User login
CAR T cells may fight fungal infections
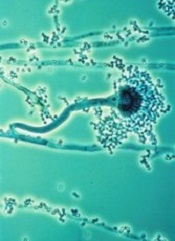
T cells modified using the Sleeping Beauty gene transfer system may help fight infections caused by invasive Aspergillus fungus.
Sleeping Beauty is already being used to create chimeric antigen receptor (CAR) T cells to treat leukemias and lymphomas.
And now, researchers have found the system may also be effective for combatting fungal infections that can be deadly for immunosuppressed patients, such as those receiving transplants to treat hematologic cancers.
“We demonstrated a new approach for Aspergillus immunotherapy based on redirecting T-cell specificity through a CAR that recognizes carbohydrate antigen on the fungal cell wall,” said study author Laurence Cooper, MD, PhD, of MD Anderson Cancer Center in Houston, Texas.
He and his colleagues described this approach in the Proceedings of the National Academy of Sciences.
Dr Cooper originally learned about Sleeping Beauty gene transfer from a study published by Perry Hackett, PhD, a professor at the University of Minnesota who created the process.
The system is named Sleeping Beauty because Dr Hackett was able to “awaken” an extinct transposon—DNA that can replicate itself and insert the copy back into the genome—and package it with a gene he wants to transfer into a plasmid. An associated transposase enzyme binds to the plasmid, cuts the transposon and gene out of the plasmid, and pastes it into the target DNA sequence.
Dr Cooper and his colleagues have found they can use this process to engineer T cells that target sugar molecules in the Aspergillus cell walls, thereby killing the fungus.
Specifically, the team adapted the pattern-recognition receptor Dectin-1 to activate T cells via chimeric CD28 and CD3-ζ (D-CAR) upon binding with carbohydrate in the cell wall of Aspergillus germlings. They used Sleeping Beauty to modify the T cells to express D-CAR.
These D-CAR+ T cells exhibited specificity for β-glucan, and this inhibited Aspergillus growth both in vitro and in vivo. Furthermore, the researchers found that treating D-CAR+ T cells with steroids did not significantly compromise antifungal activity.
“The [D-CAR+ T cells] can be manipulated in a manner suitable for human application,” Dr Cooper said, “enabling this immunology to be translated into immunotherapy.”![]()
T cells modified using the Sleeping Beauty gene transfer system may help fight infections caused by invasive Aspergillus fungus.
Sleeping Beauty is already being used to create chimeric antigen receptor (CAR) T cells to treat leukemias and lymphomas.
And now, researchers have found the system may also be effective for combatting fungal infections that can be deadly for immunosuppressed patients, such as those receiving transplants to treat hematologic cancers.
“We demonstrated a new approach for Aspergillus immunotherapy based on redirecting T-cell specificity through a CAR that recognizes carbohydrate antigen on the fungal cell wall,” said study author Laurence Cooper, MD, PhD, of MD Anderson Cancer Center in Houston, Texas.
He and his colleagues described this approach in the Proceedings of the National Academy of Sciences.
Dr Cooper originally learned about Sleeping Beauty gene transfer from a study published by Perry Hackett, PhD, a professor at the University of Minnesota who created the process.
The system is named Sleeping Beauty because Dr Hackett was able to “awaken” an extinct transposon—DNA that can replicate itself and insert the copy back into the genome—and package it with a gene he wants to transfer into a plasmid. An associated transposase enzyme binds to the plasmid, cuts the transposon and gene out of the plasmid, and pastes it into the target DNA sequence.
Dr Cooper and his colleagues have found they can use this process to engineer T cells that target sugar molecules in the Aspergillus cell walls, thereby killing the fungus.
Specifically, the team adapted the pattern-recognition receptor Dectin-1 to activate T cells via chimeric CD28 and CD3-ζ (D-CAR) upon binding with carbohydrate in the cell wall of Aspergillus germlings. They used Sleeping Beauty to modify the T cells to express D-CAR.
These D-CAR+ T cells exhibited specificity for β-glucan, and this inhibited Aspergillus growth both in vitro and in vivo. Furthermore, the researchers found that treating D-CAR+ T cells with steroids did not significantly compromise antifungal activity.
“The [D-CAR+ T cells] can be manipulated in a manner suitable for human application,” Dr Cooper said, “enabling this immunology to be translated into immunotherapy.”![]()
T cells modified using the Sleeping Beauty gene transfer system may help fight infections caused by invasive Aspergillus fungus.
Sleeping Beauty is already being used to create chimeric antigen receptor (CAR) T cells to treat leukemias and lymphomas.
And now, researchers have found the system may also be effective for combatting fungal infections that can be deadly for immunosuppressed patients, such as those receiving transplants to treat hematologic cancers.
“We demonstrated a new approach for Aspergillus immunotherapy based on redirecting T-cell specificity through a CAR that recognizes carbohydrate antigen on the fungal cell wall,” said study author Laurence Cooper, MD, PhD, of MD Anderson Cancer Center in Houston, Texas.
He and his colleagues described this approach in the Proceedings of the National Academy of Sciences.
Dr Cooper originally learned about Sleeping Beauty gene transfer from a study published by Perry Hackett, PhD, a professor at the University of Minnesota who created the process.
The system is named Sleeping Beauty because Dr Hackett was able to “awaken” an extinct transposon—DNA that can replicate itself and insert the copy back into the genome—and package it with a gene he wants to transfer into a plasmid. An associated transposase enzyme binds to the plasmid, cuts the transposon and gene out of the plasmid, and pastes it into the target DNA sequence.
Dr Cooper and his colleagues have found they can use this process to engineer T cells that target sugar molecules in the Aspergillus cell walls, thereby killing the fungus.
Specifically, the team adapted the pattern-recognition receptor Dectin-1 to activate T cells via chimeric CD28 and CD3-ζ (D-CAR) upon binding with carbohydrate in the cell wall of Aspergillus germlings. They used Sleeping Beauty to modify the T cells to express D-CAR.
These D-CAR+ T cells exhibited specificity for β-glucan, and this inhibited Aspergillus growth both in vitro and in vivo. Furthermore, the researchers found that treating D-CAR+ T cells with steroids did not significantly compromise antifungal activity.
“The [D-CAR+ T cells] can be manipulated in a manner suitable for human application,” Dr Cooper said, “enabling this immunology to be translated into immunotherapy.”![]()
Inhibitor improves survival in older AML patients
Credit: Rhoda Baer
Adding the Plk1 inhibitor volasertib to chemotherapy can prolong survival in older patients with previously untreated acute myeloid leukemia (AML), researchers have reported in Blood.
In a phase 2 study, AML patients aged 65 or older who were ineligible for intensive induction therapy had higher response and survival rates when they received volasertib plus low-dose cytarabine (LDAC), compared to LDAC alone.
However, adverse events, such as febrile neutropenia and infections, were more common with volasertib.
“These clinical trial results . . . are important and have informed future research for this rare disease, where new treatment options are greatly needed,” said study author Hartmut Döhner, MD, of the University Hospital Ulm in Germany.
“The established approach to treat younger AML patients is an intensive chemotherapy regimen, [but] older patients often cannot tolerate these chemotherapy doses and have very limited treatment options.”
To test volasertib as a potential option, the researchers enrolled and treated 87 patients with previously untreated AML who were ineligible for intensive induction therapy. Their median age was 75 years.
Patients received LDAC at 20 mg BID subcutaneously on days 1 through 10 (n=45) or LDAC plus volasertib at 350 mg intravenously on days 1 and 15, every 4 weeks (n=42). Overall, patient demographics and baseline disease characteristics were balanced between the treatment arms.
The response rate (complete response or complete response with incomplete blood count recovery) was more than doubled for patients receiving volasertib and LDAC compared to LDAC alone. The rates were 31% (13/42) and 13.3% (6/45), respectively (odds ratio, 2.91; P=0.052).
Responses in patients receiving volasertib and LDAC were observed across all genetic groups, including 5 of 14 patients with adverse genetics.
Remissions with the combination treatment appeared to be more durable than those observed with LDAC alone. The median relapse-free survival was 18.5 months and 10.0 months, respectively.
The median event-free survival was prolonged in patients receiving volasertib as well. Their event-free survival was 5.6 months, compared to 2.3 months for patients who received LDAC alone (hazard ratio 0.57,
P=0.021).
Patients who received volasertib also experienced improvements in overall survival. The median overall survival was 8.0 months for the volasertib arm and 5.2 months for the LDAC-alone arm (hazard ratio 0.63; P=0.047).
