User login
Depression in older adults: How to treat its distinct clinical manifestations
Discuss this article at http://currentpsychiatry.blogspot.com/2010/08/depression-in-older-adults.html#comments
Depression in older adults (age ≥65) can devastate their quality of life and increase the likelihood of institutionalization because of behavioral problems.1 Depression is a primary risk factor for suicide, and suicide rates are highest among those age ≥65, especially among white males.2 The burden of geriatric depression can extend to caregivers.1 Prompt recognition and treatment of depression could help minimize morbidity and reduce suffering in older adults and their caregivers.
Although geriatric depression varies in severity and presentation, common categories include:
- major depressive disorder (MDD)
- vascular depression
- dysthymia
- depression in the context of dementias, psychosis, bipolar disorder, and executive dysfunction.
Diagnoses in this population generally correspond with DSM-IV-TR criteria, but geriatric depression has distinct clinical manifestations.1,2 Compared with younger depressed patients, older adults are less likely to endorse depressed mood and more likely to report a lack of emotions.1,2 Older patients report feelings of irritability and fearfulness more often than sadness.1,2 Mood symptoms tend to be transient, reoccur frequently, and display either a diurnal pattern or multiple fluctuations in a single day.1,2 Other common presentations include loss of interest in usual activities, lack of motivation, social withdrawal, and decline in activities of daily living.1,2
Summary of recommendations
Age-specific recommendations for assessing and treating geriatric depression can be generated in part from evidence-based reviews, meta-analyses,3 and geriatric expert consensus guidelines.4 Such guidelines and recommendations often do not take into account the marked heterogeneity of medical, cognitive, and overall functioning in patients age ≥65, however, because they are based on studies of younger populations and patients with complicated issues often are excluded from studies. The recommendations in this article are based largely on findings from a National Institutes of Health (NIH)-sponsored project by Alexopoulos et al to develop consensus guidelines for managing geriatric depression and expert opinion from clinicians who treat geriatric patients.4
During your initial clinical evaluation, confirm the diagnosis and type, duration, and severity of depression. Seek to understand the biopsychosocial context of each patient’s presentation. Carefully consider your patient’s suicide risk. Hospitalization may be required if he or she is at high risk for suicide or has complex medical and social circumstances that cannot be managed adequately in an outpatient setting.5
Unipolar major depression
For unipolar, nonpsychotic geriatric depression, the NIH-Alexopoulos et al guidelines emphasize a combination of antidepressants and psychotherapy (Algorithm 1).4 Selective serotonin reuptake inhibitors (SSRIs) and venlafaxine are first-line options.4,6,7 Tricyclic antidepressants (TCAs), bupropion, and mirtazapine are alternatives.4 Among SSRIs, citalopram, escitalopram, and sertraline are preferred initial antidepressants. Fluoxetine is used less frequently.4 Paroxetine also is less commonly used because of its anticholinergic effects and because the drug inhibits cytochrome P4502D6,2 which metabolizes several medications commonly prescribed for older adults. Among TCAs, nortriptyline is preferred.4 Studies have shown that duloxetine improves depression and is safe and well-tolerated in older adults with recurrent MDD.8 Electroconvulsive therapy (ECT) is an option for treating severe or treatment-resistant unipolar major depression.9
For unipolar depression with psychotic symptoms, guidelines recommend a combination of an antidepressant and an antipsychotic or ECT.4 Atypical antipsychotics are preferred over typical antipsychotics4; risperidone, olanzapine, and quetiapine are most frequently used.4 Clinical data on aripiprazole and ziprasidone in older adults are limited. Many geriatric experts recommend continuing an antipsychotic for 6 months after symptom remission, then gradually tapering the dose.4
During acute illness, administer an anti-depressant for 6 to 12 weeks at the individually determined dose required to achieve symptom remission.6 For an older adult experiencing a first lifetime episode of major depression, continue antidepressant treatment for 1 year after remission.4 If your patient has had 2 lifetime episodes of major depression, continue the antidepressant at the same dose used to achieve remission for at least 3 years. For patients who have had ≥3 episodes of depression or whose index episode was particularly severe or involved significant suicidal thoughts or behaviors, continue maintenance treatment indefinitely.
Algorithm 1: Treatment for unipolar depression in geriatric patients
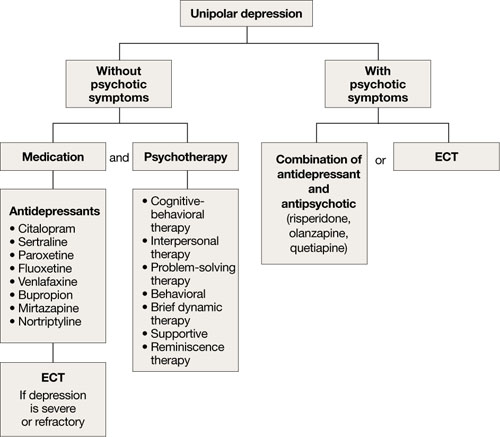
ECT: electroconvulsive therapy
Bipolar depression
Mood stabilizers such as lithium or valproate—as monotherapy or in combination with an antidepressant—are recommended to treat bipolar depression without psychotic symptoms in older adults (Algorithm 2).10 For bipolar depression with psychotic symptoms, a combination of a mood stabilizer and an atypical antipsychotic or ECT is recommended.10
Older adults’ increased sensitivity to side effects and reduced ability to tolerate lithium may limit its use and may prompt you to consider atypical antipsychotics as alternatives to other mood stabilizers. Although quetiapine and fluoxetineolanzapine combination are well studied in younger patients,11,12 there is a lack of data to support their clinical effectiveness and tolerability in older adults. Among antidepressants, SSRIs or bupropion are preferred over TCAs to prevent a switch to mania.10 Lamotrigine is an effective maintenance treatment for bipolar depressive episodes in older adults.13
Although optimal mood stabilizer and antidepressant dosing for this population has not been adequately assessed, pharmacotherapy that has been effective generally should be continued without modification for at least 6 to 12 months.10 After the patient achieves remission, gradually discontinue antidepressants while maintaining the mood stabilizer.10
Algorithm 2: Bipolar depression: Options for combination therapy
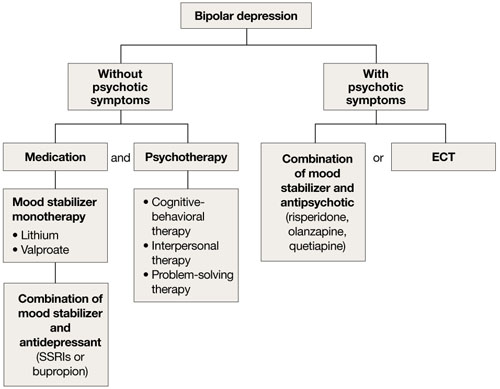
ECT: electroconvulsive therapy; SSRIs: selective serotonin reuptake inhibitors
Depression in dementia
Managing depression in dementia patients is similar to treatment in older adults without dementia,5,14 although pharmacologic agents must be carefully selected because of increased risk of side effects (Algorithm 3). American Psychiatric Association practice guidelines recommend considering antidepressants for depressed patients with dementia even if their mood disturbances do not meet DSM-IV-TR criteria for MDD.5
SSRIs’ lower side effect profile make them the preferred treatment; the selective serotonin-norepinephrine reuptake inhibitor (SNRI) venlafaxine is a second-line option.4,14 Avoid TCAs and other agents with anticholinergic side effects because of potential cardiovascular complications and cognitive side effects, unless SSRIs or SNRIs are ineffective or contraindicated.14 Recently clinicians have been reluctant to use antipsychotics in patients with dementia, because of the FDA’s “black-box” warning regarding the increased mortality risk associated with their use in this population.
When using ECT to treat depression in patients with dementia, the treatment protocol often is modified to twice-a-week, unilateral stimulus because of these patients’ increased risk of delirium.14 The safety of ECT to treat depression in patients with dementia has not been adequately assessed.14
Algorithm 3: Treating comorbid depression and dementia
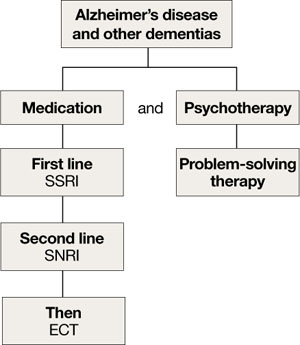
ECT: electroconvulsive therapy; SNRI: selective serotoninnorepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor
Vascular depression
The “vascular depression hypothesis” proposes that accumulation of subcortical white matter hyperintensities can disrupt frontostriatal pathways, resulting in depressive symptoms.15 This hypothesis is supported by the confluence of depression and vascular risk factors.15 Sertraline, citalopram, nortriptyline,16 and trazodone15 have been shown to reduce depressive symptoms after a stroke.
Minor depression and dysthymia
Although the efficacy of antidepressants in minor depression—depression that does not meet criteria for MDD—is not well established, expert consensus guidelines recommend SSRIs and psychotherapy, separately or in combination, for minor depression and dysthymia in older adults (Algorithm 4).4 Depression in executive dysfunction responds poorly to SSRI treatment2; however, behaviorally oriented psychotherapeutic interventions such as problem-solving therapy (PST) show promise.2
Algorithm 4: Minor depression: SSRIs plus psychotherapy
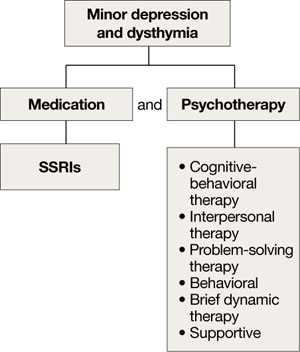
SSRIs: selective serotonin reuptake inhibitors
Comorbid medical conditions
When an older adult has a medical problem that likely contributes to depression—such as hypothyroidism—treat the condition and prescribe antidepressants simultaneously.2 However, if the medical problem likely causes depression—such as substance withdrawal—treat the condition first and prescribe antidepressants only if mood symptoms persist.2
Refractory depression
If your patient does not respond to an antidepressant trial of adequate dosage and duration, first make sure he or she is taking it correctly (Algorithm 5). After ruling out poor adherence, screen for comorbid psychiatric or medical conditions or psychosocial stressors and reassess the principal diagnosis.5
If these steps don’t address your patient’s depressive symptoms, expert consensus guidelines suggest switching to a different antidepressant:4
- If you first prescribed an SSRI, consider venlafaxine XR or bupropion SR.4,17
- If your patient initially received a TCA or bupropion, an SSRI or venlafaxine XR would be appropriate.4
- If venlafaxine XR was the first antidepressant, a SSRI is recommended.4
If your patient experienced a partial response but not full remission with the initial antidepressant, consider adding a second antidepressant or an augmenting agent:4
- If your patient first received an SSRI, adding bupropion, lithium, or nortriptyline is recommended.
- If the initial antidepressant was a TCA or bupropion, consider adding lithium or an SSRI.
- Augmenting venlafaxine XR with lithium is recommended.4
The National Institutes of Mental Health-sponsored Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study of treatment-resistant depression in mixed-age groups reported that patients who do not attain remission with an initial SSRI may respond to switching to bupropion SR or venlafaxine XR.17 Augmenting an SSRI with bupropion SR has been shown to be effective.18 In addition, consider mirtazapine augmentation,19 especially if your patient experiences insomnia or anorexia. A combination of mirtazapine and venlafaxine have better efficacy and tolerability compared with the monoamine oxidase inhibitor tranylcypromine.19 Some studies have shown augmenting SSRIs with buspirone in patients with severe depression is efficacious and safe in younger adults,20 but this practice is not well studied in older patients.
Algorithm 5: Treatment-resistant geriatric depression: Partial vs no response
SNRI: selective serotonin-norepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor; TCA: tricyclic antidepressant
Nonpharmacologic treatments
ECT is an important therapeutic intervention because of its safety, efficacy, and faster clinical response.6,7,9,21 Consider ECT for older adults with severe or psychotic major depression, acute suicidality, catatonia, or severe malnutrition caused by refusal to eat. Patients who remain significantly symptomatic after multiple medication trials, do not tolerate medications well, or have comorbid medical conditions that preclude antidepressant use also are potential candidates for ECT.5,22
ECT can be administered to many older depressed adults with relatively low complication rates. Pretreatment clinical and laboratory evaluations and consultation with medical colleagues may minimize the risk of adverse effects, including cardiovascular instability, delirium, and falls.9 Anterograde memory loss—a common concern for clinicians and patients—usually is temporary and can be reduced by modifying the ECT administration parameters, such as switching from bilateral to unilateral stimulus and spacing treatments.9 Use caution when considering ECT for patients with cardiovascular or neurologic conditions—such as myocardial infarction or cerebrovascular accident within 6 months of treatment—that may increase the risk of adverse effects. Some pharmacologic agents, such as benzodiazepines and anticonvulsant mood stabilizers, may decrease ECT’s efficacy by inhibiting seizure.22
Depressive relapse after ECT is a major clinical concern.21 Continuation ECT— within the first 6 months of remission— aims to prevent relapse of the same episode, whereas maintenance ECT—beyond the first 6 months—helps avert occurrence of new episodes.4,21 Relapse and recurrence also can be prevented with continuation or maintenance pharmacotherapy,4,21 which should be initiated immediately after the index course of ECT.21 Typically, ECT continuation/maintenance treatments are provided weekly, then gradually spaced out to once a month based on the minimum frequency that is effective for an individual patient.21
Psychotherapy for geriatric depression generally is effective.23 One-half of older patients prefer psychotherapy over pharmacotherapy.24 Efficacious psychotherapies include behavioral therapy, cognitive-behavioral therapy (CBT), PST, brief dynamic therapy, interpersonal therapy, supportive therapy, and reminiscence therapy.23 CBT has the most empiric support for treating geriatric depression.5,6
Psychotherapy alone is appropriate for mild-to-moderate depression, although severe depression requires adding medication.25 The combination of pharmacotherapy and psychotherapy appears to be more effective than either intervention alone in preventing recurrent major depression, especially when a specific psychosocial stressor has been identified.5,6 CBT, interpersonal therapy, and family-focused therapy enhance pharmacotherapy outcomes in bipolar disorder.13
The Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD) study found that in mixed-age patients, pharmacotherapy plus psychotherapy is more beneficial than medication alone in stabilizing bipolar depression.26 For older adults with executive dysfunction, research suggests that PST is more effective than other psychotherapies.27 Psychosocial interventions—such as psychoeducation for the family and caregivers, family counseling, and participation in senior citizen centers and services—are strongly recommended for many patients.4
Related Resources
- Blazer DG, Steffens DC, Koenig HG. Mood disorders. In: Blazer DG, Steffens DC, eds. The American Psychiatric Publishing textbook of geriatric psychiatry. 4th ed. Arlington, VA: American Psychiatric Publishing, Inc.; 2009:275-300.
- American Association for Geriatric Psychiatry. www.aagponline.org.
Drug Brand Names
- Aripiprazole • Abilify
- Bupropion • Wellbutrin, Zyban
- Buspirone • Buspar
- Citalopram • Celexa
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluoxetine-olanzapine • Symbyax
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid
- Mirtazapine • Remeron
- Nortriptyline • Aventyl, Pamelor
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sertraline • Zoloft
- Tranylcypromine • Parnate
- Trazodone • Desyrel
- Valproate • Depakote
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with the manufacturer of any product mentioned in this article or with manufacturers of competing products.
1. Lyketsos CG, Lee HB. Diagnosis and treatment of depression in Alzheimer’s disease. A practical update for the clinician. Dement Geriatr Cogn Disord. 2004;17(1-2):55-64.
2. Alexopoulos G. Late-life mood disorders. In: Sadavoy J, Jarvik LF, Grossberg GT, et al, eds. Comprehensive textbook of geriatric psychiatry. 3rd ed. New York, NY: W.W. Norton and Company; 2004:609-653.
3. Shanmugham B, Karp J, Drayer R, et al. Evidence-based pharmacologic interventions of geriatric depression. Psychiatr Clin North Am. 2005;28(4):821-835,viii.
4. Alexopoulos GS, Katz IR, Reynolds CF, III, et al. The expert consensus guidelines series. Pharmacotherapy of depressive disorders in older patients. Postgrad Med. 2001; Spect No Pharmacolotherapy:1–86.
5. American Psychiatric Association practice guidelines for the treatment of psychiatric disorders. Arlington, VA: American Psychiatric Association; 2006:793–794.
6. Bartels SJ, Dums AR, Oxman TE, et al. Evidence-based practice in geriatric mental health care. Psychiatr Serv. 2002;53(11):1419-1431.
7. Bartels SJ, Dums AR, Oxman TE, et al. Evidence-based practices in geriatric mental health care: an overview of systematic reviews and meta-analyses. Psychiatr Clin North Am. 2003;26(4):971-990,x–xi.
8. Raskin J, Wiltse CG, Siegal A, et al. Efficacy of duloxetine on cognition, depression, and pain in elderly patients with major depressive disorder: an 8-week, double-blind, placebo-controlled trial. Am J Psychiatry. 2007;164(6):900-909.
9. Alexopoulos GS, Young RC, Abrams RC. ECT in the high-risk geriatric patient. Convuls Ther. 1989;5(1):75-87.
10. Young RC, Gyulai L, Mulsant BH, et al. Pharmacotherapy of bipolar disorder in old age: review and recommendations. Am J Geriatr Psychiatry. 2004;12:342-357.
11. Vieta E, Calabrese JR, Goikolea JM, et al. Quetiapine monotherapy in the treatment of patients with bipolar I or II depression and a rapid-cycling disease course: a randomized, double-blind, placebo-controlled study. Bipolar Disord. 2007;9(4):413-425.
12. Corya SA, Perlis RH, Keck PE, Jr, et al. A 24-week open-label extension study of olanzapine-fluoxetine combination and olanzapine monotherapy in the treatment of bipolar depression. J Clin Psychiatry. 2006;67(5):798-806.
13. Sajatovic M, Gyulai L, Calabrese JR, et al. Maintenance treatment outcomes in older patients with bipolar I disorder. Am J Geriatr Psychiatry. 2005;13(4):305-311.
14. Lyketsos CG, Olin J. Depression in Alzheimer’s disease: overview and treatment. Biol Psychiatry. 2002;52(3):243-252.
15. Alexopoulos GS, Meyers BS, Young RC, et al. ‘Vascular depression’ hypothesis. Arch Gen Psychiatry. 1997;54(10):915-922.
16. Starkstein SE, Mizrahi R, Power BD. Antidepressant therapy in post-stroke depression. Expert Opin Pharmacother. 2008;9(8):1291-1298.
17. Rush AJ, Trivedi MH, Wisniewski SR, et al. and STAR*D Study Team. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med. 2006;354(12):1231-1242.
18. Trivedi MH, Fava M, Wisniewski SR, et al. and the STAR*D Study Team. Medication augmentation after the failure of SSRIs for depression. N Engl J Med. 2006;354(12):1243-1252.
19. McGrath PJ, Stewart JW, Fava M, et al. Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: a STAR*D report. Am J Psychiatry. 2006;163(9):1531-1541.
20. Appelberg BG, Syvälahti EK, Koskinen TE, et al. Patients with severe depression may benefit from buspirone augmentation of selective serotonin reuptake inhibitors: results from a placebo-controlled, randomized, double-blind, placebo wash-in study. J Clin Psychiatry. 2001;62(6):448-452.
21. Greenberg RM, Kellner CH. Electroconvulsive therapy: a selected review. Am J Geriatric Psychiatry. 2005;13(4):268-281.
22. Kaplan HI, Sadock BJ. Electroconvulsive therapy. In: Kaplan and Sadock’s synopsis of psychiatry. 8th ed. Philadelphia, PA: Lippincott Williams and Wilkins; 1998:1138–1143.
23. Gum A, Areán P. Current status of psychotherapy for mental disorders in the elderly. Curr Psychiatry Rep. 2004;6:32-38.
24. Unützer J, Katon W, Callahan CM, et al. Collaborative care management of late-life depression in primary care settings: a randomized controlled trial. JAMA. 2002;288:2836-2845.
25. Niederehe G, Schneider LS. Treatments for depression and anxiety in the aged. In: Nathan PE, Gorman JM, eds. A guide to treatments that work. New York, NY: Oxford University Press; 1998:270–287.
26. Miklowitz DJ, Otto MW, Frank E, et al. Psychosocial treatments for bipolar depression: a 1-year randomized trial from the Systematic Treatment Enhancement Program. Arch Gen Psychiatry. 2007;64:419-426.
27. Alexopoulos GS, Raue P, Areán P. Problem-solving therapy versus supportive therapy in geriatric major depression with executive dysfunction. Am J Geriatr Psychiatry. 2003;11:46-52.
Discuss this article at http://currentpsychiatry.blogspot.com/2010/08/depression-in-older-adults.html#comments
Depression in older adults (age ≥65) can devastate their quality of life and increase the likelihood of institutionalization because of behavioral problems.1 Depression is a primary risk factor for suicide, and suicide rates are highest among those age ≥65, especially among white males.2 The burden of geriatric depression can extend to caregivers.1 Prompt recognition and treatment of depression could help minimize morbidity and reduce suffering in older adults and their caregivers.
Although geriatric depression varies in severity and presentation, common categories include:
- major depressive disorder (MDD)
- vascular depression
- dysthymia
- depression in the context of dementias, psychosis, bipolar disorder, and executive dysfunction.
Diagnoses in this population generally correspond with DSM-IV-TR criteria, but geriatric depression has distinct clinical manifestations.1,2 Compared with younger depressed patients, older adults are less likely to endorse depressed mood and more likely to report a lack of emotions.1,2 Older patients report feelings of irritability and fearfulness more often than sadness.1,2 Mood symptoms tend to be transient, reoccur frequently, and display either a diurnal pattern or multiple fluctuations in a single day.1,2 Other common presentations include loss of interest in usual activities, lack of motivation, social withdrawal, and decline in activities of daily living.1,2
Summary of recommendations
Age-specific recommendations for assessing and treating geriatric depression can be generated in part from evidence-based reviews, meta-analyses,3 and geriatric expert consensus guidelines.4 Such guidelines and recommendations often do not take into account the marked heterogeneity of medical, cognitive, and overall functioning in patients age ≥65, however, because they are based on studies of younger populations and patients with complicated issues often are excluded from studies. The recommendations in this article are based largely on findings from a National Institutes of Health (NIH)-sponsored project by Alexopoulos et al to develop consensus guidelines for managing geriatric depression and expert opinion from clinicians who treat geriatric patients.4
During your initial clinical evaluation, confirm the diagnosis and type, duration, and severity of depression. Seek to understand the biopsychosocial context of each patient’s presentation. Carefully consider your patient’s suicide risk. Hospitalization may be required if he or she is at high risk for suicide or has complex medical and social circumstances that cannot be managed adequately in an outpatient setting.5
Unipolar major depression
For unipolar, nonpsychotic geriatric depression, the NIH-Alexopoulos et al guidelines emphasize a combination of antidepressants and psychotherapy (Algorithm 1).4 Selective serotonin reuptake inhibitors (SSRIs) and venlafaxine are first-line options.4,6,7 Tricyclic antidepressants (TCAs), bupropion, and mirtazapine are alternatives.4 Among SSRIs, citalopram, escitalopram, and sertraline are preferred initial antidepressants. Fluoxetine is used less frequently.4 Paroxetine also is less commonly used because of its anticholinergic effects and because the drug inhibits cytochrome P4502D6,2 which metabolizes several medications commonly prescribed for older adults. Among TCAs, nortriptyline is preferred.4 Studies have shown that duloxetine improves depression and is safe and well-tolerated in older adults with recurrent MDD.8 Electroconvulsive therapy (ECT) is an option for treating severe or treatment-resistant unipolar major depression.9
For unipolar depression with psychotic symptoms, guidelines recommend a combination of an antidepressant and an antipsychotic or ECT.4 Atypical antipsychotics are preferred over typical antipsychotics4; risperidone, olanzapine, and quetiapine are most frequently used.4 Clinical data on aripiprazole and ziprasidone in older adults are limited. Many geriatric experts recommend continuing an antipsychotic for 6 months after symptom remission, then gradually tapering the dose.4
During acute illness, administer an anti-depressant for 6 to 12 weeks at the individually determined dose required to achieve symptom remission.6 For an older adult experiencing a first lifetime episode of major depression, continue antidepressant treatment for 1 year after remission.4 If your patient has had 2 lifetime episodes of major depression, continue the antidepressant at the same dose used to achieve remission for at least 3 years. For patients who have had ≥3 episodes of depression or whose index episode was particularly severe or involved significant suicidal thoughts or behaviors, continue maintenance treatment indefinitely.
Algorithm 1: Treatment for unipolar depression in geriatric patients
ECT: electroconvulsive therapy
Bipolar depression
Mood stabilizers such as lithium or valproate—as monotherapy or in combination with an antidepressant—are recommended to treat bipolar depression without psychotic symptoms in older adults (Algorithm 2).10 For bipolar depression with psychotic symptoms, a combination of a mood stabilizer and an atypical antipsychotic or ECT is recommended.10
Older adults’ increased sensitivity to side effects and reduced ability to tolerate lithium may limit its use and may prompt you to consider atypical antipsychotics as alternatives to other mood stabilizers. Although quetiapine and fluoxetineolanzapine combination are well studied in younger patients,11,12 there is a lack of data to support their clinical effectiveness and tolerability in older adults. Among antidepressants, SSRIs or bupropion are preferred over TCAs to prevent a switch to mania.10 Lamotrigine is an effective maintenance treatment for bipolar depressive episodes in older adults.13
Although optimal mood stabilizer and antidepressant dosing for this population has not been adequately assessed, pharmacotherapy that has been effective generally should be continued without modification for at least 6 to 12 months.10 After the patient achieves remission, gradually discontinue antidepressants while maintaining the mood stabilizer.10
Algorithm 2: Bipolar depression: Options for combination therapy
ECT: electroconvulsive therapy; SSRIs: selective serotonin reuptake inhibitors
Depression in dementia
Managing depression in dementia patients is similar to treatment in older adults without dementia,5,14 although pharmacologic agents must be carefully selected because of increased risk of side effects (Algorithm 3). American Psychiatric Association practice guidelines recommend considering antidepressants for depressed patients with dementia even if their mood disturbances do not meet DSM-IV-TR criteria for MDD.5
SSRIs’ lower side effect profile make them the preferred treatment; the selective serotonin-norepinephrine reuptake inhibitor (SNRI) venlafaxine is a second-line option.4,14 Avoid TCAs and other agents with anticholinergic side effects because of potential cardiovascular complications and cognitive side effects, unless SSRIs or SNRIs are ineffective or contraindicated.14 Recently clinicians have been reluctant to use antipsychotics in patients with dementia, because of the FDA’s “black-box” warning regarding the increased mortality risk associated with their use in this population.
When using ECT to treat depression in patients with dementia, the treatment protocol often is modified to twice-a-week, unilateral stimulus because of these patients’ increased risk of delirium.14 The safety of ECT to treat depression in patients with dementia has not been adequately assessed.14
Algorithm 3: Treating comorbid depression and dementia
ECT: electroconvulsive therapy; SNRI: selective serotoninnorepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor
Vascular depression
The “vascular depression hypothesis” proposes that accumulation of subcortical white matter hyperintensities can disrupt frontostriatal pathways, resulting in depressive symptoms.15 This hypothesis is supported by the confluence of depression and vascular risk factors.15 Sertraline, citalopram, nortriptyline,16 and trazodone15 have been shown to reduce depressive symptoms after a stroke.
Minor depression and dysthymia
Although the efficacy of antidepressants in minor depression—depression that does not meet criteria for MDD—is not well established, expert consensus guidelines recommend SSRIs and psychotherapy, separately or in combination, for minor depression and dysthymia in older adults (Algorithm 4).4 Depression in executive dysfunction responds poorly to SSRI treatment2; however, behaviorally oriented psychotherapeutic interventions such as problem-solving therapy (PST) show promise.2
Algorithm 4: Minor depression: SSRIs plus psychotherapy
SSRIs: selective serotonin reuptake inhibitors
Comorbid medical conditions
When an older adult has a medical problem that likely contributes to depression—such as hypothyroidism—treat the condition and prescribe antidepressants simultaneously.2 However, if the medical problem likely causes depression—such as substance withdrawal—treat the condition first and prescribe antidepressants only if mood symptoms persist.2
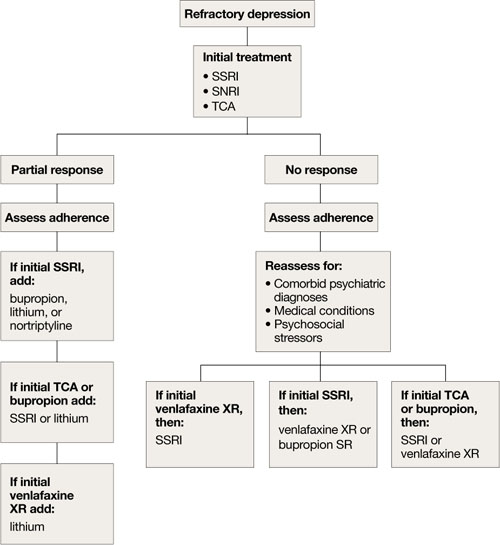
Refractory depression
If your patient does not respond to an antidepressant trial of adequate dosage and duration, first make sure he or she is taking it correctly (Algorithm 5). After ruling out poor adherence, screen for comorbid psychiatric or medical conditions or psychosocial stressors and reassess the principal diagnosis.5
If these steps don’t address your patient’s depressive symptoms, expert consensus guidelines suggest switching to a different antidepressant:4
- If you first prescribed an SSRI, consider venlafaxine XR or bupropion SR.4,17
- If your patient initially received a TCA or bupropion, an SSRI or venlafaxine XR would be appropriate.4
- If venlafaxine XR was the first antidepressant, a SSRI is recommended.4
If your patient experienced a partial response but not full remission with the initial antidepressant, consider adding a second antidepressant or an augmenting agent:4
- If your patient first received an SSRI, adding bupropion, lithium, or nortriptyline is recommended.
- If the initial antidepressant was a TCA or bupropion, consider adding lithium or an SSRI.
- Augmenting venlafaxine XR with lithium is recommended.4
The National Institutes of Mental Health-sponsored Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study of treatment-resistant depression in mixed-age groups reported that patients who do not attain remission with an initial SSRI may respond to switching to bupropion SR or venlafaxine XR.17 Augmenting an SSRI with bupropion SR has been shown to be effective.18 In addition, consider mirtazapine augmentation,19 especially if your patient experiences insomnia or anorexia. A combination of mirtazapine and venlafaxine have better efficacy and tolerability compared with the monoamine oxidase inhibitor tranylcypromine.19 Some studies have shown augmenting SSRIs with buspirone in patients with severe depression is efficacious and safe in younger adults,20 but this practice is not well studied in older patients.
Algorithm 5: Treatment-resistant geriatric depression: Partial vs no response
SNRI: selective serotonin-norepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor; TCA: tricyclic antidepressant
Nonpharmacologic treatments
ECT is an important therapeutic intervention because of its safety, efficacy, and faster clinical response.6,7,9,21 Consider ECT for older adults with severe or psychotic major depression, acute suicidality, catatonia, or severe malnutrition caused by refusal to eat. Patients who remain significantly symptomatic after multiple medication trials, do not tolerate medications well, or have comorbid medical conditions that preclude antidepressant use also are potential candidates for ECT.5,22
ECT can be administered to many older depressed adults with relatively low complication rates. Pretreatment clinical and laboratory evaluations and consultation with medical colleagues may minimize the risk of adverse effects, including cardiovascular instability, delirium, and falls.9 Anterograde memory loss—a common concern for clinicians and patients—usually is temporary and can be reduced by modifying the ECT administration parameters, such as switching from bilateral to unilateral stimulus and spacing treatments.9 Use caution when considering ECT for patients with cardiovascular or neurologic conditions—such as myocardial infarction or cerebrovascular accident within 6 months of treatment—that may increase the risk of adverse effects. Some pharmacologic agents, such as benzodiazepines and anticonvulsant mood stabilizers, may decrease ECT’s efficacy by inhibiting seizure.22
Depressive relapse after ECT is a major clinical concern.21 Continuation ECT— within the first 6 months of remission— aims to prevent relapse of the same episode, whereas maintenance ECT—beyond the first 6 months—helps avert occurrence of new episodes.4,21 Relapse and recurrence also can be prevented with continuation or maintenance pharmacotherapy,4,21 which should be initiated immediately after the index course of ECT.21 Typically, ECT continuation/maintenance treatments are provided weekly, then gradually spaced out to once a month based on the minimum frequency that is effective for an individual patient.21
Psychotherapy for geriatric depression generally is effective.23 One-half of older patients prefer psychotherapy over pharmacotherapy.24 Efficacious psychotherapies include behavioral therapy, cognitive-behavioral therapy (CBT), PST, brief dynamic therapy, interpersonal therapy, supportive therapy, and reminiscence therapy.23 CBT has the most empiric support for treating geriatric depression.5,6
Psychotherapy alone is appropriate for mild-to-moderate depression, although severe depression requires adding medication.25 The combination of pharmacotherapy and psychotherapy appears to be more effective than either intervention alone in preventing recurrent major depression, especially when a specific psychosocial stressor has been identified.5,6 CBT, interpersonal therapy, and family-focused therapy enhance pharmacotherapy outcomes in bipolar disorder.13
The Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD) study found that in mixed-age patients, pharmacotherapy plus psychotherapy is more beneficial than medication alone in stabilizing bipolar depression.26 For older adults with executive dysfunction, research suggests that PST is more effective than other psychotherapies.27 Psychosocial interventions—such as psychoeducation for the family and caregivers, family counseling, and participation in senior citizen centers and services—are strongly recommended for many patients.4
Related Resources
- Blazer DG, Steffens DC, Koenig HG. Mood disorders. In: Blazer DG, Steffens DC, eds. The American Psychiatric Publishing textbook of geriatric psychiatry. 4th ed. Arlington, VA: American Psychiatric Publishing, Inc.; 2009:275-300.
- American Association for Geriatric Psychiatry. www.aagponline.org.
Drug Brand Names
- Aripiprazole • Abilify
- Bupropion • Wellbutrin, Zyban
- Buspirone • Buspar
- Citalopram • Celexa
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluoxetine-olanzapine • Symbyax
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid
- Mirtazapine • Remeron
- Nortriptyline • Aventyl, Pamelor
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sertraline • Zoloft
- Tranylcypromine • Parnate
- Trazodone • Desyrel
- Valproate • Depakote
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with the manufacturer of any product mentioned in this article or with manufacturers of competing products.
Discuss this article at http://currentpsychiatry.blogspot.com/2010/08/depression-in-older-adults.html#comments
Depression in older adults (age ≥65) can devastate their quality of life and increase the likelihood of institutionalization because of behavioral problems.1 Depression is a primary risk factor for suicide, and suicide rates are highest among those age ≥65, especially among white males.2 The burden of geriatric depression can extend to caregivers.1 Prompt recognition and treatment of depression could help minimize morbidity and reduce suffering in older adults and their caregivers.
Although geriatric depression varies in severity and presentation, common categories include:
- major depressive disorder (MDD)
- vascular depression
- dysthymia
- depression in the context of dementias, psychosis, bipolar disorder, and executive dysfunction.
Diagnoses in this population generally correspond with DSM-IV-TR criteria, but geriatric depression has distinct clinical manifestations.1,2 Compared with younger depressed patients, older adults are less likely to endorse depressed mood and more likely to report a lack of emotions.1,2 Older patients report feelings of irritability and fearfulness more often than sadness.1,2 Mood symptoms tend to be transient, reoccur frequently, and display either a diurnal pattern or multiple fluctuations in a single day.1,2 Other common presentations include loss of interest in usual activities, lack of motivation, social withdrawal, and decline in activities of daily living.1,2
Summary of recommendations
Age-specific recommendations for assessing and treating geriatric depression can be generated in part from evidence-based reviews, meta-analyses,3 and geriatric expert consensus guidelines.4 Such guidelines and recommendations often do not take into account the marked heterogeneity of medical, cognitive, and overall functioning in patients age ≥65, however, because they are based on studies of younger populations and patients with complicated issues often are excluded from studies. The recommendations in this article are based largely on findings from a National Institutes of Health (NIH)-sponsored project by Alexopoulos et al to develop consensus guidelines for managing geriatric depression and expert opinion from clinicians who treat geriatric patients.4
During your initial clinical evaluation, confirm the diagnosis and type, duration, and severity of depression. Seek to understand the biopsychosocial context of each patient’s presentation. Carefully consider your patient’s suicide risk. Hospitalization may be required if he or she is at high risk for suicide or has complex medical and social circumstances that cannot be managed adequately in an outpatient setting.5
Unipolar major depression
For unipolar, nonpsychotic geriatric depression, the NIH-Alexopoulos et al guidelines emphasize a combination of antidepressants and psychotherapy (Algorithm 1).4 Selective serotonin reuptake inhibitors (SSRIs) and venlafaxine are first-line options.4,6,7 Tricyclic antidepressants (TCAs), bupropion, and mirtazapine are alternatives.4 Among SSRIs, citalopram, escitalopram, and sertraline are preferred initial antidepressants. Fluoxetine is used less frequently.4 Paroxetine also is less commonly used because of its anticholinergic effects and because the drug inhibits cytochrome P4502D6,2 which metabolizes several medications commonly prescribed for older adults. Among TCAs, nortriptyline is preferred.4 Studies have shown that duloxetine improves depression and is safe and well-tolerated in older adults with recurrent MDD.8 Electroconvulsive therapy (ECT) is an option for treating severe or treatment-resistant unipolar major depression.9
For unipolar depression with psychotic symptoms, guidelines recommend a combination of an antidepressant and an antipsychotic or ECT.4 Atypical antipsychotics are preferred over typical antipsychotics4; risperidone, olanzapine, and quetiapine are most frequently used.4 Clinical data on aripiprazole and ziprasidone in older adults are limited. Many geriatric experts recommend continuing an antipsychotic for 6 months after symptom remission, then gradually tapering the dose.4
During acute illness, administer an anti-depressant for 6 to 12 weeks at the individually determined dose required to achieve symptom remission.6 For an older adult experiencing a first lifetime episode of major depression, continue antidepressant treatment for 1 year after remission.4 If your patient has had 2 lifetime episodes of major depression, continue the antidepressant at the same dose used to achieve remission for at least 3 years. For patients who have had ≥3 episodes of depression or whose index episode was particularly severe or involved significant suicidal thoughts or behaviors, continue maintenance treatment indefinitely.
Algorithm 1: Treatment for unipolar depression in geriatric patients
ECT: electroconvulsive therapy
Bipolar depression
Mood stabilizers such as lithium or valproate—as monotherapy or in combination with an antidepressant—are recommended to treat bipolar depression without psychotic symptoms in older adults (Algorithm 2).10 For bipolar depression with psychotic symptoms, a combination of a mood stabilizer and an atypical antipsychotic or ECT is recommended.10
Older adults’ increased sensitivity to side effects and reduced ability to tolerate lithium may limit its use and may prompt you to consider atypical antipsychotics as alternatives to other mood stabilizers. Although quetiapine and fluoxetineolanzapine combination are well studied in younger patients,11,12 there is a lack of data to support their clinical effectiveness and tolerability in older adults. Among antidepressants, SSRIs or bupropion are preferred over TCAs to prevent a switch to mania.10 Lamotrigine is an effective maintenance treatment for bipolar depressive episodes in older adults.13
Although optimal mood stabilizer and antidepressant dosing for this population has not been adequately assessed, pharmacotherapy that has been effective generally should be continued without modification for at least 6 to 12 months.10 After the patient achieves remission, gradually discontinue antidepressants while maintaining the mood stabilizer.10
Algorithm 2: Bipolar depression: Options for combination therapy
ECT: electroconvulsive therapy; SSRIs: selective serotonin reuptake inhibitors
Depression in dementia
Managing depression in dementia patients is similar to treatment in older adults without dementia,5,14 although pharmacologic agents must be carefully selected because of increased risk of side effects (Algorithm 3). American Psychiatric Association practice guidelines recommend considering antidepressants for depressed patients with dementia even if their mood disturbances do not meet DSM-IV-TR criteria for MDD.5
SSRIs’ lower side effect profile make them the preferred treatment; the selective serotonin-norepinephrine reuptake inhibitor (SNRI) venlafaxine is a second-line option.4,14 Avoid TCAs and other agents with anticholinergic side effects because of potential cardiovascular complications and cognitive side effects, unless SSRIs or SNRIs are ineffective or contraindicated.14 Recently clinicians have been reluctant to use antipsychotics in patients with dementia, because of the FDA’s “black-box” warning regarding the increased mortality risk associated with their use in this population.
When using ECT to treat depression in patients with dementia, the treatment protocol often is modified to twice-a-week, unilateral stimulus because of these patients’ increased risk of delirium.14 The safety of ECT to treat depression in patients with dementia has not been adequately assessed.14
Algorithm 3: Treating comorbid depression and dementia
ECT: electroconvulsive therapy; SNRI: selective serotoninnorepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor
Vascular depression
The “vascular depression hypothesis” proposes that accumulation of subcortical white matter hyperintensities can disrupt frontostriatal pathways, resulting in depressive symptoms.15 This hypothesis is supported by the confluence of depression and vascular risk factors.15 Sertraline, citalopram, nortriptyline,16 and trazodone15 have been shown to reduce depressive symptoms after a stroke.
Minor depression and dysthymia
Although the efficacy of antidepressants in minor depression—depression that does not meet criteria for MDD—is not well established, expert consensus guidelines recommend SSRIs and psychotherapy, separately or in combination, for minor depression and dysthymia in older adults (Algorithm 4).4 Depression in executive dysfunction responds poorly to SSRI treatment2; however, behaviorally oriented psychotherapeutic interventions such as problem-solving therapy (PST) show promise.2
Algorithm 4: Minor depression: SSRIs plus psychotherapy
SSRIs: selective serotonin reuptake inhibitors
Comorbid medical conditions
When an older adult has a medical problem that likely contributes to depression—such as hypothyroidism—treat the condition and prescribe antidepressants simultaneously.2 However, if the medical problem likely causes depression—such as substance withdrawal—treat the condition first and prescribe antidepressants only if mood symptoms persist.2
Refractory depression
If your patient does not respond to an antidepressant trial of adequate dosage and duration, first make sure he or she is taking it correctly (Algorithm 5). After ruling out poor adherence, screen for comorbid psychiatric or medical conditions or psychosocial stressors and reassess the principal diagnosis.5
If these steps don’t address your patient’s depressive symptoms, expert consensus guidelines suggest switching to a different antidepressant:4
- If you first prescribed an SSRI, consider venlafaxine XR or bupropion SR.4,17
- If your patient initially received a TCA or bupropion, an SSRI or venlafaxine XR would be appropriate.4
- If venlafaxine XR was the first antidepressant, a SSRI is recommended.4
If your patient experienced a partial response but not full remission with the initial antidepressant, consider adding a second antidepressant or an augmenting agent:4
- If your patient first received an SSRI, adding bupropion, lithium, or nortriptyline is recommended.
- If the initial antidepressant was a TCA or bupropion, consider adding lithium or an SSRI.
- Augmenting venlafaxine XR with lithium is recommended.4
The National Institutes of Mental Health-sponsored Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study of treatment-resistant depression in mixed-age groups reported that patients who do not attain remission with an initial SSRI may respond to switching to bupropion SR or venlafaxine XR.17 Augmenting an SSRI with bupropion SR has been shown to be effective.18 In addition, consider mirtazapine augmentation,19 especially if your patient experiences insomnia or anorexia. A combination of mirtazapine and venlafaxine have better efficacy and tolerability compared with the monoamine oxidase inhibitor tranylcypromine.19 Some studies have shown augmenting SSRIs with buspirone in patients with severe depression is efficacious and safe in younger adults,20 but this practice is not well studied in older patients.
Algorithm 5: Treatment-resistant geriatric depression: Partial vs no response
SNRI: selective serotonin-norepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor; TCA: tricyclic antidepressant
Nonpharmacologic treatments
ECT is an important therapeutic intervention because of its safety, efficacy, and faster clinical response.6,7,9,21 Consider ECT for older adults with severe or psychotic major depression, acute suicidality, catatonia, or severe malnutrition caused by refusal to eat. Patients who remain significantly symptomatic after multiple medication trials, do not tolerate medications well, or have comorbid medical conditions that preclude antidepressant use also are potential candidates for ECT.5,22
ECT can be administered to many older depressed adults with relatively low complication rates. Pretreatment clinical and laboratory evaluations and consultation with medical colleagues may minimize the risk of adverse effects, including cardiovascular instability, delirium, and falls.9 Anterograde memory loss—a common concern for clinicians and patients—usually is temporary and can be reduced by modifying the ECT administration parameters, such as switching from bilateral to unilateral stimulus and spacing treatments.9 Use caution when considering ECT for patients with cardiovascular or neurologic conditions—such as myocardial infarction or cerebrovascular accident within 6 months of treatment—that may increase the risk of adverse effects. Some pharmacologic agents, such as benzodiazepines and anticonvulsant mood stabilizers, may decrease ECT’s efficacy by inhibiting seizure.22
Depressive relapse after ECT is a major clinical concern.21 Continuation ECT— within the first 6 months of remission— aims to prevent relapse of the same episode, whereas maintenance ECT—beyond the first 6 months—helps avert occurrence of new episodes.4,21 Relapse and recurrence also can be prevented with continuation or maintenance pharmacotherapy,4,21 which should be initiated immediately after the index course of ECT.21 Typically, ECT continuation/maintenance treatments are provided weekly, then gradually spaced out to once a month based on the minimum frequency that is effective for an individual patient.21
Psychotherapy for geriatric depression generally is effective.23 One-half of older patients prefer psychotherapy over pharmacotherapy.24 Efficacious psychotherapies include behavioral therapy, cognitive-behavioral therapy (CBT), PST, brief dynamic therapy, interpersonal therapy, supportive therapy, and reminiscence therapy.23 CBT has the most empiric support for treating geriatric depression.5,6
Psychotherapy alone is appropriate for mild-to-moderate depression, although severe depression requires adding medication.25 The combination of pharmacotherapy and psychotherapy appears to be more effective than either intervention alone in preventing recurrent major depression, especially when a specific psychosocial stressor has been identified.5,6 CBT, interpersonal therapy, and family-focused therapy enhance pharmacotherapy outcomes in bipolar disorder.13
The Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD) study found that in mixed-age patients, pharmacotherapy plus psychotherapy is more beneficial than medication alone in stabilizing bipolar depression.26 For older adults with executive dysfunction, research suggests that PST is more effective than other psychotherapies.27 Psychosocial interventions—such as psychoeducation for the family and caregivers, family counseling, and participation in senior citizen centers and services—are strongly recommended for many patients.4
Related Resources
- Blazer DG, Steffens DC, Koenig HG. Mood disorders. In: Blazer DG, Steffens DC, eds. The American Psychiatric Publishing textbook of geriatric psychiatry. 4th ed. Arlington, VA: American Psychiatric Publishing, Inc.; 2009:275-300.
- American Association for Geriatric Psychiatry. www.aagponline.org.
Drug Brand Names
- Aripiprazole • Abilify
- Bupropion • Wellbutrin, Zyban
- Buspirone • Buspar
- Citalopram • Celexa
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluoxetine-olanzapine • Symbyax
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid
- Mirtazapine • Remeron
- Nortriptyline • Aventyl, Pamelor
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sertraline • Zoloft
- Tranylcypromine • Parnate
- Trazodone • Desyrel
- Valproate • Depakote
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with the manufacturer of any product mentioned in this article or with manufacturers of competing products.
1. Lyketsos CG, Lee HB. Diagnosis and treatment of depression in Alzheimer’s disease. A practical update for the clinician. Dement Geriatr Cogn Disord. 2004;17(1-2):55-64.
2. Alexopoulos G. Late-life mood disorders. In: Sadavoy J, Jarvik LF, Grossberg GT, et al, eds. Comprehensive textbook of geriatric psychiatry. 3rd ed. New York, NY: W.W. Norton and Company; 2004:609-653.
3. Shanmugham B, Karp J, Drayer R, et al. Evidence-based pharmacologic interventions of geriatric depression. Psychiatr Clin North Am. 2005;28(4):821-835,viii.
4. Alexopoulos GS, Katz IR, Reynolds CF, III, et al. The expert consensus guidelines series. Pharmacotherapy of depressive disorders in older patients. Postgrad Med. 2001; Spect No Pharmacolotherapy:1–86.
5. American Psychiatric Association practice guidelines for the treatment of psychiatric disorders. Arlington, VA: American Psychiatric Association; 2006:793–794.
6. Bartels SJ, Dums AR, Oxman TE, et al. Evidence-based practice in geriatric mental health care. Psychiatr Serv. 2002;53(11):1419-1431.
7. Bartels SJ, Dums AR, Oxman TE, et al. Evidence-based practices in geriatric mental health care: an overview of systematic reviews and meta-analyses. Psychiatr Clin North Am. 2003;26(4):971-990,x–xi.
8. Raskin J, Wiltse CG, Siegal A, et al. Efficacy of duloxetine on cognition, depression, and pain in elderly patients with major depressive disorder: an 8-week, double-blind, placebo-controlled trial. Am J Psychiatry. 2007;164(6):900-909.
9. Alexopoulos GS, Young RC, Abrams RC. ECT in the high-risk geriatric patient. Convuls Ther. 1989;5(1):75-87.
10. Young RC, Gyulai L, Mulsant BH, et al. Pharmacotherapy of bipolar disorder in old age: review and recommendations. Am J Geriatr Psychiatry. 2004;12:342-357.
11. Vieta E, Calabrese JR, Goikolea JM, et al. Quetiapine monotherapy in the treatment of patients with bipolar I or II depression and a rapid-cycling disease course: a randomized, double-blind, placebo-controlled study. Bipolar Disord. 2007;9(4):413-425.
12. Corya SA, Perlis RH, Keck PE, Jr, et al. A 24-week open-label extension study of olanzapine-fluoxetine combination and olanzapine monotherapy in the treatment of bipolar depression. J Clin Psychiatry. 2006;67(5):798-806.
13. Sajatovic M, Gyulai L, Calabrese JR, et al. Maintenance treatment outcomes in older patients with bipolar I disorder. Am J Geriatr Psychiatry. 2005;13(4):305-311.
14. Lyketsos CG, Olin J. Depression in Alzheimer’s disease: overview and treatment. Biol Psychiatry. 2002;52(3):243-252.
15. Alexopoulos GS, Meyers BS, Young RC, et al. ‘Vascular depression’ hypothesis. Arch Gen Psychiatry. 1997;54(10):915-922.
16. Starkstein SE, Mizrahi R, Power BD. Antidepressant therapy in post-stroke depression. Expert Opin Pharmacother. 2008;9(8):1291-1298.
17. Rush AJ, Trivedi MH, Wisniewski SR, et al. and STAR*D Study Team. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med. 2006;354(12):1231-1242.
18. Trivedi MH, Fava M, Wisniewski SR, et al. and the STAR*D Study Team. Medication augmentation after the failure of SSRIs for depression. N Engl J Med. 2006;354(12):1243-1252.
19. McGrath PJ, Stewart JW, Fava M, et al. Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: a STAR*D report. Am J Psychiatry. 2006;163(9):1531-1541.
20. Appelberg BG, Syvälahti EK, Koskinen TE, et al. Patients with severe depression may benefit from buspirone augmentation of selective serotonin reuptake inhibitors: results from a placebo-controlled, randomized, double-blind, placebo wash-in study. J Clin Psychiatry. 2001;62(6):448-452.
21. Greenberg RM, Kellner CH. Electroconvulsive therapy: a selected review. Am J Geriatric Psychiatry. 2005;13(4):268-281.
22. Kaplan HI, Sadock BJ. Electroconvulsive therapy. In: Kaplan and Sadock’s synopsis of psychiatry. 8th ed. Philadelphia, PA: Lippincott Williams and Wilkins; 1998:1138–1143.
23. Gum A, Areán P. Current status of psychotherapy for mental disorders in the elderly. Curr Psychiatry Rep. 2004;6:32-38.
24. Unützer J, Katon W, Callahan CM, et al. Collaborative care management of late-life depression in primary care settings: a randomized controlled trial. JAMA. 2002;288:2836-2845.
25. Niederehe G, Schneider LS. Treatments for depression and anxiety in the aged. In: Nathan PE, Gorman JM, eds. A guide to treatments that work. New York, NY: Oxford University Press; 1998:270–287.
26. Miklowitz DJ, Otto MW, Frank E, et al. Psychosocial treatments for bipolar depression: a 1-year randomized trial from the Systematic Treatment Enhancement Program. Arch Gen Psychiatry. 2007;64:419-426.
27. Alexopoulos GS, Raue P, Areán P. Problem-solving therapy versus supportive therapy in geriatric major depression with executive dysfunction. Am J Geriatr Psychiatry. 2003;11:46-52.
1. Lyketsos CG, Lee HB. Diagnosis and treatment of depression in Alzheimer’s disease. A practical update for the clinician. Dement Geriatr Cogn Disord. 2004;17(1-2):55-64.
2. Alexopoulos G. Late-life mood disorders. In: Sadavoy J, Jarvik LF, Grossberg GT, et al, eds. Comprehensive textbook of geriatric psychiatry. 3rd ed. New York, NY: W.W. Norton and Company; 2004:609-653.
3. Shanmugham B, Karp J, Drayer R, et al. Evidence-based pharmacologic interventions of geriatric depression. Psychiatr Clin North Am. 2005;28(4):821-835,viii.
4. Alexopoulos GS, Katz IR, Reynolds CF, III, et al. The expert consensus guidelines series. Pharmacotherapy of depressive disorders in older patients. Postgrad Med. 2001; Spect No Pharmacolotherapy:1–86.
5. American Psychiatric Association practice guidelines for the treatment of psychiatric disorders. Arlington, VA: American Psychiatric Association; 2006:793–794.
6. Bartels SJ, Dums AR, Oxman TE, et al. Evidence-based practice in geriatric mental health care. Psychiatr Serv. 2002;53(11):1419-1431.
7. Bartels SJ, Dums AR, Oxman TE, et al. Evidence-based practices in geriatric mental health care: an overview of systematic reviews and meta-analyses. Psychiatr Clin North Am. 2003;26(4):971-990,x–xi.
8. Raskin J, Wiltse CG, Siegal A, et al. Efficacy of duloxetine on cognition, depression, and pain in elderly patients with major depressive disorder: an 8-week, double-blind, placebo-controlled trial. Am J Psychiatry. 2007;164(6):900-909.
9. Alexopoulos GS, Young RC, Abrams RC. ECT in the high-risk geriatric patient. Convuls Ther. 1989;5(1):75-87.
10. Young RC, Gyulai L, Mulsant BH, et al. Pharmacotherapy of bipolar disorder in old age: review and recommendations. Am J Geriatr Psychiatry. 2004;12:342-357.
11. Vieta E, Calabrese JR, Goikolea JM, et al. Quetiapine monotherapy in the treatment of patients with bipolar I or II depression and a rapid-cycling disease course: a randomized, double-blind, placebo-controlled study. Bipolar Disord. 2007;9(4):413-425.
12. Corya SA, Perlis RH, Keck PE, Jr, et al. A 24-week open-label extension study of olanzapine-fluoxetine combination and olanzapine monotherapy in the treatment of bipolar depression. J Clin Psychiatry. 2006;67(5):798-806.
13. Sajatovic M, Gyulai L, Calabrese JR, et al. Maintenance treatment outcomes in older patients with bipolar I disorder. Am J Geriatr Psychiatry. 2005;13(4):305-311.
14. Lyketsos CG, Olin J. Depression in Alzheimer’s disease: overview and treatment. Biol Psychiatry. 2002;52(3):243-252.
15. Alexopoulos GS, Meyers BS, Young RC, et al. ‘Vascular depression’ hypothesis. Arch Gen Psychiatry. 1997;54(10):915-922.
16. Starkstein SE, Mizrahi R, Power BD. Antidepressant therapy in post-stroke depression. Expert Opin Pharmacother. 2008;9(8):1291-1298.
17. Rush AJ, Trivedi MH, Wisniewski SR, et al. and STAR*D Study Team. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med. 2006;354(12):1231-1242.
18. Trivedi MH, Fava M, Wisniewski SR, et al. and the STAR*D Study Team. Medication augmentation after the failure of SSRIs for depression. N Engl J Med. 2006;354(12):1243-1252.
19. McGrath PJ, Stewart JW, Fava M, et al. Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: a STAR*D report. Am J Psychiatry. 2006;163(9):1531-1541.
20. Appelberg BG, Syvälahti EK, Koskinen TE, et al. Patients with severe depression may benefit from buspirone augmentation of selective serotonin reuptake inhibitors: results from a placebo-controlled, randomized, double-blind, placebo wash-in study. J Clin Psychiatry. 2001;62(6):448-452.
21. Greenberg RM, Kellner CH. Electroconvulsive therapy: a selected review. Am J Geriatric Psychiatry. 2005;13(4):268-281.
22. Kaplan HI, Sadock BJ. Electroconvulsive therapy. In: Kaplan and Sadock’s synopsis of psychiatry. 8th ed. Philadelphia, PA: Lippincott Williams and Wilkins; 1998:1138–1143.
23. Gum A, Areán P. Current status of psychotherapy for mental disorders in the elderly. Curr Psychiatry Rep. 2004;6:32-38.
24. Unützer J, Katon W, Callahan CM, et al. Collaborative care management of late-life depression in primary care settings: a randomized controlled trial. JAMA. 2002;288:2836-2845.
25. Niederehe G, Schneider LS. Treatments for depression and anxiety in the aged. In: Nathan PE, Gorman JM, eds. A guide to treatments that work. New York, NY: Oxford University Press; 1998:270–287.
26. Miklowitz DJ, Otto MW, Frank E, et al. Psychosocial treatments for bipolar depression: a 1-year randomized trial from the Systematic Treatment Enhancement Program. Arch Gen Psychiatry. 2007;64:419-426.
27. Alexopoulos GS, Raue P, Areán P. Problem-solving therapy versus supportive therapy in geriatric major depression with executive dysfunction. Am J Geriatr Psychiatry. 2003;11:46-52.
Cholesterol, mood, and vascular health: Untangling the relationship
Discuss this article at http://currentpsychiatry.blogspot.com/2010/07/cholesterol-mood-and-vascular-health.html#comments
A growing body of literature examining the putative links among cholesterol, mood disorders, and suicide has produced inconsistent findings and unclear clinical implications that may leave psychiatrists unsure of how to interpret the data. Understanding cholesterol’s role in mood disorders may be relevant to the 2 primary causes of excess deaths in patients with mood disorders: suicide and vascular disease.1

Plausible links
In the early 1990s several studies suggested a link between low cholesterol (<160 mg/dL) and unnatural deaths, including suicide.2-4 Follow-up studies confirmed associations between low cholesterol and suicide attempts, especially violent ones.5 These associations are compelling given the neurobiologic effects of cholesterol, such as a net reduction of serotonergic function (Box 1). Low cholesterol may predispose an individual to aggression, impulsivity, and violence (Table 1).6 Many studies have found that patients with mood disorders have lower cholesterol levels;7 however, other research suggests they are at increased risk of hyperlipidemia, typically hypertriglyceridemia rather than hypercholesterolemia.8
Depression. Several studies have shown an association between low cholesterol and depressive symptoms, although this finding has not been replicated in Asian subjects.9,10 Patients with manic or mixed syndromes have been found to have lower serum cholesterol,11 and individuals with major depression and bipolar disorder have lower cholesterol levels in the brain compared with healthy controls.12 Some studies have observed higher total cholesterol levels after patients receive pharmacotherapy for major depressive symptoms.13 These findings have led to speculation that low serum cholesterol in patients with mood disorders is partially a state-dependent effect of depressive illness.
Suicide. Cohort, case-control, and cross-sectional studies have linked low cholesterol to an increased risk of suicide.2,5 Individuals who attempt suicide by violent means have lower cholesterol compared with those who use less violent methods.5,14 A meta-analysis found statistically significant correlations between low cholesterol and future or past suicidal behavior; however, low cholesterol explained <0.01% of suicidal behavior.15 Studies comparing cholesterol levels of individuals following violent vs nonviolent suicide attempts have demonstrated stronger associations.15
Assessing suicide risk. Current evidence does not support considering low serum cholesterol a risk factor for suicide. One study used cholesterol as a clinical predictor of suicide,16 but this model has not been prospectively validated. As a whole, the evidence does not suggest that cholesterol levels explain a substantial portion of suicidal behaviors.
The neurobiologic effects of low cholesterol—particularly those related to serotonergic hypofunction—are thought to be mediate impulsive, aggressive, and violent behaviors that may predispose an individual to suicide.a,b The CNS contains one-fourth of the body’s free cholesterol,c which is synthesized primarily in situ.
Cholesterol improves membrane stability, reduces permeability, and may influence serotonergic function. Cholesterol depletion may impair function of 5-HT1A and 5-HT7 receptorsd,e and serotonin transporter activity.f Reduced cholesterol after treatment with simvastatin—an HMG-CoA reductase inhibitor that readily crosses the blood-brain barrier—resulted in acute (1-month) increases in serotonin transporter activity followed by subacute (>2 months) decreases.g Lower cholesterol levels may further decrease expression of serotonin receptors and cause a net reduction in serotonergic activity.
In addition, cholesterol is necessary for synapse formation and myelin production. Cholesterol depletion may have more diffuse effects on neurotransmission, such as gamma-aminobutyric acid receptors,hN-methyl-D-aspartate receptors,i opioid signaling,j and excitatory amino acids transport.k
Impulsivity associated with low serotonergic function and low total cholesterol has been suggested as a potential pathway for suicide.l Low cholesterol is associated with self-report measures of impulsivity;m however, increased impulsivity associated with lipid-lowering therapy may be temporary,n which is similar to the time-limited changes in serotonin transporter activity.g Human and animal data have suggested that low cholesterol may be linked to violent behaviors, including suicide.o
Source:
a. Vevera J, Fisar Z, Kvasnicka T, et al. Cholesterol-lowering therapy evokes time-limited changes in serotonergic transmission. Psychiatry Res. 2005;133(2-3):197-203.
b. Kaplan JR, Shively CA, Fontenot MB, et al. Demonstration of an association among dietary cholesterol, central serotonergic activity, and social behavior in monkeys. Psychosom Med. 1994;56(6):479-484.
c. Chattopadhyay A, Paila YD. Lipid-protein interactions, regulation and dysfunction of brain cholesterol. Biochem Biophys Res Commun. 2007;354(3):627-633.
d. Singh P, Paila YD, Chattopadhyay A. Differential effects of cholesterol and 7-dehydrocholesterol on the ligand binding activity of the hippocampal serotonin(1A) receptor: implications in SLOS. Biochem Biophys Res Commun. 2007;358(2):495-499.
e. Sjögren B, Hamblin MW, Svenningsson P. Cholesterol depletion reduces serotonin binding and signaling via human 5-HT(7(a)) receptors. Eur J Pharmacol. 2006;552(1-3):1-10.
f. Scanlon SM, Williams DC, Schloss P. Membrane cholesterol modulates serotonin transporter activity. Biochemistry. 2001;40(35):10507-10513.
g. Vevera J, Fisar Z, Kvasnicka T, et al. Cholesterol-lowering therapy evokes time-limited changes in serotonergic transmission. Psychiatry Res. 2005;133(2-3):197-203.
h. Sooksawate T, Simmonds MA. Effects of membrane cholesterol on the sensitivity of the GABA(A) receptor to GABA in acutely dissociated rat hippocampal neurones. Neuropharmacology. 2001;40(2):178-184.
i. Abulrob A, Tauskela JS, Mealing G, et al. Protection by cholesterol-extracting cyclodextrins: a role for N-methyl-daspartate receptor redistribution. J Neurochem. 2005;92(6):1477-1486.
j. Huang P, Xu W, Yoon SI, et al. Cholesterol reduction by methyl-beta-cyclodextrin attenuates the delta opioid receptor-mediated signaling in neuronal cells but enhances it in non-neuronal cells. Biochem Pharmacol. 2007;73(4):534-549.
k. Butchbach ME, Tian G, Guo H, et al. Association of excitatory amino acid transporters, especially EAAT2, with cholesterol-rich lipid raft microdomains: importance for excitatory amino acid transporter localization and function. J Biol Chem. 2004;279(33):34388-34396.
l. Fawcett J, Busch KA, Jacobs D, et al. Suicide: a four-pathway clinical-biochemical model. Annals N Y Acad Sci. 1997;836:288-301.
m. Garland M, Hickey D, Corvin A, et al. Total serum cholesterol in relation to psychological correlates in parasuicide. Br J Psychiatry. 2000;177:77-83.
n. Ormiston T, Wolkowitz OM, Reus VI, et al. Behavioral implications of lowering cholesterol levels: a double-blind pilot study. Psychosomatics. 2003;44(5):412-414.
o. Golomb BA. Cholesterol and violence: is there a connection? Ann Intern Med. 1998;128(6):478-487.
Table 1
Psychiatric features associated with low cholesterol*
| Symptoms |
| Anxiety, depressed mood, emotional lability, euphoria, impulsivity, irritability, suicidal ideation, aggression |
| Syndromes |
| Anorexia nervosa, bipolar disorder, borderline personality disorder, major depressive disorder, seasonal affective disorder |
| Behaviors |
| Suicide and suicide attempts, violence |
| *Small studies have suggested possible relationships with dissociative and panic disorders |
Effects of lipid-lowering agents
If there is a causal relationship between low cholesterol and mood disorders, then it stands to reason that using cholesterol-lowering drugs would increase the risk of depression and suicide. However, the data do not support that conclusion.
Many case reports have documented adverse psychiatric reactions to statins, including depression, suicidality, emotional lability, agitation, irritability, anxiety, panic, and euphoria.17 In an early analysis of primary prevention trials, patients receiving cholesterol-lowering treatment—mainly non-statins—were estimated to have twice the risk of death by suicide or violence compared with controls.3 However, a more recent meta-analysis of larger clinical trials of lipid-lowering agents including statins and observational studies did not reveal an association between lipid-lowering medications and suicide.15,18
In a large case-control study, statin users had a lower risk of depression (adjusted odds ratio [OR] 0.4, 95% confidence interval [CI], 0.2 to 0.9) than patients taking non-statin lipid-lowering drugs (adjusted OR 1.0, 95% CI, 0.5 to 2.1).19 However, statins reduced cholesterol more (30% to 50%) than non-statin drugs (10% to 20%). A clinical trial of >1,000 patients with stable coronary artery disease treated with pravastatin—an HMG-CoA reductase inhibitor with low lipophilicity that is less likely than other statins to cross the blood-brain barrier—revealed no changes in self-reported anger, impulsiveness, anxiety, or depression.20
This study did not exclude patients with psychiatric illness—who are at greatest risk of suicide—but other trials of lipid-lowering drugs did.21 As a result, the effects of lipid-lowering medications on psychiatric patients are unclear. A clinical trial is underway to assess the effects of pravastatin (low lipophilicity), simvastatin (high lipophilicity), or placebo on mood, sleep, and aggression.21
Low cholesterol: State or trait?
Much of the research linking low cholesterol, mood disorders, and suicidality could be confounded by depressed mood leading to reduced serum cholesterol. There has been considerable debate about whether low cholesterol predisposes patients to suicide or if depression independently leads to poor nutrition and therefore low cholesterol and increased suicide risk.6,22
Some researchers have suggested that depression lowers cholesterol and increases risk of suicide,23 but study designs have limited the ability to discern the directionality of the relationship. Attempts to control for depression-related malnutrition and weight loss—which lowers total cholesterol, low-density lipoprotein cholesterol (LDL-C), and high-density lipoprotein cholesterol (HDL-C)24—suggest the association may be independent of these variables.25-27 These findings suggest that cholesterol may be considered a trait marker and is not entirely state-dependent. However, multiple, large, long-term randomized controlled trials have not shown increased depression and suicide with use of lipid-lowering agents in healthy populations.20
The Figure illustrates known epidemiologic associations of low cholesterol, low serotoninergic function, and suicide and contrasts conceptual models of cholesterol as a state and a trait marker. A case can be made for cholesterol as both a state and a trait marker, and these models could overlap, with depression-induced decreases in cholesterol further mediating changes in serotonergic function and related behavioral sequelae.
Figure
Cholesterol, depression, and suicide: How are they linked? 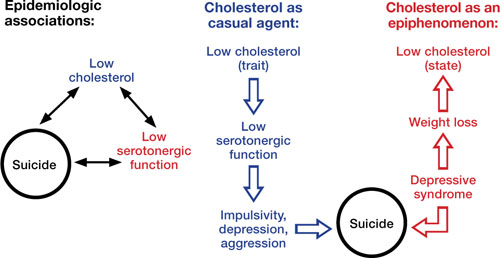
Low cholesterol may be considered a trait marker, predisposing patients to lower serotonergic function and placing them at greater risk for impulsivity, depression, aggression, and suicide. Other models suggest that lower cholesterol is a state-dependent consequence of depression, and not part of a causal chain toward suicide
Improving cardiac health
Limited epidemiologic studies suggest that patients with mood disorders may have lower levels of total cholesterol and LDL-C, but higher rates of hypertriglyceridemia compared with the general population.8 Unfortunately, psychiatric patients—who may be at increased risk of developing cardiovascular disease—may be less likely to be screened and appropriately treated for lipid abnormalities.28 To address this disparity, consider assuming an active role in assessing and managing hyperlipidemia in your patients with mood disorders. Be aware of your patients’ lipid profile and ensure that they follow monitoring recommendations.
The National Cholesterol Education Program recommends screening all adults age >20 for hyperlipidemia every 5 years using measures of total cholesterol, LDL-C, HDL-C, and triglycerides. If LDL-C or triglycerides exceed target values (Table 2), appropriate management includes recommending lifestyle changes and pharmacotherapy (Box 2).
Patients should receive a fasting lipid profile before and 12 weeks after starting any antipsychotic and semiannually thereafter.29 Consider closely monitoring lipids when patients gain weight with psychotropics. Refer patients with hyperlipidemia to a primary care physician, but in the absence of such a provider, mental health clinicians who are familiar with treatment guidelines can manage these patients.30
Closely monitor individuals with mood disorders for changes in behavior or mental status after starting a lipid-lowering agent. Consider discontinuing the drug if a patient develops an adverse reaction. If symptoms return after medication rechallenge, consider other management strategies such as an alternate lipid-lowering agent or re-emphasizing behavioral measures.
Table 2
National Cholesterol Education Program recommended LDL levels
| Risk category* | LDL goal | When to consider medications |
|---|---|---|
| CHD or CHD equivalent | <100 mg/dL | ≥130 mg/dL |
| ≥2 major risk factors | <130 mg/dL | ≥130 to 160 mg/dL (based on 10-year risk) |
| 0 or 1 risk factor | <160 mg/dL | ≥190 mg/dL |
| CHD: coronary heart disease; HDL: high-density lipoprotein; LDL: low-density lipoprotein | ||
| *Risk category is based on the presence of CHD or equivalent and major risk factors for CHD. CHD equivalents include symptomatic carotid artery disease, peripheral artery disease, and abdominal aortic aneurysm. Major risk factors include smoking, hypertension, low HDL, family history, and age. LDL levels to consider medications for those with ≥2 major risk factors vary by 10-year CHD risk | ||
| Source: National Cholesterol Education Program, Adult Treatment Panel III (ATP III) Quick Desk Reference. www.nhlbi.nih.gov/guidelines/cholesterol/atglance.htm | ||
National Cholesterol Education Program guidelines state that when a patient’s low-density lipoprotein cholesterol (LDL-C) exceeds targets (Table 2), first recommend lifestyle changes such as a diet low in saturated fat (<7% of calories) and cholesterol (<200 mg/d), weight management, and exercise. Increases in soluble fiber (10 to 25 g/d) and plant stanols/sterols also may be considered. If LDL-C levels are still too high, pharmacologic therapy such as an HMGCoA reductase inhibitor is suggested.
Treatment of elevated triglycerides (≥150 mg/dL) includes reaching the target LDL-C, intensifying a weight management program, and increasing exercise. Address quitting smoking and limiting alcohol when indicated. If triglyceride levels are ≥200 mg/dL after the LDL-C target is reached, set a secondary goal of reaching a target non-high-density lipoprotein cholesterol (HDL-C) (non-HDL-C; total cholesterol minus HDL-C) 30 mg/dL greater than the LDL goal. This can be achieved by adding an LDL-lowering drug such as a statin, nicotinic acid, or ezetimibe. When triglycerides are ≥500 mg/dL, more aggressive intervention, such as with a fibrate, omega-3 fatty acids, very low-fat diets, and exercise, is required to prevent pancreatitis.
Source: National Heart Lung and Blood Institute. National Cholesterol Education Program. www.nhlbi.nih.gov/guidelines/cholesterol/index.htm
Related Resources
- Fiedorowicz JG, Coryell WH. Cholesterol and suicide attempts: a prospective study of depressed inpatients. Psychiatry Res. 2007;152(1):11-20.
- National Cholesterol Education Program, Adult Treatment Panel III (ATP III) Quick Desk Reference. www.nhlbi.nih.gov/guidelines/cholesterol/atglance.htm.
- Executive Summary of the third report of the national Cholesterol Education Program (nCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA. 2001;285(19):2486-2497.
Drug Brand Names
- Ezetimibe • Zetia
- Pravastatin • Pravachol
- Simvastatin • Zocor
Acknowledgements
Dr. Fiedorowicz thanks Lois Warren and Miriam Weiner for their editorial assistance.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products. Dr. Fiedorowicz is supported by the national Institutes of Health (1K23MH083695-01A210), nARSAD, and the Institute for Clinical and Translational Science at the University of Iowa (3 UL1 RR024979-03S4). He has received support for participating in a colleague’s investigator-initiated project with Eli Lilly. Dr. Haynes’ research is supported by grants from the national Institutes of Health (nHLBI: HL58972 & HL14388; nCRR CTSA: 1UL1RR024979).
1. Osby U, Brandt L, Correia N, et al. Excess mortality in bipolar and unipolar disorder in Sweden. Arch Gen Psychiatry. 2001;58(9):844-850.
2. Lindberg G, Råstam L, Gullberg B, et al. Low serum cholesterol concentration and short term mortality from injuries in men and women. BMJ. 1992;305(6848):277-279.
3. Muldoon MF, Manuck SB, Matthews KA. Lowering cholesterol concentrations and mortality: a quantitative review of primary prevention trials. BMJ. 1990;301(6747):309-314.
4. Neaton JD, Blackburn H, Jacobs D, et al. Serum cholesterol level and mortality findings for men screened in the Multiple Risk Factor Intervention Trial. Multiple Risk Factor Intervention Trial Research Group. Arch Intern Med. 1992;152(7):1490-1500.
5. Fiedorowicz JG, Coryell WH. Cholesterol and suicide attempts: a prospective study of depressed inpatients. Psychiatry Res. 2007;152(1):11-20.
6. Golomb BA. Cholesterol and violence: is there a connection? Ann Intern Med. 1998;128(6):478-487.
7. Pae CU, Kim JJ, Lee SJ, et al. Aberration of cholesterol level in first-onset bipolar I patients. J Affect Disord. 2004;83(1):79-82.
8. Fiedorowicz JG, Palagummi NM, Forman-Hoffman VL, et al. Elevated prevalence of obesity, metabolic syndrome, and cardiovascular risk factors in bipolar disorder. Ann Clin Psychiatry. 2008;20(3):131-137.
9. Chung KH, Tsai SY, Lee HC. Mood symptoms and serum lipids in acute phase of bipolar disorder in Taiwan. Psychiatry Clin Neurosci. 2007;61(4):428-433.
10. Jow GM, Yang TT, Chen CL. Leptin and cholesterol levels are low in major depressive disorder, but high in schizophrenia. J Affect Disord. 2006;90(1):21-27.
11. Sagud M, Mihaljevic-Peles A, Pivac N, et al. Platelet serotonin and serum lipids in psychotic mania. J Affect Disord. 2007;97(1-3):247-251.
