User login
For overweight children, inject epinephrine in lower thigh
SAN ANTONIO – Overweight and obese children in need of epinephrine for anaphylaxis should be injected in the calf or in the lower thigh, rather than upper half of the thigh, to ensure intramuscular administration, according to findings from an ultrasound study of 93 children.
Ultrasound measurement demonstrated that the distance from skin surface to muscle depth was greater than auto-injector needle length at one quarter of the distance down the thigh in 82% of obese children vs. 25% of nonobese children. At three-quarters of the way down the thigh, this was the case in only 17% of obese children and 2% of nonobese children, Dr. Peter Arkwright reported at the annual meeting of the American Academy of Allergy, Asthma, and Immunology.
At a point midway down the calf, the skin surface to muscle depth was less than the length of the auto-injector needle in all of the children, said Dr. Arkwright of the University of Manchester (England).
Intramuscular injection, rather than subcutaneous injection, is imperative for effective delivery of epinephrine, he said, noting that this study was undertaken because of growing concerns that increasing obesity among children could make existing auto-injectors inadequate for providing intramuscular delivery in a significant proportion of patients.
Children included in the study were patients from regional pediatric allergy clinics. All were measured for height, weight, and body mass index, and all underwent ultrasound measurement at set distances down the thigh and leg. Higher weight, BMI, and waist circumference – but not age or gender – were associated with skin surface to muscle depth greater than auto-injector needle length, he noted.
"Based on our study, injecting epinephrine into the lower rather than upper thigh would be advised in overweight or obese children," he said, adding that caregivers of children at risk of anaphylaxis should be advised about the importance of administering epinephrine into the muscle in the most effective way.
For overweight and obese children, this involves injecting into the lower half of the thigh, and for very obese children it involves injecting at the middle of the calf, he said.
Dr. Arkwright reported having no disclosures.
SAN ANTONIO – Overweight and obese children in need of epinephrine for anaphylaxis should be injected in the calf or in the lower thigh, rather than upper half of the thigh, to ensure intramuscular administration, according to findings from an ultrasound study of 93 children.
Ultrasound measurement demonstrated that the distance from skin surface to muscle depth was greater than auto-injector needle length at one quarter of the distance down the thigh in 82% of obese children vs. 25% of nonobese children. At three-quarters of the way down the thigh, this was the case in only 17% of obese children and 2% of nonobese children, Dr. Peter Arkwright reported at the annual meeting of the American Academy of Allergy, Asthma, and Immunology.
At a point midway down the calf, the skin surface to muscle depth was less than the length of the auto-injector needle in all of the children, said Dr. Arkwright of the University of Manchester (England).
Intramuscular injection, rather than subcutaneous injection, is imperative for effective delivery of epinephrine, he said, noting that this study was undertaken because of growing concerns that increasing obesity among children could make existing auto-injectors inadequate for providing intramuscular delivery in a significant proportion of patients.
Children included in the study were patients from regional pediatric allergy clinics. All were measured for height, weight, and body mass index, and all underwent ultrasound measurement at set distances down the thigh and leg. Higher weight, BMI, and waist circumference – but not age or gender – were associated with skin surface to muscle depth greater than auto-injector needle length, he noted.
"Based on our study, injecting epinephrine into the lower rather than upper thigh would be advised in overweight or obese children," he said, adding that caregivers of children at risk of anaphylaxis should be advised about the importance of administering epinephrine into the muscle in the most effective way.
For overweight and obese children, this involves injecting into the lower half of the thigh, and for very obese children it involves injecting at the middle of the calf, he said.
Dr. Arkwright reported having no disclosures.
SAN ANTONIO – Overweight and obese children in need of epinephrine for anaphylaxis should be injected in the calf or in the lower thigh, rather than upper half of the thigh, to ensure intramuscular administration, according to findings from an ultrasound study of 93 children.
Ultrasound measurement demonstrated that the distance from skin surface to muscle depth was greater than auto-injector needle length at one quarter of the distance down the thigh in 82% of obese children vs. 25% of nonobese children. At three-quarters of the way down the thigh, this was the case in only 17% of obese children and 2% of nonobese children, Dr. Peter Arkwright reported at the annual meeting of the American Academy of Allergy, Asthma, and Immunology.
At a point midway down the calf, the skin surface to muscle depth was less than the length of the auto-injector needle in all of the children, said Dr. Arkwright of the University of Manchester (England).
Intramuscular injection, rather than subcutaneous injection, is imperative for effective delivery of epinephrine, he said, noting that this study was undertaken because of growing concerns that increasing obesity among children could make existing auto-injectors inadequate for providing intramuscular delivery in a significant proportion of patients.
Children included in the study were patients from regional pediatric allergy clinics. All were measured for height, weight, and body mass index, and all underwent ultrasound measurement at set distances down the thigh and leg. Higher weight, BMI, and waist circumference – but not age or gender – were associated with skin surface to muscle depth greater than auto-injector needle length, he noted.
"Based on our study, injecting epinephrine into the lower rather than upper thigh would be advised in overweight or obese children," he said, adding that caregivers of children at risk of anaphylaxis should be advised about the importance of administering epinephrine into the muscle in the most effective way.
For overweight and obese children, this involves injecting into the lower half of the thigh, and for very obese children it involves injecting at the middle of the calf, he said.
Dr. Arkwright reported having no disclosures.
AT THE AAAAI ANNUAL MEETING
EBSOS implementation improves asthma guideline compliance
SAN ANTONIO – More of the children who present to the pediatric emergency department with asthma exacerbation received recommended care when the staff had instituted a nurse-initiated, evidence-based, standardized order set, according to Dr. Moira E. Breslin.
Specifically, the percentage of patients receiving at least one dose of ipratropium bromide improved from 55.4% before implementation of the order set to 90.9% after implementation. Compliance with the recommendation of the National Asthma Guidelines that patients receive three consecutive nebulized treatments of ipratropium bromide increased from 13.5% to 40.9%, Dr. Breslin of Duke University Medical Center, Durham, N.C., reported in a poster at the annual meeting of the American Academy of Allergy, Asthma, and Immunology.
The median time to delivery of rescue medication also improved from 21 minutes to 14 minutes for first inhaled bronchodilator administration, and from 41 minutes to 19 minutes for delivery of systemic corticosteroids.
All differences were statistically significant.
The findings are based on a review of charts for 193 patients treated in the pediatric emergency department for status asthmaticus before implementation of the evidence-based standardized order set, or EBSOS, (between Feb. 23, 2009, and Feb. 22, 2012), and for 22 patients treated after implementation (between Feb. 23, 2012, and July 31, 2012).
The EBSOS for the treatment of pediatric asthma used in this study was developed and incorporated into the emergency department electronic ordering system because personnel were not consistently following national asthma treatment guidelines, according to a separate 2010 emergency department records review.
That review showed that 24% of patients admitted for status asthmaticus had not received the recommended ipratropium bromide treatment, and that only 14% of those who did receive ipratropium bromide received the recommended three consecutive doses.
Implementation of the EBSOS involved the use of an algorithm based on a validated Modified Pulmonary Index Score that allowed for triage nurse initiation of the EBSOS. The EBSOS called for continuous pulse oximetry, supplemental oxygen as needed, evaluation by a respiratory therapist, nebulized albuterol administration at 5 mg every 20 minutes for three treatments, administration of nebulized ipratropium bromide at 0.5 mg every 20 minutes for three treatments, and administration of one dose of oral prednisolone at 2 mg/kg up to a maximum of 60 mg.
"Implementation of an EBSOS improved compliance to national asthma guidelines, as evidenced by a higher proportion of pediatric emergency department patients in status asthmaticus receiving ipratropium bromide, as well as shortened time to delivery of inhaled bronchodilators and systemic steroids," Dr. Breslin concluded, noting that future analysis of this review will focus on patient-centered outcomes.
Dr. Breslin reported having no relevant financial disclosures
SAN ANTONIO – More of the children who present to the pediatric emergency department with asthma exacerbation received recommended care when the staff had instituted a nurse-initiated, evidence-based, standardized order set, according to Dr. Moira E. Breslin.
Specifically, the percentage of patients receiving at least one dose of ipratropium bromide improved from 55.4% before implementation of the order set to 90.9% after implementation. Compliance with the recommendation of the National Asthma Guidelines that patients receive three consecutive nebulized treatments of ipratropium bromide increased from 13.5% to 40.9%, Dr. Breslin of Duke University Medical Center, Durham, N.C., reported in a poster at the annual meeting of the American Academy of Allergy, Asthma, and Immunology.
The median time to delivery of rescue medication also improved from 21 minutes to 14 minutes for first inhaled bronchodilator administration, and from 41 minutes to 19 minutes for delivery of systemic corticosteroids.
All differences were statistically significant.
The findings are based on a review of charts for 193 patients treated in the pediatric emergency department for status asthmaticus before implementation of the evidence-based standardized order set, or EBSOS, (between Feb. 23, 2009, and Feb. 22, 2012), and for 22 patients treated after implementation (between Feb. 23, 2012, and July 31, 2012).
The EBSOS for the treatment of pediatric asthma used in this study was developed and incorporated into the emergency department electronic ordering system because personnel were not consistently following national asthma treatment guidelines, according to a separate 2010 emergency department records review.
That review showed that 24% of patients admitted for status asthmaticus had not received the recommended ipratropium bromide treatment, and that only 14% of those who did receive ipratropium bromide received the recommended three consecutive doses.
Implementation of the EBSOS involved the use of an algorithm based on a validated Modified Pulmonary Index Score that allowed for triage nurse initiation of the EBSOS. The EBSOS called for continuous pulse oximetry, supplemental oxygen as needed, evaluation by a respiratory therapist, nebulized albuterol administration at 5 mg every 20 minutes for three treatments, administration of nebulized ipratropium bromide at 0.5 mg every 20 minutes for three treatments, and administration of one dose of oral prednisolone at 2 mg/kg up to a maximum of 60 mg.
"Implementation of an EBSOS improved compliance to national asthma guidelines, as evidenced by a higher proportion of pediatric emergency department patients in status asthmaticus receiving ipratropium bromide, as well as shortened time to delivery of inhaled bronchodilators and systemic steroids," Dr. Breslin concluded, noting that future analysis of this review will focus on patient-centered outcomes.
Dr. Breslin reported having no relevant financial disclosures
SAN ANTONIO – More of the children who present to the pediatric emergency department with asthma exacerbation received recommended care when the staff had instituted a nurse-initiated, evidence-based, standardized order set, according to Dr. Moira E. Breslin.
Specifically, the percentage of patients receiving at least one dose of ipratropium bromide improved from 55.4% before implementation of the order set to 90.9% after implementation. Compliance with the recommendation of the National Asthma Guidelines that patients receive three consecutive nebulized treatments of ipratropium bromide increased from 13.5% to 40.9%, Dr. Breslin of Duke University Medical Center, Durham, N.C., reported in a poster at the annual meeting of the American Academy of Allergy, Asthma, and Immunology.
The median time to delivery of rescue medication also improved from 21 minutes to 14 minutes for first inhaled bronchodilator administration, and from 41 minutes to 19 minutes for delivery of systemic corticosteroids.
All differences were statistically significant.
The findings are based on a review of charts for 193 patients treated in the pediatric emergency department for status asthmaticus before implementation of the evidence-based standardized order set, or EBSOS, (between Feb. 23, 2009, and Feb. 22, 2012), and for 22 patients treated after implementation (between Feb. 23, 2012, and July 31, 2012).
The EBSOS for the treatment of pediatric asthma used in this study was developed and incorporated into the emergency department electronic ordering system because personnel were not consistently following national asthma treatment guidelines, according to a separate 2010 emergency department records review.
That review showed that 24% of patients admitted for status asthmaticus had not received the recommended ipratropium bromide treatment, and that only 14% of those who did receive ipratropium bromide received the recommended three consecutive doses.
Implementation of the EBSOS involved the use of an algorithm based on a validated Modified Pulmonary Index Score that allowed for triage nurse initiation of the EBSOS. The EBSOS called for continuous pulse oximetry, supplemental oxygen as needed, evaluation by a respiratory therapist, nebulized albuterol administration at 5 mg every 20 minutes for three treatments, administration of nebulized ipratropium bromide at 0.5 mg every 20 minutes for three treatments, and administration of one dose of oral prednisolone at 2 mg/kg up to a maximum of 60 mg.
"Implementation of an EBSOS improved compliance to national asthma guidelines, as evidenced by a higher proportion of pediatric emergency department patients in status asthmaticus receiving ipratropium bromide, as well as shortened time to delivery of inhaled bronchodilators and systemic steroids," Dr. Breslin concluded, noting that future analysis of this review will focus on patient-centered outcomes.
Dr. Breslin reported having no relevant financial disclosures
AT THE AAAAI ANNUAL MEETING
Food and milk allergies increase growth impairment risk
Earn 0.25 hours AMA PRA Category 1 credit: Read this article, and click the link at the end to take the post-test.
SAN ANTONIO – Dietary restrictions prescribed for children with food allergies may lead to growth impairment, according to findings from a review of medical records for 245 food-allergic pediatric patients.
The risk of growth impairment was greatest for children whose dietary restrictions required elimination of more than two foods and/or elimination of cow’s milk, Dr. Brian P. Vickery reported at the annual meeting of the American Academy of Allergy, Asthma, and Immunology.
After age 2 years, the food-allergic children had lower mean percentiles for weight (67.5 vs. 72.5) and a lower body mass index (57.6 vs.68.0), than did 4,584 healthy age-matched controls.
Furthermore, the 52 patients with more than two food allergies (and thus more than two food restrictions), compared with 193 patients with one or two food allergies, had significantly lower mean percentiles for height (62.2 vs. 74.8) and weight (55.3 vs. 69.2). The 66 patients with milk allergy, compared with those with other food allergies, had lower mean percentiles for weight (54.5 vs. 70.6) and BMI (48.9 vs. 58.8), according to Dr. Vickery of the University of North Carolina at Chapel Hill.
Milk-allergic children younger than 2 years of age were particularly vulnerable to growth restriction, he said during a press briefing at the meeting.
The food-allergic children in this study, who were aged 1 month to 11 years and who presented to a University of North Carolina outpatient clinic between 2007 and 2011, also were compared with 205 "disease controls," consisting of children with either cystic fibrosis or celiac disease, two conditions that are associated with impaired growth. When children passed their second birthday, the effect of food allergy on growth was very similar to the effect of celiac disease on growth, Dr. Vickery said.
The findings of this study confirm those from a smaller study, conducted more than a decade ago, that also showed that milk allergy and multiple food allergies were associated with growth impairment.
That study is "the most commonly cited previous study to address the growth of food-allergic children in the United States," Dr. Vickery noted.
"The prevalence [of food allergy] has increased over the past 10 years, so we wanted to take another look in a bigger population to kind of reassess the impact of elimination diets on growth," he said.
The current findings demonstrate that a food allergy–associated elimination diet can place children at risk of impaired growth, compared with their healthy peers, regardless of whether they are under age 2 years, or are 2-11 years old, and that after age 2, the effect of food allergy on growth is very similar to that of chronic diseases known to affect growth, he said.
"While awareness of food allergy is increasing along with the prevalence of the disease, it is important to draw attention to the important consequences of elimination diets. We feel that providers should counsel patients and caregivers about the growth-related risks of the elimination diets that are used to treat food allergy, and ensure that families are excluding only the foods that are medically required or otherwise culturally indicated, that nutritional assessment and/or supplementation is provided as needed, and that subspecialty consultation is arranged, especially for children at highest risk," he said.
Dr. Vickery reported having no relevant financial disclosures.
To earn 0.25 hours AMA PRA Category 1 credit after reading this article, take the post-test here.
Earn 0.25 hours AMA PRA Category 1 credit: Read this article, and click the link at the end to take the post-test.
SAN ANTONIO – Dietary restrictions prescribed for children with food allergies may lead to growth impairment, according to findings from a review of medical records for 245 food-allergic pediatric patients.
The risk of growth impairment was greatest for children whose dietary restrictions required elimination of more than two foods and/or elimination of cow’s milk, Dr. Brian P. Vickery reported at the annual meeting of the American Academy of Allergy, Asthma, and Immunology.
After age 2 years, the food-allergic children had lower mean percentiles for weight (67.5 vs. 72.5) and a lower body mass index (57.6 vs.68.0), than did 4,584 healthy age-matched controls.
Furthermore, the 52 patients with more than two food allergies (and thus more than two food restrictions), compared with 193 patients with one or two food allergies, had significantly lower mean percentiles for height (62.2 vs. 74.8) and weight (55.3 vs. 69.2). The 66 patients with milk allergy, compared with those with other food allergies, had lower mean percentiles for weight (54.5 vs. 70.6) and BMI (48.9 vs. 58.8), according to Dr. Vickery of the University of North Carolina at Chapel Hill.
Milk-allergic children younger than 2 years of age were particularly vulnerable to growth restriction, he said during a press briefing at the meeting.
The food-allergic children in this study, who were aged 1 month to 11 years and who presented to a University of North Carolina outpatient clinic between 2007 and 2011, also were compared with 205 "disease controls," consisting of children with either cystic fibrosis or celiac disease, two conditions that are associated with impaired growth. When children passed their second birthday, the effect of food allergy on growth was very similar to the effect of celiac disease on growth, Dr. Vickery said.
The findings of this study confirm those from a smaller study, conducted more than a decade ago, that also showed that milk allergy and multiple food allergies were associated with growth impairment.
That study is "the most commonly cited previous study to address the growth of food-allergic children in the United States," Dr. Vickery noted.
"The prevalence [of food allergy] has increased over the past 10 years, so we wanted to take another look in a bigger population to kind of reassess the impact of elimination diets on growth," he said.
The current findings demonstrate that a food allergy–associated elimination diet can place children at risk of impaired growth, compared with their healthy peers, regardless of whether they are under age 2 years, or are 2-11 years old, and that after age 2, the effect of food allergy on growth is very similar to that of chronic diseases known to affect growth, he said.
"While awareness of food allergy is increasing along with the prevalence of the disease, it is important to draw attention to the important consequences of elimination diets. We feel that providers should counsel patients and caregivers about the growth-related risks of the elimination diets that are used to treat food allergy, and ensure that families are excluding only the foods that are medically required or otherwise culturally indicated, that nutritional assessment and/or supplementation is provided as needed, and that subspecialty consultation is arranged, especially for children at highest risk," he said.
Dr. Vickery reported having no relevant financial disclosures.
To earn 0.25 hours AMA PRA Category 1 credit after reading this article, take the post-test here.
Earn 0.25 hours AMA PRA Category 1 credit: Read this article, and click the link at the end to take the post-test.
SAN ANTONIO – Dietary restrictions prescribed for children with food allergies may lead to growth impairment, according to findings from a review of medical records for 245 food-allergic pediatric patients.
The risk of growth impairment was greatest for children whose dietary restrictions required elimination of more than two foods and/or elimination of cow’s milk, Dr. Brian P. Vickery reported at the annual meeting of the American Academy of Allergy, Asthma, and Immunology.
After age 2 years, the food-allergic children had lower mean percentiles for weight (67.5 vs. 72.5) and a lower body mass index (57.6 vs.68.0), than did 4,584 healthy age-matched controls.
Furthermore, the 52 patients with more than two food allergies (and thus more than two food restrictions), compared with 193 patients with one or two food allergies, had significantly lower mean percentiles for height (62.2 vs. 74.8) and weight (55.3 vs. 69.2). The 66 patients with milk allergy, compared with those with other food allergies, had lower mean percentiles for weight (54.5 vs. 70.6) and BMI (48.9 vs. 58.8), according to Dr. Vickery of the University of North Carolina at Chapel Hill.
Milk-allergic children younger than 2 years of age were particularly vulnerable to growth restriction, he said during a press briefing at the meeting.
The food-allergic children in this study, who were aged 1 month to 11 years and who presented to a University of North Carolina outpatient clinic between 2007 and 2011, also were compared with 205 "disease controls," consisting of children with either cystic fibrosis or celiac disease, two conditions that are associated with impaired growth. When children passed their second birthday, the effect of food allergy on growth was very similar to the effect of celiac disease on growth, Dr. Vickery said.
The findings of this study confirm those from a smaller study, conducted more than a decade ago, that also showed that milk allergy and multiple food allergies were associated with growth impairment.
That study is "the most commonly cited previous study to address the growth of food-allergic children in the United States," Dr. Vickery noted.
"The prevalence [of food allergy] has increased over the past 10 years, so we wanted to take another look in a bigger population to kind of reassess the impact of elimination diets on growth," he said.
The current findings demonstrate that a food allergy–associated elimination diet can place children at risk of impaired growth, compared with their healthy peers, regardless of whether they are under age 2 years, or are 2-11 years old, and that after age 2, the effect of food allergy on growth is very similar to that of chronic diseases known to affect growth, he said.
"While awareness of food allergy is increasing along with the prevalence of the disease, it is important to draw attention to the important consequences of elimination diets. We feel that providers should counsel patients and caregivers about the growth-related risks of the elimination diets that are used to treat food allergy, and ensure that families are excluding only the foods that are medically required or otherwise culturally indicated, that nutritional assessment and/or supplementation is provided as needed, and that subspecialty consultation is arranged, especially for children at highest risk," he said.
Dr. Vickery reported having no relevant financial disclosures.
To earn 0.25 hours AMA PRA Category 1 credit after reading this article, take the post-test here.
AT THE AAAAI ANNUAL MEETING

Omalizumab shows efficacy for refractory chronic idiopathic urticaria
SAN ANTONIO – Omalizumab diminished the signs and symptoms of chronic idiopathic urticaria in a dose-dependent fashion in a phase III study of 323 patients who failed to respond adequately to H1-antihistamines.
After 12 weeks of treatment with the recombinant humanized monoclonal antibody, improvements from baseline in weekly itch-severity scores were significantly greater in patients randomized to receive three doses of either 300 mg or 150 mg every 4 weeks, compared with placebo (score change of -9.8 and -8.1 vs. -5.1, respectively). The itch-severity score in patients randomized to receive a 75-mg dose changed by -5.9 points, but this did not differ significantly from the change in the placebo group, according to Dr. Marcus Maurer of Charite-Universitatsmedizin, Berlin. The report was published online on Feb. 24 in the New England Journal of Medicine.
The findings from this international double-blind trial were reported simultaneously at the annual meeting of the American Academy of Allergy, Asthma, and Immunology.
In addition to meeting the primary 12-week response endpoint of change in weekly itch-severity score, patients receiving the 300-mg and 150-mg doses of omalizumab also experienced significant improvements, compared with patients given placebo, on all but one secondary endpoint, including change in the 7-day urticaria activity score (UAS7), change in the score for the weekly number of hives, time until a reduction from baseline of at least 5 points in the weekly itch-severity score (the MID), the proportions of patients with a UAS7 of 6 or less, the number of patients with a weekly MID response in the itch-severity score, the change from baseline in the score for the size of the largest hive, and change from baseline in the overall score on the Dermatology Life Quality Index. Those receiving 300 mg, but not those receiving 150 mg, also experienced significant improvement in the proportion of angioedema-free days from weeks 4-12 (N. Engl. J. Med. 2013 Feb. 24 [doi:10.1056/NEJMoa1215372]).
In a post hoc analysis at 12 weeks, 53%, 23%, 18%, and 10%, of those in the 300-mg, 150-mg, 75-mg, and placebo groups, respectively, were completely free of hives, and 44%, 22%, 16%, and 5%, respectively, were free of both hives and itching.
Of note, many patients experienced extremely rapid improvement, raising questions about a potential, as-yet unidentified and fundamental characteristic of this disease, study coauthors Dr. Thomas B. Casale, professor of medicine and medical microbiology and immunology and chief of allergy/immunology at Creighton University Medical Center, Omaha, Neb., and Dr. Allen P. Kaplan, clinical professor of medicine at the Medical University of South Carolina, Charleston, reported during a press briefing at AAAAI.
They explained that omalizumab, currently approved as an add-on therapy for moderate to severe persistent allergic asthma, is known to bind the allergic antibody immunoglobulin E (IgE). Many patients with chronic urticaria have an antibody that binds to a protein on the surface of histamine-containing cells, and that protein binds IgE.
"Just one injection drops one’s IgE level pretty close to zero ... and we learned that when we drop IgE to rock bottom, the protein on the cell to which it is attached also drops – and that’s the protein that the circulating antibody interacts with. The thinking was that if we drop the surface protein low enough, there’s nothing for the antibody to react with, and the hives would improve," he said.
This was, in fact, the case, as demonstrated in a phase I study of 12 patients, in which 7 patients responded dramatically, 4 responded partially, and 1 had no response. These findings led to the current phase III study, which is one of three such studies that investigators hope will lead to the drug’s approval for chronic idiopathic urticaria, because the high cost of the drug is prohibitive with respect to off-label use.
The same effect was seen in the current study.
This effect, however, takes a couple weeks to become apparent, so the rapid responses that occur in numerous patients suggest there is something more at play.
"This is working even faster than we thought, and probably there is a function of the allergic antibody that we do not yet understand," Dr. Kaplan said. "So this is going to be not only a terrific therapy for the disorder, but will lead to research in the future that probably – we hope – will elucidate the underlying abnormality of chronic urticaria beyond our current understanding of it. It’s very exciting, because it might elucidate some fundamental analogy that we don’t appreciate, and would have implications for allergic disease across the board."
The current study comprised patients aged 12-75 years who had a 6-month or longer history of chronic idiopathic urticaria, the presence of hives associated with itching for at least 8 consecutive weeks at any time prior to enrollment (despite H1-antihistamine use), a UAS7 of 16 or more during a 7-day period, a weekly itch-severity score of at least 8, a score of at least 4 on the UAS on at least one of the screening-visit days, and receipt of a licensed dose of a second-generation H1-antihistamine for at least 3 consecutive days immediately prior to the screening visit. Those with a clearly defined underlying cause for their symptoms were excluded. The doses evaluated in this study were based on a prior dose-ranging study, which showed no additional benefit with doses over 300 mg, the investigators said.
Patients continued to receive stable doses of H1-antihistamines throughout the 12-week treatment period, and were permitted to use diphenhydramine as a rescue medication.
Omalizumab was well tolerated in this study, with a similar number of adverse events occurring across the treatment groups. Serious adverse events occurred more often in the 300-mg group, with 6% of patients in that group experiencing a serious adverse event, compared with 3% of patients in the placebo group, and 1% of patients in the 150-mg and 75-mg groups, but the events were not considered to be related to the study drug.
The duration of response, however, was limited, as noted during a 16-week observation period following the initial 12-week treatment period.
Although the weekly itch-severity scores did not return to baseline during follow-up, they did increase to the levels seen in the placebo group.
The findings are nonetheless encouraging, Dr. Kaplan said, noting that this disease, which can have dramatic adverse effects on quality of life, is not uncommon, and can be difficult to treat, with about half of all patients failing to respond to standard therapy with high dose antihistamines. Alternate treatments used to treat the disease, including steroids and cyclosporine, can be effective, but can be highly toxic, he said.
"For refractory patients we have nothing that matches (omalizumab’s) combination of this kind of efficacy with low side effects, so many of us in the field kind of view this as a game changer for the patients," he said.
This study was sponsored by Genentech and Novartis Pharma. Several study authors made disclosures. A complete list of these disclosures is available with the full text of the article at NEJM.org.
recombinant humanized monoclonal antibody, itch-severity scores, Dr. Marcus Maurer, New England Journal of Medicine, American Academy of Allergy, Asthma, and Immunology, 7-day urticaria activity score,
SAN ANTONIO – Omalizumab diminished the signs and symptoms of chronic idiopathic urticaria in a dose-dependent fashion in a phase III study of 323 patients who failed to respond adequately to H1-antihistamines.
After 12 weeks of treatment with the recombinant humanized monoclonal antibody, improvements from baseline in weekly itch-severity scores were significantly greater in patients randomized to receive three doses of either 300 mg or 150 mg every 4 weeks, compared with placebo (score change of -9.8 and -8.1 vs. -5.1, respectively). The itch-severity score in patients randomized to receive a 75-mg dose changed by -5.9 points, but this did not differ significantly from the change in the placebo group, according to Dr. Marcus Maurer of Charite-Universitatsmedizin, Berlin. The report was published online on Feb. 24 in the New England Journal of Medicine.
The findings from this international double-blind trial were reported simultaneously at the annual meeting of the American Academy of Allergy, Asthma, and Immunology.
In addition to meeting the primary 12-week response endpoint of change in weekly itch-severity score, patients receiving the 300-mg and 150-mg doses of omalizumab also experienced significant improvements, compared with patients given placebo, on all but one secondary endpoint, including change in the 7-day urticaria activity score (UAS7), change in the score for the weekly number of hives, time until a reduction from baseline of at least 5 points in the weekly itch-severity score (the MID), the proportions of patients with a UAS7 of 6 or less, the number of patients with a weekly MID response in the itch-severity score, the change from baseline in the score for the size of the largest hive, and change from baseline in the overall score on the Dermatology Life Quality Index. Those receiving 300 mg, but not those receiving 150 mg, also experienced significant improvement in the proportion of angioedema-free days from weeks 4-12 (N. Engl. J. Med. 2013 Feb. 24 [doi:10.1056/NEJMoa1215372]).
In a post hoc analysis at 12 weeks, 53%, 23%, 18%, and 10%, of those in the 300-mg, 150-mg, 75-mg, and placebo groups, respectively, were completely free of hives, and 44%, 22%, 16%, and 5%, respectively, were free of both hives and itching.
Of note, many patients experienced extremely rapid improvement, raising questions about a potential, as-yet unidentified and fundamental characteristic of this disease, study coauthors Dr. Thomas B. Casale, professor of medicine and medical microbiology and immunology and chief of allergy/immunology at Creighton University Medical Center, Omaha, Neb., and Dr. Allen P. Kaplan, clinical professor of medicine at the Medical University of South Carolina, Charleston, reported during a press briefing at AAAAI.
They explained that omalizumab, currently approved as an add-on therapy for moderate to severe persistent allergic asthma, is known to bind the allergic antibody immunoglobulin E (IgE). Many patients with chronic urticaria have an antibody that binds to a protein on the surface of histamine-containing cells, and that protein binds IgE.
"Just one injection drops one’s IgE level pretty close to zero ... and we learned that when we drop IgE to rock bottom, the protein on the cell to which it is attached also drops – and that’s the protein that the circulating antibody interacts with. The thinking was that if we drop the surface protein low enough, there’s nothing for the antibody to react with, and the hives would improve," he said.
This was, in fact, the case, as demonstrated in a phase I study of 12 patients, in which 7 patients responded dramatically, 4 responded partially, and 1 had no response. These findings led to the current phase III study, which is one of three such studies that investigators hope will lead to the drug’s approval for chronic idiopathic urticaria, because the high cost of the drug is prohibitive with respect to off-label use.
The same effect was seen in the current study.
This effect, however, takes a couple weeks to become apparent, so the rapid responses that occur in numerous patients suggest there is something more at play.
"This is working even faster than we thought, and probably there is a function of the allergic antibody that we do not yet understand," Dr. Kaplan said. "So this is going to be not only a terrific therapy for the disorder, but will lead to research in the future that probably – we hope – will elucidate the underlying abnormality of chronic urticaria beyond our current understanding of it. It’s very exciting, because it might elucidate some fundamental analogy that we don’t appreciate, and would have implications for allergic disease across the board."
The current study comprised patients aged 12-75 years who had a 6-month or longer history of chronic idiopathic urticaria, the presence of hives associated with itching for at least 8 consecutive weeks at any time prior to enrollment (despite H1-antihistamine use), a UAS7 of 16 or more during a 7-day period, a weekly itch-severity score of at least 8, a score of at least 4 on the UAS on at least one of the screening-visit days, and receipt of a licensed dose of a second-generation H1-antihistamine for at least 3 consecutive days immediately prior to the screening visit. Those with a clearly defined underlying cause for their symptoms were excluded. The doses evaluated in this study were based on a prior dose-ranging study, which showed no additional benefit with doses over 300 mg, the investigators said.
Patients continued to receive stable doses of H1-antihistamines throughout the 12-week treatment period, and were permitted to use diphenhydramine as a rescue medication.
Omalizumab was well tolerated in this study, with a similar number of adverse events occurring across the treatment groups. Serious adverse events occurred more often in the 300-mg group, with 6% of patients in that group experiencing a serious adverse event, compared with 3% of patients in the placebo group, and 1% of patients in the 150-mg and 75-mg groups, but the events were not considered to be related to the study drug.
The duration of response, however, was limited, as noted during a 16-week observation period following the initial 12-week treatment period.
Although the weekly itch-severity scores did not return to baseline during follow-up, they did increase to the levels seen in the placebo group.
The findings are nonetheless encouraging, Dr. Kaplan said, noting that this disease, which can have dramatic adverse effects on quality of life, is not uncommon, and can be difficult to treat, with about half of all patients failing to respond to standard therapy with high dose antihistamines. Alternate treatments used to treat the disease, including steroids and cyclosporine, can be effective, but can be highly toxic, he said.
"For refractory patients we have nothing that matches (omalizumab’s) combination of this kind of efficacy with low side effects, so many of us in the field kind of view this as a game changer for the patients," he said.
This study was sponsored by Genentech and Novartis Pharma. Several study authors made disclosures. A complete list of these disclosures is available with the full text of the article at NEJM.org.
SAN ANTONIO – Omalizumab diminished the signs and symptoms of chronic idiopathic urticaria in a dose-dependent fashion in a phase III study of 323 patients who failed to respond adequately to H1-antihistamines.
After 12 weeks of treatment with the recombinant humanized monoclonal antibody, improvements from baseline in weekly itch-severity scores were significantly greater in patients randomized to receive three doses of either 300 mg or 150 mg every 4 weeks, compared with placebo (score change of -9.8 and -8.1 vs. -5.1, respectively). The itch-severity score in patients randomized to receive a 75-mg dose changed by -5.9 points, but this did not differ significantly from the change in the placebo group, according to Dr. Marcus Maurer of Charite-Universitatsmedizin, Berlin. The report was published online on Feb. 24 in the New England Journal of Medicine.
The findings from this international double-blind trial were reported simultaneously at the annual meeting of the American Academy of Allergy, Asthma, and Immunology.
In addition to meeting the primary 12-week response endpoint of change in weekly itch-severity score, patients receiving the 300-mg and 150-mg doses of omalizumab also experienced significant improvements, compared with patients given placebo, on all but one secondary endpoint, including change in the 7-day urticaria activity score (UAS7), change in the score for the weekly number of hives, time until a reduction from baseline of at least 5 points in the weekly itch-severity score (the MID), the proportions of patients with a UAS7 of 6 or less, the number of patients with a weekly MID response in the itch-severity score, the change from baseline in the score for the size of the largest hive, and change from baseline in the overall score on the Dermatology Life Quality Index. Those receiving 300 mg, but not those receiving 150 mg, also experienced significant improvement in the proportion of angioedema-free days from weeks 4-12 (N. Engl. J. Med. 2013 Feb. 24 [doi:10.1056/NEJMoa1215372]).
In a post hoc analysis at 12 weeks, 53%, 23%, 18%, and 10%, of those in the 300-mg, 150-mg, 75-mg, and placebo groups, respectively, were completely free of hives, and 44%, 22%, 16%, and 5%, respectively, were free of both hives and itching.
Of note, many patients experienced extremely rapid improvement, raising questions about a potential, as-yet unidentified and fundamental characteristic of this disease, study coauthors Dr. Thomas B. Casale, professor of medicine and medical microbiology and immunology and chief of allergy/immunology at Creighton University Medical Center, Omaha, Neb., and Dr. Allen P. Kaplan, clinical professor of medicine at the Medical University of South Carolina, Charleston, reported during a press briefing at AAAAI.
They explained that omalizumab, currently approved as an add-on therapy for moderate to severe persistent allergic asthma, is known to bind the allergic antibody immunoglobulin E (IgE). Many patients with chronic urticaria have an antibody that binds to a protein on the surface of histamine-containing cells, and that protein binds IgE.
"Just one injection drops one’s IgE level pretty close to zero ... and we learned that when we drop IgE to rock bottom, the protein on the cell to which it is attached also drops – and that’s the protein that the circulating antibody interacts with. The thinking was that if we drop the surface protein low enough, there’s nothing for the antibody to react with, and the hives would improve," he said.
This was, in fact, the case, as demonstrated in a phase I study of 12 patients, in which 7 patients responded dramatically, 4 responded partially, and 1 had no response. These findings led to the current phase III study, which is one of three such studies that investigators hope will lead to the drug’s approval for chronic idiopathic urticaria, because the high cost of the drug is prohibitive with respect to off-label use.
The same effect was seen in the current study.
This effect, however, takes a couple weeks to become apparent, so the rapid responses that occur in numerous patients suggest there is something more at play.
"This is working even faster than we thought, and probably there is a function of the allergic antibody that we do not yet understand," Dr. Kaplan said. "So this is going to be not only a terrific therapy for the disorder, but will lead to research in the future that probably – we hope – will elucidate the underlying abnormality of chronic urticaria beyond our current understanding of it. It’s very exciting, because it might elucidate some fundamental analogy that we don’t appreciate, and would have implications for allergic disease across the board."
The current study comprised patients aged 12-75 years who had a 6-month or longer history of chronic idiopathic urticaria, the presence of hives associated with itching for at least 8 consecutive weeks at any time prior to enrollment (despite H1-antihistamine use), a UAS7 of 16 or more during a 7-day period, a weekly itch-severity score of at least 8, a score of at least 4 on the UAS on at least one of the screening-visit days, and receipt of a licensed dose of a second-generation H1-antihistamine for at least 3 consecutive days immediately prior to the screening visit. Those with a clearly defined underlying cause for their symptoms were excluded. The doses evaluated in this study were based on a prior dose-ranging study, which showed no additional benefit with doses over 300 mg, the investigators said.
