities in cancer death rates continue to decline. The excess risk of cancer death in black men has dropped from 47% in 1990 to 21% in 2014. The black/white disparity declined similarly in women, from a peak of 20% in 1998 to 13% in 2014.
On the other hand, significant gender disparities persist for both cancer incidence and death in the US. For all cancer sites combined, the incidence rate is 20% higher in men than in women, and the cancer death rate is 40% higher in men.
Dr Brawley said the gender gap in cancer mortality largely reflects variation in the distribution of cancers that occur in men and women, much of which is due to differences in the prevalence of cancer risk factors.
The yearly “Cancer Statistics” reports have been published by ACS researchers since 1967 to inform and guide clinicians, investigators, and others in public health in prioritizing efforts to reduce the burden of cancer.
Cancer incidence data for the current report were collected by the Surveillance, Epidemiology, and End Results Program; the National Program of Cancer Registries; and the North American Association of Central Cancer Registries. Mortality data were collected by the National Center for Health Statistics. ![]()
patient and her father
Photo by Rhoda Baer
The US may see nearly 1.7 million new cancer cases in 2017 and more than 600,000 cancer-related deaths, according to a report from the American Cancer Society (ACS).
In addition to estimates for 2017, the report, “Cancer Statistics 2017,” includes the most recent data on cancer incidence, mortality, and survival in the US.
The report was published in CA: A Cancer Journal for Clinicians.
The report projects there will be 1,688,780 new cancer cases and 600,920 cancer deaths in the US this year.
This includes:
- 80,500 new cases of lymphoma and 21,210 lymphoma deaths
- 62,130 new cases of leukemia and 24,500 leukemia deaths
- 30,280 new cases of myeloma and 12,590 myeloma deaths.
The report also shows that, from 2004 to 2013, the overall cancer incidence rate was stable in women and declined by about 2% per year in men. From 2005 to 2014, the cancer death rate declined by about 1.5% annually in both men and women.
Overall, the cancer death rate dropped 25% from its peak of 215.1 (per 100,000 population) in 1991 to 161.2 (per 100,000 population) in 2014, the latest year for which data was available. This translates to about 2,143,200 fewer cancer deaths.
“The continuing drops in the cancer death rate are a powerful sign of the potential we have to reduce cancer’s deadly toll,” said Otis W. Brawley, MD, chief medical officer of the ACS.
He said the decrease in cancer death rates is the result of steady reductions in smoking and advances in early detection and treatment. The decrease is driven by decreasing death rates for the 4 major cancer sites—lung, breast, colorectal, and prostate.
The report also shows that racial disparities in cancer death rates continue to decline. The excess risk of cancer death in black men has dropped from 47% in 1990 to 21% in 2014. The black/white disparity declined similarly in women, from a peak of 20% in 1998 to 13% in 2014.
On the other hand, significant gender disparities persist for both cancer incidence and death in the US. For all cancer sites combined, the incidence rate is 20% higher in men than in women, and the cancer death rate is 40% higher in men.
Dr Brawley said the gender gap in cancer mortality largely reflects variation in the distribution of cancers that occur in men and women, much of which is due to differences in the prevalence of cancer risk factors.
The yearly “Cancer Statistics” reports have been published by ACS researchers since 1967 to inform and guide clinicians, investigators, and others in public health in prioritizing efforts to reduce the burden of cancer.
Cancer incidence data for the current report were collected by the Surveillance, Epidemiology, and End Results Program; the National Program of Cancer Registries; and the North American Association of Central Cancer Registries. Mortality data were collected by the National Center for Health Statistics. ![]()
patient and her father
Photo by Rhoda Baer
The US may see nearly 1.7 million new cancer cases in 2017 and more than 600,000 cancer-related deaths, according to a report from the American Cancer Society (ACS).
In addition to estimates for 2017, the report, “Cancer Statistics 2017,” includes the most recent data on cancer incidence, mortality, and survival in the US.
The report was published in CA: A Cancer Journal for Clinicians.
The report projects there will be 1,688,780 new cancer cases and 600,920 cancer deaths in the US this year.
This includes:
- 80,500 new cases of lymphoma and 21,210 lymphoma deaths
- 62,130 new cases of leukemia and 24,500 leukemia deaths
- 30,280 new cases of myeloma and 12,590 myeloma deaths.
The report also shows that, from 2004 to 2013, the overall cancer incidence rate was stable in women and declined by about 2% per year in men. From 2005 to 2014, the cancer death rate declined by about 1.5% annually in both men and women.
Overall, the cancer death rate dropped 25% from its peak of 215.1 (per 100,000 population) in 1991 to 161.2 (per 100,000 population) in 2014, the latest year for which data was available. This translates to about 2,143,200 fewer cancer deaths.
“The continuing drops in the cancer death rate are a powerful sign of the potential we have to reduce cancer’s deadly toll,” said Otis W. Brawley, MD, chief medical officer of the ACS.
He said the decrease in cancer death rates is the result of steady reductions in smoking and advances in early detection and treatment. The decrease is driven by decreasing death rates for the 4 major cancer sites—lung, breast, colorectal, and prostate.