12. Beasley CL, Honer WG, Bergmann K, et al. Reductions in cholesterol and synaptic markers in association cortex in mood disorders. Bipolar Disord. 2005;7(5):449-455.
13. Gabriel A. Changes in plasma cholesterol in mood disorder patients: does treatment make a difference? J Affect Disord. 2007;99(1-3):273-278.
14. Lalovic A, Levy E, Luheshi G, et al. Cholesterol content in brains of suicide completers. Int J Neuropsychopharmacol. 2007;10(2):159-166.
15. Lester D. Serum cholesterol levels and suicide: a meta-analysis. Suicide Life Threat Behav. 2002;32(3):333-346.
16. Coryell W, Schlesser M. Combined biological tests for suicide prediction. Psychiatry Res. 2007;150(2):187-191.
17. Tatley M, Savage R. Psychiatric adverse reactions with statins, fibrates and ezetimibe: implications for the use of lipid-lowering agents. Drug Saf. 2007;30(3):195-201.
18. Callréus T, Agerskov Andersen U, Hallas J, et al. Cardiovascular drugs and the risk of suicide: a nested case-control study. Eur J Clin Pharmacol. 2007;63(6):591-596.
19. Yang CC, Jick SS, Jick H. Lipid-lowering drugs and the risk of depression and suicidal behavior. Arch Intern Med. 2003;163(16):1926-1932.
20. Stewart RA, Sharples KJ, North FM, et al. Long-term assessment of psychological well-being in a randomized placebo-controlled trial of cholesterol reduction with pravastatin. The LIPID Study Investigators. Arch Intern Med. 2000;160(20):3144-3152.
21. Golomb BA, Criqui MH, White HL, et al. The UCSD Statin Study: a randomized controlled trial assessing the impact of statins on selected noncardiac outcomes. Control Clin Trials. 2004;25(2):178-202.
22. Fawcett J, Busch KA, Jacobs D, et al. Suicide: a four-pathway clinical-biochemical model. Annals N Y Acad Sci. 1997;836:288-301.
23. Law MR, Thompson SG, Wald NJ. Assessing possible hazards of reducing serum cholesterol. BMJ. 1994;308(6925):373-379.
24. Dattilo AM, Kris-Etherton PM. Effects of weight reduction on blood lipids and lipoproteins: a meta-analysis. Am J Clin Nutr. 1992;56(2):320-328.
25. Garland M, Hickey D, Corvin A, et al. Total serum cholesterol in relation to psychological correlates in parasuicide. Br J Psychiatry. 2000;177:77-83.
26. Golier JA, Marzuk PM, Leon AC, et al. Low serum cholesterol level and attempted suicide. Am J Psychiatry. 1995;152(3):419-423.
27. Kunugi H, Takei N, Aoki H, et al. Low serum cholesterol in suicide attempters. Biol Psychiatry. 1997;41(2):196-200.
28. Murray DP, Weiner M, Prabhakar M, et al. Mania and mortality: why the excess cardiovascular risk in bipolar disorder? Curr Psychiatry Rep. 2009;11(6):475-480.
29. Sernyak MJ. Implementation of monitoring and management guidelines for second-generation antipsychotics. J Clin Psychiatry. 2007;68(suppl 4):14-18.
30. Marder SR, Essock SM, Miller AL, et al. Physical health monitoring of patients with schizophrenia. Am J Psychiatry. 2004;161(8):1334-1349.
Discuss this article at http://currentpsychiatry.blogspot.com/2010/07/cholesterol-mood-and-vascular-health.html#comments
A growing body of literature examining the putative links among cholesterol, mood disorders, and suicide has produced inconsistent findings and unclear clinical implications that may leave psychiatrists unsure of how to interpret the data. Understanding cholesterol’s role in mood disorders may be relevant to the 2 primary causes of excess deaths in patients with mood disorders: suicide and vascular disease.1
Plausible links
In the early 1990s several studies suggested a link between low cholesterol (<160 mg/dL) and unnatural deaths, including suicide.2-4 Follow-up studies confirmed associations between low cholesterol and suicide attempts, especially violent ones.5 These associations are compelling given the neurobiologic effects of cholesterol, such as a net reduction of serotonergic function (Box 1). Low cholesterol may predispose an individual to aggression, impulsivity, and violence (Table 1).6 Many studies have found that patients with mood disorders have lower cholesterol levels;7 however, other research suggests they are at increased risk of hyperlipidemia, typically hypertriglyceridemia rather than hypercholesterolemia.8
Depression. Several studies have shown an association between low cholesterol and depressive symptoms, although this finding has not been replicated in Asian subjects.9,10 Patients with manic or mixed syndromes have been found to have lower serum cholesterol,11 and individuals with major depression and bipolar disorder have lower cholesterol levels in the brain compared with healthy controls.12 Some studies have observed higher total cholesterol levels after patients receive pharmacotherapy for major depressive symptoms.13 These findings have led to speculation that low serum cholesterol in patients with mood disorders is partially a state-dependent effect of depressive illness.
Suicide. Cohort, case-control, and cross-sectional studies have linked low cholesterol to an increased risk of suicide.2,5 Individuals who attempt suicide by violent means have lower cholesterol compared with those who use less violent methods.5,14 A meta-analysis found statistically significant correlations between low cholesterol and future or past suicidal behavior; however, low cholesterol explained <0.01% of suicidal behavior.15 Studies comparing cholesterol levels of individuals following violent vs nonviolent suicide attempts have demonstrated stronger associations.15
Assessing suicide risk. Current evidence does not support considering low serum cholesterol a risk factor for suicide. One study used cholesterol as a clinical predictor of suicide,16 but this model has not been prospectively validated. As a whole, the evidence does not suggest that cholesterol levels explain a substantial portion of suicidal behaviors.
The neurobiologic effects of low cholesterol—particularly those related to serotonergic hypofunction—are thought to be mediate impulsive, aggressive, and violent behaviors that may predispose an individual to suicide.a,b The CNS contains one-fourth of the body’s free cholesterol,c which is synthesized primarily in situ.
Cholesterol improves membrane stability, reduces permeability, and may influence serotonergic function. Cholesterol depletion may impair function of 5-HT1A and 5-HT7 receptorsd,e and serotonin transporter activity.f Reduced cholesterol after treatment with simvastatin—an HMG-CoA reductase inhibitor that readily crosses the blood-brain barrier—resulted in acute (1-month) increases in serotonin transporter activity followed by subacute (>2 months) decreases.g Lower cholesterol levels may further decrease expression of serotonin receptors and cause a net reduction in serotonergic activity.
In addition, cholesterol is necessary for synapse formation and myelin production. Cholesterol depletion may have more diffuse effects on neurotransmission, such as gamma-aminobutyric acid receptors,hN-methyl-D-aspartate receptors,i opioid signaling,j and excitatory amino acids transport.k
Impulsivity associated with low serotonergic function and low total cholesterol has been suggested as a potential pathway for suicide.l Low cholesterol is associated with self-report measures of impulsivity;m however, increased impulsivity associated with lipid-lowering therapy may be temporary,n which is similar to the time-limited changes in serotonin transporter activity.g Human and animal data have suggested that low cholesterol may be linked to violent behaviors, including suicide.o
Source:
a. Vevera J, Fisar Z, Kvasnicka T, et al. Cholesterol-lowering therapy evokes time-limited changes in serotonergic transmission. Psychiatry Res. 2005;133(2-3):197-203.
b. Kaplan JR, Shively CA, Fontenot MB, et al. Demonstration of an association among dietary cholesterol, central serotonergic activity, and social behavior in monkeys. Psychosom Med. 1994;56(6):479-484.
c. Chattopadhyay A, Paila YD. Lipid-protein interactions, regulation and dysfunction of brain cholesterol. Biochem Biophys Res Commun. 2007;354(3):627-633.
d. Singh P, Paila YD, Chattopadhyay A. Differential effects of cholesterol and 7-dehydrocholesterol on the ligand binding activity of the hippocampal serotonin(1A) receptor: implications in SLOS. Biochem Biophys Res Commun. 2007;358(2):495-499.
e. Sjögren B, Hamblin MW, Svenningsson P. Cholesterol depletion reduces serotonin binding and signaling via human 5-HT(7(a)) receptors. Eur J Pharmacol. 2006;552(1-3):1-10.
f. Scanlon SM, Williams DC, Schloss P. Membrane cholesterol modulates serotonin transporter activity. Biochemistry. 2001;40(35):10507-10513.
g. Vevera J, Fisar Z, Kvasnicka T, et al. Cholesterol-lowering therapy evokes time-limited changes in serotonergic transmission. Psychiatry Res. 2005;133(2-3):197-203.
h. Sooksawate T, Simmonds MA. Effects of membrane cholesterol on the sensitivity of the GABA(A) receptor to GABA in acutely dissociated rat hippocampal neurones. Neuropharmacology. 2001;40(2):178-184.
i. Abulrob A, Tauskela JS, Mealing G, et al. Protection by cholesterol-extracting cyclodextrins: a role for N-methyl-daspartate receptor redistribution. J Neurochem. 2005;92(6):1477-1486.
j. Huang P, Xu W, Yoon SI, et al. Cholesterol reduction by methyl-beta-cyclodextrin attenuates the delta opioid receptor-mediated signaling in neuronal cells but enhances it in non-neuronal cells. Biochem Pharmacol. 2007;73(4):534-549.
k. Butchbach ME, Tian G, Guo H, et al. Association of excitatory amino acid transporters, especially EAAT2, with cholesterol-rich lipid raft microdomains: importance for excitatory amino acid transporter localization and function. J Biol Chem. 2004;279(33):34388-34396.
l. Fawcett J, Busch KA, Jacobs D, et al. Suicide: a four-pathway clinical-biochemical model. Annals N Y Acad Sci. 1997;836:288-301.
m. Garland M, Hickey D, Corvin A, et al. Total serum cholesterol in relation to psychological correlates in parasuicide. Br J Psychiatry. 2000;177:77-83.
n. Ormiston T, Wolkowitz OM, Reus VI, et al. Behavioral implications of lowering cholesterol levels: a double-blind pilot study. Psychosomatics. 2003;44(5):412-414.
o. Golomb BA. Cholesterol and violence: is there a connection? Ann Intern Med. 1998;128(6):478-487.
Table 1
Psychiatric features associated with low cholesterol*
| Symptoms |
| Anxiety, depressed mood, emotional lability, euphoria, impulsivity, irritability, suicidal ideation, aggression |
| Syndromes |
| Anorexia nervosa, bipolar disorder, borderline personality disorder, major depressive disorder, seasonal affective disorder |
| Behaviors |
| Suicide and suicide attempts, violence |
| *Small studies have suggested possible relationships with dissociative and panic disorders |
Effects of lipid-lowering agents
If there is a causal relationship between low cholesterol and mood disorders, then it stands to reason that using cholesterol-lowering drugs would increase the risk of depression and suicide. However, the data do not support that conclusion.
Many case reports have documented adverse psychiatric reactions to statins, including depression, suicidality, emotional lability, agitation, irritability, anxiety, panic, and euphoria.17 In an early analysis of primary prevention trials, patients receiving cholesterol-lowering treatment—mainly non-statins—were estimated to have twice the risk of death by suicide or violence compared with controls.3 However, a more recent meta-analysis of larger clinical trials of lipid-lowering agents including statins and observational studies did not reveal an association between lipid-lowering medications and suicide.15,18
In a large case-control study, statin users had a lower risk of depression (adjusted odds ratio [OR] 0.4, 95% confidence interval [CI], 0.2 to 0.9) than patients taking non-statin lipid-lowering drugs (adjusted OR 1.0, 95% CI, 0.5 to 2.1).19 However, statins reduced cholesterol more (30% to 50%) than non-statin drugs (10% to 20%). A clinical trial of >1,000 patients with stable coronary artery disease treated with pravastatin—an HMG-CoA reductase inhibitor with low lipophilicity that is less likely than other statins to cross the blood-brain barrier—revealed no changes in self-reported anger, impulsiveness, anxiety, or depression.20
This study did not exclude patients with psychiatric illness—who are at greatest risk of suicide—but other trials of lipid-lowering drugs did.21 As a result, the effects of lipid-lowering medications on psychiatric patients are unclear. A clinical trial is underway to assess the effects of pravastatin (low lipophilicity), simvastatin (high lipophilicity), or placebo on mood, sleep, and aggression.21
Low cholesterol: State or trait?
Much of the research linking low cholesterol, mood disorders, and suicidality could be confounded by depressed mood leading to reduced serum cholesterol. There has been considerable debate about whether low cholesterol predisposes patients to suicide or if depression independently leads to poor nutrition and therefore low cholesterol and increased suicide risk.6,22
Some researchers have suggested that depression lowers cholesterol and increases risk of suicide,23 but study designs have limited the ability to discern the directionality of the relationship. Attempts to control for depression-related malnutrition and weight loss—which lowers total cholesterol, low-density lipoprotein cholesterol (LDL-C), and high-density lipoprotein cholesterol (HDL-C)24—suggest the association may be independent of these variables.25-27 These findings suggest that cholesterol may be considered a trait marker and is not entirely state-dependent. However, multiple, large, long-term randomized controlled trials have not shown increased depression and suicide with use of lipid-lowering agents in healthy populations.20
The Figure illustrates known epidemiologic associations of low cholesterol, low serotoninergic function, and suicide and contrasts conceptual models of cholesterol as a state and a trait marker. A case can be made for cholesterol as both a state and a trait marker, and these models could overlap, with depression-induced decreases in cholesterol further mediating changes in serotonergic function and related behavioral sequelae.
Figure
Cholesterol, depression, and suicide: How are they linked?
Low cholesterol may be considered a trait marker, predisposing patients to lower serotonergic function and placing them at greater risk for impulsivity, depression, aggression, and suicide. Other models suggest that lower cholesterol is a state-dependent consequence of depression, and not part of a causal chain toward suicide
Improving cardiac health
Limited epidemiologic studies suggest that patients with mood disorders may have lower levels of total cholesterol and LDL-C, but higher rates of hypertriglyceridemia compared with the general population.8 Unfortunately, psychiatric patients—who may be at increased risk of developing cardiovascular disease—may be less likely to be screened and appropriately treated for lipid abnormalities.28 To address this disparity, consider assuming an active role in assessing and managing hyperlipidemia in your patients with mood disorders. Be aware of your patients’ lipid profile and ensure that they follow monitoring recommendations.
The National Cholesterol Education Program recommends screening all adults age >20 for hyperlipidemia every 5 years using measures of total cholesterol, LDL-C, HDL-C, and triglycerides. If LDL-C or triglycerides exceed target values (Table 2), appropriate management includes recommending lifestyle changes and pharmacotherapy (Box 2).
Patients should receive a fasting lipid profile before and 12 weeks after starting any antipsychotic and semiannually thereafter.29 Consider closely monitoring lipids when patients gain weight with psychotropics. Refer patients with hyperlipidemia to a primary care physician, but in the absence of such a provider, mental health clinicians who are familiar with treatment guidelines can manage these patients.30
Closely monitor individuals with mood disorders for changes in behavior or mental status after starting a lipid-lowering agent. Consider discontinuing the drug if a patient develops an adverse reaction. If symptoms return after medication rechallenge, consider other management strategies such as an alternate lipid-lowering agent or re-emphasizing behavioral measures.
Table 2
National Cholesterol Education Program recommended LDL levels
| Risk category* | LDL goal | When to consider medications |
|---|---|---|
| CHD or CHD equivalent | <100 mg/dL | ≥130 mg/dL |
| ≥2 major risk factors | <130 mg/dL | ≥130 to 160 mg/dL (based on 10-year risk) |
| 0 or 1 risk factor | <160 mg/dL | ≥190 mg/dL |
| CHD: coronary heart disease; HDL: high-density lipoprotein; LDL: low-density lipoprotein | ||
| *Risk category is based on the presence of CHD or equivalent and major risk factors for CHD. CHD equivalents include symptomatic carotid artery disease, peripheral artery disease, and abdominal aortic aneurysm. Major risk factors include smoking, hypertension, low HDL, family history, and age. LDL levels to consider medications for those with ≥2 major risk factors vary by 10-year CHD risk | ||
| Source: National Cholesterol Education Program, Adult Treatment Panel III (ATP III) Quick Desk Reference. www.nhlbi.nih.gov/guidelines/cholesterol/atglance.htm | ||
National Cholesterol Education Program guidelines state that when a patient’s low-density lipoprotein cholesterol (LDL-C) exceeds targets (Table 2), first recommend lifestyle changes such as a diet low in saturated fat (<7% of calories) and cholesterol (<200 mg/d), weight management, and exercise. Increases in soluble fiber (10 to 25 g/d) and plant stanols/sterols also may be considered. If LDL-C levels are still too high, pharmacologic therapy such as an HMGCoA reductase inhibitor is suggested.
Treatment of elevated triglycerides (≥150 mg/dL) includes reaching the target LDL-C, intensifying a weight management program, and increasing exercise. Address quitting smoking and limiting alcohol when indicated. If triglyceride levels are ≥200 mg/dL after the LDL-C target is reached, set a secondary goal of reaching a target non-high-density lipoprotein cholesterol (HDL-C) (non-HDL-C; total cholesterol minus HDL-C) 30 mg/dL greater than the LDL goal. This can be achieved by adding an LDL-lowering drug such as a statin, nicotinic acid, or ezetimibe. When triglycerides are ≥500 mg/dL, more aggressive intervention, such as with a fibrate, omega-3 fatty acids, very low-fat diets, and exercise, is required to prevent pancreatitis.
Source: National Heart Lung and Blood Institute. National Cholesterol Education Program. www.nhlbi.nih.gov/guidelines/cholesterol/index.htm
Related Resources
- Fiedorowicz JG, Coryell WH. Cholesterol and suicide attempts: a prospective study of depressed inpatients. Psychiatry Res. 2007;152(1):11-20.
- National Cholesterol Education Program, Adult Treatment Panel III (ATP III) Quick Desk Reference. www.nhlbi.nih.gov/guidelines/cholesterol/atglance.htm.
- Executive Summary of the third report of the national Cholesterol Education Program (nCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA. 2001;285(19):2486-2497.
Drug Brand Names
- Ezetimibe • Zetia
- Pravastatin • Pravachol
- Simvastatin • Zocor
Acknowledgements
Dr. Fiedorowicz thanks Lois Warren and Miriam Weiner for their editorial assistance.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products. Dr. Fiedorowicz is supported by the national Institutes of Health (1K23MH083695-01A210), nARSAD, and the Institute for Clinical and Translational Science at the University of Iowa (3 UL1 RR024979-03S4). He has received support for participating in a colleague’s investigator-initiated project with Eli Lilly. Dr. Haynes’ research is supported by grants from the national Institutes of Health (nHLBI: HL58972 & HL14388; nCRR CTSA: 1UL1RR024979).
Discuss this article at http://currentpsychiatry.blogspot.com/2010/07/cholesterol-mood-and-vascular-health.html#comments
A growing body of literature examining the putative links among cholesterol, mood disorders, and suicide has produced inconsistent findings and unclear clinical implications that may leave psychiatrists unsure of how to interpret the data. Understanding cholesterol’s role in mood disorders may be relevant to the 2 primary causes of excess deaths in patients with mood disorders: suicide and vascular disease.1
Plausible links
In the early 1990s several studies suggested a link between low cholesterol (<160 mg/dL) and unnatural deaths, including suicide.2-4 Follow-up studies confirmed associations between low cholesterol and suicide attempts, especially violent ones.5 These associations are compelling given the neurobiologic effects of cholesterol, such as a net reduction of serotonergic function (Box 1). Low cholesterol may predispose an individual to aggression, impulsivity, and violence (Table 1).6 Many studies have found that patients with mood disorders have lower cholesterol levels;7 however, other research suggests they are at increased risk of hyperlipidemia, typically hypertriglyceridemia rather than hypercholesterolemia.8
Depression. Several studies have shown an association between low cholesterol and depressive symptoms, although this finding has not been replicated in Asian subjects.9,10 Patients with manic or mixed syndromes have been found to have lower serum cholesterol,11 and individuals with major depression and bipolar disorder have lower cholesterol levels in the brain compared with healthy controls.12 Some studies have observed higher total cholesterol levels after patients receive pharmacotherapy for major depressive symptoms.13 These findings have led to speculation that low serum cholesterol in patients with mood disorders is partially a state-dependent effect of depressive illness.
Suicide. Cohort, case-control, and cross-sectional studies have linked low cholesterol to an increased risk of suicide.2,5 Individuals who attempt suicide by violent means have lower cholesterol compared with those who use less violent methods.5,14 A meta-analysis found statistically significant correlations between low cholesterol and future or past suicidal behavior; however, low cholesterol explained <0.01% of suicidal behavior.15 Studies comparing cholesterol levels of individuals following violent vs nonviolent suicide attempts have demonstrated stronger associations.15
Assessing suicide risk. Current evidence does not support considering low serum cholesterol a risk factor for suicide. One study used cholesterol as a clinical predictor of suicide,16 but this model has not been prospectively validated. As a whole, the evidence does not suggest that cholesterol levels explain a substantial portion of suicidal behaviors.
The neurobiologic effects of low cholesterol—particularly those related to serotonergic hypofunction—are thought to be mediate impulsive, aggressive, and violent behaviors that may predispose an individual to suicide.a,b The CNS contains one-fourth of the body’s free cholesterol,c which is synthesized primarily in situ.
Cholesterol improves membrane stability, reduces permeability, and may influence serotonergic function. Cholesterol depletion may impair function of 5-HT1A and 5-HT7 receptorsd,e and serotonin transporter activity.f Reduced cholesterol after treatment with simvastatin—an HMG-CoA reductase inhibitor that readily crosses the blood-brain barrier—resulted in acute (1-month) increases in serotonin transporter activity followed by subacute (>2 months) decreases.g Lower cholesterol levels may further decrease expression of serotonin receptors and cause a net reduction in serotonergic activity.
In addition, cholesterol is necessary for synapse formation and myelin production. Cholesterol depletion may have more diffuse effects on neurotransmission, such as gamma-aminobutyric acid receptors,hN-methyl-D-aspartate receptors,i opioid signaling,j and excitatory amino acids transport.k
Impulsivity associated with low serotonergic function and low total cholesterol has been suggested as a potential pathway for suicide.l Low cholesterol is associated with self-report measures of impulsivity;m however, increased impulsivity associated with lipid-lowering therapy may be temporary,n which is similar to the time-limited changes in serotonin transporter activity.g Human and animal data have suggested that low cholesterol may be linked to violent behaviors, including suicide.o
Source:
a. Vevera J, Fisar Z, Kvasnicka T, et al. Cholesterol-lowering therapy evokes time-limited changes in serotonergic transmission. Psychiatry Res. 2005;133(2-3):197-203.
b. Kaplan JR, Shively CA, Fontenot MB, et al. Demonstration of an association among dietary cholesterol, central serotonergic activity, and social behavior in monkeys. Psychosom Med. 1994;56(6):479-484.
c. Chattopadhyay A, Paila YD. Lipid-protein interactions, regulation and dysfunction of brain cholesterol. Biochem Biophys Res Commun. 2007;354(3):627-633.
d. Singh P, Paila YD, Chattopadhyay A. Differential effects of cholesterol and 7-dehydrocholesterol on the ligand binding activity of the hippocampal serotonin(1A) receptor: implications in SLOS. Biochem Biophys Res Commun. 2007;358(2):495-499.
e. Sjögren B, Hamblin MW, Svenningsson P. Cholesterol depletion reduces serotonin binding and signaling via human 5-HT(7(a)) receptors. Eur J Pharmacol. 2006;552(1-3):1-10.
f. Scanlon SM, Williams DC, Schloss P. Membrane cholesterol modulates serotonin transporter activity. Biochemistry. 2001;40(35):10507-10513.
g. Vevera J, Fisar Z, Kvasnicka T, et al. Cholesterol-lowering therapy evokes time-limited changes in serotonergic transmission. Psychiatry Res. 2005;133(2-3):197-203.
h. Sooksawate T, Simmonds MA. Effects of membrane cholesterol on the sensitivity of the GABA(A) receptor to GABA in acutely dissociated rat hippocampal neurones. Neuropharmacology. 2001;40(2):178-184.
i. Abulrob A, Tauskela JS, Mealing G, et al. Protection by cholesterol-extracting cyclodextrins: a role for N-methyl-daspartate receptor redistribution. J Neurochem. 2005;92(6):1477-1486.
j. Huang P, Xu W, Yoon SI, et al. Cholesterol reduction by methyl-beta-cyclodextrin attenuates the delta opioid receptor-mediated signaling in neuronal cells but enhances it in non-neuronal cells. Biochem Pharmacol. 2007;73(4):534-549.
k. Butchbach ME, Tian G, Guo H, et al. Association of excitatory amino acid transporters, especially EAAT2, with cholesterol-rich lipid raft microdomains: importance for excitatory amino acid transporter localization and function. J Biol Chem. 2004;279(33):34388-34396.
l. Fawcett J, Busch KA, Jacobs D, et al. Suicide: a four-pathway clinical-biochemical model. Annals N Y Acad Sci. 1997;836:288-301.
m. Garland M, Hickey D, Corvin A, et al. Total serum cholesterol in relation to psychological correlates in parasuicide. Br J Psychiatry. 2000;177:77-83.
n. Ormiston T, Wolkowitz OM, Reus VI, et al. Behavioral implications of lowering cholesterol levels: a double-blind pilot study. Psychosomatics. 2003;44(5):412-414.
o. Golomb BA. Cholesterol and violence: is there a connection? Ann Intern Med. 1998;128(6):478-487.
Table 1
Psychiatric features associated with low cholesterol*
| Symptoms |
| Anxiety, depressed mood, emotional lability, euphoria, impulsivity, irritability, suicidal ideation, aggression |
| Syndromes |
| Anorexia nervosa, bipolar disorder, borderline personality disorder, major depressive disorder, seasonal affective disorder |
| Behaviors |
| Suicide and suicide attempts, violence |
| *Small studies have suggested possible relationships with dissociative and panic disorders |
Effects of lipid-lowering agents
If there is a causal relationship between low cholesterol and mood disorders, then it stands to reason that using cholesterol-lowering drugs would increase the risk of depression and suicide. However, the data do not support that conclusion.
Many case reports have documented adverse psychiatric reactions to statins, including depression, suicidality, emotional lability, agitation, irritability, anxiety, panic, and euphoria.17 In an early analysis of primary prevention trials, patients receiving cholesterol-lowering treatment—mainly non-statins—were estimated to have twice the risk of death by suicide or violence compared with controls.3 However, a more recent meta-analysis of larger clinical trials of lipid-lowering agents including statins and observational studies did not reveal an association between lipid-lowering medications and suicide.15,18
In a large case-control study, statin users had a lower risk of depression (adjusted odds ratio [OR] 0.4, 95% confidence interval [CI], 0.2 to 0.9) than patients taking non-statin lipid-lowering drugs (adjusted OR 1.0, 95% CI, 0.5 to 2.1).19 However, statins reduced cholesterol more (30% to 50%) than non-statin drugs (10% to 20%). A clinical trial of >1,000 patients with stable coronary artery disease treated with pravastatin—an HMG-CoA reductase inhibitor with low lipophilicity that is less likely than other statins to cross the blood-brain barrier—revealed no changes in self-reported anger, impulsiveness, anxiety, or depression.20
This study did not exclude patients with psychiatric illness—who are at greatest risk of suicide—but other trials of lipid-lowering drugs did.21 As a result, the effects of lipid-lowering medications on psychiatric patients are unclear. A clinical trial is underway to assess the effects of pravastatin (low lipophilicity), simvastatin (high lipophilicity), or placebo on mood, sleep, and aggression.21
Low cholesterol: State or trait?
Much of the research linking low cholesterol, mood disorders, and suicidality could be confounded by depressed mood leading to reduced serum cholesterol. There has been considerable debate about whether low cholesterol predisposes patients to suicide or if depression independently leads to poor nutrition and therefore low cholesterol and increased suicide risk.6,22
Some researchers have suggested that depression lowers cholesterol and increases risk of suicide,23 but study designs have limited the ability to discern the directionality of the relationship. Attempts to control for depression-related malnutrition and weight loss—which lowers total cholesterol, low-density lipoprotein cholesterol (LDL-C), and high-density lipoprotein cholesterol (HDL-C)24—suggest the association may be independent of these variables.25-27 These findings suggest that cholesterol may be considered a trait marker and is not entirely state-dependent. However, multiple, large, long-term randomized controlled trials have not shown increased depression and suicide with use of lipid-lowering agents in healthy populations.20
The Figure illustrates known epidemiologic associations of low cholesterol, low serotoninergic function, and suicide and contrasts conceptual models of cholesterol as a state and a trait marker. A case can be made for cholesterol as both a state and a trait marker, and these models could overlap, with depression-induced decreases in cholesterol further mediating changes in serotonergic function and related behavioral sequelae.
Figure
Cholesterol, depression, and suicide: How are they linked?
Low cholesterol may be considered a trait marker, predisposing patients to lower serotonergic function and placing them at greater risk for impulsivity, depression, aggression, and suicide. Other models suggest that lower cholesterol is a state-dependent consequence of depression, and not part of a causal chain toward suicide
Improving cardiac health
Limited epidemiologic studies suggest that patients with mood disorders may have lower levels of total cholesterol and LDL-C, but higher rates of hypertriglyceridemia compared with the general population.8 Unfortunately, psychiatric patients—who may be at increased risk of developing cardiovascular disease—may be less likely to be screened and appropriately treated for lipid abnormalities.28 To address this disparity, consider assuming an active role in assessing and managing hyperlipidemia in your patients with mood disorders. Be aware of your patients’ lipid profile and ensure that they follow monitoring recommendations.
The National Cholesterol Education Program recommends screening all adults age >20 for hyperlipidemia every 5 years using measures of total cholesterol, LDL-C, HDL-C, and triglycerides. If LDL-C or triglycerides exceed target values (Table 2), appropriate management includes recommending lifestyle changes and pharmacotherapy (Box 2).
Patients should receive a fasting lipid profile before and 12 weeks after starting any antipsychotic and semiannually thereafter.29 Consider closely monitoring lipids when patients gain weight with psychotropics. Refer patients with hyperlipidemia to a primary care physician, but in the absence of such a provider, mental health clinicians who are familiar with treatment guidelines can manage these patients.30
Closely monitor individuals with mood disorders for changes in behavior or mental status after starting a lipid-lowering agent. Consider discontinuing the drug if a patient develops an adverse reaction. If symptoms return after medication rechallenge, consider other management strategies such as an alternate lipid-lowering agent or re-emphasizing behavioral measures.
Table 2
National Cholesterol Education Program recommended LDL levels
| Risk category* | LDL goal | When to consider medications |
|---|---|---|
| CHD or CHD equivalent | <100 mg/dL | ≥130 mg/dL |
| ≥2 major risk factors | <130 mg/dL | ≥130 to 160 mg/dL (based on 10-year risk) |
| 0 or 1 risk factor | <160 mg/dL | ≥190 mg/dL |
| CHD: coronary heart disease; HDL: high-density lipoprotein; LDL: low-density lipoprotein | ||
| *Risk category is based on the presence of CHD or equivalent and major risk factors for CHD. CHD equivalents include symptomatic carotid artery disease, peripheral artery disease, and abdominal aortic aneurysm. Major risk factors include smoking, hypertension, low HDL, family history, and age. LDL levels to consider medications for those with ≥2 major risk factors vary by 10-year CHD risk | ||
| Source: National Cholesterol Education Program, Adult Treatment Panel III (ATP III) Quick Desk Reference. www.nhlbi.nih.gov/guidelines/cholesterol/atglance.htm | ||
National Cholesterol Education Program guidelines state that when a patient’s low-density lipoprotein cholesterol (LDL-C) exceeds targets (Table 2), first recommend lifestyle changes such as a diet low in saturated fat (<7% of calories) and cholesterol (<200 mg/d), weight management, and exercise. Increases in soluble fiber (10 to 25 g/d) and plant stanols/sterols also may be considered. If LDL-C levels are still too high, pharmacologic therapy such as an HMGCoA reductase inhibitor is suggested.
Treatment of elevated triglycerides (≥150 mg/dL) includes reaching the target LDL-C, intensifying a weight management program, and increasing exercise. Address quitting smoking and limiting alcohol when indicated. If triglyceride levels are ≥200 mg/dL after the LDL-C target is reached, set a secondary goal of reaching a target non-high-density lipoprotein cholesterol (HDL-C) (non-HDL-C; total cholesterol minus HDL-C) 30 mg/dL greater than the LDL goal. This can be achieved by adding an LDL-lowering drug such as a statin, nicotinic acid, or ezetimibe. When triglycerides are ≥500 mg/dL, more aggressive intervention, such as with a fibrate, omega-3 fatty acids, very low-fat diets, and exercise, is required to prevent pancreatitis.
Source: National Heart Lung and Blood Institute. National Cholesterol Education Program. www.nhlbi.nih.gov/guidelines/cholesterol/index.htm
Related Resources
- Fiedorowicz JG, Coryell WH. Cholesterol and suicide attempts: a prospective study of depressed inpatients. Psychiatry Res. 2007;152(1):11-20.
- National Cholesterol Education Program, Adult Treatment Panel III (ATP III) Quick Desk Reference. www.nhlbi.nih.gov/guidelines/cholesterol/atglance.htm.
- Executive Summary of the third report of the national Cholesterol Education Program (nCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA. 2001;285(19):2486-2497.
Drug Brand Names
- Ezetimibe • Zetia
- Pravastatin • Pravachol
- Simvastatin • Zocor
Acknowledgements
Dr. Fiedorowicz thanks Lois Warren and Miriam Weiner for their editorial assistance.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products. Dr. Fiedorowicz is supported by the national Institutes of Health (1K23MH083695-01A210), nARSAD, and the Institute for Clinical and Translational Science at the University of Iowa (3 UL1 RR024979-03S4). He has received support for participating in a colleague’s investigator-initiated project with Eli Lilly. Dr. Haynes’ research is supported by grants from the national Institutes of Health (nHLBI: HL58972 & HL14388; nCRR CTSA: 1UL1RR024979).
1. Osby U, Brandt L, Correia N, et al. Excess mortality in bipolar and unipolar disorder in Sweden. Arch Gen Psychiatry. 2001;58(9):844-850.
2. Lindberg G, Råstam L, Gullberg B, et al. Low serum cholesterol concentration and short term mortality from injuries in men and women. BMJ. 1992;305(6848):277-279.
3. Muldoon MF, Manuck SB, Matthews KA. Lowering cholesterol concentrations and mortality: a quantitative review of primary prevention trials. BMJ. 1990;301(6747):309-314.
4. Neaton JD, Blackburn H, Jacobs D, et al. Serum cholesterol level and mortality findings for men screened in the Multiple Risk Factor Intervention Trial. Multiple Risk Factor Intervention Trial Research Group. Arch Intern Med. 1992;152(7):1490-1500.
5. Fiedorowicz JG, Coryell WH. Cholesterol and suicide attempts: a prospective study of depressed inpatients. Psychiatry Res. 2007;152(1):11-20.
6. Golomb BA. Cholesterol and violence: is there a connection? Ann Intern Med. 1998;128(6):478-487.
7. Pae CU, Kim JJ, Lee SJ, et al. Aberration of cholesterol level in first-onset bipolar I patients. J Affect Disord. 2004;83(1):79-82.
8. Fiedorowicz JG, Palagummi NM, Forman-Hoffman VL, et al. Elevated prevalence of obesity, metabolic syndrome, and cardiovascular risk factors in bipolar disorder. Ann Clin Psychiatry. 2008;20(3):131-137.
9. Chung KH, Tsai SY, Lee HC. Mood symptoms and serum lipids in acute phase of bipolar disorder in Taiwan. Psychiatry Clin Neurosci. 2007;61(4):428-433.
10. Jow GM, Yang TT, Chen CL. Leptin and cholesterol levels are low in major depressive disorder, but high in schizophrenia. J Affect Disord. 2006;90(1):21-27.
11. Sagud M, Mihaljevic-Peles A, Pivac N, et al. Platelet serotonin and serum lipids in psychotic mania. J Affect Disord. 2007;97(1-3):247-251.
12. Beasley CL, Honer WG, Bergmann K, et al. Reductions in cholesterol and synaptic markers in association cortex in mood disorders. Bipolar Disord. 2005;7(5):449-455.
13. Gabriel A. Changes in plasma cholesterol in mood disorder patients: does treatment make a difference? J Affect Disord. 2007;99(1-3):273-278.
14. Lalovic A, Levy E, Luheshi G, et al. Cholesterol content in brains of suicide completers. Int J Neuropsychopharmacol. 2007;10(2):159-166.
15. Lester D. Serum cholesterol levels and suicide: a meta-analysis. Suicide Life Threat Behav. 2002;32(3):333-346.
16. Coryell W, Schlesser M. Combined biological tests for suicide prediction. Psychiatry Res. 2007;150(2):187-191.
17. Tatley M, Savage R. Psychiatric adverse reactions with statins, fibrates and ezetimibe: implications for the use of lipid-lowering agents. Drug Saf. 2007;30(3):195-201.
18. Callréus T, Agerskov Andersen U, Hallas J, et al. Cardiovascular drugs and the risk of suicide: a nested case-control study. Eur J Clin Pharmacol. 2007;63(6):591-596.
19. Yang CC, Jick SS, Jick H. Lipid-lowering drugs and the risk of depression and suicidal behavior. Arch Intern Med. 2003;163(16):1926-1932.
20. Stewart RA, Sharples KJ, North FM, et al. Long-term assessment of psychological well-being in a randomized placebo-controlled trial of cholesterol reduction with pravastatin. The LIPID Study Investigators. Arch Intern Med. 2000;160(20):3144-3152.
21. Golomb BA, Criqui MH, White HL, et al. The UCSD Statin Study: a randomized controlled trial assessing the impact of statins on selected noncardiac outcomes. Control Clin Trials. 2004;25(2):178-202.
22. Fawcett J, Busch KA, Jacobs D, et al. Suicide: a four-pathway clinical-biochemical model. Annals N Y Acad Sci. 1997;836:288-301.
23. Law MR, Thompson SG, Wald NJ. Assessing possible hazards of reducing serum cholesterol. BMJ. 1994;308(6925):373-379.
24. Dattilo AM, Kris-Etherton PM. Effects of weight reduction on blood lipids and lipoproteins: a meta-analysis. Am J Clin Nutr. 1992;56(2):320-328.
25. Garland M, Hickey D, Corvin A, et al. Total serum cholesterol in relation to psychological correlates in parasuicide. Br J Psychiatry. 2000;177:77-83.
26. Golier JA, Marzuk PM, Leon AC, et al. Low serum cholesterol level and attempted suicide. Am J Psychiatry. 1995;152(3):419-423.
27. Kunugi H, Takei N, Aoki H, et al. Low serum cholesterol in suicide attempters. Biol Psychiatry. 1997;41(2):196-200.
28. Murray DP, Weiner M, Prabhakar M, et al. Mania and mortality: why the excess cardiovascular risk in bipolar disorder? Curr Psychiatry Rep. 2009;11(6):475-480.
29. Sernyak MJ. Implementation of monitoring and management guidelines for second-generation antipsychotics. J Clin Psychiatry. 2007;68(suppl 4):14-18.
30. Marder SR, Essock SM, Miller AL, et al. Physical health monitoring of patients with schizophrenia. Am J Psychiatry. 2004;161(8):1334-1349.
1. Osby U, Brandt L, Correia N, et al. Excess mortality in bipolar and unipolar disorder in Sweden. Arch Gen Psychiatry. 2001;58(9):844-850.
2. Lindberg G, Råstam L, Gullberg B, et al. Low serum cholesterol concentration and short term mortality from injuries in men and women. BMJ. 1992;305(6848):277-279.
3. Muldoon MF, Manuck SB, Matthews KA. Lowering cholesterol concentrations and mortality: a quantitative review of primary prevention trials. BMJ. 1990;301(6747):309-314.
4. Neaton JD, Blackburn H, Jacobs D, et al. Serum cholesterol level and mortality findings for men screened in the Multiple Risk Factor Intervention Trial. Multiple Risk Factor Intervention Trial Research Group. Arch Intern Med. 1992;152(7):1490-1500.
5. Fiedorowicz JG, Coryell WH. Cholesterol and suicide attempts: a prospective study of depressed inpatients. Psychiatry Res. 2007;152(1):11-20.
6. Golomb BA. Cholesterol and violence: is there a connection? Ann Intern Med. 1998;128(6):478-487.
7. Pae CU, Kim JJ, Lee SJ, et al. Aberration of cholesterol level in first-onset bipolar I patients. J Affect Disord. 2004;83(1):79-82.
8. Fiedorowicz JG, Palagummi NM, Forman-Hoffman VL, et al. Elevated prevalence of obesity, metabolic syndrome, and cardiovascular risk factors in bipolar disorder. Ann Clin Psychiatry. 2008;20(3):131-137.
9. Chung KH, Tsai SY, Lee HC. Mood symptoms and serum lipids in acute phase of bipolar disorder in Taiwan. Psychiatry Clin Neurosci. 2007;61(4):428-433.
10. Jow GM, Yang TT, Chen CL. Leptin and cholesterol levels are low in major depressive disorder, but high in schizophrenia. J Affect Disord. 2006;90(1):21-27.
11. Sagud M, Mihaljevic-Peles A, Pivac N, et al. Platelet serotonin and serum lipids in psychotic mania. J Affect Disord. 2007;97(1-3):247-251.
12. Beasley CL, Honer WG, Bergmann K, et al. Reductions in cholesterol and synaptic markers in association cortex in mood disorders. Bipolar Disord. 2005;7(5):449-455.
13. Gabriel A. Changes in plasma cholesterol in mood disorder patients: does treatment make a difference? J Affect Disord. 2007;99(1-3):273-278.
14. Lalovic A, Levy E, Luheshi G, et al. Cholesterol content in brains of suicide completers. Int J Neuropsychopharmacol. 2007;10(2):159-166.
15. Lester D. Serum cholesterol levels and suicide: a meta-analysis. Suicide Life Threat Behav. 2002;32(3):333-346.
16. Coryell W, Schlesser M. Combined biological tests for suicide prediction. Psychiatry Res. 2007;150(2):187-191.
17. Tatley M, Savage R. Psychiatric adverse reactions with statins, fibrates and ezetimibe: implications for the use of lipid-lowering agents. Drug Saf. 2007;30(3):195-201.
18. Callréus T, Agerskov Andersen U, Hallas J, et al. Cardiovascular drugs and the risk of suicide: a nested case-control study. Eur J Clin Pharmacol. 2007;63(6):591-596.
19. Yang CC, Jick SS, Jick H. Lipid-lowering drugs and the risk of depression and suicidal behavior. Arch Intern Med. 2003;163(16):1926-1932.
20. Stewart RA, Sharples KJ, North FM, et al. Long-term assessment of psychological well-being in a randomized placebo-controlled trial of cholesterol reduction with pravastatin. The LIPID Study Investigators. Arch Intern Med. 2000;160(20):3144-3152.
21. Golomb BA, Criqui MH, White HL, et al. The UCSD Statin Study: a randomized controlled trial assessing the impact of statins on selected noncardiac outcomes. Control Clin Trials. 2004;25(2):178-202.
22. Fawcett J, Busch KA, Jacobs D, et al. Suicide: a four-pathway clinical-biochemical model. Annals N Y Acad Sci. 1997;836:288-301.
23. Law MR, Thompson SG, Wald NJ. Assessing possible hazards of reducing serum cholesterol. BMJ. 1994;308(6925):373-379.
24. Dattilo AM, Kris-Etherton PM. Effects of weight reduction on blood lipids and lipoproteins: a meta-analysis. Am J Clin Nutr. 1992;56(2):320-328.
25. Garland M, Hickey D, Corvin A, et al. Total serum cholesterol in relation to psychological correlates in parasuicide. Br J Psychiatry. 2000;177:77-83.
26. Golier JA, Marzuk PM, Leon AC, et al. Low serum cholesterol level and attempted suicide. Am J Psychiatry. 1995;152(3):419-423.
27. Kunugi H, Takei N, Aoki H, et al. Low serum cholesterol in suicide attempters. Biol Psychiatry. 1997;41(2):196-200.
28. Murray DP, Weiner M, Prabhakar M, et al. Mania and mortality: why the excess cardiovascular risk in bipolar disorder? Curr Psychiatry Rep. 2009;11(6):475-480.
29. Sernyak MJ. Implementation of monitoring and management guidelines for second-generation antipsychotics. J Clin Psychiatry. 2007;68(suppl 4):14-18.
30. Marder SR, Essock SM, Miller AL, et al. Physical health monitoring of patients with schizophrenia. Am J Psychiatry. 2004;161(8):1334-1349.
Treating insomnia across women’s life stages
Discuss this article at http://currentpsychiatry.blogspot.com/2010/07/treating-insomnia-in-women.html#comments
Ms. A, age 44, reports a 3-month history of forgetfulness, difficulty concentrating, and insomnia. She says she can fall asleep but wakes up multiple times during the night and feels tired during the day. She has no history of a mood or anxiety disorder or medications that might be responsible for her symptoms.
Before her current insomnia began, Ms. A could sleep for 7 to 8 hours at night. Her husband suffers from obstructive sleep apnea (OSA), and his snoring occasionally would awaken her, but she slept well overall. Ms. A cannot identify anything that could be causing her sleep complaints. She states “The weird thing is that sometimes I am not sure if I’m cold or hot” and “I sometimes wake up drenched in sweat.” She also reports recent changes in the timing of her otherwise regular menstrual flow.
Ms. A attributes her memory problems to her poor sleep. A recent audit at her company held her responsible for several accounting errors, and Ms. A is worried that she might lose her job. She denies symptoms that would suggest major depression. You are unable to elicit a history of limb movements or excessive snoring.
Compared with men, women have a 1.3- to 1.8-fold greater risk for developing insomnia.Improve sleep with group CBT for insomnia,” Current Psychiatry, April 2009.) Pharmacotherapy during pregnancy and for breast-feeding mothers is guided by evaluating the risk/benefit ratio and safety considerations.
Maintain a high index of suspicion for breathing-related sleep disorders, such as OSA,21 and RLS.22 Atypical presentations of OSA are common in pregnant or postpartum women; compared with men, women with OSA are more likely to report fatigue and less likely than to report sleepiness. Refer patients whom you think may have OSA for polysomnography.
If you suspect RLS, check for low ferritin and folate levels. Nutritional supplements may be necessary for women in high-risk groups, including those who are pregnant or have varicose veins, venous reflux, folate deficiency, uremia, diabetes, thyroid problems, peripheral neuropathy, Parkinson’s disease, or certain autoimmune disorders, such as Sjögren’s syndrome, celiac disease, and rheumatoid arthritis.23 Advise these patients to avoid caffeine.
Although indicated for treating RLS, ropinirole and pramipexole are FDA Pregnancy Category C, which means animal studies have shown adverse effects on the fetus and there are no adequate and well-controlled studies in humans, but potential benefits may warrant use of the drug in pregnant women despite risks. Opioids, carbamazepine, or gabapentin may be safer for pregnant patients.24
Insomnia during menopause
The prevalence of insomnia increases from 33% to 36% in premenopausal women to 44% to 61% in postmenopausal women.14 Hot flashes, comorbid mood disturbances, sleep-disordered breathing, and RLS contribute to increased insomnia risk in postmenopausal women (Table 3).4,14,25,26
Treatment strategy. Always inquire about sleep in perimenopausal/postmenopausal women, even when her presenting complaint is related to menstrual cycle changes or vasomotor symptoms such as hot flashes.16 Assess patients for OSA, RLS, and mood, anxiety, and cognitive symptoms.26 In addition to pharmacotherapy and behavioral therapy, treatment options include hormone replacement therapy (HRT) and herbal and dietary supplements (Table 4).27-32
Table 3
Sleep difficulties during menopause: Differential diagnoses
| Condition | Features | Findings | Other considerations |
|---|---|---|---|
| Hot flashes (prevalence: 75% to 85%)14 | Vasomotor phenomenon characterized by feelings such as ‘spreading warmth,’ diaphoresis, palpitations, nausea, and insomnia Mediated through the preoptic area of the anterior hypothalamus, which regulates temperature and sleep Increased brain norepinephrine metabolism | Discrepancies between objective (PSG) and subjective measures (surveys)4 Discrepancies between self-reported and laboratory reported sleep data might be explained by thermoregulatory differences between NREM and REM sleep24 | Nocturnal hot flashes trigger awakenings and insomnia14 Hot flashes can follow arousals and awakenings HRT is highly effective in treating hot flashes; however, data on its direct effects on sleep complaints are inconsistent |
| Primary menopausal insomnia25 | Menopausal symptoms (eg, hot flashes) trigger insomnia that persists secondary to behavioral conditioning | Increase in nocturnal skin temperature coincides with decrease in skin resistance and waking episodes in PSG | Behavioral insomnia therapies are useful adjuncts to treatment of menopause symptoms |
| Sleep-disordered breathing (OSA) | Menopause increases risk for OSA independent of body weight Redistribution of body fat with an increase in the waist-to-hip circumference ratio occurs in menopause Loss of ventilatory drive because of diminished progesterone levels | Sleep fragmentation and daytime sleepiness are common, as opposed to apneic episodes or oxygen desaturation in men | Maintain a high index of suspicion and promptly refer patients to a sleep center |
| Restless legs syndrome | Related to iron deficiency | Low ferritin and folate levels | Advise patients to avoid caffeine |
| HRT: hormone replacement therapy; NREM: non-rapid eye movement; OSA: obstructive sleep apnea; PSG: polysomnography; REM: rapid eye movement | |||
Table 4
Treating insomnia in menopausal women
| Therapy | Comments |
|---|---|
| Hormone replacement therapy (HRT) | Effective for hot flashes, insomnia,26-28 and sleep apnea29 Long-term safety is questionable4 |
| Behavioral therapy (cognitive-behavioral therapy,30 stimulus control therapy, sleep restriction therapy, sleep hygiene, hypnotherapy, biofeedback) | Limited data in menopausal women |
| Sedatives/hypnotics/antidepressants (eg, zolpidem, 10 mg; eszopiclone, 3 mg; trazodone, 75 mg; ramelteon, 8 mg; SSRIs and SNRIs) | Benzodiazepines may be useful, although not specifically evaluated in menopausal women. Risk of tolerance, dependence, and psychomotor slowing |
| Herbal and dietary supplements (Cimicifuga racemosa [Black cohosh],31 valerian | Popular alternatives to HRT; however, evidence of efficacy as treatment for insomnia is inconclusive |
| SNRIs: serotonin-norepinephrine reuptake inhibitors; SSRIs: selective serotonin reuptake inhibitors | |
Comorbid psychiatric disorders
Women have a higher prevalence of psychiatric disorders such as major depressive disorder and anxiety disorders than men.1 Women have a 10% to 25% lifetime risk of developing major depression. Three quarters of depressed patients experience insomnia.1 Recent literature suggests insomnia is a risk factor for depression,33 which emphasizes the need to screen women who present with sleep problems for depression and anxiety.
Five percent to 20% of women experience postpartum depression. Depression and insomnia are correlated to the rapid decline in estrogen and progesterone after delivery.34
Treatment strategy. Insomnia is a common presenting symptom in patients with psychiatric conditions such as mood and anxiety disorders. Treating the underlying psychiatric disorder often alleviates sleeping difficulties. However, if the insomnia is disabling, treat the psychiatric disorder and insomnia concurrently.
CASE CONTINUED: Perimenopausal insomnia
Based on her history, you diagnose Ms. A with insomnia related to general medical condition (perimenopause). There are no indications to refer her for polysomnography. You educate Ms. A about sleep hygiene and recommend that she discuss her menstrual and physical complaints with her primary care physician or gynecologist. Ms. A is not interested in HRT because she has a strong family history of endometrial cancer. You reassure Ms. A and schedule a follow-up visit in 2 months to re-evaluate her insomnia.
Related resource
- Krahn LE. Perimenopausal depression? Ask how she’s sleeping. Current Psychiatry. 2005;4(6):39-53.
Drug brand names
- Carbamazepine • Carbatrol, Tegretol, others
- Escitalopram • Lexapro
- Eszopiclone • Lunesta
- Fluoxetine • Prozac
- Gabapentin • Neurontin, Gabarone
- Paroxetine • Paxil
- Pramipexole • Mirapex
- Ramelteon • Rozerem
- Ropinirole • Requip
- Sertraline • Zoloft
- Trazodone • Desyrel
- Zolpidem • Ambien
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgements
The authors thank Dr. Namita Dhiman and Darrel E. Willoughby for their assistance with this article.
1. Krystal AD. Depression and insomnia in women. Clin Cornerstone. 2004;6(suppl 1B):S19-S28.
2. Ohayon MM. Epidemiology of insomnia: what we know and what we still need to learn. Sleep Med Rev. 2002;6(2):97-111.
3. Krishnan V, Collop NA. Gender differences in sleep disorders. Curr Opin Pulm Med. 2006;12(6):383-389.
4. Soares CN, Murray BJ. Sleep disorders in women: clinical evidence and treatment strategies. Psychiatr Clin North Am. 2006;29(4):1095-1113.
5. Ohayon M. Epidemiological study on insomnia in the general population. Sleep. 1996;19(3 suppl):S7-S15.
6. Daley M, Morin CM, LeBlanc M, et al. Insomnia and its relationship to health-care utilization, work absenteeism, productivity and accidents. Sleep Med. 2009;10(4):427-438.
7. Diagnostic and statistical manual of mental disorders, 4th ed, text revision. Washington, DC: American Psychiatric Association; 2000.
8. Ancoli-Israel S, Roth T. Characteristics of insomnia in the United States: results of the 1991 National Sleep Foundation Survey. I. Sleep. 1999;2(suppl 2):S347-S353.
9. Zhang B, Wing YK. Sex differences in insomnia: a meta-analysis. Sleep. 2006;29(1):85-93.
10. Buysse DJ, Reynolds CF, Monk TH, et al. The Pittsburgh Sleep Quality Index (PSQI): a new instrument for psychiatric research and practice. Psychiatry Res. 1989;28(2):193-213.
11. Manber R, Bootzin RR. Sleep and the menstrual cycle. Health Psychol. 1997;16:209-214.
12. Ito M, Kohsaka M, Fukuda N, et al. Effects of menstrual cycle on plasma melatonin level and sleep characteristics. Jpn J Psychiatry Neurol. 1993;47:478-479.
13. Driver HS, Dijk DJ, Werth E, et al. Sleep and the sleep electroencephalogram across the menstrual cycle in young healthy women. J Clin Endocrinol Metab. 1996;81:728-735.
14. Moline ML, Broch L, Zak R. Sleep in women across the life cycle from adulthood through menopause. Med Clin North Am. 2004;88(3):705-736.
15. Steiner M, Pearlstein T, Cohen LS, et al. Expert guidelines for the treatment of severe PMS, PMDD, and comorbidities: the role of SSRIs. J Womens Health (Larchmt). 2006;15(1):57-69.
16. Krystal AD. Insomnia in women. Clin Cornerstone. 2003;5(3):41-50.
17. Mindell JA, Jacobson BJ. Sleep disturbances during pregnancy. J Obstet Gynecol Neonatal Nurs. 2000;29(6):590-597.
18. Lee KA, Zaffke ME, McEnany G. Parity and sleep patterns during and after pregnancy. Obstet Gynecol. 2000;95(1):14-18.
19. Brunner DP, Münch M, Biedermann K, et al. Changes in sleep and sleep electroencephalogram during pregnancy. Sleep. 1994;17(7):576-582.
20. Ross LE, Murray BJ, Steiner M. Sleep and perinatal mood disorders: a critical review. J Psychiatry Neurosci. 2005;30(4):247-256.
21. Edwards N, Middleton PG, Blyton DM, et al. Sleep disordered breathing and pregnancy. Thorax. 2002;57(6):555-558.
22. Manconi M, Govoni V, De Vito A, et al. Restless legs syndrome and pregnancy. Neurology. 2004;63(6):1065-1069.
23. Lee KA, Zaffke ME, Baratte-Beebe K. Restless legs syndrome and sleep disturbance during pregnancy: the role of folate and iron. J Womens Health Gend Based Med. 2001;10(4):335-341.
24. Djokanovic N, Garcia-Bournissen F, Koren G. Medications for restless legs syndrome in pregnancy. J Obstet Gynaecol Can. 2008;30(6):505-507.
25. Freedman RR, Roehrs TA. Effects of REM sleep and ambient temperature on hot flash-induced sleep disturbance. Menopause. 2006;13(4):576-583.
26. Krystal AD, Edinger J, Wohlgemuth W, et al. Sleep in perimenopausal and postmenopausal women. Sleep Med Rev. 1998;2(4):243-253.
27. Polo-Kantola P, Erkkola R, Irjala K, et al. Effect of short-term transdermal estrogen replacement therapy on sleep: a randomized, double-blind crossover trial in postmenopausal women. Fertil Steril. 1999;71(5):873-880.
28. Watts NB, Notelovitz M, Timmons MC, et al. Comparison of oral estrogens and estrogens plus androgen on bone mineral density, menopausal symptoms, and lipid-lipoprotein profiles in surgical menopause. Obstet Gynecol. 1995;85(4):529-537.Erratum in: Obstet Gynecol 1995;85(5 Pt 1):668.
29. Boyle GJ, Murrihy R. A preliminary study of hormone replacement therapy and psychological mood states in perimenopausal women. Psychol Rep. 2001;88(1):160-170.
30. Cistulli PA, Barnes DJ, Grunstein RR, et al. Effect of short-term hormone replacement in the treatment of obstructive sleep apnoea in postmenopausal women. Thorax. 1994;49:699-702.
31. Yang CM, Spielman AJ, Glovinsky P. Nonpharmacologic strategies in the management of insomnia. Psychiatr Clin North Am. 2006;29(4):895-919.
32. Mahady GB. Black cohosh (Actaea/Cimicifuga racemosa): review of the clinical data for safety and efficacy in menopausal symptoms. Treat Endocrinol. 2005;4(3):177-184.
33. Breslau N, Roth T, Rosenthal L, et al. Sleep disturbance and psychiatric disorders: a longitudinal epidemiological study of young adults. Biol Psychiatry. 1996;39:411-418.
34. Burt VK, Stein K. Epidemiology of depression throughout the female life cycle. J Clin Psychiatry. 2002;63(suppl 7):9-15.
Discuss this article at http://currentpsychiatry.blogspot.com/2010/07/treating-insomnia-in-women.html#comments
Ms. A, age 44, reports a 3-month history of forgetfulness, difficulty concentrating, and insomnia. She says she can fall asleep but wakes up multiple times during the night and feels tired during the day. She has no history of a mood or anxiety disorder or medications that might be responsible for her symptoms.
Before her current insomnia began, Ms. A could sleep for 7 to 8 hours at night. Her husband suffers from obstructive sleep apnea (OSA), and his snoring occasionally would awaken her, but she slept well overall. Ms. A cannot identify anything that could be causing her sleep complaints. She states “The weird thing is that sometimes I am not sure if I’m cold or hot” and “I sometimes wake up drenched in sweat.” She also reports recent changes in the timing of her otherwise regular menstrual flow.
Ms. A attributes her memory problems to her poor sleep. A recent audit at her company held her responsible for several accounting errors, and Ms. A is worried that she might lose her job. She denies symptoms that would suggest major depression. You are unable to elicit a history of limb movements or excessive snoring.
Compared with men, women have a 1.3- to 1.8-fold greater risk for developing insomnia.Improve sleep with group CBT for insomnia,” Current Psychiatry, April 2009.) Pharmacotherapy during pregnancy and for breast-feeding mothers is guided by evaluating the risk/benefit ratio and safety considerations.
Maintain a high index of suspicion for breathing-related sleep disorders, such as OSA,21 and RLS.22 Atypical presentations of OSA are common in pregnant or postpartum women; compared with men, women with OSA are more likely to report fatigue and less likely than to report sleepiness. Refer patients whom you think may have OSA for polysomnography.
If you suspect RLS, check for low ferritin and folate levels. Nutritional supplements may be necessary for women in high-risk groups, including those who are pregnant or have varicose veins, venous reflux, folate deficiency, uremia, diabetes, thyroid problems, peripheral neuropathy, Parkinson’s disease, or certain autoimmune disorders, such as Sjögren’s syndrome, celiac disease, and rheumatoid arthritis.23 Advise these patients to avoid caffeine.
Although indicated for treating RLS, ropinirole and pramipexole are FDA Pregnancy Category C, which means animal studies have shown adverse effects on the fetus and there are no adequate and well-controlled studies in humans, but potential benefits may warrant use of the drug in pregnant women despite risks. Opioids, carbamazepine, or gabapentin may be safer for pregnant patients.24
Insomnia during menopause
The prevalence of insomnia increases from 33% to 36% in premenopausal women to 44% to 61% in postmenopausal women.14 Hot flashes, comorbid mood disturbances, sleep-disordered breathing, and RLS contribute to increased insomnia risk in postmenopausal women (Table 3).4,14,25,26
Treatment strategy. Always inquire about sleep in perimenopausal/postmenopausal women, even when her presenting complaint is related to menstrual cycle changes or vasomotor symptoms such as hot flashes.16 Assess patients for OSA, RLS, and mood, anxiety, and cognitive symptoms.26 In addition to pharmacotherapy and behavioral therapy, treatment options include hormone replacement therapy (HRT) and herbal and dietary supplements (Table 4).27-32
Table 3
Sleep difficulties during menopause: Differential diagnoses
| Condition | Features | Findings | Other considerations |
|---|---|---|---|
| Hot flashes (prevalence: 75% to 85%)14 | Vasomotor phenomenon characterized by feelings such as ‘spreading warmth,’ diaphoresis, palpitations, nausea, and insomnia Mediated through the preoptic area of the anterior hypothalamus, which regulates temperature and sleep Increased brain norepinephrine metabolism | Discrepancies between objective (PSG) and subjective measures (surveys)4 Discrepancies between self-reported and laboratory reported sleep data might be explained by thermoregulatory differences between NREM and REM sleep24 | Nocturnal hot flashes trigger awakenings and insomnia14 Hot flashes can follow arousals and awakenings HRT is highly effective in treating hot flashes; however, data on its direct effects on sleep complaints are inconsistent |
| Primary menopausal insomnia25 | Menopausal symptoms (eg, hot flashes) trigger insomnia that persists secondary to behavioral conditioning | Increase in nocturnal skin temperature coincides with decrease in skin resistance and waking episodes in PSG | Behavioral insomnia therapies are useful adjuncts to treatment of menopause symptoms |
| Sleep-disordered breathing (OSA) | Menopause increases risk for OSA independent of body weight Redistribution of body fat with an increase in the waist-to-hip circumference ratio occurs in menopause Loss of ventilatory drive because of diminished progesterone levels | Sleep fragmentation and daytime sleepiness are common, as opposed to apneic episodes or oxygen desaturation in men | Maintain a high index of suspicion and promptly refer patients to a sleep center |
| Restless legs syndrome | Related to iron deficiency | Low ferritin and folate levels | Advise patients to avoid caffeine |
| HRT: hormone replacement therapy; NREM: non-rapid eye movement; OSA: obstructive sleep apnea; PSG: polysomnography; REM: rapid eye movement | |||
Table 4
Treating insomnia in menopausal women
| Therapy | Comments |
|---|---|
| Hormone replacement therapy (HRT) | Effective for hot flashes, insomnia,26-28 and sleep apnea29 Long-term safety is questionable4 |
| Behavioral therapy (cognitive-behavioral therapy,30 stimulus control therapy, sleep restriction therapy, sleep hygiene, hypnotherapy, biofeedback) | Limited data in menopausal women |
| Sedatives/hypnotics/antidepressants (eg, zolpidem, 10 mg; eszopiclone, 3 mg; trazodone, 75 mg; ramelteon, 8 mg; SSRIs and SNRIs) | Benzodiazepines may be useful, although not specifically evaluated in menopausal women. Risk of tolerance, dependence, and psychomotor slowing |
| Herbal and dietary supplements (Cimicifuga racemosa [Black cohosh],31 valerian | Popular alternatives to HRT; however, evidence of efficacy as treatment for insomnia is inconclusive |
| SNRIs: serotonin-norepinephrine reuptake inhibitors; SSRIs: selective serotonin reuptake inhibitors | |
Comorbid psychiatric disorders
Women have a higher prevalence of psychiatric disorders such as major depressive disorder and anxiety disorders than men.1 Women have a 10% to 25% lifetime risk of developing major depression. Three quarters of depressed patients experience insomnia.1 Recent literature suggests insomnia is a risk factor for depression,33 which emphasizes the need to screen women who present with sleep problems for depression and anxiety.
Five percent to 20% of women experience postpartum depression. Depression and insomnia are correlated to the rapid decline in estrogen and progesterone after delivery.34
Treatment strategy. Insomnia is a common presenting symptom in patients with psychiatric conditions such as mood and anxiety disorders. Treating the underlying psychiatric disorder often alleviates sleeping difficulties. However, if the insomnia is disabling, treat the psychiatric disorder and insomnia concurrently.
CASE CONTINUED: Perimenopausal insomnia
Based on her history, you diagnose Ms. A with insomnia related to general medical condition (perimenopause). There are no indications to refer her for polysomnography. You educate Ms. A about sleep hygiene and recommend that she discuss her menstrual and physical complaints with her primary care physician or gynecologist. Ms. A is not interested in HRT because she has a strong family history of endometrial cancer. You reassure Ms. A and schedule a follow-up visit in 2 months to re-evaluate her insomnia.
Related resource
- Krahn LE. Perimenopausal depression? Ask how she’s sleeping. Current Psychiatry. 2005;4(6):39-53.
Drug brand names
- Carbamazepine • Carbatrol, Tegretol, others
- Escitalopram • Lexapro
- Eszopiclone • Lunesta
- Fluoxetine • Prozac
- Gabapentin • Neurontin, Gabarone
- Paroxetine • Paxil
- Pramipexole • Mirapex
- Ramelteon • Rozerem
- Ropinirole • Requip
- Sertraline • Zoloft
- Trazodone • Desyrel
- Zolpidem • Ambien
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgements
The authors thank Dr. Namita Dhiman and Darrel E. Willoughby for their assistance with this article.
Discuss this article at http://currentpsychiatry.blogspot.com/2010/07/treating-insomnia-in-women.html#comments
Ms. A, age 44, reports a 3-month history of forgetfulness, difficulty concentrating, and insomnia. She says she can fall asleep but wakes up multiple times during the night and feels tired during the day. She has no history of a mood or anxiety disorder or medications that might be responsible for her symptoms.
Before her current insomnia began, Ms. A could sleep for 7 to 8 hours at night. Her husband suffers from obstructive sleep apnea (OSA), and his snoring occasionally would awaken her, but she slept well overall. Ms. A cannot identify anything that could be causing her sleep complaints. She states “The weird thing is that sometimes I am not sure if I’m cold or hot” and “I sometimes wake up drenched in sweat.” She also reports recent changes in the timing of her otherwise regular menstrual flow.
Ms. A attributes her memory problems to her poor sleep. A recent audit at her company held her responsible for several accounting errors, and Ms. A is worried that she might lose her job. She denies symptoms that would suggest major depression. You are unable to elicit a history of limb movements or excessive snoring.
Compared with men, women have a 1.3- to 1.8-fold greater risk for developing insomnia.Improve sleep with group CBT for insomnia,” Current Psychiatry, April 2009.) Pharmacotherapy during pregnancy and for breast-feeding mothers is guided by evaluating the risk/benefit ratio and safety considerations.
Maintain a high index of suspicion for breathing-related sleep disorders, such as OSA,21 and RLS.22 Atypical presentations of OSA are common in pregnant or postpartum women; compared with men, women with OSA are more likely to report fatigue and less likely than to report sleepiness. Refer patients whom you think may have OSA for polysomnography.
If you suspect RLS, check for low ferritin and folate levels. Nutritional supplements may be necessary for women in high-risk groups, including those who are pregnant or have varicose veins, venous reflux, folate deficiency, uremia, diabetes, thyroid problems, peripheral neuropathy, Parkinson’s disease, or certain autoimmune disorders, such as Sjögren’s syndrome, celiac disease, and rheumatoid arthritis.23 Advise these patients to avoid caffeine.
Although indicated for treating RLS, ropinirole and pramipexole are FDA Pregnancy Category C, which means animal studies have shown adverse effects on the fetus and there are no adequate and well-controlled studies in humans, but potential benefits may warrant use of the drug in pregnant women despite risks. Opioids, carbamazepine, or gabapentin may be safer for pregnant patients.24
Insomnia during menopause
The prevalence of insomnia increases from 33% to 36% in premenopausal women to 44% to 61% in postmenopausal women.14 Hot flashes, comorbid mood disturbances, sleep-disordered breathing, and RLS contribute to increased insomnia risk in postmenopausal women (Table 3).4,14,25,26
Treatment strategy. Always inquire about sleep in perimenopausal/postmenopausal women, even when her presenting complaint is related to menstrual cycle changes or vasomotor symptoms such as hot flashes.16 Assess patients for OSA, RLS, and mood, anxiety, and cognitive symptoms.26 In addition to pharmacotherapy and behavioral therapy, treatment options include hormone replacement therapy (HRT) and herbal and dietary supplements (Table 4).27-32
Table 3
Sleep difficulties during menopause: Differential diagnoses
| Condition | Features | Findings | Other considerations |
|---|---|---|---|
| Hot flashes (prevalence: 75% to 85%)14 | Vasomotor phenomenon characterized by feelings such as ‘spreading warmth,’ diaphoresis, palpitations, nausea, and insomnia Mediated through the preoptic area of the anterior hypothalamus, which regulates temperature and sleep Increased brain norepinephrine metabolism | Discrepancies between objective (PSG) and subjective measures (surveys)4 Discrepancies between self-reported and laboratory reported sleep data might be explained by thermoregulatory differences between NREM and REM sleep24 | Nocturnal hot flashes trigger awakenings and insomnia14 Hot flashes can follow arousals and awakenings HRT is highly effective in treating hot flashes; however, data on its direct effects on sleep complaints are inconsistent |
| Primary menopausal insomnia25 | Menopausal symptoms (eg, hot flashes) trigger insomnia that persists secondary to behavioral conditioning | Increase in nocturnal skin temperature coincides with decrease in skin resistance and waking episodes in PSG | Behavioral insomnia therapies are useful adjuncts to treatment of menopause symptoms |
| Sleep-disordered breathing (OSA) | Menopause increases risk for OSA independent of body weight Redistribution of body fat with an increase in the waist-to-hip circumference ratio occurs in menopause Loss of ventilatory drive because of diminished progesterone levels | Sleep fragmentation and daytime sleepiness are common, as opposed to apneic episodes or oxygen desaturation in men | Maintain a high index of suspicion and promptly refer patients to a sleep center |
| Restless legs syndrome | Related to iron deficiency | Low ferritin and folate levels | Advise patients to avoid caffeine |
| HRT: hormone replacement therapy; NREM: non-rapid eye movement; OSA: obstructive sleep apnea; PSG: polysomnography; REM: rapid eye movement | |||
Table 4
Treating insomnia in menopausal women
| Therapy | Comments |
|---|---|
| Hormone replacement therapy (HRT) | Effective for hot flashes, insomnia,26-28 and sleep apnea29 Long-term safety is questionable4 |
| Behavioral therapy (cognitive-behavioral therapy,30 stimulus control therapy, sleep restriction therapy, sleep hygiene, hypnotherapy, biofeedback) | Limited data in menopausal women |
| Sedatives/hypnotics/antidepressants (eg, zolpidem, 10 mg; eszopiclone, 3 mg; trazodone, 75 mg; ramelteon, 8 mg; SSRIs and SNRIs) | Benzodiazepines may be useful, although not specifically evaluated in menopausal women. Risk of tolerance, dependence, and psychomotor slowing |
| Herbal and dietary supplements (Cimicifuga racemosa [Black cohosh],31 valerian | Popular alternatives to HRT; however, evidence of efficacy as treatment for insomnia is inconclusive |
| SNRIs: serotonin-norepinephrine reuptake inhibitors; SSRIs: selective serotonin reuptake inhibitors | |
Comorbid psychiatric disorders
Women have a higher prevalence of psychiatric disorders such as major depressive disorder and anxiety disorders than men.1 Women have a 10% to 25% lifetime risk of developing major depression. Three quarters of depressed patients experience insomnia.1 Recent literature suggests insomnia is a risk factor for depression,33 which emphasizes the need to screen women who present with sleep problems for depression and anxiety.
Five percent to 20% of women experience postpartum depression. Depression and insomnia are correlated to the rapid decline in estrogen and progesterone after delivery.34
Treatment strategy. Insomnia is a common presenting symptom in patients with psychiatric conditions such as mood and anxiety disorders. Treating the underlying psychiatric disorder often alleviates sleeping difficulties. However, if the insomnia is disabling, treat the psychiatric disorder and insomnia concurrently.
CASE CONTINUED: Perimenopausal insomnia
Based on her history, you diagnose Ms. A with insomnia related to general medical condition (perimenopause). There are no indications to refer her for polysomnography. You educate Ms. A about sleep hygiene and recommend that she discuss her menstrual and physical complaints with her primary care physician or gynecologist. Ms. A is not interested in HRT because she has a strong family history of endometrial cancer. You reassure Ms. A and schedule a follow-up visit in 2 months to re-evaluate her insomnia.
Related resource
- Krahn LE. Perimenopausal depression? Ask how she’s sleeping. Current Psychiatry. 2005;4(6):39-53.
Drug brand names
- Carbamazepine • Carbatrol, Tegretol, others
- Escitalopram • Lexapro
- Eszopiclone • Lunesta
- Fluoxetine • Prozac
- Gabapentin • Neurontin, Gabarone
- Paroxetine • Paxil
- Pramipexole • Mirapex
- Ramelteon • Rozerem
- Ropinirole • Requip
- Sertraline • Zoloft
- Trazodone • Desyrel
- Zolpidem • Ambien
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgements
The authors thank Dr. Namita Dhiman and Darrel E. Willoughby for their assistance with this article.
1. Krystal AD. Depression and insomnia in women. Clin Cornerstone. 2004;6(suppl 1B):S19-S28.
2. Ohayon MM. Epidemiology of insomnia: what we know and what we still need to learn. Sleep Med Rev. 2002;6(2):97-111.
3. Krishnan V, Collop NA. Gender differences in sleep disorders. Curr Opin Pulm Med. 2006;12(6):383-389.
4. Soares CN, Murray BJ. Sleep disorders in women: clinical evidence and treatment strategies. Psychiatr Clin North Am. 2006;29(4):1095-1113.
5. Ohayon M. Epidemiological study on insomnia in the general population. Sleep. 1996;19(3 suppl):S7-S15.
6. Daley M, Morin CM, LeBlanc M, et al. Insomnia and its relationship to health-care utilization, work absenteeism, productivity and accidents. Sleep Med. 2009;10(4):427-438.
7. Diagnostic and statistical manual of mental disorders, 4th ed, text revision. Washington, DC: American Psychiatric Association; 2000.
8. Ancoli-Israel S, Roth T. Characteristics of insomnia in the United States: results of the 1991 National Sleep Foundation Survey. I. Sleep. 1999;2(suppl 2):S347-S353.
9. Zhang B, Wing YK. Sex differences in insomnia: a meta-analysis. Sleep. 2006;29(1):85-93.
10. Buysse DJ, Reynolds CF, Monk TH, et al. The Pittsburgh Sleep Quality Index (PSQI): a new instrument for psychiatric research and practice. Psychiatry Res. 1989;28(2):193-213.
11. Manber R, Bootzin RR. Sleep and the menstrual cycle. Health Psychol. 1997;16:209-214.
12. Ito M, Kohsaka M, Fukuda N, et al. Effects of menstrual cycle on plasma melatonin level and sleep characteristics. Jpn J Psychiatry Neurol. 1993;47:478-479.
