User login
Preop anemia management saves blood, costs
BOSTON – A pilot anemia optimization program resulted in significant increases in day-of-surgery hemoglobin levels and reductions in RBC transfusion rates and costs in one center, but whether patient outcomes also improved is still not known.

By diagnosing anemia at the preanesthesia visit and providing anemic patients with dietary guidance and supplementation prior to cardiac surgery, blood program managers noticed a more than $360 reduction in per-patient blood-product acquisition costs, a more than $1,800 average reduction per patient in transfusion costs, and overall cost savings of more than $100,000 over 18 months, compared with historical data.
The findings were reported by Christine M. Cahill, RN, from Strong Memorial Hospital in Rochester, N.Y., and the University of Rochester (N.Y.), at AABB 2018, the annual meeting of the group formerly known as the American Association of Blood Banks.
“Anemia has been thought of as a relatively benign thing our patients live with traditionally, but what we have been finding lately is that anemia is actually more serious than we once thought, and is an independent risk factor for hospitalization, readmission, increased patient length of stay, loss of function, and diminished quality of life,” she said.
Anemia also increases the likelihood that a patient will require allogeneic transfusions and is an independent risk factor for morbidity and mortality, she added.
The pilot program, which ran from February 2016 to September 2017, was designed to test the feasibility of diagnosing anemia during a cardiology consult visit and implementing a management plan.
During the study period, 240 patients presenting for elective cardiac surgery were screened for anemia, and 58 were diagnosed as anemic, defined as a hemoglobin level of less than 12 g/dL. These patients were referred for anemia work-ups, which found that 33 patients had iron-deficient anemia and 25 had anemia from other causes. Controls were patients who underwent cardiac surgery from March to July 2015, matched by age, sex, and procedures.
Treatments for iron-deficient patients included oral iron (7 patients), intravenous iron with or without folate (20 patients), or oral folate with or without vitamin B12 (5 patients). One iron-deficient patient could not have surgery delayed for anemia management.
Of the iron-replete patients, one received oral iron and 17 received folate plus or minus vitamin B12. The remaining seven iron-replete patients were not treated for anemia.
One iron-deficient patient had a reaction to the infusion and did not receive a scheduled second dose due to the need for immediate surgery. A second patient scheduled for intravenous iron and folate broke an arm and therefore missed an intravenous infusion appointment. No other complications or reactions occurred.
Intraoperative transfusion units used in the anemia management group totaled 10, compared with 68 for controls. Postoperative transfusion units used were also significantly lower following anemia management at 13 versus 122, respectively.
The rate of RBC transfusions among patients with anemia management was 24%, compared with 60% for controls (P less than .0001). Patients in the management program also had significantly higher day-of-surgery hemoglobin, at 11.01 g/dL versus 10.16 g/dL (P less than .001), and less RBC utilization, at an average 0.40 units per patient versus 2.07 for controls (P less than .0001).
The average per patient savings in acquisition costs was $367.40, the average transfusion cost saving was $1,837, and the total cost savings over the life of the pilot program was $106,546.
The keys to success for similar programs is “to make sure you do your homework,” Ms. Cahill said. Specifically, she recommended feasibility studies, evaluation of the potential impact of infusions on the service, work flow analyses, and cost analyses. It’s also important to get high-level administrative support as well as buy-in from surgeons and patients.
Future studies should include assessment of patient outcomes, safety, and length of ICU and hospital stay, she emphasized.
The study was internally funded. Ms. Cahill reported having no conflicts of interest.
SOURCE: Cahill CM et al. AABB 2018, Abstract PBM4-ST4-22.
BOSTON – A pilot anemia optimization program resulted in significant increases in day-of-surgery hemoglobin levels and reductions in RBC transfusion rates and costs in one center, but whether patient outcomes also improved is still not known.
By diagnosing anemia at the preanesthesia visit and providing anemic patients with dietary guidance and supplementation prior to cardiac surgery, blood program managers noticed a more than $360 reduction in per-patient blood-product acquisition costs, a more than $1,800 average reduction per patient in transfusion costs, and overall cost savings of more than $100,000 over 18 months, compared with historical data.
The findings were reported by Christine M. Cahill, RN, from Strong Memorial Hospital in Rochester, N.Y., and the University of Rochester (N.Y.), at AABB 2018, the annual meeting of the group formerly known as the American Association of Blood Banks.
“Anemia has been thought of as a relatively benign thing our patients live with traditionally, but what we have been finding lately is that anemia is actually more serious than we once thought, and is an independent risk factor for hospitalization, readmission, increased patient length of stay, loss of function, and diminished quality of life,” she said.
Anemia also increases the likelihood that a patient will require allogeneic transfusions and is an independent risk factor for morbidity and mortality, she added.
The pilot program, which ran from February 2016 to September 2017, was designed to test the feasibility of diagnosing anemia during a cardiology consult visit and implementing a management plan.
During the study period, 240 patients presenting for elective cardiac surgery were screened for anemia, and 58 were diagnosed as anemic, defined as a hemoglobin level of less than 12 g/dL. These patients were referred for anemia work-ups, which found that 33 patients had iron-deficient anemia and 25 had anemia from other causes. Controls were patients who underwent cardiac surgery from March to July 2015, matched by age, sex, and procedures.
Treatments for iron-deficient patients included oral iron (7 patients), intravenous iron with or without folate (20 patients), or oral folate with or without vitamin B12 (5 patients). One iron-deficient patient could not have surgery delayed for anemia management.
Of the iron-replete patients, one received oral iron and 17 received folate plus or minus vitamin B12. The remaining seven iron-replete patients were not treated for anemia.
One iron-deficient patient had a reaction to the infusion and did not receive a scheduled second dose due to the need for immediate surgery. A second patient scheduled for intravenous iron and folate broke an arm and therefore missed an intravenous infusion appointment. No other complications or reactions occurred.
Intraoperative transfusion units used in the anemia management group totaled 10, compared with 68 for controls. Postoperative transfusion units used were also significantly lower following anemia management at 13 versus 122, respectively.
The rate of RBC transfusions among patients with anemia management was 24%, compared with 60% for controls (P less than .0001). Patients in the management program also had significantly higher day-of-surgery hemoglobin, at 11.01 g/dL versus 10.16 g/dL (P less than .001), and less RBC utilization, at an average 0.40 units per patient versus 2.07 for controls (P less than .0001).
The average per patient savings in acquisition costs was $367.40, the average transfusion cost saving was $1,837, and the total cost savings over the life of the pilot program was $106,546.
The keys to success for similar programs is “to make sure you do your homework,” Ms. Cahill said. Specifically, she recommended feasibility studies, evaluation of the potential impact of infusions on the service, work flow analyses, and cost analyses. It’s also important to get high-level administrative support as well as buy-in from surgeons and patients.
Future studies should include assessment of patient outcomes, safety, and length of ICU and hospital stay, she emphasized.
The study was internally funded. Ms. Cahill reported having no conflicts of interest.
SOURCE: Cahill CM et al. AABB 2018, Abstract PBM4-ST4-22.
BOSTON – A pilot anemia optimization program resulted in significant increases in day-of-surgery hemoglobin levels and reductions in RBC transfusion rates and costs in one center, but whether patient outcomes also improved is still not known.
By diagnosing anemia at the preanesthesia visit and providing anemic patients with dietary guidance and supplementation prior to cardiac surgery, blood program managers noticed a more than $360 reduction in per-patient blood-product acquisition costs, a more than $1,800 average reduction per patient in transfusion costs, and overall cost savings of more than $100,000 over 18 months, compared with historical data.
The findings were reported by Christine M. Cahill, RN, from Strong Memorial Hospital in Rochester, N.Y., and the University of Rochester (N.Y.), at AABB 2018, the annual meeting of the group formerly known as the American Association of Blood Banks.
“Anemia has been thought of as a relatively benign thing our patients live with traditionally, but what we have been finding lately is that anemia is actually more serious than we once thought, and is an independent risk factor for hospitalization, readmission, increased patient length of stay, loss of function, and diminished quality of life,” she said.
Anemia also increases the likelihood that a patient will require allogeneic transfusions and is an independent risk factor for morbidity and mortality, she added.
The pilot program, which ran from February 2016 to September 2017, was designed to test the feasibility of diagnosing anemia during a cardiology consult visit and implementing a management plan.
During the study period, 240 patients presenting for elective cardiac surgery were screened for anemia, and 58 were diagnosed as anemic, defined as a hemoglobin level of less than 12 g/dL. These patients were referred for anemia work-ups, which found that 33 patients had iron-deficient anemia and 25 had anemia from other causes. Controls were patients who underwent cardiac surgery from March to July 2015, matched by age, sex, and procedures.
Treatments for iron-deficient patients included oral iron (7 patients), intravenous iron with or without folate (20 patients), or oral folate with or without vitamin B12 (5 patients). One iron-deficient patient could not have surgery delayed for anemia management.
Of the iron-replete patients, one received oral iron and 17 received folate plus or minus vitamin B12. The remaining seven iron-replete patients were not treated for anemia.
One iron-deficient patient had a reaction to the infusion and did not receive a scheduled second dose due to the need for immediate surgery. A second patient scheduled for intravenous iron and folate broke an arm and therefore missed an intravenous infusion appointment. No other complications or reactions occurred.
Intraoperative transfusion units used in the anemia management group totaled 10, compared with 68 for controls. Postoperative transfusion units used were also significantly lower following anemia management at 13 versus 122, respectively.
The rate of RBC transfusions among patients with anemia management was 24%, compared with 60% for controls (P less than .0001). Patients in the management program also had significantly higher day-of-surgery hemoglobin, at 11.01 g/dL versus 10.16 g/dL (P less than .001), and less RBC utilization, at an average 0.40 units per patient versus 2.07 for controls (P less than .0001).
The average per patient savings in acquisition costs was $367.40, the average transfusion cost saving was $1,837, and the total cost savings over the life of the pilot program was $106,546.
The keys to success for similar programs is “to make sure you do your homework,” Ms. Cahill said. Specifically, she recommended feasibility studies, evaluation of the potential impact of infusions on the service, work flow analyses, and cost analyses. It’s also important to get high-level administrative support as well as buy-in from surgeons and patients.
Future studies should include assessment of patient outcomes, safety, and length of ICU and hospital stay, she emphasized.
The study was internally funded. Ms. Cahill reported having no conflicts of interest.
SOURCE: Cahill CM et al. AABB 2018, Abstract PBM4-ST4-22.
REPORTING FROM AABB 2018
Key clinical point:
Major finding: The total cost savings over the life of a pilot anemia management program was $106,546.
Study details: A case-control study with 58 patients scheduled for elective cardiac surgery and matched historical controls.
Disclosures: The study was internally funded. Ms. Cahill reported having no conflicts of interest.
Source: Cahill CM et al. AABB 2018, Abstract PBM4-ST4-22.
Scleroderma SCOT trial findings hold similar in lung disease
CHICAGO – Changes in quantitative lung CT scores for scleroderma-related interstitial lung disease independently validate the superiority of hematopoietic stem cell transplantation versus cyclophosphamide for severe systemic sclerosis, according to findings in a subset of patients from the SCOT (Scleroderma: Cyclophosphamide or Transplantation) trial.

The recently published findings from the SCOT trial showed that myeloablation followed by autologous hematopoietic stem cell transplant (HSCT) significantly improved event-free and overall survival of systemic sclerosis patients at 54 months, compared with 12 monthly treatments with intravenous cyclophosphamide (N Engl J Med. 2018;378:35-47).
In a subset of 75 patients from the SCOT trial, the investigators analyzed changes in lung parenchymal abnormalities on high-resolution CT scans between baseline and serial follow-up exams performed yearly for up to 5 years. Follow-up scans at 14, 26, 48, and 54 months in available patients at each time point showed that whole-lung quantitative interstitial lung disease (QILD) scores – a validated measure that combines various CT texture-based characteristics to determine disease extent – decreased significantly by 7% at 54 months in patients who underwent HSCT, compared with no change in those who received cyclophosphamide (CYC; P = .024), Keith M. Sullivan, MD, reported at the annual meeting of the American College of Rheumatology.
Additionally, whole-lung quantitative lung fibrosis (QLF) scores were stable (–1%) in the HSCT patients, but increased 3% in the CYC patients (P = .047), said Dr. Sullivan, a professor of medicine at Duke University, Durham, N.C.
Dr. Sullivan was the first author on the SCOT trial, and he reported the current study results on behalf of lead investigator Jonathan Goldin, MD, PhD, of the department of radiologic sciences at the University of California, Los Angeles.
“These are really kind of meaningful associations, especially since the worst of the [CYC] treatment group didn’t make it to month 54,” Dr. Sullivan said.
Quantitative scores of scleroderma-related interstitial lung disease were measured using computer-based quantitative image analysis of standardized, noncontrast, volumetric, thin-section, thoracic, high-resolution CT. The same CT machine was used for all time points (except for one subject) with careful attention to breath hold reproducibility and image quality. Baseline characteristics were not different between the HSCT and CYC groups, he noted, stressing the rigorous study design.
CT assessments were also compared for the most severe lobe in each patient and showed similar findings, with both QILD and QLF scores for that lobe improving in the HSCT patients relative to the CYC patients (P = .004 and P = .002, respectively), Dr. Sullivan said, adding that the direction of change in structural measures of QILD and QLF for both whole lung and most severe lobe CTs tracked with physiological pulmonary function tests, including forced vital capacity (FVC), forced expiratory volume in 1 second, and diffusing capacity of the lungs for carbon monoxide.
“The FVC improved while QILD decreased, and that’s what you would expect to see,” he said. “So for each of these ways of displaying data, there was an expected and sensible inverse correlation.”
Scleroderma-related interstitial lung disease is a major cause of morbidity and mortality in severe systemic sclerosis. In the wake of the SCOT trial findings, questions remained with respect to correlation between those findings and pulmonary function; if the improvements with HSCT are real and meaningful, they should have meaningful correlation with pulmonary function, and these findings demonstrate those correlates, he said.
“Changes in quantitative lung CT scoring of scleroderma lung disease provide an objective radiologic validation of the long-term benefits of transplant compared to cyclophosphamide in individuals with severe scleroderma and lung involvement. Improvement in imaging after transplant continues for up to 54 months after randomization, giving radiologic confirmation of a durable treatment benefit,” Dr. Sullivan concluded.
The investigators reported having no relevant disclosures.
