User login
Alabama, Oregon, and pediatric asthma
according to estimates from the American Lung Association.
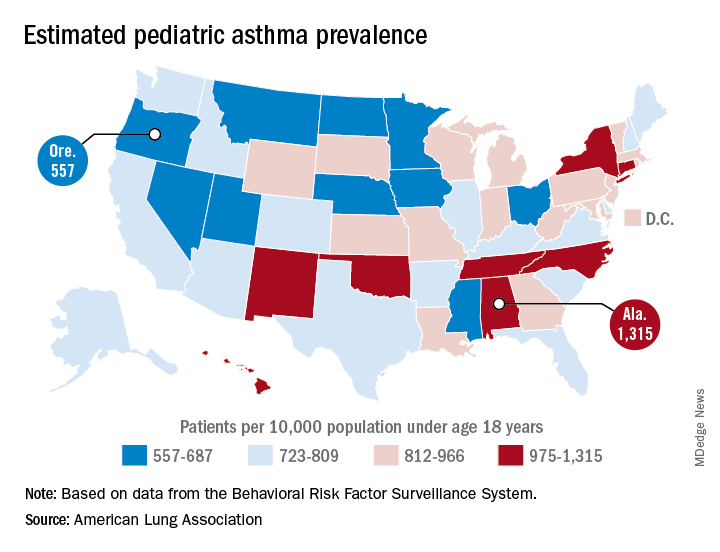
Oregon’s rate comes in at 557 per 10,000 population under the age of 18 years, just ahead of Montana at 574 per 10,000 and Iowa at 577. The prevalence of pediatric asthma in Alabama is 1,315 per 10,000, with North Carolina (1,149), Connecticut (1,107), Hawaii (1,026), and New York (1,005) joining it as members of the over-1,000 club. (MDedge News used the ALA’s estimates for persons under age 18 years with asthma in each state and Census Bureau estimates for population to calculate an unadjusted rate for each state.)
The ALA analysis was based on data from the Behavioral Risk Factor Behavioral System surveys for 2016 (31 states), 2015 (District of Columbia, Louisiana, New Hampshire, Texas), 2014 (Alabama, Maryland, North Carolina, Tennessee, West Virginia), 2012 (North Dakota and Wyoming), and 2011 (Iowa). National data were used for eight states (Alaska, Arkansas, Colorado, Delaware, Idaho, South Carolina, South Dakota, Virginia) that had no data available.
according to estimates from the American Lung Association.
Oregon’s rate comes in at 557 per 10,000 population under the age of 18 years, just ahead of Montana at 574 per 10,000 and Iowa at 577. The prevalence of pediatric asthma in Alabama is 1,315 per 10,000, with North Carolina (1,149), Connecticut (1,107), Hawaii (1,026), and New York (1,005) joining it as members of the over-1,000 club. (MDedge News used the ALA’s estimates for persons under age 18 years with asthma in each state and Census Bureau estimates for population to calculate an unadjusted rate for each state.)
The ALA analysis was based on data from the Behavioral Risk Factor Behavioral System surveys for 2016 (31 states), 2015 (District of Columbia, Louisiana, New Hampshire, Texas), 2014 (Alabama, Maryland, North Carolina, Tennessee, West Virginia), 2012 (North Dakota and Wyoming), and 2011 (Iowa). National data were used for eight states (Alaska, Arkansas, Colorado, Delaware, Idaho, South Carolina, South Dakota, Virginia) that had no data available.
according to estimates from the American Lung Association.
Oregon’s rate comes in at 557 per 10,000 population under the age of 18 years, just ahead of Montana at 574 per 10,000 and Iowa at 577. The prevalence of pediatric asthma in Alabama is 1,315 per 10,000, with North Carolina (1,149), Connecticut (1,107), Hawaii (1,026), and New York (1,005) joining it as members of the over-1,000 club. (MDedge News used the ALA’s estimates for persons under age 18 years with asthma in each state and Census Bureau estimates for population to calculate an unadjusted rate for each state.)
The ALA analysis was based on data from the Behavioral Risk Factor Behavioral System surveys for 2016 (31 states), 2015 (District of Columbia, Louisiana, New Hampshire, Texas), 2014 (Alabama, Maryland, North Carolina, Tennessee, West Virginia), 2012 (North Dakota and Wyoming), and 2011 (Iowa). National data were used for eight states (Alaska, Arkansas, Colorado, Delaware, Idaho, South Carolina, South Dakota, Virginia) that had no data available.
Pertussis vaccine at birth shows immune response, tolerability
compared with a group receiving only the hepatitis B vaccine, a randomized clinical trial from Australia has found.

“These results indicate that a birth dose of aP vaccine is immunogenic in newborns and significantly narrows the immunity gap between birth and 14 days after receipt of DTaP at 6 or 8 weeks of age, marking the critical period when infants are most vulnerable to severe pertussis infection,” reported Nicholas Wood, PhD, of the National Centre for Immunisation Research and Surveillance of Vaccine Preventable Diseases in New South Wales, Australia, and his colleagues.
“Administration of the acellular pertussis vaccine at birth has the potential to reduce severe morbidity from Bordetella pertussis infection in the first 3 months of life, especially for infants of mothers who have not received a pertussis vaccine during pregnancy,” the researchers concluded in JAMA Pediatrics.
The researchers enrolled 417 infants from Sydney, Melbourne, Adelaide, and Perth between June 2010 and March 2013 and randomized them to receive either the hepatitis B vaccine alone (n = 205) or the hepatitis B vaccine with a monovalent acellular pertussis vaccine (n = 212) within the first 5 days after birth. The randomization was stratified for mothers’ receipt of the Tdap before pregnancy.
The Centers for Disease Control and Prevention currently recommends all newborns receive the hepatitis B vaccine shortly after birth and that pregnant women receive the Tdap vaccine during each pregnancy. There is not currently a monovalent acellular pertussis vaccine licensed in the United States.
The study infants then received the hexavalent DTaP-Hib-hep B-polio vaccine and the 10-valent pneumococcal conjugate vaccine at 6 weeks, 4 months, and 6 months.
The primary outcome was detectable levels of IgG antibody to pertussis toxin and pertactin at 10 weeks old.
Of the 206 infants receiving the pertussis vaccine at birth, 93% had detectable antibodies to pertussis toxin and pertactin at 10 weeks, compared with 51% of the 193 infants who received only the hepatitis B shot (P less than .001). Geometric mean concentration for pertussis toxin IgG also was four times higher in infants who received the pertussis vaccine at birth.
Adverse events were similar in the two groups both at birth and at 32 weeks, demonstrating that the pertussis birth dose is safe and tolerable.
“More important, in this study, the prevalence of fever after receipt of the birth dose, which can mistakenly be associated with potential sepsis and result in additional investigations in the neonatal period, was similar in both the group that received the aP vaccine at birth and the control group,” the authors reported.
A remaining question is the potential impact of maternal antibodies on protection from pertussis.
“The presence of maternal pertussis antibodies at birth can negatively affect postprimary responses to pertussis, diphtheria, and diphtheria-related CRM197 conjugate vaccines with a variety of infant immunization schedules and vaccines,” the authors noted. “The clinical significance of reductions in pertussis antibody related to maternal interference will require ongoing clinical evaluation, because there are no accepted serologic correlates of protection.”
The research was funded by a Australian National Health and Medical Research Council (NHMRC) grant, and several authors received NHMRC grants. One author also was supported by a Murdoch Children’s Research Institute Career Development Award. GlaxoSmithKline provided the vaccine and conducted the serologic assays. The authors reported having no conflicts of interest.
SOURCE: Wood N et al, JAMA Pediatr. 2018 Sep 10. doi: 10.1001/jamapediatrics.2018.2349.
Pertussis is most likely to cause morbidity or kill neonates between birth and when they are given their first pertussis vaccine at 6-8 weeks of age. This is well known.
In the current study giving the acellular pertussis (aP) vaccine at birth led to “significantly higher antibody titers to pertussis antigens at 10 weeks of age,” compared with those who did not receive it. Those infants who received the birth dose of aP vaccine also had higher pertussis antibodies at 6 weeks, whether or not their mothers had received Tdap within 5 years prior to delivery.
When this study began in 2009, maternal immunization was not a well accepted concept, but this attitude has changed, in part due to the safe vaccination of pregnant women with the pandemic flu vaccine. Despite this, Centers for Disease Control and Prevention 2016 data showed that only 49% of pregnant women in the United Stated received Tdap. These rates need to increase.
Administering the aP vaccine with the existing hepatitis B vaccine at birth to infants whose mothers who did not receive Tdap during pregnancy would be a practical solution, if the aP vaccine were universally available.
But the aP vaccine currently is not available in the United States and many other countries as a standalone vaccine, and the administration of DTaP as a birth dose has been linked with “significant immune interference.” The aP vaccine could have a place in countries where it is available, and there is no maternal immunization program. Otherwise, boosting maternal immunization appears to be the primary approach for now.
Kathryn M. Edwards, MD, is the Sarah H. Sell and Cornelius Vanderbilt Chair in Pediatrics at Vanderbilt University, Nashville. She specializes in pediatric infectious diseases. These comments are a summary of her editorial accompanying the article by Wood et al. (Pediatrics. 2018 Sep 10. doi: 10.1001/jamapediatrics.2018.2363). Dr. Edwards said she had no conflicts of interest.
Pertussis is most likely to cause morbidity or kill neonates between birth and when they are given their first pertussis vaccine at 6-8 weeks of age. This is well known.
In the current study giving the acellular pertussis (aP) vaccine at birth led to “significantly higher antibody titers to pertussis antigens at 10 weeks of age,” compared with those who did not receive it. Those infants who received the birth dose of aP vaccine also had higher pertussis antibodies at 6 weeks, whether or not their mothers had received Tdap within 5 years prior to delivery.