Patients continued to receive stable doses of H1-antihistamines throughout the 12-week treatment period, and were permitted to use diphenhydramine as a rescue medication.
Omalizumab was well tolerated in this study, with a similar number of adverse events occurring across the treatment groups. Serious adverse events occurred more often in the 300-mg group, with 6% of patients in that group experiencing a serious adverse event, compared with 3% of patients in the placebo group, and 1% of patients in the 150-mg and 75-mg groups, but the events were not considered to be related to the study drug.
The duration of response, however, was limited, as noted during a 16-week observation period following the initial 12-week treatment period.
Although the weekly itch-severity scores did not return to baseline during follow-up, they did increase to the levels seen in the placebo group.
The findings are nonetheless encouraging, Dr. Kaplan said, noting that this disease, which can have dramatic adverse effects on quality of life, is not uncommon, and can be difficult to treat, with about half of all patients failing to respond to standard therapy with high dose antihistamines. Alternate treatments used to treat the disease, including steroids and cyclosporine, can be effective, but can be highly toxic, he said.
"For refractory patients we have nothing that matches (omalizumab’s) combination of this kind of efficacy with low side effects, so many of us in the field kind of view this as a game changer for the patients," he said.
This study was sponsored by Genentech and Novartis Pharma. Several study authors made disclosures. A complete list of these disclosures is available with the full text of the article at NEJM.org.
recombinant humanized monoclonal antibody, itch-severity scores, Dr. Marcus Maurer, New England Journal of Medicine, American Academy of Allergy, Asthma, and Immunology, 7-day urticaria activity score,
recombinant humanized monoclonal antibody, itch-severity scores, Dr. Marcus Maurer, New England Journal of Medicine, American Academy of Allergy, Asthma, and Immunology, 7-day urticaria activity score,
AT THE AAAAI ANNUAL MEETING
Recent recommendations on steroid-induced osteoporosis: More targeted, but more complicated
Whenever a patient begins treatment with a glucocorticoid drug, we need to think about bone loss.
The American College of Rheumatology (ACR) issued recommendations for preventing and treating glucocorticoid-induced osteoporosis in 2010.1 Compared with its previous guidelines,2 the new ones are more tailored and nuanced but may be more difficult for physicians to follow. The guidelines call for assessing fracture risk using the computer-based Fracture Risk Assessment Tool, or FRAX (www/shef.ac.uk/FRAX), developed by the World Health Organization (WHO). For those without a computer or ready access to the Web, an application of FRAX is available for download on smartphones.
In this article, my purpose is to review the new recommendations and to offer my perspective, which does not necessarily reflect the opinions of the ACR.
DESPITE EVIDENCE, MANY PATIENTS RECEIVE NO INTERVENTION
Use of glucocorticoids is the most common cause of secondary osteoporosis. During the first 6 to 12 months of use, these drugs can cause a rapid loss of bone mass due to increased bone resorption; with continued use, they cause a slower but steady decline in bone mass due to reduced bone formation.3 Epidemiologic studies have found that the risk of fractures increases with dose, starting with doses as low as 2.5 mg per day of prednisone or its equivalent.4
Numerous clinical trials have evaluated the effect of bisphosphonates and teriparatide (Forteo) on bone mass and fracture risk in patients on glucocorticoid therapy. The bisphosphonates alendronate (Fosamax) and risedronate (Actonel) have both been shown to increase bone mass and reduce vertebral fracture risk in glucocorticoid recipients.5–8 Zoledronic acid (Reclast), a parenteral bisphosphonate given in one annual dose, was shown to increase bone mass more than oral risedronate taken daily,9 and teriparatide, a formulation of parathyroid hormone, was better than alendronate.10
However, despite the known risk of fractures with glucocorticoid use and the demonstrated efficacy of available agents in preventing bone loss and fracture, many patients do not receive any intervention.11,12
WHAT HAS HAPPENED SINCE 2001?
In the interval since 2001, several guidelines for managing glucocorticoid-induced osteoporosis have been published in other countries.13–17 Broadly speaking, they recommend starting preventive drug therapy for patients at risk of fracture at the same time glucocorticoid drugs are started if the patient is expected to take glucocorticoids for more than 3 to 6 months in doses higher than 5 to 7.5 mg of prednisone or its equivalent daily.
Recommendations for patients who have been on glucocorticoids for longer than 3 to 6 months at initial evaluation have been based largely on T scores derived from dual-energy x-ray absorptiometry (DXA). Thresholds for initiating therapy have varied: the ACR in 2001 recommended preventive treatment if the T score is lower than −1.0, whereas British guidelines said −1.5 and Dutch guidelines said −2.5.
In the United States, since 2001 when the ACR published its last guidelines,2 zoledronic acid and teriparatide have been approved for use in glucocorticoid-induced osteoporosis. In addition, guideline-development methodology has evolved and now is more scientifically rigorous. Finally, a risk-assessment tool has been developed that enables a more tailored approach (see below).
FRAX (www.shef.ac.uk/FRAX)
FRAX is a tool developed by the WHO to calculate the risk of fracture. If you go to the FRAX Web site and enter the required clinical information (race, age, sex, weight, height, previous fracture, family history of a fractured hip in a parent, current smoking, use of glucocorticoids, rheumatoid arthritis, secondary osteoporosis, consumption of three or more units of alcohol per day, and bone mineral density of the femoral neck), it will tell you the patient’s 10-year absolute (not relative) risk of major osteoporotic fracture and of hip fracture.
Since FRAX was unveiled in 2008, calculation of absolute fracture risk has become the standard method for making treatment decisions in patients with low bone mass who have not yet received any fracture-preventing treatment.18 The use of clinical risk factors in FRAX increases its ability to predict risk over and above the use of bone density by itself. And glucocorticoids are one of the clinical risk factors in FRAX.
But in which patients is treatment with a bisphosphonate or teriparatide cost-effective?
Thresholds for cost-effectiveness have been developed on the basis of economic assumptions that are country-specific. In the United States, the National Osteoporosis Foundation recommends drug therapy if the 10-year absolute risk of a major osteoporotic fracture of the hip, spine (clinical, not radiographic), wrist, or humerus is greater than 20% or if the risk of a hip fracture is greater than 3%.19
At equivalent bone densities, women taking glucocorticoids are at considerably higher risk of fracture than nonusers.20 For example, consider a 65-year-old white woman, weight 59 kg, height 163 cm, no previous fractures, no parent with a fractured hip, no current smoking, no rheumatoid arthritis, no secondary osteoporosis, no excessive alcohol use, and a T score of −2.2 in the femoral neck. (Try this on the FRAX Web site.) If she does not use glucocorticoids, her 10-year risk of hip fracture is 2.0%; using glucocorticoids increases the risk to 3.6%. This is higher than the 3% National Osteoporosis Foundation guideline; thus, treatment would be recommended.
Also using FRAX, a 55-year-old white woman with a T score of −1.8 and on glucocorticoid therapy has a 67% higher risk of major osteoporotic fracture and an 80% higher risk of hip fracture.
For a third example, a white woman age 60, weight 70 kg, height 168 cm, negative for all the other risk factors but with a T score of −2.1 and on glucocorticoids has a calculated 10-year fracture risk of 2.1%, which is below the National Osteoporosis Foundation treatment threshold. However, most clinicians would probably recommend treatment for her, depending on the anticipated dose and duration of glucocorticoid therapy.
A caveat. In FRAX, glucocorticoid therapy is a categorical variable—a yes-or-no question—and yes is defined as having ever used a glucocorticoid in a dose greater than 5 mg for more than 3 months. Therefore, according to FRAX, a patient who took 5 mg of prednisone for 3 months 5 years ago has the same fracture risk as a patient on 60 mg of prednisone after a diagnosis of temporal arteritis. For this reason, the FRAX tool is likely to underestimate fracture risk, especially in patients currently taking glucocorticoids and those on higher doses of these drugs.
Kanis et al used the General Practice Research Database to adjust the fracture risk for glucocorticoid use in FRAX.21 At doses higher than 7.5 mg, the fracture risk had to be revised upward by 10% to 25% depending on the fracture site (hip vs any major osteoporotic fracture) and age (greater at age 40 than at age 90).
The underestimation of fracture risk led the ACR Expert Advisory Panel to create risk strata for major osteoporotic fractures, ie, low (< 10% risk per 10 years), medium (10%–20%), and high (> 20%) and uses these cut points to make treatment recommendations.
HOW THE 2010 GUIDELINES WERE DEVELOPED
Whereas the 2001 recommendations were based on a more informal consensus approach, the 2010 recommendations use a more scientifically rigorous methodology for guideline development, the Research and Development/University of California at Los Angeles (RAND/UCLA) Appropriateness Method. The RAND/UCLA method combines the best available scientific evidence with expert opinion to develop practice guidelines.
In drawing up the 2010 recommendations the ACR used three panels of experts. The Core Executive Panel conducted a systematic review of controlled clinical trials of therapies currently approved for treating glucocorticoid-induced osteoporosis in the United States, Canada, or the European Union. They found 53 articles meeting their inclusion criteria; an evidence report was produced that informed the development of the recommendations. This evidence report and guideline development process is available at http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)2151-4658. The Expert Advisory Panel framed the recommendations, and the Task Force Panel voted on them. The Core Executive Panel and Expert Advisory Panel constructed 48 patient-specific clinical scenarios using four variables: sex, age, race/ethnicity, and femoral neck T scores.
The members of the Task Force Panel were asked to use the evidence report and their expert judgment to vote on and rate the appropriateness of using a specific therapy in the context of each scenario on a 9-point Likert scale (1 = appropriate; 9 = not appropriate). Agreement occurred when 7 or more of the 10 panel members rated a scenario 1, 2, or 3. Disagreements were defined as 3 or more of the 10 members rating the scenario between 4 and 9 while the other members rated it lower.
Disagreements in voting were discussed in an attempt to achieve consensus, and a second vote was conducted which determined the final recommendations. If disagreement remained after the vote, no recommendation was made.
No attempt was made to assign priority of one drug over another when multiple drugs were deemed appropriate, although the final recommendations did differentiate drugs based on patient categories.
START WITH COUNSELING, ASSESSMENT

For patients starting or already on glucocorticoid therapy that is expected to last at least 3 months, the first step is to counsel them on lifestyle modifications (Table 1) and to assess their risk factors (Figure 1). Recommendations for monitoring patients receiving glucocorticoid therapy for at least 3 months are presented in Table 2.
These recommendations are based on literature review, and the strength of evidence is graded:
- Grade A—derived from multiple randomized controlled trials or a meta-analysis
- Grade B—derived from a single randomized controlled trial or nonrandomized study
- Grade C—derived from consensus, expert opinion, or case series.
This system is the same one used by the American College of Cardiology and is based on clinical trial data.22

Recommendations for calcium intake and vitamin D supplementation were graded A; all other recommendations were graded C (Tables 1 and 2). It is important to note that practices that receive a grade of C may still be accepted as standard of care, such as fall assessment and smoking cessation.
FOR POSTMENOPAUSAL WOMEN AND FOR MEN AGE 50 AND OLDER
FRAX low-risk group

Recall that “low risk” based on the new ACR guidelines means that the 10-year absolute risk of a major osteoporotic fracture, as calculated with FRAX, is less than 10%.
- If glucocorticoid use is expected to last or has already lasted at least 3 months and the dose is less than 7.5 mg/day, no pharmacologic treatment is recommended.
- If glucocorticoid use is expected to last or has already lasted at least 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended.
Comment. These are the most straightforward of the recommendations. All three bisphosphonates are recommended as treatment options if the glucocorticoid dose is at least 7.5 mg/day and the duration at least 3 months. Ibandronate (Boniva) was not included because it has no data from clinical trials.
FRAX medium-risk group
“Medium risk” means that the 10-year absolute fracture risk of major osteoporotic fractures is 10% to 20%.
- If glucocorticoid use is anticipated to last or has lasted at least 3 months and the dose is less than 7.5 mg/day, alendronate or risedronate is recommended.
- If glucocorticoid use is anticipated to last or has lasted at least 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended.
Comment. Treatment is recommended at all glucocorticoid doses for patients in the medium-risk category if the duration of glucocorticoid treatment is at least 3 months, with one difference: zoledronic acid is recommended only if the glucocorticoid dose is 7.5 mg/day or higher. This inconsistency persisted after a second round of voting by the Task Force Panel.
FRAX high-risk group
In this group, the 10-year risk of major osteoporotic fractures is higher than 20%.
- If the glucocorticoid dose is less than 5 mg/day for up to 1 month, alendronate, risedronate, or zoledronic acid is recommended.
- If the dose is 5 mg/day or more for up to 1 month, or any dose for more than 1 month, alendronate, risedronate, zoledronic acid or teriparatide is recommended.
Comment. Based on current National Osteoporosis Foundation guidelines, all patients with a 10-year risk greater than 20% are recommended for treatment for any duration and dose of glucocorticoid use. However, teriparatide is recommended only if the duration of glucocorticoid therapy is more than 1 month.
FOR PREMENOPAUSAL WOMEN AND FOR MEN YOUNGER THAN AGE 50
Use of FRAX is not appropriate in premenopausal women or in men younger than 50 years.
Younger patients with no prevalent fracture
For men younger than 50 and premenopausal women who have not had a previous fracture, data were considered inadequate to make a recommendation, and no votes were taken.
Prevalent fracture in premenopausal women of nonchildbearing potential
In premenopausal women of nonchildbearing potential who have had a fracture:
- If the glucocorticoid duration is 1 to 3 months and the dose is 5 mg/day or higher, alendronate or risedronate is recommended.
- If the duration is 1 to 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended
- If the duration is more than 3 months, alendronate, risedronate, zoledronic acid, or teriparatide is recommended.
Comment. Treatment is recommended with any of the four medications in patients with a fracture and treated with glucocorticoids for more than 3 months. For shorter-duration glucocorticoid use (1–3 months) at 5 mg/day or higher, only alendronate and risedronate are recommended. If the dose is 7.5 mg/day or higher, any bisphosphonate is recommended. Zoledronic acid was consistently differentiated by the expert panel on the basis of dose and duration of glucocorticoid use, in view of its 1-year duration of effect after one dose.
Prevalent fracture in women of childbearing potential
- If the glucocorticoid duration is 1 to 3 months, there was no consensus (ie, voting disagreements could not be resolved).
- If the glucocorticoid duration is more than 3 months and the dose is 7.5 mg/day or more, alendronate, risedronate, or teriparatide is recommended.
- If the glucocorticoid duration is more than 3 months and the dose is less than 7.5 mg/day, there was no consensus.
Comment. Childbearing potential creates further complexities because of concern about fetal toxicity with bisphosphonates. For short-term glucocorticoid therapy at any dose and for therapy longer than 3 months at less than 7.5 mg, no consensus could be reached. For therapy longer than 3 months and with 7.5 mg/day or higher, treatment is recommended but not with zoledronic acid, based on the long half-life of the drug and concern for fetal toxicity.
Additional risk stratification
The panel recommended that if the following were present, a shift to a higher fracture risk category should be considered (low to medium, or medium to high):
- High daily dose of glucocorticoid
- High cumulative glucocorticoid dose
- Declining bone mineral density on serial DXA.
These are known risk factors that increase fracture risk but would not affect fracture risk in the FRAX model.
WHAT IS NEW IN THE 2010 RECOMMENDATIONS?
Recommendations for counseling now include fall risk assessment, height measurement, 25-hydroxyvitamin D measurement, and evaluation of patients for prevalent and incident fractures using vertebral fracture assessment by DXA or radiographic imaging of the spine.
Recommended drugs now include teriparatide and zoledronic acid, while estrogen and testosterone are no longer recommended as therapies for glucocorticoid-induced osteoporosis. Ibandronate is not included, since there have been no randomized controlled trials of this bisphosphonate in glucocorticoid-induced osteoporosis.
Recommendations for treatment in 2001 were based on T scores alone, while the 2010 recommendations use an assessment of absolute fracture risk based on FRAX for postmenopausal women and for men age 50 and older.
A clinician’s guide that summarizes the ACR recommendations is available at www.rheumatology.org/practice/clinical/guidelines/.
RECOMMENDATIONS DO NOT REPLACE CLINICAL JUDGMENT
Although the 2010 recommendations were more rigorous in their development process than those of 2001, they have limitations and they should not replace clinical judgment. Rather, they are intended to provide an evidence-based approach to guide clinicians in making treatment choices in patients on glucocorticoid therapy.
CONSIDERING ABSOLUTE FRACTURE RISK IN TREATMENT DECISIONS
The 2001 ACR guidelines recommended fracture-preventing treatment in all patients starting glucocorticoid therapy at more than 5 mg/day if the planned duration of treatment was at least 3 months, and in patients on long-term glucocorticoid therapy if the T score was less than −1.0. While these guidelines were simple and easy to use, they were not specific enough to provide useful guidance in specific scenarios.
A model of absolute fracture risk was not available in 2001. A 55-year old white woman with a T score of −1.1 who smoked, who had been using 5 mg of prednisone for the last 12 months, and who had stable bone mass on serial DXA scans would have been recommended for treatment based on the 2001 recommendations. If this patient’s FRAX-calculated 10-year absolute risk of a major osteoporotic fracture is less than 10%, that would be well below the National Osteoporosis Foundation’s cost-effective treatment threshold of 20%. The new guidelines suggest no treatment is needed, since the risk category is low and the dose is less than 7.5 mg. However, if on serial DXA this patient had a significant decline in bone mass, the guidelines suggest shifting the patient to a higher risk category, ie, from low to medium risk, which would result in a recommendation in favor of treatment.
The 2010 recommendations are not as simple to use as those from 2001. They encourage using FRAX to calculate fracture risk; thus, knowledge of the strengths and limitations of FRAX is required. Access to the internet in the examination room or use of the FRAX tool on a smartphone as well as willingness to spend a minute to calculate fracture risk are needed. For those who cannot or choose not to use the FRAX tool, the ACR publication provides tables for patient risk assessment based on age and T score. However, the tables would have to be readily available in the clinic, which may not be practical.
The 2010 recommendation provide a more nuanced approach to treatment in patients on glucocorticoid therapy and are likely to change treatment decisions based on their use, just as FRAX has altered treatment decisions in patients with primary osteoporosis.23
FRAX has limitations
FRAX underestimates the effect of glucocorticoids on fracture risk because steroid use is a yes-or-no question and its weight represents the average risk in a population that has ever used steroids, most of whom were using doses between 2.5 and 7.5 mg.
The WHO recognized this limitation and suggested an upward adjustment of risk for patients on 7.5 mg or more, ranging from 10% to 25%.21 For patients on high doses of steroids, this adjustment is still likely to result in underestimation of fracture risk and undertreatment of glucocorticoid-treated patients.
The 2010 recommendations adjust for this limitation, recommending treatment in the low-risk and medium-risk categories if the glucocorticoid dose is 7.5 mg or higher. If a patient is using high daily doses of steroids or has a declining bone density, the 2010 recommendations suggest increasing the risk category from low to medium or medium to high.
FRAX risk factors are dichotomous (yes/no) and are not adjusted for dose effects such as multiple fractures (vs a single fracture), heavy smoking (vs light smoking), heavy alcohol use (6 units per day vs 3 units), or severe rheumatoid arthritis (vs mild disease). Family history of osteoporosis in the FRAX is limited to parents with a hip fracture—vertebral fractures in a family member do not count.
Since FRAX uses the bone mineral density in the hip, it underestimates fracture risk in patients with low spine density but normal hip density. It may also underestimate fracture risk in patients with declining bone mass; the 2010 recommendations suggest the clinician should increase the risk category in this situation.
LIMITATIONS OF THE GUIDELINES
The 2010 recommendations do not include several important groups in which steroids are used, including transplant recipients, children, and patients on inhaled corticosteroids. The panel thought that there were insufficient data to make recommendations for these populations, as well as for premenopausal women and men younger than 50 years who did not have a prevalent fracture. The absence of a recommendation in these situations should not be considered a recommendation for no treatment; it is an acknowledgment of a lack of evidence, a lack of consensus among experts, and the need for additional clinical trials.
For premenopausal women and men under age 50 with a fracture, the recommendations are complicated and not intuitive. Zoledronic acid is not recommended for women of non-childbearing potential with a glucocorticoid duration of 1 to 3 months unless the steroid dose is at least 7.5 mg. This recommendation was based on panel voting and consensus that giving zoledronic acid, a medication with a 1-year duration of effect, in a patient on steroids for only 1 to 3 months was not warranted.
Teriparatide was recommended only if glucocorticoids are used for at least 3 months, although anyone who already has a fracture might be considered at high enough risk to warrant anabolic therapy regardless of steroid use or duration.
Zoledronic acid was excluded in women of childbearing potential, based on panel voting and consensus that drugs given in smaller amounts over 1 year might be less harmful to a fetus than one with a longer half-life given in a larger bolus once a year.
The panel could reach no consensus on women of childbearing potential with a prevalent fracture who were using less than 7.5 mg/day of glucocorticoids. A lack of consensus was the result of insufficient data to make evidence-based decisions and a disagreement among experts on the correct treatment.
The guidelines do not address the duration of treatment with bisphosphonates, a topic of importance because of concern for the potential long-term side effects of these medications.
THE BOTTOM LINE
The 2010 recommendations add a degree of complexity, with different medications recommended on the basis of glucocorticoid dose and duration as well as patient age, menopausal status, and childbearing potential. Guideline developers and clinicians face a difficult trade-off: easy-to-follow guidelines or more targeted guidelines that are more complex and therefore more difficult to use than previous guidelines.
This criticism is reasonable. The complexity is a result of insufficient evidence from clinical trials to make more exact and user-friendly recommendations, and also a result of the RAND/UCLA methodology. In cases that lack sufficient evidence on which to make a decision, the guideline development uses voting among experts in an attempt to develop consensus. This often results in complexity, lack of consensus, or inconsistencies.
The guidelines are straightforward for postmenopausal women and men age 50 and older on at least 7.5 mg prednisone for more than 3 months.
Since there is substantial evidence that many patients on glucocorticoid therapy go untreated, the risk of fracture in this population would be substantially reduced if clinicians would adhere to the recommendations.
- Grossman JM, Gordon R, Ranganath VK, et al; American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res (Hoboken) 2010; 62:1515–1526.
- Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Arthritis Rheum 2001; 44:1496–1503.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Oral corticosteroids and fracture risk: relationship to daily and cumulative doses. Rheumatology (Oxford) 2000; 39:1383–1389.
- Saag KG, Emkey R, Schnitzer TJ, et al. Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. Glucocorticoid-Induced Osteoporosis Intervention Study Group. N Engl J Med 1998; 339:292–299.
- Cohen S, Levy RM, Keller M, et al. Risedronate therapy prevents corticosteroid-induced bone loss: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum 1999; 42:2309–2318.
- Reid DM, Hughes RA, Laan RF, et al. Efficacy and safety of daily risedronate in the treatment of corticosteroid-induced osteoporosis in men and women: a randomized trial. European Corticosteroid-Induced Osteoporosis Treatment Study. J Bone Miner Res 2000; 15:1006–1013.
- Wallach S, Cohen S, Reid DM, et al. Effects of risedronate treatment on bone density and vertebral fracture in patients on corticosteroid therapy. Calcif Tissue Int 2000; 67:277–285.
- Reid DM, Devogelaer JP, Saag K, et al; HORIZON investigators. Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet 2009; 373:1253–1263.
- Saag KG, Shane E, Boonen S, et al. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med 2007; 357:2028–2039.
- Curtis JR, Westfall AO, Allison JJ, et al. Longitudinal patterns in the prevention of osteoporosis in glucocorticoid-treated patients. Arthritis Rheum 2005; 52:2485–2494.
- Feldstein AC, Elmer PJ, Nichols GA, Herson M. Practice patterns in patients at risk for glucocorticoid-induced osteoporosis. Osteoporos Int 2005; 16:2168–2174.
- Brown JP, Josse RG; Scientific Advisory Council of the Osteoporosis Society of Canada. 2002 clinical practice guidelines for the diagnosis and management of osteoporosis in Canada. CMAJ 2002; 167(suppl 10):S1–S34.
- Devogelaer JP, Goemaere S, Boonen S, et al. Evidence-based guidelines for the prevention and treatment of glucocorticoid-induced osteoporosis: a consensus document of the Belgian Bone Club. Osteoporos Int 2006; 17:8–19.
- Gourlay M, Franceschini N, Sheyn Y. Prevention and treatment strategies for glucocorticoid-induced osteoporotic fractures. Clin Rheumatol 2007; 26:144–153.
- Nawata H, Soen S, Takayanagi R, et al; Subcommittee to Study Diagnostic Criteria for Glucocorticoid-Induced Osteoporosis. Guidelines on the management and treatment of glucocorticoid-induced osteoporosis of the Japanese Society for Bone and Mineral Research (2004). J Bone Miner Metab 2005; 23:105–109.
- Geusens PP, Lems WF, Verhaar HJ, et al. Review and evaluation of the Dutch guidelines for osteoporosis. J Eval Clin Pract 2006; 12:539–548.
- Kanis JA, Johnell O, Oden A, Johansson H, McCloskey E. FRAX and the assessment of fracture probability in men and women from the UK. Osteoporos Int 2008; 19:385–389.
- National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. Washington, DC, National Osteoporosis Foundation, 2010. http://nof.org/files/nof/public/content/file/344/upload/159.pdf. Accessed December 31, 2012.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johansson H, Oden A, McCloskey EV. Guidance for the adjustment of FRAX according to the dose of glucocorticoids. Osteoporos Int 2011; 22:809–816.
- Hunt SA, Abraham WT, Chin MH, et al; American College of Cardiology. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation 2005; 112:e154–e235.
- Dawson-Hughes B, Tosteson AN, Melton LJ, et al; National Osteoporosis Foundation Guide Committee. Implications of absolute fracture risk assessment for osteoporosis practice guidelines in the USA. Osteoporos Int 2008; 19:449–458.
Whenever a patient begins treatment with a glucocorticoid drug, we need to think about bone loss.
The American College of Rheumatology (ACR) issued recommendations for preventing and treating glucocorticoid-induced osteoporosis in 2010.1 Compared with its previous guidelines,2 the new ones are more tailored and nuanced but may be more difficult for physicians to follow. The guidelines call for assessing fracture risk using the computer-based Fracture Risk Assessment Tool, or FRAX (www/shef.ac.uk/FRAX), developed by the World Health Organization (WHO). For those without a computer or ready access to the Web, an application of FRAX is available for download on smartphones.
In this article, my purpose is to review the new recommendations and to offer my perspective, which does not necessarily reflect the opinions of the ACR.
DESPITE EVIDENCE, MANY PATIENTS RECEIVE NO INTERVENTION
Use of glucocorticoids is the most common cause of secondary osteoporosis. During the first 6 to 12 months of use, these drugs can cause a rapid loss of bone mass due to increased bone resorption; with continued use, they cause a slower but steady decline in bone mass due to reduced bone formation.3 Epidemiologic studies have found that the risk of fractures increases with dose, starting with doses as low as 2.5 mg per day of prednisone or its equivalent.4
Numerous clinical trials have evaluated the effect of bisphosphonates and teriparatide (Forteo) on bone mass and fracture risk in patients on glucocorticoid therapy. The bisphosphonates alendronate (Fosamax) and risedronate (Actonel) have both been shown to increase bone mass and reduce vertebral fracture risk in glucocorticoid recipients.5–8 Zoledronic acid (Reclast), a parenteral bisphosphonate given in one annual dose, was shown to increase bone mass more than oral risedronate taken daily,9 and teriparatide, a formulation of parathyroid hormone, was better than alendronate.10
However, despite the known risk of fractures with glucocorticoid use and the demonstrated efficacy of available agents in preventing bone loss and fracture, many patients do not receive any intervention.11,12
WHAT HAS HAPPENED SINCE 2001?
In the interval since 2001, several guidelines for managing glucocorticoid-induced osteoporosis have been published in other countries.13–17 Broadly speaking, they recommend starting preventive drug therapy for patients at risk of fracture at the same time glucocorticoid drugs are started if the patient is expected to take glucocorticoids for more than 3 to 6 months in doses higher than 5 to 7.5 mg of prednisone or its equivalent daily.
Recommendations for patients who have been on glucocorticoids for longer than 3 to 6 months at initial evaluation have been based largely on T scores derived from dual-energy x-ray absorptiometry (DXA). Thresholds for initiating therapy have varied: the ACR in 2001 recommended preventive treatment if the T score is lower than −1.0, whereas British guidelines said −1.5 and Dutch guidelines said −2.5.
In the United States, since 2001 when the ACR published its last guidelines,2 zoledronic acid and teriparatide have been approved for use in glucocorticoid-induced osteoporosis. In addition, guideline-development methodology has evolved and now is more scientifically rigorous. Finally, a risk-assessment tool has been developed that enables a more tailored approach (see below).
FRAX (www.shef.ac.uk/FRAX)
FRAX is a tool developed by the WHO to calculate the risk of fracture. If you go to the FRAX Web site and enter the required clinical information (race, age, sex, weight, height, previous fracture, family history of a fractured hip in a parent, current smoking, use of glucocorticoids, rheumatoid arthritis, secondary osteoporosis, consumption of three or more units of alcohol per day, and bone mineral density of the femoral neck), it will tell you the patient’s 10-year absolute (not relative) risk of major osteoporotic fracture and of hip fracture.
Since FRAX was unveiled in 2008, calculation of absolute fracture risk has become the standard method for making treatment decisions in patients with low bone mass who have not yet received any fracture-preventing treatment.18 The use of clinical risk factors in FRAX increases its ability to predict risk over and above the use of bone density by itself. And glucocorticoids are one of the clinical risk factors in FRAX.
But in which patients is treatment with a bisphosphonate or teriparatide cost-effective?
Thresholds for cost-effectiveness have been developed on the basis of economic assumptions that are country-specific. In the United States, the National Osteoporosis Foundation recommends drug therapy if the 10-year absolute risk of a major osteoporotic fracture of the hip, spine (clinical, not radiographic), wrist, or humerus is greater than 20% or if the risk of a hip fracture is greater than 3%.19
At equivalent bone densities, women taking glucocorticoids are at considerably higher risk of fracture than nonusers.20 For example, consider a 65-year-old white woman, weight 59 kg, height 163 cm, no previous fractures, no parent with a fractured hip, no current smoking, no rheumatoid arthritis, no secondary osteoporosis, no excessive alcohol use, and a T score of −2.2 in the femoral neck. (Try this on the FRAX Web site.) If she does not use glucocorticoids, her 10-year risk of hip fracture is 2.0%; using glucocorticoids increases the risk to 3.6%. This is higher than the 3% National Osteoporosis Foundation guideline; thus, treatment would be recommended.
Also using FRAX, a 55-year-old white woman with a T score of −1.8 and on glucocorticoid therapy has a 67% higher risk of major osteoporotic fracture and an 80% higher risk of hip fracture.
For a third example, a white woman age 60, weight 70 kg, height 168 cm, negative for all the other risk factors but with a T score of −2.1 and on glucocorticoids has a calculated 10-year fracture risk of 2.1%, which is below the National Osteoporosis Foundation treatment threshold. However, most clinicians would probably recommend treatment for her, depending on the anticipated dose and duration of glucocorticoid therapy.
A caveat. In FRAX, glucocorticoid therapy is a categorical variable—a yes-or-no question—and yes is defined as having ever used a glucocorticoid in a dose greater than 5 mg for more than 3 months. Therefore, according to FRAX, a patient who took 5 mg of prednisone for 3 months 5 years ago has the same fracture risk as a patient on 60 mg of prednisone after a diagnosis of temporal arteritis. For this reason, the FRAX tool is likely to underestimate fracture risk, especially in patients currently taking glucocorticoids and those on higher doses of these drugs.
Kanis et al used the General Practice Research Database to adjust the fracture risk for glucocorticoid use in FRAX.21 At doses higher than 7.5 mg, the fracture risk had to be revised upward by 10% to 25% depending on the fracture site (hip vs any major osteoporotic fracture) and age (greater at age 40 than at age 90).
The underestimation of fracture risk led the ACR Expert Advisory Panel to create risk strata for major osteoporotic fractures, ie, low (< 10% risk per 10 years), medium (10%–20%), and high (> 20%) and uses these cut points to make treatment recommendations.
HOW THE 2010 GUIDELINES WERE DEVELOPED
Whereas the 2001 recommendations were based on a more informal consensus approach, the 2010 recommendations use a more scientifically rigorous methodology for guideline development, the Research and Development/University of California at Los Angeles (RAND/UCLA) Appropriateness Method. The RAND/UCLA method combines the best available scientific evidence with expert opinion to develop practice guidelines.
In drawing up the 2010 recommendations the ACR used three panels of experts. The Core Executive Panel conducted a systematic review of controlled clinical trials of therapies currently approved for treating glucocorticoid-induced osteoporosis in the United States, Canada, or the European Union. They found 53 articles meeting their inclusion criteria; an evidence report was produced that informed the development of the recommendations. This evidence report and guideline development process is available at http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)2151-4658. The Expert Advisory Panel framed the recommendations, and the Task Force Panel voted on them. The Core Executive Panel and Expert Advisory Panel constructed 48 patient-specific clinical scenarios using four variables: sex, age, race/ethnicity, and femoral neck T scores.
The members of the Task Force Panel were asked to use the evidence report and their expert judgment to vote on and rate the appropriateness of using a specific therapy in the context of each scenario on a 9-point Likert scale (1 = appropriate; 9 = not appropriate). Agreement occurred when 7 or more of the 10 panel members rated a scenario 1, 2, or 3. Disagreements were defined as 3 or more of the 10 members rating the scenario between 4 and 9 while the other members rated it lower.
Disagreements in voting were discussed in an attempt to achieve consensus, and a second vote was conducted which determined the final recommendations. If disagreement remained after the vote, no recommendation was made.
No attempt was made to assign priority of one drug over another when multiple drugs were deemed appropriate, although the final recommendations did differentiate drugs based on patient categories.
START WITH COUNSELING, ASSESSMENT
For patients starting or already on glucocorticoid therapy that is expected to last at least 3 months, the first step is to counsel them on lifestyle modifications (Table 1) and to assess their risk factors (Figure 1). Recommendations for monitoring patients receiving glucocorticoid therapy for at least 3 months are presented in Table 2.
These recommendations are based on literature review, and the strength of evidence is graded:
- Grade A—derived from multiple randomized controlled trials or a meta-analysis
- Grade B—derived from a single randomized controlled trial or nonrandomized study
- Grade C—derived from consensus, expert opinion, or case series.
This system is the same one used by the American College of Cardiology and is based on clinical trial data.22
Recommendations for calcium intake and vitamin D supplementation were graded A; all other recommendations were graded C (Tables 1 and 2). It is important to note that practices that receive a grade of C may still be accepted as standard of care, such as fall assessment and smoking cessation.
FOR POSTMENOPAUSAL WOMEN AND FOR MEN AGE 50 AND OLDER
FRAX low-risk group
Recall that “low risk” based on the new ACR guidelines means that the 10-year absolute risk of a major osteoporotic fracture, as calculated with FRAX, is less than 10%.
- If glucocorticoid use is expected to last or has already lasted at least 3 months and the dose is less than 7.5 mg/day, no pharmacologic treatment is recommended.
- If glucocorticoid use is expected to last or has already lasted at least 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended.
Comment. These are the most straightforward of the recommendations. All three bisphosphonates are recommended as treatment options if the glucocorticoid dose is at least 7.5 mg/day and the duration at least 3 months. Ibandronate (Boniva) was not included because it has no data from clinical trials.
FRAX medium-risk group
“Medium risk” means that the 10-year absolute fracture risk of major osteoporotic fractures is 10% to 20%.
- If glucocorticoid use is anticipated to last or has lasted at least 3 months and the dose is less than 7.5 mg/day, alendronate or risedronate is recommended.
- If glucocorticoid use is anticipated to last or has lasted at least 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended.
Comment. Treatment is recommended at all glucocorticoid doses for patients in the medium-risk category if the duration of glucocorticoid treatment is at least 3 months, with one difference: zoledronic acid is recommended only if the glucocorticoid dose is 7.5 mg/day or higher. This inconsistency persisted after a second round of voting by the Task Force Panel.
FRAX high-risk group
In this group, the 10-year risk of major osteoporotic fractures is higher than 20%.
- If the glucocorticoid dose is less than 5 mg/day for up to 1 month, alendronate, risedronate, or zoledronic acid is recommended.
- If the dose is 5 mg/day or more for up to 1 month, or any dose for more than 1 month, alendronate, risedronate, zoledronic acid or teriparatide is recommended.
Comment. Based on current National Osteoporosis Foundation guidelines, all patients with a 10-year risk greater than 20% are recommended for treatment for any duration and dose of glucocorticoid use. However, teriparatide is recommended only if the duration of glucocorticoid therapy is more than 1 month.
FOR PREMENOPAUSAL WOMEN AND FOR MEN YOUNGER THAN AGE 50
Use of FRAX is not appropriate in premenopausal women or in men younger than 50 years.
Younger patients with no prevalent fracture
For men younger than 50 and premenopausal women who have not had a previous fracture, data were considered inadequate to make a recommendation, and no votes were taken.
Prevalent fracture in premenopausal women of nonchildbearing potential
In premenopausal women of nonchildbearing potential who have had a fracture:
- If the glucocorticoid duration is 1 to 3 months and the dose is 5 mg/day or higher, alendronate or risedronate is recommended.
- If the duration is 1 to 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended
- If the duration is more than 3 months, alendronate, risedronate, zoledronic acid, or teriparatide is recommended.