SOURCE: Goldin J et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 901.
CHICAGO – Changes in quantitative lung CT scores for scleroderma-related interstitial lung disease independently validate the superiority of hematopoietic stem cell transplantation versus cyclophosphamide for severe systemic sclerosis, according to findings in a subset of patients from the SCOT (Scleroderma: Cyclophosphamide or Transplantation) trial.
The recently published findings from the SCOT trial showed that myeloablation followed by autologous hematopoietic stem cell transplant (HSCT) significantly improved event-free and overall survival of systemic sclerosis patients at 54 months, compared with 12 monthly treatments with intravenous cyclophosphamide (N Engl J Med. 2018;378:35-47).
In a subset of 75 patients from the SCOT trial, the investigators analyzed changes in lung parenchymal abnormalities on high-resolution CT scans between baseline and serial follow-up exams performed yearly for up to 5 years. Follow-up scans at 14, 26, 48, and 54 months in available patients at each time point showed that whole-lung quantitative interstitial lung disease (QILD) scores – a validated measure that combines various CT texture-based characteristics to determine disease extent – decreased significantly by 7% at 54 months in patients who underwent HSCT, compared with no change in those who received cyclophosphamide (CYC; P = .024), Keith M. Sullivan, MD, reported at the annual meeting of the American College of Rheumatology.
Additionally, whole-lung quantitative lung fibrosis (QLF) scores were stable (–1%) in the HSCT patients, but increased 3% in the CYC patients (P = .047), said Dr. Sullivan, a professor of medicine at Duke University, Durham, N.C.
Dr. Sullivan was the first author on the SCOT trial, and he reported the current study results on behalf of lead investigator Jonathan Goldin, MD, PhD, of the department of radiologic sciences at the University of California, Los Angeles.
“These are really kind of meaningful associations, especially since the worst of the [CYC] treatment group didn’t make it to month 54,” Dr. Sullivan said.
Quantitative scores of scleroderma-related interstitial lung disease were measured using computer-based quantitative image analysis of standardized, noncontrast, volumetric, thin-section, thoracic, high-resolution CT. The same CT machine was used for all time points (except for one subject) with careful attention to breath hold reproducibility and image quality. Baseline characteristics were not different between the HSCT and CYC groups, he noted, stressing the rigorous study design.
CT assessments were also compared for the most severe lobe in each patient and showed similar findings, with both QILD and QLF scores for that lobe improving in the HSCT patients relative to the CYC patients (P = .004 and P = .002, respectively), Dr. Sullivan said, adding that the direction of change in structural measures of QILD and QLF for both whole lung and most severe lobe CTs tracked with physiological pulmonary function tests, including forced vital capacity (FVC), forced expiratory volume in 1 second, and diffusing capacity of the lungs for carbon monoxide.
“The FVC improved while QILD decreased, and that’s what you would expect to see,” he said. “So for each of these ways of displaying data, there was an expected and sensible inverse correlation.”
Scleroderma-related interstitial lung disease is a major cause of morbidity and mortality in severe systemic sclerosis. In the wake of the SCOT trial findings, questions remained with respect to correlation between those findings and pulmonary function; if the improvements with HSCT are real and meaningful, they should have meaningful correlation with pulmonary function, and these findings demonstrate those correlates, he said.
“Changes in quantitative lung CT scoring of scleroderma lung disease provide an objective radiologic validation of the long-term benefits of transplant compared to cyclophosphamide in individuals with severe scleroderma and lung involvement. Improvement in imaging after transplant continues for up to 54 months after randomization, giving radiologic confirmation of a durable treatment benefit,” Dr. Sullivan concluded.
The investigators reported having no relevant disclosures.
SOURCE: Goldin J et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 901.
CHICAGO – Changes in quantitative lung CT scores for scleroderma-related interstitial lung disease independently validate the superiority of hematopoietic stem cell transplantation versus cyclophosphamide for severe systemic sclerosis, according to findings in a subset of patients from the SCOT (Scleroderma: Cyclophosphamide or Transplantation) trial.
The recently published findings from the SCOT trial showed that myeloablation followed by autologous hematopoietic stem cell transplant (HSCT) significantly improved event-free and overall survival of systemic sclerosis patients at 54 months, compared with 12 monthly treatments with intravenous cyclophosphamide (N Engl J Med. 2018;378:35-47).
In a subset of 75 patients from the SCOT trial, the investigators analyzed changes in lung parenchymal abnormalities on high-resolution CT scans between baseline and serial follow-up exams performed yearly for up to 5 years. Follow-up scans at 14, 26, 48, and 54 months in available patients at each time point showed that whole-lung quantitative interstitial lung disease (QILD) scores – a validated measure that combines various CT texture-based characteristics to determine disease extent – decreased significantly by 7% at 54 months in patients who underwent HSCT, compared with no change in those who received cyclophosphamide (CYC; P = .024), Keith M. Sullivan, MD, reported at the annual meeting of the American College of Rheumatology.
Additionally, whole-lung quantitative lung fibrosis (QLF) scores were stable (–1%) in the HSCT patients, but increased 3% in the CYC patients (P = .047), said Dr. Sullivan, a professor of medicine at Duke University, Durham, N.C.
Dr. Sullivan was the first author on the SCOT trial, and he reported the current study results on behalf of lead investigator Jonathan Goldin, MD, PhD, of the department of radiologic sciences at the University of California, Los Angeles.
“These are really kind of meaningful associations, especially since the worst of the [CYC] treatment group didn’t make it to month 54,” Dr. Sullivan said.
Quantitative scores of scleroderma-related interstitial lung disease were measured using computer-based quantitative image analysis of standardized, noncontrast, volumetric, thin-section, thoracic, high-resolution CT. The same CT machine was used for all time points (except for one subject) with careful attention to breath hold reproducibility and image quality. Baseline characteristics were not different between the HSCT and CYC groups, he noted, stressing the rigorous study design.
CT assessments were also compared for the most severe lobe in each patient and showed similar findings, with both QILD and QLF scores for that lobe improving in the HSCT patients relative to the CYC patients (P = .004 and P = .002, respectively), Dr. Sullivan said, adding that the direction of change in structural measures of QILD and QLF for both whole lung and most severe lobe CTs tracked with physiological pulmonary function tests, including forced vital capacity (FVC), forced expiratory volume in 1 second, and diffusing capacity of the lungs for carbon monoxide.
“The FVC improved while QILD decreased, and that’s what you would expect to see,” he said. “So for each of these ways of displaying data, there was an expected and sensible inverse correlation.”
Scleroderma-related interstitial lung disease is a major cause of morbidity and mortality in severe systemic sclerosis. In the wake of the SCOT trial findings, questions remained with respect to correlation between those findings and pulmonary function; if the improvements with HSCT are real and meaningful, they should have meaningful correlation with pulmonary function, and these findings demonstrate those correlates, he said.
“Changes in quantitative lung CT scoring of scleroderma lung disease provide an objective radiologic validation of the long-term benefits of transplant compared to cyclophosphamide in individuals with severe scleroderma and lung involvement. Improvement in imaging after transplant continues for up to 54 months after randomization, giving radiologic confirmation of a durable treatment benefit,” Dr. Sullivan concluded.
The investigators reported having no relevant disclosures.
SOURCE: Goldin J et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 901.
REPORTING FROM THE ACR ANNUAL MEETING
Key clinical point: Lung CT scores remain stable or improve following hematopoietic stem cell transplantation in patients with scleroderma-related interstitial lung disease when compared against monthly cyclophosphamide treatments.
Major finding: Quantitative interstitial lung disease scores decreased by 7% at 54 months in hematopoietic stem cell transplant patients versus no change in those who received cyclophosphamide (P = .024).
Study details: A study of 75 patients from the SCOT trial.
Disclosures: The investigators reported having no relevant disclosures.
Source: Goldin J et al. Arthritis Rheumatol. 2018;70(Suppl 10): Abstract 901.
Palliative-rehab combo may improve QoL in newly diagnosed cancer patients
In patients with a new diagnosis of advanced cancer, an intervention that combined palliative care with rehabilitation helped improve quality of life, results of a randomized, single-center study suggest.
Patients had a significant improvement in their most pressing quality-of-life issues after participating in the intervention, which included individualized palliative care consultations and a patient/caregiver “school” of lectures, discussion, and physical exercise, investigators said.
These findings suggest that every patient facing an advanced cancer diagnosis should at least have an initial exploratory consultation with a specialized palliative care team, and should be offered not only the usual components of palliative care, but also cancer rehabilitation, said Lise Nottelmann, MD, of the department of oncology at Vejle Hospital in Denmark.
“We should be active as a health care system in approaching these patients and offering them this intervention, or at least a consultation exploring these aspects of quality of life,” Dr. Nottelmann said in an interview at the 2018 Palliative and Supportive Care in Oncology Symposium.
The study by Dr. Nottelmann and her colleagues, presented at the symposium, comprised 301 patients with nonresectable solid tumors, including lung, gastrointestinal, prostate, and others. Those patients were randomly allocated to the palliative rehabilitation intervention or to standard care only.
Every patient participated in two consultations with a specialized palliative care team, and then had the opportunity for individualized contact with the team in a 12-week open contact period. They were also invited to participate in the school sessions, each of which included a 20-minute lecture on topics such as physical activity and good nutrition plus a 40-minute discussion period, followed by an exercise session.
Of the patients randomized to the palliative rehabilitation intervention, 26 participated only in the initial consultations, while 59 participated in the group program, and 47 had individual consultations, Dr. Nottelmann reported.
To measure quality of life, the investigators asked patients to identify a “primary problem” that corresponded to one of 12 scales in the EORTC QLQ-C30 questionnaire related to physical and role functioning, emotional and cognitive functioning, or symptoms.
The primary endpoint of the analysis was improvement in QLQ-C30 scores at 12 weeks. The analysis was done on specific scales in the patients who identified a primary problem, combined with global QLQ-C30 scores for the remaining one-quarter of the patients who did not, Dr. Nottelmann said.
After 12 weeks, the patients in the intervention arm had a significant improvement versus the no-intervention arm as measured by a version of the EORTC QLQ-C30 questionnaire. The absolute between-group difference in scores was 3.0 (95% confidence interval, 0.0-6.0; P less than .047), according to researchers.
Starting palliative care earlier in the course of cancer, as done in this intervention, is an increasingly accepted practice, supported by large studies and recent clinical practice guidelines that recommend early integration of palliative care into the seriously ill patient’s care plan.
What was different about this intervention was the integration of rehabilitation aspects into palliative care, Dr. Nottelmann said in the interview. While not traditionally thought of as a component of palliative care, the concept of palliative rehabilitation is gaining ground, she said.
The goal of rehabilitative palliative care is to help individuals with life-limiting or terminal conditions actively self-manage their conditions so they can “live fully” and enjoy the best quality of life possible, according to Hospice UK, a national charity for hospice care in the United Kingdom.
The symposium was cosponsored by AAHPM, ASCO, ASTRO, and MASCC. Dr. Nottelmann and her colleagues reported research funding from the Danish Cancer Society. Dr. Nottelmann had no disclosures related to the presentation. One coauthor provided disclosures related to Roche, Amgen, Bayer, and Merck Sharp & Dohme.
SOURCE: Nottelmann L et al. PallOnc 2018, Abstract 75.
In patients with a new diagnosis of advanced cancer, an intervention that combined palliative care with rehabilitation helped improve quality of life, results of a randomized, single-center study suggest.
Patients had a significant improvement in their most pressing quality-of-life issues after participating in the intervention, which included individualized palliative care consultations and a patient/caregiver “school” of lectures, discussion, and physical exercise, investigators said.
These findings suggest that every patient facing an advanced cancer diagnosis should at least have an initial exploratory consultation with a specialized palliative care team, and should be offered not only the usual components of palliative care, but also cancer rehabilitation, said Lise Nottelmann, MD, of the department of oncology at Vejle Hospital in Denmark.
“We should be active as a health care system in approaching these patients and offering them this intervention, or at least a consultation exploring these aspects of quality of life,” Dr. Nottelmann said in an interview at the 2018 Palliative and Supportive Care in Oncology Symposium.
The study by Dr. Nottelmann and her colleagues, presented at the symposium, comprised 301 patients with nonresectable solid tumors, including lung, gastrointestinal, prostate, and others. Those patients were randomly allocated to the palliative rehabilitation intervention or to standard care only.
Every patient participated in two consultations with a specialized palliative care team, and then had the opportunity for individualized contact with the team in a 12-week open contact period. They were also invited to participate in the school sessions, each of which included a 20-minute lecture on topics such as physical activity and good nutrition plus a 40-minute discussion period, followed by an exercise session.
Of the patients randomized to the palliative rehabilitation intervention, 26 participated only in the initial consultations, while 59 participated in the group program, and 47 had individual consultations, Dr. Nottelmann reported.
To measure quality of life, the investigators asked patients to identify a “primary problem” that corresponded to one of 12 scales in the EORTC QLQ-C30 questionnaire related to physical and role functioning, emotional and cognitive functioning, or symptoms.
The primary endpoint of the analysis was improvement in QLQ-C30 scores at 12 weeks. The analysis was done on specific scales in the patients who identified a primary problem, combined with global QLQ-C30 scores for the remaining one-quarter of the patients who did not, Dr. Nottelmann said.
After 12 weeks, the patients in the intervention arm had a significant improvement versus the no-intervention arm as measured by a version of the EORTC QLQ-C30 questionnaire. The absolute between-group difference in scores was 3.0 (95% confidence interval, 0.0-6.0; P less than .047), according to researchers.
Starting palliative care earlier in the course of cancer, as done in this intervention, is an increasingly accepted practice, supported by large studies and recent clinical practice guidelines that recommend early integration of palliative care into the seriously ill patient’s care plan.
What was different about this intervention was the integration of rehabilitation aspects into palliative care, Dr. Nottelmann said in the interview. While not traditionally thought of as a component of palliative care, the concept of palliative rehabilitation is gaining ground, she said.
The goal of rehabilitative palliative care is to help individuals with life-limiting or terminal conditions actively self-manage their conditions so they can “live fully” and enjoy the best quality of life possible, according to Hospice UK, a national charity for hospice care in the United Kingdom.
The symposium was cosponsored by AAHPM, ASCO, ASTRO, and MASCC. Dr. Nottelmann and her colleagues reported research funding from the Danish Cancer Society. Dr. Nottelmann had no disclosures related to the presentation. One coauthor provided disclosures related to Roche, Amgen, Bayer, and Merck Sharp & Dohme.
