User login
Haplo-HSCT appears comparable to fully matched HSCT
![]()
Photo by Chad McNeeley
A retrospective study suggests that, for patients with hematologic disorders, a haploidentical hematopoietic stem cell transplant (HSCT)
can be roughly as safe and effective as a fully matched HSCT.
The study showed that, when patients received an identical conditioning regimen, graft T-cell dose, and graft-vs-host disease (GVHD) prophylaxis, haploidentical and fully matched HSCTs produced comparable results.
Patients had similar rates of overall and progression-free survival, relapse, non-relapse mortality, and chronic GVHD.
However, patients who received haploidentical transplants had higher rates of grade 2-4 acute GVHD and cytomegalovirus reactivation.
Researchers reported these results in Biology of Blood and Marrow Transplantation.
“This is the first study to compare the gold standard to a half-match using an identical protocol,” said Neal Flomenberg, MD, of Thomas Jefferson University in Philadelphia, Pennsylvania.
“The field has debated whether the differences in outcomes between full and partial matches were caused by the quality of the match or by all the procedures the patient goes through before and after the donor cells are administered. We haven’t had a clear answer.”
With that in mind, Dr Flomenberg and his colleagues compared 3-year outcome data from patients who received haploidentical HSCTs (n=50) or fully matched HSCTs (n=27), when both groups of patients were treated with a 2-step protocol.
The patients had acute myeloid leukemia (n=38), acute lymphoblastic leukemia (n=20), myelodysplastic syndromes/myeloproliferative neoplasms (n=7), non-Hodgkin lymphoma (n=11), and aplastic anemia (n=1).
The 2-step protocol
All patients received a myeloablative conditioning regimen consisting of 12 Gy of total body irradiation administered in 8 fractions over 4 days. After the last fraction, they received a fixed T-cell dose (2 x 108 cells/kg), which was followed, 2 days later, by cyclophosphamide at 60 mg/kg/day for 2 days.
Twenty-four hours after they completed cyclophosphamide, patients received CD34-selected peripheral blood stem cells from a half-matched or fully matched donor.
On day -1, patients began taking tacrolimus and mycophenolate mofetil as GVHD prophylaxis. They also received growth factor support (granulocyte-macrophage colony-stimulating factor at 250 μg/m2) starting on day +1.
In the absence of GVHD, mycophenolate mofetil was discontinued on day 28 and tacrolimus was tapered, starting on day +60 after HSCT.
Results
The researchers said that early immune recovery was comparable between the patient groups in nearly all assessed T-cell subsets. The exception was the median CD3/CD8 cell count, which was significantly higher at day 28 in the fully matched group than the haploidentical group (P=0.029).
Survival rates were comparable between the groups. The estimated 3-year overall survival was 70% in the haploidentical group and 71% in the fully matched group (P=0.81). The 3-year progression-free survival was 68% and 70%, respectively (P=0.97).
The 3-year cumulative incidence of non-relapse mortality was 10% in the haploidentical group and 4% in the fully matched group (P=0.34). The 3-year cumulative incidence of relapse was 21% and 27%, respectively (P=0.93).
The 100-day cumulative incidence of grade 2-4 acute GVHD was significantly higher in the haploidentical group than the fully matched group—40% and 8%, respectively (P<0.001). But there was no significant difference in the incidence of grade 3-4 acute GVHD—6% and 4%, respectively (P=0.49).
The cumulative incidence of chronic GVHD at 2 years was not significantly different between the haploidentical and fully matched groups—19% and 12%, respectively (P=0.47). The same was true for severe chronic GVHD—4% and 8%, respectively (P=0.49).
The cumulative incidence of cytomegalovirus reactivation was significantly higher in the haploidentical group than the fully matched group—68% and 19%, respectively (P<0.001).
There were no deaths from infections or GVHD in either group.
“The results of the current study are certainly encouraging and suggest that outcomes from a half-matched, related donor are similar to fully matched donors,” said study author Sameh Gaballa, MD, also of Thomas Jefferson University.
“It might be time to reassess whether half-matched, related transplants can be considered the best alternative donor source for patients lacking a fully matched family member donor. For that, we’ll need more evidence from a randomly controlled, prospective trial, rather than studies that look at patient data retrospectively, to help solidify our findings here.” ![]()
![]()
Photo by Chad McNeeley
A retrospective study suggests that, for patients with hematologic disorders, a haploidentical hematopoietic stem cell transplant (HSCT)
can be roughly as safe and effective as a fully matched HSCT.
The study showed that, when patients received an identical conditioning regimen, graft T-cell dose, and graft-vs-host disease (GVHD) prophylaxis, haploidentical and fully matched HSCTs produced comparable results.
Patients had similar rates of overall and progression-free survival, relapse, non-relapse mortality, and chronic GVHD.
However, patients who received haploidentical transplants had higher rates of grade 2-4 acute GVHD and cytomegalovirus reactivation.
Researchers reported these results in Biology of Blood and Marrow Transplantation.
“This is the first study to compare the gold standard to a half-match using an identical protocol,” said Neal Flomenberg, MD, of Thomas Jefferson University in Philadelphia, Pennsylvania.
“The field has debated whether the differences in outcomes between full and partial matches were caused by the quality of the match or by all the procedures the patient goes through before and after the donor cells are administered. We haven’t had a clear answer.”
With that in mind, Dr Flomenberg and his colleagues compared 3-year outcome data from patients who received haploidentical HSCTs (n=50) or fully matched HSCTs (n=27), when both groups of patients were treated with a 2-step protocol.
The patients had acute myeloid leukemia (n=38), acute lymphoblastic leukemia (n=20), myelodysplastic syndromes/myeloproliferative neoplasms (n=7), non-Hodgkin lymphoma (n=11), and aplastic anemia (n=1).
The 2-step protocol
All patients received a myeloablative conditioning regimen consisting of 12 Gy of total body irradiation administered in 8 fractions over 4 days. After the last fraction, they received a fixed T-cell dose (2 x 108 cells/kg), which was followed, 2 days later, by cyclophosphamide at 60 mg/kg/day for 2 days.
Twenty-four hours after they completed cyclophosphamide, patients received CD34-selected peripheral blood stem cells from a half-matched or fully matched donor.
On day -1, patients began taking tacrolimus and mycophenolate mofetil as GVHD prophylaxis. They also received growth factor support (granulocyte-macrophage colony-stimulating factor at 250 μg/m2) starting on day +1.
In the absence of GVHD, mycophenolate mofetil was discontinued on day 28 and tacrolimus was tapered, starting on day +60 after HSCT.
Results
The researchers said that early immune recovery was comparable between the patient groups in nearly all assessed T-cell subsets. The exception was the median CD3/CD8 cell count, which was significantly higher at day 28 in the fully matched group than the haploidentical group (P=0.029).
Survival rates were comparable between the groups. The estimated 3-year overall survival was 70% in the haploidentical group and 71% in the fully matched group (P=0.81). The 3-year progression-free survival was 68% and 70%, respectively (P=0.97).
The 3-year cumulative incidence of non-relapse mortality was 10% in the haploidentical group and 4% in the fully matched group (P=0.34). The 3-year cumulative incidence of relapse was 21% and 27%, respectively (P=0.93).
The 100-day cumulative incidence of grade 2-4 acute GVHD was significantly higher in the haploidentical group than the fully matched group—40% and 8%, respectively (P<0.001). But there was no significant difference in the incidence of grade 3-4 acute GVHD—6% and 4%, respectively (P=0.49).