Comment. Treatment is recommended with any of the four medications in patients with a fracture and treated with glucocorticoids for more than 3 months. For shorter-duration glucocorticoid use (1–3 months) at 5 mg/day or higher, only alendronate and risedronate are recommended. If the dose is 7.5 mg/day or higher, any bisphosphonate is recommended. Zoledronic acid was consistently differentiated by the expert panel on the basis of dose and duration of glucocorticoid use, in view of its 1-year duration of effect after one dose.
Prevalent fracture in women of childbearing potential
- If the glucocorticoid duration is 1 to 3 months, there was no consensus (ie, voting disagreements could not be resolved).
- If the glucocorticoid duration is more than 3 months and the dose is 7.5 mg/day or more, alendronate, risedronate, or teriparatide is recommended.
- If the glucocorticoid duration is more than 3 months and the dose is less than 7.5 mg/day, there was no consensus.
Comment. Childbearing potential creates further complexities because of concern about fetal toxicity with bisphosphonates. For short-term glucocorticoid therapy at any dose and for therapy longer than 3 months at less than 7.5 mg, no consensus could be reached. For therapy longer than 3 months and with 7.5 mg/day or higher, treatment is recommended but not with zoledronic acid, based on the long half-life of the drug and concern for fetal toxicity.
Additional risk stratification
The panel recommended that if the following were present, a shift to a higher fracture risk category should be considered (low to medium, or medium to high):
- High daily dose of glucocorticoid
- High cumulative glucocorticoid dose
- Declining bone mineral density on serial DXA.
These are known risk factors that increase fracture risk but would not affect fracture risk in the FRAX model.
WHAT IS NEW IN THE 2010 RECOMMENDATIONS?
Recommendations for counseling now include fall risk assessment, height measurement, 25-hydroxyvitamin D measurement, and evaluation of patients for prevalent and incident fractures using vertebral fracture assessment by DXA or radiographic imaging of the spine.
Recommended drugs now include teriparatide and zoledronic acid, while estrogen and testosterone are no longer recommended as therapies for glucocorticoid-induced osteoporosis. Ibandronate is not included, since there have been no randomized controlled trials of this bisphosphonate in glucocorticoid-induced osteoporosis.
Recommendations for treatment in 2001 were based on T scores alone, while the 2010 recommendations use an assessment of absolute fracture risk based on FRAX for postmenopausal women and for men age 50 and older.
A clinician’s guide that summarizes the ACR recommendations is available at www.rheumatology.org/practice/clinical/guidelines/.
RECOMMENDATIONS DO NOT REPLACE CLINICAL JUDGMENT
Although the 2010 recommendations were more rigorous in their development process than those of 2001, they have limitations and they should not replace clinical judgment. Rather, they are intended to provide an evidence-based approach to guide clinicians in making treatment choices in patients on glucocorticoid therapy.
CONSIDERING ABSOLUTE FRACTURE RISK IN TREATMENT DECISIONS
The 2001 ACR guidelines recommended fracture-preventing treatment in all patients starting glucocorticoid therapy at more than 5 mg/day if the planned duration of treatment was at least 3 months, and in patients on long-term glucocorticoid therapy if the T score was less than −1.0. While these guidelines were simple and easy to use, they were not specific enough to provide useful guidance in specific scenarios.
A model of absolute fracture risk was not available in 2001. A 55-year old white woman with a T score of −1.1 who smoked, who had been using 5 mg of prednisone for the last 12 months, and who had stable bone mass on serial DXA scans would have been recommended for treatment based on the 2001 recommendations. If this patient’s FRAX-calculated 10-year absolute risk of a major osteoporotic fracture is less than 10%, that would be well below the National Osteoporosis Foundation’s cost-effective treatment threshold of 20%. The new guidelines suggest no treatment is needed, since the risk category is low and the dose is less than 7.5 mg. However, if on serial DXA this patient had a significant decline in bone mass, the guidelines suggest shifting the patient to a higher risk category, ie, from low to medium risk, which would result in a recommendation in favor of treatment.
The 2010 recommendations are not as simple to use as those from 2001. They encourage using FRAX to calculate fracture risk; thus, knowledge of the strengths and limitations of FRAX is required. Access to the internet in the examination room or use of the FRAX tool on a smartphone as well as willingness to spend a minute to calculate fracture risk are needed. For those who cannot or choose not to use the FRAX tool, the ACR publication provides tables for patient risk assessment based on age and T score. However, the tables would have to be readily available in the clinic, which may not be practical.
The 2010 recommendation provide a more nuanced approach to treatment in patients on glucocorticoid therapy and are likely to change treatment decisions based on their use, just as FRAX has altered treatment decisions in patients with primary osteoporosis.23
FRAX has limitations
FRAX underestimates the effect of glucocorticoids on fracture risk because steroid use is a yes-or-no question and its weight represents the average risk in a population that has ever used steroids, most of whom were using doses between 2.5 and 7.5 mg.
The WHO recognized this limitation and suggested an upward adjustment of risk for patients on 7.5 mg or more, ranging from 10% to 25%.21 For patients on high doses of steroids, this adjustment is still likely to result in underestimation of fracture risk and undertreatment of glucocorticoid-treated patients.
The 2010 recommendations adjust for this limitation, recommending treatment in the low-risk and medium-risk categories if the glucocorticoid dose is 7.5 mg or higher. If a patient is using high daily doses of steroids or has a declining bone density, the 2010 recommendations suggest increasing the risk category from low to medium or medium to high.
FRAX risk factors are dichotomous (yes/no) and are not adjusted for dose effects such as multiple fractures (vs a single fracture), heavy smoking (vs light smoking), heavy alcohol use (6 units per day vs 3 units), or severe rheumatoid arthritis (vs mild disease). Family history of osteoporosis in the FRAX is limited to parents with a hip fracture—vertebral fractures in a family member do not count.
Since FRAX uses the bone mineral density in the hip, it underestimates fracture risk in patients with low spine density but normal hip density. It may also underestimate fracture risk in patients with declining bone mass; the 2010 recommendations suggest the clinician should increase the risk category in this situation.
LIMITATIONS OF THE GUIDELINES
The 2010 recommendations do not include several important groups in which steroids are used, including transplant recipients, children, and patients on inhaled corticosteroids. The panel thought that there were insufficient data to make recommendations for these populations, as well as for premenopausal women and men younger than 50 years who did not have a prevalent fracture. The absence of a recommendation in these situations should not be considered a recommendation for no treatment; it is an acknowledgment of a lack of evidence, a lack of consensus among experts, and the need for additional clinical trials.
For premenopausal women and men under age 50 with a fracture, the recommendations are complicated and not intuitive. Zoledronic acid is not recommended for women of non-childbearing potential with a glucocorticoid duration of 1 to 3 months unless the steroid dose is at least 7.5 mg. This recommendation was based on panel voting and consensus that giving zoledronic acid, a medication with a 1-year duration of effect, in a patient on steroids for only 1 to 3 months was not warranted.
Teriparatide was recommended only if glucocorticoids are used for at least 3 months, although anyone who already has a fracture might be considered at high enough risk to warrant anabolic therapy regardless of steroid use or duration.
Zoledronic acid was excluded in women of childbearing potential, based on panel voting and consensus that drugs given in smaller amounts over 1 year might be less harmful to a fetus than one with a longer half-life given in a larger bolus once a year.
The panel could reach no consensus on women of childbearing potential with a prevalent fracture who were using less than 7.5 mg/day of glucocorticoids. A lack of consensus was the result of insufficient data to make evidence-based decisions and a disagreement among experts on the correct treatment.
The guidelines do not address the duration of treatment with bisphosphonates, a topic of importance because of concern for the potential long-term side effects of these medications.
THE BOTTOM LINE
The 2010 recommendations add a degree of complexity, with different medications recommended on the basis of glucocorticoid dose and duration as well as patient age, menopausal status, and childbearing potential. Guideline developers and clinicians face a difficult trade-off: easy-to-follow guidelines or more targeted guidelines that are more complex and therefore more difficult to use than previous guidelines.
This criticism is reasonable. The complexity is a result of insufficient evidence from clinical trials to make more exact and user-friendly recommendations, and also a result of the RAND/UCLA methodology. In cases that lack sufficient evidence on which to make a decision, the guideline development uses voting among experts in an attempt to develop consensus. This often results in complexity, lack of consensus, or inconsistencies.
The guidelines are straightforward for postmenopausal women and men age 50 and older on at least 7.5 mg prednisone for more than 3 months.
Since there is substantial evidence that many patients on glucocorticoid therapy go untreated, the risk of fracture in this population would be substantially reduced if clinicians would adhere to the recommendations.
Whenever a patient begins treatment with a glucocorticoid drug, we need to think about bone loss.
The American College of Rheumatology (ACR) issued recommendations for preventing and treating glucocorticoid-induced osteoporosis in 2010.1 Compared with its previous guidelines,2 the new ones are more tailored and nuanced but may be more difficult for physicians to follow. The guidelines call for assessing fracture risk using the computer-based Fracture Risk Assessment Tool, or FRAX (www/shef.ac.uk/FRAX), developed by the World Health Organization (WHO). For those without a computer or ready access to the Web, an application of FRAX is available for download on smartphones.
In this article, my purpose is to review the new recommendations and to offer my perspective, which does not necessarily reflect the opinions of the ACR.
DESPITE EVIDENCE, MANY PATIENTS RECEIVE NO INTERVENTION
Use of glucocorticoids is the most common cause of secondary osteoporosis. During the first 6 to 12 months of use, these drugs can cause a rapid loss of bone mass due to increased bone resorption; with continued use, they cause a slower but steady decline in bone mass due to reduced bone formation.3 Epidemiologic studies have found that the risk of fractures increases with dose, starting with doses as low as 2.5 mg per day of prednisone or its equivalent.4
Numerous clinical trials have evaluated the effect of bisphosphonates and teriparatide (Forteo) on bone mass and fracture risk in patients on glucocorticoid therapy. The bisphosphonates alendronate (Fosamax) and risedronate (Actonel) have both been shown to increase bone mass and reduce vertebral fracture risk in glucocorticoid recipients.5–8 Zoledronic acid (Reclast), a parenteral bisphosphonate given in one annual dose, was shown to increase bone mass more than oral risedronate taken daily,9 and teriparatide, a formulation of parathyroid hormone, was better than alendronate.10
However, despite the known risk of fractures with glucocorticoid use and the demonstrated efficacy of available agents in preventing bone loss and fracture, many patients do not receive any intervention.11,12
WHAT HAS HAPPENED SINCE 2001?
In the interval since 2001, several guidelines for managing glucocorticoid-induced osteoporosis have been published in other countries.13–17 Broadly speaking, they recommend starting preventive drug therapy for patients at risk of fracture at the same time glucocorticoid drugs are started if the patient is expected to take glucocorticoids for more than 3 to 6 months in doses higher than 5 to 7.5 mg of prednisone or its equivalent daily.
Recommendations for patients who have been on glucocorticoids for longer than 3 to 6 months at initial evaluation have been based largely on T scores derived from dual-energy x-ray absorptiometry (DXA). Thresholds for initiating therapy have varied: the ACR in 2001 recommended preventive treatment if the T score is lower than −1.0, whereas British guidelines said −1.5 and Dutch guidelines said −2.5.
In the United States, since 2001 when the ACR published its last guidelines,2 zoledronic acid and teriparatide have been approved for use in glucocorticoid-induced osteoporosis. In addition, guideline-development methodology has evolved and now is more scientifically rigorous. Finally, a risk-assessment tool has been developed that enables a more tailored approach (see below).
FRAX (www.shef.ac.uk/FRAX)
FRAX is a tool developed by the WHO to calculate the risk of fracture. If you go to the FRAX Web site and enter the required clinical information (race, age, sex, weight, height, previous fracture, family history of a fractured hip in a parent, current smoking, use of glucocorticoids, rheumatoid arthritis, secondary osteoporosis, consumption of three or more units of alcohol per day, and bone mineral density of the femoral neck), it will tell you the patient’s 10-year absolute (not relative) risk of major osteoporotic fracture and of hip fracture.
Since FRAX was unveiled in 2008, calculation of absolute fracture risk has become the standard method for making treatment decisions in patients with low bone mass who have not yet received any fracture-preventing treatment.18 The use of clinical risk factors in FRAX increases its ability to predict risk over and above the use of bone density by itself. And glucocorticoids are one of the clinical risk factors in FRAX.
But in which patients is treatment with a bisphosphonate or teriparatide cost-effective?
Thresholds for cost-effectiveness have been developed on the basis of economic assumptions that are country-specific. In the United States, the National Osteoporosis Foundation recommends drug therapy if the 10-year absolute risk of a major osteoporotic fracture of the hip, spine (clinical, not radiographic), wrist, or humerus is greater than 20% or if the risk of a hip fracture is greater than 3%.19
At equivalent bone densities, women taking glucocorticoids are at considerably higher risk of fracture than nonusers.20 For example, consider a 65-year-old white woman, weight 59 kg, height 163 cm, no previous fractures, no parent with a fractured hip, no current smoking, no rheumatoid arthritis, no secondary osteoporosis, no excessive alcohol use, and a T score of −2.2 in the femoral neck. (Try this on the FRAX Web site.) If she does not use glucocorticoids, her 10-year risk of hip fracture is 2.0%; using glucocorticoids increases the risk to 3.6%. This is higher than the 3% National Osteoporosis Foundation guideline; thus, treatment would be recommended.
Also using FRAX, a 55-year-old white woman with a T score of −1.8 and on glucocorticoid therapy has a 67% higher risk of major osteoporotic fracture and an 80% higher risk of hip fracture.
For a third example, a white woman age 60, weight 70 kg, height 168 cm, negative for all the other risk factors but with a T score of −2.1 and on glucocorticoids has a calculated 10-year fracture risk of 2.1%, which is below the National Osteoporosis Foundation treatment threshold. However, most clinicians would probably recommend treatment for her, depending on the anticipated dose and duration of glucocorticoid therapy.
A caveat. In FRAX, glucocorticoid therapy is a categorical variable—a yes-or-no question—and yes is defined as having ever used a glucocorticoid in a dose greater than 5 mg for more than 3 months. Therefore, according to FRAX, a patient who took 5 mg of prednisone for 3 months 5 years ago has the same fracture risk as a patient on 60 mg of prednisone after a diagnosis of temporal arteritis. For this reason, the FRAX tool is likely to underestimate fracture risk, especially in patients currently taking glucocorticoids and those on higher doses of these drugs.
Kanis et al used the General Practice Research Database to adjust the fracture risk for glucocorticoid use in FRAX.21 At doses higher than 7.5 mg, the fracture risk had to be revised upward by 10% to 25% depending on the fracture site (hip vs any major osteoporotic fracture) and age (greater at age 40 than at age 90).
The underestimation of fracture risk led the ACR Expert Advisory Panel to create risk strata for major osteoporotic fractures, ie, low (< 10% risk per 10 years), medium (10%–20%), and high (> 20%) and uses these cut points to make treatment recommendations.
HOW THE 2010 GUIDELINES WERE DEVELOPED
Whereas the 2001 recommendations were based on a more informal consensus approach, the 2010 recommendations use a more scientifically rigorous methodology for guideline development, the Research and Development/University of California at Los Angeles (RAND/UCLA) Appropriateness Method. The RAND/UCLA method combines the best available scientific evidence with expert opinion to develop practice guidelines.
In drawing up the 2010 recommendations the ACR used three panels of experts. The Core Executive Panel conducted a systematic review of controlled clinical trials of therapies currently approved for treating glucocorticoid-induced osteoporosis in the United States, Canada, or the European Union. They found 53 articles meeting their inclusion criteria; an evidence report was produced that informed the development of the recommendations. This evidence report and guideline development process is available at http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)2151-4658. The Expert Advisory Panel framed the recommendations, and the Task Force Panel voted on them. The Core Executive Panel and Expert Advisory Panel constructed 48 patient-specific clinical scenarios using four variables: sex, age, race/ethnicity, and femoral neck T scores.
The members of the Task Force Panel were asked to use the evidence report and their expert judgment to vote on and rate the appropriateness of using a specific therapy in the context of each scenario on a 9-point Likert scale (1 = appropriate; 9 = not appropriate). Agreement occurred when 7 or more of the 10 panel members rated a scenario 1, 2, or 3. Disagreements were defined as 3 or more of the 10 members rating the scenario between 4 and 9 while the other members rated it lower.
Disagreements in voting were discussed in an attempt to achieve consensus, and a second vote was conducted which determined the final recommendations. If disagreement remained after the vote, no recommendation was made.
No attempt was made to assign priority of one drug over another when multiple drugs were deemed appropriate, although the final recommendations did differentiate drugs based on patient categories.
START WITH COUNSELING, ASSESSMENT
For patients starting or already on glucocorticoid therapy that is expected to last at least 3 months, the first step is to counsel them on lifestyle modifications (Table 1) and to assess their risk factors (Figure 1). Recommendations for monitoring patients receiving glucocorticoid therapy for at least 3 months are presented in Table 2.
These recommendations are based on literature review, and the strength of evidence is graded:
- Grade A—derived from multiple randomized controlled trials or a meta-analysis
- Grade B—derived from a single randomized controlled trial or nonrandomized study
- Grade C—derived from consensus, expert opinion, or case series.
This system is the same one used by the American College of Cardiology and is based on clinical trial data.22
Recommendations for calcium intake and vitamin D supplementation were graded A; all other recommendations were graded C (Tables 1 and 2). It is important to note that practices that receive a grade of C may still be accepted as standard of care, such as fall assessment and smoking cessation.
FOR POSTMENOPAUSAL WOMEN AND FOR MEN AGE 50 AND OLDER
FRAX low-risk group
Recall that “low risk” based on the new ACR guidelines means that the 10-year absolute risk of a major osteoporotic fracture, as calculated with FRAX, is less than 10%.
- If glucocorticoid use is expected to last or has already lasted at least 3 months and the dose is less than 7.5 mg/day, no pharmacologic treatment is recommended.
- If glucocorticoid use is expected to last or has already lasted at least 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended.
Comment. These are the most straightforward of the recommendations. All three bisphosphonates are recommended as treatment options if the glucocorticoid dose is at least 7.5 mg/day and the duration at least 3 months. Ibandronate (Boniva) was not included because it has no data from clinical trials.
FRAX medium-risk group
“Medium risk” means that the 10-year absolute fracture risk of major osteoporotic fractures is 10% to 20%.
- If glucocorticoid use is anticipated to last or has lasted at least 3 months and the dose is less than 7.5 mg/day, alendronate or risedronate is recommended.
- If glucocorticoid use is anticipated to last or has lasted at least 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended.
Comment. Treatment is recommended at all glucocorticoid doses for patients in the medium-risk category if the duration of glucocorticoid treatment is at least 3 months, with one difference: zoledronic acid is recommended only if the glucocorticoid dose is 7.5 mg/day or higher. This inconsistency persisted after a second round of voting by the Task Force Panel.
FRAX high-risk group
In this group, the 10-year risk of major osteoporotic fractures is higher than 20%.
- If the glucocorticoid dose is less than 5 mg/day for up to 1 month, alendronate, risedronate, or zoledronic acid is recommended.
- If the dose is 5 mg/day or more for up to 1 month, or any dose for more than 1 month, alendronate, risedronate, zoledronic acid or teriparatide is recommended.
Comment. Based on current National Osteoporosis Foundation guidelines, all patients with a 10-year risk greater than 20% are recommended for treatment for any duration and dose of glucocorticoid use. However, teriparatide is recommended only if the duration of glucocorticoid therapy is more than 1 month.
FOR PREMENOPAUSAL WOMEN AND FOR MEN YOUNGER THAN AGE 50
Use of FRAX is not appropriate in premenopausal women or in men younger than 50 years.
Younger patients with no prevalent fracture
For men younger than 50 and premenopausal women who have not had a previous fracture, data were considered inadequate to make a recommendation, and no votes were taken.
Prevalent fracture in premenopausal women of nonchildbearing potential
In premenopausal women of nonchildbearing potential who have had a fracture:
- If the glucocorticoid duration is 1 to 3 months and the dose is 5 mg/day or higher, alendronate or risedronate is recommended.
- If the duration is 1 to 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended
- If the duration is more than 3 months, alendronate, risedronate, zoledronic acid, or teriparatide is recommended.
Comment. Treatment is recommended with any of the four medications in patients with a fracture and treated with glucocorticoids for more than 3 months. For shorter-duration glucocorticoid use (1–3 months) at 5 mg/day or higher, only alendronate and risedronate are recommended. If the dose is 7.5 mg/day or higher, any bisphosphonate is recommended. Zoledronic acid was consistently differentiated by the expert panel on the basis of dose and duration of glucocorticoid use, in view of its 1-year duration of effect after one dose.
Prevalent fracture in women of childbearing potential
- If the glucocorticoid duration is 1 to 3 months, there was no consensus (ie, voting disagreements could not be resolved).
- If the glucocorticoid duration is more than 3 months and the dose is 7.5 mg/day or more, alendronate, risedronate, or teriparatide is recommended.
- If the glucocorticoid duration is more than 3 months and the dose is less than 7.5 mg/day, there was no consensus.
Comment. Childbearing potential creates further complexities because of concern about fetal toxicity with bisphosphonates. For short-term glucocorticoid therapy at any dose and for therapy longer than 3 months at less than 7.5 mg, no consensus could be reached. For therapy longer than 3 months and with 7.5 mg/day or higher, treatment is recommended but not with zoledronic acid, based on the long half-life of the drug and concern for fetal toxicity.
Additional risk stratification
The panel recommended that if the following were present, a shift to a higher fracture risk category should be considered (low to medium, or medium to high):
- High daily dose of glucocorticoid
- High cumulative glucocorticoid dose
- Declining bone mineral density on serial DXA.
These are known risk factors that increase fracture risk but would not affect fracture risk in the FRAX model.
WHAT IS NEW IN THE 2010 RECOMMENDATIONS?
Recommendations for counseling now include fall risk assessment, height measurement, 25-hydroxyvitamin D measurement, and evaluation of patients for prevalent and incident fractures using vertebral fracture assessment by DXA or radiographic imaging of the spine.
Recommended drugs now include teriparatide and zoledronic acid, while estrogen and testosterone are no longer recommended as therapies for glucocorticoid-induced osteoporosis. Ibandronate is not included, since there have been no randomized controlled trials of this bisphosphonate in glucocorticoid-induced osteoporosis.
Recommendations for treatment in 2001 were based on T scores alone, while the 2010 recommendations use an assessment of absolute fracture risk based on FRAX for postmenopausal women and for men age 50 and older.
A clinician’s guide that summarizes the ACR recommendations is available at www.rheumatology.org/practice/clinical/guidelines/.
RECOMMENDATIONS DO NOT REPLACE CLINICAL JUDGMENT
Although the 2010 recommendations were more rigorous in their development process than those of 2001, they have limitations and they should not replace clinical judgment. Rather, they are intended to provide an evidence-based approach to guide clinicians in making treatment choices in patients on glucocorticoid therapy.
CONSIDERING ABSOLUTE FRACTURE RISK IN TREATMENT DECISIONS
The 2001 ACR guidelines recommended fracture-preventing treatment in all patients starting glucocorticoid therapy at more than 5 mg/day if the planned duration of treatment was at least 3 months, and in patients on long-term glucocorticoid therapy if the T score was less than −1.0. While these guidelines were simple and easy to use, they were not specific enough to provide useful guidance in specific scenarios.
A model of absolute fracture risk was not available in 2001. A 55-year old white woman with a T score of −1.1 who smoked, who had been using 5 mg of prednisone for the last 12 months, and who had stable bone mass on serial DXA scans would have been recommended for treatment based on the 2001 recommendations. If this patient’s FRAX-calculated 10-year absolute risk of a major osteoporotic fracture is less than 10%, that would be well below the National Osteoporosis Foundation’s cost-effective treatment threshold of 20%. The new guidelines suggest no treatment is needed, since the risk category is low and the dose is less than 7.5 mg. However, if on serial DXA this patient had a significant decline in bone mass, the guidelines suggest shifting the patient to a higher risk category, ie, from low to medium risk, which would result in a recommendation in favor of treatment.
The 2010 recommendations are not as simple to use as those from 2001. They encourage using FRAX to calculate fracture risk; thus, knowledge of the strengths and limitations of FRAX is required. Access to the internet in the examination room or use of the FRAX tool on a smartphone as well as willingness to spend a minute to calculate fracture risk are needed. For those who cannot or choose not to use the FRAX tool, the ACR publication provides tables for patient risk assessment based on age and T score. However, the tables would have to be readily available in the clinic, which may not be practical.
The 2010 recommendation provide a more nuanced approach to treatment in patients on glucocorticoid therapy and are likely to change treatment decisions based on their use, just as FRAX has altered treatment decisions in patients with primary osteoporosis.23
FRAX has limitations
FRAX underestimates the effect of glucocorticoids on fracture risk because steroid use is a yes-or-no question and its weight represents the average risk in a population that has ever used steroids, most of whom were using doses between 2.5 and 7.5 mg.
The WHO recognized this limitation and suggested an upward adjustment of risk for patients on 7.5 mg or more, ranging from 10% to 25%.21 For patients on high doses of steroids, this adjustment is still likely to result in underestimation of fracture risk and undertreatment of glucocorticoid-treated patients.
The 2010 recommendations adjust for this limitation, recommending treatment in the low-risk and medium-risk categories if the glucocorticoid dose is 7.5 mg or higher. If a patient is using high daily doses of steroids or has a declining bone density, the 2010 recommendations suggest increasing the risk category from low to medium or medium to high.
FRAX risk factors are dichotomous (yes/no) and are not adjusted for dose effects such as multiple fractures (vs a single fracture), heavy smoking (vs light smoking), heavy alcohol use (6 units per day vs 3 units), or severe rheumatoid arthritis (vs mild disease). Family history of osteoporosis in the FRAX is limited to parents with a hip fracture—vertebral fractures in a family member do not count.
Since FRAX uses the bone mineral density in the hip, it underestimates fracture risk in patients with low spine density but normal hip density. It may also underestimate fracture risk in patients with declining bone mass; the 2010 recommendations suggest the clinician should increase the risk category in this situation.
LIMITATIONS OF THE GUIDELINES
The 2010 recommendations do not include several important groups in which steroids are used, including transplant recipients, children, and patients on inhaled corticosteroids. The panel thought that there were insufficient data to make recommendations for these populations, as well as for premenopausal women and men younger than 50 years who did not have a prevalent fracture. The absence of a recommendation in these situations should not be considered a recommendation for no treatment; it is an acknowledgment of a lack of evidence, a lack of consensus among experts, and the need for additional clinical trials.
For premenopausal women and men under age 50 with a fracture, the recommendations are complicated and not intuitive. Zoledronic acid is not recommended for women of non-childbearing potential with a glucocorticoid duration of 1 to 3 months unless the steroid dose is at least 7.5 mg. This recommendation was based on panel voting and consensus that giving zoledronic acid, a medication with a 1-year duration of effect, in a patient on steroids for only 1 to 3 months was not warranted.
Teriparatide was recommended only if glucocorticoids are used for at least 3 months, although anyone who already has a fracture might be considered at high enough risk to warrant anabolic therapy regardless of steroid use or duration.
Zoledronic acid was excluded in women of childbearing potential, based on panel voting and consensus that drugs given in smaller amounts over 1 year might be less harmful to a fetus than one with a longer half-life given in a larger bolus once a year.
The panel could reach no consensus on women of childbearing potential with a prevalent fracture who were using less than 7.5 mg/day of glucocorticoids. A lack of consensus was the result of insufficient data to make evidence-based decisions and a disagreement among experts on the correct treatment.
The guidelines do not address the duration of treatment with bisphosphonates, a topic of importance because of concern for the potential long-term side effects of these medications.
THE BOTTOM LINE
The 2010 recommendations add a degree of complexity, with different medications recommended on the basis of glucocorticoid dose and duration as well as patient age, menopausal status, and childbearing potential. Guideline developers and clinicians face a difficult trade-off: easy-to-follow guidelines or more targeted guidelines that are more complex and therefore more difficult to use than previous guidelines.
This criticism is reasonable. The complexity is a result of insufficient evidence from clinical trials to make more exact and user-friendly recommendations, and also a result of the RAND/UCLA methodology. In cases that lack sufficient evidence on which to make a decision, the guideline development uses voting among experts in an attempt to develop consensus. This often results in complexity, lack of consensus, or inconsistencies.
The guidelines are straightforward for postmenopausal women and men age 50 and older on at least 7.5 mg prednisone for more than 3 months.
Since there is substantial evidence that many patients on glucocorticoid therapy go untreated, the risk of fracture in this population would be substantially reduced if clinicians would adhere to the recommendations.
- Grossman JM, Gordon R, Ranganath VK, et al; American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res (Hoboken) 2010; 62:1515–1526.
- Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Arthritis Rheum 2001; 44:1496–1503.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Oral corticosteroids and fracture risk: relationship to daily and cumulative doses. Rheumatology (Oxford) 2000; 39:1383–1389.
- Saag KG, Emkey R, Schnitzer TJ, et al. Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. Glucocorticoid-Induced Osteoporosis Intervention Study Group. N Engl J Med 1998; 339:292–299.
- Cohen S, Levy RM, Keller M, et al. Risedronate therapy prevents corticosteroid-induced bone loss: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum 1999; 42:2309–2318.
- Reid DM, Hughes RA, Laan RF, et al. Efficacy and safety of daily risedronate in the treatment of corticosteroid-induced osteoporosis in men and women: a randomized trial. European Corticosteroid-Induced Osteoporosis Treatment Study. J Bone Miner Res 2000; 15:1006–1013.
- Wallach S, Cohen S, Reid DM, et al. Effects of risedronate treatment on bone density and vertebral fracture in patients on corticosteroid therapy. Calcif Tissue Int 2000; 67:277–285.
- Reid DM, Devogelaer JP, Saag K, et al; HORIZON investigators. Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet 2009; 373:1253–1263.
- Saag KG, Shane E, Boonen S, et al. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med 2007; 357:2028–2039.
- Curtis JR, Westfall AO, Allison JJ, et al. Longitudinal patterns in the prevention of osteoporosis in glucocorticoid-treated patients. Arthritis Rheum 2005; 52:2485–2494.
- Feldstein AC, Elmer PJ, Nichols GA, Herson M. Practice patterns in patients at risk for glucocorticoid-induced osteoporosis. Osteoporos Int 2005; 16:2168–2174.
- Brown JP, Josse RG; Scientific Advisory Council of the Osteoporosis Society of Canada. 2002 clinical practice guidelines for the diagnosis and management of osteoporosis in Canada. CMAJ 2002; 167(suppl 10):S1–S34.
- Devogelaer JP, Goemaere S, Boonen S, et al. Evidence-based guidelines for the prevention and treatment of glucocorticoid-induced osteoporosis: a consensus document of the Belgian Bone Club. Osteoporos Int 2006; 17:8–19.
- Gourlay M, Franceschini N, Sheyn Y. Prevention and treatment strategies for glucocorticoid-induced osteoporotic fractures. Clin Rheumatol 2007; 26:144–153.
- Nawata H, Soen S, Takayanagi R, et al; Subcommittee to Study Diagnostic Criteria for Glucocorticoid-Induced Osteoporosis. Guidelines on the management and treatment of glucocorticoid-induced osteoporosis of the Japanese Society for Bone and Mineral Research (2004). J Bone Miner Metab 2005; 23:105–109.
- Geusens PP, Lems WF, Verhaar HJ, et al. Review and evaluation of the Dutch guidelines for osteoporosis. J Eval Clin Pract 2006; 12:539–548.
- Kanis JA, Johnell O, Oden A, Johansson H, McCloskey E. FRAX and the assessment of fracture probability in men and women from the UK. Osteoporos Int 2008; 19:385–389.
- National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. Washington, DC, National Osteoporosis Foundation, 2010. http://nof.org/files/nof/public/content/file/344/upload/159.pdf. Accessed December 31, 2012.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johansson H, Oden A, McCloskey EV. Guidance for the adjustment of FRAX according to the dose of glucocorticoids. Osteoporos Int 2011; 22:809–816.
- Hunt SA, Abraham WT, Chin MH, et al; American College of Cardiology. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation 2005; 112:e154–e235.
- Dawson-Hughes B, Tosteson AN, Melton LJ, et al; National Osteoporosis Foundation Guide Committee. Implications of absolute fracture risk assessment for osteoporosis practice guidelines in the USA. Osteoporos Int 2008; 19:449–458.
- Grossman JM, Gordon R, Ranganath VK, et al; American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res (Hoboken) 2010; 62:1515–1526.
- Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Arthritis Rheum 2001; 44:1496–1503.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Oral corticosteroids and fracture risk: relationship to daily and cumulative doses. Rheumatology (Oxford) 2000; 39:1383–1389.
- Saag KG, Emkey R, Schnitzer TJ, et al. Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. Glucocorticoid-Induced Osteoporosis Intervention Study Group. N Engl J Med 1998; 339:292–299.
- Cohen S, Levy RM, Keller M, et al. Risedronate therapy prevents corticosteroid-induced bone loss: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum 1999; 42:2309–2318.
- Reid DM, Hughes RA, Laan RF, et al. Efficacy and safety of daily risedronate in the treatment of corticosteroid-induced osteoporosis in men and women: a randomized trial. European Corticosteroid-Induced Osteoporosis Treatment Study. J Bone Miner Res 2000; 15:1006–1013.
- Wallach S, Cohen S, Reid DM, et al. Effects of risedronate treatment on bone density and vertebral fracture in patients on corticosteroid therapy. Calcif Tissue Int 2000; 67:277–285.
- Reid DM, Devogelaer JP, Saag K, et al; HORIZON investigators. Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet 2009; 373:1253–1263.
- Saag KG, Shane E, Boonen S, et al. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med 2007; 357:2028–2039.
- Curtis JR, Westfall AO, Allison JJ, et al. Longitudinal patterns in the prevention of osteoporosis in glucocorticoid-treated patients. Arthritis Rheum 2005; 52:2485–2494.
- Feldstein AC, Elmer PJ, Nichols GA, Herson M. Practice patterns in patients at risk for glucocorticoid-induced osteoporosis. Osteoporos Int 2005; 16:2168–2174.
- Brown JP, Josse RG; Scientific Advisory Council of the Osteoporosis Society of Canada. 2002 clinical practice guidelines for the diagnosis and management of osteoporosis in Canada. CMAJ 2002; 167(suppl 10):S1–S34.
- Devogelaer JP, Goemaere S, Boonen S, et al. Evidence-based guidelines for the prevention and treatment of glucocorticoid-induced osteoporosis: a consensus document of the Belgian Bone Club. Osteoporos Int 2006; 17:8–19.
- Gourlay M, Franceschini N, Sheyn Y. Prevention and treatment strategies for glucocorticoid-induced osteoporotic fractures. Clin Rheumatol 2007; 26:144–153.
- Nawata H, Soen S, Takayanagi R, et al; Subcommittee to Study Diagnostic Criteria for Glucocorticoid-Induced Osteoporosis. Guidelines on the management and treatment of glucocorticoid-induced osteoporosis of the Japanese Society for Bone and Mineral Research (2004). J Bone Miner Metab 2005; 23:105–109.
- Geusens PP, Lems WF, Verhaar HJ, et al. Review and evaluation of the Dutch guidelines for osteoporosis. J Eval Clin Pract 2006; 12:539–548.
- Kanis JA, Johnell O, Oden A, Johansson H, McCloskey E. FRAX and the assessment of fracture probability in men and women from the UK. Osteoporos Int 2008; 19:385–389.
- National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. Washington, DC, National Osteoporosis Foundation, 2010. http://nof.org/files/nof/public/content/file/344/upload/159.pdf. Accessed December 31, 2012.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johansson H, Oden A, McCloskey EV. Guidance for the adjustment of FRAX according to the dose of glucocorticoids. Osteoporos Int 2011; 22:809–816.
- Hunt SA, Abraham WT, Chin MH, et al; American College of Cardiology. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation 2005; 112:e154–e235.
- Dawson-Hughes B, Tosteson AN, Melton LJ, et al; National Osteoporosis Foundation Guide Committee. Implications of absolute fracture risk assessment for osteoporosis practice guidelines in the USA. Osteoporos Int 2008; 19:449–458.
KEY POINTS
- The risk of fracture should be assessed at the start of glucocorticoid therapy.
- Factors that affect the decision to prescribe osteoporosis drugs include the patient’s risk of fractures as assessed with FRAX (www.shef.ac.uk/FRAX), the dose of glucocorticoid, and the projected duration of treatment.
- Since FRAX treats glucocorticoid use simply as a yes-or-no question, it likely underestimates the fracture risk in current users and at high doses. The estimate of risk should be adjusted upward in these situations.
When do Raynaud symptoms merit a workup for autoimmune rheumatic disease?
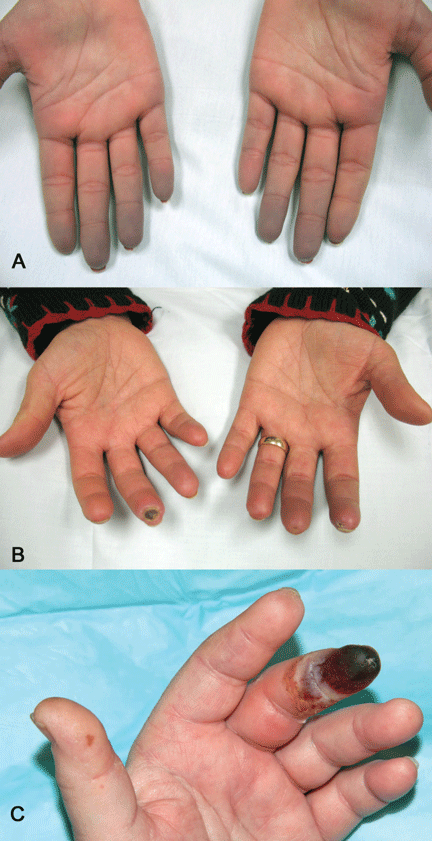
Indications that Raynaud phenomenon may be the presenting manifestation of a systemic autoimmune rheumatic disease are older age at onset (ie, over age 30), male sex, asymmetric involvement, and prolonged and painful attacks that can be severe enough to cause ischemic digital ulceration or gangrene (Figure 1).