13. Driver HS, Dijk DJ, Werth E, et al. Sleep and the sleep electroencephalogram across the menstrual cycle in young healthy women. J Clin Endocrinol Metab. 1996;81:728-735.
14. Moline ML, Broch L, Zak R. Sleep in women across the life cycle from adulthood through menopause. Med Clin North Am. 2004;88(3):705-736.
15. Steiner M, Pearlstein T, Cohen LS, et al. Expert guidelines for the treatment of severe PMS, PMDD, and comorbidities: the role of SSRIs. J Womens Health (Larchmt). 2006;15(1):57-69.
16. Krystal AD. Insomnia in women. Clin Cornerstone. 2003;5(3):41-50.
17. Mindell JA, Jacobson BJ. Sleep disturbances during pregnancy. J Obstet Gynecol Neonatal Nurs. 2000;29(6):590-597.
18. Lee KA, Zaffke ME, McEnany G. Parity and sleep patterns during and after pregnancy. Obstet Gynecol. 2000;95(1):14-18.
19. Brunner DP, Münch M, Biedermann K, et al. Changes in sleep and sleep electroencephalogram during pregnancy. Sleep. 1994;17(7):576-582.
20. Ross LE, Murray BJ, Steiner M. Sleep and perinatal mood disorders: a critical review. J Psychiatry Neurosci. 2005;30(4):247-256.
21. Edwards N, Middleton PG, Blyton DM, et al. Sleep disordered breathing and pregnancy. Thorax. 2002;57(6):555-558.
22. Manconi M, Govoni V, De Vito A, et al. Restless legs syndrome and pregnancy. Neurology. 2004;63(6):1065-1069.
23. Lee KA, Zaffke ME, Baratte-Beebe K. Restless legs syndrome and sleep disturbance during pregnancy: the role of folate and iron. J Womens Health Gend Based Med. 2001;10(4):335-341.
24. Djokanovic N, Garcia-Bournissen F, Koren G. Medications for restless legs syndrome in pregnancy. J Obstet Gynaecol Can. 2008;30(6):505-507.
25. Freedman RR, Roehrs TA. Effects of REM sleep and ambient temperature on hot flash-induced sleep disturbance. Menopause. 2006;13(4):576-583.
26. Krystal AD, Edinger J, Wohlgemuth W, et al. Sleep in perimenopausal and postmenopausal women. Sleep Med Rev. 1998;2(4):243-253.
27. Polo-Kantola P, Erkkola R, Irjala K, et al. Effect of short-term transdermal estrogen replacement therapy on sleep: a randomized, double-blind crossover trial in postmenopausal women. Fertil Steril. 1999;71(5):873-880.
28. Watts NB, Notelovitz M, Timmons MC, et al. Comparison of oral estrogens and estrogens plus androgen on bone mineral density, menopausal symptoms, and lipid-lipoprotein profiles in surgical menopause. Obstet Gynecol. 1995;85(4):529-537.Erratum in: Obstet Gynecol 1995;85(5 Pt 1):668.
29. Boyle GJ, Murrihy R. A preliminary study of hormone replacement therapy and psychological mood states in perimenopausal women. Psychol Rep. 2001;88(1):160-170.
30. Cistulli PA, Barnes DJ, Grunstein RR, et al. Effect of short-term hormone replacement in the treatment of obstructive sleep apnoea in postmenopausal women. Thorax. 1994;49:699-702.
31. Yang CM, Spielman AJ, Glovinsky P. Nonpharmacologic strategies in the management of insomnia. Psychiatr Clin North Am. 2006;29(4):895-919.
32. Mahady GB. Black cohosh (Actaea/Cimicifuga racemosa): review of the clinical data for safety and efficacy in menopausal symptoms. Treat Endocrinol. 2005;4(3):177-184.
33. Breslau N, Roth T, Rosenthal L, et al. Sleep disturbance and psychiatric disorders: a longitudinal epidemiological study of young adults. Biol Psychiatry. 1996;39:411-418.
34. Burt VK, Stein K. Epidemiology of depression throughout the female life cycle. J Clin Psychiatry. 2002;63(suppl 7):9-15.
1. Krystal AD. Depression and insomnia in women. Clin Cornerstone. 2004;6(suppl 1B):S19-S28.
2. Ohayon MM. Epidemiology of insomnia: what we know and what we still need to learn. Sleep Med Rev. 2002;6(2):97-111.
3. Krishnan V, Collop NA. Gender differences in sleep disorders. Curr Opin Pulm Med. 2006;12(6):383-389.
4. Soares CN, Murray BJ. Sleep disorders in women: clinical evidence and treatment strategies. Psychiatr Clin North Am. 2006;29(4):1095-1113.
5. Ohayon M. Epidemiological study on insomnia in the general population. Sleep. 1996;19(3 suppl):S7-S15.
6. Daley M, Morin CM, LeBlanc M, et al. Insomnia and its relationship to health-care utilization, work absenteeism, productivity and accidents. Sleep Med. 2009;10(4):427-438.
7. Diagnostic and statistical manual of mental disorders, 4th ed, text revision. Washington, DC: American Psychiatric Association; 2000.
8. Ancoli-Israel S, Roth T. Characteristics of insomnia in the United States: results of the 1991 National Sleep Foundation Survey. I. Sleep. 1999;2(suppl 2):S347-S353.
9. Zhang B, Wing YK. Sex differences in insomnia: a meta-analysis. Sleep. 2006;29(1):85-93.
10. Buysse DJ, Reynolds CF, Monk TH, et al. The Pittsburgh Sleep Quality Index (PSQI): a new instrument for psychiatric research and practice. Psychiatry Res. 1989;28(2):193-213.
11. Manber R, Bootzin RR. Sleep and the menstrual cycle. Health Psychol. 1997;16:209-214.
12. Ito M, Kohsaka M, Fukuda N, et al. Effects of menstrual cycle on plasma melatonin level and sleep characteristics. Jpn J Psychiatry Neurol. 1993;47:478-479.
13. Driver HS, Dijk DJ, Werth E, et al. Sleep and the sleep electroencephalogram across the menstrual cycle in young healthy women. J Clin Endocrinol Metab. 1996;81:728-735.
14. Moline ML, Broch L, Zak R. Sleep in women across the life cycle from adulthood through menopause. Med Clin North Am. 2004;88(3):705-736.
15. Steiner M, Pearlstein T, Cohen LS, et al. Expert guidelines for the treatment of severe PMS, PMDD, and comorbidities: the role of SSRIs. J Womens Health (Larchmt). 2006;15(1):57-69.
16. Krystal AD. Insomnia in women. Clin Cornerstone. 2003;5(3):41-50.
17. Mindell JA, Jacobson BJ. Sleep disturbances during pregnancy. J Obstet Gynecol Neonatal Nurs. 2000;29(6):590-597.
18. Lee KA, Zaffke ME, McEnany G. Parity and sleep patterns during and after pregnancy. Obstet Gynecol. 2000;95(1):14-18.
19. Brunner DP, Münch M, Biedermann K, et al. Changes in sleep and sleep electroencephalogram during pregnancy. Sleep. 1994;17(7):576-582.
20. Ross LE, Murray BJ, Steiner M. Sleep and perinatal mood disorders: a critical review. J Psychiatry Neurosci. 2005;30(4):247-256.
21. Edwards N, Middleton PG, Blyton DM, et al. Sleep disordered breathing and pregnancy. Thorax. 2002;57(6):555-558.
22. Manconi M, Govoni V, De Vito A, et al. Restless legs syndrome and pregnancy. Neurology. 2004;63(6):1065-1069.
23. Lee KA, Zaffke ME, Baratte-Beebe K. Restless legs syndrome and sleep disturbance during pregnancy: the role of folate and iron. J Womens Health Gend Based Med. 2001;10(4):335-341.
24. Djokanovic N, Garcia-Bournissen F, Koren G. Medications for restless legs syndrome in pregnancy. J Obstet Gynaecol Can. 2008;30(6):505-507.
25. Freedman RR, Roehrs TA. Effects of REM sleep and ambient temperature on hot flash-induced sleep disturbance. Menopause. 2006;13(4):576-583.
26. Krystal AD, Edinger J, Wohlgemuth W, et al. Sleep in perimenopausal and postmenopausal women. Sleep Med Rev. 1998;2(4):243-253.
27. Polo-Kantola P, Erkkola R, Irjala K, et al. Effect of short-term transdermal estrogen replacement therapy on sleep: a randomized, double-blind crossover trial in postmenopausal women. Fertil Steril. 1999;71(5):873-880.
28. Watts NB, Notelovitz M, Timmons MC, et al. Comparison of oral estrogens and estrogens plus androgen on bone mineral density, menopausal symptoms, and lipid-lipoprotein profiles in surgical menopause. Obstet Gynecol. 1995;85(4):529-537.Erratum in: Obstet Gynecol 1995;85(5 Pt 1):668.
29. Boyle GJ, Murrihy R. A preliminary study of hormone replacement therapy and psychological mood states in perimenopausal women. Psychol Rep. 2001;88(1):160-170.
30. Cistulli PA, Barnes DJ, Grunstein RR, et al. Effect of short-term hormone replacement in the treatment of obstructive sleep apnoea in postmenopausal women. Thorax. 1994;49:699-702.
31. Yang CM, Spielman AJ, Glovinsky P. Nonpharmacologic strategies in the management of insomnia. Psychiatr Clin North Am. 2006;29(4):895-919.
32. Mahady GB. Black cohosh (Actaea/Cimicifuga racemosa): review of the clinical data for safety and efficacy in menopausal symptoms. Treat Endocrinol. 2005;4(3):177-184.
33. Breslau N, Roth T, Rosenthal L, et al. Sleep disturbance and psychiatric disorders: a longitudinal epidemiological study of young adults. Biol Psychiatry. 1996;39:411-418.
34. Burt VK, Stein K. Epidemiology of depression throughout the female life cycle. J Clin Psychiatry. 2002;63(suppl 7):9-15.
Impaired physicians: How to recognize, when to report, and where to refer
From all outward appearances Dr. S, a part-time psychiatrist at an inpatient psychiatric facility and in private practice for 12 years, is living the “perfect life,” with a wife, children, and successful practice.
In retrospect, his drug addiction began insidiously. In college, Dr. S continued to use oxycodone/acetaminophen prescribed for a shoulder injury long after his pain had resolved. He began to use cocaine in residency to help him “get through” the 36-hour call days, and it “helped” him earn the chief resident position because of his heightened energy and concentration. Dr. S’ primary care physician initially prescribed him benzodiazepines for anxiety and to help him sleep. Opiates were prescribed for a musculoskeletal injury. Dr. S obtained prescriptions for these medications from multiple providers. This ultimately escalated to self-prescription using aliases. Dr. S also began to drink heavily each evening.
Dr. S disregards colleagues’ comment about his obvious mood swings, which he attributes to his stressful job and “nagging” wife, despite having a family history of bipolar disorder. He becomes enraged when his wife or friends suggest he seek help. His colleagues whisper behind his back, but for years, no one confronts him about his unpredictable and frequently inappropriate behavior. Eventually, a nurse files a sexual harassment suit against Dr. S, and a patient complains to the medical board that Dr. S exhibited sexually inappropriate behavior during a therapy session.
As physicians, recognizing impairment in our colleagues or ourselves can be difficult. The American Medical Association defines an impaired physician as one who is unable to fulfill personal or professional responsibilities because of psychiatric illness, alcoholism, or drug dependence.1 Impairment is present when a physician is unable to perform in a manner that conforms to acceptable standards of practice, exhibits serious flaws in judgment, and provides incompetent care.1-3
Recognizing when a physician is impaired, deciding whether to report him or her to the state medical board, and referring a colleague for treatment can be challenging. This article will:
- review substance abuse, cognitive decline, and other causes of impairment
- address legal and ethical issues involved in reporting a colleague to the state medical board
- provide resources for physician treatment and assistance.
Physicians and addiction
Chemical dependence is the most frequent disabling illness among physicians,4 and substance abuse is the most common form of impairment that results in discipline by a state medical board.5 An estimated 6% to 8% of physicians abuse drugs, and approximately 14% develop alcohol use disorders; these rates are comparable to those of the general population.5 Psychiatrists, emergency room physicians, and solo practitioners are 3 times more likely to abuse substances than other doctors.6 An obsessive-compulsive personality and other factors may predispose physicians to substance abuse (Table 1).7,8
Alcohol is the most commonly abused substance, followed by opiates, cocaine, and other stimulants.9 Physicians are estimated to use opiates and benzodiazepines at a rate 5 times greater than that of the general public.10,11
An often-hidden problem. Physicians frequently deny substance abuse and many are able to conceal the problem from coworkers, even as their personal lives disintegrate.1,12 Marital and relationship problems may be the first indication of impairment, which gradually spreads to other aspects of their lives (Table 2).1,2,5 A doctor’s professional performance often is the last area to be affected.1,12
Substance abuse in physicians may long go unreported. The clinician’s family may want to protect the physician’s reputation, career, and income. Colleagues may be intimidated, uncertain of their concerns, or fearful for their jobs if they report the physician’s impairment. Patients may be reluctant to report their concerns because they depend on their provider for health care, respect the physician, or deny that a doctor could have a drug or alcohol problem.5
Table 1
Physicians and substance abuse: Predisposing factors
| Obsessive-compulsive personality style |
| Family history of substance use disorders or mental illness |
| Childhood family problems |
| Personal mental illness |
| Sensation-seeking behavior |
| Denial of personal and social problems |
| Perfectionism |
| Idealism |
| Source: References 7,8 |
Table 2
Signs of substance abuse
| Frequent tardiness and absences |
| Unexplained disappearances during working hours |
| Inappropriate behavior |
| Affective lability or irritability |
| Interpersonal conflict |
| Avoidance of peers or supervisors |
| Keeping odd hours |
| Disorganization and forgetfulness |
| Diminished chart completion and work performance |
| Heavy drinking at social functions |
| Unexplained changes in weight or energy level |
| Diminished personal hygiene |
| Slurred or rapid speech |
| Frequently dilated pupils or red and watery eyes and a runny nose |
| Defensiveness, anxiety, apathy, or manipulative behavior |
| Withdrawal from long-standing relationships |
| Source: References 1,2,5 |
Screening for cognitive decline
Many people with cognitive impairment lack insight into their problem and may minimize or deny the degree of their impairment.13 The prevalence of dementia in individuals age ≥65 is 3% to 11%,14 and 18% of physicians are in this age group.15
Ethical, legal, and practical issues arise in determining who, when, and how to screen physicians for cognitive problems. Standard screening exams may not be adequately sensitive for a well-educated physician, and neuropsychological testing may be necessary to detect mild cognitive impairment.13 In addition, the cognitive, visual-spacial, reactivity, reasoning, and calculation skills required for capable medical practice vary among specialties.16
One screening option is a “360-degree review” of information obtained from peers, patients, and non-physician colleagues that the College of Physicians and Surgeons in Alberta, Canada, has incorporated into its Physician’s Achievement Review (PAR) for physicians age ≥65.17 Compiled in a confidential manner and shared with the physician, the 360-degree survey assesses his or her:
- skill and knowledge
- psychosocial functioning
- management skills
- performance
- collegiality.
In Alberta, physicians who score in the lower one-third of the survey are assessed with an on-site evaluation by physicians from his or her specialty appointed by the PAR Director of Practice Improvement.
Alternately, physicians age ≥65 could be required to undergo annual or biannual neuropsychological testing to screen for mild cognitive impairment or other evidence of cognitive decline. In the United States, any screening requirement must be structured to comply with the Age Discrimination in Employment Act.18
If a physician shows evidence of cognitive impairment, the state medical board should initiate closer scrutiny and modify or revoke privileges if indicated. Remediation programs designed to assist impaired physicians may not be effective for those with cognitive impairment because the decline in cognitive functioning associated with illnesses such as Alzheimer’s disease often is progressive.13
‘Disruptive’ physicians
Mental illnesses such as personality or affective disorders, interpersonal problems within or outside the workplace, or other stressors could lead a physician to disrupt the workplace or patient care. Numerous programs have been established across the United States to help evaluate and treat disruptive physicians. Remediation programs can help identify and offer education for “dyscompetent” physicians (see Related Resources).13
To report, or not to report
In a national survey of physicians, only 45% of respondents indicated that they had notified the state licensing board of a colleague they felt was impaired or incompetent, yet almost all (96%) indicated that these individuals should be reported.19 Any duty to report requires, at minimum, that the physician be affected by an illness that impairs his or her cognition, concentration, rapid decision making, and ability to handle emergencies or perform work functions safely.20
Shouten20 cautions that someone who is considering filing a report because of fear of liability if they don’t should balance this concern against potential liability for breaching confidentiality. If there is evidence of an imminent risk or serious harm to the physician or patients, you may be legally required to breach confidentiality. Some states require licensed health practitioners to report acts of professional misconduct, unless the information is obtained solely from directly treating the physician. These requirements apply only within the state, and only to that state’s licensees.20-22
An ethical requirement to report also must be balanced against the obligation to maintain confidentiality. Ethics are largely a matter of individual standards, and individuals’ perceived ethical duties vary.
If you are considering reporting an impaired colleague, learn the laws in your state. If the physician is from another jurisdiction, the law provides little definitive guidance. Shouten recommends focusing on clinical outcomes for the doctor and his or her patients rather than on legal liability.20
Physician health programs
Nationwide directories of physician health programs are available from the Federation of State Physician Health Programs and the Federation of State Medical Boards (see Related Resources). Some programs are affiliated with state licensing boards, some are branches of state medical societies, and some are independent. These programs provide confidential treatment and assistance to practitioners experiencing substance or alcohol abuse, mental illness, or disruptive behavior. Some institutions may offer physicians an employee assistance program.
State medical societies may provide information about accessing a physician health program. Programs sponsored by medical societies almost always are independent of state licensing boards. This arrangement allows physicians to seek help without fear of punishment or censure or revocation of their license. Noncompliance with a physician health program, however, likely will result in being reported to the medical board.
Physician health programs typically employ a rehabilitative approach. Punitive measures such as reporting physicians to the medical board usually are not pursued unless the individual does not comply with treatment and monitoring guidelines. A physician who abuses substances, for example, may be required to complete a residential treatment program, attend support group meetings such as 12-step programs, participate in individual therapy, and undergo random screening for alcohol and illicit drug use.5
Abstinence is the goal of treating clinicians who abuse substances. Physicians have better outcomes than the general population, with reported abstinence rates of 70% to 90% for those who complete treatment;23,24 75% to 85% of physicians who complete rehabilitation and comply with close monitoring and follow-up care are able to return to work.24,25 Acceptance of recovery as a lifelong process, monitoring, and self-vigilance often are necessary to achieve and maintain abstinence.5
Risk factors for relapse include:
- denial of illness
- poor stress-coping and relationship skills
- social and professional isolation
- inability to accept feedback
- complacency and overconfidence
- failure to attend support group meetings
- dysfunctional family dynamics
- feelings of self-pity, blame, and guilt.5
Treating an impaired colleague. Reid26 recommends that psychiatrists should not evaluate or treat a self-referred, potentially impaired physician unless the relationship is strictly clinical. A physician may withhold symptoms, behaviors, or problems because his or her license, malpractice case, or career are at stake.
Advise a physician who requests evaluation or treatment related to license concerns or any legal matter to seek legal counsel. Working with such physician/patients only upon referral by a lawyer, licensing board, or physicians’ health committee provides treating psychiatrists with a clear professional role, allowing them to focus solely on the physician/patient’s treatment needs.
CASE CONTINUED: Extensive help, then success
The medical director of the hospital where Dr. S works refers him to his state’s impaired physician program. After investigating the complaints by the nurse and patient, the medical board suspends Dr. S’ license and requires him to enter a substance abuse treatment program. He completes an intensive residential program for impaired physicians and achieves sobriety from drugs and alcohol. His mood disorder is successfully treated with medications and psychotherapy. The medical board requires Dr. S to have a chaperone present for all visits with patients and submit random urine drug screens once his license is provisionally restored. The medical board also requires Dr. S to undergo ongoing psychiatric care and medication monitoring. He remains abstinent from alcohol and drugs, complies with the medical board’s requirements, and enjoys a productive practice and improved relationship with his family.
Disruptive physicians
- Samenow C, Swiggart W, Spickard A Jr. Consequence of physician disruptive behavior. Tenn Med. 2007;100(11):38-40.
- Leape LL, Fromson JA. Problem doctors: is there a system-level solution? Ann Intern Med. 2006;144(2):107-115.
- Linney BJ. Confronting the disruptive physician. Physician Exec. 1997;23:55-59.
Physician evaluation
- Anfang SA, Faulkner LR, Fromson JA, et al. The American Psychiatric Association’s resource document on guidelines for psychiatric fitness-for-duty evaluations of physicians. J Am Acad Psychiatry Law. 2005;33:85-88.
- Harmon L, Pomm R. Evaluation, treatment, and monitoring of disruptive physicians’ behavior. Psychiatr Ann. 2004; 34:770-774.
Other resources
- Federation of State Physician Health Programs. www.fsphp.org.
- Federation of State Medical Boards: Directory of Physician Assessment and Remedial Education Programs. www.fsmb.org/pdf/RemEdProg.pdf.
- The Center for Professional Health, Vanderbilt University Medical Center. www.mc.vanderbilt.edu/cph.
- Vanderbilt Comprehensive Assessment Program. www.mc.vanderbilt.edu/root/vcap.
- Alcoholics Anonymous. www.aa.org.
- Narcotics Anonymous. www.na.org.
Drug Brand Name
- Oxycodone/acetaminophen • Percocet
Disclosure
The authors report no financial relationship with the manufacturer of any product mentioned in this article or with manufacturers of competing products.
1. Breiner SJ. The impaired physician. J Med Educ. 1979;54:673.-
2. Talbott GD, Gallegos KV, Angres DH. Impairment and recovery in physicians and other health professionals. In: Graham AW, Schultz TK, eds. Principles of addiction medicine. 2nd ed. Chevy Chase, MD: American Society of Addiction Medicine, Inc; 1998:1263-1277.
3. American Medical Association Council on Mental Health. The sick physician: impairment by psychiatric disorders, including alcoholism and drug dependence. JAMA. 1973;223:684-687.
4. Talbott G, Wright C. Chemical dependency in healthcare professionals. Occup Med. 1987;2:581-591.
5. Baldisseri MR. Impaired healthcare professional. Crit Care Med. 2007;35:S106-116.
6. Mansky PA. Physician health programs and the potentially impaired physician with a substance use disorder. Psychiatr Serv. 1996;47:465-467.
7. Boisaubin EV, Levine RE. Identifying and assisting the impaired physician. Am J Med Sci. 2001;322:31-36.
8. Bissel L, Jones RW. The alcoholic physician: a survey. Am J Psychiatry. 1976;133:1142-1146.
9. Robins L, Reiger D. Psychiatric disorders in America: the Epidemiologic Catchment Area Study. New York, NY: The Free Press; 1991.
10. Gallegos KV, Browne CH, Veit FW, et al. Addiction in anesthesiologists: drug access and patterns of substance abuse. QRB Qual Rev Bull. 1988;14:116-122.
11. Hughes PH, Brandenburg N, Baldwin DC, et al. Prevalence of substance abuse among U.S. physicians. JAMA. 1992;267:2333-2339.
12. Vaillant GE, Clark W, Cyrus C, et al. Prospective study of alcoholism treatment. Eight year follow-up. Am J Med. 1983;75:455-463.
13. LoboPrabhu SM, Molinari VA, Hamilton JD, et al. The aging physician with cognitive impairment: approaches to oversight, prevention and remediation. Am J Geriatr Psychiatry. 2009;17:445-454.
14. U.S. General Accounting Office. Alzheimer’s disease: estimates of prevalence in the United States. Washington, DC: U.S. General Accounting Office; 1998:98. Publication GAO/HEHS-98-16.
15. American Medical Association. Physician characteristics and distribution in the U.S., 2006. Washington, DC: American Medical Association; 2006.
16. Blasier R. The problem of the aging surgeon. Clin Orthop Relat Res. 2009;467:402-411.
17. College of Physicians and Surgeons of Alberta. Physician Achievement Review (PAR) Program. Available at: http://www.cpsa.ab.ca/Services/PARprogram/Overview.aspx. Accessed March 27, 2010.
18. The Age Discrimination in Employment Act. (Vol. Pub. L. No. 90-202, 81 Stat. 602 (Dec. 15, 1967), codified as Chapter 14 of Title 29 of the United States Code, 29 U.S.C. § 621 through 29 U.S.C. § 63), 1967.
19. Campbell EG, Regan S, Gruen RL, et al. Professionalism in medicine: results of a national survey of physicians. Ann Int Med. 2007;147:795-802.
20. Shouten R. Impaired physicians: is there a duty to report to state licensing boards? Harvard Rev Psychiatry. 2000;8:36-39.
21. Mass Gen Laws ch 112 § 5F.
22. NY PHL § 230 (11) (e).
23. Femino J, Nirenberg TD. Treatment outcome studies on physician impairment: a review of the literature. R I Med. 1994;77:345-350.
24. Alpern F, Correnti CE, Dolan TE, et al. A survey of recovering Maryland physicians. Md Med J. 1992;41:301-303.
25. Gallegos KV, Lubin BH, Bowers C, et al. Relapse and recovery: five to ten year follow-up study of chemically dependent physicians—the Georgia experience. Md Med J. 1992;41:315-319.
26. Reid W. Evaluating and treating disabled or impaired colleagues. J Psychiatr Pract. 2007;13:44-48.
From all outward appearances Dr. S, a part-time psychiatrist at an inpatient psychiatric facility and in private practice for 12 years, is living the “perfect life,” with a wife, children, and successful practice.
In retrospect, his drug addiction began insidiously. In college, Dr. S continued to use oxycodone/acetaminophen prescribed for a shoulder injury long after his pain had resolved. He began to use cocaine in residency to help him “get through” the 36-hour call days, and it “helped” him earn the chief resident position because of his heightened energy and concentration. Dr. S’ primary care physician initially prescribed him benzodiazepines for anxiety and to help him sleep. Opiates were prescribed for a musculoskeletal injury. Dr. S obtained prescriptions for these medications from multiple providers. This ultimately escalated to self-prescription using aliases. Dr. S also began to drink heavily each evening.
Dr. S disregards colleagues’ comment about his obvious mood swings, which he attributes to his stressful job and “nagging” wife, despite having a family history of bipolar disorder. He becomes enraged when his wife or friends suggest he seek help. His colleagues whisper behind his back, but for years, no one confronts him about his unpredictable and frequently inappropriate behavior. Eventually, a nurse files a sexual harassment suit against Dr. S, and a patient complains to the medical board that Dr. S exhibited sexually inappropriate behavior during a therapy session.
As physicians, recognizing impairment in our colleagues or ourselves can be difficult. The American Medical Association defines an impaired physician as one who is unable to fulfill personal or professional responsibilities because of psychiatric illness, alcoholism, or drug dependence.1 Impairment is present when a physician is unable to perform in a manner that conforms to acceptable standards of practice, exhibits serious flaws in judgment, and provides incompetent care.1-3
Recognizing when a physician is impaired, deciding whether to report him or her to the state medical board, and referring a colleague for treatment can be challenging. This article will:
- review substance abuse, cognitive decline, and other causes of impairment
- address legal and ethical issues involved in reporting a colleague to the state medical board
- provide resources for physician treatment and assistance.
Physicians and addiction
Chemical dependence is the most frequent disabling illness among physicians,4 and substance abuse is the most common form of impairment that results in discipline by a state medical board.5 An estimated 6% to 8% of physicians abuse drugs, and approximately 14% develop alcohol use disorders; these rates are comparable to those of the general population.5 Psychiatrists, emergency room physicians, and solo practitioners are 3 times more likely to abuse substances than other doctors.6 An obsessive-compulsive personality and other factors may predispose physicians to substance abuse (Table 1).7,8
Alcohol is the most commonly abused substance, followed by opiates, cocaine, and other stimulants.9 Physicians are estimated to use opiates and benzodiazepines at a rate 5 times greater than that of the general public.10,11
An often-hidden problem. Physicians frequently deny substance abuse and many are able to conceal the problem from coworkers, even as their personal lives disintegrate.1,12 Marital and relationship problems may be the first indication of impairment, which gradually spreads to other aspects of their lives (Table 2).1,2,5 A doctor’s professional performance often is the last area to be affected.1,12
Substance abuse in physicians may long go unreported. The clinician’s family may want to protect the physician’s reputation, career, and income. Colleagues may be intimidated, uncertain of their concerns, or fearful for their jobs if they report the physician’s impairment. Patients may be reluctant to report their concerns because they depend on their provider for health care, respect the physician, or deny that a doctor could have a drug or alcohol problem.5
Table 1
Physicians and substance abuse: Predisposing factors
| Obsessive-compulsive personality style |
| Family history of substance use disorders or mental illness |
| Childhood family problems |
| Personal mental illness |
| Sensation-seeking behavior |
| Denial of personal and social problems |
| Perfectionism |
| Idealism |
| Source: References 7,8 |
Table 2
Signs of substance abuse
| Frequent tardiness and absences |
| Unexplained disappearances during working hours |
| Inappropriate behavior |
| Affective lability or irritability |
| Interpersonal conflict |
| Avoidance of peers or supervisors |
| Keeping odd hours |
| Disorganization and forgetfulness |
| Diminished chart completion and work performance |
| Heavy drinking at social functions |
| Unexplained changes in weight or energy level |
| Diminished personal hygiene |
| Slurred or rapid speech |
| Frequently dilated pupils or red and watery eyes and a runny nose |
| Defensiveness, anxiety, apathy, or manipulative behavior |
| Withdrawal from long-standing relationships |
| Source: References 1,2,5 |
Screening for cognitive decline
Many people with cognitive impairment lack insight into their problem and may minimize or deny the degree of their impairment.13 The prevalence of dementia in individuals age ≥65 is 3% to 11%,14 and 18% of physicians are in this age group.15
Ethical, legal, and practical issues arise in determining who, when, and how to screen physicians for cognitive problems. Standard screening exams may not be adequately sensitive for a well-educated physician, and neuropsychological testing may be necessary to detect mild cognitive impairment.13 In addition, the cognitive, visual-spacial, reactivity, reasoning, and calculation skills required for capable medical practice vary among specialties.16
One screening option is a “360-degree review” of information obtained from peers, patients, and non-physician colleagues that the College of Physicians and Surgeons in Alberta, Canada, has incorporated into its Physician’s Achievement Review (PAR) for physicians age ≥65.17 Compiled in a confidential manner and shared with the physician, the 360-degree survey assesses his or her:
- skill and knowledge
- psychosocial functioning
- management skills
- performance
- collegiality.
In Alberta, physicians who score in the lower one-third of the survey are assessed with an on-site evaluation by physicians from his or her specialty appointed by the PAR Director of Practice Improvement.
Alternately, physicians age ≥65 could be required to undergo annual or biannual neuropsychological testing to screen for mild cognitive impairment or other evidence of cognitive decline. In the United States, any screening requirement must be structured to comply with the Age Discrimination in Employment Act.18
If a physician shows evidence of cognitive impairment, the state medical board should initiate closer scrutiny and modify or revoke privileges if indicated. Remediation programs designed to assist impaired physicians may not be effective for those with cognitive impairment because the decline in cognitive functioning associated with illnesses such as Alzheimer’s disease often is progressive.13
‘Disruptive’ physicians
Mental illnesses such as personality or affective disorders, interpersonal problems within or outside the workplace, or other stressors could lead a physician to disrupt the workplace or patient care. Numerous programs have been established across the United States to help evaluate and treat disruptive physicians. Remediation programs can help identify and offer education for “dyscompetent” physicians (see Related Resources).13
To report, or not to report
In a national survey of physicians, only 45% of respondents indicated that they had notified the state licensing board of a colleague they felt was impaired or incompetent, yet almost all (96%) indicated that these individuals should be reported.19 Any duty to report requires, at minimum, that the physician be affected by an illness that impairs his or her cognition, concentration, rapid decision making, and ability to handle emergencies or perform work functions safely.20
Shouten20 cautions that someone who is considering filing a report because of fear of liability if they don’t should balance this concern against potential liability for breaching confidentiality. If there is evidence of an imminent risk or serious harm to the physician or patients, you may be legally required to breach confidentiality. Some states require licensed health practitioners to report acts of professional misconduct, unless the information is obtained solely from directly treating the physician. These requirements apply only within the state, and only to that state’s licensees.20-22
An ethical requirement to report also must be balanced against the obligation to maintain confidentiality. Ethics are largely a matter of individual standards, and individuals’ perceived ethical duties vary.
If you are considering reporting an impaired colleague, learn the laws in your state. If the physician is from another jurisdiction, the law provides little definitive guidance. Shouten recommends focusing on clinical outcomes for the doctor and his or her patients rather than on legal liability.20
Physician health programs
Nationwide directories of physician health programs are available from the Federation of State Physician Health Programs and the Federation of State Medical Boards (see Related Resources). Some programs are affiliated with state licensing boards, some are branches of state medical societies, and some are independent. These programs provide confidential treatment and assistance to practitioners experiencing substance or alcohol abuse, mental illness, or disruptive behavior. Some institutions may offer physicians an employee assistance program.
State medical societies may provide information about accessing a physician health program. Programs sponsored by medical societies almost always are independent of state licensing boards. This arrangement allows physicians to seek help without fear of punishment or censure or revocation of their license. Noncompliance with a physician health program, however, likely will result in being reported to the medical board.
Physician health programs typically employ a rehabilitative approach. Punitive measures such as reporting physicians to the medical board usually are not pursued unless the individual does not comply with treatment and monitoring guidelines. A physician who abuses substances, for example, may be required to complete a residential treatment program, attend support group meetings such as 12-step programs, participate in individual therapy, and undergo random screening for alcohol and illicit drug use.5
Abstinence is the goal of treating clinicians who abuse substances. Physicians have better outcomes than the general population, with reported abstinence rates of 70% to 90% for those who complete treatment;23,24 75% to 85% of physicians who complete rehabilitation and comply with close monitoring and follow-up care are able to return to work.24,25 Acceptance of recovery as a lifelong process, monitoring, and self-vigilance often are necessary to achieve and maintain abstinence.5
Risk factors for relapse include:
- denial of illness
- poor stress-coping and relationship skills
- social and professional isolation
- inability to accept feedback
- complacency and overconfidence
- failure to attend support group meetings
- dysfunctional family dynamics
- feelings of self-pity, blame, and guilt.5
Treating an impaired colleague. Reid26 recommends that psychiatrists should not evaluate or treat a self-referred, potentially impaired physician unless the relationship is strictly clinical. A physician may withhold symptoms, behaviors, or problems because his or her license, malpractice case, or career are at stake.
Advise a physician who requests evaluation or treatment related to license concerns or any legal matter to seek legal counsel. Working with such physician/patients only upon referral by a lawyer, licensing board, or physicians’ health committee provides treating psychiatrists with a clear professional role, allowing them to focus solely on the physician/patient’s treatment needs.
CASE CONTINUED: Extensive help, then success
The medical director of the hospital where Dr. S works refers him to his state’s impaired physician program. After investigating the complaints by the nurse and patient, the medical board suspends Dr. S’ license and requires him to enter a substance abuse treatment program. He completes an intensive residential program for impaired physicians and achieves sobriety from drugs and alcohol. His mood disorder is successfully treated with medications and psychotherapy. The medical board requires Dr. S to have a chaperone present for all visits with patients and submit random urine drug screens once his license is provisionally restored. The medical board also requires Dr. S to undergo ongoing psychiatric care and medication monitoring. He remains abstinent from alcohol and drugs, complies with the medical board’s requirements, and enjoys a productive practice and improved relationship with his family.
Disruptive physicians
- Samenow C, Swiggart W, Spickard A Jr. Consequence of physician disruptive behavior. Tenn Med. 2007;100(11):38-40.
- Leape LL, Fromson JA. Problem doctors: is there a system-level solution? Ann Intern Med. 2006;144(2):107-115.
- Linney BJ. Confronting the disruptive physician. Physician Exec. 1997;23:55-59.
Physician evaluation
- Anfang SA, Faulkner LR, Fromson JA, et al. The American Psychiatric Association’s resource document on guidelines for psychiatric fitness-for-duty evaluations of physicians. J Am Acad Psychiatry Law. 2005;33:85-88.
- Harmon L, Pomm R. Evaluation, treatment, and monitoring of disruptive physicians’ behavior. Psychiatr Ann. 2004; 34:770-774.
Other resources
- Federation of State Physician Health Programs. www.fsphp.org.
- Federation of State Medical Boards: Directory of Physician Assessment and Remedial Education Programs. www.fsmb.org/pdf/RemEdProg.pdf.
- The Center for Professional Health, Vanderbilt University Medical Center. www.mc.vanderbilt.edu/cph.
- Vanderbilt Comprehensive Assessment Program. www.mc.vanderbilt.edu/root/vcap.
- Alcoholics Anonymous. www.aa.org.
- Narcotics Anonymous. www.na.org.
Drug Brand Name
- Oxycodone/acetaminophen • Percocet
Disclosure
The authors report no financial relationship with the manufacturer of any product mentioned in this article or with manufacturers of competing products.
From all outward appearances Dr. S, a part-time psychiatrist at an inpatient psychiatric facility and in private practice for 12 years, is living the “perfect life,” with a wife, children, and successful practice.
In retrospect, his drug addiction began insidiously. In college, Dr. S continued to use oxycodone/acetaminophen prescribed for a shoulder injury long after his pain had resolved. He began to use cocaine in residency to help him “get through” the 36-hour call days, and it “helped” him earn the chief resident position because of his heightened energy and concentration. Dr. S’ primary care physician initially prescribed him benzodiazepines for anxiety and to help him sleep. Opiates were prescribed for a musculoskeletal injury. Dr. S obtained prescriptions for these medications from multiple providers. This ultimately escalated to self-prescription using aliases. Dr. S also began to drink heavily each evening.
Dr. S disregards colleagues’ comment about his obvious mood swings, which he attributes to his stressful job and “nagging” wife, despite having a family history of bipolar disorder. He becomes enraged when his wife or friends suggest he seek help. His colleagues whisper behind his back, but for years, no one confronts him about his unpredictable and frequently inappropriate behavior. Eventually, a nurse files a sexual harassment suit against Dr. S, and a patient complains to the medical board that Dr. S exhibited sexually inappropriate behavior during a therapy session.
As physicians, recognizing impairment in our colleagues or ourselves can be difficult. The American Medical Association defines an impaired physician as one who is unable to fulfill personal or professional responsibilities because of psychiatric illness, alcoholism, or drug dependence.1 Impairment is present when a physician is unable to perform in a manner that conforms to acceptable standards of practice, exhibits serious flaws in judgment, and provides incompetent care.1-3
Recognizing when a physician is impaired, deciding whether to report him or her to the state medical board, and referring a colleague for treatment can be challenging. This article will:
- review substance abuse, cognitive decline, and other causes of impairment
- address legal and ethical issues involved in reporting a colleague to the state medical board
- provide resources for physician treatment and assistance.
Physicians and addiction
Chemical dependence is the most frequent disabling illness among physicians,4 and substance abuse is the most common form of impairment that results in discipline by a state medical board.5 An estimated 6% to 8% of physicians abuse drugs, and approximately 14% develop alcohol use disorders; these rates are comparable to those of the general population.5 Psychiatrists, emergency room physicians, and solo practitioners are 3 times more likely to abuse substances than other doctors.6 An obsessive-compulsive personality and other factors may predispose physicians to substance abuse (Table 1).7,8
Alcohol is the most commonly abused substance, followed by opiates, cocaine, and other stimulants.9 Physicians are estimated to use opiates and benzodiazepines at a rate 5 times greater than that of the general public.10,11
An often-hidden problem. Physicians frequently deny substance abuse and many are able to conceal the problem from coworkers, even as their personal lives disintegrate.1,12 Marital and relationship problems may be the first indication of impairment, which gradually spreads to other aspects of their lives (Table 2).1,2,5 A doctor’s professional performance often is the last area to be affected.1,12
Substance abuse in physicians may long go unreported. The clinician’s family may want to protect the physician’s reputation, career, and income. Colleagues may be intimidated, uncertain of their concerns, or fearful for their jobs if they report the physician’s impairment. Patients may be reluctant to report their concerns because they depend on their provider for health care, respect the physician, or deny that a doctor could have a drug or alcohol problem.5
Table 1
Physicians and substance abuse: Predisposing factors
| Obsessive-compulsive personality style |
| Family history of substance use disorders or mental illness |
| Childhood family problems |
| Personal mental illness |
| Sensation-seeking behavior |
| Denial of personal and social problems |
| Perfectionism |
| Idealism |
| Source: References 7,8 |
Table 2
Signs of substance abuse
| Frequent tardiness and absences |
| Unexplained disappearances during working hours |
| Inappropriate behavior |
| Affective lability or irritability |
| Interpersonal conflict |
| Avoidance of peers or supervisors |
| Keeping odd hours |
| Disorganization and forgetfulness |
| Diminished chart completion and work performance |
| Heavy drinking at social functions |
| Unexplained changes in weight or energy level |
| Diminished personal hygiene |
| Slurred or rapid speech |
| Frequently dilated pupils or red and watery eyes and a runny nose |
| Defensiveness, anxiety, apathy, or manipulative behavior |
| Withdrawal from long-standing relationships |
| Source: References 1,2,5 |
Screening for cognitive decline
Many people with cognitive impairment lack insight into their problem and may minimize or deny the degree of their impairment.13 The prevalence of dementia in individuals age ≥65 is 3% to 11%,14 and 18% of physicians are in this age group.15
Ethical, legal, and practical issues arise in determining who, when, and how to screen physicians for cognitive problems. Standard screening exams may not be adequately sensitive for a well-educated physician, and neuropsychological testing may be necessary to detect mild cognitive impairment.13 In addition, the cognitive, visual-spacial, reactivity, reasoning, and calculation skills required for capable medical practice vary among specialties.16
One screening option is a “360-degree review” of information obtained from peers, patients, and non-physician colleagues that the College of Physicians and Surgeons in Alberta, Canada, has incorporated into its Physician’s Achievement Review (PAR) for physicians age ≥65.17 Compiled in a confidential manner and shared with the physician, the 360-degree survey assesses his or her:
- skill and knowledge
- psychosocial functioning
- management skills
- performance
- collegiality.
In Alberta, physicians who score in the lower one-third of the survey are assessed with an on-site evaluation by physicians from his or her specialty appointed by the PAR Director of Practice Improvement.
Alternately, physicians age ≥65 could be required to undergo annual or biannual neuropsychological testing to screen for mild cognitive impairment or other evidence of cognitive decline. In the United States, any screening requirement must be structured to comply with the Age Discrimination in Employment Act.18
If a physician shows evidence of cognitive impairment, the state medical board should initiate closer scrutiny and modify or revoke privileges if indicated. Remediation programs designed to assist impaired physicians may not be effective for those with cognitive impairment because the decline in cognitive functioning associated with illnesses such as Alzheimer’s disease often is progressive.13
‘Disruptive’ physicians
Mental illnesses such as personality or affective disorders, interpersonal problems within or outside the workplace, or other stressors could lead a physician to disrupt the workplace or patient care. Numerous programs have been established across the United States to help evaluate and treat disruptive physicians. Remediation programs can help identify and offer education for “dyscompetent” physicians (see Related Resources).13
To report, or not to report
In a national survey of physicians, only 45% of respondents indicated that they had notified the state licensing board of a colleague they felt was impaired or incompetent, yet almost all (96%) indicated that these individuals should be reported.19 Any duty to report requires, at minimum, that the physician be affected by an illness that impairs his or her cognition, concentration, rapid decision making, and ability to handle emergencies or perform work functions safely.20
Shouten20 cautions that someone who is considering filing a report because of fear of liability if they don’t should balance this concern against potential liability for breaching confidentiality. If there is evidence of an imminent risk or serious harm to the physician or patients, you may be legally required to breach confidentiality. Some states require licensed health practitioners to report acts of professional misconduct, unless the information is obtained solely from directly treating the physician. These requirements apply only within the state, and only to that state’s licensees.20-22
An ethical requirement to report also must be balanced against the obligation to maintain confidentiality. Ethics are largely a matter of individual standards, and individuals’ perceived ethical duties vary.
If you are considering reporting an impaired colleague, learn the laws in your state. If the physician is from another jurisdiction, the law provides little definitive guidance. Shouten recommends focusing on clinical outcomes for the doctor and his or her patients rather than on legal liability.20
Physician health programs
Nationwide directories of physician health programs are available from the Federation of State Physician Health Programs and the Federation of State Medical Boards (see Related Resources). Some programs are affiliated with state licensing boards, some are branches of state medical societies, and some are independent. These programs provide confidential treatment and assistance to practitioners experiencing substance or alcohol abuse, mental illness, or disruptive behavior. Some institutions may offer physicians an employee assistance program.
State medical societies may provide information about accessing a physician health program. Programs sponsored by medical societies almost always are independent of state licensing boards. This arrangement allows physicians to seek help without fear of punishment or censure or revocation of their license. Noncompliance with a physician health program, however, likely will result in being reported to the medical board.
Physician health programs typically employ a rehabilitative approach. Punitive measures such as reporting physicians to the medical board usually are not pursued unless the individual does not comply with treatment and monitoring guidelines. A physician who abuses substances, for example, may be required to complete a residential treatment program, attend support group meetings such as 12-step programs, participate in individual therapy, and undergo random screening for alcohol and illicit drug use.5
Abstinence is the goal of treating clinicians who abuse substances. Physicians have better outcomes than the general population, with reported abstinence rates of 70% to 90% for those who complete treatment;23,24 75% to 85% of physicians who complete rehabilitation and comply with close monitoring and follow-up care are able to return to work.24,25 Acceptance of recovery as a lifelong process, monitoring, and self-vigilance often are necessary to achieve and maintain abstinence.5
Risk factors for relapse include:
- denial of illness
- poor stress-coping and relationship skills
- social and professional isolation
- inability to accept feedback
- complacency and overconfidence
- failure to attend support group meetings
- dysfunctional family dynamics
- feelings of self-pity, blame, and guilt.5
Treating an impaired colleague. Reid26 recommends that psychiatrists should not evaluate or treat a self-referred, potentially impaired physician unless the relationship is strictly clinical. A physician may withhold symptoms, behaviors, or problems because his or her license, malpractice case, or career are at stake.
Advise a physician who requests evaluation or treatment related to license concerns or any legal matter to seek legal counsel. Working with such physician/patients only upon referral by a lawyer, licensing board, or physicians’ health committee provides treating psychiatrists with a clear professional role, allowing them to focus solely on the physician/patient’s treatment needs.
CASE CONTINUED: Extensive help, then success
The medical director of the hospital where Dr. S works refers him to his state’s impaired physician program. After investigating the complaints by the nurse and patient, the medical board suspends Dr. S’ license and requires him to enter a substance abuse treatment program. He completes an intensive residential program for impaired physicians and achieves sobriety from drugs and alcohol. His mood disorder is successfully treated with medications and psychotherapy. The medical board requires Dr. S to have a chaperone present for all visits with patients and submit random urine drug screens once his license is provisionally restored. The medical board also requires Dr. S to undergo ongoing psychiatric care and medication monitoring. He remains abstinent from alcohol and drugs, complies with the medical board’s requirements, and enjoys a productive practice and improved relationship with his family.
Disruptive physicians
- Samenow C, Swiggart W, Spickard A Jr. Consequence of physician disruptive behavior. Tenn Med. 2007;100(11):38-40.
- Leape LL, Fromson JA. Problem doctors: is there a system-level solution? Ann Intern Med. 2006;144(2):107-115.
- Linney BJ. Confronting the disruptive physician. Physician Exec. 1997;23:55-59.
Physician evaluation
- Anfang SA, Faulkner LR, Fromson JA, et al. The American Psychiatric Association’s resource document on guidelines for psychiatric fitness-for-duty evaluations of physicians. J Am Acad Psychiatry Law. 2005;33:85-88.
- Harmon L, Pomm R. Evaluation, treatment, and monitoring of disruptive physicians’ behavior. Psychiatr Ann. 2004; 34:770-774.
Other resources
- Federation of State Physician Health Programs. www.fsphp.org.
- Federation of State Medical Boards: Directory of Physician Assessment and Remedial Education Programs. www.fsmb.org/pdf/RemEdProg.pdf.
- The Center for Professional Health, Vanderbilt University Medical Center. www.mc.vanderbilt.edu/cph.
- Vanderbilt Comprehensive Assessment Program. www.mc.vanderbilt.edu/root/vcap.
- Alcoholics Anonymous. www.aa.org.
- Narcotics Anonymous. www.na.org.
Drug Brand Name
- Oxycodone/acetaminophen • Percocet
Disclosure
The authors report no financial relationship with the manufacturer of any product mentioned in this article or with manufacturers of competing products.
1. Breiner SJ. The impaired physician. J Med Educ. 1979;54:673.-
2. Talbott GD, Gallegos KV, Angres DH. Impairment and recovery in physicians and other health professionals. In: Graham AW, Schultz TK, eds. Principles of addiction medicine. 2nd ed. Chevy Chase, MD: American Society of Addiction Medicine, Inc; 1998:1263-1277.
3. American Medical Association Council on Mental Health. The sick physician: impairment by psychiatric disorders, including alcoholism and drug dependence. JAMA. 1973;223:684-687.
4. Talbott G, Wright C. Chemical dependency in healthcare professionals. Occup Med. 1987;2:581-591.
5. Baldisseri MR. Impaired healthcare professional. Crit Care Med. 2007;35:S106-116.
6. Mansky PA. Physician health programs and the potentially impaired physician with a substance use disorder. Psychiatr Serv. 1996;47:465-467.
7. Boisaubin EV, Levine RE. Identifying and assisting the impaired physician. Am J Med Sci. 2001;322:31-36.
8. Bissel L, Jones RW. The alcoholic physician: a survey. Am J Psychiatry. 1976;133:1142-1146.
9. Robins L, Reiger D. Psychiatric disorders in America: the Epidemiologic Catchment Area Study. New York, NY: The Free Press; 1991.
10. Gallegos KV, Browne CH, Veit FW, et al. Addiction in anesthesiologists: drug access and patterns of substance abuse. QRB Qual Rev Bull. 1988;14:116-122.
11. Hughes PH, Brandenburg N, Baldwin DC, et al. Prevalence of substance abuse among U.S. physicians. JAMA. 1992;267:2333-2339.
12. Vaillant GE, Clark W, Cyrus C, et al. Prospective study of alcoholism treatment. Eight year follow-up. Am J Med. 1983;75:455-463.
13. LoboPrabhu SM, Molinari VA, Hamilton JD, et al. The aging physician with cognitive impairment: approaches to oversight, prevention and remediation. Am J Geriatr Psychiatry. 2009;17:445-454.
14. U.S. General Accounting Office. Alzheimer’s disease: estimates of prevalence in the United States. Washington, DC: U.S. General Accounting Office; 1998:98. Publication GAO/HEHS-98-16.
15. American Medical Association. Physician characteristics and distribution in the U.S., 2006. Washington, DC: American Medical Association; 2006.
16. Blasier R. The problem of the aging surgeon. Clin Orthop Relat Res. 2009;467:402-411.
17. College of Physicians and Surgeons of Alberta. Physician Achievement Review (PAR) Program. Available at: http://www.cpsa.ab.ca/Services/PARprogram/Overview.aspx. Accessed March 27, 2010.
18. The Age Discrimination in Employment Act. (Vol. Pub. L. No. 90-202, 81 Stat. 602 (Dec. 15, 1967), codified as Chapter 14 of Title 29 of the United States Code, 29 U.S.C. § 621 through 29 U.S.C. § 63), 1967.
19. Campbell EG, Regan S, Gruen RL, et al. Professionalism in medicine: results of a national survey of physicians. Ann Int Med. 2007;147:795-802.
20. Shouten R. Impaired physicians: is there a duty to report to state licensing boards? Harvard Rev Psychiatry. 2000;8:36-39.
21. Mass Gen Laws ch 112 § 5F.
22. NY PHL § 230 (11) (e).
23. Femino J, Nirenberg TD. Treatment outcome studies on physician impairment: a review of the literature. R I Med. 1994;77:345-350.
24. Alpern F, Correnti CE, Dolan TE, et al. A survey of recovering Maryland physicians. Md Med J. 1992;41:301-303.
25. Gallegos KV, Lubin BH, Bowers C, et al. Relapse and recovery: five to ten year follow-up study of chemically dependent physicians—the Georgia experience. Md Med J. 1992;41:315-319.
26. Reid W. Evaluating and treating disabled or impaired colleagues. J Psychiatr Pract. 2007;13:44-48.
1. Breiner SJ. The impaired physician. J Med Educ. 1979;54:673.-
2. Talbott GD, Gallegos KV, Angres DH. Impairment and recovery in physicians and other health professionals. In: Graham AW, Schultz TK, eds. Principles of addiction medicine. 2nd ed. Chevy Chase, MD: American Society of Addiction Medicine, Inc; 1998:1263-1277.
3. American Medical Association Council on Mental Health. The sick physician: impairment by psychiatric disorders, including alcoholism and drug dependence. JAMA. 1973;223:684-687.
4. Talbott G, Wright C. Chemical dependency in healthcare professionals. Occup Med. 1987;2:581-591.
5. Baldisseri MR. Impaired healthcare professional. Crit Care Med. 2007;35:S106-116.
6. Mansky PA. Physician health programs and the potentially impaired physician with a substance use disorder. Psychiatr Serv. 1996;47:465-467.
7. Boisaubin EV, Levine RE. Identifying and assisting the impaired physician. Am J Med Sci. 2001;322:31-36.
8. Bissel L, Jones RW. The alcoholic physician: a survey. Am J Psychiatry. 1976;133:1142-1146.
9. Robins L, Reiger D. Psychiatric disorders in America: the Epidemiologic Catchment Area Study. New York, NY: The Free Press; 1991.
10. Gallegos KV, Browne CH, Veit FW, et al. Addiction in anesthesiologists: drug access and patterns of substance abuse. QRB Qual Rev Bull. 1988;14:116-122.
11. Hughes PH, Brandenburg N, Baldwin DC, et al. Prevalence of substance abuse among U.S. physicians. JAMA. 1992;267:2333-2339.
12. Vaillant GE, Clark W, Cyrus C, et al. Prospective study of alcoholism treatment. Eight year follow-up. Am J Med. 1983;75:455-463.
13. LoboPrabhu SM, Molinari VA, Hamilton JD, et al. The aging physician with cognitive impairment: approaches to oversight, prevention and remediation. Am J Geriatr Psychiatry. 2009;17:445-454.
14. U.S. General Accounting Office. Alzheimer’s disease: estimates of prevalence in the United States. Washington, DC: U.S. General Accounting Office; 1998:98. Publication GAO/HEHS-98-16.
15. American Medical Association. Physician characteristics and distribution in the U.S., 2006. Washington, DC: American Medical Association; 2006.
16. Blasier R. The problem of the aging surgeon. Clin Orthop Relat Res. 2009;467:402-411.
17. College of Physicians and Surgeons of Alberta. Physician Achievement Review (PAR) Program. Available at: http://www.cpsa.ab.ca/Services/PARprogram/Overview.aspx. Accessed March 27, 2010.
18. The Age Discrimination in Employment Act. (Vol. Pub. L. No. 90-202, 81 Stat. 602 (Dec. 15, 1967), codified as Chapter 14 of Title 29 of the United States Code, 29 U.S.C. § 621 through 29 U.S.C. § 63), 1967.
19. Campbell EG, Regan S, Gruen RL, et al. Professionalism in medicine: results of a national survey of physicians. Ann Int Med. 2007;147:795-802.
20. Shouten R. Impaired physicians: is there a duty to report to state licensing boards? Harvard Rev Psychiatry. 2000;8:36-39.
21. Mass Gen Laws ch 112 § 5F.
22. NY PHL § 230 (11) (e).
23. Femino J, Nirenberg TD. Treatment outcome studies on physician impairment: a review of the literature. R I Med. 1994;77:345-350.
24. Alpern F, Correnti CE, Dolan TE, et al. A survey of recovering Maryland physicians. Md Med J. 1992;41:301-303.
25. Gallegos KV, Lubin BH, Bowers C, et al. Relapse and recovery: five to ten year follow-up study of chemically dependent physicians—the Georgia experience. Md Med J. 1992;41:315-319.
26. Reid W. Evaluating and treating disabled or impaired colleagues. J Psychiatr Pract. 2007;13:44-48.
Lowering risk of Alzheimer’s disease
Pharmacologic treatments for Alzheimer’s disease (AD) may improve symptoms but have not been shown to prevent AD onset. Primary prevention therefore remains the goal. Although preventing AD by managing risk factors such as age or genetics is beyond our control (Box 1), we can do something about other factors.
This article summarizes the findings of many studies that address AD prevention and includes an online-only bibliography for readers seeking an in-depth review. The evidence does not support a firm recommendation for any specific form of primary prevention and has revealed hazards associated with estrogen therapy and nonsteroidal anti-inflammatory drugs (Box 2). Most important, it suggests that you could reduce your patients’ risk of developing AD by routinely supporting their mental, physical, and social health.
The potential benefits of modifying an individual’s AD risk factors likely will depend on his or her genetic makeup, environment, and lifestyle. Even so, counseling patients to exercise more and improve their diets—such as by eating more fish, fruits, and vegetables and less saturated fat—might help protect the brain. Your ongoing efforts to manage hypertension, hypercholesterolemia, and diabetes also may reduce their AD risk.
Age remains the strongest risk factor for dementia, particularly for Alzheimer’s disease (AD).a The risk of developing AD doubles every 5 years after age 65 and approaches 50% after age 85.b
Family history is a risk factor for AD, although true familial AD accounts for <5% of cases.c When diseases show a familial pattern, either genetics, environmental factors, or both may play a role. Patients with a first-degree relative with dementia have a 10% to 30% increased risk of developing the disorder.d
Genetic factors play a role in both early-onset and late-onset AD. Early-onset AD (before age 65) accounts for 6% to 7% of cases.e From this small pool of patients, only 13% exhibit clear autosomal dominant transmission over >1 generation.f Three gene mutations have been associated with early-onset AD:
- 30% to 70% are in the presenilin-1 gene
- 10% to 15% are in the amyloid precursor protein gene
- <5% are in the presenilin-2 gene.g,h
For late-onset AD (after age 65), the strongest evidence for a genetic risk factor exists for the epsilon 4 allele of the apolipoprotein E gene (APOE e4).i This genotype has been linked to the development of AD and possibly to vascular dementia.j,k In contrast, the epsilon 2 allele of APOE may exert a protective effect in AD.l APOE e3, the most common allele, appears to play a neutral role in the development of AD.
References
a. Evans DA. The epidemiology of dementia and Alzheimer’s disease: an evolving field. J Am Geriatr Soc. 1996;44:1482-1483.
b. Jorm AF, Jolley D. The incidence of dementia: a meta-analysis. Neurology. 1998;51:728-733.
c. van Duijn CM, Clayton D, Chandra V, et al. Familial aggregation of Alzheimer’s disease and related disorders: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int J Epidemiol. 1991;20(suppl 2):S13-S20.
d. Chang JB, Wang PN, Chen WT, et al. ApoE epsilon4 allele is associated with incidental hallucinations and delusions in patients with AD. Neurology. 2004;63:1105-1107.
e. Sleegers K, Roks G, Theuns J, et al. Familial clustering and genetic risk for dementia in a genetically isolated Dutch population. Brain. 2004;127:1641-1649.
f. Schoenberg BS, Anderson DW, Haerer AF. Severe dementia. Prevalence and clinical features in a biracial US population. Arch Neurol. 1985;42:740-743.
g. Hsiung GY, Sadovnick AD. Genetics and dementia: risk factors, diagnosis and management. Alzheimers Dement. 2007;3:418-427.
h. GeneTests database. Available at: http://www.genetests.org. Accessed March 19, 2010.
i. Li H, Wetten S, Li L, et al. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch Neurol. 2008;65:45-53.
j. Graff-Radford NR, Green RC, Go RC, et al. Association between apolipoprotein E genotype and Alzheimer disease in African American subjects. Arch Neurol. 2002;59:594-600.
k. Slooter AJ, Cruts M, Hofman A, et al. The impact of APOE on myocardial infarction, stroke, and dementia: the Rotterdam Study. Neurology. 2004;62:1196-1198.
l. Tiraboschi P, Hansen LA, Masliah E, et al. Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology. 2004;62:1977-1983.
Estrogen. Before the Women’s Health Initiative (WHI) study, various trials of the effects of estrogen therapy on the development of Alzheimer’s disease (AD) in women age ≥65 showed inconsistent results. In the randomized, placebo-controlled WHI Memory Study, conjugated equine estrogen, 0.625 mg/d, plus medroxyprogesterone acetate, 2.5 mg/d, did not prevent mild cognitive impairment or improve global cognitive function and was associated with an increased risk for probable dementia.a Based on this evidence, conjugated equine estrogen with or without medroxyprogesterone is not recommended as therapy to protect cognitive function in older women.
NSAID therapy. Cytokine-mediated inflammation may play a role in neurodegenerative disorders and cognitive impairment in the elderly. Nonsteroidal anti-inflammatory drugs (NSAIDs), including cyclooxygenase-2 (COX-2) inhibitors, have been studied for a possible protective effect against AD and cognitive decline,b possibly by lowering amyloidogenic proteins.c A 1-year randomized controlled trial by the Alzheimer’s Disease Cooperative Consortium found no significant differences in cognition scores of patients treated with once-daily rofecoxib, 25 mg, or twice-daily naproxen sodium, 220 mg, when compared with placebo.d Similarly, naproxen and celecoxib did not prevent AD in the randomized, controlled Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT).e Rofecoxib has been withdrawn from the market, and celecoxib labeling carries a warning of potential for increased risk of cardiovascular events and life-threatening gastrointestinal bleeding associated with its use.
NSAIDs and COX-2 inhibitors are not recommended for the treatment or prevention of dementia or cognitive impairment. Their use for AD prevention is not supported by randomized clinical trialsd,e and they may have serious adverse effects.
References
a. Shumaker SA, Legault C, Kuller L, et al. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. JAMA. 2004;291:2947-2958.
b. Szekely CA, Breitner JC, Fitzpatrick AL, et al. NSAID use and dementia risk in the Cardiovascular Health Study: role of APOE and NSAID type. Neurology. 2008;70:17-24.
c. Weggen S, Eriksen JL, Das P, et al. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001;414:212-216.
d. Aisen PS, Schafer KA, Grundman M, et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003;289(21):2819-2826.
e. ADAPT Research Group, Martin BK, Szekely C, Brandt J, et al. Cognitive function over time in the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch Neurol. 2008;65(7):896-905.
Cardiovascular risk factors
The risk of developing AD or vascular dementia appears to be increased by conditions that damage the heart or blood vessels. Recent evidence suggests that successfully managing cardiovascular risk factors may decrease the likelihood of dementia in later life.
Hypertension is associated with a higher risk of AD and all-cause dementia. Curiously, some studies have shown that low blood pressure also increases dementia risk, suggesting a U-shaped relationship between blood pressure and cognitive decline. Systolic hypertension in midlife may be associated with dementia 20 years later.
One might assume that antihypertensive therapy would help prevent dementia, but the data are conflicting. The Systolic Hypertension in Europe (SYST-EUR) study1 showed a 53% reduction in vascular dementia or mixed dementia among patients receiving antihypertensive medication and a 60% reduction in AD. Similarly, the PROGRESS2 clinical trial of prevention of recurrent stroke by antihypertensive treatment reported a 34% reduction in a composite measure of cognitive impairment and dementia. On the other hand, cognitive function neither improved nor worsened in the Hypertension in the Very Elderly Trial (HYVET-COG),3 whether patients received blood pressure treatment or placebo.
Hyperlipidemia. Lipid metabolism likely is an important pathway in amyloid beta-protein deposition, tau phosphorylation, and disruption of synaptic plasticity and neurodegenerative endpoints. Cognitive decline and incident dementia have been associated with higher dietary intake of saturated fats, partially hydrogenated unsaturated fatty acids (trans fats), and cholesterol. Not all studies have found this association, however. This could be because serum cholesterol levels may decrease in early dementia, limiting the ability to detect an effect of hypercholesterolemia on dementia risk when measurements are made later in life.
Using statins (3-hydroxy-3-methylglutaryl–coenzyme A reductase inhibitors) to treat hypercholesterolemia has been hypothesized to impede large vessel atherosclerosis and its consequences and to trigger metabolic effects in the brain related to AD pathogenesis. Mechanisms by which statins might help prevent dementia include:
- a direct association between amyloid processing and cholesterol in the brain
- an indirect effect by decreasing the risk of stroke, as even small cerebral infarcts worsen AD severity.
Nonrandomized epidemiologic studies such as the Cardiovascular Health Study4 and MRC/BHF Heart Protection Study5 suggested that statin treatment might reduce the incidence of dementia, the degree of age-related cognitive decline, and AD’s neuropathologic burden. Large, randomized, controlled trials have not supported these observations, however. Statins failed to reduce the incidence of dementia in:
- the Heart Protection Study, testing simvastatin for 5 years in 20,536 subjects age 40 to 805
- the 3-year Preventive Study of Pravastatin in the Elderly at Risk (PROSPER) of 5,800 subjects.6
Similarly, patients receiving adjunctive atorvastatin or placebo showed no significant differences in cognition assessments after 72 weeks in the Lipitor’s Effect in Alzheimer’s Dementia (LEADe) study. This trial enrolled 640 subjects age 50 to 90 with mild-to-moderate dementia who were treated with donepezil.7 A recent Cochrane review concluded that high serum cholesterol may contribute to the development of AD and vascular dementia, but lowering cholesterol levels with statins does not prevent these problems.8
Diabetes mellitus. Diabetes and cognitive decline are closely associated. Diabetes is associated with a 50% to 100% increase in risk of AD and dementia overall and a 100% to 150% increased risk of vascular dementia. The mechanism by which diabetes increases dementia risk is uncertain but does not appear to be mediated entirely through vascular disease. High and low insulin levels may increase the risk of dementia, independent of diabetes and blood glucose. Increased peripheral insulin levels are associated with reduced brain atrophy and cognitive impairment in patients with early AD, suggesting a role for insulin signaling in AD pathophysiology. A possible relationship between insulin and beta amyloid metabolism is being studied.
Elevated postprandial plasma glucose has been associated with accelerated declines in cognitive performance.9 An inverse correlation has been noted between some cognitive measures and hemoglobin A1C levels.10 It is not clear that treating diabetes reduces the risk of dementia. In addition, in the prospective, population-based Rotterdam study, elderly patients with type 2 diabetes treated with insulin had the highest incidence of dementia.11
Tobacco smoke directly affects neuronal function, integrity, and survival. Chronic smoking has been linked to decreased global cerebral blood flow, accelerated cerebral atrophy, and ventricular enlargement.
Some studies suggest an increased risk of dementia in middle-aged and elderly smokers, possibly through a cerebrovascular mechanism such as stroke. Other studies found no association between smoking and dementia risk, and 1 suggested that nicotine may protect against AD by reducing senile plaque formation. Any protective effect of smoking would be offset by increased risks of lung cancer, chronic obstructive pulmonary disease, and vascular dementia.
The apolipoprotein E epsilon 4 (APOE e4) gene may explain, at least in part, the conflicting results of these studies. In 2 population-based cohorts,12,13 smoking was associated with memory decline in patients without, but not with, the APOE e4 genotype.
Dietary factors
Antioxidants. The brains of patients with AD contain elevated levels of endogenous antioxidants. In vitro studies show exogenous antioxidants can reduce the toxicity of beta-amyloid in brain tissue of persons with AD. These findings have led to interest in assessing the role of dietary antioxidants such as vitamins E and C for AD prevention.
High-dose alpha-tocopherol (vitamin E, 2,000 IU/d) may slow disease progression in patients with AD, but this association is not consistently found. Furthermore, a meta-analysis of 19 randomized controlled trials (RCTs) totaling >135,000 patients found an association between vitamin E doses >400 IU/d and increased all-cause mortality.14 High-dose vitamin E supplementation for primary or secondary prevention of AD may be dangerous and is not recommended.
The lack of consistent efficacy data for vitamin C in preventing or treating AD may discourage its routine use for this purpose.15
Homocysteine is a risk factor for stroke and heart disease. It also could play a role in vascular dementia through its association with large- and small-vessel disease.
Low folate and hyperhomocysteinemia have been associated with dementia or cognitive impairment, although a cause-effect relationship is not clear. In non-demented elderly populations, plasma homocysteine is inversely associated with poor performance in tests of global cognitive function, particularly in measures of psychomotor speed.
In a recent double-blind RCT, folic acid supplementation for 3 years significantly improved domains of cognitive function that tend to decline with age, especially information processing and sensorimotor speed.16 No other good evidence, however, has shown that homocysteine-lowering therapy using folic acid or other vitamin B supplements improves cognitive function or prevents cognitive decline.
Fish and omega-3 fatty acids. High total fat, saturated fat, and total cholesterol intake increases the risk for incident dementia. In epidemiologic studies, low omega-3 fatty acid serum levels have been linked to increased dementia risk.
Fish consumption may be beneficial in reducing the risk of dementia or cognitive decline. A prospective study of 815 elderly persons found 60% less risk of developing AD in those who ate ≥1 fish meal per week, compared with those who rarely or never ate fish.17 In the Framingham study, individuals who at baseline were in the top quartile of docosahexaenoic acid consumption had lower dementia rates over 9 years of follow-up.18 Results from cross-sectional and longitudinal studies have been inconsistent; some have shown that high intake of n-3 polyunsaturated fatty acids is associated with less cognitive decline,19 whereas others have not.20
Although we cannot offer unequivocal advice regarding seafood or omega-3 fatty acid intake for primary prevention of dementia without evidence from RCTs, these uncontrolled studies show promise.
Mediterranean diet (MeDi) components include abundant fruits and vegetables, fish or shellfish at least twice weekly, very limited red meat, olive oil or canola oil instead of butter or margarine, tree nuts such as walnuts or pecans, red wine in moderation, and using herbs and spices instead of salt to season food. High adherence to the MeDi has been associated with a significantly lower risk for incident AD. The MeDi may affect the risk of developing AD21 as well as subsequent disease course, with a possible dose-response relationship in lower mortality.22
Eating fruits and vegetables has been associated with improved cognitive performance22 and decreased incident dementia in elderly subjects.18
Alcohol. A U-shaped relationship exists between alcohol consumption and dementia risk. High alcohol intake is associated with clinical problem drinking and alcoholism and can lead to cognitive decline. Conversely, moderate wine consumption (250 to 500 mL/d) may be protective—compared with more or less than this amount—and is associated with approximately 50% less risk of dementia.
Alcohol use may increase the risk of dementia in persons carrying the APOE e4 allele, according to the population-based Cardiovascular Risk Factors, Aging and Dementia (CAIDE) study from Sweden.23 After an average 21 years of follow-up of 1,449 individuals, researchers found that environmental factors—such as physical inactivity, dietary fat intake, alcohol consumption, and smoking at midlife—were associated with an increased risk of dementia at age 65 to 79 in APOE e4 carriers compared with noncarriers. The study also found that physical inactivity, dietary fat intake, and smoking at midlife increase AD risk, especially among APOE e4 carriers.
In the absence of evidence from RCTs, we cannot recommend alcohol to reduce the risk of AD.
Lifestyle and activity
Three components of lifestyle—social, mental, and physical activity—are inversely associated with the risk for dementia, AD, and cognitive impairment.
Physical exercise has been thought to enhance brain neurotrophic factor and modify apoptosis. Exercise can deter stroke and microvascular disease and improve regional cerebral blood flow. In the Cardiovascular Health Study, participants who expended the highest quartile of energy had a lower risk of all-cause dementia and AD compared with participants who expended the lowest quartile of energy.24
Mental and social activity. Epidemiologic studies have shown associations between higher educational achievement and other socioeconomic factors and reduced AD risk. Advanced education is believed to represent a cognitive reserve that delays presentation of AD’s effects on memory and cognitive function, rather than providing a protective effect against accumulation of AD pathology. Higher-educated individuals appear to experience a somewhat more rapid rate of cognitive decline when AD does become apparent, perhaps because they have accumulated a greater degree of AD pathology at that point compared with less-educated persons.
Among 117 persons with dementia in the Bronx Aging Study, each additional year of formal education delayed the time of accelerated decline by 0.21 years. After accelerated decline began, each year of additional formal education was associated with a slightly faster rate of memory decline.25
The longitudinal, population-based Kungsholmen Project in Stockholm, Sweden, found an association between daily mentally stimulating activities and decreased risk of all-cause dementia.26 Similarly, higher levels of leisure activity were linked to reduced risk of all-cause dementia in a longitudinal study of 1,772 persons age ≥65 living in Manhattan, NY.27 In a randomized, single-controlled study of the long-term effects of cognitive training, elderly individuals from 6 U.S. cities showed sustained improvement in specific cognitive performance up to 5 years after training sessions began, including memory, reasoning, and speed of processing.28
It seems reasonable to encourage older patients to maintain or increase physical, cognitive, and leisure activities as well as social interaction. These interventions can improve the quality of life and lower the risk of depression, which may be a response to cognitive decline or an independent risk factor for dementia (Box 3). The Table lists “brain exercises” you can suggest to patients to increase their mental and social activity.
Head trauma. The Multi-Institutional Research in Alzheimer’s Genetic Epidemiology (MIRAGE) project found an association between AD risk and a history of head trauma, especially in persons with APOE e4 alleles.29 Conversely, the Rotterdam Study showed no change in dementia risk for persons with a history of head trauma.30
Even in the absence of conclusive evidence supporting AD prevention, protecting the head by buckling seat belts while driving, wearing helmets when participating in sports, and “fall-proofing” the home is recommended.
Depression often occurs before or as a coexisting condition with Alzheimer’s disease (AD).a Although depression has been considered a response to cognitive decline or an early manifestation of dementia,b it also could be an independent risk factor.c,d
The pathologic mechanism linking depression and subsequent dementia is not well understood. Hypotheses include an indirect neurotoxic effect of depression mediated by cortisol-induced hippocampal atrophy or lowered brain-derived neurotrophic factor levels.e Depression and dementia might share genetic links, although a cohort study of 404 individuals with AD detected no association between apolipoprotein E genotypes or alleles and depressive symptoms.f
References
a. Lupien SJ, Nair NP, Brière S, et al. Increased cortisol levels and impaired cognition in human aging: implication for depression and dementia in later life. Rev Neurosci. 1999;10(2):117-139.
b. Preuss UW, Siafarikas N, Petrucci M, et al. Depressive disorders in dementia and mild cognitive impairments: is comorbidity a cause or a risk factor? Fortschr Neurol Psychiatr. 2009;77:399-406.
c. Green RC, Cupples LA, Kurz A, et al. Depression as a risk factor for Alzheimer disease: the MIRAGE Study. Arch Neurol. 2003;60(5):753-759.
d. Ownby RL, Crocco E, Acevedo A, et al. Depression and risk for Alzheimer’s disease: systematic review, meta-analysis, and metaregression analysis. Arch Gen Psychiatry. 2006;63(5):530-538.
e. Meeks TW, Ropacki SA, Jeste DV. The neurobiology of neuropsychiatric syndromes in dementia. Curr Opin Psychiatry. 2006;19(6):581-586.
f. Craig D, Hart DJ, McIlroy SP, et al. Association analysis of apolipoprotein E genotype and risk of depressive symptoms in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2005;19(2-3):154-157.
Table
Brain exercises to suggest to patients
| Learn something new (how to play a musical instrument, a foreign language, or a new hobby) |
| Play memory games |
| Practice using the opposite hand to perform tasks you usually do with your dominant hand |
| Read, especially challenging material |
| Join a book discussion group |
| Write; if not a book or article, write a diary, letters, or emails or start your memoirs |
| Do crossword, Sudoku, or jigsaw puzzles |
| Play board games, card games, and other strategy games |
| Debate or discuss topics |
Related resource
- For an extensive bibliography of literature on Alzheimer’s disease risk factors and prevention, see this article at CurrentPsychiatry.com.
