User login
NASH liver transplant mortality differs from non-NASH
Cardiovascular complications and sepsis are two major concerns after liver transplant for nonalcoholic steatohepatitis, more so than after liver transplant for other indications.
The finding comes from what the authors called the first systematic review and meta-analysis to investigate the cumulative clinical experience of liver transplant for NASH, wrote Dr. Xiaofei Wang and colleagues in the March issue of Clinical Gastroenterology and Hepatology (doi: 10.1016/j.cgh.2013.09.023).
In their analysis, Dr. Wang, of the Affiliated Hospital of North Sichuan Medical College, Nanchong, China, and colleagues performed a search of PubMed, Embase, the Cochrane Library, and the Web of Science for studies published through Sept. 1, 2012, that looked at liver transplant for nonalcoholic fatty liver disease (NAFLD) or NASH. Reviews were excluded, as were studies in which NASH patients could not be separated out from non-NASH liver transplant recipients.
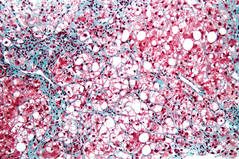
Overall, nine studies were analyzed, all of which measured outcomes after liver transplant for NASH. Other indications for transplant included primary biliary cirrhosis/primary sclerosing cholangitis, alcoholic liver disease, hepatitis C, hepatitis B, and cryptogenic cirrhosis.
The researchers found that, on the whole, survival rates at post-transplant years 1, 3, and 5 did not significantly differ between NASH patients and their non-NASH counterparts (1-year odds ratio, 0.77; 95% confidence interval, 0.59-1.00; P = .05; 3-year OR, 0.97; 95% CI, 0.67-1.40; P = .86; 5-year OR, 1.09; 95% CI, 0.77-1.56; P = .63).
Nevertheless, NASH patients registered more deaths due to "cardiovascular events" compared with non-NASH liver transplant recipients, the authors wrote (OR, 1.65; 95% CI, 1.01-2.70; P = .05).
The authors also found that NASH patients were significantly more likely to die of sepsis post transplant than were non-NASH patients (OR, 1.71; 95% CI, 1.17-2.50; P = .006).
On the other hand, NASH patients had fewer deaths caused by graft failure than did patients undergoing liver transplant for other indications (OR, 0.21; 95% CI, 0.05-0.89; P = .03).
In an attempt to explain their findings, the authors pointed out that while cardiovascular events are the top cause of non–graft-related mortality in all liver transplants, NASH patients might be especially susceptible, given that their diagnosis is "frequently associated" with cardiovascular risk factors such as obesity, diabetes, and hypertension. Indeed, these same characteristics might also predispose these patients to postoperative infection and sepsis, they added.
And regarding the finding that graft-related mortality is actually lower among NASH patients, Dr. Wang and colleagues postulated that this may be due to the "lower likelihood of disease recurrence, and this may mean lower rates of graft failure compared with other liver diseases, such as hepatitis C virus and hepatitis B virus infection."
The authors conceded that there were several limitations to their analysis, not the least being heterogeneity between included studies. Moreover, two large population-based studies using national databases were ultimately excluded due to patient duplication and poor accuracy with regard to the cause of death.
In any case, the current analysis shows that "more attention and careful consideration are required in selecting patients with NASH for liver transplant, along with aggressive management of cardiovascular complications and sepsis after transplantation."
The authors disclosed no conflicts of interest related to this study. They wrote that funding was provided by the scientific research development project of North Sichuan Medical College.
Cardiovascular complications and sepsis are two major concerns after liver transplant for nonalcoholic steatohepatitis, more so than after liver transplant for other indications.
The finding comes from what the authors called the first systematic review and meta-analysis to investigate the cumulative clinical experience of liver transplant for NASH, wrote Dr. Xiaofei Wang and colleagues in the March issue of Clinical Gastroenterology and Hepatology (doi: 10.1016/j.cgh.2013.09.023).
In their analysis, Dr. Wang, of the Affiliated Hospital of North Sichuan Medical College, Nanchong, China, and colleagues performed a search of PubMed, Embase, the Cochrane Library, and the Web of Science for studies published through Sept. 1, 2012, that looked at liver transplant for nonalcoholic fatty liver disease (NAFLD) or NASH. Reviews were excluded, as were studies in which NASH patients could not be separated out from non-NASH liver transplant recipients.
Overall, nine studies were analyzed, all of which measured outcomes after liver transplant for NASH. Other indications for transplant included primary biliary cirrhosis/primary sclerosing cholangitis, alcoholic liver disease, hepatitis C, hepatitis B, and cryptogenic cirrhosis.
The researchers found that, on the whole, survival rates at post-transplant years 1, 3, and 5 did not significantly differ between NASH patients and their non-NASH counterparts (1-year odds ratio, 0.77; 95% confidence interval, 0.59-1.00; P = .05; 3-year OR, 0.97; 95% CI, 0.67-1.40; P = .86; 5-year OR, 1.09; 95% CI, 0.77-1.56; P = .63).
Nevertheless, NASH patients registered more deaths due to "cardiovascular events" compared with non-NASH liver transplant recipients, the authors wrote (OR, 1.65; 95% CI, 1.01-2.70; P = .05).
The authors also found that NASH patients were significantly more likely to die of sepsis post transplant than were non-NASH patients (OR, 1.71; 95% CI, 1.17-2.50; P = .006).
On the other hand, NASH patients had fewer deaths caused by graft failure than did patients undergoing liver transplant for other indications (OR, 0.21; 95% CI, 0.05-0.89; P = .03).
In an attempt to explain their findings, the authors pointed out that while cardiovascular events are the top cause of non–graft-related mortality in all liver transplants, NASH patients might be especially susceptible, given that their diagnosis is "frequently associated" with cardiovascular risk factors such as obesity, diabetes, and hypertension. Indeed, these same characteristics might also predispose these patients to postoperative infection and sepsis, they added.
And regarding the finding that graft-related mortality is actually lower among NASH patients, Dr. Wang and colleagues postulated that this may be due to the "lower likelihood of disease recurrence, and this may mean lower rates of graft failure compared with other liver diseases, such as hepatitis C virus and hepatitis B virus infection."
The authors conceded that there were several limitations to their analysis, not the least being heterogeneity between included studies. Moreover, two large population-based studies using national databases were ultimately excluded due to patient duplication and poor accuracy with regard to the cause of death.
In any case, the current analysis shows that "more attention and careful consideration are required in selecting patients with NASH for liver transplant, along with aggressive management of cardiovascular complications and sepsis after transplantation."
The authors disclosed no conflicts of interest related to this study. They wrote that funding was provided by the scientific research development project of North Sichuan Medical College.
Cardiovascular complications and sepsis are two major concerns after liver transplant for nonalcoholic steatohepatitis, more so than after liver transplant for other indications.
The finding comes from what the authors called the first systematic review and meta-analysis to investigate the cumulative clinical experience of liver transplant for NASH, wrote Dr. Xiaofei Wang and colleagues in the March issue of Clinical Gastroenterology and Hepatology (doi: 10.1016/j.cgh.2013.09.023).
In their analysis, Dr. Wang, of the Affiliated Hospital of North Sichuan Medical College, Nanchong, China, and colleagues performed a search of PubMed, Embase, the Cochrane Library, and the Web of Science for studies published through Sept. 1, 2012, that looked at liver transplant for nonalcoholic fatty liver disease (NAFLD) or NASH. Reviews were excluded, as were studies in which NASH patients could not be separated out from non-NASH liver transplant recipients.
Overall, nine studies were analyzed, all of which measured outcomes after liver transplant for NASH. Other indications for transplant included primary biliary cirrhosis/primary sclerosing cholangitis, alcoholic liver disease, hepatitis C, hepatitis B, and cryptogenic cirrhosis.
The researchers found that, on the whole, survival rates at post-transplant years 1, 3, and 5 did not significantly differ between NASH patients and their non-NASH counterparts (1-year odds ratio, 0.77; 95% confidence interval, 0.59-1.00; P = .05; 3-year OR, 0.97; 95% CI, 0.67-1.40; P = .86; 5-year OR, 1.09; 95% CI, 0.77-1.56; P = .63).
Nevertheless, NASH patients registered more deaths due to "cardiovascular events" compared with non-NASH liver transplant recipients, the authors wrote (OR, 1.65; 95% CI, 1.01-2.70; P = .05).
The authors also found that NASH patients were significantly more likely to die of sepsis post transplant than were non-NASH patients (OR, 1.71; 95% CI, 1.17-2.50; P = .006).
On the other hand, NASH patients had fewer deaths caused by graft failure than did patients undergoing liver transplant for other indications (OR, 0.21; 95% CI, 0.05-0.89; P = .03).
In an attempt to explain their findings, the authors pointed out that while cardiovascular events are the top cause of non–graft-related mortality in all liver transplants, NASH patients might be especially susceptible, given that their diagnosis is "frequently associated" with cardiovascular risk factors such as obesity, diabetes, and hypertension. Indeed, these same characteristics might also predispose these patients to postoperative infection and sepsis, they added.
And regarding the finding that graft-related mortality is actually lower among NASH patients, Dr. Wang and colleagues postulated that this may be due to the "lower likelihood of disease recurrence, and this may mean lower rates of graft failure compared with other liver diseases, such as hepatitis C virus and hepatitis B virus infection."
The authors conceded that there were several limitations to their analysis, not the least being heterogeneity between included studies. Moreover, two large population-based studies using national databases were ultimately excluded due to patient duplication and poor accuracy with regard to the cause of death.
In any case, the current analysis shows that "more attention and careful consideration are required in selecting patients with NASH for liver transplant, along with aggressive management of cardiovascular complications and sepsis after transplantation."
The authors disclosed no conflicts of interest related to this study. They wrote that funding was provided by the scientific research development project of North Sichuan Medical College.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Major finding: Patients undergoing liver transplant for nonalcoholic steatohepatitis are at a greater risk for death due to sepsis and cardiovascular complications.
Data source: A meta-analysis of nine studies published through September 2012.
Disclosures: The authors disclosed no conflicts of interest related to this study. They wrote that funding was provided by the scientific research development project of North Sichuan Medical College.
Pancytopenia warning added to label of hepatitis C drug boceprevir
A warning about cases of pancytopenia linked to the use of the oral hepatitis C antiviral drug boceprevir has been added to the drug’s label, the Food and Drug Administration has announced.
There have been postmarketing reports of "serious cases" of pancytopenia in people treated with boceprevir in combination with peginterferon alfa and ribavirin, according to the FDA. This information has been added to the "Warnings and Precautions" section of the label, with the recommendation that "complete blood counts (with white blood cell differential counts) should be obtained at pretreatment, and at treatment weeks 2, 4, 8, and 12, and should be monitored closely at other time points, as clinically appropriate."
Approved in 2011, boceprevir is an HCV Ns3/4A protease inhibitor approved for the treatment of chronic hepatitis C (CHC) genotype 1 infection, in combination with peginterferon alfa and ribavirin, in adults with compensated liver disease, including cirrhosis, who are previously untreated or who have failed previous interferon and ribavirin therapy, including prior null responders. Boceprevir, in a 200-mg capsule formulation, is marketed as Victrelis by Merck Sharp & Dohme; the recommended dose is 800 mg, three times a day.
The boceprevir Medication Guide will also be updated with this information, according to the FDA.
Serious adverse events associated with boceprevir should be reported to the FDA at 800-332-1088 or at MedWatch.
A warning about cases of pancytopenia linked to the use of the oral hepatitis C antiviral drug boceprevir has been added to the drug’s label, the Food and Drug Administration has announced.
There have been postmarketing reports of "serious cases" of pancytopenia in people treated with boceprevir in combination with peginterferon alfa and ribavirin, according to the FDA. This information has been added to the "Warnings and Precautions" section of the label, with the recommendation that "complete blood counts (with white blood cell differential counts) should be obtained at pretreatment, and at treatment weeks 2, 4, 8, and 12, and should be monitored closely at other time points, as clinically appropriate."
Approved in 2011, boceprevir is an HCV Ns3/4A protease inhibitor approved for the treatment of chronic hepatitis C (CHC) genotype 1 infection, in combination with peginterferon alfa and ribavirin, in adults with compensated liver disease, including cirrhosis, who are previously untreated or who have failed previous interferon and ribavirin therapy, including prior null responders. Boceprevir, in a 200-mg capsule formulation, is marketed as Victrelis by Merck Sharp & Dohme; the recommended dose is 800 mg, three times a day.
The boceprevir Medication Guide will also be updated with this information, according to the FDA.
Serious adverse events associated with boceprevir should be reported to the FDA at 800-332-1088 or at MedWatch.
A warning about cases of pancytopenia linked to the use of the oral hepatitis C antiviral drug boceprevir has been added to the drug’s label, the Food and Drug Administration has announced.
There have been postmarketing reports of "serious cases" of pancytopenia in people treated with boceprevir in combination with peginterferon alfa and ribavirin, according to the FDA. This information has been added to the "Warnings and Precautions" section of the label, with the recommendation that "complete blood counts (with white blood cell differential counts) should be obtained at pretreatment, and at treatment weeks 2, 4, 8, and 12, and should be monitored closely at other time points, as clinically appropriate."