The cumulative incidence of chronic GVHD at 2 years was not significantly different between the haploidentical and fully matched groups—19% and 12%, respectively (P=0.47). The same was true for severe chronic GVHD—4% and 8%, respectively (P=0.49).
The cumulative incidence of cytomegalovirus reactivation was significantly higher in the haploidentical group than the fully matched group—68% and 19%, respectively (P<0.001).
There were no deaths from infections or GVHD in either group.
“The results of the current study are certainly encouraging and suggest that outcomes from a half-matched, related donor are similar to fully matched donors,” said study author Sameh Gaballa, MD, also of Thomas Jefferson University.
“It might be time to reassess whether half-matched, related transplants can be considered the best alternative donor source for patients lacking a fully matched family member donor. For that, we’ll need more evidence from a randomly controlled, prospective trial, rather than studies that look at patient data retrospectively, to help solidify our findings here.” ![]()
![]()
Photo by Chad McNeeley
A retrospective study suggests that, for patients with hematologic disorders, a haploidentical hematopoietic stem cell transplant (HSCT)
can be roughly as safe and effective as a fully matched HSCT.
The study showed that, when patients received an identical conditioning regimen, graft T-cell dose, and graft-vs-host disease (GVHD) prophylaxis, haploidentical and fully matched HSCTs produced comparable results.
Patients had similar rates of overall and progression-free survival, relapse, non-relapse mortality, and chronic GVHD.
However, patients who received haploidentical transplants had higher rates of grade 2-4 acute GVHD and cytomegalovirus reactivation.
Researchers reported these results in Biology of Blood and Marrow Transplantation.
“This is the first study to compare the gold standard to a half-match using an identical protocol,” said Neal Flomenberg, MD, of Thomas Jefferson University in Philadelphia, Pennsylvania.
“The field has debated whether the differences in outcomes between full and partial matches were caused by the quality of the match or by all the procedures the patient goes through before and after the donor cells are administered. We haven’t had a clear answer.”
With that in mind, Dr Flomenberg and his colleagues compared 3-year outcome data from patients who received haploidentical HSCTs (n=50) or fully matched HSCTs (n=27), when both groups of patients were treated with a 2-step protocol.
The patients had acute myeloid leukemia (n=38), acute lymphoblastic leukemia (n=20), myelodysplastic syndromes/myeloproliferative neoplasms (n=7), non-Hodgkin lymphoma (n=11), and aplastic anemia (n=1).
The 2-step protocol
All patients received a myeloablative conditioning regimen consisting of 12 Gy of total body irradiation administered in 8 fractions over 4 days. After the last fraction, they received a fixed T-cell dose (2 x 108 cells/kg), which was followed, 2 days later, by cyclophosphamide at 60 mg/kg/day for 2 days.
Twenty-four hours after they completed cyclophosphamide, patients received CD34-selected peripheral blood stem cells from a half-matched or fully matched donor.
On day -1, patients began taking tacrolimus and mycophenolate mofetil as GVHD prophylaxis. They also received growth factor support (granulocyte-macrophage colony-stimulating factor at 250 μg/m2) starting on day +1.
In the absence of GVHD, mycophenolate mofetil was discontinued on day 28 and tacrolimus was tapered, starting on day +60 after HSCT.
Results
The researchers said that early immune recovery was comparable between the patient groups in nearly all assessed T-cell subsets. The exception was the median CD3/CD8 cell count, which was significantly higher at day 28 in the fully matched group than the haploidentical group (P=0.029).
Survival rates were comparable between the groups. The estimated 3-year overall survival was 70% in the haploidentical group and 71% in the fully matched group (P=0.81). The 3-year progression-free survival was 68% and 70%, respectively (P=0.97).
The 3-year cumulative incidence of non-relapse mortality was 10% in the haploidentical group and 4% in the fully matched group (P=0.34). The 3-year cumulative incidence of relapse was 21% and 27%, respectively (P=0.93).
The 100-day cumulative incidence of grade 2-4 acute GVHD was significantly higher in the haploidentical group than the fully matched group—40% and 8%, respectively (P<0.001). But there was no significant difference in the incidence of grade 3-4 acute GVHD—6% and 4%, respectively (P=0.49).
The cumulative incidence of chronic GVHD at 2 years was not significantly different between the haploidentical and fully matched groups—19% and 12%, respectively (P=0.47). The same was true for severe chronic GVHD—4% and 8%, respectively (P=0.49).
The cumulative incidence of cytomegalovirus reactivation was significantly higher in the haploidentical group than the fully matched group—68% and 19%, respectively (P<0.001).
There were no deaths from infections or GVHD in either group.
“The results of the current study are certainly encouraging and suggest that outcomes from a half-matched, related donor are similar to fully matched donors,” said study author Sameh Gaballa, MD, also of Thomas Jefferson University.
“It might be time to reassess whether half-matched, related transplants can be considered the best alternative donor source for patients lacking a fully matched family member donor. For that, we’ll need more evidence from a randomly controlled, prospective trial, rather than studies that look at patient data retrospectively, to help solidify our findings here.” ![]()
Findings may inform design of new treatments for JMML

Image from the Salk Institute
Researchers have used induced pluripotent stem cells (iPSCs) to model juvenile myelomonocytic leukemia (JMML) and gain new insight into the disease.
The team noted that somatic PTPN11 mutations are known to cause JMML, germline PTPN11 defects cause Noonan syndrome (NS), and specific inherited mutations cause NS/JMML.
With their work, the researchers found that hematopoietic cells differentiated from iPSCs harboring NS/JMML-causing PTPN11 mutations recapitulate the features of JMML.
They described this work in Cell Reports.
“By studying an inherited human cancer syndrome, our study clarified early events in the development of [JMML],” said Bruce D. Gelb, MD, of the Icahn School of Medicine at Mount Sinai in New York, New York.
“More than just creating a model of a disease, we were able to prove that mechanisms seen in our model also happen in the bone marrow of people with this kind of leukemia.”
Specifically, the team found that NS/JMML myeloid cells derived from iPSCs demonstrated increased signaling through STAT5 and upregulation of 2 microRNAs—miR-223 and miR-15a.
Likewise, miR-223 and miR-15a were upregulated in 11 of 19 bone marrow samples from patients with JMML harboring PTPN11 mutations.
However, the microRNAs were not upregulated in patients without PTPN11 mutations. And when the researchers reduced miR-223’s function in NS/JMML iPSCs, they observed a normalization of myelogenesis.
“Going into the current study, experts in the field had tended to lump all forms of JMML together, but the new study was able to isolate biological changes specific to hematopoietic cells with PTPN11 mutations, which causes more severe JMML,” Dr Gelb said.
He and his colleagues also found that microRNA target gene expression levels were reduced in the iPSC-derived myeloid cells and in cells from JMML patients with PTPN11 mutations.
“Our results provide further evidence that the severity of this form of leukemia arises from the degree of changes in the gene PTPN11, altering the protein it codes for, SHP-2, and biologic pathways related to it,” Dr Gelb said. “These proteins promise to become a focus of future drug design efforts.” ![]()
Image from the Salk Institute
Researchers have used induced pluripotent stem cells (iPSCs) to model juvenile myelomonocytic leukemia (JMML) and gain new insight into the disease.
The team noted that somatic PTPN11 mutations are known to cause JMML, germline PTPN11 defects cause Noonan syndrome (NS), and specific inherited mutations cause NS/JMML.
With their work, the researchers found that hematopoietic cells differentiated from iPSCs harboring NS/JMML-causing PTPN11 mutations recapitulate the features of JMML.
They described this work in Cell Reports.