SOURCE: Nottelmann L et al. PallOnc 2018, Abstract 75.
In patients with a new diagnosis of advanced cancer, an intervention that combined palliative care with rehabilitation helped improve quality of life, results of a randomized, single-center study suggest.
Patients had a significant improvement in their most pressing quality-of-life issues after participating in the intervention, which included individualized palliative care consultations and a patient/caregiver “school” of lectures, discussion, and physical exercise, investigators said.
These findings suggest that every patient facing an advanced cancer diagnosis should at least have an initial exploratory consultation with a specialized palliative care team, and should be offered not only the usual components of palliative care, but also cancer rehabilitation, said Lise Nottelmann, MD, of the department of oncology at Vejle Hospital in Denmark.
“We should be active as a health care system in approaching these patients and offering them this intervention, or at least a consultation exploring these aspects of quality of life,” Dr. Nottelmann said in an interview at the 2018 Palliative and Supportive Care in Oncology Symposium.
The study by Dr. Nottelmann and her colleagues, presented at the symposium, comprised 301 patients with nonresectable solid tumors, including lung, gastrointestinal, prostate, and others. Those patients were randomly allocated to the palliative rehabilitation intervention or to standard care only.
Every patient participated in two consultations with a specialized palliative care team, and then had the opportunity for individualized contact with the team in a 12-week open contact period. They were also invited to participate in the school sessions, each of which included a 20-minute lecture on topics such as physical activity and good nutrition plus a 40-minute discussion period, followed by an exercise session.
Of the patients randomized to the palliative rehabilitation intervention, 26 participated only in the initial consultations, while 59 participated in the group program, and 47 had individual consultations, Dr. Nottelmann reported.
To measure quality of life, the investigators asked patients to identify a “primary problem” that corresponded to one of 12 scales in the EORTC QLQ-C30 questionnaire related to physical and role functioning, emotional and cognitive functioning, or symptoms.
The primary endpoint of the analysis was improvement in QLQ-C30 scores at 12 weeks. The analysis was done on specific scales in the patients who identified a primary problem, combined with global QLQ-C30 scores for the remaining one-quarter of the patients who did not, Dr. Nottelmann said.
After 12 weeks, the patients in the intervention arm had a significant improvement versus the no-intervention arm as measured by a version of the EORTC QLQ-C30 questionnaire. The absolute between-group difference in scores was 3.0 (95% confidence interval, 0.0-6.0; P less than .047), according to researchers.
Starting palliative care earlier in the course of cancer, as done in this intervention, is an increasingly accepted practice, supported by large studies and recent clinical practice guidelines that recommend early integration of palliative care into the seriously ill patient’s care plan.
What was different about this intervention was the integration of rehabilitation aspects into palliative care, Dr. Nottelmann said in the interview. While not traditionally thought of as a component of palliative care, the concept of palliative rehabilitation is gaining ground, she said.
The goal of rehabilitative palliative care is to help individuals with life-limiting or terminal conditions actively self-manage their conditions so they can “live fully” and enjoy the best quality of life possible, according to Hospice UK, a national charity for hospice care in the United Kingdom.
The symposium was cosponsored by AAHPM, ASCO, ASTRO, and MASCC. Dr. Nottelmann and her colleagues reported research funding from the Danish Cancer Society. Dr. Nottelmann had no disclosures related to the presentation. One coauthor provided disclosures related to Roche, Amgen, Bayer, and Merck Sharp & Dohme.
SOURCE: Nottelmann L et al. PallOnc 2018, Abstract 75.
FROM PALLONC 2018
Key clinical point: An intervention combining palliative care and rehabilitation aspects improved quality of life in patients with newly diagnosed, advanced cancers.
Major finding: Patients in the rehabilitative palliative care program had a significant improvement, compared with no intervention (absolute between-group difference in EORTC QLQ-30 scores, 3.0; 95% CI, 0.0-6.0; P less than .047).
Study details: A single-center randomized study of 301 patients with a newly diagnosed advanced solid tumor cancers.
Disclosures: Research funding came from the Danish Cancer Society. One study coauthor had disclosures related to Roche, Amgen, Bayer, and Merck Sharp & Dohme.
Source: Nottelmann L et al. 2018 Palliative and Supportive Care in Oncology Symposium Abstract 75.
Portable hematology analyzer gets FDA nod
The Food and Drug Administration has granted .

HemoScreen requires a single drop of blood and uses disposable cartridges that provide automatic sample preparation.
HemoScreen can analyze 20 standard complete blood count parameters and produces results within 5 minutes.
Study results suggested that HemoScreen provides results comparable to those of another hematology analyzer, Sysmex XE-2100 (J Clin Pathol. 2016 Aug;69[8]:720-5).
“The HemoScreen delivers lab accurate results,” Avishay Bransky, PhD, CEO of PixCell, said in a statement.
HemoScreen “would be especially useful” in physicians’ offices, emergency rooms, intensive care units, oncology clinics, and remote locations, he added.
HemoScreen makes use of a technology called viscoelastic focusing, which employs microfluidics and machine vision algorithms to analyze cells.
The Food and Drug Administration has granted .
HemoScreen requires a single drop of blood and uses disposable cartridges that provide automatic sample preparation.
HemoScreen can analyze 20 standard complete blood count parameters and produces results within 5 minutes.
Study results suggested that HemoScreen provides results comparable to those of another hematology analyzer, Sysmex XE-2100 (J Clin Pathol. 2016 Aug;69[8]:720-5).
“The HemoScreen delivers lab accurate results,” Avishay Bransky, PhD, CEO of PixCell, said in a statement.
HemoScreen “would be especially useful” in physicians’ offices, emergency rooms, intensive care units, oncology clinics, and remote locations, he added.
HemoScreen makes use of a technology called viscoelastic focusing, which employs microfluidics and machine vision algorithms to analyze cells.
The Food and Drug Administration has granted .
HemoScreen requires a single drop of blood and uses disposable cartridges that provide automatic sample preparation.
HemoScreen can analyze 20 standard complete blood count parameters and produces results within 5 minutes.
Study results suggested that HemoScreen provides results comparable to those of another hematology analyzer, Sysmex XE-2100 (J Clin Pathol. 2016 Aug;69[8]:720-5).
“The HemoScreen delivers lab accurate results,” Avishay Bransky, PhD, CEO of PixCell, said in a statement.
HemoScreen “would be especially useful” in physicians’ offices, emergency rooms, intensive care units, oncology clinics, and remote locations, he added.
HemoScreen makes use of a technology called viscoelastic focusing, which employs microfluidics and machine vision algorithms to analyze cells.
Researchers seek more sickle cell drug research
BETHESDA, MD – While there are experimental treatments such as sevuparin and gene therapy in testing for sickle cell disease (SCD), researchers said this field is lagging because of a lack of funding.

Yogen Saunthararajah, MD, of the Cleveland Clinic, described what he called a “paltry” landscape of new drugs for SCD at Sickle Cell in Focus, a conference held by the National Institutes of Health.
There are only four main approaches taken by drugs now in clinical testing for addressing the root causes of SCD, despite decades’ worth of research of the genetic and mechanistic underpinnings of this disease, he said.
“It’s pretty sad,” Dr. Saunthararajah said, referring to the quantity of efforts, not their quality.
Within days of his presentation at the conference, one of the drugs he highlighted had officially fallen out of contention. An Oct. 26 post on NIH’s Clinicaltrials.gov site said Incyte had terminated its phase 1 study of INCB059872 in SCD “due to a business decision” not to pursue this indication. Incyte confirmed that it dropped development of INCB059872 for SCD but will continue testing it for other indications, including acute myeloid leukemia (AML).
Dr. Saunthararajah said that work on another approach, using decitabine (Dacogen), for which he has done a phase 1 study, is “struggling,” because of the search for funding.
Two other approaches that Dr. Saunthararajah cited in his presentation appear to remain on track. These are gene therapy and the once-daily voxelotor treatment from Global Blood Therapeutics.
In the field of gene therapy, Sangamo Therapeutics and Sanofi’s Bioverativ in May said the Food and Drug Administration had cleared the way for them to start a phase 1/2 clinical trial for the BIVV003 product that they are developing together. This uses zinc finger nuclease (ZFN) gene-editing technology to modifying a short sequence of the BCL11A gene, with the aim of reactivating fetal hemoglobin.
Bluebird Bio’s LentiGlobin gene therapy has advanced as far as phase 3 for transfusion-dependent beta-thalassemia and phase 1/2 for SCD. In October, the European Medicines Agency accepted the company’s application for approval of LentiGlobin gene therapy for the treatment of adolescents and adults with transfusion-dependent beta-thalassemia (TDT) and a non-beta0/beta0 genotype. The company has not said when it expects to file with the FDA for approval of this treatment.
In the field of oral therapies, Global Blood Therapeutics has said it’s in discussions with the FDA about a potential accelerated approval of voxelotor. The tablet is meant to inhibit the underlying mechanism that causes sickling of red blood cells. In June, the company completed a planned review of early data from its phase 3 trial, known as the HOPE study.
“On the primary endpoint (the proportion of patients with greater than 1 g/dL increase in hemoglobin versus baseline), a statistically significant increase was demonstrated with voxelotor at both the 1,500-mg and 900-mg doses after 12 weeks of treatment versus placebo,” Global Blood Therapeutics said in an August regulatory filing with the Securities and Exchange Commission.
The company has said that voxelotor may meet the FDA’s standard for accelerated approval under Subpart H program.
In his presentation at the NIH conference, Tom Williams, MD, PhD, of KEMRI/Wellcome Trust Research Programme highlighted a recent review of experimental SCD treatments by Marilyn Jo Telen, MD, of Duke University, Durham, N.C. (Blood. 2016;127[7]:810-9).
Also among the drugs being tested for SCD is Modus Therapeutics’ sevuparin, which Dr. Williams described as being “a heparin-like molecule without the anticoagulant complications.”
The compound originated as a “passion project” of Mats Wahlgren, MD, PhD, of the Karolinska Institute, Stockholm, who developed it for the treatment of malaria, Dr. Williams said. The company that’s developing sevuparin, Modus Therapeutics, is moving it forward first as a treatment for SCD for commercial reasons, Dr. Williams said.
“They think it’s a better first target,” he said.
An ongoing study of sevuparin for painful crisis is expected to be completed in December, with data then expected to be released in the middle of 2019, Ellen K. Donnelly, PhD, chief executive officer of Modus Therapeutics, said in an interview. She cited a mix of scientific, medical and commercial reasons for her company’s decision to advance sevuparin in SCD.
“First, and most importantly, there is proof of clinical benefit of a similar molecule (the low-molecular-weight heparin called tinzaparin) in patients with sickle cell disease,” Dr. Donnelly said. “Unfortunately, there is a bleeding risk with tinzaparin that limits use of the agent for the treatment of sickle cell disease. Sevuparin does not have the bleeding risk and thus is a strong candidate for SCD.”
Dr. Donnelly also noted the emphasis that the FDA’s Division of Hematology Products has put on development of therapeutics for SCD as a reason for proceeding first with this indication. The agency and the American Society of Hematology in early October held a workshop of experts, physicians, patients, and industry collaborators focused on identifying new endpoints for clinical studies.
Still, she noted that commercial reasons did factor into this decision.
“[I]t is possible for a small company like Modus to take an asset all the way to market when developing a therapeutic for a rare disease given the need for fewer patients and smaller trials,” Dr. Donnelly wrote. “As you can see from www.clinicaltrials.gov, we were able to run our phase 2 study with a small number of sites. In addition, in SCD it is possible to get approval with only one or two confirmatory studies.”
Financial interests
Other drugs in testing for SCD include rivipansel from GlycoMimetics, for which Pfizer is leading development. The sponsors expect to complete the so-called RESET trial by 2019, according to the clinicaltrials.gov. In this study, which is intended to enroll 350 participants, patients who have vaso-occlusive crises are randomly selected for treatment with either rivipansel or placebo.
Speaking during a question-and-answer session at the NIH conference, Robert Swift, PhD, said there’s a need for inexpensive oral drugs to treat SCD. Many other options will remain beyond the finances of people living in poor countries, he said.
“We need to focus not only on the root cause, but on something that is oral and inexpensive to solve the greater sickle cell problem,” Dr. Swift said.
Large drugmakers already have hospital-based sales forces, making SCD drugs administered in this setting attractive to them, he said.
“This is partly about where money is. The drug companies are going where the money is. It’s not oral drugs to treat everybody, it’s something else,” Dr. Swift said. “So someone else is going to have to fund the basic research” into treatments that could be more broadly used.
Dr. Swift said in an interview that he has received NIH funding for developing SCD-101, an oral drug, for which a placebo-controlled crossover study is underway.
Presenters at the NIH conference, including Dr. Saunthararajah, expressed frustration about what they see as relatively little work being done on SCD despite decades of knowledge about the root causes. Like Dr. Swift, he criticized the approach taken in selecting which treatments advance in this field.
“It’s not being driven by what is the most cost effective, what the patients need the most,” Dr. Saunthararajah said. “It’s driven by what will make the most money, not just for [the] drug company, but also for the hospital and also for the physicians.”
Dr. Saunthararajah reported having patents and patent applications around decitabine/tetrahydrouridine, 5-azacytidine/tetrahydrouridine, and differentiation therapy for oncology. He has also been a consultant for EpiDestiny, Novo Nordisk, and Takeda Oncology. Dr. Williams reported having no relevant financial disclosures. Dr. Swift is a managing member of Invenux and reported equity in Mast Therapeutics and SCD Development.
This article was updated on 11/9/2018.
BETHESDA, MD – While there are experimental treatments such as sevuparin and gene therapy in testing for sickle cell disease (SCD), researchers said this field is lagging because of a lack of funding.
Yogen Saunthararajah, MD, of the Cleveland Clinic, described what he called a “paltry” landscape of new drugs for SCD at Sickle Cell in Focus, a conference held by the National Institutes of Health.
There are only four main approaches taken by drugs now in clinical testing for addressing the root causes of SCD, despite decades’ worth of research of the genetic and mechanistic underpinnings of this disease, he said.
“It’s pretty sad,” Dr. Saunthararajah said, referring to the quantity of efforts, not their quality.
