User login
LVADs for Severe Heart Failure Gradually Take Hold
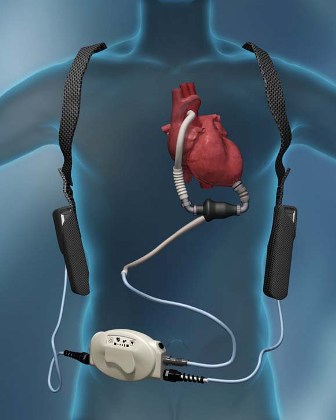
A sea change in management of severe heart failure began two and a half years ago, in January 2010, when the Food and Drug Administration approved U.S. marketing of the HeartMate II continuous-flow, left ventricular assist device for destination therapy. Patients, cardiologists, and cardiothoracic surgeons at last had a reasonably durable, effective, and relatively safe alternative to heart transplant to offer patients at the end stage of deteriorating heart function.
The Heart Mate II quickly supplanted the prior-generation, pulsatile-flow devices, and 2010 also saw a 10-fold spike in the number of left ventricular assist devices (LVADs) placed in U.S. patients as destination therapy.
During the first half of 2011, 6-month survival among U.S. patients with a continuous-flow LVAD (which means the HeartMate II, the only continuous-flow device on the U.S. market) was 89%, and 12-month survival during 2010 was 81% (J. Heart Lung Transplant 2012;31:117-26, putting the continuous-flow LVAD in at least the same ballpark as heart transplant, which has a 5-year survival of about 80% and a median survival of about 10 years in U.S. patients.
But despite estimates that many thousands, if not tens of thousands, of Americans meet the clinical criteria to qualify for placement of an LVAD, the reality is that in the 30 months since the HeartMate II became available for routine destination therapy through mid-2012, only about 4,600 were placed in U.S. patients. In addition, just slightly more than a third of those patients, roughly 1,700, received their LVAD explicitly for destination therapy.
Even in 2012, a majority of patients who received an LVAD got their device with the understanding that it was as a bridge to transplant, an LVAD placed with temporary intent to help patients survive and improve clinically until their LVAD could be swapped out and replaced by a transplanted heart.
LVAD Numbers Up, but Still Low
If the availability of the HeartMate II for destination therapy in routine practice was a revolution for the management of many patients with New York Heart Association class IV heart failure – which is how many experts in this field view the device – it has so far been a revolution played out in slow motion. In large part that’s because the field started small, and stayed small until recently. At the end of 2008, fewer than 1,000 patients had ever received a ventricular assist device or a total artificial heart. Even though the number nearly doubled in 2009, by decade’s end the cumulative total still remained under 2,000.

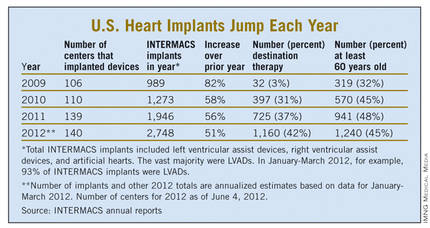
Despite the low numbers compared with the estimated need, many physicians and surgeons who specialize in advanced heart failure say they are satisfied with the pace at which continuous-flow LVADs have entered patients over the past 2 years. Though they acknowledge that the number of potential U.S. candidates for an LVAD undoubtedly is far larger than the nearly 4,600 who received one since early 2010, they note that the implantation rate grew in a steady and robust way during the past 30 months, as has the number of centers performing the surgery. With a better-than-50% year-over-year growth spurt in both 2010 and 2011, and with that pace continuing into the first months of 2012, the 140 U.S. centers that now place LVADs are on track to do nearly 3,000 this year (although only 42% were for destination therapy during January-March 2012).
"You probably don’t want it to increase too rapidly, because it is still high risk and expensive," said Dr. David E. Lanfear, a cardiologist specializing in advanced heart failure and transplantation at Henry Ford Hospital in Detroit. LVAD placements "have not reached the maximum number of patients who they could help, but you need to be cautious because it requires specialized expertise to do it properly. I wouldn’t expect that [during 2 years] it would immediately reach maximum use. It’s still in the growth phase. There are a lot of patients out there, so it will continue to grow; I’m not concerned that it’s not growing fast enough," he said in an interview.
An LVAD or a Heart Transplant?
Experts also note an important shift in attitude about the role for LVADs, a change that has brought them to the brink of replacing transplanted hearts as the default treatment for patients with end-stage heart failure.

"The current outcomes with HeartMate II have already changed the field. We published a paper last year that showed there was no difference between LVADs and transplant in costs and outcomes at 1 year (Ann. Thorac. Surg. 2011;91:1330-4)," said Dr. Mark S. Slaughter, professor of surgery and chief of thoracic and cardiovascular surgery at the University of Louisville (Ky.). Between that and the competitive mortality benefit from LVADs "we need to rethink the role of transplant. In appropriately selected patients, we can achieve outstanding long-term results with limited adverse events."
In their 2011 paper, Dr. Slaughter and his associates put the pending change more bluntly: "As outcomes for continuous-flow LVADs and heart transplantation converge, the therapy of heart transplantation could emerge as salvage therapy for major device-related complications or dysfunction or progressive right heart failure as opposed to the default option for all patients who are eligible for transplant."
Despite their huge promise, some experts have lingering concerns about the current generation of continuous-flow LVADs that make them reluctant to say that heart transplant has unquestionably stepped down from its pedestal as the gold standard treatment for patients with severe, end-stage heart failure.

"There is no question that [current continuous-flow LVADs] are very good technology. But whether they are great technology remains to be proven," said Dr. James K. Kirklin, professor of surgery and director of the division of cardiothoracic surgery at the University of Alabama at Birmingham. "We still have important concerns about things like drive-line infections and thromboembolic complications. Neither rate is terribly high, but they are high enough to be a problem if you apply LVADs to patients who are doing okay. We still have important doubts about whether we can really mimic the quality of life" achieved by transplant, he said in an interview.
LVAD placement also poses an operative risk, which may be as high as 10%, said Dr. David Taylor, a cardiologist and advanced heart failure specialist at the Cleveland Clinic. "If the risk was 2% or 3%, I’d say do it, but a risk closer to 10% makes you hesitate.
"There are a lot of really sick, elderly patients with heart failure who could unquestionably benefit from an LVAD. But as soon as you push the envelope [by treating sicker patients] you increase mortality. A large group of heart failure patients are very ill with chronic disease and feel terrible, and these are the patients where you’d see the greatest benefit, but we can’t afford to do that. We can’t afford to allocate this resource to patients with a 30% mortality risk, because that would limit our ability to extend it to other patients," Dr. Kirklin said in an interview.
"Most patients appreciate an LVAD and like it, but they still look forward to getting a transplant," said Dr. Stephen H. Bailey, director of cardiothoracic surgery at Allegheny General Hospital in Pittsburgh. "In July 2012, while the margin between LVAD and transplant is narrowing, transplant is still the gold standard for definitive treatment. Most patients still favor transplant, but that might change. It’s not quite there yet that an LVAD is equal to a transplant for the long term. It also requires getting rid of the drive line. That would be a game changer. It would also help if we could reduce gastrointestinal bleeds, but that is more of a nuisance."
Part of the bleeding problem comes from anticoagulation treatment to prevent clots from forming in the LVAD, but another facet is an acquired Von Willebrand factor deficiency caused by absent pulsatile blood flow, Dr. Bailey said. Early clinical results suggest that allowing the heart to beat every few seconds, by adjusting the LVAD’s continuous flow rate, can minimize the acquired deficiency.
Which INTERMACS Level?
The most widely used gauge physicians and surgeons have for assessing LVAD candidates is their level in a seven-step sequence created by INTERMACS (the Interagency Registry for Mechanically Assisted Circulatory Support), which subdivides a patient’s descent from advanced New York Heart Association class III heart failure through the strata of class IV disease. (See table.) The most severe INTERMACS stage, level 1 patients (also known as "crash and burn") are those in cardiogenic shock. During the past year or two, about 16% of U.S. LVAD recipients have been level 1 patients, a percentage that should ideally drop much closer to zero, experts say.

But beyond the maxim that an advanced heart failure patient should get an LVAD before reaching level 1, opinions vary on the best target.
"Everyone agrees that inotrope-dependent patients [profile 2 and 3] should get an LVAD," said Dr. Lanfear. "But you definitely should not wait until INTERMACS 1 or 2. Everyone tries hard to treat level 3 and 4 patients. Level 5 is controversial. We need more data about these clearly less- sick patients."
"INTERMACS 2 and 3 is the sweet spot. Patients in INTERMACS 4, 5, or 6 are not as motivated to be connected to a device and undergo big open heart surgery," said Dr. Bailey.
"When patients are on the cusp of inotrope dependence [before they reach level 3], I start to think about implanting," said Dr. Jeffrey J. Teuteberg, a cardiologist and associate director of the cardiac transplant program at the University of Pittsburgh. "It’s nice to get patients who are pre–inotrope dependent – profile 4, 5, or 6 – but are symptom limited and quality of life limited. For destination therapy, you want patients with function and good quality of life. Mechanical support provides more than just improved survival; it also reduces adverse events and raises quality of life."
"For level 2 patients, there is no question that an LVAD as bridge to transplant is better than continued medical treatment," said Dr. Taylor. "For level 3 patients, it’s a little trickier, but I think the majority would say that if a patient is stable and inotropic dependent, use an LVAD unless you believe a transplant will occur quickly. An LVAD would reduce the risk for developing cardiogenic shock. The patient who does the best with an LVAD is the one who is [relatively] healthy when implanted, but the patient who needs it most is the one who is literally dying."
"Everyone knows that if you take patients who are sicker [for LVADs], you’ll have trouble reaching 80% survival after 2 years. But there is not yet enough confidence in the treatment to extend it to INTERMACS level 4, 5, or 6 patients," said Dr. Kirklin. "That’s where the big potential is. The action currently is in INTERMACS 2 and 3. For patients who are INTERMACS 3, there is no question that if they can’t get a transplant they need a VAD. The sweet spot will be patients at INTERMACS 4."
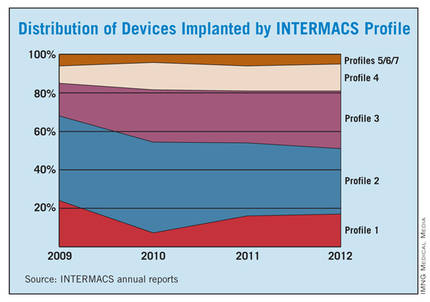
The INTERMACS registry numbers show that the field is currently stuck when it comes to INTERMACS levels, with the greatest number of LVADs going into level 2 patients, who received 34% of U.S LVADS during January-March 2012. The 34% level in early 2012 was down from 38% of LVAD recipients in 2011 and 47% in 2010, but growth in less-severe levels has been slow. At the start of 2012, 30% of LVAD recipients were at level 3, a small increase from the 27% rate in 2011 and 2010. Level 4 patients constituted 14% of LVAD recipients in early 2012, essentially unchanged from the prior 2 years, and level 5, 6, or 7 patients have consistently been a small slice of the U.S. LVAD pie, roughly 5% of recipients each year.
Beyond the INTERMACS Level
Heart failure specialists now recognize that INTERMACS level tells just part of the story.
"INTERMACS profiles depend on heart failure symptoms but not comorbidities. Severe diabetes, obstructive pulmonary disease, cardiorenal syndrome, chronic malnourishment, morbid obesity, and other factors all fall outside the INTERMACS profile criteria," said Dr. Kirklin.
He and his associates are formulating a risk assessment equation that will take comorbidities into account for a more global patient assessment. The most recently published INTERMACS registry analysis, which he first authored using data through the end of June 2011 with a total of 4,366 patients who received left ventricular support since 2006 (J. Heart Lung Transplant 2012;31:117-26), identified several comorbidity markers that each significantly linked with increased mortality. For example, a 1-unit increase in bilirubin linked with a 10% boost in mortality, a 1-unit increase in creatinine raised mortality by 16%, and a 0.5-unit increase in body surface area linked with a 48% rise in deaths. One of the strongest risk factors was being at INTERMACS level 1, which linked with a more-than-threefold higher mortality rate.
But more work must be done before the risk assessment formula is ready for clinical use. Right now, the formula "is not very reliable yet, because the maximum patient follow-up is 2 years. We need a little more follow-up," Dr. Kirklin said.
Growing the LVAD Numbers
Further growth in LVAD placements will happen on two fronts: broader use in patients at INTERMACS levels 2 and 3, and possibly level 4, where a strong consensus exists for LVAD support; and new evidence to document efficacy and safety in patients with less-severe disease at INTERMACS levels 4, 5, 6, and 7.
For the existing population, it’s a matter of physician and patient awareness. "We need to educate physicians that this is an option," said Dr. Lanfear. "Awareness is lacking. There is a lot of heart failure out there that is underrecognized and underreferred. Neither patients nor their physicians realize how sick they are, and that they are at a high enough risk to justify this."
Furthermore, there are parts of the country where the technology is underused. "In areas without a big center nearby, some physicians may not recognize patients who are sick enough to be LVAD candidates. We are trying to spread the word on who these patients are and when they should be referred."
According to Dr. Teuteberg, major warning signs that should flag patients who are potential LVAD candidates include advanced symptoms, increasing numbers of hospitalizations for heart failure, dwindling responses to ACE inhibitors and beta-blockers, increasing dosages of diuretics, a persistently high serum level of brain natriuretic peptide (BNP) despite good medical treatment, and lack of response to cardiac resynchronization therapy.
Expansion of the evidence base to show LVAD benefits in patients with INTERMACS level 5, 6, or 7 disease depends on a trial just starting, the REVIVE-IT (Randomized Evaluation of VAD Intervention Before Inotropic Therapy) study that’s set to enroll about 100 patients. But at press time, REVIVE-IT had not yet begun, posing doubts about LVAD use in the study’s targeted patients. "We haven’t really demonstrated reproducibly good survival [with LVADs] to compete with medical therapy in level 5 and 6 patients," said Dr. Kirklin. "The FDA put the study on hold while they reflected on that."
Making LVADs Better
With fast-paced technological advancement, continuous-flow LVADs will continue to evolve and improve. In June, results appeared on a new continuous-flow LVAD, the HeartWare device (Circulation 2012;125:3191-200), and last April an FDA advisory committee recommended that the agency approve the HeartWare LVAD for use as a bridge to (a trial testing the HeartWare LVAD for destination therapy is ongoing).
But no one interviewed for this article anticipates that the HeartWare LVAD will be a major advance. "Fundamentally, the major components and the implant technique are the same for the two devices," the HeartWare and the HeartMate II, said Dr. Slaughter. The HeartWare LVAD is smaller and designed to be placed completely in the pericardial space, but any clinical advantages based on these differences remain to be proved, he said in an interview.
A more meaningful improvement in LVAD design is in the works, and may reach initial clinical testing within a couple of years: a fully implantable LVAD with no transcutaneous drive line, a part that is subject to infection, prevents patients from submerging, and physically and psychologically limits patients by tethering them to equipment. "If there were one thing that could make a dramatic difference, it would be getting rid of the drive line. That is the Holy Grail for the field," said Dr. Bailey.
When a fully implantable LVAD becomes available for routine use, it will complete the LVAD revolution and help device therapy for advanced heart failure reach its full potential.
Dr. Lanfear has received research support and has received honoraria as a speaker for Thoratec, the company that markets the HeartMate II, and has received research support from HeartWare, the company developing the HeartWare LVAD. Dr. Slaughter has had contracts for services to Thoratec and HeartWare. Dr. Kirklin, Dr. Bailey, Dr. Teuteberg, and Dr. Taylor said that they had no disclosures.
*CORRECTION 8/10/12: The credit for the photo with the caption "The next LVAD in line, HeartWare, is smaller and dwells in the pericardial space" was misstated and should have been ©2012 HeartWare International, Inc.
A sea change in management of severe heart failure began two and a half years ago, in January 2010, when the Food and Drug Administration approved U.S. marketing of the HeartMate II continuous-flow, left ventricular assist device for destination therapy. Patients, cardiologists, and cardiothoracic surgeons at last had a reasonably durable, effective, and relatively safe alternative to heart transplant to offer patients at the end stage of deteriorating heart function.
The Heart Mate II quickly supplanted the prior-generation, pulsatile-flow devices, and 2010 also saw a 10-fold spike in the number of left ventricular assist devices (LVADs) placed in U.S. patients as destination therapy.
During the first half of 2011, 6-month survival among U.S. patients with a continuous-flow LVAD (which means the HeartMate II, the only continuous-flow device on the U.S. market) was 89%, and 12-month survival during 2010 was 81% (J. Heart Lung Transplant 2012;31:117-26, putting the continuous-flow LVAD in at least the same ballpark as heart transplant, which has a 5-year survival of about 80% and a median survival of about 10 years in U.S. patients.
But despite estimates that many thousands, if not tens of thousands, of Americans meet the clinical criteria to qualify for placement of an LVAD, the reality is that in the 30 months since the HeartMate II became available for routine destination therapy through mid-2012, only about 4,600 were placed in U.S. patients. In addition, just slightly more than a third of those patients, roughly 1,700, received their LVAD explicitly for destination therapy.
Even in 2012, a majority of patients who received an LVAD got their device with the understanding that it was as a bridge to transplant, an LVAD placed with temporary intent to help patients survive and improve clinically until their LVAD could be swapped out and replaced by a transplanted heart.
LVAD Numbers Up, but Still Low
If the availability of the HeartMate II for destination therapy in routine practice was a revolution for the management of many patients with New York Heart Association class IV heart failure – which is how many experts in this field view the device – it has so far been a revolution played out in slow motion. In large part that’s because the field started small, and stayed small until recently. At the end of 2008, fewer than 1,000 patients had ever received a ventricular assist device or a total artificial heart. Even though the number nearly doubled in 2009, by decade’s end the cumulative total still remained under 2,000.
Despite the low numbers compared with the estimated need, many physicians and surgeons who specialize in advanced heart failure say they are satisfied with the pace at which continuous-flow LVADs have entered patients over the past 2 years. Though they acknowledge that the number of potential U.S. candidates for an LVAD undoubtedly is far larger than the nearly 4,600 who received one since early 2010, they note that the implantation rate grew in a steady and robust way during the past 30 months, as has the number of centers performing the surgery. With a better-than-50% year-over-year growth spurt in both 2010 and 2011, and with that pace continuing into the first months of 2012, the 140 U.S. centers that now place LVADs are on track to do nearly 3,000 this year (although only 42% were for destination therapy during January-March 2012).
"You probably don’t want it to increase too rapidly, because it is still high risk and expensive," said Dr. David E. Lanfear, a cardiologist specializing in advanced heart failure and transplantation at Henry Ford Hospital in Detroit. LVAD placements "have not reached the maximum number of patients who they could help, but you need to be cautious because it requires specialized expertise to do it properly. I wouldn’t expect that [during 2 years] it would immediately reach maximum use. It’s still in the growth phase. There are a lot of patients out there, so it will continue to grow; I’m not concerned that it’s not growing fast enough," he said in an interview.