“By studying an inherited human cancer syndrome, our study clarified early events in the development of [JMML],” said Bruce D. Gelb, MD, of the Icahn School of Medicine at Mount Sinai in New York, New York.
“More than just creating a model of a disease, we were able to prove that mechanisms seen in our model also happen in the bone marrow of people with this kind of leukemia.”
Specifically, the team found that NS/JMML myeloid cells derived from iPSCs demonstrated increased signaling through STAT5 and upregulation of 2 microRNAs—miR-223 and miR-15a.
Likewise, miR-223 and miR-15a were upregulated in 11 of 19 bone marrow samples from patients with JMML harboring PTPN11 mutations.
However, the microRNAs were not upregulated in patients without PTPN11 mutations. And when the researchers reduced miR-223’s function in NS/JMML iPSCs, they observed a normalization of myelogenesis.
“Going into the current study, experts in the field had tended to lump all forms of JMML together, but the new study was able to isolate biological changes specific to hematopoietic cells with PTPN11 mutations, which causes more severe JMML,” Dr Gelb said.
He and his colleagues also found that microRNA target gene expression levels were reduced in the iPSC-derived myeloid cells and in cells from JMML patients with PTPN11 mutations.
“Our results provide further evidence that the severity of this form of leukemia arises from the degree of changes in the gene PTPN11, altering the protein it codes for, SHP-2, and biologic pathways related to it,” Dr Gelb said. “These proteins promise to become a focus of future drug design efforts.” ![]()
Image from the Salk Institute
Researchers have used induced pluripotent stem cells (iPSCs) to model juvenile myelomonocytic leukemia (JMML) and gain new insight into the disease.
The team noted that somatic PTPN11 mutations are known to cause JMML, germline PTPN11 defects cause Noonan syndrome (NS), and specific inherited mutations cause NS/JMML.
With their work, the researchers found that hematopoietic cells differentiated from iPSCs harboring NS/JMML-causing PTPN11 mutations recapitulate the features of JMML.
They described this work in Cell Reports.
“By studying an inherited human cancer syndrome, our study clarified early events in the development of [JMML],” said Bruce D. Gelb, MD, of the Icahn School of Medicine at Mount Sinai in New York, New York.
“More than just creating a model of a disease, we were able to prove that mechanisms seen in our model also happen in the bone marrow of people with this kind of leukemia.”
Specifically, the team found that NS/JMML myeloid cells derived from iPSCs demonstrated increased signaling through STAT5 and upregulation of 2 microRNAs—miR-223 and miR-15a.
Likewise, miR-223 and miR-15a were upregulated in 11 of 19 bone marrow samples from patients with JMML harboring PTPN11 mutations.
However, the microRNAs were not upregulated in patients without PTPN11 mutations. And when the researchers reduced miR-223’s function in NS/JMML iPSCs, they observed a normalization of myelogenesis.
“Going into the current study, experts in the field had tended to lump all forms of JMML together, but the new study was able to isolate biological changes specific to hematopoietic cells with PTPN11 mutations, which causes more severe JMML,” Dr Gelb said.
He and his colleagues also found that microRNA target gene expression levels were reduced in the iPSC-derived myeloid cells and in cells from JMML patients with PTPN11 mutations.
“Our results provide further evidence that the severity of this form of leukemia arises from the degree of changes in the gene PTPN11, altering the protein it codes for, SHP-2, and biologic pathways related to it,” Dr Gelb said. “These proteins promise to become a focus of future drug design efforts.” ![]()
IgA increase linked to fewer infections in CLL patients on ibrutinib
Increases in IgA levels were associated with a reduced risk of infections in 84 chronic lymphocytic leukemia (CLL) patients participating in a trial of ibrutinib 420 mg once daily.
After 28 months of ibrutinib treatment, 69 (82%) patients had developed 177 infections. Lower rates of infections were found in those who experienced an IgA increase of at least 50% from their baseline values (P = .03), reported Dr. Clare Sun of the hematology branch of the National Heart, Lung and Blood Institute in Bethesda, Md., and her associates.
At baseline, the patients’ median IgA value was 0.47 g/L; after 6 months of treatment with ibrutinib, the median IgA value was 0.74 g/L. The levels of IgA continued to rise in the next 12 months (n = 43, median increase of 45%, P less than 0001), and patients’ IgA levels at 24 months also were greater than their baseline levels (n = 28, median increase of 64%, P less than .0001).
Using serum-free light chain measures to distinguish clonal and normal B cells, researchers also found recovery of normal B cells and increases in B-cell precursors in bone marrow and in normal B cells in the peripheral blood. This growth, however, was not large enough to raise the majority of patients’ normal B cells to normal levels.
The findings suggest “ibrutinib allows for a clinically meaningful recovery of humoral immune function in patients with CLL,” Dr. Sun and her associates wrote. “The rapidity of increase in IgA suggests that pre-existing antibody-producing cells may be secreting more immunoglobulins, whilst CLL cells, which impair immunoglobulin production, are removed by ibrutinib.”
The patients also had a decline in IgG levels, however, which did not appear to have an adverse impact. The patients’ IgG levels remained stable during the first 6 months of treatment, but by 12 months they had decreased (n = 35, median reduction of 4%, P < .0006), falling further at 24 months (n = 21, median reduction of 23%, P < .0001).
Because ibrutinib may be given indefinitely, extended follow-up is needed to determine the immunologic consequences of prolonged Bruton’s tyrosine kinase inhibition, the researchers wrote.
Read the full study in Blood (2015. doi: 10.1182/blood-2015-04-639203).
Increases in IgA levels were associated with a reduced risk of infections in 84 chronic lymphocytic leukemia (CLL) patients participating in a trial of ibrutinib 420 mg once daily.
After 28 months of ibrutinib treatment, 69 (82%) patients had developed 177 infections. Lower rates of infections were found in those who experienced an IgA increase of at least 50% from their baseline values (P = .03), reported Dr. Clare Sun of the hematology branch of the National Heart, Lung and Blood Institute in Bethesda, Md., and her associates.
At baseline, the patients’ median IgA value was 0.47 g/L; after 6 months of treatment with ibrutinib, the median IgA value was 0.74 g/L. The levels of IgA continued to rise in the next 12 months (n = 43, median increase of 45%, P less than 0001), and patients’ IgA levels at 24 months also were greater than their baseline levels (n = 28, median increase of 64%, P less than .0001).
Using serum-free light chain measures to distinguish clonal and normal B cells, researchers also found recovery of normal B cells and increases in B-cell precursors in bone marrow and in normal B cells in the peripheral blood. This growth, however, was not large enough to raise the majority of patients’ normal B cells to normal levels.
The findings suggest “ibrutinib allows for a clinically meaningful recovery of humoral immune function in patients with CLL,” Dr. Sun and her associates wrote. “The rapidity of increase in IgA suggests that pre-existing antibody-producing cells may be secreting more immunoglobulins, whilst CLL cells, which impair immunoglobulin production, are removed by ibrutinib.”
The patients also had a decline in IgG levels, however, which did not appear to have an adverse impact. The patients’ IgG levels remained stable during the first 6 months of treatment, but by 12 months they had decreased (n = 35, median reduction of 4%, P < .0006), falling further at 24 months (n = 21, median reduction of 23%, P < .0001).
Because ibrutinib may be given indefinitely, extended follow-up is needed to determine the immunologic consequences of prolonged Bruton’s tyrosine kinase inhibition, the researchers wrote.
Read the full study in Blood (2015. doi: 10.1182/blood-2015-04-639203).