Within days of his presentation at the conference, one of the drugs he highlighted had officially fallen out of contention. An Oct. 26 post on NIH’s Clinicaltrials.gov site said Incyte had terminated its phase 1 study of INCB059872 in SCD “due to a business decision” not to pursue this indication. Incyte confirmed that it dropped development of INCB059872 for SCD but will continue testing it for other indications, including acute myeloid leukemia (AML).
Dr. Saunthararajah said that work on another approach, using decitabine (Dacogen), for which he has done a phase 1 study, is “struggling,” because of the search for funding.
Two other approaches that Dr. Saunthararajah cited in his presentation appear to remain on track. These are gene therapy and the once-daily voxelotor treatment from Global Blood Therapeutics.
In the field of gene therapy, Sangamo Therapeutics and Sanofi’s Bioverativ in May said the Food and Drug Administration had cleared the way for them to start a phase 1/2 clinical trial for the BIVV003 product that they are developing together. This uses zinc finger nuclease (ZFN) gene-editing technology to modifying a short sequence of the BCL11A gene, with the aim of reactivating fetal hemoglobin.
Bluebird Bio’s LentiGlobin gene therapy has advanced as far as phase 3 for transfusion-dependent beta-thalassemia and phase 1/2 for SCD. In October, the European Medicines Agency accepted the company’s application for approval of LentiGlobin gene therapy for the treatment of adolescents and adults with transfusion-dependent beta-thalassemia (TDT) and a non-beta0/beta0 genotype. The company has not said when it expects to file with the FDA for approval of this treatment.
In the field of oral therapies, Global Blood Therapeutics has said it’s in discussions with the FDA about a potential accelerated approval of voxelotor. The tablet is meant to inhibit the underlying mechanism that causes sickling of red blood cells. In June, the company completed a planned review of early data from its phase 3 trial, known as the HOPE study.
“On the primary endpoint (the proportion of patients with greater than 1 g/dL increase in hemoglobin versus baseline), a statistically significant increase was demonstrated with voxelotor at both the 1,500-mg and 900-mg doses after 12 weeks of treatment versus placebo,” Global Blood Therapeutics said in an August regulatory filing with the Securities and Exchange Commission.
The company has said that voxelotor may meet the FDA’s standard for accelerated approval under Subpart H program.
In his presentation at the NIH conference, Tom Williams, MD, PhD, of KEMRI/Wellcome Trust Research Programme highlighted a recent review of experimental SCD treatments by Marilyn Jo Telen, MD, of Duke University, Durham, N.C. (Blood. 2016;127[7]:810-9).
Also among the drugs being tested for SCD is Modus Therapeutics’ sevuparin, which Dr. Williams described as being “a heparin-like molecule without the anticoagulant complications.”
The compound originated as a “passion project” of Mats Wahlgren, MD, PhD, of the Karolinska Institute, Stockholm, who developed it for the treatment of malaria, Dr. Williams said. The company that’s developing sevuparin, Modus Therapeutics, is moving it forward first as a treatment for SCD for commercial reasons, Dr. Williams said.
“They think it’s a better first target,” he said.
An ongoing study of sevuparin for painful crisis is expected to be completed in December, with data then expected to be released in the middle of 2019, Ellen K. Donnelly, PhD, chief executive officer of Modus Therapeutics, said in an interview. She cited a mix of scientific, medical and commercial reasons for her company’s decision to advance sevuparin in SCD.
“First, and most importantly, there is proof of clinical benefit of a similar molecule (the low-molecular-weight heparin called tinzaparin) in patients with sickle cell disease,” Dr. Donnelly said. “Unfortunately, there is a bleeding risk with tinzaparin that limits use of the agent for the treatment of sickle cell disease. Sevuparin does not have the bleeding risk and thus is a strong candidate for SCD.”
Dr. Donnelly also noted the emphasis that the FDA’s Division of Hematology Products has put on development of therapeutics for SCD as a reason for proceeding first with this indication. The agency and the American Society of Hematology in early October held a workshop of experts, physicians, patients, and industry collaborators focused on identifying new endpoints for clinical studies.
Still, she noted that commercial reasons did factor into this decision.
“[I]t is possible for a small company like Modus to take an asset all the way to market when developing a therapeutic for a rare disease given the need for fewer patients and smaller trials,” Dr. Donnelly wrote. “As you can see from www.clinicaltrials.gov, we were able to run our phase 2 study with a small number of sites. In addition, in SCD it is possible to get approval with only one or two confirmatory studies.”
Financial interests
Other drugs in testing for SCD include rivipansel from GlycoMimetics, for which Pfizer is leading development. The sponsors expect to complete the so-called RESET trial by 2019, according to the clinicaltrials.gov. In this study, which is intended to enroll 350 participants, patients who have vaso-occlusive crises are randomly selected for treatment with either rivipansel or placebo.
Speaking during a question-and-answer session at the NIH conference, Robert Swift, PhD, said there’s a need for inexpensive oral drugs to treat SCD. Many other options will remain beyond the finances of people living in poor countries, he said.
“We need to focus not only on the root cause, but on something that is oral and inexpensive to solve the greater sickle cell problem,” Dr. Swift said.
Large drugmakers already have hospital-based sales forces, making SCD drugs administered in this setting attractive to them, he said.
“This is partly about where money is. The drug companies are going where the money is. It’s not oral drugs to treat everybody, it’s something else,” Dr. Swift said. “So someone else is going to have to fund the basic research” into treatments that could be more broadly used.
Dr. Swift said in an interview that he has received NIH funding for developing SCD-101, an oral drug, for which a placebo-controlled crossover study is underway.
Presenters at the NIH conference, including Dr. Saunthararajah, expressed frustration about what they see as relatively little work being done on SCD despite decades of knowledge about the root causes. Like Dr. Swift, he criticized the approach taken in selecting which treatments advance in this field.
“It’s not being driven by what is the most cost effective, what the patients need the most,” Dr. Saunthararajah said. “It’s driven by what will make the most money, not just for [the] drug company, but also for the hospital and also for the physicians.”
Dr. Saunthararajah reported having patents and patent applications around decitabine/tetrahydrouridine, 5-azacytidine/tetrahydrouridine, and differentiation therapy for oncology. He has also been a consultant for EpiDestiny, Novo Nordisk, and Takeda Oncology. Dr. Williams reported having no relevant financial disclosures. Dr. Swift is a managing member of Invenux and reported equity in Mast Therapeutics and SCD Development.
This article was updated on 11/9/2018.
BETHESDA, MD – While there are experimental treatments such as sevuparin and gene therapy in testing for sickle cell disease (SCD), researchers said this field is lagging because of a lack of funding.
Yogen Saunthararajah, MD, of the Cleveland Clinic, described what he called a “paltry” landscape of new drugs for SCD at Sickle Cell in Focus, a conference held by the National Institutes of Health.
There are only four main approaches taken by drugs now in clinical testing for addressing the root causes of SCD, despite decades’ worth of research of the genetic and mechanistic underpinnings of this disease, he said.
“It’s pretty sad,” Dr. Saunthararajah said, referring to the quantity of efforts, not their quality.
Within days of his presentation at the conference, one of the drugs he highlighted had officially fallen out of contention. An Oct. 26 post on NIH’s Clinicaltrials.gov site said Incyte had terminated its phase 1 study of INCB059872 in SCD “due to a business decision” not to pursue this indication. Incyte confirmed that it dropped development of INCB059872 for SCD but will continue testing it for other indications, including acute myeloid leukemia (AML).
Dr. Saunthararajah said that work on another approach, using decitabine (Dacogen), for which he has done a phase 1 study, is “struggling,” because of the search for funding.
Two other approaches that Dr. Saunthararajah cited in his presentation appear to remain on track. These are gene therapy and the once-daily voxelotor treatment from Global Blood Therapeutics.
In the field of gene therapy, Sangamo Therapeutics and Sanofi’s Bioverativ in May said the Food and Drug Administration had cleared the way for them to start a phase 1/2 clinical trial for the BIVV003 product that they are developing together. This uses zinc finger nuclease (ZFN) gene-editing technology to modifying a short sequence of the BCL11A gene, with the aim of reactivating fetal hemoglobin.
Bluebird Bio’s LentiGlobin gene therapy has advanced as far as phase 3 for transfusion-dependent beta-thalassemia and phase 1/2 for SCD. In October, the European Medicines Agency accepted the company’s application for approval of LentiGlobin gene therapy for the treatment of adolescents and adults with transfusion-dependent beta-thalassemia (TDT) and a non-beta0/beta0 genotype. The company has not said when it expects to file with the FDA for approval of this treatment.
In the field of oral therapies, Global Blood Therapeutics has said it’s in discussions with the FDA about a potential accelerated approval of voxelotor. The tablet is meant to inhibit the underlying mechanism that causes sickling of red blood cells. In June, the company completed a planned review of early data from its phase 3 trial, known as the HOPE study.
“On the primary endpoint (the proportion of patients with greater than 1 g/dL increase in hemoglobin versus baseline), a statistically significant increase was demonstrated with voxelotor at both the 1,500-mg and 900-mg doses after 12 weeks of treatment versus placebo,” Global Blood Therapeutics said in an August regulatory filing with the Securities and Exchange Commission.
The company has said that voxelotor may meet the FDA’s standard for accelerated approval under Subpart H program.
In his presentation at the NIH conference, Tom Williams, MD, PhD, of KEMRI/Wellcome Trust Research Programme highlighted a recent review of experimental SCD treatments by Marilyn Jo Telen, MD, of Duke University, Durham, N.C. (Blood. 2016;127[7]:810-9).
Also among the drugs being tested for SCD is Modus Therapeutics’ sevuparin, which Dr. Williams described as being “a heparin-like molecule without the anticoagulant complications.”
The compound originated as a “passion project” of Mats Wahlgren, MD, PhD, of the Karolinska Institute, Stockholm, who developed it for the treatment of malaria, Dr. Williams said. The company that’s developing sevuparin, Modus Therapeutics, is moving it forward first as a treatment for SCD for commercial reasons, Dr. Williams said.
“They think it’s a better first target,” he said.
An ongoing study of sevuparin for painful crisis is expected to be completed in December, with data then expected to be released in the middle of 2019, Ellen K. Donnelly, PhD, chief executive officer of Modus Therapeutics, said in an interview. She cited a mix of scientific, medical and commercial reasons for her company’s decision to advance sevuparin in SCD.
“First, and most importantly, there is proof of clinical benefit of a similar molecule (the low-molecular-weight heparin called tinzaparin) in patients with sickle cell disease,” Dr. Donnelly said. “Unfortunately, there is a bleeding risk with tinzaparin that limits use of the agent for the treatment of sickle cell disease. Sevuparin does not have the bleeding risk and thus is a strong candidate for SCD.”
Dr. Donnelly also noted the emphasis that the FDA’s Division of Hematology Products has put on development of therapeutics for SCD as a reason for proceeding first with this indication. The agency and the American Society of Hematology in early October held a workshop of experts, physicians, patients, and industry collaborators focused on identifying new endpoints for clinical studies.
Still, she noted that commercial reasons did factor into this decision.
“[I]t is possible for a small company like Modus to take an asset all the way to market when developing a therapeutic for a rare disease given the need for fewer patients and smaller trials,” Dr. Donnelly wrote. “As you can see from www.clinicaltrials.gov, we were able to run our phase 2 study with a small number of sites. In addition, in SCD it is possible to get approval with only one or two confirmatory studies.”
Financial interests
Other drugs in testing for SCD include rivipansel from GlycoMimetics, for which Pfizer is leading development. The sponsors expect to complete the so-called RESET trial by 2019, according to the clinicaltrials.gov. In this study, which is intended to enroll 350 participants, patients who have vaso-occlusive crises are randomly selected for treatment with either rivipansel or placebo.
Speaking during a question-and-answer session at the NIH conference, Robert Swift, PhD, said there’s a need for inexpensive oral drugs to treat SCD. Many other options will remain beyond the finances of people living in poor countries, he said.
“We need to focus not only on the root cause, but on something that is oral and inexpensive to solve the greater sickle cell problem,” Dr. Swift said.
Large drugmakers already have hospital-based sales forces, making SCD drugs administered in this setting attractive to them, he said.
“This is partly about where money is. The drug companies are going where the money is. It’s not oral drugs to treat everybody, it’s something else,” Dr. Swift said. “So someone else is going to have to fund the basic research” into treatments that could be more broadly used.
Dr. Swift said in an interview that he has received NIH funding for developing SCD-101, an oral drug, for which a placebo-controlled crossover study is underway.
Presenters at the NIH conference, including Dr. Saunthararajah, expressed frustration about what they see as relatively little work being done on SCD despite decades of knowledge about the root causes. Like Dr. Swift, he criticized the approach taken in selecting which treatments advance in this field.
“It’s not being driven by what is the most cost effective, what the patients need the most,” Dr. Saunthararajah said. “It’s driven by what will make the most money, not just for [the] drug company, but also for the hospital and also for the physicians.”
Dr. Saunthararajah reported having patents and patent applications around decitabine/tetrahydrouridine, 5-azacytidine/tetrahydrouridine, and differentiation therapy for oncology. He has also been a consultant for EpiDestiny, Novo Nordisk, and Takeda Oncology. Dr. Williams reported having no relevant financial disclosures. Dr. Swift is a managing member of Invenux and reported equity in Mast Therapeutics and SCD Development.
This article was updated on 11/9/2018.
REPORTING FROM SICKLE CELL IN FOCUS
Report details financial burden of blood cancers
with costs for acute leukemia almost tripling that amount, according to a new report from the Leukemia & Lymphoma Society (LLS).
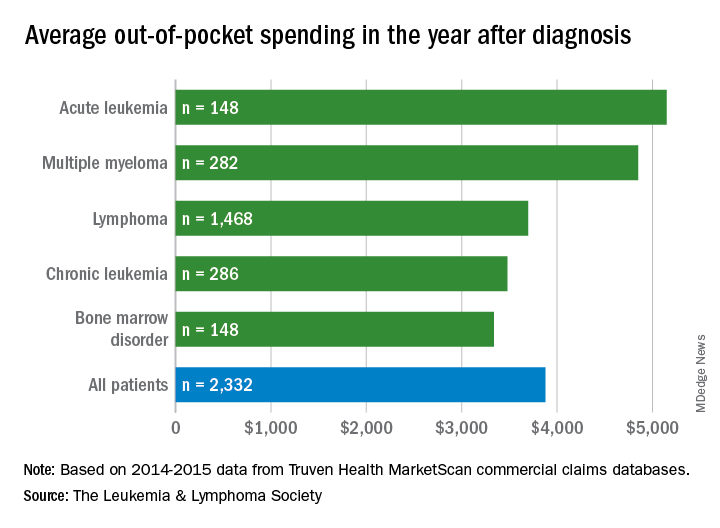
Total allowed cost – the average amount paid by the insurer and patient combined – for acute leukemia was more than $463,000 for the 12 months after initial diagnosis. Averages for the other four cancers included in the analysis came in at $214,000 for multiple myeloma, $134,000 for bone marrow disorders, $131,000 for lymphoma, and $89,000 for chronic leukemia, the LLS said.