Approved in 2011, boceprevir is an HCV Ns3/4A protease inhibitor approved for the treatment of chronic hepatitis C (CHC) genotype 1 infection, in combination with peginterferon alfa and ribavirin, in adults with compensated liver disease, including cirrhosis, who are previously untreated or who have failed previous interferon and ribavirin therapy, including prior null responders. Boceprevir, in a 200-mg capsule formulation, is marketed as Victrelis by Merck Sharp & Dohme; the recommended dose is 800 mg, three times a day.
The boceprevir Medication Guide will also be updated with this information, according to the FDA.
Serious adverse events associated with boceprevir should be reported to the FDA at 800-332-1088 or at MedWatch.
Daclatasvir-sofosbuvir combo scores in resistant hepatitis C
A combined regimen of daclatasvir and sofosbuvir achieved sustained virologic response rates greater than 90% in an industry-sponsored trial involving patients with chronic HCV, according to study published online Jan. 16 in the New England Journal of Medicine.
The once-daily oral treatment was effective against HCV type 1 among patients with any viral subtype, as well as among patients who had failed to respond to or had developed resistance to protease inhibitors. The latter group currently has no other treatment options. It also was effective against HCV types 2 and 3, said Dr. Mark S. Sulkowski of Johns Hopkins University, Baltimore, and his associates (N. Engl. J. Med. 2014;370:211-21).
In particular, the daclatasvir-sofosbuvir combination was very effective in patients "with characteristics that were previously associated with a poor response to treatment – HCV genotypes 1a and 3, the non-CC IL28B genotype, and black race," the investigators noted.
Dr. Sulkowski and his colleagues assessed the combination therapy in an open-label study involving 211 adults treated during a 17-month period at 18 medical centers across the United States.
Initially, 44 previously untreated patients with HCV 1 infection and 44 patients with HCV 2 or 3 were randomly assigned to receive daclatasvir plus sofosbuvir, with or without concurrent ribavirin, for 24 weeks. Then the study was expanded to include another 123 patients with HCV 1 infection: 82 who were previously untreated were randomly assigned to receive the combination therapy, with or without ribavirin, for 12 weeks; and the remaining 41 who had failed on telaprevir or boceprevir plus peginterferon alfa-ribavirin were assigned to receive the combination therapy, with or without ribavirin, for 24 weeks.
The primary efficacy endpoint was a sustained virologic response (defined as an HCV RNA level of less than 25 IU/mL) at week 12 after the end of treatment.
That rate was 98% (164 of 167 patients) among those who had HCV 1 infection. Of the three patients in that group classified as not having a sustained virologic response at week 12, two missed their assessment visit at 12 weeks but showed a sustained virologic response at 24 weeks, and the third was lost to follow-up.

The rate of sustained virologic response was 91% (40 of 44 patients) among those who had HCV 2 or 3 infection. Individually, the rate was 92% in patients with HCV 2 and 89% among those with HCV 3, the investigators said.
The high rates of sustained virologic response were consistent across all subgroups of patients, including 129 of the 132 (98%) who had genotype 1a; 57 of the 61 (93%) who had genotype IL28B; 25 of the 26 (96%) who were black; 9 of the 10 (90%) who were of "other" race/ethnicity; 85 of the 90 (94%) who received concurrent ribavirin; 119 of the 121 (98%) who did not receive ribavirin; and 40 of the 41 (98%) who had failed on or developed resistance to protease inhibitors.
"This represents proof of concept that a sustained virologic response can be achieved in patients in whom previous treatment with telaprevir or boceprevir had failed, including patients who have persistent HCV variants with resistance to protease inhibitors," Dr. Sulkowski and his associates said.
Two patients discontinued treatment because of adverse events. One patient developed fibromyalgia, and another had a stroke. Both had shown a sustained virologic response to the combination therapy. The most common adverse events were fatigue, headache, and nausea. The most common grade 3 or 4 laboratory abnormalities were low phosphorous levels and elevated glucose levels.
The study was supported by Bristol-Myers Squibb and Pharmasset (now Gilead). Dr. Sulkowski reported financial ties to Bristol-Myers Squibb and Gilead, in addition to other pharmaceutical companies. His associates reported ties to numerous industry sources.
A combined regimen of daclatasvir and sofosbuvir achieved sustained virologic response rates greater than 90% in an industry-sponsored trial involving patients with chronic HCV, according to study published online Jan. 16 in the New England Journal of Medicine.
The once-daily oral treatment was effective against HCV type 1 among patients with any viral subtype, as well as among patients who had failed to respond to or had developed resistance to protease inhibitors. The latter group currently has no other treatment options. It also was effective against HCV types 2 and 3, said Dr. Mark S. Sulkowski of Johns Hopkins University, Baltimore, and his associates (N. Engl. J. Med. 2014;370:211-21).
In particular, the daclatasvir-sofosbuvir combination was very effective in patients "with characteristics that were previously associated with a poor response to treatment – HCV genotypes 1a and 3, the non-CC IL28B genotype, and black race," the investigators noted.
Dr. Sulkowski and his colleagues assessed the combination therapy in an open-label study involving 211 adults treated during a 17-month period at 18 medical centers across the United States.
Initially, 44 previously untreated patients with HCV 1 infection and 44 patients with HCV 2 or 3 were randomly assigned to receive daclatasvir plus sofosbuvir, with or without concurrent ribavirin, for 24 weeks. Then the study was expanded to include another 123 patients with HCV 1 infection: 82 who were previously untreated were randomly assigned to receive the combination therapy, with or without ribavirin, for 12 weeks; and the remaining 41 who had failed on telaprevir or boceprevir plus peginterferon alfa-ribavirin were assigned to receive the combination therapy, with or without ribavirin, for 24 weeks.
The primary efficacy endpoint was a sustained virologic response (defined as an HCV RNA level of less than 25 IU/mL) at week 12 after the end of treatment.
That rate was 98% (164 of 167 patients) among those who had HCV 1 infection. Of the three patients in that group classified as not having a sustained virologic response at week 12, two missed their assessment visit at 12 weeks but showed a sustained virologic response at 24 weeks, and the third was lost to follow-up.
The rate of sustained virologic response was 91% (40 of 44 patients) among those who had HCV 2 or 3 infection. Individually, the rate was 92% in patients with HCV 2 and 89% among those with HCV 3, the investigators said.
The high rates of sustained virologic response were consistent across all subgroups of patients, including 129 of the 132 (98%) who had genotype 1a; 57 of the 61 (93%) who had genotype IL28B; 25 of the 26 (96%) who were black; 9 of the 10 (90%) who were of "other" race/ethnicity; 85 of the 90 (94%) who received concurrent ribavirin; 119 of the 121 (98%) who did not receive ribavirin; and 40 of the 41 (98%) who had failed on or developed resistance to protease inhibitors.
"This represents proof of concept that a sustained virologic response can be achieved in patients in whom previous treatment with telaprevir or boceprevir had failed, including patients who have persistent HCV variants with resistance to protease inhibitors," Dr. Sulkowski and his associates said.
Two patients discontinued treatment because of adverse events. One patient developed fibromyalgia, and another had a stroke. Both had shown a sustained virologic response to the combination therapy. The most common adverse events were fatigue, headache, and nausea. The most common grade 3 or 4 laboratory abnormalities were low phosphorous levels and elevated glucose levels.
The study was supported by Bristol-Myers Squibb and Pharmasset (now Gilead). Dr. Sulkowski reported financial ties to Bristol-Myers Squibb and Gilead, in addition to other pharmaceutical companies. His associates reported ties to numerous industry sources.
A combined regimen of daclatasvir and sofosbuvir achieved sustained virologic response rates greater than 90% in an industry-sponsored trial involving patients with chronic HCV, according to study published online Jan. 16 in the New England Journal of Medicine.
The once-daily oral treatment was effective against HCV type 1 among patients with any viral subtype, as well as among patients who had failed to respond to or had developed resistance to protease inhibitors. The latter group currently has no other treatment options. It also was effective against HCV types 2 and 3, said Dr. Mark S. Sulkowski of Johns Hopkins University, Baltimore, and his associates (N. Engl. J. Med. 2014;370:211-21).
In particular, the daclatasvir-sofosbuvir combination was very effective in patients "with characteristics that were previously associated with a poor response to treatment – HCV genotypes 1a and 3, the non-CC IL28B genotype, and black race," the investigators noted.
Dr. Sulkowski and his colleagues assessed the combination therapy in an open-label study involving 211 adults treated during a 17-month period at 18 medical centers across the United States.
Initially, 44 previously untreated patients with HCV 1 infection and 44 patients with HCV 2 or 3 were randomly assigned to receive daclatasvir plus sofosbuvir, with or without concurrent ribavirin, for 24 weeks. Then the study was expanded to include another 123 patients with HCV 1 infection: 82 who were previously untreated were randomly assigned to receive the combination therapy, with or without ribavirin, for 12 weeks; and the remaining 41 who had failed on telaprevir or boceprevir plus peginterferon alfa-ribavirin were assigned to receive the combination therapy, with or without ribavirin, for 24 weeks.
The primary efficacy endpoint was a sustained virologic response (defined as an HCV RNA level of less than 25 IU/mL) at week 12 after the end of treatment.
That rate was 98% (164 of 167 patients) among those who had HCV 1 infection. Of the three patients in that group classified as not having a sustained virologic response at week 12, two missed their assessment visit at 12 weeks but showed a sustained virologic response at 24 weeks, and the third was lost to follow-up.
The rate of sustained virologic response was 91% (40 of 44 patients) among those who had HCV 2 or 3 infection. Individually, the rate was 92% in patients with HCV 2 and 89% among those with HCV 3, the investigators said.
The high rates of sustained virologic response were consistent across all subgroups of patients, including 129 of the 132 (98%) who had genotype 1a; 57 of the 61 (93%) who had genotype IL28B; 25 of the 26 (96%) who were black; 9 of the 10 (90%) who were of "other" race/ethnicity; 85 of the 90 (94%) who received concurrent ribavirin; 119 of the 121 (98%) who did not receive ribavirin; and 40 of the 41 (98%) who had failed on or developed resistance to protease inhibitors.
"This represents proof of concept that a sustained virologic response can be achieved in patients in whom previous treatment with telaprevir or boceprevir had failed, including patients who have persistent HCV variants with resistance to protease inhibitors," Dr. Sulkowski and his associates said.
Two patients discontinued treatment because of adverse events. One patient developed fibromyalgia, and another had a stroke. Both had shown a sustained virologic response to the combination therapy. The most common adverse events were fatigue, headache, and nausea. The most common grade 3 or 4 laboratory abnormalities were low phosphorous levels and elevated glucose levels.
The study was supported by Bristol-Myers Squibb and Pharmasset (now Gilead). Dr. Sulkowski reported financial ties to Bristol-Myers Squibb and Gilead, in addition to other pharmaceutical companies. His associates reported ties to numerous industry sources.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Major finding: A total of 98% of patients with chronic HCV 1 and 91% of those with chronic HCV 2 or 3 achieved a sustained virologic response on combined daclatasvir plus sofosbuvir once-daily oral therapy.
Data Source: An open-label randomized trial involving 211 adults with chronic HCV who were treated with combined daclatasvir plus sofosbuvir for either 12 or 24 weeks.
Disclosures: The study was supported by Bristol-Myers Squibb and Pharmasset (now Gilead). Dr. Sulkowski reported financial ties to Bristol-Myers Squibb and Gilead, in addition to other pharmaceutical companies. His associates reported ties to numerous industry sources.
New website provides guidance on treating hepatitis C, with regular updates
A new website has been launched that provides the latest information to help guide clinicians on treating patients with chronic hepatitis C and will be updated regularly with developments in this rapidly changing area of medicine.
A collaboration of the American Association for the Study of Liver Diseases (AASLD) and the Infectious Diseases Society of America (IDSA) with the International Antiviral Society-USA (IAS-USA), the website – HCVguidelines.org – provides evidence-based consensus recommendations for screening and treating people with hepatitis C virus (HCV).

The recommendations were developed by a panel of 27 liver disease and infectious diseases specialists and a patient advocate and are intended for "health care providers who treat the disease and others who need updated information on the best practices," according to a statement issued by IDSA and AASLD announcing the launch of the website, which became available on Jan. 29. Examples of the information provided include guidance on the use of direct-acting HCV antivirals, with and without interferon for the most common type of HCV, genotype 1; treatment of different subgroups, such as those coinfected with HIV, those with decompensated liver disease, and liver transplant recipients; and regimens for treatment of interferon-ineligible and interferon-eligible patients.
The website is a living document and will be updated regularly with new recommendations that reflect new research, Food and Drug Administration (FDA) approvals of new drugs, changing recommendations for monitoring patients, and other developments. This aspect is "absolutely necessary in this field" because of the many recent developments and multiple drugs that are in development and approaching approval, Dr. Michael Saag, the panel cochair for IAS-USA, said during a telephone briefing held to announce the launch.
