User login
EMA wants to suspend drugs due to data manipulation

Credit: Steven Harbour
The European Medicines Agency (EMA) has recommended suspending a number of drugs that were approved in the European Union (EU) based on clinical studies conducted at GVK Biosciences in Hyderabad, India.
An inspection of the site suggested GVK Bio employees may have manipulated data from trials that took place there.
So the EMA compiled a list of drugs—which includes clopidogrel, dexamethasone, and tacrolimus, among others—that should be suspended.
However, the EMA said there is no evidence of harm or lack of effectiveness linked to the conduct of studies by GVK Bio, and patients should continue taking their medicines as prescribed.
The EMA’s opinion has been forwarded to the European Commission (EC), which will adopt a legally binding decision.
It was at the request of the EC that the EMA’s Committee for Medicinal Products for Human Use (CHMP) reviewed the drugs set to be suspended.
An inspection by the French medicines agency, Agence nationale de sécurité du médicament et des produits de santé (ANSM), in May 2014 uncovered “non-compliance with good clinical practice” at the GVK Bio site in Hyderabad.
The ANSM inspector analyzed 9 studies conducted there from 2008 to 2013 and found evidence suggesting that data from electrocardiograms (ECGs) had been manipulated. It appeared that GVK Bio employees were taking multiple ECGs of one volunteer and presenting them as ECGs of other volunteers.
The EMA said the systematic nature of these data manipulations, the extended period of time during which they took place, and the number of staff members involved casts doubt on the integrity of the way trials were performed at the Hyderabad facility and on the reliability of data generated there.
With this in mind, the CHMP looked at more than 1000 pharmaceutical forms and strengths of medicines studied at the site. For more than 300 of the medications, there was sufficient data from other sources to support the drugs’ authorization, so these will remain on the market in the EU.
For drugs that lack data from other studies, the CHMP recommended suspension unless they are of critical importance for patients because alternatives will not be able to meet patients’ needs.
Whether a medicine is critical for patients will be determined by the national authorities of EU member states, depending on the situation in their country. For drugs that are considered critical, companies developing those drugs will be given 12 months to submit additional data.
The CHMP’s recommendation will be sent to the EC for a legally binding decision that will apply to all EU member states whether or not they have taken interim measures to suspend medicines.
GVK Bio responds
GVK Bio said an internal audit conducted following the ANSM inspection suggested that standard operating procedures were followed for the 9 trials analyzed. The organization also sought the opinion of 4 independent cardiologists, who said it was difficult to determine if the ECGs belong to the same volunteer or more than one person.
GVK Bio presented this information to the ANSM, but the agency concluded that the studies did not meet good clinical practice guidelines and should be rejected.
Following subsequent meetings and analyses, the EMA came to a similar conclusion—that the overall findings cast doubt on the results of trials conducted at the Hyderabad facility from 2008 to 2014.
GVK Bio said it respects the EMA’s decision, but their recommended suspension of drugs is “unprecedented and highly disproportional” for a few reasons.
First, the ANSM said the ECGs in question were not essential to demonstrate the bioequivalence of the drugs tested, and the agency’s recommendation to reject the 9 studies was a “precautionary” measure.
The ANSM also said the observations made during the inspection should not be extrapolated to other trial-related activities at the Hyderabad site or GVK Bio’s other site in Ahmedabad.
And finally, the EMA itself said there is no evidence proving that the drugs in question are ineffective or pose a risk to human health. ![]()
Credit: Steven Harbour
The European Medicines Agency (EMA) has recommended suspending a number of drugs that were approved in the European Union (EU) based on clinical studies conducted at GVK Biosciences in Hyderabad, India.
An inspection of the site suggested GVK Bio employees may have manipulated data from trials that took place there.
So the EMA compiled a list of drugs—which includes clopidogrel, dexamethasone, and tacrolimus, among others—that should be suspended.
However, the EMA said there is no evidence of harm or lack of effectiveness linked to the conduct of studies by GVK Bio, and patients should continue taking their medicines as prescribed.
The EMA’s opinion has been forwarded to the European Commission (EC), which will adopt a legally binding decision.
It was at the request of the EC that the EMA’s Committee for Medicinal Products for Human Use (CHMP) reviewed the drugs set to be suspended.
An inspection by the French medicines agency, Agence nationale de sécurité du médicament et des produits de santé (ANSM), in May 2014 uncovered “non-compliance with good clinical practice” at the GVK Bio site in Hyderabad.
The ANSM inspector analyzed 9 studies conducted there from 2008 to 2013 and found evidence suggesting that data from electrocardiograms (ECGs) had been manipulated. It appeared that GVK Bio employees were taking multiple ECGs of one volunteer and presenting them as ECGs of other volunteers.
The EMA said the systematic nature of these data manipulations, the extended period of time during which they took place, and the number of staff members involved casts doubt on the integrity of the way trials were performed at the Hyderabad facility and on the reliability of data generated there.
With this in mind, the CHMP looked at more than 1000 pharmaceutical forms and strengths of medicines studied at the site. For more than 300 of the medications, there was sufficient data from other sources to support the drugs’ authorization, so these will remain on the market in the EU.
For drugs that lack data from other studies, the CHMP recommended suspension unless they are of critical importance for patients because alternatives will not be able to meet patients’ needs.
Whether a medicine is critical for patients will be determined by the national authorities of EU member states, depending on the situation in their country. For drugs that are considered critical, companies developing those drugs will be given 12 months to submit additional data.
The CHMP’s recommendation will be sent to the EC for a legally binding decision that will apply to all EU member states whether or not they have taken interim measures to suspend medicines.
GVK Bio responds
GVK Bio said an internal audit conducted following the ANSM inspection suggested that standard operating procedures were followed for the 9 trials analyzed. The organization also sought the opinion of 4 independent cardiologists, who said it was difficult to determine if the ECGs belong to the same volunteer or more than one person.
GVK Bio presented this information to the ANSM, but the agency concluded that the studies did not meet good clinical practice guidelines and should be rejected.
Following subsequent meetings and analyses, the EMA came to a similar conclusion—that the overall findings cast doubt on the results of trials conducted at the Hyderabad facility from 2008 to 2014.
GVK Bio said it respects the EMA’s decision, but their recommended suspension of drugs is “unprecedented and highly disproportional” for a few reasons.
First, the ANSM said the ECGs in question were not essential to demonstrate the bioequivalence of the drugs tested, and the agency’s recommendation to reject the 9 studies was a “precautionary” measure.
The ANSM also said the observations made during the inspection should not be extrapolated to other trial-related activities at the Hyderabad site or GVK Bio’s other site in Ahmedabad.
And finally, the EMA itself said there is no evidence proving that the drugs in question are ineffective or pose a risk to human health. ![]()
Credit: Steven Harbour
The European Medicines Agency (EMA) has recommended suspending a number of drugs that were approved in the European Union (EU) based on clinical studies conducted at GVK Biosciences in Hyderabad, India.
An inspection of the site suggested GVK Bio employees may have manipulated data from trials that took place there.
So the EMA compiled a list of drugs—which includes clopidogrel, dexamethasone, and tacrolimus, among others—that should be suspended.
However, the EMA said there is no evidence of harm or lack of effectiveness linked to the conduct of studies by GVK Bio, and patients should continue taking their medicines as prescribed.
The EMA’s opinion has been forwarded to the European Commission (EC), which will adopt a legally binding decision.
It was at the request of the EC that the EMA’s Committee for Medicinal Products for Human Use (CHMP) reviewed the drugs set to be suspended.
An inspection by the French medicines agency, Agence nationale de sécurité du médicament et des produits de santé (ANSM), in May 2014 uncovered “non-compliance with good clinical practice” at the GVK Bio site in Hyderabad.
The ANSM inspector analyzed 9 studies conducted there from 2008 to 2013 and found evidence suggesting that data from electrocardiograms (ECGs) had been manipulated. It appeared that GVK Bio employees were taking multiple ECGs of one volunteer and presenting them as ECGs of other volunteers.
The EMA said the systematic nature of these data manipulations, the extended period of time during which they took place, and the number of staff members involved casts doubt on the integrity of the way trials were performed at the Hyderabad facility and on the reliability of data generated there.
With this in mind, the CHMP looked at more than 1000 pharmaceutical forms and strengths of medicines studied at the site. For more than 300 of the medications, there was sufficient data from other sources to support the drugs’ authorization, so these will remain on the market in the EU.
For drugs that lack data from other studies, the CHMP recommended suspension unless they are of critical importance for patients because alternatives will not be able to meet patients’ needs.
Whether a medicine is critical for patients will be determined by the national authorities of EU member states, depending on the situation in their country. For drugs that are considered critical, companies developing those drugs will be given 12 months to submit additional data.
The CHMP’s recommendation will be sent to the EC for a legally binding decision that will apply to all EU member states whether or not they have taken interim measures to suspend medicines.
GVK Bio responds
GVK Bio said an internal audit conducted following the ANSM inspection suggested that standard operating procedures were followed for the 9 trials analyzed. The organization also sought the opinion of 4 independent cardiologists, who said it was difficult to determine if the ECGs belong to the same volunteer or more than one person.
GVK Bio presented this information to the ANSM, but the agency concluded that the studies did not meet good clinical practice guidelines and should be rejected.
Following subsequent meetings and analyses, the EMA came to a similar conclusion—that the overall findings cast doubt on the results of trials conducted at the Hyderabad facility from 2008 to 2014.
GVK Bio said it respects the EMA’s decision, but their recommended suspension of drugs is “unprecedented and highly disproportional” for a few reasons.
First, the ANSM said the ECGs in question were not essential to demonstrate the bioequivalence of the drugs tested, and the agency’s recommendation to reject the 9 studies was a “precautionary” measure.
The ANSM also said the observations made during the inspection should not be extrapolated to other trial-related activities at the Hyderabad site or GVK Bio’s other site in Ahmedabad.