The report also shows that racial disparities in cancer death rates continue to decline. The excess risk of cancer death in black men has dropped from 47% in 1990 to 21% in 2014. The black/white disparity declined similarly in women, from a peak of 20% in 1998 to 13% in 2014.
On the other hand, significant gender disparities persist for both cancer incidence and death in the US. For all cancer sites combined, the incidence rate is 20% higher in men than in women, and the cancer death rate is 40% higher in men.
Dr Brawley said the gender gap in cancer mortality largely reflects variation in the distribution of cancers that occur in men and women, much of which is due to differences in the prevalence of cancer risk factors.
The yearly “Cancer Statistics” reports have been published by ACS researchers since 1967 to inform and guide clinicians, investigators, and others in public health in prioritizing efforts to reduce the burden of cancer.
Cancer incidence data for the current report were collected by the Surveillance, Epidemiology, and End Results Program; the National Program of Cancer Registries; and the North American Association of Central Cancer Registries. Mortality data were collected by the National Center for Health Statistics. ![]()
Cancer genomic data released to public

Photo courtesy of the
National Institute of
General Medical Sciences
The American Association for Cancer Research (AACR) has announced the first public release of cancer genomic data aggregated through the AACR Project Genomics Evidence Neoplasia Information Exchange (GENIE).
The data set includes nearly 19,000 de-identified genomic records collected from patients who were treated at 8 international institutions, making it one of the largest public cancer genomic data sets released to date.
The release includes data for 59 major cancer types, including leukemias, lymphomas, and multiple myeloma.
The genomic data and a limited amount of linked clinical data for each patient can be accessed via the AACR Project GENIE cBioPortal or from Sage Bionetworks. (Users must create an account for either site to access the data.)
“We are excited to make publicly available this very large set of clinical-grade, next-generation sequencing data obtained during routine patient care,” said Charles L. Sawyers, MD, AACR Project GENIE Steering Committee chairperson.
“These data were generated as part of routine patient care and, without AACR Project GENIE, they would likely never have been shared with the global cancer research community.”
AACR Project GENIE is a multi-phase, international data-sharing project aimed at catalyzing precision oncology through the development of a registry that aggregates and links clinical-grade cancer genomic data with clinical outcomes from tens of thousands of cancer patients treated at multiple institutions.
The newly released data are fully de-identified in compliance with the Health Insurance Portability and Accountability Act (HIPAA).
The data are derived from patients whose tumors were genetically sequenced as part of their care at any of the 8 institutions that participated in the first phase of AACR Project GENIE.
The goal of releasing these data to the cancer research community is to aid new research that will accelerate the pace of progress against cancer.
According to AACR, the data can be used to validate gene signatures of drug response or prognosis, identify new patient populations for drugs that are currently available, and uncover new drug targets and biomarkers.
“I am extremely proud that the American Association for Cancer Research, as the coordinating center for AACR Project GENIE, is delivering on its promise to make these important data publicly available just over a year after unveiling the initiative,” said Margaret Foti, PhD, MD, chief executive officer of the AACR.
To expand the AACR Project GENIE registry, the consortium is accepting applications for new participating centers. Any nonprofit institution that meets certain criteria can submit an application to become a project participant.
For more information on AACR Project GENIE, visit the project website or send an email to info@aacrgenie.org. ![]()
Photo courtesy of the
National Institute of
General Medical Sciences
The American Association for Cancer Research (AACR) has announced the first public release of cancer genomic data aggregated through the AACR Project Genomics Evidence Neoplasia Information Exchange (GENIE).
The data set includes nearly 19,000 de-identified genomic records collected from patients who were treated at 8 international institutions, making it one of the largest public cancer genomic data sets released to date.
The release includes data for 59 major cancer types, including leukemias, lymphomas, and multiple myeloma.
The genomic data and a limited amount of linked clinical data for each patient can be accessed via the AACR Project GENIE cBioPortal or from Sage Bionetworks. (Users must create an account for either site to access the data.)
“We are excited to make publicly available this very large set of clinical-grade, next-generation sequencing data obtained during routine patient care,” said Charles L. Sawyers, MD, AACR Project GENIE Steering Committee chairperson.
“These data were generated as part of routine patient care and, without AACR Project GENIE, they would likely never have been shared with the global cancer research community.”
AACR Project GENIE is a multi-phase, international data-sharing project aimed at catalyzing precision oncology through the development of a registry that aggregates and links clinical-grade cancer genomic data with clinical outcomes from tens of thousands of cancer patients treated at multiple institutions.
The newly released data are fully de-identified in compliance with the Health Insurance Portability and Accountability Act (HIPAA).
The data are derived from patients whose tumors were genetically sequenced as part of their care at any of the 8 institutions that participated in the first phase of AACR Project GENIE.
The goal of releasing these data to the cancer research community is to aid new research that will accelerate the pace of progress against cancer.
According to AACR, the data can be used to validate gene signatures of drug response or prognosis, identify new patient populations for drugs that are currently available, and uncover new drug targets and biomarkers.