Hence, chronic and severe digital ischemia causing ulceration or infarction differentiates secondary from primary Raynaud phenomenon and should prompt an investigation for an autoimmune rheumatic process. When taking the history, the clinician should seek clues to an underlying autoimmune condition, such as arthralgia, heartburn, dysphagia, shortness of breath, cough, and should examine the patient for telltale signs such as puffy hands and fingers, sclerodactyly, digital pitting scars, loss of fingertip pulp tissue, telangiectasias, and calcinosis.
CLUES TO PRIMARY VS SECONDARY RAYNAUD PHENOMENON
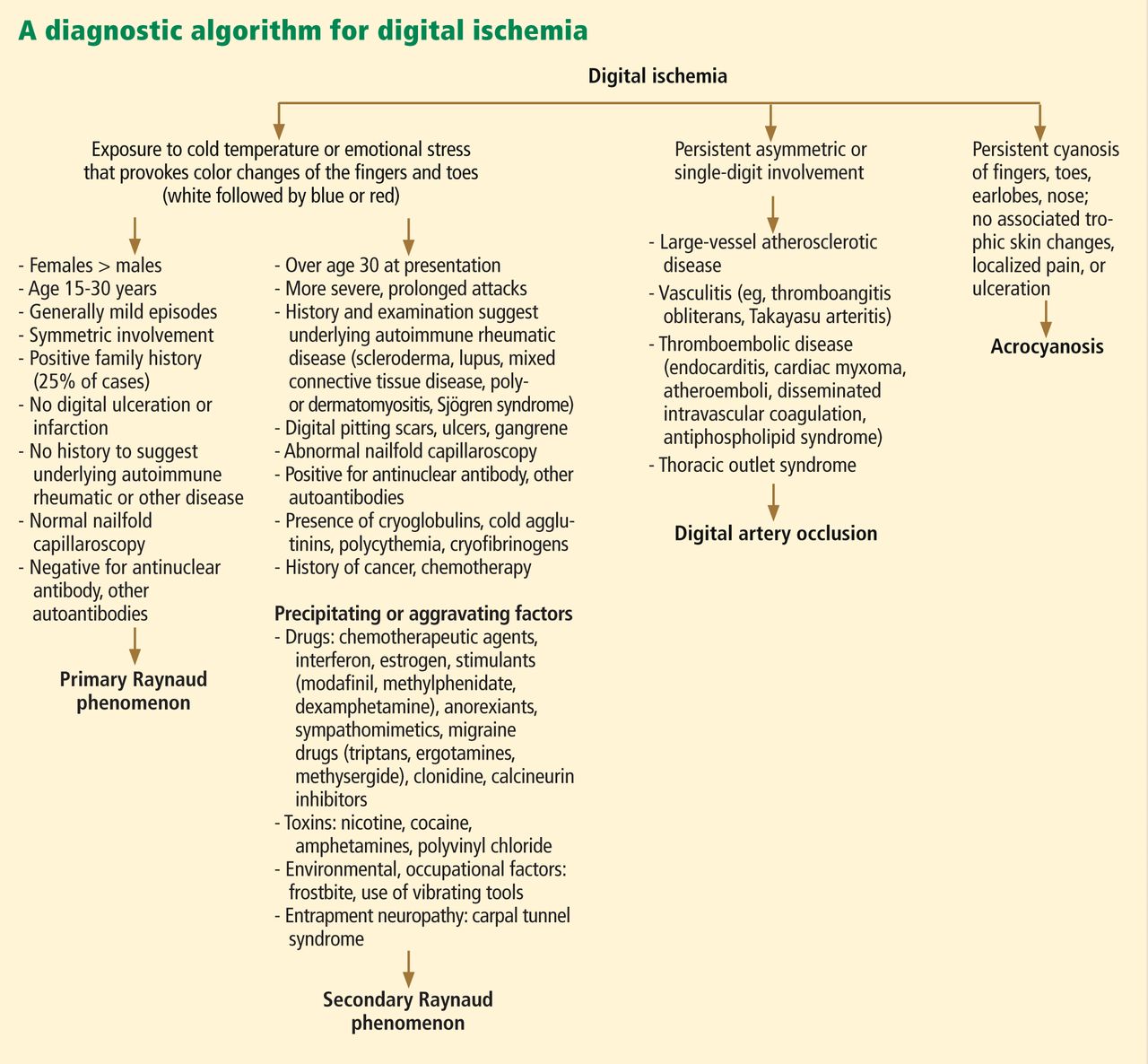
A diagnostic algorithm of digital ischemia (Figure 2) illustrates the range of presentations and possible causes. In Raynaud phenomenon, cold temperature and emotional stress provoke reversible color changes of the fingers and toes. Intense vasospasm of the digital arteries produces three well-defined phases1: white (pallor resulting from vasospasm), blue (dusky cyanosis due to deoxygenation of static venous blood) (Figure 1), and red (reactive hyperemia after the restoration of blood flow). However, only about 60% of patients have all three color changes. The attacks are associated with paresthesias, an uncomfortable feeling of coldness in the fingers, and ischemic pain.
Primary Raynaud phenomenon
Primary or idiopathic Raynaud phenomenon is seen in 5% to 10% of the general population. It more commonly affects women ages 15 to 30, is generally mild, involves the digits symmetrically, and is sometimes familial. An increase in alpha-2 adrenergic responses in the digital vessels leads to arterial vasospasm, an exaggerated physiologic response to cold temperatures.2 Geographic variability in prevalence likely represents differences in mean outdoor temperatures,3 which is in part why attacks of primary Raynaud phenomenon tend to be worse in the winter months.4
Secondary Raynaud phenomenon
Raynaud phenomenon also often occurs in certain autoimmune rheumatic diseases (secondary Raynaud phenomenon): for example, it is seen in scleroderma (90% to 95% of patients), mixed connective tissue disease (85%), systemic lupus erythematosus (40%), antisynthetase syndrome (40%), and sometimes in patients with other autoimmune rheumatic diseases. It may also be seen in hematologic disorders (cryoglobulinemia, cryofibrinogenemia, paraproteinemias, cold agglutinin disease, and polycythemia rubra vera), and it can also result from environmental and occupational exposures (frostbite, use of vibrating tools) and from exposure to certain drugs and toxins, such as polyvinyl chloride (Figure 2).
Acrocyanosis, a benign neurohormonal condition, should be included in the differential diagnosis for Raynaud phenomenon. Raynaud phenomenon is episodic, whereas acrocyanosis leads to persistent cyanosis of the acral body parts (fingers, toes) that is exacerbated by cold temperatures. However, the trophic skin changes, localized pain, and ulceration are not seen in acrocyanosis.
NAILFOLD CAPILLAROSCOPY: A KEY PART OF THE WORKUP
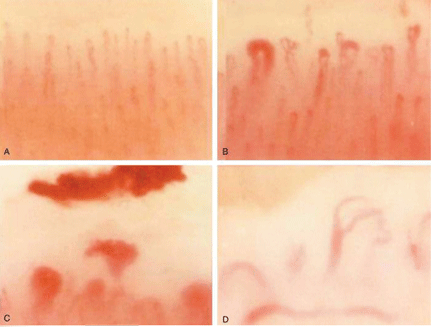
Nailfold capillaroscopy should be part of the evaluation of patients with Raynaud phenomenon (Figure 3), as it is one of the most reliable tests for distinguishing between primary and secondary Raynaud phenomenon.5 The sensitivity of the American College of Rheumatology classification criteria for systemic sclerosis increases significantly with the addition of nailfold capillary abnormalities.6,7
A stereomicroscope or videocapillaroscope is usually recommended to evaluate nailfold capillary morphology,5 but if such equipment is not available, a regular ophthalmoscope (with the lens set at 20 diopters or higher for better resolution) can serve the purpose at the bedside.8 A drop of mineral oil is placed on the nailfold to improve the image resolution, as it makes the horny layer of the cuticle transparent.
Abnormal patterns include dilated and enlarged capillary loops, disorganized capillaries, “dropouts” (avascular areas), microhemorrhages, and arborized capillaries (Figure 3).5 At no additional cost, the presence of these microvascular changes would add to the suspicion of secondary Raynaud phenomenon (negative predictive value of 93%).9 In addition, evolving capillaroscopic changes can be seen during follow-up visits, indicating the progressive nature of the microvasculopathy seen in these autoimmune rheumatic diseases.10
ADDITIONAL TESTING
If an underlying autoimmune rheumatic disease is suspected, laboratory testing should include a complete blood cell count, an erythrocyte sedimentation rate, and an antinuclear antibody (ANA) assay. If the ANA assay is negative, no further testing is usually necessary; however, a positive test should alert the clinician to consider an underlying autoimmune rheumatic process (negative predictive value of 93%).9 In a patient presenting with Raynaud phenomenon, a positive ANA test (even in the absence of other symptoms) warrants more frequent follow-up, urinalysis, and perhaps referral to a rheumatologist.
In the case of a positive ANA test, before ordering additional autoantibody tests, it is useful to consider the relevant non-Raynaud clinical manifestations. Indiscriminate ordering of a battery of autoantibodies should be avoided because of significant added cost and because it is not likely to provide additional information to guide management.
On the other hand, these more specific antibody tests may be of value in confirming the diagnosis suggested by the clinical profile of specific autoimmune rheumatic diseases, eg, anti-double-stranded DNA11 and anti-Smith12 antibodies for lupus, anti-topoisomerase I (Scl-70) and anti-centromere antibodies for scleroderma, 13 and anti-synthetase (eg, anti-Jo-1) antibodies for autoimmune myositis.14,15
- Raynaud M. On local asphyxia and symmetrical gangrene of the extremities (1862), and new research on the nature and treatment of local asphyxia of the extremities (1872).Barlow T, trans. Selected monographs (121). London: New Sydenham Society, 1988.
- Boin F, Wigley FM. Understanding, assessing and treating Raynaud’s phenomenon. Curr Opin Rheumatol 2005; 17:752–760.
- Maricq HR, Carpentier PH, Weinrich MC, et al. Geographic variation in the prevalence of Raynaud’s phenomenon: a 5-region comparison. J Rheumatol 1997; 24:879–889.
- Wigley FM. Clinical practice. Raynaud’s phenomenon. N Engl J Med 2002; 347:1001–1018.
- Cutolo M, Pizzorni C, Sulli A. Capillaroscopy. Best Pract Res Clin Rheumatol 2005; 19:437–452.
- Lonzetti LS, Joyal F, Raynauld JP, et al. Updating the American College of Rheumatology preliminary classification criteria for systemic sclerosis: addition of severe nailfold capillaroscopy abnormalities markedly increases the sensitivity for limited scleroderma. Arthritis Rheum 2001; 44:735–736.
- Hudson M, Taillefer S, Steele R, et al. Improving the sensitivity of the American College of Rheumatology classification criteria for systemic sclerosis. Clin Exp Rheumatol 2007; 25:754–757.
- Anders HJ, Sigl T, Schattenkirchner M. Differentiation between primary and secondary Raynaud’s phenomenon: a prospective study comparing nailfold capillaroscopy using an ophthalmoscope or stereomicroscope. Ann Rheum Dis 2001; 60:407–409.
- Spencer-Green G. Outcomes in primary Raynaud phenomenon: a meta-analysis of the frequency, rates, and predictors of transition to secondary diseases. Arch Intern Med 1998; 158:595–600.
- Wong ML, Highton J, Palmer DG. Sequential nailfold capillary microscopy in scleroderma and related disorders. Ann Rheum Dis 1988; 47:53–61.
- Weinstein A, Bordwell B, Stone B, Tibbetts C, Rothfield NF. Antibodies to native DNA and serum complement (C3) levels. Application to diagnosis and classification of systemic lupus erythematosus. Am J Med 1983; 74:206–216.
- Craft J. Antibodies to snRNPs in systemic lupus erythematosus. Rheum Dis Clin North Am 1992; 18:311–335.
- Weiner ES, Hildebrandt S, Senécal JL, et al. Prognostic significance of anticentromere antibodies and anti-topoisomerase I antibodies in Raynaud’s disease. A prospective study. Arthritis Rheum 1991; 34:68–77.
- Miller FW, Twitty SA, Biswas T, Plotz PH. Origin and regulation of a disease-specific autoantibody response. Antigenic epitopes, spectrotype stability, and isotype restriction of anti-Jo-1 autoantibodies. J Clin Invest 1990; 85:468–475.
- Ghirardello A, Zampieri S, Tarricone E, et al. Clinical implications of autoantibody screening in patients with autoimmune myositis. Autoimmunity 2006; 39:217–221.
Indications that Raynaud phenomenon may be the presenting manifestation of a systemic autoimmune rheumatic disease are older age at onset (ie, over age 30), male sex, asymmetric involvement, and prolonged and painful attacks that can be severe enough to cause ischemic digital ulceration or gangrene (Figure 1).
Hence, chronic and severe digital ischemia causing ulceration or infarction differentiates secondary from primary Raynaud phenomenon and should prompt an investigation for an autoimmune rheumatic process. When taking the history, the clinician should seek clues to an underlying autoimmune condition, such as arthralgia, heartburn, dysphagia, shortness of breath, cough, and should examine the patient for telltale signs such as puffy hands and fingers, sclerodactyly, digital pitting scars, loss of fingertip pulp tissue, telangiectasias, and calcinosis.
CLUES TO PRIMARY VS SECONDARY RAYNAUD PHENOMENON
A diagnostic algorithm of digital ischemia (Figure 2) illustrates the range of presentations and possible causes. In Raynaud phenomenon, cold temperature and emotional stress provoke reversible color changes of the fingers and toes. Intense vasospasm of the digital arteries produces three well-defined phases1: white (pallor resulting from vasospasm), blue (dusky cyanosis due to deoxygenation of static venous blood) (Figure 1), and red (reactive hyperemia after the restoration of blood flow). However, only about 60% of patients have all three color changes. The attacks are associated with paresthesias, an uncomfortable feeling of coldness in the fingers, and ischemic pain.
Primary Raynaud phenomenon
Primary or idiopathic Raynaud phenomenon is seen in 5% to 10% of the general population. It more commonly affects women ages 15 to 30, is generally mild, involves the digits symmetrically, and is sometimes familial. An increase in alpha-2 adrenergic responses in the digital vessels leads to arterial vasospasm, an exaggerated physiologic response to cold temperatures.2 Geographic variability in prevalence likely represents differences in mean outdoor temperatures,3 which is in part why attacks of primary Raynaud phenomenon tend to be worse in the winter months.4
Secondary Raynaud phenomenon
Raynaud phenomenon also often occurs in certain autoimmune rheumatic diseases (secondary Raynaud phenomenon): for example, it is seen in scleroderma (90% to 95% of patients), mixed connective tissue disease (85%), systemic lupus erythematosus (40%), antisynthetase syndrome (40%), and sometimes in patients with other autoimmune rheumatic diseases. It may also be seen in hematologic disorders (cryoglobulinemia, cryofibrinogenemia, paraproteinemias, cold agglutinin disease, and polycythemia rubra vera), and it can also result from environmental and occupational exposures (frostbite, use of vibrating tools) and from exposure to certain drugs and toxins, such as polyvinyl chloride (Figure 2).
Acrocyanosis, a benign neurohormonal condition, should be included in the differential diagnosis for Raynaud phenomenon. Raynaud phenomenon is episodic, whereas acrocyanosis leads to persistent cyanosis of the acral body parts (fingers, toes) that is exacerbated by cold temperatures. However, the trophic skin changes, localized pain, and ulceration are not seen in acrocyanosis.
NAILFOLD CAPILLAROSCOPY: A KEY PART OF THE WORKUP
Nailfold capillaroscopy should be part of the evaluation of patients with Raynaud phenomenon (Figure 3), as it is one of the most reliable tests for distinguishing between primary and secondary Raynaud phenomenon.5 The sensitivity of the American College of Rheumatology classification criteria for systemic sclerosis increases significantly with the addition of nailfold capillary abnormalities.6,7
A stereomicroscope or videocapillaroscope is usually recommended to evaluate nailfold capillary morphology,5 but if such equipment is not available, a regular ophthalmoscope (with the lens set at 20 diopters or higher for better resolution) can serve the purpose at the bedside.8 A drop of mineral oil is placed on the nailfold to improve the image resolution, as it makes the horny layer of the cuticle transparent.
Abnormal patterns include dilated and enlarged capillary loops, disorganized capillaries, “dropouts” (avascular areas), microhemorrhages, and arborized capillaries (Figure 3).5 At no additional cost, the presence of these microvascular changes would add to the suspicion of secondary Raynaud phenomenon (negative predictive value of 93%).9 In addition, evolving capillaroscopic changes can be seen during follow-up visits, indicating the progressive nature of the microvasculopathy seen in these autoimmune rheumatic diseases.10
ADDITIONAL TESTING
If an underlying autoimmune rheumatic disease is suspected, laboratory testing should include a complete blood cell count, an erythrocyte sedimentation rate, and an antinuclear antibody (ANA) assay. If the ANA assay is negative, no further testing is usually necessary; however, a positive test should alert the clinician to consider an underlying autoimmune rheumatic process (negative predictive value of 93%).9 In a patient presenting with Raynaud phenomenon, a positive ANA test (even in the absence of other symptoms) warrants more frequent follow-up, urinalysis, and perhaps referral to a rheumatologist.
In the case of a positive ANA test, before ordering additional autoantibody tests, it is useful to consider the relevant non-Raynaud clinical manifestations. Indiscriminate ordering of a battery of autoantibodies should be avoided because of significant added cost and because it is not likely to provide additional information to guide management.
On the other hand, these more specific antibody tests may be of value in confirming the diagnosis suggested by the clinical profile of specific autoimmune rheumatic diseases, eg, anti-double-stranded DNA11 and anti-Smith12 antibodies for lupus, anti-topoisomerase I (Scl-70) and anti-centromere antibodies for scleroderma, 13 and anti-synthetase (eg, anti-Jo-1) antibodies for autoimmune myositis.14,15
Indications that Raynaud phenomenon may be the presenting manifestation of a systemic autoimmune rheumatic disease are older age at onset (ie, over age 30), male sex, asymmetric involvement, and prolonged and painful attacks that can be severe enough to cause ischemic digital ulceration or gangrene (Figure 1).
Hence, chronic and severe digital ischemia causing ulceration or infarction differentiates secondary from primary Raynaud phenomenon and should prompt an investigation for an autoimmune rheumatic process. When taking the history, the clinician should seek clues to an underlying autoimmune condition, such as arthralgia, heartburn, dysphagia, shortness of breath, cough, and should examine the patient for telltale signs such as puffy hands and fingers, sclerodactyly, digital pitting scars, loss of fingertip pulp tissue, telangiectasias, and calcinosis.
CLUES TO PRIMARY VS SECONDARY RAYNAUD PHENOMENON
A diagnostic algorithm of digital ischemia (Figure 2) illustrates the range of presentations and possible causes. In Raynaud phenomenon, cold temperature and emotional stress provoke reversible color changes of the fingers and toes. Intense vasospasm of the digital arteries produces three well-defined phases1: white (pallor resulting from vasospasm), blue (dusky cyanosis due to deoxygenation of static venous blood) (Figure 1), and red (reactive hyperemia after the restoration of blood flow). However, only about 60% of patients have all three color changes. The attacks are associated with paresthesias, an uncomfortable feeling of coldness in the fingers, and ischemic pain.
Primary Raynaud phenomenon
Primary or idiopathic Raynaud phenomenon is seen in 5% to 10% of the general population. It more commonly affects women ages 15 to 30, is generally mild, involves the digits symmetrically, and is sometimes familial. An increase in alpha-2 adrenergic responses in the digital vessels leads to arterial vasospasm, an exaggerated physiologic response to cold temperatures.2 Geographic variability in prevalence likely represents differences in mean outdoor temperatures,3 which is in part why attacks of primary Raynaud phenomenon tend to be worse in the winter months.4
Secondary Raynaud phenomenon
Raynaud phenomenon also often occurs in certain autoimmune rheumatic diseases (secondary Raynaud phenomenon): for example, it is seen in scleroderma (90% to 95% of patients), mixed connective tissue disease (85%), systemic lupus erythematosus (40%), antisynthetase syndrome (40%), and sometimes in patients with other autoimmune rheumatic diseases. It may also be seen in hematologic disorders (cryoglobulinemia, cryofibrinogenemia, paraproteinemias, cold agglutinin disease, and polycythemia rubra vera), and it can also result from environmental and occupational exposures (frostbite, use of vibrating tools) and from exposure to certain drugs and toxins, such as polyvinyl chloride (Figure 2).
Acrocyanosis, a benign neurohormonal condition, should be included in the differential diagnosis for Raynaud phenomenon. Raynaud phenomenon is episodic, whereas acrocyanosis leads to persistent cyanosis of the acral body parts (fingers, toes) that is exacerbated by cold temperatures. However, the trophic skin changes, localized pain, and ulceration are not seen in acrocyanosis.
NAILFOLD CAPILLAROSCOPY: A KEY PART OF THE WORKUP
Nailfold capillaroscopy should be part of the evaluation of patients with Raynaud phenomenon (Figure 3), as it is one of the most reliable tests for distinguishing between primary and secondary Raynaud phenomenon.5 The sensitivity of the American College of Rheumatology classification criteria for systemic sclerosis increases significantly with the addition of nailfold capillary abnormalities.6,7
A stereomicroscope or videocapillaroscope is usually recommended to evaluate nailfold capillary morphology,5 but if such equipment is not available, a regular ophthalmoscope (with the lens set at 20 diopters or higher for better resolution) can serve the purpose at the bedside.8 A drop of mineral oil is placed on the nailfold to improve the image resolution, as it makes the horny layer of the cuticle transparent.
Abnormal patterns include dilated and enlarged capillary loops, disorganized capillaries, “dropouts” (avascular areas), microhemorrhages, and arborized capillaries (Figure 3).5 At no additional cost, the presence of these microvascular changes would add to the suspicion of secondary Raynaud phenomenon (negative predictive value of 93%).9 In addition, evolving capillaroscopic changes can be seen during follow-up visits, indicating the progressive nature of the microvasculopathy seen in these autoimmune rheumatic diseases.10
ADDITIONAL TESTING
If an underlying autoimmune rheumatic disease is suspected, laboratory testing should include a complete blood cell count, an erythrocyte sedimentation rate, and an antinuclear antibody (ANA) assay. If the ANA assay is negative, no further testing is usually necessary; however, a positive test should alert the clinician to consider an underlying autoimmune rheumatic process (negative predictive value of 93%).9 In a patient presenting with Raynaud phenomenon, a positive ANA test (even in the absence of other symptoms) warrants more frequent follow-up, urinalysis, and perhaps referral to a rheumatologist.
In the case of a positive ANA test, before ordering additional autoantibody tests, it is useful to consider the relevant non-Raynaud clinical manifestations. Indiscriminate ordering of a battery of autoantibodies should be avoided because of significant added cost and because it is not likely to provide additional information to guide management.
On the other hand, these more specific antibody tests may be of value in confirming the diagnosis suggested by the clinical profile of specific autoimmune rheumatic diseases, eg, anti-double-stranded DNA11 and anti-Smith12 antibodies for lupus, anti-topoisomerase I (Scl-70) and anti-centromere antibodies for scleroderma, 13 and anti-synthetase (eg, anti-Jo-1) antibodies for autoimmune myositis.14,15
- Raynaud M. On local asphyxia and symmetrical gangrene of the extremities (1862), and new research on the nature and treatment of local asphyxia of the extremities (1872).Barlow T, trans. Selected monographs (121). London: New Sydenham Society, 1988.
- Boin F, Wigley FM. Understanding, assessing and treating Raynaud’s phenomenon. Curr Opin Rheumatol 2005; 17:752–760.
- Maricq HR, Carpentier PH, Weinrich MC, et al. Geographic variation in the prevalence of Raynaud’s phenomenon: a 5-region comparison. J Rheumatol 1997; 24:879–889.
- Wigley FM. Clinical practice. Raynaud’s phenomenon. N Engl J Med 2002; 347:1001–1018.
- Cutolo M, Pizzorni C, Sulli A. Capillaroscopy. Best Pract Res Clin Rheumatol 2005; 19:437–452.
- Lonzetti LS, Joyal F, Raynauld JP, et al. Updating the American College of Rheumatology preliminary classification criteria for systemic sclerosis: addition of severe nailfold capillaroscopy abnormalities markedly increases the sensitivity for limited scleroderma. Arthritis Rheum 2001; 44:735–736.
- Hudson M, Taillefer S, Steele R, et al. Improving the sensitivity of the American College of Rheumatology classification criteria for systemic sclerosis. Clin Exp Rheumatol 2007; 25:754–757.
- Anders HJ, Sigl T, Schattenkirchner M. Differentiation between primary and secondary Raynaud’s phenomenon: a prospective study comparing nailfold capillaroscopy using an ophthalmoscope or stereomicroscope. Ann Rheum Dis 2001; 60:407–409.
- Spencer-Green G. Outcomes in primary Raynaud phenomenon: a meta-analysis of the frequency, rates, and predictors of transition to secondary diseases. Arch Intern Med 1998; 158:595–600.
- Wong ML, Highton J, Palmer DG. Sequential nailfold capillary microscopy in scleroderma and related disorders. Ann Rheum Dis 1988; 47:53–61.
- Weinstein A, Bordwell B, Stone B, Tibbetts C, Rothfield NF. Antibodies to native DNA and serum complement (C3) levels. Application to diagnosis and classification of systemic lupus erythematosus. Am J Med 1983; 74:206–216.
- Craft J. Antibodies to snRNPs in systemic lupus erythematosus. Rheum Dis Clin North Am 1992; 18:311–335.
- Weiner ES, Hildebrandt S, Senécal JL, et al. Prognostic significance of anticentromere antibodies and anti-topoisomerase I antibodies in Raynaud’s disease. A prospective study. Arthritis Rheum 1991; 34:68–77.
- Miller FW, Twitty SA, Biswas T, Plotz PH. Origin and regulation of a disease-specific autoantibody response. Antigenic epitopes, spectrotype stability, and isotype restriction of anti-Jo-1 autoantibodies. J Clin Invest 1990; 85:468–475.
- Ghirardello A, Zampieri S, Tarricone E, et al. Clinical implications of autoantibody screening in patients with autoimmune myositis. Autoimmunity 2006; 39:217–221.
- Raynaud M. On local asphyxia and symmetrical gangrene of the extremities (1862), and new research on the nature and treatment of local asphyxia of the extremities (1872).Barlow T, trans. Selected monographs (121). London: New Sydenham Society, 1988.
- Boin F, Wigley FM. Understanding, assessing and treating Raynaud’s phenomenon. Curr Opin Rheumatol 2005; 17:752–760.
- Maricq HR, Carpentier PH, Weinrich MC, et al. Geographic variation in the prevalence of Raynaud’s phenomenon: a 5-region comparison. J Rheumatol 1997; 24:879–889.
- Wigley FM. Clinical practice. Raynaud’s phenomenon. N Engl J Med 2002; 347:1001–1018.
- Cutolo M, Pizzorni C, Sulli A. Capillaroscopy. Best Pract Res Clin Rheumatol 2005; 19:437–452.
- Lonzetti LS, Joyal F, Raynauld JP, et al. Updating the American College of Rheumatology preliminary classification criteria for systemic sclerosis: addition of severe nailfold capillaroscopy abnormalities markedly increases the sensitivity for limited scleroderma. Arthritis Rheum 2001; 44:735–736.
- Hudson M, Taillefer S, Steele R, et al. Improving the sensitivity of the American College of Rheumatology classification criteria for systemic sclerosis. Clin Exp Rheumatol 2007; 25:754–757.
- Anders HJ, Sigl T, Schattenkirchner M. Differentiation between primary and secondary Raynaud’s phenomenon: a prospective study comparing nailfold capillaroscopy using an ophthalmoscope or stereomicroscope. Ann Rheum Dis 2001; 60:407–409.
- Spencer-Green G. Outcomes in primary Raynaud phenomenon: a meta-analysis of the frequency, rates, and predictors of transition to secondary diseases. Arch Intern Med 1998; 158:595–600.
- Wong ML, Highton J, Palmer DG. Sequential nailfold capillary microscopy in scleroderma and related disorders. Ann Rheum Dis 1988; 47:53–61.
- Weinstein A, Bordwell B, Stone B, Tibbetts C, Rothfield NF. Antibodies to native DNA and serum complement (C3) levels. Application to diagnosis and classification of systemic lupus erythematosus. Am J Med 1983; 74:206–216.
- Craft J. Antibodies to snRNPs in systemic lupus erythematosus. Rheum Dis Clin North Am 1992; 18:311–335.
- Weiner ES, Hildebrandt S, Senécal JL, et al. Prognostic significance of anticentromere antibodies and anti-topoisomerase I antibodies in Raynaud’s disease. A prospective study. Arthritis Rheum 1991; 34:68–77.
- Miller FW, Twitty SA, Biswas T, Plotz PH. Origin and regulation of a disease-specific autoantibody response. Antigenic epitopes, spectrotype stability, and isotype restriction of anti-Jo-1 autoantibodies. J Clin Invest 1990; 85:468–475.
- Ghirardello A, Zampieri S, Tarricone E, et al. Clinical implications of autoantibody screening in patients with autoimmune myositis. Autoimmunity 2006; 39:217–221.
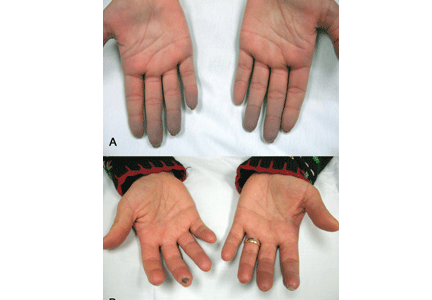
The 2012 ACR guidelines for osteoarthritis: Not a cookbook
“When I see a patient with arthritis coming in the front door, I leave by the back door.”
—Sir William Ostler
Fortunately for today’s physicians treating patients with osteoarthritis, we need not be as pessimistic as Osler was more than a century ago when he uttered his now-famous words. Still, there is no magic bullet for the contemporary clinician treating an elderly patient with osteoarthritis. Instead, there are many imperfect bullets, and choosing between them is always a balancing act between benefit and risk from various agents: nonsteroidal anti-inflammatory drugs (NSAIDs), analgesics such as acetaminophen and tramadol, opioids, and supplements such as glucosamine and chondroitin sulfate.
So there was great interest when, in 2012,1 the American College of Rheumatology (ACR) updated its previous guidelines (from 2000) on drug and nondrug therapies for osteoarthritis of the hip and the knee2 and added new recommendations on osteoarthritis of the hand.
Revising the guidelines was appropriate, since new therapies have become available. But, as the guideline authors state, with osteoarthritis, as with other diseases, guidelines cannot be a “cookbook.”
The treatment approach differs depending on the patient’s clinical presentation and on the preferences of the patient and the physician. Often, more than one approach is possible, and more than one approach may be appropriate in a given patient at a given time. The guideline authors also point out that some physicians may disagree with some of the recommendations.
I wish to review here several of the key recommendations. But I also provide some of my personal perspective and experience after 4 decades of treating patients with osteoarthritis.
HOW THE GUIDELINES WERE MADE
The new ACR guidelines were developed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach, a formal process to develop recommendations that are as evidence-based as possible.3
The authors are outstanding experts in the field of osteoarthritis from throughout the United States and Canada. Further, the recommendations were voted on by a “technical expert panel” representing the fields of rheumatology, orthopedics, physical medicine, and rehabilitation, from both academic medicine and private practice. This representation provides a balance of input from the types of clinicians frequently involved in managing osteoarthritis.
The initial literature searches for drug therapies were conducted during late 2008, and those for nonpharmacologic treatments were conducted during the second and third quarters of 2009. The goal of the literature searches was to identify the most current systematic reviews and meta-analyses that would provide reliable estimates of benefits of intervention for the prespecified clinically relevant outcomes of pain and function, as well as data on safety.
Recommendations: For or against, strong or weak—and the informed patient’s perspective
Therapies received the following possible recommendations:
- Strong recommendation to use
- Weak (or conditional) recommendation to use
- No recommendation
- Weak (or conditional) recommendation not to use
- Strong recommendation not to use.
A strong recommendation required high-quality evidence and evidence of a large difference between desirable and undesirable effects of the treatment. A conditional recommendation was based on the absence of high-quality evidence, evidence of only a small difference between desirable and undesirable effects of the treatment, or both.
One interesting feature of these recommendations is that they took into account how informed patients might themselves evaluate the data with their medical condition.
For instance, if a therapy received a strong favorable recommendation, we can assume that most informed patients would choose to receive it, and we can shape our interaction with the patient accordingly. A conditional recommendation means that most informed patients would choose the treatment—but many would not, and physicians should keep the patient’s values and preferences in mind.
I admit I had a problem with the meaning of the word “conditional” in the context of these guidelines. When evaluating a treatment, the term “weak” is readily understood and clearer. By using the word “weak,” one is making a positive statement in support of use but letting you know that the data and recommendation are weak. The word “conditional” is less readily defined and does not necessarily imply support for use.
Recommendations were drafted after discussion of the evidence at each meeting of the technical expert panel. Consensus was defined as 75% or more of the members of the panel voting to either strongly or conditionally recommend using a therapy, to either strongly or conditionally recommend not using it, or to choose not to make a recommendation on its use.
OSTEOARTHRITIS OF THE HAND: NO STRONG RECOMMENDATIONS
The technical expert panel gave no strong recommendations for any nondrug or drug treatment for osteoarthritis of the hand.
Conditional recommendations for nondrug treatments
The panel conditionally recommended the following:
- All patients with osteoarthritis of the hand should be evaluated either by their primary physician or by an occupational or physical therapist, particularly with respect to ability to perform activities of daily living.
- Assistive devices such as jar openers, key turners, and pull tabs for zippers should be recommended, as needed.
- Patients should be instructed in joint protection and in the use of thermal treatments (eg, heating pads, ultrasound devices, hot packs, and ice packs).
Comments. Appliances are often beneficial in patients who have involvement of the first carpometacarpal (trapeziometacarpal) joint. Although over-the-counter thumb splints are an option, referral to an occupational therapist for splint prescription is advantageous to achieve a comfortable fit and, importantly, for instructions to the patient on how to avoid joint trauma.
It would be unrealistic to expect primary care physicians and internists to have the expertise to make detailed recommendations about orthopedic appliances. Accordingly, referral to an occupational therapist or an orthopedist is advisable for these situations. It is important, however, that the physician be aware of what treatments are available and most effective, and of the indications for referral.
As for heat treatment, elastic stretch gloves may relieve symptoms through their warming and massaging effects.4
Some drugs for hand osteoarthritis got conditional recommendations in favor
The expert panel gave conditional recommendations in favor of:
- Topical capsaicin
- Topical NSAIDs
- Oral NSAIDs (including both nonselective and selective agents)
- Tramadol (Ultram)
- Topical rather than oral NSAIDs for patients age 75 and older.
Other drug treatments got conditional recommendations against their use
The expert panel gave conditional recommendations against using:
- Intra-articular injections, and in particular, corticosteroid injections in the trapeziometacarpal (first carpometacarpal) joint
- Opioid analgesics
- Oral methotrexate or sulfasalazine in patients with erosive inflammatory interphalangeal osteoarthritis.
No recommendation for or against
- Hydroxychloroquine.
Comments—Intra-articular injections, opioids, and oral NSAIDs
I differ with these recommendations on several points.
Although the guidelines committee conditionally recommended against using intra-articular therapies for hand osteoarthritis, I find that intra-articular corticosteroid injections are often effective, particularly in patients who have inflammatory forms of the disease, ie, “erosive inflammatory osteoarthritis.” Most nonspecialist physicians probably have limited experience in giving injections into small joints, and referral to a rheumatologist or orthopedist would be appropriate.
I disagree as well with the conditional recommendation that intra-articular corticosteroid injections not be used for involvement of the trapeziometacarpal (first carpometacarpal) joint. I find that many patients with osteoarthritis of this joint experience improvement with intra-articular corticosteroid injections.
I agree that there are limited data on the use of intra-articular hyaluronan injections in this situation and do not routinely use them in this joint.
Opioid analgesics also received a conditional recommendation against their use. The same caveats apply here as for these drugs elsewhere.5 If used, opioids should be used at the lowest dose possible and for as short a time as possible. If the physician is uncomfortable prescribing opioids for patients with osteoarthritis, referral to a pain specialist is recommended.
I disagree to some extent with the conditional recommendation that people age 75 and older should use topical rather than oral NSAIDs. I understand the recommendation, given that older people have a higher frequency of gastrointestinal, renal, and cardiac disease and are best served by avoiding NSAIDs. However, we all see patients over age 75 who are physiologically younger than their numerical age. Accordingly, I feel that the judgment of the physician plays a role in whether NSAIDs are reasonable for some older patients.
The committee recommended not using oral methotrexate or sulfasalazine in patients with erosive inflammatory interphalangeal osteoarthritis. I have used oral hydroxychloroquine off-label in such patients and find that they respond in a very rewarding fashion.
Given that this is an off-label use of hydroxychloroquine, the drug should be used only with appropriate consideration and after discussion with the patient about toxicity, especially about the risk of ocular manifestations.