And finally, the EMA itself said there is no evidence proving that the drugs in question are ineffective or pose a risk to human health. ![]()
Gains in CLL are ‘Advance of the Year’

Credit: NIH
The “transformation” of treatment for chronic lymphocytic leukemia (CLL) is the “Advance of the Year” for 2015, according to a report by the American Society of Clinical Oncology (ASCO).
The report said 4 therapies that were recently approved in the US fill a major unmet need for CLL patients—obinutuzumab and ofatumumab for patients with previously untreated CLL and idelalisib and ibrutinib for patients with relapsed or refractory CLL.
“For many older patients, especially, these drugs essentially offer the first chance at effective treatment, since the side effects of earlier options were simply too toxic for many to handle,” said Gregory Masters, MD, ASCO expert and co-executive editor of the report.
The report, “Clinical Cancer Advances 2015: ASCO’s Annual Report on Progress Against Cancer,” is available in the Journal of Clinical Oncology and on ASCO’s cancer research advocacy website, CancerProgress.net.
The report was developed under the direction of an 18-person editorial board of experts from a wide range of oncology specialties. It features:
- The top cancer research advances of the past year: Identifying major trends in cancer prevention and screening, treatment, quality of life, survivorship, and tumor biology
- A Decade in Review: Recounting improvements in cancer care since the first issue of Clinical Cancer Advances
- The 10-Year Horizon: Previewing trends likely to shape the next decade of cancer care, including genomic technology, nanomedicine, and health information technologies
- Progress in Rare Cancers: Highlighting promising early achievements in treating certain uncommon but devastating cancers.
“This has truly been a banner year for CLL and for clinical cancer research as a whole,” said ASCO President Peter P. Yu, MD. “Advances in cancer prevention and care, especially those in precision medicine, are offering stunning new possibilities for patients.” ![]()
Credit: NIH
The “transformation” of treatment for chronic lymphocytic leukemia (CLL) is the “Advance of the Year” for 2015, according to a report by the American Society of Clinical Oncology (ASCO).
The report said 4 therapies that were recently approved in the US fill a major unmet need for CLL patients—obinutuzumab and ofatumumab for patients with previously untreated CLL and idelalisib and ibrutinib for patients with relapsed or refractory CLL.
“For many older patients, especially, these drugs essentially offer the first chance at effective treatment, since the side effects of earlier options were simply too toxic for many to handle,” said Gregory Masters, MD, ASCO expert and co-executive editor of the report.
The report, “Clinical Cancer Advances 2015: ASCO’s Annual Report on Progress Against Cancer,” is available in the Journal of Clinical Oncology and on ASCO’s cancer research advocacy website, CancerProgress.net.
The report was developed under the direction of an 18-person editorial board of experts from a wide range of oncology specialties. It features:
- The top cancer research advances of the past year: Identifying major trends in cancer prevention and screening, treatment, quality of life, survivorship, and tumor biology
- A Decade in Review: Recounting improvements in cancer care since the first issue of Clinical Cancer Advances
- The 10-Year Horizon: Previewing trends likely to shape the next decade of cancer care, including genomic technology, nanomedicine, and health information technologies
- Progress in Rare Cancers: Highlighting promising early achievements in treating certain uncommon but devastating cancers.
“This has truly been a banner year for CLL and for clinical cancer research as a whole,” said ASCO President Peter P. Yu, MD. “Advances in cancer prevention and care, especially those in precision medicine, are offering stunning new possibilities for patients.” ![]()
Credit: NIH
The “transformation” of treatment for chronic lymphocytic leukemia (CLL) is the “Advance of the Year” for 2015, according to a report by the American Society of Clinical Oncology (ASCO).
The report said 4 therapies that were recently approved in the US fill a major unmet need for CLL patients—obinutuzumab and ofatumumab for patients with previously untreated CLL and idelalisib and ibrutinib for patients with relapsed or refractory CLL.
“For many older patients, especially, these drugs essentially offer the first chance at effective treatment, since the side effects of earlier options were simply too toxic for many to handle,” said Gregory Masters, MD, ASCO expert and co-executive editor of the report.
The report, “Clinical Cancer Advances 2015: ASCO’s Annual Report on Progress Against Cancer,” is available in the Journal of Clinical Oncology and on ASCO’s cancer research advocacy website, CancerProgress.net.
The report was developed under the direction of an 18-person editorial board of experts from a wide range of oncology specialties. It features:
- The top cancer research advances of the past year: Identifying major trends in cancer prevention and screening, treatment, quality of life, survivorship, and tumor biology
- A Decade in Review: Recounting improvements in cancer care since the first issue of Clinical Cancer Advances
- The 10-Year Horizon: Previewing trends likely to shape the next decade of cancer care, including genomic technology, nanomedicine, and health information technologies
- Progress in Rare Cancers: Highlighting promising early achievements in treating certain uncommon but devastating cancers.
“This has truly been a banner year for CLL and for clinical cancer research as a whole,” said ASCO President Peter P. Yu, MD. “Advances in cancer prevention and care, especially those in precision medicine, are offering stunning new possibilities for patients.” ![]()
FDA puts drug on fast track to treat secondary AML

The US Food and Drug Administration (FDA) has granted fast track designation for CPX-351, a fixed-ratio combination of cytarabine and daunorubicin inside a lipid vesicle, to treat elderly patients with secondary acute myeloid leukemia (AML).
In a phase 2 study of elderly AML patients, there was no significant difference in response or survival rates between patients who received CPX-351 and those who received cytarabine and daunorubicin.
However, CPX-351 conferred a significant response benefit among patients with poor cytogenetics and a significant survival benefit in patients with secondary AML.
“We are pleased that FDA has granted fast track status for CPX-351 for the treatment of elderly patients with secondary AML,” said Scott Jackson, Chief Executive Officer of Celator Pharmaceuticals, the company developing CPX-351.
“Our ongoing phase 3 study in these patients has completed enrollment, and we expect induction response rate data to be available in the second quarter of this year, and to have overall survival data, the primary endpoint of the study, in the first quarter of 2016.”
“If our phase 3 study comparing CPX-351 to the current standard of care is successful, the fast track designation may provide an added benefit of facilitating the [new drug application] review process.”
The FDA established the fast track designation process to expedite the review of drugs that are intended to treat serious or life-threatening conditions and that demonstrate the potential to address unmet medical needs.
The designation allows a drug’s developer to submit sections of a new drug application (NDA) on a rolling basis, so the FDA can review portions of the NDA as they are received instead of waiting for the entire NDA submission. A fast-track-designated product could be eligible for priority review if supported by clinical data at the time of NDA submission.
Phase 2 trial
In an article published in Blood last April, researchers reported results with CPX-351 in elderly patients with newly diagnosed AML. The study enrolled 126 patients who were 60 to 75 years of age.
They were randomized to receive CPX-351 (n=85) or “control” treatment consisting of cytarabine and daunorubicin (n=41). The treatment groups were well-balanced for disease and patient characteristics at baseline.
Overall, the response rate was 66.7% in the CPX-351 arm and 51.2% in the control arm (P=0.07). Among patients with adverse cytogenetics, the response rates were 77.3% and 38.5%, respectively (P=0.03). And among patients with secondary AML, response rates were 57.6% and 31.6%, respectively (P=0.06).
The median overall survival was 14.7 months in the CPX-351 arm and 12.9 months in the control arm. The median event-free survival was 6.5 months and 2.0 months, respectively. These differences were not statistically significant.
However, secondary AML patients treated with CPX-351 had significantly better overall survival than secondary AML patients in the control arm. The median overall survival was 12.1 months and 6.1 months, respectively (P=0.01). The median event-free survival was 4.5 months and 1.3 months, respectively (P=0.08).
Common adverse events included febrile neutropenia, infection, rash, diarrhea, nausea, edema, and constipation. There were minimal differences between the treatment arms in the incidence of these events.
The median time to neutrophil recovery was longer in the CPX-351 arm than in the control arm—36 days and 32 days, respectively. And the same was true for platelet recovery—37 days and 28 days, respectively.
Patients in the CPX-351 arm had a higher incidence of grade 3-4 infection than controls—70.6% and 43.9%, respectively—but not infection-related deaths—3.5% and 7.3%, respectively.
By day 60, 4.7% of patients in the CPX-351 arm and 14.6% of patients in the control arm had died. All of these deaths occurred in high-risk patients, particularly those with secondary AML. ![]()
The US Food and Drug Administration (FDA) has granted fast track designation for CPX-351, a fixed-ratio combination of cytarabine and daunorubicin inside a lipid vesicle, to treat elderly patients with secondary acute myeloid leukemia (AML).
In a phase 2 study of elderly AML patients, there was no significant difference in response or survival rates between patients who received CPX-351 and those who received cytarabine and daunorubicin.
However, CPX-351 conferred a significant response benefit among patients with poor cytogenetics and a significant survival benefit in patients with secondary AML.
“We are pleased that FDA has granted fast track status for CPX-351 for the treatment of elderly patients with secondary AML,” said Scott Jackson, Chief Executive Officer of Celator Pharmaceuticals, the company developing CPX-351.
“Our ongoing phase 3 study in these patients has completed enrollment, and we expect induction response rate data to be available in the second quarter of this year, and to have overall survival data, the primary endpoint of the study, in the first quarter of 2016.”
“If our phase 3 study comparing CPX-351 to the current standard of care is successful, the fast track designation may provide an added benefit of facilitating the [new drug application] review process.”
The FDA established the fast track designation process to expedite the review of drugs that are intended to treat serious or life-threatening conditions and that demonstrate the potential to address unmet medical needs.
The designation allows a drug’s developer to submit sections of a new drug application (NDA) on a rolling basis, so the FDA can review portions of the NDA as they are received instead of waiting for the entire NDA submission. A fast-track-designated product could be eligible for priority review if supported by clinical data at the time of NDA submission.
Phase 2 trial
In an article published in Blood last April, researchers reported results with CPX-351 in elderly patients with newly diagnosed AML. The study enrolled 126 patients who were 60 to 75 years of age.