“I am extremely proud that the American Association for Cancer Research, as the coordinating center for AACR Project GENIE, is delivering on its promise to make these important data publicly available just over a year after unveiling the initiative,” said Margaret Foti, PhD, MD, chief executive officer of the AACR.
To expand the AACR Project GENIE registry, the consortium is accepting applications for new participating centers. Any nonprofit institution that meets certain criteria can submit an application to become a project participant.
For more information on AACR Project GENIE, visit the project website or send an email to info@aacrgenie.org. ![]()
Photo courtesy of the
National Institute of
General Medical Sciences
The American Association for Cancer Research (AACR) has announced the first public release of cancer genomic data aggregated through the AACR Project Genomics Evidence Neoplasia Information Exchange (GENIE).
The data set includes nearly 19,000 de-identified genomic records collected from patients who were treated at 8 international institutions, making it one of the largest public cancer genomic data sets released to date.
The release includes data for 59 major cancer types, including leukemias, lymphomas, and multiple myeloma.
The genomic data and a limited amount of linked clinical data for each patient can be accessed via the AACR Project GENIE cBioPortal or from Sage Bionetworks. (Users must create an account for either site to access the data.)
“We are excited to make publicly available this very large set of clinical-grade, next-generation sequencing data obtained during routine patient care,” said Charles L. Sawyers, MD, AACR Project GENIE Steering Committee chairperson.
“These data were generated as part of routine patient care and, without AACR Project GENIE, they would likely never have been shared with the global cancer research community.”
AACR Project GENIE is a multi-phase, international data-sharing project aimed at catalyzing precision oncology through the development of a registry that aggregates and links clinical-grade cancer genomic data with clinical outcomes from tens of thousands of cancer patients treated at multiple institutions.
The newly released data are fully de-identified in compliance with the Health Insurance Portability and Accountability Act (HIPAA).
The data are derived from patients whose tumors were genetically sequenced as part of their care at any of the 8 institutions that participated in the first phase of AACR Project GENIE.
The goal of releasing these data to the cancer research community is to aid new research that will accelerate the pace of progress against cancer.
According to AACR, the data can be used to validate gene signatures of drug response or prognosis, identify new patient populations for drugs that are currently available, and uncover new drug targets and biomarkers.
“I am extremely proud that the American Association for Cancer Research, as the coordinating center for AACR Project GENIE, is delivering on its promise to make these important data publicly available just over a year after unveiling the initiative,” said Margaret Foti, PhD, MD, chief executive officer of the AACR.
To expand the AACR Project GENIE registry, the consortium is accepting applications for new participating centers. Any nonprofit institution that meets certain criteria can submit an application to become a project participant.
For more information on AACR Project GENIE, visit the project website or send an email to info@aacrgenie.org. ![]()
Daratumumab combo holds up across POLLUX myeloma subgroups
SAN DIEGO – Adding daratumumab (D) to lenalidomide and dexamethasone (Rd) significantly improved outcomes in relapsed and refractory multiple myeloma, even when patients had previously received lenalidomide, were refractory to bortezomib, or had high-risk tumor cytogenetics, based on updated analyses from the multicenter, randomized, phase III, open-label POLLUX trial.
The findings underscore the “significant benefit of combining daratumumab with lenalidomide and dexamethasone for relapsed or refractory multiple myeloma,” said lead investigator Philippe Moreau, MD, of University Hospital Hotel-Dieu in Nantes, France.