An LVAD or a Heart Transplant?
Experts also note an important shift in attitude about the role for LVADs, a change that has brought them to the brink of replacing transplanted hearts as the default treatment for patients with end-stage heart failure.
"The current outcomes with HeartMate II have already changed the field. We published a paper last year that showed there was no difference between LVADs and transplant in costs and outcomes at 1 year (Ann. Thorac. Surg. 2011;91:1330-4)," said Dr. Mark S. Slaughter, professor of surgery and chief of thoracic and cardiovascular surgery at the University of Louisville (Ky.). Between that and the competitive mortality benefit from LVADs "we need to rethink the role of transplant. In appropriately selected patients, we can achieve outstanding long-term results with limited adverse events."
In their 2011 paper, Dr. Slaughter and his associates put the pending change more bluntly: "As outcomes for continuous-flow LVADs and heart transplantation converge, the therapy of heart transplantation could emerge as salvage therapy for major device-related complications or dysfunction or progressive right heart failure as opposed to the default option for all patients who are eligible for transplant."
Despite their huge promise, some experts have lingering concerns about the current generation of continuous-flow LVADs that make them reluctant to say that heart transplant has unquestionably stepped down from its pedestal as the gold standard treatment for patients with severe, end-stage heart failure.
"There is no question that [current continuous-flow LVADs] are very good technology. But whether they are great technology remains to be proven," said Dr. James K. Kirklin, professor of surgery and director of the division of cardiothoracic surgery at the University of Alabama at Birmingham. "We still have important concerns about things like drive-line infections and thromboembolic complications. Neither rate is terribly high, but they are high enough to be a problem if you apply LVADs to patients who are doing okay. We still have important doubts about whether we can really mimic the quality of life" achieved by transplant, he said in an interview.
LVAD placement also poses an operative risk, which may be as high as 10%, said Dr. David Taylor, a cardiologist and advanced heart failure specialist at the Cleveland Clinic. "If the risk was 2% or 3%, I’d say do it, but a risk closer to 10% makes you hesitate.
"There are a lot of really sick, elderly patients with heart failure who could unquestionably benefit from an LVAD. But as soon as you push the envelope [by treating sicker patients] you increase mortality. A large group of heart failure patients are very ill with chronic disease and feel terrible, and these are the patients where you’d see the greatest benefit, but we can’t afford to do that. We can’t afford to allocate this resource to patients with a 30% mortality risk, because that would limit our ability to extend it to other patients," Dr. Kirklin said in an interview.
"Most patients appreciate an LVAD and like it, but they still look forward to getting a transplant," said Dr. Stephen H. Bailey, director of cardiothoracic surgery at Allegheny General Hospital in Pittsburgh. "In July 2012, while the margin between LVAD and transplant is narrowing, transplant is still the gold standard for definitive treatment. Most patients still favor transplant, but that might change. It’s not quite there yet that an LVAD is equal to a transplant for the long term. It also requires getting rid of the drive line. That would be a game changer. It would also help if we could reduce gastrointestinal bleeds, but that is more of a nuisance."
Part of the bleeding problem comes from anticoagulation treatment to prevent clots from forming in the LVAD, but another facet is an acquired Von Willebrand factor deficiency caused by absent pulsatile blood flow, Dr. Bailey said. Early clinical results suggest that allowing the heart to beat every few seconds, by adjusting the LVAD’s continuous flow rate, can minimize the acquired deficiency.
Which INTERMACS Level?
The most widely used gauge physicians and surgeons have for assessing LVAD candidates is their level in a seven-step sequence created by INTERMACS (the Interagency Registry for Mechanically Assisted Circulatory Support), which subdivides a patient’s descent from advanced New York Heart Association class III heart failure through the strata of class IV disease. (See table.) The most severe INTERMACS stage, level 1 patients (also known as "crash and burn") are those in cardiogenic shock. During the past year or two, about 16% of U.S. LVAD recipients have been level 1 patients, a percentage that should ideally drop much closer to zero, experts say.
But beyond the maxim that an advanced heart failure patient should get an LVAD before reaching level 1, opinions vary on the best target.
"Everyone agrees that inotrope-dependent patients [profile 2 and 3] should get an LVAD," said Dr. Lanfear. "But you definitely should not wait until INTERMACS 1 or 2. Everyone tries hard to treat level 3 and 4 patients. Level 5 is controversial. We need more data about these clearly less- sick patients."
"INTERMACS 2 and 3 is the sweet spot. Patients in INTERMACS 4, 5, or 6 are not as motivated to be connected to a device and undergo big open heart surgery," said Dr. Bailey.
"When patients are on the cusp of inotrope dependence [before they reach level 3], I start to think about implanting," said Dr. Jeffrey J. Teuteberg, a cardiologist and associate director of the cardiac transplant program at the University of Pittsburgh. "It’s nice to get patients who are pre–inotrope dependent – profile 4, 5, or 6 – but are symptom limited and quality of life limited. For destination therapy, you want patients with function and good quality of life. Mechanical support provides more than just improved survival; it also reduces adverse events and raises quality of life."
"For level 2 patients, there is no question that an LVAD as bridge to transplant is better than continued medical treatment," said Dr. Taylor. "For level 3 patients, it’s a little trickier, but I think the majority would say that if a patient is stable and inotropic dependent, use an LVAD unless you believe a transplant will occur quickly. An LVAD would reduce the risk for developing cardiogenic shock. The patient who does the best with an LVAD is the one who is [relatively] healthy when implanted, but the patient who needs it most is the one who is literally dying."
"Everyone knows that if you take patients who are sicker [for LVADs], you’ll have trouble reaching 80% survival after 2 years. But there is not yet enough confidence in the treatment to extend it to INTERMACS level 4, 5, or 6 patients," said Dr. Kirklin. "That’s where the big potential is. The action currently is in INTERMACS 2 and 3. For patients who are INTERMACS 3, there is no question that if they can’t get a transplant they need a VAD. The sweet spot will be patients at INTERMACS 4."
The INTERMACS registry numbers show that the field is currently stuck when it comes to INTERMACS levels, with the greatest number of LVADs going into level 2 patients, who received 34% of U.S LVADS during January-March 2012. The 34% level in early 2012 was down from 38% of LVAD recipients in 2011 and 47% in 2010, but growth in less-severe levels has been slow. At the start of 2012, 30% of LVAD recipients were at level 3, a small increase from the 27% rate in 2011 and 2010. Level 4 patients constituted 14% of LVAD recipients in early 2012, essentially unchanged from the prior 2 years, and level 5, 6, or 7 patients have consistently been a small slice of the U.S. LVAD pie, roughly 5% of recipients each year.
Beyond the INTERMACS Level
Heart failure specialists now recognize that INTERMACS level tells just part of the story.
"INTERMACS profiles depend on heart failure symptoms but not comorbidities. Severe diabetes, obstructive pulmonary disease, cardiorenal syndrome, chronic malnourishment, morbid obesity, and other factors all fall outside the INTERMACS profile criteria," said Dr. Kirklin.
He and his associates are formulating a risk assessment equation that will take comorbidities into account for a more global patient assessment. The most recently published INTERMACS registry analysis, which he first authored using data through the end of June 2011 with a total of 4,366 patients who received left ventricular support since 2006 (J. Heart Lung Transplant 2012;31:117-26), identified several comorbidity markers that each significantly linked with increased mortality. For example, a 1-unit increase in bilirubin linked with a 10% boost in mortality, a 1-unit increase in creatinine raised mortality by 16%, and a 0.5-unit increase in body surface area linked with a 48% rise in deaths. One of the strongest risk factors was being at INTERMACS level 1, which linked with a more-than-threefold higher mortality rate.
But more work must be done before the risk assessment formula is ready for clinical use. Right now, the formula "is not very reliable yet, because the maximum patient follow-up is 2 years. We need a little more follow-up," Dr. Kirklin said.
Growing the LVAD Numbers
Further growth in LVAD placements will happen on two fronts: broader use in patients at INTERMACS levels 2 and 3, and possibly level 4, where a strong consensus exists for LVAD support; and new evidence to document efficacy and safety in patients with less-severe disease at INTERMACS levels 4, 5, 6, and 7.
For the existing population, it’s a matter of physician and patient awareness. "We need to educate physicians that this is an option," said Dr. Lanfear. "Awareness is lacking. There is a lot of heart failure out there that is underrecognized and underreferred. Neither patients nor their physicians realize how sick they are, and that they are at a high enough risk to justify this."
Furthermore, there are parts of the country where the technology is underused. "In areas without a big center nearby, some physicians may not recognize patients who are sick enough to be LVAD candidates. We are trying to spread the word on who these patients are and when they should be referred."
According to Dr. Teuteberg, major warning signs that should flag patients who are potential LVAD candidates include advanced symptoms, increasing numbers of hospitalizations for heart failure, dwindling responses to ACE inhibitors and beta-blockers, increasing dosages of diuretics, a persistently high serum level of brain natriuretic peptide (BNP) despite good medical treatment, and lack of response to cardiac resynchronization therapy.
Expansion of the evidence base to show LVAD benefits in patients with INTERMACS level 5, 6, or 7 disease depends on a trial just starting, the REVIVE-IT (Randomized Evaluation of VAD Intervention Before Inotropic Therapy) study that’s set to enroll about 100 patients. But at press time, REVIVE-IT had not yet begun, posing doubts about LVAD use in the study’s targeted patients. "We haven’t really demonstrated reproducibly good survival [with LVADs] to compete with medical therapy in level 5 and 6 patients," said Dr. Kirklin. "The FDA put the study on hold while they reflected on that."
Making LVADs Better
With fast-paced technological advancement, continuous-flow LVADs will continue to evolve and improve. In June, results appeared on a new continuous-flow LVAD, the HeartWare device (Circulation 2012;125:3191-200), and last April an FDA advisory committee recommended that the agency approve the HeartWare LVAD for use as a bridge to (a trial testing the HeartWare LVAD for destination therapy is ongoing).
But no one interviewed for this article anticipates that the HeartWare LVAD will be a major advance. "Fundamentally, the major components and the implant technique are the same for the two devices," the HeartWare and the HeartMate II, said Dr. Slaughter. The HeartWare LVAD is smaller and designed to be placed completely in the pericardial space, but any clinical advantages based on these differences remain to be proved, he said in an interview.
A more meaningful improvement in LVAD design is in the works, and may reach initial clinical testing within a couple of years: a fully implantable LVAD with no transcutaneous drive line, a part that is subject to infection, prevents patients from submerging, and physically and psychologically limits patients by tethering them to equipment. "If there were one thing that could make a dramatic difference, it would be getting rid of the drive line. That is the Holy Grail for the field," said Dr. Bailey.
When a fully implantable LVAD becomes available for routine use, it will complete the LVAD revolution and help device therapy for advanced heart failure reach its full potential.
Dr. Lanfear has received research support and has received honoraria as a speaker for Thoratec, the company that markets the HeartMate II, and has received research support from HeartWare, the company developing the HeartWare LVAD. Dr. Slaughter has had contracts for services to Thoratec and HeartWare. Dr. Kirklin, Dr. Bailey, Dr. Teuteberg, and Dr. Taylor said that they had no disclosures.
*CORRECTION 8/10/12: The credit for the photo with the caption "The next LVAD in line, HeartWare, is smaller and dwells in the pericardial space" was misstated and should have been ©2012 HeartWare International, Inc.
A sea change in management of severe heart failure began two and a half years ago, in January 2010, when the Food and Drug Administration approved U.S. marketing of the HeartMate II continuous-flow, left ventricular assist device for destination therapy. Patients, cardiologists, and cardiothoracic surgeons at last had a reasonably durable, effective, and relatively safe alternative to heart transplant to offer patients at the end stage of deteriorating heart function.
The Heart Mate II quickly supplanted the prior-generation, pulsatile-flow devices, and 2010 also saw a 10-fold spike in the number of left ventricular assist devices (LVADs) placed in U.S. patients as destination therapy.
During the first half of 2011, 6-month survival among U.S. patients with a continuous-flow LVAD (which means the HeartMate II, the only continuous-flow device on the U.S. market) was 89%, and 12-month survival during 2010 was 81% (J. Heart Lung Transplant 2012;31:117-26, putting the continuous-flow LVAD in at least the same ballpark as heart transplant, which has a 5-year survival of about 80% and a median survival of about 10 years in U.S. patients.
But despite estimates that many thousands, if not tens of thousands, of Americans meet the clinical criteria to qualify for placement of an LVAD, the reality is that in the 30 months since the HeartMate II became available for routine destination therapy through mid-2012, only about 4,600 were placed in U.S. patients. In addition, just slightly more than a third of those patients, roughly 1,700, received their LVAD explicitly for destination therapy.
Even in 2012, a majority of patients who received an LVAD got their device with the understanding that it was as a bridge to transplant, an LVAD placed with temporary intent to help patients survive and improve clinically until their LVAD could be swapped out and replaced by a transplanted heart.
LVAD Numbers Up, but Still Low
If the availability of the HeartMate II for destination therapy in routine practice was a revolution for the management of many patients with New York Heart Association class IV heart failure – which is how many experts in this field view the device – it has so far been a revolution played out in slow motion. In large part that’s because the field started small, and stayed small until recently. At the end of 2008, fewer than 1,000 patients had ever received a ventricular assist device or a total artificial heart. Even though the number nearly doubled in 2009, by decade’s end the cumulative total still remained under 2,000.
Despite the low numbers compared with the estimated need, many physicians and surgeons who specialize in advanced heart failure say they are satisfied with the pace at which continuous-flow LVADs have entered patients over the past 2 years. Though they acknowledge that the number of potential U.S. candidates for an LVAD undoubtedly is far larger than the nearly 4,600 who received one since early 2010, they note that the implantation rate grew in a steady and robust way during the past 30 months, as has the number of centers performing the surgery. With a better-than-50% year-over-year growth spurt in both 2010 and 2011, and with that pace continuing into the first months of 2012, the 140 U.S. centers that now place LVADs are on track to do nearly 3,000 this year (although only 42% were for destination therapy during January-March 2012).
"You probably don’t want it to increase too rapidly, because it is still high risk and expensive," said Dr. David E. Lanfear, a cardiologist specializing in advanced heart failure and transplantation at Henry Ford Hospital in Detroit. LVAD placements "have not reached the maximum number of patients who they could help, but you need to be cautious because it requires specialized expertise to do it properly. I wouldn’t expect that [during 2 years] it would immediately reach maximum use. It’s still in the growth phase. There are a lot of patients out there, so it will continue to grow; I’m not concerned that it’s not growing fast enough," he said in an interview.
An LVAD or a Heart Transplant?
Experts also note an important shift in attitude about the role for LVADs, a change that has brought them to the brink of replacing transplanted hearts as the default treatment for patients with end-stage heart failure.
"The current outcomes with HeartMate II have already changed the field. We published a paper last year that showed there was no difference between LVADs and transplant in costs and outcomes at 1 year (Ann. Thorac. Surg. 2011;91:1330-4)," said Dr. Mark S. Slaughter, professor of surgery and chief of thoracic and cardiovascular surgery at the University of Louisville (Ky.). Between that and the competitive mortality benefit from LVADs "we need to rethink the role of transplant. In appropriately selected patients, we can achieve outstanding long-term results with limited adverse events."
In their 2011 paper, Dr. Slaughter and his associates put the pending change more bluntly: "As outcomes for continuous-flow LVADs and heart transplantation converge, the therapy of heart transplantation could emerge as salvage therapy for major device-related complications or dysfunction or progressive right heart failure as opposed to the default option for all patients who are eligible for transplant."
Despite their huge promise, some experts have lingering concerns about the current generation of continuous-flow LVADs that make them reluctant to say that heart transplant has unquestionably stepped down from its pedestal as the gold standard treatment for patients with severe, end-stage heart failure.
"There is no question that [current continuous-flow LVADs] are very good technology. But whether they are great technology remains to be proven," said Dr. James K. Kirklin, professor of surgery and director of the division of cardiothoracic surgery at the University of Alabama at Birmingham. "We still have important concerns about things like drive-line infections and thromboembolic complications. Neither rate is terribly high, but they are high enough to be a problem if you apply LVADs to patients who are doing okay. We still have important doubts about whether we can really mimic the quality of life" achieved by transplant, he said in an interview.
LVAD placement also poses an operative risk, which may be as high as 10%, said Dr. David Taylor, a cardiologist and advanced heart failure specialist at the Cleveland Clinic. "If the risk was 2% or 3%, I’d say do it, but a risk closer to 10% makes you hesitate.
"There are a lot of really sick, elderly patients with heart failure who could unquestionably benefit from an LVAD. But as soon as you push the envelope [by treating sicker patients] you increase mortality. A large group of heart failure patients are very ill with chronic disease and feel terrible, and these are the patients where you’d see the greatest benefit, but we can’t afford to do that. We can’t afford to allocate this resource to patients with a 30% mortality risk, because that would limit our ability to extend it to other patients," Dr. Kirklin said in an interview.
"Most patients appreciate an LVAD and like it, but they still look forward to getting a transplant," said Dr. Stephen H. Bailey, director of cardiothoracic surgery at Allegheny General Hospital in Pittsburgh. "In July 2012, while the margin between LVAD and transplant is narrowing, transplant is still the gold standard for definitive treatment. Most patients still favor transplant, but that might change. It’s not quite there yet that an LVAD is equal to a transplant for the long term. It also requires getting rid of the drive line. That would be a game changer. It would also help if we could reduce gastrointestinal bleeds, but that is more of a nuisance."
Part of the bleeding problem comes from anticoagulation treatment to prevent clots from forming in the LVAD, but another facet is an acquired Von Willebrand factor deficiency caused by absent pulsatile blood flow, Dr. Bailey said. Early clinical results suggest that allowing the heart to beat every few seconds, by adjusting the LVAD’s continuous flow rate, can minimize the acquired deficiency.
Which INTERMACS Level?
The most widely used gauge physicians and surgeons have for assessing LVAD candidates is their level in a seven-step sequence created by INTERMACS (the Interagency Registry for Mechanically Assisted Circulatory Support), which subdivides a patient’s descent from advanced New York Heart Association class III heart failure through the strata of class IV disease. (See table.) The most severe INTERMACS stage, level 1 patients (also known as "crash and burn") are those in cardiogenic shock. During the past year or two, about 16% of U.S. LVAD recipients have been level 1 patients, a percentage that should ideally drop much closer to zero, experts say.
But beyond the maxim that an advanced heart failure patient should get an LVAD before reaching level 1, opinions vary on the best target.
"Everyone agrees that inotrope-dependent patients [profile 2 and 3] should get an LVAD," said Dr. Lanfear. "But you definitely should not wait until INTERMACS 1 or 2. Everyone tries hard to treat level 3 and 4 patients. Level 5 is controversial. We need more data about these clearly less- sick patients."
"INTERMACS 2 and 3 is the sweet spot. Patients in INTERMACS 4, 5, or 6 are not as motivated to be connected to a device and undergo big open heart surgery," said Dr. Bailey.
