User login
Type 2 diabetes boosts risk of death in heart failure patients by 70%
VIENNA – Among patients with ischemic heart failure, type 2 diabetes increased the risk of death by 70% over 2 years.
Even patients with a prior revascularization were in danger, with a 60% increased risk of death. The findings were slightly better for patients with a preserved ejection fraction of at least 50%; type 2 diabetes increased their mortality risk by 40%, Dr. Anna Norhammar and Dr. Isabelle Johansson of Karolinska Institute, Stockholm, reported at the annual meeting of the European Society for the Study of Diabetes.
The results were drawn from the large Swedish Heart Failure Registry. It contains data on about 41,000 patients who have been treated for heart failure since 2003. Of these, 17,673 had ischemic heart failure, and 30% had both type 2 diabetes and ischemic heart failure.
These patients were younger than those without diabetes (75 vs. 77 years), and congestive heart failure of at least 6 months was present in 61% of those with diabetes and 54% of those without it. Those with diabetes had more severe heart failure, with a New York Heart Association class of III/IV in 53%, compared with 46% of those without diabetes, Dr. Norhammar reported.
Hypertension and atrial fibrillation were significantly more common among those with diabetes. A prior revascularization had occurred in 48% of those without diabetes, compared with 54% of those with diabetes.
In a multivariate model that adjusted for 29 variables, the investigators found that type 2 diabetes increased the risk of death by 70% over a median follow-up of 22 months. Prior revascularization cut the risk by 60%.
Dr. Johansson presented a separate analysis of patients who had a preserved ejection fraction of at least 50% (6,705). Of these, 1,658 had type 2 diabetes. Again, these patients were younger (76 vs. 78 years), more likely to have congestive heart failure (53% vs. 48%), and more likely to be in the NYHA class III/IV (44% vs. 39%). More of those with diabetes had concomitant ischemic heart disease, hypertension, atrial fibrillation, and valvular heart disease.
In this multivariate analysis, having type 2 diabetes increased the risk of death by 40%, Dr. Johansson said.
Dr. Norhammar and Dr. Johansson had no relevant financial disclosures.
On Twitter @alz_gal
VIENNA – Among patients with ischemic heart failure, type 2 diabetes increased the risk of death by 70% over 2 years.
Even patients with a prior revascularization were in danger, with a 60% increased risk of death. The findings were slightly better for patients with a preserved ejection fraction of at least 50%; type 2 diabetes increased their mortality risk by 40%, Dr. Anna Norhammar and Dr. Isabelle Johansson of Karolinska Institute, Stockholm, reported at the annual meeting of the European Society for the Study of Diabetes.
The results were drawn from the large Swedish Heart Failure Registry. It contains data on about 41,000 patients who have been treated for heart failure since 2003. Of these, 17,673 had ischemic heart failure, and 30% had both type 2 diabetes and ischemic heart failure.
These patients were younger than those without diabetes (75 vs. 77 years), and congestive heart failure of at least 6 months was present in 61% of those with diabetes and 54% of those without it. Those with diabetes had more severe heart failure, with a New York Heart Association class of III/IV in 53%, compared with 46% of those without diabetes, Dr. Norhammar reported.
Hypertension and atrial fibrillation were significantly more common among those with diabetes. A prior revascularization had occurred in 48% of those without diabetes, compared with 54% of those with diabetes.
In a multivariate model that adjusted for 29 variables, the investigators found that type 2 diabetes increased the risk of death by 70% over a median follow-up of 22 months. Prior revascularization cut the risk by 60%.
Dr. Johansson presented a separate analysis of patients who had a preserved ejection fraction of at least 50% (6,705). Of these, 1,658 had type 2 diabetes. Again, these patients were younger (76 vs. 78 years), more likely to have congestive heart failure (53% vs. 48%), and more likely to be in the NYHA class III/IV (44% vs. 39%). More of those with diabetes had concomitant ischemic heart disease, hypertension, atrial fibrillation, and valvular heart disease.
In this multivariate analysis, having type 2 diabetes increased the risk of death by 40%, Dr. Johansson said.
Dr. Norhammar and Dr. Johansson had no relevant financial disclosures.
On Twitter @alz_gal
VIENNA – Among patients with ischemic heart failure, type 2 diabetes increased the risk of death by 70% over 2 years.
Even patients with a prior revascularization were in danger, with a 60% increased risk of death. The findings were slightly better for patients with a preserved ejection fraction of at least 50%; type 2 diabetes increased their mortality risk by 40%, Dr. Anna Norhammar and Dr. Isabelle Johansson of Karolinska Institute, Stockholm, reported at the annual meeting of the European Society for the Study of Diabetes.
The results were drawn from the large Swedish Heart Failure Registry. It contains data on about 41,000 patients who have been treated for heart failure since 2003. Of these, 17,673 had ischemic heart failure, and 30% had both type 2 diabetes and ischemic heart failure.
These patients were younger than those without diabetes (75 vs. 77 years), and congestive heart failure of at least 6 months was present in 61% of those with diabetes and 54% of those without it. Those with diabetes had more severe heart failure, with a New York Heart Association class of III/IV in 53%, compared with 46% of those without diabetes, Dr. Norhammar reported.
Hypertension and atrial fibrillation were significantly more common among those with diabetes. A prior revascularization had occurred in 48% of those without diabetes, compared with 54% of those with diabetes.
In a multivariate model that adjusted for 29 variables, the investigators found that type 2 diabetes increased the risk of death by 70% over a median follow-up of 22 months. Prior revascularization cut the risk by 60%.
Dr. Johansson presented a separate analysis of patients who had a preserved ejection fraction of at least 50% (6,705). Of these, 1,658 had type 2 diabetes. Again, these patients were younger (76 vs. 78 years), more likely to have congestive heart failure (53% vs. 48%), and more likely to be in the NYHA class III/IV (44% vs. 39%). More of those with diabetes had concomitant ischemic heart disease, hypertension, atrial fibrillation, and valvular heart disease.
In this multivariate analysis, having type 2 diabetes increased the risk of death by 40%, Dr. Johansson said.
Dr. Norhammar and Dr. Johansson had no relevant financial disclosures.
On Twitter @alz_gal
AT EASD 2014
Key clinical point: Among patients with ischemic heart failure, type 2 diabetes is associated with an increased mortality risk.
Major finding: Type 2 diabetes increased the risk of death by 70% in patients with ischemic heart disease.
Data source: A prospective database study of 41,000 patients with heart failure.
Disclosures: Dr. Norhammar and Dr. Johansson had no relevant financial disclosures.
Wearable defibrillator impresses as bridge to ICD decision
BARCELONA – Use of the LifeVest wearable cardioverter defibrillator for 3 months is a safe and effective means of buying time to decide whether to place a permanent implantable cardioverter defibrillator for primary prevention of sudden cardiac death in potential candidates, according to the first report from a large prospective U.S. registry.
The wearable cardioverter defibrillator (WCD) simultaneously provides patient monitoring and reliable protection against fatal arrhythmias while physicians wait to see if, for example, a patient’s left ventricular ejection fraction, depressed to less than 35% immediately post-MI, will recover over the first several months or if life-threatening arrhythmias will arise warranting implantable cardioverter defibrillator (ICD) implantation, Dr. Valentina Kutyifa explained while presenting the results of the WEARIT-II registry at the annual congress of the European Society of Cardiology.

"The WCD seems to be a powerful risk-assessment tool to identify patients at a high risk for sudden cardiac death who need subsequent ICD implantation. The WCD can be used as a bridge to a decision for appropriate ICD implantation in patients with a low ejection fraction who are immediately post-MI, or following coronary revascularization, or with new-onset dilated cardiomyopathy who are at high risk for sudden cardiac death until stabilization, or who have an inherited arrhythmia or congenital disorder," said Dr. Kutyifa of the University of Rochester (N.Y.).
She reported on 2,000 U.S. patients in those categories who participated in the prospective WEARIT-II (Prospective Registry and Follow-Up of Patients Using the Wearable Defibrillator) registry. All received from their physicians a 3-month prescription for the LifeVest WCD, which is covered by Medicare and by most health plans. Of the 2,000 patients, 927 had a left ventricular ejection fraction (LVEF) of 35% or below owing to nonischemic cardiomyopathy, 805 had ischemic cardiomyopathy, and 268 had an inherited or congenital arrhythmogenic disorder, such as long QT syndrome or arrhythmogenic right ventricular dysplasia.
Participants’ median ejection fraction at enrollment was 25%, and 52% of subjects had heart failure symptoms at that time. Patients wore the WCD for a median of 22.5 hours per day, with no difference in wear time by disease etiology.
In terms of WCD safety, the inappropriate shock rate was just 0.5% in 2,000 patients during 3 months. No study deaths occurred related to unsuccessful attempts at termination of ventricular tachycardia (VT) or ventricular fibrillation (VF) by the WCD. Three patients died while wearing the WCD, and in all three cases the WCD detected asystole at the time of death.
During 90 days of WCD wear, 2.1% of patients experienced VT or VF, for an event rate of 22 per 100 person-years. The WCD administered a shock for VT/VF in 1.1% of patients. Sustained VT or VF occurred in 1.4% of patients, while 3.6% of participants experienced atrial arrhythmias or supraventricular tachycardia, with an event rate of 121 per 100 person-years.
At the end of the 3-month period of WCD use, 42% of patients with ischemic cardiomyopathy got an ICD, as did 36% of those with nonischemic cardiomyopathy and 46% who had a congenital or inherited condition. The main reason for not implanting an ICD was a boost in ejection fraction.
The WCD frequently detected arrhythmias which facilitated the decision whether to implant an ICD or not. Eighty-five percent of patients with VT/VF treated with a WCD shock got an ICD, as did 65% of those with sustained VT that terminated spontaneously during the wearable device’s detection time. Of patients with WCD-detected atrial arrhythmias, 48% received an ICD, as did 39% of those with no arrhythmias during the 3-month period.
Dr. Kutyifa characterized the WCD as helping to fill an unmet need for improved selection of patients for primary ICD therapy. In the landmark MADIT-II trial, for example, only 4% of patients received an appropriate ICD shock during 4 years of follow-up (N. Engl. J. Med. 2002;346:877-82).
Asked about the cost-effectiveness of WCD as a bridge to the ICD decision, she replied that such data weren’t included in the WEARIT-II registry.
"The up-front cost of the device may seem rather high, but if you think about the fact that if we use the WCD and are then able to make the decision to not implant an ICD in patients who don’t need it, we really need to look at the long-term savings: the cost of the ICD, battery changes, and issues of possible lead extraction. So I think long-term this management strategy would be cost-effective," the electrophysiologist said, adding that the WCD is also rentable.
At the congress-closing conference highlights overview session, Dr. Sylvia G. Priori singled out the WEARIT-II registry as one of the top developments in the field of arrhythmias presented at this year’s meeting. She noted that the WCD has been available for several years – Medicare covers it, as do most U.S. health plans – yet until now many physicians have wanted to see stronger evidence of safety and effectiveness before incorporating the WCD into their own practices. WEARIT-II, she said, provides that supporting evidence.
"This was quite an important study. It showed the device is able to recognize life-threatening arrhythmias, so it is safe, and it does not deliver an excessive number of inappropriate shocks. And there were no deaths related to the device," commented Dr. Priori of the University of Pavia (Italy).
"The take-home message is that, yes, we know there are patients where we are reluctant to immediately say that an ICD should be implanted forever, and now we should consider testing with this WCD, which has a low risk of inappropriate shocks and a good record in shocking patients out of the arrhythmia," she added.
The WEARIT-II registry is now expanding via enrollment of patients in Europe and Israel. The registry is funded by Zoll, which markets the WCD. Dr. Kutyifa reported having received research grants from the company as well as from other device manufacturers.
BARCELONA – Use of the LifeVest wearable cardioverter defibrillator for 3 months is a safe and effective means of buying time to decide whether to place a permanent implantable cardioverter defibrillator for primary prevention of sudden cardiac death in potential candidates, according to the first report from a large prospective U.S. registry.
The wearable cardioverter defibrillator (WCD) simultaneously provides patient monitoring and reliable protection against fatal arrhythmias while physicians wait to see if, for example, a patient’s left ventricular ejection fraction, depressed to less than 35% immediately post-MI, will recover over the first several months or if life-threatening arrhythmias will arise warranting implantable cardioverter defibrillator (ICD) implantation, Dr. Valentina Kutyifa explained while presenting the results of the WEARIT-II registry at the annual congress of the European Society of Cardiology.
"The WCD seems to be a powerful risk-assessment tool to identify patients at a high risk for sudden cardiac death who need subsequent ICD implantation. The WCD can be used as a bridge to a decision for appropriate ICD implantation in patients with a low ejection fraction who are immediately post-MI, or following coronary revascularization, or with new-onset dilated cardiomyopathy who are at high risk for sudden cardiac death until stabilization, or who have an inherited arrhythmia or congenital disorder," said Dr. Kutyifa of the University of Rochester (N.Y.).
She reported on 2,000 U.S. patients in those categories who participated in the prospective WEARIT-II (Prospective Registry and Follow-Up of Patients Using the Wearable Defibrillator) registry. All received from their physicians a 3-month prescription for the LifeVest WCD, which is covered by Medicare and by most health plans. Of the 2,000 patients, 927 had a left ventricular ejection fraction (LVEF) of 35% or below owing to nonischemic cardiomyopathy, 805 had ischemic cardiomyopathy, and 268 had an inherited or congenital arrhythmogenic disorder, such as long QT syndrome or arrhythmogenic right ventricular dysplasia.
Participants’ median ejection fraction at enrollment was 25%, and 52% of subjects had heart failure symptoms at that time. Patients wore the WCD for a median of 22.5 hours per day, with no difference in wear time by disease etiology.
In terms of WCD safety, the inappropriate shock rate was just 0.5% in 2,000 patients during 3 months. No study deaths occurred related to unsuccessful attempts at termination of ventricular tachycardia (VT) or ventricular fibrillation (VF) by the WCD. Three patients died while wearing the WCD, and in all three cases the WCD detected asystole at the time of death.
During 90 days of WCD wear, 2.1% of patients experienced VT or VF, for an event rate of 22 per 100 person-years. The WCD administered a shock for VT/VF in 1.1% of patients. Sustained VT or VF occurred in 1.4% of patients, while 3.6% of participants experienced atrial arrhythmias or supraventricular tachycardia, with an event rate of 121 per 100 person-years.
At the end of the 3-month period of WCD use, 42% of patients with ischemic cardiomyopathy got an ICD, as did 36% of those with nonischemic cardiomyopathy and 46% who had a congenital or inherited condition. The main reason for not implanting an ICD was a boost in ejection fraction.
The WCD frequently detected arrhythmias which facilitated the decision whether to implant an ICD or not. Eighty-five percent of patients with VT/VF treated with a WCD shock got an ICD, as did 65% of those with sustained VT that terminated spontaneously during the wearable device’s detection time. Of patients with WCD-detected atrial arrhythmias, 48% received an ICD, as did 39% of those with no arrhythmias during the 3-month period.
Dr. Kutyifa characterized the WCD as helping to fill an unmet need for improved selection of patients for primary ICD therapy. In the landmark MADIT-II trial, for example, only 4% of patients received an appropriate ICD shock during 4 years of follow-up (N. Engl. J. Med. 2002;346:877-82).