Patients receiving volasertib and LDAC had higher rates of adverse events than patients in the LDAC-alone arm. Events of note included grade 3 febrile neutropenia (38% vs 7%), grade 3 infections (38% vs 7%) and grade 3 gastrointestinal events (21% vs 7%).
Based on these results, researchers are now investigating volasertib in combination with LDAC in a randomized, double-blind, phase 3 trial for AML called POLO-AML-2. ![]()
Credit: Rhoda Baer
Adding the Plk1 inhibitor volasertib to chemotherapy can prolong survival in older patients with previously untreated acute myeloid leukemia (AML), researchers have reported in Blood.
In a phase 2 study, AML patients aged 65 or older who were ineligible for intensive induction therapy had higher response and survival rates when they received volasertib plus low-dose cytarabine (LDAC), compared to LDAC alone.
However, adverse events, such as febrile neutropenia and infections, were more common with volasertib.
“These clinical trial results . . . are important and have informed future research for this rare disease, where new treatment options are greatly needed,” said study author Hartmut Döhner, MD, of the University Hospital Ulm in Germany.
“The established approach to treat younger AML patients is an intensive chemotherapy regimen, [but] older patients often cannot tolerate these chemotherapy doses and have very limited treatment options.”
To test volasertib as a potential option, the researchers enrolled and treated 87 patients with previously untreated AML who were ineligible for intensive induction therapy. Their median age was 75 years.
Patients received LDAC at 20 mg BID subcutaneously on days 1 through 10 (n=45) or LDAC plus volasertib at 350 mg intravenously on days 1 and 15, every 4 weeks (n=42). Overall, patient demographics and baseline disease characteristics were balanced between the treatment arms.
The response rate (complete response or complete response with incomplete blood count recovery) was more than doubled for patients receiving volasertib and LDAC compared to LDAC alone. The rates were 31% (13/42) and 13.3% (6/45), respectively (odds ratio, 2.91; P=0.052).
Responses in patients receiving volasertib and LDAC were observed across all genetic groups, including 5 of 14 patients with adverse genetics.
Remissions with the combination treatment appeared to be more durable than those observed with LDAC alone. The median relapse-free survival was 18.5 months and 10.0 months, respectively.
The median event-free survival was prolonged in patients receiving volasertib as well. Their event-free survival was 5.6 months, compared to 2.3 months for patients who received LDAC alone (hazard ratio 0.57,
P=0.021).
Patients who received volasertib also experienced improvements in overall survival. The median overall survival was 8.0 months for the volasertib arm and 5.2 months for the LDAC-alone arm (hazard ratio 0.63; P=0.047).
Patients receiving volasertib and LDAC had higher rates of adverse events than patients in the LDAC-alone arm. Events of note included grade 3 febrile neutropenia (38% vs 7%), grade 3 infections (38% vs 7%) and grade 3 gastrointestinal events (21% vs 7%).
Based on these results, researchers are now investigating volasertib in combination with LDAC in a randomized, double-blind, phase 3 trial for AML called POLO-AML-2. ![]()
Credit: Rhoda Baer
Adding the Plk1 inhibitor volasertib to chemotherapy can prolong survival in older patients with previously untreated acute myeloid leukemia (AML), researchers have reported in Blood.
In a phase 2 study, AML patients aged 65 or older who were ineligible for intensive induction therapy had higher response and survival rates when they received volasertib plus low-dose cytarabine (LDAC), compared to LDAC alone.
However, adverse events, such as febrile neutropenia and infections, were more common with volasertib.
“These clinical trial results . . . are important and have informed future research for this rare disease, where new treatment options are greatly needed,” said study author Hartmut Döhner, MD, of the University Hospital Ulm in Germany.
“The established approach to treat younger AML patients is an intensive chemotherapy regimen, [but] older patients often cannot tolerate these chemotherapy doses and have very limited treatment options.”
To test volasertib as a potential option, the researchers enrolled and treated 87 patients with previously untreated AML who were ineligible for intensive induction therapy. Their median age was 75 years.
Patients received LDAC at 20 mg BID subcutaneously on days 1 through 10 (n=45) or LDAC plus volasertib at 350 mg intravenously on days 1 and 15, every 4 weeks (n=42). Overall, patient demographics and baseline disease characteristics were balanced between the treatment arms.
The response rate (complete response or complete response with incomplete blood count recovery) was more than doubled for patients receiving volasertib and LDAC compared to LDAC alone. The rates were 31% (13/42) and 13.3% (6/45), respectively (odds ratio, 2.91; P=0.052).
Responses in patients receiving volasertib and LDAC were observed across all genetic groups, including 5 of 14 patients with adverse genetics.
Remissions with the combination treatment appeared to be more durable than those observed with LDAC alone. The median relapse-free survival was 18.5 months and 10.0 months, respectively.
The median event-free survival was prolonged in patients receiving volasertib as well. Their event-free survival was 5.6 months, compared to 2.3 months for patients who received LDAC alone (hazard ratio 0.57,
P=0.021).
Patients who received volasertib also experienced improvements in overall survival. The median overall survival was 8.0 months for the volasertib arm and 5.2 months for the LDAC-alone arm (hazard ratio 0.63; P=0.047).
Patients receiving volasertib and LDAC had higher rates of adverse events than patients in the LDAC-alone arm. Events of note included grade 3 febrile neutropenia (38% vs 7%), grade 3 infections (38% vs 7%) and grade 3 gastrointestinal events (21% vs 7%).
Based on these results, researchers are now investigating volasertib in combination with LDAC in a randomized, double-blind, phase 3 trial for AML called POLO-AML-2. ![]()
Increasing AYA enrollment in cancer trials

patient and her father
Credit: Rhoda Baer
Age limits on clinical trials must be more flexible to allow more adolescent and young adult (AYA) cancer patients the opportunity to access new treatments, according to a report published in The Lancet Oncology.
The report’s authors discovered that expanding age eligibility criteria for cancer trials increased the enrollment of AYA patients and patients belonging to other age groups. But there is still room for improvement, according to the authors.
“[R]ight now, too many of our young patients are needlessly falling through the gap between pediatric and adult cancer trials,” said Lorna Fern, PhD, of University College London Hospitals in the UK.
“By encouraging doctors to take into account the full age range of patients affected by individual types of cancer, we’ve shown that it’s possible to design trials that include teenage cancer patients and, importantly, that better match the underlying biology of the disease and the people affected.”
To assess AYA enrollment in cancer trials, Dr Fern and her colleagues looked at 68,275 cancer patients aged 0 to 59 years. They were diagnosed with leukemias, lymphomas, or solid tumor malignancies between April 1, 2005, and March 31, 2010.
During this 6-year period, trial participation increased among all age groups. There was a 13% increase in participation among 15- to 19-year-olds (from 24% to 37%), a 5% increase among 20- to 24-year-olds (from 13% to 18%), and a 6% increase among 0- to 14-year-olds (from 52% to 58%).
Dr Fern and her colleagues said the rise in enrollment, particularly among AYAs, was due to increased availability and access to trials; increased awareness from healthcare professionals, patients, and the public about research; and the opening of trials with broader age limits that allow AYAs to enter trials.
In light of this study, Cancer Research UK has started asking researchers to justify age restrictions on new studies, in an effort to recruit more AYA cancer patients onto its trials.
“[I]t’s vital that effective treatments are being developed to tackle cancer across all age brackets,” said Kate Law, Cancer Research UK’s director of clinical trials.
“We now only accept age limits on our clinical trials if they are backed up by hard evidence, which will hopefully mean more young cancer patients get the chance to contribute to research and have the latest experimental treatments.” ![]()
patient and her father
Credit: Rhoda Baer
Age limits on clinical trials must be more flexible to allow more adolescent and young adult (AYA) cancer patients the opportunity to access new treatments, according to a report published in The Lancet Oncology.
The report’s authors discovered that expanding age eligibility criteria for cancer trials increased the enrollment of AYA patients and patients belonging to other age groups. But there is still room for improvement, according to the authors.
“[R]ight now, too many of our young patients are needlessly falling through the gap between pediatric and adult cancer trials,” said Lorna Fern, PhD, of University College London Hospitals in the UK.
“By encouraging doctors to take into account the full age range of patients affected by individual types of cancer, we’ve shown that it’s possible to design trials that include teenage cancer patients and, importantly, that better match the underlying biology of the disease and the people affected.”
To assess AYA enrollment in cancer trials, Dr Fern and her colleagues looked at 68,275 cancer patients aged 0 to 59 years. They were diagnosed with leukemias, lymphomas, or solid tumor malignancies between April 1, 2005, and March 31, 2010.