"When patients are on the cusp of inotrope dependence [before they reach level 3], I start to think about implanting," said Dr. Jeffrey J. Teuteberg, a cardiologist and associate director of the cardiac transplant program at the University of Pittsburgh. "It’s nice to get patients who are pre–inotrope dependent – profile 4, 5, or 6 – but are symptom limited and quality of life limited. For destination therapy, you want patients with function and good quality of life. Mechanical support provides more than just improved survival; it also reduces adverse events and raises quality of life."
"For level 2 patients, there is no question that an LVAD as bridge to transplant is better than continued medical treatment," said Dr. Taylor. "For level 3 patients, it’s a little trickier, but I think the majority would say that if a patient is stable and inotropic dependent, use an LVAD unless you believe a transplant will occur quickly. An LVAD would reduce the risk for developing cardiogenic shock. The patient who does the best with an LVAD is the one who is [relatively] healthy when implanted, but the patient who needs it most is the one who is literally dying."
"Everyone knows that if you take patients who are sicker [for LVADs], you’ll have trouble reaching 80% survival after 2 years. But there is not yet enough confidence in the treatment to extend it to INTERMACS level 4, 5, or 6 patients," said Dr. Kirklin. "That’s where the big potential is. The action currently is in INTERMACS 2 and 3. For patients who are INTERMACS 3, there is no question that if they can’t get a transplant they need a VAD. The sweet spot will be patients at INTERMACS 4."
The INTERMACS registry numbers show that the field is currently stuck when it comes to INTERMACS levels, with the greatest number of LVADs going into level 2 patients, who received 34% of U.S LVADS during January-March 2012. The 34% level in early 2012 was down from 38% of LVAD recipients in 2011 and 47% in 2010, but growth in less-severe levels has been slow. At the start of 2012, 30% of LVAD recipients were at level 3, a small increase from the 27% rate in 2011 and 2010. Level 4 patients constituted 14% of LVAD recipients in early 2012, essentially unchanged from the prior 2 years, and level 5, 6, or 7 patients have consistently been a small slice of the U.S. LVAD pie, roughly 5% of recipients each year.
Beyond the INTERMACS Level
Heart failure specialists now recognize that INTERMACS level tells just part of the story.
"INTERMACS profiles depend on heart failure symptoms but not comorbidities. Severe diabetes, obstructive pulmonary disease, cardiorenal syndrome, chronic malnourishment, morbid obesity, and other factors all fall outside the INTERMACS profile criteria," said Dr. Kirklin.
He and his associates are formulating a risk assessment equation that will take comorbidities into account for a more global patient assessment. The most recently published INTERMACS registry analysis, which he first authored using data through the end of June 2011 with a total of 4,366 patients who received left ventricular support since 2006 (J. Heart Lung Transplant 2012;31:117-26), identified several comorbidity markers that each significantly linked with increased mortality. For example, a 1-unit increase in bilirubin linked with a 10% boost in mortality, a 1-unit increase in creatinine raised mortality by 16%, and a 0.5-unit increase in body surface area linked with a 48% rise in deaths. One of the strongest risk factors was being at INTERMACS level 1, which linked with a more-than-threefold higher mortality rate.
But more work must be done before the risk assessment formula is ready for clinical use. Right now, the formula "is not very reliable yet, because the maximum patient follow-up is 2 years. We need a little more follow-up," Dr. Kirklin said.
Growing the LVAD Numbers
Further growth in LVAD placements will happen on two fronts: broader use in patients at INTERMACS levels 2 and 3, and possibly level 4, where a strong consensus exists for LVAD support; and new evidence to document efficacy and safety in patients with less-severe disease at INTERMACS levels 4, 5, 6, and 7.
For the existing population, it’s a matter of physician and patient awareness. "We need to educate physicians that this is an option," said Dr. Lanfear. "Awareness is lacking. There is a lot of heart failure out there that is underrecognized and underreferred. Neither patients nor their physicians realize how sick they are, and that they are at a high enough risk to justify this."
Furthermore, there are parts of the country where the technology is underused. "In areas without a big center nearby, some physicians may not recognize patients who are sick enough to be LVAD candidates. We are trying to spread the word on who these patients are and when they should be referred."
According to Dr. Teuteberg, major warning signs that should flag patients who are potential LVAD candidates include advanced symptoms, increasing numbers of hospitalizations for heart failure, dwindling responses to ACE inhibitors and beta-blockers, increasing dosages of diuretics, a persistently high serum level of brain natriuretic peptide (BNP) despite good medical treatment, and lack of response to cardiac resynchronization therapy.
Expansion of the evidence base to show LVAD benefits in patients with INTERMACS level 5, 6, or 7 disease depends on a trial just starting, the REVIVE-IT (Randomized Evaluation of VAD Intervention Before Inotropic Therapy) study that’s set to enroll about 100 patients. But at press time, REVIVE-IT had not yet begun, posing doubts about LVAD use in the study’s targeted patients. "We haven’t really demonstrated reproducibly good survival [with LVADs] to compete with medical therapy in level 5 and 6 patients," said Dr. Kirklin. "The FDA put the study on hold while they reflected on that."
Making LVADs Better
With fast-paced technological advancement, continuous-flow LVADs will continue to evolve and improve. In June, results appeared on a new continuous-flow LVAD, the HeartWare device (Circulation 2012;125:3191-200), and last April an FDA advisory committee recommended that the agency approve the HeartWare LVAD for use as a bridge to (a trial testing the HeartWare LVAD for destination therapy is ongoing).
But no one interviewed for this article anticipates that the HeartWare LVAD will be a major advance. "Fundamentally, the major components and the implant technique are the same for the two devices," the HeartWare and the HeartMate II, said Dr. Slaughter. The HeartWare LVAD is smaller and designed to be placed completely in the pericardial space, but any clinical advantages based on these differences remain to be proved, he said in an interview.
A more meaningful improvement in LVAD design is in the works, and may reach initial clinical testing within a couple of years: a fully implantable LVAD with no transcutaneous drive line, a part that is subject to infection, prevents patients from submerging, and physically and psychologically limits patients by tethering them to equipment. "If there were one thing that could make a dramatic difference, it would be getting rid of the drive line. That is the Holy Grail for the field," said Dr. Bailey.
When a fully implantable LVAD becomes available for routine use, it will complete the LVAD revolution and help device therapy for advanced heart failure reach its full potential.
Dr. Lanfear has received research support and has received honoraria as a speaker for Thoratec, the company that markets the HeartMate II, and has received research support from HeartWare, the company developing the HeartWare LVAD. Dr. Slaughter has had contracts for services to Thoratec and HeartWare. Dr. Kirklin, Dr. Bailey, Dr. Teuteberg, and Dr. Taylor said that they had no disclosures.
*CORRECTION 8/10/12: The credit for the photo with the caption "The next LVAD in line, HeartWare, is smaller and dwells in the pericardial space" was misstated and should have been ©2012 HeartWare International, Inc.
Forgoing Defibrillation Testing at ICD Insertion Found to Be Safe
Patients who did not undergo defibrillation testing during the insertion of their first implantable cardioverter defibrillator had outcomes similar to those who did undergo defibrillation testing of the device in the largest study to date comparing the two approaches, which was reported online August 1 in the Journal of the American College of Cardiology.

This finding supports a strategy of omitting defibrillation testing in most such patients – a strategy that clinicians are already adopting in increasing numbers, said Dr. Michele Brignole, chief of cardiology at Ospedale del Tigullio, Lavagna, Italy, and his associates.
Defibrillation testing has been considered a standard procedure at ICD insertion "to ensure adequate sensing of ventricular fibrillation, appropriate connection of high-voltage electrodes, and the ability of the device to terminate VF with a shock. Nevertheless, implant techniques and technology have evolved in recent years, and deviations from this clinical practice are frequent," the investigators noted.
To assess the safety of omitting this step in the implantation process, Dr. Brignole and his colleagues performed SAFE-ICD (Safety of Two Strategies of ICD Management at Implantation), a prospective study of 2,120 consecutive procedures in adults at 41 Italian medical centers. The treating physicians were allowed either to perform or not perform defibrillation testing according to their standard practice; patients were followed for 2 years.
The frequency of performing defibrillation testing varied widely among the different medical centers, with some of them conducting the test in all patients and others doing so in no patients. Overall, 836 study subjects (39%) underwent defibrillation testing during insertion of their ICD, and the remaining 1,284 (61%) did not.
The primary end point was a composite of severe implant-related complications periprocedurally plus serious events during follow-up, such as sudden cardiac death or resuscitation after delivery of ineffective but appropriate ICD shocks.
This end point was reached in 18 patients who underwent defibrillation testing and 16 who did not. The estimated yearly incidence of this composite end point was 1.15% with defibrillation testing and 0.68% without it, a "negligible" difference.
In addition, 2-year all-cause mortality was not significantly lower for patients who underwent defibrillation testing (12.9%) than for those who did not (14.6%).
During follow-up, the devices delivered appropriate and effective shocks in a similar proportion of patients in the two study groups.
These findings indicate that "the clinical relevance of defibrillation testing is limited, thus supporting the practice of omitting [it] at implant," Dr. Brignole and his associates said (J. Am. Coll. Cardiol. 2012 Aug. 1 [doi:10.1016/j.jacc.2012.05.014]).
ICD recipients "are very well protected from sudden cardiac death irrespective of performing defibrillation testing or not," the authors said. Moreover, performing defibrillation testing is not likely to decrease the rate of sudden cardiac death to a clinically relevant degree, below the already low 1% rate observed in this study population, they added.
The strengths of this study included its large population that represented the general ICD population in Western countries, its very low (3%) dropout rate, and the use of any commercially available ICD devices.
However, the study was limited in that the unexpectedly low incidence of sudden cardiac death may have been insufficient to show a true difference between the two study groups.
The observational design of SAFE-ICD, unlike that of a randomized clinical trial, "does not allow us to draw a definitive conclusion" as to the safety of omitting defibrillation testing. But there is such a large, prospective, multicenter, randomized clinical trial – SIMPLE (Shockless Implant Evaluation), taking place now – that should provide a definitive answer, the researchers said.
This study was funded by Boston Scientific. One of Dr. Brignole’s associates is an employee of Boston Scientific, and others reported ties to Boston Scientific and Medtronic. Dr. Estes reported ties to Boston Scientific, Medtronic, and St. Jude Medical.
The study by Dr. Brignole and colleagues makes a "meaningful contribution to the collective evidence that routine determination of defibrillation thresholds has more risks than benefits for many patients at the time of initial ICD insertion," said Dr. N.A. Mark Estes III.
However, the absence of randomization resulted in confounding differences in the clinical profiles of the patients who were enrolled in the two strategies, he added. For example, patients who underwent defibrillation testing had lower rates of atrial fibrillation, New York Heart Association class III and IV heart failure, and use of diuretics and digoxin. In addition, as the authors pointed out, the lower-than-anticipated number of patients reaching the primary end point rendered the study underpowered to detect a difference between the two groups.
Thus, this observational study cannot provide definitive proof that omitting this step is clinically justified, so it is not yet time to abandon the practice. "Implanting physicians will have to decide on the basis of the best available data, their experience, and judgment whether to omit defibrillation testing selectively in low-risk patients," Dr. Estes noted.
Dr. Estes is professor of medicine at Tufts University and director of the cardiac arrhythmia center at Tufts Medical Center, Boston. He reported ties to Boston Scientific, Medtronic, and St. Jude Medical. These remarks were taken from his editorial comment accompanying Dr. Brignole’s report (J. Am. Coll. Cardiol. 2012 Aug. 1 [doi:10.1016/j.jacc.2012.05.016]).
The study by Dr. Brignole and colleagues makes a "meaningful contribution to the collective evidence that routine determination of defibrillation thresholds has more risks than benefits for many patients at the time of initial ICD insertion," said Dr. N.A. Mark Estes III.
However, the absence of randomization resulted in confounding differences in the clinical profiles of the patients who were enrolled in the two strategies, he added. For example, patients who underwent defibrillation testing had lower rates of atrial fibrillation, New York Heart Association class III and IV heart failure, and use of diuretics and digoxin. In addition, as the authors pointed out, the lower-than-anticipated number of patients reaching the primary end point rendered the study underpowered to detect a difference between the two groups.
Thus, this observational study cannot provide definitive proof that omitting this step is clinically justified, so it is not yet time to abandon the practice. "Implanting physicians will have to decide on the basis of the best available data, their experience, and judgment whether to omit defibrillation testing selectively in low-risk patients," Dr. Estes noted.
Dr. Estes is professor of medicine at Tufts University and director of the cardiac arrhythmia center at Tufts Medical Center, Boston. He reported ties to Boston Scientific, Medtronic, and St. Jude Medical. These remarks were taken from his editorial comment accompanying Dr. Brignole’s report (J. Am. Coll. Cardiol. 2012 Aug. 1 [doi:10.1016/j.jacc.2012.05.016]).
The study by Dr. Brignole and colleagues makes a "meaningful contribution to the collective evidence that routine determination of defibrillation thresholds has more risks than benefits for many patients at the time of initial ICD insertion," said Dr. N.A. Mark Estes III.
However, the absence of randomization resulted in confounding differences in the clinical profiles of the patients who were enrolled in the two strategies, he added. For example, patients who underwent defibrillation testing had lower rates of atrial fibrillation, New York Heart Association class III and IV heart failure, and use of diuretics and digoxin. In addition, as the authors pointed out, the lower-than-anticipated number of patients reaching the primary end point rendered the study underpowered to detect a difference between the two groups.
Thus, this observational study cannot provide definitive proof that omitting this step is clinically justified, so it is not yet time to abandon the practice. "Implanting physicians will have to decide on the basis of the best available data, their experience, and judgment whether to omit defibrillation testing selectively in low-risk patients," Dr. Estes noted.
Dr. Estes is professor of medicine at Tufts University and director of the cardiac arrhythmia center at Tufts Medical Center, Boston. He reported ties to Boston Scientific, Medtronic, and St. Jude Medical. These remarks were taken from his editorial comment accompanying Dr. Brignole’s report (J. Am. Coll. Cardiol. 2012 Aug. 1 [doi:10.1016/j.jacc.2012.05.016]).
Patients who did not undergo defibrillation testing during the insertion of their first implantable cardioverter defibrillator had outcomes similar to those who did undergo defibrillation testing of the device in the largest study to date comparing the two approaches, which was reported online August 1 in the Journal of the American College of Cardiology.
This finding supports a strategy of omitting defibrillation testing in most such patients – a strategy that clinicians are already adopting in increasing numbers, said Dr. Michele Brignole, chief of cardiology at Ospedale del Tigullio, Lavagna, Italy, and his associates.
Defibrillation testing has been considered a standard procedure at ICD insertion "to ensure adequate sensing of ventricular fibrillation, appropriate connection of high-voltage electrodes, and the ability of the device to terminate VF with a shock. Nevertheless, implant techniques and technology have evolved in recent years, and deviations from this clinical practice are frequent," the investigators noted.
To assess the safety of omitting this step in the implantation process, Dr. Brignole and his colleagues performed SAFE-ICD (Safety of Two Strategies of ICD Management at Implantation), a prospective study of 2,120 consecutive procedures in adults at 41 Italian medical centers. The treating physicians were allowed either to perform or not perform defibrillation testing according to their standard practice; patients were followed for 2 years.
The frequency of performing defibrillation testing varied widely among the different medical centers, with some of them conducting the test in all patients and others doing so in no patients. Overall, 836 study subjects (39%) underwent defibrillation testing during insertion of their ICD, and the remaining 1,284 (61%) did not.
The primary end point was a composite of severe implant-related complications periprocedurally plus serious events during follow-up, such as sudden cardiac death or resuscitation after delivery of ineffective but appropriate ICD shocks.
This end point was reached in 18 patients who underwent defibrillation testing and 16 who did not. The estimated yearly incidence of this composite end point was 1.15% with defibrillation testing and 0.68% without it, a "negligible" difference.
In addition, 2-year all-cause mortality was not significantly lower for patients who underwent defibrillation testing (12.9%) than for those who did not (14.6%).
During follow-up, the devices delivered appropriate and effective shocks in a similar proportion of patients in the two study groups.
These findings indicate that "the clinical relevance of defibrillation testing is limited, thus supporting the practice of omitting [it] at implant," Dr. Brignole and his associates said (J. Am. Coll. Cardiol. 2012 Aug. 1 [doi:10.1016/j.jacc.2012.05.014]).
ICD recipients "are very well protected from sudden cardiac death irrespective of performing defibrillation testing or not," the authors said. Moreover, performing defibrillation testing is not likely to decrease the rate of sudden cardiac death to a clinically relevant degree, below the already low 1% rate observed in this study population, they added.
The strengths of this study included its large population that represented the general ICD population in Western countries, its very low (3%) dropout rate, and the use of any commercially available ICD devices.
However, the study was limited in that the unexpectedly low incidence of sudden cardiac death may have been insufficient to show a true difference between the two study groups.
The observational design of SAFE-ICD, unlike that of a randomized clinical trial, "does not allow us to draw a definitive conclusion" as to the safety of omitting defibrillation testing. But there is such a large, prospective, multicenter, randomized clinical trial – SIMPLE (Shockless Implant Evaluation), taking place now – that should provide a definitive answer, the researchers said.
This study was funded by Boston Scientific. One of Dr. Brignole’s associates is an employee of Boston Scientific, and others reported ties to Boston Scientific and Medtronic. Dr. Estes reported ties to Boston Scientific, Medtronic, and St. Jude Medical.
Patients who did not undergo defibrillation testing during the insertion of their first implantable cardioverter defibrillator had outcomes similar to those who did undergo defibrillation testing of the device in the largest study to date comparing the two approaches, which was reported online August 1 in the Journal of the American College of Cardiology.
This finding supports a strategy of omitting defibrillation testing in most such patients – a strategy that clinicians are already adopting in increasing numbers, said Dr. Michele Brignole, chief of cardiology at Ospedale del Tigullio, Lavagna, Italy, and his associates.
Defibrillation testing has been considered a standard procedure at ICD insertion "to ensure adequate sensing of ventricular fibrillation, appropriate connection of high-voltage electrodes, and the ability of the device to terminate VF with a shock. Nevertheless, implant techniques and technology have evolved in recent years, and deviations from this clinical practice are frequent," the investigators noted.
To assess the safety of omitting this step in the implantation process, Dr. Brignole and his colleagues performed SAFE-ICD (Safety of Two Strategies of ICD Management at Implantation), a prospective study of 2,120 consecutive procedures in adults at 41 Italian medical centers. The treating physicians were allowed either to perform or not perform defibrillation testing according to their standard practice; patients were followed for 2 years.
The frequency of performing defibrillation testing varied widely among the different medical centers, with some of them conducting the test in all patients and others doing so in no patients. Overall, 836 study subjects (39%) underwent defibrillation testing during insertion of their ICD, and the remaining 1,284 (61%) did not.