Increases in IgA levels were associated with a reduced risk of infections in 84 chronic lymphocytic leukemia (CLL) patients participating in a trial of ibrutinib 420 mg once daily.
After 28 months of ibrutinib treatment, 69 (82%) patients had developed 177 infections. Lower rates of infections were found in those who experienced an IgA increase of at least 50% from their baseline values (P = .03), reported Dr. Clare Sun of the hematology branch of the National Heart, Lung and Blood Institute in Bethesda, Md., and her associates.
At baseline, the patients’ median IgA value was 0.47 g/L; after 6 months of treatment with ibrutinib, the median IgA value was 0.74 g/L. The levels of IgA continued to rise in the next 12 months (n = 43, median increase of 45%, P less than 0001), and patients’ IgA levels at 24 months also were greater than their baseline levels (n = 28, median increase of 64%, P less than .0001).
Using serum-free light chain measures to distinguish clonal and normal B cells, researchers also found recovery of normal B cells and increases in B-cell precursors in bone marrow and in normal B cells in the peripheral blood. This growth, however, was not large enough to raise the majority of patients’ normal B cells to normal levels.
The findings suggest “ibrutinib allows for a clinically meaningful recovery of humoral immune function in patients with CLL,” Dr. Sun and her associates wrote. “The rapidity of increase in IgA suggests that pre-existing antibody-producing cells may be secreting more immunoglobulins, whilst CLL cells, which impair immunoglobulin production, are removed by ibrutinib.”
The patients also had a decline in IgG levels, however, which did not appear to have an adverse impact. The patients’ IgG levels remained stable during the first 6 months of treatment, but by 12 months they had decreased (n = 35, median reduction of 4%, P < .0006), falling further at 24 months (n = 21, median reduction of 23%, P < .0001).
Because ibrutinib may be given indefinitely, extended follow-up is needed to determine the immunologic consequences of prolonged Bruton’s tyrosine kinase inhibition, the researchers wrote.
Read the full study in Blood (2015. doi: 10.1182/blood-2015-04-639203).
FROM BLOOD
Case suggests GSIs could treat Notch-mutated ALL
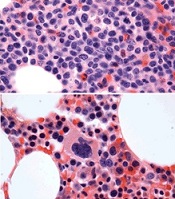
before (top) and after 7 weeks
of treatment (bottom)
© Knoechel et al.
Results of a case study suggest a gamma-secretase inhibitor (GSI) can be effective against Notch-mutated acute lymphoblastic leukemia (ALL).
The patient, who had early T-cell precursor ALL (ETP-ALL), achieved a complete hematologic response to treatment with BMS-906024, a GSI with anti-Notch
activity.
The patient was then able to proceed to hematopoietic stem cell transplant and was leukemia-free at last follow-up.
The researchers said this suggests that GSIs might hold promise for treating ALL and other cancers characterized by Notch mutations.
Birgit Knoechel, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, and her colleagues described this case study in Cold Spring Harbor Molecular Case Studies.
The patient was a 53-year-old male with ETP-ALL who had failed previous rounds of chemotherapy and was then enrolled in a clinical trial of BMS-906024.
The patient began to show immediate improvement after starting treatment with the GSI. After 3 cycles, he went on to transplant and has since been leukemia-free—for 19 months so far.
To determine the genetic basis for the patient’s response to BMS-906024, researchers performed targeted and whole-exome sequencing on his leukemic cells.
They identified 4 potential mutations driving disease progression, including a novel mutation in the NOTCH1 gene that resulted in hyperactive signaling. This mutated gene copy was also duplicated in the cancer genome, resulting in elevated expression.
However, the NOTCH1 mutation, along with 2 of the other mutations, were absent in the remission bone marrow.
The researchers also cultured the patient’s leukemic cells to determine the molecular response to treatment.
Cells treated with BMS-906024 had greatly reduced levels of mutated NOTCH1 protein. RNA sequencing demonstrated that Notch target genes were sensitive to the treatment.
The MYC oncogene, on the other hand, was not sensitive to BMS-906024.
Epigenetic analysis revealed that the enhancer driving MYC expression in the leukemic cells was not Notch-dependent, but rather BRD4-dependent, suggesting another possible therapeutic option for MYC-expressing tumors. ![]()
before (top) and after 7 weeks
of treatment (bottom)
© Knoechel et al.
Results of a case study suggest a gamma-secretase inhibitor (GSI) can be effective against Notch-mutated acute lymphoblastic leukemia (ALL).
The patient, who had early T-cell precursor ALL (ETP-ALL), achieved a complete hematologic response to treatment with BMS-906024, a GSI with anti-Notch
activity.
The patient was then able to proceed to hematopoietic stem cell transplant and was leukemia-free at last follow-up.
The researchers said this suggests that GSIs might hold promise for treating ALL and other cancers characterized by Notch mutations.
Birgit Knoechel, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, and her colleagues described this case study in Cold Spring Harbor Molecular Case Studies.
The patient was a 53-year-old male with ETP-ALL who had failed previous rounds of chemotherapy and was then enrolled in a clinical trial of BMS-906024.
The patient began to show immediate improvement after starting treatment with the GSI. After 3 cycles, he went on to transplant and has since been leukemia-free—for 19 months so far.
To determine the genetic basis for the patient’s response to BMS-906024, researchers performed targeted and whole-exome sequencing on his leukemic cells.
They identified 4 potential mutations driving disease progression, including a novel mutation in the NOTCH1 gene that resulted in hyperactive signaling. This mutated gene copy was also duplicated in the cancer genome, resulting in elevated expression.
However, the NOTCH1 mutation, along with 2 of the other mutations, were absent in the remission bone marrow.
The researchers also cultured the patient’s leukemic cells to determine the molecular response to treatment.
Cells treated with BMS-906024 had greatly reduced levels of mutated NOTCH1 protein. RNA sequencing demonstrated that Notch target genes were sensitive to the treatment.
The MYC oncogene, on the other hand, was not sensitive to BMS-906024.
Epigenetic analysis revealed that the enhancer driving MYC expression in the leukemic cells was not Notch-dependent, but rather BRD4-dependent, suggesting another possible therapeutic option for MYC-expressing tumors. ![]()
before (top) and after 7 weeks
of treatment (bottom)
© Knoechel et al.
Results of a case study suggest a gamma-secretase inhibitor (GSI) can be effective against Notch-mutated acute lymphoblastic leukemia (ALL).
The patient, who had early T-cell precursor ALL (ETP-ALL), achieved a complete hematologic response to treatment with BMS-906024, a GSI with anti-Notch
activity.
The patient was then able to proceed to hematopoietic stem cell transplant and was leukemia-free at last follow-up.
The researchers said this suggests that GSIs might hold promise for treating ALL and other cancers characterized by Notch mutations.
Birgit Knoechel, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, and her colleagues described this case study in Cold Spring Harbor Molecular Case Studies.
The patient was a 53-year-old male with ETP-ALL who had failed previous rounds of chemotherapy and was then enrolled in a clinical trial of BMS-906024.
The patient began to show immediate improvement after starting treatment with the GSI. After 3 cycles, he went on to transplant and has since been leukemia-free—for 19 months so far.
To determine the genetic basis for the patient’s response to BMS-906024, researchers performed targeted and whole-exome sequencing on his leukemic cells.
They identified 4 potential mutations driving disease progression, including a novel mutation in the NOTCH1 gene that resulted in hyperactive signaling. This mutated gene copy was also duplicated in the cancer genome, resulting in elevated expression.
However, the NOTCH1 mutation, along with 2 of the other mutations, were absent in the remission bone marrow.