During this 6-year period, trial participation increased among all age groups. There was a 13% increase in participation among 15- to 19-year-olds (from 24% to 37%), a 5% increase among 20- to 24-year-olds (from 13% to 18%), and a 6% increase among 0- to 14-year-olds (from 52% to 58%).
Dr Fern and her colleagues said the rise in enrollment, particularly among AYAs, was due to increased availability and access to trials; increased awareness from healthcare professionals, patients, and the public about research; and the opening of trials with broader age limits that allow AYAs to enter trials.
In light of this study, Cancer Research UK has started asking researchers to justify age restrictions on new studies, in an effort to recruit more AYA cancer patients onto its trials.
“[I]t’s vital that effective treatments are being developed to tackle cancer across all age brackets,” said Kate Law, Cancer Research UK’s director of clinical trials.
“We now only accept age limits on our clinical trials if they are backed up by hard evidence, which will hopefully mean more young cancer patients get the chance to contribute to research and have the latest experimental treatments.” ![]()
patient and her father
Credit: Rhoda Baer
Age limits on clinical trials must be more flexible to allow more adolescent and young adult (AYA) cancer patients the opportunity to access new treatments, according to a report published in The Lancet Oncology.
The report’s authors discovered that expanding age eligibility criteria for cancer trials increased the enrollment of AYA patients and patients belonging to other age groups. But there is still room for improvement, according to the authors.
“[R]ight now, too many of our young patients are needlessly falling through the gap between pediatric and adult cancer trials,” said Lorna Fern, PhD, of University College London Hospitals in the UK.
“By encouraging doctors to take into account the full age range of patients affected by individual types of cancer, we’ve shown that it’s possible to design trials that include teenage cancer patients and, importantly, that better match the underlying biology of the disease and the people affected.”
To assess AYA enrollment in cancer trials, Dr Fern and her colleagues looked at 68,275 cancer patients aged 0 to 59 years. They were diagnosed with leukemias, lymphomas, or solid tumor malignancies between April 1, 2005, and March 31, 2010.
During this 6-year period, trial participation increased among all age groups. There was a 13% increase in participation among 15- to 19-year-olds (from 24% to 37%), a 5% increase among 20- to 24-year-olds (from 13% to 18%), and a 6% increase among 0- to 14-year-olds (from 52% to 58%).
Dr Fern and her colleagues said the rise in enrollment, particularly among AYAs, was due to increased availability and access to trials; increased awareness from healthcare professionals, patients, and the public about research; and the opening of trials with broader age limits that allow AYAs to enter trials.
In light of this study, Cancer Research UK has started asking researchers to justify age restrictions on new studies, in an effort to recruit more AYA cancer patients onto its trials.
“[I]t’s vital that effective treatments are being developed to tackle cancer across all age brackets,” said Kate Law, Cancer Research UK’s director of clinical trials.
“We now only accept age limits on our clinical trials if they are backed up by hard evidence, which will hopefully mean more young cancer patients get the chance to contribute to research and have the latest experimental treatments.” ![]()
CAR T-cell therapy gets breakthrough designation

The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for the T-cell therapy CTL019 to treat adults and children with relapsed or refractory acute lymphoblastic leukemia (ALL).
The therapy consists of a patient’s own T cells genetically engineered to produce chimeric antigen receptors (CARs) directed against CD19.
CTL019 is the first personalized cellular therapy for cancer to receive breakthrough designation from the FDA.
Breakthrough designation is intended to expedite the development and review of new medicines that treat serious or life-threatening conditions, if the therapy has demonstrated substantial improvement over available therapies.
The breakthrough designation for CTL019 is based on early trial results in adults and children with ALL.
At ASH 2013, researchers presented results of the first 27 ALL patients (22 children and 5 adults) treated with CTL019. Eighty-nine percent of the patients achieved a complete response to the treatment. Six patients relapsed during follow-up, which ranged from 2 months to 18 months.
There was also a high rate of toxicity, particularly cytokine release syndrome, but this was resolved via treatment with the IL-6 agonist tocilizumab.
The first pediatric ALL patient to receive CTL019 celebrated the second anniversary of her cancer remission in May. And the first adult patient remains in remission 1 year after receiving the therapy.
“Our early findings reveal tremendous promise for a desperate group of patients, many of whom have been able to return to their normal lives at school and work after receiving this new, personalized immunotherapy,” said Carl June, MD, of the University of Pennsylvania.
“Receiving the FDA’s breakthrough designation is an essential step in our work with Novartis to expand this therapy to patients across the world who desperately need new options to help them fight this disease.”
In August 2012, the University of Pennsylvania announced an exclusive global research and licensing agreement with Novartis to further study, develop, and commercialize personalized CAR T-cell therapies for the treatment of cancers.
Trials of CTL019 began in the summer of 2010 in patients with relapsed and refractory chronic lymphocytic leukemia, and they are now underway for patients with ALL, non-Hodgkin lymphoma, and myeloma.
CTL019 cells are a patients’ own T cells genetically engineered to express an anti-CD19 scFv coupled to CD3ζ signaling and 4-1BB co-stimulatory domains. The cells are activated and expanded ex vivo with anti-CD3 and anti-CD28 beads, then infused into patients. ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for the T-cell therapy CTL019 to treat adults and children with relapsed or refractory acute lymphoblastic leukemia (ALL).
The therapy consists of a patient’s own T cells genetically engineered to produce chimeric antigen receptors (CARs) directed against CD19.
CTL019 is the first personalized cellular therapy for cancer to receive breakthrough designation from the FDA.
Breakthrough designation is intended to expedite the development and review of new medicines that treat serious or life-threatening conditions, if the therapy has demonstrated substantial improvement over available therapies.
The breakthrough designation for CTL019 is based on early trial results in adults and children with ALL.
At ASH 2013, researchers presented results of the first 27 ALL patients (22 children and 5 adults) treated with CTL019. Eighty-nine percent of the patients achieved a complete response to the treatment. Six patients relapsed during follow-up, which ranged from 2 months to 18 months.
There was also a high rate of toxicity, particularly cytokine release syndrome, but this was resolved via treatment with the IL-6 agonist tocilizumab.
The first pediatric ALL patient to receive CTL019 celebrated the second anniversary of her cancer remission in May. And the first adult patient remains in remission 1 year after receiving the therapy.
“Our early findings reveal tremendous promise for a desperate group of patients, many of whom have been able to return to their normal lives at school and work after receiving this new, personalized immunotherapy,” said Carl June, MD, of the University of Pennsylvania.
“Receiving the FDA’s breakthrough designation is an essential step in our work with Novartis to expand this therapy to patients across the world who desperately need new options to help them fight this disease.”
In August 2012, the University of Pennsylvania announced an exclusive global research and licensing agreement with Novartis to further study, develop, and commercialize personalized CAR T-cell therapies for the treatment of cancers.
Trials of CTL019 began in the summer of 2010 in patients with relapsed and refractory chronic lymphocytic leukemia, and they are now underway for patients with ALL, non-Hodgkin lymphoma, and myeloma.
CTL019 cells are a patients’ own T cells genetically engineered to express an anti-CD19 scFv coupled to CD3ζ signaling and 4-1BB co-stimulatory domains. The cells are activated and expanded ex vivo with anti-CD3 and anti-CD28 beads, then infused into patients. ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for the T-cell therapy CTL019 to treat adults and children with relapsed or refractory acute lymphoblastic leukemia (ALL).
The therapy consists of a patient’s own T cells genetically engineered to produce chimeric antigen receptors (CARs) directed against CD19.
CTL019 is the first personalized cellular therapy for cancer to receive breakthrough designation from the FDA.
Breakthrough designation is intended to expedite the development and review of new medicines that treat serious or life-threatening conditions, if the therapy has demonstrated substantial improvement over available therapies.
The breakthrough designation for CTL019 is based on early trial results in adults and children with ALL.
At ASH 2013, researchers presented results of the first 27 ALL patients (22 children and 5 adults) treated with CTL019. Eighty-nine percent of the patients achieved a complete response to the treatment. Six patients relapsed during follow-up, which ranged from 2 months to 18 months.
There was also a high rate of toxicity, particularly cytokine release syndrome, but this was resolved via treatment with the IL-6 agonist tocilizumab.
The first pediatric ALL patient to receive CTL019 celebrated the second anniversary of her cancer remission in May. And the first adult patient remains in remission 1 year after receiving the therapy.
“Our early findings reveal tremendous promise for a desperate group of patients, many of whom have been able to return to their normal lives at school and work after receiving this new, personalized immunotherapy,” said Carl June, MD, of the University of Pennsylvania.