They were randomized to receive CPX-351 (n=85) or “control” treatment consisting of cytarabine and daunorubicin (n=41). The treatment groups were well-balanced for disease and patient characteristics at baseline.
Overall, the response rate was 66.7% in the CPX-351 arm and 51.2% in the control arm (P=0.07). Among patients with adverse cytogenetics, the response rates were 77.3% and 38.5%, respectively (P=0.03). And among patients with secondary AML, response rates were 57.6% and 31.6%, respectively (P=0.06).
The median overall survival was 14.7 months in the CPX-351 arm and 12.9 months in the control arm. The median event-free survival was 6.5 months and 2.0 months, respectively. These differences were not statistically significant.
However, secondary AML patients treated with CPX-351 had significantly better overall survival than secondary AML patients in the control arm. The median overall survival was 12.1 months and 6.1 months, respectively (P=0.01). The median event-free survival was 4.5 months and 1.3 months, respectively (P=0.08).
Common adverse events included febrile neutropenia, infection, rash, diarrhea, nausea, edema, and constipation. There were minimal differences between the treatment arms in the incidence of these events.
The median time to neutrophil recovery was longer in the CPX-351 arm than in the control arm—36 days and 32 days, respectively. And the same was true for platelet recovery—37 days and 28 days, respectively.
Patients in the CPX-351 arm had a higher incidence of grade 3-4 infection than controls—70.6% and 43.9%, respectively—but not infection-related deaths—3.5% and 7.3%, respectively.
By day 60, 4.7% of patients in the CPX-351 arm and 14.6% of patients in the control arm had died. All of these deaths occurred in high-risk patients, particularly those with secondary AML. ![]()
The US Food and Drug Administration (FDA) has granted fast track designation for CPX-351, a fixed-ratio combination of cytarabine and daunorubicin inside a lipid vesicle, to treat elderly patients with secondary acute myeloid leukemia (AML).
In a phase 2 study of elderly AML patients, there was no significant difference in response or survival rates between patients who received CPX-351 and those who received cytarabine and daunorubicin.
However, CPX-351 conferred a significant response benefit among patients with poor cytogenetics and a significant survival benefit in patients with secondary AML.
“We are pleased that FDA has granted fast track status for CPX-351 for the treatment of elderly patients with secondary AML,” said Scott Jackson, Chief Executive Officer of Celator Pharmaceuticals, the company developing CPX-351.
“Our ongoing phase 3 study in these patients has completed enrollment, and we expect induction response rate data to be available in the second quarter of this year, and to have overall survival data, the primary endpoint of the study, in the first quarter of 2016.”
“If our phase 3 study comparing CPX-351 to the current standard of care is successful, the fast track designation may provide an added benefit of facilitating the [new drug application] review process.”
The FDA established the fast track designation process to expedite the review of drugs that are intended to treat serious or life-threatening conditions and that demonstrate the potential to address unmet medical needs.
The designation allows a drug’s developer to submit sections of a new drug application (NDA) on a rolling basis, so the FDA can review portions of the NDA as they are received instead of waiting for the entire NDA submission. A fast-track-designated product could be eligible for priority review if supported by clinical data at the time of NDA submission.
Phase 2 trial
In an article published in Blood last April, researchers reported results with CPX-351 in elderly patients with newly diagnosed AML. The study enrolled 126 patients who were 60 to 75 years of age.
They were randomized to receive CPX-351 (n=85) or “control” treatment consisting of cytarabine and daunorubicin (n=41). The treatment groups were well-balanced for disease and patient characteristics at baseline.
Overall, the response rate was 66.7% in the CPX-351 arm and 51.2% in the control arm (P=0.07). Among patients with adverse cytogenetics, the response rates were 77.3% and 38.5%, respectively (P=0.03). And among patients with secondary AML, response rates were 57.6% and 31.6%, respectively (P=0.06).
The median overall survival was 14.7 months in the CPX-351 arm and 12.9 months in the control arm. The median event-free survival was 6.5 months and 2.0 months, respectively. These differences were not statistically significant.
However, secondary AML patients treated with CPX-351 had significantly better overall survival than secondary AML patients in the control arm. The median overall survival was 12.1 months and 6.1 months, respectively (P=0.01). The median event-free survival was 4.5 months and 1.3 months, respectively (P=0.08).
Common adverse events included febrile neutropenia, infection, rash, diarrhea, nausea, edema, and constipation. There were minimal differences between the treatment arms in the incidence of these events.
The median time to neutrophil recovery was longer in the CPX-351 arm than in the control arm—36 days and 32 days, respectively. And the same was true for platelet recovery—37 days and 28 days, respectively.
Patients in the CPX-351 arm had a higher incidence of grade 3-4 infection than controls—70.6% and 43.9%, respectively—but not infection-related deaths—3.5% and 7.3%, respectively.
By day 60, 4.7% of patients in the CPX-351 arm and 14.6% of patients in the control arm had died. All of these deaths occurred in high-risk patients, particularly those with secondary AML. ![]()
EC supports continued use of ponatinib

Credit: Rhoda Baer
The European Commission (EC) has concluded that ponatinib (Iclusig) should continue to be prescribed in accordance with its already approved indications.
After trial results suggested the drug poses an increased risk of thrombotic events, the European Medicines Agency’s (EMA) Pharmacovigilance Risk Assessment Committee (PRAC) conducted a review of available ponatinib data.
Results of that review suggested the benefits of ponatinib outweigh the risks. So the committee said the drug should be prescribed as indicated.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) recently endorsed this recommendation, and, now, the EC has followed suit. The EC’s decision is legally binding.
Ponatinib is approved in the European Union to treat adults with chronic phase, accelerated phase, or blast phase chronic myeloid leukemia who are resistant to dasatinib or nilotinib, who are intolerant to dasatinib or nilotinib and for whom subsequent treatment with imatinib is not clinically appropriate, or who have the T315I mutation.
The drug is also approved to treat adults with Philadelphia-chromosome positive acute lymphoblastic leukemia who are resistant to dasatinib, who cannot tolerate dasatinib and subsequent treatment with imatinib is not clinically appropriate, or who have the T315I mutation.
In October 2013, extended follow-up data from the PACE trial revealed that ponatinib-treated patients had a higher incidence of arterial and venous thrombotic events than was observed when the drug first gained approval. So one ponatinib trial was discontinued, and the rest were placed on partial clinical hold.
Soon after, ponatinib was pulled from the US market. The drug ultimately returned to the marketplace with new recommendations designed to decrease the risk of thrombotic events.
The EMA also revised its recommendations for ponatinib—discouraging use of the drug in certain patients, providing advice for managing comorbidities, and suggesting patient monitoring—but kept the drug on the market.
In October 2014, the PRAC concluded its 11-month review of ponatinib data, confirming that the benefit-risk profile of the drug was favorable in its approved indications and recommending that the indications remain unchanged.
However, the PRAC also said the risk of vascular occlusive events with ponatinib is likely dose-related. So the committee recommended that healthcare professionals monitor ponatinib-treated patients and consider dose reductions or discontinuing the drug in certain patients.
The CHMP endorsed these recommendations, and, now, the EC has as well. This is a legally binding decision for ponatinib to continue to be prescribed in Europe in accordance with its already approved indications.
Ponatinib is being developed by ARIAD Pharmaceuticals, Inc. ![]()
Credit: Rhoda Baer
The European Commission (EC) has concluded that ponatinib (Iclusig) should continue to be prescribed in accordance with its already approved indications.
After trial results suggested the drug poses an increased risk of thrombotic events, the European Medicines Agency’s (EMA) Pharmacovigilance Risk Assessment Committee (PRAC) conducted a review of available ponatinib data.
Results of that review suggested the benefits of ponatinib outweigh the risks. So the committee said the drug should be prescribed as indicated.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) recently endorsed this recommendation, and, now, the EC has followed suit. The EC’s decision is legally binding.
Ponatinib is approved in the European Union to treat adults with chronic phase, accelerated phase, or blast phase chronic myeloid leukemia who are resistant to dasatinib or nilotinib, who are intolerant to dasatinib or nilotinib and for whom subsequent treatment with imatinib is not clinically appropriate, or who have the T315I mutation.
The drug is also approved to treat adults with Philadelphia-chromosome positive acute lymphoblastic leukemia who are resistant to dasatinib, who cannot tolerate dasatinib and subsequent treatment with imatinib is not clinically appropriate, or who have the T315I mutation.
In October 2013, extended follow-up data from the PACE trial revealed that ponatinib-treated patients had a higher incidence of arterial and venous thrombotic events than was observed when the drug first gained approval. So one ponatinib trial was discontinued, and the rest were placed on partial clinical hold.
Soon after, ponatinib was pulled from the US market. The drug ultimately returned to the marketplace with new recommendations designed to decrease the risk of thrombotic events.
The EMA also revised its recommendations for ponatinib—discouraging use of the drug in certain patients, providing advice for managing comorbidities, and suggesting patient monitoring—but kept the drug on the market.
In October 2014, the PRAC concluded its 11-month review of ponatinib data, confirming that the benefit-risk profile of the drug was favorable in its approved indications and recommending that the indications remain unchanged.
However, the PRAC also said the risk of vascular occlusive events with ponatinib is likely dose-related. So the committee recommended that healthcare professionals monitor ponatinib-treated patients and consider dose reductions or discontinuing the drug in certain patients.
The CHMP endorsed these recommendations, and, now, the EC has as well. This is a legally binding decision for ponatinib to continue to be prescribed in Europe in accordance with its already approved indications.
Ponatinib is being developed by ARIAD Pharmaceuticals, Inc. ![]()
Credit: Rhoda Baer
The European Commission (EC) has concluded that ponatinib (Iclusig) should continue to be prescribed in accordance with its already approved indications.
After trial results suggested the drug poses an increased risk of thrombotic events, the European Medicines Agency’s (EMA) Pharmacovigilance Risk Assessment Committee (PRAC) conducted a review of available ponatinib data.