Changes can be made weekly or monthly, "so that clinicians can have confidence that, when they go to this website, they will get the most accurate and up-to-date guidance on the treatment of hepatitis C," added Dr. Saag, professor of medicine and director of the division of infectious diseases at the University of Alabama at Birmingham.
During the briefing, Dr. Henry Masur of the National Institutes of Health, Bethesda, Md., pointed out that, for the first time, treatments that cure hepatitis C are available, which are more effective than previous treatments and easier to tolerate. "What we’re eager to see is that these drugs are used by practitioners to treat the millions of patients who have hepatitis C both in North America and elsewhere ... and it’s important that clinicians have access to guidance on how best to use these drugs as new trials are quickly done and new information becomes available."
An estimated 3-4 million people in the United States are infected with HCV and could develop chronic liver disease; the cure rate for chronic HCV is now close to 95% with currently available treatments.
Noting that the next-generation antivirals have the potential to cure most patients with hepatitis C, Dr. David Thomas, professor of medicine and chair of the division of infectious diseases at Johns Hopkins University, Baltimore, said that the website is expected to be used not only by clinicians who are well versed in prescribing antiviral drugs, "but also by many who are inexperienced or even new to the treatment of hepatitis C." This includes gastroenterologists and infectious disease clinicians who have not been treating patients with hepatitis C and may include primary care physicians as well, he added. Dr. Thomas was the panel cochair for IDSA.
Recent developments in the treatment of hepatitis C include the FDA approvals of sofosbuvir and simeprevir late last year for treating chronic HCV, considered major advances in treatment.
The website is HCVguidelines.org. More information on hepatitis C is available at www.idsociety.org/HCV.
A new website has been launched that provides the latest information to help guide clinicians on treating patients with chronic hepatitis C and will be updated regularly with developments in this rapidly changing area of medicine.
A collaboration of the American Association for the Study of Liver Diseases (AASLD) and the Infectious Diseases Society of America (IDSA) with the International Antiviral Society-USA (IAS-USA), the website – HCVguidelines.org – provides evidence-based consensus recommendations for screening and treating people with hepatitis C virus (HCV).
The recommendations were developed by a panel of 27 liver disease and infectious diseases specialists and a patient advocate and are intended for "health care providers who treat the disease and others who need updated information on the best practices," according to a statement issued by IDSA and AASLD announcing the launch of the website, which became available on Jan. 29. Examples of the information provided include guidance on the use of direct-acting HCV antivirals, with and without interferon for the most common type of HCV, genotype 1; treatment of different subgroups, such as those coinfected with HIV, those with decompensated liver disease, and liver transplant recipients; and regimens for treatment of interferon-ineligible and interferon-eligible patients.
The website is a living document and will be updated regularly with new recommendations that reflect new research, Food and Drug Administration (FDA) approvals of new drugs, changing recommendations for monitoring patients, and other developments. This aspect is "absolutely necessary in this field" because of the many recent developments and multiple drugs that are in development and approaching approval, Dr. Michael Saag, the panel cochair for IAS-USA, said during a telephone briefing held to announce the launch.
Changes can be made weekly or monthly, "so that clinicians can have confidence that, when they go to this website, they will get the most accurate and up-to-date guidance on the treatment of hepatitis C," added Dr. Saag, professor of medicine and director of the division of infectious diseases at the University of Alabama at Birmingham.
During the briefing, Dr. Henry Masur of the National Institutes of Health, Bethesda, Md., pointed out that, for the first time, treatments that cure hepatitis C are available, which are more effective than previous treatments and easier to tolerate. "What we’re eager to see is that these drugs are used by practitioners to treat the millions of patients who have hepatitis C both in North America and elsewhere ... and it’s important that clinicians have access to guidance on how best to use these drugs as new trials are quickly done and new information becomes available."
An estimated 3-4 million people in the United States are infected with HCV and could develop chronic liver disease; the cure rate for chronic HCV is now close to 95% with currently available treatments.
Noting that the next-generation antivirals have the potential to cure most patients with hepatitis C, Dr. David Thomas, professor of medicine and chair of the division of infectious diseases at Johns Hopkins University, Baltimore, said that the website is expected to be used not only by clinicians who are well versed in prescribing antiviral drugs, "but also by many who are inexperienced or even new to the treatment of hepatitis C." This includes gastroenterologists and infectious disease clinicians who have not been treating patients with hepatitis C and may include primary care physicians as well, he added. Dr. Thomas was the panel cochair for IDSA.
Recent developments in the treatment of hepatitis C include the FDA approvals of sofosbuvir and simeprevir late last year for treating chronic HCV, considered major advances in treatment.
The website is HCVguidelines.org. More information on hepatitis C is available at www.idsociety.org/HCV.
A new website has been launched that provides the latest information to help guide clinicians on treating patients with chronic hepatitis C and will be updated regularly with developments in this rapidly changing area of medicine.
A collaboration of the American Association for the Study of Liver Diseases (AASLD) and the Infectious Diseases Society of America (IDSA) with the International Antiviral Society-USA (IAS-USA), the website – HCVguidelines.org – provides evidence-based consensus recommendations for screening and treating people with hepatitis C virus (HCV).
The recommendations were developed by a panel of 27 liver disease and infectious diseases specialists and a patient advocate and are intended for "health care providers who treat the disease and others who need updated information on the best practices," according to a statement issued by IDSA and AASLD announcing the launch of the website, which became available on Jan. 29. Examples of the information provided include guidance on the use of direct-acting HCV antivirals, with and without interferon for the most common type of HCV, genotype 1; treatment of different subgroups, such as those coinfected with HIV, those with decompensated liver disease, and liver transplant recipients; and regimens for treatment of interferon-ineligible and interferon-eligible patients.
The website is a living document and will be updated regularly with new recommendations that reflect new research, Food and Drug Administration (FDA) approvals of new drugs, changing recommendations for monitoring patients, and other developments. This aspect is "absolutely necessary in this field" because of the many recent developments and multiple drugs that are in development and approaching approval, Dr. Michael Saag, the panel cochair for IAS-USA, said during a telephone briefing held to announce the launch.
Changes can be made weekly or monthly, "so that clinicians can have confidence that, when they go to this website, they will get the most accurate and up-to-date guidance on the treatment of hepatitis C," added Dr. Saag, professor of medicine and director of the division of infectious diseases at the University of Alabama at Birmingham.
During the briefing, Dr. Henry Masur of the National Institutes of Health, Bethesda, Md., pointed out that, for the first time, treatments that cure hepatitis C are available, which are more effective than previous treatments and easier to tolerate. "What we’re eager to see is that these drugs are used by practitioners to treat the millions of patients who have hepatitis C both in North America and elsewhere ... and it’s important that clinicians have access to guidance on how best to use these drugs as new trials are quickly done and new information becomes available."
An estimated 3-4 million people in the United States are infected with HCV and could develop chronic liver disease; the cure rate for chronic HCV is now close to 95% with currently available treatments.
Noting that the next-generation antivirals have the potential to cure most patients with hepatitis C, Dr. David Thomas, professor of medicine and chair of the division of infectious diseases at Johns Hopkins University, Baltimore, said that the website is expected to be used not only by clinicians who are well versed in prescribing antiviral drugs, "but also by many who are inexperienced or even new to the treatment of hepatitis C." This includes gastroenterologists and infectious disease clinicians who have not been treating patients with hepatitis C and may include primary care physicians as well, he added. Dr. Thomas was the panel cochair for IDSA.
Recent developments in the treatment of hepatitis C include the FDA approvals of sofosbuvir and simeprevir late last year for treating chronic HCV, considered major advances in treatment.
The website is HCVguidelines.org. More information on hepatitis C is available at www.idsociety.org/HCV.

The price and cost of hepatitis C treatment
Chronic hepatitis C virus (HCV) infection is a major cause of liver disease affecting 180 million people globally. HCV infection, both silent and slow in its revelation, and devastating in its complications, has been described as a "viral time bomb." In December 2013, the Food and Drug Administration approved sofosbuvir, a nucleotide NS5B polymerase inhibitor, given compelling evidence that when taken with pegylated interferon-alfa and ribavirin for genotype 1 or 4 (for 12 weeks) or with ribavirin for genotype 2 (for 12 weeks) or 3 (for 24 weeks), sustained virologic response (SVR) rates are excellent, with very limited side effects.
The excitement has been tempered by the knowledge that sofosbuvir alone is priced at approximately $80,000 for a 12-week course. This cost is beyond the means of many individuals in countries with affluent economies, even with insurance and/or patient assistance programs, and vastly beyond those of developing countries – where HCV infection is most burdensome. It should be noted that the cost of therapy with high SVR rates may greatly offset the price required to manage the long-term complications and indirect effects of chronic HCV infection. And further, since the treatment is shorter than in the past and since the regimen has far fewer side effects, the overall cost of therapy may be similar to that of first-generation protease inhibitor–based regimens that constituted the standard of care for genotype 1 previously. With that said, idealized studies have shown that generic sofosbuvir–based regimens could be priced substantially lower.

The query "why must it cost so much?" articulates traditional concerns of the public and its advocates who feel confusion, cynicism, and anger at corporate strategies. As physicians and fellow citizens, armed with both Hippocratic and democratic principles, we immediately identify with these positions. Yet, we would also urge restraint in attempting traditional actions, such as government fiat, that would seek to arbitrarily lower price.
The concepts "price" and "cost" are commonly conflated: The former is the amount a buyer, or third party, is willing to pay the seller; the latter is the amount it takes to bring that good or service to market. The pharmaceutical industry is straddled with high fixed costs of production (research and development) and aggressive FDA regulatory control over efficacy and safety. Prices attempt to recapture cost and justify the risk of initial investment. Generic drug replication does not embody these extreme costs that are partially reflected in lower prices.
Nevertheless, it is hoped that price reduction through traditional market measures will be achieved, i.e., to create "cooperation without coercion." Perhaps price reduction will be achieved when competitive products reach the market in the near future. Perhaps price reduction will be achieved by aggressive negotiation of cost with third-party payers. Sofosbuvir retains patent protection until 2029; thus, generics are not readily possible outside of "compulsory licensure" – the ability of governments to violate patent law in periods of national necessity. Such intervention, or the threat of it, has occurred in many countries, including the United States. We believe it is unlikely to occur in this case.
The HCV market, estimated at greater than $10 billion annually in sales, provides a strong incentive for drug development – producing the multitude of agents in various stages of development for FDA approval. The competition of multiple sellers for buyers will drive down price and facilitate quality. An extreme example of this can be seen in countries practicing socialized medicine, which effectively have a single buyer, so there is greater leverage for price reduction.
Compulsory licensure, while based upon sound moral intentions, would stifle future innovation by removing profit incentive and creating an unstable economic atmosphere. It is informative to review the vaccine market, in which economic instability has led to fewer sellers, either from attrition or conglomeration. The ultimate consequence is decreased innovation, competition, and drug availability.
Corporations are not moral agents. Similarly so, government has no agency, but rather should be considered an instrument through which a moral citizenry can achieve agreed upon objectives. Aggressive education and collaboration amongst physicians and their patients, purchasing groups, and public lobbying agencies to collectively negotiate reasonable price reduction are encouraged – to realize the potential we have as buyers en masse.
Pharmaceutical profits may be conspicuous, but nevertheless, it is that very incentive of profit that brings such powerful drugs so quickly to the market in the first place. Patients can neither benefit from nor feel the loss of drugs that never make it to market. A morality of intention, absolved of comprehensive economic parameters, while psychologically enticing and proximally politically advantageous, would lead to greater long-term harm for the very patients we have vowed to protect.
We hope that price and cost issues will not prohibit large numbers of HCV-afflicted patients from receiving the current therapies and other highly efficacious regimens that will soon be forthcoming.
Dr. Christopher Moore is a fellow in transplant hepatology, and Dr. Steven Flamm is chief of transplantation hepatology and professor of medicine and surgery, at Northwestern University Feinberg School of Medicine, Chicago. Dr. Moore reported having no conflicts of interest. Dr. Flamm is a speaker for Vertex and Gilead; does research and consults for Abbvie, Vertex, and other companies; and does research for Boehringer Ingelheim.
Chronic hepatitis C virus (HCV) infection is a major cause of liver disease affecting 180 million people globally. HCV infection, both silent and slow in its revelation, and devastating in its complications, has been described as a "viral time bomb." In December 2013, the Food and Drug Administration approved sofosbuvir, a nucleotide NS5B polymerase inhibitor, given compelling evidence that when taken with pegylated interferon-alfa and ribavirin for genotype 1 or 4 (for 12 weeks) or with ribavirin for genotype 2 (for 12 weeks) or 3 (for 24 weeks), sustained virologic response (SVR) rates are excellent, with very limited side effects.