Drug brand names
- Atorvastatin • Lipitor
- Celecoxib • Celebrex
- Donepezil • Aricept
- Medroxyprogesterone • Provera
- Pravastatin • Pravachol
- Rofecoxib • Vioxx
- Simvastatin • Zocor
Disclosures
Dr. Bassil reports no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
Dr. Grossberg receives research/grant support from and is a consultant to Bristol-Myers Squibb, Forest Pharmaceuticals, Novartis, Pfizer Inc., and Wyeth Pharmaceuticals. He also receives research/grant support from Baxter.
1. Forette F, Seux ML, Staessen JA, et al. The prevention of dementia with antihypertensive treatment: new evidence from the systolic hypertension in Europe (Syst-Eur) study. Arch Intern Med. 2002;162:2046-2052.
2. Tzourio C, Anderson C, Chapman N, et al. Effects of blood pressure lowering with perindopril and indapamide therapy on dementia and cognitive decline in patients with cerebrovascular disease. Arch Intern Med. 2003;163:1069-1075.
3. Peters R, Beckett N, Forette F. Incident dementia and blood pressure lowering in the Hypertension in the Very Elderly Trial cognitive function assessment (HYVET-COG). Lancet Neurol. 2008;7(8):683-689.
4. Rea TD, Breitner JC, Psaty BM, et al. Statin use and the risk of incident dementia: the Cardiovascular Health Study. Arch Neurol. 2005;62:1047-1051.
5. Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:7-22.
6. Kulbertus H, Scheen AJ. [The PROSPER Study (PROspective study of pravastatin in the elderly at risk)]. Rev Med Liege. 2002;57(12):809-813.
7. Feldman HH, Doody RS, Kivipelto M, et al. Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADe. Neurology. 2010;74(12):956-964.
8. McGuinness B, Bullock R, Craig D, et al. Statins for the treatment of Alzheimer’s disease and dementia (protocol). Cochrane Database Syst Rev. 2009;1:CD007514.-
9. Abbatecola AM, Rizzo MR, Barbieri M, et al. Postprandial plasmaglucose excursions and cognitive functioning in aged type 2 diabetics. Neurology. 2006;67:235-240.
10. Munshi M, Grande L, Hayes M, et al. Cognitive dysfunction is associated with poor diabetes control in older adults. Diabetes Care. 2006;29:1794-1799.
11. Ott A, Stolk RP, van Harskamp F, et al. Diabetes mellitus and the risk of dementia. The Rotterdam study. Neurology. 1999;53:1937-1942.
12. Reitz C, Luchsinger J, Tang MX, et al. Effect of smoking and time on cognitive function in the elderly without dementia. Neurology. 2005;65:870-875.
13. Reitz C, den Heijer T, van Duijn C, et al. Relation between smoking and risk of dementia and Alzheimer disease: the Rotterdam Study. Neurology. 2007;69:998-1005.
14. Miller ER, III, Pastor-Barriuso R, Dalal D, et al. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med. 2005;142(1):37-46.
15. Boothby LA, Doering PL. Vitamin C and vitamin E for Alzheimer’s disease. Ann Pharmacother. 2005;39(12):2073-2080.
16. Durga J, van Boxtel MP, Schouten EG, et al. Effect of 3-year folic acid supplementation on cognitive function in older adults in the FACIT trial: a randomised, double blind, controlled trial. Lancet. 2007;369:208-216.
17. Morris MC, Evans DA, Bienias JL, et al. Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch Neurol. 2003;60:940-946.
18. Schaefer EJ, Bongard V, Beiser AS, et al. Plasma phosphatidylcholine docosahexaenoic acid content and risk of dementia and Alzheimer disease: the Framingham Heart Study. Arch Neurol. 2006;63:1545-1550.
19. Kalmijn S, Launer LJ, Ott A, et al. Dietary fat intake and the risk of incident dementia in the Rotterdam Study. Ann Neurol. 1997;42:776-782.
20. van Gelder BM, Tijhuis M, Kalmijn S, et al. Fish consumption, n-3 fatty acids, and subsequent 5-y cognitive decline in elderly men: the Zutphen Elderly Study. Am J Clin Nutr. 2007;85:1142-1147.
21. Solfrizzi V, Capurso C, Panza F. Adherence to a Mediterranean dietary pattern and risk of Alzheimer’s disease. Ann Neurol. 2006;60:620.-
22. Scarmeas N, Luchsinger JA, Mayeux R, et al. Mediterranean diet and Alzheimer disease mortality. Neurology. 2007;69(11):1084-1093.
23. Kivipelto M, Rovio S, Ngandu T, et al. Apolipoprotein E epsilon4 magnifies lifestyle risks for dementia: a population-based study. J Cell Mol Med. 2008;12(6B):2762-2771.
24. Podewils LJ, Guallar E, Kuller LH, et al. Physical activity, APOE genotype and dementia risk: findings from the Cardiovascular Health Cognition Study. Am J Epidemiol. 2005;161:639-651.
25. Hall CB, Derby C, LeValley A, et al. Education delays accelerated decline on a memory test in persons who develop dementia. Neurology. 2007;69:1657-1664.
26. Wang HX, Karp A, Winblad B, et al. Late-life engagement in social and leisure activities is associated with a decreased risk of dementia: a longitudinal study from the Kungsholmen Project. Am J Epidemiol. 2002;155:1081-1087.
27. Scarmeas N, Levy G, Tang MX, et al. Influence of leisure activity on the incidence of Alzheimer’s disease. Neurology. 2001;57:2236-2242.
28. Willis SL, Tennstedt SL, Marsiske M, et al. Long-term effects of cognitive training on everyday functional outcomes in older adults. JAMA. 2006;296:2805-2814.
29. Guo Z, Cupples LA, Kurz A, et al. Head injury and the risk of AD in the MIRAGE study. Neurology. 2000;54:1316-1323.
30. Ruitenberg A, van Swieten JC, Witteman JC, et al. Alcohol consumption and risk of dementia: the Rotterdam Study. Lancet. 2002;359:281-286.
Pharmacologic treatments for Alzheimer’s disease (AD) may improve symptoms but have not been shown to prevent AD onset. Primary prevention therefore remains the goal. Although preventing AD by managing risk factors such as age or genetics is beyond our control (Box 1), we can do something about other factors.
This article summarizes the findings of many studies that address AD prevention and includes an online-only bibliography for readers seeking an in-depth review. The evidence does not support a firm recommendation for any specific form of primary prevention and has revealed hazards associated with estrogen therapy and nonsteroidal anti-inflammatory drugs (Box 2). Most important, it suggests that you could reduce your patients’ risk of developing AD by routinely supporting their mental, physical, and social health.
The potential benefits of modifying an individual’s AD risk factors likely will depend on his or her genetic makeup, environment, and lifestyle. Even so, counseling patients to exercise more and improve their diets—such as by eating more fish, fruits, and vegetables and less saturated fat—might help protect the brain. Your ongoing efforts to manage hypertension, hypercholesterolemia, and diabetes also may reduce their AD risk.
Age remains the strongest risk factor for dementia, particularly for Alzheimer’s disease (AD).a The risk of developing AD doubles every 5 years after age 65 and approaches 50% after age 85.b
Family history is a risk factor for AD, although true familial AD accounts for <5% of cases.c When diseases show a familial pattern, either genetics, environmental factors, or both may play a role. Patients with a first-degree relative with dementia have a 10% to 30% increased risk of developing the disorder.d
Genetic factors play a role in both early-onset and late-onset AD. Early-onset AD (before age 65) accounts for 6% to 7% of cases.e From this small pool of patients, only 13% exhibit clear autosomal dominant transmission over >1 generation.f Three gene mutations have been associated with early-onset AD:
- 30% to 70% are in the presenilin-1 gene
- 10% to 15% are in the amyloid precursor protein gene
- <5% are in the presenilin-2 gene.g,h
For late-onset AD (after age 65), the strongest evidence for a genetic risk factor exists for the epsilon 4 allele of the apolipoprotein E gene (APOE e4).i This genotype has been linked to the development of AD and possibly to vascular dementia.j,k In contrast, the epsilon 2 allele of APOE may exert a protective effect in AD.l APOE e3, the most common allele, appears to play a neutral role in the development of AD.
References
a. Evans DA. The epidemiology of dementia and Alzheimer’s disease: an evolving field. J Am Geriatr Soc. 1996;44:1482-1483.
b. Jorm AF, Jolley D. The incidence of dementia: a meta-analysis. Neurology. 1998;51:728-733.
c. van Duijn CM, Clayton D, Chandra V, et al. Familial aggregation of Alzheimer’s disease and related disorders: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int J Epidemiol. 1991;20(suppl 2):S13-S20.
d. Chang JB, Wang PN, Chen WT, et al. ApoE epsilon4 allele is associated with incidental hallucinations and delusions in patients with AD. Neurology. 2004;63:1105-1107.
e. Sleegers K, Roks G, Theuns J, et al. Familial clustering and genetic risk for dementia in a genetically isolated Dutch population. Brain. 2004;127:1641-1649.
f. Schoenberg BS, Anderson DW, Haerer AF. Severe dementia. Prevalence and clinical features in a biracial US population. Arch Neurol. 1985;42:740-743.
g. Hsiung GY, Sadovnick AD. Genetics and dementia: risk factors, diagnosis and management. Alzheimers Dement. 2007;3:418-427.
h. GeneTests database. Available at: http://www.genetests.org. Accessed March 19, 2010.
i. Li H, Wetten S, Li L, et al. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch Neurol. 2008;65:45-53.
j. Graff-Radford NR, Green RC, Go RC, et al. Association between apolipoprotein E genotype and Alzheimer disease in African American subjects. Arch Neurol. 2002;59:594-600.
k. Slooter AJ, Cruts M, Hofman A, et al. The impact of APOE on myocardial infarction, stroke, and dementia: the Rotterdam Study. Neurology. 2004;62:1196-1198.
l. Tiraboschi P, Hansen LA, Masliah E, et al. Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology. 2004;62:1977-1983.
Estrogen. Before the Women’s Health Initiative (WHI) study, various trials of the effects of estrogen therapy on the development of Alzheimer’s disease (AD) in women age ≥65 showed inconsistent results. In the randomized, placebo-controlled WHI Memory Study, conjugated equine estrogen, 0.625 mg/d, plus medroxyprogesterone acetate, 2.5 mg/d, did not prevent mild cognitive impairment or improve global cognitive function and was associated with an increased risk for probable dementia.a Based on this evidence, conjugated equine estrogen with or without medroxyprogesterone is not recommended as therapy to protect cognitive function in older women.
NSAID therapy. Cytokine-mediated inflammation may play a role in neurodegenerative disorders and cognitive impairment in the elderly. Nonsteroidal anti-inflammatory drugs (NSAIDs), including cyclooxygenase-2 (COX-2) inhibitors, have been studied for a possible protective effect against AD and cognitive decline,b possibly by lowering amyloidogenic proteins.c A 1-year randomized controlled trial by the Alzheimer’s Disease Cooperative Consortium found no significant differences in cognition scores of patients treated with once-daily rofecoxib, 25 mg, or twice-daily naproxen sodium, 220 mg, when compared with placebo.d Similarly, naproxen and celecoxib did not prevent AD in the randomized, controlled Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT).e Rofecoxib has been withdrawn from the market, and celecoxib labeling carries a warning of potential for increased risk of cardiovascular events and life-threatening gastrointestinal bleeding associated with its use.
NSAIDs and COX-2 inhibitors are not recommended for the treatment or prevention of dementia or cognitive impairment. Their use for AD prevention is not supported by randomized clinical trialsd,e and they may have serious adverse effects.
References
a. Shumaker SA, Legault C, Kuller L, et al. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. JAMA. 2004;291:2947-2958.
b. Szekely CA, Breitner JC, Fitzpatrick AL, et al. NSAID use and dementia risk in the Cardiovascular Health Study: role of APOE and NSAID type. Neurology. 2008;70:17-24.
c. Weggen S, Eriksen JL, Das P, et al. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001;414:212-216.
d. Aisen PS, Schafer KA, Grundman M, et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003;289(21):2819-2826.
e. ADAPT Research Group, Martin BK, Szekely C, Brandt J, et al. Cognitive function over time in the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch Neurol. 2008;65(7):896-905.
Cardiovascular risk factors
The risk of developing AD or vascular dementia appears to be increased by conditions that damage the heart or blood vessels. Recent evidence suggests that successfully managing cardiovascular risk factors may decrease the likelihood of dementia in later life.
Hypertension is associated with a higher risk of AD and all-cause dementia. Curiously, some studies have shown that low blood pressure also increases dementia risk, suggesting a U-shaped relationship between blood pressure and cognitive decline. Systolic hypertension in midlife may be associated with dementia 20 years later.
One might assume that antihypertensive therapy would help prevent dementia, but the data are conflicting. The Systolic Hypertension in Europe (SYST-EUR) study1 showed a 53% reduction in vascular dementia or mixed dementia among patients receiving antihypertensive medication and a 60% reduction in AD. Similarly, the PROGRESS2 clinical trial of prevention of recurrent stroke by antihypertensive treatment reported a 34% reduction in a composite measure of cognitive impairment and dementia. On the other hand, cognitive function neither improved nor worsened in the Hypertension in the Very Elderly Trial (HYVET-COG),3 whether patients received blood pressure treatment or placebo.
Hyperlipidemia. Lipid metabolism likely is an important pathway in amyloid beta-protein deposition, tau phosphorylation, and disruption of synaptic plasticity and neurodegenerative endpoints. Cognitive decline and incident dementia have been associated with higher dietary intake of saturated fats, partially hydrogenated unsaturated fatty acids (trans fats), and cholesterol. Not all studies have found this association, however. This could be because serum cholesterol levels may decrease in early dementia, limiting the ability to detect an effect of hypercholesterolemia on dementia risk when measurements are made later in life.
Using statins (3-hydroxy-3-methylglutaryl–coenzyme A reductase inhibitors) to treat hypercholesterolemia has been hypothesized to impede large vessel atherosclerosis and its consequences and to trigger metabolic effects in the brain related to AD pathogenesis. Mechanisms by which statins might help prevent dementia include:
- a direct association between amyloid processing and cholesterol in the brain
- an indirect effect by decreasing the risk of stroke, as even small cerebral infarcts worsen AD severity.
Nonrandomized epidemiologic studies such as the Cardiovascular Health Study4 and MRC/BHF Heart Protection Study5 suggested that statin treatment might reduce the incidence of dementia, the degree of age-related cognitive decline, and AD’s neuropathologic burden. Large, randomized, controlled trials have not supported these observations, however. Statins failed to reduce the incidence of dementia in:
- the Heart Protection Study, testing simvastatin for 5 years in 20,536 subjects age 40 to 805
- the 3-year Preventive Study of Pravastatin in the Elderly at Risk (PROSPER) of 5,800 subjects.6
Similarly, patients receiving adjunctive atorvastatin or placebo showed no significant differences in cognition assessments after 72 weeks in the Lipitor’s Effect in Alzheimer’s Dementia (LEADe) study. This trial enrolled 640 subjects age 50 to 90 with mild-to-moderate dementia who were treated with donepezil.7 A recent Cochrane review concluded that high serum cholesterol may contribute to the development of AD and vascular dementia, but lowering cholesterol levels with statins does not prevent these problems.8
Diabetes mellitus. Diabetes and cognitive decline are closely associated. Diabetes is associated with a 50% to 100% increase in risk of AD and dementia overall and a 100% to 150% increased risk of vascular dementia. The mechanism by which diabetes increases dementia risk is uncertain but does not appear to be mediated entirely through vascular disease. High and low insulin levels may increase the risk of dementia, independent of diabetes and blood glucose. Increased peripheral insulin levels are associated with reduced brain atrophy and cognitive impairment in patients with early AD, suggesting a role for insulin signaling in AD pathophysiology. A possible relationship between insulin and beta amyloid metabolism is being studied.
Elevated postprandial plasma glucose has been associated with accelerated declines in cognitive performance.9 An inverse correlation has been noted between some cognitive measures and hemoglobin A1C levels.10 It is not clear that treating diabetes reduces the risk of dementia. In addition, in the prospective, population-based Rotterdam study, elderly patients with type 2 diabetes treated with insulin had the highest incidence of dementia.11
Tobacco smoke directly affects neuronal function, integrity, and survival. Chronic smoking has been linked to decreased global cerebral blood flow, accelerated cerebral atrophy, and ventricular enlargement.
Some studies suggest an increased risk of dementia in middle-aged and elderly smokers, possibly through a cerebrovascular mechanism such as stroke. Other studies found no association between smoking and dementia risk, and 1 suggested that nicotine may protect against AD by reducing senile plaque formation. Any protective effect of smoking would be offset by increased risks of lung cancer, chronic obstructive pulmonary disease, and vascular dementia.
The apolipoprotein E epsilon 4 (APOE e4) gene may explain, at least in part, the conflicting results of these studies. In 2 population-based cohorts,12,13 smoking was associated with memory decline in patients without, but not with, the APOE e4 genotype.
Dietary factors
Antioxidants. The brains of patients with AD contain elevated levels of endogenous antioxidants. In vitro studies show exogenous antioxidants can reduce the toxicity of beta-amyloid in brain tissue of persons with AD. These findings have led to interest in assessing the role of dietary antioxidants such as vitamins E and C for AD prevention.
High-dose alpha-tocopherol (vitamin E, 2,000 IU/d) may slow disease progression in patients with AD, but this association is not consistently found. Furthermore, a meta-analysis of 19 randomized controlled trials (RCTs) totaling >135,000 patients found an association between vitamin E doses >400 IU/d and increased all-cause mortality.14 High-dose vitamin E supplementation for primary or secondary prevention of AD may be dangerous and is not recommended.
The lack of consistent efficacy data for vitamin C in preventing or treating AD may discourage its routine use for this purpose.15
Homocysteine is a risk factor for stroke and heart disease. It also could play a role in vascular dementia through its association with large- and small-vessel disease.
Low folate and hyperhomocysteinemia have been associated with dementia or cognitive impairment, although a cause-effect relationship is not clear. In non-demented elderly populations, plasma homocysteine is inversely associated with poor performance in tests of global cognitive function, particularly in measures of psychomotor speed.
In a recent double-blind RCT, folic acid supplementation for 3 years significantly improved domains of cognitive function that tend to decline with age, especially information processing and sensorimotor speed.16 No other good evidence, however, has shown that homocysteine-lowering therapy using folic acid or other vitamin B supplements improves cognitive function or prevents cognitive decline.
Fish and omega-3 fatty acids. High total fat, saturated fat, and total cholesterol intake increases the risk for incident dementia. In epidemiologic studies, low omega-3 fatty acid serum levels have been linked to increased dementia risk.
Fish consumption may be beneficial in reducing the risk of dementia or cognitive decline. A prospective study of 815 elderly persons found 60% less risk of developing AD in those who ate ≥1 fish meal per week, compared with those who rarely or never ate fish.17 In the Framingham study, individuals who at baseline were in the top quartile of docosahexaenoic acid consumption had lower dementia rates over 9 years of follow-up.18 Results from cross-sectional and longitudinal studies have been inconsistent; some have shown that high intake of n-3 polyunsaturated fatty acids is associated with less cognitive decline,19 whereas others have not.20
Although we cannot offer unequivocal advice regarding seafood or omega-3 fatty acid intake for primary prevention of dementia without evidence from RCTs, these uncontrolled studies show promise.
Mediterranean diet (MeDi) components include abundant fruits and vegetables, fish or shellfish at least twice weekly, very limited red meat, olive oil or canola oil instead of butter or margarine, tree nuts such as walnuts or pecans, red wine in moderation, and using herbs and spices instead of salt to season food. High adherence to the MeDi has been associated with a significantly lower risk for incident AD. The MeDi may affect the risk of developing AD21 as well as subsequent disease course, with a possible dose-response relationship in lower mortality.22
Eating fruits and vegetables has been associated with improved cognitive performance22 and decreased incident dementia in elderly subjects.18
Alcohol. A U-shaped relationship exists between alcohol consumption and dementia risk. High alcohol intake is associated with clinical problem drinking and alcoholism and can lead to cognitive decline. Conversely, moderate wine consumption (250 to 500 mL/d) may be protective—compared with more or less than this amount—and is associated with approximately 50% less risk of dementia.
Alcohol use may increase the risk of dementia in persons carrying the APOE e4 allele, according to the population-based Cardiovascular Risk Factors, Aging and Dementia (CAIDE) study from Sweden.23 After an average 21 years of follow-up of 1,449 individuals, researchers found that environmental factors—such as physical inactivity, dietary fat intake, alcohol consumption, and smoking at midlife—were associated with an increased risk of dementia at age 65 to 79 in APOE e4 carriers compared with noncarriers. The study also found that physical inactivity, dietary fat intake, and smoking at midlife increase AD risk, especially among APOE e4 carriers.
In the absence of evidence from RCTs, we cannot recommend alcohol to reduce the risk of AD.
Lifestyle and activity
Three components of lifestyle—social, mental, and physical activity—are inversely associated with the risk for dementia, AD, and cognitive impairment.
Physical exercise has been thought to enhance brain neurotrophic factor and modify apoptosis. Exercise can deter stroke and microvascular disease and improve regional cerebral blood flow. In the Cardiovascular Health Study, participants who expended the highest quartile of energy had a lower risk of all-cause dementia and AD compared with participants who expended the lowest quartile of energy.24
Mental and social activity. Epidemiologic studies have shown associations between higher educational achievement and other socioeconomic factors and reduced AD risk. Advanced education is believed to represent a cognitive reserve that delays presentation of AD’s effects on memory and cognitive function, rather than providing a protective effect against accumulation of AD pathology. Higher-educated individuals appear to experience a somewhat more rapid rate of cognitive decline when AD does become apparent, perhaps because they have accumulated a greater degree of AD pathology at that point compared with less-educated persons.
Among 117 persons with dementia in the Bronx Aging Study, each additional year of formal education delayed the time of accelerated decline by 0.21 years. After accelerated decline began, each year of additional formal education was associated with a slightly faster rate of memory decline.25
The longitudinal, population-based Kungsholmen Project in Stockholm, Sweden, found an association between daily mentally stimulating activities and decreased risk of all-cause dementia.26 Similarly, higher levels of leisure activity were linked to reduced risk of all-cause dementia in a longitudinal study of 1,772 persons age ≥65 living in Manhattan, NY.27 In a randomized, single-controlled study of the long-term effects of cognitive training, elderly individuals from 6 U.S. cities showed sustained improvement in specific cognitive performance up to 5 years after training sessions began, including memory, reasoning, and speed of processing.28
It seems reasonable to encourage older patients to maintain or increase physical, cognitive, and leisure activities as well as social interaction. These interventions can improve the quality of life and lower the risk of depression, which may be a response to cognitive decline or an independent risk factor for dementia (Box 3). The Table lists “brain exercises” you can suggest to patients to increase their mental and social activity.
Head trauma. The Multi-Institutional Research in Alzheimer’s Genetic Epidemiology (MIRAGE) project found an association between AD risk and a history of head trauma, especially in persons with APOE e4 alleles.29 Conversely, the Rotterdam Study showed no change in dementia risk for persons with a history of head trauma.30
Even in the absence of conclusive evidence supporting AD prevention, protecting the head by buckling seat belts while driving, wearing helmets when participating in sports, and “fall-proofing” the home is recommended.
Depression often occurs before or as a coexisting condition with Alzheimer’s disease (AD).a Although depression has been considered a response to cognitive decline or an early manifestation of dementia,b it also could be an independent risk factor.c,d
The pathologic mechanism linking depression and subsequent dementia is not well understood. Hypotheses include an indirect neurotoxic effect of depression mediated by cortisol-induced hippocampal atrophy or lowered brain-derived neurotrophic factor levels.e Depression and dementia might share genetic links, although a cohort study of 404 individuals with AD detected no association between apolipoprotein E genotypes or alleles and depressive symptoms.f
References
a. Lupien SJ, Nair NP, Brière S, et al. Increased cortisol levels and impaired cognition in human aging: implication for depression and dementia in later life. Rev Neurosci. 1999;10(2):117-139.
b. Preuss UW, Siafarikas N, Petrucci M, et al. Depressive disorders in dementia and mild cognitive impairments: is comorbidity a cause or a risk factor? Fortschr Neurol Psychiatr. 2009;77:399-406.
c. Green RC, Cupples LA, Kurz A, et al. Depression as a risk factor for Alzheimer disease: the MIRAGE Study. Arch Neurol. 2003;60(5):753-759.
d. Ownby RL, Crocco E, Acevedo A, et al. Depression and risk for Alzheimer’s disease: systematic review, meta-analysis, and metaregression analysis. Arch Gen Psychiatry. 2006;63(5):530-538.
e. Meeks TW, Ropacki SA, Jeste DV. The neurobiology of neuropsychiatric syndromes in dementia. Curr Opin Psychiatry. 2006;19(6):581-586.
f. Craig D, Hart DJ, McIlroy SP, et al. Association analysis of apolipoprotein E genotype and risk of depressive symptoms in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2005;19(2-3):154-157.
Table
Brain exercises to suggest to patients
| Learn something new (how to play a musical instrument, a foreign language, or a new hobby) |
| Play memory games |
| Practice using the opposite hand to perform tasks you usually do with your dominant hand |
| Read, especially challenging material |
| Join a book discussion group |
| Write; if not a book or article, write a diary, letters, or emails or start your memoirs |
| Do crossword, Sudoku, or jigsaw puzzles |
| Play board games, card games, and other strategy games |
| Debate or discuss topics |
Related resource
- For an extensive bibliography of literature on Alzheimer’s disease risk factors and prevention, see this article at CurrentPsychiatry.com.
Drug brand names
- Atorvastatin • Lipitor
- Celecoxib • Celebrex
- Donepezil • Aricept
- Medroxyprogesterone • Provera
- Pravastatin • Pravachol
- Rofecoxib • Vioxx
- Simvastatin • Zocor
Disclosures
Dr. Bassil reports no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
Dr. Grossberg receives research/grant support from and is a consultant to Bristol-Myers Squibb, Forest Pharmaceuticals, Novartis, Pfizer Inc., and Wyeth Pharmaceuticals. He also receives research/grant support from Baxter.
Pharmacologic treatments for Alzheimer’s disease (AD) may improve symptoms but have not been shown to prevent AD onset. Primary prevention therefore remains the goal. Although preventing AD by managing risk factors such as age or genetics is beyond our control (Box 1), we can do something about other factors.
This article summarizes the findings of many studies that address AD prevention and includes an online-only bibliography for readers seeking an in-depth review. The evidence does not support a firm recommendation for any specific form of primary prevention and has revealed hazards associated with estrogen therapy and nonsteroidal anti-inflammatory drugs (Box 2). Most important, it suggests that you could reduce your patients’ risk of developing AD by routinely supporting their mental, physical, and social health.
The potential benefits of modifying an individual’s AD risk factors likely will depend on his or her genetic makeup, environment, and lifestyle. Even so, counseling patients to exercise more and improve their diets—such as by eating more fish, fruits, and vegetables and less saturated fat—might help protect the brain. Your ongoing efforts to manage hypertension, hypercholesterolemia, and diabetes also may reduce their AD risk.
Age remains the strongest risk factor for dementia, particularly for Alzheimer’s disease (AD).a The risk of developing AD doubles every 5 years after age 65 and approaches 50% after age 85.b
Family history is a risk factor for AD, although true familial AD accounts for <5% of cases.c When diseases show a familial pattern, either genetics, environmental factors, or both may play a role. Patients with a first-degree relative with dementia have a 10% to 30% increased risk of developing the disorder.d
Genetic factors play a role in both early-onset and late-onset AD. Early-onset AD (before age 65) accounts for 6% to 7% of cases.e From this small pool of patients, only 13% exhibit clear autosomal dominant transmission over >1 generation.f Three gene mutations have been associated with early-onset AD:
- 30% to 70% are in the presenilin-1 gene
- 10% to 15% are in the amyloid precursor protein gene
- <5% are in the presenilin-2 gene.g,h
For late-onset AD (after age 65), the strongest evidence for a genetic risk factor exists for the epsilon 4 allele of the apolipoprotein E gene (APOE e4).i This genotype has been linked to the development of AD and possibly to vascular dementia.j,k In contrast, the epsilon 2 allele of APOE may exert a protective effect in AD.l APOE e3, the most common allele, appears to play a neutral role in the development of AD.
References
a. Evans DA. The epidemiology of dementia and Alzheimer’s disease: an evolving field. J Am Geriatr Soc. 1996;44:1482-1483.
b. Jorm AF, Jolley D. The incidence of dementia: a meta-analysis. Neurology. 1998;51:728-733.
c. van Duijn CM, Clayton D, Chandra V, et al. Familial aggregation of Alzheimer’s disease and related disorders: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int J Epidemiol. 1991;20(suppl 2):S13-S20.
d. Chang JB, Wang PN, Chen WT, et al. ApoE epsilon4 allele is associated with incidental hallucinations and delusions in patients with AD. Neurology. 2004;63:1105-1107.
e. Sleegers K, Roks G, Theuns J, et al. Familial clustering and genetic risk for dementia in a genetically isolated Dutch population. Brain. 2004;127:1641-1649.
f. Schoenberg BS, Anderson DW, Haerer AF. Severe dementia. Prevalence and clinical features in a biracial US population. Arch Neurol. 1985;42:740-743.
g. Hsiung GY, Sadovnick AD. Genetics and dementia: risk factors, diagnosis and management. Alzheimers Dement. 2007;3:418-427.
h. GeneTests database. Available at: http://www.genetests.org. Accessed March 19, 2010.
i. Li H, Wetten S, Li L, et al. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch Neurol. 2008;65:45-53.
j. Graff-Radford NR, Green RC, Go RC, et al. Association between apolipoprotein E genotype and Alzheimer disease in African American subjects. Arch Neurol. 2002;59:594-600.
k. Slooter AJ, Cruts M, Hofman A, et al. The impact of APOE on myocardial infarction, stroke, and dementia: the Rotterdam Study. Neurology. 2004;62:1196-1198.
l. Tiraboschi P, Hansen LA, Masliah E, et al. Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology. 2004;62:1977-1983.
Estrogen. Before the Women’s Health Initiative (WHI) study, various trials of the effects of estrogen therapy on the development of Alzheimer’s disease (AD) in women age ≥65 showed inconsistent results. In the randomized, placebo-controlled WHI Memory Study, conjugated equine estrogen, 0.625 mg/d, plus medroxyprogesterone acetate, 2.5 mg/d, did not prevent mild cognitive impairment or improve global cognitive function and was associated with an increased risk for probable dementia.a Based on this evidence, conjugated equine estrogen with or without medroxyprogesterone is not recommended as therapy to protect cognitive function in older women.
NSAID therapy. Cytokine-mediated inflammation may play a role in neurodegenerative disorders and cognitive impairment in the elderly. Nonsteroidal anti-inflammatory drugs (NSAIDs), including cyclooxygenase-2 (COX-2) inhibitors, have been studied for a possible protective effect against AD and cognitive decline,b possibly by lowering amyloidogenic proteins.c A 1-year randomized controlled trial by the Alzheimer’s Disease Cooperative Consortium found no significant differences in cognition scores of patients treated with once-daily rofecoxib, 25 mg, or twice-daily naproxen sodium, 220 mg, when compared with placebo.d Similarly, naproxen and celecoxib did not prevent AD in the randomized, controlled Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT).e Rofecoxib has been withdrawn from the market, and celecoxib labeling carries a warning of potential for increased risk of cardiovascular events and life-threatening gastrointestinal bleeding associated with its use.
NSAIDs and COX-2 inhibitors are not recommended for the treatment or prevention of dementia or cognitive impairment. Their use for AD prevention is not supported by randomized clinical trialsd,e and they may have serious adverse effects.
References
a. Shumaker SA, Legault C, Kuller L, et al. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. JAMA. 2004;291:2947-2958.
b. Szekely CA, Breitner JC, Fitzpatrick AL, et al. NSAID use and dementia risk in the Cardiovascular Health Study: role of APOE and NSAID type. Neurology. 2008;70:17-24.
c. Weggen S, Eriksen JL, Das P, et al. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001;414:212-216.
d. Aisen PS, Schafer KA, Grundman M, et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003;289(21):2819-2826.
e. ADAPT Research Group, Martin BK, Szekely C, Brandt J, et al. Cognitive function over time in the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch Neurol. 2008;65(7):896-905.
Cardiovascular risk factors
The risk of developing AD or vascular dementia appears to be increased by conditions that damage the heart or blood vessels. Recent evidence suggests that successfully managing cardiovascular risk factors may decrease the likelihood of dementia in later life.
Hypertension is associated with a higher risk of AD and all-cause dementia. Curiously, some studies have shown that low blood pressure also increases dementia risk, suggesting a U-shaped relationship between blood pressure and cognitive decline. Systolic hypertension in midlife may be associated with dementia 20 years later.
One might assume that antihypertensive therapy would help prevent dementia, but the data are conflicting. The Systolic Hypertension in Europe (SYST-EUR) study1 showed a 53% reduction in vascular dementia or mixed dementia among patients receiving antihypertensive medication and a 60% reduction in AD. Similarly, the PROGRESS2 clinical trial of prevention of recurrent stroke by antihypertensive treatment reported a 34% reduction in a composite measure of cognitive impairment and dementia. On the other hand, cognitive function neither improved nor worsened in the Hypertension in the Very Elderly Trial (HYVET-COG),3 whether patients received blood pressure treatment or placebo.
Hyperlipidemia. Lipid metabolism likely is an important pathway in amyloid beta-protein deposition, tau phosphorylation, and disruption of synaptic plasticity and neurodegenerative endpoints. Cognitive decline and incident dementia have been associated with higher dietary intake of saturated fats, partially hydrogenated unsaturated fatty acids (trans fats), and cholesterol. Not all studies have found this association, however. This could be because serum cholesterol levels may decrease in early dementia, limiting the ability to detect an effect of hypercholesterolemia on dementia risk when measurements are made later in life.
Using statins (3-hydroxy-3-methylglutaryl–coenzyme A reductase inhibitors) to treat hypercholesterolemia has been hypothesized to impede large vessel atherosclerosis and its consequences and to trigger metabolic effects in the brain related to AD pathogenesis. Mechanisms by which statins might help prevent dementia include:
- a direct association between amyloid processing and cholesterol in the brain
- an indirect effect by decreasing the risk of stroke, as even small cerebral infarcts worsen AD severity.
Nonrandomized epidemiologic studies such as the Cardiovascular Health Study4 and MRC/BHF Heart Protection Study5 suggested that statin treatment might reduce the incidence of dementia, the degree of age-related cognitive decline, and AD’s neuropathologic burden. Large, randomized, controlled trials have not supported these observations, however. Statins failed to reduce the incidence of dementia in:
- the Heart Protection Study, testing simvastatin for 5 years in 20,536 subjects age 40 to 805
- the 3-year Preventive Study of Pravastatin in the Elderly at Risk (PROSPER) of 5,800 subjects.6
Similarly, patients receiving adjunctive atorvastatin or placebo showed no significant differences in cognition assessments after 72 weeks in the Lipitor’s Effect in Alzheimer’s Dementia (LEADe) study. This trial enrolled 640 subjects age 50 to 90 with mild-to-moderate dementia who were treated with donepezil.7 A recent Cochrane review concluded that high serum cholesterol may contribute to the development of AD and vascular dementia, but lowering cholesterol levels with statins does not prevent these problems.8
Diabetes mellitus. Diabetes and cognitive decline are closely associated. Diabetes is associated with a 50% to 100% increase in risk of AD and dementia overall and a 100% to 150% increased risk of vascular dementia. The mechanism by which diabetes increases dementia risk is uncertain but does not appear to be mediated entirely through vascular disease. High and low insulin levels may increase the risk of dementia, independent of diabetes and blood glucose. Increased peripheral insulin levels are associated with reduced brain atrophy and cognitive impairment in patients with early AD, suggesting a role for insulin signaling in AD pathophysiology. A possible relationship between insulin and beta amyloid metabolism is being studied.
Elevated postprandial plasma glucose has been associated with accelerated declines in cognitive performance.9 An inverse correlation has been noted between some cognitive measures and hemoglobin A1C levels.10 It is not clear that treating diabetes reduces the risk of dementia. In addition, in the prospective, population-based Rotterdam study, elderly patients with type 2 diabetes treated with insulin had the highest incidence of dementia.11
Tobacco smoke directly affects neuronal function, integrity, and survival. Chronic smoking has been linked to decreased global cerebral blood flow, accelerated cerebral atrophy, and ventricular enlargement.
Some studies suggest an increased risk of dementia in middle-aged and elderly smokers, possibly through a cerebrovascular mechanism such as stroke. Other studies found no association between smoking and dementia risk, and 1 suggested that nicotine may protect against AD by reducing senile plaque formation. Any protective effect of smoking would be offset by increased risks of lung cancer, chronic obstructive pulmonary disease, and vascular dementia.
The apolipoprotein E epsilon 4 (APOE e4) gene may explain, at least in part, the conflicting results of these studies. In 2 population-based cohorts,12,13 smoking was associated with memory decline in patients without, but not with, the APOE e4 genotype.
Dietary factors
Antioxidants. The brains of patients with AD contain elevated levels of endogenous antioxidants. In vitro studies show exogenous antioxidants can reduce the toxicity of beta-amyloid in brain tissue of persons with AD. These findings have led to interest in assessing the role of dietary antioxidants such as vitamins E and C for AD prevention.
High-dose alpha-tocopherol (vitamin E, 2,000 IU/d) may slow disease progression in patients with AD, but this association is not consistently found. Furthermore, a meta-analysis of 19 randomized controlled trials (RCTs) totaling >135,000 patients found an association between vitamin E doses >400 IU/d and increased all-cause mortality.14 High-dose vitamin E supplementation for primary or secondary prevention of AD may be dangerous and is not recommended.
The lack of consistent efficacy data for vitamin C in preventing or treating AD may discourage its routine use for this purpose.15
Homocysteine is a risk factor for stroke and heart disease. It also could play a role in vascular dementia through its association with large- and small-vessel disease.
Low folate and hyperhomocysteinemia have been associated with dementia or cognitive impairment, although a cause-effect relationship is not clear. In non-demented elderly populations, plasma homocysteine is inversely associated with poor performance in tests of global cognitive function, particularly in measures of psychomotor speed.
In a recent double-blind RCT, folic acid supplementation for 3 years significantly improved domains of cognitive function that tend to decline with age, especially information processing and sensorimotor speed.16 No other good evidence, however, has shown that homocysteine-lowering therapy using folic acid or other vitamin B supplements improves cognitive function or prevents cognitive decline.
Fish and omega-3 fatty acids. High total fat, saturated fat, and total cholesterol intake increases the risk for incident dementia. In epidemiologic studies, low omega-3 fatty acid serum levels have been linked to increased dementia risk.
Fish consumption may be beneficial in reducing the risk of dementia or cognitive decline. A prospective study of 815 elderly persons found 60% less risk of developing AD in those who ate ≥1 fish meal per week, compared with those who rarely or never ate fish.17 In the Framingham study, individuals who at baseline were in the top quartile of docosahexaenoic acid consumption had lower dementia rates over 9 years of follow-up.18 Results from cross-sectional and longitudinal studies have been inconsistent; some have shown that high intake of n-3 polyunsaturated fatty acids is associated with less cognitive decline,19 whereas others have not.20
Although we cannot offer unequivocal advice regarding seafood or omega-3 fatty acid intake for primary prevention of dementia without evidence from RCTs, these uncontrolled studies show promise.
Mediterranean diet (MeDi) components include abundant fruits and vegetables, fish or shellfish at least twice weekly, very limited red meat, olive oil or canola oil instead of butter or margarine, tree nuts such as walnuts or pecans, red wine in moderation, and using herbs and spices instead of salt to season food. High adherence to the MeDi has been associated with a significantly lower risk for incident AD. The MeDi may affect the risk of developing AD21 as well as subsequent disease course, with a possible dose-response relationship in lower mortality.22
Eating fruits and vegetables has been associated with improved cognitive performance22 and decreased incident dementia in elderly subjects.18
Alcohol. A U-shaped relationship exists between alcohol consumption and dementia risk. High alcohol intake is associated with clinical problem drinking and alcoholism and can lead to cognitive decline. Conversely, moderate wine consumption (250 to 500 mL/d) may be protective—compared with more or less than this amount—and is associated with approximately 50% less risk of dementia.
Alcohol use may increase the risk of dementia in persons carrying the APOE e4 allele, according to the population-based Cardiovascular Risk Factors, Aging and Dementia (CAIDE) study from Sweden.23 After an average 21 years of follow-up of 1,449 individuals, researchers found that environmental factors—such as physical inactivity, dietary fat intake, alcohol consumption, and smoking at midlife—were associated with an increased risk of dementia at age 65 to 79 in APOE e4 carriers compared with noncarriers. The study also found that physical inactivity, dietary fat intake, and smoking at midlife increase AD risk, especially among APOE e4 carriers.
In the absence of evidence from RCTs, we cannot recommend alcohol to reduce the risk of AD.
Lifestyle and activity
Three components of lifestyle—social, mental, and physical activity—are inversely associated with the risk for dementia, AD, and cognitive impairment.
Physical exercise has been thought to enhance brain neurotrophic factor and modify apoptosis. Exercise can deter stroke and microvascular disease and improve regional cerebral blood flow. In the Cardiovascular Health Study, participants who expended the highest quartile of energy had a lower risk of all-cause dementia and AD compared with participants who expended the lowest quartile of energy.24
Mental and social activity. Epidemiologic studies have shown associations between higher educational achievement and other socioeconomic factors and reduced AD risk. Advanced education is believed to represent a cognitive reserve that delays presentation of AD’s effects on memory and cognitive function, rather than providing a protective effect against accumulation of AD pathology. Higher-educated individuals appear to experience a somewhat more rapid rate of cognitive decline when AD does become apparent, perhaps because they have accumulated a greater degree of AD pathology at that point compared with less-educated persons.
Among 117 persons with dementia in the Bronx Aging Study, each additional year of formal education delayed the time of accelerated decline by 0.21 years. After accelerated decline began, each year of additional formal education was associated with a slightly faster rate of memory decline.25
The longitudinal, population-based Kungsholmen Project in Stockholm, Sweden, found an association between daily mentally stimulating activities and decreased risk of all-cause dementia.26 Similarly, higher levels of leisure activity were linked to reduced risk of all-cause dementia in a longitudinal study of 1,772 persons age ≥65 living in Manhattan, NY.27 In a randomized, single-controlled study of the long-term effects of cognitive training, elderly individuals from 6 U.S. cities showed sustained improvement in specific cognitive performance up to 5 years after training sessions began, including memory, reasoning, and speed of processing.28
It seems reasonable to encourage older patients to maintain or increase physical, cognitive, and leisure activities as well as social interaction. These interventions can improve the quality of life and lower the risk of depression, which may be a response to cognitive decline or an independent risk factor for dementia (Box 3). The Table lists “brain exercises” you can suggest to patients to increase their mental and social activity.
Head trauma. The Multi-Institutional Research in Alzheimer’s Genetic Epidemiology (MIRAGE) project found an association between AD risk and a history of head trauma, especially in persons with APOE e4 alleles.29 Conversely, the Rotterdam Study showed no change in dementia risk for persons with a history of head trauma.30
Even in the absence of conclusive evidence supporting AD prevention, protecting the head by buckling seat belts while driving, wearing helmets when participating in sports, and “fall-proofing” the home is recommended.
Depression often occurs before or as a coexisting condition with Alzheimer’s disease (AD).a Although depression has been considered a response to cognitive decline or an early manifestation of dementia,b it also could be an independent risk factor.c,d
The pathologic mechanism linking depression and subsequent dementia is not well understood. Hypotheses include an indirect neurotoxic effect of depression mediated by cortisol-induced hippocampal atrophy or lowered brain-derived neurotrophic factor levels.e Depression and dementia might share genetic links, although a cohort study of 404 individuals with AD detected no association between apolipoprotein E genotypes or alleles and depressive symptoms.f
References
a. Lupien SJ, Nair NP, Brière S, et al. Increased cortisol levels and impaired cognition in human aging: implication for depression and dementia in later life. Rev Neurosci. 1999;10(2):117-139.
b. Preuss UW, Siafarikas N, Petrucci M, et al. Depressive disorders in dementia and mild cognitive impairments: is comorbidity a cause or a risk factor? Fortschr Neurol Psychiatr. 2009;77:399-406.
c. Green RC, Cupples LA, Kurz A, et al. Depression as a risk factor for Alzheimer disease: the MIRAGE Study. Arch Neurol. 2003;60(5):753-759.
d. Ownby RL, Crocco E, Acevedo A, et al. Depression and risk for Alzheimer’s disease: systematic review, meta-analysis, and metaregression analysis. Arch Gen Psychiatry. 2006;63(5):530-538.
e. Meeks TW, Ropacki SA, Jeste DV. The neurobiology of neuropsychiatric syndromes in dementia. Curr Opin Psychiatry. 2006;19(6):581-586.
f. Craig D, Hart DJ, McIlroy SP, et al. Association analysis of apolipoprotein E genotype and risk of depressive symptoms in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2005;19(2-3):154-157.
Table
Brain exercises to suggest to patients
| Learn something new (how to play a musical instrument, a foreign language, or a new hobby) |
| Play memory games |
| Practice using the opposite hand to perform tasks you usually do with your dominant hand |
| Read, especially challenging material |
| Join a book discussion group |
| Write; if not a book or article, write a diary, letters, or emails or start your memoirs |
| Do crossword, Sudoku, or jigsaw puzzles |
| Play board games, card games, and other strategy games |
| Debate or discuss topics |
Related resource
- For an extensive bibliography of literature on Alzheimer’s disease risk factors and prevention, see this article at CurrentPsychiatry.com.
Drug brand names
- Atorvastatin • Lipitor
- Celecoxib • Celebrex
- Donepezil • Aricept
- Medroxyprogesterone • Provera
- Pravastatin • Pravachol
- Rofecoxib • Vioxx
- Simvastatin • Zocor
Disclosures
Dr. Bassil reports no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
Dr. Grossberg receives research/grant support from and is a consultant to Bristol-Myers Squibb, Forest Pharmaceuticals, Novartis, Pfizer Inc., and Wyeth Pharmaceuticals. He also receives research/grant support from Baxter.
1. Forette F, Seux ML, Staessen JA, et al. The prevention of dementia with antihypertensive treatment: new evidence from the systolic hypertension in Europe (Syst-Eur) study. Arch Intern Med. 2002;162:2046-2052.
2. Tzourio C, Anderson C, Chapman N, et al. Effects of blood pressure lowering with perindopril and indapamide therapy on dementia and cognitive decline in patients with cerebrovascular disease. Arch Intern Med. 2003;163:1069-1075.
3. Peters R, Beckett N, Forette F. Incident dementia and blood pressure lowering in the Hypertension in the Very Elderly Trial cognitive function assessment (HYVET-COG). Lancet Neurol. 2008;7(8):683-689.
4. Rea TD, Breitner JC, Psaty BM, et al. Statin use and the risk of incident dementia: the Cardiovascular Health Study. Arch Neurol. 2005;62:1047-1051.
5. Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:7-22.
6. Kulbertus H, Scheen AJ. [The PROSPER Study (PROspective study of pravastatin in the elderly at risk)]. Rev Med Liege. 2002;57(12):809-813.
7. Feldman HH, Doody RS, Kivipelto M, et al. Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADe. Neurology. 2010;74(12):956-964.
8. McGuinness B, Bullock R, Craig D, et al. Statins for the treatment of Alzheimer’s disease and dementia (protocol). Cochrane Database Syst Rev. 2009;1:CD007514.-
9. Abbatecola AM, Rizzo MR, Barbieri M, et al. Postprandial plasmaglucose excursions and cognitive functioning in aged type 2 diabetics. Neurology. 2006;67:235-240.
10. Munshi M, Grande L, Hayes M, et al. Cognitive dysfunction is associated with poor diabetes control in older adults. Diabetes Care. 2006;29:1794-1799.
11. Ott A, Stolk RP, van Harskamp F, et al. Diabetes mellitus and the risk of dementia. The Rotterdam study. Neurology. 1999;53:1937-1942.
12. Reitz C, Luchsinger J, Tang MX, et al. Effect of smoking and time on cognitive function in the elderly without dementia. Neurology. 2005;65:870-875.
13. Reitz C, den Heijer T, van Duijn C, et al. Relation between smoking and risk of dementia and Alzheimer disease: the Rotterdam Study. Neurology. 2007;69:998-1005.
14. Miller ER, III, Pastor-Barriuso R, Dalal D, et al. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med. 2005;142(1):37-46.
15. Boothby LA, Doering PL. Vitamin C and vitamin E for Alzheimer’s disease. Ann Pharmacother. 2005;39(12):2073-2080.
16. Durga J, van Boxtel MP, Schouten EG, et al. Effect of 3-year folic acid supplementation on cognitive function in older adults in the FACIT trial: a randomised, double blind, controlled trial. Lancet. 2007;369:208-216.
17. Morris MC, Evans DA, Bienias JL, et al. Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch Neurol. 2003;60:940-946.
18. Schaefer EJ, Bongard V, Beiser AS, et al. Plasma phosphatidylcholine docosahexaenoic acid content and risk of dementia and Alzheimer disease: the Framingham Heart Study. Arch Neurol. 2006;63:1545-1550.
19. Kalmijn S, Launer LJ, Ott A, et al. Dietary fat intake and the risk of incident dementia in the Rotterdam Study. Ann Neurol. 1997;42:776-782.
20. van Gelder BM, Tijhuis M, Kalmijn S, et al. Fish consumption, n-3 fatty acids, and subsequent 5-y cognitive decline in elderly men: the Zutphen Elderly Study. Am J Clin Nutr. 2007;85:1142-1147.
21. Solfrizzi V, Capurso C, Panza F. Adherence to a Mediterranean dietary pattern and risk of Alzheimer’s disease. Ann Neurol. 2006;60:620.-
22. Scarmeas N, Luchsinger JA, Mayeux R, et al. Mediterranean diet and Alzheimer disease mortality. Neurology. 2007;69(11):1084-1093.
23. Kivipelto M, Rovio S, Ngandu T, et al. Apolipoprotein E epsilon4 magnifies lifestyle risks for dementia: a population-based study. J Cell Mol Med. 2008;12(6B):2762-2771.
24. Podewils LJ, Guallar E, Kuller LH, et al. Physical activity, APOE genotype and dementia risk: findings from the Cardiovascular Health Cognition Study. Am J Epidemiol. 2005;161:639-651.
25. Hall CB, Derby C, LeValley A, et al. Education delays accelerated decline on a memory test in persons who develop dementia. Neurology. 2007;69:1657-1664.
26. Wang HX, Karp A, Winblad B, et al. Late-life engagement in social and leisure activities is associated with a decreased risk of dementia: a longitudinal study from the Kungsholmen Project. Am J Epidemiol. 2002;155:1081-1087.
27. Scarmeas N, Levy G, Tang MX, et al. Influence of leisure activity on the incidence of Alzheimer’s disease. Neurology. 2001;57:2236-2242.
28. Willis SL, Tennstedt SL, Marsiske M, et al. Long-term effects of cognitive training on everyday functional outcomes in older adults. JAMA. 2006;296:2805-2814.
29. Guo Z, Cupples LA, Kurz A, et al. Head injury and the risk of AD in the MIRAGE study. Neurology. 2000;54:1316-1323.
30. Ruitenberg A, van Swieten JC, Witteman JC, et al. Alcohol consumption and risk of dementia: the Rotterdam Study. Lancet. 2002;359:281-286.
1. Forette F, Seux ML, Staessen JA, et al. The prevention of dementia with antihypertensive treatment: new evidence from the systolic hypertension in Europe (Syst-Eur) study. Arch Intern Med. 2002;162:2046-2052.
2. Tzourio C, Anderson C, Chapman N, et al. Effects of blood pressure lowering with perindopril and indapamide therapy on dementia and cognitive decline in patients with cerebrovascular disease. Arch Intern Med. 2003;163:1069-1075.
3. Peters R, Beckett N, Forette F. Incident dementia and blood pressure lowering in the Hypertension in the Very Elderly Trial cognitive function assessment (HYVET-COG). Lancet Neurol. 2008;7(8):683-689.
4. Rea TD, Breitner JC, Psaty BM, et al. Statin use and the risk of incident dementia: the Cardiovascular Health Study. Arch Neurol. 2005;62:1047-1051.
5. Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:7-22.
6. Kulbertus H, Scheen AJ. [The PROSPER Study (PROspective study of pravastatin in the elderly at risk)]. Rev Med Liege. 2002;57(12):809-813.
7. Feldman HH, Doody RS, Kivipelto M, et al. Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADe. Neurology. 2010;74(12):956-964.
8. McGuinness B, Bullock R, Craig D, et al. Statins for the treatment of Alzheimer’s disease and dementia (protocol). Cochrane Database Syst Rev. 2009;1:CD007514.-
9. Abbatecola AM, Rizzo MR, Barbieri M, et al. Postprandial plasmaglucose excursions and cognitive functioning in aged type 2 diabetics. Neurology. 2006;67:235-240.
10. Munshi M, Grande L, Hayes M, et al. Cognitive dysfunction is associated with poor diabetes control in older adults. Diabetes Care. 2006;29:1794-1799.
11. Ott A, Stolk RP, van Harskamp F, et al. Diabetes mellitus and the risk of dementia. The Rotterdam study. Neurology. 1999;53:1937-1942.
12. Reitz C, Luchsinger J, Tang MX, et al. Effect of smoking and time on cognitive function in the elderly without dementia. Neurology. 2005;65:870-875.
13. Reitz C, den Heijer T, van Duijn C, et al. Relation between smoking and risk of dementia and Alzheimer disease: the Rotterdam Study. Neurology. 2007;69:998-1005.
14. Miller ER, III, Pastor-Barriuso R, Dalal D, et al. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med. 2005;142(1):37-46.
15. Boothby LA, Doering PL. Vitamin C and vitamin E for Alzheimer’s disease. Ann Pharmacother. 2005;39(12):2073-2080.
16. Durga J, van Boxtel MP, Schouten EG, et al. Effect of 3-year folic acid supplementation on cognitive function in older adults in the FACIT trial: a randomised, double blind, controlled trial. Lancet. 2007;369:208-216.
17. Morris MC, Evans DA, Bienias JL, et al. Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch Neurol. 2003;60:940-946.
18. Schaefer EJ, Bongard V, Beiser AS, et al. Plasma phosphatidylcholine docosahexaenoic acid content and risk of dementia and Alzheimer disease: the Framingham Heart Study. Arch Neurol. 2006;63:1545-1550.
19. Kalmijn S, Launer LJ, Ott A, et al. Dietary fat intake and the risk of incident dementia in the Rotterdam Study. Ann Neurol. 1997;42:776-782.
20. van Gelder BM, Tijhuis M, Kalmijn S, et al. Fish consumption, n-3 fatty acids, and subsequent 5-y cognitive decline in elderly men: the Zutphen Elderly Study. Am J Clin Nutr. 2007;85:1142-1147.
21. Solfrizzi V, Capurso C, Panza F. Adherence to a Mediterranean dietary pattern and risk of Alzheimer’s disease. Ann Neurol. 2006;60:620.-
22. Scarmeas N, Luchsinger JA, Mayeux R, et al. Mediterranean diet and Alzheimer disease mortality. Neurology. 2007;69(11):1084-1093.
23. Kivipelto M, Rovio S, Ngandu T, et al. Apolipoprotein E epsilon4 magnifies lifestyle risks for dementia: a population-based study. J Cell Mol Med. 2008;12(6B):2762-2771.
24. Podewils LJ, Guallar E, Kuller LH, et al. Physical activity, APOE genotype and dementia risk: findings from the Cardiovascular Health Cognition Study. Am J Epidemiol. 2005;161:639-651.
25. Hall CB, Derby C, LeValley A, et al. Education delays accelerated decline on a memory test in persons who develop dementia. Neurology. 2007;69:1657-1664.
26. Wang HX, Karp A, Winblad B, et al. Late-life engagement in social and leisure activities is associated with a decreased risk of dementia: a longitudinal study from the Kungsholmen Project. Am J Epidemiol. 2002;155:1081-1087.
27. Scarmeas N, Levy G, Tang MX, et al. Influence of leisure activity on the incidence of Alzheimer’s disease. Neurology. 2001;57:2236-2242.
28. Willis SL, Tennstedt SL, Marsiske M, et al. Long-term effects of cognitive training on everyday functional outcomes in older adults. JAMA. 2006;296:2805-2814.
29. Guo Z, Cupples LA, Kurz A, et al. Head injury and the risk of AD in the MIRAGE study. Neurology. 2000;54:1316-1323.
30. Ruitenberg A, van Swieten JC, Witteman JC, et al. Alcohol consumption and risk of dementia: the Rotterdam Study. Lancet. 2002;359:281-286.
Do beta blockers cause depression?
Dr. Muzyk is a clinical pharmacist, Duke University Medical Center, and Dr. Galiardi is assistant professor of psychiatry and behavioral sciences and assistant clinical professor of medicine, Duke University School of Medicine, Durham, NC.
Principal Source: van Melle JP, Verbeek DEP, van den Berg MP, et al. Beta-blockers and depression after myocardial infarction: a multicenter prospective study. J Am Coll Cardiol. 2006;48(11):2209-2214.
- Although patients with cardiovascular disease are at increased risk for developing depression, there is no convincing evidence that adding beta blockers will further increase their risk.
- Initiating beta-blocker therapy at the lowest possible dose and slowly titrating the dose over time could minimize adverse effects such as fatigue and sexual side effects.
- If a patient taking beta blockers develops signs of major depression, carefully evaluate and treat symptoms with appropriate psychotherapy, psychotropics, and monitoring.
Beyond their well-known role for treating cardiovascular disease, beta adrenergic receptor antagonists—beta blockers—are used for a variety of medical conditions, including coronary artery disease, hypertension, migraines, and tremor. Their usefulness makes them 1 of the most commonly prescribed medication classes. Unfortunately, their increased use comes with increased reports of depression. Being able to sort fact from fiction will help guide your care for patients taking beta blockers who report new or worsening depressive symptoms.
Does research support a link?
First reported in the 1960s, beta blocker-induced depression was thought to result from the drugs’ antagonistic effect on norepinephrine at ß1 post-synaptic brain receptors. Prompted by case reports of a possible association between beta blockers and depression, 2 prescription database reviews found that patients taking beta blockers were more likely to receive a concurrent antidepressant prescription than patients prescribed other cardiovascular and diabetic medications.1,2 However, these reviews had major limitations, such as inadequately defined methods for defining depression and lack of control for potential confounding factors.
Mechanistically, peripheral effects of beta blockers on the heart and kidneys lead to decreased chronotropy and inotropy as well as lower blood pressure. These cardiovascular and hemodynamic changes could cause fatigue, decreased energy, and sexual dysfunction that may be interpreted as symptoms of new-onset depression.
Researchers found that beta-blocker use was not associated with depression in a case-control study examining 4,302 New Jersey Medicaid records.3 Also, because most patients in this study received propranolol, the authors were unable to confirm a long-held belief that highly lipophilic beta blockers (such as propranolol, metoprolol, and timolol) are more likely than hydrophilic beta blockers such as atenolol to produce depression.
A retrospective cohort study analyzed 381 patients from 2 myocardial infarction (MI) trials who had been assessed for depressive symptoms and severity.4 Researchers matched 254 subjects taking beta blockers during hospitalization for MI with 127 subjects not taking beta blockers. Patients in the study were well balanced on multiple baseline characteristics, including demographics, history of depression, and left ventricular ejection fraction, although those who did not take beta blockers had a significantly higher incidence of chronic obstructive pulmonary disease, digoxin use, and pre-MI beta-blocker use. Researchers assessed depressive symptoms using the Beck Depression Inventory (BDI) at baseline and 3, 6, and 12 months post-MI and identified patients with depression using a Composite International Diagnostic Interview. They found no statistically significant difference in BDI scores between beta-blockers users and nonusers at discharge and at 3, 6, and 12 months post-MI after accounting for potential confounding factors, including:
- contraindications for beta-blocker use (other than history of depression)
- indicators and risk factors for cardiac disease
- baseline depressive symptoms
- benzodiazepine use.
In fact, after controlling for baseline depression, researchers found that beta-blocker users demonstrated significantly lower BDI scores 3 months post-MI than nonusers. Based on these results, the authors concluded that clinicians should not be deterred from prescribing beta blockers because the drugs’ benefit in reducing morbidity and mortality in cardiovascular disease greatly outweighs the risk—if any—of new-onset depression associated with beta-blocker use.
Two additional studies reported no significant difference in the incidence of depression between patients who received beta blockers and those who received other antihypertensives or placebo.5,6 Future studies assessing depression among subjects randomized to beta blockers vs placebo would be helpful, though withholding beta blockers in some cardiac conditions is not justifiable, and such studies may not be feasible.
Treatment for psychiatric patients
Evidence supports beta-blocker use in coronary artery disease and congestive heart failure. Although patients with these conditions are at increased risk for developing depression,7 there is little evidence that their risk will be further increased by adding beta blockers (Table),3-6 Although patients taking beta blockers report a higher incidence of fatigue and sexual side effects—which could be interpreted as related to depression—studies do not support an association between these medications and depression. As with any medication, initiate beta-blocker therapy with the lowest possible dose and titrate slowly to minimize side effects. Any patient who develops signs and symptoms of major depression should be thoroughly evaluated and treated with appropriate psychotherapy, psychotropics, and careful monitoring.
Table
Beta blockers and depression: Is there a link?
| Study | Methods | Results |
|---|---|---|
| Bright et al, 19923 | Case-control study of 4,302 patients with new-onset depression | Beta-blocker use was not associated with depression after controlling for confounding factors, although depressed patients were more likely to receive beta blockers |
| van Melle et al, 20064 | A prospective study of post-myocardial infarction patients; 254 taking beta blockers, 127 controls | No significant differences in depressive symptoms or incidence of depressive disorder between beta-blocker users and nonusers |
| Gerstman et al, 19965 | New users of propranolol (n=704) other beta blockers (n=587), angiotensin-converting enzyme inhibitors (n=976), calcium channel blockers (n=742), and diuretics (n=773) | Depression occurred no more frequently among beta-blocker users than other subjects |
| Ko et al, 20026 | Quantitative review of randomized trials that tested beta blockers in myocardial infarction, heart failure, and hypertension | Beta-blocker therapy was not associated with a significant absolute annual increase in risk of depressive symptoms (6 per 1,000 patients; 95% confidence interval, -7 to 19) |
- Rivelli S, Jiang W. Depression and ischemic heart disease: what have we learned from clinical trials? Curr Opin Cardiol. 2007;22(4):286-291.
- National guideline clearinghouse. Secondary prevention of coronary artery disease. www.guideline.gov/summary/summary.aspx?doc_id=14585.
Drug brand names
- Atenolol • Tenormin
- Digoxin • Lanoxin
- Metoprolol • Lopressor, Toprol-XL
- Propranolol • Inderal
- Timolol • Blocadren
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Avorn J, Everitt D, Weiss S. Increased antidepressant use in patients prescribed beta-blockers. JAMA. 1986;256:357-360.
2. Thiessen B, Wallace S, Blackburn J, et al. Increased prescribing of antidepressants subsequent to beta-blocker therapy. Arch Intern Med. 1990;150:2286-2290.
3. Bright R, Everitt D. Beta-blockers and depression. Evidence against an association. JAMA. 1992;267(13):1783-1787.
4. van Melle JP, Verbeek DEP, van den Berg MP, et al. Beta-blockers and depression after myocardial infarction: a multicenter prospective study. J Am Coll Cardiol. 2006;48(11):2209-2214.
5. Gerstman B, Jolson HM, Bauer M, et al. The incidence of depression in new users of beta-blockers and selected antihypertensives. J Clin Epidemiol. 1996;49(7):809-815.
6. Ko D, Hebert P, Coffey C, et al. Beta-blockers therapy and symptoms of depression, fatigue, and sexual dysfunction. JAMA. 2002;288(3):351-357.
7. Pozuelo L, Tesar G, Zhang J, et al. Depression and heart disease: what do we know, and where are we headed? Cleve Clin J Med. 2009;76(1):59-70.
Andrew J. Muzyk, PharmD
Jane P. Gagliardi, MD
Robert M. McCarron, DO
Series Editor
Andrew J. Muzyk, PharmD
Jane P. Gagliardi, MD
Robert M. McCarron, DO
Series Editor
Andrew J. Muzyk, PharmD
Jane P. Gagliardi, MD
Robert M. McCarron, DO
Series Editor
Dr. Muzyk is a clinical pharmacist, Duke University Medical Center, and Dr. Galiardi is assistant professor of psychiatry and behavioral sciences and assistant clinical professor of medicine, Duke University School of Medicine, Durham, NC.
Principal Source: van Melle JP, Verbeek DEP, van den Berg MP, et al. Beta-blockers and depression after myocardial infarction: a multicenter prospective study. J Am Coll Cardiol. 2006;48(11):2209-2214.
- Although patients with cardiovascular disease are at increased risk for developing depression, there is no convincing evidence that adding beta blockers will further increase their risk.
- Initiating beta-blocker therapy at the lowest possible dose and slowly titrating the dose over time could minimize adverse effects such as fatigue and sexual side effects.
- If a patient taking beta blockers develops signs of major depression, carefully evaluate and treat symptoms with appropriate psychotherapy, psychotropics, and monitoring.
Beyond their well-known role for treating cardiovascular disease, beta adrenergic receptor antagonists—beta blockers—are used for a variety of medical conditions, including coronary artery disease, hypertension, migraines, and tremor. Their usefulness makes them 1 of the most commonly prescribed medication classes. Unfortunately, their increased use comes with increased reports of depression. Being able to sort fact from fiction will help guide your care for patients taking beta blockers who report new or worsening depressive symptoms.
Does research support a link?
First reported in the 1960s, beta blocker-induced depression was thought to result from the drugs’ antagonistic effect on norepinephrine at ß1 post-synaptic brain receptors. Prompted by case reports of a possible association between beta blockers and depression, 2 prescription database reviews found that patients taking beta blockers were more likely to receive a concurrent antidepressant prescription than patients prescribed other cardiovascular and diabetic medications.1,2 However, these reviews had major limitations, such as inadequately defined methods for defining depression and lack of control for potential confounding factors.
Mechanistically, peripheral effects of beta blockers on the heart and kidneys lead to decreased chronotropy and inotropy as well as lower blood pressure. These cardiovascular and hemodynamic changes could cause fatigue, decreased energy, and sexual dysfunction that may be interpreted as symptoms of new-onset depression.
Researchers found that beta-blocker use was not associated with depression in a case-control study examining 4,302 New Jersey Medicaid records.3 Also, because most patients in this study received propranolol, the authors were unable to confirm a long-held belief that highly lipophilic beta blockers (such as propranolol, metoprolol, and timolol) are more likely than hydrophilic beta blockers such as atenolol to produce depression.
A retrospective cohort study analyzed 381 patients from 2 myocardial infarction (MI) trials who had been assessed for depressive symptoms and severity.4 Researchers matched 254 subjects taking beta blockers during hospitalization for MI with 127 subjects not taking beta blockers. Patients in the study were well balanced on multiple baseline characteristics, including demographics, history of depression, and left ventricular ejection fraction, although those who did not take beta blockers had a significantly higher incidence of chronic obstructive pulmonary disease, digoxin use, and pre-MI beta-blocker use. Researchers assessed depressive symptoms using the Beck Depression Inventory (BDI) at baseline and 3, 6, and 12 months post-MI and identified patients with depression using a Composite International Diagnostic Interview. They found no statistically significant difference in BDI scores between beta-blockers users and nonusers at discharge and at 3, 6, and 12 months post-MI after accounting for potential confounding factors, including:
- contraindications for beta-blocker use (other than history of depression)
- indicators and risk factors for cardiac disease
- baseline depressive symptoms
- benzodiazepine use.
In fact, after controlling for baseline depression, researchers found that beta-blocker users demonstrated significantly lower BDI scores 3 months post-MI than nonusers. Based on these results, the authors concluded that clinicians should not be deterred from prescribing beta blockers because the drugs’ benefit in reducing morbidity and mortality in cardiovascular disease greatly outweighs the risk—if any—of new-onset depression associated with beta-blocker use.
Two additional studies reported no significant difference in the incidence of depression between patients who received beta blockers and those who received other antihypertensives or placebo.5,6 Future studies assessing depression among subjects randomized to beta blockers vs placebo would be helpful, though withholding beta blockers in some cardiac conditions is not justifiable, and such studies may not be feasible.
Treatment for psychiatric patients
Evidence supports beta-blocker use in coronary artery disease and congestive heart failure. Although patients with these conditions are at increased risk for developing depression,7 there is little evidence that their risk will be further increased by adding beta blockers (Table),3-6 Although patients taking beta blockers report a higher incidence of fatigue and sexual side effects—which could be interpreted as related to depression—studies do not support an association between these medications and depression. As with any medication, initiate beta-blocker therapy with the lowest possible dose and titrate slowly to minimize side effects. Any patient who develops signs and symptoms of major depression should be thoroughly evaluated and treated with appropriate psychotherapy, psychotropics, and careful monitoring.
Table
Beta blockers and depression: Is there a link?
| Study | Methods | Results |
|---|---|---|
| Bright et al, 19923 | Case-control study of 4,302 patients with new-onset depression | Beta-blocker use was not associated with depression after controlling for confounding factors, although depressed patients were more likely to receive beta blockers |
| van Melle et al, 20064 | A prospective study of post-myocardial infarction patients; 254 taking beta blockers, 127 controls | No significant differences in depressive symptoms or incidence of depressive disorder between beta-blocker users and nonusers |
| Gerstman et al, 19965 | New users of propranolol (n=704) other beta blockers (n=587), angiotensin-converting enzyme inhibitors (n=976), calcium channel blockers (n=742), and diuretics (n=773) | Depression occurred no more frequently among beta-blocker users than other subjects |
| Ko et al, 20026 | Quantitative review of randomized trials that tested beta blockers in myocardial infarction, heart failure, and hypertension | Beta-blocker therapy was not associated with a significant absolute annual increase in risk of depressive symptoms (6 per 1,000 patients; 95% confidence interval, -7 to 19) |
- Rivelli S, Jiang W. Depression and ischemic heart disease: what have we learned from clinical trials? Curr Opin Cardiol. 2007;22(4):286-291.
- National guideline clearinghouse. Secondary prevention of coronary artery disease. www.guideline.gov/summary/summary.aspx?doc_id=14585.
Drug brand names
- Atenolol • Tenormin
- Digoxin • Lanoxin
- Metoprolol • Lopressor, Toprol-XL
- Propranolol • Inderal
- Timolol • Blocadren
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Muzyk is a clinical pharmacist, Duke University Medical Center, and Dr. Galiardi is assistant professor of psychiatry and behavioral sciences and assistant clinical professor of medicine, Duke University School of Medicine, Durham, NC.
Principal Source: van Melle JP, Verbeek DEP, van den Berg MP, et al. Beta-blockers and depression after myocardial infarction: a multicenter prospective study. J Am Coll Cardiol. 2006;48(11):2209-2214.
- Although patients with cardiovascular disease are at increased risk for developing depression, there is no convincing evidence that adding beta blockers will further increase their risk.
- Initiating beta-blocker therapy at the lowest possible dose and slowly titrating the dose over time could minimize adverse effects such as fatigue and sexual side effects.
- If a patient taking beta blockers develops signs of major depression, carefully evaluate and treat symptoms with appropriate psychotherapy, psychotropics, and monitoring.
Beyond their well-known role for treating cardiovascular disease, beta adrenergic receptor antagonists—beta blockers—are used for a variety of medical conditions, including coronary artery disease, hypertension, migraines, and tremor. Their usefulness makes them 1 of the most commonly prescribed medication classes. Unfortunately, their increased use comes with increased reports of depression. Being able to sort fact from fiction will help guide your care for patients taking beta blockers who report new or worsening depressive symptoms.
Does research support a link?
First reported in the 1960s, beta blocker-induced depression was thought to result from the drugs’ antagonistic effect on norepinephrine at ß1 post-synaptic brain receptors. Prompted by case reports of a possible association between beta blockers and depression, 2 prescription database reviews found that patients taking beta blockers were more likely to receive a concurrent antidepressant prescription than patients prescribed other cardiovascular and diabetic medications.1,2 However, these reviews had major limitations, such as inadequately defined methods for defining depression and lack of control for potential confounding factors.
Mechanistically, peripheral effects of beta blockers on the heart and kidneys lead to decreased chronotropy and inotropy as well as lower blood pressure. These cardiovascular and hemodynamic changes could cause fatigue, decreased energy, and sexual dysfunction that may be interpreted as symptoms of new-onset depression.
Researchers found that beta-blocker use was not associated with depression in a case-control study examining 4,302 New Jersey Medicaid records.3 Also, because most patients in this study received propranolol, the authors were unable to confirm a long-held belief that highly lipophilic beta blockers (such as propranolol, metoprolol, and timolol) are more likely than hydrophilic beta blockers such as atenolol to produce depression.
A retrospective cohort study analyzed 381 patients from 2 myocardial infarction (MI) trials who had been assessed for depressive symptoms and severity.4 Researchers matched 254 subjects taking beta blockers during hospitalization for MI with 127 subjects not taking beta blockers. Patients in the study were well balanced on multiple baseline characteristics, including demographics, history of depression, and left ventricular ejection fraction, although those who did not take beta blockers had a significantly higher incidence of chronic obstructive pulmonary disease, digoxin use, and pre-MI beta-blocker use. Researchers assessed depressive symptoms using the Beck Depression Inventory (BDI) at baseline and 3, 6, and 12 months post-MI and identified patients with depression using a Composite International Diagnostic Interview. They found no statistically significant difference in BDI scores between beta-blockers users and nonusers at discharge and at 3, 6, and 12 months post-MI after accounting for potential confounding factors, including:
- contraindications for beta-blocker use (other than history of depression)
- indicators and risk factors for cardiac disease
- baseline depressive symptoms
- benzodiazepine use.
In fact, after controlling for baseline depression, researchers found that beta-blocker users demonstrated significantly lower BDI scores 3 months post-MI than nonusers. Based on these results, the authors concluded that clinicians should not be deterred from prescribing beta blockers because the drugs’ benefit in reducing morbidity and mortality in cardiovascular disease greatly outweighs the risk—if any—of new-onset depression associated with beta-blocker use.
Two additional studies reported no significant difference in the incidence of depression between patients who received beta blockers and those who received other antihypertensives or placebo.5,6 Future studies assessing depression among subjects randomized to beta blockers vs placebo would be helpful, though withholding beta blockers in some cardiac conditions is not justifiable, and such studies may not be feasible.
Treatment for psychiatric patients
Evidence supports beta-blocker use in coronary artery disease and congestive heart failure. Although patients with these conditions are at increased risk for developing depression,7 there is little evidence that their risk will be further increased by adding beta blockers (Table),3-6 Although patients taking beta blockers report a higher incidence of fatigue and sexual side effects—which could be interpreted as related to depression—studies do not support an association between these medications and depression. As with any medication, initiate beta-blocker therapy with the lowest possible dose and titrate slowly to minimize side effects. Any patient who develops signs and symptoms of major depression should be thoroughly evaluated and treated with appropriate psychotherapy, psychotropics, and careful monitoring.