The researchers also cultured the patient’s leukemic cells to determine the molecular response to treatment.
Cells treated with BMS-906024 had greatly reduced levels of mutated NOTCH1 protein. RNA sequencing demonstrated that Notch target genes were sensitive to the treatment.
The MYC oncogene, on the other hand, was not sensitive to BMS-906024.
Epigenetic analysis revealed that the enhancer driving MYC expression in the leukemic cells was not Notch-dependent, but rather BRD4-dependent, suggesting another possible therapeutic option for MYC-expressing tumors. ![]()
Study reveals tumor suppressor in AML
The protein-coding gene hnRNP K acts as a tumor suppressor in acute myeloid leukemia (AML), according to research published in Cancer Cell.
Investigators found that AML patients who carried a partial deletion of chromosome 9 also experienced a significant decrease in hnRNP K expression.
This deletion, 9q21.32, along with the decreased levels of hnRNP K, led to reduced survival and increased tumor formation.
“Our data implicates hnRNP K in the development of blood disorders and suggests it acts as a tumor suppressor,” said Sean Post, PhD, of The University of Texas MD Anderson Cancer Center in Houston.
“Both in vivo and in vitro results indicate that hnRNP K achieves this through regulation of key genetic pathways. Our study found that hnRNP K expression must be maintained for proper cellular regulation and to prevent tumor formation.”
Dr Post and his colleagues examined hnRNP K’s role in tumorigenesis by generating a mouse model harboring an Hnrnpk knockout allele.
They found that Hnrnpk haploinsufficiency resulted in reduced survival, increased tumor formation, genomic instability, and the development of transplantable hematopoietic neoplasms with myeloproliferation.
“Our findings showed that Hnrnpk haploinsufficiency led to tumor development by deregulating cell proliferation and differentiation programs through control of key cellular pathways, which suggests these pathways may be exploited by targeted therapies,” Dr Post said.
Specifically, he and his colleagues found that reduced hnRNP K expression attenuated p21 activation, downregulated C/EBP levels, and activated STAT3 signaling.
The investigators also analyzed samples from AML patients who harbored 9q21.32 and found a significant decrease in hnRNP K expression.
“It was clear that these changes in AML patients with the 9q21.32 deletion resulted in a tumor suppressor gene involved in blood cancer development,” Dr Post said. ![]()
The protein-coding gene hnRNP K acts as a tumor suppressor in acute myeloid leukemia (AML), according to research published in Cancer Cell.
Investigators found that AML patients who carried a partial deletion of chromosome 9 also experienced a significant decrease in hnRNP K expression.
This deletion, 9q21.32, along with the decreased levels of hnRNP K, led to reduced survival and increased tumor formation.
“Our data implicates hnRNP K in the development of blood disorders and suggests it acts as a tumor suppressor,” said Sean Post, PhD, of The University of Texas MD Anderson Cancer Center in Houston.
“Both in vivo and in vitro results indicate that hnRNP K achieves this through regulation of key genetic pathways. Our study found that hnRNP K expression must be maintained for proper cellular regulation and to prevent tumor formation.”
Dr Post and his colleagues examined hnRNP K’s role in tumorigenesis by generating a mouse model harboring an Hnrnpk knockout allele.
They found that Hnrnpk haploinsufficiency resulted in reduced survival, increased tumor formation, genomic instability, and the development of transplantable hematopoietic neoplasms with myeloproliferation.
“Our findings showed that Hnrnpk haploinsufficiency led to tumor development by deregulating cell proliferation and differentiation programs through control of key cellular pathways, which suggests these pathways may be exploited by targeted therapies,” Dr Post said.
Specifically, he and his colleagues found that reduced hnRNP K expression attenuated p21 activation, downregulated C/EBP levels, and activated STAT3 signaling.
The investigators also analyzed samples from AML patients who harbored 9q21.32 and found a significant decrease in hnRNP K expression.
“It was clear that these changes in AML patients with the 9q21.32 deletion resulted in a tumor suppressor gene involved in blood cancer development,” Dr Post said. ![]()
The protein-coding gene hnRNP K acts as a tumor suppressor in acute myeloid leukemia (AML), according to research published in Cancer Cell.
Investigators found that AML patients who carried a partial deletion of chromosome 9 also experienced a significant decrease in hnRNP K expression.
This deletion, 9q21.32, along with the decreased levels of hnRNP K, led to reduced survival and increased tumor formation.
“Our data implicates hnRNP K in the development of blood disorders and suggests it acts as a tumor suppressor,” said Sean Post, PhD, of The University of Texas MD Anderson Cancer Center in Houston.
“Both in vivo and in vitro results indicate that hnRNP K achieves this through regulation of key genetic pathways. Our study found that hnRNP K expression must be maintained for proper cellular regulation and to prevent tumor formation.”
Dr Post and his colleagues examined hnRNP K’s role in tumorigenesis by generating a mouse model harboring an Hnrnpk knockout allele.
They found that Hnrnpk haploinsufficiency resulted in reduced survival, increased tumor formation, genomic instability, and the development of transplantable hematopoietic neoplasms with myeloproliferation.
“Our findings showed that Hnrnpk haploinsufficiency led to tumor development by deregulating cell proliferation and differentiation programs through control of key cellular pathways, which suggests these pathways may be exploited by targeted therapies,” Dr Post said.
Specifically, he and his colleagues found that reduced hnRNP K expression attenuated p21 activation, downregulated C/EBP levels, and activated STAT3 signaling.
The investigators also analyzed samples from AML patients who harbored 9q21.32 and found a significant decrease in hnRNP K expression.
“It was clear that these changes in AML patients with the 9q21.32 deletion resulted in a tumor suppressor gene involved in blood cancer development,” Dr Post said. ![]()
MHM: Novel agents, combos show promise for relapsed/refractory CLL
CHICAGO – Consider using novel agents in chronic lymphocytic leukemia (CLL) patients who are refractory to treatment or who relapse within 2 years of first-line therapy, Dr. John G. Gribben said.

Ibrutinib or idelalisib/rituximab, or other novel agent combinations within clinical trials are his choice in these patients, thus fitness for therapy becomes irrelevant, he said at the American Society of Hematology Meeting on Hematologic Malignancies.
“Of course, I’m particularly excited by the results in CLL of ABT-199. We saw at [the International Workshop on CLL] last week that there are increasing numbers of patients who are on this therapy in combination with anti-CD20 monoclonal antibodies who are achieving minimal residual disease (MRD) eradication and are able to stop that therapy, unlike what we’ve been seeing,” he said. “I have a patient at my own center now who was treated with ABT-199 plus obinutuzumab within a clinical trial – a 17p deletion patient refractory to seven previous lines of therapy. Had a donor, I was trying to get him to transplant, and now he’s MRD-negative and off therapy, having had 6 months of therapy with ABT-199 and obinutuzumab. [This is] a very exciting combination.”
Other novel-novel agent combinations are also being looked at, he noted.
In CLL patients without 17p deletions who progress later than 2 years after initial chemotherapy, novel agents can be considered, as could alternative approaches with chemotherapy.
“But of course, in the setting of p53 deletions or mutations, then ibrutinib or idelalisib/rituximab, or novel agents like ABT-199 within clinical trials become the treatment approach to be thinking about,” he said.
CHICAGO – Consider using novel agents in chronic lymphocytic leukemia (CLL) patients who are refractory to treatment or who relapse within 2 years of first-line therapy, Dr. John G. Gribben said.
Ibrutinib or idelalisib/rituximab, or other novel agent combinations within clinical trials are his choice in these patients, thus fitness for therapy becomes irrelevant, he said at the American Society of Hematology Meeting on Hematologic Malignancies.