The cost figures are drawn from claims data for 2,332 patients diagnosed in 2014.
Differences in out-of-pocket (OOP) costs were smaller, with the average for all patients at almost $3,900 in the year after diagnosis and acute leukemia coming in the highest at $5,100. Over time, however, OOP costs for multiple myeloma patients became the highest, totaling $9,100 for the 3 years after diagnosis, compared with $8,800 for acute leukemia and an average of less than $7,800 for the other blood cancers, the LLS said in the report, which was prepared by the actuarial firm Milliman.
OOP costs also varied by the type of plan. Patients in high-deductible plans averaged nearly $5,400 for the first year after diagnosis, compared with $3,300 for those with traditional insurance, the LLS noted. For acute leukemia, the OOP costs of high-deductible plans were more than twice as high as those of traditional plans.
The study was based on data for adults aged 18-64 years from the Truven Health MarketScan commercial claims databases for the years from 2013 to 2016. The LLS received support for the study from Pfizer, Genentech, and Amgen.
with costs for acute leukemia almost tripling that amount, according to a new report from the Leukemia & Lymphoma Society (LLS).
Total allowed cost – the average amount paid by the insurer and patient combined – for acute leukemia was more than $463,000 for the 12 months after initial diagnosis. Averages for the other four cancers included in the analysis came in at $214,000 for multiple myeloma, $134,000 for bone marrow disorders, $131,000 for lymphoma, and $89,000 for chronic leukemia, the LLS said.
The cost figures are drawn from claims data for 2,332 patients diagnosed in 2014.
Differences in out-of-pocket (OOP) costs were smaller, with the average for all patients at almost $3,900 in the year after diagnosis and acute leukemia coming in the highest at $5,100. Over time, however, OOP costs for multiple myeloma patients became the highest, totaling $9,100 for the 3 years after diagnosis, compared with $8,800 for acute leukemia and an average of less than $7,800 for the other blood cancers, the LLS said in the report, which was prepared by the actuarial firm Milliman.
OOP costs also varied by the type of plan. Patients in high-deductible plans averaged nearly $5,400 for the first year after diagnosis, compared with $3,300 for those with traditional insurance, the LLS noted. For acute leukemia, the OOP costs of high-deductible plans were more than twice as high as those of traditional plans.
The study was based on data for adults aged 18-64 years from the Truven Health MarketScan commercial claims databases for the years from 2013 to 2016. The LLS received support for the study from Pfizer, Genentech, and Amgen.
with costs for acute leukemia almost tripling that amount, according to a new report from the Leukemia & Lymphoma Society (LLS).
Total allowed cost – the average amount paid by the insurer and patient combined – for acute leukemia was more than $463,000 for the 12 months after initial diagnosis. Averages for the other four cancers included in the analysis came in at $214,000 for multiple myeloma, $134,000 for bone marrow disorders, $131,000 for lymphoma, and $89,000 for chronic leukemia, the LLS said.
The cost figures are drawn from claims data for 2,332 patients diagnosed in 2014.
Differences in out-of-pocket (OOP) costs were smaller, with the average for all patients at almost $3,900 in the year after diagnosis and acute leukemia coming in the highest at $5,100. Over time, however, OOP costs for multiple myeloma patients became the highest, totaling $9,100 for the 3 years after diagnosis, compared with $8,800 for acute leukemia and an average of less than $7,800 for the other blood cancers, the LLS said in the report, which was prepared by the actuarial firm Milliman.
OOP costs also varied by the type of plan. Patients in high-deductible plans averaged nearly $5,400 for the first year after diagnosis, compared with $3,300 for those with traditional insurance, the LLS noted. For acute leukemia, the OOP costs of high-deductible plans were more than twice as high as those of traditional plans.
The study was based on data for adults aged 18-64 years from the Truven Health MarketScan commercial claims databases for the years from 2013 to 2016. The LLS received support for the study from Pfizer, Genentech, and Amgen.
FDA approves second pegfilgrastim biosimilar
The Food and Drug Administration has approved a second biosimilar to pegfilgrastim (Neulasta) to decrease the chance of infection in patients with nonmyeloid cancer who are receiving myelosuppressive chemotherapy and are at risk of febrile neutropenia.
Approval of pegfilgrastim-cbqv, previously known as CHS-1701, was based on analyses establishing biosimilarity, including pharmacokinetic, pharmacodynamic, and immunogenicity studies. Clinical trial results were presented at the 2017 ASCO Annual Meeting.
The most common adverse reactions with pegfilgrastim-cbqv are bone pain and pain in extremities.
The FDA approved the first pegfilgrastim biosimilar, pegfilgrastim-jmdb (Fulphila) in June.
Pegfilgrastim-cbqv will be marketed as Udenyca by Coherus BioSciences.
“Udenyca’s robust clinical package includes a dedicated immunogenicity similarity study in over 300 healthy subjects,” Barbara Finck, MD, chief medical officer of Coherus BioSciences, said in a press release.
“In support of that study, and as part of our commitment to ensuring patient safety, we deployed a battery of sensitive immunogenicity assays. This effort not only supported the biosimilarity of Udenyca but also advanced the understanding of the immunogenic response of pegfilgrastim products.”
Coherus BioSciences plans to provide details about pricing and the launch of pegfilgrastim-cbqv during an earnings call on Nov. 8.
The Food and Drug Administration has approved a second biosimilar to pegfilgrastim (Neulasta) to decrease the chance of infection in patients with nonmyeloid cancer who are receiving myelosuppressive chemotherapy and are at risk of febrile neutropenia.
Approval of pegfilgrastim-cbqv, previously known as CHS-1701, was based on analyses establishing biosimilarity, including pharmacokinetic, pharmacodynamic, and immunogenicity studies. Clinical trial results were presented at the 2017 ASCO Annual Meeting.
The most common adverse reactions with pegfilgrastim-cbqv are bone pain and pain in extremities.
The FDA approved the first pegfilgrastim biosimilar, pegfilgrastim-jmdb (Fulphila) in June.
Pegfilgrastim-cbqv will be marketed as Udenyca by Coherus BioSciences.
“Udenyca’s robust clinical package includes a dedicated immunogenicity similarity study in over 300 healthy subjects,” Barbara Finck, MD, chief medical officer of Coherus BioSciences, said in a press release.
“In support of that study, and as part of our commitment to ensuring patient safety, we deployed a battery of sensitive immunogenicity assays. This effort not only supported the biosimilarity of Udenyca but also advanced the understanding of the immunogenic response of pegfilgrastim products.”
Coherus BioSciences plans to provide details about pricing and the launch of pegfilgrastim-cbqv during an earnings call on Nov. 8.
The Food and Drug Administration has approved a second biosimilar to pegfilgrastim (Neulasta) to decrease the chance of infection in patients with nonmyeloid cancer who are receiving myelosuppressive chemotherapy and are at risk of febrile neutropenia.
Approval of pegfilgrastim-cbqv, previously known as CHS-1701, was based on analyses establishing biosimilarity, including pharmacokinetic, pharmacodynamic, and immunogenicity studies. Clinical trial results were presented at the 2017 ASCO Annual Meeting.
The most common adverse reactions with pegfilgrastim-cbqv are bone pain and pain in extremities.
The FDA approved the first pegfilgrastim biosimilar, pegfilgrastim-jmdb (Fulphila) in June.
Pegfilgrastim-cbqv will be marketed as Udenyca by Coherus BioSciences.
“Udenyca’s robust clinical package includes a dedicated immunogenicity similarity study in over 300 healthy subjects,” Barbara Finck, MD, chief medical officer of Coherus BioSciences, said in a press release.
“In support of that study, and as part of our commitment to ensuring patient safety, we deployed a battery of sensitive immunogenicity assays. This effort not only supported the biosimilarity of Udenyca but also advanced the understanding of the immunogenic response of pegfilgrastim products.”
Coherus BioSciences plans to provide details about pricing and the launch of pegfilgrastim-cbqv during an earnings call on Nov. 8.
Platelet transfusion threshold matters for preterm infants
A lower threshold for platelet transfusions in preterm infants with severe thrombocytopenia is associated with significantly lower incidence of death and major bleeding, compared with a higher threshold, a new study suggests.
A new major bleeding episode or death occurred in 26% of infants in the high-threshold group, compared with 19% in the low-threshold group, representing a 57% higher risk of poor outcomes even after researchers adjusted for gestational age and intrauterine growth restriction (odds ratio, 1.57; P = .02).
Researchers reported the results of a trial in 660 infants with a mean gestational age of 26.6 weeks, who were randomized to a platelet infusion either at a high platelet–count threshold of 50,000/mm3 or a low threshold of 25,000/mm3.
“Although retrospective studies have suggested that platelet transfusions may cause harm in neonates independently of the disease process, data from randomized controlled trials to support this are lacking,” Anna Curley, MD, of the National Maternity Hospital in Dublin and her coauthors reported in the New England Journal of Medicine.
The rates of minor or worse bleeding were similar between the two groups, and the percentage of infants surviving with bronchopulmonary dysplasia at 36 weeks of corrected age was higher in the high-threshold group (63% vs. 54%; OR, 1.54).
The rates of serious adverse events, not including major bleeding, were similar between the high- and low-threshold groups.
The outcomes of transfusions were not influenced by other factors such as intrauterine growth restriction, gestational age, or postnatal age at randomization.
“Our trial highlights the importance of trials of platelet transfusion involving patients with conditions other than haematological malignancies,” the authors wrote.
However they acknowledged that the reasons for the differences in mortality and outcomes between the two study groups were unknown.
“Platelets have recognized immunological and inflammatory effects, outside of effects on hemostasis,” they wrote. “The effect of transfusing adult platelets to a delicately balance neonatal hemostatic system with relatively hypofunctional platelets is also poorly understood.”
The study was supported by the National Health Service Blood and Transplant Research and Development Committee and other foundations. Authors reported financial disclosures related to Sanquin and Cerus.
SOURCE: Curley A et al. N Engl J Med. 2018 Nov 2. doi: 10.1056/NEJMoa1807320.
A lower threshold for platelet transfusions in preterm infants with severe thrombocytopenia is associated with significantly lower incidence of death and major bleeding, compared with a higher threshold, a new study suggests.
A new major bleeding episode or death occurred in 26% of infants in the high-threshold group, compared with 19% in the low-threshold group, representing a 57% higher risk of poor outcomes even after researchers adjusted for gestational age and intrauterine growth restriction (odds ratio, 1.57; P = .02).
Researchers reported the results of a trial in 660 infants with a mean gestational age of 26.6 weeks, who were randomized to a platelet infusion either at a high platelet–count threshold of 50,000/mm3 or a low threshold of 25,000/mm3.
“Although retrospective studies have suggested that platelet transfusions may cause harm in neonates independently of the disease process, data from randomized controlled trials to support this are lacking,” Anna Curley, MD, of the National Maternity Hospital in Dublin and her coauthors reported in the New England Journal of Medicine.
The rates of minor or worse bleeding were similar between the two groups, and the percentage of infants surviving with bronchopulmonary dysplasia at 36 weeks of corrected age was higher in the high-threshold group (63% vs. 54%; OR, 1.54).
The rates of serious adverse events, not including major bleeding, were similar between the high- and low-threshold groups.
The outcomes of transfusions were not influenced by other factors such as intrauterine growth restriction, gestational age, or postnatal age at randomization.
“Our trial highlights the importance of trials of platelet transfusion involving patients with conditions other than haematological malignancies,” the authors wrote.
However they acknowledged that the reasons for the differences in mortality and outcomes between the two study groups were unknown.
“Platelets have recognized immunological and inflammatory effects, outside of effects on hemostasis,” they wrote. “The effect of transfusing adult platelets to a delicately balance neonatal hemostatic system with relatively hypofunctional platelets is also poorly understood.”
The study was supported by the National Health Service Blood and Transplant Research and Development Committee and other foundations. Authors reported financial disclosures related to Sanquin and Cerus.
SOURCE: Curley A et al. N Engl J Med. 2018 Nov 2. doi: 10.1056/NEJMoa1807320.
A lower threshold for platelet transfusions in preterm infants with severe thrombocytopenia is associated with significantly lower incidence of death and major bleeding, compared with a higher threshold, a new study suggests.
A new major bleeding episode or death occurred in 26% of infants in the high-threshold group, compared with 19% in the low-threshold group, representing a 57% higher risk of poor outcomes even after researchers adjusted for gestational age and intrauterine growth restriction (odds ratio, 1.57; P = .02).
Researchers reported the results of a trial in 660 infants with a mean gestational age of 26.6 weeks, who were randomized to a platelet infusion either at a high platelet–count threshold of 50,000/mm3 or a low threshold of 25,000/mm3.
“Although retrospective studies have suggested that platelet transfusions may cause harm in neonates independently of the disease process, data from randomized controlled trials to support this are lacking,” Anna Curley, MD, of the National Maternity Hospital in Dublin and her coauthors reported in the New England Journal of Medicine.
The rates of minor or worse bleeding were similar between the two groups, and the percentage of infants surviving with bronchopulmonary dysplasia at 36 weeks of corrected age was higher in the high-threshold group (63% vs. 54%; OR, 1.54).
The rates of serious adverse events, not including major bleeding, were similar between the high- and low-threshold groups.
The outcomes of transfusions were not influenced by other factors such as intrauterine growth restriction, gestational age, or postnatal age at randomization.
“Our trial highlights the importance of trials of platelet transfusion involving patients with conditions other than haematological malignancies,” the authors wrote.
However they acknowledged that the reasons for the differences in mortality and outcomes between the two study groups were unknown.
“Platelets have recognized immunological and inflammatory effects, outside of effects on hemostasis,” they wrote. “The effect of transfusing adult platelets to a delicately balance neonatal hemostatic system with relatively hypofunctional platelets is also poorly understood.”