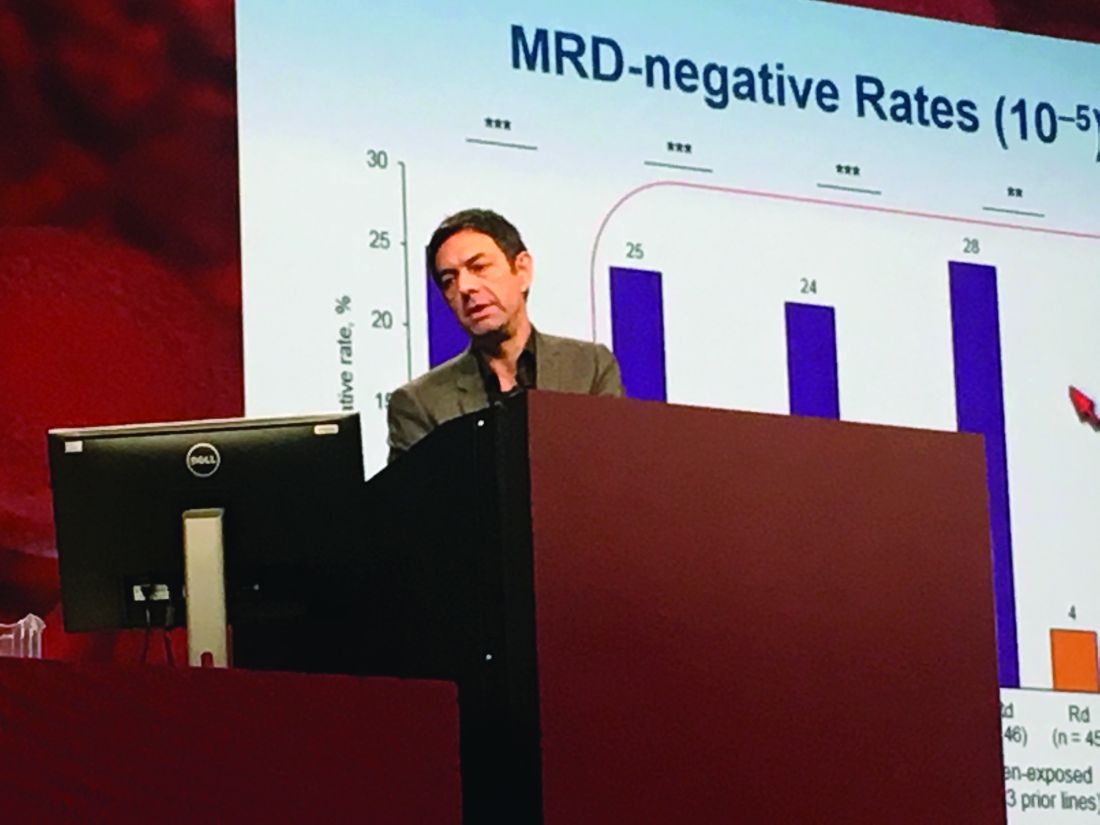
Among a large subgroup of 524 POLLUX patients who had received one to three prior lines of therapy, estimated median progression-free survival (PFS) has not been reached in the daratumumab, lenalidomide, and dexamethasone (DRd) arm, versus 18.4 months in the lenalidomide and dexamethasone (Rd) arm (hazard ratio, 0.36; 95% CI: 0.26 to 0.49; P less than .0001), Dr. Moreau said at the annual meeting of the American Society of Hematology.
That means adding daratumumab to Rd led to a 64% reduction in the risk of disease progression or death among patients with relapsed or refractory multiple myeloma, he noted. Fully 77% of DRd patients were alive without having progressed at 18 months, and responses “continued to deepen in the DRd group with longer follow-up,” he added.
Additional analyses supported the use of DRd in relapsed or refractory multiple myeloma, “irrespective of prior lenalidomide treatment or bortezomib refractoriness,” Dr. Moreau continued. He reported that DRd significantly improved PFS over Rd alone not only among 445 lenalidomide-naive patients (HR, 0.37; P less than .0001), but also among 91 lenalidomide-exposed patients (HR, 0.45; P = .04), 140 patients who were refractory to their most recent line of therapy (HR, 0.45; P = .001), and 99 bortezomib- refractory patients (HR 0.51; P = .02).
Daratumumab (Darzalex), a human CD38 IgG1k monoclonal antibody, was first approved as monotherapy for multiple myeloma in patients who had received at least three prior lines of therapy or had double-refractory disease. In 2016, results from the twin POLLUX and CASTOR studies won daratumumab a Food and Drug Administration breakthrough designation status for use with Rd in patients who had received at least one prior line.
The POLLUX trial included 569 patients with multiple myeloma who had received a median of 1 and up to 11 prior lines of therapy. Patients were randomized to either Rd alone or to Rd plus intravenous daratumumab (16 mg/kg) once a week during the first two 28-day treatment cycles, every 2 weeks during cycles 3-6, and once only on day 1 of subsequent cycles.
POLLUX patients were fairly heavily pretreated, Dr. Moreau noted. Thirteen percent had received three prior lines of therapy, 86% had received a proteasome inhibitor, 18% had received lenalidomide, 21% were refractory to bortezomib, and 28% were refractory to their most recent line of therapy.
Researchers performed “stringent, unbiased” assessments of minimal residual disease (MRD) negativity not only when a complete response was suspected, but also 3 and 6 months later, Dr. Moreau said. He emphasized that rates of MRD negativity in lenalidomide-exposed, bortezomib-refractory subgroups in POLLUX almost exactly matched those in the intent-to-treat population (25% on DRd vs. 6% on Rd; P less than .0001).
A total of 17% of DRd patients and 25% of Rd patients had high-risk cytogenetic profiles, and DRd performed well in these individuals, Dr. Moreau reported. Fully 85% of all evaluable high-risk patients had at least a partial response to DRd, and 33% had a complete response, versus only 67% and 6% of high-risk Rd patients, respectively. Among patients with standard-risk cytogenetics, rates of best overall response were 95% on DRd and 82% on Rd, and rates of complete response were 52% on DRd and 24% on Rd.
POLLUX yielded no new safety signals for DRd, Dr. Moreau said. Rates of primary and secondary malignancies were less than 2%. Neutropenia, the most common adverse effect, was managed by interrupting treatment, reducing the dose of lenalidomide, and administering growth factor.
Janssen Research & Development funded the study. Dr. Moreau had no relevant financial disclosures.
SAN DIEGO – Adding daratumumab (D) to lenalidomide and dexamethasone (Rd) significantly improved outcomes in relapsed and refractory multiple myeloma, even when patients had previously received lenalidomide, were refractory to bortezomib, or had high-risk tumor cytogenetics, based on updated analyses from the multicenter, randomized, phase III, open-label POLLUX trial.