Table
Beta blockers and depression: Is there a link?
| Study | Methods | Results |
|---|---|---|
| Bright et al, 19923 | Case-control study of 4,302 patients with new-onset depression | Beta-blocker use was not associated with depression after controlling for confounding factors, although depressed patients were more likely to receive beta blockers |
| van Melle et al, 20064 | A prospective study of post-myocardial infarction patients; 254 taking beta blockers, 127 controls | No significant differences in depressive symptoms or incidence of depressive disorder between beta-blocker users and nonusers |
| Gerstman et al, 19965 | New users of propranolol (n=704) other beta blockers (n=587), angiotensin-converting enzyme inhibitors (n=976), calcium channel blockers (n=742), and diuretics (n=773) | Depression occurred no more frequently among beta-blocker users than other subjects |
| Ko et al, 20026 | Quantitative review of randomized trials that tested beta blockers in myocardial infarction, heart failure, and hypertension | Beta-blocker therapy was not associated with a significant absolute annual increase in risk of depressive symptoms (6 per 1,000 patients; 95% confidence interval, -7 to 19) |
- Rivelli S, Jiang W. Depression and ischemic heart disease: what have we learned from clinical trials? Curr Opin Cardiol. 2007;22(4):286-291.
- National guideline clearinghouse. Secondary prevention of coronary artery disease. www.guideline.gov/summary/summary.aspx?doc_id=14585.
Drug brand names
- Atenolol • Tenormin
- Digoxin • Lanoxin
- Metoprolol • Lopressor, Toprol-XL
- Propranolol • Inderal
- Timolol • Blocadren
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Avorn J, Everitt D, Weiss S. Increased antidepressant use in patients prescribed beta-blockers. JAMA. 1986;256:357-360.
2. Thiessen B, Wallace S, Blackburn J, et al. Increased prescribing of antidepressants subsequent to beta-blocker therapy. Arch Intern Med. 1990;150:2286-2290.
3. Bright R, Everitt D. Beta-blockers and depression. Evidence against an association. JAMA. 1992;267(13):1783-1787.
4. van Melle JP, Verbeek DEP, van den Berg MP, et al. Beta-blockers and depression after myocardial infarction: a multicenter prospective study. J Am Coll Cardiol. 2006;48(11):2209-2214.
5. Gerstman B, Jolson HM, Bauer M, et al. The incidence of depression in new users of beta-blockers and selected antihypertensives. J Clin Epidemiol. 1996;49(7):809-815.
6. Ko D, Hebert P, Coffey C, et al. Beta-blockers therapy and symptoms of depression, fatigue, and sexual dysfunction. JAMA. 2002;288(3):351-357.
7. Pozuelo L, Tesar G, Zhang J, et al. Depression and heart disease: what do we know, and where are we headed? Cleve Clin J Med. 2009;76(1):59-70.
1. Avorn J, Everitt D, Weiss S. Increased antidepressant use in patients prescribed beta-blockers. JAMA. 1986;256:357-360.
2. Thiessen B, Wallace S, Blackburn J, et al. Increased prescribing of antidepressants subsequent to beta-blocker therapy. Arch Intern Med. 1990;150:2286-2290.
3. Bright R, Everitt D. Beta-blockers and depression. Evidence against an association. JAMA. 1992;267(13):1783-1787.
4. van Melle JP, Verbeek DEP, van den Berg MP, et al. Beta-blockers and depression after myocardial infarction: a multicenter prospective study. J Am Coll Cardiol. 2006;48(11):2209-2214.
5. Gerstman B, Jolson HM, Bauer M, et al. The incidence of depression in new users of beta-blockers and selected antihypertensives. J Clin Epidemiol. 1996;49(7):809-815.
6. Ko D, Hebert P, Coffey C, et al. Beta-blockers therapy and symptoms of depression, fatigue, and sexual dysfunction. JAMA. 2002;288(3):351-357.
7. Pozuelo L, Tesar G, Zhang J, et al. Depression and heart disease: what do we know, and where are we headed? Cleve Clin J Med. 2009;76(1):59-70.
Sex-related differences in antidepressant response: When to adjust treatment
With a history of panic disorder, perfectionistic tendencies, and depression, Ms. C, age 32, presents 29 weeks into her first pregnancy with a chief complaint that “the Zoloft is not working; my sadness and anxiety are increased and I feel dizzy, like when I miss a dose.” For the past 7 years, she has done well on sertraline, 50 mg/d; she has had no depressive symptoms and experienced minimal to manageable anxiety. Ms. C has found psychotherapy helpful for the last 2 years, including during her pregnancy.
After discussion with her obstetrician, Ms. C remained on sertraline through her early pregnancy. She did well until several weeks ago, when she noticed a return of sadness and incessant worry. She resumed an old habit of excessively cleaning her home. Ms. C denies missing doses but states she has the physical feeling as if she were—a lightheadedness that she clearly distinguishes from pregnancy symptoms.
Both men and women respond well to antidepressants, yet there are notable differences between the 2. Understanding why men and women may differ in response to antidepressants helps clinicians better tailor their treatment choice and dosing.
This article outlines some of differences—and lack thereof—in response rates to antidepressants. Our discussion of why these differences may occur is framed in the context of pharmacokinetics, pharmacodynamics, and the influence of gonadal hormones on antidepressant-related neurotransmitter systems. The second section focuses on major reproductive phases of adult women (the menstrual cycle, pregnancy, postpartum, and menopause) and how antidepressant response rates can influence clinical decision making, such as antidepressant timing, dose, and choice of potential adjunct treatments.
What the evidence says
Most studies look at sex differences in response to a single antidepressant, but several comparing sex differences among classes have produced fascinating results (Table 1). One of the most robust and replicated findings—although not universally reproduced1—is that compared with men, women are more likely to respond to selective serotonin reuptake inhibitors (SSRIs) than to tricyclic antidepressants (TCAs).2-4 Because of this and the fact that SSRIs are so commonly used, this article primarily will address SSRIs in women.
Initially, however, in reviewing non-SSRI anti depressants, monoamine oxidase inhibitors (MAOIs) are reported to produce a superior response in women than in men.5 Women are more likely to have atypical depression symptoms, which MAOIs often treat better than other antidepressants. In contrast, a recent meta-analysis of TCAs6 found no sex response difference within the class. However, 1 study reported women may be slower to respond to TCAs than men.2
Studies on the newer and more frequently prescribed antidepressants reveal some interesting sex differences. Although smaller studies initially did not find a sex difference in SSRIs,5,7 when response rates to citalopram were compared in 2,876 subjects in Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study, women were more likely to reach remission and response than men.8 Younger women—generally those age <50—respond better to SSRIs than women age ≥50.2,3,9
There are less data concerning newer non-SSRI antidepressants. In the second stage of the STAR*D trial, when subjects who did not respond to citalopram were randomly assigned venlafaxine, bupropion, or sertraline, there was no sex difference in response.10 Pooled analysis of randomized controlled trials specifically looking at remission rates between the sexes for venlafaxine,9 bupropion,11 or duloxetine12 found no difference between men and women, regardless of age. No published sex differences in antidepressant response were found for mirtazapine.
Numerous studies have detailed sex differences in antidepressant pharmacokinetics (Box 1) and pharmacodynamics (Box 2), as well as human sexual dimorphism of the serotonergic system. Estrogen’s influence on the serotonergic system (Box 3) may be a component of men and women’s different responses to antidepressants, particularly across reproductive phases.
Table 1
Sex differences in antidepressant response
| Class | Response: Male vs female |
|---|---|
| Monoamine oxidase inhibitors | M |
| Serotonin-norepinephrine reuptake inhibitors | M=F |
| Selective serotonin reuptake inhibitors | Age <50: M< F Age ≥50: M=F |
| Tricyclic antidepressants | M=F |
| References 1-12 | |
Medical literature has documented gender differences in antidepressant absorption, distribution, metabolism, and elimination.a-c Compared with men, women—especially premenopausal women—have slower gastric emptyingd and small bowel and colonic transit times.e,f Also, because antidepressants generally are lipophilic,a,g a lower ratio of lean muscle to adipose tissue in women compared with men may result in a greater volume of drug distribution (Vd).
Sex differences also have been reported in hepatic enzyme activity and may affect clinical response. Most medications, including antidepressants, undergo phase I metabolism, commonly via the cytochrome P450 (CYP450) pathway, and/or phase II conjugation reactions. Generally, phase I oxidative metabolism appears to be greater in women than in men; in contrast, phase II conjugation activity appears to be greater in men than in women.h
Lower CYP1A2 activity in womeni along with gonadal steroid inhibition of CYP1A2j,k may explain why clomipramine metabolic clearance is reduced in young womenl and mean steady state plasma levels of fluvoxamine are almost double in women than in men for the same dose.m In theory, greater CYP3A4 activity in womeni has the potential to accelerate metabolism and/or decrease plasma levels of some commonly used antidepressants metabolized via CYP3A4, such as nefazodone and (to some extent) sertraline and citalopram. In contrast, CYP2D6 and CYP2C9 do not show sex differences in metabolism.
Differences in antidepressant blood levels, however, are difficult to base solely on CYP metabolic route differences. Sex differences in plasma antidepressant levels likely reflect a summation of several sex-associated pharmacokinetic processes and may impact one of many factors that contribute to the small observed difference in antidepressant efficacy between men and women.
References
a. Yonkers KA, Kando JC, Cole JO, et al. Gender differences in pharmacokinetics and pharmacodynamics of psychotropic medication. Am J Psychiatry. 1992;149(5):587-595.
b. Kando JC, Yonkers KA, Cole JO. Gender as a risk factor for adverse events to medications. Drugs. 1995;50(1):1-6.
c. Bies RR, Bigos KL, Pollock BG. Gender differences in the pharmacokinetics and pharmacodynamics of antidepressants. J Gend Specif Med. 2003;6(3):12-20.
d. Hutson WR, Roehrkasse RL, Wald A. Influence of gender and menopause on gastric emptying and motility. Gastroenterology. 1989;96(1):11-17.
e. Sadik R, Abrahamsson H, Stotzer PO. Gender differences in gut transit shown with a newly developed radiological procedure. Scand J Gastroenterol. 2003;38(1):36-42.
f. Lorena SL, Tinois E, Hirata ES, et al. [Scintigraphic study of gastric emptying and intragastric distribution of a solid meal: gender differences]. Arq Gastroenterol. 2000;37(2):102-106.
g. Greenblatt DJ, Divoll M, Abernethy DR, et al. Physiologic changes in old age: relation to altered drug disposition. J Am Geriatr Soc. 1982;30(11 suppl):S6-10.
h. Anderson GD. Gender differences in pharmacological response. Int Rev Neurobiol. 2008;83:1-10.
i. Anderson GD. Sex and racial differences in pharmacological response: where is the evidence? Pharmacogenetics, pharmacokinetics, and pharmacodynamics. J Womens Health (Larchmt). 2005;14(1):19-29.
j. Lane JD, Steege JF, Rupp SL, et al. Menstrual cycle effects on caffeine elimination in the human female. Eur J Clin Pharmacol. 1992;43(5):543-546.
k. Pollock BG, Wylie M, Stack JA, et al. Inhibition of caffeine metabolism by estrogen replacement therapy in postmenopausal women. J Clin Pharmacol. 1999;39(9):936-940.
l. Gex-Fabry M, Balant-Gorgia AE, Balant LP, et al. Clomipramine metabolism. Model-based analysis of variability factors from drug monitoring data. Clin Pharmacokinet. 1990;19(3):241-255.
m. Hartter S, Wetzel H, Hammes E, et al. Nonlinear pharmacokinetics of fluvoxamine and gender differences. Ther Drug Monit. 1998;20(4):446-449.
Sexual dimorphisms in the localization and concentration of endogenous neurotransmitters such as serotonin and dopamine and their degradative enzymes and transporters have the potential to clinically affect antidepressant pharmacodynamics (eg, drug-receptor interactions).
Recent investigations report sex differences in some key monoaminergic enzymes in the brain, notably monoamine oxidase-A (MAO)a,b and catechol-O-methyltransferase (COMT).c-e
For example, estrogen has been found to inhibit MAO,f which is potentially clinically relevant in light of the finding that women respond better than men to MAO inhibitors. COMT—which is responsible for metabolism of norepinephrine, epinephrine, and dopamine—is down regulated by estradiole,g likely accounting for some sex effects. Recently, the sexually dimorphic effect of a COMT polymorphism was associated with a poorer fluoxetine response in men treated for major depression.h
References
a. Domschke K, Hohoff C, Mortensen LS, et al. Monoamine oxidase A variant influences antidepressant treatment response in female patients with major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(1):224-228.
b. Yu YW, Tsai SJ, Hong CJ, et al. Association study of a monoamine oxidase a gene promoter polymorphism with major depressive disorder and antidepressant response. Neuropsychopharmacology. 2005;30(9):1719-1723.
c. Baune BT, Hohoff C, Berger K, et al. Association of the COMT val158met variant with antidepressant treatment response in major depression. Neuropsychopharmacology. 2008;33(4):924-932.
d. Harrison PJ, Tunbridge EM. Catechol-O-methyltransferase (COMT): a gene contributing to sex differences in brain function, and to sexual dimorphism in the predisposition to psychiatric disorders. Neuropsychopharmacology. 2008;33(13):3037-3045.
e. Jiang H, Xie T, Ramsden DB, et al. Human catechol-O-methyltransferase down-regulation by estradiol. Neuropharmacology. 2003;45(7):1011-1018.
f. Luine VN, Khylchevskaya RI, McEwen BS. Effect of gonadal steroids on activities of monoamine oxidase and choline acetylase in rat brain. Brain Res. 1975;86(2):293-306.
g. Xie T, Ho SL, Ramsden D. Characterization and implications of estrogenic down-regulation of human catechol-O-methyltransferase gene transcription. Mol Pharmacol. 1999;56(1):31-38.
h. Tsai SJ, Gau YT, Hong CJ, et al. Sexually dimorphic effect of catechol-O-methyltransferase val158met polymorphism on clinical response to fluoxetine in major depressive patients. J Affect Disord. 2009;113(1-2):183-187.
Change across reproductive phases
In contrast to men, women’s estrogen and progesterone status varies widely across a woman’s reproductive lifecycle (menstrual cycle, pregnancy, postpartum, premenopause vs post menopause). In men and women, androgen levels—including testosterone—tend to remain at steady levels, and then slowly decline with age.
Menstrual cycle. Hormone-related changes associated with the menstrual cycle may affect antidepressant absorption and distribution. During the luteal phase—second half of the menstrual cycle post-ovulation—and pregnancy, increased progesterone concentrations are associated with slowed gastrointestinal transit time13,14 compared with the follicular phase (preovulation).
Premenstrually, at the end of luteal phase, reduced serum antidepressant levels have been associated with breakthrough depressive symptoms.15,16 In these case reports, serum antidepressant levels returned to baseline and depressive symptoms resolved after menses ended. It is possible that women may be at increased risk of symptom recurrence before menses because of hormonally driven changes in drug absorption, distribution, and metabolism. Increased dosing of sertraline in the luteal phase has helped reduce premenstrual exacerbation of depression.17
Pregnancy. Dose requirements for the SSRIs citalopram, escitalopram, and sertraline,18 the serotonin-norepinephrine reuptake inhibitor venlafaxine,19 and the TCAs nortriptyline, clomipramine, and imipramine20 increase during the second half of pregnancy. This appears to be the result of increased drug metabolism. Altered cytochrome P450 (CYP450) enzymatic activity in pregnancy—likely mediated by elevated estrogen and progesterone—may have clinical effects on drug levels and treatment response. Studies indicate that CYP3A4—and possibly CYP2D6—are induced during pregnancy.21,22 Dose increases are necessary in two-thirds of pregnant women on antidepressant monotherapy, typically after 20 weeks gestation18,20,23 to treat symptom recurrence or maintain euthymia.
During pregnancy, drug elimination may increase because of higher renal blood flow and glomerular filtration rate (GFR).24 This could reduce blood levels of water-soluble active metabolites of some TCAs. Pregnancy-associated reductions in intestinal motility and gastric pH alone do not change medication bioavailability. Increased body fat could increase the volume of drug distribution for antidepressants, and, in theory, create a dilutional drop in free drug concentration, but this likely would have only a minor effect.
The range of antidepressant effectiveness among pregnant patients is wide, which reflects individual differences in pharmacokinetics and pharmacodynamics.25 Because we cannot predict which women will require dose changes during pregnancy or postpartum, patients should be monitored frequently for depressive symptom recurrence. Dose adjustments may be necessary to prevent relapse (eg, when net metabolism is increased) or pronounced side effects (eg, when net metabolism is reduced).18,26
When prescribing antidepressants for pregnant women, a personalized discussion of the risks and benefits with each patient in the context of her psychiatric history, the developing fetus, and her value system is warranted. The potential consequences of antidepressant effect on patient and fetus, or lack there of, continues to be an evolving area; long-term data on prenatal exposure are limited.
Postpartum. The postpartum period—when depression can hit 10% to 15% of new mothers27—entails rapid shifts in many factors that may influence antidepressant response. Levels of gonadal hormones such as estrogen and progesterone decline, plasma volume contracts, and hepatic enzymatic metabolism and GFR return to pre-pregnancy levels. Together these changes may result in increased antidepressant blood levels postpartum, especially when the dosage used during pregnancy is held constant.19
The postpartum period is associated with a high risk for depression onset or worsening and is a time of great hormonal and pharmacokinetic change. Accordingly, a postpartum woman should be followed closely for changes in response and adverse effects, and her antidepressant dosage adjusted. Breastfeeding is a critical consideration in the postpartum. Meltzer-Brody et al28 provide a discussion of postpartum depression and what to tell patients who breast-feed.
Menopause. Despite evidence that reproductive-age women may respond better to SSRIs than men, the same findings have not been reproduced in postmenopausal women. For example, compared with men, postmenopausal women had no significant difference in SSRI treatment response in primary care clinics. In contrast, the same postmenopausal women had a significantly worse treatment response than premenopausal women.29
In considering why SSRI response among women would differ depending on reproductive stage or hormonal status, researchers examined the effect of estrogen on antidepressant response with the use of estrogen therapy (ET). As detailed in Box 3, estrogen has many serotonergic-enhancing properties. Early studies with TCAs and a retrospective analysis of SSRIs did not demonstrate improved antidepressant effect with the addition of ET in depressed women.30,31 In contrast, recent studies have demonstrated better SSRI response—regardless of which medication was used—in postmenopausal women on ET or ET with progesterone, compared with postmenopausal women taking placebo.32,33 Perhaps explaining the discrepancy, in a randomized, placebo-controlled trial, Rasgon et al34 found transdermal estrogen shortened time to response to sertraline in postmenopausal women, although it did not improve end response rate.
Human sexual dimorphism of the serotonergic system has been described for many years,a,b including estrogen’s sexually dimorphic effects on the brain.c Sex steroid receptors are found in mood-processing brain regions in men and womend and may influence sex differences in antidepressant response.
Estrogen has been found to augment serotonergic activitye by increasing serotonin synthesis and decreasing serotonin reuptakef as well as increasing serotonin 5-HT2A binding sites.g Estrogen therapy has been shown to increase the number of sites available for active transport of 5-HT into brain cells.h
References
a. Biver F, Lotstra F, Monclus M, et al. Sex difference in 5HT2 receptor in the living human brain. Neurosci Lett. 1996;204(1-2):25-28.
b. Nishizawa S, Benkelfat C, Young SN, et al. Differences between males and females in rates of serotonin synthesis in human brain. Proc Natl Acad Sci U S A. 1997;94(10):5308-5313.
c. Rubinow DR, Schmidt PJ, Roca CA. Estrogen-serotonin interactions: implications for affective regulation. Biol Psychiatry. 1998;44(9):839-850.
d. McEwen BS, Alves SE, Bulloch K, et al. Ovarian steroids and the brain: implications for cognition and aging. Neurology. 1997;48(5 suppl 7):S8-15.
e. Halbreich U, Rojansky N, Palter S, et al. Estrogen augments serotonergic activity in postmenopausal women. Biol Psychiatry. 1995;37(7):434-441.
f. Shors TJ, Leuner B. Estrogen-mediated effects on depression and memory formation in females. J Affect Disord. 2003;74(1):85-96.
g. Kendall DA, Stancel GM, Enna SJ. The influence of sex hormones on antidepressant-induced alterations in neurotransmitter receptor binding. J Neurosci. 1982;2(3):354-360.
h. Sherwin BB, Suranyi-Cadotte BE. Up-regulatory effect of estrogen on platelet 3H-imipramine binding sites in surgically menopausal women. Biol Psychiatry. 1990;28(4):339-348.
CASE CONTINUED: Dosage increase
After a detailed discussion with her psychiatrist about the potential benefits, known risks, and possible alternatives to using and increasing sertraline in pregnancy, Ms. C agrees to a dosage increase to 75 mg/d. Within 2 weeks she reports decreased anxiety and depression. Her depression remits for the remainder of the pregnancy and she gives birth to a full-term healthy infant. Ms. C’s sertraline dose is held at 75 mg/d during the early postpartum period, as she experienced no side effects at that dose, then reduced to 50 mg/d after a period of sustained euthymia.
Related resources
- Rubinow DR, Schmidt PJ, Roca CA. Estrogen-serotonin interactions: implications for affective regulation. Biol Psychiatry. 1998;44(9):839-850.
- Cohen LS, Nonacs RM, eds. Mood and anxiety disorders during pregnancy and postpartum. Arlington, VA: American Psychiatric Publishing, Inc.; 2005.
- Kornstein SG, Schatzberg AF, Thase ME, et al. Gender differences in treatment response to sertraline versus imipramine in chronic depression. Am J Psychiatry. 2000;157(9):1445-1452.
Drug brand names
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Clomipramine • Anafranil
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Estradiol • Estrace, Climara, others
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Imipramine • Tofranil
- Mirtazapine • Remeron
- Nefazodone • Serzone
- Nortriptyline • Pamelor
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosures
Dr. Marsh receives grant/research support from the University of Massachusetts.
Dr. Deligiannidis receives grant/research support from the Worcester Foundation for Biomedical Research and Forest Research Institute.
Acknowledgement
Dr. Deligiannidis’ contribution to this article was supported by the University of Massachusetts Medical School Department of Psychiatry and the University of Massachusetts Medical School Center for Psychopharmacologic Research and Treatment.
1. Parker G, Parker K, Austin MP, et al. Gender differences in response to differing antidepressant drug classes: two negative studies. Psychol Med. 2003;33(8):1473-1477.
2. Kornstein SG, Schatzberg AF, Thase ME, et al. Gender differences in treatment response to sertraline versus imipramine in chronic depression. Am J Psychiatry. 2000;157(9):1445-1452.
3. Baca E, Garcia-Garcia M, Porras-Chavarino A. Gender differences in treatment response to sertraline versus imipramine in patients with nonmelancholic depressive disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(1):57-65.
4. Joyce PR, Mulder RT, Luty SE, et al. Melancholia: definitions, risk factors, personality, neuroendocrine markers and differential antidepressant response. Aust N Z J Psychiatry. 2002;36(3):376-383.
5. Quitkin FM, Stewart JW, McGrath PJ, et al. Are there differences between women’s and men’s antidepressant responses? Am J Psychiatry. 2002;159(11):1848-1854.
6. Wohlfarth T, Storosum JG, Elferink AJ, et al. Response to tricyclic antidepressants: independent of gender? Am J Psychiatry. 2004;161(2):370-372.
7. Hildebrandt MG, Steyerberg EW, Stage KB, et al. Are gender differences important for the clinical effects of antidepressants? Am J Psychiatry. 2003;160(9):1643-1650.
8. Young EA, Kornstein SG, Marcus SM, et al. Sex differences in response to citalopram: a STAR*D report. J Psychiatr Res. 2009;43(5):503-511.
9. Thase ME, Entsuah R, Cantillon M, et al. Relative antidepressant efficacy of venlafaxine and SSRIs: sex-age interactions. J Womens Health (Larchmt). 2005;14(7):609-616.
10. Rush AJ, Wisniewski SR, Warden D, et al. Selecting among second-step antidepressant medication monotherapies: predictive value of clinical, demographic, or first-step treatment features. Arch Gen Psychiatry. 2008;65(8):870-880.
11. Papakostas GI, Kornstein SG, Clayton AH, et al. Relative antidepressant efficacy of bupropion and the selective serotonin reuptake inhibitors in major depressive disorder: gender-age interactions. Int Clin Psychopharmacol. 2007;22(4):226-229.
12. Kornstein SG, Wohlreich MM, Mallinckrodt CH, et al. Duloxetine efficacy for major depressive disorder in male vs. female patients: data from 7 randomized, double-blind, placebo-controlled trials. J Clin Psychiatry. 2006;67(5):761-770.
13. Wald A, Van Thiel DH, Hoechstetter L, et al. Effect of pregnancy on gastrointestinal transit. Dig Dis Sci. 1982;27(11):1015-1018.
14. Datz FL, Christian PE, Moore J. Gender-related differences in gastric emptying. J Nucl Med. 1987;28(7):1204-1207.
15. Kimmel L, Gonsalvcs D, Youngs D, et al. Fluctuating levels of antidepressants premenstrually. J Psychosom Obstet Gynaecol. 1992;13:277-280.
16. Jensvold MF, Halbrich U, Hamilton JA. Psycho-pharmacology and women: sex, gender and hormones. Washington, DC: American Psychiatric Press, Inc.; 1996.
17. Miller MN, Newell CL, Miller BE, et al. Variable dosing of sertraline for premenstrual exacerbation of depression: a pilot study. J Womens Health (Larchmt). 2008;17(6):993-997.
18. Sit DK, Perel JM, Helsel JC. Changes in antidepressant metabolism and dosing across pregnancy and early postpartum. J Clin Psychiatry. 2008;69(4):652-658.
19. Klier CM, Mossaheb N, Saria A, et al. Pharmacokinetics and elimination of quetiapine, venlafaxine, and trazodone during pregnancy and postpartum. J Clin Psychopharmacol. 2007;27(6):720-722.
20. Wisner KL, Perel JM, Wheeler SB. Tricyclic dose requirements across pregnancy. Am J Psychiatry. 1993;150(10):1541-1542.
21. Wadelius M, Darj E, Frenne G, et al. Induction of CYP2D6 in pregnancy. Clin Pharmacol Ther. 1997;62(4):400-407.
22. Anderson GD. Pregnancy-induced changes in pharmacokinetics: a mechanistic-based approach. Clin Pharmacokinet. 2005;44(10):989-1008.
23. Hostetter A, Stowe ZN, Strader JR, Jr, et al. Dose of selective serotonin uptake inhibitors across pregnancy: clinical implications. Depress Anxiety. 2000;11(2):51-57.
24. Dunlop W. Serial changes in renal haemodynamics during normal human pregnancy. Br J Obstet Gynaecol. 1981;88(1):1-9.
25. Freeman MP, Nolan PE, Jr, Davis MF, et al. Pharmacokinetics of sertraline across pregnancy and postpartum. J Clin Psychopharmacol. 2008;28(6):646-653.
26. Wisner KL, Perel JM, Peindl KS, et al. Effects of the postpartum period on nortriptyline pharmacokinetics. Psychopharmacol Bull. 1997;33(2):243-248.
27. O’Hara MW, Schlechte JA, Lewis DA, et al. Controlled prospective study of postpartum mood disorders: psychological, environmental, and hormonal variables. J Abnorm Psychol. 1991;100(1):63-73.
28. Meltzer-Brody S, Payne J, Rubinow DR. Postpartum depression: what to tell patients who breast-feed. Current Psychiatry. 2008;7(5):87-95.
29. Pinto-Meza A, Usall J, Serrano-Blanco A, et al. Gender differences in response to antidepressant treatment prescribed in primary care. Does menopause make a difference? J Affect Disord. 2006;93(1-3):53-60.
30. Amsterdam J, Garcia-Espana F, Fawcett J, et al. Fluoxetine efficacy in menopausal women with and without estrogen replacement. J Affect Disord. 1999;55(1):11-17.
31. Shapira B, Oppenheim G, Zohar J, et al. Lack of efficacy of estrogen supplementation to imipramine in resistant female depressives. Biol Psychiatry. 1985;20(5):576-579.
32. Schneider LS, Small GW, Clary CM. Estrogen replacement therapy and antidepressant response to sertraline in older depressed women. Am J Geriatr Psychiatry. 2001;9(4):393-399.
33. Zanardi R, Rossini D, Magri L, et al. Response to SSRIs and role of the hormonal therapy in post-menopausal depression. Eur Neuropsychopharmacol. 2007;17(6-7):400-405.
34. Rasgon NL, Dunkin J, Fairbanks L, et al. Estrogen and response to sertraline in postmenopausal women with major depressive disorder: a pilot study. J Psychiatr Res. 2007;41(3-4):338-343.
With a history of panic disorder, perfectionistic tendencies, and depression, Ms. C, age 32, presents 29 weeks into her first pregnancy with a chief complaint that “the Zoloft is not working; my sadness and anxiety are increased and I feel dizzy, like when I miss a dose.” For the past 7 years, she has done well on sertraline, 50 mg/d; she has had no depressive symptoms and experienced minimal to manageable anxiety. Ms. C has found psychotherapy helpful for the last 2 years, including during her pregnancy.
After discussion with her obstetrician, Ms. C remained on sertraline through her early pregnancy. She did well until several weeks ago, when she noticed a return of sadness and incessant worry. She resumed an old habit of excessively cleaning her home. Ms. C denies missing doses but states she has the physical feeling as if she were—a lightheadedness that she clearly distinguishes from pregnancy symptoms.
Both men and women respond well to antidepressants, yet there are notable differences between the 2. Understanding why men and women may differ in response to antidepressants helps clinicians better tailor their treatment choice and dosing.
This article outlines some of differences—and lack thereof—in response rates to antidepressants. Our discussion of why these differences may occur is framed in the context of pharmacokinetics, pharmacodynamics, and the influence of gonadal hormones on antidepressant-related neurotransmitter systems. The second section focuses on major reproductive phases of adult women (the menstrual cycle, pregnancy, postpartum, and menopause) and how antidepressant response rates can influence clinical decision making, such as antidepressant timing, dose, and choice of potential adjunct treatments.
What the evidence says
Most studies look at sex differences in response to a single antidepressant, but several comparing sex differences among classes have produced fascinating results (Table 1). One of the most robust and replicated findings—although not universally reproduced1—is that compared with men, women are more likely to respond to selective serotonin reuptake inhibitors (SSRIs) than to tricyclic antidepressants (TCAs).2-4 Because of this and the fact that SSRIs are so commonly used, this article primarily will address SSRIs in women.
Initially, however, in reviewing non-SSRI anti depressants, monoamine oxidase inhibitors (MAOIs) are reported to produce a superior response in women than in men.5 Women are more likely to have atypical depression symptoms, which MAOIs often treat better than other antidepressants. In contrast, a recent meta-analysis of TCAs6 found no sex response difference within the class. However, 1 study reported women may be slower to respond to TCAs than men.2
Studies on the newer and more frequently prescribed antidepressants reveal some interesting sex differences. Although smaller studies initially did not find a sex difference in SSRIs,5,7 when response rates to citalopram were compared in 2,876 subjects in Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study, women were more likely to reach remission and response than men.8 Younger women—generally those age <50—respond better to SSRIs than women age ≥50.2,3,9
There are less data concerning newer non-SSRI antidepressants. In the second stage of the STAR*D trial, when subjects who did not respond to citalopram were randomly assigned venlafaxine, bupropion, or sertraline, there was no sex difference in response.10 Pooled analysis of randomized controlled trials specifically looking at remission rates between the sexes for venlafaxine,9 bupropion,11 or duloxetine12 found no difference between men and women, regardless of age. No published sex differences in antidepressant response were found for mirtazapine.
Numerous studies have detailed sex differences in antidepressant pharmacokinetics (Box 1) and pharmacodynamics (Box 2), as well as human sexual dimorphism of the serotonergic system. Estrogen’s influence on the serotonergic system (Box 3) may be a component of men and women’s different responses to antidepressants, particularly across reproductive phases.
Table 1
Sex differences in antidepressant response
| Class | Response: Male vs female |
|---|---|
| Monoamine oxidase inhibitors | M |
| Serotonin-norepinephrine reuptake inhibitors | M=F |
| Selective serotonin reuptake inhibitors | Age <50: M< F Age ≥50: M=F |
| Tricyclic antidepressants | M=F |
| References 1-12 | |
Medical literature has documented gender differences in antidepressant absorption, distribution, metabolism, and elimination.a-c Compared with men, women—especially premenopausal women—have slower gastric emptyingd and small bowel and colonic transit times.e,f Also, because antidepressants generally are lipophilic,a,g a lower ratio of lean muscle to adipose tissue in women compared with men may result in a greater volume of drug distribution (Vd).
Sex differences also have been reported in hepatic enzyme activity and may affect clinical response. Most medications, including antidepressants, undergo phase I metabolism, commonly via the cytochrome P450 (CYP450) pathway, and/or phase II conjugation reactions. Generally, phase I oxidative metabolism appears to be greater in women than in men; in contrast, phase II conjugation activity appears to be greater in men than in women.h
Lower CYP1A2 activity in womeni along with gonadal steroid inhibition of CYP1A2j,k may explain why clomipramine metabolic clearance is reduced in young womenl and mean steady state plasma levels of fluvoxamine are almost double in women than in men for the same dose.m In theory, greater CYP3A4 activity in womeni has the potential to accelerate metabolism and/or decrease plasma levels of some commonly used antidepressants metabolized via CYP3A4, such as nefazodone and (to some extent) sertraline and citalopram. In contrast, CYP2D6 and CYP2C9 do not show sex differences in metabolism.
Differences in antidepressant blood levels, however, are difficult to base solely on CYP metabolic route differences. Sex differences in plasma antidepressant levels likely reflect a summation of several sex-associated pharmacokinetic processes and may impact one of many factors that contribute to the small observed difference in antidepressant efficacy between men and women.
References
a. Yonkers KA, Kando JC, Cole JO, et al. Gender differences in pharmacokinetics and pharmacodynamics of psychotropic medication. Am J Psychiatry. 1992;149(5):587-595.
b. Kando JC, Yonkers KA, Cole JO. Gender as a risk factor for adverse events to medications. Drugs. 1995;50(1):1-6.
c. Bies RR, Bigos KL, Pollock BG. Gender differences in the pharmacokinetics and pharmacodynamics of antidepressants. J Gend Specif Med. 2003;6(3):12-20.
d. Hutson WR, Roehrkasse RL, Wald A. Influence of gender and menopause on gastric emptying and motility. Gastroenterology. 1989;96(1):11-17.
e. Sadik R, Abrahamsson H, Stotzer PO. Gender differences in gut transit shown with a newly developed radiological procedure. Scand J Gastroenterol. 2003;38(1):36-42.
f. Lorena SL, Tinois E, Hirata ES, et al. [Scintigraphic study of gastric emptying and intragastric distribution of a solid meal: gender differences]. Arq Gastroenterol. 2000;37(2):102-106.
g. Greenblatt DJ, Divoll M, Abernethy DR, et al. Physiologic changes in old age: relation to altered drug disposition. J Am Geriatr Soc. 1982;30(11 suppl):S6-10.
h. Anderson GD. Gender differences in pharmacological response. Int Rev Neurobiol. 2008;83:1-10.
i. Anderson GD. Sex and racial differences in pharmacological response: where is the evidence? Pharmacogenetics, pharmacokinetics, and pharmacodynamics. J Womens Health (Larchmt). 2005;14(1):19-29.
j. Lane JD, Steege JF, Rupp SL, et al. Menstrual cycle effects on caffeine elimination in the human female. Eur J Clin Pharmacol. 1992;43(5):543-546.
k. Pollock BG, Wylie M, Stack JA, et al. Inhibition of caffeine metabolism by estrogen replacement therapy in postmenopausal women. J Clin Pharmacol. 1999;39(9):936-940.
l. Gex-Fabry M, Balant-Gorgia AE, Balant LP, et al. Clomipramine metabolism. Model-based analysis of variability factors from drug monitoring data. Clin Pharmacokinet. 1990;19(3):241-255.
m. Hartter S, Wetzel H, Hammes E, et al. Nonlinear pharmacokinetics of fluvoxamine and gender differences. Ther Drug Monit. 1998;20(4):446-449.
Sexual dimorphisms in the localization and concentration of endogenous neurotransmitters such as serotonin and dopamine and their degradative enzymes and transporters have the potential to clinically affect antidepressant pharmacodynamics (eg, drug-receptor interactions).
Recent investigations report sex differences in some key monoaminergic enzymes in the brain, notably monoamine oxidase-A (MAO)a,b and catechol-O-methyltransferase (COMT).c-e
For example, estrogen has been found to inhibit MAO,f which is potentially clinically relevant in light of the finding that women respond better than men to MAO inhibitors. COMT—which is responsible for metabolism of norepinephrine, epinephrine, and dopamine—is down regulated by estradiole,g likely accounting for some sex effects. Recently, the sexually dimorphic effect of a COMT polymorphism was associated with a poorer fluoxetine response in men treated for major depression.h
References
a. Domschke K, Hohoff C, Mortensen LS, et al. Monoamine oxidase A variant influences antidepressant treatment response in female patients with major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(1):224-228.
b. Yu YW, Tsai SJ, Hong CJ, et al. Association study of a monoamine oxidase a gene promoter polymorphism with major depressive disorder and antidepressant response. Neuropsychopharmacology. 2005;30(9):1719-1723.
c. Baune BT, Hohoff C, Berger K, et al. Association of the COMT val158met variant with antidepressant treatment response in major depression. Neuropsychopharmacology. 2008;33(4):924-932.
d. Harrison PJ, Tunbridge EM. Catechol-O-methyltransferase (COMT): a gene contributing to sex differences in brain function, and to sexual dimorphism in the predisposition to psychiatric disorders. Neuropsychopharmacology. 2008;33(13):3037-3045.
e. Jiang H, Xie T, Ramsden DB, et al. Human catechol-O-methyltransferase down-regulation by estradiol. Neuropharmacology. 2003;45(7):1011-1018.
f. Luine VN, Khylchevskaya RI, McEwen BS. Effect of gonadal steroids on activities of monoamine oxidase and choline acetylase in rat brain. Brain Res. 1975;86(2):293-306.
g. Xie T, Ho SL, Ramsden D. Characterization and implications of estrogenic down-regulation of human catechol-O-methyltransferase gene transcription. Mol Pharmacol. 1999;56(1):31-38.
h. Tsai SJ, Gau YT, Hong CJ, et al. Sexually dimorphic effect of catechol-O-methyltransferase val158met polymorphism on clinical response to fluoxetine in major depressive patients. J Affect Disord. 2009;113(1-2):183-187.
Change across reproductive phases
In contrast to men, women’s estrogen and progesterone status varies widely across a woman’s reproductive lifecycle (menstrual cycle, pregnancy, postpartum, premenopause vs post menopause). In men and women, androgen levels—including testosterone—tend to remain at steady levels, and then slowly decline with age.
Menstrual cycle. Hormone-related changes associated with the menstrual cycle may affect antidepressant absorption and distribution. During the luteal phase—second half of the menstrual cycle post-ovulation—and pregnancy, increased progesterone concentrations are associated with slowed gastrointestinal transit time13,14 compared with the follicular phase (preovulation).
Premenstrually, at the end of luteal phase, reduced serum antidepressant levels have been associated with breakthrough depressive symptoms.15,16 In these case reports, serum antidepressant levels returned to baseline and depressive symptoms resolved after menses ended. It is possible that women may be at increased risk of symptom recurrence before menses because of hormonally driven changes in drug absorption, distribution, and metabolism. Increased dosing of sertraline in the luteal phase has helped reduce premenstrual exacerbation of depression.17
Pregnancy. Dose requirements for the SSRIs citalopram, escitalopram, and sertraline,18 the serotonin-norepinephrine reuptake inhibitor venlafaxine,19 and the TCAs nortriptyline, clomipramine, and imipramine20 increase during the second half of pregnancy. This appears to be the result of increased drug metabolism. Altered cytochrome P450 (CYP450) enzymatic activity in pregnancy—likely mediated by elevated estrogen and progesterone—may have clinical effects on drug levels and treatment response. Studies indicate that CYP3A4—and possibly CYP2D6—are induced during pregnancy.21,22 Dose increases are necessary in two-thirds of pregnant women on antidepressant monotherapy, typically after 20 weeks gestation18,20,23 to treat symptom recurrence or maintain euthymia.
During pregnancy, drug elimination may increase because of higher renal blood flow and glomerular filtration rate (GFR).24 This could reduce blood levels of water-soluble active metabolites of some TCAs. Pregnancy-associated reductions in intestinal motility and gastric pH alone do not change medication bioavailability. Increased body fat could increase the volume of drug distribution for antidepressants, and, in theory, create a dilutional drop in free drug concentration, but this likely would have only a minor effect.
The range of antidepressant effectiveness among pregnant patients is wide, which reflects individual differences in pharmacokinetics and pharmacodynamics.25 Because we cannot predict which women will require dose changes during pregnancy or postpartum, patients should be monitored frequently for depressive symptom recurrence. Dose adjustments may be necessary to prevent relapse (eg, when net metabolism is increased) or pronounced side effects (eg, when net metabolism is reduced).18,26
When prescribing antidepressants for pregnant women, a personalized discussion of the risks and benefits with each patient in the context of her psychiatric history, the developing fetus, and her value system is warranted. The potential consequences of antidepressant effect on patient and fetus, or lack there of, continues to be an evolving area; long-term data on prenatal exposure are limited.
Postpartum. The postpartum period—when depression can hit 10% to 15% of new mothers27—entails rapid shifts in many factors that may influence antidepressant response. Levels of gonadal hormones such as estrogen and progesterone decline, plasma volume contracts, and hepatic enzymatic metabolism and GFR return to pre-pregnancy levels. Together these changes may result in increased antidepressant blood levels postpartum, especially when the dosage used during pregnancy is held constant.19
The postpartum period is associated with a high risk for depression onset or worsening and is a time of great hormonal and pharmacokinetic change. Accordingly, a postpartum woman should be followed closely for changes in response and adverse effects, and her antidepressant dosage adjusted. Breastfeeding is a critical consideration in the postpartum. Meltzer-Brody et al28 provide a discussion of postpartum depression and what to tell patients who breast-feed.
Menopause. Despite evidence that reproductive-age women may respond better to SSRIs than men, the same findings have not been reproduced in postmenopausal women. For example, compared with men, postmenopausal women had no significant difference in SSRI treatment response in primary care clinics. In contrast, the same postmenopausal women had a significantly worse treatment response than premenopausal women.29
In considering why SSRI response among women would differ depending on reproductive stage or hormonal status, researchers examined the effect of estrogen on antidepressant response with the use of estrogen therapy (ET). As detailed in Box 3, estrogen has many serotonergic-enhancing properties. Early studies with TCAs and a retrospective analysis of SSRIs did not demonstrate improved antidepressant effect with the addition of ET in depressed women.30,31 In contrast, recent studies have demonstrated better SSRI response—regardless of which medication was used—in postmenopausal women on ET or ET with progesterone, compared with postmenopausal women taking placebo.32,33 Perhaps explaining the discrepancy, in a randomized, placebo-controlled trial, Rasgon et al34 found transdermal estrogen shortened time to response to sertraline in postmenopausal women, although it did not improve end response rate.
Human sexual dimorphism of the serotonergic system has been described for many years,a,b including estrogen’s sexually dimorphic effects on the brain.c Sex steroid receptors are found in mood-processing brain regions in men and womend and may influence sex differences in antidepressant response.
Estrogen has been found to augment serotonergic activitye by increasing serotonin synthesis and decreasing serotonin reuptakef as well as increasing serotonin 5-HT2A binding sites.g Estrogen therapy has been shown to increase the number of sites available for active transport of 5-HT into brain cells.h
References
a. Biver F, Lotstra F, Monclus M, et al. Sex difference in 5HT2 receptor in the living human brain. Neurosci Lett. 1996;204(1-2):25-28.
b. Nishizawa S, Benkelfat C, Young SN, et al. Differences between males and females in rates of serotonin synthesis in human brain. Proc Natl Acad Sci U S A. 1997;94(10):5308-5313.
c. Rubinow DR, Schmidt PJ, Roca CA. Estrogen-serotonin interactions: implications for affective regulation. Biol Psychiatry. 1998;44(9):839-850.
d. McEwen BS, Alves SE, Bulloch K, et al. Ovarian steroids and the brain: implications for cognition and aging. Neurology. 1997;48(5 suppl 7):S8-15.
e. Halbreich U, Rojansky N, Palter S, et al. Estrogen augments serotonergic activity in postmenopausal women. Biol Psychiatry. 1995;37(7):434-441.
f. Shors TJ, Leuner B. Estrogen-mediated effects on depression and memory formation in females. J Affect Disord. 2003;74(1):85-96.
g. Kendall DA, Stancel GM, Enna SJ. The influence of sex hormones on antidepressant-induced alterations in neurotransmitter receptor binding. J Neurosci. 1982;2(3):354-360.
h. Sherwin BB, Suranyi-Cadotte BE. Up-regulatory effect of estrogen on platelet 3H-imipramine binding sites in surgically menopausal women. Biol Psychiatry. 1990;28(4):339-348.
CASE CONTINUED: Dosage increase
After a detailed discussion with her psychiatrist about the potential benefits, known risks, and possible alternatives to using and increasing sertraline in pregnancy, Ms. C agrees to a dosage increase to 75 mg/d. Within 2 weeks she reports decreased anxiety and depression. Her depression remits for the remainder of the pregnancy and she gives birth to a full-term healthy infant. Ms. C’s sertraline dose is held at 75 mg/d during the early postpartum period, as she experienced no side effects at that dose, then reduced to 50 mg/d after a period of sustained euthymia.
Related resources
- Rubinow DR, Schmidt PJ, Roca CA. Estrogen-serotonin interactions: implications for affective regulation. Biol Psychiatry. 1998;44(9):839-850.
- Cohen LS, Nonacs RM, eds. Mood and anxiety disorders during pregnancy and postpartum. Arlington, VA: American Psychiatric Publishing, Inc.; 2005.
- Kornstein SG, Schatzberg AF, Thase ME, et al. Gender differences in treatment response to sertraline versus imipramine in chronic depression. Am J Psychiatry. 2000;157(9):1445-1452.
Drug brand names
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Clomipramine • Anafranil
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Estradiol • Estrace, Climara, others
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Imipramine • Tofranil
- Mirtazapine • Remeron
- Nefazodone • Serzone
- Nortriptyline • Pamelor
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosures
Dr. Marsh receives grant/research support from the University of Massachusetts.
Dr. Deligiannidis receives grant/research support from the Worcester Foundation for Biomedical Research and Forest Research Institute.
Acknowledgement
Dr. Deligiannidis’ contribution to this article was supported by the University of Massachusetts Medical School Department of Psychiatry and the University of Massachusetts Medical School Center for Psychopharmacologic Research and Treatment.
With a history of panic disorder, perfectionistic tendencies, and depression, Ms. C, age 32, presents 29 weeks into her first pregnancy with a chief complaint that “the Zoloft is not working; my sadness and anxiety are increased and I feel dizzy, like when I miss a dose.” For the past 7 years, she has done well on sertraline, 50 mg/d; she has had no depressive symptoms and experienced minimal to manageable anxiety. Ms. C has found psychotherapy helpful for the last 2 years, including during her pregnancy.
After discussion with her obstetrician, Ms. C remained on sertraline through her early pregnancy. She did well until several weeks ago, when she noticed a return of sadness and incessant worry. She resumed an old habit of excessively cleaning her home. Ms. C denies missing doses but states she has the physical feeling as if she were—a lightheadedness that she clearly distinguishes from pregnancy symptoms.
Both men and women respond well to antidepressants, yet there are notable differences between the 2. Understanding why men and women may differ in response to antidepressants helps clinicians better tailor their treatment choice and dosing.
This article outlines some of differences—and lack thereof—in response rates to antidepressants. Our discussion of why these differences may occur is framed in the context of pharmacokinetics, pharmacodynamics, and the influence of gonadal hormones on antidepressant-related neurotransmitter systems. The second section focuses on major reproductive phases of adult women (the menstrual cycle, pregnancy, postpartum, and menopause) and how antidepressant response rates can influence clinical decision making, such as antidepressant timing, dose, and choice of potential adjunct treatments.
What the evidence says
Most studies look at sex differences in response to a single antidepressant, but several comparing sex differences among classes have produced fascinating results (Table 1). One of the most robust and replicated findings—although not universally reproduced1—is that compared with men, women are more likely to respond to selective serotonin reuptake inhibitors (SSRIs) than to tricyclic antidepressants (TCAs).2-4 Because of this and the fact that SSRIs are so commonly used, this article primarily will address SSRIs in women.
Initially, however, in reviewing non-SSRI anti depressants, monoamine oxidase inhibitors (MAOIs) are reported to produce a superior response in women than in men.5 Women are more likely to have atypical depression symptoms, which MAOIs often treat better than other antidepressants. In contrast, a recent meta-analysis of TCAs6 found no sex response difference within the class. However, 1 study reported women may be slower to respond to TCAs than men.2
Studies on the newer and more frequently prescribed antidepressants reveal some interesting sex differences. Although smaller studies initially did not find a sex difference in SSRIs,5,7 when response rates to citalopram were compared in 2,876 subjects in Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study, women were more likely to reach remission and response than men.8 Younger women—generally those age <50—respond better to SSRIs than women age ≥50.2,3,9
There are less data concerning newer non-SSRI antidepressants. In the second stage of the STAR*D trial, when subjects who did not respond to citalopram were randomly assigned venlafaxine, bupropion, or sertraline, there was no sex difference in response.10 Pooled analysis of randomized controlled trials specifically looking at remission rates between the sexes for venlafaxine,9 bupropion,11 or duloxetine12 found no difference between men and women, regardless of age. No published sex differences in antidepressant response were found for mirtazapine.
Numerous studies have detailed sex differences in antidepressant pharmacokinetics (Box 1) and pharmacodynamics (Box 2), as well as human sexual dimorphism of the serotonergic system. Estrogen’s influence on the serotonergic system (Box 3) may be a component of men and women’s different responses to antidepressants, particularly across reproductive phases.
Table 1
Sex differences in antidepressant response
| Class | Response: Male vs female |
|---|---|
| Monoamine oxidase inhibitors | M |
| Serotonin-norepinephrine reuptake inhibitors | M=F |
| Selective serotonin reuptake inhibitors | Age <50: M< F Age ≥50: M=F |
| Tricyclic antidepressants | M=F |
| References 1-12 | |
Medical literature has documented gender differences in antidepressant absorption, distribution, metabolism, and elimination.a-c Compared with men, women—especially premenopausal women—have slower gastric emptyingd and small bowel and colonic transit times.e,f Also, because antidepressants generally are lipophilic,a,g a lower ratio of lean muscle to adipose tissue in women compared with men may result in a greater volume of drug distribution (Vd).
Sex differences also have been reported in hepatic enzyme activity and may affect clinical response. Most medications, including antidepressants, undergo phase I metabolism, commonly via the cytochrome P450 (CYP450) pathway, and/or phase II conjugation reactions. Generally, phase I oxidative metabolism appears to be greater in women than in men; in contrast, phase II conjugation activity appears to be greater in men than in women.h
Lower CYP1A2 activity in womeni along with gonadal steroid inhibition of CYP1A2j,k may explain why clomipramine metabolic clearance is reduced in young womenl and mean steady state plasma levels of fluvoxamine are almost double in women than in men for the same dose.m In theory, greater CYP3A4 activity in womeni has the potential to accelerate metabolism and/or decrease plasma levels of some commonly used antidepressants metabolized via CYP3A4, such as nefazodone and (to some extent) sertraline and citalopram. In contrast, CYP2D6 and CYP2C9 do not show sex differences in metabolism.
Differences in antidepressant blood levels, however, are difficult to base solely on CYP metabolic route differences. Sex differences in plasma antidepressant levels likely reflect a summation of several sex-associated pharmacokinetic processes and may impact one of many factors that contribute to the small observed difference in antidepressant efficacy between men and women.
References
a. Yonkers KA, Kando JC, Cole JO, et al. Gender differences in pharmacokinetics and pharmacodynamics of psychotropic medication. Am J Psychiatry. 1992;149(5):587-595.
b. Kando JC, Yonkers KA, Cole JO. Gender as a risk factor for adverse events to medications. Drugs. 1995;50(1):1-6.
c. Bies RR, Bigos KL, Pollock BG. Gender differences in the pharmacokinetics and pharmacodynamics of antidepressants. J Gend Specif Med. 2003;6(3):12-20.
d. Hutson WR, Roehrkasse RL, Wald A. Influence of gender and menopause on gastric emptying and motility. Gastroenterology. 1989;96(1):11-17.
e. Sadik R, Abrahamsson H, Stotzer PO. Gender differences in gut transit shown with a newly developed radiological procedure. Scand J Gastroenterol. 2003;38(1):36-42.
f. Lorena SL, Tinois E, Hirata ES, et al. [Scintigraphic study of gastric emptying and intragastric distribution of a solid meal: gender differences]. Arq Gastroenterol. 2000;37(2):102-106.
g. Greenblatt DJ, Divoll M, Abernethy DR, et al. Physiologic changes in old age: relation to altered drug disposition. J Am Geriatr Soc. 1982;30(11 suppl):S6-10.
h. Anderson GD. Gender differences in pharmacological response. Int Rev Neurobiol. 2008;83:1-10.
i. Anderson GD. Sex and racial differences in pharmacological response: where is the evidence? Pharmacogenetics, pharmacokinetics, and pharmacodynamics. J Womens Health (Larchmt). 2005;14(1):19-29.
j. Lane JD, Steege JF, Rupp SL, et al. Menstrual cycle effects on caffeine elimination in the human female. Eur J Clin Pharmacol. 1992;43(5):543-546.
k. Pollock BG, Wylie M, Stack JA, et al. Inhibition of caffeine metabolism by estrogen replacement therapy in postmenopausal women. J Clin Pharmacol. 1999;39(9):936-940.
l. Gex-Fabry M, Balant-Gorgia AE, Balant LP, et al. Clomipramine metabolism. Model-based analysis of variability factors from drug monitoring data. Clin Pharmacokinet. 1990;19(3):241-255.
m. Hartter S, Wetzel H, Hammes E, et al. Nonlinear pharmacokinetics of fluvoxamine and gender differences. Ther Drug Monit. 1998;20(4):446-449.
Sexual dimorphisms in the localization and concentration of endogenous neurotransmitters such as serotonin and dopamine and their degradative enzymes and transporters have the potential to clinically affect antidepressant pharmacodynamics (eg, drug-receptor interactions).
Recent investigations report sex differences in some key monoaminergic enzymes in the brain, notably monoamine oxidase-A (MAO)a,b and catechol-O-methyltransferase (COMT).c-e
For example, estrogen has been found to inhibit MAO,f which is potentially clinically relevant in light of the finding that women respond better than men to MAO inhibitors. COMT—which is responsible for metabolism of norepinephrine, epinephrine, and dopamine—is down regulated by estradiole,g likely accounting for some sex effects. Recently, the sexually dimorphic effect of a COMT polymorphism was associated with a poorer fluoxetine response in men treated for major depression.h
References
a. Domschke K, Hohoff C, Mortensen LS, et al. Monoamine oxidase A variant influences antidepressant treatment response in female patients with major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(1):224-228.
b. Yu YW, Tsai SJ, Hong CJ, et al. Association study of a monoamine oxidase a gene promoter polymorphism with major depressive disorder and antidepressant response. Neuropsychopharmacology. 2005;30(9):1719-1723.
c. Baune BT, Hohoff C, Berger K, et al. Association of the COMT val158met variant with antidepressant treatment response in major depression. Neuropsychopharmacology. 2008;33(4):924-932.
d. Harrison PJ, Tunbridge EM. Catechol-O-methyltransferase (COMT): a gene contributing to sex differences in brain function, and to sexual dimorphism in the predisposition to psychiatric disorders. Neuropsychopharmacology. 2008;33(13):3037-3045.
e. Jiang H, Xie T, Ramsden DB, et al. Human catechol-O-methyltransferase down-regulation by estradiol. Neuropharmacology. 2003;45(7):1011-1018.
f. Luine VN, Khylchevskaya RI, McEwen BS. Effect of gonadal steroids on activities of monoamine oxidase and choline acetylase in rat brain. Brain Res. 1975;86(2):293-306.
g. Xie T, Ho SL, Ramsden D. Characterization and implications of estrogenic down-regulation of human catechol-O-methyltransferase gene transcription. Mol Pharmacol. 1999;56(1):31-38.
h. Tsai SJ, Gau YT, Hong CJ, et al. Sexually dimorphic effect of catechol-O-methyltransferase val158met polymorphism on clinical response to fluoxetine in major depressive patients. J Affect Disord. 2009;113(1-2):183-187.
Change across reproductive phases
In contrast to men, women’s estrogen and progesterone status varies widely across a woman’s reproductive lifecycle (menstrual cycle, pregnancy, postpartum, premenopause vs post menopause). In men and women, androgen levels—including testosterone—tend to remain at steady levels, and then slowly decline with age.
Menstrual cycle. Hormone-related changes associated with the menstrual cycle may affect antidepressant absorption and distribution. During the luteal phase—second half of the menstrual cycle post-ovulation—and pregnancy, increased progesterone concentrations are associated with slowed gastrointestinal transit time13,14 compared with the follicular phase (preovulation).
Premenstrually, at the end of luteal phase, reduced serum antidepressant levels have been associated with breakthrough depressive symptoms.15,16 In these case reports, serum antidepressant levels returned to baseline and depressive symptoms resolved after menses ended. It is possible that women may be at increased risk of symptom recurrence before menses because of hormonally driven changes in drug absorption, distribution, and metabolism. Increased dosing of sertraline in the luteal phase has helped reduce premenstrual exacerbation of depression.17
Pregnancy. Dose requirements for the SSRIs citalopram, escitalopram, and sertraline,18 the serotonin-norepinephrine reuptake inhibitor venlafaxine,19 and the TCAs nortriptyline, clomipramine, and imipramine20 increase during the second half of pregnancy. This appears to be the result of increased drug metabolism. Altered cytochrome P450 (CYP450) enzymatic activity in pregnancy—likely mediated by elevated estrogen and progesterone—may have clinical effects on drug levels and treatment response. Studies indicate that CYP3A4—and possibly CYP2D6—are induced during pregnancy.21,22 Dose increases are necessary in two-thirds of pregnant women on antidepressant monotherapy, typically after 20 weeks gestation18,20,23 to treat symptom recurrence or maintain euthymia.
During pregnancy, drug elimination may increase because of higher renal blood flow and glomerular filtration rate (GFR).24 This could reduce blood levels of water-soluble active metabolites of some TCAs. Pregnancy-associated reductions in intestinal motility and gastric pH alone do not change medication bioavailability. Increased body fat could increase the volume of drug distribution for antidepressants, and, in theory, create a dilutional drop in free drug concentration, but this likely would have only a minor effect.
The range of antidepressant effectiveness among pregnant patients is wide, which reflects individual differences in pharmacokinetics and pharmacodynamics.25 Because we cannot predict which women will require dose changes during pregnancy or postpartum, patients should be monitored frequently for depressive symptom recurrence. Dose adjustments may be necessary to prevent relapse (eg, when net metabolism is increased) or pronounced side effects (eg, when net metabolism is reduced).18,26
When prescribing antidepressants for pregnant women, a personalized discussion of the risks and benefits with each patient in the context of her psychiatric history, the developing fetus, and her value system is warranted. The potential consequences of antidepressant effect on patient and fetus, or lack there of, continues to be an evolving area; long-term data on prenatal exposure are limited.
Postpartum. The postpartum period—when depression can hit 10% to 15% of new mothers27—entails rapid shifts in many factors that may influence antidepressant response. Levels of gonadal hormones such as estrogen and progesterone decline, plasma volume contracts, and hepatic enzymatic metabolism and GFR return to pre-pregnancy levels. Together these changes may result in increased antidepressant blood levels postpartum, especially when the dosage used during pregnancy is held constant.19
The postpartum period is associated with a high risk for depression onset or worsening and is a time of great hormonal and pharmacokinetic change. Accordingly, a postpartum woman should be followed closely for changes in response and adverse effects, and her antidepressant dosage adjusted. Breastfeeding is a critical consideration in the postpartum. Meltzer-Brody et al28 provide a discussion of postpartum depression and what to tell patients who breast-feed.
Menopause. Despite evidence that reproductive-age women may respond better to SSRIs than men, the same findings have not been reproduced in postmenopausal women. For example, compared with men, postmenopausal women had no significant difference in SSRI treatment response in primary care clinics. In contrast, the same postmenopausal women had a significantly worse treatment response than premenopausal women.29
In considering why SSRI response among women would differ depending on reproductive stage or hormonal status, researchers examined the effect of estrogen on antidepressant response with the use of estrogen therapy (ET). As detailed in Box 3, estrogen has many serotonergic-enhancing properties. Early studies with TCAs and a retrospective analysis of SSRIs did not demonstrate improved antidepressant effect with the addition of ET in depressed women.30,31 In contrast, recent studies have demonstrated better SSRI response—regardless of which medication was used—in postmenopausal women on ET or ET with progesterone, compared with postmenopausal women taking placebo.32,33 Perhaps explaining the discrepancy, in a randomized, placebo-controlled trial, Rasgon et al34 found transdermal estrogen shortened time to response to sertraline in postmenopausal women, although it did not improve end response rate.
Human sexual dimorphism of the serotonergic system has been described for many years,a,b including estrogen’s sexually dimorphic effects on the brain.c Sex steroid receptors are found in mood-processing brain regions in men and womend and may influence sex differences in antidepressant response.
Estrogen has been found to augment serotonergic activitye by increasing serotonin synthesis and decreasing serotonin reuptakef as well as increasing serotonin 5-HT2A binding sites.g Estrogen therapy has been shown to increase the number of sites available for active transport of 5-HT into brain cells.h
References
a. Biver F, Lotstra F, Monclus M, et al. Sex difference in 5HT2 receptor in the living human brain. Neurosci Lett. 1996;204(1-2):25-28.
b. Nishizawa S, Benkelfat C, Young SN, et al. Differences between males and females in rates of serotonin synthesis in human brain. Proc Natl Acad Sci U S A. 1997;94(10):5308-5313.
c. Rubinow DR, Schmidt PJ, Roca CA. Estrogen-serotonin interactions: implications for affective regulation. Biol Psychiatry. 1998;44(9):839-850.
d. McEwen BS, Alves SE, Bulloch K, et al. Ovarian steroids and the brain: implications for cognition and aging. Neurology. 1997;48(5 suppl 7):S8-15.
e. Halbreich U, Rojansky N, Palter S, et al. Estrogen augments serotonergic activity in postmenopausal women. Biol Psychiatry. 1995;37(7):434-441.
f. Shors TJ, Leuner B. Estrogen-mediated effects on depression and memory formation in females. J Affect Disord. 2003;74(1):85-96.
g. Kendall DA, Stancel GM, Enna SJ. The influence of sex hormones on antidepressant-induced alterations in neurotransmitter receptor binding. J Neurosci. 1982;2(3):354-360.
h. Sherwin BB, Suranyi-Cadotte BE. Up-regulatory effect of estrogen on platelet 3H-imipramine binding sites in surgically menopausal women. Biol Psychiatry. 1990;28(4):339-348.
CASE CONTINUED: Dosage increase
After a detailed discussion with her psychiatrist about the potential benefits, known risks, and possible alternatives to using and increasing sertraline in pregnancy, Ms. C agrees to a dosage increase to 75 mg/d. Within 2 weeks she reports decreased anxiety and depression. Her depression remits for the remainder of the pregnancy and she gives birth to a full-term healthy infant. Ms. C’s sertraline dose is held at 75 mg/d during the early postpartum period, as she experienced no side effects at that dose, then reduced to 50 mg/d after a period of sustained euthymia.
Related resources
- Rubinow DR, Schmidt PJ, Roca CA. Estrogen-serotonin interactions: implications for affective regulation. Biol Psychiatry. 1998;44(9):839-850.
- Cohen LS, Nonacs RM, eds. Mood and anxiety disorders during pregnancy and postpartum. Arlington, VA: American Psychiatric Publishing, Inc.; 2005.
- Kornstein SG, Schatzberg AF, Thase ME, et al. Gender differences in treatment response to sertraline versus imipramine in chronic depression. Am J Psychiatry. 2000;157(9):1445-1452.
Drug brand names
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Clomipramine • Anafranil
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Estradiol • Estrace, Climara, others
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Imipramine • Tofranil
- Mirtazapine • Remeron
- Nefazodone • Serzone
- Nortriptyline • Pamelor
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosures
Dr. Marsh receives grant/research support from the University of Massachusetts.
Dr. Deligiannidis receives grant/research support from the Worcester Foundation for Biomedical Research and Forest Research Institute.
Acknowledgement
Dr. Deligiannidis’ contribution to this article was supported by the University of Massachusetts Medical School Department of Psychiatry and the University of Massachusetts Medical School Center for Psychopharmacologic Research and Treatment.
1. Parker G, Parker K, Austin MP, et al. Gender differences in response to differing antidepressant drug classes: two negative studies. Psychol Med. 2003;33(8):1473-1477.
2. Kornstein SG, Schatzberg AF, Thase ME, et al. Gender differences in treatment response to sertraline versus imipramine in chronic depression. Am J Psychiatry. 2000;157(9):1445-1452.
3. Baca E, Garcia-Garcia M, Porras-Chavarino A. Gender differences in treatment response to sertraline versus imipramine in patients with nonmelancholic depressive disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(1):57-65.
4. Joyce PR, Mulder RT, Luty SE, et al. Melancholia: definitions, risk factors, personality, neuroendocrine markers and differential antidepressant response. Aust N Z J Psychiatry. 2002;36(3):376-383.
5. Quitkin FM, Stewart JW, McGrath PJ, et al. Are there differences between women’s and men’s antidepressant responses? Am J Psychiatry. 2002;159(11):1848-1854.
6. Wohlfarth T, Storosum JG, Elferink AJ, et al. Response to tricyclic antidepressants: independent of gender? Am J Psychiatry. 2004;161(2):370-372.
7. Hildebrandt MG, Steyerberg EW, Stage KB, et al. Are gender differences important for the clinical effects of antidepressants? Am J Psychiatry. 2003;160(9):1643-1650.
8. Young EA, Kornstein SG, Marcus SM, et al. Sex differences in response to citalopram: a STAR*D report. J Psychiatr Res. 2009;43(5):503-511.
9. Thase ME, Entsuah R, Cantillon M, et al. Relative antidepressant efficacy of venlafaxine and SSRIs: sex-age interactions. J Womens Health (Larchmt). 2005;14(7):609-616.
10. Rush AJ, Wisniewski SR, Warden D, et al. Selecting among second-step antidepressant medication monotherapies: predictive value of clinical, demographic, or first-step treatment features. Arch Gen Psychiatry. 2008;65(8):870-880.
11. Papakostas GI, Kornstein SG, Clayton AH, et al. Relative antidepressant efficacy of bupropion and the selective serotonin reuptake inhibitors in major depressive disorder: gender-age interactions. Int Clin Psychopharmacol. 2007;22(4):226-229.
12. Kornstein SG, Wohlreich MM, Mallinckrodt CH, et al. Duloxetine efficacy for major depressive disorder in male vs. female patients: data from 7 randomized, double-blind, placebo-controlled trials. J Clin Psychiatry. 2006;67(5):761-770.
13. Wald A, Van Thiel DH, Hoechstetter L, et al. Effect of pregnancy on gastrointestinal transit. Dig Dis Sci. 1982;27(11):1015-1018.
14. Datz FL, Christian PE, Moore J. Gender-related differences in gastric emptying. J Nucl Med. 1987;28(7):1204-1207.
15. Kimmel L, Gonsalvcs D, Youngs D, et al. Fluctuating levels of antidepressants premenstrually. J Psychosom Obstet Gynaecol. 1992;13:277-280.
16. Jensvold MF, Halbrich U, Hamilton JA. Psycho-pharmacology and women: sex, gender and hormones. Washington, DC: American Psychiatric Press, Inc.; 1996.
17. Miller MN, Newell CL, Miller BE, et al. Variable dosing of sertraline for premenstrual exacerbation of depression: a pilot study. J Womens Health (Larchmt). 2008;17(6):993-997.
18. Sit DK, Perel JM, Helsel JC. Changes in antidepressant metabolism and dosing across pregnancy and early postpartum. J Clin Psychiatry. 2008;69(4):652-658.
19. Klier CM, Mossaheb N, Saria A, et al. Pharmacokinetics and elimination of quetiapine, venlafaxine, and trazodone during pregnancy and postpartum. J Clin Psychopharmacol. 2007;27(6):720-722.
20. Wisner KL, Perel JM, Wheeler SB. Tricyclic dose requirements across pregnancy. Am J Psychiatry. 1993;150(10):1541-1542.
21. Wadelius M, Darj E, Frenne G, et al. Induction of CYP2D6 in pregnancy. Clin Pharmacol Ther. 1997;62(4):400-407.
22. Anderson GD. Pregnancy-induced changes in pharmacokinetics: a mechanistic-based approach. Clin Pharmacokinet. 2005;44(10):989-1008.
23. Hostetter A, Stowe ZN, Strader JR, Jr, et al. Dose of selective serotonin uptake inhibitors across pregnancy: clinical implications. Depress Anxiety. 2000;11(2):51-57.
24. Dunlop W. Serial changes in renal haemodynamics during normal human pregnancy. Br J Obstet Gynaecol. 1981;88(1):1-9.
25. Freeman MP, Nolan PE, Jr, Davis MF, et al. Pharmacokinetics of sertraline across pregnancy and postpartum. J Clin Psychopharmacol. 2008;28(6):646-653.
26. Wisner KL, Perel JM, Peindl KS, et al. Effects of the postpartum period on nortriptyline pharmacokinetics. Psychopharmacol Bull. 1997;33(2):243-248.
27. O’Hara MW, Schlechte JA, Lewis DA, et al. Controlled prospective study of postpartum mood disorders: psychological, environmental, and hormonal variables. J Abnorm Psychol. 1991;100(1):63-73.
28. Meltzer-Brody S, Payne J, Rubinow DR. Postpartum depression: what to tell patients who breast-feed. Current Psychiatry. 2008;7(5):87-95.
29. Pinto-Meza A, Usall J, Serrano-Blanco A, et al. Gender differences in response to antidepressant treatment prescribed in primary care. Does menopause make a difference? J Affect Disord. 2006;93(1-3):53-60.
30. Amsterdam J, Garcia-Espana F, Fawcett J, et al. Fluoxetine efficacy in menopausal women with and without estrogen replacement. J Affect Disord. 1999;55(1):11-17.
31. Shapira B, Oppenheim G, Zohar J, et al. Lack of efficacy of estrogen supplementation to imipramine in resistant female depressives. Biol Psychiatry. 1985;20(5):576-579.
32. Schneider LS, Small GW, Clary CM. Estrogen replacement therapy and antidepressant response to sertraline in older depressed women. Am J Geriatr Psychiatry. 2001;9(4):393-399.
33. Zanardi R, Rossini D, Magri L, et al. Response to SSRIs and role of the hormonal therapy in post-menopausal depression. Eur Neuropsychopharmacol. 2007;17(6-7):400-405.
34. Rasgon NL, Dunkin J, Fairbanks L, et al. Estrogen and response to sertraline in postmenopausal women with major depressive disorder: a pilot study. J Psychiatr Res. 2007;41(3-4):338-343.
1. Parker G, Parker K, Austin MP, et al. Gender differences in response to differing antidepressant drug classes: two negative studies. Psychol Med. 2003;33(8):1473-1477.
2. Kornstein SG, Schatzberg AF, Thase ME, et al. Gender differences in treatment response to sertraline versus imipramine in chronic depression. Am J Psychiatry. 2000;157(9):1445-1452.
3. Baca E, Garcia-Garcia M, Porras-Chavarino A. Gender differences in treatment response to sertraline versus imipramine in patients with nonmelancholic depressive disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(1):57-65.
4. Joyce PR, Mulder RT, Luty SE, et al. Melancholia: definitions, risk factors, personality, neuroendocrine markers and differential antidepressant response. Aust N Z J Psychiatry. 2002;36(3):376-383.
5. Quitkin FM, Stewart JW, McGrath PJ, et al. Are there differences between women’s and men’s antidepressant responses? Am J Psychiatry. 2002;159(11):1848-1854.
6. Wohlfarth T, Storosum JG, Elferink AJ, et al. Response to tricyclic antidepressants: independent of gender? Am J Psychiatry. 2004;161(2):370-372.
7. Hildebrandt MG, Steyerberg EW, Stage KB, et al. Are gender differences important for the clinical effects of antidepressants? Am J Psychiatry. 2003;160(9):1643-1650.
8. Young EA, Kornstein SG, Marcus SM, et al. Sex differences in response to citalopram: a STAR*D report. J Psychiatr Res. 2009;43(5):503-511.
9. Thase ME, Entsuah R, Cantillon M, et al. Relative antidepressant efficacy of venlafaxine and SSRIs: sex-age interactions. J Womens Health (Larchmt). 2005;14(7):609-616.
10. Rush AJ, Wisniewski SR, Warden D, et al. Selecting among second-step antidepressant medication monotherapies: predictive value of clinical, demographic, or first-step treatment features. Arch Gen Psychiatry. 2008;65(8):870-880.
11. Papakostas GI, Kornstein SG, Clayton AH, et al. Relative antidepressant efficacy of bupropion and the selective serotonin reuptake inhibitors in major depressive disorder: gender-age interactions. Int Clin Psychopharmacol. 2007;22(4):226-229.
12. Kornstein SG, Wohlreich MM, Mallinckrodt CH, et al. Duloxetine efficacy for major depressive disorder in male vs. female patients: data from 7 randomized, double-blind, placebo-controlled trials. J Clin Psychiatry. 2006;67(5):761-770.
13. Wald A, Van Thiel DH, Hoechstetter L, et al. Effect of pregnancy on gastrointestinal transit. Dig Dis Sci. 1982;27(11):1015-1018.
14. Datz FL, Christian PE, Moore J. Gender-related differences in gastric emptying. J Nucl Med. 1987;28(7):1204-1207.
15. Kimmel L, Gonsalvcs D, Youngs D, et al. Fluctuating levels of antidepressants premenstrually. J Psychosom Obstet Gynaecol. 1992;13:277-280.
16. Jensvold MF, Halbrich U, Hamilton JA. Psycho-pharmacology and women: sex, gender and hormones. Washington, DC: American Psychiatric Press, Inc.; 1996.
17. Miller MN, Newell CL, Miller BE, et al. Variable dosing of sertraline for premenstrual exacerbation of depression: a pilot study. J Womens Health (Larchmt). 2008;17(6):993-997.
18. Sit DK, Perel JM, Helsel JC. Changes in antidepressant metabolism and dosing across pregnancy and early postpartum. J Clin Psychiatry. 2008;69(4):652-658.
19. Klier CM, Mossaheb N, Saria A, et al. Pharmacokinetics and elimination of quetiapine, venlafaxine, and trazodone during pregnancy and postpartum. J Clin Psychopharmacol. 2007;27(6):720-722.
20. Wisner KL, Perel JM, Wheeler SB. Tricyclic dose requirements across pregnancy. Am J Psychiatry. 1993;150(10):1541-1542.
21. Wadelius M, Darj E, Frenne G, et al. Induction of CYP2D6 in pregnancy. Clin Pharmacol Ther. 1997;62(4):400-407.
22. Anderson GD. Pregnancy-induced changes in pharmacokinetics: a mechanistic-based approach. Clin Pharmacokinet. 2005;44(10):989-1008.
23. Hostetter A, Stowe ZN, Strader JR, Jr, et al. Dose of selective serotonin uptake inhibitors across pregnancy: clinical implications. Depress Anxiety. 2000;11(2):51-57.
24. Dunlop W. Serial changes in renal haemodynamics during normal human pregnancy. Br J Obstet Gynaecol. 1981;88(1):1-9.
25. Freeman MP, Nolan PE, Jr, Davis MF, et al. Pharmacokinetics of sertraline across pregnancy and postpartum. J Clin Psychopharmacol. 2008;28(6):646-653.
26. Wisner KL, Perel JM, Peindl KS, et al. Effects of the postpartum period on nortriptyline pharmacokinetics. Psychopharmacol Bull. 1997;33(2):243-248.