Asked about the cost-effectiveness of WCD as a bridge to the ICD decision, she replied that such data weren’t included in the WEARIT-II registry.
"The up-front cost of the device may seem rather high, but if you think about the fact that if we use the WCD and are then able to make the decision to not implant an ICD in patients who don’t need it, we really need to look at the long-term savings: the cost of the ICD, battery changes, and issues of possible lead extraction. So I think long-term this management strategy would be cost-effective," the electrophysiologist said, adding that the WCD is also rentable.
At the congress-closing conference highlights overview session, Dr. Sylvia G. Priori singled out the WEARIT-II registry as one of the top developments in the field of arrhythmias presented at this year’s meeting. She noted that the WCD has been available for several years – Medicare covers it, as do most U.S. health plans – yet until now many physicians have wanted to see stronger evidence of safety and effectiveness before incorporating the WCD into their own practices. WEARIT-II, she said, provides that supporting evidence.
"This was quite an important study. It showed the device is able to recognize life-threatening arrhythmias, so it is safe, and it does not deliver an excessive number of inappropriate shocks. And there were no deaths related to the device," commented Dr. Priori of the University of Pavia (Italy).
"The take-home message is that, yes, we know there are patients where we are reluctant to immediately say that an ICD should be implanted forever, and now we should consider testing with this WCD, which has a low risk of inappropriate shocks and a good record in shocking patients out of the arrhythmia," she added.
The WEARIT-II registry is now expanding via enrollment of patients in Europe and Israel. The registry is funded by Zoll, which markets the WCD. Dr. Kutyifa reported having received research grants from the company as well as from other device manufacturers.
BARCELONA – Use of the LifeVest wearable cardioverter defibrillator for 3 months is a safe and effective means of buying time to decide whether to place a permanent implantable cardioverter defibrillator for primary prevention of sudden cardiac death in potential candidates, according to the first report from a large prospective U.S. registry.
The wearable cardioverter defibrillator (WCD) simultaneously provides patient monitoring and reliable protection against fatal arrhythmias while physicians wait to see if, for example, a patient’s left ventricular ejection fraction, depressed to less than 35% immediately post-MI, will recover over the first several months or if life-threatening arrhythmias will arise warranting implantable cardioverter defibrillator (ICD) implantation, Dr. Valentina Kutyifa explained while presenting the results of the WEARIT-II registry at the annual congress of the European Society of Cardiology.
"The WCD seems to be a powerful risk-assessment tool to identify patients at a high risk for sudden cardiac death who need subsequent ICD implantation. The WCD can be used as a bridge to a decision for appropriate ICD implantation in patients with a low ejection fraction who are immediately post-MI, or following coronary revascularization, or with new-onset dilated cardiomyopathy who are at high risk for sudden cardiac death until stabilization, or who have an inherited arrhythmia or congenital disorder," said Dr. Kutyifa of the University of Rochester (N.Y.).
She reported on 2,000 U.S. patients in those categories who participated in the prospective WEARIT-II (Prospective Registry and Follow-Up of Patients Using the Wearable Defibrillator) registry. All received from their physicians a 3-month prescription for the LifeVest WCD, which is covered by Medicare and by most health plans. Of the 2,000 patients, 927 had a left ventricular ejection fraction (LVEF) of 35% or below owing to nonischemic cardiomyopathy, 805 had ischemic cardiomyopathy, and 268 had an inherited or congenital arrhythmogenic disorder, such as long QT syndrome or arrhythmogenic right ventricular dysplasia.
Participants’ median ejection fraction at enrollment was 25%, and 52% of subjects had heart failure symptoms at that time. Patients wore the WCD for a median of 22.5 hours per day, with no difference in wear time by disease etiology.
In terms of WCD safety, the inappropriate shock rate was just 0.5% in 2,000 patients during 3 months. No study deaths occurred related to unsuccessful attempts at termination of ventricular tachycardia (VT) or ventricular fibrillation (VF) by the WCD. Three patients died while wearing the WCD, and in all three cases the WCD detected asystole at the time of death.
During 90 days of WCD wear, 2.1% of patients experienced VT or VF, for an event rate of 22 per 100 person-years. The WCD administered a shock for VT/VF in 1.1% of patients. Sustained VT or VF occurred in 1.4% of patients, while 3.6% of participants experienced atrial arrhythmias or supraventricular tachycardia, with an event rate of 121 per 100 person-years.
At the end of the 3-month period of WCD use, 42% of patients with ischemic cardiomyopathy got an ICD, as did 36% of those with nonischemic cardiomyopathy and 46% who had a congenital or inherited condition. The main reason for not implanting an ICD was a boost in ejection fraction.
The WCD frequently detected arrhythmias which facilitated the decision whether to implant an ICD or not. Eighty-five percent of patients with VT/VF treated with a WCD shock got an ICD, as did 65% of those with sustained VT that terminated spontaneously during the wearable device’s detection time. Of patients with WCD-detected atrial arrhythmias, 48% received an ICD, as did 39% of those with no arrhythmias during the 3-month period.
Dr. Kutyifa characterized the WCD as helping to fill an unmet need for improved selection of patients for primary ICD therapy. In the landmark MADIT-II trial, for example, only 4% of patients received an appropriate ICD shock during 4 years of follow-up (N. Engl. J. Med. 2002;346:877-82).
Asked about the cost-effectiveness of WCD as a bridge to the ICD decision, she replied that such data weren’t included in the WEARIT-II registry.
"The up-front cost of the device may seem rather high, but if you think about the fact that if we use the WCD and are then able to make the decision to not implant an ICD in patients who don’t need it, we really need to look at the long-term savings: the cost of the ICD, battery changes, and issues of possible lead extraction. So I think long-term this management strategy would be cost-effective," the electrophysiologist said, adding that the WCD is also rentable.
At the congress-closing conference highlights overview session, Dr. Sylvia G. Priori singled out the WEARIT-II registry as one of the top developments in the field of arrhythmias presented at this year’s meeting. She noted that the WCD has been available for several years – Medicare covers it, as do most U.S. health plans – yet until now many physicians have wanted to see stronger evidence of safety and effectiveness before incorporating the WCD into their own practices. WEARIT-II, she said, provides that supporting evidence.
"This was quite an important study. It showed the device is able to recognize life-threatening arrhythmias, so it is safe, and it does not deliver an excessive number of inappropriate shocks. And there were no deaths related to the device," commented Dr. Priori of the University of Pavia (Italy).
"The take-home message is that, yes, we know there are patients where we are reluctant to immediately say that an ICD should be implanted forever, and now we should consider testing with this WCD, which has a low risk of inappropriate shocks and a good record in shocking patients out of the arrhythmia," she added.
The WEARIT-II registry is now expanding via enrollment of patients in Europe and Israel. The registry is funded by Zoll, which markets the WCD. Dr. Kutyifa reported having received research grants from the company as well as from other device manufacturers.
AT THE ESC CONGRESS 2014
Key clinical point: The temporary use of a commercially available wearable cardioverter defibrillator buys physicians time to decide whether or not to take the big step of permanent implantable cardioverter defibrillator placement in potential candidates for whom questions exist.
Major finding: During 3 months use of the LifeVest wearable cardioverter defibrillator, the vest detected VT/VF and administered an arrhythmia-ending shock in 1.1% of device wearers with an LVEF of 35% or less.
Data source: The prospective WEARIT-II registry includes to date 2,000 patients who are potential candidates for prophylactic implantation of a permanent cardioverter defibrillator for primary prevention of sudden cardiac arrest.
Disclosures: The presenter reported receiving research grant support from Zoll, which funded the WEARIT-II registry.

Beta-blockers no help for heart failure with atrial fib
Beta-blocker therapy doesn’t reduce all-cause mortality, cardiovascular mortality, cardiovascular hospitalization, or stroke in patients with heart failure who also have atrial fibrillation, Dipak Kotecha, Ph.D., reported at the annual congress of the European Society of Cardiology in Barcelona.
In a meta-analysis that used the "arduous" process of analyzing individual-patient data from all the high-quality randomized controlled trials available, Dr. Kotecha and his associates found no evidence that beta-blockers prevent adverse clinical events in this patient population. "Beta-blockers should no longer be regarded as standard therapy to improve prognosis" in patients with concomitant heart failure (HF) and atrial fibrillation (AF), he said.
In contrast, the drugs are effective and are strongly recommended for patients with HF who are in sinus rhythm, Dr. Kotecha said in a paper presented at the meeting and simultaneously published online Sept. 2 (Lancet 2014 [doi:10.1016/S0140-6736(14)61373-8]).
Heart failure and atrial fibrillation are common disorders, and both are becoming more prevalent. They often coexist, and an estimated 14%-50% of patients who have symptomatic HF also have AF.
At present, both European and American guidelines advise the use of beta-blockers for symptomatic heart failure without regard to AF status, based on trials that predominantly enrolled patients in sinus rhythm. There have been concerns about the drugs’ efficacy in certain subgroups of patients, including those with AF. "Patients with AF are often prescribed beta-blockers for both prognostic benefit in HF and heart-rate control, although there is little and underpowered evidence for efficacy in terms of clinical outcomes," he noted.
Dr. Kotecha and his associates in the Beta-Blockers in Heart Failure Collaborative Group – a multinational organization "formed to provide definitive answers to open questions about HF and beta-blocker therapy" and to provide clinicians with clear guidance – reviewed data from 18,254 study participants who were followed for a mean of 1.5 years, which allowed a robust and adequately powered analysis of this issue. They included "only unconfounded head-to-head trials with recruitment of more than 300 patients and a planned follow-up of more than 6 months."
A total of 3,066 (17%) of the study participants had AF as well as heart failure, and 633 of them died during follow-up. "A consistent benefit of beta-blockers versus placebo was noted for all death or hospital admission outcomes in patients with sinus rhythm, but the differences were not significant in patients with AF," said Dr. Kotecha of the University of Birmingham (England) Centre for Cardiovascular Sciences.
"The substantial benefit identified in patients with sinus rhythm should not be extrapolated to patients with AF," he said.
It is important to note that beta-blockers did appear to be safe, with no increase in mortality or adverse events, compared with placebo. "This should reassure clinicians, particularly for patients with another indication for beta-blockers, such as MI or the need for rate control of rapid AF with ongoing symptoms," he added.
This study was funded by Menarini Farmaceutica Internazionale, and GlaxoSmithKline provided data extraction support. Neither pharmaceutical group had a role in data analysis or manuscript preparation. Dr. Kotecha is supported by the U.K. National Institute for Health Research and reported receiving grants and honoraria from Menarini. His associates reported ties to numerous device and pharmaceutical sources.
Beta-blocker therapy doesn’t reduce all-cause mortality, cardiovascular mortality, cardiovascular hospitalization, or stroke in patients with heart failure who also have atrial fibrillation, Dipak Kotecha, Ph.D., reported at the annual congress of the European Society of Cardiology in Barcelona.
In a meta-analysis that used the "arduous" process of analyzing individual-patient data from all the high-quality randomized controlled trials available, Dr. Kotecha and his associates found no evidence that beta-blockers prevent adverse clinical events in this patient population. "Beta-blockers should no longer be regarded as standard therapy to improve prognosis" in patients with concomitant heart failure (HF) and atrial fibrillation (AF), he said.
In contrast, the drugs are effective and are strongly recommended for patients with HF who are in sinus rhythm, Dr. Kotecha said in a paper presented at the meeting and simultaneously published online Sept. 2 (Lancet 2014 [doi:10.1016/S0140-6736(14)61373-8]).
Heart failure and atrial fibrillation are common disorders, and both are becoming more prevalent. They often coexist, and an estimated 14%-50% of patients who have symptomatic HF also have AF.
At present, both European and American guidelines advise the use of beta-blockers for symptomatic heart failure without regard to AF status, based on trials that predominantly enrolled patients in sinus rhythm. There have been concerns about the drugs’ efficacy in certain subgroups of patients, including those with AF. "Patients with AF are often prescribed beta-blockers for both prognostic benefit in HF and heart-rate control, although there is little and underpowered evidence for efficacy in terms of clinical outcomes," he noted.
Dr. Kotecha and his associates in the Beta-Blockers in Heart Failure Collaborative Group – a multinational organization "formed to provide definitive answers to open questions about HF and beta-blocker therapy" and to provide clinicians with clear guidance – reviewed data from 18,254 study participants who were followed for a mean of 1.5 years, which allowed a robust and adequately powered analysis of this issue. They included "only unconfounded head-to-head trials with recruitment of more than 300 patients and a planned follow-up of more than 6 months."
A total of 3,066 (17%) of the study participants had AF as well as heart failure, and 633 of them died during follow-up. "A consistent benefit of beta-blockers versus placebo was noted for all death or hospital admission outcomes in patients with sinus rhythm, but the differences were not significant in patients with AF," said Dr. Kotecha of the University of Birmingham (England) Centre for Cardiovascular Sciences.
"The substantial benefit identified in patients with sinus rhythm should not be extrapolated to patients with AF," he said.
It is important to note that beta-blockers did appear to be safe, with no increase in mortality or adverse events, compared with placebo. "This should reassure clinicians, particularly for patients with another indication for beta-blockers, such as MI or the need for rate control of rapid AF with ongoing symptoms," he added.
This study was funded by Menarini Farmaceutica Internazionale, and GlaxoSmithKline provided data extraction support. Neither pharmaceutical group had a role in data analysis or manuscript preparation. Dr. Kotecha is supported by the U.K. National Institute for Health Research and reported receiving grants and honoraria from Menarini. His associates reported ties to numerous device and pharmaceutical sources.
Beta-blocker therapy doesn’t reduce all-cause mortality, cardiovascular mortality, cardiovascular hospitalization, or stroke in patients with heart failure who also have atrial fibrillation, Dipak Kotecha, Ph.D., reported at the annual congress of the European Society of Cardiology in Barcelona.
In a meta-analysis that used the "arduous" process of analyzing individual-patient data from all the high-quality randomized controlled trials available, Dr. Kotecha and his associates found no evidence that beta-blockers prevent adverse clinical events in this patient population. "Beta-blockers should no longer be regarded as standard therapy to improve prognosis" in patients with concomitant heart failure (HF) and atrial fibrillation (AF), he said.
In contrast, the drugs are effective and are strongly recommended for patients with HF who are in sinus rhythm, Dr. Kotecha said in a paper presented at the meeting and simultaneously published online Sept. 2 (Lancet 2014 [doi:10.1016/S0140-6736(14)61373-8]).
Heart failure and atrial fibrillation are common disorders, and both are becoming more prevalent. They often coexist, and an estimated 14%-50% of patients who have symptomatic HF also have AF.
At present, both European and American guidelines advise the use of beta-blockers for symptomatic heart failure without regard to AF status, based on trials that predominantly enrolled patients in sinus rhythm. There have been concerns about the drugs’ efficacy in certain subgroups of patients, including those with AF. "Patients with AF are often prescribed beta-blockers for both prognostic benefit in HF and heart-rate control, although there is little and underpowered evidence for efficacy in terms of clinical outcomes," he noted.
Dr. Kotecha and his associates in the Beta-Blockers in Heart Failure Collaborative Group – a multinational organization "formed to provide definitive answers to open questions about HF and beta-blocker therapy" and to provide clinicians with clear guidance – reviewed data from 18,254 study participants who were followed for a mean of 1.5 years, which allowed a robust and adequately powered analysis of this issue. They included "only unconfounded head-to-head trials with recruitment of more than 300 patients and a planned follow-up of more than 6 months."