The excitement has been tempered by the knowledge that sofosbuvir alone is priced at approximately $80,000 for a 12-week course. This cost is beyond the means of many individuals in countries with affluent economies, even with insurance and/or patient assistance programs, and vastly beyond those of developing countries – where HCV infection is most burdensome. It should be noted that the cost of therapy with high SVR rates may greatly offset the price required to manage the long-term complications and indirect effects of chronic HCV infection. And further, since the treatment is shorter than in the past and since the regimen has far fewer side effects, the overall cost of therapy may be similar to that of first-generation protease inhibitor–based regimens that constituted the standard of care for genotype 1 previously. With that said, idealized studies have shown that generic sofosbuvir–based regimens could be priced substantially lower.
The query "why must it cost so much?" articulates traditional concerns of the public and its advocates who feel confusion, cynicism, and anger at corporate strategies. As physicians and fellow citizens, armed with both Hippocratic and democratic principles, we immediately identify with these positions. Yet, we would also urge restraint in attempting traditional actions, such as government fiat, that would seek to arbitrarily lower price.
The concepts "price" and "cost" are commonly conflated: The former is the amount a buyer, or third party, is willing to pay the seller; the latter is the amount it takes to bring that good or service to market. The pharmaceutical industry is straddled with high fixed costs of production (research and development) and aggressive FDA regulatory control over efficacy and safety. Prices attempt to recapture cost and justify the risk of initial investment. Generic drug replication does not embody these extreme costs that are partially reflected in lower prices.
Nevertheless, it is hoped that price reduction through traditional market measures will be achieved, i.e., to create "cooperation without coercion." Perhaps price reduction will be achieved when competitive products reach the market in the near future. Perhaps price reduction will be achieved by aggressive negotiation of cost with third-party payers. Sofosbuvir retains patent protection until 2029; thus, generics are not readily possible outside of "compulsory licensure" – the ability of governments to violate patent law in periods of national necessity. Such intervention, or the threat of it, has occurred in many countries, including the United States. We believe it is unlikely to occur in this case.
The HCV market, estimated at greater than $10 billion annually in sales, provides a strong incentive for drug development – producing the multitude of agents in various stages of development for FDA approval. The competition of multiple sellers for buyers will drive down price and facilitate quality. An extreme example of this can be seen in countries practicing socialized medicine, which effectively have a single buyer, so there is greater leverage for price reduction.
Compulsory licensure, while based upon sound moral intentions, would stifle future innovation by removing profit incentive and creating an unstable economic atmosphere. It is informative to review the vaccine market, in which economic instability has led to fewer sellers, either from attrition or conglomeration. The ultimate consequence is decreased innovation, competition, and drug availability.
Corporations are not moral agents. Similarly so, government has no agency, but rather should be considered an instrument through which a moral citizenry can achieve agreed upon objectives. Aggressive education and collaboration amongst physicians and their patients, purchasing groups, and public lobbying agencies to collectively negotiate reasonable price reduction are encouraged – to realize the potential we have as buyers en masse.
Pharmaceutical profits may be conspicuous, but nevertheless, it is that very incentive of profit that brings such powerful drugs so quickly to the market in the first place. Patients can neither benefit from nor feel the loss of drugs that never make it to market. A morality of intention, absolved of comprehensive economic parameters, while psychologically enticing and proximally politically advantageous, would lead to greater long-term harm for the very patients we have vowed to protect.
We hope that price and cost issues will not prohibit large numbers of HCV-afflicted patients from receiving the current therapies and other highly efficacious regimens that will soon be forthcoming.
Dr. Christopher Moore is a fellow in transplant hepatology, and Dr. Steven Flamm is chief of transplantation hepatology and professor of medicine and surgery, at Northwestern University Feinberg School of Medicine, Chicago. Dr. Moore reported having no conflicts of interest. Dr. Flamm is a speaker for Vertex and Gilead; does research and consults for Abbvie, Vertex, and other companies; and does research for Boehringer Ingelheim.
Chronic hepatitis C virus (HCV) infection is a major cause of liver disease affecting 180 million people globally. HCV infection, both silent and slow in its revelation, and devastating in its complications, has been described as a "viral time bomb." In December 2013, the Food and Drug Administration approved sofosbuvir, a nucleotide NS5B polymerase inhibitor, given compelling evidence that when taken with pegylated interferon-alfa and ribavirin for genotype 1 or 4 (for 12 weeks) or with ribavirin for genotype 2 (for 12 weeks) or 3 (for 24 weeks), sustained virologic response (SVR) rates are excellent, with very limited side effects.
The excitement has been tempered by the knowledge that sofosbuvir alone is priced at approximately $80,000 for a 12-week course. This cost is beyond the means of many individuals in countries with affluent economies, even with insurance and/or patient assistance programs, and vastly beyond those of developing countries – where HCV infection is most burdensome. It should be noted that the cost of therapy with high SVR rates may greatly offset the price required to manage the long-term complications and indirect effects of chronic HCV infection. And further, since the treatment is shorter than in the past and since the regimen has far fewer side effects, the overall cost of therapy may be similar to that of first-generation protease inhibitor–based regimens that constituted the standard of care for genotype 1 previously. With that said, idealized studies have shown that generic sofosbuvir–based regimens could be priced substantially lower.
The query "why must it cost so much?" articulates traditional concerns of the public and its advocates who feel confusion, cynicism, and anger at corporate strategies. As physicians and fellow citizens, armed with both Hippocratic and democratic principles, we immediately identify with these positions. Yet, we would also urge restraint in attempting traditional actions, such as government fiat, that would seek to arbitrarily lower price.
The concepts "price" and "cost" are commonly conflated: The former is the amount a buyer, or third party, is willing to pay the seller; the latter is the amount it takes to bring that good or service to market. The pharmaceutical industry is straddled with high fixed costs of production (research and development) and aggressive FDA regulatory control over efficacy and safety. Prices attempt to recapture cost and justify the risk of initial investment. Generic drug replication does not embody these extreme costs that are partially reflected in lower prices.
Nevertheless, it is hoped that price reduction through traditional market measures will be achieved, i.e., to create "cooperation without coercion." Perhaps price reduction will be achieved when competitive products reach the market in the near future. Perhaps price reduction will be achieved by aggressive negotiation of cost with third-party payers. Sofosbuvir retains patent protection until 2029; thus, generics are not readily possible outside of "compulsory licensure" – the ability of governments to violate patent law in periods of national necessity. Such intervention, or the threat of it, has occurred in many countries, including the United States. We believe it is unlikely to occur in this case.
The HCV market, estimated at greater than $10 billion annually in sales, provides a strong incentive for drug development – producing the multitude of agents in various stages of development for FDA approval. The competition of multiple sellers for buyers will drive down price and facilitate quality. An extreme example of this can be seen in countries practicing socialized medicine, which effectively have a single buyer, so there is greater leverage for price reduction.
Compulsory licensure, while based upon sound moral intentions, would stifle future innovation by removing profit incentive and creating an unstable economic atmosphere. It is informative to review the vaccine market, in which economic instability has led to fewer sellers, either from attrition or conglomeration. The ultimate consequence is decreased innovation, competition, and drug availability.
Corporations are not moral agents. Similarly so, government has no agency, but rather should be considered an instrument through which a moral citizenry can achieve agreed upon objectives. Aggressive education and collaboration amongst physicians and their patients, purchasing groups, and public lobbying agencies to collectively negotiate reasonable price reduction are encouraged – to realize the potential we have as buyers en masse.
Pharmaceutical profits may be conspicuous, but nevertheless, it is that very incentive of profit that brings such powerful drugs so quickly to the market in the first place. Patients can neither benefit from nor feel the loss of drugs that never make it to market. A morality of intention, absolved of comprehensive economic parameters, while psychologically enticing and proximally politically advantageous, would lead to greater long-term harm for the very patients we have vowed to protect.
We hope that price and cost issues will not prohibit large numbers of HCV-afflicted patients from receiving the current therapies and other highly efficacious regimens that will soon be forthcoming.
Dr. Christopher Moore is a fellow in transplant hepatology, and Dr. Steven Flamm is chief of transplantation hepatology and professor of medicine and surgery, at Northwestern University Feinberg School of Medicine, Chicago. Dr. Moore reported having no conflicts of interest. Dr. Flamm is a speaker for Vertex and Gilead; does research and consults for Abbvie, Vertex, and other companies; and does research for Boehringer Ingelheim.

Triple-antiviral therapy achieves 94% response without interferon, ribavirin
Both a 12-week and a 24-week regimen of three oral antiviral drugs achieved sustained viral remissions at 12 weeks in up to 94% of treatment-naive patients with genotype 1 hepatitis C virus infections.
The three oral antivirals included daclatasvir, an NS5A replication complex inhibitor; asunaprevir, an NS3 protease inhibitor; and BMS-791325, an investigational selective non-nucleoside polymerase inhibitor. A key point of the study is its proof that short-duration treatment – even without interferon or ribavirin – can result in sustained remission, Dr. Gregory T. Everson and his colleagues wrote in the February issue of Gastroenterology (doi.org/10.1053/j.gastro.2013.10.057). "Shorter treatment durations are preferable because they may improve patient compliance. In this study, treatment periods of both 12 and 24 weeks yielded high SVR (sustained viral remission) rates, suggesting no advantage for extending treatment duration to 24 weeks."
Source: American Gastroenterological Association
Further, the triple-antiviral combination apparently controlled viral replication without using interferon or ribavirin, thus avoiding their side effects. The current regimen for treatment-naive patients with genotype 1 hepatitis C virus infections calls for a 48-week treatment with peginterferon and ribavirin with telaprevir or boceprevir.
"Ribavirin contributes to anemia and it is teratogenic; thus, effective treatments without ribavirin are desirable," wrote Dr. Everson of the University of Colorado, Denver, and his coauthors. "This interferon- and ribavirin-free regimen did not alter hemoglobin levels in a clinically meaningfully manner, as evidenced by no grade 1 or higher hemoglobin reductions and no adverse events of anemia."
Dr. Everson and his colleagues gave the regimen for periods of 12 and 24 weeks and with two different doses of BMS-791325, resulting in a four-way randomized study. Groups 1 and 2 were treated for 12 weeks with the combination of daclatasvir 60 mg once daily, asunaprevir 200 mg twice daily, and BMS-791325 (twice daily 75 mg or 150 mg). Groups 3 and 4 had exactly the same two-treatment regimen, but took the drugs for 24 weeks. After treatment, there was a 48-week follow-up period.
Among the 66 patients in the study, the average age was about 50 years old, and the mean HCV-RNA level was 6 log10 IU/mL. Most (75%) had HCV genotype 1a; the rest had 1b. All were treatment naive. Four patients withdrew before the study’s end; none of the withdrawals were for adverse events.
In groups 1 and 2 (75 mg BMS-791325, twice daily for 12 and 24 weeks), the viral load decreased rapidly. By week 4, all of these patients achieved a HCV-RNA level of less than 25 IU/mL; 97% maintained that level through the end of treatment. In a modified intent-to-treat analysis, 94% achieved a sustained viral response by week 12.
In groups 3 and 4 (150 mg BMS-791325, twice daily for 12 or 24 weeks), HCV-RNA levels also fell rapidly in the first month, By week 4, all of those in the 12-week group and all but one in the 24-week group had levels of less than 25 IU/mL, which were sustained through the end of treatment. In the intent-to-treat analysis, 91% overall had an HCV-RNA load of less than 25 IU/mL by the end of treatment.
Overall, there were three treatment failures: one each in groups 3 and 4 had viral breakthrough, and one in group 4 experienced a relapse at week 4.
Both breakthrough patients were given peginterferon-alfa/ribavirin in addition to the direct-acting antivirals. After 16 weeks, one discontinued treatment because of interferon-related cerebral vasoconstriction. That patient had an undetectable HCV-RNA level at the end of treatment, but relapsed by post treatment week 4. The other patient had a sustained viral response.
The study was sponsored by Bristol-Myers Squibb. Dr. Everson listed financial and research relationships with numerous pharmaceutical companies, including Bristol-Myers. His coauthors also declared relationships with numerous drug manufacturers, including Bristol-Myers.
This critical proof-of-concept study demonstrates that a combination of three direct-acting antivirals (an NS5A inhibitor + NS3PI + NNPI) without ribavirin can achieve sustained viral response at 12 weeks at rates of 94% in a genotype 1 treatment-naive (TN) population. The data are a foundation for multiple large, randomized, phase III trials using this regimen in TN patients, treatment failure, cirrhosis, HIV coinfection, and other genotypes, and thus, it is important to the development of interferon- and ribavirin-free therapies for HCV.
The authors have selected a TN population without advanced fibrosis for this regimen so far, and it remains to be seen whether these high sustained viral remission (SVR) rates can be duplicated in cirrhosis and those with prior treatment failure. It may also show reduced efficacy when studied in more difficult-to-treat patients such as with protease inhibitor failure, HIV coinfection, and other genotypes. However, the importance of this study is that the lack of efficacy seen in genotype 1a with the dual therapy published in 2012 was successfully overcome simply by adding an non-nucleoside polymerase inhibitor (N. Engl. J. Med. 2012;366:216-24). The implications may extend to other interferon-free, direct-acting antiviral regimens in development. An NS5B nucleotide polymerase inhibitor may not be required for the backbone of therapy, as it appears to be the based on sofosbuvir studies (N. Engl. J. Med. 2013;368:34-44; 1878-87). Rather, a best-in-class drug, here daclatasvir for the NS5A class, may be added to two other relatively weak compounds and still have greater than 90% SVR rates as an interferon-free regimen.