27. O’Hara MW, Schlechte JA, Lewis DA, et al. Controlled prospective study of postpartum mood disorders: psychological, environmental, and hormonal variables. J Abnorm Psychol. 1991;100(1):63-73.
28. Meltzer-Brody S, Payne J, Rubinow DR. Postpartum depression: what to tell patients who breast-feed. Current Psychiatry. 2008;7(5):87-95.
29. Pinto-Meza A, Usall J, Serrano-Blanco A, et al. Gender differences in response to antidepressant treatment prescribed in primary care. Does menopause make a difference? J Affect Disord. 2006;93(1-3):53-60.
30. Amsterdam J, Garcia-Espana F, Fawcett J, et al. Fluoxetine efficacy in menopausal women with and without estrogen replacement. J Affect Disord. 1999;55(1):11-17.
31. Shapira B, Oppenheim G, Zohar J, et al. Lack of efficacy of estrogen supplementation to imipramine in resistant female depressives. Biol Psychiatry. 1985;20(5):576-579.
32. Schneider LS, Small GW, Clary CM. Estrogen replacement therapy and antidepressant response to sertraline in older depressed women. Am J Geriatr Psychiatry. 2001;9(4):393-399.
33. Zanardi R, Rossini D, Magri L, et al. Response to SSRIs and role of the hormonal therapy in post-menopausal depression. Eur Neuropsychopharmacol. 2007;17(6-7):400-405.
34. Rasgon NL, Dunkin J, Fairbanks L, et al. Estrogen and response to sertraline in postmenopausal women with major depressive disorder: a pilot study. J Psychiatr Res. 2007;41(3-4):338-343.
Antidepressants in bipolar disorder: 7 myths and realities
Few topics are as controversial as the role of antidepressants for patients with bipolar disorder. Although depression usually is the predominant, most enduring mood state in bipolar disorder, clinicians often face uncertainty about using antidepressants because of concerns about safety and efficacy. Whether and when to use antidepressants for bipolar depression hinges on complex parameters that preclude any single, simple rule.
Rather than asking if antidepressants are useful or detrimental for depressed patients with bipolar disorder, a more practical question might be: Under what circumstances are antidepressants likely to be beneficial, deleterious, or ineffective for an individual patient? Because “real world” patients often have idiosyncrasies that defy practice guidelines’ generic treatment recommendations, clinicians who practice in the proverbial trenches need strategies to tailor treatments to each patient that are informed—but not dictated—by evidence-based research.
Early suspicions
Concerns that antidepressants might precipitate mania were first described with tricyclic antidepressant (TCA) use in Europe in the 1960s. After bupropion and selective serotonin reuptake inhibitors (SSRIs) emerged, some clinicians believed they posed a lesser risk for this phenomenon compared with TCAs1,2 or monoamine oxidase inhibitors (MAOIs).3
Antidepressants’ potential to induce short-term mania/hypomania following acute exposure has been weighed against the longer-term risk for worsening illness course by increasing frequency of subsequent episodes (so-called cycle acceleration). In the 1980s, some researchers suggested that rapid cycling might—at least in some instances—represent an iatrogenic phenomenon caused by long-term antidepressant use. These issues remain controversial, but more than 20 years of research suggest that antidepressants induce mania or accelerate cycling in a smaller minority of bipolar disorder patients than was once thought.
Table 1 and Table 2 summarize findings from randomized controlled studies that have examined antidepressants’ efficacy for acute bipolar depression. Except for a study of fluoxetine plus olanzapine,4 no large-scale placebo-controlled trial has demonstrated superior antidepressant response to a mood stabilizer plus antidepressant compared with a mood stabilizer alone.
Table 1
Antidepressants for bipolar depression: SSRIs and SNRIs*
| Acute efficacy | Reported switch risk |
|---|---|
| Fluoxetine (SSRI) | |
| 86% response rate after 3 weeks in 6-week double-blind randomized comparison with imipramine or placeboa | 0% |
| 38% response rate after 8 weeks of placebo-controlled monotherapy in bipolar II or NOS subjectsb | 0% |
| 56% response rate over 8 weeks in combination with olanzapine; significantly better than placebo plus olanzapine (30%)c | 6% |
| Paroxetine (SSRI) | |
| Same as placebo when added to an antimanic drug (STEP-BD) for up to 26 weeksd | 10.1% (reported only jointly for paroxetine or bupropion) |
| 36% response rate (no different from placebo) when coadministered with therapeutically dosed lithium over 10 weekse | 7% |
| Same as divalproex plus lithium when coadministered with divalproex or lithium over 6 weeks (actual response rates not reported)f | 0% |
| 43% response (coadministered with lithium, divalproex, or carbamazepine) over 6 weeksg | 3% (not statistically significantly different from venlafaxine comparison arm) |
| Sertraline (SSRI) | |
| 41% improved (comparable to rates seen with bupropion [33%] or venlafaxine [36%] when coadministered with a mood stabilizer over 10 weeks)h | 12% |
| Venlafaxine (SNRI) | |
| 36% improved (comparable to rates seen with bupropion [33%] or sertraline [41%]) when coadministered with a mood stabilizer over 10 weeksh | 6% |
| 48% response (coadministered with lithium, divalproex, or carbamazepine) over 6 weeksg | 13% (not statistically significantly different from paroxetine comparison arm) |
| *No data are available for citalopram, desvenlafaxine, duloxetine, escitalopram, fluvoxamine, or milnacipran | |
| NOS: not otherwise specified; SNRI: serotonin/norepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor; STEP-BD: Systematic Treatment Enhancement Program for Bipolar Disorder | |
| Source: References a. Cohn JB, Collins G, Ashbrook E, et al. A comparison of fluoxetine, imipramine and placebo in patients with bipolar depressive disorder. Int Clin Psychopharmaol. 1989;4:313-322. b. Amsterdam JD, Shults J. Fluoxetine monotherapy of bipolar type II and bipolar NOS major depression: a double-blind, placebo-substitution, continuation study. Int Clin Psychopharmacol. 2005;20:257-264. c. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry. 2003;60:1079-1088. d. Sachs GS, Nierenberg AA, Calabrese JR, et al. Effectiveness of adjunctive antidepressant treatment for bipolar depression. N Engl J Med. 2007;356:1711-1722. e. Nemeroff CB, Evans DL, Gyulai L, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry. 2001;158:906-912. f. Young LT, Joffe RT, Robb JC, et al. Double-blind comparison of addition of a second mood stabilizer versus an antidepressant to an initial mood stabilizer for treatment of patients with bipolar depression. Am J Psychiatry. 2000;157:124-126. g. Vieta E, Martinez-Aran A, Goikolea JM. A randomized trial comparing paroxetine and venlafaxine in the treatment of bipolar depressed patients taking mood stabilizers. J Clin Psychiatry. 2002;63:508-512. h. Leverich GS, Altshuler LL, Frye MA, et al. Risk of switch in mood polarity to hypomania or mania in patients with bipolar depression during acute and continuation trials of venlafaxine, sertraline, and bupropion as adjuncts to mood stabilizers. Am J Psychiatry. 2006;163:232-239. | |
Table 2
Antidepressants for bipolar depression: MAOIs, TCAs, and bupropion*
| Acute efficacy | Reported switch risk |
|---|---|
| Tranylcypromine (MAOI) | |
| 81% response (monotherapy) in bipolar I (n=24) or bipolar II (n=32) patients over 16 weeksa | 21% |
| 75% response among imipramine nonresponders (n=12)b | 17% |
| Moclobemide (MAOI) | |
| 46% response over 8 weeks in 156 bipolar patients (some, but not all, took concomitant mood stabilizers), not significantly different from imipramine comparatorc | 4% |
| Imipramine (TCA) | |
| 57% response rate after 3 weeks in a 6-week double-blind randomized comparison with fluoxetine or placebod | Not reported |
| 48% response (monotherapy) in bipolar I (n=24) or bipolar II (N=32) patients over 16 weeksa | 24% |
| 53% response over 8 weeks in 156 bipolar patients (some, but not all, took concomitant mood stabilizers), not significantly different from moclobemide comparatorc | 11% |
| 41% (coadministered with therapeutically dosed lithium)e | 8% |
| Desipramine (TCA) | |
| 50% (5/10) response rate (coadministered with a mood stabilizer over 8 weeks)f | 50% |
| Bupropion | |
| 55% response (5/9) (coadministered with a mood stabilizer over 8 weeks)f | 11% |
| 33% response rate (coadministered with mood stabilizers over 10 weeks)g | 20% |
| *No data are available for isocarboxazid, mirtazapine, nefazodone, phenelzine, or selegiline transdermal | |
| MAOI: monoamine oxidase inhibitor; TCA: tricyclic antidepressant | |
| Source: References a. Himmelhoch JM, Thase ME, Mallinger AG, et al. Tranylcypromine versus imipramine in anergic bipolar depression. Am J Psychiatry. 1991;148:910-916. b. Thase ME, Malinger AG, McKnight D, et al. Treatment of imipramine-resistant recurrent depressions, IV: a double-blind crossover study of tranylcypromine for anergic bipolar depression. Am J Psychiatry. 1992;149:195-198. c. Silverstone T. Moclobemide vs. imipramine in bipolar depression: a multicentre double-blind clinical trial. Acta Psychiatr Scand. 2001;104:104-109. d. Cohn JB, Collins G, Ashbrook E, et al. A comparison of fluoxetine, imipramine and placebo in patients with bipolar depressive disorder. Int Clin Psychopharmaol. 1989;4:313-322. e. Nemeroff CB, Evans DL, Gyulai L, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry. 2001;158:906-912. f. Sachs GS, Lafer B, Stoll AL, et al. A double blind trial of bupropion versus desipramine for bipolar depression. J Clin Psychiatry. 1994;55:391-393. g. Leverich GS, Altshuler LL, Frye MA, et al. Risk of switch in mood polarity to hypomania or mania in patients with bipolar depression during acute and continuation trials of venlafaxine, sertraline, and bupropion as adjuncts to mood stabilizers. Am J Psychiatry. 2006;163:232-239. | |
MYTH 1: Antidepressant-induced mania is a highly prevalent, widespread problem.
Reality: Although some might argue that the precise relative risk of antidepressant-induced mania or hypomania is unknown (eg, considering intervening factors such as the natural illness course), recent literature suggests that the emergence of mania or hypomania can be reasonably attributed to antidepressant use in no more than 10% to 25% of patients with bipolar disorder.5,6 Part of the difficulty in estimating the true prevalence of antidepressant-induced mania involves variability and inconsistency in defining mania induction.
A recent consensus statement proposed a graduated series of definitions for treatment-emergent affective switch:7
- “Definite” switch involves fulfilling DSM-IV syndromic criteria for a manic, hypomanic, or mixed episode for at least 2 days, within 8 weeks of antidepressant introduction.
- “Likely” switches call for at least 2 DSM-IV mania or hypomania symptoms plus a Young Mania Rating Scale (YMRS) score >12, occurring for at least 2 days, within 12 weeks of antidepressant introduction.
- “Possible” switches require a “clear change” in mood or energy with a YMRS score >8, persisting ≥4 hours over 2 days, occurring within 12 weeks of antidepressant initiation.
Adverse effects such as agitation typically diminish or remit with dosage reductions or drug cessation, whereas true antidepressant-induced polarity switches persist even after the medication is discontinued. Moreover, it is often difficult—if not impossible—to know with certainty when a polarity switch results from treatment effects vs the natural illness course. In my experience, true manic or hypomanic syndromes soon after antidepressant exposure are less common than heterogeneous, nonspecific symptoms such as agitation, anxiety, insomnia, or worsening depression (ie, lack of efficacy).
MYTH 2: Antidepressant response rates are lower in bipolar depression.
Reality: It is difficult to draw broad conclusions about antidepressant response rates in unipolar vs bipolar depression because:
- few direct comparisons have been reported
- all relevant studies are retrospective
- small sample sizes in most studies may not have satisfactorily controlled for factors that could predispose to mood destabilization (Table 3).
Table 3
What increases risk of antidepressant-induced mania?
| Factor | Findings |
|---|---|
| History of antidepressant-induced mania or hypomania | Confers an approximate 2- to 5-fold increased risk for subsequent antidepressant-induced mania/hypomania, regardless of antidepressanta |
| Recent mania preceding current depressive episode | Higher risk for antidepressant-associated mania if current depressive episode was preceded by manic phaseb |
| Bipolar I vs bipolar II subtype | Greater risk for switch in bipolar Ic,d |
| Comorbid alcohol or substance use disorder | 5- to 7-fold increased risk for antidepressant-associated maniae |
| Noradrenergic vs serotonergic antidepressants | Possible higher risk for mania induction with TCAs or SNRIs than with bupropionf or SSRIsg |
| Concurrent mania symptoms during a depressive episode | Mild or subthreshold mania symptoms during a depressive episode increase risk for maniah,i |
| Hyperthymic temperamental traits | Associated with increased likelihood of antidepressant-induced maniaj |
| SNRIs: serotonin/norepinephrine reuptake inhibitors; SSRIs: selective serotonin reuptake inhibitors; TCAs: tricyclic antidepressants | |
| Source: References a. Truman CJ, Goldberg JF, Ghaemi SN, et al. Self-reported history of manic/hypomanic switch associated with antidepressant use: data from the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). J Clin Psychiatry. 2007;68:1472-1479. b. MacQueen GM, Young LT, Marriott M, et al. Previous mood state predicts response and switch rates in patients with bipolar depression. Acta Psychiatr Scand. 2002;105:414-418. c. Himmelhoch JM, Thase ME, Mallinger AG, et al. Tranylcypromine versus imipramine in anergic bipolar depression. Am J Psychiatry. 1991;148:910-916. d. Altshuler LL, Suppes T, Black DO, et al. Lower switch rate in depressed patients with bipolar II than bipolar I disorder treated adjunctively with second-generation antidepressants. Am J Psychiatry. 2006;163:313-315. e. Goldberg JF, Truman CJ. Antidepressant-induced mania: an overview of current controversies. Bipolar Disord. 2003;5:407-420. f. Sachs GS, Lafer B, Stoll AL, et al. A double blind trial of bupropion versus desipramine for bipolar depression. J Clin Psychiatry. 1994;55:391-393. g. Peet M. Induction of mania with selective serotonin re-uptake inhibitors and tricyclic antidepressants. Br J Psychiatry. 1994;164:549-550. h. Frye MA, Hellmann G, McElroy SL, et al. Correlates of treatment-emergent mania associated with antidepressant treatment in bipolar depression. Am J Psychiatry. 2009;166:164-172. i. Bottlender R, Rudolf D, Strauss A, et al. Mood-stabilisers reduce the risk of developing antidepressant-induced maniform states in acute treatment of bipolar I depressed patients. J Affect Disord. 2001;63:79-83. j. Henry C, Sorbara F, Lacoste J, et al. Antidepressant-induced mania in bipolar patients: identification of risk factors. J Clin Psychiatry. 2001;62:249-255. | |
A retrospective review of bipolar (n=41) and unipolar (n=37) depressed patients by Ghaemi et al8 found no significant difference in acute nonresponse rates between the groups. Similarly, Bottlender et al9 found no differences in treatment response when comparing naturalistic antidepressant outcomes for 50 unipolar and 50 bipolar patients matched for age, sex, and illness duration. Comparable antidepressant response outcomes also were reported in a retrospective study of 2,032 unipolar and bipolar inpatients conducted by Möller et al,10 and between unipolar (n=31) vs bipolar II (n=17) depressed patients receiving venlafaxine monotherapy for 6 weeks.11
Antidepressant response may depend on factors such as episode chronicity or the number of failed medication trials within a given episode, regardless of illness polarity. This was suggested by the remarkably low response rates after 2 failed initial antidepressant treatments in the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) unipolar depression trials. In my experience, antidepressant efficacy is more often a function of factors in addition to polarity, including:
- illness severity
- chronicity
- psychiatric, medical, or substance use comorbidity
- psychosocial skills, such as the capacity to tolerate distress, utilize effective coping techniques, and maintain appropriate relationships with others.
MYTH 3: Most antidepressants have been systematically studied for treatment of depression in bipolar disorder.
Reality: Only paroxetine,12,13 bupropion,12 and imipramine13 have been studied in randomized, large-scale, adequately powered placebo-controlled trials. Studies of other antidepressants suffer from small sample sizes (inadequate statistical power), lack of placebo controls, or failure to control for possible confounding factors, such as lack of stratification for bipolar I vs II subtype or presence vs absence of rapid cycling.
One large randomized trial showed comparable antidepressant efficacy with a mood stabilizer plus adjunctive venlafaxine (43%) vs sertraline (55%) vs bupropion (49%) over 10 weeks,14 but the lack of a mood stabilizer monotherapy comparison group limits the ability to anticipate whether adjunctive antidepressants increase response or remission rates more than mood stabilizers alone. Adjunctive imipramine,13 paroxetine,12,13,15 and bupropion12 yield no greater improvement in depressive symptoms than is seen with optimally dosed mood stabilizers alone.
Mirtazapine, a serotonergic/noradrenergic antidepressant that is sometimes prescribed off-label as a sleep aid, has not been systematically studied for safety or efficacy in bipolar depression. In case reports, mirtazapine has induced mania in patients with unipolar depression.16-18 Using mirtazapine to counteract insomnia may be safer in patients with unipolar depression than in those with bipolar disorder. Because poor sleep is a core feature of mania, be certain to differentiate complaints that reflect simple insomnia from a loss of need for sleep:
- daytime fatigue is more common in insomnia than loss of need for sleep
- nocturnal hyperactivity is more often associated with loss of need for sleep.
Using an antidepressant to treat sleep problems that may derive from emerging mania or hypomania runs counter to basic pharmacodynamic principles and may pose greater risk than benefit.
Generally, using a medication that has been studied for treating a specific clinical entity such as bipolar depression is preferable to using one that has not. Avoid medications that have multiple negative placebo-controlled trials—such as paroxetine—unless you have evidence of efficacy in an individual patient.
MYTH 4: Risk for inducing mania is higher with noradrenergic antidepressants.
Reality: This popular belief arose from a unifying hypothesis offered by Sachs et al1 and Leverich et al14 to explain higher rates of mania following treatment with desipramine than bupropion,1 SSRIs compared with TCAs,2 or venlafaxine compared with bupropion or sertraline.14 However, while plausible, this hypothesis does not fully account for the putative noradrenergic properties of bupropion—presumably via increased pre-synaptic norepinephrine outflow, rather than noradrenergic reuptake inhibition19—which reportedly has a lower risk of switching than desipramine1 or venlafaxine.14
The risk for venlafaxine monotherapy to induce mania or hypomania in patients with bipolar II depression has been reported to be nonexistent11 or no higher than seen with lithium.20 Also, some noradrenergic agents, such as duloxetine, have not been shown to induce mania in major depression,21 although duloxetine’s potential to destabilize mood is unknown because of the absence of data in bipolar disorder. Finally, although large-scale clinical trials have not examined the safety and efficacy of the noradrenergic reuptake inhibitor atomoxetine, several case reports have suggested its potential for inducing mania or hypomania.22,23
Likely, all-or-none admonitions against using noradrenergic antidepressants are oversimplifications.
MYTH 5: Coadministering an antimanic mood stabilizer reliably prevents antidepressant-induced mania.
Reality: Most practice guidelines advise administering antimanic mood stabilizers before initiating an antidepressant. Clinicians widely interpret this recommendation as reinforcing the assumption that a mood stabilizer will diminish mania risk when introducing an antidepressant. (Less often, clinicians interpret it as meaning that a mood stabilizer itself may provide antidepressant efficacy.) In fact, whether (and which) antimanic agents mitigate the risk for antidepressant-induced mania has received little empirical study. The largest dataset on this topic—the randomized controlled data from Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD)12—found that the risk for treatment-emergent manic switch with paroxetine or bupropion was almost identical (about 10%) with or without an FDA-approved antimanic agent.
In a retrospective study, Henry et al6 found that cotherapy with lithium but not divalproex or carbamazepine protects against antidepressant-induced mania, and that switch rates to mania were the same whether or not an antidepressant was taken with an anticonvulsant. In a naturalistic retrospective study (n=158), Bottlender et al24 revealed that mood stabilizers (lithium, carbamazepine, or divalproex) prevented switches from depression to mania during treatment with TCAs but not SSRIs or MAOIs.
I favor incorporating lithium or other antimanic agents in the regimens of patients with bipolar depression not primarily to guard against antidepressant-induced mania but more for pharmacodynamic synergy—complementary mechanisms of action that collectively may produce more substantial antidepressant effects—especially when the patient’s illness course has included manic or hypomanic features in the preceding year.
MYTH 6: Antidepressants cause or worsen rapid cycling.
Reality: Wehr et al25 reported that antidepressants may accelerate cycling frequency (ie, inter-episode durations become shorter) in a small subgroup (N=10) of patients. By contrast, use of TCAs was not more likely in the weeks preceding shifts from depression to mania or hypomania in a 14-year follow-up study of bipolar rapid cycling from the NIMH Collaborative Depression Study.26 In fact, rapid-cycling patients spent more weeks depressed when taking lithium without a TCA than with 1.
Findings from STEP-BD indicate that prospectively observed rapid cycling, as defined by DSM-IV criteria, is relatively rare, although subjects taking antidepressants often had multiple episodes per year.27 These naturalistic data could suggest that antidepressant use leads to more depressive episodes, or that more depressive episodes lead to more antidepressant use. Causal relationships cannot be inferred from the nonrandomized study design. Nevertheless, antidepressant use was not associated with reduced depressive episodes over 1 year.
I believe that, in general, antidepressants are unlikely to improve a truly rapid-cycling illness course. In this scenario, a more “panoramic” understanding of the need to treat multiple relapses and polarity changes over time likely warrants using multiple anti-cycling agents. Rapid cycling is treated over the course of 1 year, rather than 1 episode.
MYTH 7: Antidepressants should never be used without a mood stabilizer for bipolar depression.
Reality: This admonition is widely cited as a general recommendation from modern practice guidelines; however, it mainly pertains to depression treatment in patients with bipolar I disorder, for whom most controlled trial data exist. For example, relatively high rates of treatment-emergent mania have been reported with TCA or MAOI monotherapy in bipolar I disorder patients (Table 2). Yet for bipolar II disorder, controlled trials demonstrate superior outcomes with venlafaxine monotherapy compared with lithium monotherapy, with no increase in mood destabilization.20
Neither the safety nor the efficacy of antidepressants with vs without mood stabilizers has been studied systematically in cyclothymic or mood disorder patients who may fall within the so-called bipolar spectrum but have never met DSM-IV criteria for a lifetime manic or hypomanic episode (ie, bipolar disorder not otherwise specified). Extrapolation from findings based on bipolar I disorder patients may not be valid for all bipolar subtypes.
Clinical strategies
In constructing a rationale-based approach to bipolar depression, consider these steps:
Step 1: Assess candidacy for antidepressant use. A number of key features can help you delineate the current illness state and context in which depressive symptoms arise—features that may influence you patient’s vulnerability to mood destabilization, and therefore are pertinent for gauging the likelihood that antidepressants may help or harm (Table 4).
Step 2: Consider mood stabilizers with antidepressant properties. Determine whether your patient is taking any mood stabilizers that possess robust antidepressant properties, or whether it may be beneficial to introduce one of these agents before initiating adjunctive antidepressants. Mood stabilizers with antidepressant efficacy are compelling options for patients presenting with any of the features listed in the right-hand column of Table 4, as well as those with:
- psychotic features
- marked agitation
- multiple prior antidepressant nonresponses
- high depression recurrence rates regardless of episode duration (ie, cyclicity, irrespective of ≥4 discrete episodes per year).
Table 4
Assessing antidepressant candidacy in bipolar depression
| Favors antidepressant use | Discourages antidepressant use |
|---|---|
| Bipolar II disorder | Bipolar I disordera |
| Depressed (non-mixed) states | Mixed manic and depressive featuresb,c |
| Absence of rapid cycling | Presence of rapid cyclingd,e |
| Absence of recent mania or hypomania (preceding 2 to 3 months) | Mania or hypomania in past 2 to 3 monthsf |
| Absence of comorbid alcohol or substance use disorder | Presence of comorbid alcohol or substance use disorderg,h |
| Prior favorable antidepressant response | Suboptimal responses to prior antidepressants |
| No history of antidepressant-induced mania or hypomania | History of antidepressant-induced mania or hypomaniai |
| Source: References a. Altshuler LL, Suppes T, Black DO, et al. Lower switch rate in depressed patients with bipolar II than bipolar I disorder treated adjunctively with second-generation antidepressants. Am J Psychiatry. 2006;163:313-315. b. Frye MA, Hellmann G, McElroy SL, et al. Correlates of treatment-emergent mania associated with antidepressant treatment in bipolar depression. Am J Psychiatry. 2009;166:164-172. c. Goldberg JF, Perlis RH, Ghaemi SN, et al. Adjunctive antidepressant use and symptomatic recovery among bipolar depressed patients with concomitant manic symptoms: findings from the STEP-BD. Am J Psychiatry. 2007;164(9):1348-1355. d. Schneck CD, Miklowitz DJ, Miyahara S, et al. The prospective course of rapid-cycling bipolar disorder: findings from the STEP-BD. Am J Psychiatry. 2008;165:370-377. e. Ghaemi SN, Ostacher MM, El-Mallakh RS, et al. Antidepressant discontinuation in bipolar depression: a STEP-BD randomized clinical trial of long-term effectiveness and safety. J Clin Psychiatry. In press. f. MacQueen GM, Young LT, Marriott M, et al. Previous mood state predicts response and switch rates in patients with bipolar depression. Acta Psychiatr Scand. 2002;105:414-418. g. Goldberg JF, Whiteside JE. The association between substance abuse and antidepressant-induced mania in bipolar disorder: a preliminary study. J Clin Psychiatry. 2002;63:791-795. h. Manwani SG, Pardo TB, Albanese MJ, et al. Substance use disorder and other predictors of antidepressant-induced mania: a retrospective chart review. J Clin Psychiatry. 2006;67:1341-1345. i. Truman CJ, Goldberg JF, Ghaemi SN, et al. Self-reported history of manic/hypomanic switch associated with antidepressant use: data from the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). J Clin Psychiatry. 2007;68:1472-1479. | |
Prospective mood charting may help to establish the latter, in which case recurrence (rather than polarity) may cause waxing and waning depressed mood states.
Psychotropic agents or combinations that have shown to be effective for bipolar depression (supported by at least 1 randomized controlled trial) without destabilizing mood include quetiapine, olanzapine, olanzapine-fluoxetine combination, lamotrigine, and lithium plus lamotrigine. Those with some—but less robustly demonstrated—antidepressant action include lithium, divalproex, and carbamazepine. Other than quetiapine and olanzapine, second-generation antipsychotics have not demonstrated antidepressant effects in bipolar depression.
In general, optimize therapy with 1 or more mood stabilizers with antidepressant properties before deciding it is necessary to add antidepressants.
Step 3: Use antidepressants in suitable patients. For patients with no risk factors for mood destabilization from antidepressants (Table 3), these drugs may be worth incorporating, keeping in mind the following guiding principles:
- In patients with bipolar I depression, it is preferable to add an antidepressant to an antimanic mood stabilizer (ie, lithium, divalproex, carbamazepine, or an antipsychotic) rather than prescribing antidepressant monotherapy. There is greater diversity of opinion about the safety of antidepressant monotherapy for bipolar II depression.
- Consider using antidepressants that have at least 1 positive randomized controlled trial in bipolar disorder and low risk for mood destabilization (bupropion,12,14 sertraline,14 fluoxetine,4,5 tranylcypromine,3,28 or venlafaxine in bipolar II depression20) before using those with reported increased risk for inducing mania or hypomania (TCAs1,2 or venlafaxine in bipolar I depression14), multiple negative controlled trials (paroxetine12,13), or no controlled data in bipolar depression (citalopram, escitalopram, fluvoxamine, mirtazapine, duloxetine, desvenlafaxine, nefazodone, and selegiline transdermal). Combinations of antidepressants have not been adequately studied in bipolar depression.
- The optimal duration of antidepressant therapy is unknown. However, longer-term treatment may be worthwhile in patients who show robust acute antidepressant response and experience infrequent mania or hypomania. Long-term antidepressant use is less compelling in patients with a poor initial response29 or rapid cycling.30 Abrupt antidepressant cessation also may induce mania, potentially by disrupting homeostasis.31 In the absence of rapid cycling, manic/hypomanic features, or worsening suicidal features, and in the presence of an unequivocal acute response and a greater predisposition to depression than mania, it is reasonable to continue an antidepressant indefinitely until new signs of mania or hypomania emerge.
- Emerging signs of mania or hypomania should signal the need to discontinue the antidepressant. Dosage reductions alone may not diminish emerging manic or hypomanic symptoms, and “counterbalancing” maneuvers (ie, adding antimanic agents while continuing an antidepressant) may not effectively stabilize mood.
Step 4: Consider novel strategies. In the absence of a response to the strategy outlined above—particularly among poor candidates for continued antidepressant therapy—other novel strategies have support from at least 1 randomized controlled trial, including pramipexole,32,33 modafinil,34 riluzole,35 and n-acetyl cysteine.36 Other interventions worth considering include:
- adjunctive thyroid hormone
- cognitive therapy
- light therapy (if a seasonal component is evident)
- electroconvulsive therapy.
Related resources
- Goldberg JF. Treating depression in bipolar disorder. http://thedoctorschannel.com/video/3077.html.
- Goldberg JF. Pharmacologic treatment of acute mania.http://thedoctorschannel.com/video/3032.html.
Drug brand names
- Atomoxetine • Strattera
- Bupropion • Wellbutrin
- Carbamazepine • Tegretol, Equetro
- Citalopram • Celexa
- Desipramine • Norpramin
- Desvenlafaxine • Pristiq
- Divalproex • Depakote, Depakene
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Imipramine • Tofranil
- Isocarboxazid • Marplan
- Lamotrigine • Lamictal
- Lithium • Lithobid, Eskalith
- Milnacipran • Ixel, Savella
- Mirtazapine • Remeron
- Moclobemide • Aurorix, Manerix
- Modafinil • Provigil
- Nefazodone • Serzone
- Olanzapine • Zyprexa
- Olanzapine-fluoxetine • Symbyax
- Paroxetine • Paxil
- Phenelzine • Nardil
- Pramipexole • Mirapex
- Quetiapine • Seroquel
- Riluzole • Rilutek
- Selegiline transdermal • EMSAM
- Sertraline • Zoloft
- Tranylcypromine • Parnate
- Venlafaxine • Effexor
Disclosures
Dr. Goldberg is a consultant to Eli Lilly and Company and a speaker for AstraZeneca, Eli Lilly and Company, GlaxoSmithKline, Merck, and Pfizer Inc., and has received speaking honoraria from Janssen-Cilag.
1. Sachs GS, Lafer B, Stoll AL, et al. A double blind trial of bupropion versus desipramine for bipolar depression. J Clin Psychiatry. 1994;55:391-393.
2. Peet M. Induction of mania with selective serotonin re-uptake inhibitors and tricyclic antidepressants. Br J Psychiatry. 1994;164:549-550.
3. Himmelhoch JM, Thase ME, Mallinger AG, et al. Tranylcypromine versus imipramine in anergic bipolar depression. Am J Psychiatry. 1991;148:910-916.
4. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry. 2003;60:1079-1088.
5. Goldberg JF, Truman CJ. Antidepressant-induced mania: an overview of current controversies. Bipolar Disord. 2003;5:407-420.
6. Henry C, Sorbara F, Lacoste J, et al. Antidepressant-induced mania in bipolar patients: identification of risk factors. J Clin Psychiatry. 2001;62:249-255.
7. Tohen M, Frank E, Bowden CL, et al. The International Society for Bipolar Disorders (ISBD) Task Force report on the nomenclature of course and outcome in bipolar disorders. Bipolar Disord. 2009;11:453-473.
8. Ghaemi SN, Rosenquist KJ, Ko JY, et al. Antidepressant treatment in bipolar versus unipolar depression. Am J Psychiatry. 2004;161:163-165.
9. Bottlender R, Rudolf D, Jäger M, et al. Are bipolar I depressive patients less responsive to treatment with antidepressants than unipolar depressive patients? Results from a case control study. Eur Psychiatry. 2002;17:200-205.
10. Möller HJ, Bottlender R, Grunze H, et al. Are antidepressants less effective in the acute treatment of bipolar I compared to unipolar depression? J Affect Disord. 2001;67(1-3):141-146.
11. Amsterdam J. Efficacy and safety of venlafaxine in the treatment of bipolar II major depressive episode. J Clin Psychopharmacol. 1998;18:313-317.
12. Sachs GS, Nierenberg AA, Calabrese JR, et al. Effectiveness of adjunctive antidepressant treatment for bipolar depression. N Engl J Med. 2007;356:1711-1722.
13. Nemeroff CB, Evans DL, Gyulai L, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry. 2001;158:906-912.
14. Leverich GS, Altshuler LL, Frye MA, et al. Risk of switch in mood polarity to hypomania or mania in patients with bipolar depression during acute and continuation trials of venlafaxine, sertraline, and bupropion as adjuncts to mood stabilizers. Am J Psychiatry. 2006;163:232-239.
15. Young LT, Joffe RT, Robb JC, et al. Double-blind comparison of addition of a second mood stabilizer versus an antidepressant to an initial mood stabilizer for treatment of patients with bipolar depression. Am J Psychiatry. 2000;157:124-126.
16. Soutullo CA, McElroy SL, Keck PE, Jr. Hypomania associated with mirtazapine augmentation of sertraline. J Clin Psychiatry. 1998;59(6):320.-
17. Bhanji NH, Margolese HC, Saint-Laurent M, et al. Dysphoric mania induced by high-dose mirtazapine: a case for “norepinephrine syndrome”? Int Clin Psychopharmacol. 2002;17(6):319-322.
18. Goyal N, Sinha VK. Mirtazapine-induced manic switch in adolescent unipolar depression. Aust N Z J Psychiatry. 2008;42(12):1070-1071.
19. Dong J, Blier P. Modification of norepinephrine and serotonin, but not dopamine, neuron firing by sustained bupropion treatment. Psychopharmacol (Berl). 2001;155:52-57.
20. Amsterdam JD, Wang CH, Shwarz M, et al. Venlafaxine versus lithium monotherapy of rapid and non-rapid cycling patients with bipolar II major depressive disorder: a randomized, parallel group, open-label trial. J Affect Disord. 2009;112(1-3):219-230.
21. Dunner DL, D’Souza DN, Kajdasz DK, et al. Is treatment-associated mania rare with duloxetine: secondary analysis of controlled trials in non-bipolar depression. J Affect Disord. 2005;87:115-119.
22. Henderson TA. Mania induction associated with atomoxetine. J Clin Psychopharmacol. 2004;24(5):567-568.
23. Henderson TA, Hartman K. Aggression, mania, and hypomania induction associated with atomoxetine. Pediatrics. 2004;114(3):895-896.
24. Bottlender R, Rudolf D, Strauss A, et al. Mood-stabilisers reduce the risk of developing antidepressant-induced maniform states in acute treatment of bipolar I depressed patients. J Affect Disord. 2001;63:79-83.
25. Wehr TA, Sack DA, Rosenthal NE, et al. Rapid cycling affective disorder: contributing factors and treatment responses in 51 patients. Am J Psychiatry. 1988;145:179-184.
26. Coryell W, Solomon D, Turvey C, et al. The long-term course of rapid-cycling bipolar disorder. Arch Gen Psychiatry. 2003;60:914-920.
27. Schneck CD, Miklowitz DJ, Miyahara S, et al. The prospective course of rapid-cycling bipolar disorder: findings from the STEP-BD. Am J Psychiatry. 2008;165:370-377.
28. Thase ME, Malinger AG, McKnight D, et al. Treatment of imipramine-resistant recurrent depressions, IV: a double-blind crossover study of tranylcypromine for anergic bipolar depression. Am J Psychiatry. 1992;149:195-198.
29. Altshuler LL, Post RM, Hellemann G, et al. Impact of antidepressant continuation after acute positive or partial treatment response for bipolar depression: a blinded, randomized study. J Clin Psychiatry. 2009;70(4):450-457.
30. Ghaemi SN, Ostacher MM, El-Mallakh RS, et al. Antidepressant discontinuation in bipolar depression: a STEP-BD randomized clinical trial of long-term effectiveness and safety. J Clin Psychiatry. In press.
31. Goldstein TR, Frye MA, Denicoff KD, et al. Antidepressant discontinuation-related mania: critical prospective observation and theoretical implications in bipolar disorder. J Clin Psychiatry. 1999;60(8):563-567.
32. Goldberg JF, Burdick KE, Endick CE. A preliminary randomized, double-blind, placebo-controlled trial of pramipexole added to mood stabilizers for treatment-resistant bipolar depression. Am J Psychiatry. 2004;161:564-566.
33. Zarate CA, Jr, Payne JL, Singh J, et al. Pramipexole for bipolar II depression: a placebo-controlled proof of concept study. Biol Psychiatry. 2004;56:54-60.
34. Frye MA, Grunze H, Suppes T, et al. A placebo-controlled evaluation of adjunctive modafinil in the treatment of bipolar depression. Am J Psychiatry. 2007;164(8):1242-1249.
35. Zarate CA, Jr, Quiroz JA, Singh JB, et al. An open-label trial of the glutamate-modulating agent riluzole in combination with lithium for the treatment of bipolar depression. Biol Psychiatry. 2005;57(4):430-432.
36. Berk M, Copolov DL, Dean O, et al. N-acetyl cysteine for depressive symptoms in bipolar disorder—a double-blind, randomized placebo-controlled trial. Biol Psychiatry. 2008;64(6):468-475.
Few topics are as controversial as the role of antidepressants for patients with bipolar disorder. Although depression usually is the predominant, most enduring mood state in bipolar disorder, clinicians often face uncertainty about using antidepressants because of concerns about safety and efficacy. Whether and when to use antidepressants for bipolar depression hinges on complex parameters that preclude any single, simple rule.
Rather than asking if antidepressants are useful or detrimental for depressed patients with bipolar disorder, a more practical question might be: Under what circumstances are antidepressants likely to be beneficial, deleterious, or ineffective for an individual patient? Because “real world” patients often have idiosyncrasies that defy practice guidelines’ generic treatment recommendations, clinicians who practice in the proverbial trenches need strategies to tailor treatments to each patient that are informed—but not dictated—by evidence-based research.
Early suspicions
Concerns that antidepressants might precipitate mania were first described with tricyclic antidepressant (TCA) use in Europe in the 1960s. After bupropion and selective serotonin reuptake inhibitors (SSRIs) emerged, some clinicians believed they posed a lesser risk for this phenomenon compared with TCAs1,2 or monoamine oxidase inhibitors (MAOIs).3
Antidepressants’ potential to induce short-term mania/hypomania following acute exposure has been weighed against the longer-term risk for worsening illness course by increasing frequency of subsequent episodes (so-called cycle acceleration). In the 1980s, some researchers suggested that rapid cycling might—at least in some instances—represent an iatrogenic phenomenon caused by long-term antidepressant use. These issues remain controversial, but more than 20 years of research suggest that antidepressants induce mania or accelerate cycling in a smaller minority of bipolar disorder patients than was once thought.
Table 1 and Table 2 summarize findings from randomized controlled studies that have examined antidepressants’ efficacy for acute bipolar depression. Except for a study of fluoxetine plus olanzapine,4 no large-scale placebo-controlled trial has demonstrated superior antidepressant response to a mood stabilizer plus antidepressant compared with a mood stabilizer alone.
Table 1
Antidepressants for bipolar depression: SSRIs and SNRIs*
| Acute efficacy | Reported switch risk |
|---|---|
| Fluoxetine (SSRI) | |
| 86% response rate after 3 weeks in 6-week double-blind randomized comparison with imipramine or placeboa | 0% |
| 38% response rate after 8 weeks of placebo-controlled monotherapy in bipolar II or NOS subjectsb | 0% |
| 56% response rate over 8 weeks in combination with olanzapine; significantly better than placebo plus olanzapine (30%)c | 6% |
| Paroxetine (SSRI) | |
| Same as placebo when added to an antimanic drug (STEP-BD) for up to 26 weeksd | 10.1% (reported only jointly for paroxetine or bupropion) |
| 36% response rate (no different from placebo) when coadministered with therapeutically dosed lithium over 10 weekse | 7% |
| Same as divalproex plus lithium when coadministered with divalproex or lithium over 6 weeks (actual response rates not reported)f | 0% |
| 43% response (coadministered with lithium, divalproex, or carbamazepine) over 6 weeksg | 3% (not statistically significantly different from venlafaxine comparison arm) |
| Sertraline (SSRI) | |
| 41% improved (comparable to rates seen with bupropion [33%] or venlafaxine [36%] when coadministered with a mood stabilizer over 10 weeks)h | 12% |
| Venlafaxine (SNRI) | |
| 36% improved (comparable to rates seen with bupropion [33%] or sertraline [41%]) when coadministered with a mood stabilizer over 10 weeksh | 6% |
| 48% response (coadministered with lithium, divalproex, or carbamazepine) over 6 weeksg | 13% (not statistically significantly different from paroxetine comparison arm) |
| *No data are available for citalopram, desvenlafaxine, duloxetine, escitalopram, fluvoxamine, or milnacipran | |
| NOS: not otherwise specified; SNRI: serotonin/norepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor; STEP-BD: Systematic Treatment Enhancement Program for Bipolar Disorder | |
| Source: References a. Cohn JB, Collins G, Ashbrook E, et al. A comparison of fluoxetine, imipramine and placebo in patients with bipolar depressive disorder. Int Clin Psychopharmaol. 1989;4:313-322. b. Amsterdam JD, Shults J. Fluoxetine monotherapy of bipolar type II and bipolar NOS major depression: a double-blind, placebo-substitution, continuation study. Int Clin Psychopharmacol. 2005;20:257-264. c. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry. 2003;60:1079-1088. d. Sachs GS, Nierenberg AA, Calabrese JR, et al. Effectiveness of adjunctive antidepressant treatment for bipolar depression. N Engl J Med. 2007;356:1711-1722. e. Nemeroff CB, Evans DL, Gyulai L, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry. 2001;158:906-912. f. Young LT, Joffe RT, Robb JC, et al. Double-blind comparison of addition of a second mood stabilizer versus an antidepressant to an initial mood stabilizer for treatment of patients with bipolar depression. Am J Psychiatry. 2000;157:124-126. g. Vieta E, Martinez-Aran A, Goikolea JM. A randomized trial comparing paroxetine and venlafaxine in the treatment of bipolar depressed patients taking mood stabilizers. J Clin Psychiatry. 2002;63:508-512. h. Leverich GS, Altshuler LL, Frye MA, et al. Risk of switch in mood polarity to hypomania or mania in patients with bipolar depression during acute and continuation trials of venlafaxine, sertraline, and bupropion as adjuncts to mood stabilizers. Am J Psychiatry. 2006;163:232-239. | |
Table 2
Antidepressants for bipolar depression: MAOIs, TCAs, and bupropion*
| Acute efficacy | Reported switch risk |
|---|---|
| Tranylcypromine (MAOI) | |
| 81% response (monotherapy) in bipolar I (n=24) or bipolar II (n=32) patients over 16 weeksa | 21% |
| 75% response among imipramine nonresponders (n=12)b | 17% |
| Moclobemide (MAOI) | |
| 46% response over 8 weeks in 156 bipolar patients (some, but not all, took concomitant mood stabilizers), not significantly different from imipramine comparatorc | 4% |
| Imipramine (TCA) | |
| 57% response rate after 3 weeks in a 6-week double-blind randomized comparison with fluoxetine or placebod | Not reported |
| 48% response (monotherapy) in bipolar I (n=24) or bipolar II (N=32) patients over 16 weeksa | 24% |
| 53% response over 8 weeks in 156 bipolar patients (some, but not all, took concomitant mood stabilizers), not significantly different from moclobemide comparatorc | 11% |
| 41% (coadministered with therapeutically dosed lithium)e | 8% |
| Desipramine (TCA) | |
| 50% (5/10) response rate (coadministered with a mood stabilizer over 8 weeks)f | 50% |
| Bupropion | |
| 55% response (5/9) (coadministered with a mood stabilizer over 8 weeks)f | 11% |
| 33% response rate (coadministered with mood stabilizers over 10 weeks)g | 20% |
| *No data are available for isocarboxazid, mirtazapine, nefazodone, phenelzine, or selegiline transdermal | |
| MAOI: monoamine oxidase inhibitor; TCA: tricyclic antidepressant | |
| Source: References a. Himmelhoch JM, Thase ME, Mallinger AG, et al. Tranylcypromine versus imipramine in anergic bipolar depression. Am J Psychiatry. 1991;148:910-916. b. Thase ME, Malinger AG, McKnight D, et al. Treatment of imipramine-resistant recurrent depressions, IV: a double-blind crossover study of tranylcypromine for anergic bipolar depression. Am J Psychiatry. 1992;149:195-198. c. Silverstone T. Moclobemide vs. imipramine in bipolar depression: a multicentre double-blind clinical trial. Acta Psychiatr Scand. 2001;104:104-109. d. Cohn JB, Collins G, Ashbrook E, et al. A comparison of fluoxetine, imipramine and placebo in patients with bipolar depressive disorder. Int Clin Psychopharmaol. 1989;4:313-322. e. Nemeroff CB, Evans DL, Gyulai L, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry. 2001;158:906-912. f. Sachs GS, Lafer B, Stoll AL, et al. A double blind trial of bupropion versus desipramine for bipolar depression. J Clin Psychiatry. 1994;55:391-393. g. Leverich GS, Altshuler LL, Frye MA, et al. Risk of switch in mood polarity to hypomania or mania in patients with bipolar depression during acute and continuation trials of venlafaxine, sertraline, and bupropion as adjuncts to mood stabilizers. Am J Psychiatry. 2006;163:232-239. | |
MYTH 1: Antidepressant-induced mania is a highly prevalent, widespread problem.
Reality: Although some might argue that the precise relative risk of antidepressant-induced mania or hypomania is unknown (eg, considering intervening factors such as the natural illness course), recent literature suggests that the emergence of mania or hypomania can be reasonably attributed to antidepressant use in no more than 10% to 25% of patients with bipolar disorder.5,6 Part of the difficulty in estimating the true prevalence of antidepressant-induced mania involves variability and inconsistency in defining mania induction.
A recent consensus statement proposed a graduated series of definitions for treatment-emergent affective switch:7
- “Definite” switch involves fulfilling DSM-IV syndromic criteria for a manic, hypomanic, or mixed episode for at least 2 days, within 8 weeks of antidepressant introduction.
- “Likely” switches call for at least 2 DSM-IV mania or hypomania symptoms plus a Young Mania Rating Scale (YMRS) score >12, occurring for at least 2 days, within 12 weeks of antidepressant introduction.
- “Possible” switches require a “clear change” in mood or energy with a YMRS score >8, persisting ≥4 hours over 2 days, occurring within 12 weeks of antidepressant initiation.
Adverse effects such as agitation typically diminish or remit with dosage reductions or drug cessation, whereas true antidepressant-induced polarity switches persist even after the medication is discontinued. Moreover, it is often difficult—if not impossible—to know with certainty when a polarity switch results from treatment effects vs the natural illness course. In my experience, true manic or hypomanic syndromes soon after antidepressant exposure are less common than heterogeneous, nonspecific symptoms such as agitation, anxiety, insomnia, or worsening depression (ie, lack of efficacy).
MYTH 2: Antidepressant response rates are lower in bipolar depression.
Reality: It is difficult to draw broad conclusions about antidepressant response rates in unipolar vs bipolar depression because:
- few direct comparisons have been reported
- all relevant studies are retrospective
- small sample sizes in most studies may not have satisfactorily controlled for factors that could predispose to mood destabilization (Table 3).
Table 3
What increases risk of antidepressant-induced mania?
| Factor | Findings |
|---|---|
| History of antidepressant-induced mania or hypomania | Confers an approximate 2- to 5-fold increased risk for subsequent antidepressant-induced mania/hypomania, regardless of antidepressanta |
| Recent mania preceding current depressive episode | Higher risk for antidepressant-associated mania if current depressive episode was preceded by manic phaseb |
| Bipolar I vs bipolar II subtype | Greater risk for switch in bipolar Ic,d |
| Comorbid alcohol or substance use disorder | 5- to 7-fold increased risk for antidepressant-associated maniae |
| Noradrenergic vs serotonergic antidepressants | Possible higher risk for mania induction with TCAs or SNRIs than with bupropionf or SSRIsg |
| Concurrent mania symptoms during a depressive episode | Mild or subthreshold mania symptoms during a depressive episode increase risk for maniah,i |
| Hyperthymic temperamental traits | Associated with increased likelihood of antidepressant-induced maniaj |
| SNRIs: serotonin/norepinephrine reuptake inhibitors; SSRIs: selective serotonin reuptake inhibitors; TCAs: tricyclic antidepressants | |
| Source: References a. Truman CJ, Goldberg JF, Ghaemi SN, et al. Self-reported history of manic/hypomanic switch associated with antidepressant use: data from the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). J Clin Psychiatry. 2007;68:1472-1479. b. MacQueen GM, Young LT, Marriott M, et al. Previous mood state predicts response and switch rates in patients with bipolar depression. Acta Psychiatr Scand. 2002;105:414-418. c. Himmelhoch JM, Thase ME, Mallinger AG, et al. Tranylcypromine versus imipramine in anergic bipolar depression. Am J Psychiatry. 1991;148:910-916. d. Altshuler LL, Suppes T, Black DO, et al. Lower switch rate in depressed patients with bipolar II than bipolar I disorder treated adjunctively with second-generation antidepressants. Am J Psychiatry. 2006;163:313-315. e. Goldberg JF, Truman CJ. Antidepressant-induced mania: an overview of current controversies. Bipolar Disord. 2003;5:407-420. f. Sachs GS, Lafer B, Stoll AL, et al. A double blind trial of bupropion versus desipramine for bipolar depression. J Clin Psychiatry. 1994;55:391-393. g. Peet M. Induction of mania with selective serotonin re-uptake inhibitors and tricyclic antidepressants. Br J Psychiatry. 1994;164:549-550. h. Frye MA, Hellmann G, McElroy SL, et al. Correlates of treatment-emergent mania associated with antidepressant treatment in bipolar depression. Am J Psychiatry. 2009;166:164-172. i. Bottlender R, Rudolf D, Strauss A, et al. Mood-stabilisers reduce the risk of developing antidepressant-induced maniform states in acute treatment of bipolar I depressed patients. J Affect Disord. 2001;63:79-83. j. Henry C, Sorbara F, Lacoste J, et al. Antidepressant-induced mania in bipolar patients: identification of risk factors. J Clin Psychiatry. 2001;62:249-255. | |
A retrospective review of bipolar (n=41) and unipolar (n=37) depressed patients by Ghaemi et al8 found no significant difference in acute nonresponse rates between the groups. Similarly, Bottlender et al9 found no differences in treatment response when comparing naturalistic antidepressant outcomes for 50 unipolar and 50 bipolar patients matched for age, sex, and illness duration. Comparable antidepressant response outcomes also were reported in a retrospective study of 2,032 unipolar and bipolar inpatients conducted by Möller et al,10 and between unipolar (n=31) vs bipolar II (n=17) depressed patients receiving venlafaxine monotherapy for 6 weeks.11
Antidepressant response may depend on factors such as episode chronicity or the number of failed medication trials within a given episode, regardless of illness polarity. This was suggested by the remarkably low response rates after 2 failed initial antidepressant treatments in the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) unipolar depression trials. In my experience, antidepressant efficacy is more often a function of factors in addition to polarity, including:
- illness severity
- chronicity
- psychiatric, medical, or substance use comorbidity
- psychosocial skills, such as the capacity to tolerate distress, utilize effective coping techniques, and maintain appropriate relationships with others.
MYTH 3: Most antidepressants have been systematically studied for treatment of depression in bipolar disorder.
Reality: Only paroxetine,12,13 bupropion,12 and imipramine13 have been studied in randomized, large-scale, adequately powered placebo-controlled trials. Studies of other antidepressants suffer from small sample sizes (inadequate statistical power), lack of placebo controls, or failure to control for possible confounding factors, such as lack of stratification for bipolar I vs II subtype or presence vs absence of rapid cycling.
One large randomized trial showed comparable antidepressant efficacy with a mood stabilizer plus adjunctive venlafaxine (43%) vs sertraline (55%) vs bupropion (49%) over 10 weeks,14 but the lack of a mood stabilizer monotherapy comparison group limits the ability to anticipate whether adjunctive antidepressants increase response or remission rates more than mood stabilizers alone. Adjunctive imipramine,13 paroxetine,12,13,15 and bupropion12 yield no greater improvement in depressive symptoms than is seen with optimally dosed mood stabilizers alone.
Mirtazapine, a serotonergic/noradrenergic antidepressant that is sometimes prescribed off-label as a sleep aid, has not been systematically studied for safety or efficacy in bipolar depression. In case reports, mirtazapine has induced mania in patients with unipolar depression.16-18 Using mirtazapine to counteract insomnia may be safer in patients with unipolar depression than in those with bipolar disorder. Because poor sleep is a core feature of mania, be certain to differentiate complaints that reflect simple insomnia from a loss of need for sleep:
- daytime fatigue is more common in insomnia than loss of need for sleep
- nocturnal hyperactivity is more often associated with loss of need for sleep.
Using an antidepressant to treat sleep problems that may derive from emerging mania or hypomania runs counter to basic pharmacodynamic principles and may pose greater risk than benefit.
Generally, using a medication that has been studied for treating a specific clinical entity such as bipolar depression is preferable to using one that has not. Avoid medications that have multiple negative placebo-controlled trials—such as paroxetine—unless you have evidence of efficacy in an individual patient.
MYTH 4: Risk for inducing mania is higher with noradrenergic antidepressants.
Reality: This popular belief arose from a unifying hypothesis offered by Sachs et al1 and Leverich et al14 to explain higher rates of mania following treatment with desipramine than bupropion,1 SSRIs compared with TCAs,2 or venlafaxine compared with bupropion or sertraline.14 However, while plausible, this hypothesis does not fully account for the putative noradrenergic properties of bupropion—presumably via increased pre-synaptic norepinephrine outflow, rather than noradrenergic reuptake inhibition19—which reportedly has a lower risk of switching than desipramine1 or venlafaxine.14
The risk for venlafaxine monotherapy to induce mania or hypomania in patients with bipolar II depression has been reported to be nonexistent11 or no higher than seen with lithium.20 Also, some noradrenergic agents, such as duloxetine, have not been shown to induce mania in major depression,21 although duloxetine’s potential to destabilize mood is unknown because of the absence of data in bipolar disorder. Finally, although large-scale clinical trials have not examined the safety and efficacy of the noradrenergic reuptake inhibitor atomoxetine, several case reports have suggested its potential for inducing mania or hypomania.22,23
Likely, all-or-none admonitions against using noradrenergic antidepressants are oversimplifications.
MYTH 5: Coadministering an antimanic mood stabilizer reliably prevents antidepressant-induced mania.
Reality: Most practice guidelines advise administering antimanic mood stabilizers before initiating an antidepressant. Clinicians widely interpret this recommendation as reinforcing the assumption that a mood stabilizer will diminish mania risk when introducing an antidepressant. (Less often, clinicians interpret it as meaning that a mood stabilizer itself may provide antidepressant efficacy.) In fact, whether (and which) antimanic agents mitigate the risk for antidepressant-induced mania has received little empirical study. The largest dataset on this topic—the randomized controlled data from Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD)12—found that the risk for treatment-emergent manic switch with paroxetine or bupropion was almost identical (about 10%) with or without an FDA-approved antimanic agent.
In a retrospective study, Henry et al6 found that cotherapy with lithium but not divalproex or carbamazepine protects against antidepressant-induced mania, and that switch rates to mania were the same whether or not an antidepressant was taken with an anticonvulsant. In a naturalistic retrospective study (n=158), Bottlender et al24 revealed that mood stabilizers (lithium, carbamazepine, or divalproex) prevented switches from depression to mania during treatment with TCAs but not SSRIs or MAOIs.
I favor incorporating lithium or other antimanic agents in the regimens of patients with bipolar depression not primarily to guard against antidepressant-induced mania but more for pharmacodynamic synergy—complementary mechanisms of action that collectively may produce more substantial antidepressant effects—especially when the patient’s illness course has included manic or hypomanic features in the preceding year.
MYTH 6: Antidepressants cause or worsen rapid cycling.
Reality: Wehr et al25 reported that antidepressants may accelerate cycling frequency (ie, inter-episode durations become shorter) in a small subgroup (N=10) of patients. By contrast, use of TCAs was not more likely in the weeks preceding shifts from depression to mania or hypomania in a 14-year follow-up study of bipolar rapid cycling from the NIMH Collaborative Depression Study.26 In fact, rapid-cycling patients spent more weeks depressed when taking lithium without a TCA than with 1.
Findings from STEP-BD indicate that prospectively observed rapid cycling, as defined by DSM-IV criteria, is relatively rare, although subjects taking antidepressants often had multiple episodes per year.27 These naturalistic data could suggest that antidepressant use leads to more depressive episodes, or that more depressive episodes lead to more antidepressant use. Causal relationships cannot be inferred from the nonrandomized study design. Nevertheless, antidepressant use was not associated with reduced depressive episodes over 1 year.
I believe that, in general, antidepressants are unlikely to improve a truly rapid-cycling illness course. In this scenario, a more “panoramic” understanding of the need to treat multiple relapses and polarity changes over time likely warrants using multiple anti-cycling agents. Rapid cycling is treated over the course of 1 year, rather than 1 episode.
MYTH 7: Antidepressants should never be used without a mood stabilizer for bipolar depression.
Reality: This admonition is widely cited as a general recommendation from modern practice guidelines; however, it mainly pertains to depression treatment in patients with bipolar I disorder, for whom most controlled trial data exist. For example, relatively high rates of treatment-emergent mania have been reported with TCA or MAOI monotherapy in bipolar I disorder patients (Table 2). Yet for bipolar II disorder, controlled trials demonstrate superior outcomes with venlafaxine monotherapy compared with lithium monotherapy, with no increase in mood destabilization.20
Neither the safety nor the efficacy of antidepressants with vs without mood stabilizers has been studied systematically in cyclothymic or mood disorder patients who may fall within the so-called bipolar spectrum but have never met DSM-IV criteria for a lifetime manic or hypomanic episode (ie, bipolar disorder not otherwise specified). Extrapolation from findings based on bipolar I disorder patients may not be valid for all bipolar subtypes.
Clinical strategies
In constructing a rationale-based approach to bipolar depression, consider these steps:
Step 1: Assess candidacy for antidepressant use. A number of key features can help you delineate the current illness state and context in which depressive symptoms arise—features that may influence you patient’s vulnerability to mood destabilization, and therefore are pertinent for gauging the likelihood that antidepressants may help or harm (Table 4).
Step 2: Consider mood stabilizers with antidepressant properties. Determine whether your patient is taking any mood stabilizers that possess robust antidepressant properties, or whether it may be beneficial to introduce one of these agents before initiating adjunctive antidepressants. Mood stabilizers with antidepressant efficacy are compelling options for patients presenting with any of the features listed in the right-hand column of Table 4, as well as those with:
- psychotic features
- marked agitation
- multiple prior antidepressant nonresponses
- high depression recurrence rates regardless of episode duration (ie, cyclicity, irrespective of ≥4 discrete episodes per year).
Table 4
Assessing antidepressant candidacy in bipolar depression
| Favors antidepressant use | Discourages antidepressant use |
|---|---|
| Bipolar II disorder | Bipolar I disordera |
| Depressed (non-mixed) states | Mixed manic and depressive featuresb,c |
| Absence of rapid cycling | Presence of rapid cyclingd,e |
| Absence of recent mania or hypomania (preceding 2 to 3 months) | Mania or hypomania in past 2 to 3 monthsf |
| Absence of comorbid alcohol or substance use disorder | Presence of comorbid alcohol or substance use disorderg,h |
| Prior favorable antidepressant response | Suboptimal responses to prior antidepressants |
| No history of antidepressant-induced mania or hypomania | History of antidepressant-induced mania or hypomaniai |
| Source: References a. Altshuler LL, Suppes T, Black DO, et al. Lower switch rate in depressed patients with bipolar II than bipolar I disorder treated adjunctively with second-generation antidepressants. Am J Psychiatry. 2006;163:313-315. b. Frye MA, Hellmann G, McElroy SL, et al. Correlates of treatment-emergent mania associated with antidepressant treatment in bipolar depression. Am J Psychiatry. 2009;166:164-172. c. Goldberg JF, Perlis RH, Ghaemi SN, et al. Adjunctive antidepressant use and symptomatic recovery among bipolar depressed patients with concomitant manic symptoms: findings from the STEP-BD. Am J Psychiatry. 2007;164(9):1348-1355. d. Schneck CD, Miklowitz DJ, Miyahara S, et al. The prospective course of rapid-cycling bipolar disorder: findings from the STEP-BD. Am J Psychiatry. 2008;165:370-377. e. Ghaemi SN, Ostacher MM, El-Mallakh RS, et al. Antidepressant discontinuation in bipolar depression: a STEP-BD randomized clinical trial of long-term effectiveness and safety. J Clin Psychiatry. In press. f. MacQueen GM, Young LT, Marriott M, et al. Previous mood state predicts response and switch rates in patients with bipolar depression. Acta Psychiatr Scand. 2002;105:414-418. g. Goldberg JF, Whiteside JE. The association between substance abuse and antidepressant-induced mania in bipolar disorder: a preliminary study. J Clin Psychiatry. 2002;63:791-795. h. Manwani SG, Pardo TB, Albanese MJ, et al. Substance use disorder and other predictors of antidepressant-induced mania: a retrospective chart review. J Clin Psychiatry. 2006;67:1341-1345. i. Truman CJ, Goldberg JF, Ghaemi SN, et al. Self-reported history of manic/hypomanic switch associated with antidepressant use: data from the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). J Clin Psychiatry. 2007;68:1472-1479. | |
Prospective mood charting may help to establish the latter, in which case recurrence (rather than polarity) may cause waxing and waning depressed mood states.
Psychotropic agents or combinations that have shown to be effective for bipolar depression (supported by at least 1 randomized controlled trial) without destabilizing mood include quetiapine, olanzapine, olanzapine-fluoxetine combination, lamotrigine, and lithium plus lamotrigine. Those with some—but less robustly demonstrated—antidepressant action include lithium, divalproex, and carbamazepine. Other than quetiapine and olanzapine, second-generation antipsychotics have not demonstrated antidepressant effects in bipolar depression.
In general, optimize therapy with 1 or more mood stabilizers with antidepressant properties before deciding it is necessary to add antidepressants.
Step 3: Use antidepressants in suitable patients. For patients with no risk factors for mood destabilization from antidepressants (Table 3), these drugs may be worth incorporating, keeping in mind the following guiding principles:
- In patients with bipolar I depression, it is preferable to add an antidepressant to an antimanic mood stabilizer (ie, lithium, divalproex, carbamazepine, or an antipsychotic) rather than prescribing antidepressant monotherapy. There is greater diversity of opinion about the safety of antidepressant monotherapy for bipolar II depression.
- Consider using antidepressants that have at least 1 positive randomized controlled trial in bipolar disorder and low risk for mood destabilization (bupropion,12,14 sertraline,14 fluoxetine,4,5 tranylcypromine,3,28 or venlafaxine in bipolar II depression20) before using those with reported increased risk for inducing mania or hypomania (TCAs1,2 or venlafaxine in bipolar I depression14), multiple negative controlled trials (paroxetine12,13), or no controlled data in bipolar depression (citalopram, escitalopram, fluvoxamine, mirtazapine, duloxetine, desvenlafaxine, nefazodone, and selegiline transdermal). Combinations of antidepressants have not been adequately studied in bipolar depression.
- The optimal duration of antidepressant therapy is unknown. However, longer-term treatment may be worthwhile in patients who show robust acute antidepressant response and experience infrequent mania or hypomania. Long-term antidepressant use is less compelling in patients with a poor initial response29 or rapid cycling.30 Abrupt antidepressant cessation also may induce mania, potentially by disrupting homeostasis.31 In the absence of rapid cycling, manic/hypomanic features, or worsening suicidal features, and in the presence of an unequivocal acute response and a greater predisposition to depression than mania, it is reasonable to continue an antidepressant indefinitely until new signs of mania or hypomania emerge.
- Emerging signs of mania or hypomania should signal the need to discontinue the antidepressant. Dosage reductions alone may not diminish emerging manic or hypomanic symptoms, and “counterbalancing” maneuvers (ie, adding antimanic agents while continuing an antidepressant) may not effectively stabilize mood.
Step 4: Consider novel strategies. In the absence of a response to the strategy outlined above—particularly among poor candidates for continued antidepressant therapy—other novel strategies have support from at least 1 randomized controlled trial, including pramipexole,32,33 modafinil,34 riluzole,35 and n-acetyl cysteine.36 Other interventions worth considering include:
- adjunctive thyroid hormone
- cognitive therapy
- light therapy (if a seasonal component is evident)
- electroconvulsive therapy.
Related resources
- Goldberg JF. Treating depression in bipolar disorder. http://thedoctorschannel.com/video/3077.html.
- Goldberg JF. Pharmacologic treatment of acute mania.http://thedoctorschannel.com/video/3032.html.
Drug brand names
- Atomoxetine • Strattera
- Bupropion • Wellbutrin
- Carbamazepine • Tegretol, Equetro
- Citalopram • Celexa
- Desipramine • Norpramin
- Desvenlafaxine • Pristiq
- Divalproex • Depakote, Depakene
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Imipramine • Tofranil
- Isocarboxazid • Marplan
- Lamotrigine • Lamictal
- Lithium • Lithobid, Eskalith
- Milnacipran • Ixel, Savella
- Mirtazapine • Remeron
- Moclobemide • Aurorix, Manerix
- Modafinil • Provigil
- Nefazodone • Serzone
- Olanzapine • Zyprexa
- Olanzapine-fluoxetine • Symbyax
- Paroxetine • Paxil
- Phenelzine • Nardil
- Pramipexole • Mirapex
- Quetiapine • Seroquel
- Riluzole • Rilutek
- Selegiline transdermal • EMSAM
- Sertraline • Zoloft
- Tranylcypromine • Parnate
- Venlafaxine • Effexor
Disclosures
Dr. Goldberg is a consultant to Eli Lilly and Company and a speaker for AstraZeneca, Eli Lilly and Company, GlaxoSmithKline, Merck, and Pfizer Inc., and has received speaking honoraria from Janssen-Cilag.
Few topics are as controversial as the role of antidepressants for patients with bipolar disorder. Although depression usually is the predominant, most enduring mood state in bipolar disorder, clinicians often face uncertainty about using antidepressants because of concerns about safety and efficacy. Whether and when to use antidepressants for bipolar depression hinges on complex parameters that preclude any single, simple rule.
Rather than asking if antidepressants are useful or detrimental for depressed patients with bipolar disorder, a more practical question might be: Under what circumstances are antidepressants likely to be beneficial, deleterious, or ineffective for an individual patient? Because “real world” patients often have idiosyncrasies that defy practice guidelines’ generic treatment recommendations, clinicians who practice in the proverbial trenches need strategies to tailor treatments to each patient that are informed—but not dictated—by evidence-based research.
Early suspicions
Concerns that antidepressants might precipitate mania were first described with tricyclic antidepressant (TCA) use in Europe in the 1960s. After bupropion and selective serotonin reuptake inhibitors (SSRIs) emerged, some clinicians believed they posed a lesser risk for this phenomenon compared with TCAs1,2 or monoamine oxidase inhibitors (MAOIs).3
Antidepressants’ potential to induce short-term mania/hypomania following acute exposure has been weighed against the longer-term risk for worsening illness course by increasing frequency of subsequent episodes (so-called cycle acceleration). In the 1980s, some researchers suggested that rapid cycling might—at least in some instances—represent an iatrogenic phenomenon caused by long-term antidepressant use. These issues remain controversial, but more than 20 years of research suggest that antidepressants induce mania or accelerate cycling in a smaller minority of bipolar disorder patients than was once thought.
Table 1 and Table 2 summarize findings from randomized controlled studies that have examined antidepressants’ efficacy for acute bipolar depression. Except for a study of fluoxetine plus olanzapine,4 no large-scale placebo-controlled trial has demonstrated superior antidepressant response to a mood stabilizer plus antidepressant compared with a mood stabilizer alone.
Table 1
Antidepressants for bipolar depression: SSRIs and SNRIs*
| Acute efficacy | Reported switch risk |
|---|---|
| Fluoxetine (SSRI) | |
| 86% response rate after 3 weeks in 6-week double-blind randomized comparison with imipramine or placeboa | 0% |
| 38% response rate after 8 weeks of placebo-controlled monotherapy in bipolar II or NOS subjectsb | 0% |
| 56% response rate over 8 weeks in combination with olanzapine; significantly better than placebo plus olanzapine (30%)c | 6% |
| Paroxetine (SSRI) | |
| Same as placebo when added to an antimanic drug (STEP-BD) for up to 26 weeksd | 10.1% (reported only jointly for paroxetine or bupropion) |
| 36% response rate (no different from placebo) when coadministered with therapeutically dosed lithium over 10 weekse | 7% |
| Same as divalproex plus lithium when coadministered with divalproex or lithium over 6 weeks (actual response rates not reported)f | 0% |
| 43% response (coadministered with lithium, divalproex, or carbamazepine) over 6 weeksg | 3% (not statistically significantly different from venlafaxine comparison arm) |
| Sertraline (SSRI) | |
| 41% improved (comparable to rates seen with bupropion [33%] or venlafaxine [36%] when coadministered with a mood stabilizer over 10 weeks)h | 12% |
| Venlafaxine (SNRI) | |
| 36% improved (comparable to rates seen with bupropion [33%] or sertraline [41%]) when coadministered with a mood stabilizer over 10 weeksh | 6% |
| 48% response (coadministered with lithium, divalproex, or carbamazepine) over 6 weeksg | 13% (not statistically significantly different from paroxetine comparison arm) |
| *No data are available for citalopram, desvenlafaxine, duloxetine, escitalopram, fluvoxamine, or milnacipran | |
| NOS: not otherwise specified; SNRI: serotonin/norepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor; STEP-BD: Systematic Treatment Enhancement Program for Bipolar Disorder | |
| Source: References a. Cohn JB, Collins G, Ashbrook E, et al. A comparison of fluoxetine, imipramine and placebo in patients with bipolar depressive disorder. Int Clin Psychopharmaol. 1989;4:313-322. b. Amsterdam JD, Shults J. Fluoxetine monotherapy of bipolar type II and bipolar NOS major depression: a double-blind, placebo-substitution, continuation study. Int Clin Psychopharmacol. 2005;20:257-264. c. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry. 2003;60:1079-1088. d. Sachs GS, Nierenberg AA, Calabrese JR, et al. Effectiveness of adjunctive antidepressant treatment for bipolar depression. N Engl J Med. 2007;356:1711-1722. e. Nemeroff CB, Evans DL, Gyulai L, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry. 2001;158:906-912. f. Young LT, Joffe RT, Robb JC, et al. Double-blind comparison of addition of a second mood stabilizer versus an antidepressant to an initial mood stabilizer for treatment of patients with bipolar depression. Am J Psychiatry. 2000;157:124-126. g. Vieta E, Martinez-Aran A, Goikolea JM. A randomized trial comparing paroxetine and venlafaxine in the treatment of bipolar depressed patients taking mood stabilizers. J Clin Psychiatry. 2002;63:508-512. h. Leverich GS, Altshuler LL, Frye MA, et al. Risk of switch in mood polarity to hypomania or mania in patients with bipolar depression during acute and continuation trials of venlafaxine, sertraline, and bupropion as adjuncts to mood stabilizers. Am J Psychiatry. 2006;163:232-239. | |
Table 2
Antidepressants for bipolar depression: MAOIs, TCAs, and bupropion*
| Acute efficacy | Reported switch risk |
|---|---|
| Tranylcypromine (MAOI) | |
| 81% response (monotherapy) in bipolar I (n=24) or bipolar II (n=32) patients over 16 weeksa | 21% |
| 75% response among imipramine nonresponders (n=12)b | 17% |
| Moclobemide (MAOI) | |
| 46% response over 8 weeks in 156 bipolar patients (some, but not all, took concomitant mood stabilizers), not significantly different from imipramine comparatorc | 4% |
| Imipramine (TCA) | |
| 57% response rate after 3 weeks in a 6-week double-blind randomized comparison with fluoxetine or placebod | Not reported |
| 48% response (monotherapy) in bipolar I (n=24) or bipolar II (N=32) patients over 16 weeksa | 24% |
| 53% response over 8 weeks in 156 bipolar patients (some, but not all, took concomitant mood stabilizers), not significantly different from moclobemide comparatorc | 11% |
| 41% (coadministered with therapeutically dosed lithium)e | 8% |
| Desipramine (TCA) | |
| 50% (5/10) response rate (coadministered with a mood stabilizer over 8 weeks)f | 50% |
| Bupropion | |
| 55% response (5/9) (coadministered with a mood stabilizer over 8 weeks)f | 11% |
| 33% response rate (coadministered with mood stabilizers over 10 weeks)g | 20% |
| *No data are available for isocarboxazid, mirtazapine, nefazodone, phenelzine, or selegiline transdermal | |
| MAOI: monoamine oxidase inhibitor; TCA: tricyclic antidepressant | |
| Source: References a. Himmelhoch JM, Thase ME, Mallinger AG, et al. Tranylcypromine versus imipramine in anergic bipolar depression. Am J Psychiatry. 1991;148:910-916. b. Thase ME, Malinger AG, McKnight D, et al. Treatment of imipramine-resistant recurrent depressions, IV: a double-blind crossover study of tranylcypromine for anergic bipolar depression. Am J Psychiatry. 1992;149:195-198. c. Silverstone T. Moclobemide vs. imipramine in bipolar depression: a multicentre double-blind clinical trial. Acta Psychiatr Scand. 2001;104:104-109. d. Cohn JB, Collins G, Ashbrook E, et al. A comparison of fluoxetine, imipramine and placebo in patients with bipolar depressive disorder. Int Clin Psychopharmaol. 1989;4:313-322. e. Nemeroff CB, Evans DL, Gyulai L, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry. 2001;158:906-912. f. Sachs GS, Lafer B, Stoll AL, et al. A double blind trial of bupropion versus desipramine for bipolar depression. J Clin Psychiatry. 1994;55:391-393. g. Leverich GS, Altshuler LL, Frye MA, et al. Risk of switch in mood polarity to hypomania or mania in patients with bipolar depression during acute and continuation trials of venlafaxine, sertraline, and bupropion as adjuncts to mood stabilizers. Am J Psychiatry. 2006;163:232-239. | |
MYTH 1: Antidepressant-induced mania is a highly prevalent, widespread problem.
Reality: Although some might argue that the precise relative risk of antidepressant-induced mania or hypomania is unknown (eg, considering intervening factors such as the natural illness course), recent literature suggests that the emergence of mania or hypomania can be reasonably attributed to antidepressant use in no more than 10% to 25% of patients with bipolar disorder.5,6 Part of the difficulty in estimating the true prevalence of antidepressant-induced mania involves variability and inconsistency in defining mania induction.
A recent consensus statement proposed a graduated series of definitions for treatment-emergent affective switch:7
- “Definite” switch involves fulfilling DSM-IV syndromic criteria for a manic, hypomanic, or mixed episode for at least 2 days, within 8 weeks of antidepressant introduction.
- “Likely” switches call for at least 2 DSM-IV mania or hypomania symptoms plus a Young Mania Rating Scale (YMRS) score >12, occurring for at least 2 days, within 12 weeks of antidepressant introduction.
- “Possible” switches require a “clear change” in mood or energy with a YMRS score >8, persisting ≥4 hours over 2 days, occurring within 12 weeks of antidepressant initiation.