The findings underscore the “significant benefit of combining daratumumab with lenalidomide and dexamethasone for relapsed or refractory multiple myeloma,” said lead investigator Philippe Moreau, MD, of University Hospital Hotel-Dieu in Nantes, France.
Among a large subgroup of 524 POLLUX patients who had received one to three prior lines of therapy, estimated median progression-free survival (PFS) has not been reached in the daratumumab, lenalidomide, and dexamethasone (DRd) arm, versus 18.4 months in the lenalidomide and dexamethasone (Rd) arm (hazard ratio, 0.36; 95% CI: 0.26 to 0.49; P less than .0001), Dr. Moreau said at the annual meeting of the American Society of Hematology.
That means adding daratumumab to Rd led to a 64% reduction in the risk of disease progression or death among patients with relapsed or refractory multiple myeloma, he noted. Fully 77% of DRd patients were alive without having progressed at 18 months, and responses “continued to deepen in the DRd group with longer follow-up,” he added.
Additional analyses supported the use of DRd in relapsed or refractory multiple myeloma, “irrespective of prior lenalidomide treatment or bortezomib refractoriness,” Dr. Moreau continued. He reported that DRd significantly improved PFS over Rd alone not only among 445 lenalidomide-naive patients (HR, 0.37; P less than .0001), but also among 91 lenalidomide-exposed patients (HR, 0.45; P = .04), 140 patients who were refractory to their most recent line of therapy (HR, 0.45; P = .001), and 99 bortezomib- refractory patients (HR 0.51; P = .02).
Daratumumab (Darzalex), a human CD38 IgG1k monoclonal antibody, was first approved as monotherapy for multiple myeloma in patients who had received at least three prior lines of therapy or had double-refractory disease. In 2016, results from the twin POLLUX and CASTOR studies won daratumumab a Food and Drug Administration breakthrough designation status for use with Rd in patients who had received at least one prior line.
The POLLUX trial included 569 patients with multiple myeloma who had received a median of 1 and up to 11 prior lines of therapy. Patients were randomized to either Rd alone or to Rd plus intravenous daratumumab (16 mg/kg) once a week during the first two 28-day treatment cycles, every 2 weeks during cycles 3-6, and once only on day 1 of subsequent cycles.
POLLUX patients were fairly heavily pretreated, Dr. Moreau noted. Thirteen percent had received three prior lines of therapy, 86% had received a proteasome inhibitor, 18% had received lenalidomide, 21% were refractory to bortezomib, and 28% were refractory to their most recent line of therapy.
Researchers performed “stringent, unbiased” assessments of minimal residual disease (MRD) negativity not only when a complete response was suspected, but also 3 and 6 months later, Dr. Moreau said. He emphasized that rates of MRD negativity in lenalidomide-exposed, bortezomib-refractory subgroups in POLLUX almost exactly matched those in the intent-to-treat population (25% on DRd vs. 6% on Rd; P less than .0001).
A total of 17% of DRd patients and 25% of Rd patients had high-risk cytogenetic profiles, and DRd performed well in these individuals, Dr. Moreau reported. Fully 85% of all evaluable high-risk patients had at least a partial response to DRd, and 33% had a complete response, versus only 67% and 6% of high-risk Rd patients, respectively. Among patients with standard-risk cytogenetics, rates of best overall response were 95% on DRd and 82% on Rd, and rates of complete response were 52% on DRd and 24% on Rd.
POLLUX yielded no new safety signals for DRd, Dr. Moreau said. Rates of primary and secondary malignancies were less than 2%. Neutropenia, the most common adverse effect, was managed by interrupting treatment, reducing the dose of lenalidomide, and administering growth factor.
Janssen Research & Development funded the study. Dr. Moreau had no relevant financial disclosures.
SAN DIEGO – Adding daratumumab (D) to lenalidomide and dexamethasone (Rd) significantly improved outcomes in relapsed and refractory multiple myeloma, even when patients had previously received lenalidomide, were refractory to bortezomib, or had high-risk tumor cytogenetics, based on updated analyses from the multicenter, randomized, phase III, open-label POLLUX trial.
The findings underscore the “significant benefit of combining daratumumab with lenalidomide and dexamethasone for relapsed or refractory multiple myeloma,” said lead investigator Philippe Moreau, MD, of University Hospital Hotel-Dieu in Nantes, France.
Among a large subgroup of 524 POLLUX patients who had received one to three prior lines of therapy, estimated median progression-free survival (PFS) has not been reached in the daratumumab, lenalidomide, and dexamethasone (DRd) arm, versus 18.4 months in the lenalidomide and dexamethasone (Rd) arm (hazard ratio, 0.36; 95% CI: 0.26 to 0.49; P less than .0001), Dr. Moreau said at the annual meeting of the American Society of Hematology.
That means adding daratumumab to Rd led to a 64% reduction in the risk of disease progression or death among patients with relapsed or refractory multiple myeloma, he noted. Fully 77% of DRd patients were alive without having progressed at 18 months, and responses “continued to deepen in the DRd group with longer follow-up,” he added.