OSTEOARTHRITIS OF THE KNEE
Some nondrug therapies got strong recommendations
The expert panel strongly recommended:
- Exercise (aerobic, resistance, land-based, and aquatic)
- Weight loss (for patients who are overweight).
Other nondrug therapies got conditional recommendations
The panel conditionally recommended:
- Self-management programs
- Manual therapy in combination with supervised exercise
- Psychosocial interventions
- Medially directed patellar taping
- Medially wedged insoles (if the patient has lateral compartment osteoarthritis)
- Laterally wedged subtalar strapped insoles (if the patient has medial compartment osteoarthritis)
- Heat therapy
- Walking aids, as needed
- Tai chi
- Chinese acupuncture
- Transcutaneous electrical nerve stimulation.
Comments. The ACR panel appropriately noted that Chinese acupuncture or transcutaneous electrical stimulation should be recommended only if the patient has chronic moderate to severe pain and is a candidate for total knee arthroplasty but is unwilling to undergo the procedure or has comorbid medical conditions that rule out surgery.
Nondrug therapies for knee osteoarthritis that got no recommendation for or against
- Balance exercise
- Laterally wedged insoles
- Manual therapy alone
- Knee braces
- Laterally directed patellar taping.
Comments. It was somewhat surprising that there were no recommendations about laterally wedged insoles or knee braces. Laterally wedged insoles have been recommended for patients who have medial compartment knee osteoarthritis6; being thinner at the instep and thicker at the outer edge of the foot, they reduce load on the medial aspect of the knee. One has to be cautious in using knee wedging in patients who have concomitant ankle or hip angle deformities, lest these joints be compromised.
Some of these treatments would be out of the realm of the nonspecialist physician.
Conditional recommendations for initial drug therapy for knee osteoarthritis
The panel conditionally recommended that patients who have osteoarthritis of the knee use one of the following:
- Acetaminophen (contained in Tylenol and a host of other products)
- Oral NSAIDs
- Topical NSAIDs (with a strong recommendation for topical NSAIDs rather than oral NSAIDs in patients age 75 and older)
- Tramadol
- Intra-articular corticosteroid injections.
Comments. In the past, it was recommended that acetaminophen in full doses of up to 4,000 mg per day be considered.7 Current dogma, however, is that doses of acetaminophen should not exceed 3,000 mg per day to avoid damaging the liver. This concern led the US Food and Drug Administration (FDA) in 2011 to advise that the maximum daily dose be limited.8 The ACR panel recommended that patients be counseled to avoid all other products that contain acetaminophen, which is especially cogent, given the presence of this agent in many over-the-counter medications.9
The panel conditionally recommended that people age 75 and older use topical rather than oral NSAIDs. As mentioned earlier, a specific age limit does not take into account that many people age 75 and older may actually be physiologically younger than some in their 50s or 60s. Accordingly, it is recommended that the physician use judgment in this regard so that NSAIDs will not be denied to patients for whom they might be of significant value.
Strong recommendation for gastric protection in patients at risk on NSAIDs
If a patient with knee osteoarthritis has a history of a symptomatic or complicated upper gastrointestinal ulcer but has not had an upper gastrointestinal bleed in the past year and the physician chooses to prescribe an oral NSAID, the expert panel strongly recommended using either a cyclooxygenase (COX)-2-selective inhibitor or a nonselective NSAID in combination with a proton pump inhibitor.
Comment. The suggestion that patients who have had a complicated upper gastrointestinal ulcer in the past year could be considered for treatment with a COX-2-selective inhibitor or nonselective NSAID in combination with a proton pump inhibitor seemed a bit aggressive. My own inclination would be to avoid both nonselective and selective inhibitors in this situation. Alternative agents such as acetaminophen in full doses, tramadol, intra-articular hyaluronan injections, and intra-articular corticosteroid injections seem preferable with respect to safety in such patients.
The suggestion that a proton pump inhibitor be used whenever an NSAID is given for chronic management of knee or hip osteoarthritis is reasonable.10,11 Although some studies have suggested that chronic use of proton pump inhibitors may predispose to osteopenia or osteoporosis, others have not, and gastric protection should be considered in patients at gastrointestinal risk.
Strong recommendation against ibuprofen in patients taking aspirin
The ACR panel strongly recommended that ibuprofen (Advil) not be prescribed to patients with knee osteoarthritis who are using aspirin in low doses for cardioprotection, and strongly recommended using another nonselective NSAID plus a proton pump inhibitor instead. The panel also strongly recommended against using a COX-2-selective inhibitor in this situation.12,13
Comment. The rationale for these recommendations is that ibuprofen may render aspirin ineffective as a cardioprotective agent. Ibuprofen interferes with the aspirin-binding site on platelets, so that the protective effect of aspirin is lost.14,15 Celecoxib (Celebrex)16 and diclofenac (Voltaren) have binding sites different from that of aspirin, although the ACR recommends against using COX-2-selective inhibitors such as celecoxib in the situation and gives no recommendation about other NSAIDs.
No recommendations for or against
The panel issued no recommendations for or against the following treatments for patients with knee osteoarthritis:
- Intra-articular hyaluronan injections
- Duloxetine (Cymbalta)
- Opioid analgesics.
Comments on knee injections
Intra-articular injections of corticosteroids or hyaluronan are commonly used for knee osteoarthritis. As noted, corticosteroid injections received a conditional recommendation, while hyaluronan injections received no recommendation for or against.
How often to inject corticosteroids? In general, too-frequent injection of corticosteroids is to be avoided, in view of the risk of promoting joint breakdown. There is no “magic” number of injections that is safe, although more than 4 per year in the same joint should generally be avoided. In some situations, however, repeat injections may be reasonable if alternative therapies are associated with higher risk.
Raynauld et al,17 in a randomized, double-blind, placebo-controlled trial, demonstrated that intra-articular corticosteroid injections at 3-month intervals for 2 years were not deleterious to knees.
My philosophy is generally not to inject on a regular basis, but to be selective and be guided by the patient’s clinical condition and response to prior injections.
Are hyaluronan injections effective? Although experts differ in their enthusiasm for intra-articular hyaluronan injections in the knee, I have found that many patients benefit from this treatment. Multiple studies have found it efficacious and safe overall.18–21 However, some systematic reviews have called its efficacy into question.7
Although differences in efficacy have been noted, this therapy was approved as being useful in patients with knee osteoarthritis in the Osteoarthritis Research Society International (OARSI) recommendations.7 The effect sizes were smaller in later assessments.22
Hyaluronan injections do not pose the risk of joint breakdown that corticosteroid injections do, but their clinical efficacy is not as dramatic. Adverse reactions to most intraarticular hyaluronans are limited, with slight increases in pain and stiffness after injection. Significant inflammatory reactions characterized as “postinjection flares” are more commonly seen with high-molecular-weight crosslinked preparations. These reactions can be severe and can mimic joint infection clinically. Joint aspiration with synovial fluid analysis and culture may be necessary to exclude infection. Response to aspiration and nonsteroidal inflammatory agents or intra-articular corticosteroids is usually excellent.
Ultrasonographic guidance. As with intraarticular injections in other areas, ultrasonographic guidance is becoming more common, as it allows for more accurate drug administration.
Pes anserine bursitis must be ruled out as a cause of the patient’s knee symptoms—misdiagnosis is not uncommon. The bursa is located on the medial aspect of the tibia, and inflammation of the bursa is a common cause of pain in this area. Local steroid injection is extremely effective in symptomatic therapy. Physical therapy and NSAIDs may be adequate to treat milder cases.
Conditional recommendation against glucosamine, chondroitin, capsaicin
The ACR panel conditionally recommended that patients with knee osteoarthritis not use:
- Chondroitin sulfate
- Glucosamine
- Topical capsaicin.
Comment. Evidence is mixed about the efficacy of glucosamine and chondroitin sulfate, which are so-called nutraceuticals. Some studies found them useful23–25 but some did not,26 and a meta-analysis concluded that they do not help.27 The OARSI guidelines published in 2008 stated that these agents may relieve symptoms of osteoarthritis of the knee.7 The OARSI update published in 2010 found that glucosamine was effective, but less so than in previous studies.22 If glucosamine is effective, some studies suggest that glucosamine sulfate is more effective than glucosamine hydrochloride.22
The same OARSI review revealed that chondroitin sulfate relieved pain but with heterogeneous, dissimilar effect sizes. Of interest was the finding that the 5-year incidence of total knee replacement was lower in patients treated with glucosamine sulfate 1,500 mg/day than with placebo. Also, the rate of decline of joint space narrowing was reported to be reduced in chondroitin sulfate-treated patients.22
In practice, a conditional recommendation against a treatment means that most informed patients would not want the treatment, but some would. Accordingly, if patients still want to take chondroitin or glucosamine after being informed of the limited evidence of benefit, I feel a trial of their use is reasonable.
OSTEOARTHRITIS OF THE HIP
Indications for therapy of osteoarthritis of the hip are similar to those for osteoarthritis of the knee.
As in the knee, nonpharmacologic therapies are important. Loss of weight for overweight patients is extremely important; supervised exercise is especially valuable. Use of canes or crutches as needed is conditionally recommended.
Pharmacologic management is similar to that of osteoarthritis of the knee, with particular use of acetaminophen, NSAIDs, tramadol, and intra-articular corticosteroid injections.
Comment. Intra-articular injection of corticosteroids into the hip would be out of the realm of most nonspecialist practices. Although some rheumatologists are expert in such injections, this treatment is generally best left to an orthopedist or invasive radiologist. The use of ultrasonographic guidance is becoming more frequent, with many rheumatologists having developed expertise in this approach to the knee and the hip. Since most studies were in patients with osteoarthritis of the knee, fewer data are available as to the efficacy of these agents in patients with hip osteoarthritis.
Fewer data are available also with respect to the benefit of chondroitin sulfate and glucosamine in patients with osteoarthritis of the hip. Total joint replacement is extremely effective if conservative therapy does not help.
FIRST, DO NO HARM
Guidelines from the ACR,1,2 the European League Against Rheumatism (EULAR),28,29 the American Academy of Orthopedic Surgeons (AAOS),30 and the OARSI7,22 all differ somewhat, owing to the different evidence available at the time each guideline was developed and to different geographic and cultural backgrounds.
The compositions of these various panels also differ sufficiently to affect their overall recommendations. For example, the EULAR panel consisted of only rheumatologists and an orthopedic surgeon; for the hand osteoarthritis recommendations they added a physiatrist and two allied health professionals.28,29 The OARSI panel included two primary care physicians in addition to rheumatologists and an orthopedic surgeon.7 The ACR was the only professional society to include primary care physicians, physiatrists, and geriatricians along with rheumatologists, an orthopedic surgeon, and physical and occupational therapists.
Although it is to be expected that there will not be universal agreement on all points of management of osteoarthritis by diverse groups, it is essential that input from all these experts representing various subspecialties be recognized. Therapeutic approaches will vary depending on patient characteristics and the experience of the treating physician. As long as therapy is based on reasonable supportive data, beneficial effects can be anticipated. Therapies that received conditional recommendations are not to be discounted if a reasonable percent of patients respond in positive fashion. Obviously, strong recommendations are more likely to be universally accepted since the likelihood that they will be beneficial is stronger.
In any approach to therapy, the caveat primum non nocere—first, do no harm—must always be kept in mind.
- Hochberg MC, Altman RD, April KT, et al; American College of Rheumatology. American College of Rheumatology 2012 recommendations for the use of non-pharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care Res (Hoboken) 2012; 64:465–474.
- American College of Rheumatology Subcommittee on Osteoarthritis Guidelines. Recommendations for the medical management of osteoarthritis of the hip and knee: 2000 update. Arthritis Rheum 2000; 43:1905–1915.
- Atkins D, Best D, Briss PA, et al; GRADE Working Group. Grading quality of evidence and strength of recommendations. BMJ 2004; 328:1490.
- Askari A, Moskowitz RW, Ryan C. Stretch gloves. A study of objective and subjective effectiveness in arthritis of the hands. Arthritis Rheum 1974; 17:263–265.
- Chou R, Fanciullo GJ, Fine PG, et al; American Pain Society-American Academy of Pain Medicine Opioids Guidelines Panel. Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. J Pain 2009; 10:113–130.
- Fang MA, Taylor CE, Nouvong A, Masih S, Kao KC, Perell KL. Effects of footwear on medial compartment knee osteoarthritis. J Rehabil Res Dev 2006; 43:427–434.
- Zhang W, Moskowitz RW, Nuki G, et al. OARSI recommendations for the management of hip and knee osteoarthritis, Part II: OARSI evidence-based, expert consensus guidelines. Osteoarthritis Cartilage 2008; 16:137–162.
- US Food and Drug Administration (FDA). FDA Drug Safety Communication: Prescription Acetaminophen Products to be Limited to 325 mg Per Dosage Unit; Boxed Warning Will Highlight Potential for Severe Liver Failure. January 13, 2011. http://www.fda.gov/Drugs/DrugSafety/ucm239821.htm. Accessed November 28, 2012.
- Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med 2010; 77:19–27.
- Bolten WW. Rational use of nonsteroidal anti-inflammatory drugs and proton pump inhibitors in combination for rheumatic diseases. Orthopedic Research and Reviews 2010; 2:75–84.
- Graham DY, Agrawal NM, Campbell DR, et al; NSAID-Associated Gastric Ulcer Prevention Study Group. Ulcer prevention in long-term users of nonsteroidal anti-inflammatory drugs: results of a double-blind, randomized, multicenter, active- and placebo-controlled study of misoprostol vs lansoprazole. Arch Intern Med 2002; 162:169–175.
- American College of Rheumatology Ad Hoc Group on Use of Selective and Nonselective Nonsteroidal Antiinflammatory Drugs. Recommendations for use of selective and nonselective nonsteroidal antiinflammatory drugs: an American College of Rheumatology white paper. Arthritis Rheum 2008; 59:1058–1073.
- Antman EM, Bennett JS, Daugherty A, Furberg C, Roberts H, Taubert KA; American Heart Association. Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart Association. Circulation 2007; 115:1634–1642.
- Ellison J, Dager W. Recent FDA warning of the concomitant use of aspirin and ibuprofen and the effects on platelet aggregation. Prev Cardiol 2007; 10:61–63.
- Schuijt MP, Huntjens-Fleuren HW, de Metz M, Vollaard EJ. The interaction of ibuprofen and diclofenac with aspirin in healthy volunteers. Br J Pharmacol 2009; 157:931–934.
- Wilner KD, Rushing M, Walden C, et al. Celecoxib does not affect the antiplatelet activity of aspirin in healthy volunteers. J Clin Pharmacol 2002; 42:1027–1030.
- Raynauld JP, Buckland-Wright C, Ward R, et al. Safety and efficacy of long-term intraarticular steroid injections in osteoarthritis of the knee: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 2003; 48:370–377.
- Berenbaum F, Grifka J, Cazzaniga S, et al. A randomised, double-blind, controlled trial comparing two intra-articular hyaluronic acid preparations differing by their molecular weight in symptomatic knee osteoarthritis. Ann Rheum Dis 2012; 71:1454–1460.
- Colen S, van den Bekerom MP, Mulier M, Haverkamp D. Hyaluronic acid in the treatment of knee osteoarthritis: a systematic review and meta-analysis with emphasis on the efficacy of different products. BioDrugs 2012; 26:257–268.
- Wang CT, Lin J, Chang CJ, Lin YT, Hou SM. Therapeutic effects of hyaluronic acid on osteoarthritis of the knee. A meta-analysis of randomized controlled trials. J Bone Joint Surg Am 2004; 86-A:538–545.
- Rutjes AW, Jüni P, da Costa BR, Trelle S, Nüesch E, Reichenbach S. Visco-supplementation for osteoarthritis of the knee: a systematic review and meta-analysis. Ann Intern Med 2012; 157:180–191.
- Zhang W, Nuki G, Moskowitz RW, et al. OARSI recommendations for the management of hip and knee osteoarthritis: part III: changes in evidence following systematic cumulative update of research published through January 2009. Osteoarthritis Cartilage 2010; 18:476–499.
- Zhang W, Moskowitz RW, Nuki G, et al. OARSI recommendations for the management of hip and knee osteoarthritis, part I: critical appraisal of existing treat-ment guidelines and systematic review of current research evidence. Osteoarthritis Cartilage 2007; 15:981–1000.
- Towheed TE, Maxwell L, Anastassiades TP, et al. Glucosamine therapy for treating osteoarthritis. Cochrane Database Syst Rev 2005; 2:CD002946.
- Vlad SC, LaValley MP, McAlindon TE, Felson DT. Glucosamine for pain in osteoarthritis: why do trial results differ? Arthritis Rheum 2007; 56:2267–2277.
- Clegg DO, Reda DJ, Harris CL, et al. Glucosamine, chondroitin sulfate, and the two in combination for painful knee osteoarthritis. N Engl J Med 2006; 354:795–808.
- Wandel S, Jüni P, Tendal B, et al. Effects of glucosamine, chondroitin, or placebo in patients with osteoarthritis of hip or knee: network meta-analysis. BMJ 2010; 341:c4675.
- Jordan KM, Arden NK, Doherty M, Bannwarth B, Bijlsma JW, Dieppe P, et al. EULAR recommendations 2003: an evidence based approach to the management of knee osteoarthritis: report of a Task Force of the EULAR Standing Committee for International Clinical Studies Including Therapeutic Trials (ESCISIT). Ann Rheum Dis 2003;62:1145–55. Studies Including Therapeutic Trials (ESCISIT). Ann Rheum Dis 2005; 64:669–681.
- Zhang W, Doherty M, Leeb BF, et al. EULAR evidence based recommendations for the management of hand osteoarthritis: report of a Task Force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis 2007; 66:377–388.
- American Academy of Orthopaedic Surgeons. Treatment of osteoarthritis of the knee (non-arthroplasty). Rosemont, IL: American Academy of Orthopaedic Surgeons; 2008.
“When I see a patient with arthritis coming in the front door, I leave by the back door.”
—Sir William Ostler
Fortunately for today’s physicians treating patients with osteoarthritis, we need not be as pessimistic as Osler was more than a century ago when he uttered his now-famous words. Still, there is no magic bullet for the contemporary clinician treating an elderly patient with osteoarthritis. Instead, there are many imperfect bullets, and choosing between them is always a balancing act between benefit and risk from various agents: nonsteroidal anti-inflammatory drugs (NSAIDs), analgesics such as acetaminophen and tramadol, opioids, and supplements such as glucosamine and chondroitin sulfate.
So there was great interest when, in 2012,1 the American College of Rheumatology (ACR) updated its previous guidelines (from 2000) on drug and nondrug therapies for osteoarthritis of the hip and the knee2 and added new recommendations on osteoarthritis of the hand.
Revising the guidelines was appropriate, since new therapies have become available. But, as the guideline authors state, with osteoarthritis, as with other diseases, guidelines cannot be a “cookbook.”
The treatment approach differs depending on the patient’s clinical presentation and on the preferences of the patient and the physician. Often, more than one approach is possible, and more than one approach may be appropriate in a given patient at a given time. The guideline authors also point out that some physicians may disagree with some of the recommendations.
I wish to review here several of the key recommendations. But I also provide some of my personal perspective and experience after 4 decades of treating patients with osteoarthritis.
HOW THE GUIDELINES WERE MADE
The new ACR guidelines were developed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach, a formal process to develop recommendations that are as evidence-based as possible.3
The authors are outstanding experts in the field of osteoarthritis from throughout the United States and Canada. Further, the recommendations were voted on by a “technical expert panel” representing the fields of rheumatology, orthopedics, physical medicine, and rehabilitation, from both academic medicine and private practice. This representation provides a balance of input from the types of clinicians frequently involved in managing osteoarthritis.
The initial literature searches for drug therapies were conducted during late 2008, and those for nonpharmacologic treatments were conducted during the second and third quarters of 2009. The goal of the literature searches was to identify the most current systematic reviews and meta-analyses that would provide reliable estimates of benefits of intervention for the prespecified clinically relevant outcomes of pain and function, as well as data on safety.
Recommendations: For or against, strong or weak—and the informed patient’s perspective
Therapies received the following possible recommendations:
- Strong recommendation to use
- Weak (or conditional) recommendation to use
- No recommendation
- Weak (or conditional) recommendation not to use
- Strong recommendation not to use.
A strong recommendation required high-quality evidence and evidence of a large difference between desirable and undesirable effects of the treatment. A conditional recommendation was based on the absence of high-quality evidence, evidence of only a small difference between desirable and undesirable effects of the treatment, or both.
One interesting feature of these recommendations is that they took into account how informed patients might themselves evaluate the data with their medical condition.
For instance, if a therapy received a strong favorable recommendation, we can assume that most informed patients would choose to receive it, and we can shape our interaction with the patient accordingly. A conditional recommendation means that most informed patients would choose the treatment—but many would not, and physicians should keep the patient’s values and preferences in mind.
I admit I had a problem with the meaning of the word “conditional” in the context of these guidelines. When evaluating a treatment, the term “weak” is readily understood and clearer. By using the word “weak,” one is making a positive statement in support of use but letting you know that the data and recommendation are weak. The word “conditional” is less readily defined and does not necessarily imply support for use.
Recommendations were drafted after discussion of the evidence at each meeting of the technical expert panel. Consensus was defined as 75% or more of the members of the panel voting to either strongly or conditionally recommend using a therapy, to either strongly or conditionally recommend not using it, or to choose not to make a recommendation on its use.
OSTEOARTHRITIS OF THE HAND: NO STRONG RECOMMENDATIONS
The technical expert panel gave no strong recommendations for any nondrug or drug treatment for osteoarthritis of the hand.
Conditional recommendations for nondrug treatments
The panel conditionally recommended the following:
- All patients with osteoarthritis of the hand should be evaluated either by their primary physician or by an occupational or physical therapist, particularly with respect to ability to perform activities of daily living.
- Assistive devices such as jar openers, key turners, and pull tabs for zippers should be recommended, as needed.
- Patients should be instructed in joint protection and in the use of thermal treatments (eg, heating pads, ultrasound devices, hot packs, and ice packs).
Comments. Appliances are often beneficial in patients who have involvement of the first carpometacarpal (trapeziometacarpal) joint. Although over-the-counter thumb splints are an option, referral to an occupational therapist for splint prescription is advantageous to achieve a comfortable fit and, importantly, for instructions to the patient on how to avoid joint trauma.
It would be unrealistic to expect primary care physicians and internists to have the expertise to make detailed recommendations about orthopedic appliances. Accordingly, referral to an occupational therapist or an orthopedist is advisable for these situations. It is important, however, that the physician be aware of what treatments are available and most effective, and of the indications for referral.
As for heat treatment, elastic stretch gloves may relieve symptoms through their warming and massaging effects.4
Some drugs for hand osteoarthritis got conditional recommendations in favor
The expert panel gave conditional recommendations in favor of:
- Topical capsaicin
- Topical NSAIDs
- Oral NSAIDs (including both nonselective and selective agents)
- Tramadol (Ultram)
- Topical rather than oral NSAIDs for patients age 75 and older.
Other drug treatments got conditional recommendations against their use
The expert panel gave conditional recommendations against using:
- Intra-articular injections, and in particular, corticosteroid injections in the trapeziometacarpal (first carpometacarpal) joint
- Opioid analgesics
- Oral methotrexate or sulfasalazine in patients with erosive inflammatory interphalangeal osteoarthritis.
No recommendation for or against
- Hydroxychloroquine.
Comments—Intra-articular injections, opioids, and oral NSAIDs
I differ with these recommendations on several points.
Although the guidelines committee conditionally recommended against using intra-articular therapies for hand osteoarthritis, I find that intra-articular corticosteroid injections are often effective, particularly in patients who have inflammatory forms of the disease, ie, “erosive inflammatory osteoarthritis.” Most nonspecialist physicians probably have limited experience in giving injections into small joints, and referral to a rheumatologist or orthopedist would be appropriate.
I disagree as well with the conditional recommendation that intra-articular corticosteroid injections not be used for involvement of the trapeziometacarpal (first carpometacarpal) joint. I find that many patients with osteoarthritis of this joint experience improvement with intra-articular corticosteroid injections.
I agree that there are limited data on the use of intra-articular hyaluronan injections in this situation and do not routinely use them in this joint.
Opioid analgesics also received a conditional recommendation against their use. The same caveats apply here as for these drugs elsewhere.5 If used, opioids should be used at the lowest dose possible and for as short a time as possible. If the physician is uncomfortable prescribing opioids for patients with osteoarthritis, referral to a pain specialist is recommended.
I disagree to some extent with the conditional recommendation that people age 75 and older should use topical rather than oral NSAIDs. I understand the recommendation, given that older people have a higher frequency of gastrointestinal, renal, and cardiac disease and are best served by avoiding NSAIDs. However, we all see patients over age 75 who are physiologically younger than their numerical age. Accordingly, I feel that the judgment of the physician plays a role in whether NSAIDs are reasonable for some older patients.
The committee recommended not using oral methotrexate or sulfasalazine in patients with erosive inflammatory interphalangeal osteoarthritis. I have used oral hydroxychloroquine off-label in such patients and find that they respond in a very rewarding fashion.
Given that this is an off-label use of hydroxychloroquine, the drug should be used only with appropriate consideration and after discussion with the patient about toxicity, especially about the risk of ocular manifestations.
OSTEOARTHRITIS OF THE KNEE
Some nondrug therapies got strong recommendations
The expert panel strongly recommended:
- Exercise (aerobic, resistance, land-based, and aquatic)
- Weight loss (for patients who are overweight).
Other nondrug therapies got conditional recommendations
The panel conditionally recommended:
- Self-management programs
- Manual therapy in combination with supervised exercise
- Psychosocial interventions
- Medially directed patellar taping
- Medially wedged insoles (if the patient has lateral compartment osteoarthritis)
- Laterally wedged subtalar strapped insoles (if the patient has medial compartment osteoarthritis)
- Heat therapy
- Walking aids, as needed
- Tai chi
- Chinese acupuncture
- Transcutaneous electrical nerve stimulation.
Comments. The ACR panel appropriately noted that Chinese acupuncture or transcutaneous electrical stimulation should be recommended only if the patient has chronic moderate to severe pain and is a candidate for total knee arthroplasty but is unwilling to undergo the procedure or has comorbid medical conditions that rule out surgery.
Nondrug therapies for knee osteoarthritis that got no recommendation for or against
- Balance exercise
- Laterally wedged insoles
- Manual therapy alone
- Knee braces
- Laterally directed patellar taping.
Comments. It was somewhat surprising that there were no recommendations about laterally wedged insoles or knee braces. Laterally wedged insoles have been recommended for patients who have medial compartment knee osteoarthritis6; being thinner at the instep and thicker at the outer edge of the foot, they reduce load on the medial aspect of the knee. One has to be cautious in using knee wedging in patients who have concomitant ankle or hip angle deformities, lest these joints be compromised.
Some of these treatments would be out of the realm of the nonspecialist physician.
Conditional recommendations for initial drug therapy for knee osteoarthritis
The panel conditionally recommended that patients who have osteoarthritis of the knee use one of the following:
- Acetaminophen (contained in Tylenol and a host of other products)
- Oral NSAIDs
- Topical NSAIDs (with a strong recommendation for topical NSAIDs rather than oral NSAIDs in patients age 75 and older)
- Tramadol
- Intra-articular corticosteroid injections.
Comments. In the past, it was recommended that acetaminophen in full doses of up to 4,000 mg per day be considered.7 Current dogma, however, is that doses of acetaminophen should not exceed 3,000 mg per day to avoid damaging the liver. This concern led the US Food and Drug Administration (FDA) in 2011 to advise that the maximum daily dose be limited.8 The ACR panel recommended that patients be counseled to avoid all other products that contain acetaminophen, which is especially cogent, given the presence of this agent in many over-the-counter medications.9
The panel conditionally recommended that people age 75 and older use topical rather than oral NSAIDs. As mentioned earlier, a specific age limit does not take into account that many people age 75 and older may actually be physiologically younger than some in their 50s or 60s. Accordingly, it is recommended that the physician use judgment in this regard so that NSAIDs will not be denied to patients for whom they might be of significant value.
Strong recommendation for gastric protection in patients at risk on NSAIDs
If a patient with knee osteoarthritis has a history of a symptomatic or complicated upper gastrointestinal ulcer but has not had an upper gastrointestinal bleed in the past year and the physician chooses to prescribe an oral NSAID, the expert panel strongly recommended using either a cyclooxygenase (COX)-2-selective inhibitor or a nonselective NSAID in combination with a proton pump inhibitor.
Comment. The suggestion that patients who have had a complicated upper gastrointestinal ulcer in the past year could be considered for treatment with a COX-2-selective inhibitor or nonselective NSAID in combination with a proton pump inhibitor seemed a bit aggressive. My own inclination would be to avoid both nonselective and selective inhibitors in this situation. Alternative agents such as acetaminophen in full doses, tramadol, intra-articular hyaluronan injections, and intra-articular corticosteroid injections seem preferable with respect to safety in such patients.
The suggestion that a proton pump inhibitor be used whenever an NSAID is given for chronic management of knee or hip osteoarthritis is reasonable.10,11 Although some studies have suggested that chronic use of proton pump inhibitors may predispose to osteopenia or osteoporosis, others have not, and gastric protection should be considered in patients at gastrointestinal risk.
Strong recommendation against ibuprofen in patients taking aspirin
The ACR panel strongly recommended that ibuprofen (Advil) not be prescribed to patients with knee osteoarthritis who are using aspirin in low doses for cardioprotection, and strongly recommended using another nonselective NSAID plus a proton pump inhibitor instead. The panel also strongly recommended against using a COX-2-selective inhibitor in this situation.12,13
Comment. The rationale for these recommendations is that ibuprofen may render aspirin ineffective as a cardioprotective agent. Ibuprofen interferes with the aspirin-binding site on platelets, so that the protective effect of aspirin is lost.14,15 Celecoxib (Celebrex)16 and diclofenac (Voltaren) have binding sites different from that of aspirin, although the ACR recommends against using COX-2-selective inhibitors such as celecoxib in the situation and gives no recommendation about other NSAIDs.
No recommendations for or against
The panel issued no recommendations for or against the following treatments for patients with knee osteoarthritis:
- Intra-articular hyaluronan injections
- Duloxetine (Cymbalta)
- Opioid analgesics.
Comments on knee injections
Intra-articular injections of corticosteroids or hyaluronan are commonly used for knee osteoarthritis. As noted, corticosteroid injections received a conditional recommendation, while hyaluronan injections received no recommendation for or against.
How often to inject corticosteroids? In general, too-frequent injection of corticosteroids is to be avoided, in view of the risk of promoting joint breakdown. There is no “magic” number of injections that is safe, although more than 4 per year in the same joint should generally be avoided. In some situations, however, repeat injections may be reasonable if alternative therapies are associated with higher risk.
Raynauld et al,17 in a randomized, double-blind, placebo-controlled trial, demonstrated that intra-articular corticosteroid injections at 3-month intervals for 2 years were not deleterious to knees.
My philosophy is generally not to inject on a regular basis, but to be selective and be guided by the patient’s clinical condition and response to prior injections.
Are hyaluronan injections effective? Although experts differ in their enthusiasm for intra-articular hyaluronan injections in the knee, I have found that many patients benefit from this treatment. Multiple studies have found it efficacious and safe overall.18–21 However, some systematic reviews have called its efficacy into question.7
Although differences in efficacy have been noted, this therapy was approved as being useful in patients with knee osteoarthritis in the Osteoarthritis Research Society International (OARSI) recommendations.7 The effect sizes were smaller in later assessments.22
Hyaluronan injections do not pose the risk of joint breakdown that corticosteroid injections do, but their clinical efficacy is not as dramatic. Adverse reactions to most intraarticular hyaluronans are limited, with slight increases in pain and stiffness after injection. Significant inflammatory reactions characterized as “postinjection flares” are more commonly seen with high-molecular-weight crosslinked preparations. These reactions can be severe and can mimic joint infection clinically. Joint aspiration with synovial fluid analysis and culture may be necessary to exclude infection. Response to aspiration and nonsteroidal inflammatory agents or intra-articular corticosteroids is usually excellent.
Ultrasonographic guidance. As with intraarticular injections in other areas, ultrasonographic guidance is becoming more common, as it allows for more accurate drug administration.
Pes anserine bursitis must be ruled out as a cause of the patient’s knee symptoms—misdiagnosis is not uncommon. The bursa is located on the medial aspect of the tibia, and inflammation of the bursa is a common cause of pain in this area. Local steroid injection is extremely effective in symptomatic therapy. Physical therapy and NSAIDs may be adequate to treat milder cases.
Conditional recommendation against glucosamine, chondroitin, capsaicin
The ACR panel conditionally recommended that patients with knee osteoarthritis not use:
- Chondroitin sulfate
- Glucosamine
- Topical capsaicin.
Comment. Evidence is mixed about the efficacy of glucosamine and chondroitin sulfate, which are so-called nutraceuticals. Some studies found them useful23–25 but some did not,26 and a meta-analysis concluded that they do not help.27 The OARSI guidelines published in 2008 stated that these agents may relieve symptoms of osteoarthritis of the knee.7 The OARSI update published in 2010 found that glucosamine was effective, but less so than in previous studies.22 If glucosamine is effective, some studies suggest that glucosamine sulfate is more effective than glucosamine hydrochloride.22
The same OARSI review revealed that chondroitin sulfate relieved pain but with heterogeneous, dissimilar effect sizes. Of interest was the finding that the 5-year incidence of total knee replacement was lower in patients treated with glucosamine sulfate 1,500 mg/day than with placebo. Also, the rate of decline of joint space narrowing was reported to be reduced in chondroitin sulfate-treated patients.22
In practice, a conditional recommendation against a treatment means that most informed patients would not want the treatment, but some would. Accordingly, if patients still want to take chondroitin or glucosamine after being informed of the limited evidence of benefit, I feel a trial of their use is reasonable.
OSTEOARTHRITIS OF THE HIP
Indications for therapy of osteoarthritis of the hip are similar to those for osteoarthritis of the knee.
As in the knee, nonpharmacologic therapies are important. Loss of weight for overweight patients is extremely important; supervised exercise is especially valuable. Use of canes or crutches as needed is conditionally recommended.
Pharmacologic management is similar to that of osteoarthritis of the knee, with particular use of acetaminophen, NSAIDs, tramadol, and intra-articular corticosteroid injections.
Comment. Intra-articular injection of corticosteroids into the hip would be out of the realm of most nonspecialist practices. Although some rheumatologists are expert in such injections, this treatment is generally best left to an orthopedist or invasive radiologist. The use of ultrasonographic guidance is becoming more frequent, with many rheumatologists having developed expertise in this approach to the knee and the hip. Since most studies were in patients with osteoarthritis of the knee, fewer data are available as to the efficacy of these agents in patients with hip osteoarthritis.
Fewer data are available also with respect to the benefit of chondroitin sulfate and glucosamine in patients with osteoarthritis of the hip. Total joint replacement is extremely effective if conservative therapy does not help.
FIRST, DO NO HARM
Guidelines from the ACR,1,2 the European League Against Rheumatism (EULAR),28,29 the American Academy of Orthopedic Surgeons (AAOS),30 and the OARSI7,22 all differ somewhat, owing to the different evidence available at the time each guideline was developed and to different geographic and cultural backgrounds.
The compositions of these various panels also differ sufficiently to affect their overall recommendations. For example, the EULAR panel consisted of only rheumatologists and an orthopedic surgeon; for the hand osteoarthritis recommendations they added a physiatrist and two allied health professionals.28,29 The OARSI panel included two primary care physicians in addition to rheumatologists and an orthopedic surgeon.7 The ACR was the only professional society to include primary care physicians, physiatrists, and geriatricians along with rheumatologists, an orthopedic surgeon, and physical and occupational therapists.
Although it is to be expected that there will not be universal agreement on all points of management of osteoarthritis by diverse groups, it is essential that input from all these experts representing various subspecialties be recognized. Therapeutic approaches will vary depending on patient characteristics and the experience of the treating physician. As long as therapy is based on reasonable supportive data, beneficial effects can be anticipated. Therapies that received conditional recommendations are not to be discounted if a reasonable percent of patients respond in positive fashion. Obviously, strong recommendations are more likely to be universally accepted since the likelihood that they will be beneficial is stronger.
In any approach to therapy, the caveat primum non nocere—first, do no harm—must always be kept in mind.
“When I see a patient with arthritis coming in the front door, I leave by the back door.”
—Sir William Ostler
Fortunately for today’s physicians treating patients with osteoarthritis, we need not be as pessimistic as Osler was more than a century ago when he uttered his now-famous words. Still, there is no magic bullet for the contemporary clinician treating an elderly patient with osteoarthritis. Instead, there are many imperfect bullets, and choosing between them is always a balancing act between benefit and risk from various agents: nonsteroidal anti-inflammatory drugs (NSAIDs), analgesics such as acetaminophen and tramadol, opioids, and supplements such as glucosamine and chondroitin sulfate.
So there was great interest when, in 2012,1 the American College of Rheumatology (ACR) updated its previous guidelines (from 2000) on drug and nondrug therapies for osteoarthritis of the hip and the knee2 and added new recommendations on osteoarthritis of the hand.
Revising the guidelines was appropriate, since new therapies have become available. But, as the guideline authors state, with osteoarthritis, as with other diseases, guidelines cannot be a “cookbook.”
The treatment approach differs depending on the patient’s clinical presentation and on the preferences of the patient and the physician. Often, more than one approach is possible, and more than one approach may be appropriate in a given patient at a given time. The guideline authors also point out that some physicians may disagree with some of the recommendations.
I wish to review here several of the key recommendations. But I also provide some of my personal perspective and experience after 4 decades of treating patients with osteoarthritis.