Adverse effects such as agitation typically diminish or remit with dosage reductions or drug cessation, whereas true antidepressant-induced polarity switches persist even after the medication is discontinued. Moreover, it is often difficult—if not impossible—to know with certainty when a polarity switch results from treatment effects vs the natural illness course. In my experience, true manic or hypomanic syndromes soon after antidepressant exposure are less common than heterogeneous, nonspecific symptoms such as agitation, anxiety, insomnia, or worsening depression (ie, lack of efficacy).
MYTH 2: Antidepressant response rates are lower in bipolar depression.
Reality: It is difficult to draw broad conclusions about antidepressant response rates in unipolar vs bipolar depression because:
- few direct comparisons have been reported
- all relevant studies are retrospective
- small sample sizes in most studies may not have satisfactorily controlled for factors that could predispose to mood destabilization (Table 3).
Table 3
What increases risk of antidepressant-induced mania?
| Factor | Findings |
|---|---|
| History of antidepressant-induced mania or hypomania | Confers an approximate 2- to 5-fold increased risk for subsequent antidepressant-induced mania/hypomania, regardless of antidepressanta |
| Recent mania preceding current depressive episode | Higher risk for antidepressant-associated mania if current depressive episode was preceded by manic phaseb |
| Bipolar I vs bipolar II subtype | Greater risk for switch in bipolar Ic,d |
| Comorbid alcohol or substance use disorder | 5- to 7-fold increased risk for antidepressant-associated maniae |
| Noradrenergic vs serotonergic antidepressants | Possible higher risk for mania induction with TCAs or SNRIs than with bupropionf or SSRIsg |
| Concurrent mania symptoms during a depressive episode | Mild or subthreshold mania symptoms during a depressive episode increase risk for maniah,i |
| Hyperthymic temperamental traits | Associated with increased likelihood of antidepressant-induced maniaj |
| SNRIs: serotonin/norepinephrine reuptake inhibitors; SSRIs: selective serotonin reuptake inhibitors; TCAs: tricyclic antidepressants | |
| Source: References a. Truman CJ, Goldberg JF, Ghaemi SN, et al. Self-reported history of manic/hypomanic switch associated with antidepressant use: data from the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). J Clin Psychiatry. 2007;68:1472-1479. b. MacQueen GM, Young LT, Marriott M, et al. Previous mood state predicts response and switch rates in patients with bipolar depression. Acta Psychiatr Scand. 2002;105:414-418. c. Himmelhoch JM, Thase ME, Mallinger AG, et al. Tranylcypromine versus imipramine in anergic bipolar depression. Am J Psychiatry. 1991;148:910-916. d. Altshuler LL, Suppes T, Black DO, et al. Lower switch rate in depressed patients with bipolar II than bipolar I disorder treated adjunctively with second-generation antidepressants. Am J Psychiatry. 2006;163:313-315. e. Goldberg JF, Truman CJ. Antidepressant-induced mania: an overview of current controversies. Bipolar Disord. 2003;5:407-420. f. Sachs GS, Lafer B, Stoll AL, et al. A double blind trial of bupropion versus desipramine for bipolar depression. J Clin Psychiatry. 1994;55:391-393. g. Peet M. Induction of mania with selective serotonin re-uptake inhibitors and tricyclic antidepressants. Br J Psychiatry. 1994;164:549-550. h. Frye MA, Hellmann G, McElroy SL, et al. Correlates of treatment-emergent mania associated with antidepressant treatment in bipolar depression. Am J Psychiatry. 2009;166:164-172. i. Bottlender R, Rudolf D, Strauss A, et al. Mood-stabilisers reduce the risk of developing antidepressant-induced maniform states in acute treatment of bipolar I depressed patients. J Affect Disord. 2001;63:79-83. j. Henry C, Sorbara F, Lacoste J, et al. Antidepressant-induced mania in bipolar patients: identification of risk factors. J Clin Psychiatry. 2001;62:249-255. | |
A retrospective review of bipolar (n=41) and unipolar (n=37) depressed patients by Ghaemi et al8 found no significant difference in acute nonresponse rates between the groups. Similarly, Bottlender et al9 found no differences in treatment response when comparing naturalistic antidepressant outcomes for 50 unipolar and 50 bipolar patients matched for age, sex, and illness duration. Comparable antidepressant response outcomes also were reported in a retrospective study of 2,032 unipolar and bipolar inpatients conducted by Möller et al,10 and between unipolar (n=31) vs bipolar II (n=17) depressed patients receiving venlafaxine monotherapy for 6 weeks.11
Antidepressant response may depend on factors such as episode chronicity or the number of failed medication trials within a given episode, regardless of illness polarity. This was suggested by the remarkably low response rates after 2 failed initial antidepressant treatments in the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) unipolar depression trials. In my experience, antidepressant efficacy is more often a function of factors in addition to polarity, including:
- illness severity
- chronicity
- psychiatric, medical, or substance use comorbidity
- psychosocial skills, such as the capacity to tolerate distress, utilize effective coping techniques, and maintain appropriate relationships with others.
MYTH 3: Most antidepressants have been systematically studied for treatment of depression in bipolar disorder.
Reality: Only paroxetine,12,13 bupropion,12 and imipramine13 have been studied in randomized, large-scale, adequately powered placebo-controlled trials. Studies of other antidepressants suffer from small sample sizes (inadequate statistical power), lack of placebo controls, or failure to control for possible confounding factors, such as lack of stratification for bipolar I vs II subtype or presence vs absence of rapid cycling.
One large randomized trial showed comparable antidepressant efficacy with a mood stabilizer plus adjunctive venlafaxine (43%) vs sertraline (55%) vs bupropion (49%) over 10 weeks,14 but the lack of a mood stabilizer monotherapy comparison group limits the ability to anticipate whether adjunctive antidepressants increase response or remission rates more than mood stabilizers alone. Adjunctive imipramine,13 paroxetine,12,13,15 and bupropion12 yield no greater improvement in depressive symptoms than is seen with optimally dosed mood stabilizers alone.
Mirtazapine, a serotonergic/noradrenergic antidepressant that is sometimes prescribed off-label as a sleep aid, has not been systematically studied for safety or efficacy in bipolar depression. In case reports, mirtazapine has induced mania in patients with unipolar depression.16-18 Using mirtazapine to counteract insomnia may be safer in patients with unipolar depression than in those with bipolar disorder. Because poor sleep is a core feature of mania, be certain to differentiate complaints that reflect simple insomnia from a loss of need for sleep:
- daytime fatigue is more common in insomnia than loss of need for sleep
- nocturnal hyperactivity is more often associated with loss of need for sleep.
Using an antidepressant to treat sleep problems that may derive from emerging mania or hypomania runs counter to basic pharmacodynamic principles and may pose greater risk than benefit.
Generally, using a medication that has been studied for treating a specific clinical entity such as bipolar depression is preferable to using one that has not. Avoid medications that have multiple negative placebo-controlled trials—such as paroxetine—unless you have evidence of efficacy in an individual patient.
MYTH 4: Risk for inducing mania is higher with noradrenergic antidepressants.
Reality: This popular belief arose from a unifying hypothesis offered by Sachs et al1 and Leverich et al14 to explain higher rates of mania following treatment with desipramine than bupropion,1 SSRIs compared with TCAs,2 or venlafaxine compared with bupropion or sertraline.14 However, while plausible, this hypothesis does not fully account for the putative noradrenergic properties of bupropion—presumably via increased pre-synaptic norepinephrine outflow, rather than noradrenergic reuptake inhibition19—which reportedly has a lower risk of switching than desipramine1 or venlafaxine.14
The risk for venlafaxine monotherapy to induce mania or hypomania in patients with bipolar II depression has been reported to be nonexistent11 or no higher than seen with lithium.20 Also, some noradrenergic agents, such as duloxetine, have not been shown to induce mania in major depression,21 although duloxetine’s potential to destabilize mood is unknown because of the absence of data in bipolar disorder. Finally, although large-scale clinical trials have not examined the safety and efficacy of the noradrenergic reuptake inhibitor atomoxetine, several case reports have suggested its potential for inducing mania or hypomania.22,23
Likely, all-or-none admonitions against using noradrenergic antidepressants are oversimplifications.
MYTH 5: Coadministering an antimanic mood stabilizer reliably prevents antidepressant-induced mania.
Reality: Most practice guidelines advise administering antimanic mood stabilizers before initiating an antidepressant. Clinicians widely interpret this recommendation as reinforcing the assumption that a mood stabilizer will diminish mania risk when introducing an antidepressant. (Less often, clinicians interpret it as meaning that a mood stabilizer itself may provide antidepressant efficacy.) In fact, whether (and which) antimanic agents mitigate the risk for antidepressant-induced mania has received little empirical study. The largest dataset on this topic—the randomized controlled data from Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD)12—found that the risk for treatment-emergent manic switch with paroxetine or bupropion was almost identical (about 10%) with or without an FDA-approved antimanic agent.
In a retrospective study, Henry et al6 found that cotherapy with lithium but not divalproex or carbamazepine protects against antidepressant-induced mania, and that switch rates to mania were the same whether or not an antidepressant was taken with an anticonvulsant. In a naturalistic retrospective study (n=158), Bottlender et al24 revealed that mood stabilizers (lithium, carbamazepine, or divalproex) prevented switches from depression to mania during treatment with TCAs but not SSRIs or MAOIs.
I favor incorporating lithium or other antimanic agents in the regimens of patients with bipolar depression not primarily to guard against antidepressant-induced mania but more for pharmacodynamic synergy—complementary mechanisms of action that collectively may produce more substantial antidepressant effects—especially when the patient’s illness course has included manic or hypomanic features in the preceding year.
MYTH 6: Antidepressants cause or worsen rapid cycling.
Reality: Wehr et al25 reported that antidepressants may accelerate cycling frequency (ie, inter-episode durations become shorter) in a small subgroup (N=10) of patients. By contrast, use of TCAs was not more likely in the weeks preceding shifts from depression to mania or hypomania in a 14-year follow-up study of bipolar rapid cycling from the NIMH Collaborative Depression Study.26 In fact, rapid-cycling patients spent more weeks depressed when taking lithium without a TCA than with 1.
Findings from STEP-BD indicate that prospectively observed rapid cycling, as defined by DSM-IV criteria, is relatively rare, although subjects taking antidepressants often had multiple episodes per year.27 These naturalistic data could suggest that antidepressant use leads to more depressive episodes, or that more depressive episodes lead to more antidepressant use. Causal relationships cannot be inferred from the nonrandomized study design. Nevertheless, antidepressant use was not associated with reduced depressive episodes over 1 year.
I believe that, in general, antidepressants are unlikely to improve a truly rapid-cycling illness course. In this scenario, a more “panoramic” understanding of the need to treat multiple relapses and polarity changes over time likely warrants using multiple anti-cycling agents. Rapid cycling is treated over the course of 1 year, rather than 1 episode.
MYTH 7: Antidepressants should never be used without a mood stabilizer for bipolar depression.
Reality: This admonition is widely cited as a general recommendation from modern practice guidelines; however, it mainly pertains to depression treatment in patients with bipolar I disorder, for whom most controlled trial data exist. For example, relatively high rates of treatment-emergent mania have been reported with TCA or MAOI monotherapy in bipolar I disorder patients (Table 2). Yet for bipolar II disorder, controlled trials demonstrate superior outcomes with venlafaxine monotherapy compared with lithium monotherapy, with no increase in mood destabilization.20
Neither the safety nor the efficacy of antidepressants with vs without mood stabilizers has been studied systematically in cyclothymic or mood disorder patients who may fall within the so-called bipolar spectrum but have never met DSM-IV criteria for a lifetime manic or hypomanic episode (ie, bipolar disorder not otherwise specified). Extrapolation from findings based on bipolar I disorder patients may not be valid for all bipolar subtypes.
Clinical strategies
In constructing a rationale-based approach to bipolar depression, consider these steps:
Step 1: Assess candidacy for antidepressant use. A number of key features can help you delineate the current illness state and context in which depressive symptoms arise—features that may influence you patient’s vulnerability to mood destabilization, and therefore are pertinent for gauging the likelihood that antidepressants may help or harm (Table 4).
Step 2: Consider mood stabilizers with antidepressant properties. Determine whether your patient is taking any mood stabilizers that possess robust antidepressant properties, or whether it may be beneficial to introduce one of these agents before initiating adjunctive antidepressants. Mood stabilizers with antidepressant efficacy are compelling options for patients presenting with any of the features listed in the right-hand column of Table 4, as well as those with:
- psychotic features
- marked agitation
- multiple prior antidepressant nonresponses
- high depression recurrence rates regardless of episode duration (ie, cyclicity, irrespective of ≥4 discrete episodes per year).
Table 4
Assessing antidepressant candidacy in bipolar depression
| Favors antidepressant use | Discourages antidepressant use |
|---|---|
| Bipolar II disorder | Bipolar I disordera |
| Depressed (non-mixed) states | Mixed manic and depressive featuresb,c |
| Absence of rapid cycling | Presence of rapid cyclingd,e |
| Absence of recent mania or hypomania (preceding 2 to 3 months) | Mania or hypomania in past 2 to 3 monthsf |
| Absence of comorbid alcohol or substance use disorder | Presence of comorbid alcohol or substance use disorderg,h |
| Prior favorable antidepressant response | Suboptimal responses to prior antidepressants |
| No history of antidepressant-induced mania or hypomania | History of antidepressant-induced mania or hypomaniai |
| Source: References a. Altshuler LL, Suppes T, Black DO, et al. Lower switch rate in depressed patients with bipolar II than bipolar I disorder treated adjunctively with second-generation antidepressants. Am J Psychiatry. 2006;163:313-315. b. Frye MA, Hellmann G, McElroy SL, et al. Correlates of treatment-emergent mania associated with antidepressant treatment in bipolar depression. Am J Psychiatry. 2009;166:164-172. c. Goldberg JF, Perlis RH, Ghaemi SN, et al. Adjunctive antidepressant use and symptomatic recovery among bipolar depressed patients with concomitant manic symptoms: findings from the STEP-BD. Am J Psychiatry. 2007;164(9):1348-1355. d. Schneck CD, Miklowitz DJ, Miyahara S, et al. The prospective course of rapid-cycling bipolar disorder: findings from the STEP-BD. Am J Psychiatry. 2008;165:370-377. e. Ghaemi SN, Ostacher MM, El-Mallakh RS, et al. Antidepressant discontinuation in bipolar depression: a STEP-BD randomized clinical trial of long-term effectiveness and safety. J Clin Psychiatry. In press. f. MacQueen GM, Young LT, Marriott M, et al. Previous mood state predicts response and switch rates in patients with bipolar depression. Acta Psychiatr Scand. 2002;105:414-418. g. Goldberg JF, Whiteside JE. The association between substance abuse and antidepressant-induced mania in bipolar disorder: a preliminary study. J Clin Psychiatry. 2002;63:791-795. h. Manwani SG, Pardo TB, Albanese MJ, et al. Substance use disorder and other predictors of antidepressant-induced mania: a retrospective chart review. J Clin Psychiatry. 2006;67:1341-1345. i. Truman CJ, Goldberg JF, Ghaemi SN, et al. Self-reported history of manic/hypomanic switch associated with antidepressant use: data from the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). J Clin Psychiatry. 2007;68:1472-1479. | |
Prospective mood charting may help to establish the latter, in which case recurrence (rather than polarity) may cause waxing and waning depressed mood states.
Psychotropic agents or combinations that have shown to be effective for bipolar depression (supported by at least 1 randomized controlled trial) without destabilizing mood include quetiapine, olanzapine, olanzapine-fluoxetine combination, lamotrigine, and lithium plus lamotrigine. Those with some—but less robustly demonstrated—antidepressant action include lithium, divalproex, and carbamazepine. Other than quetiapine and olanzapine, second-generation antipsychotics have not demonstrated antidepressant effects in bipolar depression.
In general, optimize therapy with 1 or more mood stabilizers with antidepressant properties before deciding it is necessary to add antidepressants.
Step 3: Use antidepressants in suitable patients. For patients with no risk factors for mood destabilization from antidepressants (Table 3), these drugs may be worth incorporating, keeping in mind the following guiding principles:
- In patients with bipolar I depression, it is preferable to add an antidepressant to an antimanic mood stabilizer (ie, lithium, divalproex, carbamazepine, or an antipsychotic) rather than prescribing antidepressant monotherapy. There is greater diversity of opinion about the safety of antidepressant monotherapy for bipolar II depression.
- Consider using antidepressants that have at least 1 positive randomized controlled trial in bipolar disorder and low risk for mood destabilization (bupropion,12,14 sertraline,14 fluoxetine,4,5 tranylcypromine,3,28 or venlafaxine in bipolar II depression20) before using those with reported increased risk for inducing mania or hypomania (TCAs1,2 or venlafaxine in bipolar I depression14), multiple negative controlled trials (paroxetine12,13), or no controlled data in bipolar depression (citalopram, escitalopram, fluvoxamine, mirtazapine, duloxetine, desvenlafaxine, nefazodone, and selegiline transdermal). Combinations of antidepressants have not been adequately studied in bipolar depression.
- The optimal duration of antidepressant therapy is unknown. However, longer-term treatment may be worthwhile in patients who show robust acute antidepressant response and experience infrequent mania or hypomania. Long-term antidepressant use is less compelling in patients with a poor initial response29 or rapid cycling.30 Abrupt antidepressant cessation also may induce mania, potentially by disrupting homeostasis.31 In the absence of rapid cycling, manic/hypomanic features, or worsening suicidal features, and in the presence of an unequivocal acute response and a greater predisposition to depression than mania, it is reasonable to continue an antidepressant indefinitely until new signs of mania or hypomania emerge.
- Emerging signs of mania or hypomania should signal the need to discontinue the antidepressant. Dosage reductions alone may not diminish emerging manic or hypomanic symptoms, and “counterbalancing” maneuvers (ie, adding antimanic agents while continuing an antidepressant) may not effectively stabilize mood.
Step 4: Consider novel strategies. In the absence of a response to the strategy outlined above—particularly among poor candidates for continued antidepressant therapy—other novel strategies have support from at least 1 randomized controlled trial, including pramipexole,32,33 modafinil,34 riluzole,35 and n-acetyl cysteine.36 Other interventions worth considering include:
- adjunctive thyroid hormone
- cognitive therapy
- light therapy (if a seasonal component is evident)
- electroconvulsive therapy.
Related resources
- Goldberg JF. Treating depression in bipolar disorder. http://thedoctorschannel.com/video/3077.html.
- Goldberg JF. Pharmacologic treatment of acute mania.http://thedoctorschannel.com/video/3032.html.
Drug brand names
- Atomoxetine • Strattera
- Bupropion • Wellbutrin
- Carbamazepine • Tegretol, Equetro
- Citalopram • Celexa
- Desipramine • Norpramin
- Desvenlafaxine • Pristiq
- Divalproex • Depakote, Depakene
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Imipramine • Tofranil
- Isocarboxazid • Marplan
- Lamotrigine • Lamictal
- Lithium • Lithobid, Eskalith
- Milnacipran • Ixel, Savella
- Mirtazapine • Remeron
- Moclobemide • Aurorix, Manerix
- Modafinil • Provigil
- Nefazodone • Serzone
- Olanzapine • Zyprexa
- Olanzapine-fluoxetine • Symbyax
- Paroxetine • Paxil
- Phenelzine • Nardil
- Pramipexole • Mirapex
- Quetiapine • Seroquel
- Riluzole • Rilutek
- Selegiline transdermal • EMSAM
- Sertraline • Zoloft
- Tranylcypromine • Parnate
- Venlafaxine • Effexor
Disclosures
Dr. Goldberg is a consultant to Eli Lilly and Company and a speaker for AstraZeneca, Eli Lilly and Company, GlaxoSmithKline, Merck, and Pfizer Inc., and has received speaking honoraria from Janssen-Cilag.
1. Sachs GS, Lafer B, Stoll AL, et al. A double blind trial of bupropion versus desipramine for bipolar depression. J Clin Psychiatry. 1994;55:391-393.
2. Peet M. Induction of mania with selective serotonin re-uptake inhibitors and tricyclic antidepressants. Br J Psychiatry. 1994;164:549-550.
3. Himmelhoch JM, Thase ME, Mallinger AG, et al. Tranylcypromine versus imipramine in anergic bipolar depression. Am J Psychiatry. 1991;148:910-916.
4. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry. 2003;60:1079-1088.
5. Goldberg JF, Truman CJ. Antidepressant-induced mania: an overview of current controversies. Bipolar Disord. 2003;5:407-420.
6. Henry C, Sorbara F, Lacoste J, et al. Antidepressant-induced mania in bipolar patients: identification of risk factors. J Clin Psychiatry. 2001;62:249-255.
7. Tohen M, Frank E, Bowden CL, et al. The International Society for Bipolar Disorders (ISBD) Task Force report on the nomenclature of course and outcome in bipolar disorders. Bipolar Disord. 2009;11:453-473.
8. Ghaemi SN, Rosenquist KJ, Ko JY, et al. Antidepressant treatment in bipolar versus unipolar depression. Am J Psychiatry. 2004;161:163-165.
9. Bottlender R, Rudolf D, Jäger M, et al. Are bipolar I depressive patients less responsive to treatment with antidepressants than unipolar depressive patients? Results from a case control study. Eur Psychiatry. 2002;17:200-205.
10. Möller HJ, Bottlender R, Grunze H, et al. Are antidepressants less effective in the acute treatment of bipolar I compared to unipolar depression? J Affect Disord. 2001;67(1-3):141-146.
11. Amsterdam J. Efficacy and safety of venlafaxine in the treatment of bipolar II major depressive episode. J Clin Psychopharmacol. 1998;18:313-317.
12. Sachs GS, Nierenberg AA, Calabrese JR, et al. Effectiveness of adjunctive antidepressant treatment for bipolar depression. N Engl J Med. 2007;356:1711-1722.
13. Nemeroff CB, Evans DL, Gyulai L, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry. 2001;158:906-912.
14. Leverich GS, Altshuler LL, Frye MA, et al. Risk of switch in mood polarity to hypomania or mania in patients with bipolar depression during acute and continuation trials of venlafaxine, sertraline, and bupropion as adjuncts to mood stabilizers. Am J Psychiatry. 2006;163:232-239.
15. Young LT, Joffe RT, Robb JC, et al. Double-blind comparison of addition of a second mood stabilizer versus an antidepressant to an initial mood stabilizer for treatment of patients with bipolar depression. Am J Psychiatry. 2000;157:124-126.
16. Soutullo CA, McElroy SL, Keck PE, Jr. Hypomania associated with mirtazapine augmentation of sertraline. J Clin Psychiatry. 1998;59(6):320.-
17. Bhanji NH, Margolese HC, Saint-Laurent M, et al. Dysphoric mania induced by high-dose mirtazapine: a case for “norepinephrine syndrome”? Int Clin Psychopharmacol. 2002;17(6):319-322.
18. Goyal N, Sinha VK. Mirtazapine-induced manic switch in adolescent unipolar depression. Aust N Z J Psychiatry. 2008;42(12):1070-1071.
19. Dong J, Blier P. Modification of norepinephrine and serotonin, but not dopamine, neuron firing by sustained bupropion treatment. Psychopharmacol (Berl). 2001;155:52-57.
20. Amsterdam JD, Wang CH, Shwarz M, et al. Venlafaxine versus lithium monotherapy of rapid and non-rapid cycling patients with bipolar II major depressive disorder: a randomized, parallel group, open-label trial. J Affect Disord. 2009;112(1-3):219-230.
21. Dunner DL, D’Souza DN, Kajdasz DK, et al. Is treatment-associated mania rare with duloxetine: secondary analysis of controlled trials in non-bipolar depression. J Affect Disord. 2005;87:115-119.
22. Henderson TA. Mania induction associated with atomoxetine. J Clin Psychopharmacol. 2004;24(5):567-568.
23. Henderson TA, Hartman K. Aggression, mania, and hypomania induction associated with atomoxetine. Pediatrics. 2004;114(3):895-896.
24. Bottlender R, Rudolf D, Strauss A, et al. Mood-stabilisers reduce the risk of developing antidepressant-induced maniform states in acute treatment of bipolar I depressed patients. J Affect Disord. 2001;63:79-83.
25. Wehr TA, Sack DA, Rosenthal NE, et al. Rapid cycling affective disorder: contributing factors and treatment responses in 51 patients. Am J Psychiatry. 1988;145:179-184.
26. Coryell W, Solomon D, Turvey C, et al. The long-term course of rapid-cycling bipolar disorder. Arch Gen Psychiatry. 2003;60:914-920.
27. Schneck CD, Miklowitz DJ, Miyahara S, et al. The prospective course of rapid-cycling bipolar disorder: findings from the STEP-BD. Am J Psychiatry. 2008;165:370-377.
28. Thase ME, Malinger AG, McKnight D, et al. Treatment of imipramine-resistant recurrent depressions, IV: a double-blind crossover study of tranylcypromine for anergic bipolar depression. Am J Psychiatry. 1992;149:195-198.
29. Altshuler LL, Post RM, Hellemann G, et al. Impact of antidepressant continuation after acute positive or partial treatment response for bipolar depression: a blinded, randomized study. J Clin Psychiatry. 2009;70(4):450-457.
30. Ghaemi SN, Ostacher MM, El-Mallakh RS, et al. Antidepressant discontinuation in bipolar depression: a STEP-BD randomized clinical trial of long-term effectiveness and safety. J Clin Psychiatry. In press.
31. Goldstein TR, Frye MA, Denicoff KD, et al. Antidepressant discontinuation-related mania: critical prospective observation and theoretical implications in bipolar disorder. J Clin Psychiatry. 1999;60(8):563-567.
32. Goldberg JF, Burdick KE, Endick CE. A preliminary randomized, double-blind, placebo-controlled trial of pramipexole added to mood stabilizers for treatment-resistant bipolar depression. Am J Psychiatry. 2004;161:564-566.
33. Zarate CA, Jr, Payne JL, Singh J, et al. Pramipexole for bipolar II depression: a placebo-controlled proof of concept study. Biol Psychiatry. 2004;56:54-60.
34. Frye MA, Grunze H, Suppes T, et al. A placebo-controlled evaluation of adjunctive modafinil in the treatment of bipolar depression. Am J Psychiatry. 2007;164(8):1242-1249.
35. Zarate CA, Jr, Quiroz JA, Singh JB, et al. An open-label trial of the glutamate-modulating agent riluzole in combination with lithium for the treatment of bipolar depression. Biol Psychiatry. 2005;57(4):430-432.
36. Berk M, Copolov DL, Dean O, et al. N-acetyl cysteine for depressive symptoms in bipolar disorder—a double-blind, randomized placebo-controlled trial. Biol Psychiatry. 2008;64(6):468-475.
1. Sachs GS, Lafer B, Stoll AL, et al. A double blind trial of bupropion versus desipramine for bipolar depression. J Clin Psychiatry. 1994;55:391-393.
2. Peet M. Induction of mania with selective serotonin re-uptake inhibitors and tricyclic antidepressants. Br J Psychiatry. 1994;164:549-550.
3. Himmelhoch JM, Thase ME, Mallinger AG, et al. Tranylcypromine versus imipramine in anergic bipolar depression. Am J Psychiatry. 1991;148:910-916.
4. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry. 2003;60:1079-1088.
5. Goldberg JF, Truman CJ. Antidepressant-induced mania: an overview of current controversies. Bipolar Disord. 2003;5:407-420.
6. Henry C, Sorbara F, Lacoste J, et al. Antidepressant-induced mania in bipolar patients: identification of risk factors. J Clin Psychiatry. 2001;62:249-255.
7. Tohen M, Frank E, Bowden CL, et al. The International Society for Bipolar Disorders (ISBD) Task Force report on the nomenclature of course and outcome in bipolar disorders. Bipolar Disord. 2009;11:453-473.
8. Ghaemi SN, Rosenquist KJ, Ko JY, et al. Antidepressant treatment in bipolar versus unipolar depression. Am J Psychiatry. 2004;161:163-165.
9. Bottlender R, Rudolf D, Jäger M, et al. Are bipolar I depressive patients less responsive to treatment with antidepressants than unipolar depressive patients? Results from a case control study. Eur Psychiatry. 2002;17:200-205.
10. Möller HJ, Bottlender R, Grunze H, et al. Are antidepressants less effective in the acute treatment of bipolar I compared to unipolar depression? J Affect Disord. 2001;67(1-3):141-146.
11. Amsterdam J. Efficacy and safety of venlafaxine in the treatment of bipolar II major depressive episode. J Clin Psychopharmacol. 1998;18:313-317.
12. Sachs GS, Nierenberg AA, Calabrese JR, et al. Effectiveness of adjunctive antidepressant treatment for bipolar depression. N Engl J Med. 2007;356:1711-1722.
13. Nemeroff CB, Evans DL, Gyulai L, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry. 2001;158:906-912.
14. Leverich GS, Altshuler LL, Frye MA, et al. Risk of switch in mood polarity to hypomania or mania in patients with bipolar depression during acute and continuation trials of venlafaxine, sertraline, and bupropion as adjuncts to mood stabilizers. Am J Psychiatry. 2006;163:232-239.
15. Young LT, Joffe RT, Robb JC, et al. Double-blind comparison of addition of a second mood stabilizer versus an antidepressant to an initial mood stabilizer for treatment of patients with bipolar depression. Am J Psychiatry. 2000;157:124-126.
16. Soutullo CA, McElroy SL, Keck PE, Jr. Hypomania associated with mirtazapine augmentation of sertraline. J Clin Psychiatry. 1998;59(6):320.-
17. Bhanji NH, Margolese HC, Saint-Laurent M, et al. Dysphoric mania induced by high-dose mirtazapine: a case for “norepinephrine syndrome”? Int Clin Psychopharmacol. 2002;17(6):319-322.
18. Goyal N, Sinha VK. Mirtazapine-induced manic switch in adolescent unipolar depression. Aust N Z J Psychiatry. 2008;42(12):1070-1071.
19. Dong J, Blier P. Modification of norepinephrine and serotonin, but not dopamine, neuron firing by sustained bupropion treatment. Psychopharmacol (Berl). 2001;155:52-57.
20. Amsterdam JD, Wang CH, Shwarz M, et al. Venlafaxine versus lithium monotherapy of rapid and non-rapid cycling patients with bipolar II major depressive disorder: a randomized, parallel group, open-label trial. J Affect Disord. 2009;112(1-3):219-230.
21. Dunner DL, D’Souza DN, Kajdasz DK, et al. Is treatment-associated mania rare with duloxetine: secondary analysis of controlled trials in non-bipolar depression. J Affect Disord. 2005;87:115-119.
22. Henderson TA. Mania induction associated with atomoxetine. J Clin Psychopharmacol. 2004;24(5):567-568.
23. Henderson TA, Hartman K. Aggression, mania, and hypomania induction associated with atomoxetine. Pediatrics. 2004;114(3):895-896.
24. Bottlender R, Rudolf D, Strauss A, et al. Mood-stabilisers reduce the risk of developing antidepressant-induced maniform states in acute treatment of bipolar I depressed patients. J Affect Disord. 2001;63:79-83.
25. Wehr TA, Sack DA, Rosenthal NE, et al. Rapid cycling affective disorder: contributing factors and treatment responses in 51 patients. Am J Psychiatry. 1988;145:179-184.
26. Coryell W, Solomon D, Turvey C, et al. The long-term course of rapid-cycling bipolar disorder. Arch Gen Psychiatry. 2003;60:914-920.
27. Schneck CD, Miklowitz DJ, Miyahara S, et al. The prospective course of rapid-cycling bipolar disorder: findings from the STEP-BD. Am J Psychiatry. 2008;165:370-377.
28. Thase ME, Malinger AG, McKnight D, et al. Treatment of imipramine-resistant recurrent depressions, IV: a double-blind crossover study of tranylcypromine for anergic bipolar depression. Am J Psychiatry. 1992;149:195-198.
29. Altshuler LL, Post RM, Hellemann G, et al. Impact of antidepressant continuation after acute positive or partial treatment response for bipolar depression: a blinded, randomized study. J Clin Psychiatry. 2009;70(4):450-457.
30. Ghaemi SN, Ostacher MM, El-Mallakh RS, et al. Antidepressant discontinuation in bipolar depression: a STEP-BD randomized clinical trial of long-term effectiveness and safety. J Clin Psychiatry. In press.
31. Goldstein TR, Frye MA, Denicoff KD, et al. Antidepressant discontinuation-related mania: critical prospective observation and theoretical implications in bipolar disorder. J Clin Psychiatry. 1999;60(8):563-567.
32. Goldberg JF, Burdick KE, Endick CE. A preliminary randomized, double-blind, placebo-controlled trial of pramipexole added to mood stabilizers for treatment-resistant bipolar depression. Am J Psychiatry. 2004;161:564-566.
33. Zarate CA, Jr, Payne JL, Singh J, et al. Pramipexole for bipolar II depression: a placebo-controlled proof of concept study. Biol Psychiatry. 2004;56:54-60.
34. Frye MA, Grunze H, Suppes T, et al. A placebo-controlled evaluation of adjunctive modafinil in the treatment of bipolar depression. Am J Psychiatry. 2007;164(8):1242-1249.
35. Zarate CA, Jr, Quiroz JA, Singh JB, et al. An open-label trial of the glutamate-modulating agent riluzole in combination with lithium for the treatment of bipolar depression. Biol Psychiatry. 2005;57(4):430-432.
36. Berk M, Copolov DL, Dean O, et al. N-acetyl cysteine for depressive symptoms in bipolar disorder—a double-blind, randomized placebo-controlled trial. Biol Psychiatry. 2008;64(6):468-475.
How to treat PTSD in patients with comorbid mood disorders
Major depressive disorder (MDD) and bipolar spectrum disorders are associated with some symptoms of—and fully defined—posttraumatic stress disorder (PTSD). Many traumatic experiences can lead to this comorbidity, the most common being exposure to or witnessing combat for men and rape and sexual molestation for women.1
Trauma has major prognostic and treatment implications for affectively ill patients, including those whose symptoms do not meet PTSD’s full diagnostic criteria. This article aims to help clinicians by:
- presenting evidence characterizing the overlap between affective disorders and PTSD
- reviewing evidence that the bipolar spectrum may be broader than generally thought, an insight that affects PTSD treatment
- making a case for routine PTSD screening for all patients with affective illnesses
- recommending PTSD treatments tailored to the patient’s comorbid affective disorder.
Overlap of trauma and affective illness
PTSD is remarkably comorbid with mood disorders. Americans with MDD and bipolar disorder (BPD) are 7 and 9.4 times, respectively, more likely to meet criteria for PTSD than persons in the general population, according to odds ratios Kessler et al2 calculated from the National Comorbidity Survey database.
I have never seen a patient with PTSD who did not also meet criteria for an affective disorder. The concurrence of PTSD and MDD is not the product of overlapping diagnostic criteria. Rather, evidence indicates these are distinct diagnostic entities.3 A review of diagnostic criteria for PTSD and hypomania/mania leads to the same conclusion.
Bipolar spectrum disorders
DSM-IV-TR assumes that mood disorders fall neatly into boxes. Other data (Table 1)4–8 indicate that these disorders fall along a continuum or—more conservatively—that the scope of bipolarity is much wider than DSM-IV-TR recognizes. This is a controversial topic, and the individual clinician’s position could impact how one manages PTSD patients.
Table 1
Evidence of bipolar spectrum features in major depressive episodes
| Study | Design | Conclusion |
|---|---|---|
| Akiskal and Mallya, 19874 | 200 community mental health clinic patients diagnosed as having MDD | 50% could be classified as having a bipolar disorder |
| Benazzi, 19975 | 203 consecutively presenting patients with depression | 45% met criteria for bipolar II disorder |
| Akiskal and Benazzi, 20056 | 563 consecutive patients presenting with a DSM-IV-diagnosed MDE | 58% showed features of bipolar II disorder |
| Akiskal et al, 20067 | 493 patients in a French national study presenting with MDE | 65% were determined to fall along the ‘bipolar spectrum’ |
| Rabakowski et al, 20058 | 880 Polish outpatients presenting with MDE | 40% met criteria for bipolar disorder |
| MDD: major depressive disorder; MDE: major depressive episode | ||
In this article, I include bipolar I disorder, bipolar II disorder, and mixed depression within the “bipolar spectrum disorders.” If one accepts this—and I do—it follows that 50% to 70% of all major depressive episodes (MDEs) are bipolar in nature.4–9 Depending on your practice setting, you may see a higher or lower base rate of bipolar spectrum disorders.
Mixed depression is not recognized in DSM-IV-TR, and the purpose of this article is not to defend its inclusion as a bipolar spectrum phenomenon. A proposed definition of mixed depression9 requires the presence of an MDE contaminated by ≥3 features of hypomania or mania, without euphoria or inflated self-esteem/grandiosity (Table 2).10
Some experts believe episodes of hypomania and mania frequently occur in the illness course of persons with mixed depression; indeed, mixed depression is a predictor of a bipolar course. It is observed in outpatient9 and inpatient settings.11 Common forms of mixed depression feature combinations of irritability, psychomotor agitation (mild to severe), increased talkativeness (which may fall short of frank pressured speech), racing or “crowded” thoughts (or “mental overactivity”), and distractibility. Other than increased self-esteem/grandiosity, any symptoms within DSM-IV-TR criterion B for a hypomanic or manic episode may be seen in mixed depression. Psychosis is an exclusion criterion for mixed depression.
Mixed depression responds poorly to antidepressant monotherapy. Validation studies suggest that mixed depression is a bipolar variant, as determined by its capacity to predict a bipolar course and its association with a family history of bipolar disorder and age of onset.9
Table 2
Diagnostic characteristics of a hypomanic episode, DSM-IV-TR criteria A and B
| A. A distinct period of persistently elevated, expansive, or irritable mood, lasting throughout at least 4 days, that is clearly different from the usual nondepressed mood. |
| B. During the period of mood disturbance, 3 or more of the following symptoms have persisted (4 if the mood is only irritable) and have been present to a significant degree: 1) inflated self-esteem or grandiosity 2) decreased need for sleep (eg, feels rested after only 3 hours of sleep) 3) more talkative than usual or pressure to keep talking 4) flight of ideas or subjective experience that thoughts are racing 5) distractibility (ie, attention too easily drawn to unimportant or irrelevant external stimuli) 6) increase in goal-directed activity (either socially, at work or school, or sexually) or psychomotor agitation 7) excessive involvement in pleasurable activities that have a high potential for painful consequences (eg, the person engages in unrestrained buying sprees, sexual indiscretions, or foolish business investments). |
| Source: Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000 |
PTSD risk in affective illness
An adolescent sample. A preliminary cross-sectional study conducted by our group indicates that adolescents with affective disorders may have a much higher risk of developing PTSD than psychiatric comparison subjects.12 We used modules from the Structured Clinical Interview for DSM-IV (SCID) to screen for intra-episode psychopathology (as opposed to lifetime prevalence of disorders) in 79 adolescents with MDD, 34 with BPD as defined in the DSM-IV-TR, and 26 with neither affective disorder (psychiatric controls). We found:
- 38.2% of subjects with BPD met criteria for PTSD, compared with 13.9% of those with MDD (OR 4.9; P =.001)
- 3.8% of adolescents without a mood disorder met criteria for PTSD.
We also found that comorbid PTSD was associated with a 4.5-fold higher risk of a suicide attempt, even after we controlled for BPD diagnosis. When we controlled for the presence of other concurrent anxiety disorders, the likelihood of an adolescent with PTSD having attempted suicide remained significant (OR 3.4; P=.023). This finding suggests that PTSD is an independent risk factor for a suicide attempt.
An adult sample. We then focused on adults meeting criteria for MDD or BPD. In a study of 187 consecutively presenting affectively ill patients, we used the SCID to screen for multiple anxiety disorders including PTSD.13 Lifetime—as opposed to intra-episode—PTSD prevalence was 23.8% among the 118 patients with MDD and 62.3% among the 69 patients with BPD. A patient with BPD was 5 times more likely to have PTSD than a patient with MDD (OR 5.3; P < .0001). The most common cause of trauma leading to PTSD was sexual molestation or rape as a child or adolescent in this predominantly female Latino population.
Populations at risk for PTSD
The prevalence of PTSD in clinical samples varies, depending on the population studied. For instance, women are at much higher risk for developing PTSD than men, even in comparisons where men are exposed to a greater number of traumatic events and analyses control for differences in the prevalence of sexual abuse. The gender difference is greater if the trauma occurs during childhood.14 Essentially all patients in our adolescent and adult studies developed PTSD in response to childhood or adolescent sexual trauma.12,13
A population exposed to a high rate of violent crime would be expected to show a higher PTSD prevalence than one exposed to substantially less violence. The base rate of PTSD also is much higher in affectively ill patients than in the general population.
An analysis by Otto et al15 found a 16% lifetime prevalence of concomitant PTSD in 1,214 persons with BPD (not the manifold forms within the bipolar spectrum). Oquendo et al16 reported a 25.7% lifetime prevalence of PTSD in 230 patients with a history of MDD. Other epidemiologic2 and clinical studies12,13 suggest a considerably higher base rate of PTSD among persons with bipolar disorders than those with MDD.
The method of ascertaining the presence of this disorder may be another variable affecting the reported PTSD prevalence. Persistent avoidance—including “efforts to avoid thoughts, feelings, or conversations associated with the trauma”—is a diagnostic feature of PTSD.10 Researchers and clinicians who do not intentionally screen patients for PTSD are not likely to detect it. Determining the true prevalence of PTSD requires empathic inquiry about exposure to traumatic events.
PTSD screening
Humans are remarkably resilient, and most persons exposed to major trauma are thought not to develop PTSD. However, in my experience, because PTSD appears to be common among persons with affective illness, determining whether such patients have been traumatized is important for prognosis and treatment selection.
To get started, you could create a 1-page form to record traumatic events and identify features of PTSD according to DSM-IV-TR criteria (Checklist).10 PTSD screening without a form can become second nature with practice; an experienced clinician can screen a traumatized patient for the disorder within 3 to 5 minutes.
When screening for a history of trauma, ask patients in a straightforward manner if they have:
- been victims of violent crimes
- witnessed violent crimes
- been exposed to events in which people could have suffered grave injury
- experienced emotional, physical, or sexual abuse.
A person who has experienced emotional abuse but not physical or sexual abuse cannot meet DSM-IV-TR criterion A and therefore does not meet full criteria for PTSD. Many emotionally abused persons meet criteria B through F, however, and they are most reasonably managed similarly to persons who also meet criterion A. When formulating a treatment plan, I recommend using clinical judgment rather than rigid adherence to DSM-IV-TR.
Checklist
DSM-IV-TR diagnostic criteria for posttraumatic stress disorder
| Criterion A. The person has been exposed to a traumatic event in which both of the following have been present: | |
| □ | 1. The person has experienced, witnessed, or been confronted with an event or events that involve actual or threatened death or serious injury, or a threat to the physical integrity of oneself or others |
| □ | 2. The person’s response involved intense fear, helplessness, or horror |
| Criterion B. The traumatic event is persistently re-experienced in at least 1 of the following ways: | |
| □ | 1. Recurrent and intrusive distressing recollections of the event, including images, thoughts, or perceptions |
| □ | 2. Recurrent distressing dreams of the event |
| □ | 3. Acting or feeling as if the traumatic event were recurring (includes a sense of reliving the experience, illusions, hallucinations, and dissociative flashback episodes, including those that occur upon awakening or when intoxicated) |
| □ | 4. Intense psychological distress at exposure to internal or external cues that symbolize or resemble an aspect of the traumatic event |
| □ | 5. Physiologic reactivity upon exposure to internal or external cues that symbolize or resemble an aspect of the traumatic event |
| Criterion C. Persistent avoidance of stimuli associated with the trauma and numbing of general responsiveness (not present before the trauma), as indicated by at least 3 of the following: | |
| □ | 1. Efforts to avoid thoughts, feelings, or conversations associated with the trauma |
| □ | 2. Efforts to avoid activities, places, or people that arouse recollections of the trauma |
| □ | 3. Inability to recall an important aspect of the trauma |
| □ | 4. Markedly diminished interest or participation in significant activities |
| □ | 5. Feeling of detachment or estrangement from others |
| □ | 6. Restricted range of affect |
| □ | 7. Sense of foreshortened future |
| Criterion D. Persistent symptoms of increasing arousal (not present before the trauma), indicated by at least 2 of the following: | |
| □ | 1. Difficulty falling or staying asleep |
| □ | 2. Irritability or outbursts of anger |
| □ | 3. Difficulty concentrating |
| □ | 4. Hypervigilance |
| □ | 5. Exaggerated startle response |
| □ | Criterion E. Duration of disturbance (symptoms in B, C, and D) is >1 month |
| □ | Criterion F. Disturbance causes clinically significant distress or impairment in social, occupational, or other important areas of functioning |
| Source: Adapted from Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000 | |
Treating PTSD in depression
Pharmacotherapy and psychotherapeutic interventions are important to PTSD patients’ recovery. Limited resources often prevent these patients from receiving expert psychotherapeutic intervention, however, leaving pharmacotherapy as the mainstay of treatment. This is unfortunate, because psychological interventions may be sufficient and preferred in some instances (Box).17–20
Pharmacotherapy for comorbid MDD. Selective serotonin reuptake inhibitors (SSRIs) and venlafaxine are first-line interventions for PTSD in depressed patients who do not meet criteria for a bipolar spectrum disorder. Placebo-controlled studies suggest that sertraline,21,22 fluoxetine,23 and paroxetine,24 are effective; doses higher than those used to treat depression may be required. Extended-release venlafaxine25 in dosages similar to those needed for depressive disorders also can be effective. Bupropion does not appear to be beneficial in treating PTSD.
The monoamine oxidase inhibitor phenelzine was long used successfully in treating PTSD but for the most part has been replaced by SSRIs. Because of its associated dietary restrictions, risk of hypertensive crises, and other side effects, phenelzine probably is best reserved for patients who do not respond to treatment with SSRIs or venlafaxine.
Pharmacotherapy for comorbid bipolar spectrum. If one accepts that most patients meeting criteria for MDE have a bipolar spectrum disorder, then most affectively ill patients with PTSD would need to be treated as if they have bipolar disorder. Oddly enough, this creates difficulties for the use of not only antidepressants and benzodiazepines, but also mood stabilizers:
- Patients with BPD and comorbid anxiety disorders, including PTSD, may be resistant to mood stabilizers.26,27
- Antidepressants can precipitate hypomanic or manic switches or onset of mixed hypomania, a mixed state, or rapid cycling in patients with a bipolar spectrum disorder.28–30
- Benzodiazepines do not appear to relieve acute or chronic PTSD-related distress, and discontinuation could cause rebound symptoms.31
Because no outcome studies have addressed PTSD management in patients with bipolar spectrum disorders, clinicians must rely on their judgment when formulating treatment plans. One strategy is to treat patients with mood stabilizers, then leave well enough alone if both the mood and anxiety symptoms remit (which is possible but unlikely in my experience). I often start treatment for the bipolar spectrum disorder and co-existing PTSD using mood stabilizers (including atypical antipsychotics) and prazosin, an α-1antagonist originally used for treating hypertension.
Prazosin can help diminish nightmares, dreams, and other painful recollections of trauma.32,33 The drug does not affect time to sleep onset. It also has been reported to reduce avoidance behavior and hyperarousal, such as irritability and anger.34 This has been my experience.
Cognitive-behavioral therapy (CBT) involving prolonged exposure (PE) to trauma-related stimuli has been shown to be effective for posttraumatic stress disorder (PTSD) in controlled studies.17,18 PE is an individual CBT designed to help patients process traumatic events and reduce psychological distress. It involves education about reactions to trauma, relaxation techniques, imaginal reliving of the trauma, exposure to cues associated with the trauma, and cognitive restructuring.
Administering D-cycloserine before behavioral treatment sessions facilitates fear extinction, and its use to enhance prolonged PE constitutes state-of-the-art treatment.19 Eye movement desensitization and reprocessing also may be effective.18,20
PE is a reasonable first-line treatment for PTSD patients with comorbid bipolar spectrum disorders when PTSD symptoms persist after pharmacologic treatment for the bipolar spectrum disorder. PE also is a first-line treatment for PTSD in patients with comorbid major depressive disorder. Barriers to PE treatment include its cost and finding professionals who are expert in its use.
Prazosin to treat PTSD-related symptoms in children or adolescents has not been studied, but it can be useful in adults over a wide range of doses. As little as 1 mg at bedtime may confer benefit, although the mean prazosin dose in an 8-week, placebo-controlled study of 40 combat veterans was 13.3 mg in the evening.33
I often initiate prazosin treatment as follows:
- 1 mg on the first night of treatment
- 2 mg on the second night
- 3 mg on the third night
- then, if tolerated, 1 mg upon waking, 1 mg 8 hours later, and 3 mg at bedtime. I then slowly adjust the dose schedule based on the patient’s needs, such as minimizing painful re-experiencing of the trauma. Reducing avoidance and hyperarousal also are reasonable targets. For example, when using prazosin to treat extremely angry men with PTSD stemming from exposure to violent crimes, I have observed that even “murderous” rage ceases with prazosin treatment, only to reappear when prazosin is discontinued.
In treating approximately 100 patients with prazosin, I have not exceeded 16 mg/d. Dosages used for treating hypertension usually are 5 to 20 mg/d. When using prazosin, I always:
- warn patients that faintness or fainting is a side effect and record this caveat in their chart
- obtain sitting and standing blood pressure and pulse before starting treatment and subsequently
- ask patients if they feel dizzy when changing posture before and after initiating treatment.
Most of my PTSD patients are suffering so much that they are willing to accept the risk of fainting associated with prazosin use. For PTSD comorbid with severe panic disorder,12,13 I find that a benzodiazepine with antipanic properties such as alprazolam or clonazepam often works well in conjunction with prazosin.
Some patients with bipolar spectrum disorders might benefit from the addition of an SSRI after mood stabilizer treatment proves effective. However, I have never managed a patient in this manner, and like my own treatment strategy, this has not been subjected to rigorous empiric inquiry. In my view, psychological treatment is much preferred to antidepressant therapy.
Related resource
- Benazzi F. Bipolar disorder—focus on bipolar II disorder and mixed depression. Lancet. 2007;369:935-945.
Drug brand names
- Alprazolam • Xanax
- Bupropion • Wellbutrin
- Clonazepam • Klonopin
- D-cycloserine • Seromycin
- Fluoxetine • Prozac
- Paroxetine • Paxil
- Phenelzine • Nardil
- Prazosin • Minipress
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosure
Dr. Dilsaver reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: results from the National Comorbidity-Replication (NCS-R). JAMA. 2003;289:3095-3105.
2. Kessler RC, Sonnega A, Bromet E, et al. Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry. 1995;52:1048-1060.
3. Franklin CL, Zimmerman M. Posttraumatic stress disorder and major depressive disorder: investigating the role of overlapping symptoms in diagnostic comorbidity. J Nerv Ment Dis. 2001;189:548-551.
4. Akiskal HS, Mallya G. Criteria for the “soft” bipolar spectrum: treatment implications. Psychopharmacol Bull. 1987;23:68-73.
5. Benazzi F. Prevalence of bipolar II disorder in outpatient depression: a 203-case study in a private practice. J Affect Disord. 1997;43:163-164.
6. Akiskal HS, Benazzi F. Optimizing the detection of bipolar II in outpatient private practice: toward a systematization of clinical diagnostic wisdom. J Clin Psychiatry. 2005;66:914-921.
7. Akiskal HS, Akiskal KK, Lancrenon S, et al. Validating the soft bipolar spectrum in the French National EPIDEP study: the prominence of BP-II. J Affect Disord. 2006;96:207-213.
8. Rabakowski JK, Suwalska D, Lojko D, et al. Bipolar disorders among Polish psychiatric outpatients treated for major depression. J Affect Disord. 2005;84:141-147.
9. Benazzi F. Bipolar disorder—focus on bipolar II disorder and mixed depression. Lancet. 2007;369:935-945.
10. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
11. Maj M, Pirozzi R, Magliano, et al. Agitated ‘unipolar’ major depression: prevalence, phenomenology, and outcome. J Clin Psychiatry. 2006;67:712-719.
12. Dilsaver SC, Benazzi F, Akiskal HS, et al. Post-traumatic stress disorder among adolescents with bipolar disorder and its relationship to suicidality. Bipolar Disord. 2007;9:649-655.
13. Dilsaver SC, Benazzi F, Akiskal KK, et al. Differential patterns of lifetime multiple anxiety disorder comorbidity between Latino adults with bipolar I and major depressive disorders. Bull Menninger Clinic. 2008;72:130-148.
14. Stein MB, Walker JR, Forde DR. Gender differences in susceptibility to posttraumatic stress disorder. Behav Res Ther. 2000;38:619-628.
15. Otto MW, Perlman CA, Wernicke R, et al. Posttraumatic stress disorder in patients with bipolar disorder: a review of prevalence, correlates, and treatment strategies. Bipolar Disord. 2004;6:470-479.
16. Oquendo M, Brent DA, Birmaher B, et al. Posttraumatic stress disorder comorbid with major depression: factors mediating the association with suicidal behavior. Am J Psychiatry. 2005;162:560-566.
17. Schnurr PP, Friedman MJ, Engel CC, et al. Cognitive behavioral therapy for posttraumatic stress disorder in women: a randomized-controlled trial. JAMA. 2007;297:820-830.
18. Bisson J, Andrew M. Psychological treatment for posttraumatic stress disorder (PTSD). Cochrane Database Syst Rev. 2005;CD003388.-
19. Cukor J, Spitalnick J, Difede J, et al. Emerging treatments for PTSD. Clin Psychol Rev. 2009;29(8):715-726.
20. Hogberg G, Pagani M, Sundin O, et al. Treatment of posttraumatic stress disorder with eye movement desensitization and reprocessing: outcome is stable in 35-month follow-up. Psychiatry Res. 2008;159(1-2):101-108.
21. Brady K, Pearlstein T, Asnis GM, et al. Efficacy and safety of sertraline treatment of posttraumatic stress disorder: a randomized controlled trial. JAMA. 2000;283:1837-1844.
22. Friedman MJ, Marmar CR, Baker DG, et al. Randomized, double-blind comparison of sertraline and placebo for posttraumatic stress disorder in a Department of Veterans Affairs setting. J Clin Psychiatry. 2007;68:711-720.
23. Martenyi F, Brown EB, Zhang H, et al. Fluoxetine versus placebo in posttraumatic stress disorder. J Clin Psychiatry. 2002;63:199-206.
24. Tucker P, Zaninelli R, Yehuda R, et al. Paroxetine in the treatment of chronic posttraumatic stress disorder: results of a placebo-controlled, flexible-dosage trial. J Clin Psychiatry. 2001;62:860-868.
25. Pae CU, Lim HK, Ajwani N, et al. Extended-release formulation of venlafaxine in the treatment of post-traumatic stress disorder. Expert Rev Neurother. 2007;7:603-615.
26. Simon NM, Otto MW, Weiss RD, et al. Pharmacotherapy for bipolar disorder and comorbid conditions: baseline data from the STEP-BD. J Clin Psychopharmacol. 2004;24(5):512-520.
27. Quarantini LC, Miranda-Scippa A, Nery-Fernandes F, et al. The impact of comorbid posttraumatic stress disorder on bipolar patients. Affect Disord. 2009; [Epub ahead of print].
28. Henry C, Sorbara F, Lacoste J, et al. Antidepressant induced mania in bipolar patients: identification and risk factors. J Clin Psychiatry. 2001;62:249-255.
29. Gao K, Kemp DE, Gonocy SJ, et al. Treatment-emergent mania/hypomania during antidepressant monotherapy in patients with rapid cycling bipolar disorder. Bipolar Disord. 2008;10:907-915.
30. Dilsaver SC, Swann AC. Mixed mania: apparent induction by a tricyclic antidepressant in five consecutively treated patients with bipolar depression. Biol Psychiatry. 1995;1:60-62.
31. Braun P, Greenberg D, Dasberg H, et al. Core symptoms of posttraumatic stress disorder unimproved by alprazolam treatment. J Clin Psychiatry. 1990;51:236-238.
32. Taylor FB, Martin P, Thompson C, et al. Prazosin effects on objective sleep measures and clinical symptoms in civilian trauma posttraumatic stress disorder: a placebo-controlled study. Biol Psychiatry. 2008;63:629-632.
33. Raskind MA, Perskind ER, Hoff DJ, et al. A parallel group placebo controlled study of prazosin for trauma nightmares and sleep disturbance in combat veterans with posttraumatic stress disorder. Biol Psychiatry. 2007;61:928-934.
34. Taylor FB, Lowe K, Thompson C, et al. Daytime prazosin reduces psychological distress to trauma specific cues in civilian trauma posttraumatic stress disorder. Biol Psychiatry. 2006;59:577-581.
Major depressive disorder (MDD) and bipolar spectrum disorders are associated with some symptoms of—and fully defined—posttraumatic stress disorder (PTSD). Many traumatic experiences can lead to this comorbidity, the most common being exposure to or witnessing combat for men and rape and sexual molestation for women.1
Trauma has major prognostic and treatment implications for affectively ill patients, including those whose symptoms do not meet PTSD’s full diagnostic criteria. This article aims to help clinicians by:
- presenting evidence characterizing the overlap between affective disorders and PTSD
- reviewing evidence that the bipolar spectrum may be broader than generally thought, an insight that affects PTSD treatment
- making a case for routine PTSD screening for all patients with affective illnesses
- recommending PTSD treatments tailored to the patient’s comorbid affective disorder.
Overlap of trauma and affective illness
PTSD is remarkably comorbid with mood disorders. Americans with MDD and bipolar disorder (BPD) are 7 and 9.4 times, respectively, more likely to meet criteria for PTSD than persons in the general population, according to odds ratios Kessler et al2 calculated from the National Comorbidity Survey database.
I have never seen a patient with PTSD who did not also meet criteria for an affective disorder. The concurrence of PTSD and MDD is not the product of overlapping diagnostic criteria. Rather, evidence indicates these are distinct diagnostic entities.3 A review of diagnostic criteria for PTSD and hypomania/mania leads to the same conclusion.
Bipolar spectrum disorders
DSM-IV-TR assumes that mood disorders fall neatly into boxes. Other data (Table 1)4–8 indicate that these disorders fall along a continuum or—more conservatively—that the scope of bipolarity is much wider than DSM-IV-TR recognizes. This is a controversial topic, and the individual clinician’s position could impact how one manages PTSD patients.
Table 1
Evidence of bipolar spectrum features in major depressive episodes
| Study | Design | Conclusion |
|---|---|---|
| Akiskal and Mallya, 19874 | 200 community mental health clinic patients diagnosed as having MDD | 50% could be classified as having a bipolar disorder |
| Benazzi, 19975 | 203 consecutively presenting patients with depression | 45% met criteria for bipolar II disorder |
| Akiskal and Benazzi, 20056 | 563 consecutive patients presenting with a DSM-IV-diagnosed MDE | 58% showed features of bipolar II disorder |
| Akiskal et al, 20067 | 493 patients in a French national study presenting with MDE | 65% were determined to fall along the ‘bipolar spectrum’ |
| Rabakowski et al, 20058 | 880 Polish outpatients presenting with MDE | 40% met criteria for bipolar disorder |
| MDD: major depressive disorder; MDE: major depressive episode | ||
In this article, I include bipolar I disorder, bipolar II disorder, and mixed depression within the “bipolar spectrum disorders.” If one accepts this—and I do—it follows that 50% to 70% of all major depressive episodes (MDEs) are bipolar in nature.4–9 Depending on your practice setting, you may see a higher or lower base rate of bipolar spectrum disorders.
Mixed depression is not recognized in DSM-IV-TR, and the purpose of this article is not to defend its inclusion as a bipolar spectrum phenomenon. A proposed definition of mixed depression9 requires the presence of an MDE contaminated by ≥3 features of hypomania or mania, without euphoria or inflated self-esteem/grandiosity (Table 2).10
Some experts believe episodes of hypomania and mania frequently occur in the illness course of persons with mixed depression; indeed, mixed depression is a predictor of a bipolar course. It is observed in outpatient9 and inpatient settings.11 Common forms of mixed depression feature combinations of irritability, psychomotor agitation (mild to severe), increased talkativeness (which may fall short of frank pressured speech), racing or “crowded” thoughts (or “mental overactivity”), and distractibility. Other than increased self-esteem/grandiosity, any symptoms within DSM-IV-TR criterion B for a hypomanic or manic episode may be seen in mixed depression. Psychosis is an exclusion criterion for mixed depression.
Mixed depression responds poorly to antidepressant monotherapy. Validation studies suggest that mixed depression is a bipolar variant, as determined by its capacity to predict a bipolar course and its association with a family history of bipolar disorder and age of onset.9
Table 2
Diagnostic characteristics of a hypomanic episode, DSM-IV-TR criteria A and B
| A. A distinct period of persistently elevated, expansive, or irritable mood, lasting throughout at least 4 days, that is clearly different from the usual nondepressed mood. |
| B. During the period of mood disturbance, 3 or more of the following symptoms have persisted (4 if the mood is only irritable) and have been present to a significant degree: 1) inflated self-esteem or grandiosity 2) decreased need for sleep (eg, feels rested after only 3 hours of sleep) 3) more talkative than usual or pressure to keep talking 4) flight of ideas or subjective experience that thoughts are racing 5) distractibility (ie, attention too easily drawn to unimportant or irrelevant external stimuli) 6) increase in goal-directed activity (either socially, at work or school, or sexually) or psychomotor agitation 7) excessive involvement in pleasurable activities that have a high potential for painful consequences (eg, the person engages in unrestrained buying sprees, sexual indiscretions, or foolish business investments). |
| Source: Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000 |
PTSD risk in affective illness
An adolescent sample. A preliminary cross-sectional study conducted by our group indicates that adolescents with affective disorders may have a much higher risk of developing PTSD than psychiatric comparison subjects.12 We used modules from the Structured Clinical Interview for DSM-IV (SCID) to screen for intra-episode psychopathology (as opposed to lifetime prevalence of disorders) in 79 adolescents with MDD, 34 with BPD as defined in the DSM-IV-TR, and 26 with neither affective disorder (psychiatric controls). We found:
- 38.2% of subjects with BPD met criteria for PTSD, compared with 13.9% of those with MDD (OR 4.9; P =.001)
- 3.8% of adolescents without a mood disorder met criteria for PTSD.
We also found that comorbid PTSD was associated with a 4.5-fold higher risk of a suicide attempt, even after we controlled for BPD diagnosis. When we controlled for the presence of other concurrent anxiety disorders, the likelihood of an adolescent with PTSD having attempted suicide remained significant (OR 3.4; P=.023). This finding suggests that PTSD is an independent risk factor for a suicide attempt.
An adult sample. We then focused on adults meeting criteria for MDD or BPD. In a study of 187 consecutively presenting affectively ill patients, we used the SCID to screen for multiple anxiety disorders including PTSD.13 Lifetime—as opposed to intra-episode—PTSD prevalence was 23.8% among the 118 patients with MDD and 62.3% among the 69 patients with BPD. A patient with BPD was 5 times more likely to have PTSD than a patient with MDD (OR 5.3; P < .0001). The most common cause of trauma leading to PTSD was sexual molestation or rape as a child or adolescent in this predominantly female Latino population.
Populations at risk for PTSD
The prevalence of PTSD in clinical samples varies, depending on the population studied. For instance, women are at much higher risk for developing PTSD than men, even in comparisons where men are exposed to a greater number of traumatic events and analyses control for differences in the prevalence of sexual abuse. The gender difference is greater if the trauma occurs during childhood.14 Essentially all patients in our adolescent and adult studies developed PTSD in response to childhood or adolescent sexual trauma.12,13
A population exposed to a high rate of violent crime would be expected to show a higher PTSD prevalence than one exposed to substantially less violence. The base rate of PTSD also is much higher in affectively ill patients than in the general population.
An analysis by Otto et al15 found a 16% lifetime prevalence of concomitant PTSD in 1,214 persons with BPD (not the manifold forms within the bipolar spectrum). Oquendo et al16 reported a 25.7% lifetime prevalence of PTSD in 230 patients with a history of MDD. Other epidemiologic2 and clinical studies12,13 suggest a considerably higher base rate of PTSD among persons with bipolar disorders than those with MDD.
The method of ascertaining the presence of this disorder may be another variable affecting the reported PTSD prevalence. Persistent avoidance—including “efforts to avoid thoughts, feelings, or conversations associated with the trauma”—is a diagnostic feature of PTSD.10 Researchers and clinicians who do not intentionally screen patients for PTSD are not likely to detect it. Determining the true prevalence of PTSD requires empathic inquiry about exposure to traumatic events.
PTSD screening
Humans are remarkably resilient, and most persons exposed to major trauma are thought not to develop PTSD. However, in my experience, because PTSD appears to be common among persons with affective illness, determining whether such patients have been traumatized is important for prognosis and treatment selection.
To get started, you could create a 1-page form to record traumatic events and identify features of PTSD according to DSM-IV-TR criteria (Checklist).10 PTSD screening without a form can become second nature with practice; an experienced clinician can screen a traumatized patient for the disorder within 3 to 5 minutes.
When screening for a history of trauma, ask patients in a straightforward manner if they have:
- been victims of violent crimes
- witnessed violent crimes
- been exposed to events in which people could have suffered grave injury
- experienced emotional, physical, or sexual abuse.
A person who has experienced emotional abuse but not physical or sexual abuse cannot meet DSM-IV-TR criterion A and therefore does not meet full criteria for PTSD. Many emotionally abused persons meet criteria B through F, however, and they are most reasonably managed similarly to persons who also meet criterion A. When formulating a treatment plan, I recommend using clinical judgment rather than rigid adherence to DSM-IV-TR.
Checklist
DSM-IV-TR diagnostic criteria for posttraumatic stress disorder
| Criterion A. The person has been exposed to a traumatic event in which both of the following have been present: | |
| □ | 1. The person has experienced, witnessed, or been confronted with an event or events that involve actual or threatened death or serious injury, or a threat to the physical integrity of oneself or others |
| □ | 2. The person’s response involved intense fear, helplessness, or horror |
| Criterion B. The traumatic event is persistently re-experienced in at least 1 of the following ways: | |
| □ | 1. Recurrent and intrusive distressing recollections of the event, including images, thoughts, or perceptions |
| □ | 2. Recurrent distressing dreams of the event |
| □ | 3. Acting or feeling as if the traumatic event were recurring (includes a sense of reliving the experience, illusions, hallucinations, and dissociative flashback episodes, including those that occur upon awakening or when intoxicated) |
| □ | 4. Intense psychological distress at exposure to internal or external cues that symbolize or resemble an aspect of the traumatic event |
| □ | 5. Physiologic reactivity upon exposure to internal or external cues that symbolize or resemble an aspect of the traumatic event |
| Criterion C. Persistent avoidance of stimuli associated with the trauma and numbing of general responsiveness (not present before the trauma), as indicated by at least 3 of the following: | |
| □ | 1. Efforts to avoid thoughts, feelings, or conversations associated with the trauma |
| □ | 2. Efforts to avoid activities, places, or people that arouse recollections of the trauma |
| □ | 3. Inability to recall an important aspect of the trauma |
| □ | 4. Markedly diminished interest or participation in significant activities |
| □ | 5. Feeling of detachment or estrangement from others |
| □ | 6. Restricted range of affect |
| □ | 7. Sense of foreshortened future |
| Criterion D. Persistent symptoms of increasing arousal (not present before the trauma), indicated by at least 2 of the following: | |
| □ | 1. Difficulty falling or staying asleep |
| □ | 2. Irritability or outbursts of anger |
| □ | 3. Difficulty concentrating |
| □ | 4. Hypervigilance |
| □ | 5. Exaggerated startle response |
| □ | Criterion E. Duration of disturbance (symptoms in B, C, and D) is >1 month |
| □ | Criterion F. Disturbance causes clinically significant distress or impairment in social, occupational, or other important areas of functioning |
| Source: Adapted from Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000 | |
Treating PTSD in depression
Pharmacotherapy and psychotherapeutic interventions are important to PTSD patients’ recovery. Limited resources often prevent these patients from receiving expert psychotherapeutic intervention, however, leaving pharmacotherapy as the mainstay of treatment. This is unfortunate, because psychological interventions may be sufficient and preferred in some instances (Box).17–20
Pharmacotherapy for comorbid MDD. Selective serotonin reuptake inhibitors (SSRIs) and venlafaxine are first-line interventions for PTSD in depressed patients who do not meet criteria for a bipolar spectrum disorder. Placebo-controlled studies suggest that sertraline,21,22 fluoxetine,23 and paroxetine,24 are effective; doses higher than those used to treat depression may be required. Extended-release venlafaxine25 in dosages similar to those needed for depressive disorders also can be effective. Bupropion does not appear to be beneficial in treating PTSD.
The monoamine oxidase inhibitor phenelzine was long used successfully in treating PTSD but for the most part has been replaced by SSRIs. Because of its associated dietary restrictions, risk of hypertensive crises, and other side effects, phenelzine probably is best reserved for patients who do not respond to treatment with SSRIs or venlafaxine.
Pharmacotherapy for comorbid bipolar spectrum. If one accepts that most patients meeting criteria for MDE have a bipolar spectrum disorder, then most affectively ill patients with PTSD would need to be treated as if they have bipolar disorder. Oddly enough, this creates difficulties for the use of not only antidepressants and benzodiazepines, but also mood stabilizers:
- Patients with BPD and comorbid anxiety disorders, including PTSD, may be resistant to mood stabilizers.26,27
- Antidepressants can precipitate hypomanic or manic switches or onset of mixed hypomania, a mixed state, or rapid cycling in patients with a bipolar spectrum disorder.28–30
- Benzodiazepines do not appear to relieve acute or chronic PTSD-related distress, and discontinuation could cause rebound symptoms.31
Because no outcome studies have addressed PTSD management in patients with bipolar spectrum disorders, clinicians must rely on their judgment when formulating treatment plans. One strategy is to treat patients with mood stabilizers, then leave well enough alone if both the mood and anxiety symptoms remit (which is possible but unlikely in my experience). I often start treatment for the bipolar spectrum disorder and co-existing PTSD using mood stabilizers (including atypical antipsychotics) and prazosin, an α-1antagonist originally used for treating hypertension.
Prazosin can help diminish nightmares, dreams, and other painful recollections of trauma.32,33 The drug does not affect time to sleep onset. It also has been reported to reduce avoidance behavior and hyperarousal, such as irritability and anger.34 This has been my experience.
Cognitive-behavioral therapy (CBT) involving prolonged exposure (PE) to trauma-related stimuli has been shown to be effective for posttraumatic stress disorder (PTSD) in controlled studies.17,18 PE is an individual CBT designed to help patients process traumatic events and reduce psychological distress. It involves education about reactions to trauma, relaxation techniques, imaginal reliving of the trauma, exposure to cues associated with the trauma, and cognitive restructuring.
Administering D-cycloserine before behavioral treatment sessions facilitates fear extinction, and its use to enhance prolonged PE constitutes state-of-the-art treatment.19 Eye movement desensitization and reprocessing also may be effective.18,20
PE is a reasonable first-line treatment for PTSD patients with comorbid bipolar spectrum disorders when PTSD symptoms persist after pharmacologic treatment for the bipolar spectrum disorder. PE also is a first-line treatment for PTSD in patients with comorbid major depressive disorder. Barriers to PE treatment include its cost and finding professionals who are expert in its use.
Prazosin to treat PTSD-related symptoms in children or adolescents has not been studied, but it can be useful in adults over a wide range of doses. As little as 1 mg at bedtime may confer benefit, although the mean prazosin dose in an 8-week, placebo-controlled study of 40 combat veterans was 13.3 mg in the evening.33
I often initiate prazosin treatment as follows:
- 1 mg on the first night of treatment
- 2 mg on the second night
- 3 mg on the third night
- then, if tolerated, 1 mg upon waking, 1 mg 8 hours later, and 3 mg at bedtime. I then slowly adjust the dose schedule based on the patient’s needs, such as minimizing painful re-experiencing of the trauma. Reducing avoidance and hyperarousal also are reasonable targets. For example, when using prazosin to treat extremely angry men with PTSD stemming from exposure to violent crimes, I have observed that even “murderous” rage ceases with prazosin treatment, only to reappear when prazosin is discontinued.
In treating approximately 100 patients with prazosin, I have not exceeded 16 mg/d. Dosages used for treating hypertension usually are 5 to 20 mg/d. When using prazosin, I always:
- warn patients that faintness or fainting is a side effect and record this caveat in their chart
- obtain sitting and standing blood pressure and pulse before starting treatment and subsequently
- ask patients if they feel dizzy when changing posture before and after initiating treatment.
Most of my PTSD patients are suffering so much that they are willing to accept the risk of fainting associated with prazosin use. For PTSD comorbid with severe panic disorder,12,13 I find that a benzodiazepine with antipanic properties such as alprazolam or clonazepam often works well in conjunction with prazosin.
Some patients with bipolar spectrum disorders might benefit from the addition of an SSRI after mood stabilizer treatment proves effective. However, I have never managed a patient in this manner, and like my own treatment strategy, this has not been subjected to rigorous empiric inquiry. In my view, psychological treatment is much preferred to antidepressant therapy.
Related resource
- Benazzi F. Bipolar disorder—focus on bipolar II disorder and mixed depression. Lancet. 2007;369:935-945.
Drug brand names
- Alprazolam • Xanax
- Bupropion • Wellbutrin
- Clonazepam • Klonopin
- D-cycloserine • Seromycin
- Fluoxetine • Prozac
- Paroxetine • Paxil
- Phenelzine • Nardil
- Prazosin • Minipress
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosure
Dr. Dilsaver reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Major depressive disorder (MDD) and bipolar spectrum disorders are associated with some symptoms of—and fully defined—posttraumatic stress disorder (PTSD). Many traumatic experiences can lead to this comorbidity, the most common being exposure to or witnessing combat for men and rape and sexual molestation for women.1
Trauma has major prognostic and treatment implications for affectively ill patients, including those whose symptoms do not meet PTSD’s full diagnostic criteria. This article aims to help clinicians by:
- presenting evidence characterizing the overlap between affective disorders and PTSD
- reviewing evidence that the bipolar spectrum may be broader than generally thought, an insight that affects PTSD treatment
- making a case for routine PTSD screening for all patients with affective illnesses
- recommending PTSD treatments tailored to the patient’s comorbid affective disorder.
Overlap of trauma and affective illness
PTSD is remarkably comorbid with mood disorders. Americans with MDD and bipolar disorder (BPD) are 7 and 9.4 times, respectively, more likely to meet criteria for PTSD than persons in the general population, according to odds ratios Kessler et al2 calculated from the National Comorbidity Survey database.
I have never seen a patient with PTSD who did not also meet criteria for an affective disorder. The concurrence of PTSD and MDD is not the product of overlapping diagnostic criteria. Rather, evidence indicates these are distinct diagnostic entities.3 A review of diagnostic criteria for PTSD and hypomania/mania leads to the same conclusion.
Bipolar spectrum disorders
DSM-IV-TR assumes that mood disorders fall neatly into boxes. Other data (Table 1)4–8 indicate that these disorders fall along a continuum or—more conservatively—that the scope of bipolarity is much wider than DSM-IV-TR recognizes. This is a controversial topic, and the individual clinician’s position could impact how one manages PTSD patients.
Table 1
Evidence of bipolar spectrum features in major depressive episodes
| Study | Design | Conclusion |
|---|---|---|
| Akiskal and Mallya, 19874 | 200 community mental health clinic patients diagnosed as having MDD | 50% could be classified as having a bipolar disorder |
| Benazzi, 19975 | 203 consecutively presenting patients with depression | 45% met criteria for bipolar II disorder |
| Akiskal and Benazzi, 20056 | 563 consecutive patients presenting with a DSM-IV-diagnosed MDE | 58% showed features of bipolar II disorder |
| Akiskal et al, 20067 | 493 patients in a French national study presenting with MDE | 65% were determined to fall along the ‘bipolar spectrum’ |
| Rabakowski et al, 20058 | 880 Polish outpatients presenting with MDE | 40% met criteria for bipolar disorder |
| MDD: major depressive disorder; MDE: major depressive episode | ||
In this article, I include bipolar I disorder, bipolar II disorder, and mixed depression within the “bipolar spectrum disorders.” If one accepts this—and I do—it follows that 50% to 70% of all major depressive episodes (MDEs) are bipolar in nature.4–9 Depending on your practice setting, you may see a higher or lower base rate of bipolar spectrum disorders.