The primary end point was a composite of severe implant-related complications periprocedurally plus serious events during follow-up, such as sudden cardiac death or resuscitation after delivery of ineffective but appropriate ICD shocks.
This end point was reached in 18 patients who underwent defibrillation testing and 16 who did not. The estimated yearly incidence of this composite end point was 1.15% with defibrillation testing and 0.68% without it, a "negligible" difference.
In addition, 2-year all-cause mortality was not significantly lower for patients who underwent defibrillation testing (12.9%) than for those who did not (14.6%).
During follow-up, the devices delivered appropriate and effective shocks in a similar proportion of patients in the two study groups.
These findings indicate that "the clinical relevance of defibrillation testing is limited, thus supporting the practice of omitting [it] at implant," Dr. Brignole and his associates said (J. Am. Coll. Cardiol. 2012 Aug. 1 [doi:10.1016/j.jacc.2012.05.014]).
ICD recipients "are very well protected from sudden cardiac death irrespective of performing defibrillation testing or not," the authors said. Moreover, performing defibrillation testing is not likely to decrease the rate of sudden cardiac death to a clinically relevant degree, below the already low 1% rate observed in this study population, they added.
The strengths of this study included its large population that represented the general ICD population in Western countries, its very low (3%) dropout rate, and the use of any commercially available ICD devices.
However, the study was limited in that the unexpectedly low incidence of sudden cardiac death may have been insufficient to show a true difference between the two study groups.
The observational design of SAFE-ICD, unlike that of a randomized clinical trial, "does not allow us to draw a definitive conclusion" as to the safety of omitting defibrillation testing. But there is such a large, prospective, multicenter, randomized clinical trial – SIMPLE (Shockless Implant Evaluation), taking place now – that should provide a definitive answer, the researchers said.
This study was funded by Boston Scientific. One of Dr. Brignole’s associates is an employee of Boston Scientific, and others reported ties to Boston Scientific and Medtronic. Dr. Estes reported ties to Boston Scientific, Medtronic, and St. Jude Medical.
FROM THE JOURNAL OF THE AMERICAN COLLEGE OF CARDIOLOGY
Major Finding: The annual incidence of the primary end point – sudden cardiac death or severe complications either periprocedurally or during follow-up – was 1.15% with defibrillation testing and 0.68% without it.
Data Source: SAFE-ICD was a prospective observational study of 836 adults who underwent defibrillation testing at ICD implantation and 1,284 who did not, and who were followed for sudden cardiac death and other adverse outcomes for 2 years.
Disclosures: This study was funded by Boston Scientific. One of Dr. Brignole’s associates is an employee of Boston Scientific, and others reported ties to Boston Scientific and Medtronic.

Exercise Cuts Depressive Symptoms in Heart Failure Patients
Aerobic exercise for 90-120 minutes per week modestly reduces depressive symptoms in patients with heart failure, according to a report in the Aug. 1 issue of JAMA.
After 3 months of a supervised exercise program, patients who exercised showed a small but significant reduction in depression scores, compared with patients who received usual care. After an additional 9 months of home exercise, that difference persisted.

The difference was characterized as modest, "and the clinical significance of this small improvement is not known," said James A. Blumenthal, Ph.D., of Duke University, Durham, N.C., and his associates.
However, because the difference was consistent over a full year, it appears to be robust and "is likely to be associated with better social functioning and higher quality of life," they noted.
As many as 40% of patients with heart failure have been reported to have comorbid depression, and as many as 75% of heart failure patients have been reported to have elevated depressive symptoms. But few randomized trials have examined treatment of depression in this patient population.
HF-ACTION (Heart Failure–A Controlled Trial Investigating Outcomes of Exercise Training) found that adding exercise to usual care modestly reduced rates of death from any cause and hospitalization for any cause. Dr. Blumenthal and his colleagues reported on an ancillary study to HF-ACTION in which they further assessed the effects of exercise on depressive symptoms.
The study subjects were 2,322 patients whose HF failed to improve after at least 6 weeks of optimal therapy at 82 medical centers in the United States, Canada, and France. All the subjects had undergone exercise stress testing and had completed the Beck Depression Inventory II at baseline in 2003-2007. The median age was 59 years (range, 19-91 years).
The BDI-II is a self-reported measure of depressive symptoms with a possible score ranging from 0 (no depressive symptoms) to 63 (maximal depressive symptoms). A score of 14 or higher reflects clinically significant depressive symptoms. A total of 28% of the study subjects scored in this range at baseline.
A total of 1,164 subjects were randomly assigned to usual care (the control group) and 1,158 to usual care plus three supervised sessions per week on a treadmill or stationary cycle for 3 months. The exercise group was then given the exercise equipment of their choice to take home and was encouraged to increase to 90 min/week of exercise until they reached 1 year from baseline.
At 3 months, the mean BDI-II score was 8.95 for the exercise group and 9.70 for the control group. At 1 year, the mean scores were 8.86 and 9.54, respectively. Both of these differences are statistically significant, although small, the investigators said (JAMA 2012;308:465-74).
The results were more robust for the subgroup of 653 patients who scored 14 or higher on the BDI-II at baseline. At 3 months, the mean score in the exercise group was 16.66, compared with a mean score of 17.98 in the control group. At 1 year, the mean scores were 15.85 and 17.34, respectively.
In addition, the amount of exercise was inversely associated with the reduction in depressive symptoms. "Compared with a participant reporting no exercise, a participant reporting 90 minutes of exercise per week could be expected to have a 1.55-point lower BDI-II score at 3 months and a 1.67-point lower BDI-II score at 12 months," Dr. Blumenthal and his associates said.
Such a difference "is comparable with placebo-control trials involving patients with major depressive disorder," they added.
"We also observed that elevated depressive symptoms were associated with more than a 20% increase in risk for all-cause mortality and hospitalizations and that the increased risk was independent of antidepressant use and established risk factors in patients with heart failure, including age and disease severity.
"These data add to the evidence suggesting that elevated depressive symptoms, without necessarily meeting diagnostic criteria for major depressive disorder, are associated with increased risk for adverse clinical events," the researchers said.
The study findings also support the recent recommendation by the American Heart Association that all patients with cardiac disease be routinely assessed for depression, they added.
The study results also demonstrated that HF patients whose depression worsened over time were at particularly increased risk for hospitalization and death. This highlights the need to not only reduce depressive symptoms but also to prevent the worsening of existing depressive symptoms, Dr. Blumenthal and his colleagues said.
The study had two important limitations.
First, only about 40% of the patients assigned to exercise were fully adherent to the program and actually exercised 90-120 minutes per week. And 40%-50% of the patients in the control reported that they had actually exercised to some degree during follow-up. Such unintended crossover might have adversely affected the study findings.
Second, the study could not rule out the possibility that patients with more severe depressive symptoms were less likely to exercise, "so it is not clear whether exercise resulted in less depression or if depression resulted in less exercise," the researchers said.
Aerobic exercise for 90-120 minutes per week modestly reduces depressive symptoms in patients with heart failure, according to a report in the Aug. 1 issue of JAMA.
After 3 months of a supervised exercise program, patients who exercised showed a small but significant reduction in depression scores, compared with patients who received usual care. After an additional 9 months of home exercise, that difference persisted.
The difference was characterized as modest, "and the clinical significance of this small improvement is not known," said James A. Blumenthal, Ph.D., of Duke University, Durham, N.C., and his associates.
However, because the difference was consistent over a full year, it appears to be robust and "is likely to be associated with better social functioning and higher quality of life," they noted.
As many as 40% of patients with heart failure have been reported to have comorbid depression, and as many as 75% of heart failure patients have been reported to have elevated depressive symptoms. But few randomized trials have examined treatment of depression in this patient population.
HF-ACTION (Heart Failure–A Controlled Trial Investigating Outcomes of Exercise Training) found that adding exercise to usual care modestly reduced rates of death from any cause and hospitalization for any cause. Dr. Blumenthal and his colleagues reported on an ancillary study to HF-ACTION in which they further assessed the effects of exercise on depressive symptoms.
The study subjects were 2,322 patients whose HF failed to improve after at least 6 weeks of optimal therapy at 82 medical centers in the United States, Canada, and France. All the subjects had undergone exercise stress testing and had completed the Beck Depression Inventory II at baseline in 2003-2007. The median age was 59 years (range, 19-91 years).
The BDI-II is a self-reported measure of depressive symptoms with a possible score ranging from 0 (no depressive symptoms) to 63 (maximal depressive symptoms). A score of 14 or higher reflects clinically significant depressive symptoms. A total of 28% of the study subjects scored in this range at baseline.
A total of 1,164 subjects were randomly assigned to usual care (the control group) and 1,158 to usual care plus three supervised sessions per week on a treadmill or stationary cycle for 3 months. The exercise group was then given the exercise equipment of their choice to take home and was encouraged to increase to 90 min/week of exercise until they reached 1 year from baseline.
At 3 months, the mean BDI-II score was 8.95 for the exercise group and 9.70 for the control group. At 1 year, the mean scores were 8.86 and 9.54, respectively. Both of these differences are statistically significant, although small, the investigators said (JAMA 2012;308:465-74).
The results were more robust for the subgroup of 653 patients who scored 14 or higher on the BDI-II at baseline. At 3 months, the mean score in the exercise group was 16.66, compared with a mean score of 17.98 in the control group. At 1 year, the mean scores were 15.85 and 17.34, respectively.
In addition, the amount of exercise was inversely associated with the reduction in depressive symptoms. "Compared with a participant reporting no exercise, a participant reporting 90 minutes of exercise per week could be expected to have a 1.55-point lower BDI-II score at 3 months and a 1.67-point lower BDI-II score at 12 months," Dr. Blumenthal and his associates said.
Such a difference "is comparable with placebo-control trials involving patients with major depressive disorder," they added.
"We also observed that elevated depressive symptoms were associated with more than a 20% increase in risk for all-cause mortality and hospitalizations and that the increased risk was independent of antidepressant use and established risk factors in patients with heart failure, including age and disease severity.
"These data add to the evidence suggesting that elevated depressive symptoms, without necessarily meeting diagnostic criteria for major depressive disorder, are associated with increased risk for adverse clinical events," the researchers said.
The study findings also support the recent recommendation by the American Heart Association that all patients with cardiac disease be routinely assessed for depression, they added.
The study results also demonstrated that HF patients whose depression worsened over time were at particularly increased risk for hospitalization and death. This highlights the need to not only reduce depressive symptoms but also to prevent the worsening of existing depressive symptoms, Dr. Blumenthal and his colleagues said.
The study had two important limitations.
First, only about 40% of the patients assigned to exercise were fully adherent to the program and actually exercised 90-120 minutes per week. And 40%-50% of the patients in the control reported that they had actually exercised to some degree during follow-up. Such unintended crossover might have adversely affected the study findings.
Second, the study could not rule out the possibility that patients with more severe depressive symptoms were less likely to exercise, "so it is not clear whether exercise resulted in less depression or if depression resulted in less exercise," the researchers said.
Aerobic exercise for 90-120 minutes per week modestly reduces depressive symptoms in patients with heart failure, according to a report in the Aug. 1 issue of JAMA.
After 3 months of a supervised exercise program, patients who exercised showed a small but significant reduction in depression scores, compared with patients who received usual care. After an additional 9 months of home exercise, that difference persisted.
The difference was characterized as modest, "and the clinical significance of this small improvement is not known," said James A. Blumenthal, Ph.D., of Duke University, Durham, N.C., and his associates.
However, because the difference was consistent over a full year, it appears to be robust and "is likely to be associated with better social functioning and higher quality of life," they noted.
As many as 40% of patients with heart failure have been reported to have comorbid depression, and as many as 75% of heart failure patients have been reported to have elevated depressive symptoms. But few randomized trials have examined treatment of depression in this patient population.
HF-ACTION (Heart Failure–A Controlled Trial Investigating Outcomes of Exercise Training) found that adding exercise to usual care modestly reduced rates of death from any cause and hospitalization for any cause. Dr. Blumenthal and his colleagues reported on an ancillary study to HF-ACTION in which they further assessed the effects of exercise on depressive symptoms.
The study subjects were 2,322 patients whose HF failed to improve after at least 6 weeks of optimal therapy at 82 medical centers in the United States, Canada, and France. All the subjects had undergone exercise stress testing and had completed the Beck Depression Inventory II at baseline in 2003-2007. The median age was 59 years (range, 19-91 years).
The BDI-II is a self-reported measure of depressive symptoms with a possible score ranging from 0 (no depressive symptoms) to 63 (maximal depressive symptoms). A score of 14 or higher reflects clinically significant depressive symptoms. A total of 28% of the study subjects scored in this range at baseline.
A total of 1,164 subjects were randomly assigned to usual care (the control group) and 1,158 to usual care plus three supervised sessions per week on a treadmill or stationary cycle for 3 months. The exercise group was then given the exercise equipment of their choice to take home and was encouraged to increase to 90 min/week of exercise until they reached 1 year from baseline.
At 3 months, the mean BDI-II score was 8.95 for the exercise group and 9.70 for the control group. At 1 year, the mean scores were 8.86 and 9.54, respectively. Both of these differences are statistically significant, although small, the investigators said (JAMA 2012;308:465-74).
The results were more robust for the subgroup of 653 patients who scored 14 or higher on the BDI-II at baseline. At 3 months, the mean score in the exercise group was 16.66, compared with a mean score of 17.98 in the control group. At 1 year, the mean scores were 15.85 and 17.34, respectively.
In addition, the amount of exercise was inversely associated with the reduction in depressive symptoms. "Compared with a participant reporting no exercise, a participant reporting 90 minutes of exercise per week could be expected to have a 1.55-point lower BDI-II score at 3 months and a 1.67-point lower BDI-II score at 12 months," Dr. Blumenthal and his associates said.
Such a difference "is comparable with placebo-control trials involving patients with major depressive disorder," they added.
"We also observed that elevated depressive symptoms were associated with more than a 20% increase in risk for all-cause mortality and hospitalizations and that the increased risk was independent of antidepressant use and established risk factors in patients with heart failure, including age and disease severity.
"These data add to the evidence suggesting that elevated depressive symptoms, without necessarily meeting diagnostic criteria for major depressive disorder, are associated with increased risk for adverse clinical events," the researchers said.
The study findings also support the recent recommendation by the American Heart Association that all patients with cardiac disease be routinely assessed for depression, they added.
The study results also demonstrated that HF patients whose depression worsened over time were at particularly increased risk for hospitalization and death. This highlights the need to not only reduce depressive symptoms but also to prevent the worsening of existing depressive symptoms, Dr. Blumenthal and his colleagues said.
The study had two important limitations.
First, only about 40% of the patients assigned to exercise were fully adherent to the program and actually exercised 90-120 minutes per week. And 40%-50% of the patients in the control reported that they had actually exercised to some degree during follow-up. Such unintended crossover might have adversely affected the study findings.
Second, the study could not rule out the possibility that patients with more severe depressive symptoms were less likely to exercise, "so it is not clear whether exercise resulted in less depression or if depression resulted in less exercise," the researchers said.
FROM JAMA
Major Finding: Mean scores on the BDI-II were modestly but significantly lower in 1,158 heart failure patients who exercised on a treadmill or stationary bike for 90 min/week than for 1,164 controls.
Data Source: An ancillary study of the HF-ACTION randomized clinical trial involving HF patients at 82 medical centers who were followed at 3 and 12 months.
Disclosures: This study was funded by the National Heart, Lung, and Blood Institute. Dr. Blumenthal reported no conflicts of interest, and an associate reported ties to numerous industry sources.

Fitness Curbs Mortality in Men With Diabetes
HOUSTON – Physical fitness nearly halved the risk of death in men with type 2 diabetes, regardless of whether they had left ventricular hypertrophy, a longitudinal study of 866 patients found.
During a follow-up period as long as 24 years (with a median 9-year follow-up), 236 men who had left ventricular hypertrophy and were within the lower 50th percentile of physical fitness were 20% more likely to die, compared with 225 men in a reference group who did not have left ventricular hypertrophy and also had a low level of fitness. The difference in mortality risk between these two groups did not reach statistical significance.
In contrast, compared with the reference group, the risk of death was 41% lower in 218 men who were physically fit (in the upper 50th percentile) and did not have left ventricular hypertrophy and 43% lower in 187 men who were fit and did have left ventricular hypertrophy, Dr. Khaled Alswat and his associates reported in a poster presentation at the annual meeting of the Endocrine Society.
The risk reductions in the two fit groups were statistically significant.
"We, as doctors, do a lot of testing, and one of those tests should be an exercise test" for patients with diabetes who have an increased risk for heart disease, said Dr. Alswat, an endocrinology fellow at Veterans Affairs Medical Center and George Washington University, Washington. An exercise stress test provides objective measures that physicians can use to work with patients on improving their physical fitness, he said in an interview.
The patients underwent a standardized exercise stress test and echocardiographic evaluation at the VA Medical Center during 1986-2011. Those who had a peak exercise capacity of at least six metabolic equivalence tasks (METs) were considered fit, and the rest were defined as having a low level of fitness (within the lower 50th percentile).
Left ventricular hypertrophy was defined by a left ventricular mass index, calculated by dividing left ventricular mass by height in meters to the power of 2.7. A left ventricular mass index greater than 48 g/m2.7 indicated left ventricular hypertrophy.
During follow-up, 346 patients died, for an annual death rate of 4%.
Smoking significantly increased mortality risk by 54%, a multivariate Cox proportional hazards analysis showed. The study controlled for the potential influences of age, body mass index, hypertension, smoking, and medications.
The adjusted mortality risk declined by 17% for every one-MET increase in fitness, Dr. Alswat reported.
Previous studies had shown that higher exercise capacity was associated with lower mortality risk in people with diabetes, but it had not been clear whether this applies to people with diabetes and left ventricular hypertrophy, he said.
Dr. Alswat reported having no relevant financial disclosures.
HOUSTON – Physical fitness nearly halved the risk of death in men with type 2 diabetes, regardless of whether they had left ventricular hypertrophy, a longitudinal study of 866 patients found.
During a follow-up period as long as 24 years (with a median 9-year follow-up), 236 men who had left ventricular hypertrophy and were within the lower 50th percentile of physical fitness were 20% more likely to die, compared with 225 men in a reference group who did not have left ventricular hypertrophy and also had a low level of fitness. The difference in mortality risk between these two groups did not reach statistical significance.
In contrast, compared with the reference group, the risk of death was 41% lower in 218 men who were physically fit (in the upper 50th percentile) and did not have left ventricular hypertrophy and 43% lower in 187 men who were fit and did have left ventricular hypertrophy, Dr. Khaled Alswat and his associates reported in a poster presentation at the annual meeting of the Endocrine Society.
The risk reductions in the two fit groups were statistically significant.
"We, as doctors, do a lot of testing, and one of those tests should be an exercise test" for patients with diabetes who have an increased risk for heart disease, said Dr. Alswat, an endocrinology fellow at Veterans Affairs Medical Center and George Washington University, Washington. An exercise stress test provides objective measures that physicians can use to work with patients on improving their physical fitness, he said in an interview.