Additional analyses supported the use of DRd in relapsed or refractory multiple myeloma, “irrespective of prior lenalidomide treatment or bortezomib refractoriness,” Dr. Moreau continued. He reported that DRd significantly improved PFS over Rd alone not only among 445 lenalidomide-naive patients (HR, 0.37; P less than .0001), but also among 91 lenalidomide-exposed patients (HR, 0.45; P = .04), 140 patients who were refractory to their most recent line of therapy (HR, 0.45; P = .001), and 99 bortezomib- refractory patients (HR 0.51; P = .02).
Daratumumab (Darzalex), a human CD38 IgG1k monoclonal antibody, was first approved as monotherapy for multiple myeloma in patients who had received at least three prior lines of therapy or had double-refractory disease. In 2016, results from the twin POLLUX and CASTOR studies won daratumumab a Food and Drug Administration breakthrough designation status for use with Rd in patients who had received at least one prior line.
The POLLUX trial included 569 patients with multiple myeloma who had received a median of 1 and up to 11 prior lines of therapy. Patients were randomized to either Rd alone or to Rd plus intravenous daratumumab (16 mg/kg) once a week during the first two 28-day treatment cycles, every 2 weeks during cycles 3-6, and once only on day 1 of subsequent cycles.
POLLUX patients were fairly heavily pretreated, Dr. Moreau noted. Thirteen percent had received three prior lines of therapy, 86% had received a proteasome inhibitor, 18% had received lenalidomide, 21% were refractory to bortezomib, and 28% were refractory to their most recent line of therapy.
Researchers performed “stringent, unbiased” assessments of minimal residual disease (MRD) negativity not only when a complete response was suspected, but also 3 and 6 months later, Dr. Moreau said. He emphasized that rates of MRD negativity in lenalidomide-exposed, bortezomib-refractory subgroups in POLLUX almost exactly matched those in the intent-to-treat population (25% on DRd vs. 6% on Rd; P less than .0001).
A total of 17% of DRd patients and 25% of Rd patients had high-risk cytogenetic profiles, and DRd performed well in these individuals, Dr. Moreau reported. Fully 85% of all evaluable high-risk patients had at least a partial response to DRd, and 33% had a complete response, versus only 67% and 6% of high-risk Rd patients, respectively. Among patients with standard-risk cytogenetics, rates of best overall response were 95% on DRd and 82% on Rd, and rates of complete response were 52% on DRd and 24% on Rd.
POLLUX yielded no new safety signals for DRd, Dr. Moreau said. Rates of primary and secondary malignancies were less than 2%. Neutropenia, the most common adverse effect, was managed by interrupting treatment, reducing the dose of lenalidomide, and administering growth factor.
Janssen Research & Development funded the study. Dr. Moreau had no relevant financial disclosures.
AT ASH 2016
Key clinical point: Adding daratumumab (D) to lenalidomide and dexamethasone (Rd) significantly improved outcomes in patients with relapsed and refractory multiple myeloma, regardless of factors such as bortezomib refractoriness, lenalidomide exposure, or high-risk tumor cytogenetics.
Major finding: DRd significantly improved PFS over Rd not only among 445 lenalidomide-naive patients (HR, 0.37; P less than .0001), but also among 91 lenalidomide-exposed patients (HR, 0.45; P = .04), 140 patients who were refractory to their most recent line of therapy (HR, 0.45; P = .001), and 99 bortezomib-refractory patients (HR 0.51; P = .02).
Data source: POLLUX, a multicenter, randomized, phase III, open-label trial of 569 patients with multiple myeloma who had received one or more previous lines of therapy.
Disclosures: Janssen Research & Development funded the study. Dr. Moreau had no relevant financial disclosures.
Intervention relieves distress in cancer patients
chemotherapy
Photo by Rhoda Baer
Results of a small study suggest a single dose of the hallucinogenic drug psilocybin, when combined with counseling, can significantly lessen psychological distress in cancer patients for months at a time.
The study showed that psychological counseling and a single dose of psilocybin brought relief from distress that lasted for more than 6 months in a majority of the subjects monitored.
This was based on clinical evaluation scores for anxiety and depression.
“Our results represent the strongest evidence to date of a clinical benefit from psilocybin therapy, with the potential to transform care for patients with cancer-related psychological distress,” said study author Stephen Ross, MD, of New York University School of Medicine in New York, New York.
“If larger clinical trials prove successful, then we could ultimately have available a safe, effective, and inexpensive medication—dispensed under strict control—to alleviate the distress that increases suicide rates among cancer patients.”
Dr Ross and his colleagues reported the results of their study in the Journal of Psychopharmacology alongside a related study and 11 accompanying editorials.
Dr Ross’s study included 29 patients with cancer-related anxiety and depression. Their mean age was 56, and 62% were female. Ninety percent were Caucasian, and 10% were classified as “other” race.
Patients had breast cancer (31%), reproductive cancers (28%), digestive cancers (17%), leukemia/lymphoma (14%), and other cancers (10%).