“Receiving the FDA’s breakthrough designation is an essential step in our work with Novartis to expand this therapy to patients across the world who desperately need new options to help them fight this disease.”
In August 2012, the University of Pennsylvania announced an exclusive global research and licensing agreement with Novartis to further study, develop, and commercialize personalized CAR T-cell therapies for the treatment of cancers.
Trials of CTL019 began in the summer of 2010 in patients with relapsed and refractory chronic lymphocytic leukemia, and they are now underway for patients with ALL, non-Hodgkin lymphoma, and myeloma.
CTL019 cells are a patients’ own T cells genetically engineered to express an anti-CD19 scFv coupled to CD3ζ signaling and 4-1BB co-stimulatory domains. The cells are activated and expanded ex vivo with anti-CD3 and anti-CD28 beads, then infused into patients. ![]()
Biochemical cascade can lead to MPNs

Credit: Indiana University
Preclinical research has revealed a cascade of molecular events in the bone marrow that produce high levels of inflammation, disrupt hematopoiesis, and lead to the development of myeloproliferative neoplasms (MPNs).
The discovery points the way to potential new strategies for treating MPNs and leukemias and further illuminates the relationship between inflammation and cancer, according to Nadia Carlesso, MD, PhD, of the Indiana University School of Medicine in Indianapolis.
Dr Carlesso and her colleagues described the discovery in Cell Stem Cell.
The team used a mouse model to elucidate the role of Notch in hematopoiesis. And they found that loss of Notch function in the microenvironment causes a chain of molecular events that result in excess production of inflammatory factors.
“Some of these inflammatory molecules are cytokines that induce uncontrolled proliferation of myeloid cells and lead to myeloproliferative disorders,” Dr Carlesso said. “[However,] loss of Notch has to occur in specific cells of the bone marrow microenvironment, like endothelial cells, to really be capable to trigger such a high inflammatory status.”
Specifically, Dr Carlesso and her colleagues showed that Notch signaling represses expression of the microRNA miR-155 by promoting binding of RBPJ, a nonredundant downstream effector of the canonical Notch signaling cascade, to the miR-155 promoter.
Loss of Notch/RBPJ signaling upregulates miR-155 in bone marrow endothelial cells. And this leads to miR-155-mediated targeting of the NF-kB inhibitor kB-Ras1, NF-kB activation, increased proinflammatory cytokine production, and the development of an MPN-like disorder.
But when the researchers deleted miR-155 in the stroma of RBPJ_/_ mice, they were able to prevent cytokine induction and the MPN-like disease.
The team also discovered elevated levels of miR-155 in samples from humans with MPNs. This suggests that developing drugs to target the inflammatory reaction at key points could be a promising strategy to limit the development of MPNs in humans.
Dr Carlesso noted that a key finding of this research was that the molecular cascade leading to inflammation was not occurring directly in hematopoietic stem cells but in cells of the bone marrow microenvironment.
“This work indicates that we need to target not only the tumor cells but also the inflammatory microenvironment that surrounds them and may contribute to their generation,” she said. “We believe that this combined strategy will be more effective in preventing myeloproliferative disease progression and transformation in acute leukemias.”
Dr Carlesso also pointed out that, because Notch is an oncogene, it is often targeted by therapies for other types of cancer. But this research suggests targeting Notch can have adverse effects on hematopoiesis, and clinicians should be aware of this risk. ![]()
Credit: Indiana University
Preclinical research has revealed a cascade of molecular events in the bone marrow that produce high levels of inflammation, disrupt hematopoiesis, and lead to the development of myeloproliferative neoplasms (MPNs).
The discovery points the way to potential new strategies for treating MPNs and leukemias and further illuminates the relationship between inflammation and cancer, according to Nadia Carlesso, MD, PhD, of the Indiana University School of Medicine in Indianapolis.
Dr Carlesso and her colleagues described the discovery in Cell Stem Cell.
The team used a mouse model to elucidate the role of Notch in hematopoiesis. And they found that loss of Notch function in the microenvironment causes a chain of molecular events that result in excess production of inflammatory factors.
“Some of these inflammatory molecules are cytokines that induce uncontrolled proliferation of myeloid cells and lead to myeloproliferative disorders,” Dr Carlesso said. “[However,] loss of Notch has to occur in specific cells of the bone marrow microenvironment, like endothelial cells, to really be capable to trigger such a high inflammatory status.”
Specifically, Dr Carlesso and her colleagues showed that Notch signaling represses expression of the microRNA miR-155 by promoting binding of RBPJ, a nonredundant downstream effector of the canonical Notch signaling cascade, to the miR-155 promoter.
Loss of Notch/RBPJ signaling upregulates miR-155 in bone marrow endothelial cells. And this leads to miR-155-mediated targeting of the NF-kB inhibitor kB-Ras1, NF-kB activation, increased proinflammatory cytokine production, and the development of an MPN-like disorder.
But when the researchers deleted miR-155 in the stroma of RBPJ_/_ mice, they were able to prevent cytokine induction and the MPN-like disease.
The team also discovered elevated levels of miR-155 in samples from humans with MPNs. This suggests that developing drugs to target the inflammatory reaction at key points could be a promising strategy to limit the development of MPNs in humans.
Dr Carlesso noted that a key finding of this research was that the molecular cascade leading to inflammation was not occurring directly in hematopoietic stem cells but in cells of the bone marrow microenvironment.
“This work indicates that we need to target not only the tumor cells but also the inflammatory microenvironment that surrounds them and may contribute to their generation,” she said. “We believe that this combined strategy will be more effective in preventing myeloproliferative disease progression and transformation in acute leukemias.”
Dr Carlesso also pointed out that, because Notch is an oncogene, it is often targeted by therapies for other types of cancer. But this research suggests targeting Notch can have adverse effects on hematopoiesis, and clinicians should be aware of this risk. ![]()
Credit: Indiana University
Preclinical research has revealed a cascade of molecular events in the bone marrow that produce high levels of inflammation, disrupt hematopoiesis, and lead to the development of myeloproliferative neoplasms (MPNs).
The discovery points the way to potential new strategies for treating MPNs and leukemias and further illuminates the relationship between inflammation and cancer, according to Nadia Carlesso, MD, PhD, of the Indiana University School of Medicine in Indianapolis.
Dr Carlesso and her colleagues described the discovery in Cell Stem Cell.
The team used a mouse model to elucidate the role of Notch in hematopoiesis. And they found that loss of Notch function in the microenvironment causes a chain of molecular events that result in excess production of inflammatory factors.
“Some of these inflammatory molecules are cytokines that induce uncontrolled proliferation of myeloid cells and lead to myeloproliferative disorders,” Dr Carlesso said. “[However,] loss of Notch has to occur in specific cells of the bone marrow microenvironment, like endothelial cells, to really be capable to trigger such a high inflammatory status.”
Specifically, Dr Carlesso and her colleagues showed that Notch signaling represses expression of the microRNA miR-155 by promoting binding of RBPJ, a nonredundant downstream effector of the canonical Notch signaling cascade, to the miR-155 promoter.
Loss of Notch/RBPJ signaling upregulates miR-155 in bone marrow endothelial cells. And this leads to miR-155-mediated targeting of the NF-kB inhibitor kB-Ras1, NF-kB activation, increased proinflammatory cytokine production, and the development of an MPN-like disorder.
But when the researchers deleted miR-155 in the stroma of RBPJ_/_ mice, they were able to prevent cytokine induction and the MPN-like disease.
The team also discovered elevated levels of miR-155 in samples from humans with MPNs. This suggests that developing drugs to target the inflammatory reaction at key points could be a promising strategy to limit the development of MPNs in humans.
Dr Carlesso noted that a key finding of this research was that the molecular cascade leading to inflammation was not occurring directly in hematopoietic stem cells but in cells of the bone marrow microenvironment.
“This work indicates that we need to target not only the tumor cells but also the inflammatory microenvironment that surrounds them and may contribute to their generation,” she said. “We believe that this combined strategy will be more effective in preventing myeloproliferative disease progression and transformation in acute leukemias.”
Dr Carlesso also pointed out that, because Notch is an oncogene, it is often targeted by therapies for other types of cancer. But this research suggests targeting Notch can have adverse effects on hematopoiesis, and clinicians should be aware of this risk. ![]()
Adipose-derived SCs can resist methotrexate

A new study indicates that adipose-derived stem cells (ASCs) are highly resistant to the chemotherapy drug methotrexate (MTX).
Cultured ASCs and tissue samples that included ASCs were able to withstand exposure to MTX quite well. The drug had little or no effect on ASC viability, division, senescence, or differentiation.