HOW THE GUIDELINES WERE MADE
The new ACR guidelines were developed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach, a formal process to develop recommendations that are as evidence-based as possible.3
The authors are outstanding experts in the field of osteoarthritis from throughout the United States and Canada. Further, the recommendations were voted on by a “technical expert panel” representing the fields of rheumatology, orthopedics, physical medicine, and rehabilitation, from both academic medicine and private practice. This representation provides a balance of input from the types of clinicians frequently involved in managing osteoarthritis.
The initial literature searches for drug therapies were conducted during late 2008, and those for nonpharmacologic treatments were conducted during the second and third quarters of 2009. The goal of the literature searches was to identify the most current systematic reviews and meta-analyses that would provide reliable estimates of benefits of intervention for the prespecified clinically relevant outcomes of pain and function, as well as data on safety.
Recommendations: For or against, strong or weak—and the informed patient’s perspective
Therapies received the following possible recommendations:
- Strong recommendation to use
- Weak (or conditional) recommendation to use
- No recommendation
- Weak (or conditional) recommendation not to use
- Strong recommendation not to use.
A strong recommendation required high-quality evidence and evidence of a large difference between desirable and undesirable effects of the treatment. A conditional recommendation was based on the absence of high-quality evidence, evidence of only a small difference between desirable and undesirable effects of the treatment, or both.
One interesting feature of these recommendations is that they took into account how informed patients might themselves evaluate the data with their medical condition.
For instance, if a therapy received a strong favorable recommendation, we can assume that most informed patients would choose to receive it, and we can shape our interaction with the patient accordingly. A conditional recommendation means that most informed patients would choose the treatment—but many would not, and physicians should keep the patient’s values and preferences in mind.
I admit I had a problem with the meaning of the word “conditional” in the context of these guidelines. When evaluating a treatment, the term “weak” is readily understood and clearer. By using the word “weak,” one is making a positive statement in support of use but letting you know that the data and recommendation are weak. The word “conditional” is less readily defined and does not necessarily imply support for use.
Recommendations were drafted after discussion of the evidence at each meeting of the technical expert panel. Consensus was defined as 75% or more of the members of the panel voting to either strongly or conditionally recommend using a therapy, to either strongly or conditionally recommend not using it, or to choose not to make a recommendation on its use.
OSTEOARTHRITIS OF THE HAND: NO STRONG RECOMMENDATIONS
The technical expert panel gave no strong recommendations for any nondrug or drug treatment for osteoarthritis of the hand.
Conditional recommendations for nondrug treatments
The panel conditionally recommended the following:
- All patients with osteoarthritis of the hand should be evaluated either by their primary physician or by an occupational or physical therapist, particularly with respect to ability to perform activities of daily living.
- Assistive devices such as jar openers, key turners, and pull tabs for zippers should be recommended, as needed.
- Patients should be instructed in joint protection and in the use of thermal treatments (eg, heating pads, ultrasound devices, hot packs, and ice packs).
Comments. Appliances are often beneficial in patients who have involvement of the first carpometacarpal (trapeziometacarpal) joint. Although over-the-counter thumb splints are an option, referral to an occupational therapist for splint prescription is advantageous to achieve a comfortable fit and, importantly, for instructions to the patient on how to avoid joint trauma.
It would be unrealistic to expect primary care physicians and internists to have the expertise to make detailed recommendations about orthopedic appliances. Accordingly, referral to an occupational therapist or an orthopedist is advisable for these situations. It is important, however, that the physician be aware of what treatments are available and most effective, and of the indications for referral.
As for heat treatment, elastic stretch gloves may relieve symptoms through their warming and massaging effects.4
Some drugs for hand osteoarthritis got conditional recommendations in favor
The expert panel gave conditional recommendations in favor of:
- Topical capsaicin
- Topical NSAIDs
- Oral NSAIDs (including both nonselective and selective agents)
- Tramadol (Ultram)
- Topical rather than oral NSAIDs for patients age 75 and older.
Other drug treatments got conditional recommendations against their use
The expert panel gave conditional recommendations against using:
- Intra-articular injections, and in particular, corticosteroid injections in the trapeziometacarpal (first carpometacarpal) joint
- Opioid analgesics
- Oral methotrexate or sulfasalazine in patients with erosive inflammatory interphalangeal osteoarthritis.
No recommendation for or against
- Hydroxychloroquine.
Comments—Intra-articular injections, opioids, and oral NSAIDs
I differ with these recommendations on several points.
Although the guidelines committee conditionally recommended against using intra-articular therapies for hand osteoarthritis, I find that intra-articular corticosteroid injections are often effective, particularly in patients who have inflammatory forms of the disease, ie, “erosive inflammatory osteoarthritis.” Most nonspecialist physicians probably have limited experience in giving injections into small joints, and referral to a rheumatologist or orthopedist would be appropriate.
I disagree as well with the conditional recommendation that intra-articular corticosteroid injections not be used for involvement of the trapeziometacarpal (first carpometacarpal) joint. I find that many patients with osteoarthritis of this joint experience improvement with intra-articular corticosteroid injections.
I agree that there are limited data on the use of intra-articular hyaluronan injections in this situation and do not routinely use them in this joint.
Opioid analgesics also received a conditional recommendation against their use. The same caveats apply here as for these drugs elsewhere.5 If used, opioids should be used at the lowest dose possible and for as short a time as possible. If the physician is uncomfortable prescribing opioids for patients with osteoarthritis, referral to a pain specialist is recommended.
I disagree to some extent with the conditional recommendation that people age 75 and older should use topical rather than oral NSAIDs. I understand the recommendation, given that older people have a higher frequency of gastrointestinal, renal, and cardiac disease and are best served by avoiding NSAIDs. However, we all see patients over age 75 who are physiologically younger than their numerical age. Accordingly, I feel that the judgment of the physician plays a role in whether NSAIDs are reasonable for some older patients.
The committee recommended not using oral methotrexate or sulfasalazine in patients with erosive inflammatory interphalangeal osteoarthritis. I have used oral hydroxychloroquine off-label in such patients and find that they respond in a very rewarding fashion.
Given that this is an off-label use of hydroxychloroquine, the drug should be used only with appropriate consideration and after discussion with the patient about toxicity, especially about the risk of ocular manifestations.
OSTEOARTHRITIS OF THE KNEE
Some nondrug therapies got strong recommendations
The expert panel strongly recommended:
- Exercise (aerobic, resistance, land-based, and aquatic)
- Weight loss (for patients who are overweight).
Other nondrug therapies got conditional recommendations
The panel conditionally recommended:
- Self-management programs
- Manual therapy in combination with supervised exercise
- Psychosocial interventions
- Medially directed patellar taping
- Medially wedged insoles (if the patient has lateral compartment osteoarthritis)
- Laterally wedged subtalar strapped insoles (if the patient has medial compartment osteoarthritis)
- Heat therapy
- Walking aids, as needed
- Tai chi
- Chinese acupuncture
- Transcutaneous electrical nerve stimulation.
Comments. The ACR panel appropriately noted that Chinese acupuncture or transcutaneous electrical stimulation should be recommended only if the patient has chronic moderate to severe pain and is a candidate for total knee arthroplasty but is unwilling to undergo the procedure or has comorbid medical conditions that rule out surgery.
Nondrug therapies for knee osteoarthritis that got no recommendation for or against
- Balance exercise
- Laterally wedged insoles
- Manual therapy alone
- Knee braces
- Laterally directed patellar taping.
Comments. It was somewhat surprising that there were no recommendations about laterally wedged insoles or knee braces. Laterally wedged insoles have been recommended for patients who have medial compartment knee osteoarthritis6; being thinner at the instep and thicker at the outer edge of the foot, they reduce load on the medial aspect of the knee. One has to be cautious in using knee wedging in patients who have concomitant ankle or hip angle deformities, lest these joints be compromised.
Some of these treatments would be out of the realm of the nonspecialist physician.
Conditional recommendations for initial drug therapy for knee osteoarthritis
The panel conditionally recommended that patients who have osteoarthritis of the knee use one of the following:
- Acetaminophen (contained in Tylenol and a host of other products)
- Oral NSAIDs
- Topical NSAIDs (with a strong recommendation for topical NSAIDs rather than oral NSAIDs in patients age 75 and older)
- Tramadol
- Intra-articular corticosteroid injections.
Comments. In the past, it was recommended that acetaminophen in full doses of up to 4,000 mg per day be considered.7 Current dogma, however, is that doses of acetaminophen should not exceed 3,000 mg per day to avoid damaging the liver. This concern led the US Food and Drug Administration (FDA) in 2011 to advise that the maximum daily dose be limited.8 The ACR panel recommended that patients be counseled to avoid all other products that contain acetaminophen, which is especially cogent, given the presence of this agent in many over-the-counter medications.9
The panel conditionally recommended that people age 75 and older use topical rather than oral NSAIDs. As mentioned earlier, a specific age limit does not take into account that many people age 75 and older may actually be physiologically younger than some in their 50s or 60s. Accordingly, it is recommended that the physician use judgment in this regard so that NSAIDs will not be denied to patients for whom they might be of significant value.
Strong recommendation for gastric protection in patients at risk on NSAIDs
If a patient with knee osteoarthritis has a history of a symptomatic or complicated upper gastrointestinal ulcer but has not had an upper gastrointestinal bleed in the past year and the physician chooses to prescribe an oral NSAID, the expert panel strongly recommended using either a cyclooxygenase (COX)-2-selective inhibitor or a nonselective NSAID in combination with a proton pump inhibitor.
Comment. The suggestion that patients who have had a complicated upper gastrointestinal ulcer in the past year could be considered for treatment with a COX-2-selective inhibitor or nonselective NSAID in combination with a proton pump inhibitor seemed a bit aggressive. My own inclination would be to avoid both nonselective and selective inhibitors in this situation. Alternative agents such as acetaminophen in full doses, tramadol, intra-articular hyaluronan injections, and intra-articular corticosteroid injections seem preferable with respect to safety in such patients.
The suggestion that a proton pump inhibitor be used whenever an NSAID is given for chronic management of knee or hip osteoarthritis is reasonable.10,11 Although some studies have suggested that chronic use of proton pump inhibitors may predispose to osteopenia or osteoporosis, others have not, and gastric protection should be considered in patients at gastrointestinal risk.
Strong recommendation against ibuprofen in patients taking aspirin
The ACR panel strongly recommended that ibuprofen (Advil) not be prescribed to patients with knee osteoarthritis who are using aspirin in low doses for cardioprotection, and strongly recommended using another nonselective NSAID plus a proton pump inhibitor instead. The panel also strongly recommended against using a COX-2-selective inhibitor in this situation.12,13
Comment. The rationale for these recommendations is that ibuprofen may render aspirin ineffective as a cardioprotective agent. Ibuprofen interferes with the aspirin-binding site on platelets, so that the protective effect of aspirin is lost.14,15 Celecoxib (Celebrex)16 and diclofenac (Voltaren) have binding sites different from that of aspirin, although the ACR recommends against using COX-2-selective inhibitors such as celecoxib in the situation and gives no recommendation about other NSAIDs.
No recommendations for or against
The panel issued no recommendations for or against the following treatments for patients with knee osteoarthritis:
- Intra-articular hyaluronan injections
- Duloxetine (Cymbalta)
- Opioid analgesics.
Comments on knee injections
Intra-articular injections of corticosteroids or hyaluronan are commonly used for knee osteoarthritis. As noted, corticosteroid injections received a conditional recommendation, while hyaluronan injections received no recommendation for or against.
How often to inject corticosteroids? In general, too-frequent injection of corticosteroids is to be avoided, in view of the risk of promoting joint breakdown. There is no “magic” number of injections that is safe, although more than 4 per year in the same joint should generally be avoided. In some situations, however, repeat injections may be reasonable if alternative therapies are associated with higher risk.
Raynauld et al,17 in a randomized, double-blind, placebo-controlled trial, demonstrated that intra-articular corticosteroid injections at 3-month intervals for 2 years were not deleterious to knees.
My philosophy is generally not to inject on a regular basis, but to be selective and be guided by the patient’s clinical condition and response to prior injections.
Are hyaluronan injections effective? Although experts differ in their enthusiasm for intra-articular hyaluronan injections in the knee, I have found that many patients benefit from this treatment. Multiple studies have found it efficacious and safe overall.18–21 However, some systematic reviews have called its efficacy into question.7
Although differences in efficacy have been noted, this therapy was approved as being useful in patients with knee osteoarthritis in the Osteoarthritis Research Society International (OARSI) recommendations.7 The effect sizes were smaller in later assessments.22
Hyaluronan injections do not pose the risk of joint breakdown that corticosteroid injections do, but their clinical efficacy is not as dramatic. Adverse reactions to most intraarticular hyaluronans are limited, with slight increases in pain and stiffness after injection. Significant inflammatory reactions characterized as “postinjection flares” are more commonly seen with high-molecular-weight crosslinked preparations. These reactions can be severe and can mimic joint infection clinically. Joint aspiration with synovial fluid analysis and culture may be necessary to exclude infection. Response to aspiration and nonsteroidal inflammatory agents or intra-articular corticosteroids is usually excellent.
Ultrasonographic guidance. As with intraarticular injections in other areas, ultrasonographic guidance is becoming more common, as it allows for more accurate drug administration.
Pes anserine bursitis must be ruled out as a cause of the patient’s knee symptoms—misdiagnosis is not uncommon. The bursa is located on the medial aspect of the tibia, and inflammation of the bursa is a common cause of pain in this area. Local steroid injection is extremely effective in symptomatic therapy. Physical therapy and NSAIDs may be adequate to treat milder cases.
Conditional recommendation against glucosamine, chondroitin, capsaicin
The ACR panel conditionally recommended that patients with knee osteoarthritis not use:
- Chondroitin sulfate
- Glucosamine
- Topical capsaicin.
Comment. Evidence is mixed about the efficacy of glucosamine and chondroitin sulfate, which are so-called nutraceuticals. Some studies found them useful23–25 but some did not,26 and a meta-analysis concluded that they do not help.27 The OARSI guidelines published in 2008 stated that these agents may relieve symptoms of osteoarthritis of the knee.7 The OARSI update published in 2010 found that glucosamine was effective, but less so than in previous studies.22 If glucosamine is effective, some studies suggest that glucosamine sulfate is more effective than glucosamine hydrochloride.22
The same OARSI review revealed that chondroitin sulfate relieved pain but with heterogeneous, dissimilar effect sizes. Of interest was the finding that the 5-year incidence of total knee replacement was lower in patients treated with glucosamine sulfate 1,500 mg/day than with placebo. Also, the rate of decline of joint space narrowing was reported to be reduced in chondroitin sulfate-treated patients.22
In practice, a conditional recommendation against a treatment means that most informed patients would not want the treatment, but some would. Accordingly, if patients still want to take chondroitin or glucosamine after being informed of the limited evidence of benefit, I feel a trial of their use is reasonable.
OSTEOARTHRITIS OF THE HIP
Indications for therapy of osteoarthritis of the hip are similar to those for osteoarthritis of the knee.
As in the knee, nonpharmacologic therapies are important. Loss of weight for overweight patients is extremely important; supervised exercise is especially valuable. Use of canes or crutches as needed is conditionally recommended.
Pharmacologic management is similar to that of osteoarthritis of the knee, with particular use of acetaminophen, NSAIDs, tramadol, and intra-articular corticosteroid injections.
Comment. Intra-articular injection of corticosteroids into the hip would be out of the realm of most nonspecialist practices. Although some rheumatologists are expert in such injections, this treatment is generally best left to an orthopedist or invasive radiologist. The use of ultrasonographic guidance is becoming more frequent, with many rheumatologists having developed expertise in this approach to the knee and the hip. Since most studies were in patients with osteoarthritis of the knee, fewer data are available as to the efficacy of these agents in patients with hip osteoarthritis.
Fewer data are available also with respect to the benefit of chondroitin sulfate and glucosamine in patients with osteoarthritis of the hip. Total joint replacement is extremely effective if conservative therapy does not help.
FIRST, DO NO HARM
Guidelines from the ACR,1,2 the European League Against Rheumatism (EULAR),28,29 the American Academy of Orthopedic Surgeons (AAOS),30 and the OARSI7,22 all differ somewhat, owing to the different evidence available at the time each guideline was developed and to different geographic and cultural backgrounds.
The compositions of these various panels also differ sufficiently to affect their overall recommendations. For example, the EULAR panel consisted of only rheumatologists and an orthopedic surgeon; for the hand osteoarthritis recommendations they added a physiatrist and two allied health professionals.28,29 The OARSI panel included two primary care physicians in addition to rheumatologists and an orthopedic surgeon.7 The ACR was the only professional society to include primary care physicians, physiatrists, and geriatricians along with rheumatologists, an orthopedic surgeon, and physical and occupational therapists.
Although it is to be expected that there will not be universal agreement on all points of management of osteoarthritis by diverse groups, it is essential that input from all these experts representing various subspecialties be recognized. Therapeutic approaches will vary depending on patient characteristics and the experience of the treating physician. As long as therapy is based on reasonable supportive data, beneficial effects can be anticipated. Therapies that received conditional recommendations are not to be discounted if a reasonable percent of patients respond in positive fashion. Obviously, strong recommendations are more likely to be universally accepted since the likelihood that they will be beneficial is stronger.
In any approach to therapy, the caveat primum non nocere—first, do no harm—must always be kept in mind.
- Hochberg MC, Altman RD, April KT, et al; American College of Rheumatology. American College of Rheumatology 2012 recommendations for the use of non-pharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care Res (Hoboken) 2012; 64:465–474.
- American College of Rheumatology Subcommittee on Osteoarthritis Guidelines. Recommendations for the medical management of osteoarthritis of the hip and knee: 2000 update. Arthritis Rheum 2000; 43:1905–1915.
- Atkins D, Best D, Briss PA, et al; GRADE Working Group. Grading quality of evidence and strength of recommendations. BMJ 2004; 328:1490.
- Askari A, Moskowitz RW, Ryan C. Stretch gloves. A study of objective and subjective effectiveness in arthritis of the hands. Arthritis Rheum 1974; 17:263–265.
- Chou R, Fanciullo GJ, Fine PG, et al; American Pain Society-American Academy of Pain Medicine Opioids Guidelines Panel. Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. J Pain 2009; 10:113–130.
- Fang MA, Taylor CE, Nouvong A, Masih S, Kao KC, Perell KL. Effects of footwear on medial compartment knee osteoarthritis. J Rehabil Res Dev 2006; 43:427–434.
- Zhang W, Moskowitz RW, Nuki G, et al. OARSI recommendations for the management of hip and knee osteoarthritis, Part II: OARSI evidence-based, expert consensus guidelines. Osteoarthritis Cartilage 2008; 16:137–162.
- US Food and Drug Administration (FDA). FDA Drug Safety Communication: Prescription Acetaminophen Products to be Limited to 325 mg Per Dosage Unit; Boxed Warning Will Highlight Potential for Severe Liver Failure. January 13, 2011. http://www.fda.gov/Drugs/DrugSafety/ucm239821.htm. Accessed November 28, 2012.
- Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med 2010; 77:19–27.
- Bolten WW. Rational use of nonsteroidal anti-inflammatory drugs and proton pump inhibitors in combination for rheumatic diseases. Orthopedic Research and Reviews 2010; 2:75–84.
- Graham DY, Agrawal NM, Campbell DR, et al; NSAID-Associated Gastric Ulcer Prevention Study Group. Ulcer prevention in long-term users of nonsteroidal anti-inflammatory drugs: results of a double-blind, randomized, multicenter, active- and placebo-controlled study of misoprostol vs lansoprazole. Arch Intern Med 2002; 162:169–175.
- American College of Rheumatology Ad Hoc Group on Use of Selective and Nonselective Nonsteroidal Antiinflammatory Drugs. Recommendations for use of selective and nonselective nonsteroidal antiinflammatory drugs: an American College of Rheumatology white paper. Arthritis Rheum 2008; 59:1058–1073.
- Antman EM, Bennett JS, Daugherty A, Furberg C, Roberts H, Taubert KA; American Heart Association. Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart Association. Circulation 2007; 115:1634–1642.
- Ellison J, Dager W. Recent FDA warning of the concomitant use of aspirin and ibuprofen and the effects on platelet aggregation. Prev Cardiol 2007; 10:61–63.
- Schuijt MP, Huntjens-Fleuren HW, de Metz M, Vollaard EJ. The interaction of ibuprofen and diclofenac with aspirin in healthy volunteers. Br J Pharmacol 2009; 157:931–934.
- Wilner KD, Rushing M, Walden C, et al. Celecoxib does not affect the antiplatelet activity of aspirin in healthy volunteers. J Clin Pharmacol 2002; 42:1027–1030.
- Raynauld JP, Buckland-Wright C, Ward R, et al. Safety and efficacy of long-term intraarticular steroid injections in osteoarthritis of the knee: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 2003; 48:370–377.
- Berenbaum F, Grifka J, Cazzaniga S, et al. A randomised, double-blind, controlled trial comparing two intra-articular hyaluronic acid preparations differing by their molecular weight in symptomatic knee osteoarthritis. Ann Rheum Dis 2012; 71:1454–1460.
- Colen S, van den Bekerom MP, Mulier M, Haverkamp D. Hyaluronic acid in the treatment of knee osteoarthritis: a systematic review and meta-analysis with emphasis on the efficacy of different products. BioDrugs 2012; 26:257–268.
- Wang CT, Lin J, Chang CJ, Lin YT, Hou SM. Therapeutic effects of hyaluronic acid on osteoarthritis of the knee. A meta-analysis of randomized controlled trials. J Bone Joint Surg Am 2004; 86-A:538–545.
- Rutjes AW, Jüni P, da Costa BR, Trelle S, Nüesch E, Reichenbach S. Visco-supplementation for osteoarthritis of the knee: a systematic review and meta-analysis. Ann Intern Med 2012; 157:180–191.
- Zhang W, Nuki G, Moskowitz RW, et al. OARSI recommendations for the management of hip and knee osteoarthritis: part III: changes in evidence following systematic cumulative update of research published through January 2009. Osteoarthritis Cartilage 2010; 18:476–499.
- Zhang W, Moskowitz RW, Nuki G, et al. OARSI recommendations for the management of hip and knee osteoarthritis, part I: critical appraisal of existing treat-ment guidelines and systematic review of current research evidence. Osteoarthritis Cartilage 2007; 15:981–1000.
- Towheed TE, Maxwell L, Anastassiades TP, et al. Glucosamine therapy for treating osteoarthritis. Cochrane Database Syst Rev 2005; 2:CD002946.
- Vlad SC, LaValley MP, McAlindon TE, Felson DT. Glucosamine for pain in osteoarthritis: why do trial results differ? Arthritis Rheum 2007; 56:2267–2277.
- Clegg DO, Reda DJ, Harris CL, et al. Glucosamine, chondroitin sulfate, and the two in combination for painful knee osteoarthritis. N Engl J Med 2006; 354:795–808.
- Wandel S, Jüni P, Tendal B, et al. Effects of glucosamine, chondroitin, or placebo in patients with osteoarthritis of hip or knee: network meta-analysis. BMJ 2010; 341:c4675.
- Jordan KM, Arden NK, Doherty M, Bannwarth B, Bijlsma JW, Dieppe P, et al. EULAR recommendations 2003: an evidence based approach to the management of knee osteoarthritis: report of a Task Force of the EULAR Standing Committee for International Clinical Studies Including Therapeutic Trials (ESCISIT). Ann Rheum Dis 2003;62:1145–55. Studies Including Therapeutic Trials (ESCISIT). Ann Rheum Dis 2005; 64:669–681.
- Zhang W, Doherty M, Leeb BF, et al. EULAR evidence based recommendations for the management of hand osteoarthritis: report of a Task Force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis 2007; 66:377–388.
- American Academy of Orthopaedic Surgeons. Treatment of osteoarthritis of the knee (non-arthroplasty). Rosemont, IL: American Academy of Orthopaedic Surgeons; 2008.
- Hochberg MC, Altman RD, April KT, et al; American College of Rheumatology. American College of Rheumatology 2012 recommendations for the use of non-pharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care Res (Hoboken) 2012; 64:465–474.
- American College of Rheumatology Subcommittee on Osteoarthritis Guidelines. Recommendations for the medical management of osteoarthritis of the hip and knee: 2000 update. Arthritis Rheum 2000; 43:1905–1915.
- Atkins D, Best D, Briss PA, et al; GRADE Working Group. Grading quality of evidence and strength of recommendations. BMJ 2004; 328:1490.
- Askari A, Moskowitz RW, Ryan C. Stretch gloves. A study of objective and subjective effectiveness in arthritis of the hands. Arthritis Rheum 1974; 17:263–265.
- Chou R, Fanciullo GJ, Fine PG, et al; American Pain Society-American Academy of Pain Medicine Opioids Guidelines Panel. Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. J Pain 2009; 10:113–130.
- Fang MA, Taylor CE, Nouvong A, Masih S, Kao KC, Perell KL. Effects of footwear on medial compartment knee osteoarthritis. J Rehabil Res Dev 2006; 43:427–434.
- Zhang W, Moskowitz RW, Nuki G, et al. OARSI recommendations for the management of hip and knee osteoarthritis, Part II: OARSI evidence-based, expert consensus guidelines. Osteoarthritis Cartilage 2008; 16:137–162.
- US Food and Drug Administration (FDA). FDA Drug Safety Communication: Prescription Acetaminophen Products to be Limited to 325 mg Per Dosage Unit; Boxed Warning Will Highlight Potential for Severe Liver Failure. January 13, 2011. http://www.fda.gov/Drugs/DrugSafety/ucm239821.htm. Accessed November 28, 2012.
- Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med 2010; 77:19–27.
- Bolten WW. Rational use of nonsteroidal anti-inflammatory drugs and proton pump inhibitors in combination for rheumatic diseases. Orthopedic Research and Reviews 2010; 2:75–84.
- Graham DY, Agrawal NM, Campbell DR, et al; NSAID-Associated Gastric Ulcer Prevention Study Group. Ulcer prevention in long-term users of nonsteroidal anti-inflammatory drugs: results of a double-blind, randomized, multicenter, active- and placebo-controlled study of misoprostol vs lansoprazole. Arch Intern Med 2002; 162:169–175.
- American College of Rheumatology Ad Hoc Group on Use of Selective and Nonselective Nonsteroidal Antiinflammatory Drugs. Recommendations for use of selective and nonselective nonsteroidal antiinflammatory drugs: an American College of Rheumatology white paper. Arthritis Rheum 2008; 59:1058–1073.
- Antman EM, Bennett JS, Daugherty A, Furberg C, Roberts H, Taubert KA; American Heart Association. Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart Association. Circulation 2007; 115:1634–1642.
- Ellison J, Dager W. Recent FDA warning of the concomitant use of aspirin and ibuprofen and the effects on platelet aggregation. Prev Cardiol 2007; 10:61–63.
- Schuijt MP, Huntjens-Fleuren HW, de Metz M, Vollaard EJ. The interaction of ibuprofen and diclofenac with aspirin in healthy volunteers. Br J Pharmacol 2009; 157:931–934.
- Wilner KD, Rushing M, Walden C, et al. Celecoxib does not affect the antiplatelet activity of aspirin in healthy volunteers. J Clin Pharmacol 2002; 42:1027–1030.
- Raynauld JP, Buckland-Wright C, Ward R, et al. Safety and efficacy of long-term intraarticular steroid injections in osteoarthritis of the knee: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 2003; 48:370–377.
- Berenbaum F, Grifka J, Cazzaniga S, et al. A randomised, double-blind, controlled trial comparing two intra-articular hyaluronic acid preparations differing by their molecular weight in symptomatic knee osteoarthritis. Ann Rheum Dis 2012; 71:1454–1460.
- Colen S, van den Bekerom MP, Mulier M, Haverkamp D. Hyaluronic acid in the treatment of knee osteoarthritis: a systematic review and meta-analysis with emphasis on the efficacy of different products. BioDrugs 2012; 26:257–268.
- Wang CT, Lin J, Chang CJ, Lin YT, Hou SM. Therapeutic effects of hyaluronic acid on osteoarthritis of the knee. A meta-analysis of randomized controlled trials. J Bone Joint Surg Am 2004; 86-A:538–545.
- Rutjes AW, Jüni P, da Costa BR, Trelle S, Nüesch E, Reichenbach S. Visco-supplementation for osteoarthritis of the knee: a systematic review and meta-analysis. Ann Intern Med 2012; 157:180–191.
- Zhang W, Nuki G, Moskowitz RW, et al. OARSI recommendations for the management of hip and knee osteoarthritis: part III: changes in evidence following systematic cumulative update of research published through January 2009. Osteoarthritis Cartilage 2010; 18:476–499.
- Zhang W, Moskowitz RW, Nuki G, et al. OARSI recommendations for the management of hip and knee osteoarthritis, part I: critical appraisal of existing treat-ment guidelines and systematic review of current research evidence. Osteoarthritis Cartilage 2007; 15:981–1000.
- Towheed TE, Maxwell L, Anastassiades TP, et al. Glucosamine therapy for treating osteoarthritis. Cochrane Database Syst Rev 2005; 2:CD002946.
- Vlad SC, LaValley MP, McAlindon TE, Felson DT. Glucosamine for pain in osteoarthritis: why do trial results differ? Arthritis Rheum 2007; 56:2267–2277.
- Clegg DO, Reda DJ, Harris CL, et al. Glucosamine, chondroitin sulfate, and the two in combination for painful knee osteoarthritis. N Engl J Med 2006; 354:795–808.
- Wandel S, Jüni P, Tendal B, et al. Effects of glucosamine, chondroitin, or placebo in patients with osteoarthritis of hip or knee: network meta-analysis. BMJ 2010; 341:c4675.
- Jordan KM, Arden NK, Doherty M, Bannwarth B, Bijlsma JW, Dieppe P, et al. EULAR recommendations 2003: an evidence based approach to the management of knee osteoarthritis: report of a Task Force of the EULAR Standing Committee for International Clinical Studies Including Therapeutic Trials (ESCISIT). Ann Rheum Dis 2003;62:1145–55. Studies Including Therapeutic Trials (ESCISIT). Ann Rheum Dis 2005; 64:669–681.
- Zhang W, Doherty M, Leeb BF, et al. EULAR evidence based recommendations for the management of hand osteoarthritis: report of a Task Force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis 2007; 66:377–388.
- American Academy of Orthopaedic Surgeons. Treatment of osteoarthritis of the knee (non-arthroplasty). Rosemont, IL: American Academy of Orthopaedic Surgeons; 2008.
Lupus in Hispanics: A matter of serious concern
Some diseases are either more serious or more frequent in US Hispanics, and systemic lupus erythematosus is one of them. This fact has not yet diffused to all providers, many of whom will be the ones dealing with these individuals when the disease first emerges.
In order to raise physicians’ awareness of this situation, we will briefly review here the salient features of lupus in US Hispanics and its short-term and long-term impact.
HISPANICS ARE THE LARGEST MINORITY IN THE UNITED STATES
Over the last 30 years, the Hispanic population in the United States has increased to the point that it is now the largest US minority group, and the fastest-growing. In the 2010 US census, Hispanics surpassed the 50 million mark.1 Physicians and health care providers are becoming familiar with this growing population and its ailments, but more needs to be done to familiarize them with specific conditions that are more frequent and more serious in US Hispanics.
No population-based study has yet defined the prevalence and incidence of lupus in US Hispanics. However, on the basis of hospital and outpatient visits in regions in which Hispanics make up a large part of the population, it has been inferred that this group has a higher frequency of lupus, probably as high as in African Americans.
Likewise, clinicians taking care of these patients have suspected that lupus is more severe in US Hispanics than in non-Hispanic Caucasians, but this was documented and brought to general attention only with the publication of reports from the Lupus in Minorities: Nature versus Nurture (LUMINA) study.2
LUMINA, a longitudinal study
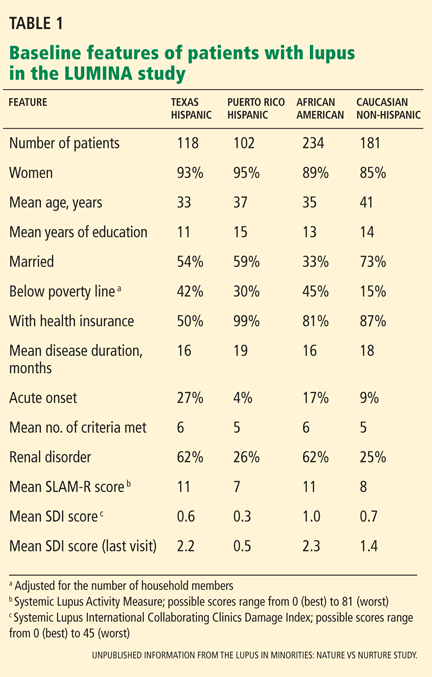
LUMINA is a longitudinal study of 640 patients with lupus from four populations: Hispanic from Texas, Hispanic from Puerto Rico, African American, and Caucasian non-Hispanic (Table 1). At the time of recruitment, patients were at least 16 years old and had had lupus for 5 years or less. They come in for periodic visits to the University of Alabama at Birmingham, the University of Texas Health Science Center at Houston, and the University of Puerto Rico Medical Sciences Campus. Recruitment began in 1994 and finished in 2007. Follow-up ranges from 1 to 14 years, with a mean of 4.5 years.
LUMINA is supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases, the National Institutes of Health General Clinical Research Centers program, the National Center for Research Resources Clinical Research Infrastructure Initiative, the Mary Kirkland Center for Lupus Research Scholars Program, and Rheuminations Inc (New York, NY).
The purpose of the study is to shed light on the interplay of genetics and environment in this disease and, in the process, to raise awareness about the problem of lupus in Hispanics. In fact, much of the information in the following sections is from the LUMINA study.
HISPANICS ARE NOT A HOMOGENEOUS GROUP
In the United States, the term Hispanic describes anyone whose origin goes back to a Spanish-speaking country. However, US Hispanics are not a homogeneous racial group: they differ in genetics, culture, and problems.
The largest US Hispanic subgroup and the one more likely to be seen by US physicians is Hispanics of Mexican origin, who account for 66% of all US Hispanics. This group has a higher percentage of Amerindian genes than those of Puerto Rican ancestry.3 LUMINA researchers analyzed the DNA of 492 patients and found the following mixtures of genes3:
- Hispanics in Texas (mostly of Mexican origin): 48% Amerindian, 18% African, 34% European
- Hispanics from Puerto Rico: 20% Amerindian, 45% African, 35% European
- African Americans: 0% Amerindian, 79% African, 21% European
- Non-Hispanic Caucasians: 10% Amerindian, 18% African, 72% European.
Latin Americans of mixed European and Amerindian ancestry (which includes Aztec, Mayan, Quechuan, Aymaran, and other Central and South American groups) are called mestizos. Not all people in Latin America are mestizos: some are of European, African, or Asian ancestry, but in the United States they are all called Hispanics.
LUPUS DIFFERS AMONG SUBGROUPS
LUMINA research has revealed that lupus is heterogeneous also among US Hispanic subgroups. When people from Puerto Rico get lupus, it is generally less serious and devastating than in those from Mexico or Central America. Since US Hispanics of Mexican or Central American origin possess more Amerindian genes, this observation supports the notion that these genes are important contributors to the occurrence and expression of the disease.
Amerindian genes contribute to a greater susceptibility to lupus,4,5 although there is an interplay between genetic and nongenetic factors in the etiology and expression.6 Lupus starts at a younger age in Hispanics of predominantly Amerindian ancestry than in non-Hispanic Caucasians, and the onset is more likely to be acute.7
Renal involvement in these patients8 and mestizos from Latin America is rather common, probably as common as it is in US African Americans, and it tends to develop earlier than in non-Hispanic Caucasians.9 Amerindian ancestral genes, like African genes, contribute to the occurrence of renal disease in lupus patients.4 Furthermore, once nephritis ensues, end-stage renal disease occurs more often in US Hispanic and African American than in non-Hispanic Caucasian children, as demonstrated by Hiraki et al10 using national databases, and the same is true in adults, as shown in the LUMINA cohort.11
Other potentially serious manifestations of the disease are also more common, including hematologic and central nervous system manifestations. Not surprisingly, then, these patients show a higher degree of disease activity, both early in the course of the disease12,13 and over time.14
Table 1 compares the demographic and clinical features of LUMINA patients according to ethnicity. By and large, Hispanics from Texas have lower levels of education and income (comparable with levels in African Americans), and this can adversely affect the disease course by limiting these patients’ access to adequate care.15
DISEASE ACTIVITY AND ORGAN DAMAGE ARE GREATER IN HISPANICS
Disease activity in lupus reflects the ongoing immune-mediated inflammatory process. In LUMINA patients, regardless of the time at which disease activity was ascertained, it was higher in Hispanics from Texas and in African Americans than in non-Hispanic Caucasians and in Hispanics from Puerto Rico.7,12,16–18 Similar findings were seen in the Grupo Latinoamericano de Estudio de Lupus (GLADEL) cohort,13 in which mestizos and Hispanics of mixed African and European ancestry had higher maximum disease activity scores than non-Hispanic Caucasians.13
In addition, organ damage in lupus—the irreversible changes that occur in organ systems as a consequence of the disease or its treatments (eg, glucocorticoids, immunosuppressive drugs)—is more severe and develops sooner in Hispanics from Texas than in other groups.6,18,19 Using multivariate analysis, LUMINA investigators19 estimated the hazard ratio for the time until organ damage appeared for various risk factors, with values of 1 or greater indicating a shorter time and lower values indicating a longer time. Being a Hispanic from Texas carried a hazard ratio of 2.11 (95% confidence interval 1.15–3.88).