A total of 3,066 (17%) of the study participants had AF as well as heart failure, and 633 of them died during follow-up. "A consistent benefit of beta-blockers versus placebo was noted for all death or hospital admission outcomes in patients with sinus rhythm, but the differences were not significant in patients with AF," said Dr. Kotecha of the University of Birmingham (England) Centre for Cardiovascular Sciences.
"The substantial benefit identified in patients with sinus rhythm should not be extrapolated to patients with AF," he said.
It is important to note that beta-blockers did appear to be safe, with no increase in mortality or adverse events, compared with placebo. "This should reassure clinicians, particularly for patients with another indication for beta-blockers, such as MI or the need for rate control of rapid AF with ongoing symptoms," he added.
This study was funded by Menarini Farmaceutica Internazionale, and GlaxoSmithKline provided data extraction support. Neither pharmaceutical group had a role in data analysis or manuscript preparation. Dr. Kotecha is supported by the U.K. National Institute for Health Research and reported receiving grants and honoraria from Menarini. His associates reported ties to numerous device and pharmaceutical sources.
FROM THE ESC CONGRESS 2014
Key clinical point: The benefit of beta-blockers in patients with HF and sinus rhythm cannot be extended to those with HF and atrial fibrillation.
Major finding: A consistent benefit of beta-blockers versus placebo was noted for all-cause mortality, CV mortality, CV hospitalization, and stroke in patients with sinus rhythm, but the differences were not significant in patients with AF.
Data source: A metaanalysis of all large high-quality randomized controlled trials assessing mortality and CV outcomes for 18,254 adults with HF, including 3,066 with concomitant AF, who received either beta-blockers or placebo and were followed for a mean of 1.5 years.
Disclosures: This study was funded by Menarini Farmaceutica Internazionale, and GlaxoSmithKline provided data extraction support. Neither pharmaceutical group had a role in data analysis or manuscript preparation. Dr. Kotecha is supported by the U.K. National Institute for Health Research and reported receiving grants and honoraria from Menarini. His associates reported ties to numerous device and pharmaceutical sources.
Prednisolone, immunotherapy ineffective for most tuberculous pericarditis
Neither standard anti-inflammatory therapy using prednisolone nor an experimental immunotherapy using Mycobacterium indicus pranii injections reduced the composite outcome of death, cardiac tamponade requiring pericardiocentesis, or constrictive pericarditis in an international clinical trial of adults with tuberculous pericarditis, a study showed.
However, prednisolone was beneficial for one component of this composite outcome – lowering the rate of constrictive pericarditis – compared with placebo, Dr. Bongani M. Mayosi reported at the annual congress of the European Society of Cardiology in Barcelona. His report was simultaneously presented at the meeting and published online Sept. 2 (N. Engl. J. Med. 2014 [doi:10.1056/NEJMoa1407380]).
Glucocorticoids are thought to attenuate the inflammatory response in patients with tuberculous pericarditis, and are recommended as adjunctive therapy in current American and World Health Organization treatment guidelines. But the studies on which these recommendations are based had "very small" numbers of events and patients, and the treatment effect was minimal, leading European expert groups to advise against using the drugs in this patient population. In addition, glucocorticoids may raise the risk of cancer in patients who are coinfected with HIV, which is common in the regions of sub-Saharan Africa and Asia where tuberculous pericarditis is most frequent, said Dr. Mayosi of the department of medicine, Old Groote Schuur Hospital, Cape Town, South Africa.
The 1,400 participants in the Investigation of the Management of Pericarditis (IMPI) trial had pericardial effusion confirmed by echocardiography and evidence of definite or probable tuberculous pericarditis; two-thirds also had concomitant HIV infection. They were treated at 19 hospitals across 8 countries in Africa during a 5-year period. All received background antimicrobial therapy for tuberculosis, and all patients with HIV received antiretrovirals according to WHO guidelines.
The participants first were randomly assigned to receive prednisolone (706 patients) or placebo (694 patients) in tapering doses for 6 weeks. In the second phase of the study, 1,250 of these participants were then randomly assigned to receive five intradermal injections of either heat-killed M. indicus pranii or placebo at intervals over the course of 3 months. This nonpathogenic, rapidly growing mycobacterium species has been shown to suppress inflammation in patients with leprosy.
The primary efficacy outcome was a composite of death, cardiac tamponade requiring pericardiocentesis, or constrictive pericarditis. After a median follow-up of 636 days, there were 14.3 such events per 100 patient-years in the prednisolone group and 14.8 in the placebo group, a nonsignificant difference.
When each component of this composite outcome was considered individually, prednisolone did not improve the death rate or the rate of cardiac tamponade, compared with placebo, but did reduce the rate of constrictive pericarditis (4.4%, vs 7.8% with placebo), and thus the rate of hospitalization (20.7%, vs 25.2% with placebo). "This finding is important because pericardiectomy, the definitive treatment for chronic pericardial constriction, is associated with high perioperative mortality and morbidity, and cardiac surgery is not widely available in Africa," Dr. Mayosi said.
Prednisolone also raised the rate of opportunistic infections, chiefly candidiasis, compared with placebo. And it markedly increased the rate of cancer, primarily Kaposi’s sarcoma, in patients coinfected with HIV (1.05 cases per 100 person-years, vs. 0.32 cases with placebo).
In the M. indicus pranii comparison, the active treatment was no different from placebo with regard to the composite outcome or any secondary outcomes, and that portion of the trial was halted early for futility. Like prednisolone, this experimental agent raised the rate of cancer in HIV-positive patients (0.92 cases per 100 person-years, vs. 0.24 cases with placebo) – an adverse event that has not been reported previously with M. indicus pranii.
In addition, significantly more patients who received M. indicus pranii (41.4%) than placebo (2.9%) developed injection-site reactions. Fifteen percent of patients given the active injections developed abscesses at the injection site, compared with only 1% of those given placebo injections.
The IMPI trial was supported by the Canadian Institutes of Health Research, the Canadian Network and Centre for Trials Internationally, the Population Health Research Institute, the South African Medical Research Council, the Lily and Ernst Hausmann Research Trust, and Cadila Pharma (India). Cadila donated the study drugs but had no role in the design or conduct of the study or in data analysis. The authors had no relevant financial conflicts of interest to disclose.
These findings clearly suggest that adjunctive glucocorticoids should not be used routinely in patients with tuberculous pericarditis, which may surprise many clinicians. But because they prevent constrictive pericarditis and reduce hospitalizations, the drugs may be appropriate for patients at highest risk for complications, such as those with large effusions, high levels of inflammatory cells or markers in the pericardial fluid, and early signs of constriction.
The use of glucocorticoids should be curtailed in patients coinfected with HIV unless the risk of constrictive pericarditis is high, since these drugs increase the risk of cancer in this patient population.
Dr. Richard E. Chaisson and Dr. Wendy S. Post of Johns Hopkins University, Baltimore, made these remarks in an editorial accompanying Dr. Mayosi’s report (N. Engl. J. Med. 2014 Sept. 2 [doi:10.1056/NEJMe1409356]. Dr. Chaisson reported receiving support from Merck.
These findings clearly suggest that adjunctive glucocorticoids should not be used routinely in patients with tuberculous pericarditis, which may surprise many clinicians. But because they prevent constrictive pericarditis and reduce hospitalizations, the drugs may be appropriate for patients at highest risk for complications, such as those with large effusions, high levels of inflammatory cells or markers in the pericardial fluid, and early signs of constriction.
The use of glucocorticoids should be curtailed in patients coinfected with HIV unless the risk of constrictive pericarditis is high, since these drugs increase the risk of cancer in this patient population.
Dr. Richard E. Chaisson and Dr. Wendy S. Post of Johns Hopkins University, Baltimore, made these remarks in an editorial accompanying Dr. Mayosi’s report (N. Engl. J. Med. 2014 Sept. 2 [doi:10.1056/NEJMe1409356]. Dr. Chaisson reported receiving support from Merck.
These findings clearly suggest that adjunctive glucocorticoids should not be used routinely in patients with tuberculous pericarditis, which may surprise many clinicians. But because they prevent constrictive pericarditis and reduce hospitalizations, the drugs may be appropriate for patients at highest risk for complications, such as those with large effusions, high levels of inflammatory cells or markers in the pericardial fluid, and early signs of constriction.
The use of glucocorticoids should be curtailed in patients coinfected with HIV unless the risk of constrictive pericarditis is high, since these drugs increase the risk of cancer in this patient population.
Dr. Richard E. Chaisson and Dr. Wendy S. Post of Johns Hopkins University, Baltimore, made these remarks in an editorial accompanying Dr. Mayosi’s report (N. Engl. J. Med. 2014 Sept. 2 [doi:10.1056/NEJMe1409356]. Dr. Chaisson reported receiving support from Merck.
Neither standard anti-inflammatory therapy using prednisolone nor an experimental immunotherapy using Mycobacterium indicus pranii injections reduced the composite outcome of death, cardiac tamponade requiring pericardiocentesis, or constrictive pericarditis in an international clinical trial of adults with tuberculous pericarditis, a study showed.
However, prednisolone was beneficial for one component of this composite outcome – lowering the rate of constrictive pericarditis – compared with placebo, Dr. Bongani M. Mayosi reported at the annual congress of the European Society of Cardiology in Barcelona. His report was simultaneously presented at the meeting and published online Sept. 2 (N. Engl. J. Med. 2014 [doi:10.1056/NEJMoa1407380]).
Glucocorticoids are thought to attenuate the inflammatory response in patients with tuberculous pericarditis, and are recommended as adjunctive therapy in current American and World Health Organization treatment guidelines. But the studies on which these recommendations are based had "very small" numbers of events and patients, and the treatment effect was minimal, leading European expert groups to advise against using the drugs in this patient population. In addition, glucocorticoids may raise the risk of cancer in patients who are coinfected with HIV, which is common in the regions of sub-Saharan Africa and Asia where tuberculous pericarditis is most frequent, said Dr. Mayosi of the department of medicine, Old Groote Schuur Hospital, Cape Town, South Africa.
The 1,400 participants in the Investigation of the Management of Pericarditis (IMPI) trial had pericardial effusion confirmed by echocardiography and evidence of definite or probable tuberculous pericarditis; two-thirds also had concomitant HIV infection. They were treated at 19 hospitals across 8 countries in Africa during a 5-year period. All received background antimicrobial therapy for tuberculosis, and all patients with HIV received antiretrovirals according to WHO guidelines.
The participants first were randomly assigned to receive prednisolone (706 patients) or placebo (694 patients) in tapering doses for 6 weeks. In the second phase of the study, 1,250 of these participants were then randomly assigned to receive five intradermal injections of either heat-killed M. indicus pranii or placebo at intervals over the course of 3 months. This nonpathogenic, rapidly growing mycobacterium species has been shown to suppress inflammation in patients with leprosy.
The primary efficacy outcome was a composite of death, cardiac tamponade requiring pericardiocentesis, or constrictive pericarditis. After a median follow-up of 636 days, there were 14.3 such events per 100 patient-years in the prednisolone group and 14.8 in the placebo group, a nonsignificant difference.
When each component of this composite outcome was considered individually, prednisolone did not improve the death rate or the rate of cardiac tamponade, compared with placebo, but did reduce the rate of constrictive pericarditis (4.4%, vs 7.8% with placebo), and thus the rate of hospitalization (20.7%, vs 25.2% with placebo). "This finding is important because pericardiectomy, the definitive treatment for chronic pericardial constriction, is associated with high perioperative mortality and morbidity, and cardiac surgery is not widely available in Africa," Dr. Mayosi said.
Prednisolone also raised the rate of opportunistic infections, chiefly candidiasis, compared with placebo. And it markedly increased the rate of cancer, primarily Kaposi’s sarcoma, in patients coinfected with HIV (1.05 cases per 100 person-years, vs. 0.32 cases with placebo).
In the M. indicus pranii comparison, the active treatment was no different from placebo with regard to the composite outcome or any secondary outcomes, and that portion of the trial was halted early for futility. Like prednisolone, this experimental agent raised the rate of cancer in HIV-positive patients (0.92 cases per 100 person-years, vs. 0.24 cases with placebo) – an adverse event that has not been reported previously with M. indicus pranii.
In addition, significantly more patients who received M. indicus pranii (41.4%) than placebo (2.9%) developed injection-site reactions. Fifteen percent of patients given the active injections developed abscesses at the injection site, compared with only 1% of those given placebo injections.
The IMPI trial was supported by the Canadian Institutes of Health Research, the Canadian Network and Centre for Trials Internationally, the Population Health Research Institute, the South African Medical Research Council, the Lily and Ernst Hausmann Research Trust, and Cadila Pharma (India). Cadila donated the study drugs but had no role in the design or conduct of the study or in data analysis. The authors had no relevant financial conflicts of interest to disclose.
Neither standard anti-inflammatory therapy using prednisolone nor an experimental immunotherapy using Mycobacterium indicus pranii injections reduced the composite outcome of death, cardiac tamponade requiring pericardiocentesis, or constrictive pericarditis in an international clinical trial of adults with tuberculous pericarditis, a study showed.
However, prednisolone was beneficial for one component of this composite outcome – lowering the rate of constrictive pericarditis – compared with placebo, Dr. Bongani M. Mayosi reported at the annual congress of the European Society of Cardiology in Barcelona. His report was simultaneously presented at the meeting and published online Sept. 2 (N. Engl. J. Med. 2014 [doi:10.1056/NEJMoa1407380]).
Glucocorticoids are thought to attenuate the inflammatory response in patients with tuberculous pericarditis, and are recommended as adjunctive therapy in current American and World Health Organization treatment guidelines. But the studies on which these recommendations are based had "very small" numbers of events and patients, and the treatment effect was minimal, leading European expert groups to advise against using the drugs in this patient population. In addition, glucocorticoids may raise the risk of cancer in patients who are coinfected with HIV, which is common in the regions of sub-Saharan Africa and Asia where tuberculous pericarditis is most frequent, said Dr. Mayosi of the department of medicine, Old Groote Schuur Hospital, Cape Town, South Africa.
The 1,400 participants in the Investigation of the Management of Pericarditis (IMPI) trial had pericardial effusion confirmed by echocardiography and evidence of definite or probable tuberculous pericarditis; two-thirds also had concomitant HIV infection. They were treated at 19 hospitals across 8 countries in Africa during a 5-year period. All received background antimicrobial therapy for tuberculosis, and all patients with HIV received antiretrovirals according to WHO guidelines.
The participants first were randomly assigned to receive prednisolone (706 patients) or placebo (694 patients) in tapering doses for 6 weeks. In the second phase of the study, 1,250 of these participants were then randomly assigned to receive five intradermal injections of either heat-killed M. indicus pranii or placebo at intervals over the course of 3 months. This nonpathogenic, rapidly growing mycobacterium species has been shown to suppress inflammation in patients with leprosy.
The primary efficacy outcome was a composite of death, cardiac tamponade requiring pericardiocentesis, or constrictive pericarditis. After a median follow-up of 636 days, there were 14.3 such events per 100 patient-years in the prednisolone group and 14.8 in the placebo group, a nonsignificant difference.