The researchers believe these findings could prove significant for cancer patients, particularly children with acute lymphoblastic leukemia.
“Kids undergo chemotherapy at such an important time, when they should be growing, but, instead, they are introduced to this very harsh environment where bone cells are damaged with these drugs,” said study author Olivia Beane, a graduate student at Brown University in Providence, Rhode Island.
“That leads to major long-term side effects, including osteoporosis and bone defects. If we found a stem cell that was resistant to the chemotherapeutic agent and could promote bone growth by becoming bone itself, then maybe they wouldn’t have these issues.”
Beane’s work, which appears in Experimental Cell Research, grew out of more basic research. She was originally looking for chemicals that could help purify ASCs from mixed cell cultures to encourage their proliferation.
Among other things, she tried chemotherapy drugs, speculating that ASCs might withstand a drug that other cells could not. The idea that this could help cancer patients did not come until later.
To explore potential cancer applications, Beane and her colleagues exposed pure human ASC cultures, stromal vascular fraction (SVF) tissue samples (which include ASCs and several other cell types), and cultures of human fibroblast cells to medically relevant concentrations of chemotherapy drugs for 24 hours.
The researchers then measured how those cell populations fared over the next 10 days. They also measured the ability of MTX-exposed ASCs, both alone and in SVF, to proliferate and differentiate.
In contrast to the fibroblast controls, the ASCs withstood a variety of MTX doses. The drug had little or no effect on ASC viability, cell division, senescence, or differentiation. ASCs also resisted vincristine to an extent, but they could not withstand exposure to cytarabine or etoposide.
The SVF tissue samples withstood MTX doses well too. That is significant, according to the researchers, because the tissue would be clinically useful if an ASC-based therapy were ever developed for cancer patients. Hypothetically, fresh SVF could be harvested from the fat of a donor and injected into bone tissue, delivering ASCs to the site.
To understand why ASCs resist MTX, the researchers conducted further tests. MTX shuts down DNA biosynthesis by binding the protein dihydrofolate reductase so it is unavailable to assist in that essential task.
The testing showed that ASCs ramped up dihydrofolate reductase levels upon exposure to the drug. They produced enough to overcome a clinically relevant dose of MTX.
Now, the researchers are eager to see if they can translate these findings to deliver a medical benefit for cancer patients. To that end, the team is planning several more experiments.
One is to test ASC survival and performance after 48- and 72-hour exposures to MTX. Another is to begin examining how the cells fare in mouse models of chemotherapy. The researchers plan to directly compare ASCs and bone marrow-derived stem cells exposed to various chemotherapies. ![]()
A new study indicates that adipose-derived stem cells (ASCs) are highly resistant to the chemotherapy drug methotrexate (MTX).
Cultured ASCs and tissue samples that included ASCs were able to withstand exposure to MTX quite well. The drug had little or no effect on ASC viability, division, senescence, or differentiation.
The researchers believe these findings could prove significant for cancer patients, particularly children with acute lymphoblastic leukemia.
“Kids undergo chemotherapy at such an important time, when they should be growing, but, instead, they are introduced to this very harsh environment where bone cells are damaged with these drugs,” said study author Olivia Beane, a graduate student at Brown University in Providence, Rhode Island.
“That leads to major long-term side effects, including osteoporosis and bone defects. If we found a stem cell that was resistant to the chemotherapeutic agent and could promote bone growth by becoming bone itself, then maybe they wouldn’t have these issues.”
Beane’s work, which appears in Experimental Cell Research, grew out of more basic research. She was originally looking for chemicals that could help purify ASCs from mixed cell cultures to encourage their proliferation.
Among other things, she tried chemotherapy drugs, speculating that ASCs might withstand a drug that other cells could not. The idea that this could help cancer patients did not come until later.
To explore potential cancer applications, Beane and her colleagues exposed pure human ASC cultures, stromal vascular fraction (SVF) tissue samples (which include ASCs and several other cell types), and cultures of human fibroblast cells to medically relevant concentrations of chemotherapy drugs for 24 hours.
The researchers then measured how those cell populations fared over the next 10 days. They also measured the ability of MTX-exposed ASCs, both alone and in SVF, to proliferate and differentiate.
In contrast to the fibroblast controls, the ASCs withstood a variety of MTX doses. The drug had little or no effect on ASC viability, cell division, senescence, or differentiation. ASCs also resisted vincristine to an extent, but they could not withstand exposure to cytarabine or etoposide.
The SVF tissue samples withstood MTX doses well too. That is significant, according to the researchers, because the tissue would be clinically useful if an ASC-based therapy were ever developed for cancer patients. Hypothetically, fresh SVF could be harvested from the fat of a donor and injected into bone tissue, delivering ASCs to the site.
To understand why ASCs resist MTX, the researchers conducted further tests. MTX shuts down DNA biosynthesis by binding the protein dihydrofolate reductase so it is unavailable to assist in that essential task.
The testing showed that ASCs ramped up dihydrofolate reductase levels upon exposure to the drug. They produced enough to overcome a clinically relevant dose of MTX.
Now, the researchers are eager to see if they can translate these findings to deliver a medical benefit for cancer patients. To that end, the team is planning several more experiments.
One is to test ASC survival and performance after 48- and 72-hour exposures to MTX. Another is to begin examining how the cells fare in mouse models of chemotherapy. The researchers plan to directly compare ASCs and bone marrow-derived stem cells exposed to various chemotherapies. ![]()
A new study indicates that adipose-derived stem cells (ASCs) are highly resistant to the chemotherapy drug methotrexate (MTX).
Cultured ASCs and tissue samples that included ASCs were able to withstand exposure to MTX quite well. The drug had little or no effect on ASC viability, division, senescence, or differentiation.
The researchers believe these findings could prove significant for cancer patients, particularly children with acute lymphoblastic leukemia.
“Kids undergo chemotherapy at such an important time, when they should be growing, but, instead, they are introduced to this very harsh environment where bone cells are damaged with these drugs,” said study author Olivia Beane, a graduate student at Brown University in Providence, Rhode Island.
“That leads to major long-term side effects, including osteoporosis and bone defects. If we found a stem cell that was resistant to the chemotherapeutic agent and could promote bone growth by becoming bone itself, then maybe they wouldn’t have these issues.”
Beane’s work, which appears in Experimental Cell Research, grew out of more basic research. She was originally looking for chemicals that could help purify ASCs from mixed cell cultures to encourage their proliferation.
Among other things, she tried chemotherapy drugs, speculating that ASCs might withstand a drug that other cells could not. The idea that this could help cancer patients did not come until later.
To explore potential cancer applications, Beane and her colleagues exposed pure human ASC cultures, stromal vascular fraction (SVF) tissue samples (which include ASCs and several other cell types), and cultures of human fibroblast cells to medically relevant concentrations of chemotherapy drugs for 24 hours.
The researchers then measured how those cell populations fared over the next 10 days. They also measured the ability of MTX-exposed ASCs, both alone and in SVF, to proliferate and differentiate.
In contrast to the fibroblast controls, the ASCs withstood a variety of MTX doses. The drug had little or no effect on ASC viability, cell division, senescence, or differentiation. ASCs also resisted vincristine to an extent, but they could not withstand exposure to cytarabine or etoposide.
The SVF tissue samples withstood MTX doses well too. That is significant, according to the researchers, because the tissue would be clinically useful if an ASC-based therapy were ever developed for cancer patients. Hypothetically, fresh SVF could be harvested from the fat of a donor and injected into bone tissue, delivering ASCs to the site.
To understand why ASCs resist MTX, the researchers conducted further tests. MTX shuts down DNA biosynthesis by binding the protein dihydrofolate reductase so it is unavailable to assist in that essential task.
The testing showed that ASCs ramped up dihydrofolate reductase levels upon exposure to the drug. They produced enough to overcome a clinically relevant dose of MTX.
Now, the researchers are eager to see if they can translate these findings to deliver a medical benefit for cancer patients. To that end, the team is planning several more experiments.
One is to test ASC survival and performance after 48- and 72-hour exposures to MTX. Another is to begin examining how the cells fare in mouse models of chemotherapy. The researchers plan to directly compare ASCs and bone marrow-derived stem cells exposed to various chemotherapies. ![]()
EC expands indication for ofatumumab

Credit: Linda Bartlett
The European Commission (EC) has expanded the indication for the anti-CD20 monoclonal antibody ofatumumab (Arzerra).
The EC granted conditional approval for ofatumumab in combination with chlorambucil or bendamustine to treat patients with chronic lymphocytic leukemia (CLL) who have not received prior therapy and are not eligible for fludarabine-based therapy.