The study was supported by the National Health Service Blood and Transplant Research and Development Committee and other foundations. Authors reported financial disclosures related to Sanquin and Cerus.
SOURCE: Curley A et al. N Engl J Med. 2018 Nov 2. doi: 10.1056/NEJMoa1807320.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point:
Major finding: The odds of new major bleeding or death were 57% higher in preterm infants who received a platelet transfusion at a higher threshold of 50,000 per mm3 than at a lower threshold of 25,000 mm3 (P = .02).Study details: Randomized study in 660 preterm infants with severe thrombocytopenia.
Disclosures: The study was supported by the National Health Service Blood and Transplant Research and Development Committee, and other foundations. Authors reported financial disclosures related to Sanquin and Cerus.
Source: Curley A et al. N Engl J Med. 2018 Nov 2. doi: 10.1056/NEJMoa1807320.
Primary care needs pile up for sickle cell patients
BETHESDA, MD. – With many people surviving well into adulthood with sickle cell disease (SCD) because of advances in treatment, there’s a strong need for more primary care to address chronic conditions, such as obesity and the complications of the blood disorder, researchers said.
People who have lived for decades with SCD may be at higher risk for renal disease while still needing the same routine vaccinations and screening for colon, prostate, and lung cancer that the general population receives, Sophie Lanzkron, MD, of Johns Hopkins University, Baltimore, said at Sickle Cell in Focus, a conference held by the National Institutes of Health.
And obesity is an additional a concern in treating people with SCD, Dr. Lanzkron said.
“It is really hard to have a conversation for 20 minutes with a patient about pain, talk about what you’re going to do about their sickle cell disease, and then address all of” their routine health needs, she said.
People with SCD seem less likely to get renal transplants, but those with end-stage kidney disease should be encouraged to be evaluated for them, Dr. Lanzkron advised.
Older data had suggested that patients with SCD who underwent renal transplant didn’t do as well as everyone else who underwent the procedure, but new data have changed that approach. “There’s some additional data in the modern era suggesting that the outcomes for people who undergo transplant with sickle cell disease are the same as for those who undergo it with diabetes,” Dr. Lanzkron said.
She highlighted one newer study in which the kidney transplant survival rate was 73.1% among individuals with SCD, compared with 74.1% for those with diabetes (Nephrol Dial Transplant. 2013 Apr;28[4]:1039-46).

It’s unclear what the average life expectancy is at this time for someone with SCD, Dr. Lanzkron said. Research looking at death certificate data suggests a median age of death in the mid-40s, but there are limitations to this work given it may exclude many older people with SCD, she said.
“We’re hopeful that people are living into their 50s and 60s, but we don’t have a lot of great data,” she said.
One of the organizers of the NIH conference said she hoped that Dr. Lanzkron’s presentation would draw attention to the need for primary care for people with SCD. Maintaining a healthy lifestyle is particularly important for this group because they likely have had complications from the disease, as well as issues seen with normal aging, Swee Lay Thein, MBBS, of the National Heart, Lung, and Blood Institute, said in an interview.
“This is a key message for many patients with sickle cell disease,” Dr. Thein said. “It’s important to hook up with a primary care physician.”
Dr. Thein cited a recent paper, which reported on four people who had lived into their 80s with sickle cell disease. The paper said their longevity was aided by factors such as being nonsmokers, abstaining from alcohol or drinking it only on occasionally, and maintaining a normal body mass index (Blood. 2016 Nov 10;128[19]:2367-9).
Additionally, the patients had close ties with relatives. The paper said that one patient was married with a helpful husband. Others in this octogenarian set had maintained close ties with their children.
“A common factor for all of the four patients in their 80s was that they had a healthy lifestyle and very strong family support,” Dr. Thein said.
Dr. Lanzkron has been an investigator for trials sponsored by Pfizer, Global Blood Therapeutics, and Ironwood.
BETHESDA, MD. – With many people surviving well into adulthood with sickle cell disease (SCD) because of advances in treatment, there’s a strong need for more primary care to address chronic conditions, such as obesity and the complications of the blood disorder, researchers said.
People who have lived for decades with SCD may be at higher risk for renal disease while still needing the same routine vaccinations and screening for colon, prostate, and lung cancer that the general population receives, Sophie Lanzkron, MD, of Johns Hopkins University, Baltimore, said at Sickle Cell in Focus, a conference held by the National Institutes of Health.
And obesity is an additional a concern in treating people with SCD, Dr. Lanzkron said.
“It is really hard to have a conversation for 20 minutes with a patient about pain, talk about what you’re going to do about their sickle cell disease, and then address all of” their routine health needs, she said.
People with SCD seem less likely to get renal transplants, but those with end-stage kidney disease should be encouraged to be evaluated for them, Dr. Lanzkron advised.
Older data had suggested that patients with SCD who underwent renal transplant didn’t do as well as everyone else who underwent the procedure, but new data have changed that approach. “There’s some additional data in the modern era suggesting that the outcomes for people who undergo transplant with sickle cell disease are the same as for those who undergo it with diabetes,” Dr. Lanzkron said.
She highlighted one newer study in which the kidney transplant survival rate was 73.1% among individuals with SCD, compared with 74.1% for those with diabetes (Nephrol Dial Transplant. 2013 Apr;28[4]:1039-46).
It’s unclear what the average life expectancy is at this time for someone with SCD, Dr. Lanzkron said. Research looking at death certificate data suggests a median age of death in the mid-40s, but there are limitations to this work given it may exclude many older people with SCD, she said.
“We’re hopeful that people are living into their 50s and 60s, but we don’t have a lot of great data,” she said.
One of the organizers of the NIH conference said she hoped that Dr. Lanzkron’s presentation would draw attention to the need for primary care for people with SCD. Maintaining a healthy lifestyle is particularly important for this group because they likely have had complications from the disease, as well as issues seen with normal aging, Swee Lay Thein, MBBS, of the National Heart, Lung, and Blood Institute, said in an interview.
“This is a key message for many patients with sickle cell disease,” Dr. Thein said. “It’s important to hook up with a primary care physician.”
Dr. Thein cited a recent paper, which reported on four people who had lived into their 80s with sickle cell disease. The paper said their longevity was aided by factors such as being nonsmokers, abstaining from alcohol or drinking it only on occasionally, and maintaining a normal body mass index (Blood. 2016 Nov 10;128[19]:2367-9).
Additionally, the patients had close ties with relatives. The paper said that one patient was married with a helpful husband. Others in this octogenarian set had maintained close ties with their children.
“A common factor for all of the four patients in their 80s was that they had a healthy lifestyle and very strong family support,” Dr. Thein said.
Dr. Lanzkron has been an investigator for trials sponsored by Pfizer, Global Blood Therapeutics, and Ironwood.
BETHESDA, MD. – With many people surviving well into adulthood with sickle cell disease (SCD) because of advances in treatment, there’s a strong need for more primary care to address chronic conditions, such as obesity and the complications of the blood disorder, researchers said.
People who have lived for decades with SCD may be at higher risk for renal disease while still needing the same routine vaccinations and screening for colon, prostate, and lung cancer that the general population receives, Sophie Lanzkron, MD, of Johns Hopkins University, Baltimore, said at Sickle Cell in Focus, a conference held by the National Institutes of Health.
And obesity is an additional a concern in treating people with SCD, Dr. Lanzkron said.
“It is really hard to have a conversation for 20 minutes with a patient about pain, talk about what you’re going to do about their sickle cell disease, and then address all of” their routine health needs, she said.
People with SCD seem less likely to get renal transplants, but those with end-stage kidney disease should be encouraged to be evaluated for them, Dr. Lanzkron advised.
Older data had suggested that patients with SCD who underwent renal transplant didn’t do as well as everyone else who underwent the procedure, but new data have changed that approach. “There’s some additional data in the modern era suggesting that the outcomes for people who undergo transplant with sickle cell disease are the same as for those who undergo it with diabetes,” Dr. Lanzkron said.
She highlighted one newer study in which the kidney transplant survival rate was 73.1% among individuals with SCD, compared with 74.1% for those with diabetes (Nephrol Dial Transplant. 2013 Apr;28[4]:1039-46).
It’s unclear what the average life expectancy is at this time for someone with SCD, Dr. Lanzkron said. Research looking at death certificate data suggests a median age of death in the mid-40s, but there are limitations to this work given it may exclude many older people with SCD, she said.
“We’re hopeful that people are living into their 50s and 60s, but we don’t have a lot of great data,” she said.
One of the organizers of the NIH conference said she hoped that Dr. Lanzkron’s presentation would draw attention to the need for primary care for people with SCD. Maintaining a healthy lifestyle is particularly important for this group because they likely have had complications from the disease, as well as issues seen with normal aging, Swee Lay Thein, MBBS, of the National Heart, Lung, and Blood Institute, said in an interview.
“This is a key message for many patients with sickle cell disease,” Dr. Thein said. “It’s important to hook up with a primary care physician.”
Dr. Thein cited a recent paper, which reported on four people who had lived into their 80s with sickle cell disease. The paper said their longevity was aided by factors such as being nonsmokers, abstaining from alcohol or drinking it only on occasionally, and maintaining a normal body mass index (Blood. 2016 Nov 10;128[19]:2367-9).
Additionally, the patients had close ties with relatives. The paper said that one patient was married with a helpful husband. Others in this octogenarian set had maintained close ties with their children.
“A common factor for all of the four patients in their 80s was that they had a healthy lifestyle and very strong family support,” Dr. Thein said.
Dr. Lanzkron has been an investigator for trials sponsored by Pfizer, Global Blood Therapeutics, and Ironwood.
EXPERT ANALYSIS FROM SICKLE CELL IN FOCUS
Prescription Drug Benefits and Survival in Myeloma Among Medicare Beneficiaries
Study Overview
Objective. To investigate the relationship between prescription drug coverage, receipt of active myeloma therapy, and overall survival (OS) among Medicare beneficiaries with multiple myeloma.
Design. Case-control and retrospective cohort archival data research.
Setting and participants. Authors examined SEER-Medicare registry and extracted patients with histologically confirmed multiple myeloma diagnosed in the period 2006 to 2011. Availability of complete Medicare part A/B claims from 1 year before diagnosis until December 2013 was required for analysis. Patients with Medicare advantage or managed care plans did not have claims data available and hence were excluded. Beneficiaries with a diagnosis of diffuse large B-cell lymphoma (DLBCL), who typically receive parenteral drugs for lymphoma therapy, were used as a control cohort.
Main outcome measures. Association between prescription drug coverage status and OS was the primary outcome measure of interest. Authors reported 3-year restricted survival time (RMST) ratios to compare OS among the beneficiaries with different prescription drug coverages. Receipt of active myeloma therapy among beneficiaries was also studied. Relative risk, adjusting for patient and disease-related characteristics, was reported to examine receipt of active myeloma therapy.
Results. Records of 9755 Medicare beneficiaries were evaluated. Of these, 1460 (15%) had no prescription coverage at diagnosis, 3283 (34%) had part D plan prescription benefits, 3607 (37%) had sponsored prescription coverage through an employer, federal employer, or veterans plan, and 1405 (14%) had a Medicaid prescription plan. Beneficiaries without coverage had fewer comorbidities, including anemia, neuropathy, or renal disease, than those with part D prescription coverage or Medicaid. Of those without any prescription drug coverage, 41% obtained prescription plan coverage after diagnosis of myeloma by the following January. Conversely, only 19% of patients with DLBCL and no coverage obtained a prescription plan.
Patients with myeloma were followed for 4.9 years and median survival was 2.3 years, with a 3-year OS rate of 43.1% (95% confidence interval [CI], 42.1%-44.1%). Relative to the group without coverage, survival was 16% longer in the Medicare part D group and sponsored plan group (RMST 1.16; 95% CI, 1.12-1.21). Medicaid/Medicare dual beneficiaries had worse OS in both myeloma and DLBCL consistent with poor performance status and unfavorable baseline comorbidities. However, among patients with myeloma, Medicaid/Medicare dual beneficiaries had better survival (RMST 1.08; 95% CI, 1.03-1.13) compared to the group without coverage. There was no difference in OS for those with or without prescription drug coverage in the DLBCL cohort.
There were significant differences in treatment of myeloma based on types of prescription drug coverage. Due to increasing use of bortezomib following its approval by the U.S. Food and Drug Administration (FDA), parenteral chemotherapy use doubled from 24% to 48% from 2006 to 2011, and utility of active myeloma care increased from 88% to 91%. Medicare part D plan enrollees were 6% more likely to receive active myeloma care, and both Medicaid group and sponsored plan group beneficiaries were equally likely to receive active myeloma care compared to beneficiaries without prescription coverage. Medicaid enrollees were less likely to receive parenteral therapy.
Conclusion. Medicare beneficiaries with prescription drug coverage and multiple myeloma are more likely to receive myeloma therapy and have longer OS compared to those without prescription drug coverage.
Commentary
First-line therapy of multiple myeloma has evolved over the past 2 decades. Parenteral agents such as vincristine, adriamycin, dexamethasone, and cyclophosphamide and oral therapy with melphalan and prednisone were the mainstay of treatment in the past. In the past decade, the arrival of oral therapy using thalidomide or lenalidomide and parenteral therapy using bortezomib has increased OS in patients with myeloma. Most recently, a combination of lenalidomide, bortezomib, and dexamethasone has emerged as one of the frontline therapies of choice.1 Incorporation of bortezomib or an oral immunomodulatory drug is almost universal in first-line therapy.
Oral antineoplastic therapy is increasingly being approved by the FDA and being utilized in the community. During the period 2016-2018, more than half the new FDA-approved oncology drugs were in oral formulation.2 As such, access to these agents is crucial in cancer therapy. The cost of oral therapy in patients without prescription drug coverage is sometimes more than $10,000 per month, which represents a significant impediment to its adoption. Forty-three states and Washington, DC, have enacted drug parity laws that require patients to pay no more for an oral cancer treatment than they would for an infusion. However, currently there is no such federal law, and Medicare beneficiaries must participate either through part D, state Medicaid, or a sponsored program to obtain prescription drug coverage. Despite being enrolled in part D, many beneficiaries fall into the “doughnut hole” (the requirement of Part D beneficiaries with high prescription drug expenses to pay more once the total cost of their medicines reaches a certain threshold) for prescription drugs at the time of need. From 2019 onward, enrollees will see significant, yet sometimes still insufficient, coverage benefits due to ending of the doughnut hole.3 Only a very limited number of oral chemotherapy agents are covered through Medicare part B, and of those covered, only oral melphalan is used for myeloma.