Results of that review suggested the benefits of ponatinib outweigh the risks. So the committee said the drug should be prescribed as indicated.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) recently endorsed this recommendation, and, now, the EC has followed suit. The EC’s decision is legally binding.
Ponatinib is approved in the European Union to treat adults with chronic phase, accelerated phase, or blast phase chronic myeloid leukemia who are resistant to dasatinib or nilotinib, who are intolerant to dasatinib or nilotinib and for whom subsequent treatment with imatinib is not clinically appropriate, or who have the T315I mutation.
The drug is also approved to treat adults with Philadelphia-chromosome positive acute lymphoblastic leukemia who are resistant to dasatinib, who cannot tolerate dasatinib and subsequent treatment with imatinib is not clinically appropriate, or who have the T315I mutation.
In October 2013, extended follow-up data from the PACE trial revealed that ponatinib-treated patients had a higher incidence of arterial and venous thrombotic events than was observed when the drug first gained approval. So one ponatinib trial was discontinued, and the rest were placed on partial clinical hold.
Soon after, ponatinib was pulled from the US market. The drug ultimately returned to the marketplace with new recommendations designed to decrease the risk of thrombotic events.
The EMA also revised its recommendations for ponatinib—discouraging use of the drug in certain patients, providing advice for managing comorbidities, and suggesting patient monitoring—but kept the drug on the market.
In October 2014, the PRAC concluded its 11-month review of ponatinib data, confirming that the benefit-risk profile of the drug was favorable in its approved indications and recommending that the indications remain unchanged.
However, the PRAC also said the risk of vascular occlusive events with ponatinib is likely dose-related. So the committee recommended that healthcare professionals monitor ponatinib-treated patients and consider dose reductions or discontinuing the drug in certain patients.
The CHMP endorsed these recommendations, and, now, the EC has as well. This is a legally binding decision for ponatinib to continue to be prescribed in Europe in accordance with its already approved indications.
Ponatinib is being developed by ARIAD Pharmaceuticals, Inc. ![]()
Label changes report new side effects for hematology drugs

Credit: CDC
Several hematology drugs approved by the US Food and Drug Administration (FDA) have recently undergone label changes to reflect newly reported adverse events.
Changes have been made to the labels for the JAK1/2 inhibitor ruxolitinib (Jakafi), the anti-CD20 monoclonal antibody obinutuzumab (Gazyva), the factor Xa inhibitor rivaroxaban (Xarelto), and the hematopoietic stem cell mobilizer plerixafor (Mozobil).
Plerixafor
Plerixafor is FDA-approved for use in combination with granulocyte-colony stimulating factor to mobilize hematopoietic stem cells to the peripheral blood for collection and subsequent autologous transplantation in patients with non-Hodgkin lymphoma and multiple myeloma.
The product’s label was changed to include a new entry under the “Adverse Reactions” heading. Postmarketing experience suggested the drug may cause abnormal dreams and nightmares.
Rivaroxaban
Rivaroxaban is a factor Xa inhibitor that’s FDA-approved to reduce the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, for the treatment of deep vein thrombosis (DVT) and pulmonary embolism (PE), to reduce the risk of recurrent DVT and PE, and to prevent DVT, which may lead to PE, in patients undergoing knee or hip replacement surgery.
Postmarketing experience has led to two changes to the “Adverse Reactions” section of rivaroxaban’s label. Thrombocytopenia has been added as an adverse reaction, and the term “cytolytic hepatitis” has been replaced with “hepatitis (including hepatocellular injury).”
Obinutuzumab
Obinutuzumab is a CD20-directed cytolytic antibody that is FDA-approved in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia.
The “Warnings and Precautions” section of obinutuzumab’s label has been changed to reflect that fatal infections have been reported in patients who received the drug.
The label has also been changed to coincide with changes in trial data. The label now states that obinutuzumab caused grade 3 or 4 neutropenia in 33% of patients and grade 3 or 4 thrombocytopenia in 10% of patients.
Ruxolitinib
Ruxolitinib is a JAK1/JAK2 inhibitor that’s FDA-approved to treat patients with polycythemia vera (PV) who cannot tolerate or don’t respond to hydroxyurea, as well as patients with intermediate or high-risk myelofibrosis.
Ruxolitinib’s label now includes a warning that symptoms of myeloproliferative neoplasms may return about a week after discontinuing treatment. The label also advises healthcare professionals to discourage patients form interrupting or discontinuing ruxolitinib without consulting their physician.
In addition, a warning about the risk of non-melanoma skin cancer associated with ruxolitinib, as well as advice for informing patients of this risk, have been added to ruxolitinib’s label.
The label has undergone significant changes in sections 6.1, “Clinical Trials Experience in Myelofibrosis” and 6.2 “Clinical Trial Experience in Polycythemia Vera.” It now includes additional information on the risk of thrombocytopenia, anemia, and neutropenia.
Under the “Special Populations” heading, recommendations were added to reduce the drug’s dose in patients with PV and moderate or severe renal impairment, as well as PV patients with hepatic impairment. ![]()
Credit: CDC
Several hematology drugs approved by the US Food and Drug Administration (FDA) have recently undergone label changes to reflect newly reported adverse events.
Changes have been made to the labels for the JAK1/2 inhibitor ruxolitinib (Jakafi), the anti-CD20 monoclonal antibody obinutuzumab (Gazyva), the factor Xa inhibitor rivaroxaban (Xarelto), and the hematopoietic stem cell mobilizer plerixafor (Mozobil).
Plerixafor
Plerixafor is FDA-approved for use in combination with granulocyte-colony stimulating factor to mobilize hematopoietic stem cells to the peripheral blood for collection and subsequent autologous transplantation in patients with non-Hodgkin lymphoma and multiple myeloma.
The product’s label was changed to include a new entry under the “Adverse Reactions” heading. Postmarketing experience suggested the drug may cause abnormal dreams and nightmares.
Rivaroxaban
Rivaroxaban is a factor Xa inhibitor that’s FDA-approved to reduce the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, for the treatment of deep vein thrombosis (DVT) and pulmonary embolism (PE), to reduce the risk of recurrent DVT and PE, and to prevent DVT, which may lead to PE, in patients undergoing knee or hip replacement surgery.
Postmarketing experience has led to two changes to the “Adverse Reactions” section of rivaroxaban’s label. Thrombocytopenia has been added as an adverse reaction, and the term “cytolytic hepatitis” has been replaced with “hepatitis (including hepatocellular injury).”
Obinutuzumab
Obinutuzumab is a CD20-directed cytolytic antibody that is FDA-approved in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia.
The “Warnings and Precautions” section of obinutuzumab’s label has been changed to reflect that fatal infections have been reported in patients who received the drug.
The label has also been changed to coincide with changes in trial data. The label now states that obinutuzumab caused grade 3 or 4 neutropenia in 33% of patients and grade 3 or 4 thrombocytopenia in 10% of patients.
Ruxolitinib
Ruxolitinib is a JAK1/JAK2 inhibitor that’s FDA-approved to treat patients with polycythemia vera (PV) who cannot tolerate or don’t respond to hydroxyurea, as well as patients with intermediate or high-risk myelofibrosis.
Ruxolitinib’s label now includes a warning that symptoms of myeloproliferative neoplasms may return about a week after discontinuing treatment. The label also advises healthcare professionals to discourage patients form interrupting or discontinuing ruxolitinib without consulting their physician.
In addition, a warning about the risk of non-melanoma skin cancer associated with ruxolitinib, as well as advice for informing patients of this risk, have been added to ruxolitinib’s label.
The label has undergone significant changes in sections 6.1, “Clinical Trials Experience in Myelofibrosis” and 6.2 “Clinical Trial Experience in Polycythemia Vera.” It now includes additional information on the risk of thrombocytopenia, anemia, and neutropenia.
Under the “Special Populations” heading, recommendations were added to reduce the drug’s dose in patients with PV and moderate or severe renal impairment, as well as PV patients with hepatic impairment. ![]()
Credit: CDC
Several hematology drugs approved by the US Food and Drug Administration (FDA) have recently undergone label changes to reflect newly reported adverse events.
Changes have been made to the labels for the JAK1/2 inhibitor ruxolitinib (Jakafi), the anti-CD20 monoclonal antibody obinutuzumab (Gazyva), the factor Xa inhibitor rivaroxaban (Xarelto), and the hematopoietic stem cell mobilizer plerixafor (Mozobil).
Plerixafor
Plerixafor is FDA-approved for use in combination with granulocyte-colony stimulating factor to mobilize hematopoietic stem cells to the peripheral blood for collection and subsequent autologous transplantation in patients with non-Hodgkin lymphoma and multiple myeloma.
The product’s label was changed to include a new entry under the “Adverse Reactions” heading. Postmarketing experience suggested the drug may cause abnormal dreams and nightmares.
Rivaroxaban
Rivaroxaban is a factor Xa inhibitor that’s FDA-approved to reduce the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, for the treatment of deep vein thrombosis (DVT) and pulmonary embolism (PE), to reduce the risk of recurrent DVT and PE, and to prevent DVT, which may lead to PE, in patients undergoing knee or hip replacement surgery.
Postmarketing experience has led to two changes to the “Adverse Reactions” section of rivaroxaban’s label. Thrombocytopenia has been added as an adverse reaction, and the term “cytolytic hepatitis” has been replaced with “hepatitis (including hepatocellular injury).”
Obinutuzumab
Obinutuzumab is a CD20-directed cytolytic antibody that is FDA-approved in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia.
The “Warnings and Precautions” section of obinutuzumab’s label has been changed to reflect that fatal infections have been reported in patients who received the drug.
The label has also been changed to coincide with changes in trial data. The label now states that obinutuzumab caused grade 3 or 4 neutropenia in 33% of patients and grade 3 or 4 thrombocytopenia in 10% of patients.