In 2010, the EC granted ofatumumab conditional approval to treat CLL patients who are refractory to fludarabine and alemtuzumab.
Ofatumumab received conditional approval because the drug’s benefits appear to outweigh the risks it poses. Ofatumumab will not receive full approval until the drug’s developers, GlaxoSmithKline and GenMab, submit results of additional research to the EC.
Trial results
The EC’s new approval of ofatumumab, for previously untreated CLL patients, is based on results from 2 trials, COMPLEMENT 1 and OMB115991.
The phase 3 COMPLEMENT 1 trial was a comparison of ofatumumab plus chlorambucil (n=221) with chlorambucil alone (n=226) in CLL patients for whom fludarabine-based treatment was considered inappropriate.
In this study, ofatumumab plus chlorambucil improved progression-free survival compared to chlorambucil alone. The median times were 22.4 months and 13.1 months, respectively, and the hazard ratio was 0.57 (P<0.001).
In the phase 2 trial known as OMB115991, researchers evaluated ofatumumab in combination with bendamustine in 44 patients with previously untreated CLL for whom fludarabine-based treatment was considered inappropriate.
The combination elicited an overall response rate of 95% and a complete response rate of 43%.
The overall safety profile of ofatumumab in CLL (previously untreated and relapsed/refractory) is based on data from 511 patients in clinical trials.
This includes 250 patients with relapsed/refractory CLL who were treated with ofatumumab alone and 261 patients with previously untreated CLL who were treated in combination with an alkylating agent.
The most common adverse effects associated with ofatumumab were infusion reactions, neutropenia, anemia, febrile neutropenia, thrombocytopenia, leukopenia, lower respiratory tract infection (including pneumonia), upper respiratory tract infection, sepsis (including neutropenic sepsis and septic shock), herpes virus infection, and urinary tract infection. ![]()
Credit: Linda Bartlett
The European Commission (EC) has expanded the indication for the anti-CD20 monoclonal antibody ofatumumab (Arzerra).
The EC granted conditional approval for ofatumumab in combination with chlorambucil or bendamustine to treat patients with chronic lymphocytic leukemia (CLL) who have not received prior therapy and are not eligible for fludarabine-based therapy.
In 2010, the EC granted ofatumumab conditional approval to treat CLL patients who are refractory to fludarabine and alemtuzumab.
Ofatumumab received conditional approval because the drug’s benefits appear to outweigh the risks it poses. Ofatumumab will not receive full approval until the drug’s developers, GlaxoSmithKline and GenMab, submit results of additional research to the EC.
Trial results
The EC’s new approval of ofatumumab, for previously untreated CLL patients, is based on results from 2 trials, COMPLEMENT 1 and OMB115991.
The phase 3 COMPLEMENT 1 trial was a comparison of ofatumumab plus chlorambucil (n=221) with chlorambucil alone (n=226) in CLL patients for whom fludarabine-based treatment was considered inappropriate.
In this study, ofatumumab plus chlorambucil improved progression-free survival compared to chlorambucil alone. The median times were 22.4 months and 13.1 months, respectively, and the hazard ratio was 0.57 (P<0.001).
In the phase 2 trial known as OMB115991, researchers evaluated ofatumumab in combination with bendamustine in 44 patients with previously untreated CLL for whom fludarabine-based treatment was considered inappropriate.
The combination elicited an overall response rate of 95% and a complete response rate of 43%.
The overall safety profile of ofatumumab in CLL (previously untreated and relapsed/refractory) is based on data from 511 patients in clinical trials.
This includes 250 patients with relapsed/refractory CLL who were treated with ofatumumab alone and 261 patients with previously untreated CLL who were treated in combination with an alkylating agent.
The most common adverse effects associated with ofatumumab were infusion reactions, neutropenia, anemia, febrile neutropenia, thrombocytopenia, leukopenia, lower respiratory tract infection (including pneumonia), upper respiratory tract infection, sepsis (including neutropenic sepsis and septic shock), herpes virus infection, and urinary tract infection. ![]()
Credit: Linda Bartlett
The European Commission (EC) has expanded the indication for the anti-CD20 monoclonal antibody ofatumumab (Arzerra).
The EC granted conditional approval for ofatumumab in combination with chlorambucil or bendamustine to treat patients with chronic lymphocytic leukemia (CLL) who have not received prior therapy and are not eligible for fludarabine-based therapy.
In 2010, the EC granted ofatumumab conditional approval to treat CLL patients who are refractory to fludarabine and alemtuzumab.
Ofatumumab received conditional approval because the drug’s benefits appear to outweigh the risks it poses. Ofatumumab will not receive full approval until the drug’s developers, GlaxoSmithKline and GenMab, submit results of additional research to the EC.
Trial results
The EC’s new approval of ofatumumab, for previously untreated CLL patients, is based on results from 2 trials, COMPLEMENT 1 and OMB115991.
The phase 3 COMPLEMENT 1 trial was a comparison of ofatumumab plus chlorambucil (n=221) with chlorambucil alone (n=226) in CLL patients for whom fludarabine-based treatment was considered inappropriate.
In this study, ofatumumab plus chlorambucil improved progression-free survival compared to chlorambucil alone. The median times were 22.4 months and 13.1 months, respectively, and the hazard ratio was 0.57 (P<0.001).
In the phase 2 trial known as OMB115991, researchers evaluated ofatumumab in combination with bendamustine in 44 patients with previously untreated CLL for whom fludarabine-based treatment was considered inappropriate.
The combination elicited an overall response rate of 95% and a complete response rate of 43%.
The overall safety profile of ofatumumab in CLL (previously untreated and relapsed/refractory) is based on data from 511 patients in clinical trials.
This includes 250 patients with relapsed/refractory CLL who were treated with ofatumumab alone and 261 patients with previously untreated CLL who were treated in combination with an alkylating agent.
The most common adverse effects associated with ofatumumab were infusion reactions, neutropenia, anemia, febrile neutropenia, thrombocytopenia, leukopenia, lower respiratory tract infection (including pneumonia), upper respiratory tract infection, sepsis (including neutropenic sepsis and septic shock), herpes virus infection, and urinary tract infection.
FDA grants "breakthrough" status to investigational T-cell therapy for relapsed/refractory ALL
A chimeric antigen receptor therapy in phase I studies for relapsed/refractory acute lymphoblastic leukemia has been granted "breakthrough therapy" status by the Food and Drug Administration, which will expedite the development and review of the product as a result, Novartis has announced.
The investigational T-cell therapy – CTL019 – is now being researched in phase I studies of pediatric and adult patients with relapsed/refractory ALL. CTL019 "uses CAR [chimeric antigen receptor] technology to reprogram a patient’s own T cells to ‘hunt’ cancer cells" that express specific proteins [CD19]. "After they have been reprogrammed, the T cells (now called CTL019) are reintroduced into the patient’s blood; they proliferate and bind to the targeted CD19+ cancer cells and destroy them," according to the statement issued by Novartis on July 7.
Researchers at the University of Pennsylvania, Philadelphia, are conducting phase I/II studies of CTL019 and are working with Novartis to study and develop CAR T-cell treatments as cancer treatments; they have submitted the filing for the breakthrough therapy status, the statement said.
If a drug is designated as breakthrough therapy, the FDA will expedite the development and review of such drug.
The breakthrough therapy designation was created by the 2012 FDA Safety and Innovation Act. The FDA defines a breakthrough therapy as a drug that is intended to treat a serious or life-threatening disease or condition, alone or in combination with other drugs, and where "preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development," according to the FDA website.
"Because of uncertainty of clinical trials, there is no guarantee that CTL019 will ever be commercially available anywhere in the world," the Novartis statement added.
A chimeric antigen receptor therapy in phase I studies for relapsed/refractory acute lymphoblastic leukemia has been granted "breakthrough therapy" status by the Food and Drug Administration, which will expedite the development and review of the product as a result, Novartis has announced.
The investigational T-cell therapy – CTL019 – is now being researched in phase I studies of pediatric and adult patients with relapsed/refractory ALL. CTL019 "uses CAR [chimeric antigen receptor] technology to reprogram a patient’s own T cells to ‘hunt’ cancer cells" that express specific proteins [CD19]. "After they have been reprogrammed, the T cells (now called CTL019) are reintroduced into the patient’s blood; they proliferate and bind to the targeted CD19+ cancer cells and destroy them," according to the statement issued by Novartis on July 7.
Researchers at the University of Pennsylvania, Philadelphia, are conducting phase I/II studies of CTL019 and are working with Novartis to study and develop CAR T-cell treatments as cancer treatments; they have submitted the filing for the breakthrough therapy status, the statement said.