The patients underwent a standardized exercise stress test and echocardiographic evaluation at the VA Medical Center during 1986-2011. Those who had a peak exercise capacity of at least six metabolic equivalence tasks (METs) were considered fit, and the rest were defined as having a low level of fitness (within the lower 50th percentile).
Left ventricular hypertrophy was defined by a left ventricular mass index, calculated by dividing left ventricular mass by height in meters to the power of 2.7. A left ventricular mass index greater than 48 g/m2.7 indicated left ventricular hypertrophy.
During follow-up, 346 patients died, for an annual death rate of 4%.
Smoking significantly increased mortality risk by 54%, a multivariate Cox proportional hazards analysis showed. The study controlled for the potential influences of age, body mass index, hypertension, smoking, and medications.
The adjusted mortality risk declined by 17% for every one-MET increase in fitness, Dr. Alswat reported.
Previous studies had shown that higher exercise capacity was associated with lower mortality risk in people with diabetes, but it had not been clear whether this applies to people with diabetes and left ventricular hypertrophy, he said.
Dr. Alswat reported having no relevant financial disclosures.
HOUSTON – Physical fitness nearly halved the risk of death in men with type 2 diabetes, regardless of whether they had left ventricular hypertrophy, a longitudinal study of 866 patients found.
During a follow-up period as long as 24 years (with a median 9-year follow-up), 236 men who had left ventricular hypertrophy and were within the lower 50th percentile of physical fitness were 20% more likely to die, compared with 225 men in a reference group who did not have left ventricular hypertrophy and also had a low level of fitness. The difference in mortality risk between these two groups did not reach statistical significance.
In contrast, compared with the reference group, the risk of death was 41% lower in 218 men who were physically fit (in the upper 50th percentile) and did not have left ventricular hypertrophy and 43% lower in 187 men who were fit and did have left ventricular hypertrophy, Dr. Khaled Alswat and his associates reported in a poster presentation at the annual meeting of the Endocrine Society.
The risk reductions in the two fit groups were statistically significant.
"We, as doctors, do a lot of testing, and one of those tests should be an exercise test" for patients with diabetes who have an increased risk for heart disease, said Dr. Alswat, an endocrinology fellow at Veterans Affairs Medical Center and George Washington University, Washington. An exercise stress test provides objective measures that physicians can use to work with patients on improving their physical fitness, he said in an interview.
The patients underwent a standardized exercise stress test and echocardiographic evaluation at the VA Medical Center during 1986-2011. Those who had a peak exercise capacity of at least six metabolic equivalence tasks (METs) were considered fit, and the rest were defined as having a low level of fitness (within the lower 50th percentile).
Left ventricular hypertrophy was defined by a left ventricular mass index, calculated by dividing left ventricular mass by height in meters to the power of 2.7. A left ventricular mass index greater than 48 g/m2.7 indicated left ventricular hypertrophy.
During follow-up, 346 patients died, for an annual death rate of 4%.
Smoking significantly increased mortality risk by 54%, a multivariate Cox proportional hazards analysis showed. The study controlled for the potential influences of age, body mass index, hypertension, smoking, and medications.
The adjusted mortality risk declined by 17% for every one-MET increase in fitness, Dr. Alswat reported.
Previous studies had shown that higher exercise capacity was associated with lower mortality risk in people with diabetes, but it had not been clear whether this applies to people with diabetes and left ventricular hypertrophy, he said.
Dr. Alswat reported having no relevant financial disclosures.
AT THE ANNUAL MEETING OF THE ENDOCRINE SOCIETY
Major Finding: Among men with diabetes, physical fitness decreased mortality risk by 43% in those with (and by 41% in those without) left ventricular hypertrophy compared with men without left ventricular hypertrophy who had a low level of fitness.
Data Source: Longitudinal study of 866 men who underwent exercise stress tests and echocardiographic evaluations at one institution from 1986 to 2011 and were followed for a median of 9 years.
Disclosures: Dr. Alswat reported having no relevant financial disclosures.

2-D Echo Is Inadequate Cardiomyopathy Screen in Childhood Cancer Survivors
Transthoracic two-dimensional echocardiography appears to be inadequate for identifying cardiomyopathy in adults who survive childhood cancer, according to a cross-sectional study published online July 16 in the Journal of Clinical Oncology.
Compared with cardiac magnetic resonance imaging (CMRI), which is considered the reference standard to which other cardiac imaging techniques are compared, 2-D echocardiography had a sensitivity of only 25% and a false-negative rate of 75% in identifying cardiomyopathy in a study of 134 adult survivors of childhood cancer, said Dr. Gregory T. Armstrong of the department of epidemiology and cancer control at St. Jude Children’s Research Hospital, Memphis, and his associates.
In these relatively young and apparently healthy study subjects who had never been diagnosed as having any cardiac abnormality, nearly half (48%) were found to have the reduced cardiac mass indicative of cancer therapy–related injury. And fully 11% of subjects who were judged to have a normal ejection fraction (EF) on 2-D echocardiography were actually proved to have an EF of less than 50% on CMRI, the researchers noted.
That number easily could have been higher, but there happened to be a low absolute number of patients (16) with this degree of EF impairment in the small cohort, they pointed out.
Adults who survive childhood cancer are at risk for cardiomyopathy because of their exposure to chemotherapy and radiotherapy. Current guidelines recommend screening such adults by transthoracic 2-D echocardiography because it is noninvasive, widely available, and less expensive than other techniques.
However, the quality of the acoustic windows obtained on 2-D echo varies widely, and the method depends on geometric assumptions that may not be valid in patients who have dilated or remodeled ventricles. Three-dimensional echocardiography yields somewhat more accurate results but is not as widely available. CMRI is the most accurate noninvasive imaging technique, but is more expensive and is even less widely available, Dr. Armstrong and his colleagues explained.
They assessed the accuracy of 2-D and 3-D echocardiography against CMRI as a screen for cardiomyopathy in a longitudinal cohort of 134 adults who had been treated at St. Jude’s for childhood cancer 18-38 years earlier. All had received chest-directed radiotherapy and/or anthracycline chemotherapy, both of which are known to impair cardiac function during treatment and to raise the risk of reduced left ventricular function later in life.
The most common pediatric malignancies were acute lymphoblastic leukemia (44 subjects) and Hodgkin’s lymphoma (37 subjects).
The median age at echocardiographic screening in adulthood was 39 years (range, 22-53 years).
Of the study subjects, 20 were unable to complete CMRI for a variety of reasons. Future studies that compare imaging techniques should take into consideration this relatively high noncompletion rate (15%) for CMRI, especially in cost-benefit analyses, Dr. Armstrong and his colleagues said (J. Clin. Oncol. 2012 July 16 [doi:10.1200/JCO.2011.40.3584]).
In the remaining 114 subjects, 2-D echocardiography consistently overestimated left ventricular ejection fraction (LVEF) and underestimated both end-systolic and end-diastolic ventricular volumes.
In all, 16 subjects were identified as having markedly decreased LVEF (50% or more) by CMRI, but only 4 of them were so identified by 2-D echocardiography and only 11 of them by 3-D echocardiography.
Compared with CMRI, the sensitivity of 2-D echocardiography was only 25%; that of 3-D echo was better but still inadequate, at only 53%. And false-negative rates were high with both 2-D echocardiography (75%) and 3-D echocardiography (47%).
Of particular concern was the finding that on CMRI, 32% of the study subjects had an LVEF that was well below normal. The rate in the subgroup of patients who had received both chest irradiation and anthracycline during childhood cancer treatment was even higher, at 42%.
A total of 48% of the study subjects had a cardiac mass that was at least 2 standard deviations below normal for their age and sex, a clear sign of cardiotoxicity from their childhood cancer treatment. "Notably, even patients who received less than 150 mg/m2 of anthracyclines had a high prevalence of reduced EF (27%), stroke volume (29%), or cardiac mass (56%)," the investigators said.
Estimates derived from Medicare data suggest that at roughly $449 each, CMRI examinations cost about $217 more than does echocardiography ($232 each). Given the high rate of cardiomyopathy discovered in this cohort, and the poor sensitivity of echocardiography as a screening tool, this cost difference may be small enough to warrant a switch in the current screening recommendations from echocardiography to CMRI.
The additional cost of a CMRI-only screening strategy per case of cardiotoxicity correctly identified would be only $1,973, they noted.
The study findings suggest that in this high-risk patient population that was exposed to cardiotoxic therapy during childhood, "consideration should be given to referring survivors with an EF of 50%-59% on [2-D echocardiography] for comprehensive cardiology assessment that includes cardiac history, symptom index, and examination; biomarker assessment; consideration of [CMRI]; functional assessment by treadmill testing; and possibly medical therapy to prevent progression of disease," Dr. Armstrong and his associates said.
This study was supported by the American Society of Clinical Oncology and the American Lebanese-Syrian Associated Charities. Dr. Armstrong’s associates reported ties to General Electric and Philips Healthcare.
Transthoracic two-dimensional echocardiography appears to be inadequate for identifying cardiomyopathy in adults who survive childhood cancer, according to a cross-sectional study published online July 16 in the Journal of Clinical Oncology.
Compared with cardiac magnetic resonance imaging (CMRI), which is considered the reference standard to which other cardiac imaging techniques are compared, 2-D echocardiography had a sensitivity of only 25% and a false-negative rate of 75% in identifying cardiomyopathy in a study of 134 adult survivors of childhood cancer, said Dr. Gregory T. Armstrong of the department of epidemiology and cancer control at St. Jude Children’s Research Hospital, Memphis, and his associates.
In these relatively young and apparently healthy study subjects who had never been diagnosed as having any cardiac abnormality, nearly half (48%) were found to have the reduced cardiac mass indicative of cancer therapy–related injury. And fully 11% of subjects who were judged to have a normal ejection fraction (EF) on 2-D echocardiography were actually proved to have an EF of less than 50% on CMRI, the researchers noted.
That number easily could have been higher, but there happened to be a low absolute number of patients (16) with this degree of EF impairment in the small cohort, they pointed out.
Adults who survive childhood cancer are at risk for cardiomyopathy because of their exposure to chemotherapy and radiotherapy. Current guidelines recommend screening such adults by transthoracic 2-D echocardiography because it is noninvasive, widely available, and less expensive than other techniques.
However, the quality of the acoustic windows obtained on 2-D echo varies widely, and the method depends on geometric assumptions that may not be valid in patients who have dilated or remodeled ventricles. Three-dimensional echocardiography yields somewhat more accurate results but is not as widely available. CMRI is the most accurate noninvasive imaging technique, but is more expensive and is even less widely available, Dr. Armstrong and his colleagues explained.
They assessed the accuracy of 2-D and 3-D echocardiography against CMRI as a screen for cardiomyopathy in a longitudinal cohort of 134 adults who had been treated at St. Jude’s for childhood cancer 18-38 years earlier. All had received chest-directed radiotherapy and/or anthracycline chemotherapy, both of which are known to impair cardiac function during treatment and to raise the risk of reduced left ventricular function later in life.
The most common pediatric malignancies were acute lymphoblastic leukemia (44 subjects) and Hodgkin’s lymphoma (37 subjects).
The median age at echocardiographic screening in adulthood was 39 years (range, 22-53 years).
Of the study subjects, 20 were unable to complete CMRI for a variety of reasons. Future studies that compare imaging techniques should take into consideration this relatively high noncompletion rate (15%) for CMRI, especially in cost-benefit analyses, Dr. Armstrong and his colleagues said (J. Clin. Oncol. 2012 July 16 [doi:10.1200/JCO.2011.40.3584]).
In the remaining 114 subjects, 2-D echocardiography consistently overestimated left ventricular ejection fraction (LVEF) and underestimated both end-systolic and end-diastolic ventricular volumes.
In all, 16 subjects were identified as having markedly decreased LVEF (50% or more) by CMRI, but only 4 of them were so identified by 2-D echocardiography and only 11 of them by 3-D echocardiography.
Compared with CMRI, the sensitivity of 2-D echocardiography was only 25%; that of 3-D echo was better but still inadequate, at only 53%. And false-negative rates were high with both 2-D echocardiography (75%) and 3-D echocardiography (47%).
Of particular concern was the finding that on CMRI, 32% of the study subjects had an LVEF that was well below normal. The rate in the subgroup of patients who had received both chest irradiation and anthracycline during childhood cancer treatment was even higher, at 42%.
A total of 48% of the study subjects had a cardiac mass that was at least 2 standard deviations below normal for their age and sex, a clear sign of cardiotoxicity from their childhood cancer treatment. "Notably, even patients who received less than 150 mg/m2 of anthracyclines had a high prevalence of reduced EF (27%), stroke volume (29%), or cardiac mass (56%)," the investigators said.
Estimates derived from Medicare data suggest that at roughly $449 each, CMRI examinations cost about $217 more than does echocardiography ($232 each). Given the high rate of cardiomyopathy discovered in this cohort, and the poor sensitivity of echocardiography as a screening tool, this cost difference may be small enough to warrant a switch in the current screening recommendations from echocardiography to CMRI.
The additional cost of a CMRI-only screening strategy per case of cardiotoxicity correctly identified would be only $1,973, they noted.
The study findings suggest that in this high-risk patient population that was exposed to cardiotoxic therapy during childhood, "consideration should be given to referring survivors with an EF of 50%-59% on [2-D echocardiography] for comprehensive cardiology assessment that includes cardiac history, symptom index, and examination; biomarker assessment; consideration of [CMRI]; functional assessment by treadmill testing; and possibly medical therapy to prevent progression of disease," Dr. Armstrong and his associates said.
This study was supported by the American Society of Clinical Oncology and the American Lebanese-Syrian Associated Charities. Dr. Armstrong’s associates reported ties to General Electric and Philips Healthcare.
Transthoracic two-dimensional echocardiography appears to be inadequate for identifying cardiomyopathy in adults who survive childhood cancer, according to a cross-sectional study published online July 16 in the Journal of Clinical Oncology.
Compared with cardiac magnetic resonance imaging (CMRI), which is considered the reference standard to which other cardiac imaging techniques are compared, 2-D echocardiography had a sensitivity of only 25% and a false-negative rate of 75% in identifying cardiomyopathy in a study of 134 adult survivors of childhood cancer, said Dr. Gregory T. Armstrong of the department of epidemiology and cancer control at St. Jude Children’s Research Hospital, Memphis, and his associates.
In these relatively young and apparently healthy study subjects who had never been diagnosed as having any cardiac abnormality, nearly half (48%) were found to have the reduced cardiac mass indicative of cancer therapy–related injury. And fully 11% of subjects who were judged to have a normal ejection fraction (EF) on 2-D echocardiography were actually proved to have an EF of less than 50% on CMRI, the researchers noted.
That number easily could have been higher, but there happened to be a low absolute number of patients (16) with this degree of EF impairment in the small cohort, they pointed out.
Adults who survive childhood cancer are at risk for cardiomyopathy because of their exposure to chemotherapy and radiotherapy. Current guidelines recommend screening such adults by transthoracic 2-D echocardiography because it is noninvasive, widely available, and less expensive than other techniques.
However, the quality of the acoustic windows obtained on 2-D echo varies widely, and the method depends on geometric assumptions that may not be valid in patients who have dilated or remodeled ventricles. Three-dimensional echocardiography yields somewhat more accurate results but is not as widely available. CMRI is the most accurate noninvasive imaging technique, but is more expensive and is even less widely available, Dr. Armstrong and his colleagues explained.
They assessed the accuracy of 2-D and 3-D echocardiography against CMRI as a screen for cardiomyopathy in a longitudinal cohort of 134 adults who had been treated at St. Jude’s for childhood cancer 18-38 years earlier. All had received chest-directed radiotherapy and/or anthracycline chemotherapy, both of which are known to impair cardiac function during treatment and to raise the risk of reduced left ventricular function later in life.
The most common pediatric malignancies were acute lymphoblastic leukemia (44 subjects) and Hodgkin’s lymphoma (37 subjects).
The median age at echocardiographic screening in adulthood was 39 years (range, 22-53 years).
Of the study subjects, 20 were unable to complete CMRI for a variety of reasons. Future studies that compare imaging techniques should take into consideration this relatively high noncompletion rate (15%) for CMRI, especially in cost-benefit analyses, Dr. Armstrong and his colleagues said (J. Clin. Oncol. 2012 July 16 [doi:10.1200/JCO.2011.40.3584]).
In the remaining 114 subjects, 2-D echocardiography consistently overestimated left ventricular ejection fraction (LVEF) and underestimated both end-systolic and end-diastolic ventricular volumes.
In all, 16 subjects were identified as having markedly decreased LVEF (50% or more) by CMRI, but only 4 of them were so identified by 2-D echocardiography and only 11 of them by 3-D echocardiography.
Compared with CMRI, the sensitivity of 2-D echocardiography was only 25%; that of 3-D echo was better but still inadequate, at only 53%. And false-negative rates were high with both 2-D echocardiography (75%) and 3-D echocardiography (47%).
Of particular concern was the finding that on CMRI, 32% of the study subjects had an LVEF that was well below normal. The rate in the subgroup of patients who had received both chest irradiation and anthracycline during childhood cancer treatment was even higher, at 42%.
A total of 48% of the study subjects had a cardiac mass that was at least 2 standard deviations below normal for their age and sex, a clear sign of cardiotoxicity from their childhood cancer treatment. "Notably, even patients who received less than 150 mg/m2 of anthracyclines had a high prevalence of reduced EF (27%), stroke volume (29%), or cardiac mass (56%)," the investigators said.
Estimates derived from Medicare data suggest that at roughly $449 each, CMRI examinations cost about $217 more than does echocardiography ($232 each). Given the high rate of cardiomyopathy discovered in this cohort, and the poor sensitivity of echocardiography as a screening tool, this cost difference may be small enough to warrant a switch in the current screening recommendations from echocardiography to CMRI.
The additional cost of a CMRI-only screening strategy per case of cardiotoxicity correctly identified would be only $1,973, they noted.
The study findings suggest that in this high-risk patient population that was exposed to cardiotoxic therapy during childhood, "consideration should be given to referring survivors with an EF of 50%-59% on [2-D echocardiography] for comprehensive cardiology assessment that includes cardiac history, symptom index, and examination; biomarker assessment; consideration of [CMRI]; functional assessment by treadmill testing; and possibly medical therapy to prevent progression of disease," Dr. Armstrong and his associates said.
This study was supported by the American Society of Clinical Oncology and the American Lebanese-Syrian Associated Charities. Dr. Armstrong’s associates reported ties to General Electric and Philips Healthcare.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Major Finding: Compared with cardiac MRI, 2-D echocardiography had only a 25% sensitivity at identifying cardiomyopathy and a 75% false-negative rate, whereas 3-D echo had only a 53% sensitivity and a 47% false-negative rate.
Data Source: A cross-sectional study of simultaneous assessment of cardiac structure and function using 2-D echo, 3-D echo, and CMRI in 134 adult survivors of childhood cancer who had no apparent cardiotoxicity from their cancer treatment.