When each component of this composite outcome was considered individually, prednisolone did not improve the death rate or the rate of cardiac tamponade, compared with placebo, but did reduce the rate of constrictive pericarditis (4.4%, vs 7.8% with placebo), and thus the rate of hospitalization (20.7%, vs 25.2% with placebo). "This finding is important because pericardiectomy, the definitive treatment for chronic pericardial constriction, is associated with high perioperative mortality and morbidity, and cardiac surgery is not widely available in Africa," Dr. Mayosi said.
Prednisolone also raised the rate of opportunistic infections, chiefly candidiasis, compared with placebo. And it markedly increased the rate of cancer, primarily Kaposi’s sarcoma, in patients coinfected with HIV (1.05 cases per 100 person-years, vs. 0.32 cases with placebo).
In the M. indicus pranii comparison, the active treatment was no different from placebo with regard to the composite outcome or any secondary outcomes, and that portion of the trial was halted early for futility. Like prednisolone, this experimental agent raised the rate of cancer in HIV-positive patients (0.92 cases per 100 person-years, vs. 0.24 cases with placebo) – an adverse event that has not been reported previously with M. indicus pranii.
In addition, significantly more patients who received M. indicus pranii (41.4%) than placebo (2.9%) developed injection-site reactions. Fifteen percent of patients given the active injections developed abscesses at the injection site, compared with only 1% of those given placebo injections.
The IMPI trial was supported by the Canadian Institutes of Health Research, the Canadian Network and Centre for Trials Internationally, the Population Health Research Institute, the South African Medical Research Council, the Lily and Ernst Hausmann Research Trust, and Cadila Pharma (India). Cadila donated the study drugs but had no role in the design or conduct of the study or in data analysis. The authors had no relevant financial conflicts of interest to disclose.
FROM THE ESC CONGRESS 2014
Key clinical point: Although treatment with either prednisolone or M. indicus pranii did not improve overall outcomes in patients with tuberculosis pericarditis, prednisone did reduce the risk of developing constrictive pericarditis and hospitalizations.
Major finding: After a median follow-up of 636 days, the primary efficacy outcome – a composite of death, cardiac tamponade requiring pericardiocentesis, or constrictive pericarditis – occurred in 14.3 cases per 100 patient-years in the prednisolone group, compared with 14.8 in the placebo group.
Data source: IMPI, a randomized controlled trial in 1,400 adults with presumed tuberculous pericarditis who were treated at 19 hospitals in 8 African countries with 6 weeks of either prednisolone or placebo, followed by 3 months of M. indicus pranii or placebo injections.
Disclosures: The IMPI trial was supported by the Canadian Institutes of Health Research, the Canadian Network and Centre for Trials Internationally, the Population Health Research Institute, the South African Medical Research Council, the Lily and Ernst Hausmann Research Trust, and Cadila Pharma (India). Cadila donated the study drugs but had no role in the design or conduct of the study or in data analysis. The authors had no relevant financial conflicts of interest to disclose.
LCZ696 surpasses enalapril for heart failure
BARCELONA – A new, dual-agent formulation for treating heart failure patients with reduced ejection fraction showed a dramatic benefit for cutting the rate of cardiovascular death and hospitalizations in an international, controlled, pivotal trial with more than 8,000 patients.
The superiority of the new compound, which combines the novel neprilysin-inhibitor drug sacubitril with the established angiotensin receptor (ARB) blocking drug valsartan, was so strong that it immediately loomed as the new cornerstone-drug of choice for heart-failure patients with reduced ejection fraction pending regulatory approvals, said Dr. Milton Packer at the annual congress of the European Society of Cardiology.

Treatment with the new combination agent, currently known as LCZ696, doubled the cardiovascular-death benefit of the ACE inhibitor enalapril, while simultaneously edging out enalapril for safety and tolerability. Neprilysin is an endopeptidase that degrades several endogenous vasoactive peptides. Inhibiting this degradation process by treatment with LCZ696 boosts levels of these peptides and counters the neurohormonal overactivation that characterizes heart failure and leads to vasoconstriction, sodium retention, and maladaptive remodeling.
"This robust finding provides strong evidence supporting use of LCZ696 as the treatment of choice for heart failure instead of an ACE inhibitor or ARB" alone, said Dr. Packer, professor and chairman of clinical sciences at UT Southwestern Medical Center in Dallas. "The intent of this trial was to provide persuasive evidence that LCZ696 should replace current use of ACE inhibitors and ARBs as the cornerstone of heart failure treatment," said Dr. Packer, coprincipal investigator of the study.
Especially striking to several heart failure experts who heard the study results was the range of clinically important benefits seen when the new combination substituted for enalapril.

"This is the first study in a long time where we saw not only a reduction in the combined endpoint of cardiovascular death and hospitalization, but also significant effect on each of these two endpoints individually as well as on all-cause mortality," commented Dr. Mariell Jessup, professor of medicine at the University of Pennsylvania in Philadelphia. The new trial "was really trying to upset the foundation of heart failure treatment that we’ve used for the last 2 decades, and it succeeded. We were hoping for a positive outcome; what we got was remarkable. It very convincingly showed that this drug has incredible promise," she said in an interview.
"This will be an important change," commented Dr. John G.F. Cleland, professor of cardiology at the University of Hull, U.K. "The science and medicine will make it hard not to switch everyone with low ejection fraction heart failure [and other features that mirror the study’s enrollment criteria] from an ACE inhibitor to this the moment it becomes available," he said in an interview. "This is now the drug of choice for any heart failure patient with persistently elevated natriuretic peptide and left ventricular systolic dysfunction."
The Prospective Comparison of ARNI [angiotensin receptor–neprilysin inhibitor] with ACEI to Determine Impact on Global Mortality and Morbidity in Heart Failure (PARADIGM-HF) trial randomized 8,399 patients with heart failure and a left ventricular ejection fraction of 40% of less to treatment with either 200 mg b.i.d. of LCZ696 or 10 mg b.i.d. enalapril plus other standard heart failure medications, including a beta-blocker (in 93%), a diuretic (in 80%) and a mineralocorticoid antagonist such as spironolactone (in about 55%). About 70% of enrolled patients had New York Heart Association class II heart failure, and another 24% had class III heart failure.
After a median treatment duration of 27 months, the primary combined endpoint of cardiovascular death or first hospitalization for heart failure occurred in 22% of the patients treated with LCZ696 and in 27% of those treated with enalapril, a 20% relative risk reduction that was statistically significant, and which showed a number needed to treat of 21 to prevent one of these primary endpoints. LCZ696 treatment also linked with a significant reduction in all-cause death, and a statistically significant and clinically meaningful improvement in patient quality of life as measured using the Kansas City Cardiomyopathy Questionnaire after patients had been on treatment for 8 months. Online publication of the full results occurred concurrent with Dr. Packer’s report at the meeting (N.Engl. J. Med 2014;DOI: 10.1056/NEJMoa1409077).
While Dr. Packer highlighted the positive impact of LCZ696 on quality of life, he also stressed that the primary impact of treatment with LCZ696 is not to make patients feel better. "The major benefit of this drug is to change the natural course and rate of progression of heart failure. It should be used in patients with mild heart failure to reduce progression and death," he said.
"By switching patients from an ACE inhibitor to LCZ696 you can prolong patient survival. It is exciting to have a new drug" that can confer as many benefits as LCZ696, Dr. Jessup said.
PARADIGM-HF was sponsored by Novartis. Dr. Packer has been a consultant to Novartis as well as several other companies. Dr. Jessup had no disclosures. Dr. Cleland has received research support and honoraria from Sorin, St. Jude, and Medtronic.
Twitter: @mitchelzoler
PARADIGM-HF is the first contemporary trial to test the concept of substituting a new agent for an established heart-failure drug rather than adding a new compound to already established treatments. It was a large trial that enrolled patients with an elevated level of natriuretic peptide, used very relevant primary and secondary outcomes, and had a modest discontinuation rate during 27 months of treatment. The result showed a significant, 20% relative risk reduction in patients who were well treated for heart failure based on current guidelines, and the outcomes showed consistent results across all prespecified subgroups studied.
The investigational drug also showed good safety for renal function and potassium levels. Although patients who received LCZ696 had significantly more hypotensive episodes than patients in the enalapril comparator group, the investigational arm had no significant excess of treatment discontinuations. Patients who received LCZ696 also had a small increase in episodes of angioedema, but the difference did no reach statistical significance.
The trial used a clever run-in design that screened out patients who were intolerant of either enalapril or the study drug, a step that excluded 12% of the initial patient group.
The magnitude of the treatment effect and the number needed to treat suggests that LCZ696 will be a cost-effective treatment for the types of patients studied. Future studies should explore the efficacy of this drug combination in patients with heart failure with preserved ejection fraction, and in patients with acute decompensated heart failure, and a study should also compare LCZ696 with the safety and efficacy of a direct renin inhibitor such as aliskiren.
Michel Komajda, M.D., is professor of cardiology at University Pierre and Marie Curie in Paris, and is past president of the European Society of Cardiology. He has been a consultant to or speaker on behalf of Novartis and several other companies. He made these comments as the designated discussant for PARADIGM-HF.
PARADIGM-HF is the first contemporary trial to test the concept of substituting a new agent for an established heart-failure drug rather than adding a new compound to already established treatments. It was a large trial that enrolled patients with an elevated level of natriuretic peptide, used very relevant primary and secondary outcomes, and had a modest discontinuation rate during 27 months of treatment. The result showed a significant, 20% relative risk reduction in patients who were well treated for heart failure based on current guidelines, and the outcomes showed consistent results across all prespecified subgroups studied.
The investigational drug also showed good safety for renal function and potassium levels. Although patients who received LCZ696 had significantly more hypotensive episodes than patients in the enalapril comparator group, the investigational arm had no significant excess of treatment discontinuations. Patients who received LCZ696 also had a small increase in episodes of angioedema, but the difference did no reach statistical significance.
The trial used a clever run-in design that screened out patients who were intolerant of either enalapril or the study drug, a step that excluded 12% of the initial patient group.
The magnitude of the treatment effect and the number needed to treat suggests that LCZ696 will be a cost-effective treatment for the types of patients studied. Future studies should explore the efficacy of this drug combination in patients with heart failure with preserved ejection fraction, and in patients with acute decompensated heart failure, and a study should also compare LCZ696 with the safety and efficacy of a direct renin inhibitor such as aliskiren.
Michel Komajda, M.D., is professor of cardiology at University Pierre and Marie Curie in Paris, and is past president of the European Society of Cardiology. He has been a consultant to or speaker on behalf of Novartis and several other companies. He made these comments as the designated discussant for PARADIGM-HF.
PARADIGM-HF is the first contemporary trial to test the concept of substituting a new agent for an established heart-failure drug rather than adding a new compound to already established treatments. It was a large trial that enrolled patients with an elevated level of natriuretic peptide, used very relevant primary and secondary outcomes, and had a modest discontinuation rate during 27 months of treatment. The result showed a significant, 20% relative risk reduction in patients who were well treated for heart failure based on current guidelines, and the outcomes showed consistent results across all prespecified subgroups studied.
The investigational drug also showed good safety for renal function and potassium levels. Although patients who received LCZ696 had significantly more hypotensive episodes than patients in the enalapril comparator group, the investigational arm had no significant excess of treatment discontinuations. Patients who received LCZ696 also had a small increase in episodes of angioedema, but the difference did no reach statistical significance.
The trial used a clever run-in design that screened out patients who were intolerant of either enalapril or the study drug, a step that excluded 12% of the initial patient group.
The magnitude of the treatment effect and the number needed to treat suggests that LCZ696 will be a cost-effective treatment for the types of patients studied. Future studies should explore the efficacy of this drug combination in patients with heart failure with preserved ejection fraction, and in patients with acute decompensated heart failure, and a study should also compare LCZ696 with the safety and efficacy of a direct renin inhibitor such as aliskiren.
Michel Komajda, M.D., is professor of cardiology at University Pierre and Marie Curie in Paris, and is past president of the European Society of Cardiology. He has been a consultant to or speaker on behalf of Novartis and several other companies. He made these comments as the designated discussant for PARADIGM-HF.
BARCELONA – A new, dual-agent formulation for treating heart failure patients with reduced ejection fraction showed a dramatic benefit for cutting the rate of cardiovascular death and hospitalizations in an international, controlled, pivotal trial with more than 8,000 patients.
The superiority of the new compound, which combines the novel neprilysin-inhibitor drug sacubitril with the established angiotensin receptor (ARB) blocking drug valsartan, was so strong that it immediately loomed as the new cornerstone-drug of choice for heart-failure patients with reduced ejection fraction pending regulatory approvals, said Dr. Milton Packer at the annual congress of the European Society of Cardiology.
Treatment with the new combination agent, currently known as LCZ696, doubled the cardiovascular-death benefit of the ACE inhibitor enalapril, while simultaneously edging out enalapril for safety and tolerability. Neprilysin is an endopeptidase that degrades several endogenous vasoactive peptides. Inhibiting this degradation process by treatment with LCZ696 boosts levels of these peptides and counters the neurohormonal overactivation that characterizes heart failure and leads to vasoconstriction, sodium retention, and maladaptive remodeling.
"This robust finding provides strong evidence supporting use of LCZ696 as the treatment of choice for heart failure instead of an ACE inhibitor or ARB" alone, said Dr. Packer, professor and chairman of clinical sciences at UT Southwestern Medical Center in Dallas. "The intent of this trial was to provide persuasive evidence that LCZ696 should replace current use of ACE inhibitors and ARBs as the cornerstone of heart failure treatment," said Dr. Packer, coprincipal investigator of the study.
Especially striking to several heart failure experts who heard the study results was the range of clinically important benefits seen when the new combination substituted for enalapril.
"This is the first study in a long time where we saw not only a reduction in the combined endpoint of cardiovascular death and hospitalization, but also significant effect on each of these two endpoints individually as well as on all-cause mortality," commented Dr. Mariell Jessup, professor of medicine at the University of Pennsylvania in Philadelphia. The new trial "was really trying to upset the foundation of heart failure treatment that we’ve used for the last 2 decades, and it succeeded. We were hoping for a positive outcome; what we got was remarkable. It very convincingly showed that this drug has incredible promise," she said in an interview.
"This will be an important change," commented Dr. John G.F. Cleland, professor of cardiology at the University of Hull, U.K. "The science and medicine will make it hard not to switch everyone with low ejection fraction heart failure [and other features that mirror the study’s enrollment criteria] from an ACE inhibitor to this the moment it becomes available," he said in an interview. "This is now the drug of choice for any heart failure patient with persistently elevated natriuretic peptide and left ventricular systolic dysfunction."
The Prospective Comparison of ARNI [angiotensin receptor–neprilysin inhibitor] with ACEI to Determine Impact on Global Mortality and Morbidity in Heart Failure (PARADIGM-HF) trial randomized 8,399 patients with heart failure and a left ventricular ejection fraction of 40% of less to treatment with either 200 mg b.i.d. of LCZ696 or 10 mg b.i.d. enalapril plus other standard heart failure medications, including a beta-blocker (in 93%), a diuretic (in 80%) and a mineralocorticoid antagonist such as spironolactone (in about 55%). About 70% of enrolled patients had New York Heart Association class II heart failure, and another 24% had class III heart failure.