The authors have acknowledged multiple limitations of their investigation, including possible unobserved clinical differences between beneficiaries. SEER-Medicare registry has limitations in obtaining individual level data and may not contain specific results of cytogenetics, laboratory risk markers, and response to therapy, which are important to determine overall outcome. A prospective evaluation may be more suitable to assess these variables independently or through a multivariate analysis in determining receipt of therapy on OS, although such a study is currently not feasible.
The indicator of active myeloma care was defined as 2 or more outpatient physician visits or receipt of parenteral chemotherapy. This definition is somewhat suboptimal, as often patients with myeloma are under surveillance and may not necessarily be receiving active treatment. Moreover, the exact prescription pattern of lenalidomide, the most active first-line oral therapy, could not be captured from this retrospective registry review. Therefore, definitive conclusions regarding use of lenalidomide and thalidomide and receipt of therapy in this population cannot be made.
A significant improvement in OS has been established using maintenance lenalidomide following high-dose chemotherapy and stem cell transplantation.4 Only 5% of this study population received stem cell transplantation. This may be due to a median age of 77 years at diagnosis in the group studied, higher than the 66 to 70 years previously published.5 Stem cell transplantation is now commonly being used even in the older population. The 3-year survival of 83% following stem cell transplantation in myeloma patients aged 75 to 84 years was nearly identical to that of the younger population.6 Since stem cell transplantation is feasible in older Medicare beneficiaries and maintenance lenalidomide for 2 years following transplant improves survival, the option of providing maintenance therapy with oral lenalidomide must be made available to Medicare beneficiaries. Due to a very limited use of transplantation in this study, the impact of oral lenalidomide maintenance in OS cannot be judged.
Of the patients reviewed in this study, 6% had a listed diagnosis of plasmacytoma. These individuals typically are treated with radiation therapy only. It is unclear if these patients also received any systemic myeloma therapy or if they ever progressed to myeloma. Availability of prescription drug coverage may not be relevant to this group. Also, the authors reported that part D participants were less likely to receive classic cytotoxic chemotherapy. This may be somewhat irrelevant in Medicare beneficiaries with a median age of 77 years for current practice, as frontline induction with old classic cytotoxic chemotherapy is less commonly used in this population.
Investigators have appropriately recognized a lack of ability to discern whether inferior survival in the group without prescription drug coverage was the result of not receiving therapy at all or inability to receive oral immunomodulatory drugs. There would have been little reason for not proceeding to parenteral therapy. As noted, 41% of beneficiaries without coverage at diagnosis subsequently obtained coverage but continued to have significantly worse survival. Cause of death, including whether related to myeloma, was not reported. The authors suggest that early separation of survival curves could therefore be reflective of suboptimal first-line therapy that lacked oral immunomodulatory drugs. During the study period 2006-2011, first-line use of lenalidomide was common.
Median survival of patients with myeloma in this study was only 27 months. According to the American Cancer Society, in 2018 median survival for stage I myeloma has not been reached, stage II myeloma is 83 months, and stage III myeloma is 43 months. A robust and dynamic landscape in myeloma therapy prevents a clear attribution to individual agents, whether oral or parenteral, in improving OS. Thus, 3-year RMST, while appropriate for 2006-2011, may not be relevant today.
Applications for Clinical Practice
The oncology community routinely encounters difficulty in initiating therapy using oral agents rapidly after diagnosis of myeloma. The retrospective data analyzed in the current study suggests that delay in initiating or unavailability of oral agents may adversely impact OS. The common approach of initiating parenteral therapy while awaiting approvals from payers or charity programs and subsequently adding oral therapy when available has not been studied in assessing OS. The oncology community should initiate plans to obtain prescription drug coverage through either Medicare part D, Medicaid, a sponsored plan, or financial assistance charity programs as soon as possible after diagnosis of myeloma. Moreover, continuation of these prescription drug plans should be strongly considered throughout the course of myeloma, as subsequent lines of treatment will quite likely involve other active and approved oral agents, such as pomalidomide, ixazomib, and panobinostat, besides other supportive therapy.
One of the mechanisms to obtain prescription drug coverage includes enrollment in state Medicaid programs for those who are eligible. Currently, 17 states have not yet adopted Medicaid expansion under the Affordable Care Act. Expansion of Medicaid in these states could increase availability of prescription drug benefits. In this study, 15.8% of Medicare and Medicaid dual enrollees with access to oral agents at low or no cost did not receive myeloma care, slightly higher than the 13.1% with no prescription drug coverage. Lower utilization in this population may be explained based on differences in comorbidities or socioeconomic conditions rather than availability of a prescription plan.
The incidence of myeloma is expected to be higher in Medicare beneficiaries, and according to one estimate, in 2030 and beyond nearly 75% of diagnosed myeloma patients will be aged 64 to 84 years, an increase from nearly 66% today.7 Changing demographics, increasing oral therapy options, and patient convenience demand attention to providing prescription drug coverage to all Medicare beneficiaries. This study lends support to that demand.
—Rakesh Gaur, MD, MPH, FACP, Cancer and Blood Center at Kansas Institute of Medicine, Lenexa, KS
1. Durie BG, Hoering A, Abidi MH, et al. Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem-cell transplant (SWOG S0777): a randomized, open-label, phase 3 trial. Lancet. 2017;389(10068):519-527.
2. U.S. Food and Drug Administration. Hematology/Oncology (Cancer) Approvals & Safety Notifications. www.fda.gov/drugs/informationondrugs/approveddrugs/ucm279174.htm. Accessed October 11, 2018.
3. Dusetzina SB, Keating NL. Mind the gap: Why closing the doughnut hole is insufficient for increasing Medicare beneficiary access to oral chemotherapy. J Clin Oncol. 2016;34:375-380.
4. McCarthy PL, Holstein SA, Petrucci MT, et al. Lenalidomide maintenance after autologous stem-cell transplantation in newly diagnosed multiple myeloma: a meta-analysis. J Clin Oncol. 2017;35:3279-3289.
5. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78:21–33.
6. Dong N, McKiernan P, Samuel D, et al. Autologous stem cell transplantation in multiple myeloma patients over age 75 [abstract]. J Clin Oncol. 2018;36(suppl): 8025.
7. Rosenberg PS, Barker KA, Anderson WF. Future distribution of multiple myeloma in the United States by sex, age, and race/ethnicity. Blood. 2015;125:410–412.
Study Overview
Objective. To investigate the relationship between prescription drug coverage, receipt of active myeloma therapy, and overall survival (OS) among Medicare beneficiaries with multiple myeloma.
Design. Case-control and retrospective cohort archival data research.
Setting and participants. Authors examined SEER-Medicare registry and extracted patients with histologically confirmed multiple myeloma diagnosed in the period 2006 to 2011. Availability of complete Medicare part A/B claims from 1 year before diagnosis until December 2013 was required for analysis. Patients with Medicare advantage or managed care plans did not have claims data available and hence were excluded. Beneficiaries with a diagnosis of diffuse large B-cell lymphoma (DLBCL), who typically receive parenteral drugs for lymphoma therapy, were used as a control cohort.
Main outcome measures. Association between prescription drug coverage status and OS was the primary outcome measure of interest. Authors reported 3-year restricted survival time (RMST) ratios to compare OS among the beneficiaries with different prescription drug coverages. Receipt of active myeloma therapy among beneficiaries was also studied. Relative risk, adjusting for patient and disease-related characteristics, was reported to examine receipt of active myeloma therapy.
Results. Records of 9755 Medicare beneficiaries were evaluated. Of these, 1460 (15%) had no prescription coverage at diagnosis, 3283 (34%) had part D plan prescription benefits, 3607 (37%) had sponsored prescription coverage through an employer, federal employer, or veterans plan, and 1405 (14%) had a Medicaid prescription plan. Beneficiaries without coverage had fewer comorbidities, including anemia, neuropathy, or renal disease, than those with part D prescription coverage or Medicaid. Of those without any prescription drug coverage, 41% obtained prescription plan coverage after diagnosis of myeloma by the following January. Conversely, only 19% of patients with DLBCL and no coverage obtained a prescription plan.
Patients with myeloma were followed for 4.9 years and median survival was 2.3 years, with a 3-year OS rate of 43.1% (95% confidence interval [CI], 42.1%-44.1%). Relative to the group without coverage, survival was 16% longer in the Medicare part D group and sponsored plan group (RMST 1.16; 95% CI, 1.12-1.21). Medicaid/Medicare dual beneficiaries had worse OS in both myeloma and DLBCL consistent with poor performance status and unfavorable baseline comorbidities. However, among patients with myeloma, Medicaid/Medicare dual beneficiaries had better survival (RMST 1.08; 95% CI, 1.03-1.13) compared to the group without coverage. There was no difference in OS for those with or without prescription drug coverage in the DLBCL cohort.
There were significant differences in treatment of myeloma based on types of prescription drug coverage. Due to increasing use of bortezomib following its approval by the U.S. Food and Drug Administration (FDA), parenteral chemotherapy use doubled from 24% to 48% from 2006 to 2011, and utility of active myeloma care increased from 88% to 91%. Medicare part D plan enrollees were 6% more likely to receive active myeloma care, and both Medicaid group and sponsored plan group beneficiaries were equally likely to receive active myeloma care compared to beneficiaries without prescription coverage. Medicaid enrollees were less likely to receive parenteral therapy.
Conclusion. Medicare beneficiaries with prescription drug coverage and multiple myeloma are more likely to receive myeloma therapy and have longer OS compared to those without prescription drug coverage.
Commentary
First-line therapy of multiple myeloma has evolved over the past 2 decades. Parenteral agents such as vincristine, adriamycin, dexamethasone, and cyclophosphamide and oral therapy with melphalan and prednisone were the mainstay of treatment in the past. In the past decade, the arrival of oral therapy using thalidomide or lenalidomide and parenteral therapy using bortezomib has increased OS in patients with myeloma. Most recently, a combination of lenalidomide, bortezomib, and dexamethasone has emerged as one of the frontline therapies of choice.1 Incorporation of bortezomib or an oral immunomodulatory drug is almost universal in first-line therapy.
Oral antineoplastic therapy is increasingly being approved by the FDA and being utilized in the community. During the period 2016-2018, more than half the new FDA-approved oncology drugs were in oral formulation.2 As such, access to these agents is crucial in cancer therapy. The cost of oral therapy in patients without prescription drug coverage is sometimes more than $10,000 per month, which represents a significant impediment to its adoption. Forty-three states and Washington, DC, have enacted drug parity laws that require patients to pay no more for an oral cancer treatment than they would for an infusion. However, currently there is no such federal law, and Medicare beneficiaries must participate either through part D, state Medicaid, or a sponsored program to obtain prescription drug coverage. Despite being enrolled in part D, many beneficiaries fall into the “doughnut hole” (the requirement of Part D beneficiaries with high prescription drug expenses to pay more once the total cost of their medicines reaches a certain threshold) for prescription drugs at the time of need. From 2019 onward, enrollees will see significant, yet sometimes still insufficient, coverage benefits due to ending of the doughnut hole.3 Only a very limited number of oral chemotherapy agents are covered through Medicare part B, and of those covered, only oral melphalan is used for myeloma.
The authors have acknowledged multiple limitations of their investigation, including possible unobserved clinical differences between beneficiaries. SEER-Medicare registry has limitations in obtaining individual level data and may not contain specific results of cytogenetics, laboratory risk markers, and response to therapy, which are important to determine overall outcome. A prospective evaluation may be more suitable to assess these variables independently or through a multivariate analysis in determining receipt of therapy on OS, although such a study is currently not feasible.
The indicator of active myeloma care was defined as 2 or more outpatient physician visits or receipt of parenteral chemotherapy. This definition is somewhat suboptimal, as often patients with myeloma are under surveillance and may not necessarily be receiving active treatment. Moreover, the exact prescription pattern of lenalidomide, the most active first-line oral therapy, could not be captured from this retrospective registry review. Therefore, definitive conclusions regarding use of lenalidomide and thalidomide and receipt of therapy in this population cannot be made.
A significant improvement in OS has been established using maintenance lenalidomide following high-dose chemotherapy and stem cell transplantation.4 Only 5% of this study population received stem cell transplantation. This may be due to a median age of 77 years at diagnosis in the group studied, higher than the 66 to 70 years previously published.5 Stem cell transplantation is now commonly being used even in the older population. The 3-year survival of 83% following stem cell transplantation in myeloma patients aged 75 to 84 years was nearly identical to that of the younger population.6 Since stem cell transplantation is feasible in older Medicare beneficiaries and maintenance lenalidomide for 2 years following transplant improves survival, the option of providing maintenance therapy with oral lenalidomide must be made available to Medicare beneficiaries. Due to a very limited use of transplantation in this study, the impact of oral lenalidomide maintenance in OS cannot be judged.
Of the patients reviewed in this study, 6% had a listed diagnosis of plasmacytoma. These individuals typically are treated with radiation therapy only. It is unclear if these patients also received any systemic myeloma therapy or if they ever progressed to myeloma. Availability of prescription drug coverage may not be relevant to this group. Also, the authors reported that part D participants were less likely to receive classic cytotoxic chemotherapy. This may be somewhat irrelevant in Medicare beneficiaries with a median age of 77 years for current practice, as frontline induction with old classic cytotoxic chemotherapy is less commonly used in this population.
Investigators have appropriately recognized a lack of ability to discern whether inferior survival in the group without prescription drug coverage was the result of not receiving therapy at all or inability to receive oral immunomodulatory drugs. There would have been little reason for not proceeding to parenteral therapy. As noted, 41% of beneficiaries without coverage at diagnosis subsequently obtained coverage but continued to have significantly worse survival. Cause of death, including whether related to myeloma, was not reported. The authors suggest that early separation of survival curves could therefore be reflective of suboptimal first-line therapy that lacked oral immunomodulatory drugs. During the study period 2006-2011, first-line use of lenalidomide was common.
Median survival of patients with myeloma in this study was only 27 months. According to the American Cancer Society, in 2018 median survival for stage I myeloma has not been reached, stage II myeloma is 83 months, and stage III myeloma is 43 months. A robust and dynamic landscape in myeloma therapy prevents a clear attribution to individual agents, whether oral or parenteral, in improving OS. Thus, 3-year RMST, while appropriate for 2006-2011, may not be relevant today.