This would pave the way for many compounds to be competitive without the nucleotide polymerase inhibitor backbone regimen, in both genotype 1a and 1b patients. However, with the promise of a 12-week regimen of sofosbuvir and ledipasvir as a single, fixed-dose combination for all patient types (Gilead Sciences press release, Dec. 18, 2013: "Gilead announces SVR12 rates from three phase III studies evaluating once-daily fixed-dose combination of sofosbuvir and ledipasvir for genotype 1 hepatitis C patients"), a competitor would need to show similar efficacy and tolerability. Even if not proven in all patient populations, this would lead to a price competitive market that would provide the patient, physician, and payer with multiple choices. Although this may not be as important in the United States, it will be critical to many resource-limited countries where price will be a primary constraint.
Dr. Paul Pockros is director, Liver Disease Center Scripps Clinic, and clinical director of research, Scripps Translational Science Institute, both in La Jolla, Calif. He does research, speaks, and consults for Bristol-Myers Squibb, Gilead, Genetech, and Janssen. He also does research and consults for Boehringer Ingelheim and Novartis.
This critical proof-of-concept study demonstrates that a combination of three direct-acting antivirals (an NS5A inhibitor + NS3PI + NNPI) without ribavirin can achieve sustained viral response at 12 weeks at rates of 94% in a genotype 1 treatment-naive (TN) population. The data are a foundation for multiple large, randomized, phase III trials using this regimen in TN patients, treatment failure, cirrhosis, HIV coinfection, and other genotypes, and thus, it is important to the development of interferon- and ribavirin-free therapies for HCV.
The authors have selected a TN population without advanced fibrosis for this regimen so far, and it remains to be seen whether these high sustained viral remission (SVR) rates can be duplicated in cirrhosis and those with prior treatment failure. It may also show reduced efficacy when studied in more difficult-to-treat patients such as with protease inhibitor failure, HIV coinfection, and other genotypes. However, the importance of this study is that the lack of efficacy seen in genotype 1a with the dual therapy published in 2012 was successfully overcome simply by adding an non-nucleoside polymerase inhibitor (N. Engl. J. Med. 2012;366:216-24). The implications may extend to other interferon-free, direct-acting antiviral regimens in development. An NS5B nucleotide polymerase inhibitor may not be required for the backbone of therapy, as it appears to be the based on sofosbuvir studies (N. Engl. J. Med. 2013;368:34-44; 1878-87). Rather, a best-in-class drug, here daclatasvir for the NS5A class, may be added to two other relatively weak compounds and still have greater than 90% SVR rates as an interferon-free regimen.
This would pave the way for many compounds to be competitive without the nucleotide polymerase inhibitor backbone regimen, in both genotype 1a and 1b patients. However, with the promise of a 12-week regimen of sofosbuvir and ledipasvir as a single, fixed-dose combination for all patient types (Gilead Sciences press release, Dec. 18, 2013: "Gilead announces SVR12 rates from three phase III studies evaluating once-daily fixed-dose combination of sofosbuvir and ledipasvir for genotype 1 hepatitis C patients"), a competitor would need to show similar efficacy and tolerability. Even if not proven in all patient populations, this would lead to a price competitive market that would provide the patient, physician, and payer with multiple choices. Although this may not be as important in the United States, it will be critical to many resource-limited countries where price will be a primary constraint.
Dr. Paul Pockros is director, Liver Disease Center Scripps Clinic, and clinical director of research, Scripps Translational Science Institute, both in La Jolla, Calif. He does research, speaks, and consults for Bristol-Myers Squibb, Gilead, Genetech, and Janssen. He also does research and consults for Boehringer Ingelheim and Novartis.
This critical proof-of-concept study demonstrates that a combination of three direct-acting antivirals (an NS5A inhibitor + NS3PI + NNPI) without ribavirin can achieve sustained viral response at 12 weeks at rates of 94% in a genotype 1 treatment-naive (TN) population. The data are a foundation for multiple large, randomized, phase III trials using this regimen in TN patients, treatment failure, cirrhosis, HIV coinfection, and other genotypes, and thus, it is important to the development of interferon- and ribavirin-free therapies for HCV.
The authors have selected a TN population without advanced fibrosis for this regimen so far, and it remains to be seen whether these high sustained viral remission (SVR) rates can be duplicated in cirrhosis and those with prior treatment failure. It may also show reduced efficacy when studied in more difficult-to-treat patients such as with protease inhibitor failure, HIV coinfection, and other genotypes. However, the importance of this study is that the lack of efficacy seen in genotype 1a with the dual therapy published in 2012 was successfully overcome simply by adding an non-nucleoside polymerase inhibitor (N. Engl. J. Med. 2012;366:216-24). The implications may extend to other interferon-free, direct-acting antiviral regimens in development. An NS5B nucleotide polymerase inhibitor may not be required for the backbone of therapy, as it appears to be the based on sofosbuvir studies (N. Engl. J. Med. 2013;368:34-44; 1878-87). Rather, a best-in-class drug, here daclatasvir for the NS5A class, may be added to two other relatively weak compounds and still have greater than 90% SVR rates as an interferon-free regimen.
This would pave the way for many compounds to be competitive without the nucleotide polymerase inhibitor backbone regimen, in both genotype 1a and 1b patients. However, with the promise of a 12-week regimen of sofosbuvir and ledipasvir as a single, fixed-dose combination for all patient types (Gilead Sciences press release, Dec. 18, 2013: "Gilead announces SVR12 rates from three phase III studies evaluating once-daily fixed-dose combination of sofosbuvir and ledipasvir for genotype 1 hepatitis C patients"), a competitor would need to show similar efficacy and tolerability. Even if not proven in all patient populations, this would lead to a price competitive market that would provide the patient, physician, and payer with multiple choices. Although this may not be as important in the United States, it will be critical to many resource-limited countries where price will be a primary constraint.
Dr. Paul Pockros is director, Liver Disease Center Scripps Clinic, and clinical director of research, Scripps Translational Science Institute, both in La Jolla, Calif. He does research, speaks, and consults for Bristol-Myers Squibb, Gilead, Genetech, and Janssen. He also does research and consults for Boehringer Ingelheim and Novartis.
Both a 12-week and a 24-week regimen of three oral antiviral drugs achieved sustained viral remissions at 12 weeks in up to 94% of treatment-naive patients with genotype 1 hepatitis C virus infections.
The three oral antivirals included daclatasvir, an NS5A replication complex inhibitor; asunaprevir, an NS3 protease inhibitor; and BMS-791325, an investigational selective non-nucleoside polymerase inhibitor. A key point of the study is its proof that short-duration treatment – even without interferon or ribavirin – can result in sustained remission, Dr. Gregory T. Everson and his colleagues wrote in the February issue of Gastroenterology (doi.org/10.1053/j.gastro.2013.10.057). "Shorter treatment durations are preferable because they may improve patient compliance. In this study, treatment periods of both 12 and 24 weeks yielded high SVR (sustained viral remission) rates, suggesting no advantage for extending treatment duration to 24 weeks."
Source: American Gastroenterological Association
Further, the triple-antiviral combination apparently controlled viral replication without using interferon or ribavirin, thus avoiding their side effects. The current regimen for treatment-naive patients with genotype 1 hepatitis C virus infections calls for a 48-week treatment with peginterferon and ribavirin with telaprevir or boceprevir.
"Ribavirin contributes to anemia and it is teratogenic; thus, effective treatments without ribavirin are desirable," wrote Dr. Everson of the University of Colorado, Denver, and his coauthors. "This interferon- and ribavirin-free regimen did not alter hemoglobin levels in a clinically meaningfully manner, as evidenced by no grade 1 or higher hemoglobin reductions and no adverse events of anemia."
Dr. Everson and his colleagues gave the regimen for periods of 12 and 24 weeks and with two different doses of BMS-791325, resulting in a four-way randomized study. Groups 1 and 2 were treated for 12 weeks with the combination of daclatasvir 60 mg once daily, asunaprevir 200 mg twice daily, and BMS-791325 (twice daily 75 mg or 150 mg). Groups 3 and 4 had exactly the same two-treatment regimen, but took the drugs for 24 weeks. After treatment, there was a 48-week follow-up period.
Among the 66 patients in the study, the average age was about 50 years old, and the mean HCV-RNA level was 6 log10 IU/mL. Most (75%) had HCV genotype 1a; the rest had 1b. All were treatment naive. Four patients withdrew before the study’s end; none of the withdrawals were for adverse events.
In groups 1 and 2 (75 mg BMS-791325, twice daily for 12 and 24 weeks), the viral load decreased rapidly. By week 4, all of these patients achieved a HCV-RNA level of less than 25 IU/mL; 97% maintained that level through the end of treatment. In a modified intent-to-treat analysis, 94% achieved a sustained viral response by week 12.
In groups 3 and 4 (150 mg BMS-791325, twice daily for 12 or 24 weeks), HCV-RNA levels also fell rapidly in the first month, By week 4, all of those in the 12-week group and all but one in the 24-week group had levels of less than 25 IU/mL, which were sustained through the end of treatment. In the intent-to-treat analysis, 91% overall had an HCV-RNA load of less than 25 IU/mL by the end of treatment.
Overall, there were three treatment failures: one each in groups 3 and 4 had viral breakthrough, and one in group 4 experienced a relapse at week 4.
Both breakthrough patients were given peginterferon-alfa/ribavirin in addition to the direct-acting antivirals. After 16 weeks, one discontinued treatment because of interferon-related cerebral vasoconstriction. That patient had an undetectable HCV-RNA level at the end of treatment, but relapsed by post treatment week 4. The other patient had a sustained viral response.
The study was sponsored by Bristol-Myers Squibb. Dr. Everson listed financial and research relationships with numerous pharmaceutical companies, including Bristol-Myers. His coauthors also declared relationships with numerous drug manufacturers, including Bristol-Myers.
Both a 12-week and a 24-week regimen of three oral antiviral drugs achieved sustained viral remissions at 12 weeks in up to 94% of treatment-naive patients with genotype 1 hepatitis C virus infections.
The three oral antivirals included daclatasvir, an NS5A replication complex inhibitor; asunaprevir, an NS3 protease inhibitor; and BMS-791325, an investigational selective non-nucleoside polymerase inhibitor. A key point of the study is its proof that short-duration treatment – even without interferon or ribavirin – can result in sustained remission, Dr. Gregory T. Everson and his colleagues wrote in the February issue of Gastroenterology (doi.org/10.1053/j.gastro.2013.10.057). "Shorter treatment durations are preferable because they may improve patient compliance. In this study, treatment periods of both 12 and 24 weeks yielded high SVR (sustained viral remission) rates, suggesting no advantage for extending treatment duration to 24 weeks."
Source: American Gastroenterological Association
Further, the triple-antiviral combination apparently controlled viral replication without using interferon or ribavirin, thus avoiding their side effects. The current regimen for treatment-naive patients with genotype 1 hepatitis C virus infections calls for a 48-week treatment with peginterferon and ribavirin with telaprevir or boceprevir.
"Ribavirin contributes to anemia and it is teratogenic; thus, effective treatments without ribavirin are desirable," wrote Dr. Everson of the University of Colorado, Denver, and his coauthors. "This interferon- and ribavirin-free regimen did not alter hemoglobin levels in a clinically meaningfully manner, as evidenced by no grade 1 or higher hemoglobin reductions and no adverse events of anemia."
Dr. Everson and his colleagues gave the regimen for periods of 12 and 24 weeks and with two different doses of BMS-791325, resulting in a four-way randomized study. Groups 1 and 2 were treated for 12 weeks with the combination of daclatasvir 60 mg once daily, asunaprevir 200 mg twice daily, and BMS-791325 (twice daily 75 mg or 150 mg). Groups 3 and 4 had exactly the same two-treatment regimen, but took the drugs for 24 weeks. After treatment, there was a 48-week follow-up period.
Among the 66 patients in the study, the average age was about 50 years old, and the mean HCV-RNA level was 6 log10 IU/mL. Most (75%) had HCV genotype 1a; the rest had 1b. All were treatment naive. Four patients withdrew before the study’s end; none of the withdrawals were for adverse events.
In groups 1 and 2 (75 mg BMS-791325, twice daily for 12 and 24 weeks), the viral load decreased rapidly. By week 4, all of these patients achieved a HCV-RNA level of less than 25 IU/mL; 97% maintained that level through the end of treatment. In a modified intent-to-treat analysis, 94% achieved a sustained viral response by week 12.