Ruxolitinib
Ruxolitinib is a JAK1/JAK2 inhibitor that’s FDA-approved to treat patients with polycythemia vera (PV) who cannot tolerate or don’t respond to hydroxyurea, as well as patients with intermediate or high-risk myelofibrosis.
Ruxolitinib’s label now includes a warning that symptoms of myeloproliferative neoplasms may return about a week after discontinuing treatment. The label also advises healthcare professionals to discourage patients form interrupting or discontinuing ruxolitinib without consulting their physician.
In addition, a warning about the risk of non-melanoma skin cancer associated with ruxolitinib, as well as advice for informing patients of this risk, have been added to ruxolitinib’s label.
The label has undergone significant changes in sections 6.1, “Clinical Trials Experience in Myelofibrosis” and 6.2 “Clinical Trial Experience in Polycythemia Vera.” It now includes additional information on the risk of thrombocytopenia, anemia, and neutropenia.
Under the “Special Populations” heading, recommendations were added to reduce the drug’s dose in patients with PV and moderate or severe renal impairment, as well as PV patients with hepatic impairment. ![]()
Update on therapies for lymphoproliferative disorders
Click on the PDF icon at the top of this introduction to read the full article.
Click on the PDF icon at the top of this introduction to read the full article.
Click on the PDF icon at the top of this introduction to read the full article.
NHS cuts 5 blood cancer drugs from CDF, adds 1
Credit: Steven Harbour
The National Health Service (NHS) has increased the budget for England’s Cancer Drugs Fund (CDF) and added a new drug to treat 2 hematologic malignancies, but 5 other blood cancer drugs will be removed from the fund in March.
The budget for the CDF will grow from £200 million in 2013/14 to £280 million in 2014/15.
However, 16 drugs (for 25 different indications) will no longer be offered through the fund as of March 12, 2015.
Still, the NHS said it has taken steps to ensure patients can receive appropriate treatment.
Review leads to cuts
A national panel of oncologists, pharmacists, and patient representatives independently reviewed the drug indications currently available through the CDF, plus new applications.
They evaluated the clinical benefit, survival, quality of life, toxicity, and safety associated with each treatment, as well as the level of unmet need and the median cost per patient. In cases where the high cost of a drug would lead to its exclusion from CDF, manufacturers were given an opportunity to reduce prices.
The result of the review is that 59 of the 84 most effective currently approved indications of drugs will rollover into the CDF next year, creating room for new drug indications that will be funded for the first time.
These are panitumumab for bowel cancer, ibrutinib for mantle cell lymphoma, and ibrutinib for chronic lymphocytic leukemia.
However, 16 drugs, including 5 blood cancer drugs—bendamustine, bortezomib, bosutinib, dasatinib, and ofatumumab—will no longer be offered through the CDF.
Following these changes, the NHS will put 4 measures in place to ensure patients can receive appropriate treatment. First, any patient currently receiving a drug through the CDF will continue to receive it, regardless of whether it remains in the CDF.
Second, drugs that are the only therapy for the cancer in question will remain available through the CDF. Third, if the CDF panel removes a drug for a particular indication, some patients may instead be able to receive it in another line of therapy or receive an alternative CDF-approved drug.
And finally, clinicians can apply for their patient to receive a drug not available through the CDF on an exceptional basis.
Cuts to blood cancer drugs
The full list of cuts to the CDF is available on the NHS website, but the following list includes all drugs for hematologic malignancies that will no longer be available. These drugs will still be available for other indications, however.
- Bendamustine for the treatment of low-grade lymphoma that is refractory to rituximab alone or in combination.
- Bortezomib for the treatment of:
- relapsed/refractory mantle cell lymphoma after 1 or more prior chemotherapies or stem cell transplant
- relapsed multiple myeloma patients with a previous partial response or complete response of 6 months or more with bortezomib
- relapsed Waldenstrom’s macroglobulinemia patients who received previous treatment with alkylating agents and purine analogues.
- Bosutinib for the treatment of:
- blast crisis chronic myeloid leukemia (CML) that is refractory to nilotinib or dasatinib if dasatinib was accessed via a clinical trial or via its current approved CDF indication
- blast crisis CML where there is treatment intolerance, specifically, significant intolerance to dasatinib (grade 3 or 4 adverse events) if dasatinib was accessed via its current approved CDF indication.
- Dasatinib for the treatment of lymphoid, blast crisis CML that is refractory to, significantly intolerant of, or resistant to prior therapy including imatinib (grade 3 or 4 adverse events); also when used as the 2nd- or 3rd-line treatment.
- Ofatumumab for the treatment of CML as the 2nd- or 3rd-line indication and if the patient is refractory to treatment with fludarabine in combination and/or alemtuzumab or if treatment with fludarabine in combination and/or alemtuzumab is contraindicated.
More about the CDF and the NHS
The CDF—set up in 2010 and currently due to run until March 2016—is money the government has set aside to pay for cancer drugs that haven’t been approved by the National Institute for Health and Care Excellence (NICE) and aren’t available within the NHS in England. Most cancer drugs are routinely funded outside of the CDF.
NHS England said it is working with cancer charities, the pharmaceutical industry, and NICE to create a sustainable model for the commissioning of chemotherapy. The agency has also updated its procedures for evaluating drugs in the CDF, in an effort to ensure sustainability.
In addition, NHS England has set up an appeals process by which pharmaceutical companies can challenge the decision-making process.
And a newly assembled national taskforce, headed by Harpal Kumar, chief executive of Cancer Research UK, is set to produce a refreshed, 5-year cancer plan for the NHS. ![]()
Credit: Steven Harbour
The National Health Service (NHS) has increased the budget for England’s Cancer Drugs Fund (CDF) and added a new drug to treat 2 hematologic malignancies, but 5 other blood cancer drugs will be removed from the fund in March.
The budget for the CDF will grow from £200 million in 2013/14 to £280 million in 2014/15.
However, 16 drugs (for 25 different indications) will no longer be offered through the fund as of March 12, 2015.
Still, the NHS said it has taken steps to ensure patients can receive appropriate treatment.
Review leads to cuts
A national panel of oncologists, pharmacists, and patient representatives independently reviewed the drug indications currently available through the CDF, plus new applications.
They evaluated the clinical benefit, survival, quality of life, toxicity, and safety associated with each treatment, as well as the level of unmet need and the median cost per patient. In cases where the high cost of a drug would lead to its exclusion from CDF, manufacturers were given an opportunity to reduce prices.
The result of the review is that 59 of the 84 most effective currently approved indications of drugs will rollover into the CDF next year, creating room for new drug indications that will be funded for the first time.
These are panitumumab for bowel cancer, ibrutinib for mantle cell lymphoma, and ibrutinib for chronic lymphocytic leukemia.
However, 16 drugs, including 5 blood cancer drugs—bendamustine, bortezomib, bosutinib, dasatinib, and ofatumumab—will no longer be offered through the CDF.
Following these changes, the NHS will put 4 measures in place to ensure patients can receive appropriate treatment. First, any patient currently receiving a drug through the CDF will continue to receive it, regardless of whether it remains in the CDF.
Second, drugs that are the only therapy for the cancer in question will remain available through the CDF. Third, if the CDF panel removes a drug for a particular indication, some patients may instead be able to receive it in another line of therapy or receive an alternative CDF-approved drug.
And finally, clinicians can apply for their patient to receive a drug not available through the CDF on an exceptional basis.
Cuts to blood cancer drugs
The full list of cuts to the CDF is available on the NHS website, but the following list includes all drugs for hematologic malignancies that will no longer be available. These drugs will still be available for other indications, however.
- Bendamustine for the treatment of low-grade lymphoma that is refractory to rituximab alone or in combination.
- Bortezomib for the treatment of:
- relapsed/refractory mantle cell lymphoma after 1 or more prior chemotherapies or stem cell transplant
- relapsed multiple myeloma patients with a previous partial response or complete response of 6 months or more with bortezomib
- relapsed Waldenstrom’s macroglobulinemia patients who received previous treatment with alkylating agents and purine analogues.
- Bosutinib for the treatment of:
- blast crisis chronic myeloid leukemia (CML) that is refractory to nilotinib or dasatinib if dasatinib was accessed via a clinical trial or via its current approved CDF indication
- blast crisis CML where there is treatment intolerance, specifically, significant intolerance to dasatinib (grade 3 or 4 adverse events) if dasatinib was accessed via its current approved CDF indication.
- Dasatinib for the treatment of lymphoid, blast crisis CML that is refractory to, significantly intolerant of, or resistant to prior therapy including imatinib (grade 3 or 4 adverse events); also when used as the 2nd- or 3rd-line treatment.
- Ofatumumab for the treatment of CML as the 2nd- or 3rd-line indication and if the patient is refractory to treatment with fludarabine in combination and/or alemtuzumab or if treatment with fludarabine in combination and/or alemtuzumab is contraindicated.
More about the CDF and the NHS
The CDF—set up in 2010 and currently due to run until March 2016—is money the government has set aside to pay for cancer drugs that haven’t been approved by the National Institute for Health and Care Excellence (NICE) and aren’t available within the NHS in England. Most cancer drugs are routinely funded outside of the CDF.
NHS England said it is working with cancer charities, the pharmaceutical industry, and NICE to create a sustainable model for the commissioning of chemotherapy. The agency has also updated its procedures for evaluating drugs in the CDF, in an effort to ensure sustainability.
In addition, NHS England has set up an appeals process by which pharmaceutical companies can challenge the decision-making process.
And a newly assembled national taskforce, headed by Harpal Kumar, chief executive of Cancer Research UK, is set to produce a refreshed, 5-year cancer plan for the NHS. ![]()
Credit: Steven Harbour
The National Health Service (NHS) has increased the budget for England’s Cancer Drugs Fund (CDF) and added a new drug to treat 2 hematologic malignancies, but 5 other blood cancer drugs will be removed from the fund in March.
The budget for the CDF will grow from £200 million in 2013/14 to £280 million in 2014/15.