Because organ damage is an important and independent predictor of further damage20 and death,21 physicians need to take this disease quite seriously and try to prevent damage early in people at risk. To achieve that, the need to control disease activity must be balanced against the risk of overtreatment, as the important contribution of glucocorticoids to organ damage is well recognized.22
HISPANICS HAVE MORE COMORBIDITIES
Obesity, hypertension, diabetes, and metabolic syndrome are more common in US Hispanics, particularly those of Amerindian ancestry, than in the majority population of non-Hispanic Caucasians.23,24 The potential deleterious effects of glucocorticoids in patients already predisposed to these conditions need to be considered, balancing adequate disease control against the potential adverse effects.22
QUALITY OF LIFE IS WORSE WITH LUPUS
Whether it is measured with a generic instrument such as the Short Form 36 (SF-36), as it was in LUMINA,25 or with a disease-specific tool such as the Lupus-Pro, quality of life is significantly worsened by lupus. Furthermore, Fernandez et al26 found that a low level of health-related quality of life, as measured by the SF-6D version of the SF-36, was predictive of poor outcomes in LUMINA patients.
POVERTY, NOT ETHNICITY, ACCOUNTS FOR HIGHER MORTALITY RATE
As yet, we have no population-based data comparing survival in US Hispanic patients with lupus vs that of other population groups.
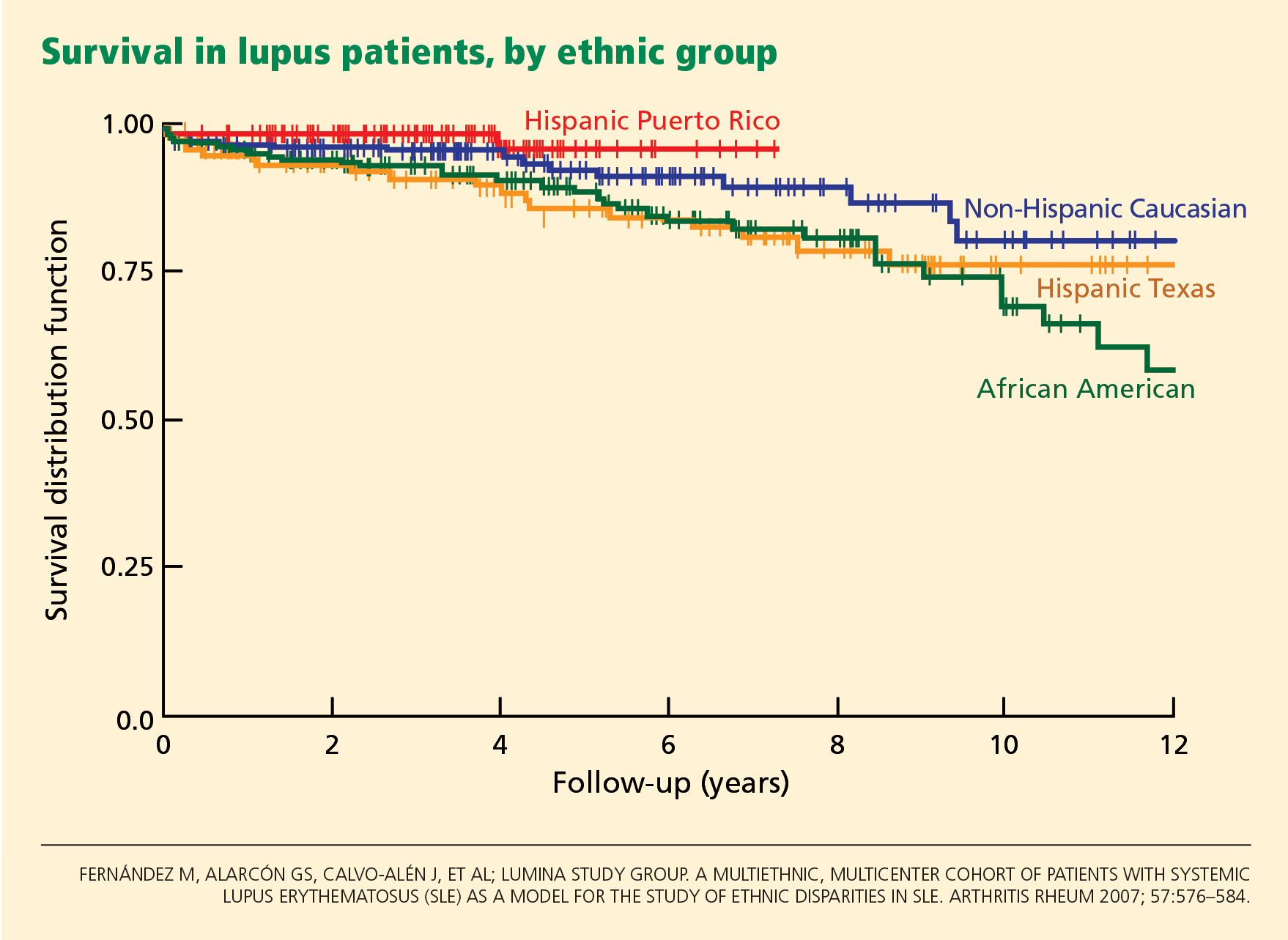
At first inspection, data from LUMINA indicate that Hispanics of primarily Amerindian ancestry have a lower survival rate than patients in other ethnic groups (Figure 1).6 However, when all other factors are taken into consideration, poverty, not ethnicity, is the major contributing factor (Table 2).6,27
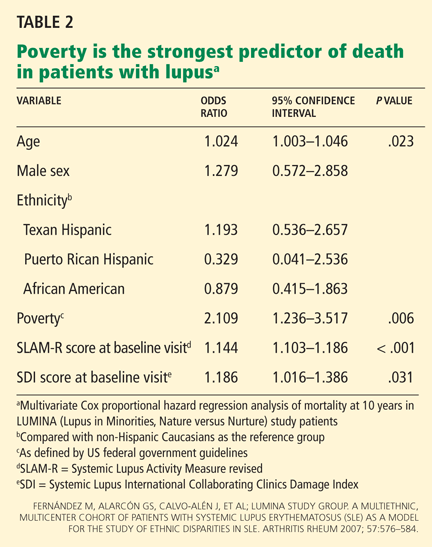
This finding illustrates the important interplay between genetic and nongenetic factors in the course and final outcome of lupus, as already alluded to, although the exact relationship between them is not clear. It remains to be determined whether poverty is only a proxy for other population characteristics such as illiteracy, limited access to specialized care, limited access to medications, or cultural beliefs that may interfere with proper care.
ANTIMALARIAL DRUGS INCREASE SURVIVAL
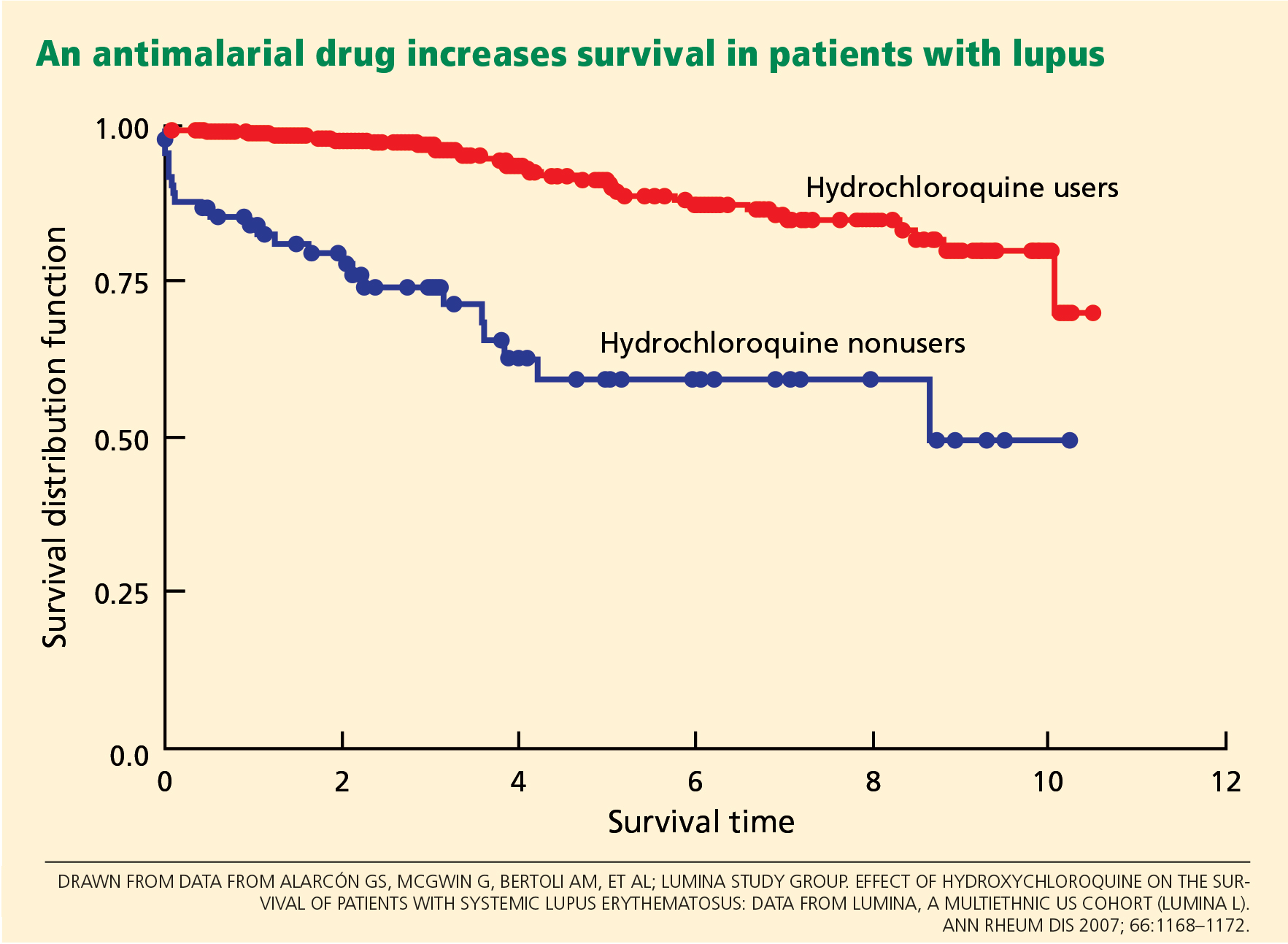
Using statistical analysis that adjusts for confounding by indication, we and others28–30 have shown that antimalarial drugs exert an independent and important protective effect on survival in lupus (Figure 2).
Important also is the protective effect of antimalarials on organ damage and the possibility of using them from disease outset in Hispanic patients at risk of early and rapid damage accrual,11 renal damage, and even lupus nephritis.31,32 This has very practical implications for the adequate and prompt management of these Hispanic patients.
PRACTICAL IMPLICATIONS
Lupus in US Hispanics is a serious disease with devastating consequences. Prompt diagnosis is paramount to prevent early organ damage and to prolong survival.
The disease may present in many different and unexpected ways, but joint pain, sun-sensitive rashes, renal involvement, cytopenias, and other manifestations should prompt the clinician to consider lupus in the differential diagnosis. Patients are often dismissed as having “arthritis” without being asked about other manifestations that may suggest a systemic connective tissue disease such as lupus. The same goes for skin rashes or unusual central nervous system manifestations.
The diagnosis of lupus is clinical, but some laboratory studies are essential to rule in or rule out renal or hematologic abnormalities and determine the level of disease activity. Tests usually ordered in patients suspected of having lupus include antinuclear antibody, complement levels, a complete blood cell count and differential, and a urinalysis. The need for additional tests depends on the results of the tests listed.
Once the disease is diagnosed, treatment should be tailored to the severity and type of clinical manifestations present. In general, glucocorticoids should be used at the smallest possible dose, antimalarials should be prescribed from the outset to all patients (following current guidelines in order to avoid ocular toxicity),33 and immunosuppressants and other treatments should be considered in certain instances. In parallel, consideration should be given to sun protection, adequate exercise, tobacco avoidance, osteoporosis and atherosclerosis prevention, planned conception, and compliance.
The goal in these people at risk is to control their lupus manifestations without causing undue damage, to preserve their quality of life, and to prevent an early demise.
- Humes KR, Jones NA, Ramirez RR. Overview of race and Hispanic origin: 2010. 2010 Census briefs; 2011. http://www.census.gov/prod/cen2010/briefs/c2010br-02.pdf. Accessed October 20, 2012.
- Reveille JD, Moulds JM, Ahn C, et al; for the LUMINA study Group. Systemic lupus erythematosus in three ethnic groups. I. The effects of HLA class II, C4, and CR1 alleles, socioeconomic factors, and ethnicity and disease onset. Arthritis Rheum 1998; 41:1161–1172.
- Alarcón GS, Beasley TM, Roseman JM, et al; LUMINA Study Group. Ethnic disparities in health and disease: the need to account for ancestral admixture when estimating the genetic contribution to both (LUMINA XXVI) (Letter). Lupus 2005; 14:867–868.
- Alarcón GS, Bastian HM, Beasley TM, et al; LUMINA Study Group. Systemic lupus erythematosus in a multi-ethnic cohort (LUMINA) XXXII: [corrected] contributions of admixture and socioeconomic status to renal involvement. Lupus 2006; 15:26–31.
- Sanchez E, Webb RD, Rasmussen A, et al. Genetically determined Amerindian ancestry correlates with increased frequency of risk alleles for systemic lupus erythematosus. Arthritis Rheum 2010; 62:3722–3729.
- Fernández M, Alarcón GS, Calvo-Alén J, et al; LUMINA Study Group. A multiethnic, multicenter cohort of patients with systemic lupus erythematosus (SLE) as a model for the study of ethnic disparities in SLE. Arthritis Rheum 2007; 57:576–584.
- Alarcón GS, Friedman AW, Straaton KV, et al. Systemic lupus erythematosus in three ethnic groups: III. A comparison of characteristics early in the natural history of the LUMINA cohort. LUpus in MInority populations: NAture vs Nurture. Lupus 1999; 8:197–209.
- Bastian HM, Alarcón GS, Roseman JM, et al; LUMINA Study Group. Systemic lupus erythematosus in a multiethnic US cohort (LUMINA) XL II: factors predictive of new or worsening proteinuria. Rheumatology (Oxford) 2007; 46:683–689.
- Burgos PI, McGwin G, Pons-Estel GJ, Reveille JD, Alarcón GS, Vilá LM. US patients of Hispanic and African ancestry develop lupus nephritis early in the disease course: data from LUMINA, a multiethnic US cohort (LUMINA LXXIV). Ann Rheum Dis 2011; 70:393–394.
- Hiraki LT, Lu B, Alexander SR, et al. End-stage renal disease due to lupus nephritis among children in the US, 1995–2006. Arthritis Rheum 2011; 63:1988–1997.
- Pons-Estel GJ, Alarcón GS, McGwin G, et al. Protective effect of hydroxychloroquine on renal damage in patients with lupus nephritis: LXV, data from a multiethnic US cohort. Arthritis Rheum 2009; 61:830–839.
- Alarcón GS, Roseman J, Bartolucci AA, et al. Systemic lupus erythematosus in three ethnic groups: II. Features predictive of disease activity early in its course. LUMINA Study Group. Lupus in minority populations, nature versus nurture. Arthritis Rheum 1998; 41:1173–1180.
- Pons-Estel BA, Catoggio LJ, Cardiel MH, et al; Grupo Latinoamericano de Estudio del Lupus. The GLADEL multinational Latin American prospective inception cohort of 1,214 patients with systemic lupus erythematosus: ethnic and disease heterogeneity among “Hispanics.” Medicine (Baltimore) 2004; 83:1–17.
- Alarcón GS, Calvo-Alén J, McGwin G, et al; LUMINA Study Group. Systemic lupus erythematosus in a multiethnic cohort: LUMINA XXXV. Predictive factors of high disease activity over time. Ann Rheum Dis 2006; 65:1168–1174.
- Vilá LM, Alarcón GS, McGwin G, Bastian HM, Fessler BJ, Reveille JD; Lumina Study Group. Systemic lupus erythematosus in a multiethnic US cohort, XXXVII: association of lymphopenia with clinical manifestations, serologic abnormalities, disease activity, and damage accrual. Arthritis Rheum 2006; 55:799–806.
- Zhang J, González LA, Roseman JM, Vilá LM, Reveille JD, Alárcon GS. Predictors of the rate of change in disease activity over time in LUMINA, a multiethnic US cohort of patients with systemic lupus erythematosus: LUMINA LXX. Lupus 2010; 19:727–733.
- Vilá LM, Alarcón GS, McGwin G, et al; LUMINA Study Group. Early clinical manifestations, disease activity and damage of systemic lupus erythematosus among two distinct US Hispanic subpopulations. Rheumatology (Oxford) 2004; 43:358–363.
- Gladman D, Ginzler E, Goldsmith C, et al. The development and initial validation of the Systemic Lupus International Collaborating Clinics/American College of Rheumatology damage index for systemic lupus erythematosus. Arthritis Rheum 1996; 39:363–369.
- Toloza SM, Roseman JM, Alarcón GS, et al. Systemic lupus erythematosus in a multiethnic US cohort (LUMINA): XXII. Predictors of time to the occurrence of initial damage. Arthritis Rheum 2004; 50:3177–3186.
- Alarcón GS, Roseman JM, McGwin G, et al; LUMINA Study Group. Systemic lupus erythematosus in three ethnic groups. XX. Damage as a predictor of further damage. Rheumatology (Oxford) 2004; 43:202–205.
- Alarcón GS, McGwin G, Bastian HM, et al. Systemic lupus erythematosus in three ethnic groups. VII [correction of VIII]. Predictors of early mortality in the LUMINA cohort. LUMINA Study Group. Arthritis Rheum 2001; 45:191–202.
- Ruiz-Irastorza G, Danza A, Khamashta M. Glucocorticoid use and abuse in SLE. Rheumatology (Oxford) 2012 E-pub ahead of print.
- Jordan HT, Tabaei BP, Nash D, Angell SY, Chamany S, Kerker B. Metabolic syndrome among adults in New York City, 2004 New York City Health and Nutrition Examination Survey. Prev Chronic Dis 2012; 9:E04.
- Matthews KA, Sowers MF, Derby CA, et al. Ethnic differences in cardiovascular risk factor burden among middle-aged women: Study of Women’s Health Across the Nation (SWAN). Am Heart J 2005; 149:1066–1073.
- Alarcón GS, McGwin G, Uribe A, et al. Systemic lupus erythematosus in a multiethnic lupus cohort (LUMINA). XVII. Predictors of selfreported health-related quality of life early in the disease course. Arthritis Rheum 2004; 51:465–474.
- Fernández M, Alarcón GS, McGwin G, et al; LUMINA Study Group. Using the Short Form 6D, as an overall measure of health, to predict damage accrual and mortality in patients with systemic lupus erythematosus: XLVII, results from a multiethnic US cohort. Arthritis Rheum 2007; 57:986–992.
- Durán S, Apte M, Alarcón GSLUMINA Study Group. Poverty, not ethnicity, accounts for the differential mortality rates among lupus patients of various ethnic groups. J Natl Med Assoc 2007; 99:1196–1198.
- Ruiz-Irastorza G, Egurbide MV, Pijoan JI, et al. Effect of antimalarials on thrombosis and survival in patients with systemic lupus erythematosus. Lupus 2006; 15:577–583.
- Alarcón GS, McGwin G, Bertoli AM, et al; LUMINA Study Group. Effect of hydroxychloroquine on the survival of patients with systemic lupus erythematosus: data from LUMINA, a multiethnic US cohort (LUMINA L). Ann Rheum Dis 2007; 66:1168–1172.
- Shinjo SK, Bonfá E, Wojdyla D, et al; Grupo Latino Americano de Estudio del Lupus Eritematoso (Gladel). Antimalarial treatment may have a time-dependent effect on lupus survival: data from a multinational Latin American inception cohort. Arthritis Rheum 2010; 62:855–862.
- Fessler BJ, Alarcón GS, McGwin G, et al; LUMINA Study Group. Systemic lupus erythematosus in three ethnic groups: XVI. Association of hydroxychloroquine use with reduced risk of damage accrual. Arthritis Rheum 2005; 52:1473–1480.
- Pons-Estel GJ, Alarcón GS, Hachuel L, et al. Antimalarials have a protective effect against the development of renal disease in Latin American SLE patients. The 9th International Congress on SLE June 24–27, 2010, Vancouver, Canada. Lupus 2010; 19(suppl 1):31–32.
- Ruiz-Irastorza G, Ramos-Casals M, Brito-Zeron P, Khamashta MA. Clinical efficacy and side effects of antimalarials in systemic lupus erythematosus: a systematic review. Ann Rheum Dis 2010; 69:20–28.
Some diseases are either more serious or more frequent in US Hispanics, and systemic lupus erythematosus is one of them. This fact has not yet diffused to all providers, many of whom will be the ones dealing with these individuals when the disease first emerges.
In order to raise physicians’ awareness of this situation, we will briefly review here the salient features of lupus in US Hispanics and its short-term and long-term impact.
HISPANICS ARE THE LARGEST MINORITY IN THE UNITED STATES
Over the last 30 years, the Hispanic population in the United States has increased to the point that it is now the largest US minority group, and the fastest-growing. In the 2010 US census, Hispanics surpassed the 50 million mark.1 Physicians and health care providers are becoming familiar with this growing population and its ailments, but more needs to be done to familiarize them with specific conditions that are more frequent and more serious in US Hispanics.
No population-based study has yet defined the prevalence and incidence of lupus in US Hispanics. However, on the basis of hospital and outpatient visits in regions in which Hispanics make up a large part of the population, it has been inferred that this group has a higher frequency of lupus, probably as high as in African Americans.
Likewise, clinicians taking care of these patients have suspected that lupus is more severe in US Hispanics than in non-Hispanic Caucasians, but this was documented and brought to general attention only with the publication of reports from the Lupus in Minorities: Nature versus Nurture (LUMINA) study.2
LUMINA, a longitudinal study
LUMINA is a longitudinal study of 640 patients with lupus from four populations: Hispanic from Texas, Hispanic from Puerto Rico, African American, and Caucasian non-Hispanic (Table 1). At the time of recruitment, patients were at least 16 years old and had had lupus for 5 years or less. They come in for periodic visits to the University of Alabama at Birmingham, the University of Texas Health Science Center at Houston, and the University of Puerto Rico Medical Sciences Campus. Recruitment began in 1994 and finished in 2007. Follow-up ranges from 1 to 14 years, with a mean of 4.5 years.
LUMINA is supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases, the National Institutes of Health General Clinical Research Centers program, the National Center for Research Resources Clinical Research Infrastructure Initiative, the Mary Kirkland Center for Lupus Research Scholars Program, and Rheuminations Inc (New York, NY).
The purpose of the study is to shed light on the interplay of genetics and environment in this disease and, in the process, to raise awareness about the problem of lupus in Hispanics. In fact, much of the information in the following sections is from the LUMINA study.
HISPANICS ARE NOT A HOMOGENEOUS GROUP
In the United States, the term Hispanic describes anyone whose origin goes back to a Spanish-speaking country. However, US Hispanics are not a homogeneous racial group: they differ in genetics, culture, and problems.
The largest US Hispanic subgroup and the one more likely to be seen by US physicians is Hispanics of Mexican origin, who account for 66% of all US Hispanics. This group has a higher percentage of Amerindian genes than those of Puerto Rican ancestry.3 LUMINA researchers analyzed the DNA of 492 patients and found the following mixtures of genes3:
- Hispanics in Texas (mostly of Mexican origin): 48% Amerindian, 18% African, 34% European
- Hispanics from Puerto Rico: 20% Amerindian, 45% African, 35% European
- African Americans: 0% Amerindian, 79% African, 21% European
- Non-Hispanic Caucasians: 10% Amerindian, 18% African, 72% European.
Latin Americans of mixed European and Amerindian ancestry (which includes Aztec, Mayan, Quechuan, Aymaran, and other Central and South American groups) are called mestizos. Not all people in Latin America are mestizos: some are of European, African, or Asian ancestry, but in the United States they are all called Hispanics.
LUPUS DIFFERS AMONG SUBGROUPS
LUMINA research has revealed that lupus is heterogeneous also among US Hispanic subgroups. When people from Puerto Rico get lupus, it is generally less serious and devastating than in those from Mexico or Central America. Since US Hispanics of Mexican or Central American origin possess more Amerindian genes, this observation supports the notion that these genes are important contributors to the occurrence and expression of the disease.
Amerindian genes contribute to a greater susceptibility to lupus,4,5 although there is an interplay between genetic and nongenetic factors in the etiology and expression.6 Lupus starts at a younger age in Hispanics of predominantly Amerindian ancestry than in non-Hispanic Caucasians, and the onset is more likely to be acute.7
Renal involvement in these patients8 and mestizos from Latin America is rather common, probably as common as it is in US African Americans, and it tends to develop earlier than in non-Hispanic Caucasians.9 Amerindian ancestral genes, like African genes, contribute to the occurrence of renal disease in lupus patients.4 Furthermore, once nephritis ensues, end-stage renal disease occurs more often in US Hispanic and African American than in non-Hispanic Caucasian children, as demonstrated by Hiraki et al10 using national databases, and the same is true in adults, as shown in the LUMINA cohort.11
Other potentially serious manifestations of the disease are also more common, including hematologic and central nervous system manifestations. Not surprisingly, then, these patients show a higher degree of disease activity, both early in the course of the disease12,13 and over time.14
Table 1 compares the demographic and clinical features of LUMINA patients according to ethnicity. By and large, Hispanics from Texas have lower levels of education and income (comparable with levels in African Americans), and this can adversely affect the disease course by limiting these patients’ access to adequate care.15
DISEASE ACTIVITY AND ORGAN DAMAGE ARE GREATER IN HISPANICS
Disease activity in lupus reflects the ongoing immune-mediated inflammatory process. In LUMINA patients, regardless of the time at which disease activity was ascertained, it was higher in Hispanics from Texas and in African Americans than in non-Hispanic Caucasians and in Hispanics from Puerto Rico.7,12,16–18 Similar findings were seen in the Grupo Latinoamericano de Estudio de Lupus (GLADEL) cohort,13 in which mestizos and Hispanics of mixed African and European ancestry had higher maximum disease activity scores than non-Hispanic Caucasians.13
In addition, organ damage in lupus—the irreversible changes that occur in organ systems as a consequence of the disease or its treatments (eg, glucocorticoids, immunosuppressive drugs)—is more severe and develops sooner in Hispanics from Texas than in other groups.6,18,19 Using multivariate analysis, LUMINA investigators19 estimated the hazard ratio for the time until organ damage appeared for various risk factors, with values of 1 or greater indicating a shorter time and lower values indicating a longer time. Being a Hispanic from Texas carried a hazard ratio of 2.11 (95% confidence interval 1.15–3.88).
Because organ damage is an important and independent predictor of further damage20 and death,21 physicians need to take this disease quite seriously and try to prevent damage early in people at risk. To achieve that, the need to control disease activity must be balanced against the risk of overtreatment, as the important contribution of glucocorticoids to organ damage is well recognized.22
HISPANICS HAVE MORE COMORBIDITIES
Obesity, hypertension, diabetes, and metabolic syndrome are more common in US Hispanics, particularly those of Amerindian ancestry, than in the majority population of non-Hispanic Caucasians.23,24 The potential deleterious effects of glucocorticoids in patients already predisposed to these conditions need to be considered, balancing adequate disease control against the potential adverse effects.22
QUALITY OF LIFE IS WORSE WITH LUPUS
Whether it is measured with a generic instrument such as the Short Form 36 (SF-36), as it was in LUMINA,25 or with a disease-specific tool such as the Lupus-Pro, quality of life is significantly worsened by lupus. Furthermore, Fernandez et al26 found that a low level of health-related quality of life, as measured by the SF-6D version of the SF-36, was predictive of poor outcomes in LUMINA patients.
POVERTY, NOT ETHNICITY, ACCOUNTS FOR HIGHER MORTALITY RATE
As yet, we have no population-based data comparing survival in US Hispanic patients with lupus vs that of other population groups.
At first inspection, data from LUMINA indicate that Hispanics of primarily Amerindian ancestry have a lower survival rate than patients in other ethnic groups (Figure 1).6 However, when all other factors are taken into consideration, poverty, not ethnicity, is the major contributing factor (Table 2).6,27
This finding illustrates the important interplay between genetic and nongenetic factors in the course and final outcome of lupus, as already alluded to, although the exact relationship between them is not clear. It remains to be determined whether poverty is only a proxy for other population characteristics such as illiteracy, limited access to specialized care, limited access to medications, or cultural beliefs that may interfere with proper care.
ANTIMALARIAL DRUGS INCREASE SURVIVAL
Using statistical analysis that adjusts for confounding by indication, we and others28–30 have shown that antimalarial drugs exert an independent and important protective effect on survival in lupus (Figure 2).
Important also is the protective effect of antimalarials on organ damage and the possibility of using them from disease outset in Hispanic patients at risk of early and rapid damage accrual,11 renal damage, and even lupus nephritis.31,32 This has very practical implications for the adequate and prompt management of these Hispanic patients.
PRACTICAL IMPLICATIONS
Lupus in US Hispanics is a serious disease with devastating consequences. Prompt diagnosis is paramount to prevent early organ damage and to prolong survival.
The disease may present in many different and unexpected ways, but joint pain, sun-sensitive rashes, renal involvement, cytopenias, and other manifestations should prompt the clinician to consider lupus in the differential diagnosis. Patients are often dismissed as having “arthritis” without being asked about other manifestations that may suggest a systemic connective tissue disease such as lupus. The same goes for skin rashes or unusual central nervous system manifestations.
The diagnosis of lupus is clinical, but some laboratory studies are essential to rule in or rule out renal or hematologic abnormalities and determine the level of disease activity. Tests usually ordered in patients suspected of having lupus include antinuclear antibody, complement levels, a complete blood cell count and differential, and a urinalysis. The need for additional tests depends on the results of the tests listed.
Once the disease is diagnosed, treatment should be tailored to the severity and type of clinical manifestations present. In general, glucocorticoids should be used at the smallest possible dose, antimalarials should be prescribed from the outset to all patients (following current guidelines in order to avoid ocular toxicity),33 and immunosuppressants and other treatments should be considered in certain instances. In parallel, consideration should be given to sun protection, adequate exercise, tobacco avoidance, osteoporosis and atherosclerosis prevention, planned conception, and compliance.
The goal in these people at risk is to control their lupus manifestations without causing undue damage, to preserve their quality of life, and to prevent an early demise.
Some diseases are either more serious or more frequent in US Hispanics, and systemic lupus erythematosus is one of them. This fact has not yet diffused to all providers, many of whom will be the ones dealing with these individuals when the disease first emerges.
In order to raise physicians’ awareness of this situation, we will briefly review here the salient features of lupus in US Hispanics and its short-term and long-term impact.
HISPANICS ARE THE LARGEST MINORITY IN THE UNITED STATES
Over the last 30 years, the Hispanic population in the United States has increased to the point that it is now the largest US minority group, and the fastest-growing. In the 2010 US census, Hispanics surpassed the 50 million mark.1 Physicians and health care providers are becoming familiar with this growing population and its ailments, but more needs to be done to familiarize them with specific conditions that are more frequent and more serious in US Hispanics.
No population-based study has yet defined the prevalence and incidence of lupus in US Hispanics. However, on the basis of hospital and outpatient visits in regions in which Hispanics make up a large part of the population, it has been inferred that this group has a higher frequency of lupus, probably as high as in African Americans.
Likewise, clinicians taking care of these patients have suspected that lupus is more severe in US Hispanics than in non-Hispanic Caucasians, but this was documented and brought to general attention only with the publication of reports from the Lupus in Minorities: Nature versus Nurture (LUMINA) study.2
LUMINA, a longitudinal study
LUMINA is a longitudinal study of 640 patients with lupus from four populations: Hispanic from Texas, Hispanic from Puerto Rico, African American, and Caucasian non-Hispanic (Table 1). At the time of recruitment, patients were at least 16 years old and had had lupus for 5 years or less. They come in for periodic visits to the University of Alabama at Birmingham, the University of Texas Health Science Center at Houston, and the University of Puerto Rico Medical Sciences Campus. Recruitment began in 1994 and finished in 2007. Follow-up ranges from 1 to 14 years, with a mean of 4.5 years.
LUMINA is supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases, the National Institutes of Health General Clinical Research Centers program, the National Center for Research Resources Clinical Research Infrastructure Initiative, the Mary Kirkland Center for Lupus Research Scholars Program, and Rheuminations Inc (New York, NY).
The purpose of the study is to shed light on the interplay of genetics and environment in this disease and, in the process, to raise awareness about the problem of lupus in Hispanics. In fact, much of the information in the following sections is from the LUMINA study.
HISPANICS ARE NOT A HOMOGENEOUS GROUP
In the United States, the term Hispanic describes anyone whose origin goes back to a Spanish-speaking country. However, US Hispanics are not a homogeneous racial group: they differ in genetics, culture, and problems.
The largest US Hispanic subgroup and the one more likely to be seen by US physicians is Hispanics of Mexican origin, who account for 66% of all US Hispanics. This group has a higher percentage of Amerindian genes than those of Puerto Rican ancestry.3 LUMINA researchers analyzed the DNA of 492 patients and found the following mixtures of genes3:
- Hispanics in Texas (mostly of Mexican origin): 48% Amerindian, 18% African, 34% European
- Hispanics from Puerto Rico: 20% Amerindian, 45% African, 35% European
- African Americans: 0% Amerindian, 79% African, 21% European
- Non-Hispanic Caucasians: 10% Amerindian, 18% African, 72% European.
Latin Americans of mixed European and Amerindian ancestry (which includes Aztec, Mayan, Quechuan, Aymaran, and other Central and South American groups) are called mestizos. Not all people in Latin America are mestizos: some are of European, African, or Asian ancestry, but in the United States they are all called Hispanics.
LUPUS DIFFERS AMONG SUBGROUPS
LUMINA research has revealed that lupus is heterogeneous also among US Hispanic subgroups. When people from Puerto Rico get lupus, it is generally less serious and devastating than in those from Mexico or Central America. Since US Hispanics of Mexican or Central American origin possess more Amerindian genes, this observation supports the notion that these genes are important contributors to the occurrence and expression of the disease.
Amerindian genes contribute to a greater susceptibility to lupus,4,5 although there is an interplay between genetic and nongenetic factors in the etiology and expression.6 Lupus starts at a younger age in Hispanics of predominantly Amerindian ancestry than in non-Hispanic Caucasians, and the onset is more likely to be acute.7
Renal involvement in these patients8 and mestizos from Latin America is rather common, probably as common as it is in US African Americans, and it tends to develop earlier than in non-Hispanic Caucasians.9 Amerindian ancestral genes, like African genes, contribute to the occurrence of renal disease in lupus patients.4 Furthermore, once nephritis ensues, end-stage renal disease occurs more often in US Hispanic and African American than in non-Hispanic Caucasian children, as demonstrated by Hiraki et al10 using national databases, and the same is true in adults, as shown in the LUMINA cohort.11
Other potentially serious manifestations of the disease are also more common, including hematologic and central nervous system manifestations. Not surprisingly, then, these patients show a higher degree of disease activity, both early in the course of the disease12,13 and over time.14
Table 1 compares the demographic and clinical features of LUMINA patients according to ethnicity. By and large, Hispanics from Texas have lower levels of education and income (comparable with levels in African Americans), and this can adversely affect the disease course by limiting these patients’ access to adequate care.15
DISEASE ACTIVITY AND ORGAN DAMAGE ARE GREATER IN HISPANICS
Disease activity in lupus reflects the ongoing immune-mediated inflammatory process. In LUMINA patients, regardless of the time at which disease activity was ascertained, it was higher in Hispanics from Texas and in African Americans than in non-Hispanic Caucasians and in Hispanics from Puerto Rico.7,12,16–18 Similar findings were seen in the Grupo Latinoamericano de Estudio de Lupus (GLADEL) cohort,13 in which mestizos and Hispanics of mixed African and European ancestry had higher maximum disease activity scores than non-Hispanic Caucasians.13
In addition, organ damage in lupus—the irreversible changes that occur in organ systems as a consequence of the disease or its treatments (eg, glucocorticoids, immunosuppressive drugs)—is more severe and develops sooner in Hispanics from Texas than in other groups.6,18,19 Using multivariate analysis, LUMINA investigators19 estimated the hazard ratio for the time until organ damage appeared for various risk factors, with values of 1 or greater indicating a shorter time and lower values indicating a longer time. Being a Hispanic from Texas carried a hazard ratio of 2.11 (95% confidence interval 1.15–3.88).
Because organ damage is an important and independent predictor of further damage20 and death,21 physicians need to take this disease quite seriously and try to prevent damage early in people at risk. To achieve that, the need to control disease activity must be balanced against the risk of overtreatment, as the important contribution of glucocorticoids to organ damage is well recognized.22
HISPANICS HAVE MORE COMORBIDITIES
Obesity, hypertension, diabetes, and metabolic syndrome are more common in US Hispanics, particularly those of Amerindian ancestry, than in the majority population of non-Hispanic Caucasians.23,24 The potential deleterious effects of glucocorticoids in patients already predisposed to these conditions need to be considered, balancing adequate disease control against the potential adverse effects.22
QUALITY OF LIFE IS WORSE WITH LUPUS
Whether it is measured with a generic instrument such as the Short Form 36 (SF-36), as it was in LUMINA,25 or with a disease-specific tool such as the Lupus-Pro, quality of life is significantly worsened by lupus. Furthermore, Fernandez et al26 found that a low level of health-related quality of life, as measured by the SF-6D version of the SF-36, was predictive of poor outcomes in LUMINA patients.
POVERTY, NOT ETHNICITY, ACCOUNTS FOR HIGHER MORTALITY RATE
As yet, we have no population-based data comparing survival in US Hispanic patients with lupus vs that of other population groups.