“Of course, I’m particularly excited by the results in CLL of ABT-199. We saw at [the International Workshop on CLL] last week that there are increasing numbers of patients who are on this therapy in combination with anti-CD20 monoclonal antibodies who are achieving minimal residual disease (MRD) eradication and are able to stop that therapy, unlike what we’ve been seeing,” he said. “I have a patient at my own center now who was treated with ABT-199 plus obinutuzumab within a clinical trial – a 17p deletion patient refractory to seven previous lines of therapy. Had a donor, I was trying to get him to transplant, and now he’s MRD-negative and off therapy, having had 6 months of therapy with ABT-199 and obinutuzumab. [This is] a very exciting combination.”
Other novel-novel agent combinations are also being looked at, he noted.
In CLL patients without 17p deletions who progress later than 2 years after initial chemotherapy, novel agents can be considered, as could alternative approaches with chemotherapy.
“But of course, in the setting of p53 deletions or mutations, then ibrutinib or idelalisib/rituximab, or novel agents like ABT-199 within clinical trials become the treatment approach to be thinking about,” he said.
CHICAGO – Consider using novel agents in chronic lymphocytic leukemia (CLL) patients who are refractory to treatment or who relapse within 2 years of first-line therapy, Dr. John G. Gribben said.
Ibrutinib or idelalisib/rituximab, or other novel agent combinations within clinical trials are his choice in these patients, thus fitness for therapy becomes irrelevant, he said at the American Society of Hematology Meeting on Hematologic Malignancies.
“Of course, I’m particularly excited by the results in CLL of ABT-199. We saw at [the International Workshop on CLL] last week that there are increasing numbers of patients who are on this therapy in combination with anti-CD20 monoclonal antibodies who are achieving minimal residual disease (MRD) eradication and are able to stop that therapy, unlike what we’ve been seeing,” he said. “I have a patient at my own center now who was treated with ABT-199 plus obinutuzumab within a clinical trial – a 17p deletion patient refractory to seven previous lines of therapy. Had a donor, I was trying to get him to transplant, and now he’s MRD-negative and off therapy, having had 6 months of therapy with ABT-199 and obinutuzumab. [This is] a very exciting combination.”
Other novel-novel agent combinations are also being looked at, he noted.
In CLL patients without 17p deletions who progress later than 2 years after initial chemotherapy, novel agents can be considered, as could alternative approaches with chemotherapy.
“But of course, in the setting of p53 deletions or mutations, then ibrutinib or idelalisib/rituximab, or novel agents like ABT-199 within clinical trials become the treatment approach to be thinking about,” he said.
EXPERT ANALYSIS FROM MHM 2015

MHM: Novel agents, combos show promise for relapsed/refractory CLL
CHICAGO – Consider using novel agents in chronic lymphocytic leukemia (CLL) patients who are refractory to treatment or who relapse within 2 years of first-line therapy, Dr. John G. Gribben said.
Ibrutinib or idelalisib/rituximab, or other novel agent combinations within clinical trials are his choice in these patients, thus fitness for therapy becomes irrelevant, he said at the American Society of Hematology Meeting on Hematologic Malignancies.
“Of course, I’m particularly excited by the results in CLL of ABT-199. We saw at [the International Workshop on CLL] last week that there are increasing numbers of patients who are on this therapy in combination with anti-CD20 monoclonal antibodies who are achieving minimal residual disease (MRD) eradication and are able to stop that therapy, unlike what we’ve been seeing,” he said. “I have a patient at my own center now who was treated with ABT-199 plus obinutuzumab within a clinical trial – a 17p deletion patient refractory to seven previous lines of therapy. Had a donor, I was trying to get him to transplant, and now he’s MRD-negative and off therapy, having had 6 months of therapy with ABT-199 and obinutuzumab. [This is] a very exciting combination.”
Other novel-novel agent combinations are also being looked at, he noted.
In CLL patients without 17p deletions who progress later than 2 years after initial chemotherapy, novel agents can be considered, as could alternative approaches with chemotherapy.
“But of course, in the setting of p53 deletions or mutations, then ibrutinib or idelalisib/rituximab, or novel agents like ABT-199 within clinical trials become the treatment approach to be thinking about,” he said.
CHICAGO – Consider using novel agents in chronic lymphocytic leukemia (CLL) patients who are refractory to treatment or who relapse within 2 years of first-line therapy, Dr. John G. Gribben said.
Ibrutinib or idelalisib/rituximab, or other novel agent combinations within clinical trials are his choice in these patients, thus fitness for therapy becomes irrelevant, he said at the American Society of Hematology Meeting on Hematologic Malignancies.
“Of course, I’m particularly excited by the results in CLL of ABT-199. We saw at [the International Workshop on CLL] last week that there are increasing numbers of patients who are on this therapy in combination with anti-CD20 monoclonal antibodies who are achieving minimal residual disease (MRD) eradication and are able to stop that therapy, unlike what we’ve been seeing,” he said. “I have a patient at my own center now who was treated with ABT-199 plus obinutuzumab within a clinical trial – a 17p deletion patient refractory to seven previous lines of therapy. Had a donor, I was trying to get him to transplant, and now he’s MRD-negative and off therapy, having had 6 months of therapy with ABT-199 and obinutuzumab. [This is] a very exciting combination.”
Other novel-novel agent combinations are also being looked at, he noted.
In CLL patients without 17p deletions who progress later than 2 years after initial chemotherapy, novel agents can be considered, as could alternative approaches with chemotherapy.
“But of course, in the setting of p53 deletions or mutations, then ibrutinib or idelalisib/rituximab, or novel agents like ABT-199 within clinical trials become the treatment approach to be thinking about,” he said.
CHICAGO – Consider using novel agents in chronic lymphocytic leukemia (CLL) patients who are refractory to treatment or who relapse within 2 years of first-line therapy, Dr. John G. Gribben said.
Ibrutinib or idelalisib/rituximab, or other novel agent combinations within clinical trials are his choice in these patients, thus fitness for therapy becomes irrelevant, he said at the American Society of Hematology Meeting on Hematologic Malignancies.
“Of course, I’m particularly excited by the results in CLL of ABT-199. We saw at [the International Workshop on CLL] last week that there are increasing numbers of patients who are on this therapy in combination with anti-CD20 monoclonal antibodies who are achieving minimal residual disease (MRD) eradication and are able to stop that therapy, unlike what we’ve been seeing,” he said. “I have a patient at my own center now who was treated with ABT-199 plus obinutuzumab within a clinical trial – a 17p deletion patient refractory to seven previous lines of therapy. Had a donor, I was trying to get him to transplant, and now he’s MRD-negative and off therapy, having had 6 months of therapy with ABT-199 and obinutuzumab. [This is] a very exciting combination.”
Other novel-novel agent combinations are also being looked at, he noted.
In CLL patients without 17p deletions who progress later than 2 years after initial chemotherapy, novel agents can be considered, as could alternative approaches with chemotherapy.
“But of course, in the setting of p53 deletions or mutations, then ibrutinib or idelalisib/rituximab, or novel agents like ABT-199 within clinical trials become the treatment approach to be thinking about,” he said.