However, 16 drugs (for 25 different indications) will no longer be offered through the fund as of March 12, 2015.
Still, the NHS said it has taken steps to ensure patients can receive appropriate treatment.
Review leads to cuts
A national panel of oncologists, pharmacists, and patient representatives independently reviewed the drug indications currently available through the CDF, plus new applications.
They evaluated the clinical benefit, survival, quality of life, toxicity, and safety associated with each treatment, as well as the level of unmet need and the median cost per patient. In cases where the high cost of a drug would lead to its exclusion from CDF, manufacturers were given an opportunity to reduce prices.
The result of the review is that 59 of the 84 most effective currently approved indications of drugs will rollover into the CDF next year, creating room for new drug indications that will be funded for the first time.
These are panitumumab for bowel cancer, ibrutinib for mantle cell lymphoma, and ibrutinib for chronic lymphocytic leukemia.
However, 16 drugs, including 5 blood cancer drugs—bendamustine, bortezomib, bosutinib, dasatinib, and ofatumumab—will no longer be offered through the CDF.
Following these changes, the NHS will put 4 measures in place to ensure patients can receive appropriate treatment. First, any patient currently receiving a drug through the CDF will continue to receive it, regardless of whether it remains in the CDF.
Second, drugs that are the only therapy for the cancer in question will remain available through the CDF. Third, if the CDF panel removes a drug for a particular indication, some patients may instead be able to receive it in another line of therapy or receive an alternative CDF-approved drug.
And finally, clinicians can apply for their patient to receive a drug not available through the CDF on an exceptional basis.
Cuts to blood cancer drugs
The full list of cuts to the CDF is available on the NHS website, but the following list includes all drugs for hematologic malignancies that will no longer be available. These drugs will still be available for other indications, however.
- Bendamustine for the treatment of low-grade lymphoma that is refractory to rituximab alone or in combination.
- Bortezomib for the treatment of:
- relapsed/refractory mantle cell lymphoma after 1 or more prior chemotherapies or stem cell transplant
- relapsed multiple myeloma patients with a previous partial response or complete response of 6 months or more with bortezomib
- relapsed Waldenstrom’s macroglobulinemia patients who received previous treatment with alkylating agents and purine analogues.
- Bosutinib for the treatment of:
- blast crisis chronic myeloid leukemia (CML) that is refractory to nilotinib or dasatinib if dasatinib was accessed via a clinical trial or via its current approved CDF indication
- blast crisis CML where there is treatment intolerance, specifically, significant intolerance to dasatinib (grade 3 or 4 adverse events) if dasatinib was accessed via its current approved CDF indication.
- Dasatinib for the treatment of lymphoid, blast crisis CML that is refractory to, significantly intolerant of, or resistant to prior therapy including imatinib (grade 3 or 4 adverse events); also when used as the 2nd- or 3rd-line treatment.
- Ofatumumab for the treatment of CML as the 2nd- or 3rd-line indication and if the patient is refractory to treatment with fludarabine in combination and/or alemtuzumab or if treatment with fludarabine in combination and/or alemtuzumab is contraindicated.
More about the CDF and the NHS
The CDF—set up in 2010 and currently due to run until March 2016—is money the government has set aside to pay for cancer drugs that haven’t been approved by the National Institute for Health and Care Excellence (NICE) and aren’t available within the NHS in England. Most cancer drugs are routinely funded outside of the CDF.
NHS England said it is working with cancer charities, the pharmaceutical industry, and NICE to create a sustainable model for the commissioning of chemotherapy. The agency has also updated its procedures for evaluating drugs in the CDF, in an effort to ensure sustainability.
In addition, NHS England has set up an appeals process by which pharmaceutical companies can challenge the decision-making process.
And a newly assembled national taskforce, headed by Harpal Kumar, chief executive of Cancer Research UK, is set to produce a refreshed, 5-year cancer plan for the NHS. ![]()
Drug can increase survival in poor-risk AML
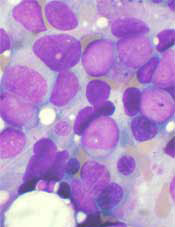
In a phase 2 trial, an investigational drug conferred a significant improvement in survival when used as salvage therapy in poor-risk patients with acute myeloid leukemia (AML).
On the other hand, the drug did not provide any significant improvements over control treatment (investigator’s choice) in the entire population of AML patients in first relapse.
The drug, CPX-351, is a fixed-ratio combination of cytarabine and daunorubicin inside a lipid vesicle.
Researchers reported these results with CPX-351 in Cancer. The study was funded by Celator Pharmaceuticals, the company developing CPX-351, and the Leukemia and Lymphoma Society.
“Patients with first-relapse AML have generally a poor prognosis, with a limited likelihood of response following salvage treatment,” said study author Jorge Cortes, MD, of the MD Anderson Cancer Center in Houston, Texas.
“This is particularly true for patients classified by the EPI [European Prognostic Index] as poor-risk upon entering the trial.”
For this trial, Dr Cortes and his colleagues evaluated 125 patients, ages 18 to 65, from 35 centers in the US, Canada, and Europe. Patients had AML in first relapse after an initial complete remission lasting a month or longer.
They were stratified per the EPI into favorable-, intermediate-, and poor-risk groups based on the duration of their first complete remission, cytogenetics, age, and transplant history. Most patients (68%) were in the poor-risk group.
Patients were randomized 2:1 to receive CPX-351 (100 units/m2 on days 1, 3, and 5 by 90-minute infusion) or the investigators’ choice of first salvage chemotherapy. The control treatment was usually based on cytarabine and an anthracycline, often with one or more additional agents.
Patient characteristics were largely well-balanced between the treatment arms. However, the CPX-351 group had a younger median age (52 years vs 56 years), more patients with secondary AML (12.3% vs 6.8%), and a higher rate of prior transplant (27.2% vs 15.9%).
Response and survival
Response rates were higher in the CPX-351 arm than in the control arm. The rates of complete response were 37% and 31.8%, respectively. And the rates of complete response with incomplete count recovery were 12.3% and 9.1%, respectively.
However, there was no significant difference in event-free survival (EFS) or overall survival (OS) between the treatment arms.
The median EFS was 4 months in the CPX-351 arm and 1.4 months in the control arm (hazard ratio[HR]=0.66, P=0.08). And the median OS was 8.5 months and 6.3 months, respectively (HR=0.75, P=0.19).
In the poor-risk population, there was no significant improvement in EFS with CPX-351, but there was a significant improvement in OS.
The median EFS was 1.8 months in the CPX-351 arm and 1.2 months in the control arm (HR=0.63, P=0.08). And the median OS was 6.5 months and 4.2 months, respectively (HR=0.55, P=0.02).
Safety and early mortality
The early mortality rate was similar between the treatment arms at 30 days—7.4% in the CPX-351 arm and 4.5% in the control arm—and at 60 days—14.8% and 15.9%, respectively. However, at 90 days, deaths were more frequent in the control arm—21.4% and 37.9%.
Patients in the CPX-351 arm had slower neutrophil recovery than those in the control arm—42 days and 34 days, respectively. The same was true for platelet recovery—45 days and 35 days, respectively.
Delayed hematologic recovery was associated with more infection-related events, such as febrile neutropenia, bacteremia, pneumonia, sepsis, urinary tract infection, pyrexia, and cellulitis.
The researchers believe these results, along with the previously published results from a phase 2 trial of CPX-351 in patients newly diagnosed with AML, support the phase 3 study of CPX-351 as a first-line therapy in older patients with high-risk (secondary) AML.
“We were very pleased to see promising response rates in this difficult-to-treat population,” Dr Cortes said. “And we eagerly await results from Celator’s pivotal phase 3 study of CPX-351, which could fulfill a considerable unmet need in AML.” ![]()
In a phase 2 trial, an investigational drug conferred a significant improvement in survival when used as salvage therapy in poor-risk patients with acute myeloid leukemia (AML).
On the other hand, the drug did not provide any significant improvements over control treatment (investigator’s choice) in the entire population of AML patients in first relapse.
The drug, CPX-351, is a fixed-ratio combination of cytarabine and daunorubicin inside a lipid vesicle.
Researchers reported these results with CPX-351 in Cancer. The study was funded by Celator Pharmaceuticals, the company developing CPX-351, and the Leukemia and Lymphoma Society.
“Patients with first-relapse AML have generally a poor prognosis, with a limited likelihood of response following salvage treatment,” said study author Jorge Cortes, MD, of the MD Anderson Cancer Center in Houston, Texas.
“This is particularly true for patients classified by the EPI [European Prognostic Index] as poor-risk upon entering the trial.”
For this trial, Dr Cortes and his colleagues evaluated 125 patients, ages 18 to 65, from 35 centers in the US, Canada, and Europe. Patients had AML in first relapse after an initial complete remission lasting a month or longer.
They were stratified per the EPI into favorable-, intermediate-, and poor-risk groups based on the duration of their first complete remission, cytogenetics, age, and transplant history. Most patients (68%) were in the poor-risk group.
Patients were randomized 2:1 to receive CPX-351 (100 units/m2 on days 1, 3, and 5 by 90-minute infusion) or the investigators’ choice of first salvage chemotherapy. The control treatment was usually based on cytarabine and an anthracycline, often with one or more additional agents.
Patient characteristics were largely well-balanced between the treatment arms. However, the CPX-351 group had a younger median age (52 years vs 56 years), more patients with secondary AML (12.3% vs 6.8%), and a higher rate of prior transplant (27.2% vs 15.9%).
Response and survival
Response rates were higher in the CPX-351 arm than in the control arm. The rates of complete response were 37% and 31.8%, respectively. And the rates of complete response with incomplete count recovery were 12.3% and 9.1%, respectively.
However, there was no significant difference in event-free survival (EFS) or overall survival (OS) between the treatment arms.
The median EFS was 4 months in the CPX-351 arm and 1.4 months in the control arm (hazard ratio[HR]=0.66, P=0.08). And the median OS was 8.5 months and 6.3 months, respectively (HR=0.75, P=0.19).