When this study began in 2009, maternal immunization was not a well accepted concept, but this attitude has changed, in part due to the safe vaccination of pregnant women with the pandemic flu vaccine. Despite this, Centers for Disease Control and Prevention 2016 data showed that only 49% of pregnant women in the United Stated received Tdap. These rates need to increase.
Administering the aP vaccine with the existing hepatitis B vaccine at birth to infants whose mothers who did not receive Tdap during pregnancy would be a practical solution, if the aP vaccine were universally available.
But the aP vaccine currently is not available in the United States and many other countries as a standalone vaccine, and the administration of DTaP as a birth dose has been linked with “significant immune interference.” The aP vaccine could have a place in countries where it is available, and there is no maternal immunization program. Otherwise, boosting maternal immunization appears to be the primary approach for now.
Kathryn M. Edwards, MD, is the Sarah H. Sell and Cornelius Vanderbilt Chair in Pediatrics at Vanderbilt University, Nashville. She specializes in pediatric infectious diseases. These comments are a summary of her editorial accompanying the article by Wood et al. (Pediatrics. 2018 Sep 10. doi: 10.1001/jamapediatrics.2018.2363). Dr. Edwards said she had no conflicts of interest.
Pertussis is most likely to cause morbidity or kill neonates between birth and when they are given their first pertussis vaccine at 6-8 weeks of age. This is well known.
In the current study giving the acellular pertussis (aP) vaccine at birth led to “significantly higher antibody titers to pertussis antigens at 10 weeks of age,” compared with those who did not receive it. Those infants who received the birth dose of aP vaccine also had higher pertussis antibodies at 6 weeks, whether or not their mothers had received Tdap within 5 years prior to delivery.
When this study began in 2009, maternal immunization was not a well accepted concept, but this attitude has changed, in part due to the safe vaccination of pregnant women with the pandemic flu vaccine. Despite this, Centers for Disease Control and Prevention 2016 data showed that only 49% of pregnant women in the United Stated received Tdap. These rates need to increase.
Administering the aP vaccine with the existing hepatitis B vaccine at birth to infants whose mothers who did not receive Tdap during pregnancy would be a practical solution, if the aP vaccine were universally available.
But the aP vaccine currently is not available in the United States and many other countries as a standalone vaccine, and the administration of DTaP as a birth dose has been linked with “significant immune interference.” The aP vaccine could have a place in countries where it is available, and there is no maternal immunization program. Otherwise, boosting maternal immunization appears to be the primary approach for now.
Kathryn M. Edwards, MD, is the Sarah H. Sell and Cornelius Vanderbilt Chair in Pediatrics at Vanderbilt University, Nashville. She specializes in pediatric infectious diseases. These comments are a summary of her editorial accompanying the article by Wood et al. (Pediatrics. 2018 Sep 10. doi: 10.1001/jamapediatrics.2018.2363). Dr. Edwards said she had no conflicts of interest.
compared with a group receiving only the hepatitis B vaccine, a randomized clinical trial from Australia has found.
“These results indicate that a birth dose of aP vaccine is immunogenic in newborns and significantly narrows the immunity gap between birth and 14 days after receipt of DTaP at 6 or 8 weeks of age, marking the critical period when infants are most vulnerable to severe pertussis infection,” reported Nicholas Wood, PhD, of the National Centre for Immunisation Research and Surveillance of Vaccine Preventable Diseases in New South Wales, Australia, and his colleagues.
“Administration of the acellular pertussis vaccine at birth has the potential to reduce severe morbidity from Bordetella pertussis infection in the first 3 months of life, especially for infants of mothers who have not received a pertussis vaccine during pregnancy,” the researchers concluded in JAMA Pediatrics.
The researchers enrolled 417 infants from Sydney, Melbourne, Adelaide, and Perth between June 2010 and March 2013 and randomized them to receive either the hepatitis B vaccine alone (n = 205) or the hepatitis B vaccine with a monovalent acellular pertussis vaccine (n = 212) within the first 5 days after birth. The randomization was stratified for mothers’ receipt of the Tdap before pregnancy.
The Centers for Disease Control and Prevention currently recommends all newborns receive the hepatitis B vaccine shortly after birth and that pregnant women receive the Tdap vaccine during each pregnancy. There is not currently a monovalent acellular pertussis vaccine licensed in the United States.
The study infants then received the hexavalent DTaP-Hib-hep B-polio vaccine and the 10-valent pneumococcal conjugate vaccine at 6 weeks, 4 months, and 6 months.
The primary outcome was detectable levels of IgG antibody to pertussis toxin and pertactin at 10 weeks old.
Of the 206 infants receiving the pertussis vaccine at birth, 93% had detectable antibodies to pertussis toxin and pertactin at 10 weeks, compared with 51% of the 193 infants who received only the hepatitis B shot (P less than .001). Geometric mean concentration for pertussis toxin IgG also was four times higher in infants who received the pertussis vaccine at birth.
Adverse events were similar in the two groups both at birth and at 32 weeks, demonstrating that the pertussis birth dose is safe and tolerable.
“More important, in this study, the prevalence of fever after receipt of the birth dose, which can mistakenly be associated with potential sepsis and result in additional investigations in the neonatal period, was similar in both the group that received the aP vaccine at birth and the control group,” the authors reported.
A remaining question is the potential impact of maternal antibodies on protection from pertussis.
“The presence of maternal pertussis antibodies at birth can negatively affect postprimary responses to pertussis, diphtheria, and diphtheria-related CRM197 conjugate vaccines with a variety of infant immunization schedules and vaccines,” the authors noted. “The clinical significance of reductions in pertussis antibody related to maternal interference will require ongoing clinical evaluation, because there are no accepted serologic correlates of protection.”
The research was funded by a Australian National Health and Medical Research Council (NHMRC) grant, and several authors received NHMRC grants. One author also was supported by a Murdoch Children’s Research Institute Career Development Award. GlaxoSmithKline provided the vaccine and conducted the serologic assays. The authors reported having no conflicts of interest.
SOURCE: Wood N et al, JAMA Pediatr. 2018 Sep 10. doi: 10.1001/jamapediatrics.2018.2349.
compared with a group receiving only the hepatitis B vaccine, a randomized clinical trial from Australia has found.
“These results indicate that a birth dose of aP vaccine is immunogenic in newborns and significantly narrows the immunity gap between birth and 14 days after receipt of DTaP at 6 or 8 weeks of age, marking the critical period when infants are most vulnerable to severe pertussis infection,” reported Nicholas Wood, PhD, of the National Centre for Immunisation Research and Surveillance of Vaccine Preventable Diseases in New South Wales, Australia, and his colleagues.
“Administration of the acellular pertussis vaccine at birth has the potential to reduce severe morbidity from Bordetella pertussis infection in the first 3 months of life, especially for infants of mothers who have not received a pertussis vaccine during pregnancy,” the researchers concluded in JAMA Pediatrics.
The researchers enrolled 417 infants from Sydney, Melbourne, Adelaide, and Perth between June 2010 and March 2013 and randomized them to receive either the hepatitis B vaccine alone (n = 205) or the hepatitis B vaccine with a monovalent acellular pertussis vaccine (n = 212) within the first 5 days after birth. The randomization was stratified for mothers’ receipt of the Tdap before pregnancy.
The Centers for Disease Control and Prevention currently recommends all newborns receive the hepatitis B vaccine shortly after birth and that pregnant women receive the Tdap vaccine during each pregnancy. There is not currently a monovalent acellular pertussis vaccine licensed in the United States.
The study infants then received the hexavalent DTaP-Hib-hep B-polio vaccine and the 10-valent pneumococcal conjugate vaccine at 6 weeks, 4 months, and 6 months.
The primary outcome was detectable levels of IgG antibody to pertussis toxin and pertactin at 10 weeks old.
Of the 206 infants receiving the pertussis vaccine at birth, 93% had detectable antibodies to pertussis toxin and pertactin at 10 weeks, compared with 51% of the 193 infants who received only the hepatitis B shot (P less than .001). Geometric mean concentration for pertussis toxin IgG also was four times higher in infants who received the pertussis vaccine at birth.
Adverse events were similar in the two groups both at birth and at 32 weeks, demonstrating that the pertussis birth dose is safe and tolerable.
“More important, in this study, the prevalence of fever after receipt of the birth dose, which can mistakenly be associated with potential sepsis and result in additional investigations in the neonatal period, was similar in both the group that received the aP vaccine at birth and the control group,” the authors reported.
A remaining question is the potential impact of maternal antibodies on protection from pertussis.
“The presence of maternal pertussis antibodies at birth can negatively affect postprimary responses to pertussis, diphtheria, and diphtheria-related CRM197 conjugate vaccines with a variety of infant immunization schedules and vaccines,” the authors noted. “The clinical significance of reductions in pertussis antibody related to maternal interference will require ongoing clinical evaluation, because there are no accepted serologic correlates of protection.”
The research was funded by a Australian National Health and Medical Research Council (NHMRC) grant, and several authors received NHMRC grants. One author also was supported by a Murdoch Children’s Research Institute Career Development Award. GlaxoSmithKline provided the vaccine and conducted the serologic assays. The authors reported having no conflicts of interest.
SOURCE: Wood N et al, JAMA Pediatr. 2018 Sep 10. doi: 10.1001/jamapediatrics.2018.2349.
FROM JAMA PEDIATRICS
Key clinical point: A monovalent acellular pertussis vaccine dose at birth appears safe, tolerable, and effective.
Major finding: 93% of 212 newborns receiving an acellular pertussis vaccine at birth showed antibodies against pertussis toxin and pertactin at 10 weeks, compared with 51% of 205 newborns without the birth dose.
Study details: The findings are based on a randomized controlled trial involving 417 healthy term newborns in four Australian cities from June 2010 to March 2013.
Disclosures: The research was funded by an Australian National Health and Medical Research Council (NHMRC) grant, and several authors received NHMRC grants. One author also was supported by a Murdoch Children’s Research Institute Career Development Award. GlaxoSmithKline provided the vaccine and conducted the serologic assays. The authors reporting having no conflicts of interest.