EXPERT ANALYSIS FROM MHM 2015

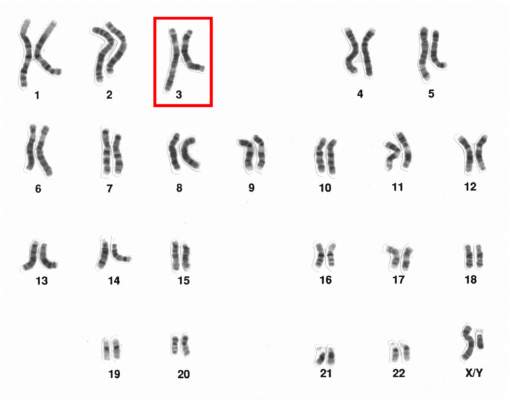
Chromosome 3 abnormalities linked to poor CML outcomes
Chromosome 3 abnormalities, specifically 3q26.2 rearrangements, were associated with treatment resistance and poor prognosis in patients with chronic myelogenous leukemia (CML), report Dr. Wei Wang and coauthors of the department of hematopathology at the University of Texas MD Anderson Cancer Center in Houston.
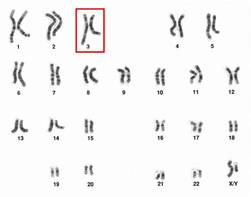
A study of 2,013 CML patients found that just 6% of those with 3q26.2 abnormalities achieved complete cytogenetic response during the course of tyrosine kinase inhibitor (TKI) treatment. Patients with other chromosome 3 abnormalities had a significantly better response rate of 42% the investigators found.
Additionally, patients with 3q26.2 chromosome rearrangements had significantly worse survival rates than those with abnormalities involving other chromosomes, with 2-year overall survival rates of 22% and 60%, respectively.
The lack of response to TKI treatment “raises the issue of how to manage these patients,” Dr. Wang and associates said in the report.
“TKIs themselves are not sufficient to control the disease with 3q26.2 abnormalities,” they added. “Intensive therapy, stem cell transplantation, or investigational therapy targeted to EVI1 should be considered,” concluded the authors, who declared that they had no competing financial interests.
Read the full article in Blood.
Chromosome 3 abnormalities, specifically 3q26.2 rearrangements, were associated with treatment resistance and poor prognosis in patients with chronic myelogenous leukemia (CML), report Dr. Wei Wang and coauthors of the department of hematopathology at the University of Texas MD Anderson Cancer Center in Houston.
A study of 2,013 CML patients found that just 6% of those with 3q26.2 abnormalities achieved complete cytogenetic response during the course of tyrosine kinase inhibitor (TKI) treatment. Patients with other chromosome 3 abnormalities had a significantly better response rate of 42% the investigators found.
Additionally, patients with 3q26.2 chromosome rearrangements had significantly worse survival rates than those with abnormalities involving other chromosomes, with 2-year overall survival rates of 22% and 60%, respectively.
The lack of response to TKI treatment “raises the issue of how to manage these patients,” Dr. Wang and associates said in the report.
“TKIs themselves are not sufficient to control the disease with 3q26.2 abnormalities,” they added. “Intensive therapy, stem cell transplantation, or investigational therapy targeted to EVI1 should be considered,” concluded the authors, who declared that they had no competing financial interests.
Read the full article in Blood.
Chromosome 3 abnormalities, specifically 3q26.2 rearrangements, were associated with treatment resistance and poor prognosis in patients with chronic myelogenous leukemia (CML), report Dr. Wei Wang and coauthors of the department of hematopathology at the University of Texas MD Anderson Cancer Center in Houston.
A study of 2,013 CML patients found that just 6% of those with 3q26.2 abnormalities achieved complete cytogenetic response during the course of tyrosine kinase inhibitor (TKI) treatment. Patients with other chromosome 3 abnormalities had a significantly better response rate of 42% the investigators found.
Additionally, patients with 3q26.2 chromosome rearrangements had significantly worse survival rates than those with abnormalities involving other chromosomes, with 2-year overall survival rates of 22% and 60%, respectively.
The lack of response to TKI treatment “raises the issue of how to manage these patients,” Dr. Wang and associates said in the report.
“TKIs themselves are not sufficient to control the disease with 3q26.2 abnormalities,” they added. “Intensive therapy, stem cell transplantation, or investigational therapy targeted to EVI1 should be considered,” concluded the authors, who declared that they had no competing financial interests.
Read the full article in Blood.
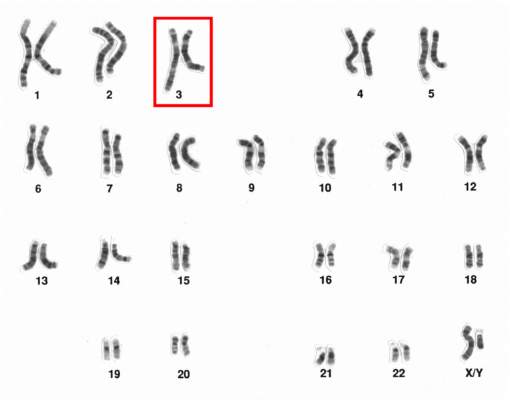
Chromosome 3 abnormalities linked to poor CML outcomes
Chromosome 3 abnormalities, specifically 3q26.2 rearrangements, were associated with treatment resistance and poor prognosis in patients with chronic myelogenous leukemia (CML), report Dr. Wei Wang and coauthors of the department of hematopathology at the University of Texas MD Anderson Cancer Center in Houston.
A study of 2,013 CML patients found that just 6% of those with 3q26.2 abnormalities achieved complete cytogenetic response during the course of tyrosine kinase inhibitor (TKI) treatment. Patients with other chromosome 3 abnormalities had a significantly better response rate of 42% the investigators found.
Additionally, patients with 3q26.2 chromosome rearrangements had significantly worse survival rates than those with abnormalities involving other chromosomes, with 2-year overall survival rates of 22% and 60%, respectively.
The lack of response to TKI treatment “raises the issue of how to manage these patients,” Dr. Wang and associates said in the report.
“TKIs themselves are not sufficient to control the disease with 3q26.2 abnormalities,” they added. “Intensive therapy, stem cell transplantation, or investigational therapy targeted to EVI1 should be considered,” concluded the authors, who declared that they had no competing financial interests.
Read the full article in Blood.
Chromosome 3 abnormalities, specifically 3q26.2 rearrangements, were associated with treatment resistance and poor prognosis in patients with chronic myelogenous leukemia (CML), report Dr. Wei Wang and coauthors of the department of hematopathology at the University of Texas MD Anderson Cancer Center in Houston.
A study of 2,013 CML patients found that just 6% of those with 3q26.2 abnormalities achieved complete cytogenetic response during the course of tyrosine kinase inhibitor (TKI) treatment. Patients with other chromosome 3 abnormalities had a significantly better response rate of 42% the investigators found.
Additionally, patients with 3q26.2 chromosome rearrangements had significantly worse survival rates than those with abnormalities involving other chromosomes, with 2-year overall survival rates of 22% and 60%, respectively.
The lack of response to TKI treatment “raises the issue of how to manage these patients,” Dr. Wang and associates said in the report.
“TKIs themselves are not sufficient to control the disease with 3q26.2 abnormalities,” they added. “Intensive therapy, stem cell transplantation, or investigational therapy targeted to EVI1 should be considered,” concluded the authors, who declared that they had no competing financial interests.
Read the full article in Blood.
Chromosome 3 abnormalities, specifically 3q26.2 rearrangements, were associated with treatment resistance and poor prognosis in patients with chronic myelogenous leukemia (CML), report Dr. Wei Wang and coauthors of the department of hematopathology at the University of Texas MD Anderson Cancer Center in Houston.
A study of 2,013 CML patients found that just 6% of those with 3q26.2 abnormalities achieved complete cytogenetic response during the course of tyrosine kinase inhibitor (TKI) treatment. Patients with other chromosome 3 abnormalities had a significantly better response rate of 42% the investigators found.