At first inspection, data from LUMINA indicate that Hispanics of primarily Amerindian ancestry have a lower survival rate than patients in other ethnic groups (Figure 1).6 However, when all other factors are taken into consideration, poverty, not ethnicity, is the major contributing factor (Table 2).6,27
This finding illustrates the important interplay between genetic and nongenetic factors in the course and final outcome of lupus, as already alluded to, although the exact relationship between them is not clear. It remains to be determined whether poverty is only a proxy for other population characteristics such as illiteracy, limited access to specialized care, limited access to medications, or cultural beliefs that may interfere with proper care.
ANTIMALARIAL DRUGS INCREASE SURVIVAL
Using statistical analysis that adjusts for confounding by indication, we and others28–30 have shown that antimalarial drugs exert an independent and important protective effect on survival in lupus (Figure 2).
Important also is the protective effect of antimalarials on organ damage and the possibility of using them from disease outset in Hispanic patients at risk of early and rapid damage accrual,11 renal damage, and even lupus nephritis.31,32 This has very practical implications for the adequate and prompt management of these Hispanic patients.
PRACTICAL IMPLICATIONS
Lupus in US Hispanics is a serious disease with devastating consequences. Prompt diagnosis is paramount to prevent early organ damage and to prolong survival.
The disease may present in many different and unexpected ways, but joint pain, sun-sensitive rashes, renal involvement, cytopenias, and other manifestations should prompt the clinician to consider lupus in the differential diagnosis. Patients are often dismissed as having “arthritis” without being asked about other manifestations that may suggest a systemic connective tissue disease such as lupus. The same goes for skin rashes or unusual central nervous system manifestations.
The diagnosis of lupus is clinical, but some laboratory studies are essential to rule in or rule out renal or hematologic abnormalities and determine the level of disease activity. Tests usually ordered in patients suspected of having lupus include antinuclear antibody, complement levels, a complete blood cell count and differential, and a urinalysis. The need for additional tests depends on the results of the tests listed.
Once the disease is diagnosed, treatment should be tailored to the severity and type of clinical manifestations present. In general, glucocorticoids should be used at the smallest possible dose, antimalarials should be prescribed from the outset to all patients (following current guidelines in order to avoid ocular toxicity),33 and immunosuppressants and other treatments should be considered in certain instances. In parallel, consideration should be given to sun protection, adequate exercise, tobacco avoidance, osteoporosis and atherosclerosis prevention, planned conception, and compliance.
The goal in these people at risk is to control their lupus manifestations without causing undue damage, to preserve their quality of life, and to prevent an early demise.
- Humes KR, Jones NA, Ramirez RR. Overview of race and Hispanic origin: 2010. 2010 Census briefs; 2011. http://www.census.gov/prod/cen2010/briefs/c2010br-02.pdf. Accessed October 20, 2012.
- Reveille JD, Moulds JM, Ahn C, et al; for the LUMINA study Group. Systemic lupus erythematosus in three ethnic groups. I. The effects of HLA class II, C4, and CR1 alleles, socioeconomic factors, and ethnicity and disease onset. Arthritis Rheum 1998; 41:1161–1172.
- Alarcón GS, Beasley TM, Roseman JM, et al; LUMINA Study Group. Ethnic disparities in health and disease: the need to account for ancestral admixture when estimating the genetic contribution to both (LUMINA XXVI) (Letter). Lupus 2005; 14:867–868.
- Alarcón GS, Bastian HM, Beasley TM, et al; LUMINA Study Group. Systemic lupus erythematosus in a multi-ethnic cohort (LUMINA) XXXII: [corrected] contributions of admixture and socioeconomic status to renal involvement. Lupus 2006; 15:26–31.
- Sanchez E, Webb RD, Rasmussen A, et al. Genetically determined Amerindian ancestry correlates with increased frequency of risk alleles for systemic lupus erythematosus. Arthritis Rheum 2010; 62:3722–3729.
- Fernández M, Alarcón GS, Calvo-Alén J, et al; LUMINA Study Group. A multiethnic, multicenter cohort of patients with systemic lupus erythematosus (SLE) as a model for the study of ethnic disparities in SLE. Arthritis Rheum 2007; 57:576–584.
- Alarcón GS, Friedman AW, Straaton KV, et al. Systemic lupus erythematosus in three ethnic groups: III. A comparison of characteristics early in the natural history of the LUMINA cohort. LUpus in MInority populations: NAture vs Nurture. Lupus 1999; 8:197–209.
- Bastian HM, Alarcón GS, Roseman JM, et al; LUMINA Study Group. Systemic lupus erythematosus in a multiethnic US cohort (LUMINA) XL II: factors predictive of new or worsening proteinuria. Rheumatology (Oxford) 2007; 46:683–689.
- Burgos PI, McGwin G, Pons-Estel GJ, Reveille JD, Alarcón GS, Vilá LM. US patients of Hispanic and African ancestry develop lupus nephritis early in the disease course: data from LUMINA, a multiethnic US cohort (LUMINA LXXIV). Ann Rheum Dis 2011; 70:393–394.
- Hiraki LT, Lu B, Alexander SR, et al. End-stage renal disease due to lupus nephritis among children in the US, 1995–2006. Arthritis Rheum 2011; 63:1988–1997.
- Pons-Estel GJ, Alarcón GS, McGwin G, et al. Protective effect of hydroxychloroquine on renal damage in patients with lupus nephritis: LXV, data from a multiethnic US cohort. Arthritis Rheum 2009; 61:830–839.
- Alarcón GS, Roseman J, Bartolucci AA, et al. Systemic lupus erythematosus in three ethnic groups: II. Features predictive of disease activity early in its course. LUMINA Study Group. Lupus in minority populations, nature versus nurture. Arthritis Rheum 1998; 41:1173–1180.
- Pons-Estel BA, Catoggio LJ, Cardiel MH, et al; Grupo Latinoamericano de Estudio del Lupus. The GLADEL multinational Latin American prospective inception cohort of 1,214 patients with systemic lupus erythematosus: ethnic and disease heterogeneity among “Hispanics.” Medicine (Baltimore) 2004; 83:1–17.
- Alarcón GS, Calvo-Alén J, McGwin G, et al; LUMINA Study Group. Systemic lupus erythematosus in a multiethnic cohort: LUMINA XXXV. Predictive factors of high disease activity over time. Ann Rheum Dis 2006; 65:1168–1174.
- Vilá LM, Alarcón GS, McGwin G, Bastian HM, Fessler BJ, Reveille JD; Lumina Study Group. Systemic lupus erythematosus in a multiethnic US cohort, XXXVII: association of lymphopenia with clinical manifestations, serologic abnormalities, disease activity, and damage accrual. Arthritis Rheum 2006; 55:799–806.
- Zhang J, González LA, Roseman JM, Vilá LM, Reveille JD, Alárcon GS. Predictors of the rate of change in disease activity over time in LUMINA, a multiethnic US cohort of patients with systemic lupus erythematosus: LUMINA LXX. Lupus 2010; 19:727–733.
- Vilá LM, Alarcón GS, McGwin G, et al; LUMINA Study Group. Early clinical manifestations, disease activity and damage of systemic lupus erythematosus among two distinct US Hispanic subpopulations. Rheumatology (Oxford) 2004; 43:358–363.
- Gladman D, Ginzler E, Goldsmith C, et al. The development and initial validation of the Systemic Lupus International Collaborating Clinics/American College of Rheumatology damage index for systemic lupus erythematosus. Arthritis Rheum 1996; 39:363–369.
- Toloza SM, Roseman JM, Alarcón GS, et al. Systemic lupus erythematosus in a multiethnic US cohort (LUMINA): XXII. Predictors of time to the occurrence of initial damage. Arthritis Rheum 2004; 50:3177–3186.
- Alarcón GS, Roseman JM, McGwin G, et al; LUMINA Study Group. Systemic lupus erythematosus in three ethnic groups. XX. Damage as a predictor of further damage. Rheumatology (Oxford) 2004; 43:202–205.
- Alarcón GS, McGwin G, Bastian HM, et al. Systemic lupus erythematosus in three ethnic groups. VII [correction of VIII]. Predictors of early mortality in the LUMINA cohort. LUMINA Study Group. Arthritis Rheum 2001; 45:191–202.
- Ruiz-Irastorza G, Danza A, Khamashta M. Glucocorticoid use and abuse in SLE. Rheumatology (Oxford) 2012 E-pub ahead of print.
- Jordan HT, Tabaei BP, Nash D, Angell SY, Chamany S, Kerker B. Metabolic syndrome among adults in New York City, 2004 New York City Health and Nutrition Examination Survey. Prev Chronic Dis 2012; 9:E04.
- Matthews KA, Sowers MF, Derby CA, et al. Ethnic differences in cardiovascular risk factor burden among middle-aged women: Study of Women’s Health Across the Nation (SWAN). Am Heart J 2005; 149:1066–1073.
- Alarcón GS, McGwin G, Uribe A, et al. Systemic lupus erythematosus in a multiethnic lupus cohort (LUMINA). XVII. Predictors of selfreported health-related quality of life early in the disease course. Arthritis Rheum 2004; 51:465–474.
- Fernández M, Alarcón GS, McGwin G, et al; LUMINA Study Group. Using the Short Form 6D, as an overall measure of health, to predict damage accrual and mortality in patients with systemic lupus erythematosus: XLVII, results from a multiethnic US cohort. Arthritis Rheum 2007; 57:986–992.
- Durán S, Apte M, Alarcón GSLUMINA Study Group. Poverty, not ethnicity, accounts for the differential mortality rates among lupus patients of various ethnic groups. J Natl Med Assoc 2007; 99:1196–1198.
- Ruiz-Irastorza G, Egurbide MV, Pijoan JI, et al. Effect of antimalarials on thrombosis and survival in patients with systemic lupus erythematosus. Lupus 2006; 15:577–583.
- Alarcón GS, McGwin G, Bertoli AM, et al; LUMINA Study Group. Effect of hydroxychloroquine on the survival of patients with systemic lupus erythematosus: data from LUMINA, a multiethnic US cohort (LUMINA L). Ann Rheum Dis 2007; 66:1168–1172.
- Shinjo SK, Bonfá E, Wojdyla D, et al; Grupo Latino Americano de Estudio del Lupus Eritematoso (Gladel). Antimalarial treatment may have a time-dependent effect on lupus survival: data from a multinational Latin American inception cohort. Arthritis Rheum 2010; 62:855–862.
- Fessler BJ, Alarcón GS, McGwin G, et al; LUMINA Study Group. Systemic lupus erythematosus in three ethnic groups: XVI. Association of hydroxychloroquine use with reduced risk of damage accrual. Arthritis Rheum 2005; 52:1473–1480.
- Pons-Estel GJ, Alarcón GS, Hachuel L, et al. Antimalarials have a protective effect against the development of renal disease in Latin American SLE patients. The 9th International Congress on SLE June 24–27, 2010, Vancouver, Canada. Lupus 2010; 19(suppl 1):31–32.
- Ruiz-Irastorza G, Ramos-Casals M, Brito-Zeron P, Khamashta MA. Clinical efficacy and side effects of antimalarials in systemic lupus erythematosus: a systematic review. Ann Rheum Dis 2010; 69:20–28.
- Humes KR, Jones NA, Ramirez RR. Overview of race and Hispanic origin: 2010. 2010 Census briefs; 2011. http://www.census.gov/prod/cen2010/briefs/c2010br-02.pdf. Accessed October 20, 2012.
- Reveille JD, Moulds JM, Ahn C, et al; for the LUMINA study Group. Systemic lupus erythematosus in three ethnic groups. I. The effects of HLA class II, C4, and CR1 alleles, socioeconomic factors, and ethnicity and disease onset. Arthritis Rheum 1998; 41:1161–1172.
- Alarcón GS, Beasley TM, Roseman JM, et al; LUMINA Study Group. Ethnic disparities in health and disease: the need to account for ancestral admixture when estimating the genetic contribution to both (LUMINA XXVI) (Letter). Lupus 2005; 14:867–868.
- Alarcón GS, Bastian HM, Beasley TM, et al; LUMINA Study Group. Systemic lupus erythematosus in a multi-ethnic cohort (LUMINA) XXXII: [corrected] contributions of admixture and socioeconomic status to renal involvement. Lupus 2006; 15:26–31.
- Sanchez E, Webb RD, Rasmussen A, et al. Genetically determined Amerindian ancestry correlates with increased frequency of risk alleles for systemic lupus erythematosus. Arthritis Rheum 2010; 62:3722–3729.
- Fernández M, Alarcón GS, Calvo-Alén J, et al; LUMINA Study Group. A multiethnic, multicenter cohort of patients with systemic lupus erythematosus (SLE) as a model for the study of ethnic disparities in SLE. Arthritis Rheum 2007; 57:576–584.
- Alarcón GS, Friedman AW, Straaton KV, et al. Systemic lupus erythematosus in three ethnic groups: III. A comparison of characteristics early in the natural history of the LUMINA cohort. LUpus in MInority populations: NAture vs Nurture. Lupus 1999; 8:197–209.
- Bastian HM, Alarcón GS, Roseman JM, et al; LUMINA Study Group. Systemic lupus erythematosus in a multiethnic US cohort (LUMINA) XL II: factors predictive of new or worsening proteinuria. Rheumatology (Oxford) 2007; 46:683–689.
- Burgos PI, McGwin G, Pons-Estel GJ, Reveille JD, Alarcón GS, Vilá LM. US patients of Hispanic and African ancestry develop lupus nephritis early in the disease course: data from LUMINA, a multiethnic US cohort (LUMINA LXXIV). Ann Rheum Dis 2011; 70:393–394.
- Hiraki LT, Lu B, Alexander SR, et al. End-stage renal disease due to lupus nephritis among children in the US, 1995–2006. Arthritis Rheum 2011; 63:1988–1997.
- Pons-Estel GJ, Alarcón GS, McGwin G, et al. Protective effect of hydroxychloroquine on renal damage in patients with lupus nephritis: LXV, data from a multiethnic US cohort. Arthritis Rheum 2009; 61:830–839.
- Alarcón GS, Roseman J, Bartolucci AA, et al. Systemic lupus erythematosus in three ethnic groups: II. Features predictive of disease activity early in its course. LUMINA Study Group. Lupus in minority populations, nature versus nurture. Arthritis Rheum 1998; 41:1173–1180.
- Pons-Estel BA, Catoggio LJ, Cardiel MH, et al; Grupo Latinoamericano de Estudio del Lupus. The GLADEL multinational Latin American prospective inception cohort of 1,214 patients with systemic lupus erythematosus: ethnic and disease heterogeneity among “Hispanics.” Medicine (Baltimore) 2004; 83:1–17.
- Alarcón GS, Calvo-Alén J, McGwin G, et al; LUMINA Study Group. Systemic lupus erythematosus in a multiethnic cohort: LUMINA XXXV. Predictive factors of high disease activity over time. Ann Rheum Dis 2006; 65:1168–1174.
- Vilá LM, Alarcón GS, McGwin G, Bastian HM, Fessler BJ, Reveille JD; Lumina Study Group. Systemic lupus erythematosus in a multiethnic US cohort, XXXVII: association of lymphopenia with clinical manifestations, serologic abnormalities, disease activity, and damage accrual. Arthritis Rheum 2006; 55:799–806.
- Zhang J, González LA, Roseman JM, Vilá LM, Reveille JD, Alárcon GS. Predictors of the rate of change in disease activity over time in LUMINA, a multiethnic US cohort of patients with systemic lupus erythematosus: LUMINA LXX. Lupus 2010; 19:727–733.
- Vilá LM, Alarcón GS, McGwin G, et al; LUMINA Study Group. Early clinical manifestations, disease activity and damage of systemic lupus erythematosus among two distinct US Hispanic subpopulations. Rheumatology (Oxford) 2004; 43:358–363.
- Gladman D, Ginzler E, Goldsmith C, et al. The development and initial validation of the Systemic Lupus International Collaborating Clinics/American College of Rheumatology damage index for systemic lupus erythematosus. Arthritis Rheum 1996; 39:363–369.
- Toloza SM, Roseman JM, Alarcón GS, et al. Systemic lupus erythematosus in a multiethnic US cohort (LUMINA): XXII. Predictors of time to the occurrence of initial damage. Arthritis Rheum 2004; 50:3177–3186.
- Alarcón GS, Roseman JM, McGwin G, et al; LUMINA Study Group. Systemic lupus erythematosus in three ethnic groups. XX. Damage as a predictor of further damage. Rheumatology (Oxford) 2004; 43:202–205.
- Alarcón GS, McGwin G, Bastian HM, et al. Systemic lupus erythematosus in three ethnic groups. VII [correction of VIII]. Predictors of early mortality in the LUMINA cohort. LUMINA Study Group. Arthritis Rheum 2001; 45:191–202.
- Ruiz-Irastorza G, Danza A, Khamashta M. Glucocorticoid use and abuse in SLE. Rheumatology (Oxford) 2012 E-pub ahead of print.
- Jordan HT, Tabaei BP, Nash D, Angell SY, Chamany S, Kerker B. Metabolic syndrome among adults in New York City, 2004 New York City Health and Nutrition Examination Survey. Prev Chronic Dis 2012; 9:E04.
- Matthews KA, Sowers MF, Derby CA, et al. Ethnic differences in cardiovascular risk factor burden among middle-aged women: Study of Women’s Health Across the Nation (SWAN). Am Heart J 2005; 149:1066–1073.
- Alarcón GS, McGwin G, Uribe A, et al. Systemic lupus erythematosus in a multiethnic lupus cohort (LUMINA). XVII. Predictors of selfreported health-related quality of life early in the disease course. Arthritis Rheum 2004; 51:465–474.
- Fernández M, Alarcón GS, McGwin G, et al; LUMINA Study Group. Using the Short Form 6D, as an overall measure of health, to predict damage accrual and mortality in patients with systemic lupus erythematosus: XLVII, results from a multiethnic US cohort. Arthritis Rheum 2007; 57:986–992.
- Durán S, Apte M, Alarcón GSLUMINA Study Group. Poverty, not ethnicity, accounts for the differential mortality rates among lupus patients of various ethnic groups. J Natl Med Assoc 2007; 99:1196–1198.
- Ruiz-Irastorza G, Egurbide MV, Pijoan JI, et al. Effect of antimalarials on thrombosis and survival in patients with systemic lupus erythematosus. Lupus 2006; 15:577–583.
- Alarcón GS, McGwin G, Bertoli AM, et al; LUMINA Study Group. Effect of hydroxychloroquine on the survival of patients with systemic lupus erythematosus: data from LUMINA, a multiethnic US cohort (LUMINA L). Ann Rheum Dis 2007; 66:1168–1172.
- Shinjo SK, Bonfá E, Wojdyla D, et al; Grupo Latino Americano de Estudio del Lupus Eritematoso (Gladel). Antimalarial treatment may have a time-dependent effect on lupus survival: data from a multinational Latin American inception cohort. Arthritis Rheum 2010; 62:855–862.
- Fessler BJ, Alarcón GS, McGwin G, et al; LUMINA Study Group. Systemic lupus erythematosus in three ethnic groups: XVI. Association of hydroxychloroquine use with reduced risk of damage accrual. Arthritis Rheum 2005; 52:1473–1480.
- Pons-Estel GJ, Alarcón GS, Hachuel L, et al. Antimalarials have a protective effect against the development of renal disease in Latin American SLE patients. The 9th International Congress on SLE June 24–27, 2010, Vancouver, Canada. Lupus 2010; 19(suppl 1):31–32.
- Ruiz-Irastorza G, Ramos-Casals M, Brito-Zeron P, Khamashta MA. Clinical efficacy and side effects of antimalarials in systemic lupus erythematosus: a systematic review. Ann Rheum Dis 2010; 69:20–28.
KEY POINTS
- Amerindian genes contribute to a greater susceptibility to lupus, although there is an interplay between genetic and nongenetic factors in its etiology and expression.
- In large studies, disease activity and organ damage were greater in African Americans and in Hispanics from Texas than in Caucasians and Hispanics from Puerto Rico.
- Hispanics of primarily Amerindian ancestry (which includes Aztec, Mayan, Quechuan, Aymaran, and other Central and South American groups) have a lower survival rate than patients in other ethnic groups, but poverty is the responsible factor.
- The need to control disease activity with corticosteroids must be balanced against the risk of overtreatment and organ damage.
- Antimalarial drugs such as chloroquine and hydroxychloroquine should be prescribed from the outset to all patients with lupus, according to current guidelines designed to avoid ocular toxicity.
A woman with a swollen uvula
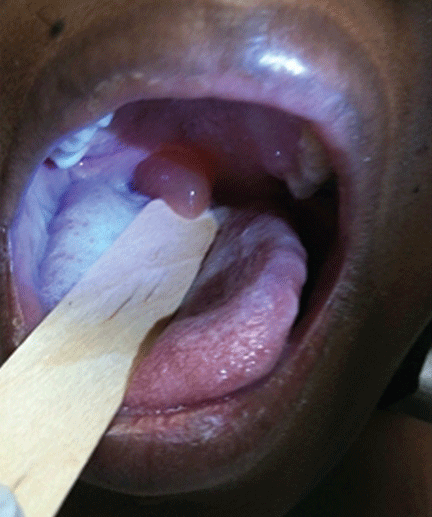
A 39-year-old woman on patient-controlled analgesia with morphine after cesarean delivery suddenly developed shortness of breath. On examination, the uvula was notably edematous, pale, and translucent, with no sign of erythema (Figure 1). The previous night, when the morphine was started, she had mild pruritus, which responded to treatment with oral diphenhydramine (Benadryl).
Given the extent of the uvular edema, emergency intubation was performed, and epinephrine and corticosteroids were given.
Q: Which is the most likely diagnosis at this point?
- Hereditary angioedema
- Infection causing epiglottitis masquerading as uvular swelling
- Opioid-induced uvular hydrops
- Myxedematous infiltration due to hypothyroidism
A: Opioid-induced uvular hydrops is the most likely diagnosis in this case, although it is rare. The most common side effect of opioids is constipation; others include lethargy, delirium, and sedation.
Hereditary angioedema is unlikely in this patient, as it usually presents in childhood or adolescence and there is usually a family history. Also, her cesarean delivery was done under regional anesthesia, which requires no oral or uvular manipulation and so cannot cause uvular swelling. In the absence of pain, fever, or signs and symptoms of pharyngitis, infection was unlikely. And she had no history of hypothyroidism.
UVULAR HYDROPS
This condition may be caused by opioid-induced direct degranulation of mast cells and basophils, inciting an inflammatory response.
Differential diagnoses include:
- Hereditary angioedema
- Effects of other drugs (angiotensin-converting enzyme inhibitors, cocaine, non-steroidal anti-inflammatory drugs)
- Infection (Haemophilus influenzae, Streptococcus pneumoniae)
- Trauma (intubation during surgery)
- Myxedematous infiltration due to hypothyroidism
- Granulomatous infiltration due to sarcoidosis.
When the patient’s condition has been stabilized, several outpatient tests may help narrow the differential diagnosis:
- Serum complement levels, of which the most reliable and cost-effective screening test for hereditary angioedema is a serum C4 level
- A serum thyroid-stimulating hormone level to rule out hypothyroidism
- Skin and lymph node biopsy (if skin lesions or lymphadenopathy is present), and chest radiograph to rule out sarcoidosis
- A urine drug screen and a skin-prick test for opioids (even though a negative skin test does not exclude opiate sensitivity).
OUR PATIENT’S COURSE
Our patient was discharged and underwent further outpatient evaluation. At discharge, she was advised to avoid opioids in the future.
SUGGESTED READING
Grigoriadou S, Longhurst HJ. Clinical Immunology Review Series: An approach to the patient with angio-oedema. Clin Exp Immunol 2009; 155:367–377.
Marx JA, Hockberger RS, Walls RM. Urticaria and angioedema. In:Marx JA, Hockberger RS, Walls RM, et aleditors. Rosen’s Emergency Medicine. 7th ed. Philadelphia, PA: Mosby/Elsevier, 2010.
Morgan BP. Hereditary angioedema—therapies old and new. N Engl J Med 2010; 363:581–583.
Neustein SM. Acute uvular edema after regional anesthesia. J Clin Anesth 2007; 19:365–366.
A 39-year-old woman on patient-controlled analgesia with morphine after cesarean delivery suddenly developed shortness of breath. On examination, the uvula was notably edematous, pale, and translucent, with no sign of erythema (Figure 1). The previous night, when the morphine was started, she had mild pruritus, which responded to treatment with oral diphenhydramine (Benadryl).
Given the extent of the uvular edema, emergency intubation was performed, and epinephrine and corticosteroids were given.
Q: Which is the most likely diagnosis at this point?
- Hereditary angioedema
- Infection causing epiglottitis masquerading as uvular swelling
- Opioid-induced uvular hydrops
- Myxedematous infiltration due to hypothyroidism
A: Opioid-induced uvular hydrops is the most likely diagnosis in this case, although it is rare. The most common side effect of opioids is constipation; others include lethargy, delirium, and sedation.
Hereditary angioedema is unlikely in this patient, as it usually presents in childhood or adolescence and there is usually a family history. Also, her cesarean delivery was done under regional anesthesia, which requires no oral or uvular manipulation and so cannot cause uvular swelling. In the absence of pain, fever, or signs and symptoms of pharyngitis, infection was unlikely. And she had no history of hypothyroidism.
UVULAR HYDROPS
This condition may be caused by opioid-induced direct degranulation of mast cells and basophils, inciting an inflammatory response.
Differential diagnoses include:
- Hereditary angioedema
- Effects of other drugs (angiotensin-converting enzyme inhibitors, cocaine, non-steroidal anti-inflammatory drugs)
- Infection (Haemophilus influenzae, Streptococcus pneumoniae)
- Trauma (intubation during surgery)
- Myxedematous infiltration due to hypothyroidism
- Granulomatous infiltration due to sarcoidosis.
When the patient’s condition has been stabilized, several outpatient tests may help narrow the differential diagnosis:
- Serum complement levels, of which the most reliable and cost-effective screening test for hereditary angioedema is a serum C4 level
- A serum thyroid-stimulating hormone level to rule out hypothyroidism
- Skin and lymph node biopsy (if skin lesions or lymphadenopathy is present), and chest radiograph to rule out sarcoidosis
- A urine drug screen and a skin-prick test for opioids (even though a negative skin test does not exclude opiate sensitivity).
OUR PATIENT’S COURSE
Our patient was discharged and underwent further outpatient evaluation. At discharge, she was advised to avoid opioids in the future.
A 39-year-old woman on patient-controlled analgesia with morphine after cesarean delivery suddenly developed shortness of breath. On examination, the uvula was notably edematous, pale, and translucent, with no sign of erythema (Figure 1). The previous night, when the morphine was started, she had mild pruritus, which responded to treatment with oral diphenhydramine (Benadryl).
Given the extent of the uvular edema, emergency intubation was performed, and epinephrine and corticosteroids were given.
Q: Which is the most likely diagnosis at this point?
- Hereditary angioedema
- Infection causing epiglottitis masquerading as uvular swelling
- Opioid-induced uvular hydrops
- Myxedematous infiltration due to hypothyroidism
A: Opioid-induced uvular hydrops is the most likely diagnosis in this case, although it is rare. The most common side effect of opioids is constipation; others include lethargy, delirium, and sedation.
Hereditary angioedema is unlikely in this patient, as it usually presents in childhood or adolescence and there is usually a family history. Also, her cesarean delivery was done under regional anesthesia, which requires no oral or uvular manipulation and so cannot cause uvular swelling. In the absence of pain, fever, or signs and symptoms of pharyngitis, infection was unlikely. And she had no history of hypothyroidism.
UVULAR HYDROPS
This condition may be caused by opioid-induced direct degranulation of mast cells and basophils, inciting an inflammatory response.
Differential diagnoses include:
- Hereditary angioedema
- Effects of other drugs (angiotensin-converting enzyme inhibitors, cocaine, non-steroidal anti-inflammatory drugs)
- Infection (Haemophilus influenzae, Streptococcus pneumoniae)
- Trauma (intubation during surgery)
- Myxedematous infiltration due to hypothyroidism
- Granulomatous infiltration due to sarcoidosis.
When the patient’s condition has been stabilized, several outpatient tests may help narrow the differential diagnosis:
- Serum complement levels, of which the most reliable and cost-effective screening test for hereditary angioedema is a serum C4 level
- A serum thyroid-stimulating hormone level to rule out hypothyroidism
- Skin and lymph node biopsy (if skin lesions or lymphadenopathy is present), and chest radiograph to rule out sarcoidosis
- A urine drug screen and a skin-prick test for opioids (even though a negative skin test does not exclude opiate sensitivity).
OUR PATIENT’S COURSE
Our patient was discharged and underwent further outpatient evaluation. At discharge, she was advised to avoid opioids in the future.
SUGGESTED READING
Grigoriadou S, Longhurst HJ. Clinical Immunology Review Series: An approach to the patient with angio-oedema. Clin Exp Immunol 2009; 155:367–377.
Marx JA, Hockberger RS, Walls RM. Urticaria and angioedema. In:Marx JA, Hockberger RS, Walls RM, et aleditors. Rosen’s Emergency Medicine. 7th ed. Philadelphia, PA: Mosby/Elsevier, 2010.
Morgan BP. Hereditary angioedema—therapies old and new. N Engl J Med 2010; 363:581–583.
Neustein SM. Acute uvular edema after regional anesthesia. J Clin Anesth 2007; 19:365–366.
SUGGESTED READING
Grigoriadou S, Longhurst HJ. Clinical Immunology Review Series: An approach to the patient with angio-oedema. Clin Exp Immunol 2009; 155:367–377.
Marx JA, Hockberger RS, Walls RM. Urticaria and angioedema. In:Marx JA, Hockberger RS, Walls RM, et aleditors. Rosen’s Emergency Medicine. 7th ed. Philadelphia, PA: Mosby/Elsevier, 2010.
Morgan BP. Hereditary angioedema—therapies old and new. N Engl J Med 2010; 363:581–583.
Neustein SM. Acute uvular edema after regional anesthesia. J Clin Anesth 2007; 19:365–366.
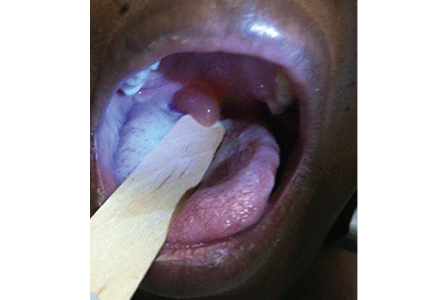
The ‘T’ in ITP remains

The “I” has changed its meaning, the “P” is not necessary to make the diagnosis, and the syndrome is not strikingly common in adults. But the disease formerly known as idiopathic thrombocytopenic purpura (ITP) remains important for internists to diagnose because thrombocytopenia is extremely common.
ITP has long been recognized in children (in whom it is often self-limited but severe) and in adults (in whom it is often more insidious and chronic, with a wider differential diagnosis).
The “I” used to stand for “idiopathic” but now it stands for “immune,” following a half decade of work by many investigators. The seminal experimental work of Dr. William J. Harrington and others while the former was a hematology fellow with Carl Moore at Washington University is the stuff of legend and ethical debate. Harrington had himself injected with a pint of serum from a patient with ITP and nearly died, demonstrating that the patient’s blood contained a substance or substances capable of inducing reversible profound thrombocytopenia in the recipient.1 Today, this classic experiment would probably not be performed, nor would the many more infusions that Harrington subsequently gave himself, other physicians, and support staff.2
Understanding the immunologic basis for ITP has led to treatments that are usually but not uniformly successful. Prednisone remains the main initial therapy, but its myriad side effects have led to the strategy of turning sooner to other therapies, such as intravenous immunoglobulin, splenectomy, rituximab (Rituxan), and, most recently, thrombopoietin agonists, in order to control the disease or even put it into remission.
Treatment decisions are often assigned to the hematologist or rheumatologist, but recognizing ITP and distinguishing it from other causes of thrombocytopenia remain the province of primary care providers, as discussed by Thota et al in this issue of the Journal.3 The diagnosis of ITP does not require the presence of purpura, which most adults ITP patients probably do not have, nor does it always require a bone marrow biopsy. It is important that ITP be distinguished from thrombocytopenia that is induced by drugs (particularly heparin) and myelodysplastic and other marrow processes (potential clues being other cytopenias, an unexplained elevated mean corpuscular volume, or constitutional symptoms). Undiagnosed thyroid disease and HIV infection should be tested for routinely, once drug-associated and other obvious causes are excluded.
- Harrington WJ, Minnich V, Hollingsworth JW, Moore CV. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J Lab Clin Med 1961; 38:1–10.
- Altman LK. Who Goes First? The Story Of Self-Experimentation. Berkeley, CA; University of California Press, 1987.
- Thota S, Kistangari G, Daw H, Spiro T. Immune thrombocytopenia in adults: an update. Cleve Clin J Med 2012; 79:641–650.
The “I” has changed its meaning, the “P” is not necessary to make the diagnosis, and the syndrome is not strikingly common in adults. But the disease formerly known as idiopathic thrombocytopenic purpura (ITP) remains important for internists to diagnose because thrombocytopenia is extremely common.
ITP has long been recognized in children (in whom it is often self-limited but severe) and in adults (in whom it is often more insidious and chronic, with a wider differential diagnosis).
The “I” used to stand for “idiopathic” but now it stands for “immune,” following a half decade of work by many investigators. The seminal experimental work of Dr. William J. Harrington and others while the former was a hematology fellow with Carl Moore at Washington University is the stuff of legend and ethical debate. Harrington had himself injected with a pint of serum from a patient with ITP and nearly died, demonstrating that the patient’s blood contained a substance or substances capable of inducing reversible profound thrombocytopenia in the recipient.1 Today, this classic experiment would probably not be performed, nor would the many more infusions that Harrington subsequently gave himself, other physicians, and support staff.2
Understanding the immunologic basis for ITP has led to treatments that are usually but not uniformly successful. Prednisone remains the main initial therapy, but its myriad side effects have led to the strategy of turning sooner to other therapies, such as intravenous immunoglobulin, splenectomy, rituximab (Rituxan), and, most recently, thrombopoietin agonists, in order to control the disease or even put it into remission.
Treatment decisions are often assigned to the hematologist or rheumatologist, but recognizing ITP and distinguishing it from other causes of thrombocytopenia remain the province of primary care providers, as discussed by Thota et al in this issue of the Journal.3 The diagnosis of ITP does not require the presence of purpura, which most adults ITP patients probably do not have, nor does it always require a bone marrow biopsy. It is important that ITP be distinguished from thrombocytopenia that is induced by drugs (particularly heparin) and myelodysplastic and other marrow processes (potential clues being other cytopenias, an unexplained elevated mean corpuscular volume, or constitutional symptoms). Undiagnosed thyroid disease and HIV infection should be tested for routinely, once drug-associated and other obvious causes are excluded.
The “I” has changed its meaning, the “P” is not necessary to make the diagnosis, and the syndrome is not strikingly common in adults. But the disease formerly known as idiopathic thrombocytopenic purpura (ITP) remains important for internists to diagnose because thrombocytopenia is extremely common.
ITP has long been recognized in children (in whom it is often self-limited but severe) and in adults (in whom it is often more insidious and chronic, with a wider differential diagnosis).
The “I” used to stand for “idiopathic” but now it stands for “immune,” following a half decade of work by many investigators. The seminal experimental work of Dr. William J. Harrington and others while the former was a hematology fellow with Carl Moore at Washington University is the stuff of legend and ethical debate. Harrington had himself injected with a pint of serum from a patient with ITP and nearly died, demonstrating that the patient’s blood contained a substance or substances capable of inducing reversible profound thrombocytopenia in the recipient.1 Today, this classic experiment would probably not be performed, nor would the many more infusions that Harrington subsequently gave himself, other physicians, and support staff.2
Understanding the immunologic basis for ITP has led to treatments that are usually but not uniformly successful. Prednisone remains the main initial therapy, but its myriad side effects have led to the strategy of turning sooner to other therapies, such as intravenous immunoglobulin, splenectomy, rituximab (Rituxan), and, most recently, thrombopoietin agonists, in order to control the disease or even put it into remission.
Treatment decisions are often assigned to the hematologist or rheumatologist, but recognizing ITP and distinguishing it from other causes of thrombocytopenia remain the province of primary care providers, as discussed by Thota et al in this issue of the Journal.3 The diagnosis of ITP does not require the presence of purpura, which most adults ITP patients probably do not have, nor does it always require a bone marrow biopsy. It is important that ITP be distinguished from thrombocytopenia that is induced by drugs (particularly heparin) and myelodysplastic and other marrow processes (potential clues being other cytopenias, an unexplained elevated mean corpuscular volume, or constitutional symptoms). Undiagnosed thyroid disease and HIV infection should be tested for routinely, once drug-associated and other obvious causes are excluded.
- Harrington WJ, Minnich V, Hollingsworth JW, Moore CV. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J Lab Clin Med 1961; 38:1–10.
- Altman LK. Who Goes First? The Story Of Self-Experimentation. Berkeley, CA; University of California Press, 1987.
- Thota S, Kistangari G, Daw H, Spiro T. Immune thrombocytopenia in adults: an update. Cleve Clin J Med 2012; 79:641–650.
- Harrington WJ, Minnich V, Hollingsworth JW, Moore CV. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J Lab Clin Med 1961; 38:1–10.
- Altman LK. Who Goes First? The Story Of Self-Experimentation. Berkeley, CA; University of California Press, 1987.
- Thota S, Kistangari G, Daw H, Spiro T. Immune thrombocytopenia in adults: an update. Cleve Clin J Med 2012; 79:641–650.