After a median treatment duration of 27 months, the primary combined endpoint of cardiovascular death or first hospitalization for heart failure occurred in 22% of the patients treated with LCZ696 and in 27% of those treated with enalapril, a 20% relative risk reduction that was statistically significant, and which showed a number needed to treat of 21 to prevent one of these primary endpoints. LCZ696 treatment also linked with a significant reduction in all-cause death, and a statistically significant and clinically meaningful improvement in patient quality of life as measured using the Kansas City Cardiomyopathy Questionnaire after patients had been on treatment for 8 months. Online publication of the full results occurred concurrent with Dr. Packer’s report at the meeting (N.Engl. J. Med 2014;DOI: 10.1056/NEJMoa1409077).
While Dr. Packer highlighted the positive impact of LCZ696 on quality of life, he also stressed that the primary impact of treatment with LCZ696 is not to make patients feel better. "The major benefit of this drug is to change the natural course and rate of progression of heart failure. It should be used in patients with mild heart failure to reduce progression and death," he said.
"By switching patients from an ACE inhibitor to LCZ696 you can prolong patient survival. It is exciting to have a new drug" that can confer as many benefits as LCZ696, Dr. Jessup said.
PARADIGM-HF was sponsored by Novartis. Dr. Packer has been a consultant to Novartis as well as several other companies. Dr. Jessup had no disclosures. Dr. Cleland has received research support and honoraria from Sorin, St. Jude, and Medtronic.
Twitter: @mitchelzoler
BARCELONA – A new, dual-agent formulation for treating heart failure patients with reduced ejection fraction showed a dramatic benefit for cutting the rate of cardiovascular death and hospitalizations in an international, controlled, pivotal trial with more than 8,000 patients.
The superiority of the new compound, which combines the novel neprilysin-inhibitor drug sacubitril with the established angiotensin receptor (ARB) blocking drug valsartan, was so strong that it immediately loomed as the new cornerstone-drug of choice for heart-failure patients with reduced ejection fraction pending regulatory approvals, said Dr. Milton Packer at the annual congress of the European Society of Cardiology.
Treatment with the new combination agent, currently known as LCZ696, doubled the cardiovascular-death benefit of the ACE inhibitor enalapril, while simultaneously edging out enalapril for safety and tolerability. Neprilysin is an endopeptidase that degrades several endogenous vasoactive peptides. Inhibiting this degradation process by treatment with LCZ696 boosts levels of these peptides and counters the neurohormonal overactivation that characterizes heart failure and leads to vasoconstriction, sodium retention, and maladaptive remodeling.
"This robust finding provides strong evidence supporting use of LCZ696 as the treatment of choice for heart failure instead of an ACE inhibitor or ARB" alone, said Dr. Packer, professor and chairman of clinical sciences at UT Southwestern Medical Center in Dallas. "The intent of this trial was to provide persuasive evidence that LCZ696 should replace current use of ACE inhibitors and ARBs as the cornerstone of heart failure treatment," said Dr. Packer, coprincipal investigator of the study.
Especially striking to several heart failure experts who heard the study results was the range of clinically important benefits seen when the new combination substituted for enalapril.
"This is the first study in a long time where we saw not only a reduction in the combined endpoint of cardiovascular death and hospitalization, but also significant effect on each of these two endpoints individually as well as on all-cause mortality," commented Dr. Mariell Jessup, professor of medicine at the University of Pennsylvania in Philadelphia. The new trial "was really trying to upset the foundation of heart failure treatment that we’ve used for the last 2 decades, and it succeeded. We were hoping for a positive outcome; what we got was remarkable. It very convincingly showed that this drug has incredible promise," she said in an interview.
"This will be an important change," commented Dr. John G.F. Cleland, professor of cardiology at the University of Hull, U.K. "The science and medicine will make it hard not to switch everyone with low ejection fraction heart failure [and other features that mirror the study’s enrollment criteria] from an ACE inhibitor to this the moment it becomes available," he said in an interview. "This is now the drug of choice for any heart failure patient with persistently elevated natriuretic peptide and left ventricular systolic dysfunction."
The Prospective Comparison of ARNI [angiotensin receptor–neprilysin inhibitor] with ACEI to Determine Impact on Global Mortality and Morbidity in Heart Failure (PARADIGM-HF) trial randomized 8,399 patients with heart failure and a left ventricular ejection fraction of 40% of less to treatment with either 200 mg b.i.d. of LCZ696 or 10 mg b.i.d. enalapril plus other standard heart failure medications, including a beta-blocker (in 93%), a diuretic (in 80%) and a mineralocorticoid antagonist such as spironolactone (in about 55%). About 70% of enrolled patients had New York Heart Association class II heart failure, and another 24% had class III heart failure.
After a median treatment duration of 27 months, the primary combined endpoint of cardiovascular death or first hospitalization for heart failure occurred in 22% of the patients treated with LCZ696 and in 27% of those treated with enalapril, a 20% relative risk reduction that was statistically significant, and which showed a number needed to treat of 21 to prevent one of these primary endpoints. LCZ696 treatment also linked with a significant reduction in all-cause death, and a statistically significant and clinically meaningful improvement in patient quality of life as measured using the Kansas City Cardiomyopathy Questionnaire after patients had been on treatment for 8 months. Online publication of the full results occurred concurrent with Dr. Packer’s report at the meeting (N.Engl. J. Med 2014;DOI: 10.1056/NEJMoa1409077).
While Dr. Packer highlighted the positive impact of LCZ696 on quality of life, he also stressed that the primary impact of treatment with LCZ696 is not to make patients feel better. "The major benefit of this drug is to change the natural course and rate of progression of heart failure. It should be used in patients with mild heart failure to reduce progression and death," he said.
"By switching patients from an ACE inhibitor to LCZ696 you can prolong patient survival. It is exciting to have a new drug" that can confer as many benefits as LCZ696, Dr. Jessup said.
PARADIGM-HF was sponsored by Novartis. Dr. Packer has been a consultant to Novartis as well as several other companies. Dr. Jessup had no disclosures. Dr. Cleland has received research support and honoraria from Sorin, St. Jude, and Medtronic.
Twitter: @mitchelzoler
AT THE ESC CONGRESS 2014
Key clinical point: Substituting LCZ696 for enalapril in heart failure patients was linked with significant improvements in survival, heart failure hospitalization, and quality of life.
Major finding: LCZ696 was associated with a 20% relative risk reduction in cardiovascular death and heart-failure hospitalization compared with enalapril in heart failure patients.
Data source: PARADIGM-HF, which assessed 8,399 patients randomized at 1,043 centers in 47 countries.
Disclosures: PARADIGM-HF was sponsored by Novartis. Dr. Packer has been a consultant to Novartis as well as several other companies. Dr. Jessup had no disclosures. Dr. Cleland has received research support and honoraria from Sorin, St. Jude, and Medtronic.

SIGNIFY: Ivabradine no help in stable CAD
Adding ivabradine to standard background therapy did not improve outcomes in a large, randomized, placebo-controlled trial of more than 19,000 patients who had stable coronary artery disease without clinical heart failure.
At 3 months, the mean heart rate was 60.7 beats per minute in 9,550 patients in the Study Assessing the Morbidity-Mortality Benefits of the If Inhibitor Ivabradine in Patients with Coronary Artery Disease (SIGNIFY) who were assigned to receive ivabradine, compared with 70.6 beats per minute in 9,552 patients who received placebo. At a median of 27.8 months, the percentage of patients reaching a composite endpoint of death from cardiovascular causes or nonfatal myocardial infarction was 6.8% and 6.4% in the groups, respectively (hazard ratio,1.08), Dr. Kim Fox of Royal Brompton Hospital, London and colleagues reported at the 2014 European Society of Cardiology Congress.
The differences between the ivabradine and placebo groups were not statistically significant, but ivabradine was associated with a nonsignificant increase in the incidence of the primary end point in patients with Canadian Cardiovascular Society class II angina or higher (7.6% vs. 6.5%, hazard ratio, 1.18), the investigators said.
The findings were simultaneously published online Aug. 31 in the New England Journal of Medicine (N. Engl. J. Med 2014 Aug. 30[doi:10.1056/NEJMoa1406430].
Study participants, who were recruited from 1,139 centers in 51 countries between October 12, 2009 and April 30, 2012, were at least 55 years of age with documented and treated stable coronary artery disease, but no evidence of clinical heart failure. They also had to be in sinus rhythm, have a resting heart rate of 70 beats per minute or more on two consecutive readings, and have at least one major or two minor adverse prognostic factors.
All patients completed a 2- to 4-week placebo run-in phase then were randomized to ivabradine at a dose of 7.5 mg twice daily (or 5 mg twice daily for those over age 74 years) or placebo in addition to stable background therapy prescribed according to contemporary guidelines. Dosing was adjusted as needed to 5, 7.5, or 10 mg twice daily based on heart rate and bradycardia. The target heart rate was 55-60 beats per minute, and the mean study-drug dose throughout the trial was 8.2 mg twice daily in the ivabradine group, and 9.5 mg twice daily in the placebo group.
Adverse events occurred in significantly more patients in the ivabradine group (73.3% vs. 66.9%). Bradycardia, in particular, was increased in the ivabradine group (7.9% vs. 1.2% for symptomatic bradycardia, and 11.0% vs. 1.3% for asymptomatic bradycardia. Atrial fibrillation and phosphenes were also increased in the ivabradine group (5.3 vs. 3.8% and 5.4% vs. 0.5%, respectively).
Serious adverse events were also significantly more common in the ivabradine group, with 37.6% and 35.4% of patients in the groups, respectively, experiencing a serious adverse event. These events were classified as cardiac disorders in 19.0% and 16.7% of patients in the groups, respectively.
The SIGNIFY findings contrast with those from previous post hoc analyses that suggested ivabradine improves outcomes in patients with stable coronary artery disease.
"In addition, in patients with heart failure, the reduction in heart rate with ivabradine has been shown to improve clinical outcomes, beyond the improvements observed with beta-blockers," the investigators said.
In fact, on the basis of findings from the phase III Systolic Heart Failure Treatment with the If Inhibitor Ivabradine Trial (SHIFT), the U.S. Food and Drug Administration recently granted priority review designation for ivabradine for the treatment of chronic heart failure.
Ivabradine lowers heart rate without affecting blood pressure or left ventricular systolic function, by inhibiting the If, or pacemaker, current in the sinoatrial node. It is approved in numerous countries outside the United States and is used for reducing angina symptoms and for chronic heart failure.
A number of hypotheses may explain the SIGNIFY findings.
"It is possible that ivabradine decreased the heart rate too much or that there may be a J-shaped curve for the relationship between heart rate and outcome," the investigators said, explaining that "ivabradine may have unintended effects (e.g., adjustment of the doses of other heart-rate lowering agents) that may have affected the potential benefits of the lowering of heart rate with ivabradine."
Further, heart rate-reducing antianginal agents may have no effect on outcomes in patients with stable coronary artery disease, they said.
The findings may also reflect the fact that an elevated heart rate is due to different pathophysiological mechanisms in those with heart failure and those with stable coronary artery disease, they noted.
"Given that the primary cardiovascular effect of ivabradine is to reduce heart rate, these results suggest that an elevated heart rate is only a marker of risk – but not a modifiable determinant of outcomes- in patients who have stable coronary artery disease without clinical heart failure," they concluded.
This study was funded by Servier. Dr. Fox reported receiving personal fees and/or nonfinancial support from Servier, AstraZeneca, TaurX, Armgo, Broadview Ventures, and CellAegis. She also is director of Heart Research Lt. and Vesalius Trials Ltd. Detailed disclosures for all study authors are available at NEJM.org.
The increased incidence of the primary endpoint among patients with Canadian Cardiovascular Society Class II or higher levels of angina in the SIGNIFY trial was surprising and warrants further study to ascertain whether this angina subgroup is one in which caution should be exercised.
In the meantime, caution should indeed be exercised with respect to the use of ivabradine (by physicians who have access to the drug) in those with more severe forms of angina, and adjustment of beta-blocker doses to effective levels should be considered before initiating ivabradine.
The experience from the trial of ivabradine in heart failure suggests that nearly 60% of patients were receiving inadequate doses of beta-blockers and that the majority of benefit with ivabradine was among patients who could not take beta-blockers or who were taking a lower dose. Whether this holds true for patients with angina is unknown, but a cautious approach may be reasonable pending better understanding of the matter.
More therapies are needed for patients with chronic angina – particularly in the United States, both to improve symptoms and to improve quality of life.
What we may need to consider is the level at which an individual patient might be willing to trade some potential risk of major nonfatal cardiovascular events for less angina and a better quality of life.
Dr. E. Magnus Ohman and Dr. Karen P. Alexander of Duke University, Durham, N.C., made these remarks in an accompanying editorial (N. Engl. J. Med. 2014, Aug. 31[doi: 10.1056/NEJMe1409369]). Dr. Ohman reported receiving grant support and/or consulting fees from Abiomed, AstraZeneca, Daiichi Sankyo, Eli Lilly, Gilead, Janssen, Pozen, Sanofi Aventis, The Medicines Company, and WebMD. Dr. Alexander reported receiving grant support from Gilead.
The increased incidence of the primary endpoint among patients with Canadian Cardiovascular Society Class II or higher levels of angina in the SIGNIFY trial was surprising and warrants further study to ascertain whether this angina subgroup is one in which caution should be exercised.
In the meantime, caution should indeed be exercised with respect to the use of ivabradine (by physicians who have access to the drug) in those with more severe forms of angina, and adjustment of beta-blocker doses to effective levels should be considered before initiating ivabradine.
The experience from the trial of ivabradine in heart failure suggests that nearly 60% of patients were receiving inadequate doses of beta-blockers and that the majority of benefit with ivabradine was among patients who could not take beta-blockers or who were taking a lower dose. Whether this holds true for patients with angina is unknown, but a cautious approach may be reasonable pending better understanding of the matter.
More therapies are needed for patients with chronic angina – particularly in the United States, both to improve symptoms and to improve quality of life.
What we may need to consider is the level at which an individual patient might be willing to trade some potential risk of major nonfatal cardiovascular events for less angina and a better quality of life.
Dr. E. Magnus Ohman and Dr. Karen P. Alexander of Duke University, Durham, N.C., made these remarks in an accompanying editorial (N. Engl. J. Med. 2014, Aug. 31[doi: 10.1056/NEJMe1409369]). Dr. Ohman reported receiving grant support and/or consulting fees from Abiomed, AstraZeneca, Daiichi Sankyo, Eli Lilly, Gilead, Janssen, Pozen, Sanofi Aventis, The Medicines Company, and WebMD. Dr. Alexander reported receiving grant support from Gilead.
The increased incidence of the primary endpoint among patients with Canadian Cardiovascular Society Class II or higher levels of angina in the SIGNIFY trial was surprising and warrants further study to ascertain whether this angina subgroup is one in which caution should be exercised.
In the meantime, caution should indeed be exercised with respect to the use of ivabradine (by physicians who have access to the drug) in those with more severe forms of angina, and adjustment of beta-blocker doses to effective levels should be considered before initiating ivabradine.
The experience from the trial of ivabradine in heart failure suggests that nearly 60% of patients were receiving inadequate doses of beta-blockers and that the majority of benefit with ivabradine was among patients who could not take beta-blockers or who were taking a lower dose. Whether this holds true for patients with angina is unknown, but a cautious approach may be reasonable pending better understanding of the matter.
More therapies are needed for patients with chronic angina – particularly in the United States, both to improve symptoms and to improve quality of life.
What we may need to consider is the level at which an individual patient might be willing to trade some potential risk of major nonfatal cardiovascular events for less angina and a better quality of life.