Additionally, patients with 3q26.2 chromosome rearrangements had significantly worse survival rates than those with abnormalities involving other chromosomes, with 2-year overall survival rates of 22% and 60%, respectively.
The lack of response to TKI treatment “raises the issue of how to manage these patients,” Dr. Wang and associates said in the report.
“TKIs themselves are not sufficient to control the disease with 3q26.2 abnormalities,” they added. “Intensive therapy, stem cell transplantation, or investigational therapy targeted to EVI1 should be considered,” concluded the authors, who declared that they had no competing financial interests.
Read the full article in Blood.
New chimeric CD19 antibody may reduce MRD in ALL

NEW YORK—Researchers have developed a pharmaceutical-grade, third-generation, CD19-specific antibody that reduced minimal residual disease (MRD) in pediatric patients with B-cell precursor acute lymphoblastic leukemia (BCP-ALL).
This chimerized, Fc-optimized antibody—4G7SDIE—was used on a compassionate-need basis in 14 patients with relapsed or refractory BCP-ALL. Nine of the patients had prior stem cell transplants.
Ursula JE Seidel, a PhD candidate at University Children’s Hospital Tubingen in Germany, discussed early results with the new antibody (poster B144) during the inaugural CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference.
Patients received 4G7SDIE infusions ranging from 5 mg/m2 to 50 mg/m2 twice a week for a year or longer.
They rarely experienced fever, nausea, or headache, according to the investigators, and all had B-cell depletion.
“The good thing about this antibody is it has a very low toxicity profile,” Seidel noted.
Upon discontinuation of therapy, B-cell counts recovered rapidly to normal levels.
The researchers followed the patients for a median of 543 days after transplant (range, 208–1137) and a median of 720 days after administration of 4G7SDIE (range, 264–1115).
Nine of the 14 patients had a reduction in MRD by 1 log or more, 2 of whom were receiving additional therapy with tyrosine kinase inhibitors.
Five patients had a reduction in MRD below the quantifiable level, and 2 patients became MRD-negative.
Six patients relapsed, and 5 of them died from relapsed disease. Two patients died of sepsis or chemotoxicity while in complete molecular remission. And 6 patients remain in complete molecular remission.
Functional characterization of 4G7SDIE
Through analysis of cells from healthy volunteers and BCP-ALL blasts of untreated and treated patients, the researchers determined that 4G7SDIE mediates enhanced antibody‑dependent cellular cytotoxicity through its improved capability to recruit FcγRIIIa-bearing effector cells.
They identified natural killer cells and γδ T cells as the main effector cells. And they determined that the FcγRIIIa-V158F polymorphism did not influence the effect of 4G7SDIE-mediated antibody‑dependent cellular cytotoxicity.
The researchers believe that the promising anti-leukemic effects of 4G7SDIE both in vitro and in vivo call for additional exploration. They are currently planning a phase 1/2 study to further assess the therapeutic activity of 4G7SDIE. ![]()
NEW YORK—Researchers have developed a pharmaceutical-grade, third-generation, CD19-specific antibody that reduced minimal residual disease (MRD) in pediatric patients with B-cell precursor acute lymphoblastic leukemia (BCP-ALL).
This chimerized, Fc-optimized antibody—4G7SDIE—was used on a compassionate-need basis in 14 patients with relapsed or refractory BCP-ALL. Nine of the patients had prior stem cell transplants.
Ursula JE Seidel, a PhD candidate at University Children’s Hospital Tubingen in Germany, discussed early results with the new antibody (poster B144) during the inaugural CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference.
Patients received 4G7SDIE infusions ranging from 5 mg/m2 to 50 mg/m2 twice a week for a year or longer.
They rarely experienced fever, nausea, or headache, according to the investigators, and all had B-cell depletion.
“The good thing about this antibody is it has a very low toxicity profile,” Seidel noted.
Upon discontinuation of therapy, B-cell counts recovered rapidly to normal levels.
The researchers followed the patients for a median of 543 days after transplant (range, 208–1137) and a median of 720 days after administration of 4G7SDIE (range, 264–1115).
Nine of the 14 patients had a reduction in MRD by 1 log or more, 2 of whom were receiving additional therapy with tyrosine kinase inhibitors.
Five patients had a reduction in MRD below the quantifiable level, and 2 patients became MRD-negative.
Six patients relapsed, and 5 of them died from relapsed disease. Two patients died of sepsis or chemotoxicity while in complete molecular remission. And 6 patients remain in complete molecular remission.
Functional characterization of 4G7SDIE
Through analysis of cells from healthy volunteers and BCP-ALL blasts of untreated and treated patients, the researchers determined that 4G7SDIE mediates enhanced antibody‑dependent cellular cytotoxicity through its improved capability to recruit FcγRIIIa-bearing effector cells.
They identified natural killer cells and γδ T cells as the main effector cells. And they determined that the FcγRIIIa-V158F polymorphism did not influence the effect of 4G7SDIE-mediated antibody‑dependent cellular cytotoxicity.
The researchers believe that the promising anti-leukemic effects of 4G7SDIE both in vitro and in vivo call for additional exploration. They are currently planning a phase 1/2 study to further assess the therapeutic activity of 4G7SDIE. ![]()
NEW YORK—Researchers have developed a pharmaceutical-grade, third-generation, CD19-specific antibody that reduced minimal residual disease (MRD) in pediatric patients with B-cell precursor acute lymphoblastic leukemia (BCP-ALL).
This chimerized, Fc-optimized antibody—4G7SDIE—was used on a compassionate-need basis in 14 patients with relapsed or refractory BCP-ALL. Nine of the patients had prior stem cell transplants.
Ursula JE Seidel, a PhD candidate at University Children’s Hospital Tubingen in Germany, discussed early results with the new antibody (poster B144) during the inaugural CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference.
Patients received 4G7SDIE infusions ranging from 5 mg/m2 to 50 mg/m2 twice a week for a year or longer.
They rarely experienced fever, nausea, or headache, according to the investigators, and all had B-cell depletion.
“The good thing about this antibody is it has a very low toxicity profile,” Seidel noted.
Upon discontinuation of therapy, B-cell counts recovered rapidly to normal levels.
The researchers followed the patients for a median of 543 days after transplant (range, 208–1137) and a median of 720 days after administration of 4G7SDIE (range, 264–1115).
Nine of the 14 patients had a reduction in MRD by 1 log or more, 2 of whom were receiving additional therapy with tyrosine kinase inhibitors.
Five patients had a reduction in MRD below the quantifiable level, and 2 patients became MRD-negative.
Six patients relapsed, and 5 of them died from relapsed disease. Two patients died of sepsis or chemotoxicity while in complete molecular remission. And 6 patients remain in complete molecular remission.
Functional characterization of 4G7SDIE
Through analysis of cells from healthy volunteers and BCP-ALL blasts of untreated and treated patients, the researchers determined that 4G7SDIE mediates enhanced antibody‑dependent cellular cytotoxicity through its improved capability to recruit FcγRIIIa-bearing effector cells.
They identified natural killer cells and γδ T cells as the main effector cells. And they determined that the FcγRIIIa-V158F polymorphism did not influence the effect of 4G7SDIE-mediated antibody‑dependent cellular cytotoxicity.
The researchers believe that the promising anti-leukemic effects of 4G7SDIE both in vitro and in vivo call for additional exploration. They are currently planning a phase 1/2 study to further assess the therapeutic activity of 4G7SDIE. ![]()