Disclosures: This study was supported by the American Society of Clinical Oncology and the American Lebanese-Syrian Associated Charities. Dr. Armstrong’s associates reported ties to General Electric and Philips Healthcare.
Studies Clash on Cardiac Effects of TKIs in Kidney Cancer
CHICAGO – Take your pick: The tyrosine kinase inhibitors sunitinib and sorafenib do/do not appear to have significant cardiac toxicity when used in adjuvant therapy for renal cell carcinoma.
Conflicting studies presented at the annual meeting of the American Society of Clinical Oncology suggest that – for now at least – it’s a toss-up.

A cardiac substudy of the phase III ECOG (Eastern Cooperative Oncology Group) E2805 ASSURE (Adjuvant Sunitinib or Sorafenib for Unfavorable Renal Carcinoma) trial, comparing either sunitinib (Sutent) or sorafenib (Nexavar) with placebo in patients with resected renal cell carcinoma (RCC), showed that neither TKI was associated with significant declines in left ventricular ejection fraction (LVEF) or other cardiac adverse events when compared with placebo, said Dr. Naomi B. Haas of the University of Pennsylvania, Philadelphia.
Left ventricular dysfunction that did occur with the TKIs was reversible, and ischemic events were uncommon and not clearly linked to therapy, she added.
"The implications for patients: Further prospective study on the effects of these agents is needed in patients who have preexisting cardiac dysfunction. This was a well population we were looking at," said Dr. Haas.
However, a retrospective study by Dr. Phillip S. Hall and colleagues at Stanford (Calif.) University found evidence of significant cardiac toxicity in patients with metastatic renal cell carcinoma that was treated with both agents and with other targeted therapies at their institution.
"Cardiovascular toxicity is an important adverse event related to targeted-therapy administration. Close monitoring for the development of CV toxicity with the use of these agents should become standard of care, as early detection of asymptomatic patients could preempt symptomatic toxicity and reduce treatment-related morbidity and mortality," they wrote in a poster presentation.
TKIs on Trial
Previous studies, most of them retrospective, have reported cardiac dysfunction with TKI use ranging from 1% to 28%. The proposed mechanism of action is through the metabolic dysfunction of cardiac myocytes, Dr. Haas said.
She and her coinvestigators in the ECOG E2805 ASSURE trial looked at data from a cardiac substudy, and asked whether either sorafenib or sunitinib was associated with a decline in LVEF, clinically significant heart failure (HF) or other effects, using multigated acquisition scans (MUGA) at baseline and at 3, 6, and 12 months (study end) or at the end of treatment.
There were nine cases of the primary end point (a decline in LVEF of 16% or greater from baseline) among 397 patients on sunitinib, seven among 394 patients on sorafenib, and five among 502 patients on placebo. The respective event rates were 2.3%, 1.8%, and 1.0%; these differences were not clinically significant.
The numbers for other cardiac events – including LVEF decline of 16% or more below the institutional level of normal occurring after 6 months, or a grade 2 or 3 left ventricular systolic or diastolic dysfunction – were also similar among the groups, occurring in 12, 11, and 11 patients, respectively.
"Looking at the data as they stand, it on the face of it is very reassuring, with the primary end point being met in a very small proportion of patients," commented the invited discussant Dr. Tim Eisen, professor of oncology at the University of Cambridge (England).
He pointed out, however, that new cardiac events were seen in the study past 6 months of therapy, which indicated that investigators should continue to monitor patients for cardiotoxicities throughout the course of therapy and in follow-up.
TKIs in Practice
The Stanford investigators looked at the incidence of hypertension, left ventricular dysfunction, changes in serum markers of cardiovascular toxicity, and heart failure in 159 patients with metastatic RCC who were treated from 2004 through 2011. They found that 116 of 159 patients (73%) developed cardiovascular toxicities.
"Sunitinib was the most frequently used and most common offending agent, with 66 of 101 sunitinib-treated patients (65%) developing a form of CV toxicity, or 32 of 101 (32%) excluding hypertension. However, it was notable that CV toxicity was observed in 68%, 66%, and 51% of patients treated with bevacizumab, sorafenib, and pazopanib as well," the investigators wrote.
They noted that there were fewer toxicities with mTOR (mammalian target of rapamycin) inhibitors than with TKIs, but the sample sizes were small.
The ECOG E2805 trial was supported by the National Cancer Institute. The Stanford study was internally funded. Dr. Haas reported having a consulting or advisory role to Boehringer Ingelheim, Dendreon, Novartis, and Pfizer, and receiving research funding from GlaxoSmithKline. Dr. Hall reported having no relevant disclosures. Dr. Eisen has received honoraria and serves in a consulting or advisory role to Astellas and AVEO.
Cardiovascular toxicity, kidney cancer
CHICAGO – Take your pick: The tyrosine kinase inhibitors sunitinib and sorafenib do/do not appear to have significant cardiac toxicity when used in adjuvant therapy for renal cell carcinoma.
Conflicting studies presented at the annual meeting of the American Society of Clinical Oncology suggest that – for now at least – it’s a toss-up.
A cardiac substudy of the phase III ECOG (Eastern Cooperative Oncology Group) E2805 ASSURE (Adjuvant Sunitinib or Sorafenib for Unfavorable Renal Carcinoma) trial, comparing either sunitinib (Sutent) or sorafenib (Nexavar) with placebo in patients with resected renal cell carcinoma (RCC), showed that neither TKI was associated with significant declines in left ventricular ejection fraction (LVEF) or other cardiac adverse events when compared with placebo, said Dr. Naomi B. Haas of the University of Pennsylvania, Philadelphia.
Left ventricular dysfunction that did occur with the TKIs was reversible, and ischemic events were uncommon and not clearly linked to therapy, she added.
"The implications for patients: Further prospective study on the effects of these agents is needed in patients who have preexisting cardiac dysfunction. This was a well population we were looking at," said Dr. Haas.
However, a retrospective study by Dr. Phillip S. Hall and colleagues at Stanford (Calif.) University found evidence of significant cardiac toxicity in patients with metastatic renal cell carcinoma that was treated with both agents and with other targeted therapies at their institution.
"Cardiovascular toxicity is an important adverse event related to targeted-therapy administration. Close monitoring for the development of CV toxicity with the use of these agents should become standard of care, as early detection of asymptomatic patients could preempt symptomatic toxicity and reduce treatment-related morbidity and mortality," they wrote in a poster presentation.
TKIs on Trial
Previous studies, most of them retrospective, have reported cardiac dysfunction with TKI use ranging from 1% to 28%. The proposed mechanism of action is through the metabolic dysfunction of cardiac myocytes, Dr. Haas said.
She and her coinvestigators in the ECOG E2805 ASSURE trial looked at data from a cardiac substudy, and asked whether either sorafenib or sunitinib was associated with a decline in LVEF, clinically significant heart failure (HF) or other effects, using multigated acquisition scans (MUGA) at baseline and at 3, 6, and 12 months (study end) or at the end of treatment.
There were nine cases of the primary end point (a decline in LVEF of 16% or greater from baseline) among 397 patients on sunitinib, seven among 394 patients on sorafenib, and five among 502 patients on placebo. The respective event rates were 2.3%, 1.8%, and 1.0%; these differences were not clinically significant.
The numbers for other cardiac events – including LVEF decline of 16% or more below the institutional level of normal occurring after 6 months, or a grade 2 or 3 left ventricular systolic or diastolic dysfunction – were also similar among the groups, occurring in 12, 11, and 11 patients, respectively.
"Looking at the data as they stand, it on the face of it is very reassuring, with the primary end point being met in a very small proportion of patients," commented the invited discussant Dr. Tim Eisen, professor of oncology at the University of Cambridge (England).
He pointed out, however, that new cardiac events were seen in the study past 6 months of therapy, which indicated that investigators should continue to monitor patients for cardiotoxicities throughout the course of therapy and in follow-up.
TKIs in Practice
The Stanford investigators looked at the incidence of hypertension, left ventricular dysfunction, changes in serum markers of cardiovascular toxicity, and heart failure in 159 patients with metastatic RCC who were treated from 2004 through 2011. They found that 116 of 159 patients (73%) developed cardiovascular toxicities.
"Sunitinib was the most frequently used and most common offending agent, with 66 of 101 sunitinib-treated patients (65%) developing a form of CV toxicity, or 32 of 101 (32%) excluding hypertension. However, it was notable that CV toxicity was observed in 68%, 66%, and 51% of patients treated with bevacizumab, sorafenib, and pazopanib as well," the investigators wrote.
They noted that there were fewer toxicities with mTOR (mammalian target of rapamycin) inhibitors than with TKIs, but the sample sizes were small.
The ECOG E2805 trial was supported by the National Cancer Institute. The Stanford study was internally funded. Dr. Haas reported having a consulting or advisory role to Boehringer Ingelheim, Dendreon, Novartis, and Pfizer, and receiving research funding from GlaxoSmithKline. Dr. Hall reported having no relevant disclosures. Dr. Eisen has received honoraria and serves in a consulting or advisory role to Astellas and AVEO.
CHICAGO – Take your pick: The tyrosine kinase inhibitors sunitinib and sorafenib do/do not appear to have significant cardiac toxicity when used in adjuvant therapy for renal cell carcinoma.
Conflicting studies presented at the annual meeting of the American Society of Clinical Oncology suggest that – for now at least – it’s a toss-up.
A cardiac substudy of the phase III ECOG (Eastern Cooperative Oncology Group) E2805 ASSURE (Adjuvant Sunitinib or Sorafenib for Unfavorable Renal Carcinoma) trial, comparing either sunitinib (Sutent) or sorafenib (Nexavar) with placebo in patients with resected renal cell carcinoma (RCC), showed that neither TKI was associated with significant declines in left ventricular ejection fraction (LVEF) or other cardiac adverse events when compared with placebo, said Dr. Naomi B. Haas of the University of Pennsylvania, Philadelphia.
Left ventricular dysfunction that did occur with the TKIs was reversible, and ischemic events were uncommon and not clearly linked to therapy, she added.
"The implications for patients: Further prospective study on the effects of these agents is needed in patients who have preexisting cardiac dysfunction. This was a well population we were looking at," said Dr. Haas.
However, a retrospective study by Dr. Phillip S. Hall and colleagues at Stanford (Calif.) University found evidence of significant cardiac toxicity in patients with metastatic renal cell carcinoma that was treated with both agents and with other targeted therapies at their institution.
"Cardiovascular toxicity is an important adverse event related to targeted-therapy administration. Close monitoring for the development of CV toxicity with the use of these agents should become standard of care, as early detection of asymptomatic patients could preempt symptomatic toxicity and reduce treatment-related morbidity and mortality," they wrote in a poster presentation.
TKIs on Trial
Previous studies, most of them retrospective, have reported cardiac dysfunction with TKI use ranging from 1% to 28%. The proposed mechanism of action is through the metabolic dysfunction of cardiac myocytes, Dr. Haas said.
She and her coinvestigators in the ECOG E2805 ASSURE trial looked at data from a cardiac substudy, and asked whether either sorafenib or sunitinib was associated with a decline in LVEF, clinically significant heart failure (HF) or other effects, using multigated acquisition scans (MUGA) at baseline and at 3, 6, and 12 months (study end) or at the end of treatment.
There were nine cases of the primary end point (a decline in LVEF of 16% or greater from baseline) among 397 patients on sunitinib, seven among 394 patients on sorafenib, and five among 502 patients on placebo. The respective event rates were 2.3%, 1.8%, and 1.0%; these differences were not clinically significant.
The numbers for other cardiac events – including LVEF decline of 16% or more below the institutional level of normal occurring after 6 months, or a grade 2 or 3 left ventricular systolic or diastolic dysfunction – were also similar among the groups, occurring in 12, 11, and 11 patients, respectively.
"Looking at the data as they stand, it on the face of it is very reassuring, with the primary end point being met in a very small proportion of patients," commented the invited discussant Dr. Tim Eisen, professor of oncology at the University of Cambridge (England).
He pointed out, however, that new cardiac events were seen in the study past 6 months of therapy, which indicated that investigators should continue to monitor patients for cardiotoxicities throughout the course of therapy and in follow-up.
TKIs in Practice
The Stanford investigators looked at the incidence of hypertension, left ventricular dysfunction, changes in serum markers of cardiovascular toxicity, and heart failure in 159 patients with metastatic RCC who were treated from 2004 through 2011. They found that 116 of 159 patients (73%) developed cardiovascular toxicities.
"Sunitinib was the most frequently used and most common offending agent, with 66 of 101 sunitinib-treated patients (65%) developing a form of CV toxicity, or 32 of 101 (32%) excluding hypertension. However, it was notable that CV toxicity was observed in 68%, 66%, and 51% of patients treated with bevacizumab, sorafenib, and pazopanib as well," the investigators wrote.
They noted that there were fewer toxicities with mTOR (mammalian target of rapamycin) inhibitors than with TKIs, but the sample sizes were small.
The ECOG E2805 trial was supported by the National Cancer Institute. The Stanford study was internally funded. Dr. Haas reported having a consulting or advisory role to Boehringer Ingelheim, Dendreon, Novartis, and Pfizer, and receiving research funding from GlaxoSmithKline. Dr. Hall reported having no relevant disclosures. Dr. Eisen has received honoraria and serves in a consulting or advisory role to Astellas and AVEO.
Cardiovascular toxicity, kidney cancer
Cardiovascular toxicity, kidney cancer
AT THE ANNUAL MEETING OF THE AMERICAN SOCIETY OF CLINICAL ONCOLOGY
Major Finding: Whereas LVEF declines of 16% or greater from baseline were seen in 1.8%-2.3% of kidney cancer patients treated with sunitinib or sorafenib in a randomized trial, most patients on targeted therapies, including sunitinib and sorafenib, developed cardiovascular toxicities, including hypertension, in a single-center study.
Data Source: Investigators from the ECOG E2805 trial and Stanford University presented prospective and retrospective findings, respectively.
Disclosures: The ECOG E2805 trial was supported by the National Cancer Institute. Dr. Haas reported having a consulting or advisory role to Boehringer Ingelheim, Dendreon, Novartis, and Pfizer, and receiving research funding from GlaxoSmithKline. Dr. Hall reported having no relevant disclosures. Dr. Eisen has received honoraria and serves in a consulting or advisory role to Astellas and AVEO.
FDA Announces Recall of Cardiac Diagnostic Tests
Certain lots of Alere Triage cardiac diagnostic tests have been recalled because their use could result in an increase in false-positive or false-negative results, the Food and Drug Administration announced on July 11.
The recall may affect laboratory supplies: The statement says that there may not be enough of these products unaffected by the recall to meet the demand in all laboratories.
The recalled products – used to aid in the diagnosis of heart failure, myocardial infarction, and other conditions – are the Triage CardioProfiler Panel PN 97100CP, Triage Cardiac Panel PN 97000HS, Triage Profiler SOB Panel PN 97300, Triage BNP PN 98000XR, and Triage D-dimer PN 98100, according to a letter issued by the manufacturer, Alere San Diego.
As many as 98,100 test kits may be defective, the FDA statement said.
"There have been reports of patients receiving inappropriate clinical management which may have been due to such erroneous results," and the product "may cause serious adverse health consequences, including death," according to the FDA statement. Quality control tests may not detect false-positive and false-negative results within lots, which are unpredictable, the statement said. For example, some lots affected by the recall provide a troponin I result that is greater than 0.05 ng/mL, which additional testing determines is lower than 0.05 ng/mL.
The manufacturer is advising that the affected product be discarded, and that unaffected lots or other methods of performing these analyses be used instead.
The lot numbers of the recalled products are available at www.alere.com/assets/articles/TriageProductRecallLotsMay22.pdf.
The recall was initiated in May.
The recall notice is available at www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm311405.htm. The manufacturer can be contacted at 877-308-8287. Adverse events associated with any of these products should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch/.
Certain lots of Alere Triage cardiac diagnostic tests have been recalled because their use could result in an increase in false-positive or false-negative results, the Food and Drug Administration announced on July 11.
The recall may affect laboratory supplies: The statement says that there may not be enough of these products unaffected by the recall to meet the demand in all laboratories.
The recalled products – used to aid in the diagnosis of heart failure, myocardial infarction, and other conditions – are the Triage CardioProfiler Panel PN 97100CP, Triage Cardiac Panel PN 97000HS, Triage Profiler SOB Panel PN 97300, Triage BNP PN 98000XR, and Triage D-dimer PN 98100, according to a letter issued by the manufacturer, Alere San Diego.
As many as 98,100 test kits may be defective, the FDA statement said.
"There have been reports of patients receiving inappropriate clinical management which may have been due to such erroneous results," and the product "may cause serious adverse health consequences, including death," according to the FDA statement. Quality control tests may not detect false-positive and false-negative results within lots, which are unpredictable, the statement said. For example, some lots affected by the recall provide a troponin I result that is greater than 0.05 ng/mL, which additional testing determines is lower than 0.05 ng/mL.
The manufacturer is advising that the affected product be discarded, and that unaffected lots or other methods of performing these analyses be used instead.
The lot numbers of the recalled products are available at www.alere.com/assets/articles/TriageProductRecallLotsMay22.pdf.
The recall was initiated in May.
The recall notice is available at www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm311405.htm. The manufacturer can be contacted at 877-308-8287. Adverse events associated with any of these products should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch/.
Certain lots of Alere Triage cardiac diagnostic tests have been recalled because their use could result in an increase in false-positive or false-negative results, the Food and Drug Administration announced on July 11.
The recall may affect laboratory supplies: The statement says that there may not be enough of these products unaffected by the recall to meet the demand in all laboratories.
The recalled products – used to aid in the diagnosis of heart failure, myocardial infarction, and other conditions – are the Triage CardioProfiler Panel PN 97100CP, Triage Cardiac Panel PN 97000HS, Triage Profiler SOB Panel PN 97300, Triage BNP PN 98000XR, and Triage D-dimer PN 98100, according to a letter issued by the manufacturer, Alere San Diego.
As many as 98,100 test kits may be defective, the FDA statement said.
"There have been reports of patients receiving inappropriate clinical management which may have been due to such erroneous results," and the product "may cause serious adverse health consequences, including death," according to the FDA statement. Quality control tests may not detect false-positive and false-negative results within lots, which are unpredictable, the statement said. For example, some lots affected by the recall provide a troponin I result that is greater than 0.05 ng/mL, which additional testing determines is lower than 0.05 ng/mL.
The manufacturer is advising that the affected product be discarded, and that unaffected lots or other methods of performing these analyses be used instead.
The lot numbers of the recalled products are available at www.alere.com/assets/articles/TriageProductRecallLotsMay22.pdf.
The recall was initiated in May.
The recall notice is available at www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm311405.htm. The manufacturer can be contacted at 877-308-8287. Adverse events associated with any of these products should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch/.
What's the Dose?
Physicians struggle every day to pick the right drug dosage for the treatment and prevention of disease. For the acute illnesses, efficacy is evident within hours or days. For the prevention of chronic disease, however, the outcome is uncertain at best. Therefore, we rely on randomized clinical trials to provide evidence that a specific drug and dosage are safe and effective.