Dr. E. Magnus Ohman and Dr. Karen P. Alexander of Duke University, Durham, N.C., made these remarks in an accompanying editorial (N. Engl. J. Med. 2014, Aug. 31[doi: 10.1056/NEJMe1409369]). Dr. Ohman reported receiving grant support and/or consulting fees from Abiomed, AstraZeneca, Daiichi Sankyo, Eli Lilly, Gilead, Janssen, Pozen, Sanofi Aventis, The Medicines Company, and WebMD. Dr. Alexander reported receiving grant support from Gilead.
Adding ivabradine to standard background therapy did not improve outcomes in a large, randomized, placebo-controlled trial of more than 19,000 patients who had stable coronary artery disease without clinical heart failure.
At 3 months, the mean heart rate was 60.7 beats per minute in 9,550 patients in the Study Assessing the Morbidity-Mortality Benefits of the If Inhibitor Ivabradine in Patients with Coronary Artery Disease (SIGNIFY) who were assigned to receive ivabradine, compared with 70.6 beats per minute in 9,552 patients who received placebo. At a median of 27.8 months, the percentage of patients reaching a composite endpoint of death from cardiovascular causes or nonfatal myocardial infarction was 6.8% and 6.4% in the groups, respectively (hazard ratio,1.08), Dr. Kim Fox of Royal Brompton Hospital, London and colleagues reported at the 2014 European Society of Cardiology Congress.
The differences between the ivabradine and placebo groups were not statistically significant, but ivabradine was associated with a nonsignificant increase in the incidence of the primary end point in patients with Canadian Cardiovascular Society class II angina or higher (7.6% vs. 6.5%, hazard ratio, 1.18), the investigators said.
The findings were simultaneously published online Aug. 31 in the New England Journal of Medicine (N. Engl. J. Med 2014 Aug. 30[doi:10.1056/NEJMoa1406430].
Study participants, who were recruited from 1,139 centers in 51 countries between October 12, 2009 and April 30, 2012, were at least 55 years of age with documented and treated stable coronary artery disease, but no evidence of clinical heart failure. They also had to be in sinus rhythm, have a resting heart rate of 70 beats per minute or more on two consecutive readings, and have at least one major or two minor adverse prognostic factors.
All patients completed a 2- to 4-week placebo run-in phase then were randomized to ivabradine at a dose of 7.5 mg twice daily (or 5 mg twice daily for those over age 74 years) or placebo in addition to stable background therapy prescribed according to contemporary guidelines. Dosing was adjusted as needed to 5, 7.5, or 10 mg twice daily based on heart rate and bradycardia. The target heart rate was 55-60 beats per minute, and the mean study-drug dose throughout the trial was 8.2 mg twice daily in the ivabradine group, and 9.5 mg twice daily in the placebo group.
Adverse events occurred in significantly more patients in the ivabradine group (73.3% vs. 66.9%). Bradycardia, in particular, was increased in the ivabradine group (7.9% vs. 1.2% for symptomatic bradycardia, and 11.0% vs. 1.3% for asymptomatic bradycardia. Atrial fibrillation and phosphenes were also increased in the ivabradine group (5.3 vs. 3.8% and 5.4% vs. 0.5%, respectively).
Serious adverse events were also significantly more common in the ivabradine group, with 37.6% and 35.4% of patients in the groups, respectively, experiencing a serious adverse event. These events were classified as cardiac disorders in 19.0% and 16.7% of patients in the groups, respectively.
The SIGNIFY findings contrast with those from previous post hoc analyses that suggested ivabradine improves outcomes in patients with stable coronary artery disease.
"In addition, in patients with heart failure, the reduction in heart rate with ivabradine has been shown to improve clinical outcomes, beyond the improvements observed with beta-blockers," the investigators said.
In fact, on the basis of findings from the phase III Systolic Heart Failure Treatment with the If Inhibitor Ivabradine Trial (SHIFT), the U.S. Food and Drug Administration recently granted priority review designation for ivabradine for the treatment of chronic heart failure.
Ivabradine lowers heart rate without affecting blood pressure or left ventricular systolic function, by inhibiting the If, or pacemaker, current in the sinoatrial node. It is approved in numerous countries outside the United States and is used for reducing angina symptoms and for chronic heart failure.
A number of hypotheses may explain the SIGNIFY findings.
"It is possible that ivabradine decreased the heart rate too much or that there may be a J-shaped curve for the relationship between heart rate and outcome," the investigators said, explaining that "ivabradine may have unintended effects (e.g., adjustment of the doses of other heart-rate lowering agents) that may have affected the potential benefits of the lowering of heart rate with ivabradine."
Further, heart rate-reducing antianginal agents may have no effect on outcomes in patients with stable coronary artery disease, they said.
The findings may also reflect the fact that an elevated heart rate is due to different pathophysiological mechanisms in those with heart failure and those with stable coronary artery disease, they noted.
"Given that the primary cardiovascular effect of ivabradine is to reduce heart rate, these results suggest that an elevated heart rate is only a marker of risk – but not a modifiable determinant of outcomes- in patients who have stable coronary artery disease without clinical heart failure," they concluded.
This study was funded by Servier. Dr. Fox reported receiving personal fees and/or nonfinancial support from Servier, AstraZeneca, TaurX, Armgo, Broadview Ventures, and CellAegis. She also is director of Heart Research Lt. and Vesalius Trials Ltd. Detailed disclosures for all study authors are available at NEJM.org.
Adding ivabradine to standard background therapy did not improve outcomes in a large, randomized, placebo-controlled trial of more than 19,000 patients who had stable coronary artery disease without clinical heart failure.
At 3 months, the mean heart rate was 60.7 beats per minute in 9,550 patients in the Study Assessing the Morbidity-Mortality Benefits of the If Inhibitor Ivabradine in Patients with Coronary Artery Disease (SIGNIFY) who were assigned to receive ivabradine, compared with 70.6 beats per minute in 9,552 patients who received placebo. At a median of 27.8 months, the percentage of patients reaching a composite endpoint of death from cardiovascular causes or nonfatal myocardial infarction was 6.8% and 6.4% in the groups, respectively (hazard ratio,1.08), Dr. Kim Fox of Royal Brompton Hospital, London and colleagues reported at the 2014 European Society of Cardiology Congress.
The differences between the ivabradine and placebo groups were not statistically significant, but ivabradine was associated with a nonsignificant increase in the incidence of the primary end point in patients with Canadian Cardiovascular Society class II angina or higher (7.6% vs. 6.5%, hazard ratio, 1.18), the investigators said.
The findings were simultaneously published online Aug. 31 in the New England Journal of Medicine (N. Engl. J. Med 2014 Aug. 30[doi:10.1056/NEJMoa1406430].
Study participants, who were recruited from 1,139 centers in 51 countries between October 12, 2009 and April 30, 2012, were at least 55 years of age with documented and treated stable coronary artery disease, but no evidence of clinical heart failure. They also had to be in sinus rhythm, have a resting heart rate of 70 beats per minute or more on two consecutive readings, and have at least one major or two minor adverse prognostic factors.
All patients completed a 2- to 4-week placebo run-in phase then were randomized to ivabradine at a dose of 7.5 mg twice daily (or 5 mg twice daily for those over age 74 years) or placebo in addition to stable background therapy prescribed according to contemporary guidelines. Dosing was adjusted as needed to 5, 7.5, or 10 mg twice daily based on heart rate and bradycardia. The target heart rate was 55-60 beats per minute, and the mean study-drug dose throughout the trial was 8.2 mg twice daily in the ivabradine group, and 9.5 mg twice daily in the placebo group.
Adverse events occurred in significantly more patients in the ivabradine group (73.3% vs. 66.9%). Bradycardia, in particular, was increased in the ivabradine group (7.9% vs. 1.2% for symptomatic bradycardia, and 11.0% vs. 1.3% for asymptomatic bradycardia. Atrial fibrillation and phosphenes were also increased in the ivabradine group (5.3 vs. 3.8% and 5.4% vs. 0.5%, respectively).
Serious adverse events were also significantly more common in the ivabradine group, with 37.6% and 35.4% of patients in the groups, respectively, experiencing a serious adverse event. These events were classified as cardiac disorders in 19.0% and 16.7% of patients in the groups, respectively.
The SIGNIFY findings contrast with those from previous post hoc analyses that suggested ivabradine improves outcomes in patients with stable coronary artery disease.
"In addition, in patients with heart failure, the reduction in heart rate with ivabradine has been shown to improve clinical outcomes, beyond the improvements observed with beta-blockers," the investigators said.
In fact, on the basis of findings from the phase III Systolic Heart Failure Treatment with the If Inhibitor Ivabradine Trial (SHIFT), the U.S. Food and Drug Administration recently granted priority review designation for ivabradine for the treatment of chronic heart failure.
Ivabradine lowers heart rate without affecting blood pressure or left ventricular systolic function, by inhibiting the If, or pacemaker, current in the sinoatrial node. It is approved in numerous countries outside the United States and is used for reducing angina symptoms and for chronic heart failure.
A number of hypotheses may explain the SIGNIFY findings.
"It is possible that ivabradine decreased the heart rate too much or that there may be a J-shaped curve for the relationship between heart rate and outcome," the investigators said, explaining that "ivabradine may have unintended effects (e.g., adjustment of the doses of other heart-rate lowering agents) that may have affected the potential benefits of the lowering of heart rate with ivabradine."
Further, heart rate-reducing antianginal agents may have no effect on outcomes in patients with stable coronary artery disease, they said.
The findings may also reflect the fact that an elevated heart rate is due to different pathophysiological mechanisms in those with heart failure and those with stable coronary artery disease, they noted.
"Given that the primary cardiovascular effect of ivabradine is to reduce heart rate, these results suggest that an elevated heart rate is only a marker of risk – but not a modifiable determinant of outcomes- in patients who have stable coronary artery disease without clinical heart failure," they concluded.
This study was funded by Servier. Dr. Fox reported receiving personal fees and/or nonfinancial support from Servier, AstraZeneca, TaurX, Armgo, Broadview Ventures, and CellAegis. She also is director of Heart Research Lt. and Vesalius Trials Ltd. Detailed disclosures for all study authors are available at NEJM.org.
FROM THE ESC CONGRESS 2014
Key clinical point: The investigational drug ivabradine did not improve outcomes in stable CAD patients with no heart failure.
Major finding: No significant difference was seen between ivabradine and placebo; HR for composite endpoint of death from cardiovascular causes or nonfatal MI, 1.08.
Data source: SIGNIFY, a randomized, double blind, placebo-controlled trial in 19.102 patients.
Disclosures: This study was funded by Servier. Dr. Fox reported receiving personal fees and/or nonfinancial support from Servier, AstraZeneca, TaurX, Armgo, Broadview Ventures, and CellAegis. She also is director of Heart Research Lt. and Vesalius Trials Ltd. Detailed disclosures for all study authors are available at NEJM.org.
VIDEO: New dual heart-failure formulation scores several benefits
BARCELONA – It’s been a while since physicians had a new treatment for heart failure patients with reduced ejection fraction that makes them feel better, stay out of hospital, and live longer. But the investigational drug LCZ696 did just that in PARADIGM-HF, a controlled, pivotal trial with more than 8,000 patients, Dr. John McMurray said at the annual congress of the European Society of Cardiology.
In addition to robustly beating the comparator drug enalapril with reduced rates of both cardiovascular deaths and the combined endpoint of cardiovascular deaths and heart-failure hospitalizations, the new LCZ696 combination drug boosted patients’ quality of life in several clinically meaningful ways. The magnitude of the quality of life effect matched that seen in prior, successful, heart-failure treatment trials, Dr. McMurray, a professor of medical cardiology at the University of Glasgow, said in an interview.
He stressed that the combination of sacubitril, a new drug that inhibits neprilysin and thereby boosts levels of endogenous vasoactive peptides, and the well-established angiotensin-receptor blocker valsartan helped patients with milder, New York Heart Association class II heart failure, roughly 70% of enrolled patients. Activity in these less severely ill patients means that treatment with LCZ696 can "keep these patients stable, feeling well, functional, and out of the hospital as well as improve their survival," he said.
PARADIGM-HF was sponsored by Novartis, the company developing LCZ696. Dr. McMurray said that he has received travel support from Novartis.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Twitter: @mitchelzoler
BARCELONA – It’s been a while since physicians had a new treatment for heart failure patients with reduced ejection fraction that makes them feel better, stay out of hospital, and live longer. But the investigational drug LCZ696 did just that in PARADIGM-HF, a controlled, pivotal trial with more than 8,000 patients, Dr. John McMurray said at the annual congress of the European Society of Cardiology.
In addition to robustly beating the comparator drug enalapril with reduced rates of both cardiovascular deaths and the combined endpoint of cardiovascular deaths and heart-failure hospitalizations, the new LCZ696 combination drug boosted patients’ quality of life in several clinically meaningful ways. The magnitude of the quality of life effect matched that seen in prior, successful, heart-failure treatment trials, Dr. McMurray, a professor of medical cardiology at the University of Glasgow, said in an interview.
He stressed that the combination of sacubitril, a new drug that inhibits neprilysin and thereby boosts levels of endogenous vasoactive peptides, and the well-established angiotensin-receptor blocker valsartan helped patients with milder, New York Heart Association class II heart failure, roughly 70% of enrolled patients. Activity in these less severely ill patients means that treatment with LCZ696 can "keep these patients stable, feeling well, functional, and out of the hospital as well as improve their survival," he said.
PARADIGM-HF was sponsored by Novartis, the company developing LCZ696. Dr. McMurray said that he has received travel support from Novartis.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Twitter: @mitchelzoler
BARCELONA – It’s been a while since physicians had a new treatment for heart failure patients with reduced ejection fraction that makes them feel better, stay out of hospital, and live longer. But the investigational drug LCZ696 did just that in PARADIGM-HF, a controlled, pivotal trial with more than 8,000 patients, Dr. John McMurray said at the annual congress of the European Society of Cardiology.
In addition to robustly beating the comparator drug enalapril with reduced rates of both cardiovascular deaths and the combined endpoint of cardiovascular deaths and heart-failure hospitalizations, the new LCZ696 combination drug boosted patients’ quality of life in several clinically meaningful ways. The magnitude of the quality of life effect matched that seen in prior, successful, heart-failure treatment trials, Dr. McMurray, a professor of medical cardiology at the University of Glasgow, said in an interview.
He stressed that the combination of sacubitril, a new drug that inhibits neprilysin and thereby boosts levels of endogenous vasoactive peptides, and the well-established angiotensin-receptor blocker valsartan helped patients with milder, New York Heart Association class II heart failure, roughly 70% of enrolled patients. Activity in these less severely ill patients means that treatment with LCZ696 can "keep these patients stable, feeling well, functional, and out of the hospital as well as improve their survival," he said.
PARADIGM-HF was sponsored by Novartis, the company developing LCZ696. Dr. McMurray said that he has received travel support from Novartis.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Twitter: @mitchelzoler
AT ESC 2014

Many acute HF patients overuse emergency departments
Approximately one-third of patients who present to the emergency department with acute heart failure syndrome are frequent ED users, and they account for "an enormous burden of health care utilization and expenditures," according to a report published online Aug. 19 in Circulation: Cardiovascular Quality and Outcomes.