All patients had been diagnosed as suffering from serious psychological distress related to their disease.
Treatment
Half of the patients were randomly assigned to receive a 0.3 mg/kg dose of psilocybin, and half received a vitamin placebo (250 mg of niacin) known to produce a “rush” that mimics a hallucinogenic drug experience.
Approximately half way through the study’s monitoring period (after 7 weeks), all patients switched treatments. Those who initially received psilocybin took a single dose of niacin, and vice-versa. Neither patients nor researchers knew who had first received psilocybin or placebo.
All patients were provided with tailored counseling from a psychiatrist, psychologist, nurse, or social worker. And the patients were monitored for side effects and improvements in their mental state.
Safety
The researchers said there were no serious adverse events (AEs), either medical or psychiatric, that were attributed to psilocybin.
The most common medical AEs that were attributable to psilocybin were non-clinically significant elevations in blood pressure and heart rate (76%), headaches/migraines (28%), and nausea (14%).
The most common psychiatric AEs attributable to psilocybin were transient anxiety (17%) and transient psychotic-like symptoms (7%; 1 case of transient paranoid ideation and 1 case of transient thought disorder).
Efficacy
The researchers said that, prior to the crossover, psilocybin produced immediate, substantial, and sustained improvements in anxiety and depression.
Specifically, patients who received psilocybin first had significant improvements in responses on the Hospital Anxiety and Depression Scale and the Beck Depression Inventory, when compared to patients who received niacin first.
The differences were significant 1 day after the patients’ first session and 7 weeks after the first session (P≤0.01 for all).
At the 6.5-month follow-up, 60% to 80% of participants continued with clinically significant reductions in depression or anxiety.
The researchers said a key finding of this study was that improvements in clinical evaluation scores for anxiety and depression lasted for the study’s extended monitoring period, which was 8 months for those who took psilocybin first.
Patients also reported post-psilocybin improvements in their quality of life, such as going out more, greater energy, getting along better with family members, and doing well at work. Some reported variations of spirituality, unusual peacefulness, and increased feelings of altruism.
“Our study showed that psilocybin facilitated experiences that drove reductions in psychological distress,” said study author Anthony Bossis, PhD, of New York University School of Medicine. “And if it’s true for cancer care, then it could apply to other stressful medical conditions.”
He cautioned that patients should not consume psilocybin on their own or without supervision from a physician and a trained counselor.
“Psilocybin therapy may not work for everyone,” he noted. “And some groups, such as people with schizophrenia, as well as adolescents, should not be treated with it.” ![]()
chemotherapy
Photo by Rhoda Baer
Results of a small study suggest a single dose of the hallucinogenic drug psilocybin, when combined with counseling, can significantly lessen psychological distress in cancer patients for months at a time.
The study showed that psychological counseling and a single dose of psilocybin brought relief from distress that lasted for more than 6 months in a majority of the subjects monitored.
This was based on clinical evaluation scores for anxiety and depression.
“Our results represent the strongest evidence to date of a clinical benefit from psilocybin therapy, with the potential to transform care for patients with cancer-related psychological distress,” said study author Stephen Ross, MD, of New York University School of Medicine in New York, New York.
“If larger clinical trials prove successful, then we could ultimately have available a safe, effective, and inexpensive medication—dispensed under strict control—to alleviate the distress that increases suicide rates among cancer patients.”
Dr Ross and his colleagues reported the results of their study in the Journal of Psychopharmacology alongside a related study and 11 accompanying editorials.
Dr Ross’s study included 29 patients with cancer-related anxiety and depression. Their mean age was 56, and 62% were female. Ninety percent were Caucasian, and 10% were classified as “other” race.
Patients had breast cancer (31%), reproductive cancers (28%), digestive cancers (17%), leukemia/lymphoma (14%), and other cancers (10%).
All patients had been diagnosed as suffering from serious psychological distress related to their disease.
Treatment
Half of the patients were randomly assigned to receive a 0.3 mg/kg dose of psilocybin, and half received a vitamin placebo (250 mg of niacin) known to produce a “rush” that mimics a hallucinogenic drug experience.
Approximately half way through the study’s monitoring period (after 7 weeks), all patients switched treatments. Those who initially received psilocybin took a single dose of niacin, and vice-versa. Neither patients nor researchers knew who had first received psilocybin or placebo.
All patients were provided with tailored counseling from a psychiatrist, psychologist, nurse, or social worker. And the patients were monitored for side effects and improvements in their mental state.
Safety
The researchers said there were no serious adverse events (AEs), either medical or psychiatric, that were attributed to psilocybin.
The most common medical AEs that were attributable to psilocybin were non-clinically significant elevations in blood pressure and heart rate (76%), headaches/migraines (28%), and nausea (14%).
The most common psychiatric AEs attributable to psilocybin were transient anxiety (17%) and transient psychotic-like symptoms (7%; 1 case of transient paranoid ideation and 1 case of transient thought disorder).
Efficacy
The researchers said that, prior to the crossover, psilocybin produced immediate, substantial, and sustained improvements in anxiety and depression.