Source: Wood N et al. JAMA Pediatr. 2018 Sep. 10. doi: 10.1001/jamapediatrics.2018.2349.
What’s The Impact of Occult HBV in Chronic HCV?
The reported prevalence of occult hepatitis B infection (OBI) varies widely: from < 1% to as high as 89.5% in HIV patients. Among patients with chronic hepatitis, the prevalence—again—ranges widely, from 0% to 52% but is highest in patients with chronic hepatitis C (CHC).
The clinical impact of OBI on patients with CHC has been extensively investigated, say researchers from the Institute of Liver and Biliary Sciences in New Delhi, India, but the available data are conflicting. In fact, when they conducted their study to assess the prevalence of OBI and evaluate its impact on clinical outcomes and response to antiviral therapy in CHC, the findings were “largely inconclusive.”
The study included 80 patients, of whom 32 (40%) had seropositive OBI. Hepatitis C virus genotype information was available for 59 patients, revealing that genotype 3 was most common.
However, analysis of clinical, biochemical, histopathologic and treatment response based on seropositivity and semiquantitative estimate of anti-HBc did not yield statistically significant results. Plasma samples of 14 were reactive for anti-HBc, 12 for anti-HBs, and 6 for both antibodies. Hepatitis B virus DNA (34 IU/mL) was detected in the plasma sample of only 1 patient by quantitative polymerase chain reaction. Therefore, the researchers say, the prevalence of OBI was 1.25%.
Anti-HBc total antibody levels did not influence clinical outcomes and response to directly acting antiviral therapy. Nor did genotype make a significant difference: 90.7% of genotype 3 patients and 92.8% of genotype 1 patients attained sustained virologic response.
More prospective studies should be conducted, the researchers urge, to further explore “this seemingly enigmatic issue.”
Source:
Bhatia M, Gupta E, Choudhary MC, Jindal A, Sarin SK. J Lab Physicians. 2018;10(3):304-308.
doi: 10.4103/JLP.JLP_12_18.
The reported prevalence of occult hepatitis B infection (OBI) varies widely: from < 1% to as high as 89.5% in HIV patients. Among patients with chronic hepatitis, the prevalence—again—ranges widely, from 0% to 52% but is highest in patients with chronic hepatitis C (CHC).
The clinical impact of OBI on patients with CHC has been extensively investigated, say researchers from the Institute of Liver and Biliary Sciences in New Delhi, India, but the available data are conflicting. In fact, when they conducted their study to assess the prevalence of OBI and evaluate its impact on clinical outcomes and response to antiviral therapy in CHC, the findings were “largely inconclusive.”
The study included 80 patients, of whom 32 (40%) had seropositive OBI. Hepatitis C virus genotype information was available for 59 patients, revealing that genotype 3 was most common.
However, analysis of clinical, biochemical, histopathologic and treatment response based on seropositivity and semiquantitative estimate of anti-HBc did not yield statistically significant results. Plasma samples of 14 were reactive for anti-HBc, 12 for anti-HBs, and 6 for both antibodies. Hepatitis B virus DNA (34 IU/mL) was detected in the plasma sample of only 1 patient by quantitative polymerase chain reaction. Therefore, the researchers say, the prevalence of OBI was 1.25%.
Anti-HBc total antibody levels did not influence clinical outcomes and response to directly acting antiviral therapy. Nor did genotype make a significant difference: 90.7% of genotype 3 patients and 92.8% of genotype 1 patients attained sustained virologic response.
More prospective studies should be conducted, the researchers urge, to further explore “this seemingly enigmatic issue.”
Source:
Bhatia M, Gupta E, Choudhary MC, Jindal A, Sarin SK. J Lab Physicians. 2018;10(3):304-308.
doi: 10.4103/JLP.JLP_12_18.
The reported prevalence of occult hepatitis B infection (OBI) varies widely: from < 1% to as high as 89.5% in HIV patients. Among patients with chronic hepatitis, the prevalence—again—ranges widely, from 0% to 52% but is highest in patients with chronic hepatitis C (CHC).
The clinical impact of OBI on patients with CHC has been extensively investigated, say researchers from the Institute of Liver and Biliary Sciences in New Delhi, India, but the available data are conflicting. In fact, when they conducted their study to assess the prevalence of OBI and evaluate its impact on clinical outcomes and response to antiviral therapy in CHC, the findings were “largely inconclusive.”
The study included 80 patients, of whom 32 (40%) had seropositive OBI. Hepatitis C virus genotype information was available for 59 patients, revealing that genotype 3 was most common.
However, analysis of clinical, biochemical, histopathologic and treatment response based on seropositivity and semiquantitative estimate of anti-HBc did not yield statistically significant results. Plasma samples of 14 were reactive for anti-HBc, 12 for anti-HBs, and 6 for both antibodies. Hepatitis B virus DNA (34 IU/mL) was detected in the plasma sample of only 1 patient by quantitative polymerase chain reaction. Therefore, the researchers say, the prevalence of OBI was 1.25%.
Anti-HBc total antibody levels did not influence clinical outcomes and response to directly acting antiviral therapy. Nor did genotype make a significant difference: 90.7% of genotype 3 patients and 92.8% of genotype 1 patients attained sustained virologic response.
More prospective studies should be conducted, the researchers urge, to further explore “this seemingly enigmatic issue.”
Source:
Bhatia M, Gupta E, Choudhary MC, Jindal A, Sarin SK. J Lab Physicians. 2018;10(3):304-308.
doi: 10.4103/JLP.JLP_12_18.
ASCO updates guidance on prophylaxis for adults with cancer-related immunosuppression
Fluoroquinolones are recommended for adults with cancer-related immunosuppression if they are at high risk of infection, according to an updated clinical practice guideline on antimicrobial prophylaxis.
By contrast, patients with solid tumors are not routinely recommended to receive antibiotic prophylaxis, according to the guideline, developed by the American Society of Clinical Oncology (ASCO) with the Infectious Diseases Society of America (IDSA).
The guideline includes antibacterial, antifungal, and antiviral prophylaxis recommendations, along with additional precautions such as hand hygiene that may reduce infection risk.
Released in the Journal of Clinical Oncology, the updated guidelines were developed by an expert panel cochaired by Christopher R. Flowers, MD of Emory University, Atlanta, and Randy A. Taplitz, MD of the University of California, San Diego, Health.
For the most part, the panel endorsed the previous ASCO recommendations, published in 2013. However, the panel considered six new high-quality studies and six new or updated meta-analyses to make modifications and add some new recommendations.
Fluoroquinolones, in the 2013 guideline, were recommended over trimethoprim-sulfamethoxazole because of fewer adverse events leading to treatment discontinuation. Panelists for the new guidelines said they continued to support that recommendation, based on an updated literature review.
That review showed significant reductions in both febrile neutropenia incidence and all-cause mortality, not only for patients at high risk of febrile neutropenia or profound, protracted neutropenia but also for lower-risk patients with solid tumors, they said.
However, the benefits did not sufficiently outweigh the harms to justify recommending fluoroquinolone prophylaxis for all patients with solid tumors or lymphoma, according to the report from the expert panel.
Those harms could include antibiotic-associated adverse effects, emergence of resistance, and Clostridium difficile infections, they said.
Accordingly, they recommended fluoroquinolone prophylaxis for the high-risk patients, including most patients with acute myeloid leukemia/myelodysplastic syndromes (AML/MDS) or those undergoing hematopoietic stem-cell transplantation (HSCT).
Similarly, the panel recommended that high-risk patients should receive antifungal prophylaxis with an oral triazole or parenteral echinocandin, while prophylaxis would not be routinely recommended for solid tumor patients.
By contrast, all patients undergoing chemotherapy for malignancy should receive yearly influenza vaccination with an inactivated quadrivalent vaccine, the panel said in its antiviral prophylaxis recommendations.
Family members, household contacts, and health care providers also should receive influenza vaccinations, said the panel, endorsing recommendations from the Centers for Disease Control and Prevention that were also cited in the 2013 ASCO guidelines.
Health care workers should follow hand hygiene and respiratory hygiene/cough etiquette to reduce risk of pathogen transmission, the panel said, endorsing CDC recommendations cited in the previous guideline.
However, the panel said they recommend against interventions such as neutropenic diet, footwear exchange, nutritional supplements, and surgical masks.
“Evidence of clinical benefit is lacking” for those interventions, they said.
Participants in the expert panel disclosed potential conflicts of interest related to Merck, Chimerix, GlyPharma Therapeutic, Pfizer, Cidara Therapeutics, Celgene, Astellas Pharma, Gilead Sciences, and Allergan, among other entities.
SOURCE: Taplitz RA et al. J Clin Oncol. 2018 Sept 4. doi: 10.1200/JCO.18.00374.
Fluoroquinolones are recommended for adults with cancer-related immunosuppression if they are at high risk of infection, according to an updated clinical practice guideline on antimicrobial prophylaxis.
By contrast, patients with solid tumors are not routinely recommended to receive antibiotic prophylaxis, according to the guideline, developed by the American Society of Clinical Oncology (ASCO) with the Infectious Diseases Society of America (IDSA).
The guideline includes antibacterial, antifungal, and antiviral prophylaxis recommendations, along with additional precautions such as hand hygiene that may reduce infection risk.
Released in the Journal of Clinical Oncology, the updated guidelines were developed by an expert panel cochaired by Christopher R. Flowers, MD of Emory University, Atlanta, and Randy A. Taplitz, MD of the University of California, San Diego, Health.
For the most part, the panel endorsed the previous ASCO recommendations, published in 2013. However, the panel considered six new high-quality studies and six new or updated meta-analyses to make modifications and add some new recommendations.
Fluoroquinolones, in the 2013 guideline, were recommended over trimethoprim-sulfamethoxazole because of fewer adverse events leading to treatment discontinuation. Panelists for the new guidelines said they continued to support that recommendation, based on an updated literature review.