Applications for Clinical Practice
The oncology community routinely encounters difficulty in initiating therapy using oral agents rapidly after diagnosis of myeloma. The retrospective data analyzed in the current study suggests that delay in initiating or unavailability of oral agents may adversely impact OS. The common approach of initiating parenteral therapy while awaiting approvals from payers or charity programs and subsequently adding oral therapy when available has not been studied in assessing OS. The oncology community should initiate plans to obtain prescription drug coverage through either Medicare part D, Medicaid, a sponsored plan, or financial assistance charity programs as soon as possible after diagnosis of myeloma. Moreover, continuation of these prescription drug plans should be strongly considered throughout the course of myeloma, as subsequent lines of treatment will quite likely involve other active and approved oral agents, such as pomalidomide, ixazomib, and panobinostat, besides other supportive therapy.
One of the mechanisms to obtain prescription drug coverage includes enrollment in state Medicaid programs for those who are eligible. Currently, 17 states have not yet adopted Medicaid expansion under the Affordable Care Act. Expansion of Medicaid in these states could increase availability of prescription drug benefits. In this study, 15.8% of Medicare and Medicaid dual enrollees with access to oral agents at low or no cost did not receive myeloma care, slightly higher than the 13.1% with no prescription drug coverage. Lower utilization in this population may be explained based on differences in comorbidities or socioeconomic conditions rather than availability of a prescription plan.
The incidence of myeloma is expected to be higher in Medicare beneficiaries, and according to one estimate, in 2030 and beyond nearly 75% of diagnosed myeloma patients will be aged 64 to 84 years, an increase from nearly 66% today.7 Changing demographics, increasing oral therapy options, and patient convenience demand attention to providing prescription drug coverage to all Medicare beneficiaries. This study lends support to that demand.
—Rakesh Gaur, MD, MPH, FACP, Cancer and Blood Center at Kansas Institute of Medicine, Lenexa, KS
Study Overview
Objective. To investigate the relationship between prescription drug coverage, receipt of active myeloma therapy, and overall survival (OS) among Medicare beneficiaries with multiple myeloma.
Design. Case-control and retrospective cohort archival data research.
Setting and participants. Authors examined SEER-Medicare registry and extracted patients with histologically confirmed multiple myeloma diagnosed in the period 2006 to 2011. Availability of complete Medicare part A/B claims from 1 year before diagnosis until December 2013 was required for analysis. Patients with Medicare advantage or managed care plans did not have claims data available and hence were excluded. Beneficiaries with a diagnosis of diffuse large B-cell lymphoma (DLBCL), who typically receive parenteral drugs for lymphoma therapy, were used as a control cohort.
Main outcome measures. Association between prescription drug coverage status and OS was the primary outcome measure of interest. Authors reported 3-year restricted survival time (RMST) ratios to compare OS among the beneficiaries with different prescription drug coverages. Receipt of active myeloma therapy among beneficiaries was also studied. Relative risk, adjusting for patient and disease-related characteristics, was reported to examine receipt of active myeloma therapy.
Results. Records of 9755 Medicare beneficiaries were evaluated. Of these, 1460 (15%) had no prescription coverage at diagnosis, 3283 (34%) had part D plan prescription benefits, 3607 (37%) had sponsored prescription coverage through an employer, federal employer, or veterans plan, and 1405 (14%) had a Medicaid prescription plan. Beneficiaries without coverage had fewer comorbidities, including anemia, neuropathy, or renal disease, than those with part D prescription coverage or Medicaid. Of those without any prescription drug coverage, 41% obtained prescription plan coverage after diagnosis of myeloma by the following January. Conversely, only 19% of patients with DLBCL and no coverage obtained a prescription plan.
Patients with myeloma were followed for 4.9 years and median survival was 2.3 years, with a 3-year OS rate of 43.1% (95% confidence interval [CI], 42.1%-44.1%). Relative to the group without coverage, survival was 16% longer in the Medicare part D group and sponsored plan group (RMST 1.16; 95% CI, 1.12-1.21). Medicaid/Medicare dual beneficiaries had worse OS in both myeloma and DLBCL consistent with poor performance status and unfavorable baseline comorbidities. However, among patients with myeloma, Medicaid/Medicare dual beneficiaries had better survival (RMST 1.08; 95% CI, 1.03-1.13) compared to the group without coverage. There was no difference in OS for those with or without prescription drug coverage in the DLBCL cohort.
There were significant differences in treatment of myeloma based on types of prescription drug coverage. Due to increasing use of bortezomib following its approval by the U.S. Food and Drug Administration (FDA), parenteral chemotherapy use doubled from 24% to 48% from 2006 to 2011, and utility of active myeloma care increased from 88% to 91%. Medicare part D plan enrollees were 6% more likely to receive active myeloma care, and both Medicaid group and sponsored plan group beneficiaries were equally likely to receive active myeloma care compared to beneficiaries without prescription coverage. Medicaid enrollees were less likely to receive parenteral therapy.
Conclusion. Medicare beneficiaries with prescription drug coverage and multiple myeloma are more likely to receive myeloma therapy and have longer OS compared to those without prescription drug coverage.
Commentary
First-line therapy of multiple myeloma has evolved over the past 2 decades. Parenteral agents such as vincristine, adriamycin, dexamethasone, and cyclophosphamide and oral therapy with melphalan and prednisone were the mainstay of treatment in the past. In the past decade, the arrival of oral therapy using thalidomide or lenalidomide and parenteral therapy using bortezomib has increased OS in patients with myeloma. Most recently, a combination of lenalidomide, bortezomib, and dexamethasone has emerged as one of the frontline therapies of choice.1 Incorporation of bortezomib or an oral immunomodulatory drug is almost universal in first-line therapy.
Oral antineoplastic therapy is increasingly being approved by the FDA and being utilized in the community. During the period 2016-2018, more than half the new FDA-approved oncology drugs were in oral formulation.2 As such, access to these agents is crucial in cancer therapy. The cost of oral therapy in patients without prescription drug coverage is sometimes more than $10,000 per month, which represents a significant impediment to its adoption. Forty-three states and Washington, DC, have enacted drug parity laws that require patients to pay no more for an oral cancer treatment than they would for an infusion. However, currently there is no such federal law, and Medicare beneficiaries must participate either through part D, state Medicaid, or a sponsored program to obtain prescription drug coverage. Despite being enrolled in part D, many beneficiaries fall into the “doughnut hole” (the requirement of Part D beneficiaries with high prescription drug expenses to pay more once the total cost of their medicines reaches a certain threshold) for prescription drugs at the time of need. From 2019 onward, enrollees will see significant, yet sometimes still insufficient, coverage benefits due to ending of the doughnut hole.3 Only a very limited number of oral chemotherapy agents are covered through Medicare part B, and of those covered, only oral melphalan is used for myeloma.
The authors have acknowledged multiple limitations of their investigation, including possible unobserved clinical differences between beneficiaries. SEER-Medicare registry has limitations in obtaining individual level data and may not contain specific results of cytogenetics, laboratory risk markers, and response to therapy, which are important to determine overall outcome. A prospective evaluation may be more suitable to assess these variables independently or through a multivariate analysis in determining receipt of therapy on OS, although such a study is currently not feasible.
The indicator of active myeloma care was defined as 2 or more outpatient physician visits or receipt of parenteral chemotherapy. This definition is somewhat suboptimal, as often patients with myeloma are under surveillance and may not necessarily be receiving active treatment. Moreover, the exact prescription pattern of lenalidomide, the most active first-line oral therapy, could not be captured from this retrospective registry review. Therefore, definitive conclusions regarding use of lenalidomide and thalidomide and receipt of therapy in this population cannot be made.
A significant improvement in OS has been established using maintenance lenalidomide following high-dose chemotherapy and stem cell transplantation.4 Only 5% of this study population received stem cell transplantation. This may be due to a median age of 77 years at diagnosis in the group studied, higher than the 66 to 70 years previously published.5 Stem cell transplantation is now commonly being used even in the older population. The 3-year survival of 83% following stem cell transplantation in myeloma patients aged 75 to 84 years was nearly identical to that of the younger population.6 Since stem cell transplantation is feasible in older Medicare beneficiaries and maintenance lenalidomide for 2 years following transplant improves survival, the option of providing maintenance therapy with oral lenalidomide must be made available to Medicare beneficiaries. Due to a very limited use of transplantation in this study, the impact of oral lenalidomide maintenance in OS cannot be judged.
Of the patients reviewed in this study, 6% had a listed diagnosis of plasmacytoma. These individuals typically are treated with radiation therapy only. It is unclear if these patients also received any systemic myeloma therapy or if they ever progressed to myeloma. Availability of prescription drug coverage may not be relevant to this group. Also, the authors reported that part D participants were less likely to receive classic cytotoxic chemotherapy. This may be somewhat irrelevant in Medicare beneficiaries with a median age of 77 years for current practice, as frontline induction with old classic cytotoxic chemotherapy is less commonly used in this population.
Investigators have appropriately recognized a lack of ability to discern whether inferior survival in the group without prescription drug coverage was the result of not receiving therapy at all or inability to receive oral immunomodulatory drugs. There would have been little reason for not proceeding to parenteral therapy. As noted, 41% of beneficiaries without coverage at diagnosis subsequently obtained coverage but continued to have significantly worse survival. Cause of death, including whether related to myeloma, was not reported. The authors suggest that early separation of survival curves could therefore be reflective of suboptimal first-line therapy that lacked oral immunomodulatory drugs. During the study period 2006-2011, first-line use of lenalidomide was common.
Median survival of patients with myeloma in this study was only 27 months. According to the American Cancer Society, in 2018 median survival for stage I myeloma has not been reached, stage II myeloma is 83 months, and stage III myeloma is 43 months. A robust and dynamic landscape in myeloma therapy prevents a clear attribution to individual agents, whether oral or parenteral, in improving OS. Thus, 3-year RMST, while appropriate for 2006-2011, may not be relevant today.
Applications for Clinical Practice
The oncology community routinely encounters difficulty in initiating therapy using oral agents rapidly after diagnosis of myeloma. The retrospective data analyzed in the current study suggests that delay in initiating or unavailability of oral agents may adversely impact OS. The common approach of initiating parenteral therapy while awaiting approvals from payers or charity programs and subsequently adding oral therapy when available has not been studied in assessing OS. The oncology community should initiate plans to obtain prescription drug coverage through either Medicare part D, Medicaid, a sponsored plan, or financial assistance charity programs as soon as possible after diagnosis of myeloma. Moreover, continuation of these prescription drug plans should be strongly considered throughout the course of myeloma, as subsequent lines of treatment will quite likely involve other active and approved oral agents, such as pomalidomide, ixazomib, and panobinostat, besides other supportive therapy.
One of the mechanisms to obtain prescription drug coverage includes enrollment in state Medicaid programs for those who are eligible. Currently, 17 states have not yet adopted Medicaid expansion under the Affordable Care Act. Expansion of Medicaid in these states could increase availability of prescription drug benefits. In this study, 15.8% of Medicare and Medicaid dual enrollees with access to oral agents at low or no cost did not receive myeloma care, slightly higher than the 13.1% with no prescription drug coverage. Lower utilization in this population may be explained based on differences in comorbidities or socioeconomic conditions rather than availability of a prescription plan.
The incidence of myeloma is expected to be higher in Medicare beneficiaries, and according to one estimate, in 2030 and beyond nearly 75% of diagnosed myeloma patients will be aged 64 to 84 years, an increase from nearly 66% today.7 Changing demographics, increasing oral therapy options, and patient convenience demand attention to providing prescription drug coverage to all Medicare beneficiaries. This study lends support to that demand.
—Rakesh Gaur, MD, MPH, FACP, Cancer and Blood Center at Kansas Institute of Medicine, Lenexa, KS
1. Durie BG, Hoering A, Abidi MH, et al. Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem-cell transplant (SWOG S0777): a randomized, open-label, phase 3 trial. Lancet. 2017;389(10068):519-527.
2. U.S. Food and Drug Administration. Hematology/Oncology (Cancer) Approvals & Safety Notifications. www.fda.gov/drugs/informationondrugs/approveddrugs/ucm279174.htm. Accessed October 11, 2018.
3. Dusetzina SB, Keating NL. Mind the gap: Why closing the doughnut hole is insufficient for increasing Medicare beneficiary access to oral chemotherapy. J Clin Oncol. 2016;34:375-380.
4. McCarthy PL, Holstein SA, Petrucci MT, et al. Lenalidomide maintenance after autologous stem-cell transplantation in newly diagnosed multiple myeloma: a meta-analysis. J Clin Oncol. 2017;35:3279-3289.
5. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78:21–33.
6. Dong N, McKiernan P, Samuel D, et al. Autologous stem cell transplantation in multiple myeloma patients over age 75 [abstract]. J Clin Oncol. 2018;36(suppl): 8025.
7. Rosenberg PS, Barker KA, Anderson WF. Future distribution of multiple myeloma in the United States by sex, age, and race/ethnicity. Blood. 2015;125:410–412.
1. Durie BG, Hoering A, Abidi MH, et al. Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem-cell transplant (SWOG S0777): a randomized, open-label, phase 3 trial. Lancet. 2017;389(10068):519-527.
2. U.S. Food and Drug Administration. Hematology/Oncology (Cancer) Approvals & Safety Notifications. www.fda.gov/drugs/informationondrugs/approveddrugs/ucm279174.htm. Accessed October 11, 2018.
3. Dusetzina SB, Keating NL. Mind the gap: Why closing the doughnut hole is insufficient for increasing Medicare beneficiary access to oral chemotherapy. J Clin Oncol. 2016;34:375-380.
4. McCarthy PL, Holstein SA, Petrucci MT, et al. Lenalidomide maintenance after autologous stem-cell transplantation in newly diagnosed multiple myeloma: a meta-analysis. J Clin Oncol. 2017;35:3279-3289.
5. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78:21–33.
6. Dong N, McKiernan P, Samuel D, et al. Autologous stem cell transplantation in multiple myeloma patients over age 75 [abstract]. J Clin Oncol. 2018;36(suppl): 8025.
7. Rosenberg PS, Barker KA, Anderson WF. Future distribution of multiple myeloma in the United States by sex, age, and race/ethnicity. Blood. 2015;125:410–412.