In the poor-risk population, there was no significant improvement in EFS with CPX-351, but there was a significant improvement in OS.
The median EFS was 1.8 months in the CPX-351 arm and 1.2 months in the control arm (HR=0.63, P=0.08). And the median OS was 6.5 months and 4.2 months, respectively (HR=0.55, P=0.02).
Safety and early mortality
The early mortality rate was similar between the treatment arms at 30 days—7.4% in the CPX-351 arm and 4.5% in the control arm—and at 60 days—14.8% and 15.9%, respectively. However, at 90 days, deaths were more frequent in the control arm—21.4% and 37.9%.
Patients in the CPX-351 arm had slower neutrophil recovery than those in the control arm—42 days and 34 days, respectively. The same was true for platelet recovery—45 days and 35 days, respectively.
Delayed hematologic recovery was associated with more infection-related events, such as febrile neutropenia, bacteremia, pneumonia, sepsis, urinary tract infection, pyrexia, and cellulitis.
The researchers believe these results, along with the previously published results from a phase 2 trial of CPX-351 in patients newly diagnosed with AML, support the phase 3 study of CPX-351 as a first-line therapy in older patients with high-risk (secondary) AML.
“We were very pleased to see promising response rates in this difficult-to-treat population,” Dr Cortes said. “And we eagerly await results from Celator’s pivotal phase 3 study of CPX-351, which could fulfill a considerable unmet need in AML.” ![]()
In a phase 2 trial, an investigational drug conferred a significant improvement in survival when used as salvage therapy in poor-risk patients with acute myeloid leukemia (AML).
On the other hand, the drug did not provide any significant improvements over control treatment (investigator’s choice) in the entire population of AML patients in first relapse.
The drug, CPX-351, is a fixed-ratio combination of cytarabine and daunorubicin inside a lipid vesicle.
Researchers reported these results with CPX-351 in Cancer. The study was funded by Celator Pharmaceuticals, the company developing CPX-351, and the Leukemia and Lymphoma Society.
“Patients with first-relapse AML have generally a poor prognosis, with a limited likelihood of response following salvage treatment,” said study author Jorge Cortes, MD, of the MD Anderson Cancer Center in Houston, Texas.
“This is particularly true for patients classified by the EPI [European Prognostic Index] as poor-risk upon entering the trial.”
For this trial, Dr Cortes and his colleagues evaluated 125 patients, ages 18 to 65, from 35 centers in the US, Canada, and Europe. Patients had AML in first relapse after an initial complete remission lasting a month or longer.
They were stratified per the EPI into favorable-, intermediate-, and poor-risk groups based on the duration of their first complete remission, cytogenetics, age, and transplant history. Most patients (68%) were in the poor-risk group.
Patients were randomized 2:1 to receive CPX-351 (100 units/m2 on days 1, 3, and 5 by 90-minute infusion) or the investigators’ choice of first salvage chemotherapy. The control treatment was usually based on cytarabine and an anthracycline, often with one or more additional agents.
Patient characteristics were largely well-balanced between the treatment arms. However, the CPX-351 group had a younger median age (52 years vs 56 years), more patients with secondary AML (12.3% vs 6.8%), and a higher rate of prior transplant (27.2% vs 15.9%).
Response and survival
Response rates were higher in the CPX-351 arm than in the control arm. The rates of complete response were 37% and 31.8%, respectively. And the rates of complete response with incomplete count recovery were 12.3% and 9.1%, respectively.
However, there was no significant difference in event-free survival (EFS) or overall survival (OS) between the treatment arms.
The median EFS was 4 months in the CPX-351 arm and 1.4 months in the control arm (hazard ratio[HR]=0.66, P=0.08). And the median OS was 8.5 months and 6.3 months, respectively (HR=0.75, P=0.19).
In the poor-risk population, there was no significant improvement in EFS with CPX-351, but there was a significant improvement in OS.
The median EFS was 1.8 months in the CPX-351 arm and 1.2 months in the control arm (HR=0.63, P=0.08). And the median OS was 6.5 months and 4.2 months, respectively (HR=0.55, P=0.02).
Safety and early mortality
The early mortality rate was similar between the treatment arms at 30 days—7.4% in the CPX-351 arm and 4.5% in the control arm—and at 60 days—14.8% and 15.9%, respectively. However, at 90 days, deaths were more frequent in the control arm—21.4% and 37.9%.
Patients in the CPX-351 arm had slower neutrophil recovery than those in the control arm—42 days and 34 days, respectively. The same was true for platelet recovery—45 days and 35 days, respectively.
Delayed hematologic recovery was associated with more infection-related events, such as febrile neutropenia, bacteremia, pneumonia, sepsis, urinary tract infection, pyrexia, and cellulitis.
The researchers believe these results, along with the previously published results from a phase 2 trial of CPX-351 in patients newly diagnosed with AML, support the phase 3 study of CPX-351 as a first-line therapy in older patients with high-risk (secondary) AML.
“We were very pleased to see promising response rates in this difficult-to-treat population,” Dr Cortes said. “And we eagerly await results from Celator’s pivotal phase 3 study of CPX-351, which could fulfill a considerable unmet need in AML.”
Study reveals potential strategy to treat AML

Credit: Lance Liotta
Researchers have discovered that interactions between two molecules—STAT3 and PRL-3—may provide a therapeutic target for acute myeloid leukemia (AML).
The team found evidence to suggest that the STAT3-PRL-3 regulatory loop contributes to the development of AML.
Chng Wee Joo, MB ChB, PhD, of the National University Cancer Institute in Singapore, and his colleagues reported these findings in Experimental Hematology.
The researchers discovered that STAT3, a transcription factor, binds and promotes the production of PRL-3 in cells. A decrease in STAT3 levels led to a corresponding decrease in the levels of PRL-3 and diminished the malignant properties of leukemic cells.
The team therefore concluded that a disruption of this regulatory loop may offer an attractive anti-AML therapeutic strategy. Furthermore, PRL-3 has the potential to be used as a biomarker in personalized therapy for AML patients.
The group was the first to report that the PRL-3 protein is overexpressed in 47% of bone marrow samples from AML patients. In addition, cellular levels of STAT3 were found to be elevated in about 50% of AML cases.
The researchers created a core STAT3 signature by analyzing datasets in the scientific literature. And they found that STAT3 core signature was significantly enriched in AML cases with high PRL-3 expression.
“Earlier studies on PRL-3 have been conducted in other cancers, but only in recent years has attention been turned to the significance of PRL-3 in blood cancer,” Dr Chng said.
“Previously, the mechanism by which PRL-3 is regulated in AML has also not been fully elucidated. This study reveals a novel connection between these two important oncogenes for the first time and also shows that the STAT3-PRL-3 regulatory loop contributes to the pathogenesis of AML.”
The researchers are now looking into methods to target the STAT3-PRL-3 pathway in AML, which could open up new avenues to treat AML patients with high expression of PRL-3 and offer an attractive anti-leukemia therapeutic strategy.
Credit: Lance Liotta
Researchers have discovered that interactions between two molecules—STAT3 and PRL-3—may provide a therapeutic target for acute myeloid leukemia (AML).
The team found evidence to suggest that the STAT3-PRL-3 regulatory loop contributes to the development of AML.
Chng Wee Joo, MB ChB, PhD, of the National University Cancer Institute in Singapore, and his colleagues reported these findings in Experimental Hematology.
The researchers discovered that STAT3, a transcription factor, binds and promotes the production of PRL-3 in cells. A decrease in STAT3 levels led to a corresponding decrease in the levels of PRL-3 and diminished the malignant properties of leukemic cells.
The team therefore concluded that a disruption of this regulatory loop may offer an attractive anti-AML therapeutic strategy. Furthermore, PRL-3 has the potential to be used as a biomarker in personalized therapy for AML patients.
The group was the first to report that the PRL-3 protein is overexpressed in 47% of bone marrow samples from AML patients. In addition, cellular levels of STAT3 were found to be elevated in about 50% of AML cases.
The researchers created a core STAT3 signature by analyzing datasets in the scientific literature. And they found that STAT3 core signature was significantly enriched in AML cases with high PRL-3 expression.
“Earlier studies on PRL-3 have been conducted in other cancers, but only in recent years has attention been turned to the significance of PRL-3 in blood cancer,” Dr Chng said.
“Previously, the mechanism by which PRL-3 is regulated in AML has also not been fully elucidated. This study reveals a novel connection between these two important oncogenes for the first time and also shows that the STAT3-PRL-3 regulatory loop contributes to the pathogenesis of AML.”
The researchers are now looking into methods to target the STAT3-PRL-3 pathway in AML, which could open up new avenues to treat AML patients with high expression of PRL-3 and offer an attractive anti-leukemia therapeutic strategy.
Credit: Lance Liotta
Researchers have discovered that interactions between two molecules—STAT3 and PRL-3—may provide a therapeutic target for acute myeloid leukemia (AML).
The team found evidence to suggest that the STAT3-PRL-3 regulatory loop contributes to the development of AML.
Chng Wee Joo, MB ChB, PhD, of the National University Cancer Institute in Singapore, and his colleagues reported these findings in Experimental Hematology.
The researchers discovered that STAT3, a transcription factor, binds and promotes the production of PRL-3 in cells. A decrease in STAT3 levels led to a corresponding decrease in the levels of PRL-3 and diminished the malignant properties of leukemic cells.
The team therefore concluded that a disruption of this regulatory loop may offer an attractive anti-AML therapeutic strategy. Furthermore, PRL-3 has the potential to be used as a biomarker in personalized therapy for AML patients.
The group was the first to report that the PRL-3 protein is overexpressed in 47% of bone marrow samples from AML patients. In addition, cellular levels of STAT3 were found to be elevated in about 50% of AML cases.
The researchers created a core STAT3 signature by analyzing datasets in the scientific literature. And they found that STAT3 core signature was significantly enriched in AML cases with high PRL-3 expression.