That review showed significant reductions in both febrile neutropenia incidence and all-cause mortality, not only for patients at high risk of febrile neutropenia or profound, protracted neutropenia but also for lower-risk patients with solid tumors, they said.
However, the benefits did not sufficiently outweigh the harms to justify recommending fluoroquinolone prophylaxis for all patients with solid tumors or lymphoma, according to the report from the expert panel.
Those harms could include antibiotic-associated adverse effects, emergence of resistance, and Clostridium difficile infections, they said.
Accordingly, they recommended fluoroquinolone prophylaxis for the high-risk patients, including most patients with acute myeloid leukemia/myelodysplastic syndromes (AML/MDS) or those undergoing hematopoietic stem-cell transplantation (HSCT).
Similarly, the panel recommended that high-risk patients should receive antifungal prophylaxis with an oral triazole or parenteral echinocandin, while prophylaxis would not be routinely recommended for solid tumor patients.
By contrast, all patients undergoing chemotherapy for malignancy should receive yearly influenza vaccination with an inactivated quadrivalent vaccine, the panel said in its antiviral prophylaxis recommendations.
Family members, household contacts, and health care providers also should receive influenza vaccinations, said the panel, endorsing recommendations from the Centers for Disease Control and Prevention that were also cited in the 2013 ASCO guidelines.
Health care workers should follow hand hygiene and respiratory hygiene/cough etiquette to reduce risk of pathogen transmission, the panel said, endorsing CDC recommendations cited in the previous guideline.
However, the panel said they recommend against interventions such as neutropenic diet, footwear exchange, nutritional supplements, and surgical masks.
“Evidence of clinical benefit is lacking” for those interventions, they said.
Participants in the expert panel disclosed potential conflicts of interest related to Merck, Chimerix, GlyPharma Therapeutic, Pfizer, Cidara Therapeutics, Celgene, Astellas Pharma, Gilead Sciences, and Allergan, among other entities.
SOURCE: Taplitz RA et al. J Clin Oncol. 2018 Sept 4. doi: 10.1200/JCO.18.00374.
Fluoroquinolones are recommended for adults with cancer-related immunosuppression if they are at high risk of infection, according to an updated clinical practice guideline on antimicrobial prophylaxis.
By contrast, patients with solid tumors are not routinely recommended to receive antibiotic prophylaxis, according to the guideline, developed by the American Society of Clinical Oncology (ASCO) with the Infectious Diseases Society of America (IDSA).
The guideline includes antibacterial, antifungal, and antiviral prophylaxis recommendations, along with additional precautions such as hand hygiene that may reduce infection risk.
Released in the Journal of Clinical Oncology, the updated guidelines were developed by an expert panel cochaired by Christopher R. Flowers, MD of Emory University, Atlanta, and Randy A. Taplitz, MD of the University of California, San Diego, Health.
For the most part, the panel endorsed the previous ASCO recommendations, published in 2013. However, the panel considered six new high-quality studies and six new or updated meta-analyses to make modifications and add some new recommendations.
Fluoroquinolones, in the 2013 guideline, were recommended over trimethoprim-sulfamethoxazole because of fewer adverse events leading to treatment discontinuation. Panelists for the new guidelines said they continued to support that recommendation, based on an updated literature review.
That review showed significant reductions in both febrile neutropenia incidence and all-cause mortality, not only for patients at high risk of febrile neutropenia or profound, protracted neutropenia but also for lower-risk patients with solid tumors, they said.
However, the benefits did not sufficiently outweigh the harms to justify recommending fluoroquinolone prophylaxis for all patients with solid tumors or lymphoma, according to the report from the expert panel.
Those harms could include antibiotic-associated adverse effects, emergence of resistance, and Clostridium difficile infections, they said.
Accordingly, they recommended fluoroquinolone prophylaxis for the high-risk patients, including most patients with acute myeloid leukemia/myelodysplastic syndromes (AML/MDS) or those undergoing hematopoietic stem-cell transplantation (HSCT).
Similarly, the panel recommended that high-risk patients should receive antifungal prophylaxis with an oral triazole or parenteral echinocandin, while prophylaxis would not be routinely recommended for solid tumor patients.
By contrast, all patients undergoing chemotherapy for malignancy should receive yearly influenza vaccination with an inactivated quadrivalent vaccine, the panel said in its antiviral prophylaxis recommendations.
Family members, household contacts, and health care providers also should receive influenza vaccinations, said the panel, endorsing recommendations from the Centers for Disease Control and Prevention that were also cited in the 2013 ASCO guidelines.
Health care workers should follow hand hygiene and respiratory hygiene/cough etiquette to reduce risk of pathogen transmission, the panel said, endorsing CDC recommendations cited in the previous guideline.
However, the panel said they recommend against interventions such as neutropenic diet, footwear exchange, nutritional supplements, and surgical masks.
“Evidence of clinical benefit is lacking” for those interventions, they said.
Participants in the expert panel disclosed potential conflicts of interest related to Merck, Chimerix, GlyPharma Therapeutic, Pfizer, Cidara Therapeutics, Celgene, Astellas Pharma, Gilead Sciences, and Allergan, among other entities.
SOURCE: Taplitz RA et al. J Clin Oncol. 2018 Sept 4. doi: 10.1200/JCO.18.00374.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Novartis nabs first CAR T approval in Canada
the first chimeric antigen receptor (CAR) T-cell therapy to receive regulatory approval in Canada.
Tisagenlecleucel is approved to treat patients aged 3-25 years who have B-cell acute lymphoblastic leukemia (ALL) and relapsed after allogenic stem cell transplant (SCT) or are otherwise ineligible for SCT, have experienced second or later relapse, or have refractory disease.
Tisagenlecleucel is also approved in Canada to treat adults who have received two or more lines of systemic therapy and have relapsed or refractory diffuse large B-cell lymphoma (DLBCL) not otherwise specified, high-grade B-cell lymphoma, or DLBCL arising from follicular lymphoma.
Novartis, the company marketing tisagenlecleucel, said it is working with qualified treatment centers in Canada to prepare for the delivery of tisagenlecleucel. Certification and training are underway at these centers and Novartis is enhancing manufacturing capacity to meet patient needs.
Tisagenlecleucel has been studied in a pair of phase 2 trials – JULIET and ELIANA.
JULIET enrolled 165 adults with relapsed/refractory DLBCL, 111 of whom received a single infusion of tisagenlecleucel.
The overall response rate was 52% and the complete response (CR) rate was 40%. The median duration of response was not reached with a median follow-up of 13.9 months. At last follow-up, none of the responders had gone on to SCT.
The 12-month overall survival (OS) rate was 49%; the median OS was 11.7 months. The median OS was not reached for patients in CR.
Within 8 weeks of tisagenlecleucel infusion, 22% of patients had developed grade 3/4 cytokine release syndrome.
These results were presented at the 2018 annual congress of the European Hematology Association in June.
The ELIANA trial included 75 children and young adults with relapsed/refractory ALL. All patients received a single infusion of tisagenlecleucel, and 72 received lymphodepleting chemotherapy.
The median duration of follow-up was 13.1 months. The overall remission rate was 81%, with 60% of patients achieving a CR and 21% achieving CR with incomplete hematologic recovery. All patients whose best response was CR with incomplete hematologic recovery were negative for minimal residual disease. The median duration of response was not met.
Eight patients proceeded to SCT while in remission. At last follow-up, four were still in remission, and four had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the OS rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
About 95% of patients had adverse events thought to be related to tisagenlecleucel. The incidence of treatment-related grade 3/4 adverse eventss was 73% (N Engl J Med 2018; 378:439-48).
the first chimeric antigen receptor (CAR) T-cell therapy to receive regulatory approval in Canada.
Tisagenlecleucel is approved to treat patients aged 3-25 years who have B-cell acute lymphoblastic leukemia (ALL) and relapsed after allogenic stem cell transplant (SCT) or are otherwise ineligible for SCT, have experienced second or later relapse, or have refractory disease.
Tisagenlecleucel is also approved in Canada to treat adults who have received two or more lines of systemic therapy and have relapsed or refractory diffuse large B-cell lymphoma (DLBCL) not otherwise specified, high-grade B-cell lymphoma, or DLBCL arising from follicular lymphoma.
Novartis, the company marketing tisagenlecleucel, said it is working with qualified treatment centers in Canada to prepare for the delivery of tisagenlecleucel. Certification and training are underway at these centers and Novartis is enhancing manufacturing capacity to meet patient needs.
Tisagenlecleucel has been studied in a pair of phase 2 trials – JULIET and ELIANA.
JULIET enrolled 165 adults with relapsed/refractory DLBCL, 111 of whom received a single infusion of tisagenlecleucel.
The overall response rate was 52% and the complete response (CR) rate was 40%. The median duration of response was not reached with a median follow-up of 13.9 months. At last follow-up, none of the responders had gone on to SCT.
The 12-month overall survival (OS) rate was 49%; the median OS was 11.7 months. The median OS was not reached for patients in CR.
Within 8 weeks of tisagenlecleucel infusion, 22% of patients had developed grade 3/4 cytokine release syndrome.
These results were presented at the 2018 annual congress of the European Hematology Association in June.
The ELIANA trial included 75 children and young adults with relapsed/refractory ALL. All patients received a single infusion of tisagenlecleucel, and 72 received lymphodepleting chemotherapy.
The median duration of follow-up was 13.1 months. The overall remission rate was 81%, with 60% of patients achieving a CR and 21% achieving CR with incomplete hematologic recovery. All patients whose best response was CR with incomplete hematologic recovery were negative for minimal residual disease. The median duration of response was not met.
Eight patients proceeded to SCT while in remission. At last follow-up, four were still in remission, and four had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the OS rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
About 95% of patients had adverse events thought to be related to tisagenlecleucel. The incidence of treatment-related grade 3/4 adverse eventss was 73% (N Engl J Med 2018; 378:439-48).
the first chimeric antigen receptor (CAR) T-cell therapy to receive regulatory approval in Canada.