Unfortunately, because of the limited average follow-up of 3-5 years, randomized clinical trials (RCTs) do not provide efficacy and safety information for lifetime therapy that is often advocated for the prevention of chronic disease.
For both the patient and physician, the side effects become the deciding factor. The physician usually chooses the smallest dose in order to avoid toxicity and presumably to achieve some benefit. The patient takes the drug irregularly at best.
As an example, consider the appropriate dosage for statin therapy for the prevention of atherosclerotic cardiovascular disease. Although numerous RCTs have defined the effective dose of a number of statins, recent trends in therapeutics have advocated that rather than using the dose that was used in RCTs, clinicians should increase the dose in order to reach a specific LDL cholesterol blood level.
Choosing the dosage of a drug in an RCT is a less-than-perfect exercise. Here’s how it usually goes:
Phase I trials – often based on pharmacokinetic data derived from animal studies – examine the physiological characteristics of the drug in healthy human volunteers in order to determine an effective and safe dosage prior to a phase II trial.
Phase II trials are larger; they usually examine the effect of several different dosages on a target population, and are focused not on physiological effects but on clinical outcomes and safety, in order to choose the best dosage for a phase III study. Because of their small size, these phase II studies are underpowered and prone to providing misleading dose choices.
Nevertheless, one or two doses are chosen to be used in the definitive phase III RCT, which includes enough patients to provide proof of benefit and safety of the drug based solely on its effect on mortality and morbidity.
Information is often collected in regard to the physiological effects of the drug on, for example, LDL cholesterol (in the case of statins) or heart rate (in the case of beta-blocking drugs). The proof of benefit, however, is determined by clinical outcomes, not on the physiological or "surrogate" measurements.
In the process of designing an RCT, we often make presumptions about mechanisms and will identify certain parameters that theoretically provide insight into the presumed benefit. However, many of the drugs we use have physiological effects that extend beyond the specific therapeutic target. We often remain ignorant about the mechanism by which drugs express their benefit long after their proof of benefit is demonstrated.
Statins, for instance, have a variety of pleiotropic effects. One of the most interesting is their ability to modulate inflammation, a process that is thought to be central to the progression of atherosclerotic disease. Although we presume that their effect is on LDL cholesterol, that presumption may be incorrect. Similarly, beta-blockers have well-known effects on heart rate and blood pressure, but their effect on modulating the up-regulated sympathetic nervous system in heart failure has presumed importance well beyond their effect on heart rate and blood pressure.
It is tempting to make presumptions about the effect of a drug intervention on the basis of surrogate measures like heart rate or LDL cholesterol effects, but their mechanisms of action on mortality and morbidity of disease may be unrelated to that measure.
RCTs have come a long way from relying on "surrogate" end points as the basis for making therapeutic decisions. More than 20 years ago, the CAST (Cardiac Arrhythmia Suppression Trial) was the watershed RCT that excluded the surrogate as a measure of therapeutic efficacy (J. Am. Coll. Cardiol. 1991;18:14-9). At a time when ventricular premature contraction (VPC) suppression was the "mantra" to prevent sudden death, CAST examined the pharmacologic suppression of VPCs in post–MI patients and found that, as the drugs decreased ventricular ectopy, mortality increased.
The use of the seemingly appropriate and obvious "surrogate" of LDL cholesterol lowering as a measure of therapeutic efficacy may be just as illusory. As enticing as surrogates are, the contemporary drive to lower LDL cholesterol may be as misdirected as the target to decrease the frequency of VPCs to prevent sudden death.
Like many things in life and science, things may not be what they seem.
Dr. Goldstein, the medical editor of Cardiology News, is a professor of medicine at Wayne State University and division head emeritus of cardiovascular medicine at Henry Ford Hospital, both in Detroit. He is on data safety monitoring committees for the National Institutes of Health and several pharmaceutical companies.
Physicians struggle every day to pick the right drug dosage for the treatment and prevention of disease. For the acute illnesses, efficacy is evident within hours or days. For the prevention of chronic disease, however, the outcome is uncertain at best. Therefore, we rely on randomized clinical trials to provide evidence that a specific drug and dosage are safe and effective.
Unfortunately, because of the limited average follow-up of 3-5 years, randomized clinical trials (RCTs) do not provide efficacy and safety information for lifetime therapy that is often advocated for the prevention of chronic disease.
For both the patient and physician, the side effects become the deciding factor. The physician usually chooses the smallest dose in order to avoid toxicity and presumably to achieve some benefit. The patient takes the drug irregularly at best.
As an example, consider the appropriate dosage for statin therapy for the prevention of atherosclerotic cardiovascular disease. Although numerous RCTs have defined the effective dose of a number of statins, recent trends in therapeutics have advocated that rather than using the dose that was used in RCTs, clinicians should increase the dose in order to reach a specific LDL cholesterol blood level.
Choosing the dosage of a drug in an RCT is a less-than-perfect exercise. Here’s how it usually goes:
Phase I trials – often based on pharmacokinetic data derived from animal studies – examine the physiological characteristics of the drug in healthy human volunteers in order to determine an effective and safe dosage prior to a phase II trial.
Phase II trials are larger; they usually examine the effect of several different dosages on a target population, and are focused not on physiological effects but on clinical outcomes and safety, in order to choose the best dosage for a phase III study. Because of their small size, these phase II studies are underpowered and prone to providing misleading dose choices.
Nevertheless, one or two doses are chosen to be used in the definitive phase III RCT, which includes enough patients to provide proof of benefit and safety of the drug based solely on its effect on mortality and morbidity.
Information is often collected in regard to the physiological effects of the drug on, for example, LDL cholesterol (in the case of statins) or heart rate (in the case of beta-blocking drugs). The proof of benefit, however, is determined by clinical outcomes, not on the physiological or "surrogate" measurements.
In the process of designing an RCT, we often make presumptions about mechanisms and will identify certain parameters that theoretically provide insight into the presumed benefit. However, many of the drugs we use have physiological effects that extend beyond the specific therapeutic target. We often remain ignorant about the mechanism by which drugs express their benefit long after their proof of benefit is demonstrated.
Statins, for instance, have a variety of pleiotropic effects. One of the most interesting is their ability to modulate inflammation, a process that is thought to be central to the progression of atherosclerotic disease. Although we presume that their effect is on LDL cholesterol, that presumption may be incorrect. Similarly, beta-blockers have well-known effects on heart rate and blood pressure, but their effect on modulating the up-regulated sympathetic nervous system in heart failure has presumed importance well beyond their effect on heart rate and blood pressure.
It is tempting to make presumptions about the effect of a drug intervention on the basis of surrogate measures like heart rate or LDL cholesterol effects, but their mechanisms of action on mortality and morbidity of disease may be unrelated to that measure.
RCTs have come a long way from relying on "surrogate" end points as the basis for making therapeutic decisions. More than 20 years ago, the CAST (Cardiac Arrhythmia Suppression Trial) was the watershed RCT that excluded the surrogate as a measure of therapeutic efficacy (J. Am. Coll. Cardiol. 1991;18:14-9). At a time when ventricular premature contraction (VPC) suppression was the "mantra" to prevent sudden death, CAST examined the pharmacologic suppression of VPCs in post–MI patients and found that, as the drugs decreased ventricular ectopy, mortality increased.
The use of the seemingly appropriate and obvious "surrogate" of LDL cholesterol lowering as a measure of therapeutic efficacy may be just as illusory. As enticing as surrogates are, the contemporary drive to lower LDL cholesterol may be as misdirected as the target to decrease the frequency of VPCs to prevent sudden death.
Like many things in life and science, things may not be what they seem.
Dr. Goldstein, the medical editor of Cardiology News, is a professor of medicine at Wayne State University and division head emeritus of cardiovascular medicine at Henry Ford Hospital, both in Detroit. He is on data safety monitoring committees for the National Institutes of Health and several pharmaceutical companies.
Physicians struggle every day to pick the right drug dosage for the treatment and prevention of disease. For the acute illnesses, efficacy is evident within hours or days. For the prevention of chronic disease, however, the outcome is uncertain at best. Therefore, we rely on randomized clinical trials to provide evidence that a specific drug and dosage are safe and effective.
Unfortunately, because of the limited average follow-up of 3-5 years, randomized clinical trials (RCTs) do not provide efficacy and safety information for lifetime therapy that is often advocated for the prevention of chronic disease.
For both the patient and physician, the side effects become the deciding factor. The physician usually chooses the smallest dose in order to avoid toxicity and presumably to achieve some benefit. The patient takes the drug irregularly at best.
As an example, consider the appropriate dosage for statin therapy for the prevention of atherosclerotic cardiovascular disease. Although numerous RCTs have defined the effective dose of a number of statins, recent trends in therapeutics have advocated that rather than using the dose that was used in RCTs, clinicians should increase the dose in order to reach a specific LDL cholesterol blood level.
Choosing the dosage of a drug in an RCT is a less-than-perfect exercise. Here’s how it usually goes:
Phase I trials – often based on pharmacokinetic data derived from animal studies – examine the physiological characteristics of the drug in healthy human volunteers in order to determine an effective and safe dosage prior to a phase II trial.
Phase II trials are larger; they usually examine the effect of several different dosages on a target population, and are focused not on physiological effects but on clinical outcomes and safety, in order to choose the best dosage for a phase III study. Because of their small size, these phase II studies are underpowered and prone to providing misleading dose choices.
Nevertheless, one or two doses are chosen to be used in the definitive phase III RCT, which includes enough patients to provide proof of benefit and safety of the drug based solely on its effect on mortality and morbidity.
Information is often collected in regard to the physiological effects of the drug on, for example, LDL cholesterol (in the case of statins) or heart rate (in the case of beta-blocking drugs). The proof of benefit, however, is determined by clinical outcomes, not on the physiological or "surrogate" measurements.
In the process of designing an RCT, we often make presumptions about mechanisms and will identify certain parameters that theoretically provide insight into the presumed benefit. However, many of the drugs we use have physiological effects that extend beyond the specific therapeutic target. We often remain ignorant about the mechanism by which drugs express their benefit long after their proof of benefit is demonstrated.
Statins, for instance, have a variety of pleiotropic effects. One of the most interesting is their ability to modulate inflammation, a process that is thought to be central to the progression of atherosclerotic disease. Although we presume that their effect is on LDL cholesterol, that presumption may be incorrect. Similarly, beta-blockers have well-known effects on heart rate and blood pressure, but their effect on modulating the up-regulated sympathetic nervous system in heart failure has presumed importance well beyond their effect on heart rate and blood pressure.
It is tempting to make presumptions about the effect of a drug intervention on the basis of surrogate measures like heart rate or LDL cholesterol effects, but their mechanisms of action on mortality and morbidity of disease may be unrelated to that measure.
RCTs have come a long way from relying on "surrogate" end points as the basis for making therapeutic decisions. More than 20 years ago, the CAST (Cardiac Arrhythmia Suppression Trial) was the watershed RCT that excluded the surrogate as a measure of therapeutic efficacy (J. Am. Coll. Cardiol. 1991;18:14-9). At a time when ventricular premature contraction (VPC) suppression was the "mantra" to prevent sudden death, CAST examined the pharmacologic suppression of VPCs in post–MI patients and found that, as the drugs decreased ventricular ectopy, mortality increased.
The use of the seemingly appropriate and obvious "surrogate" of LDL cholesterol lowering as a measure of therapeutic efficacy may be just as illusory. As enticing as surrogates are, the contemporary drive to lower LDL cholesterol may be as misdirected as the target to decrease the frequency of VPCs to prevent sudden death.
Like many things in life and science, things may not be what they seem.
Dr. Goldstein, the medical editor of Cardiology News, is a professor of medicine at Wayne State University and division head emeritus of cardiovascular medicine at Henry Ford Hospital, both in Detroit. He is on data safety monitoring committees for the National Institutes of Health and several pharmaceutical companies.
ICDs' Mortality Benefit Persists Up to 12 Years
BOSTON – More than a decade’s worth of follow-up of participants in the SCD-HeFT trial confirms that implantable cardioverter defibrillators in patients with moderate heart failure and reduced left ventricular systolic function can significantly reduce mortality, Dr. Jeanne Poole reported at the annual meeting of the Heart Rhythm Society.
ICD therapy was most beneficial in patients with New York Heart Association (NYHA) class II disease and ischemic heart failure, reported Dr. Poole, professor of medicine and director of the arrhythmia service and electrophysiology laboratory at the University of Washington in Seattle.
But as the original analysis of SCD-HeFT (Sudden Cardiac Death in Heart Failure Trial) showed, ICDs did not appear to benefit patients with NYHA class III disease, for whom cardiac resynchronization therapy (CRT) was not available at the time of enrollment (N. Engl. J. Med. 2005;352:225-37). Additionally, ICDs benefited patients with ischemic, but not nonischemic, heart failure, Dr. Poole noted.
Despite the significant reduction in mortality seen in some patients, "the mortality we observed at median follow-up of 11 years is substantial and reflects the reality of patients diagnosed at least a decade ago with heart failure," she said at a late-breaking abstracts session.
The SCD-HeFT trial was designed to see whether amiodarone (Cordarone, Pacerone) or a single-lead ICD, programmed conservatively to shock only, could reduce all-cause mortality compared with placebo in patients with ischemic or nonischemic NYHA class II-III heart failure with ejection fraction 35% or less.
In all, 829 patients were assigned to receive ICDs, 845 to amiodarone, and 847 to placebo during 1997-2001. The trial ended in October 2003.
An intention-to-treat analysis at 5 years (median follow-up 45.5 months) showed that although amiodarone was no better than placebo at preventing deaths, ICD treatment was associated with a 7.2% absolute risk reduction (hazard ratio, 0.77; P = .007).
The current analysis carried follow-up out an additional 5 or more years. The investigators contacted the 148 original trial enrollment sites asking for data on the patients. Two of the sites reported that all of the patients enrolled there had died, 110 others provided mortality data (89 included clinical or arrhythmia data), and 36 sites did not respond or chose not to participate.
Mortality data were available for 2,294 of the original 2,521 participants (91%).
The 12-year all-cause mortality for patients randomized to ICD treatment was 59%, compared with 64% for patients randomized to placebo (HR, 0.87; P = .028), translating into an absolute risk reduction of 5%.
Among patients with NYHA class II heart failure at enrollment, the all-cause mortality rate was significantly lower than among patients originally assigned to placebo (HR, 0.76; P = .001). However, patients with class III disease at enrollment did no better than did controls (HR, 1.06).
Similarly, patients with an ischemic heart failure etiology did better than did placebo patients (HR, 0.81; P = .001), but those with nonischemic origin did not.
Consistent with the observations in the original trial, amiodarone did not confer a survival benefit compared with placebo.
Study limitations include vital status determination on only 91% of the original participants, limited data on new ICD implants during follow-up, and limited data on long-term use of amiodarone. Additionally, "long-term mortality for patients in the original randomized treatment groups may have been confounded by multiple clinical and advanced heart failure therapies after SCD-HeFT was completed," Dr. Poole noted.
Dr. Christine M. Albert, director of the center for arrhythmia prevention at Brigham and Women’s Hospital in Boston, said in an interview that the long-term data show that clinicians need better tools than just ejection fraction for determining which patients with heart failure are most at risk and could benefit from more aggressive interventions.
"SCD-HeFT showed a 5% absolute difference. It would be nice to find a group of indicators that would tell you who is really going to be at risk for arrhythmic death but live 10 years with their heart failure. Unfortunately, because they don’t have the updated information about therapy, it’s difficult to make a lot of interpretation of their results," she said.
Dr. Albert comoderated the session in which the data were presented, but was not involved in the study.
The study was funded by the National Heart, Lung, and Blood Institute with a subsidiary grant for St. Jude Medical Corp., maker of the ICD used in the study. Dr. Poole disclosed being on the speakers bureau for St. Jude Medical, Medtronic Inc., and Boston Scientific Corp. Dr. Albert disclosed receiving research support from St. Jude Medical.
BOSTON – More than a decade’s worth of follow-up of participants in the SCD-HeFT trial confirms that implantable cardioverter defibrillators in patients with moderate heart failure and reduced left ventricular systolic function can significantly reduce mortality, Dr. Jeanne Poole reported at the annual meeting of the Heart Rhythm Society.
ICD therapy was most beneficial in patients with New York Heart Association (NYHA) class II disease and ischemic heart failure, reported Dr. Poole, professor of medicine and director of the arrhythmia service and electrophysiology laboratory at the University of Washington in Seattle.
But as the original analysis of SCD-HeFT (Sudden Cardiac Death in Heart Failure Trial) showed, ICDs did not appear to benefit patients with NYHA class III disease, for whom cardiac resynchronization therapy (CRT) was not available at the time of enrollment (N. Engl. J. Med. 2005;352:225-37). Additionally, ICDs benefited patients with ischemic, but not nonischemic, heart failure, Dr. Poole noted.
Despite the significant reduction in mortality seen in some patients, "the mortality we observed at median follow-up of 11 years is substantial and reflects the reality of patients diagnosed at least a decade ago with heart failure," she said at a late-breaking abstracts session.
The SCD-HeFT trial was designed to see whether amiodarone (Cordarone, Pacerone) or a single-lead ICD, programmed conservatively to shock only, could reduce all-cause mortality compared with placebo in patients with ischemic or nonischemic NYHA class II-III heart failure with ejection fraction 35% or less.
In all, 829 patients were assigned to receive ICDs, 845 to amiodarone, and 847 to placebo during 1997-2001. The trial ended in October 2003.
An intention-to-treat analysis at 5 years (median follow-up 45.5 months) showed that although amiodarone was no better than placebo at preventing deaths, ICD treatment was associated with a 7.2% absolute risk reduction (hazard ratio, 0.77; P = .007).
The current analysis carried follow-up out an additional 5 or more years. The investigators contacted the 148 original trial enrollment sites asking for data on the patients. Two of the sites reported that all of the patients enrolled there had died, 110 others provided mortality data (89 included clinical or arrhythmia data), and 36 sites did not respond or chose not to participate.
Mortality data were available for 2,294 of the original 2,521 participants (91%).
The 12-year all-cause mortality for patients randomized to ICD treatment was 59%, compared with 64% for patients randomized to placebo (HR, 0.87; P = .028), translating into an absolute risk reduction of 5%.
Among patients with NYHA class II heart failure at enrollment, the all-cause mortality rate was significantly lower than among patients originally assigned to placebo (HR, 0.76; P = .001). However, patients with class III disease at enrollment did no better than did controls (HR, 1.06).
Similarly, patients with an ischemic heart failure etiology did better than did placebo patients (HR, 0.81; P = .001), but those with nonischemic origin did not.
Consistent with the observations in the original trial, amiodarone did not confer a survival benefit compared with placebo.
Study limitations include vital status determination on only 91% of the original participants, limited data on new ICD implants during follow-up, and limited data on long-term use of amiodarone. Additionally, "long-term mortality for patients in the original randomized treatment groups may have been confounded by multiple clinical and advanced heart failure therapies after SCD-HeFT was completed," Dr. Poole noted.