Frequent ED use for this condition reflects a failure in secondary prevention and is considered to be preventable with good-quality longitudinal management, said Dr. Kohei Hasegawa of the department of emergency medicine, Massachusetts General Hospital and Harvard Medical School, Boston.
Dr. Hasegawa and his colleagues assessed the frequency of ED usage by analyzing information from two large databases, focusing on 113,033 adults who made 175,491 ED visits for acute HF syndrome in California and Florida during a 1-year period. Most of these patients (69%) required only one ED visit during that time, but the remaining 31% were frequent users (overusers) who had at least two visits, accounted for more than half of all ED visits and hospitalizations for this condition, and accounted for 58% of all near-fatal events.
In this study, if recurrent ED visits after the index visit had been prevented by better quality longitudinal care, "up to 62,458 ED visits and 53,234 hospitalizations would have been saved annually in [these] two states alone. In terms of expenditures, this would have saved $1.06 billion, in Florida alone," Dr. Hasegawa and his associates said (Circ. Cardiovasc. Qual. Outcomes 2014 Aug. 19 [doi: 10.1161/Circoutcomes.114.000949]).
Frequent users were more likely to be male than female; to be black or Hispanic than other races/ethnicities; to have several markers of low socioeconomic status such as low income and Medicaid insurance; and to have several comorbid conditions such as chronic pulmonary disease, renal failure, diabetes, depression, and drug abuse.
"Although the pathway through which socioeconomic status and health care–related factors affect health care utilization is undoubtedly complex, studies in other chronic conditions suggest that less self-management education and limited access to preventive care in this population might lead to a heavier reliance on episodic treatment in the ED," the investigators said.
This study was supported in part by the Eleanor and Miles Shore Fellowship Program and the Honjo International Scholarship Foundation. Dr. Hasegawa and his associates reported no financial conflicts of interest.
Approximately one-third of patients who present to the emergency department with acute heart failure syndrome are frequent ED users, and they account for "an enormous burden of health care utilization and expenditures," according to a report published online Aug. 19 in Circulation: Cardiovascular Quality and Outcomes.
Frequent ED use for this condition reflects a failure in secondary prevention and is considered to be preventable with good-quality longitudinal management, said Dr. Kohei Hasegawa of the department of emergency medicine, Massachusetts General Hospital and Harvard Medical School, Boston.
Dr. Hasegawa and his colleagues assessed the frequency of ED usage by analyzing information from two large databases, focusing on 113,033 adults who made 175,491 ED visits for acute HF syndrome in California and Florida during a 1-year period. Most of these patients (69%) required only one ED visit during that time, but the remaining 31% were frequent users (overusers) who had at least two visits, accounted for more than half of all ED visits and hospitalizations for this condition, and accounted for 58% of all near-fatal events.
In this study, if recurrent ED visits after the index visit had been prevented by better quality longitudinal care, "up to 62,458 ED visits and 53,234 hospitalizations would have been saved annually in [these] two states alone. In terms of expenditures, this would have saved $1.06 billion, in Florida alone," Dr. Hasegawa and his associates said (Circ. Cardiovasc. Qual. Outcomes 2014 Aug. 19 [doi: 10.1161/Circoutcomes.114.000949]).
Frequent users were more likely to be male than female; to be black or Hispanic than other races/ethnicities; to have several markers of low socioeconomic status such as low income and Medicaid insurance; and to have several comorbid conditions such as chronic pulmonary disease, renal failure, diabetes, depression, and drug abuse.
"Although the pathway through which socioeconomic status and health care–related factors affect health care utilization is undoubtedly complex, studies in other chronic conditions suggest that less self-management education and limited access to preventive care in this population might lead to a heavier reliance on episodic treatment in the ED," the investigators said.
This study was supported in part by the Eleanor and Miles Shore Fellowship Program and the Honjo International Scholarship Foundation. Dr. Hasegawa and his associates reported no financial conflicts of interest.
Approximately one-third of patients who present to the emergency department with acute heart failure syndrome are frequent ED users, and they account for "an enormous burden of health care utilization and expenditures," according to a report published online Aug. 19 in Circulation: Cardiovascular Quality and Outcomes.
Frequent ED use for this condition reflects a failure in secondary prevention and is considered to be preventable with good-quality longitudinal management, said Dr. Kohei Hasegawa of the department of emergency medicine, Massachusetts General Hospital and Harvard Medical School, Boston.
Dr. Hasegawa and his colleagues assessed the frequency of ED usage by analyzing information from two large databases, focusing on 113,033 adults who made 175,491 ED visits for acute HF syndrome in California and Florida during a 1-year period. Most of these patients (69%) required only one ED visit during that time, but the remaining 31% were frequent users (overusers) who had at least two visits, accounted for more than half of all ED visits and hospitalizations for this condition, and accounted for 58% of all near-fatal events.
In this study, if recurrent ED visits after the index visit had been prevented by better quality longitudinal care, "up to 62,458 ED visits and 53,234 hospitalizations would have been saved annually in [these] two states alone. In terms of expenditures, this would have saved $1.06 billion, in Florida alone," Dr. Hasegawa and his associates said (Circ. Cardiovasc. Qual. Outcomes 2014 Aug. 19 [doi: 10.1161/Circoutcomes.114.000949]).
Frequent users were more likely to be male than female; to be black or Hispanic than other races/ethnicities; to have several markers of low socioeconomic status such as low income and Medicaid insurance; and to have several comorbid conditions such as chronic pulmonary disease, renal failure, diabetes, depression, and drug abuse.
"Although the pathway through which socioeconomic status and health care–related factors affect health care utilization is undoubtedly complex, studies in other chronic conditions suggest that less self-management education and limited access to preventive care in this population might lead to a heavier reliance on episodic treatment in the ED," the investigators said.
This study was supported in part by the Eleanor and Miles Shore Fellowship Program and the Honjo International Scholarship Foundation. Dr. Hasegawa and his associates reported no financial conflicts of interest.
FROM CIRCULATION: CARDIOVASCULAR QUALITY AND OUTCOMES
Key clinical point: Better preventive care in acute heart failure syndrome patients could cut costly emergency department visits.
Major finding: Most acute heart failure syndrome patients (69%) required only one emergency department visit during the 1-year study, but the remaining 31% were frequent users (overusers) who accounted for more than half of all ED visits and hospitalizations for this condition and for 58% of all near-fatal events.
Data source: A retrospective cohort study of 113,033 adults in California and Florida who presented to an emergency department with acute HF syndrome during a 1-year period.
Disclosures: This study was supported in part by the Eleanor and Miles Shore Fellowship Program and the Honjo International Scholarship Foundation. Dr. Hasegawa and his associates reported no financial conflicts of interest.
Frozen or powdered? Anticoagulation options in trauma are expanding
SAN DIEGO – In the next 5-10 years, reaching for a powdered form of plasma may become the normative first-line treatment for trauma patients who present with severe bleeding. That’s because the current standard of administering fresh frozen plasma is riddled with problems, Dr. Martin Schreiber said at the University of California, San Diego, Critical Care Summer Session.
For one thing, frozen plasma takes 35 minutes to thaw. "That’s a problem, and the clotting factor function of plasma deteriorates as you freeze it and thaw it," said Dr. Schreiber, professor of surgery at Oregon Health & Science University, Portland. "Also, you need it in large volumes and that’s not good for patients with congestive heart failure on Coumadin, and the availability is limited, especially in rural settings."

Enter lyophilized plasma (LP), a process developed by HemCon Medical Technologies in which whole blood is sterilely removed, and the plasma component is separated and turned into a powder. The powdered plasma is returned and reconstituted prior to transfusion. "You can put this stuff as a powder on a shelf in nearly any environment," Dr. Schreiber said. "It’s good for at least 3 years, it survives a broad range of temperatures, and you can restore it to plasma within a couple of minutes."
An initial study of LP showed encouraging results with the use of a freeze-dried form of plasma for resuscitation (J. Trauma 2008;65[5]:975-85). A later study by researchers including Dr. Schreiber evaluated the effects of the lyophilization process on plasma clotting factor levels in swine, by adding the antioxidant ascorbic acid (vitamin C) to the reconstitution solution, and by comparing the efficacy of LP with that of fresh frozen plasma and that of plasma and packed red blood cells in a 1:1 ratio (Arch. Surg. 2009;144[9]:829-34). "What we found was that if we gave LP with packed red blood cells in a 1:1 ratio we had 14% less blood loss than if it’s given as FFP with packed cells, which was significant," Dr. Schreiber said.
"The LP was better in terms of stopping hemorrhage. We also noticed that with the vitamin C, we suppressed inflammation and got reduced IL-6 [interleukin 6] expression. Now, the Germans, the Dutch, and the French are using LP in their military settings. Our special forces people are also using it. It’s under current development for common use in your hospital in 5-10 years. I think this stuff is good anywhere. With LP we can always maintain a 1:1 ratio and we don’t have to worry about the thawing process."
Tranexamic acid, a synthetic derivative of lysine, is another anticoagulant therapy that is likely to be used with increasing frequency, he predicted. This agent "binds plasminogen so plasminogen can’t break down fibrin so you can’t get fibrinolysis," said Dr. Schreiber, who has been deployed three times as a combat surgeon.
"This drug has been around forever and is extremely inexpensive. It’s approved by the FDA for use in tooth extraction and the oral form is approved for menorrhagia." Tranexamic acid has also been studied in 53 prospective, randomized studies involving some 3,800 subjects, mostly cardiac patients. "They show that if you use tranexamic acid, you use less blood. It reduces the amount of blood necessary for transfusing people in high-bleeding settings, but no difference in mortality, thrombotic events, myocardial infarction, or stroke. It does not seem to produce a hypercoagulable state."

One study of tranexamic acid use by British surgeons during Afghanistan combat operations found that soldiers who received tranexamic acid were seven times more likely to live, compared with those who did not receive the agent (Arch. Surg. 2012;147:113-19). "There was a survival of 85% in tranexamic acid group, compared with about 70% in those who did not receive it," said Dr. Schreiber. "This has resulted in a change in practice in civilian trauma centers where it is being used widely."
Another anticoagulant being used is the prothrombin complex concentrate known as Kcentra, which contains all four vitamin K–dependent coagulation factors. Distributed by CSL Behring, Kcentra is approved for warfarin reversal in adult patients with acute major bleeding and for those who require emergency surgery. The max dose is 50 units/kg. "That’s about $4,445 for a 70-kg person," Dr. Schreiber said. "Why is it so good? It’s rapidly available, you don’t have to give a lot of fluid, there’s no infectious risk, and you can very rapidly increase coagulation factor function. This is where we’re headed in the future for trauma patients."
Dr. Schreiber said that he had no relevant financial conflicts to disclose.
On Twitter @dougbrunk
SAN DIEGO – In the next 5-10 years, reaching for a powdered form of plasma may become the normative first-line treatment for trauma patients who present with severe bleeding. That’s because the current standard of administering fresh frozen plasma is riddled with problems, Dr. Martin Schreiber said at the University of California, San Diego, Critical Care Summer Session.
For one thing, frozen plasma takes 35 minutes to thaw. "That’s a problem, and the clotting factor function of plasma deteriorates as you freeze it and thaw it," said Dr. Schreiber, professor of surgery at Oregon Health & Science University, Portland. "Also, you need it in large volumes and that’s not good for patients with congestive heart failure on Coumadin, and the availability is limited, especially in rural settings."
Enter lyophilized plasma (LP), a process developed by HemCon Medical Technologies in which whole blood is sterilely removed, and the plasma component is separated and turned into a powder. The powdered plasma is returned and reconstituted prior to transfusion. "You can put this stuff as a powder on a shelf in nearly any environment," Dr. Schreiber said. "It’s good for at least 3 years, it survives a broad range of temperatures, and you can restore it to plasma within a couple of minutes."
An initial study of LP showed encouraging results with the use of a freeze-dried form of plasma for resuscitation (J. Trauma 2008;65[5]:975-85). A later study by researchers including Dr. Schreiber evaluated the effects of the lyophilization process on plasma clotting factor levels in swine, by adding the antioxidant ascorbic acid (vitamin C) to the reconstitution solution, and by comparing the efficacy of LP with that of fresh frozen plasma and that of plasma and packed red blood cells in a 1:1 ratio (Arch. Surg. 2009;144[9]:829-34). "What we found was that if we gave LP with packed red blood cells in a 1:1 ratio we had 14% less blood loss than if it’s given as FFP with packed cells, which was significant," Dr. Schreiber said.
"The LP was better in terms of stopping hemorrhage. We also noticed that with the vitamin C, we suppressed inflammation and got reduced IL-6 [interleukin 6] expression. Now, the Germans, the Dutch, and the French are using LP in their military settings. Our special forces people are also using it. It’s under current development for common use in your hospital in 5-10 years. I think this stuff is good anywhere. With LP we can always maintain a 1:1 ratio and we don’t have to worry about the thawing process."
Tranexamic acid, a synthetic derivative of lysine, is another anticoagulant therapy that is likely to be used with increasing frequency, he predicted. This agent "binds plasminogen so plasminogen can’t break down fibrin so you can’t get fibrinolysis," said Dr. Schreiber, who has been deployed three times as a combat surgeon.
"This drug has been around forever and is extremely inexpensive. It’s approved by the FDA for use in tooth extraction and the oral form is approved for menorrhagia." Tranexamic acid has also been studied in 53 prospective, randomized studies involving some 3,800 subjects, mostly cardiac patients. "They show that if you use tranexamic acid, you use less blood. It reduces the amount of blood necessary for transfusing people in high-bleeding settings, but no difference in mortality, thrombotic events, myocardial infarction, or stroke. It does not seem to produce a hypercoagulable state."
One study of tranexamic acid use by British surgeons during Afghanistan combat operations found that soldiers who received tranexamic acid were seven times more likely to live, compared with those who did not receive the agent (Arch. Surg. 2012;147:113-19). "There was a survival of 85% in tranexamic acid group, compared with about 70% in those who did not receive it," said Dr. Schreiber. "This has resulted in a change in practice in civilian trauma centers where it is being used widely."
Another anticoagulant being used is the prothrombin complex concentrate known as Kcentra, which contains all four vitamin K–dependent coagulation factors. Distributed by CSL Behring, Kcentra is approved for warfarin reversal in adult patients with acute major bleeding and for those who require emergency surgery. The max dose is 50 units/kg. "That’s about $4,445 for a 70-kg person," Dr. Schreiber said. "Why is it so good? It’s rapidly available, you don’t have to give a lot of fluid, there’s no infectious risk, and you can very rapidly increase coagulation factor function. This is where we’re headed in the future for trauma patients."
Dr. Schreiber said that he had no relevant financial conflicts to disclose.
On Twitter @dougbrunk
SAN DIEGO – In the next 5-10 years, reaching for a powdered form of plasma may become the normative first-line treatment for trauma patients who present with severe bleeding. That’s because the current standard of administering fresh frozen plasma is riddled with problems, Dr. Martin Schreiber said at the University of California, San Diego, Critical Care Summer Session.
For one thing, frozen plasma takes 35 minutes to thaw. "That’s a problem, and the clotting factor function of plasma deteriorates as you freeze it and thaw it," said Dr. Schreiber, professor of surgery at Oregon Health & Science University, Portland. "Also, you need it in large volumes and that’s not good for patients with congestive heart failure on Coumadin, and the availability is limited, especially in rural settings."