Tisagenlecleucel is approved to treat patients aged 3-25 years who have B-cell acute lymphoblastic leukemia (ALL) and relapsed after allogenic stem cell transplant (SCT) or are otherwise ineligible for SCT, have experienced second or later relapse, or have refractory disease.
Tisagenlecleucel is also approved in Canada to treat adults who have received two or more lines of systemic therapy and have relapsed or refractory diffuse large B-cell lymphoma (DLBCL) not otherwise specified, high-grade B-cell lymphoma, or DLBCL arising from follicular lymphoma.
Novartis, the company marketing tisagenlecleucel, said it is working with qualified treatment centers in Canada to prepare for the delivery of tisagenlecleucel. Certification and training are underway at these centers and Novartis is enhancing manufacturing capacity to meet patient needs.
Tisagenlecleucel has been studied in a pair of phase 2 trials – JULIET and ELIANA.
JULIET enrolled 165 adults with relapsed/refractory DLBCL, 111 of whom received a single infusion of tisagenlecleucel.
The overall response rate was 52% and the complete response (CR) rate was 40%. The median duration of response was not reached with a median follow-up of 13.9 months. At last follow-up, none of the responders had gone on to SCT.
The 12-month overall survival (OS) rate was 49%; the median OS was 11.7 months. The median OS was not reached for patients in CR.
Within 8 weeks of tisagenlecleucel infusion, 22% of patients had developed grade 3/4 cytokine release syndrome.
These results were presented at the 2018 annual congress of the European Hematology Association in June.
The ELIANA trial included 75 children and young adults with relapsed/refractory ALL. All patients received a single infusion of tisagenlecleucel, and 72 received lymphodepleting chemotherapy.
The median duration of follow-up was 13.1 months. The overall remission rate was 81%, with 60% of patients achieving a CR and 21% achieving CR with incomplete hematologic recovery. All patients whose best response was CR with incomplete hematologic recovery were negative for minimal residual disease. The median duration of response was not met.
Eight patients proceeded to SCT while in remission. At last follow-up, four were still in remission, and four had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the OS rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
About 95% of patients had adverse events thought to be related to tisagenlecleucel. The incidence of treatment-related grade 3/4 adverse eventss was 73% (N Engl J Med 2018; 378:439-48).
How Is the Colorectal Cancer Control Program Doing?
The CDC developed the Colorectal Cancer Control Program (CRCCP) to provide direct colorectal cancer (CRC) screening services to low-income, uninsured, or underinsured populations known to have low CRC screening rates. However, early evaluators found the program was insufficient to detect impact at the state level. In response to those findings, the CDC redesigned CRCCP and funded a new 5-year grant period beginning in 2015. How did the program fare this time? CDC researchers say it “shows promise.”
The CRCCP funds 23 states, 6 universities, and 1 tribal organization to partner with health care systems, implementing evidence-based interventions (EBIs). In this study, the researchers analyzed data reported by 387 of 413 clinics of varying sizes, representing 3,438 providers, and serving a screening-eligible population of 722,925 patients.
The researchers say their evaluation suggests that the CRCCP is working as intended: Program reach was measurable and “substantial,” clinics enhanced EBIs in place or implemented new ones, and the overall average screening rate rose.
At baseline, the screening rate was low (43%), and lowest in rural clinics—although evidence indicates that death rates for CRC are highest among people living in rural areas. In the first year, the overall screening rate increased by 4.4 percentage points. Still, that 47.3% is “much lower” than the commonly cited 67.3% from the 2016 Behavioral Risk Factor Surveillance System, the researchers note. They add, though, that the results confirm that grantees are working with clinics serving the intended populations and indicate the significant gap in CRC screening rates between those reached by the CRCCP and the US population overall.
Many clinics had ≥ 1 EBI or supporting activity (SA) already in place. Grantees used CRCCP resources to implement new or to enhance EBIs in 95% of the clinics, most often patient reminder activities and provider assessment and feedback. Most of the clinics used CRCCP resources for SAs, such as small media and provider education. Only 12% of clinics used resources for supporting community health workers. However, nearly half the clinics conducted planning activities for future implementation of community health workers and patient navigators.
Nearly 80% of the clinics reported having a CRC screening champion, 73% had a CRC screening policy, and 50% had either 3 or 4 EBIs in place at the end of the first year—all factors that the researchers suggest may support greater screening rate increases.
The CDC developed the Colorectal Cancer Control Program (CRCCP) to provide direct colorectal cancer (CRC) screening services to low-income, uninsured, or underinsured populations known to have low CRC screening rates. However, early evaluators found the program was insufficient to detect impact at the state level. In response to those findings, the CDC redesigned CRCCP and funded a new 5-year grant period beginning in 2015. How did the program fare this time? CDC researchers say it “shows promise.”
The CRCCP funds 23 states, 6 universities, and 1 tribal organization to partner with health care systems, implementing evidence-based interventions (EBIs). In this study, the researchers analyzed data reported by 387 of 413 clinics of varying sizes, representing 3,438 providers, and serving a screening-eligible population of 722,925 patients.
The researchers say their evaluation suggests that the CRCCP is working as intended: Program reach was measurable and “substantial,” clinics enhanced EBIs in place or implemented new ones, and the overall average screening rate rose.
At baseline, the screening rate was low (43%), and lowest in rural clinics—although evidence indicates that death rates for CRC are highest among people living in rural areas. In the first year, the overall screening rate increased by 4.4 percentage points. Still, that 47.3% is “much lower” than the commonly cited 67.3% from the 2016 Behavioral Risk Factor Surveillance System, the researchers note. They add, though, that the results confirm that grantees are working with clinics serving the intended populations and indicate the significant gap in CRC screening rates between those reached by the CRCCP and the US population overall.
Many clinics had ≥ 1 EBI or supporting activity (SA) already in place. Grantees used CRCCP resources to implement new or to enhance EBIs in 95% of the clinics, most often patient reminder activities and provider assessment and feedback. Most of the clinics used CRCCP resources for SAs, such as small media and provider education. Only 12% of clinics used resources for supporting community health workers. However, nearly half the clinics conducted planning activities for future implementation of community health workers and patient navigators.
Nearly 80% of the clinics reported having a CRC screening champion, 73% had a CRC screening policy, and 50% had either 3 or 4 EBIs in place at the end of the first year—all factors that the researchers suggest may support greater screening rate increases.
The CDC developed the Colorectal Cancer Control Program (CRCCP) to provide direct colorectal cancer (CRC) screening services to low-income, uninsured, or underinsured populations known to have low CRC screening rates. However, early evaluators found the program was insufficient to detect impact at the state level. In response to those findings, the CDC redesigned CRCCP and funded a new 5-year grant period beginning in 2015. How did the program fare this time? CDC researchers say it “shows promise.”
The CRCCP funds 23 states, 6 universities, and 1 tribal organization to partner with health care systems, implementing evidence-based interventions (EBIs). In this study, the researchers analyzed data reported by 387 of 413 clinics of varying sizes, representing 3,438 providers, and serving a screening-eligible population of 722,925 patients.
The researchers say their evaluation suggests that the CRCCP is working as intended: Program reach was measurable and “substantial,” clinics enhanced EBIs in place or implemented new ones, and the overall average screening rate rose.
At baseline, the screening rate was low (43%), and lowest in rural clinics—although evidence indicates that death rates for CRC are highest among people living in rural areas. In the first year, the overall screening rate increased by 4.4 percentage points. Still, that 47.3% is “much lower” than the commonly cited 67.3% from the 2016 Behavioral Risk Factor Surveillance System, the researchers note. They add, though, that the results confirm that grantees are working with clinics serving the intended populations and indicate the significant gap in CRC screening rates between those reached by the CRCCP and the US population overall.
Many clinics had ≥ 1 EBI or supporting activity (SA) already in place. Grantees used CRCCP resources to implement new or to enhance EBIs in 95% of the clinics, most often patient reminder activities and provider assessment and feedback. Most of the clinics used CRCCP resources for SAs, such as small media and provider education. Only 12% of clinics used resources for supporting community health workers. However, nearly half the clinics conducted planning activities for future implementation of community health workers and patient navigators.
Nearly 80% of the clinics reported having a CRC screening champion, 73% had a CRC screening policy, and 50% had either 3 or 4 EBIs in place at the end of the first year—all factors that the researchers suggest may support greater screening rate increases.
RESONATE-2 update: First-line ibrutinib has sustained efficacy in older CLL patients
In older patients with chronic lymphocytic leukemia (CLL), first-line treatment with ibrutinib resulted in a long-term progression-free survival benefit versus chemotherapy, according to extended follow-up results of a phase 3 trial.
The quality of response to ibrutinib continued to improve over time in the study, including a substantial increase in the proportion of patients achieving complete response, the updated results of the RESONATE-2 trial show.
Rates of serious adverse events decreased over time in the study, while common reasons for initiating treatment, such as marrow failure and disease symptoms, all improved to a greater extent than with chlorambucil, reported Paul M. Barr, MD, of the University of Rochester (N.Y.) and colleagues.
“These data support the use of ibrutinib in the first-line treatment of CLL as a chemotherapy-free option that can be taken continuously, achieving long-term disease control for the majority of patients, including those with high-risk features,” Dr. Barr and coauthors said in the journal Haematologica.
Previously reported primary results of the RESONATE-2 trial, which showed an 84% reduction in risk of death for ibrutinib versus chlorambucil with a median follow-up of 18 months, led to the approval of ibrutinib for first-line CLL treatment, the authors said.
The study included 269 patients with untreated CLL or small lymphocytic lymphoma who had active disease and were at least 65 years of age. They were randomized 1:1 to ibrutinib or chlorambucil.
Out of 136 ibrutinib-treated patients, 107 (79%) remained on therapy at this extended analysis, which had a median follow-up of 29 months.