If a drug is designated as breakthrough therapy, the FDA will expedite the development and review of such drug.
The breakthrough therapy designation was created by the 2012 FDA Safety and Innovation Act. The FDA defines a breakthrough therapy as a drug that is intended to treat a serious or life-threatening disease or condition, alone or in combination with other drugs, and where "preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development," according to the FDA website.
"Because of uncertainty of clinical trials, there is no guarantee that CTL019 will ever be commercially available anywhere in the world," the Novartis statement added.
A chimeric antigen receptor therapy in phase I studies for relapsed/refractory acute lymphoblastic leukemia has been granted "breakthrough therapy" status by the Food and Drug Administration, which will expedite the development and review of the product as a result, Novartis has announced.
The investigational T-cell therapy – CTL019 – is now being researched in phase I studies of pediatric and adult patients with relapsed/refractory ALL. CTL019 "uses CAR [chimeric antigen receptor] technology to reprogram a patient’s own T cells to ‘hunt’ cancer cells" that express specific proteins [CD19]. "After they have been reprogrammed, the T cells (now called CTL019) are reintroduced into the patient’s blood; they proliferate and bind to the targeted CD19+ cancer cells and destroy them," according to the statement issued by Novartis on July 7.
Researchers at the University of Pennsylvania, Philadelphia, are conducting phase I/II studies of CTL019 and are working with Novartis to study and develop CAR T-cell treatments as cancer treatments; they have submitted the filing for the breakthrough therapy status, the statement said.
If a drug is designated as breakthrough therapy, the FDA will expedite the development and review of such drug.
The breakthrough therapy designation was created by the 2012 FDA Safety and Innovation Act. The FDA defines a breakthrough therapy as a drug that is intended to treat a serious or life-threatening disease or condition, alone or in combination with other drugs, and where "preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development," according to the FDA website.
"Because of uncertainty of clinical trials, there is no guarantee that CTL019 will ever be commercially available anywhere in the world," the Novartis statement added.
FDA grants "breakthrough" status to investigational T-cell therapy for relapsed/refractory ALL
A chimeric antigen receptor therapy in phase I studies for relapsed/refractory acute lymphoblastic leukemia has been granted "breakthrough therapy" status by the Food and Drug Administration, which will expedite the development and review of the product as a result, Novartis has announced.
The investigational T-cell therapy – CTL019 – is now being researched in phase I studies of pediatric and adult patients with relapsed/refractory ALL. CTL019 "uses CAR [chimeric antigen receptor] technology to reprogram a patient’s own T cells to ‘hunt’ cancer cells" that express specific proteins [CD19]. "After they have been reprogrammed, the T cells (now called CTL019) are reintroduced into the patient’s blood; they proliferate and bind to the targeted CD19+ cancer cells and destroy them," according to the statement issued by Novartis on July 7.
Researchers at the University of Pennsylvania, Philadelphia, are conducting phase I/II studies of CTL019 and are working with Novartis to study and develop CAR T-cell treatments as cancer treatments; they have submitted the filing for the breakthrough therapy status, the statement said.
If a drug is designated as breakthrough therapy, the FDA will expedite the development and review of such drug.
The breakthrough therapy designation was created by the 2012 FDA Safety and Innovation Act. The FDA defines a breakthrough therapy as a drug that is intended to treat a serious or life-threatening disease or condition, alone or in combination with other drugs, and where "preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development," according to the FDA website.
"Because of uncertainty of clinical trials, there is no guarantee that CTL019 will ever be commercially available anywhere in the world," the Novartis statement added.
A chimeric antigen receptor therapy in phase I studies for relapsed/refractory acute lymphoblastic leukemia has been granted "breakthrough therapy" status by the Food and Drug Administration, which will expedite the development and review of the product as a result, Novartis has announced.
The investigational T-cell therapy – CTL019 – is now being researched in phase I studies of pediatric and adult patients with relapsed/refractory ALL. CTL019 "uses CAR [chimeric antigen receptor] technology to reprogram a patient’s own T cells to ‘hunt’ cancer cells" that express specific proteins [CD19]. "After they have been reprogrammed, the T cells (now called CTL019) are reintroduced into the patient’s blood; they proliferate and bind to the targeted CD19+ cancer cells and destroy them," according to the statement issued by Novartis on July 7.
Researchers at the University of Pennsylvania, Philadelphia, are conducting phase I/II studies of CTL019 and are working with Novartis to study and develop CAR T-cell treatments as cancer treatments; they have submitted the filing for the breakthrough therapy status, the statement said.
If a drug is designated as breakthrough therapy, the FDA will expedite the development and review of such drug.
The breakthrough therapy designation was created by the 2012 FDA Safety and Innovation Act. The FDA defines a breakthrough therapy as a drug that is intended to treat a serious or life-threatening disease or condition, alone or in combination with other drugs, and where "preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development," according to the FDA website.
"Because of uncertainty of clinical trials, there is no guarantee that CTL019 will ever be commercially available anywhere in the world," the Novartis statement added.
A chimeric antigen receptor therapy in phase I studies for relapsed/refractory acute lymphoblastic leukemia has been granted "breakthrough therapy" status by the Food and Drug Administration, which will expedite the development and review of the product as a result, Novartis has announced.
The investigational T-cell therapy – CTL019 – is now being researched in phase I studies of pediatric and adult patients with relapsed/refractory ALL. CTL019 "uses CAR [chimeric antigen receptor] technology to reprogram a patient’s own T cells to ‘hunt’ cancer cells" that express specific proteins [CD19]. "After they have been reprogrammed, the T cells (now called CTL019) are reintroduced into the patient’s blood; they proliferate and bind to the targeted CD19+ cancer cells and destroy them," according to the statement issued by Novartis on July 7.
Researchers at the University of Pennsylvania, Philadelphia, are conducting phase I/II studies of CTL019 and are working with Novartis to study and develop CAR T-cell treatments as cancer treatments; they have submitted the filing for the breakthrough therapy status, the statement said.
If a drug is designated as breakthrough therapy, the FDA will expedite the development and review of such drug.
The breakthrough therapy designation was created by the 2012 FDA Safety and Innovation Act. The FDA defines a breakthrough therapy as a drug that is intended to treat a serious or life-threatening disease or condition, alone or in combination with other drugs, and where "preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development," according to the FDA website.
"Because of uncertainty of clinical trials, there is no guarantee that CTL019 will ever be commercially available anywhere in the world," the Novartis statement added.
Study validates drug’s efficacy in CLL/SLL
MILAN—Results of the phase 3 RESONATE trial suggest ibrutinib can improve response and survival rates in patients with relapsed or refractory chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), when compared to ofatumumab.
Ibrutinib conferred these benefits irrespective of baseline clinical characteristics or molecular features, including 17p deletion.
Atrial fibrillation and bleeding-related events were more common with ibrutinib. But the rate of serious adverse events was similar between the treatment arms.
About 86% of patients remained on ibrutinib at last analysis, and roughly 29% of patients initially randomized to ofatumumab crossed over to the ibrutinib arm after disease progression.
“This study undoubtedly confirms that ibrutinib is a very effective agent—as a single-agent—in relapsed CLL patients,” said investigator Peter Hillmen, MD, PhD, of The Leeds Teaching Hospitals in the UK.
Dr Hillmen presented these results at the 19th Congress of the European Hematology Association (EHA) as abstract S693. The RESONATE trial was sponsored by Pharmacyclics and Janssen, the companies developing ibrutinib.
The trial included 391 patients with relapsed or refractory CLL/SLL. They were randomized to receive oral ibrutinib at 420 mg once daily until progression or unacceptable toxicity (n=195) or intravenous ofatumumab at an initial dose of 300 mg, followed by 11 doses of 2000 mg (n=196). Patients in the ofatumumab arm were allowed to cross over to ibrutinib if they progressed (n=57).
The median age in both treatment arms was 67. Overall, roughly 50% of patients had received 3 or more prior therapies, including purine analogs, alkylating agents, and anti-CD20 antibodies. The proportion of patients with del17p was similar between the treatment arms—32% in the ibrutinib arm and 33% in the ofatumumab arm.
Response and survival
At the time of interim analysis, patients’ median time on study was 9.4 months. The best overall response among evaluable patients was 78% in the ibrutinib arm and 11% in the ofatumumab arm, according to an independent review committee.
In addition, ibrutinib significantly prolonged progression-free survival (PFS). The median PFS was 8.1 months in the ofatumumab arm and was not reached in the ibrutinib arm (P<0.0001). The improvement in PFS represents a 78% reduction in the risk of progression or death.