Specifically, patients who received psilocybin first had significant improvements in responses on the Hospital Anxiety and Depression Scale and the Beck Depression Inventory, when compared to patients who received niacin first.
The differences were significant 1 day after the patients’ first session and 7 weeks after the first session (P≤0.01 for all).
At the 6.5-month follow-up, 60% to 80% of participants continued with clinically significant reductions in depression or anxiety.
The researchers said a key finding of this study was that improvements in clinical evaluation scores for anxiety and depression lasted for the study’s extended monitoring period, which was 8 months for those who took psilocybin first.
Patients also reported post-psilocybin improvements in their quality of life, such as going out more, greater energy, getting along better with family members, and doing well at work. Some reported variations of spirituality, unusual peacefulness, and increased feelings of altruism.
“Our study showed that psilocybin facilitated experiences that drove reductions in psychological distress,” said study author Anthony Bossis, PhD, of New York University School of Medicine. “And if it’s true for cancer care, then it could apply to other stressful medical conditions.”
He cautioned that patients should not consume psilocybin on their own or without supervision from a physician and a trained counselor.
“Psilocybin therapy may not work for everyone,” he noted. “And some groups, such as people with schizophrenia, as well as adolescents, should not be treated with it.” ![]()
chemotherapy
Photo by Rhoda Baer
Results of a small study suggest a single dose of the hallucinogenic drug psilocybin, when combined with counseling, can significantly lessen psychological distress in cancer patients for months at a time.
The study showed that psychological counseling and a single dose of psilocybin brought relief from distress that lasted for more than 6 months in a majority of the subjects monitored.
This was based on clinical evaluation scores for anxiety and depression.
“Our results represent the strongest evidence to date of a clinical benefit from psilocybin therapy, with the potential to transform care for patients with cancer-related psychological distress,” said study author Stephen Ross, MD, of New York University School of Medicine in New York, New York.
“If larger clinical trials prove successful, then we could ultimately have available a safe, effective, and inexpensive medication—dispensed under strict control—to alleviate the distress that increases suicide rates among cancer patients.”
Dr Ross and his colleagues reported the results of their study in the Journal of Psychopharmacology alongside a related study and 11 accompanying editorials.
Dr Ross’s study included 29 patients with cancer-related anxiety and depression. Their mean age was 56, and 62% were female. Ninety percent were Caucasian, and 10% were classified as “other” race.
Patients had breast cancer (31%), reproductive cancers (28%), digestive cancers (17%), leukemia/lymphoma (14%), and other cancers (10%).
All patients had been diagnosed as suffering from serious psychological distress related to their disease.
Treatment
Half of the patients were randomly assigned to receive a 0.3 mg/kg dose of psilocybin, and half received a vitamin placebo (250 mg of niacin) known to produce a “rush” that mimics a hallucinogenic drug experience.
Approximately half way through the study’s monitoring period (after 7 weeks), all patients switched treatments. Those who initially received psilocybin took a single dose of niacin, and vice-versa. Neither patients nor researchers knew who had first received psilocybin or placebo.
All patients were provided with tailored counseling from a psychiatrist, psychologist, nurse, or social worker. And the patients were monitored for side effects and improvements in their mental state.
Safety
The researchers said there were no serious adverse events (AEs), either medical or psychiatric, that were attributed to psilocybin.
The most common medical AEs that were attributable to psilocybin were non-clinically significant elevations in blood pressure and heart rate (76%), headaches/migraines (28%), and nausea (14%).
The most common psychiatric AEs attributable to psilocybin were transient anxiety (17%) and transient psychotic-like symptoms (7%; 1 case of transient paranoid ideation and 1 case of transient thought disorder).
Efficacy
The researchers said that, prior to the crossover, psilocybin produced immediate, substantial, and sustained improvements in anxiety and depression.
Specifically, patients who received psilocybin first had significant improvements in responses on the Hospital Anxiety and Depression Scale and the Beck Depression Inventory, when compared to patients who received niacin first.
The differences were significant 1 day after the patients’ first session and 7 weeks after the first session (P≤0.01 for all).
At the 6.5-month follow-up, 60% to 80% of participants continued with clinically significant reductions in depression or anxiety.
The researchers said a key finding of this study was that improvements in clinical evaluation scores for anxiety and depression lasted for the study’s extended monitoring period, which was 8 months for those who took psilocybin first.
Patients also reported post-psilocybin improvements in their quality of life, such as going out more, greater energy, getting along better with family members, and doing well at work. Some reported variations of spirituality, unusual peacefulness, and increased feelings of altruism.
“Our study showed that psilocybin facilitated experiences that drove reductions in psychological distress,” said study author Anthony Bossis, PhD, of New York University School of Medicine. “And if it’s true for cancer care, then it could apply to other stressful medical conditions.”
He cautioned that patients should not consume psilocybin on their own or without supervision from a physician and a trained counselor.
“Psilocybin therapy may not work for everyone,” he noted. “And some groups, such as people with schizophrenia, as well as adolescents, should not be treated with it.”