The extended analysis also showed an 88% reduction in risk of progression or death for those patients randomized to ibrutinib (P less than .0001), with significant improvements in subgroups evaluated, which include groups typically considered high risk, according to Dr. Barr and colleagues.
The rate of complete response improved over time in ibrutinib-treated patients, from 7% at 12 months, to 15% at 24 months, and to 18% with a maximum of 36 months’ follow-up, they said.
The overall response rate for ibrutinib was 92% in this extended analysis, with comparable findings in high-risk subgroups, including those with del(11q) at 100% and unmutated IGHV at 95%, according to the report.
Lymphadenopathy improved in most ibrutinib-treated patients, with complete resolution in 42% versus 7% with chlorambucil. Splenomegaly improved by at least 50% in 95% of ibrutinib-treated patients versus 52% for chlorambucil, with complete resolution in 56% of ibrutinib-treated patients and 22% of chlorambucil-treated patients.
Adverse events of grade 3 or greater were generally seen more often in the first year of ibrutinib therapy and decreased over time. Rates of grade 3 or greater neutropenia, anemia, and thrombocytopenia were 8.1%, 5.9%, and 2.2%, respectively, in the first 12 months of treatment; those decreased to 0%, 1%, and 0% in the third year.
The rate of atrial fibrillation increased from 6% in the primary analysis to 10% in extended follow-up; however, investigators said ibrutinib dose reductions and discontinuations because of this adverse effect were uncommon and less frequent with extended treatment.
“Atrial fibrillation therefore appears manageable and does not frequently necessitate ibrutinib discontinuation,” they concluded.
The study was supported by Pharmacyclics, an AbbVie company, and by grants from the National Institutes of Health and the MD Anderson Moon Shot Program in CLL. Pharmacyclics designed the study and performed analysis of the data. Several study authors reported funding from various companies, including Pharmacyclics.
SOURCE: Barr PM, et al. Haematologica. 2018;103(9):1502-10.
In older patients with chronic lymphocytic leukemia (CLL), first-line treatment with ibrutinib resulted in a long-term progression-free survival benefit versus chemotherapy, according to extended follow-up results of a phase 3 trial.
The quality of response to ibrutinib continued to improve over time in the study, including a substantial increase in the proportion of patients achieving complete response, the updated results of the RESONATE-2 trial show.
Rates of serious adverse events decreased over time in the study, while common reasons for initiating treatment, such as marrow failure and disease symptoms, all improved to a greater extent than with chlorambucil, reported Paul M. Barr, MD, of the University of Rochester (N.Y.) and colleagues.
“These data support the use of ibrutinib in the first-line treatment of CLL as a chemotherapy-free option that can be taken continuously, achieving long-term disease control for the majority of patients, including those with high-risk features,” Dr. Barr and coauthors said in the journal Haematologica.
Previously reported primary results of the RESONATE-2 trial, which showed an 84% reduction in risk of death for ibrutinib versus chlorambucil with a median follow-up of 18 months, led to the approval of ibrutinib for first-line CLL treatment, the authors said.
The study included 269 patients with untreated CLL or small lymphocytic lymphoma who had active disease and were at least 65 years of age. They were randomized 1:1 to ibrutinib or chlorambucil.
Out of 136 ibrutinib-treated patients, 107 (79%) remained on therapy at this extended analysis, which had a median follow-up of 29 months.
The extended analysis also showed an 88% reduction in risk of progression or death for those patients randomized to ibrutinib (P less than .0001), with significant improvements in subgroups evaluated, which include groups typically considered high risk, according to Dr. Barr and colleagues.
The rate of complete response improved over time in ibrutinib-treated patients, from 7% at 12 months, to 15% at 24 months, and to 18% with a maximum of 36 months’ follow-up, they said.
The overall response rate for ibrutinib was 92% in this extended analysis, with comparable findings in high-risk subgroups, including those with del(11q) at 100% and unmutated IGHV at 95%, according to the report.
Lymphadenopathy improved in most ibrutinib-treated patients, with complete resolution in 42% versus 7% with chlorambucil. Splenomegaly improved by at least 50% in 95% of ibrutinib-treated patients versus 52% for chlorambucil, with complete resolution in 56% of ibrutinib-treated patients and 22% of chlorambucil-treated patients.
Adverse events of grade 3 or greater were generally seen more often in the first year of ibrutinib therapy and decreased over time. Rates of grade 3 or greater neutropenia, anemia, and thrombocytopenia were 8.1%, 5.9%, and 2.2%, respectively, in the first 12 months of treatment; those decreased to 0%, 1%, and 0% in the third year.
The rate of atrial fibrillation increased from 6% in the primary analysis to 10% in extended follow-up; however, investigators said ibrutinib dose reductions and discontinuations because of this adverse effect were uncommon and less frequent with extended treatment.
“Atrial fibrillation therefore appears manageable and does not frequently necessitate ibrutinib discontinuation,” they concluded.
The study was supported by Pharmacyclics, an AbbVie company, and by grants from the National Institutes of Health and the MD Anderson Moon Shot Program in CLL. Pharmacyclics designed the study and performed analysis of the data. Several study authors reported funding from various companies, including Pharmacyclics.
SOURCE: Barr PM, et al. Haematologica. 2018;103(9):1502-10.
In older patients with chronic lymphocytic leukemia (CLL), first-line treatment with ibrutinib resulted in a long-term progression-free survival benefit versus chemotherapy, according to extended follow-up results of a phase 3 trial.
The quality of response to ibrutinib continued to improve over time in the study, including a substantial increase in the proportion of patients achieving complete response, the updated results of the RESONATE-2 trial show.
Rates of serious adverse events decreased over time in the study, while common reasons for initiating treatment, such as marrow failure and disease symptoms, all improved to a greater extent than with chlorambucil, reported Paul M. Barr, MD, of the University of Rochester (N.Y.) and colleagues.
“These data support the use of ibrutinib in the first-line treatment of CLL as a chemotherapy-free option that can be taken continuously, achieving long-term disease control for the majority of patients, including those with high-risk features,” Dr. Barr and coauthors said in the journal Haematologica.
Previously reported primary results of the RESONATE-2 trial, which showed an 84% reduction in risk of death for ibrutinib versus chlorambucil with a median follow-up of 18 months, led to the approval of ibrutinib for first-line CLL treatment, the authors said.
The study included 269 patients with untreated CLL or small lymphocytic lymphoma who had active disease and were at least 65 years of age. They were randomized 1:1 to ibrutinib or chlorambucil.
Out of 136 ibrutinib-treated patients, 107 (79%) remained on therapy at this extended analysis, which had a median follow-up of 29 months.
The extended analysis also showed an 88% reduction in risk of progression or death for those patients randomized to ibrutinib (P less than .0001), with significant improvements in subgroups evaluated, which include groups typically considered high risk, according to Dr. Barr and colleagues.
The rate of complete response improved over time in ibrutinib-treated patients, from 7% at 12 months, to 15% at 24 months, and to 18% with a maximum of 36 months’ follow-up, they said.
The overall response rate for ibrutinib was 92% in this extended analysis, with comparable findings in high-risk subgroups, including those with del(11q) at 100% and unmutated IGHV at 95%, according to the report.
Lymphadenopathy improved in most ibrutinib-treated patients, with complete resolution in 42% versus 7% with chlorambucil. Splenomegaly improved by at least 50% in 95% of ibrutinib-treated patients versus 52% for chlorambucil, with complete resolution in 56% of ibrutinib-treated patients and 22% of chlorambucil-treated patients.
Adverse events of grade 3 or greater were generally seen more often in the first year of ibrutinib therapy and decreased over time. Rates of grade 3 or greater neutropenia, anemia, and thrombocytopenia were 8.1%, 5.9%, and 2.2%, respectively, in the first 12 months of treatment; those decreased to 0%, 1%, and 0% in the third year.
The rate of atrial fibrillation increased from 6% in the primary analysis to 10% in extended follow-up; however, investigators said ibrutinib dose reductions and discontinuations because of this adverse effect were uncommon and less frequent with extended treatment.
“Atrial fibrillation therefore appears manageable and does not frequently necessitate ibrutinib discontinuation,” they concluded.
The study was supported by Pharmacyclics, an AbbVie company, and by grants from the National Institutes of Health and the MD Anderson Moon Shot Program in CLL. Pharmacyclics designed the study and performed analysis of the data. Several study authors reported funding from various companies, including Pharmacyclics.
SOURCE: Barr PM, et al. Haematologica. 2018;103(9):1502-10.
FROM HAEMATOLOGICA
Key clinical point:
Major finding: There was an 88% reduction in risk of progression-free survival events for those patients randomized to ibrutinib (P less than .0001).
Study details: Extended phase 3 results from the RESONATE-2 trial, including 269 older patients with untreated CLL or small lymphocytic lymphoma.
Disclosures: This study was supported by Pharmacyclics, an AbbVie company, and by grants from the National Institutes of Health and the MD Anderson Moon Shot Program in CLL. Pharmacyclics designed the study and performed analysis of the data.
Source: Barr PM et al. Haematologica. 2018;103(9):1502-10.
England green-lights coverage of one CAR T-cell therapy
The National Health Service (NHS) of England has announced that tisagenlecleucel (Kymriah), a chimeric antigen receptor (CAR) T-cell therapy, will soon be available for certain leukemia patients.
, and patients could potentially begin receiving the treatment within weeks.
NHS England struck a deal with Novartis to lower the price of tisagenlecleucel, which costs around £282,000 per patient at its full list price. The discount offered to the NHS is confidential.
Tisagenlecleucel was recently approved by the European Commission (EC) to treat patients up to 25 years of age who have B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post transplant, or in second or later relapse.
The EC also approved tisagenlecleucel to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have received two or more lines of systemic therapy.
However, tisagenlecleucel will be available only for ALL patients in England, at least initially. A decision has not been made regarding funding for tisagenlecleucel in DLBCL, and Novartis previously decided to launch tisagenlecleucel in ALL first.