Dr Hillmen noted that PFS favored ibrutinib regardless of baseline characteristics such as refractoriness to purine analogs, del17p, age, gender, Rai stage, bulky disease, number of prior treatments, del11q, B2 microglobulin, and IgVH mutation status.
Ibrutinib significantly prolonged overall survival (OS) as well. The median OS was not reached in either arm, but the hazard ratio was 0.434 (P=0.0049). The improvement in OS represents a 56% reduction in the risk of death in patients treated with ibrutinib.
Adverse events
Dr Hillmen pointed out that the median treatment duration was 8.6 months for ibrutinib and 5.3 months for ofatumumab, and this difference confounds the assessment of side effects.
Nevertheless, nearly all patients in both treatment arms experienced adverse events—99% in the ibrutinib arm and 98% in the ofatumumab arm. Grade 3/4 events occurred in 51% and 39% of patients, respectively.
Atrial fibrillation of any grade was more common in the ibrutinib arm (n=10) than in the ofatumumab arm (n=1), but 5 of the ibrutinib-treated patients had a prior history of atrial fibrillation. Bleeding-related events were also more common with ibrutinib (44% vs 12%), as were diarrhea (48% vs 18%) and arthralgia (17% vs 7%).
Events more common in the ofatumumab arm included infusion-related reactions (28% vs 0%), peripheral sensory neuropathy (13% vs 4%), urticaria (6% vs 1%), night sweats (13% vs 5%), and pruritus (9% vs 4%).
MILAN—Results of the phase 3 RESONATE trial suggest ibrutinib can improve response and survival rates in patients with relapsed or refractory chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), when compared to ofatumumab.
Ibrutinib conferred these benefits irrespective of baseline clinical characteristics or molecular features, including 17p deletion.
Atrial fibrillation and bleeding-related events were more common with ibrutinib. But the rate of serious adverse events was similar between the treatment arms.
About 86% of patients remained on ibrutinib at last analysis, and roughly 29% of patients initially randomized to ofatumumab crossed over to the ibrutinib arm after disease progression.
“This study undoubtedly confirms that ibrutinib is a very effective agent—as a single-agent—in relapsed CLL patients,” said investigator Peter Hillmen, MD, PhD, of The Leeds Teaching Hospitals in the UK.
Dr Hillmen presented these results at the 19th Congress of the European Hematology Association (EHA) as abstract S693. The RESONATE trial was sponsored by Pharmacyclics and Janssen, the companies developing ibrutinib.
The trial included 391 patients with relapsed or refractory CLL/SLL. They were randomized to receive oral ibrutinib at 420 mg once daily until progression or unacceptable toxicity (n=195) or intravenous ofatumumab at an initial dose of 300 mg, followed by 11 doses of 2000 mg (n=196). Patients in the ofatumumab arm were allowed to cross over to ibrutinib if they progressed (n=57).
The median age in both treatment arms was 67. Overall, roughly 50% of patients had received 3 or more prior therapies, including purine analogs, alkylating agents, and anti-CD20 antibodies. The proportion of patients with del17p was similar between the treatment arms—32% in the ibrutinib arm and 33% in the ofatumumab arm.
Response and survival
At the time of interim analysis, patients’ median time on study was 9.4 months. The best overall response among evaluable patients was 78% in the ibrutinib arm and 11% in the ofatumumab arm, according to an independent review committee.
In addition, ibrutinib significantly prolonged progression-free survival (PFS). The median PFS was 8.1 months in the ofatumumab arm and was not reached in the ibrutinib arm (P<0.0001). The improvement in PFS represents a 78% reduction in the risk of progression or death.
Dr Hillmen noted that PFS favored ibrutinib regardless of baseline characteristics such as refractoriness to purine analogs, del17p, age, gender, Rai stage, bulky disease, number of prior treatments, del11q, B2 microglobulin, and IgVH mutation status.
Ibrutinib significantly prolonged overall survival (OS) as well. The median OS was not reached in either arm, but the hazard ratio was 0.434 (P=0.0049). The improvement in OS represents a 56% reduction in the risk of death in patients treated with ibrutinib.
Adverse events
Dr Hillmen pointed out that the median treatment duration was 8.6 months for ibrutinib and 5.3 months for ofatumumab, and this difference confounds the assessment of side effects.
Nevertheless, nearly all patients in both treatment arms experienced adverse events—99% in the ibrutinib arm and 98% in the ofatumumab arm. Grade 3/4 events occurred in 51% and 39% of patients, respectively.
Atrial fibrillation of any grade was more common in the ibrutinib arm (n=10) than in the ofatumumab arm (n=1), but 5 of the ibrutinib-treated patients had a prior history of atrial fibrillation. Bleeding-related events were also more common with ibrutinib (44% vs 12%), as were diarrhea (48% vs 18%) and arthralgia (17% vs 7%).
Events more common in the ofatumumab arm included infusion-related reactions (28% vs 0%), peripheral sensory neuropathy (13% vs 4%), urticaria (6% vs 1%), night sweats (13% vs 5%), and pruritus (9% vs 4%).
MILAN—Results of the phase 3 RESONATE trial suggest ibrutinib can improve response and survival rates in patients with relapsed or refractory chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), when compared to ofatumumab.
Ibrutinib conferred these benefits irrespective of baseline clinical characteristics or molecular features, including 17p deletion.
Atrial fibrillation and bleeding-related events were more common with ibrutinib. But the rate of serious adverse events was similar between the treatment arms.
About 86% of patients remained on ibrutinib at last analysis, and roughly 29% of patients initially randomized to ofatumumab crossed over to the ibrutinib arm after disease progression.
“This study undoubtedly confirms that ibrutinib is a very effective agent—as a single-agent—in relapsed CLL patients,” said investigator Peter Hillmen, MD, PhD, of The Leeds Teaching Hospitals in the UK.
Dr Hillmen presented these results at the 19th Congress of the European Hematology Association (EHA) as abstract S693. The RESONATE trial was sponsored by Pharmacyclics and Janssen, the companies developing ibrutinib.
The trial included 391 patients with relapsed or refractory CLL/SLL. They were randomized to receive oral ibrutinib at 420 mg once daily until progression or unacceptable toxicity (n=195) or intravenous ofatumumab at an initial dose of 300 mg, followed by 11 doses of 2000 mg (n=196). Patients in the ofatumumab arm were allowed to cross over to ibrutinib if they progressed (n=57).
The median age in both treatment arms was 67. Overall, roughly 50% of patients had received 3 or more prior therapies, including purine analogs, alkylating agents, and anti-CD20 antibodies. The proportion of patients with del17p was similar between the treatment arms—32% in the ibrutinib arm and 33% in the ofatumumab arm.
Response and survival
At the time of interim analysis, patients’ median time on study was 9.4 months. The best overall response among evaluable patients was 78% in the ibrutinib arm and 11% in the ofatumumab arm, according to an independent review committee.
In addition, ibrutinib significantly prolonged progression-free survival (PFS). The median PFS was 8.1 months in the ofatumumab arm and was not reached in the ibrutinib arm (P<0.0001). The improvement in PFS represents a 78% reduction in the risk of progression or death.
Dr Hillmen noted that PFS favored ibrutinib regardless of baseline characteristics such as refractoriness to purine analogs, del17p, age, gender, Rai stage, bulky disease, number of prior treatments, del11q, B2 microglobulin, and IgVH mutation status.
Ibrutinib significantly prolonged overall survival (OS) as well. The median OS was not reached in either arm, but the hazard ratio was 0.434 (P=0.0049). The improvement in OS represents a 56% reduction in the risk of death in patients treated with ibrutinib.
Adverse events
Dr Hillmen pointed out that the median treatment duration was 8.6 months for ibrutinib and 5.3 months for ofatumumab, and this difference confounds the assessment of side effects.
Nevertheless, nearly all patients in both treatment arms experienced adverse events—99% in the ibrutinib arm and 98% in the ofatumumab arm. Grade 3/4 events occurred in 51% and 39% of patients, respectively.
Atrial fibrillation of any grade was more common in the ibrutinib arm (n=10) than in the ofatumumab arm (n=1), but 5 of the ibrutinib-treated patients had a prior history of atrial fibrillation. Bleeding-related events were also more common with ibrutinib (44% vs 12%), as were diarrhea (48% vs 18%) and arthralgia (17% vs 7%).
Events more common in the ofatumumab arm included infusion-related reactions (28% vs 0%), peripheral sensory neuropathy (13% vs 4%), urticaria (6% vs 1%), night sweats (13% vs 5%), and pruritus (9% vs 4%).