Enter lyophilized plasma (LP), a process developed by HemCon Medical Technologies in which whole blood is sterilely removed, and the plasma component is separated and turned into a powder. The powdered plasma is returned and reconstituted prior to transfusion. "You can put this stuff as a powder on a shelf in nearly any environment," Dr. Schreiber said. "It’s good for at least 3 years, it survives a broad range of temperatures, and you can restore it to plasma within a couple of minutes."
An initial study of LP showed encouraging results with the use of a freeze-dried form of plasma for resuscitation (J. Trauma 2008;65[5]:975-85). A later study by researchers including Dr. Schreiber evaluated the effects of the lyophilization process on plasma clotting factor levels in swine, by adding the antioxidant ascorbic acid (vitamin C) to the reconstitution solution, and by comparing the efficacy of LP with that of fresh frozen plasma and that of plasma and packed red blood cells in a 1:1 ratio (Arch. Surg. 2009;144[9]:829-34). "What we found was that if we gave LP with packed red blood cells in a 1:1 ratio we had 14% less blood loss than if it’s given as FFP with packed cells, which was significant," Dr. Schreiber said.
"The LP was better in terms of stopping hemorrhage. We also noticed that with the vitamin C, we suppressed inflammation and got reduced IL-6 [interleukin 6] expression. Now, the Germans, the Dutch, and the French are using LP in their military settings. Our special forces people are also using it. It’s under current development for common use in your hospital in 5-10 years. I think this stuff is good anywhere. With LP we can always maintain a 1:1 ratio and we don’t have to worry about the thawing process."
Tranexamic acid, a synthetic derivative of lysine, is another anticoagulant therapy that is likely to be used with increasing frequency, he predicted. This agent "binds plasminogen so plasminogen can’t break down fibrin so you can’t get fibrinolysis," said Dr. Schreiber, who has been deployed three times as a combat surgeon.
"This drug has been around forever and is extremely inexpensive. It’s approved by the FDA for use in tooth extraction and the oral form is approved for menorrhagia." Tranexamic acid has also been studied in 53 prospective, randomized studies involving some 3,800 subjects, mostly cardiac patients. "They show that if you use tranexamic acid, you use less blood. It reduces the amount of blood necessary for transfusing people in high-bleeding settings, but no difference in mortality, thrombotic events, myocardial infarction, or stroke. It does not seem to produce a hypercoagulable state."
One study of tranexamic acid use by British surgeons during Afghanistan combat operations found that soldiers who received tranexamic acid were seven times more likely to live, compared with those who did not receive the agent (Arch. Surg. 2012;147:113-19). "There was a survival of 85% in tranexamic acid group, compared with about 70% in those who did not receive it," said Dr. Schreiber. "This has resulted in a change in practice in civilian trauma centers where it is being used widely."
Another anticoagulant being used is the prothrombin complex concentrate known as Kcentra, which contains all four vitamin K–dependent coagulation factors. Distributed by CSL Behring, Kcentra is approved for warfarin reversal in adult patients with acute major bleeding and for those who require emergency surgery. The max dose is 50 units/kg. "That’s about $4,445 for a 70-kg person," Dr. Schreiber said. "Why is it so good? It’s rapidly available, you don’t have to give a lot of fluid, there’s no infectious risk, and you can very rapidly increase coagulation factor function. This is where we’re headed in the future for trauma patients."
Dr. Schreiber said that he had no relevant financial conflicts to disclose.
On Twitter @dougbrunk
EXPERT ANALYSIS AT THE UCSD CRITICAL CARE SUMMER SESSION
Reassuring results on cardiovascular safety of DPP-4 inhibitors
CHICAGO – Treatment with a dipeptidyl peptidase-4 inhibitor in real-world community practice doesn’t increase the risk of cardiovascular events in type 2 diabetic patients; in fact, the opposite was true in a very large population-based cohort study.
The study, hailed by audience members as "a tour de force" and "a really excellent study," involved 39,769 type 2 diabetes patients who initiated treatment with a dipeptidyl peptidase-4 inhibitor (DPP-4i), either as monotherapy or, far more commonly, on top of metformin, and another 39,769 patients with type 2 diabetes on metformin who initiated combination therapy with a non–DPP-4i hypoglycemic agent. The two groups were closely matched using propensity scores that incorporated 50 variables, including comorbid conditions, medications, hemoglobin A1c, and health care utilization.
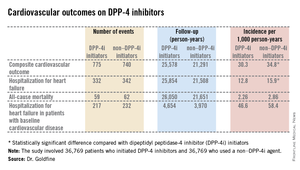
The primary endpoint was a composite cardiovascular outcome comprising acute MI, stroke, coronary revascularization, and hospitalization for heart failure. The rate was 30.3 per 1,000 person-years in patients after initiating DPP-4i therapy and 34.76 per 1,000 in controls who initiated treatment with a non–DPP-4i, for a statistically significant 13% relative risk reduction, Dr. Allison B. Goldfine reported at a joint meeting of the International Congress of Endocrinology and the Endocrine Society.
Also noteworthy was the finding that patients who initiated treatment with a DPP-4i had a 19% lower subsequent risk of hospitalization for heart failure than did those who initiated therapy with another glucose-lowering agent (see chart). This statistically significant reduction in heart failure hospitalization was particularly reassuring. Earlier, the roughly 16,500-subject, randomized, SAVOR-TIMI 53 trial showed a significant 27% increased risk of hospitalization for heart failure in patients assigned to saxagliptin, compared with placebo-treated controls (N. Engl. J. Med. 2013;369:1317-26).
Moreover, the EXAMINE study, another randomized trial, albeit smaller, reported a nonsignificant 19% increased relative risk of hospitalization for heart failure, compared with placebo (N. Engl. J. Med. 2013;369:1327-35). These findings raised a red flag at the Food and Drug Administration, which sought additional information.
Both SAVOR-TIMI 53 and EXAMINE involved type 2 diabetes patients with known cardiovascular disease at enrollment. In the new population-based cohort study, 7,293 of the 39,769 patient pairs had baseline cardiovascular disease. Reassuringly, those with baseline cardiovascular disease had a 12% relative risk reduction for the composite cardiovascular endpoint and a 16% lower risk of hospitalization for heart failure after going on a DPP-4i, compared with controls who initiated treatment with a non–DPP-4i. Both risk reductions barely missed achieving statistical significance because of insufficient patient numbers, said Dr. Goldfine, head of the section of clinical, behavioral, and outcomes research at the Joslin Diabetes Center, Boston, and an endocrinologist at Harvard University.

To investigate the additional possibility that going on DPP-4i therapy might somehow predispose to mild heart failure not severe enough to result in hospitalization, she and her coinvestigators looked at new use of loop diuretics among matched patient pairs not on that medication class at baseline. The risk of getting a new prescription for a loop diuretic during follow-up turned out to be lower by a statistically significant 26% in DPP-4i users, compared with nonusers.
The study utilized the UnitedHealthcare insurance claims database for 2005-2012. From an initial population of 4.35 million patients with type 2 diabetes, Dr. Goldfine and her coworkers utilized propensity scores to whittle down to a study population of just less than 40,000 very closely matched patient pairs.
The great majority of DPP-4i users in the study were on sitagliptin (Januvia), the first-in-class agent to reach the market.
The study was funded by the National Institutes of Health. Dr. Goldfine reported serving as an investigator on studies funded by Daiichi Sankyo, NovoNordisk, and other companies.
CHICAGO – Treatment with a dipeptidyl peptidase-4 inhibitor in real-world community practice doesn’t increase the risk of cardiovascular events in type 2 diabetic patients; in fact, the opposite was true in a very large population-based cohort study.
The study, hailed by audience members as "a tour de force" and "a really excellent study," involved 39,769 type 2 diabetes patients who initiated treatment with a dipeptidyl peptidase-4 inhibitor (DPP-4i), either as monotherapy or, far more commonly, on top of metformin, and another 39,769 patients with type 2 diabetes on metformin who initiated combination therapy with a non–DPP-4i hypoglycemic agent. The two groups were closely matched using propensity scores that incorporated 50 variables, including comorbid conditions, medications, hemoglobin A1c, and health care utilization.
The primary endpoint was a composite cardiovascular outcome comprising acute MI, stroke, coronary revascularization, and hospitalization for heart failure. The rate was 30.3 per 1,000 person-years in patients after initiating DPP-4i therapy and 34.76 per 1,000 in controls who initiated treatment with a non–DPP-4i, for a statistically significant 13% relative risk reduction, Dr. Allison B. Goldfine reported at a joint meeting of the International Congress of Endocrinology and the Endocrine Society.
Also noteworthy was the finding that patients who initiated treatment with a DPP-4i had a 19% lower subsequent risk of hospitalization for heart failure than did those who initiated therapy with another glucose-lowering agent (see chart). This statistically significant reduction in heart failure hospitalization was particularly reassuring. Earlier, the roughly 16,500-subject, randomized, SAVOR-TIMI 53 trial showed a significant 27% increased risk of hospitalization for heart failure in patients assigned to saxagliptin, compared with placebo-treated controls (N. Engl. J. Med. 2013;369:1317-26).
Moreover, the EXAMINE study, another randomized trial, albeit smaller, reported a nonsignificant 19% increased relative risk of hospitalization for heart failure, compared with placebo (N. Engl. J. Med. 2013;369:1327-35). These findings raised a red flag at the Food and Drug Administration, which sought additional information.
Both SAVOR-TIMI 53 and EXAMINE involved type 2 diabetes patients with known cardiovascular disease at enrollment. In the new population-based cohort study, 7,293 of the 39,769 patient pairs had baseline cardiovascular disease. Reassuringly, those with baseline cardiovascular disease had a 12% relative risk reduction for the composite cardiovascular endpoint and a 16% lower risk of hospitalization for heart failure after going on a DPP-4i, compared with controls who initiated treatment with a non–DPP-4i. Both risk reductions barely missed achieving statistical significance because of insufficient patient numbers, said Dr. Goldfine, head of the section of clinical, behavioral, and outcomes research at the Joslin Diabetes Center, Boston, and an endocrinologist at Harvard University.
To investigate the additional possibility that going on DPP-4i therapy might somehow predispose to mild heart failure not severe enough to result in hospitalization, she and her coinvestigators looked at new use of loop diuretics among matched patient pairs not on that medication class at baseline. The risk of getting a new prescription for a loop diuretic during follow-up turned out to be lower by a statistically significant 26% in DPP-4i users, compared with nonusers.
The study utilized the UnitedHealthcare insurance claims database for 2005-2012. From an initial population of 4.35 million patients with type 2 diabetes, Dr. Goldfine and her coworkers utilized propensity scores to whittle down to a study population of just less than 40,000 very closely matched patient pairs.
The great majority of DPP-4i users in the study were on sitagliptin (Januvia), the first-in-class agent to reach the market.
The study was funded by the National Institutes of Health. Dr. Goldfine reported serving as an investigator on studies funded by Daiichi Sankyo, NovoNordisk, and other companies.
CHICAGO – Treatment with a dipeptidyl peptidase-4 inhibitor in real-world community practice doesn’t increase the risk of cardiovascular events in type 2 diabetic patients; in fact, the opposite was true in a very large population-based cohort study.
The study, hailed by audience members as "a tour de force" and "a really excellent study," involved 39,769 type 2 diabetes patients who initiated treatment with a dipeptidyl peptidase-4 inhibitor (DPP-4i), either as monotherapy or, far more commonly, on top of metformin, and another 39,769 patients with type 2 diabetes on metformin who initiated combination therapy with a non–DPP-4i hypoglycemic agent. The two groups were closely matched using propensity scores that incorporated 50 variables, including comorbid conditions, medications, hemoglobin A1c, and health care utilization.
The primary endpoint was a composite cardiovascular outcome comprising acute MI, stroke, coronary revascularization, and hospitalization for heart failure. The rate was 30.3 per 1,000 person-years in patients after initiating DPP-4i therapy and 34.76 per 1,000 in controls who initiated treatment with a non–DPP-4i, for a statistically significant 13% relative risk reduction, Dr. Allison B. Goldfine reported at a joint meeting of the International Congress of Endocrinology and the Endocrine Society.
Also noteworthy was the finding that patients who initiated treatment with a DPP-4i had a 19% lower subsequent risk of hospitalization for heart failure than did those who initiated therapy with another glucose-lowering agent (see chart). This statistically significant reduction in heart failure hospitalization was particularly reassuring. Earlier, the roughly 16,500-subject, randomized, SAVOR-TIMI 53 trial showed a significant 27% increased risk of hospitalization for heart failure in patients assigned to saxagliptin, compared with placebo-treated controls (N. Engl. J. Med. 2013;369:1317-26).
Moreover, the EXAMINE study, another randomized trial, albeit smaller, reported a nonsignificant 19% increased relative risk of hospitalization for heart failure, compared with placebo (N. Engl. J. Med. 2013;369:1327-35). These findings raised a red flag at the Food and Drug Administration, which sought additional information.
Both SAVOR-TIMI 53 and EXAMINE involved type 2 diabetes patients with known cardiovascular disease at enrollment. In the new population-based cohort study, 7,293 of the 39,769 patient pairs had baseline cardiovascular disease. Reassuringly, those with baseline cardiovascular disease had a 12% relative risk reduction for the composite cardiovascular endpoint and a 16% lower risk of hospitalization for heart failure after going on a DPP-4i, compared with controls who initiated treatment with a non–DPP-4i. Both risk reductions barely missed achieving statistical significance because of insufficient patient numbers, said Dr. Goldfine, head of the section of clinical, behavioral, and outcomes research at the Joslin Diabetes Center, Boston, and an endocrinologist at Harvard University.
To investigate the additional possibility that going on DPP-4i therapy might somehow predispose to mild heart failure not severe enough to result in hospitalization, she and her coinvestigators looked at new use of loop diuretics among matched patient pairs not on that medication class at baseline. The risk of getting a new prescription for a loop diuretic during follow-up turned out to be lower by a statistically significant 26% in DPP-4i users, compared with nonusers.
The study utilized the UnitedHealthcare insurance claims database for 2005-2012. From an initial population of 4.35 million patients with type 2 diabetes, Dr. Goldfine and her coworkers utilized propensity scores to whittle down to a study population of just less than 40,000 very closely matched patient pairs.
The great majority of DPP-4i users in the study were on sitagliptin (Januvia), the first-in-class agent to reach the market.
The study was funded by the National Institutes of Health. Dr. Goldfine reported serving as an investigator on studies funded by Daiichi Sankyo, NovoNordisk, and other companies.
AT ICE/ENDO 2014
Key clinical point: Prescribing a DPP-4 inhibitor in type 2 diabetes appears safe from a cardiovascular risk standpoint in community practice, contrary to earlier concerns.
Major finding: Patients with type 2 diabetes who initiated therapy with a DPP-4 inhibitor had a 13% reduction in the subsequent risk of a composite cardiovascular endpoint and a 19% reduction in hospitalization for heart failure, compared with closely-matched patients who initiated treatment with hypoglycemic agents from other classes.
Data source: A population-based cohort study of 39,769 pairs of adults with type 2 diabetes. Half initiated treatment with a dipeptidyl peptidase-4 inhibitor, the other half with a drug from a different class of antihyperglycemic agents.
Disclosures: The study was sponsored by the National Institutes of Health. Dr. Goldfine reported serving as an investigator on studies funded by Daiichi Sankyo, NovoNordisk, and other companies.