“It’s fantastic news for children and young people with this form of leukemia that CAR T-cell therapy will be made available on the NHS, making them the first in Europe to have routine access to this exciting new type of immunotherapy,” said Charles Swanton, Cancer Research UK’s chief clinician.
The first three NHS hospitals to go through the international accreditation process for the provision of tisagenlecleucel are in London, Manchester, and Newcastle. Subject to passing accreditation requirements, the first treatments could begin in a matter of weeks.
Another CAR T-cell therapy, axicabtagene ciloleucel (Yescarta), has not fared as well as in England. The National Institute for Health and Care Excellence (NICE) recently issued draft guidance recommending against the use of axicabtagene ciloleucel in England.
Axicabtagene ciloleucel was approved by the EC to treat patients with relapsed/refractory DLBCL or primary mediastinal B-cell lymphoma who have received two or more lines of systemic therapy. However, NICE said it isn’t clear how much of a benefit axicabtagene ciloleucel may provide over salvage chemotherapy. NICE also said the price of axicabtagene ciloleucel is too high for the therapy to be considered a cost-effective use of NHS resources, and the therapy does not meet the criteria for inclusion in the Cancer Drugs Fund.
The National Health Service (NHS) of England has announced that tisagenlecleucel (Kymriah), a chimeric antigen receptor (CAR) T-cell therapy, will soon be available for certain leukemia patients.
, and patients could potentially begin receiving the treatment within weeks.
NHS England struck a deal with Novartis to lower the price of tisagenlecleucel, which costs around £282,000 per patient at its full list price. The discount offered to the NHS is confidential.
Tisagenlecleucel was recently approved by the European Commission (EC) to treat patients up to 25 years of age who have B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post transplant, or in second or later relapse.
The EC also approved tisagenlecleucel to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have received two or more lines of systemic therapy.
However, tisagenlecleucel will be available only for ALL patients in England, at least initially. A decision has not been made regarding funding for tisagenlecleucel in DLBCL, and Novartis previously decided to launch tisagenlecleucel in ALL first.
“It’s fantastic news for children and young people with this form of leukemia that CAR T-cell therapy will be made available on the NHS, making them the first in Europe to have routine access to this exciting new type of immunotherapy,” said Charles Swanton, Cancer Research UK’s chief clinician.
The first three NHS hospitals to go through the international accreditation process for the provision of tisagenlecleucel are in London, Manchester, and Newcastle. Subject to passing accreditation requirements, the first treatments could begin in a matter of weeks.
Another CAR T-cell therapy, axicabtagene ciloleucel (Yescarta), has not fared as well as in England. The National Institute for Health and Care Excellence (NICE) recently issued draft guidance recommending against the use of axicabtagene ciloleucel in England.
Axicabtagene ciloleucel was approved by the EC to treat patients with relapsed/refractory DLBCL or primary mediastinal B-cell lymphoma who have received two or more lines of systemic therapy. However, NICE said it isn’t clear how much of a benefit axicabtagene ciloleucel may provide over salvage chemotherapy. NICE also said the price of axicabtagene ciloleucel is too high for the therapy to be considered a cost-effective use of NHS resources, and the therapy does not meet the criteria for inclusion in the Cancer Drugs Fund.
The National Health Service (NHS) of England has announced that tisagenlecleucel (Kymriah), a chimeric antigen receptor (CAR) T-cell therapy, will soon be available for certain leukemia patients.
, and patients could potentially begin receiving the treatment within weeks.
NHS England struck a deal with Novartis to lower the price of tisagenlecleucel, which costs around £282,000 per patient at its full list price. The discount offered to the NHS is confidential.
Tisagenlecleucel was recently approved by the European Commission (EC) to treat patients up to 25 years of age who have B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post transplant, or in second or later relapse.
The EC also approved tisagenlecleucel to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have received two or more lines of systemic therapy.
However, tisagenlecleucel will be available only for ALL patients in England, at least initially. A decision has not been made regarding funding for tisagenlecleucel in DLBCL, and Novartis previously decided to launch tisagenlecleucel in ALL first.
“It’s fantastic news for children and young people with this form of leukemia that CAR T-cell therapy will be made available on the NHS, making them the first in Europe to have routine access to this exciting new type of immunotherapy,” said Charles Swanton, Cancer Research UK’s chief clinician.
The first three NHS hospitals to go through the international accreditation process for the provision of tisagenlecleucel are in London, Manchester, and Newcastle. Subject to passing accreditation requirements, the first treatments could begin in a matter of weeks.
Another CAR T-cell therapy, axicabtagene ciloleucel (Yescarta), has not fared as well as in England. The National Institute for Health and Care Excellence (NICE) recently issued draft guidance recommending against the use of axicabtagene ciloleucel in England.
Axicabtagene ciloleucel was approved by the EC to treat patients with relapsed/refractory DLBCL or primary mediastinal B-cell lymphoma who have received two or more lines of systemic therapy. However, NICE said it isn’t clear how much of a benefit axicabtagene ciloleucel may provide over salvage chemotherapy. NICE also said the price of axicabtagene ciloleucel is too high for the therapy to be considered a cost-effective use of NHS resources, and the therapy does not meet the criteria for inclusion in the Cancer Drugs Fund.
Researchers find drug target in anaplastic large-cell lymphoma
Preclinical research indicates that TYK2 inhibitors could be effective in treating anaplastic large-cell lymphoma (ALCL).

Researchers found evidence to suggest that TYK2 “is highly expressed in all cases of human ALCL.”
The team also discovered that TYK2 inhibition induces apoptosis in human ALCL cells, and it delays tumor onset, and prolongs survival in a mouse model of ALCL.
Olaf Merkel, PhD, of the Medical University of Vienna in Austria, and his colleagues detailed these findings in Leukemia.
The researchers said their analyses suggest TYK2 is expressed in all types of ALCL, regardless of ALK status, and TYK2 mediates the same anti-apoptotic response across ALCLs.
“Therefore, we could consider TYK2 signaling as the Achilles’ heel of ALCL, as, in all patients we have analyzed, the tumor cells relied on this activity to support the essential survival signal,” Dr. Merkel said in a statement.
He and his colleagues found that disrupting TYK2 – either via gene knockdown or with small-molecule TYK2 inhibitors – induced apoptosis in human ALCL cells in vitro.
In a mouse model of NPM-ALK-induced lymphoma, Tyk2 deletion slowed the rate of tumor growth and significantly prolonged survival. The median survival was 53.3 weeks in mice with Tyk2 deletion and 16.0 weeks in control mice (P less than .0001).
Additional experiments in human ALCL cell lines showed that “TYK2 is activated by autocrine production of IL-10 and IL-22 and by interaction with specific receptors expressed by the cells,” the researchers said.
They also found that “activated TYK2 leads to STAT1 and STAT3 phosphorylation, activated expression of MCL1, and aberrant ALCL cell survival.”
Taking these findings together, the researchers concluded that TYK2 inhibitors could be effective for treating ALCL.
“We are looking forward to TYK2 inhibitors becoming available,” said study coauthor Lukas Kenner, MD, of the Medical University of Vienna. “[I]n the more rare lymphomas, we urgently need better therapies.”
The researchers received grant funding from various organizations but reported having no conflicts of interest.
SOURCE: Prutsch N et al. Leukemia. 2018 Aug 21. doi: 10.1038/s41375-018-0239-1.
Preclinical research indicates that TYK2 inhibitors could be effective in treating anaplastic large-cell lymphoma (ALCL).
Researchers found evidence to suggest that TYK2 “is highly expressed in all cases of human ALCL.”
The team also discovered that TYK2 inhibition induces apoptosis in human ALCL cells, and it delays tumor onset, and prolongs survival in a mouse model of ALCL.
Olaf Merkel, PhD, of the Medical University of Vienna in Austria, and his colleagues detailed these findings in Leukemia.
The researchers said their analyses suggest TYK2 is expressed in all types of ALCL, regardless of ALK status, and TYK2 mediates the same anti-apoptotic response across ALCLs.
“Therefore, we could consider TYK2 signaling as the Achilles’ heel of ALCL, as, in all patients we have analyzed, the tumor cells relied on this activity to support the essential survival signal,” Dr. Merkel said in a statement.
He and his colleagues found that disrupting TYK2 – either via gene knockdown or with small-molecule TYK2 inhibitors – induced apoptosis in human ALCL cells in vitro.
In a mouse model of NPM-ALK-induced lymphoma, Tyk2 deletion slowed the rate of tumor growth and significantly prolonged survival. The median survival was 53.3 weeks in mice with Tyk2 deletion and 16.0 weeks in control mice (P less than .0001).
Additional experiments in human ALCL cell lines showed that “TYK2 is activated by autocrine production of IL-10 and IL-22 and by interaction with specific receptors expressed by the cells,” the researchers said.
They also found that “activated TYK2 leads to STAT1 and STAT3 phosphorylation, activated expression of MCL1, and aberrant ALCL cell survival.”
Taking these findings together, the researchers concluded that TYK2 inhibitors could be effective for treating ALCL.
“We are looking forward to TYK2 inhibitors becoming available,” said study coauthor Lukas Kenner, MD, of the Medical University of Vienna. “[I]n the more rare lymphomas, we urgently need better therapies.”
The researchers received grant funding from various organizations but reported having no conflicts of interest.
SOURCE: Prutsch N et al. Leukemia. 2018 Aug 21. doi: 10.1038/s41375-018-0239-1.
Preclinical research indicates that TYK2 inhibitors could be effective in treating anaplastic large-cell lymphoma (ALCL).
Researchers found evidence to suggest that TYK2 “is highly expressed in all cases of human ALCL.”
The team also discovered that TYK2 inhibition induces apoptosis in human ALCL cells, and it delays tumor onset, and prolongs survival in a mouse model of ALCL.
Olaf Merkel, PhD, of the Medical University of Vienna in Austria, and his colleagues detailed these findings in Leukemia.