“Earlier studies on PRL-3 have been conducted in other cancers, but only in recent years has attention been turned to the significance of PRL-3 in blood cancer,” Dr Chng said.
“Previously, the mechanism by which PRL-3 is regulated in AML has also not been fully elucidated. This study reveals a novel connection between these two important oncogenes for the first time and also shows that the STAT3-PRL-3 regulatory loop contributes to the pathogenesis of AML.”
The researchers are now looking into methods to target the STAT3-PRL-3 pathway in AML, which could open up new avenues to treat AML patients with high expression of PRL-3 and offer an attractive anti-leukemia therapeutic strategy.
Survey reveals cancer survivors’ unmet needs

patient and her father
Credit: Rhoda Baer
New research shows that, even decades after being cured, many cancer survivors face challenges resulting from their disease and its treatment.
A survey of more than 1500 cancer survivors revealed 16 themes of challenges or unmet needs, such as physical dysfunction, financial problems, a lack of education about cancer survival, and anxiety about cancer recurrence.
Mary Ann Burg, PhD, of the University of Central Florida in Orlando, and her colleagues reported these findings in Cancer.
To assess the unmet needs of cancer survivors, the researchers evaluated responses from an American Cancer Society survey in which subjects responded to the open-ended question, “Please tell us about any needs you have now as a cancer survivor that are not being met to your satisfaction.”
There were a total of 1514 respondents who were 2, 5, or 10 years from cancer diagnosis. They were 24 to 97 years of age, 65.4% were female, and 24.8% were racial/ethnic minorities (black and Hispanic/Latino).
“This study was unique in that it gave a very large sample of cancer survivors a real voice to express their needs and concerns,” Dr Burg said.
The researchers found that the number and type of challenges/unmet needs were not associated with a subject’s time since cancer treatment, although older cancer survivors tended to report fewer unmet needs than younger survivors.
Sixteen themes of challenges/unmet needs emerged from respondents’ answers, with physical issues being the most common. About 38% of respondents reported physical issues, such as pain, symptoms, and sexual dysfunction.
About 20% reported financial problems, such as issues with insurance and the affordability of needed services and products. About 20% also said they had needs related to unanswered questions and a lack of knowledge about what to expect as a cancer survivor, including guidance on follow-up care and cancer risks, causes, and prevention.
About 16% of respondents cited issues relating to personal control (a lack of physical and social autonomy). And about 16% described flaws and constraints in the healthcare system that affected early detection, diagnosis, treatment, follow-up care, and continuity of care.
About 14% of respondents reported a lack of resources (such as supplies, equipment, and medications), and about 14% cited emotional and mental health issues (such as fear of cancer recurrence, depression, and anxiety).
About 13% of respondents said they lacked social support (such as access to support groups), and 10% reported issues relating to societal perceptions of cancer survivors (such as discrimination and misinformation).
About 9% of respondents expressed the need to talk about or explain the cancer experience with their physician, friends, and family. And about 9% cited a lack of trust in healthcare providers.
Other themes included the wish for more effective cancer treatments (3.5%), body image issues such as feeling unattractive or losing trust in the body (3.5%), issues with the “survivor” identity (3.1%), trouble obtaining or maintaining appropriate employment (2.3%), and existential issues, such as finding meaning in the cancer experience (0.6%).
“Overall, we found that cancer survivors are often caught off guard by the lingering problems they experience after cancer treatment,” Dr Burg said. “In the wake of cancer, many survivors feel they have lost a sense of personal control, have reduced quality of life, and are frustrated that these problems are not sufficiently addressed within the medical care system.”
She added that this study points to several areas in which we might work to improve the situation, including raising public awareness of cancer survivors’ problems, promoting honest professional communication about the side effects of cancer and its treatment, and coordinating medical care resources to help survivors and their families cope with lingering challenges.
patient and her father
Credit: Rhoda Baer
New research shows that, even decades after being cured, many cancer survivors face challenges resulting from their disease and its treatment.
A survey of more than 1500 cancer survivors revealed 16 themes of challenges or unmet needs, such as physical dysfunction, financial problems, a lack of education about cancer survival, and anxiety about cancer recurrence.
Mary Ann Burg, PhD, of the University of Central Florida in Orlando, and her colleagues reported these findings in Cancer.
To assess the unmet needs of cancer survivors, the researchers evaluated responses from an American Cancer Society survey in which subjects responded to the open-ended question, “Please tell us about any needs you have now as a cancer survivor that are not being met to your satisfaction.”
There were a total of 1514 respondents who were 2, 5, or 10 years from cancer diagnosis. They were 24 to 97 years of age, 65.4% were female, and 24.8% were racial/ethnic minorities (black and Hispanic/Latino).
“This study was unique in that it gave a very large sample of cancer survivors a real voice to express their needs and concerns,” Dr Burg said.
The researchers found that the number and type of challenges/unmet needs were not associated with a subject’s time since cancer treatment, although older cancer survivors tended to report fewer unmet needs than younger survivors.
Sixteen themes of challenges/unmet needs emerged from respondents’ answers, with physical issues being the most common. About 38% of respondents reported physical issues, such as pain, symptoms, and sexual dysfunction.
About 20% reported financial problems, such as issues with insurance and the affordability of needed services and products. About 20% also said they had needs related to unanswered questions and a lack of knowledge about what to expect as a cancer survivor, including guidance on follow-up care and cancer risks, causes, and prevention.
About 16% of respondents cited issues relating to personal control (a lack of physical and social autonomy). And about 16% described flaws and constraints in the healthcare system that affected early detection, diagnosis, treatment, follow-up care, and continuity of care.
About 14% of respondents reported a lack of resources (such as supplies, equipment, and medications), and about 14% cited emotional and mental health issues (such as fear of cancer recurrence, depression, and anxiety).
About 13% of respondents said they lacked social support (such as access to support groups), and 10% reported issues relating to societal perceptions of cancer survivors (such as discrimination and misinformation).
About 9% of respondents expressed the need to talk about or explain the cancer experience with their physician, friends, and family. And about 9% cited a lack of trust in healthcare providers.
Other themes included the wish for more effective cancer treatments (3.5%), body image issues such as feeling unattractive or losing trust in the body (3.5%), issues with the “survivor” identity (3.1%), trouble obtaining or maintaining appropriate employment (2.3%), and existential issues, such as finding meaning in the cancer experience (0.6%).
“Overall, we found that cancer survivors are often caught off guard by the lingering problems they experience after cancer treatment,” Dr Burg said. “In the wake of cancer, many survivors feel they have lost a sense of personal control, have reduced quality of life, and are frustrated that these problems are not sufficiently addressed within the medical care system.”
She added that this study points to several areas in which we might work to improve the situation, including raising public awareness of cancer survivors’ problems, promoting honest professional communication about the side effects of cancer and its treatment, and coordinating medical care resources to help survivors and their families cope with lingering challenges.
patient and her father
Credit: Rhoda Baer
New research shows that, even decades after being cured, many cancer survivors face challenges resulting from their disease and its treatment.
A survey of more than 1500 cancer survivors revealed 16 themes of challenges or unmet needs, such as physical dysfunction, financial problems, a lack of education about cancer survival, and anxiety about cancer recurrence.
Mary Ann Burg, PhD, of the University of Central Florida in Orlando, and her colleagues reported these findings in Cancer.
To assess the unmet needs of cancer survivors, the researchers evaluated responses from an American Cancer Society survey in which subjects responded to the open-ended question, “Please tell us about any needs you have now as a cancer survivor that are not being met to your satisfaction.”
There were a total of 1514 respondents who were 2, 5, or 10 years from cancer diagnosis. They were 24 to 97 years of age, 65.4% were female, and 24.8% were racial/ethnic minorities (black and Hispanic/Latino).
“This study was unique in that it gave a very large sample of cancer survivors a real voice to express their needs and concerns,” Dr Burg said.
The researchers found that the number and type of challenges/unmet needs were not associated with a subject’s time since cancer treatment, although older cancer survivors tended to report fewer unmet needs than younger survivors.
Sixteen themes of challenges/unmet needs emerged from respondents’ answers, with physical issues being the most common. About 38% of respondents reported physical issues, such as pain, symptoms, and sexual dysfunction.
About 20% reported financial problems, such as issues with insurance and the affordability of needed services and products. About 20% also said they had needs related to unanswered questions and a lack of knowledge about what to expect as a cancer survivor, including guidance on follow-up care and cancer risks, causes, and prevention.
About 16% of respondents cited issues relating to personal control (a lack of physical and social autonomy). And about 16% described flaws and constraints in the healthcare system that affected early detection, diagnosis, treatment, follow-up care, and continuity of care.
About 14% of respondents reported a lack of resources (such as supplies, equipment, and medications), and about 14% cited emotional and mental health issues (such as fear of cancer recurrence, depression, and anxiety).
About 13% of respondents said they lacked social support (such as access to support groups), and 10% reported issues relating to societal perceptions of cancer survivors (such as discrimination and misinformation).
About 9% of respondents expressed the need to talk about or explain the cancer experience with their physician, friends, and family. And about 9% cited a lack of trust in healthcare providers.
Other themes included the wish for more effective cancer treatments (3.5%), body image issues such as feeling unattractive or losing trust in the body (3.5%), issues with the “survivor” identity (3.1%), trouble obtaining or maintaining appropriate employment (2.3%), and existential issues, such as finding meaning in the cancer experience (0.6%).
“Overall, we found that cancer survivors are often caught off guard by the lingering problems they experience after cancer treatment,” Dr Burg said. “In the wake of cancer, many survivors feel they have lost a sense of personal control, have reduced quality of life, and are frustrated that these problems are not sufficiently addressed within the medical care system.”
She added that this study points to several areas in which we might work to improve the situation, including raising public awareness of cancer survivors’ problems, promoting honest professional communication about the side effects of cancer and its treatment, and coordinating medical care resources to help survivors and their families cope with lingering challenges.