In groups 3 and 4 (150 mg BMS-791325, twice daily for 12 or 24 weeks), HCV-RNA levels also fell rapidly in the first month, By week 4, all of those in the 12-week group and all but one in the 24-week group had levels of less than 25 IU/mL, which were sustained through the end of treatment. In the intent-to-treat analysis, 91% overall had an HCV-RNA load of less than 25 IU/mL by the end of treatment.
Overall, there were three treatment failures: one each in groups 3 and 4 had viral breakthrough, and one in group 4 experienced a relapse at week 4.
Both breakthrough patients were given peginterferon-alfa/ribavirin in addition to the direct-acting antivirals. After 16 weeks, one discontinued treatment because of interferon-related cerebral vasoconstriction. That patient had an undetectable HCV-RNA level at the end of treatment, but relapsed by post treatment week 4. The other patient had a sustained viral response.
The study was sponsored by Bristol-Myers Squibb. Dr. Everson listed financial and research relationships with numerous pharmaceutical companies, including Bristol-Myers. His coauthors also declared relationships with numerous drug manufacturers, including Bristol-Myers.
Major finding: By week 4, patients achieved a HCV-RNA level of less than 25 IU/mL; 97% maintained that level through the end of treatment. In a modified intent-to-treat analysis, 94% achieved a sustained viral response by week 12.
Data source: A trial of 66 treatment-naive patients randomized to one of four antiviral regimens that differed by dose of BMS-791325, an investigational selective non-nucleoside polymerase inhibitor.
Disclosures: The study was sponsored by Bristol-Myers Squibb. Dr. Everson listed financial and research relationships with numerous pharmaceutical companies, including Bristol-Myers Squibb. His coauthors also declared relationships with numerous drug manufacturers, including Bristol-Myers.
FLINT trial halted early because of positive results
The phase II clinical trial of first-in-class farnesoid X receptor agonist obeticholic acid as a treatment for nonalcoholic steatohepatitis was stopped early because the trial endpoint was met with high significance, Intercept Pharmaceuticals announced Jan. 9.
The National Institute of Diabetes and Digestive and Kidney Diseases, which sponsored and conducted the FLINT trial, said in its statement that "OCA [obeticholic acid] has a significant beneficial effect on the liver damage due to NASH."
An interim analysis conducted by the Data Safety Monitoring Board of the trial found a highly statistically significant improvement in the primary endpoint, defined as "a decrease in the NAFLD Activity Score of at least two points with no worsening of fibrosis, as compared to placebo," the company said in a statement. The predetermined significance threshold for ending the trial was P less than .0031, but early results exceeded expectations with a significance of P = .0024.
An estimated 6 million adults in the United States have advanced liver fibrosis or cirrhosis caused bynonalcoholic steatohepatitis (NASH), said Dr. Mark Pruzanski, CEO of Intercept.
"This disease has seemingly come out of nowhere and has become an epidemic," he said. "On its current trajectory, NASH is expected to be the No. 1 indication for liver transplant over the next decade."
The FLINT trial, sponsored and conducted by the National Institute of Diabetes and Digestive and Kidney Diseases, studied the efficacy of a 25-mg. oral dose of obeticholic acid (OCA) in approximately half of 283 biopsy-confirmed NASH patients over a period of 72 weeks. Patients were randomized to receive either a 25-mg dose of OCA or placebo for the treatment period.
Participants were 18 years of age or older, with histologic evidence of nonalcoholic steatohepatitis, based on a liver biopsy, obtained no more than 90 days prior to randomization and a NAFLD activity score of 4 or greater with at least one in each component of the NAFLD activity score (steatosis scored 0-3; ballooning degeneration scored 0-2; and lobular inflammation scored 0-3). End of study biopsies were conducted in patients after the 72-week treatment period.
As specified in the protocol, a 24-week follow-up period will start, and all FLINT participants will stop taking the drug or placebo no later than Jan. 20.
Lipid derangements involving elevated total [cholesterol] and LDL cholesterol and reduced HDL cholesterol have been seen in OCA, compared with placebo-treated patients. While lipid abnormalities are not uncommon in NASH, researchers will be checking to see if lipids return to pretreatment levels during this 24-week follow-up period.
During this period, both FLINT investigators and trial participants will remain blinded to treatment. The NIDDK expects to present the results of the trial in the fourth quarter of 2014.
OCA has not only been studied as a promising treatment for NASH. A 2013 study also found that OCA increased insulin sensitivity and reduced markers of liver inflammation and fibrosis in patients with type 2 diabetes and nonalcoholic fatty liver disease. Additionally, OCA is currently in a phase III trial for the treatment of primary biliary cirrhosis.
A concurrent trial to study OCA as a treatment for NASH is underway in Japan, added Dr. Pruzanski, with top-line results expected in 2015. Dr. Pruzanski said he hopes the trial’s unexpected success will help pave the path to FDA approval, pending final NIDDK analyses and publication of the results.
The FLINT trial was sponsored and conducted by the National Institute of Diabetes and Digestive and Kidney Diseases.
Current data suggest that nonalcoholic fatty liver disease (NAFLD) is present in 30%-40% of adults in the United States, with a subset of this population having a more aggressive form of fatty liver called nonalcoholic steatohepatitis (NASH), which may progress to cirrhosis. NASH is currently the third-leading cause for liver transplantation and is predicted to become the most common within 10-15 years (Gastroenterology 2011;141:1249-53).
Most NAFLD patients are overweight or obese and have underlying insulin resistance or diabetes mellitus. Comorbid illness is also common, with cardiovascular disease being the leading cause of death (Clin. Gastroenterol. Hepatol. 2012;10:837-58). Treatment for this condition is predicated on losing weight via diet and exercise, with 7%-10% weight loss being associated with improvement in underlying liver disease. Unfortunately, it is quite difficult for most patients to lose and maintain this weight level, so attention has turned to pharmacotherapy as a possible adjunct to diet and exercise. To date, pioglitazone and vitamin E have shown the most promise but are not universally effective, and they are linked to unwanted side effects or potential disease associations (Clin. Gastroenterol. Hepatol. 2012;10:837-58).

|
| Dr. Stephen A. Harrison |
While these data are provocative, caution is needed until more complete data are presented. While the study met its primary endpoint, the report did not detail the source of the two-point improvement. NAS is based on an eight-point scale, with three points possible for the degree of steatosis, three points for lobular inflammation, and two points for ballooning degeneration of hepatocytes. Improvement in steatosis alone does not necessarily constitute improvement in NASH, the subset of disease that may progress in severity. It is important to know what percentage of patients actually resolved NASH or improved lobular inflammation or ballooning degeneration over placebo. Given that NASH patients are at increased risk for cardiovascular disease, it is concerning that serum cholesterol levels worsened. Details on LDL particle size and concentration as well as HDL subfractions are needed.
Additionally, further information on the tolerability and safety of OCA among NASH patients is eagerly awaited. While these preliminary data are promising, more details are needed before we can fully embrace this drug to treat NASH.
Dr. Stephen A. Harrison, COL, USA, MC, is director of medical education and associate dean, San Antonio Uniformed Services Health Education Consortium; chief of hepatology, Brooke Army Medical Center, San Antonio; professor of medicine, Uniformed Services University of the Health Sciences; and GI Consultant to the Office of the Surgeon General. He is an adviser to Genentech, Merck, and Nimbus Discovery, and has been on the speakers bureaus for Merck and Vertex within the past 12 months.
Current data suggest that nonalcoholic fatty liver disease (NAFLD) is present in 30%-40% of adults in the United States, with a subset of this population having a more aggressive form of fatty liver called nonalcoholic steatohepatitis (NASH), which may progress to cirrhosis. NASH is currently the third-leading cause for liver transplantation and is predicted to become the most common within 10-15 years (Gastroenterology 2011;141:1249-53).
Most NAFLD patients are overweight or obese and have underlying insulin resistance or diabetes mellitus. Comorbid illness is also common, with cardiovascular disease being the leading cause of death (Clin. Gastroenterol. Hepatol. 2012;10:837-58). Treatment for this condition is predicated on losing weight via diet and exercise, with 7%-10% weight loss being associated with improvement in underlying liver disease. Unfortunately, it is quite difficult for most patients to lose and maintain this weight level, so attention has turned to pharmacotherapy as a possible adjunct to diet and exercise. To date, pioglitazone and vitamin E have shown the most promise but are not universally effective, and they are linked to unwanted side effects or potential disease associations (Clin. Gastroenterol. Hepatol. 2012;10:837-58).
|
|
| Dr. Stephen A. Harrison |
While these data are provocative, caution is needed until more complete data are presented. While the study met its primary endpoint, the report did not detail the source of the two-point improvement. NAS is based on an eight-point scale, with three points possible for the degree of steatosis, three points for lobular inflammation, and two points for ballooning degeneration of hepatocytes. Improvement in steatosis alone does not necessarily constitute improvement in NASH, the subset of disease that may progress in severity. It is important to know what percentage of patients actually resolved NASH or improved lobular inflammation or ballooning degeneration over placebo. Given that NASH patients are at increased risk for cardiovascular disease, it is concerning that serum cholesterol levels worsened. Details on LDL particle size and concentration as well as HDL subfractions are needed.
Additionally, further information on the tolerability and safety of OCA among NASH patients is eagerly awaited. While these preliminary data are promising, more details are needed before we can fully embrace this drug to treat NASH.
Dr. Stephen A. Harrison, COL, USA, MC, is director of medical education and associate dean, San Antonio Uniformed Services Health Education Consortium; chief of hepatology, Brooke Army Medical Center, San Antonio; professor of medicine, Uniformed Services University of the Health Sciences; and GI Consultant to the Office of the Surgeon General. He is an adviser to Genentech, Merck, and Nimbus Discovery, and has been on the speakers bureaus for Merck and Vertex within the past 12 months.
Current data suggest that nonalcoholic fatty liver disease (NAFLD) is present in 30%-40% of adults in the United States, with a subset of this population having a more aggressive form of fatty liver called nonalcoholic steatohepatitis (NASH), which may progress to cirrhosis. NASH is currently the third-leading cause for liver transplantation and is predicted to become the most common within 10-15 years (Gastroenterology 2011;141:1249-53).
Most NAFLD patients are overweight or obese and have underlying insulin resistance or diabetes mellitus. Comorbid illness is also common, with cardiovascular disease being the leading cause of death (Clin. Gastroenterol. Hepatol. 2012;10:837-58). Treatment for this condition is predicated on losing weight via diet and exercise, with 7%-10% weight loss being associated with improvement in underlying liver disease. Unfortunately, it is quite difficult for most patients to lose and maintain this weight level, so attention has turned to pharmacotherapy as a possible adjunct to diet and exercise. To date, pioglitazone and vitamin E have shown the most promise but are not universally effective, and they are linked to unwanted side effects or potential disease associations (Clin. Gastroenterol. Hepatol. 2012;10:837-58).
|
|
| Dr. Stephen A. Harrison |
While these data are provocative, caution is needed until more complete data are presented. While the study met its primary endpoint, the report did not detail the source of the two-point improvement. NAS is based on an eight-point scale, with three points possible for the degree of steatosis, three points for lobular inflammation, and two points for ballooning degeneration of hepatocytes. Improvement in steatosis alone does not necessarily constitute improvement in NASH, the subset of disease that may progress in severity. It is important to know what percentage of patients actually resolved NASH or improved lobular inflammation or ballooning degeneration over placebo. Given that NASH patients are at increased risk for cardiovascular disease, it is concerning that serum cholesterol levels worsened. Details on LDL particle size and concentration as well as HDL subfractions are needed.
Additionally, further information on the tolerability and safety of OCA among NASH patients is eagerly awaited. While these preliminary data are promising, more details are needed before we can fully embrace this drug to treat NASH.
Dr. Stephen A. Harrison, COL, USA, MC, is director of medical education and associate dean, San Antonio Uniformed Services Health Education Consortium; chief of hepatology, Brooke Army Medical Center, San Antonio; professor of medicine, Uniformed Services University of the Health Sciences; and GI Consultant to the Office of the Surgeon General. He is an adviser to Genentech, Merck, and Nimbus Discovery, and has been on the speakers bureaus for Merck and Vertex within the past 12 months.
The phase II clinical trial of first-in-class farnesoid X receptor agonist obeticholic acid as a treatment for nonalcoholic steatohepatitis was stopped early because the trial endpoint was met with high significance, Intercept Pharmaceuticals announced Jan. 9.
The National Institute of Diabetes and Digestive and Kidney Diseases, which sponsored and conducted the FLINT trial, said in its statement that "OCA [obeticholic acid] has a significant beneficial effect on the liver damage due to NASH."
An interim analysis conducted by the Data Safety Monitoring Board of the trial found a highly statistically significant improvement in the primary endpoint, defined as "a decrease in the NAFLD Activity Score of at least two points with no worsening of fibrosis, as compared to placebo," the company said in a statement. The predetermined significance threshold for ending the trial was P less than .0031, but early results exceeded expectations with a significance of P = .0024.
An estimated 6 million adults in the United States have advanced liver fibrosis or cirrhosis caused bynonalcoholic steatohepatitis (NASH), said Dr. Mark Pruzanski, CEO of Intercept.