The researchers said their analyses suggest TYK2 is expressed in all types of ALCL, regardless of ALK status, and TYK2 mediates the same anti-apoptotic response across ALCLs.
“Therefore, we could consider TYK2 signaling as the Achilles’ heel of ALCL, as, in all patients we have analyzed, the tumor cells relied on this activity to support the essential survival signal,” Dr. Merkel said in a statement.
He and his colleagues found that disrupting TYK2 – either via gene knockdown or with small-molecule TYK2 inhibitors – induced apoptosis in human ALCL cells in vitro.
In a mouse model of NPM-ALK-induced lymphoma, Tyk2 deletion slowed the rate of tumor growth and significantly prolonged survival. The median survival was 53.3 weeks in mice with Tyk2 deletion and 16.0 weeks in control mice (P less than .0001).
Additional experiments in human ALCL cell lines showed that “TYK2 is activated by autocrine production of IL-10 and IL-22 and by interaction with specific receptors expressed by the cells,” the researchers said.
They also found that “activated TYK2 leads to STAT1 and STAT3 phosphorylation, activated expression of MCL1, and aberrant ALCL cell survival.”
Taking these findings together, the researchers concluded that TYK2 inhibitors could be effective for treating ALCL.
“We are looking forward to TYK2 inhibitors becoming available,” said study coauthor Lukas Kenner, MD, of the Medical University of Vienna. “[I]n the more rare lymphomas, we urgently need better therapies.”
The researchers received grant funding from various organizations but reported having no conflicts of interest.
SOURCE: Prutsch N et al. Leukemia. 2018 Aug 21. doi: 10.1038/s41375-018-0239-1.
FROM LEUKEMIA
Key clinical point:
Major finding: TYK2 was expressed in all types of ALCL studied and mediated the same anti-apoptotic response across ALCLs.
Study details: A preclinical study of mouse and human ALCL cell lines.
Disclosures: The researchers received grant funding from various organizations but reported having no conflicts of interest.
Source: Prutsch N et al. Leukemia. 2018 Aug 21. doi: 10.1038/s41375-018-0239-1.
Rituximab/lenalidomide similar to rituximab/chemotherapy for follicular lymphoma
Rituximab plus lenalidomide had efficacy similar to that of rituximab plus chemotherapy in treatment of follicular lymphoma, according to results from a phase 3 trial.
RELEVANCE (NCT01476787) was a multicenter, international, randomized, open-label trial designed to determine the superiority of rituximab/lenalidomide over rituximab/chemotherapy.
This trial randomized 1,030 patients with previously untreated follicular lymphoma to receive either rituximab plus lenalidomide (n = 513) or rituximab plus chemotherapy (n = 517) for 18 cycles; both groups then went on to receive rituximab maintenance therapy for 12 cycles. The total duration of treatment was 120 weeks. The median age of the combined groups was 59 years. The study was published in the New England Journal of Medicine.
One of the coprimary endpoints was complete response (confirmed or unconfirmed) by the end of the treatment period; the other was progression-free survival, which was planned to be assessed through three analyses, including two interim analyses, the first of which was reported in this study.
After a median follow-up of 37.9 months, the rates of coprimary endpoints were similar between the two groups. Complete response (confirmed or unconfirmed) was seen in 48% of the rituximab/lenalidomide group (95% confidence interval [CI], 44-53) and in 53% of the rituximab/chemotherapy group (95% CI, 49-57; P = .13). The hazard ratio for progression or death from any cause was 1.10 (95% CI, 0.85-1.43; P = .48).
In the subgroup analyses, the efficacy of rituximab plus chemotherapy was greater in low-risk patients (based on Follicular Lymphoma International Prognostic Index scores) and in patients whose follicular lymphoma was Ann Arbor stage I or II, whereas efficacy of rituximab/lenalidomide was independent of prognostic factors.
Safety was the biggest area of difference, with some events being more common in one group than in the other. For example, cutaneous reactions, diarrhea, rash, and myalgia were more common with rituximab/lenalidomide treatment, whereas anemia, fatigue, nausea, and febrile neutropenia were more common with rituximab/chemotherapy treatment. Among grade 3 or 4 events, cutaneous reactions were more common with rituximab/lenalidomide, and grade 3 or 4 neutropenia was more common with rituximab/chemotherapy.
“Overall, both treatment groups showed good outcomes, and a median has not yet been reached for either progression-free survival or overall survival,” the study authors wrote.
The RELEVANCE trial was sponsored by Celgene and the Lymphoma Academic Research Organisation. The study authors reported various disclosures, including financial ties to Celgene.
SOURCE: Morschhauser F et al. N Engl J Med. 2018;379:934-47.
Rituximab plus lenalidomide had efficacy similar to that of rituximab plus chemotherapy in treatment of follicular lymphoma, according to results from a phase 3 trial.
RELEVANCE (NCT01476787) was a multicenter, international, randomized, open-label trial designed to determine the superiority of rituximab/lenalidomide over rituximab/chemotherapy.
This trial randomized 1,030 patients with previously untreated follicular lymphoma to receive either rituximab plus lenalidomide (n = 513) or rituximab plus chemotherapy (n = 517) for 18 cycles; both groups then went on to receive rituximab maintenance therapy for 12 cycles. The total duration of treatment was 120 weeks. The median age of the combined groups was 59 years. The study was published in the New England Journal of Medicine.
One of the coprimary endpoints was complete response (confirmed or unconfirmed) by the end of the treatment period; the other was progression-free survival, which was planned to be assessed through three analyses, including two interim analyses, the first of which was reported in this study.
After a median follow-up of 37.9 months, the rates of coprimary endpoints were similar between the two groups. Complete response (confirmed or unconfirmed) was seen in 48% of the rituximab/lenalidomide group (95% confidence interval [CI], 44-53) and in 53% of the rituximab/chemotherapy group (95% CI, 49-57; P = .13). The hazard ratio for progression or death from any cause was 1.10 (95% CI, 0.85-1.43; P = .48).
In the subgroup analyses, the efficacy of rituximab plus chemotherapy was greater in low-risk patients (based on Follicular Lymphoma International Prognostic Index scores) and in patients whose follicular lymphoma was Ann Arbor stage I or II, whereas efficacy of rituximab/lenalidomide was independent of prognostic factors.
Safety was the biggest area of difference, with some events being more common in one group than in the other. For example, cutaneous reactions, diarrhea, rash, and myalgia were more common with rituximab/lenalidomide treatment, whereas anemia, fatigue, nausea, and febrile neutropenia were more common with rituximab/chemotherapy treatment. Among grade 3 or 4 events, cutaneous reactions were more common with rituximab/lenalidomide, and grade 3 or 4 neutropenia was more common with rituximab/chemotherapy.
“Overall, both treatment groups showed good outcomes, and a median has not yet been reached for either progression-free survival or overall survival,” the study authors wrote.
The RELEVANCE trial was sponsored by Celgene and the Lymphoma Academic Research Organisation. The study authors reported various disclosures, including financial ties to Celgene.
SOURCE: Morschhauser F et al. N Engl J Med. 2018;379:934-47.
Rituximab plus lenalidomide had efficacy similar to that of rituximab plus chemotherapy in treatment of follicular lymphoma, according to results from a phase 3 trial.
RELEVANCE (NCT01476787) was a multicenter, international, randomized, open-label trial designed to determine the superiority of rituximab/lenalidomide over rituximab/chemotherapy.
This trial randomized 1,030 patients with previously untreated follicular lymphoma to receive either rituximab plus lenalidomide (n = 513) or rituximab plus chemotherapy (n = 517) for 18 cycles; both groups then went on to receive rituximab maintenance therapy for 12 cycles. The total duration of treatment was 120 weeks. The median age of the combined groups was 59 years. The study was published in the New England Journal of Medicine.
One of the coprimary endpoints was complete response (confirmed or unconfirmed) by the end of the treatment period; the other was progression-free survival, which was planned to be assessed through three analyses, including two interim analyses, the first of which was reported in this study.
After a median follow-up of 37.9 months, the rates of coprimary endpoints were similar between the two groups. Complete response (confirmed or unconfirmed) was seen in 48% of the rituximab/lenalidomide group (95% confidence interval [CI], 44-53) and in 53% of the rituximab/chemotherapy group (95% CI, 49-57; P = .13). The hazard ratio for progression or death from any cause was 1.10 (95% CI, 0.85-1.43; P = .48).
In the subgroup analyses, the efficacy of rituximab plus chemotherapy was greater in low-risk patients (based on Follicular Lymphoma International Prognostic Index scores) and in patients whose follicular lymphoma was Ann Arbor stage I or II, whereas efficacy of rituximab/lenalidomide was independent of prognostic factors.
Safety was the biggest area of difference, with some events being more common in one group than in the other. For example, cutaneous reactions, diarrhea, rash, and myalgia were more common with rituximab/lenalidomide treatment, whereas anemia, fatigue, nausea, and febrile neutropenia were more common with rituximab/chemotherapy treatment. Among grade 3 or 4 events, cutaneous reactions were more common with rituximab/lenalidomide, and grade 3 or 4 neutropenia was more common with rituximab/chemotherapy.
“Overall, both treatment groups showed good outcomes, and a median has not yet been reached for either progression-free survival or overall survival,” the study authors wrote.
The RELEVANCE trial was sponsored by Celgene and the Lymphoma Academic Research Organisation. The study authors reported various disclosures, including financial ties to Celgene.
SOURCE: Morschhauser F et al. N Engl J Med. 2018;379:934-47.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point:
Major finding: Complete responses were seen in 48% of rituximab/lenalidomide patients versus 53% in the rituximab/chemotherapy patients (P = .13).
Study details: A phase 3 superiority trial of 1,030 patients with previously untreated follicular lymphoma.
Disclosures: Celgene and the Lymphoma Academic Research Organization funded the study. The authors reported various disclosures, including financial ties to Celgene.
Source: Morschhauser F et al. N Engl J Med. 2018;379:934-47.