Dr. Christine M. Albert, director of the center for arrhythmia prevention at Brigham and Women’s Hospital in Boston, said in an interview that the long-term data show that clinicians need better tools than just ejection fraction for determining which patients with heart failure are most at risk and could benefit from more aggressive interventions.
"SCD-HeFT showed a 5% absolute difference. It would be nice to find a group of indicators that would tell you who is really going to be at risk for arrhythmic death but live 10 years with their heart failure. Unfortunately, because they don’t have the updated information about therapy, it’s difficult to make a lot of interpretation of their results," she said.
Dr. Albert comoderated the session in which the data were presented, but was not involved in the study.
The study was funded by the National Heart, Lung, and Blood Institute with a subsidiary grant for St. Jude Medical Corp., maker of the ICD used in the study. Dr. Poole disclosed being on the speakers bureau for St. Jude Medical, Medtronic Inc., and Boston Scientific Corp. Dr. Albert disclosed receiving research support from St. Jude Medical.
BOSTON – More than a decade’s worth of follow-up of participants in the SCD-HeFT trial confirms that implantable cardioverter defibrillators in patients with moderate heart failure and reduced left ventricular systolic function can significantly reduce mortality, Dr. Jeanne Poole reported at the annual meeting of the Heart Rhythm Society.
ICD therapy was most beneficial in patients with New York Heart Association (NYHA) class II disease and ischemic heart failure, reported Dr. Poole, professor of medicine and director of the arrhythmia service and electrophysiology laboratory at the University of Washington in Seattle.
But as the original analysis of SCD-HeFT (Sudden Cardiac Death in Heart Failure Trial) showed, ICDs did not appear to benefit patients with NYHA class III disease, for whom cardiac resynchronization therapy (CRT) was not available at the time of enrollment (N. Engl. J. Med. 2005;352:225-37). Additionally, ICDs benefited patients with ischemic, but not nonischemic, heart failure, Dr. Poole noted.
Despite the significant reduction in mortality seen in some patients, "the mortality we observed at median follow-up of 11 years is substantial and reflects the reality of patients diagnosed at least a decade ago with heart failure," she said at a late-breaking abstracts session.
The SCD-HeFT trial was designed to see whether amiodarone (Cordarone, Pacerone) or a single-lead ICD, programmed conservatively to shock only, could reduce all-cause mortality compared with placebo in patients with ischemic or nonischemic NYHA class II-III heart failure with ejection fraction 35% or less.
In all, 829 patients were assigned to receive ICDs, 845 to amiodarone, and 847 to placebo during 1997-2001. The trial ended in October 2003.
An intention-to-treat analysis at 5 years (median follow-up 45.5 months) showed that although amiodarone was no better than placebo at preventing deaths, ICD treatment was associated with a 7.2% absolute risk reduction (hazard ratio, 0.77; P = .007).
The current analysis carried follow-up out an additional 5 or more years. The investigators contacted the 148 original trial enrollment sites asking for data on the patients. Two of the sites reported that all of the patients enrolled there had died, 110 others provided mortality data (89 included clinical or arrhythmia data), and 36 sites did not respond or chose not to participate.
Mortality data were available for 2,294 of the original 2,521 participants (91%).
The 12-year all-cause mortality for patients randomized to ICD treatment was 59%, compared with 64% for patients randomized to placebo (HR, 0.87; P = .028), translating into an absolute risk reduction of 5%.
Among patients with NYHA class II heart failure at enrollment, the all-cause mortality rate was significantly lower than among patients originally assigned to placebo (HR, 0.76; P = .001). However, patients with class III disease at enrollment did no better than did controls (HR, 1.06).
Similarly, patients with an ischemic heart failure etiology did better than did placebo patients (HR, 0.81; P = .001), but those with nonischemic origin did not.
Consistent with the observations in the original trial, amiodarone did not confer a survival benefit compared with placebo.
Study limitations include vital status determination on only 91% of the original participants, limited data on new ICD implants during follow-up, and limited data on long-term use of amiodarone. Additionally, "long-term mortality for patients in the original randomized treatment groups may have been confounded by multiple clinical and advanced heart failure therapies after SCD-HeFT was completed," Dr. Poole noted.
Dr. Christine M. Albert, director of the center for arrhythmia prevention at Brigham and Women’s Hospital in Boston, said in an interview that the long-term data show that clinicians need better tools than just ejection fraction for determining which patients with heart failure are most at risk and could benefit from more aggressive interventions.
"SCD-HeFT showed a 5% absolute difference. It would be nice to find a group of indicators that would tell you who is really going to be at risk for arrhythmic death but live 10 years with their heart failure. Unfortunately, because they don’t have the updated information about therapy, it’s difficult to make a lot of interpretation of their results," she said.
Dr. Albert comoderated the session in which the data were presented, but was not involved in the study.
The study was funded by the National Heart, Lung, and Blood Institute with a subsidiary grant for St. Jude Medical Corp., maker of the ICD used in the study. Dr. Poole disclosed being on the speakers bureau for St. Jude Medical, Medtronic Inc., and Boston Scientific Corp. Dr. Albert disclosed receiving research support from St. Jude Medical.
FROM THE ANNUAL MEETING OF THE HEART RHYTHM SOCIETY
Major Finding: The 12-year all-cause mortality rate for patients randomized to ICD in the SCD-HeFT trial was 59%, compared with 64% for patients randomized to placebo (HR, 0.87; P = .028), translating into an absolute risk reduction of 5%.
Data Source: Study was a follow-up of mortality data on patients originally enrolled in a prospective randomized trial.
Disclosures: The study was funded by the NHLBI with a subsidiary grant for St. Jude Medical Corp, maker of the ICD used in the study. Dr. Poole disclosed being on the speakers bureaus for St. Jude Medical, Medtronic, and Boston Scientific. Dr. Albert disclosed receiving research support from St. Jude Medical.
Shift From Atrial Overdrive Pacing for AF Prevention Urged
BOSTON – Continuous atrial overdrive pacing does not prevent the development of new atrial fibrillation and is not a useful feature of pacemakers for patients with no history of AF, a researcher said at the annual meeting of the Heart Rhythm Society.
A secondary analysis of data from the Asymptomatic Atrial Fibrillation and Stroke Evaluation in Pacemaker Patients and the Atrial Fibrillation Reduction Atrial Pacing Trial (ASSERT) of atrial pacing in older patients with no history of AF showed that continuous atrial overdrive pacing (CAOP) had "no discernible effect" on the incidence of new atrial tachyarrhythmia, AF longer than 6 minutes, or AF burden, reported Dr. Stefan Hohnloser, director of clinical electrophysiology at J.W. Goethe University in Frankfurt, Germany.

Atrial preventive pacing "was associated with a high rate of crossover to alternate pacing mode, an increase in AF burden in patients with minimal ventricular pacing, more false-positive detection of atrial fibrillation by the pacemaker, and more frequent pacemaker generator replacement," Dr. Hohnloser said.
There have been 24 small or moderately sized studies of atrial preventive pacing in more than 10,000 patients, yet the results of those studies have been muddied by relatively short follow-up, the use of many different devices from different manufacturers, different pacing algorithms, and variations in atrial lead placements, hence the rationale for the secondary goal of ASSERT, he said.
ASSERT was a randomized study of 2,343 patients aged 65 and older with a history of hypertension but no history of AF or prior use of a vitamin K antagonist. The primary hypothesis that subclinical AF detected by pacemakers or implantable cardioverter defibrillators (ICDs) could predict increased risk of stroke or systemic embolism was borne out by the results.
The same could not be said, however, for the secondary hypothesis that CAOP could prevent development of AF and clinical end points, Dr. Hohnloser said.
After a 3-month run-in period to determine the presence or absence of subclinical AF, patients were randomized in a single-blind fashion to have the CAOP feature of their devices switched on or kept off. Patients were followed every 3 months. Independent adjudicators read all device-stored electrograms longer than 6 minutes.
Over 2.5 years of follow-up, there were no significant differences between the CAOP-on or -off groups in time to atrial tachyarrhythmia or to a composite clinical end point of stroke, myocardial infarction, cardiovascular death, systemic embolism, or heart-failure hospitalization, he said.
A subgroup analysis showed that atrial lead position, atrioventricular node disease with or without sinus node disease, sinus node disease alone, or history of heart failure were not significant predictors of treatment effect by randomization. However, patients who spent less than the median time in ventricular pacing at 6 months (59%) had significantly more of the primary outcome events, compared with patients who spent more than 59% of the time in ventricular pacing, he reported.
In all, 11.4% of patients assigned to CAOP on at study entry were crossed over to CAOP off, compared with only 1.0% of patients in the off group who were crossed over to continuous atrial overdrive pacing, a significant difference.
One or more false-positive AF detections occurred in 23% of patients with continuous pacing, compared with 7.7% of those with it turned off, for a significant relative risk of 2.99.

Pacemaker generator replacement was required in 4.4% of patients with CAOP on, compared with 2.5% of those with it off (relative risk, 1.70; P = .02). This result was expected, Dr. Hohnloser said, because of the extra workload on the pacemakers in continuous overdrive.
The results confirm that patients not already in atrial fibrillation do not appear to benefit from CAOP, but the study did not address whether the strategy benefits patients who already have AF, commented Dr. Richard I. Fogel from the St. Vincent Medical Group, Indianapolis, in an interview.
"The question didn’t address whether it decreases the atrial fibrillation burden. But I think it’s very clear that if you don’t have atrial fibrillation and you have had it, you shouldn’t use this algorithm. Although you have to wonder whether there aren’t some subsets of patients who might benefit," he said. Dr. Fogel moderated the late-breaking abstracts session but was not involved in the study.
The ASSERT trial was funded by St. Jude Medical. Dr. Hohnloser disclosed serving as a consultant, member of the steering committee, and speakers bureau member for St. Jude Medical and other companies. Dr. Fogel disclosed that he has received grants for clinical research and grants for educational activities from St. Jude Medical, Medtronic, and Guidant and owns stock in Medtronic and Guidant.
BOSTON – Continuous atrial overdrive pacing does not prevent the development of new atrial fibrillation and is not a useful feature of pacemakers for patients with no history of AF, a researcher said at the annual meeting of the Heart Rhythm Society.
A secondary analysis of data from the Asymptomatic Atrial Fibrillation and Stroke Evaluation in Pacemaker Patients and the Atrial Fibrillation Reduction Atrial Pacing Trial (ASSERT) of atrial pacing in older patients with no history of AF showed that continuous atrial overdrive pacing (CAOP) had "no discernible effect" on the incidence of new atrial tachyarrhythmia, AF longer than 6 minutes, or AF burden, reported Dr. Stefan Hohnloser, director of clinical electrophysiology at J.W. Goethe University in Frankfurt, Germany.
Atrial preventive pacing "was associated with a high rate of crossover to alternate pacing mode, an increase in AF burden in patients with minimal ventricular pacing, more false-positive detection of atrial fibrillation by the pacemaker, and more frequent pacemaker generator replacement," Dr. Hohnloser said.
There have been 24 small or moderately sized studies of atrial preventive pacing in more than 10,000 patients, yet the results of those studies have been muddied by relatively short follow-up, the use of many different devices from different manufacturers, different pacing algorithms, and variations in atrial lead placements, hence the rationale for the secondary goal of ASSERT, he said.
ASSERT was a randomized study of 2,343 patients aged 65 and older with a history of hypertension but no history of AF or prior use of a vitamin K antagonist. The primary hypothesis that subclinical AF detected by pacemakers or implantable cardioverter defibrillators (ICDs) could predict increased risk of stroke or systemic embolism was borne out by the results.
The same could not be said, however, for the secondary hypothesis that CAOP could prevent development of AF and clinical end points, Dr. Hohnloser said.
After a 3-month run-in period to determine the presence or absence of subclinical AF, patients were randomized in a single-blind fashion to have the CAOP feature of their devices switched on or kept off. Patients were followed every 3 months. Independent adjudicators read all device-stored electrograms longer than 6 minutes.
Over 2.5 years of follow-up, there were no significant differences between the CAOP-on or -off groups in time to atrial tachyarrhythmia or to a composite clinical end point of stroke, myocardial infarction, cardiovascular death, systemic embolism, or heart-failure hospitalization, he said.
A subgroup analysis showed that atrial lead position, atrioventricular node disease with or without sinus node disease, sinus node disease alone, or history of heart failure were not significant predictors of treatment effect by randomization. However, patients who spent less than the median time in ventricular pacing at 6 months (59%) had significantly more of the primary outcome events, compared with patients who spent more than 59% of the time in ventricular pacing, he reported.
In all, 11.4% of patients assigned to CAOP on at study entry were crossed over to CAOP off, compared with only 1.0% of patients in the off group who were crossed over to continuous atrial overdrive pacing, a significant difference.
One or more false-positive AF detections occurred in 23% of patients with continuous pacing, compared with 7.7% of those with it turned off, for a significant relative risk of 2.99.
Pacemaker generator replacement was required in 4.4% of patients with CAOP on, compared with 2.5% of those with it off (relative risk, 1.70; P = .02). This result was expected, Dr. Hohnloser said, because of the extra workload on the pacemakers in continuous overdrive.
The results confirm that patients not already in atrial fibrillation do not appear to benefit from CAOP, but the study did not address whether the strategy benefits patients who already have AF, commented Dr. Richard I. Fogel from the St. Vincent Medical Group, Indianapolis, in an interview.
"The question didn’t address whether it decreases the atrial fibrillation burden. But I think it’s very clear that if you don’t have atrial fibrillation and you have had it, you shouldn’t use this algorithm. Although you have to wonder whether there aren’t some subsets of patients who might benefit," he said. Dr. Fogel moderated the late-breaking abstracts session but was not involved in the study.
The ASSERT trial was funded by St. Jude Medical. Dr. Hohnloser disclosed serving as a consultant, member of the steering committee, and speakers bureau member for St. Jude Medical and other companies. Dr. Fogel disclosed that he has received grants for clinical research and grants for educational activities from St. Jude Medical, Medtronic, and Guidant and owns stock in Medtronic and Guidant.
BOSTON – Continuous atrial overdrive pacing does not prevent the development of new atrial fibrillation and is not a useful feature of pacemakers for patients with no history of AF, a researcher said at the annual meeting of the Heart Rhythm Society.
A secondary analysis of data from the Asymptomatic Atrial Fibrillation and Stroke Evaluation in Pacemaker Patients and the Atrial Fibrillation Reduction Atrial Pacing Trial (ASSERT) of atrial pacing in older patients with no history of AF showed that continuous atrial overdrive pacing (CAOP) had "no discernible effect" on the incidence of new atrial tachyarrhythmia, AF longer than 6 minutes, or AF burden, reported Dr. Stefan Hohnloser, director of clinical electrophysiology at J.W. Goethe University in Frankfurt, Germany.
Atrial preventive pacing "was associated with a high rate of crossover to alternate pacing mode, an increase in AF burden in patients with minimal ventricular pacing, more false-positive detection of atrial fibrillation by the pacemaker, and more frequent pacemaker generator replacement," Dr. Hohnloser said.
There have been 24 small or moderately sized studies of atrial preventive pacing in more than 10,000 patients, yet the results of those studies have been muddied by relatively short follow-up, the use of many different devices from different manufacturers, different pacing algorithms, and variations in atrial lead placements, hence the rationale for the secondary goal of ASSERT, he said.
ASSERT was a randomized study of 2,343 patients aged 65 and older with a history of hypertension but no history of AF or prior use of a vitamin K antagonist. The primary hypothesis that subclinical AF detected by pacemakers or implantable cardioverter defibrillators (ICDs) could predict increased risk of stroke or systemic embolism was borne out by the results.
The same could not be said, however, for the secondary hypothesis that CAOP could prevent development of AF and clinical end points, Dr. Hohnloser said.
After a 3-month run-in period to determine the presence or absence of subclinical AF, patients were randomized in a single-blind fashion to have the CAOP feature of their devices switched on or kept off. Patients were followed every 3 months. Independent adjudicators read all device-stored electrograms longer than 6 minutes.
Over 2.5 years of follow-up, there were no significant differences between the CAOP-on or -off groups in time to atrial tachyarrhythmia or to a composite clinical end point of stroke, myocardial infarction, cardiovascular death, systemic embolism, or heart-failure hospitalization, he said.
A subgroup analysis showed that atrial lead position, atrioventricular node disease with or without sinus node disease, sinus node disease alone, or history of heart failure were not significant predictors of treatment effect by randomization. However, patients who spent less than the median time in ventricular pacing at 6 months (59%) had significantly more of the primary outcome events, compared with patients who spent more than 59% of the time in ventricular pacing, he reported.
In all, 11.4% of patients assigned to CAOP on at study entry were crossed over to CAOP off, compared with only 1.0% of patients in the off group who were crossed over to continuous atrial overdrive pacing, a significant difference.
One or more false-positive AF detections occurred in 23% of patients with continuous pacing, compared with 7.7% of those with it turned off, for a significant relative risk of 2.99.
Pacemaker generator replacement was required in 4.4% of patients with CAOP on, compared with 2.5% of those with it off (relative risk, 1.70; P = .02). This result was expected, Dr. Hohnloser said, because of the extra workload on the pacemakers in continuous overdrive.
The results confirm that patients not already in atrial fibrillation do not appear to benefit from CAOP, but the study did not address whether the strategy benefits patients who already have AF, commented Dr. Richard I. Fogel from the St. Vincent Medical Group, Indianapolis, in an interview.
"The question didn’t address whether it decreases the atrial fibrillation burden. But I think it’s very clear that if you don’t have atrial fibrillation and you have had it, you shouldn’t use this algorithm. Although you have to wonder whether there aren’t some subsets of patients who might benefit," he said. Dr. Fogel moderated the late-breaking abstracts session but was not involved in the study.
The ASSERT trial was funded by St. Jude Medical. Dr. Hohnloser disclosed serving as a consultant, member of the steering committee, and speakers bureau member for St. Jude Medical and other companies. Dr. Fogel disclosed that he has received grants for clinical research and grants for educational activities from St. Jude Medical, Medtronic, and Guidant and owns stock in Medtronic and Guidant.
FROM THE ANNUAL MEETING OF THE HEART RHYTHM SOCIETY
Major Finding: Over 2.5 years of follow-up, there were no significant differences in time to atrial tachyarrhythmia or to a composite clinical end point of stroke, MI, cardiovascular death, systemic embolism, or heart failure hospitalization between patients with no history of atrial fibrillation who had pacemakers/ICDs set to continuous atrial overdrive pacing on or off.
Data Source: The substudy was a secondary analysis of data from the randomized, prospective ASSERT trial.
Disclosures: ASSERT was funded by St. Jude Medical. Dr. Hohnloser disclosed serving as a consultant, member of the steering committee, and speakers bureau member for St. Jude Medical and other companies. Dr. Fogel disclosed that he has received grants for clinical research and grants for educational activities from St. Jude Medical, Medtronic, and Guidant and owns stock in Medtronic and Guidant.