"This disease has seemingly come out of nowhere and has become an epidemic," he said. "On its current trajectory, NASH is expected to be the No. 1 indication for liver transplant over the next decade."
The FLINT trial, sponsored and conducted by the National Institute of Diabetes and Digestive and Kidney Diseases, studied the efficacy of a 25-mg. oral dose of obeticholic acid (OCA) in approximately half of 283 biopsy-confirmed NASH patients over a period of 72 weeks. Patients were randomized to receive either a 25-mg dose of OCA or placebo for the treatment period.
Participants were 18 years of age or older, with histologic evidence of nonalcoholic steatohepatitis, based on a liver biopsy, obtained no more than 90 days prior to randomization and a NAFLD activity score of 4 or greater with at least one in each component of the NAFLD activity score (steatosis scored 0-3; ballooning degeneration scored 0-2; and lobular inflammation scored 0-3). End of study biopsies were conducted in patients after the 72-week treatment period.
As specified in the protocol, a 24-week follow-up period will start, and all FLINT participants will stop taking the drug or placebo no later than Jan. 20.
Lipid derangements involving elevated total [cholesterol] and LDL cholesterol and reduced HDL cholesterol have been seen in OCA, compared with placebo-treated patients. While lipid abnormalities are not uncommon in NASH, researchers will be checking to see if lipids return to pretreatment levels during this 24-week follow-up period.
During this period, both FLINT investigators and trial participants will remain blinded to treatment. The NIDDK expects to present the results of the trial in the fourth quarter of 2014.
OCA has not only been studied as a promising treatment for NASH. A 2013 study also found that OCA increased insulin sensitivity and reduced markers of liver inflammation and fibrosis in patients with type 2 diabetes and nonalcoholic fatty liver disease. Additionally, OCA is currently in a phase III trial for the treatment of primary biliary cirrhosis.
A concurrent trial to study OCA as a treatment for NASH is underway in Japan, added Dr. Pruzanski, with top-line results expected in 2015. Dr. Pruzanski said he hopes the trial’s unexpected success will help pave the path to FDA approval, pending final NIDDK analyses and publication of the results.
The FLINT trial was sponsored and conducted by the National Institute of Diabetes and Digestive and Kidney Diseases.
The phase II clinical trial of first-in-class farnesoid X receptor agonist obeticholic acid as a treatment for nonalcoholic steatohepatitis was stopped early because the trial endpoint was met with high significance, Intercept Pharmaceuticals announced Jan. 9.
The National Institute of Diabetes and Digestive and Kidney Diseases, which sponsored and conducted the FLINT trial, said in its statement that "OCA [obeticholic acid] has a significant beneficial effect on the liver damage due to NASH."
An interim analysis conducted by the Data Safety Monitoring Board of the trial found a highly statistically significant improvement in the primary endpoint, defined as "a decrease in the NAFLD Activity Score of at least two points with no worsening of fibrosis, as compared to placebo," the company said in a statement. The predetermined significance threshold for ending the trial was P less than .0031, but early results exceeded expectations with a significance of P = .0024.
An estimated 6 million adults in the United States have advanced liver fibrosis or cirrhosis caused bynonalcoholic steatohepatitis (NASH), said Dr. Mark Pruzanski, CEO of Intercept.
"This disease has seemingly come out of nowhere and has become an epidemic," he said. "On its current trajectory, NASH is expected to be the No. 1 indication for liver transplant over the next decade."
The FLINT trial, sponsored and conducted by the National Institute of Diabetes and Digestive and Kidney Diseases, studied the efficacy of a 25-mg. oral dose of obeticholic acid (OCA) in approximately half of 283 biopsy-confirmed NASH patients over a period of 72 weeks. Patients were randomized to receive either a 25-mg dose of OCA or placebo for the treatment period.
Participants were 18 years of age or older, with histologic evidence of nonalcoholic steatohepatitis, based on a liver biopsy, obtained no more than 90 days prior to randomization and a NAFLD activity score of 4 or greater with at least one in each component of the NAFLD activity score (steatosis scored 0-3; ballooning degeneration scored 0-2; and lobular inflammation scored 0-3). End of study biopsies were conducted in patients after the 72-week treatment period.
As specified in the protocol, a 24-week follow-up period will start, and all FLINT participants will stop taking the drug or placebo no later than Jan. 20.
Lipid derangements involving elevated total [cholesterol] and LDL cholesterol and reduced HDL cholesterol have been seen in OCA, compared with placebo-treated patients. While lipid abnormalities are not uncommon in NASH, researchers will be checking to see if lipids return to pretreatment levels during this 24-week follow-up period.
During this period, both FLINT investigators and trial participants will remain blinded to treatment. The NIDDK expects to present the results of the trial in the fourth quarter of 2014.
OCA has not only been studied as a promising treatment for NASH. A 2013 study also found that OCA increased insulin sensitivity and reduced markers of liver inflammation and fibrosis in patients with type 2 diabetes and nonalcoholic fatty liver disease. Additionally, OCA is currently in a phase III trial for the treatment of primary biliary cirrhosis.
A concurrent trial to study OCA as a treatment for NASH is underway in Japan, added Dr. Pruzanski, with top-line results expected in 2015. Dr. Pruzanski said he hopes the trial’s unexpected success will help pave the path to FDA approval, pending final NIDDK analyses and publication of the results.
The FLINT trial was sponsored and conducted by the National Institute of Diabetes and Digestive and Kidney Diseases.
FROM A TELECONFERENCE HELD BY INTERCEPT PHARMACEUTICALS
Major finding: Treatment for nonalcoholic steatohepatitis with obeticholic acid resulted in a highly statistically significant improvement (P = .0024) for the primary endpoint.
Data source: A multicenter, double-blind, placebo-controlled clinical trial assessing the safety and efficacy of a 25-mg oral dose of first-in-class FXR-agonist OCA administered daily to biopsy-proven adult NASH patients over a 72-week treatment period.
Disclosures: The FLINT trial was sponsored by the National Institute of Diabetes and Digestive and Kidney Diseases.
Telaprevir label changes include switch to twice-daily dosing
The label of the hepatitis C antiviral drug telaprevir has been changed to include new dosing recommendations and contraindications, described in a "Dear Healthcare Professional" letter issued by the manufacturer.
Telaprevir is a hepatitis C virus (HCV) NS3/4A protease inhibitor approved in 2011 for the treatment of genotype 1 chronic HCV infection in adults with compensated liver disease, in combination with peginterferon alfa and ribavirin. It is marketed as Incivek, by Vertex Pharmaceuticals.
The recommended dose is now 1,125 mg (three 375-mg tablets) taken orally twice a day, 10-14 hours apart with food that is not low fat, according to the Oct. 28 letter. "This change in dosing frequency was based on data from a clinical trial demonstrating similar sustained virologic response and safety profile between the twice-daily dosing and the every-8-hour dosing," the letter stated.
In addition, carbamazepine, phenobarbital, and phenytoin are now on the list of contraindicated drugs, based on a pharmacokinetics study that found coadministration of these drugs with telaprevir "has the potential to lower exposure and, thus, may result in a loss of efficacy" of telaprevir, according to the letter.
New twice-daily-dosing packaging will be available at the end of January 2014. Until then, the current packaging that contains six tablets per blister strip – with three blister sections containing two tablets each – can be used for the twice-daily recommendation. Two tablets from one blister section and one tablet from another section can be taken for the three-pill dose, administered twice a day, the letter pointed out.
The updated label is available at http://pi.vrtx.com/files/uspi_telaprevir.pdf.
Vertex can be contacted for questions at 877-824-4281. Serious adverse events associated with telaprevir should be reported to Vertex at that number, or to the Food and Drug Administration at 800-332-1088 or at www.fda.gov/medwatch.
The label of the hepatitis C antiviral drug telaprevir has been changed to include new dosing recommendations and contraindications, described in a "Dear Healthcare Professional" letter issued by the manufacturer.
Telaprevir is a hepatitis C virus (HCV) NS3/4A protease inhibitor approved in 2011 for the treatment of genotype 1 chronic HCV infection in adults with compensated liver disease, in combination with peginterferon alfa and ribavirin. It is marketed as Incivek, by Vertex Pharmaceuticals.
The recommended dose is now 1,125 mg (three 375-mg tablets) taken orally twice a day, 10-14 hours apart with food that is not low fat, according to the Oct. 28 letter. "This change in dosing frequency was based on data from a clinical trial demonstrating similar sustained virologic response and safety profile between the twice-daily dosing and the every-8-hour dosing," the letter stated.
In addition, carbamazepine, phenobarbital, and phenytoin are now on the list of contraindicated drugs, based on a pharmacokinetics study that found coadministration of these drugs with telaprevir "has the potential to lower exposure and, thus, may result in a loss of efficacy" of telaprevir, according to the letter.
New twice-daily-dosing packaging will be available at the end of January 2014. Until then, the current packaging that contains six tablets per blister strip – with three blister sections containing two tablets each – can be used for the twice-daily recommendation. Two tablets from one blister section and one tablet from another section can be taken for the three-pill dose, administered twice a day, the letter pointed out.
The updated label is available at http://pi.vrtx.com/files/uspi_telaprevir.pdf.
Vertex can be contacted for questions at 877-824-4281. Serious adverse events associated with telaprevir should be reported to Vertex at that number, or to the Food and Drug Administration at 800-332-1088 or at www.fda.gov/medwatch.
The label of the hepatitis C antiviral drug telaprevir has been changed to include new dosing recommendations and contraindications, described in a "Dear Healthcare Professional" letter issued by the manufacturer.
Telaprevir is a hepatitis C virus (HCV) NS3/4A protease inhibitor approved in 2011 for the treatment of genotype 1 chronic HCV infection in adults with compensated liver disease, in combination with peginterferon alfa and ribavirin. It is marketed as Incivek, by Vertex Pharmaceuticals.
The recommended dose is now 1,125 mg (three 375-mg tablets) taken orally twice a day, 10-14 hours apart with food that is not low fat, according to the Oct. 28 letter. "This change in dosing frequency was based on data from a clinical trial demonstrating similar sustained virologic response and safety profile between the twice-daily dosing and the every-8-hour dosing," the letter stated.
In addition, carbamazepine, phenobarbital, and phenytoin are now on the list of contraindicated drugs, based on a pharmacokinetics study that found coadministration of these drugs with telaprevir "has the potential to lower exposure and, thus, may result in a loss of efficacy" of telaprevir, according to the letter.
New twice-daily-dosing packaging will be available at the end of January 2014. Until then, the current packaging that contains six tablets per blister strip – with three blister sections containing two tablets each – can be used for the twice-daily recommendation. Two tablets from one blister section and one tablet from another section can be taken for the three-pill dose, administered twice a day, the letter pointed out.
The updated label is available at http://pi.vrtx.com/files/uspi_telaprevir.pdf.
Vertex can be contacted for questions at 877-824-4281. Serious adverse events associated with telaprevir should be reported to Vertex at that number, or to the Food and Drug Administration at 800-332-1088 or at www.fda.gov/medwatch.
FDA approves sofosbuvir for chronic hepatitis C
The Food and Drug Administration has approved sofosbuvir, a first-in-its-class antiviral, to treat chronic hepatitis C infection.
Sofosbuvir is a nucleotide analogue inhibitor of the hepatitis C virus (HCV) NS5B polymerase enzyme, which plays an important role in HCV replication. It is taken orally once a day at a 400-mg dose. It will be marketed as Sovaldi by Gilead Sciences.
The drug is approved for two chronic hepatitis C indications: In combination with pegylated interferon and ribavirin for treatment-naïve adults with genotype 1 and 4 infections, and in combination with ribavirin for adults with genotypes 2 and 3 infection.
The second indication is the first approval of an interferon-free regimen for the treatment for chronic hepatitis C.
"Today’s approval represents a significant shift in the treatment paradigm for some patients with chronic hepatitis C," Dr. Edward Cox, director of the Office of Antimicrobial Products in the FDA Center for Drug Evaluation and Research, said in a statement.
This is "truly a historic moment," Dr. Demetre Daskalakis, medical director of the HIV program at Mt. Sinai School of Medicine, New York, said at an FDA advisory committee meeting on the drug held Oct. 25. "I can’t wait to get this drug into the clinic. We are all excited," he added. The Antiviral Drugs Advisory Committee voted unanimously that day to recommend approval for sofosbuvir.
Sofosbuvir was approved based on data from six clinical trials consisting of 1,947 patients – both treatment-naïve and treatment experienced – some of whom were also HIV positive.
The most common side effects reported in clinical study participants treated with sofosbuvir and ribavirin were fatigue and headache. In participants treated with sofosbuvir, ribavirin, and peginterferon-alfa, the most common side effects reported were fatigue, headache, nausea, insomnia, and anemia, according to the FDA.
According to Gilead, sofosbuvir is also on the verge of receiving marketing approval in the European Union. Sofosbuvir is the second drug approved by the FDA in the past two weeks to treat chronic HCV infection. Simeprevir was approved Nov. 22.
Elizabeth Mechcatie contributed to this report.
The Food and Drug Administration has approved sofosbuvir, a first-in-its-class antiviral, to treat chronic hepatitis C infection.
Sofosbuvir is a nucleotide analogue inhibitor of the hepatitis C virus (HCV) NS5B polymerase enzyme, which plays an important role in HCV replication. It is taken orally once a day at a 400-mg dose. It will be marketed as Sovaldi by Gilead Sciences.
The drug is approved for two chronic hepatitis C indications: In combination with pegylated interferon and ribavirin for treatment-naïve adults with genotype 1 and 4 infections, and in combination with ribavirin for adults with genotypes 2 and 3 infection.
The second indication is the first approval of an interferon-free regimen for the treatment for chronic hepatitis C.
"Today’s approval represents a significant shift in the treatment paradigm for some patients with chronic hepatitis C," Dr. Edward Cox, director of the Office of Antimicrobial Products in the FDA Center for Drug Evaluation and Research, said in a statement.
This is "truly a historic moment," Dr. Demetre Daskalakis, medical director of the HIV program at Mt. Sinai School of Medicine, New York, said at an FDA advisory committee meeting on the drug held Oct. 25. "I can’t wait to get this drug into the clinic. We are all excited," he added. The Antiviral Drugs Advisory Committee voted unanimously that day to recommend approval for sofosbuvir.
Sofosbuvir was approved based on data from six clinical trials consisting of 1,947 patients – both treatment-naïve and treatment experienced – some of whom were also HIV positive.
The most common side effects reported in clinical study participants treated with sofosbuvir and ribavirin were fatigue and headache. In participants treated with sofosbuvir, ribavirin, and peginterferon-alfa, the most common side effects reported were fatigue, headache, nausea, insomnia, and anemia, according to the FDA.
According to Gilead, sofosbuvir is also on the verge of receiving marketing approval in the European Union. Sofosbuvir is the second drug approved by the FDA in the past two weeks to treat chronic HCV infection. Simeprevir was approved Nov. 22.
Elizabeth Mechcatie contributed to this report.
The Food and Drug Administration has approved sofosbuvir, a first-in-its-class antiviral, to treat chronic hepatitis C infection.
Sofosbuvir is a nucleotide analogue inhibitor of the hepatitis C virus (HCV) NS5B polymerase enzyme, which plays an important role in HCV replication. It is taken orally once a day at a 400-mg dose. It will be marketed as Sovaldi by Gilead Sciences.
The drug is approved for two chronic hepatitis C indications: In combination with pegylated interferon and ribavirin for treatment-naïve adults with genotype 1 and 4 infections, and in combination with ribavirin for adults with genotypes 2 and 3 infection.
The second indication is the first approval of an interferon-free regimen for the treatment for chronic hepatitis C.
"Today’s approval represents a significant shift in the treatment paradigm for some patients with chronic hepatitis C," Dr. Edward Cox, director of the Office of Antimicrobial Products in the FDA Center for Drug Evaluation and Research, said in a statement.
This is "truly a historic moment," Dr. Demetre Daskalakis, medical director of the HIV program at Mt. Sinai School of Medicine, New York, said at an FDA advisory committee meeting on the drug held Oct. 25. "I can’t wait to get this drug into the clinic. We are all excited," he added. The Antiviral Drugs Advisory Committee voted unanimously that day to recommend approval for sofosbuvir.
Sofosbuvir was approved based on data from six clinical trials consisting of 1,947 patients – both treatment-naïve and treatment experienced – some of whom were also HIV positive.
The most common side effects reported in clinical study participants treated with sofosbuvir and ribavirin were fatigue and headache. In participants treated with sofosbuvir, ribavirin, and peginterferon-alfa, the most common side effects reported were fatigue, headache, nausea, insomnia, and anemia, according to the FDA.
According to Gilead, sofosbuvir is also on the verge of receiving marketing approval in the European Union. Sofosbuvir is the second drug approved by the FDA in the past two weeks to treat chronic HCV infection. Simeprevir was approved Nov. 22.
Elizabeth Mechcatie contributed to this report.
Once-a-day HCV protease inhibitor approved for treating hepatitis C
Simeprevir, a hepatitis C virus protease inhibitor, has been approved as a treatment for chronic hepatitis C in patients who have compensated liver disease, including cirrhosis, the Food and Drug Administration announced.
For patients with HCV genotype 1 infections, simeprevir’s approved dose is a 150-mg capsule taken once a day with food, in combination with peginterferon-alfa and ribavirin for 24-48 weeks. Before treatment, clinicians should screen patients with genotype 1a HCV infections for the presence of the NS3 Q80K polymorphism, which was associated with lower response rates in studies, "and to consider alternative therapy if the strain is detected," according to the FDA’s Nov. 22 statement announcing the approval.
Simeprevir is the third protease inhibitor to be approved by the FDA for treatment of hepatitis C. Boceprevir (Victrelis) and telaprevir (Incivek) were approved in 2011 and have been the standard of care, but they require taking 6-12 pills a day. Simeprevir is an NS3/4A protease inhibitor, which blocks the viral protease enzyme that enables HCV to replicate in host cells, according to Janssen Pharmaceuticals, which will market the drug as Olysio.
Simeprevir’s simpler dosing regimen, effectiveness, and favorable risk-benefit profile in clinical trials were cited by the FDA’s antiviral drug advisory committee in its unanimous recommendation to approve the drug at a meeting on Oct. 24.
Approval is based on five trials of 2,026 treatment-naive and treatment-experienced patients with chronic hepatitis C who were randomized to simeprevir plus peginterferon-alfa and ribavirin (PR) or to placebo plus PR. The primary endpoint was sustained virologic response rate at 12 weeks after treatment was planned to end (SVR12). SVR was defined as an HCV RNA level that was undetectable or detected only below a prespecified value.
Among the treatment-naive patients, the SVR12 rate was 80% for those treated with simeprevir plus PR and 50% in those who received placebo plus PR. In a study of treatment-experienced patients, 79% of those treated with the simeprevir regimen and 37% of those on peginterferon-alfa and ribavirin achieved SVR12.
In another study of treatment-experienced patients that included prior relapsers, partial responders, and null responders (those who did not respond to previous treatments at all), the addition of simeprevir to PR improved response rates when compared with treatment with PR alone, according to the FDA.
Rash, pruritus, and nausea were the most common adverse effects associated with treatment in the studies, the FDA said. There were cases of serious photosensitivity that required hospitalization, and the prescribing information recommends that patients use sun protection and limit sun exposure during treatment. The prescribing information notes that dosing recommendations cannot be made at this time for patients with moderate to severe hepatic impairment or those of East Asian descent. At the FDA panel meeting, company officials said that the appropriate dose in this population was being investigated.
Simeprevir was approved for treatment of genotype 1 HCV infection in September 2013 in Japan, where it is marketed as Sovriad (at a lower 100-mg dose), and in November in Canada, where it is marketed as Galexos, according to Janssen. Simeprevir is under review in Europe for the treatment of genotype 1 or 4 HCV.
Imminent approval of sofosbuvir, another HCV treatment with a different mechanism of action, is expected.
Simeprevir, a hepatitis C virus protease inhibitor, has been approved as a treatment for chronic hepatitis C in patients who have compensated liver disease, including cirrhosis, the Food and Drug Administration announced.
For patients with HCV genotype 1 infections, simeprevir’s approved dose is a 150-mg capsule taken once a day with food, in combination with peginterferon-alfa and ribavirin for 24-48 weeks. Before treatment, clinicians should screen patients with genotype 1a HCV infections for the presence of the NS3 Q80K polymorphism, which was associated with lower response rates in studies, "and to consider alternative therapy if the strain is detected," according to the FDA’s Nov. 22 statement announcing the approval.
Simeprevir is the third protease inhibitor to be approved by the FDA for treatment of hepatitis C. Boceprevir (Victrelis) and telaprevir (Incivek) were approved in 2011 and have been the standard of care, but they require taking 6-12 pills a day. Simeprevir is an NS3/4A protease inhibitor, which blocks the viral protease enzyme that enables HCV to replicate in host cells, according to Janssen Pharmaceuticals, which will market the drug as Olysio.
Simeprevir’s simpler dosing regimen, effectiveness, and favorable risk-benefit profile in clinical trials were cited by the FDA’s antiviral drug advisory committee in its unanimous recommendation to approve the drug at a meeting on Oct. 24.
Approval is based on five trials of 2,026 treatment-naive and treatment-experienced patients with chronic hepatitis C who were randomized to simeprevir plus peginterferon-alfa and ribavirin (PR) or to placebo plus PR. The primary endpoint was sustained virologic response rate at 12 weeks after treatment was planned to end (SVR12). SVR was defined as an HCV RNA level that was undetectable or detected only below a prespecified value.
Among the treatment-naive patients, the SVR12 rate was 80% for those treated with simeprevir plus PR and 50% in those who received placebo plus PR. In a study of treatment-experienced patients, 79% of those treated with the simeprevir regimen and 37% of those on peginterferon-alfa and ribavirin achieved SVR12.
In another study of treatment-experienced patients that included prior relapsers, partial responders, and null responders (those who did not respond to previous treatments at all), the addition of simeprevir to PR improved response rates when compared with treatment with PR alone, according to the FDA.
Rash, pruritus, and nausea were the most common adverse effects associated with treatment in the studies, the FDA said. There were cases of serious photosensitivity that required hospitalization, and the prescribing information recommends that patients use sun protection and limit sun exposure during treatment. The prescribing information notes that dosing recommendations cannot be made at this time for patients with moderate to severe hepatic impairment or those of East Asian descent. At the FDA panel meeting, company officials said that the appropriate dose in this population was being investigated.
Simeprevir was approved for treatment of genotype 1 HCV infection in September 2013 in Japan, where it is marketed as Sovriad (at a lower 100-mg dose), and in November in Canada, where it is marketed as Galexos, according to Janssen. Simeprevir is under review in Europe for the treatment of genotype 1 or 4 HCV.
Imminent approval of sofosbuvir, another HCV treatment with a different mechanism of action, is expected.
Simeprevir, a hepatitis C virus protease inhibitor, has been approved as a treatment for chronic hepatitis C in patients who have compensated liver disease, including cirrhosis, the Food and Drug Administration announced.
For patients with HCV genotype 1 infections, simeprevir’s approved dose is a 150-mg capsule taken once a day with food, in combination with peginterferon-alfa and ribavirin for 24-48 weeks. Before treatment, clinicians should screen patients with genotype 1a HCV infections for the presence of the NS3 Q80K polymorphism, which was associated with lower response rates in studies, "and to consider alternative therapy if the strain is detected," according to the FDA’s Nov. 22 statement announcing the approval.
Simeprevir is the third protease inhibitor to be approved by the FDA for treatment of hepatitis C. Boceprevir (Victrelis) and telaprevir (Incivek) were approved in 2011 and have been the standard of care, but they require taking 6-12 pills a day. Simeprevir is an NS3/4A protease inhibitor, which blocks the viral protease enzyme that enables HCV to replicate in host cells, according to Janssen Pharmaceuticals, which will market the drug as Olysio.
Simeprevir’s simpler dosing regimen, effectiveness, and favorable risk-benefit profile in clinical trials were cited by the FDA’s antiviral drug advisory committee in its unanimous recommendation to approve the drug at a meeting on Oct. 24.
Approval is based on five trials of 2,026 treatment-naive and treatment-experienced patients with chronic hepatitis C who were randomized to simeprevir plus peginterferon-alfa and ribavirin (PR) or to placebo plus PR. The primary endpoint was sustained virologic response rate at 12 weeks after treatment was planned to end (SVR12). SVR was defined as an HCV RNA level that was undetectable or detected only below a prespecified value.
Among the treatment-naive patients, the SVR12 rate was 80% for those treated with simeprevir plus PR and 50% in those who received placebo plus PR. In a study of treatment-experienced patients, 79% of those treated with the simeprevir regimen and 37% of those on peginterferon-alfa and ribavirin achieved SVR12.
In another study of treatment-experienced patients that included prior relapsers, partial responders, and null responders (those who did not respond to previous treatments at all), the addition of simeprevir to PR improved response rates when compared with treatment with PR alone, according to the FDA.
Rash, pruritus, and nausea were the most common adverse effects associated with treatment in the studies, the FDA said. There were cases of serious photosensitivity that required hospitalization, and the prescribing information recommends that patients use sun protection and limit sun exposure during treatment. The prescribing information notes that dosing recommendations cannot be made at this time for patients with moderate to severe hepatic impairment or those of East Asian descent. At the FDA panel meeting, company officials said that the appropriate dose in this population was being investigated.
Simeprevir was approved for treatment of genotype 1 HCV infection in September 2013 in Japan, where it is marketed as Sovriad (at a lower 100-mg dose), and in November in Canada, where it is marketed as Galexos, according to Janssen. Simeprevir is under review in Europe for the treatment of genotype 1 or 4 HCV.
Imminent approval of sofosbuvir, another HCV treatment with a different mechanism of action, is expected.