Mixed depression is not recognized in DSM-IV-TR, and the purpose of this article is not to defend its inclusion as a bipolar spectrum phenomenon. A proposed definition of mixed depression9 requires the presence of an MDE contaminated by ≥3 features of hypomania or mania, without euphoria or inflated self-esteem/grandiosity (Table 2).10
Some experts believe episodes of hypomania and mania frequently occur in the illness course of persons with mixed depression; indeed, mixed depression is a predictor of a bipolar course. It is observed in outpatient9 and inpatient settings.11 Common forms of mixed depression feature combinations of irritability, psychomotor agitation (mild to severe), increased talkativeness (which may fall short of frank pressured speech), racing or “crowded” thoughts (or “mental overactivity”), and distractibility. Other than increased self-esteem/grandiosity, any symptoms within DSM-IV-TR criterion B for a hypomanic or manic episode may be seen in mixed depression. Psychosis is an exclusion criterion for mixed depression.
Mixed depression responds poorly to antidepressant monotherapy. Validation studies suggest that mixed depression is a bipolar variant, as determined by its capacity to predict a bipolar course and its association with a family history of bipolar disorder and age of onset.9
Table 2
Diagnostic characteristics of a hypomanic episode, DSM-IV-TR criteria A and B
| A. A distinct period of persistently elevated, expansive, or irritable mood, lasting throughout at least 4 days, that is clearly different from the usual nondepressed mood. |
| B. During the period of mood disturbance, 3 or more of the following symptoms have persisted (4 if the mood is only irritable) and have been present to a significant degree: 1) inflated self-esteem or grandiosity 2) decreased need for sleep (eg, feels rested after only 3 hours of sleep) 3) more talkative than usual or pressure to keep talking 4) flight of ideas or subjective experience that thoughts are racing 5) distractibility (ie, attention too easily drawn to unimportant or irrelevant external stimuli) 6) increase in goal-directed activity (either socially, at work or school, or sexually) or psychomotor agitation 7) excessive involvement in pleasurable activities that have a high potential for painful consequences (eg, the person engages in unrestrained buying sprees, sexual indiscretions, or foolish business investments). |
| Source: Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000 |
PTSD risk in affective illness
An adolescent sample. A preliminary cross-sectional study conducted by our group indicates that adolescents with affective disorders may have a much higher risk of developing PTSD than psychiatric comparison subjects.12 We used modules from the Structured Clinical Interview for DSM-IV (SCID) to screen for intra-episode psychopathology (as opposed to lifetime prevalence of disorders) in 79 adolescents with MDD, 34 with BPD as defined in the DSM-IV-TR, and 26 with neither affective disorder (psychiatric controls). We found:
- 38.2% of subjects with BPD met criteria for PTSD, compared with 13.9% of those with MDD (OR 4.9; P =.001)
- 3.8% of adolescents without a mood disorder met criteria for PTSD.
We also found that comorbid PTSD was associated with a 4.5-fold higher risk of a suicide attempt, even after we controlled for BPD diagnosis. When we controlled for the presence of other concurrent anxiety disorders, the likelihood of an adolescent with PTSD having attempted suicide remained significant (OR 3.4; P=.023). This finding suggests that PTSD is an independent risk factor for a suicide attempt.
An adult sample. We then focused on adults meeting criteria for MDD or BPD. In a study of 187 consecutively presenting affectively ill patients, we used the SCID to screen for multiple anxiety disorders including PTSD.13 Lifetime—as opposed to intra-episode—PTSD prevalence was 23.8% among the 118 patients with MDD and 62.3% among the 69 patients with BPD. A patient with BPD was 5 times more likely to have PTSD than a patient with MDD (OR 5.3; P < .0001). The most common cause of trauma leading to PTSD was sexual molestation or rape as a child or adolescent in this predominantly female Latino population.
Populations at risk for PTSD
The prevalence of PTSD in clinical samples varies, depending on the population studied. For instance, women are at much higher risk for developing PTSD than men, even in comparisons where men are exposed to a greater number of traumatic events and analyses control for differences in the prevalence of sexual abuse. The gender difference is greater if the trauma occurs during childhood.14 Essentially all patients in our adolescent and adult studies developed PTSD in response to childhood or adolescent sexual trauma.12,13
A population exposed to a high rate of violent crime would be expected to show a higher PTSD prevalence than one exposed to substantially less violence. The base rate of PTSD also is much higher in affectively ill patients than in the general population.
An analysis by Otto et al15 found a 16% lifetime prevalence of concomitant PTSD in 1,214 persons with BPD (not the manifold forms within the bipolar spectrum). Oquendo et al16 reported a 25.7% lifetime prevalence of PTSD in 230 patients with a history of MDD. Other epidemiologic2 and clinical studies12,13 suggest a considerably higher base rate of PTSD among persons with bipolar disorders than those with MDD.
The method of ascertaining the presence of this disorder may be another variable affecting the reported PTSD prevalence. Persistent avoidance—including “efforts to avoid thoughts, feelings, or conversations associated with the trauma”—is a diagnostic feature of PTSD.10 Researchers and clinicians who do not intentionally screen patients for PTSD are not likely to detect it. Determining the true prevalence of PTSD requires empathic inquiry about exposure to traumatic events.
PTSD screening
Humans are remarkably resilient, and most persons exposed to major trauma are thought not to develop PTSD. However, in my experience, because PTSD appears to be common among persons with affective illness, determining whether such patients have been traumatized is important for prognosis and treatment selection.
To get started, you could create a 1-page form to record traumatic events and identify features of PTSD according to DSM-IV-TR criteria (Checklist).10 PTSD screening without a form can become second nature with practice; an experienced clinician can screen a traumatized patient for the disorder within 3 to 5 minutes.
When screening for a history of trauma, ask patients in a straightforward manner if they have:
- been victims of violent crimes
- witnessed violent crimes
- been exposed to events in which people could have suffered grave injury
- experienced emotional, physical, or sexual abuse.
A person who has experienced emotional abuse but not physical or sexual abuse cannot meet DSM-IV-TR criterion A and therefore does not meet full criteria for PTSD. Many emotionally abused persons meet criteria B through F, however, and they are most reasonably managed similarly to persons who also meet criterion A. When formulating a treatment plan, I recommend using clinical judgment rather than rigid adherence to DSM-IV-TR.
Checklist
DSM-IV-TR diagnostic criteria for posttraumatic stress disorder
| Criterion A. The person has been exposed to a traumatic event in which both of the following have been present: | |
| □ | 1. The person has experienced, witnessed, or been confronted with an event or events that involve actual or threatened death or serious injury, or a threat to the physical integrity of oneself or others |
| □ | 2. The person’s response involved intense fear, helplessness, or horror |
| Criterion B. The traumatic event is persistently re-experienced in at least 1 of the following ways: | |
| □ | 1. Recurrent and intrusive distressing recollections of the event, including images, thoughts, or perceptions |
| □ | 2. Recurrent distressing dreams of the event |
| □ | 3. Acting or feeling as if the traumatic event were recurring (includes a sense of reliving the experience, illusions, hallucinations, and dissociative flashback episodes, including those that occur upon awakening or when intoxicated) |
| □ | 4. Intense psychological distress at exposure to internal or external cues that symbolize or resemble an aspect of the traumatic event |
| □ | 5. Physiologic reactivity upon exposure to internal or external cues that symbolize or resemble an aspect of the traumatic event |
| Criterion C. Persistent avoidance of stimuli associated with the trauma and numbing of general responsiveness (not present before the trauma), as indicated by at least 3 of the following: | |
| □ | 1. Efforts to avoid thoughts, feelings, or conversations associated with the trauma |
| □ | 2. Efforts to avoid activities, places, or people that arouse recollections of the trauma |
| □ | 3. Inability to recall an important aspect of the trauma |
| □ | 4. Markedly diminished interest or participation in significant activities |
| □ | 5. Feeling of detachment or estrangement from others |
| □ | 6. Restricted range of affect |
| □ | 7. Sense of foreshortened future |
| Criterion D. Persistent symptoms of increasing arousal (not present before the trauma), indicated by at least 2 of the following: | |
| □ | 1. Difficulty falling or staying asleep |
| □ | 2. Irritability or outbursts of anger |
| □ | 3. Difficulty concentrating |
| □ | 4. Hypervigilance |
| □ | 5. Exaggerated startle response |
| □ | Criterion E. Duration of disturbance (symptoms in B, C, and D) is >1 month |
| □ | Criterion F. Disturbance causes clinically significant distress or impairment in social, occupational, or other important areas of functioning |
| Source: Adapted from Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000 | |
Treating PTSD in depression
Pharmacotherapy and psychotherapeutic interventions are important to PTSD patients’ recovery. Limited resources often prevent these patients from receiving expert psychotherapeutic intervention, however, leaving pharmacotherapy as the mainstay of treatment. This is unfortunate, because psychological interventions may be sufficient and preferred in some instances (Box).17–20
Pharmacotherapy for comorbid MDD. Selective serotonin reuptake inhibitors (SSRIs) and venlafaxine are first-line interventions for PTSD in depressed patients who do not meet criteria for a bipolar spectrum disorder. Placebo-controlled studies suggest that sertraline,21,22 fluoxetine,23 and paroxetine,24 are effective; doses higher than those used to treat depression may be required. Extended-release venlafaxine25 in dosages similar to those needed for depressive disorders also can be effective. Bupropion does not appear to be beneficial in treating PTSD.
The monoamine oxidase inhibitor phenelzine was long used successfully in treating PTSD but for the most part has been replaced by SSRIs. Because of its associated dietary restrictions, risk of hypertensive crises, and other side effects, phenelzine probably is best reserved for patients who do not respond to treatment with SSRIs or venlafaxine.
Pharmacotherapy for comorbid bipolar spectrum. If one accepts that most patients meeting criteria for MDE have a bipolar spectrum disorder, then most affectively ill patients with PTSD would need to be treated as if they have bipolar disorder. Oddly enough, this creates difficulties for the use of not only antidepressants and benzodiazepines, but also mood stabilizers:
- Patients with BPD and comorbid anxiety disorders, including PTSD, may be resistant to mood stabilizers.26,27
- Antidepressants can precipitate hypomanic or manic switches or onset of mixed hypomania, a mixed state, or rapid cycling in patients with a bipolar spectrum disorder.28–30
- Benzodiazepines do not appear to relieve acute or chronic PTSD-related distress, and discontinuation could cause rebound symptoms.31
Because no outcome studies have addressed PTSD management in patients with bipolar spectrum disorders, clinicians must rely on their judgment when formulating treatment plans. One strategy is to treat patients with mood stabilizers, then leave well enough alone if both the mood and anxiety symptoms remit (which is possible but unlikely in my experience). I often start treatment for the bipolar spectrum disorder and co-existing PTSD using mood stabilizers (including atypical antipsychotics) and prazosin, an α-1antagonist originally used for treating hypertension.
Prazosin can help diminish nightmares, dreams, and other painful recollections of trauma.32,33 The drug does not affect time to sleep onset. It also has been reported to reduce avoidance behavior and hyperarousal, such as irritability and anger.34 This has been my experience.
Cognitive-behavioral therapy (CBT) involving prolonged exposure (PE) to trauma-related stimuli has been shown to be effective for posttraumatic stress disorder (PTSD) in controlled studies.17,18 PE is an individual CBT designed to help patients process traumatic events and reduce psychological distress. It involves education about reactions to trauma, relaxation techniques, imaginal reliving of the trauma, exposure to cues associated with the trauma, and cognitive restructuring.
Administering D-cycloserine before behavioral treatment sessions facilitates fear extinction, and its use to enhance prolonged PE constitutes state-of-the-art treatment.19 Eye movement desensitization and reprocessing also may be effective.18,20
PE is a reasonable first-line treatment for PTSD patients with comorbid bipolar spectrum disorders when PTSD symptoms persist after pharmacologic treatment for the bipolar spectrum disorder. PE also is a first-line treatment for PTSD in patients with comorbid major depressive disorder. Barriers to PE treatment include its cost and finding professionals who are expert in its use.
Prazosin to treat PTSD-related symptoms in children or adolescents has not been studied, but it can be useful in adults over a wide range of doses. As little as 1 mg at bedtime may confer benefit, although the mean prazosin dose in an 8-week, placebo-controlled study of 40 combat veterans was 13.3 mg in the evening.33
I often initiate prazosin treatment as follows:
- 1 mg on the first night of treatment
- 2 mg on the second night
- 3 mg on the third night
- then, if tolerated, 1 mg upon waking, 1 mg 8 hours later, and 3 mg at bedtime. I then slowly adjust the dose schedule based on the patient’s needs, such as minimizing painful re-experiencing of the trauma. Reducing avoidance and hyperarousal also are reasonable targets. For example, when using prazosin to treat extremely angry men with PTSD stemming from exposure to violent crimes, I have observed that even “murderous” rage ceases with prazosin treatment, only to reappear when prazosin is discontinued.
In treating approximately 100 patients with prazosin, I have not exceeded 16 mg/d. Dosages used for treating hypertension usually are 5 to 20 mg/d. When using prazosin, I always:
- warn patients that faintness or fainting is a side effect and record this caveat in their chart
- obtain sitting and standing blood pressure and pulse before starting treatment and subsequently
- ask patients if they feel dizzy when changing posture before and after initiating treatment.
Most of my PTSD patients are suffering so much that they are willing to accept the risk of fainting associated with prazosin use. For PTSD comorbid with severe panic disorder,12,13 I find that a benzodiazepine with antipanic properties such as alprazolam or clonazepam often works well in conjunction with prazosin.
Some patients with bipolar spectrum disorders might benefit from the addition of an SSRI after mood stabilizer treatment proves effective. However, I have never managed a patient in this manner, and like my own treatment strategy, this has not been subjected to rigorous empiric inquiry. In my view, psychological treatment is much preferred to antidepressant therapy.
Related resource
- Benazzi F. Bipolar disorder—focus on bipolar II disorder and mixed depression. Lancet. 2007;369:935-945.
Drug brand names
- Alprazolam • Xanax
- Bupropion • Wellbutrin
- Clonazepam • Klonopin
- D-cycloserine • Seromycin
- Fluoxetine • Prozac
- Paroxetine • Paxil
- Phenelzine • Nardil
- Prazosin • Minipress
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosure
Dr. Dilsaver reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: results from the National Comorbidity-Replication (NCS-R). JAMA. 2003;289:3095-3105.
2. Kessler RC, Sonnega A, Bromet E, et al. Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry. 1995;52:1048-1060.
3. Franklin CL, Zimmerman M. Posttraumatic stress disorder and major depressive disorder: investigating the role of overlapping symptoms in diagnostic comorbidity. J Nerv Ment Dis. 2001;189:548-551.
4. Akiskal HS, Mallya G. Criteria for the “soft” bipolar spectrum: treatment implications. Psychopharmacol Bull. 1987;23:68-73.
5. Benazzi F. Prevalence of bipolar II disorder in outpatient depression: a 203-case study in a private practice. J Affect Disord. 1997;43:163-164.
6. Akiskal HS, Benazzi F. Optimizing the detection of bipolar II in outpatient private practice: toward a systematization of clinical diagnostic wisdom. J Clin Psychiatry. 2005;66:914-921.
7. Akiskal HS, Akiskal KK, Lancrenon S, et al. Validating the soft bipolar spectrum in the French National EPIDEP study: the prominence of BP-II. J Affect Disord. 2006;96:207-213.
8. Rabakowski JK, Suwalska D, Lojko D, et al. Bipolar disorders among Polish psychiatric outpatients treated for major depression. J Affect Disord. 2005;84:141-147.
9. Benazzi F. Bipolar disorder—focus on bipolar II disorder and mixed depression. Lancet. 2007;369:935-945.
10. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
11. Maj M, Pirozzi R, Magliano, et al. Agitated ‘unipolar’ major depression: prevalence, phenomenology, and outcome. J Clin Psychiatry. 2006;67:712-719.
12. Dilsaver SC, Benazzi F, Akiskal HS, et al. Post-traumatic stress disorder among adolescents with bipolar disorder and its relationship to suicidality. Bipolar Disord. 2007;9:649-655.
13. Dilsaver SC, Benazzi F, Akiskal KK, et al. Differential patterns of lifetime multiple anxiety disorder comorbidity between Latino adults with bipolar I and major depressive disorders. Bull Menninger Clinic. 2008;72:130-148.
14. Stein MB, Walker JR, Forde DR. Gender differences in susceptibility to posttraumatic stress disorder. Behav Res Ther. 2000;38:619-628.
15. Otto MW, Perlman CA, Wernicke R, et al. Posttraumatic stress disorder in patients with bipolar disorder: a review of prevalence, correlates, and treatment strategies. Bipolar Disord. 2004;6:470-479.
16. Oquendo M, Brent DA, Birmaher B, et al. Posttraumatic stress disorder comorbid with major depression: factors mediating the association with suicidal behavior. Am J Psychiatry. 2005;162:560-566.
17. Schnurr PP, Friedman MJ, Engel CC, et al. Cognitive behavioral therapy for posttraumatic stress disorder in women: a randomized-controlled trial. JAMA. 2007;297:820-830.
18. Bisson J, Andrew M. Psychological treatment for posttraumatic stress disorder (PTSD). Cochrane Database Syst Rev. 2005;CD003388.-
19. Cukor J, Spitalnick J, Difede J, et al. Emerging treatments for PTSD. Clin Psychol Rev. 2009;29(8):715-726.
20. Hogberg G, Pagani M, Sundin O, et al. Treatment of posttraumatic stress disorder with eye movement desensitization and reprocessing: outcome is stable in 35-month follow-up. Psychiatry Res. 2008;159(1-2):101-108.
21. Brady K, Pearlstein T, Asnis GM, et al. Efficacy and safety of sertraline treatment of posttraumatic stress disorder: a randomized controlled trial. JAMA. 2000;283:1837-1844.
22. Friedman MJ, Marmar CR, Baker DG, et al. Randomized, double-blind comparison of sertraline and placebo for posttraumatic stress disorder in a Department of Veterans Affairs setting. J Clin Psychiatry. 2007;68:711-720.
23. Martenyi F, Brown EB, Zhang H, et al. Fluoxetine versus placebo in posttraumatic stress disorder. J Clin Psychiatry. 2002;63:199-206.
24. Tucker P, Zaninelli R, Yehuda R, et al. Paroxetine in the treatment of chronic posttraumatic stress disorder: results of a placebo-controlled, flexible-dosage trial. J Clin Psychiatry. 2001;62:860-868.
25. Pae CU, Lim HK, Ajwani N, et al. Extended-release formulation of venlafaxine in the treatment of post-traumatic stress disorder. Expert Rev Neurother. 2007;7:603-615.
26. Simon NM, Otto MW, Weiss RD, et al. Pharmacotherapy for bipolar disorder and comorbid conditions: baseline data from the STEP-BD. J Clin Psychopharmacol. 2004;24(5):512-520.
27. Quarantini LC, Miranda-Scippa A, Nery-Fernandes F, et al. The impact of comorbid posttraumatic stress disorder on bipolar patients. Affect Disord. 2009; [Epub ahead of print].
28. Henry C, Sorbara F, Lacoste J, et al. Antidepressant induced mania in bipolar patients: identification and risk factors. J Clin Psychiatry. 2001;62:249-255.
29. Gao K, Kemp DE, Gonocy SJ, et al. Treatment-emergent mania/hypomania during antidepressant monotherapy in patients with rapid cycling bipolar disorder. Bipolar Disord. 2008;10:907-915.
30. Dilsaver SC, Swann AC. Mixed mania: apparent induction by a tricyclic antidepressant in five consecutively treated patients with bipolar depression. Biol Psychiatry. 1995;1:60-62.
31. Braun P, Greenberg D, Dasberg H, et al. Core symptoms of posttraumatic stress disorder unimproved by alprazolam treatment. J Clin Psychiatry. 1990;51:236-238.
32. Taylor FB, Martin P, Thompson C, et al. Prazosin effects on objective sleep measures and clinical symptoms in civilian trauma posttraumatic stress disorder: a placebo-controlled study. Biol Psychiatry. 2008;63:629-632.
33. Raskind MA, Perskind ER, Hoff DJ, et al. A parallel group placebo controlled study of prazosin for trauma nightmares and sleep disturbance in combat veterans with posttraumatic stress disorder. Biol Psychiatry. 2007;61:928-934.
34. Taylor FB, Lowe K, Thompson C, et al. Daytime prazosin reduces psychological distress to trauma specific cues in civilian trauma posttraumatic stress disorder. Biol Psychiatry. 2006;59:577-581.
1. Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: results from the National Comorbidity-Replication (NCS-R). JAMA. 2003;289:3095-3105.
2. Kessler RC, Sonnega A, Bromet E, et al. Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry. 1995;52:1048-1060.
3. Franklin CL, Zimmerman M. Posttraumatic stress disorder and major depressive disorder: investigating the role of overlapping symptoms in diagnostic comorbidity. J Nerv Ment Dis. 2001;189:548-551.
4. Akiskal HS, Mallya G. Criteria for the “soft” bipolar spectrum: treatment implications. Psychopharmacol Bull. 1987;23:68-73.
5. Benazzi F. Prevalence of bipolar II disorder in outpatient depression: a 203-case study in a private practice. J Affect Disord. 1997;43:163-164.
6. Akiskal HS, Benazzi F. Optimizing the detection of bipolar II in outpatient private practice: toward a systematization of clinical diagnostic wisdom. J Clin Psychiatry. 2005;66:914-921.
7. Akiskal HS, Akiskal KK, Lancrenon S, et al. Validating the soft bipolar spectrum in the French National EPIDEP study: the prominence of BP-II. J Affect Disord. 2006;96:207-213.
8. Rabakowski JK, Suwalska D, Lojko D, et al. Bipolar disorders among Polish psychiatric outpatients treated for major depression. J Affect Disord. 2005;84:141-147.
9. Benazzi F. Bipolar disorder—focus on bipolar II disorder and mixed depression. Lancet. 2007;369:935-945.
10. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
11. Maj M, Pirozzi R, Magliano, et al. Agitated ‘unipolar’ major depression: prevalence, phenomenology, and outcome. J Clin Psychiatry. 2006;67:712-719.
12. Dilsaver SC, Benazzi F, Akiskal HS, et al. Post-traumatic stress disorder among adolescents with bipolar disorder and its relationship to suicidality. Bipolar Disord. 2007;9:649-655.
13. Dilsaver SC, Benazzi F, Akiskal KK, et al. Differential patterns of lifetime multiple anxiety disorder comorbidity between Latino adults with bipolar I and major depressive disorders. Bull Menninger Clinic. 2008;72:130-148.
14. Stein MB, Walker JR, Forde DR. Gender differences in susceptibility to posttraumatic stress disorder. Behav Res Ther. 2000;38:619-628.
15. Otto MW, Perlman CA, Wernicke R, et al. Posttraumatic stress disorder in patients with bipolar disorder: a review of prevalence, correlates, and treatment strategies. Bipolar Disord. 2004;6:470-479.
16. Oquendo M, Brent DA, Birmaher B, et al. Posttraumatic stress disorder comorbid with major depression: factors mediating the association with suicidal behavior. Am J Psychiatry. 2005;162:560-566.
17. Schnurr PP, Friedman MJ, Engel CC, et al. Cognitive behavioral therapy for posttraumatic stress disorder in women: a randomized-controlled trial. JAMA. 2007;297:820-830.
18. Bisson J, Andrew M. Psychological treatment for posttraumatic stress disorder (PTSD). Cochrane Database Syst Rev. 2005;CD003388.-
19. Cukor J, Spitalnick J, Difede J, et al. Emerging treatments for PTSD. Clin Psychol Rev. 2009;29(8):715-726.
20. Hogberg G, Pagani M, Sundin O, et al. Treatment of posttraumatic stress disorder with eye movement desensitization and reprocessing: outcome is stable in 35-month follow-up. Psychiatry Res. 2008;159(1-2):101-108.
21. Brady K, Pearlstein T, Asnis GM, et al. Efficacy and safety of sertraline treatment of posttraumatic stress disorder: a randomized controlled trial. JAMA. 2000;283:1837-1844.
22. Friedman MJ, Marmar CR, Baker DG, et al. Randomized, double-blind comparison of sertraline and placebo for posttraumatic stress disorder in a Department of Veterans Affairs setting. J Clin Psychiatry. 2007;68:711-720.
23. Martenyi F, Brown EB, Zhang H, et al. Fluoxetine versus placebo in posttraumatic stress disorder. J Clin Psychiatry. 2002;63:199-206.
24. Tucker P, Zaninelli R, Yehuda R, et al. Paroxetine in the treatment of chronic posttraumatic stress disorder: results of a placebo-controlled, flexible-dosage trial. J Clin Psychiatry. 2001;62:860-868.
25. Pae CU, Lim HK, Ajwani N, et al. Extended-release formulation of venlafaxine in the treatment of post-traumatic stress disorder. Expert Rev Neurother. 2007;7:603-615.
26. Simon NM, Otto MW, Weiss RD, et al. Pharmacotherapy for bipolar disorder and comorbid conditions: baseline data from the STEP-BD. J Clin Psychopharmacol. 2004;24(5):512-520.
27. Quarantini LC, Miranda-Scippa A, Nery-Fernandes F, et al. The impact of comorbid posttraumatic stress disorder on bipolar patients. Affect Disord. 2009; [Epub ahead of print].
28. Henry C, Sorbara F, Lacoste J, et al. Antidepressant induced mania in bipolar patients: identification and risk factors. J Clin Psychiatry. 2001;62:249-255.
29. Gao K, Kemp DE, Gonocy SJ, et al. Treatment-emergent mania/hypomania during antidepressant monotherapy in patients with rapid cycling bipolar disorder. Bipolar Disord. 2008;10:907-915.
30. Dilsaver SC, Swann AC. Mixed mania: apparent induction by a tricyclic antidepressant in five consecutively treated patients with bipolar depression. Biol Psychiatry. 1995;1:60-62.
31. Braun P, Greenberg D, Dasberg H, et al. Core symptoms of posttraumatic stress disorder unimproved by alprazolam treatment. J Clin Psychiatry. 1990;51:236-238.
32. Taylor FB, Martin P, Thompson C, et al. Prazosin effects on objective sleep measures and clinical symptoms in civilian trauma posttraumatic stress disorder: a placebo-controlled study. Biol Psychiatry. 2008;63:629-632.
33. Raskind MA, Perskind ER, Hoff DJ, et al. A parallel group placebo controlled study of prazosin for trauma nightmares and sleep disturbance in combat veterans with posttraumatic stress disorder. Biol Psychiatry. 2007;61:928-934.
34. Taylor FB, Lowe K, Thompson C, et al. Daytime prazosin reduces psychological distress to trauma specific cues in civilian trauma posttraumatic stress disorder. Biol Psychiatry. 2006;59:577-581.
Alcohol withdrawal: When to choose an adjunctive anticonvulsant
Benzodiazepines are the mainstay of alcohol detoxification treatment, with extensive evidence supporting their efficacy and relative safety.1 The risk of benzodiazepine-alcohol interaction, however, and psychomotor and cognitive impairments associated with benzodiazepine use may limit early rehabilitation efforts in hospitalized patients.2 Cross-tolerance with alcohol also limits benzodiazepines’ potential benefit in outpatients with substance use disorders.
Adding anticonvulsants to acute benzodiazepine therapy has been shown to decrease alcohol withdrawal symptom severity, reduce seizure risk, and support recovery, particularly in patients with multiple alcohol withdrawal episodes. After detoxification, long-term anticonvulsant use may reduce relapse risk by decreasing post-cessation craving, without abuse liability.3
Although not all studies endorse adding anticonvulsants to benzodiazepines for managing alcohol withdrawal syndrome (AWS),4 we present 3 cases in which anticonvulsants were used successfully as adjuncts to lorazepam. Valproic acid, levetiracetam, and gabapentin offer advantages in acute and long-term therapy of alcohol dependence with efficacy in AWS, low abuse potential, benign safety profile, and mood-stabilizing properties.
Neurobiologic rationale
AWS manifests as a cluster of clinical symptoms including delirium tremens (DTs) and seizures (Table 1). Its pathophysiology can be explained by alcohol’s agonist effect on the gamma-aminobutyric acid (GABA) system and antagonist effect on the glutamatergic system (Table 2).5
Chronic alcohol intake leads to neuroadaptation in the brain in the form of down-regulation of GABAA receptors and upregulation of N-methyl-D-aspartate receptors. During alcohol withdrawal, this neuroadaptation leads to a decrease in central GABA activity and an increase in glutamate activity, resulting in hyperexcitation, anxiety, and seizures.6
Little data exist regarding time to relapse after detoxification in alcohol-dependent patients. One theory—called “protracted withdrawal syndrome” (Table 1)—suggests that abstinent alcoholics return to drinking because of the same, but attenuated, neuroadaptations that trigger acute AWS.7
Advantages of adjunct therapy. Ntais et al8 evaluated benzodiazepines’ effectiveness and safety in treating AWS in a clinical review of 57 randomized, controlled trials totaling 4,051 patients. Benzodiazepines showed similar success rates as other drugs (relative risk [RR] 1.00) or anticonvulsants in particular (RR 0.88), as measured by changes in Clinical Institute Withdrawal Assessment for Alcohol (CIWA-Ar) scores at the end of treatment. Benzodiazepines also offered significant benefit for seizure control compared with nonanticonvulsants (RR 0.23), but less when compared with anti convulsants (RR 1.99).
Although the literature does not support anticonvulsant use for monotherapy in AWS, anticonvulsants show potential as adjunctive therapy. Valproic acid, levetiracetam, and gabapentin offer unique mechanisms of action (Table 3) and demonstrate advantages over benzodiazepine monotherapy for AWS. Adjunctive use of valproic acid,8,9 levetiracetam,10 and gabapentin11,12 in detoxification also has demonstrated efficacy in reducing risk of relapse and delaying relapse.
The neurobiologic rationale for using anticonvulsants in acute AWS is speculative, but these agents appear to:
- inhibit “kindling” (neuronal changes that may be associated with repeated intoxications)
- facilitate GABAergic mechanisms.9
Table 1
Alcohol withdrawal: Acute vs long-term symptoms
| Alcohol withdrawal syndrome | Protracted withdrawal syndrome | |
|---|---|---|
| Description | Cluster of symptoms in alcohol-dependent persons after heavy or prolonged alcohol use has lessened or ceased | Constellation of symptoms lasting weeks to months after alcohol use ends |
| Presentation | Develops during acute detoxification period and lasts 5 to 7 days | Develops after 5- to 7-day acute detoxification period and may persist for 1 year |
| Symptoms | Mild: insomnia, tremor, anxiety, GI upset, headache, diaphoresis, palpitations, anorexia Severe: alcoholic hallucinosis Seizures (generalized tonic-clonic) occur in up to 25% of withdrawal episodes, usually within 24 hours after alcohol cessation Delirium tremens (characterized by hallucinations, disorientation, tachycardia, hypertension, low-grade fever, agitation, and diaphoresis) occurs in up to 5% of patients undergoing withdrawal, may be delayed 4 to 5 days, and has mortality rates reaching 15% | Sleep disruption; anxiety; depressive symptoms; irritability; increased breathing rate, body temperature, blood pressure, and pulse |
| GI: gastrointestinal | ||
| Source: Click here for a bibliography | ||
Table 2
How alcohol affects GABA and glutamate neurotransmitters
| GABA | Glutamate | |
|---|---|---|
| GABA, the brain’s primary inhibitory neurotransmitter, renders nerve cells less sensitive to further signaling | Glutamate, the brain’s major excitatory neurotransmitter, renders nerve cells more sensitive to further signaling | |
| Alcohol facilitates the inhibitory function of the GABAA receptor, allowing more GABA to traverse the receptor, and leading to alcohol’s intoxicating effects | Alcohol seems to inhibit the excitatory function of the NMDA glutamate receptor, believed to play a role in memory, learning, and generation of seizures | |
| During alcohol withdrawal, brain GABA concentrations fall below normal and GABAA receptor sensitivity may be reduced | Long-term alcohol exposure produces an adaptive increase in the function of NMDA receptors and results in development of glutamate-NMDA supersensitivity | |
| In the absence of alcohol, the resulting decrease in inhibitory function may contribute to symptoms of CNS hyperactivity associated with acute and protracted alcohol withdrawal | Acute alcohol withdrawal activates glutamate systems, leading to autonomic nervous system hyperactivity; alcohol withdrawal seizures are associated with increased NMDA receptor function | |
| GABA: gamma-aminobutyric acid; NMDA: N-methyl-D-aspartate | ||
| Source: Click here for a bibliography | ||
Table 3
Mechanisms of action of benzodiazepines vs 3 anticonvulsants
| Agent | Mechanism of action |
|---|---|
| Benzodiazepines | Activate GABAA chloride ionophore, increasing affinity of GABAA receptor for GABA and augmenting frequency of chloride channel openinga |
| Valproic acid | GABA modulation and possibly second messenger systems; may inhibit Na1+ and/or Ca2+ channel, thereby boosting GABA and glutamate actionb |
| Levetiracetam | Decreases high voltage activated Ca2+ channels; unique binding site (synaptic vesicle protein SV2A) is thought to be involved in calcium-dependent regulation of neurotransmitter vesicle exocytosisc |
| Gabapentin | GABA analog; unique binding site (Ca2+ channel subunit in brain) decreases calcium influx and inhibits release of excitatory amino acids and monoaminesd |
| GABA: gamma-aminobutyric acid | |
| Source: Click here for a bibliography | |
CASE REPORT 1: Valproic acid for alcohol overdose
After attempting suicide with an alcohol overdose, Ms. J, age 45, is transferred from the emergency room (ER) to our psychiatry consult service 10 hours after admission. Her symptoms include nausea, tremor, headaches, agitation, disorientation, and auditory hallucinations.
Medical history reveals 25 years of alcohol dependence, multiple hospitalizations for withdrawal, and many failed attempts to quit. Ms. J reports consuming an average of 16 drink equivalents (eg, 12 oz beers) daily but denies illicit drug use.
Lab values on admission include blood alcohol concentration (BAC) 290 mg/dL (0.29%), mean corpuscular volume (MCV) 96 fL, gamma-glutamyltransferase (GGT) 164 U/L, aspartate aminotransferase (AST) 43 U/L, alanine aminotransferase (ALT) 31 U/L, and alkaline phosphatase (ALP) 151 U/L. Urine drug screen, acetaminophen, salicylate, vitamin B1 (thiamine), B12 (cyanocobalamin), B9 (folate), and electrolytes (including magnesium) are normal.
We assess alcohol withdrawal severity using the CIWA-Ar (Click here to view/download a copy of this scale). Ms. J’s initial score is 17, indicating a risk of moderate alcohol withdrawal if untreated.
In the ER, Ms. J is placed on a symptom-triggered benzodiazepine detoxification protocol with lorazepam. We add IV valproic acid, 1,250 mg (based on 20 mg/kg body weight)13 divided into 2 doses over the first 24 hours, then maintain IV valproic acid at 500 mg twice daily (Table 4). Within 12 hours of starting combination therapy, Ms. J scores 7 on the CIWA-Ar—indicating mild withdrawal—with subsequent scores <5. She scores 0 with no residual withdrawal symptoms within 36 hours.
Ms. J requires lorazepam, 7 mg, during the 10 hours before valproic acid is added. She requires only 2 mg lorazepam over the next 3 days and reports no side effects related to IV valproic acid. At discharge, Ms. J begins extended-release oral valproic acid, 1,250 mg (based on 25 mg/kg body weight)13 once daily for 2 weeks, until she can obtain outpatient follow-up.
Table 4
Benzodiazepines and anticonvulsants for alcohol detoxification
| Benzodiazepines | Valproic acid | Levetiracetam | Gabapentin | |
|---|---|---|---|---|
| Loading dose | None | 20 mg/kg of body weight, divided into 2 doses for first 24 hours | 1,500 mg IV once daily | 400 mg PO qid |
| Maintenance dose | Day 1: 2 mg tid Day 2: 2 mg morning, 1 mg afternoon, 2 mg evening Day 3: 1 mg tid Day 4: 1 mg bid Day 5: 1 mg Day 6: none | 500 mg IV bid | Either 500 mg IV tid or 1,000 mg PO bid after 2 to 3 days of treatment | 1,200 mg PO tid |
| Side effects | Impaired consciousness, respiratory depression, hypotension | Dizziness, drowsiness, hair loss/thinning, nausea, tremor, weight gain | Somnolence, asthenia, dizziness, coordination difficulties | Somnolence, dizziness, ataxia, fatigue |
| Drug interactions | ↑ BZ: cimetidine, oral contraceptives, ethanol (acute), disulfiram, isoniazid, propranolol ↓ BZ: rifampin, ethanol (chronic) | ↑ VPA: aspirin, felbamate, fluoxetine, isoniazid ↓ VPA: carbamazepine, lamotrigine, phenobarbital, phenytoin, ritonavir | None | ↓ GBP 20%: antacids |
| BZ: benzodiazepine; GBP: gabapentin; PO: per os (by mouth); VPA: valproic acid | ||||
| Source: Click here for a bibliography | ||||
Less lorazepam needed
Adjunctive anticonvulsants can reduce the amount of lorazepam required during detoxification.14,15 Compared with benzodiazepine monotherapy, the advantages of combination therapy—particularly in outpatient alcohol withdrawal treatment and relapse prevention—include:
- minimal interaction with alcohol (avoiding increased psychomotor deficits, cognitive impairment, and intoxication)15
- lower abuse potential
- possible efficacy in mood stabilization before, during, and after withdrawal (Table 5).16
Given the risk of seizures during AWS, anticonvulsants seem to make empirical sense. One study reported a 1% incidence of withdrawal-related seizures in 545 alcohol-dependent inpatients treated with valproic acid.17 Another case series of 37 patients found no acute sequelae when valproic acid was used for AWS.18
Anticonvulsants such as valproic acid may reduce the frequency and severity of alcohol relapse, whereas benzodiazepines may increase relapse risk.19 During a 6-week trial, patients receiving valproic acid maintenance therapy had greater abstinence rates and improved drinking outcomes compared with detoxification-only groups.9
One disadvantage of valproic acid is potential hepatotoxicity, an important consideration in patients with liver damage. Fortunately, Ms. J’s AST and ALT values remained within normal limits during valproic acid treatment.
Table 5
Pharmacologic profiles of benzodiazepines vs 3 anticonvulsants
| Benzodiazepines | Valproic acid | Levetiracetam | Gabapentin | |
|---|---|---|---|---|
| Metabolism | CYP 2C19: diazepam CYP 3A3/4: alprazolam, clonazepam, diazepam, triazolam Phase II only: lorazepam, temazepam, oxazepam | >95% hepatic, of which <20% occurs via CYP isoenzymes | Not extensively metabolized; renal clearance; not involved with hepatic CYP isoenzymes | Not metabolized; secreted via kidneys as unchanged drug |
| Sedation | Mild to moderate | Mild to moderate | Mild to moderate | Moderate to severe |
| Synergistic effects with alcohol | Yes | No | No | No |
| Paradoxical disinhibition | Yes | No | No | No |
| Risk of addiction in outpatient therapy | Yes | No | No | No |
| CYP: cytochrome P450 | ||||
| Source: Click here for a bibliography | ||||
CASE REPORT 2: Levetiracetam for withdrawal seizures
Mr. H, age 42, presents to the ER after suffering a seizure. His medical history includes hypertension, alcohol dependence, and seizures during alcohol withdrawal. He denies a history of psychiatric illness, and his family history is unknown. He is noncompliant with hypertension treatment, which includes clonidine. Mr. H reports his usual alcohol consumption as a 6-pack of beer nightly during the week and a 12-pack nightly on weekends. He says his last drink was 4 days before admission.
Mr. H scores 19 on the CIWA-Ar, placing him at risk for moderate withdrawal. Head CT shows diffuse atrophy, without evidence of an acute intracranial process. BAC is zero on admission, and urine drug screen is negative. Amylase, lipase, and lactate dehydrogenase (LDH) levels suggest acute pancreatitis. AST is elevated to 131 U/L, ALT is elevated to 42 U/L, but MCV is within normal limits.
The psychiatric service is consulted on day 2 of admission, and we prescribe levetiracetam, 500 mg IV every 8 hours.20 IV lorazepam also is available as needed: 1 mg every 8 hours for the first 2 days, then 1 mg every 12 hours for 2 days, then 1 mg every 24 hours. The patient’s CIWA-Ar score is 9 on days 2 and 3 of admission, followed by scores consistently between 2 and 3 after scheduled levetiracetam administration. Mr. H requires 3 mg of lorazepam the remainder of his hospitalization. He is discharged on day 7 with a CIWA-Ar score of 2, and reports no adverse effects related to levetiracetam. He leaves the hospital with a 2-week prescription for oral levetiracetam, 500 mg tid.
Advantages of levetiracetam
Levetiracetam is FDA-approved for adjunctive treatment of adults with partial-onset seizures.21 Successful AWS treatment with adjunctive levetiracetam has been supported by few but promising studies.10,20 Potential advantages of levetiracetam in detoxification include:
- a lack of GABAergic properties, which limits the risk of intoxication or respiratory insufficiency when combined with alcohol21
- low drug-drug interaction risk because of nonhepatic metabolism and primary renal excretion.22,23
We selected levetiracetam for Mr. H because of his history of alcohol withdrawal seizures and acute pancreatitis. Anticonvulsants may be more effective than lorazepam in reducing the risk of alcohol withdrawal seizures,24 and we felt valproic acid might not be safe for him because of its low but real risk of pancreatitis.13 We based our levetiracetam dosing on a small open-label trial20 and product information for treating adults with partial-onset seizures.25
Studies also demonstrate levetiracetam’s potential for relapse prevention during outpatient therapy. In a 10-week trial, levetiracetam decreased the number of standard drinks in alcohol-dependent patients from 5.3 to 1.7 per day.10 This was a small open trial, however, and large controlled trials support the usefulness of other, FDA-approved medications—including disulfiram, naltrexone, and acamprosate—for alcohol relapse prevention.
CASE REPORT 3: Gabapentin for acute withdrawal
Mr. B, age 38, presents to the ER after a 13-day alcohol binge. He has been drinking increasing amounts of alcohol over 6 weeks. Three months earlier, Mr. B was admitted for alcohol withdrawal treatment and received 49 mg of lorazepam over 3 days. This resulted in his transfer from the step-down unit to the intensive care unit for increased agitation, possibly caused by paradoxical disinhibition from excessive lorazepam use.26
Mr. B’s medical history is significant for alcohol-induced seizures, DTs, traumatic brain injury related to craniotomy, and right arm amputation. Mr. B drinks approximately 24 beers per day. He denies tobacco use but admits to past use of cocaine, marijuana, and heroin.
On admission, Mr. B’s BAC is 360 mg/dL (0.36%), AST is elevated at 72 U/L, ALT at 42 U/L, and LDH significantly elevated at 384 U/L. Urine drug screen is negative, and his CIWA-Ar score is 23. His score of –1 on the Richmond Agitation and Sedation Scale (RASS)27 correlates with very mild sedation.
Guided by Bonnet et al28 and clinical experience, we start Mr. B on gabapentin, 1,200 mg tid, and IV lorazepam, 2 mg every 8 hours as needed for breakthrough withdrawal. We decrease lorazepam by 50% every other day until Mr. B is discharged. On days 2, 3, and 4, Mr. B’s CIWA-Ar scores are 6, 9, and 2, respectively. His RASS score drops from –1 on days 1 and 2 to 0 until discharge, indicating an alert and calm state.
Mr. B requires a total of 2 mg of lorazepam throughout hospitalization. He finishes alcohol detoxification on day 4 and is discharged with a prescription for gabapentin, 1,200 mg tid. Two weeks later, when he is admitted to a 28-day inpatient alcohol rehabilitation unit, Mr. B has not relapsed.
More abstinent days
Gabapentin is FDA-approved as adjunctive therapy for partial seizures. Off-label, it has been generally efficacious as an adjunct in alcohol detoxification.29-32 We chose adjunctive anticonvulsant therapy for Mr. B because of his history of alcohol-induced seizures. We chose gabapentin instead of valproic acid because of Mr. B’s liver damage and gabapentin’s lack of hepatic metabolism.
Gabapentin may reduce alcohol consumption and craving in alcohol-dependent patients. By increasing the number of abstinent days, gabapentin may help patients maintain abstinence.33 Gabapentin does not appear to interact clinically with alcohol, causing neither sedation nor synergistic effects.34 Its relative lack of abuse potential may be valuable in outpatient alcohol withdrawal treatment and in maintaining alcohol abstinence after detoxification.
Related resource
- Asplund CA, Aaronson JW, Aaronson HE. 3 regimens for alcohol withdrawal and detoxification. J Fam Pract. 2004;53(7):545-554. www.jfponline.com/Pages.asp?AID=1730.
Drug brand names
- Acamprosate • Campral
- Alprazolam • Xanax
- Carbamazepine • Carbatrol
- Cimetidine • Tagamet
- Clonazepam • Klonopin
- Clonidine • Catapres
- Diazepam • Valium
- Disulfiram • Antabuse
- Felbamate • Felbatol
- Fluoxetine • Prozac
- Gabapentin • Neurontin
- Isoniazid • Nydrazid
- Lamotrigine • Lamictal
- Levetiracetam • Keppra
- Lorazepam • Ativan
- Naltrexone • ReVia, Vivitrol
- Oxazepam • Serax
- Phenobarbital • Luminal
- Phenytoin • Dilantin
- Propranolol • Inderal
- Rifampin • Rifadin
- Ritonavir • Norvir
- Temazepam • Restoril
- Triazolam • Halcion
- Valproic acid • Depakote, Depakene
Disclosures
Dr. Spiegel is a speaker for Pfizer Inc. and GlaxoSmithKline.
Dr. Radac reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
The authors thank Rishi Laroia, MD, Robert Swanson, MD, and Adam W. Coe, MD for their contributions to this article.
1. Mayo-Smith MF, Cushman P, Hill AJ, et al. Pharmacological management of alcohol withdrawal. JAMA. 1997;278:144-151.
2. Myrick H, Brady KT, Malcom R. Divalproex in the treatment of alcohol withdrawal—statistical data included. Am J Drug Alcohol Abuse. 2000;26(1):155-160.
3. Johnson BA, Swift RM, Addolorato G, et al. Safety and efficacy of GABAergic medications for treating alcoholism. Alcohol Clin Exp Res. 2005;29(2):248-254.
4. Lum E, Gorman SK, Slavik RS. Valproic acid management of acute alcohol withdrawal. Ann Pharmacother. 2006;40(3):441-448.
5. Trevisan LA, Boutros N, Petrakis IL, et al. Complications of alcohol withdrawal: pathophysiological insights. Alcohol Health Res World. 1998;22(1):61-66.
6. Esel E. Neurobiology of alcohol withdrawal inhibitory and excitatory neurotransmitters. Turk Psikiyatri Derg. 2006;7(2):129-138.
7. Myrick H, Brady KT. The use of divalproex in the treatment of addictive disorders. Psychopharmacol Bull. 2003;37(suppl 2):89-97.
8. Brady KT, Myrick H, Henderson S, et al. The use of divalproex in alcohol relapse prevention: a pilot study. Drug Alcohol Depend. 2002;67(3):323-330.
9. Longo L, Campbell T, Hubatch S. Divalproex sodium (Depakote) for alcohol withdrawal and relapse prevention. J Addict Dis. 2002;21(2):55-64.
10. Sarid-Segal O, Piechniczek-Buczek J, Knapp C, et al. The effects of levetiracetam on alcohol consumption in alcohol-dependent subjects: an open label study. Am J Drug Alcohol Abuse. 2008;34(4):441-447.
11. American Psychiatric Association. Treatments of psychiatric disorders: a task force report of the American Psychiatric Association. Washington, DC: American Psychiatric Association Press; 1989:187.
12. Mason BJ, Light JM, Williams LD, et al. Proof-of-concept human laboratory study for protracted abstinence in alcohol dependence: effects of gabapentin. Addict Biol. 2009;14(1):73-83.
13. Physicians’ Desk Reference 2009. 63rd ed. Montvale, NJ: Physicians’ Desk Reference; 2008:423-431.
14. Malcolm R, Ballenger JC, Sturgis ET, et al. Double-blind controlled trial comparing carbamazepine to oxazepam treatment of alcohol withdrawal. Am J Psychiatry. 1989;146:617-621.
15. Myrick H, Anton R, Voronin K, et al. A double-blind evaluation of gabapentin on alcohol effects and drinking in a clinical laboratory paradigm. Alcohol Clin Exp Res. 2007;31(2):221-227.
16. Malcolm R, Myrick H, Brady KT, et al. Update on anticonvulsants for the treatment of alcohol withdrawal. Am J Addict. 2001;10(suppl):16-23.
17. Davis LL, Ryan W, Adinoff B, et al. Comprehensive review of the psychiatric uses of valproate. J Clin Psychopharm. 2000;20(1 suppl 1):1S-17S.
18. Rosenthal RN, Perkel C, Singh P, et al. A pilot open randomized trial of valproate and phenobarbital in the treatment of acute alcohol withdrawal. Am J Addict. 1998;7:189-197.
19. Book SW, Myrick H. Novel anticonvulsants in the treatment of alcoholism. Expert Opin Investig Drugs. 2005;14(4):371-376.
20. Krebs M, Leopold K, Richter C, et al. Levetiracetam for the treatment of alcohol withdrawal syndrome: an open-label pilot trial. J Clin Psychopharmacol. 2006;26(3):347-349.
21. LaRoche SM, Helmers SL. The new antiepileptic drugs: scientific review. JAMA. 2004;291:605-614.
22. Chabolla DR, Harnois DM, Meschia JF. Levetiracetam monotherapy for liver transplant patients with seizures. Transplant Proc. 2003;35:1480-1481.
23. Paul F, Meencke HJ. Levetiracetam in focal epilepsy and hepatic porphyria: a case report. Epilepsia. 2004;45:559-560.
24. Ntais C, Pakos E, Kyzas P, et al. Benzodiazepines for alcohol withdrawal. Cochrane Database Syst Rev. 2005;(3):CD005063.-
25. Physicians’ Desk Reference 2009. 63rd ed. Montvale, NJ: Physicians’ Desk Reference; 2008:3131-3143.
26. Saias T, Gallarda T. Paradoxical aggressive reactions to benzodiazepine use: a review. Encephale. 2008;34(4):330-336.
27. Sessler CN, Gosnell MS, Grap MJ, et al. The Richmond Agitation–Sedation Scale: validity and reliability in adult intensive care unit patients. Am J Respir Crit Care Med. 2002;166:1338-1344.
28. Bonnet U, Banger M, Leweke FM, et al. Treatment of acute alcohol withdrawal with gabapentin: results from a controlled two-center trial. J Clin Psychopharmacol. 2003;23(5):514-519.
29. Myrick H, Malcolm R, Brady KT. Gabapentin treatment of alcohol withdrawal. Am J Psychiatry. 1998;155:1626.-
30. Bonnet U, Banger M, Leweke FM, et al. Treatment of alcohol withdrawal syndrome with gabapentin. Pharmacopsychiatry. 1999;32:107-109.
31. Mariani JJ, Rosenthal RN, Tross S, et al. A randomized, open-label, controlled trial of gabapentin and phenobarbital in the treatment of alcohol withdrawal. Am J Addict. 2006;15(1):76-84.
32. Voris J, Smith NL, Rao SM, et al. Gabapentin for the treatment of ethanol withdrawal. Subst Abus. 2003;24(2):129-132.
33. Furieri FA, Nakamura-Palacios EM. Gabapentin reduces alcohol consumption and craving: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2007;68(11):1691-1700.
34. Bisaga A, Evans SM. The acute effects of gabapentin in combination with alcohol in heavy drinkers. Drug Alcohol Depend. 2006;83(1):25-32.
Benzodiazepines are the mainstay of alcohol detoxification treatment, with extensive evidence supporting their efficacy and relative safety.1 The risk of benzodiazepine-alcohol interaction, however, and psychomotor and cognitive impairments associated with benzodiazepine use may limit early rehabilitation efforts in hospitalized patients.2 Cross-tolerance with alcohol also limits benzodiazepines’ potential benefit in outpatients with substance use disorders.
Adding anticonvulsants to acute benzodiazepine therapy has been shown to decrease alcohol withdrawal symptom severity, reduce seizure risk, and support recovery, particularly in patients with multiple alcohol withdrawal episodes. After detoxification, long-term anticonvulsant use may reduce relapse risk by decreasing post-cessation craving, without abuse liability.3
Although not all studies endorse adding anticonvulsants to benzodiazepines for managing alcohol withdrawal syndrome (AWS),4 we present 3 cases in which anticonvulsants were used successfully as adjuncts to lorazepam. Valproic acid, levetiracetam, and gabapentin offer advantages in acute and long-term therapy of alcohol dependence with efficacy in AWS, low abuse potential, benign safety profile, and mood-stabilizing properties.
Neurobiologic rationale
AWS manifests as a cluster of clinical symptoms including delirium tremens (DTs) and seizures (Table 1). Its pathophysiology can be explained by alcohol’s agonist effect on the gamma-aminobutyric acid (GABA) system and antagonist effect on the glutamatergic system (Table 2).5
Chronic alcohol intake leads to neuroadaptation in the brain in the form of down-regulation of GABAA receptors and upregulation of N-methyl-D-aspartate receptors. During alcohol withdrawal, this neuroadaptation leads to a decrease in central GABA activity and an increase in glutamate activity, resulting in hyperexcitation, anxiety, and seizures.6
Little data exist regarding time to relapse after detoxification in alcohol-dependent patients. One theory—called “protracted withdrawal syndrome” (Table 1)—suggests that abstinent alcoholics return to drinking because of the same, but attenuated, neuroadaptations that trigger acute AWS.7
Advantages of adjunct therapy. Ntais et al8 evaluated benzodiazepines’ effectiveness and safety in treating AWS in a clinical review of 57 randomized, controlled trials totaling 4,051 patients. Benzodiazepines showed similar success rates as other drugs (relative risk [RR] 1.00) or anticonvulsants in particular (RR 0.88), as measured by changes in Clinical Institute Withdrawal Assessment for Alcohol (CIWA-Ar) scores at the end of treatment. Benzodiazepines also offered significant benefit for seizure control compared with nonanticonvulsants (RR 0.23), but less when compared with anti convulsants (RR 1.99).
Although the literature does not support anticonvulsant use for monotherapy in AWS, anticonvulsants show potential as adjunctive therapy. Valproic acid, levetiracetam, and gabapentin offer unique mechanisms of action (Table 3) and demonstrate advantages over benzodiazepine monotherapy for AWS. Adjunctive use of valproic acid,8,9 levetiracetam,10 and gabapentin11,12 in detoxification also has demonstrated efficacy in reducing risk of relapse and delaying relapse.
The neurobiologic rationale for using anticonvulsants in acute AWS is speculative, but these agents appear to:
- inhibit “kindling” (neuronal changes that may be associated with repeated intoxications)
- facilitate GABAergic mechanisms.9
Table 1
Alcohol withdrawal: Acute vs long-term symptoms
| Alcohol withdrawal syndrome | Protracted withdrawal syndrome | |
|---|---|---|
| Description | Cluster of symptoms in alcohol-dependent persons after heavy or prolonged alcohol use has lessened or ceased | Constellation of symptoms lasting weeks to months after alcohol use ends |
| Presentation | Develops during acute detoxification period and lasts 5 to 7 days | Develops after 5- to 7-day acute detoxification period and may persist for 1 year |
| Symptoms | Mild: insomnia, tremor, anxiety, GI upset, headache, diaphoresis, palpitations, anorexia Severe: alcoholic hallucinosis Seizures (generalized tonic-clonic) occur in up to 25% of withdrawal episodes, usually within 24 hours after alcohol cessation Delirium tremens (characterized by hallucinations, disorientation, tachycardia, hypertension, low-grade fever, agitation, and diaphoresis) occurs in up to 5% of patients undergoing withdrawal, may be delayed 4 to 5 days, and has mortality rates reaching 15% | Sleep disruption; anxiety; depressive symptoms; irritability; increased breathing rate, body temperature, blood pressure, and pulse |
| GI: gastrointestinal | ||
| Source: Click here for a bibliography | ||
Table 2
How alcohol affects GABA and glutamate neurotransmitters
| GABA | Glutamate | |
|---|---|---|
| GABA, the brain’s primary inhibitory neurotransmitter, renders nerve cells less sensitive to further signaling | Glutamate, the brain’s major excitatory neurotransmitter, renders nerve cells more sensitive to further signaling | |
| Alcohol facilitates the inhibitory function of the GABAA receptor, allowing more GABA to traverse the receptor, and leading to alcohol’s intoxicating effects | Alcohol seems to inhibit the excitatory function of the NMDA glutamate receptor, believed to play a role in memory, learning, and generation of seizures | |
| During alcohol withdrawal, brain GABA concentrations fall below normal and GABAA receptor sensitivity may be reduced | Long-term alcohol exposure produces an adaptive increase in the function of NMDA receptors and results in development of glutamate-NMDA supersensitivity | |
| In the absence of alcohol, the resulting decrease in inhibitory function may contribute to symptoms of CNS hyperactivity associated with acute and protracted alcohol withdrawal | Acute alcohol withdrawal activates glutamate systems, leading to autonomic nervous system hyperactivity; alcohol withdrawal seizures are associated with increased NMDA receptor function | |
| GABA: gamma-aminobutyric acid; NMDA: N-methyl-D-aspartate | ||
| Source: Click here for a bibliography | ||
Table 3
Mechanisms of action of benzodiazepines vs 3 anticonvulsants
| Agent | Mechanism of action |
|---|---|
| Benzodiazepines | Activate GABAA chloride ionophore, increasing affinity of GABAA receptor for GABA and augmenting frequency of chloride channel openinga |
| Valproic acid | GABA modulation and possibly second messenger systems; may inhibit Na1+ and/or Ca2+ channel, thereby boosting GABA and glutamate actionb |
| Levetiracetam | Decreases high voltage activated Ca2+ channels; unique binding site (synaptic vesicle protein SV2A) is thought to be involved in calcium-dependent regulation of neurotransmitter vesicle exocytosisc |
| Gabapentin | GABA analog; unique binding site (Ca2+ channel subunit in brain) decreases calcium influx and inhibits release of excitatory amino acids and monoaminesd |
| GABA: gamma-aminobutyric acid | |
| Source: Click here for a bibliography | |
CASE REPORT 1: Valproic acid for alcohol overdose
After attempting suicide with an alcohol overdose, Ms. J, age 45, is transferred from the emergency room (ER) to our psychiatry consult service 10 hours after admission. Her symptoms include nausea, tremor, headaches, agitation, disorientation, and auditory hallucinations.
Medical history reveals 25 years of alcohol dependence, multiple hospitalizations for withdrawal, and many failed attempts to quit. Ms. J reports consuming an average of 16 drink equivalents (eg, 12 oz beers) daily but denies illicit drug use.
Lab values on admission include blood alcohol concentration (BAC) 290 mg/dL (0.29%), mean corpuscular volume (MCV) 96 fL, gamma-glutamyltransferase (GGT) 164 U/L, aspartate aminotransferase (AST) 43 U/L, alanine aminotransferase (ALT) 31 U/L, and alkaline phosphatase (ALP) 151 U/L. Urine drug screen, acetaminophen, salicylate, vitamin B1 (thiamine), B12 (cyanocobalamin), B9 (folate), and electrolytes (including magnesium) are normal.
We assess alcohol withdrawal severity using the CIWA-Ar (Click here to view/download a copy of this scale). Ms. J’s initial score is 17, indicating a risk of moderate alcohol withdrawal if untreated.
In the ER, Ms. J is placed on a symptom-triggered benzodiazepine detoxification protocol with lorazepam. We add IV valproic acid, 1,250 mg (based on 20 mg/kg body weight)13 divided into 2 doses over the first 24 hours, then maintain IV valproic acid at 500 mg twice daily (Table 4). Within 12 hours of starting combination therapy, Ms. J scores 7 on the CIWA-Ar—indicating mild withdrawal—with subsequent scores <5. She scores 0 with no residual withdrawal symptoms within 36 hours.
Ms. J requires lorazepam, 7 mg, during the 10 hours before valproic acid is added. She requires only 2 mg lorazepam over the next 3 days and reports no side effects related to IV valproic acid. At discharge, Ms. J begins extended-release oral valproic acid, 1,250 mg (based on 25 mg/kg body weight)13 once daily for 2 weeks, until she can obtain outpatient follow-up.
Table 4
Benzodiazepines and anticonvulsants for alcohol detoxification
| Benzodiazepines | Valproic acid | Levetiracetam | Gabapentin | |
|---|---|---|---|---|
| Loading dose | None | 20 mg/kg of body weight, divided into 2 doses for first 24 hours | 1,500 mg IV once daily | 400 mg PO qid |
| Maintenance dose | Day 1: 2 mg tid Day 2: 2 mg morning, 1 mg afternoon, 2 mg evening Day 3: 1 mg tid Day 4: 1 mg bid Day 5: 1 mg Day 6: none | 500 mg IV bid | Either 500 mg IV tid or 1,000 mg PO bid after 2 to 3 days of treatment | 1,200 mg PO tid |
| Side effects | Impaired consciousness, respiratory depression, hypotension | Dizziness, drowsiness, hair loss/thinning, nausea, tremor, weight gain | Somnolence, asthenia, dizziness, coordination difficulties | Somnolence, dizziness, ataxia, fatigue |
| Drug interactions | ↑ BZ: cimetidine, oral contraceptives, ethanol (acute), disulfiram, isoniazid, propranolol ↓ BZ: rifampin, ethanol (chronic) | ↑ VPA: aspirin, felbamate, fluoxetine, isoniazid ↓ VPA: carbamazepine, lamotrigine, phenobarbital, phenytoin, ritonavir | None | ↓ GBP 20%: antacids |
| BZ: benzodiazepine; GBP: gabapentin; PO: per os (by mouth); VPA: valproic acid | ||||
| Source: Click here for a bibliography | ||||
Less lorazepam needed
Adjunctive anticonvulsants can reduce the amount of lorazepam required during detoxification.14,15 Compared with benzodiazepine monotherapy, the advantages of combination therapy—particularly in outpatient alcohol withdrawal treatment and relapse prevention—include:
- minimal interaction with alcohol (avoiding increased psychomotor deficits, cognitive impairment, and intoxication)15
- lower abuse potential
- possible efficacy in mood stabilization before, during, and after withdrawal (Table 5).16
Given the risk of seizures during AWS, anticonvulsants seem to make empirical sense. One study reported a 1% incidence of withdrawal-related seizures in 545 alcohol-dependent inpatients treated with valproic acid.17 Another case series of 37 patients found no acute sequelae when valproic acid was used for AWS.18
Anticonvulsants such as valproic acid may reduce the frequency and severity of alcohol relapse, whereas benzodiazepines may increase relapse risk.19 During a 6-week trial, patients receiving valproic acid maintenance therapy had greater abstinence rates and improved drinking outcomes compared with detoxification-only groups.9
One disadvantage of valproic acid is potential hepatotoxicity, an important consideration in patients with liver damage. Fortunately, Ms. J’s AST and ALT values remained within normal limits during valproic acid treatment.
Table 5
Pharmacologic profiles of benzodiazepines vs 3 anticonvulsants
| Benzodiazepines | Valproic acid | Levetiracetam | Gabapentin | |
|---|---|---|---|---|
| Metabolism | CYP 2C19: diazepam CYP 3A3/4: alprazolam, clonazepam, diazepam, triazolam Phase II only: lorazepam, temazepam, oxazepam | >95% hepatic, of which <20% occurs via CYP isoenzymes | Not extensively metabolized; renal clearance; not involved with hepatic CYP isoenzymes | Not metabolized; secreted via kidneys as unchanged drug |
| Sedation | Mild to moderate | Mild to moderate | Mild to moderate | Moderate to severe |
| Synergistic effects with alcohol | Yes | No | No | No |
| Paradoxical disinhibition | Yes | No | No | No |
| Risk of addiction in outpatient therapy | Yes | No | No | No |
| CYP: cytochrome P450 | ||||
| Source: Click here for a bibliography | ||||
CASE REPORT 2: Levetiracetam for withdrawal seizures
Mr. H, age 42, presents to the ER after suffering a seizure. His medical history includes hypertension, alcohol dependence, and seizures during alcohol withdrawal. He denies a history of psychiatric illness, and his family history is unknown. He is noncompliant with hypertension treatment, which includes clonidine. Mr. H reports his usual alcohol consumption as a 6-pack of beer nightly during the week and a 12-pack nightly on weekends. He says his last drink was 4 days before admission.
Mr. H scores 19 on the CIWA-Ar, placing him at risk for moderate withdrawal. Head CT shows diffuse atrophy, without evidence of an acute intracranial process. BAC is zero on admission, and urine drug screen is negative. Amylase, lipase, and lactate dehydrogenase (LDH) levels suggest acute pancreatitis. AST is elevated to 131 U/L, ALT is elevated to 42 U/L, but MCV is within normal limits.
The psychiatric service is consulted on day 2 of admission, and we prescribe levetiracetam, 500 mg IV every 8 hours.20 IV lorazepam also is available as needed: 1 mg every 8 hours for the first 2 days, then 1 mg every 12 hours for 2 days, then 1 mg every 24 hours. The patient’s CIWA-Ar score is 9 on days 2 and 3 of admission, followed by scores consistently between 2 and 3 after scheduled levetiracetam administration. Mr. H requires 3 mg of lorazepam the remainder of his hospitalization. He is discharged on day 7 with a CIWA-Ar score of 2, and reports no adverse effects related to levetiracetam. He leaves the hospital with a 2-week prescription for oral levetiracetam, 500 mg tid.
Advantages of levetiracetam
Levetiracetam is FDA-approved for adjunctive treatment of adults with partial-onset seizures.21 Successful AWS treatment with adjunctive levetiracetam has been supported by few but promising studies.10,20 Potential advantages of levetiracetam in detoxification include:
- a lack of GABAergic properties, which limits the risk of intoxication or respiratory insufficiency when combined with alcohol21
- low drug-drug interaction risk because of nonhepatic metabolism and primary renal excretion.22,23
We selected levetiracetam for Mr. H because of his history of alcohol withdrawal seizures and acute pancreatitis. Anticonvulsants may be more effective than lorazepam in reducing the risk of alcohol withdrawal seizures,24 and we felt valproic acid might not be safe for him because of its low but real risk of pancreatitis.13 We based our levetiracetam dosing on a small open-label trial20 and product information for treating adults with partial-onset seizures.25
Studies also demonstrate levetiracetam’s potential for relapse prevention during outpatient therapy. In a 10-week trial, levetiracetam decreased the number of standard drinks in alcohol-dependent patients from 5.3 to 1.7 per day.10 This was a small open trial, however, and large controlled trials support the usefulness of other, FDA-approved medications—including disulfiram, naltrexone, and acamprosate—for alcohol relapse prevention.
CASE REPORT 3: Gabapentin for acute withdrawal
Mr. B, age 38, presents to the ER after a 13-day alcohol binge. He has been drinking increasing amounts of alcohol over 6 weeks. Three months earlier, Mr. B was admitted for alcohol withdrawal treatment and received 49 mg of lorazepam over 3 days. This resulted in his transfer from the step-down unit to the intensive care unit for increased agitation, possibly caused by paradoxical disinhibition from excessive lorazepam use.26
Mr. B’s medical history is significant for alcohol-induced seizures, DTs, traumatic brain injury related to craniotomy, and right arm amputation. Mr. B drinks approximately 24 beers per day. He denies tobacco use but admits to past use of cocaine, marijuana, and heroin.
On admission, Mr. B’s BAC is 360 mg/dL (0.36%), AST is elevated at 72 U/L, ALT at 42 U/L, and LDH significantly elevated at 384 U/L. Urine drug screen is negative, and his CIWA-Ar score is 23. His score of –1 on the Richmond Agitation and Sedation Scale (RASS)27 correlates with very mild sedation.
Guided by Bonnet et al28 and clinical experience, we start Mr. B on gabapentin, 1,200 mg tid, and IV lorazepam, 2 mg every 8 hours as needed for breakthrough withdrawal. We decrease lorazepam by 50% every other day until Mr. B is discharged. On days 2, 3, and 4, Mr. B’s CIWA-Ar scores are 6, 9, and 2, respectively. His RASS score drops from –1 on days 1 and 2 to 0 until discharge, indicating an alert and calm state.
Mr. B requires a total of 2 mg of lorazepam throughout hospitalization. He finishes alcohol detoxification on day 4 and is discharged with a prescription for gabapentin, 1,200 mg tid. Two weeks later, when he is admitted to a 28-day inpatient alcohol rehabilitation unit, Mr. B has not relapsed.
More abstinent days
Gabapentin is FDA-approved as adjunctive therapy for partial seizures. Off-label, it has been generally efficacious as an adjunct in alcohol detoxification.29-32 We chose adjunctive anticonvulsant therapy for Mr. B because of his history of alcohol-induced seizures. We chose gabapentin instead of valproic acid because of Mr. B’s liver damage and gabapentin’s lack of hepatic metabolism.
Gabapentin may reduce alcohol consumption and craving in alcohol-dependent patients. By increasing the number of abstinent days, gabapentin may help patients maintain abstinence.33 Gabapentin does not appear to interact clinically with alcohol, causing neither sedation nor synergistic effects.34 Its relative lack of abuse potential may be valuable in outpatient alcohol withdrawal treatment and in maintaining alcohol abstinence after detoxification.
Related resource
- Asplund CA, Aaronson JW, Aaronson HE. 3 regimens for alcohol withdrawal and detoxification. J Fam Pract. 2004;53(7):545-554. www.jfponline.com/Pages.asp?AID=1730.
Drug brand names
- Acamprosate • Campral
- Alprazolam • Xanax
- Carbamazepine • Carbatrol
- Cimetidine • Tagamet
- Clonazepam • Klonopin
- Clonidine • Catapres
- Diazepam • Valium
- Disulfiram • Antabuse
- Felbamate • Felbatol
- Fluoxetine • Prozac
- Gabapentin • Neurontin
- Isoniazid • Nydrazid
- Lamotrigine • Lamictal
- Levetiracetam • Keppra
- Lorazepam • Ativan
- Naltrexone • ReVia, Vivitrol
- Oxazepam • Serax
- Phenobarbital • Luminal
- Phenytoin • Dilantin
- Propranolol • Inderal
- Rifampin • Rifadin
- Ritonavir • Norvir
- Temazepam • Restoril
- Triazolam • Halcion
- Valproic acid • Depakote, Depakene
Disclosures
Dr. Spiegel is a speaker for Pfizer Inc. and GlaxoSmithKline.
Dr. Radac reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
The authors thank Rishi Laroia, MD, Robert Swanson, MD, and Adam W. Coe, MD for their contributions to this article.
Benzodiazepines are the mainstay of alcohol detoxification treatment, with extensive evidence supporting their efficacy and relative safety.1 The risk of benzodiazepine-alcohol interaction, however, and psychomotor and cognitive impairments associated with benzodiazepine use may limit early rehabilitation efforts in hospitalized patients.2 Cross-tolerance with alcohol also limits benzodiazepines’ potential benefit in outpatients with substance use disorders.
Adding anticonvulsants to acute benzodiazepine therapy has been shown to decrease alcohol withdrawal symptom severity, reduce seizure risk, and support recovery, particularly in patients with multiple alcohol withdrawal episodes. After detoxification, long-term anticonvulsant use may reduce relapse risk by decreasing post-cessation craving, without abuse liability.3
Although not all studies endorse adding anticonvulsants to benzodiazepines for managing alcohol withdrawal syndrome (AWS),4 we present 3 cases in which anticonvulsants were used successfully as adjuncts to lorazepam. Valproic acid, levetiracetam, and gabapentin offer advantages in acute and long-term therapy of alcohol dependence with efficacy in AWS, low abuse potential, benign safety profile, and mood-stabilizing properties.
Neurobiologic rationale
AWS manifests as a cluster of clinical symptoms including delirium tremens (DTs) and seizures (Table 1). Its pathophysiology can be explained by alcohol’s agonist effect on the gamma-aminobutyric acid (GABA) system and antagonist effect on the glutamatergic system (Table 2).5
Chronic alcohol intake leads to neuroadaptation in the brain in the form of down-regulation of GABAA receptors and upregulation of N-methyl-D-aspartate receptors. During alcohol withdrawal, this neuroadaptation leads to a decrease in central GABA activity and an increase in glutamate activity, resulting in hyperexcitation, anxiety, and seizures.6
Little data exist regarding time to relapse after detoxification in alcohol-dependent patients. One theory—called “protracted withdrawal syndrome” (Table 1)—suggests that abstinent alcoholics return to drinking because of the same, but attenuated, neuroadaptations that trigger acute AWS.7
Advantages of adjunct therapy. Ntais et al8 evaluated benzodiazepines’ effectiveness and safety in treating AWS in a clinical review of 57 randomized, controlled trials totaling 4,051 patients. Benzodiazepines showed similar success rates as other drugs (relative risk [RR] 1.00) or anticonvulsants in particular (RR 0.88), as measured by changes in Clinical Institute Withdrawal Assessment for Alcohol (CIWA-Ar) scores at the end of treatment. Benzodiazepines also offered significant benefit for seizure control compared with nonanticonvulsants (RR 0.23), but less when compared with anti convulsants (RR 1.99).
Although the literature does not support anticonvulsant use for monotherapy in AWS, anticonvulsants show potential as adjunctive therapy. Valproic acid, levetiracetam, and gabapentin offer unique mechanisms of action (Table 3) and demonstrate advantages over benzodiazepine monotherapy for AWS. Adjunctive use of valproic acid,8,9 levetiracetam,10 and gabapentin11,12 in detoxification also has demonstrated efficacy in reducing risk of relapse and delaying relapse.
The neurobiologic rationale for using anticonvulsants in acute AWS is speculative, but these agents appear to:
- inhibit “kindling” (neuronal changes that may be associated with repeated intoxications)
- facilitate GABAergic mechanisms.9
Table 1
Alcohol withdrawal: Acute vs long-term symptoms
| Alcohol withdrawal syndrome | Protracted withdrawal syndrome | |
|---|---|---|
| Description | Cluster of symptoms in alcohol-dependent persons after heavy or prolonged alcohol use has lessened or ceased | Constellation of symptoms lasting weeks to months after alcohol use ends |
| Presentation | Develops during acute detoxification period and lasts 5 to 7 days | Develops after 5- to 7-day acute detoxification period and may persist for 1 year |
| Symptoms | Mild: insomnia, tremor, anxiety, GI upset, headache, diaphoresis, palpitations, anorexia Severe: alcoholic hallucinosis Seizures (generalized tonic-clonic) occur in up to 25% of withdrawal episodes, usually within 24 hours after alcohol cessation Delirium tremens (characterized by hallucinations, disorientation, tachycardia, hypertension, low-grade fever, agitation, and diaphoresis) occurs in up to 5% of patients undergoing withdrawal, may be delayed 4 to 5 days, and has mortality rates reaching 15% | Sleep disruption; anxiety; depressive symptoms; irritability; increased breathing rate, body temperature, blood pressure, and pulse |
| GI: gastrointestinal | ||
| Source: Click here for a bibliography | ||
Table 2
How alcohol affects GABA and glutamate neurotransmitters
| GABA | Glutamate | |
|---|---|---|
| GABA, the brain’s primary inhibitory neurotransmitter, renders nerve cells less sensitive to further signaling | Glutamate, the brain’s major excitatory neurotransmitter, renders nerve cells more sensitive to further signaling | |
| Alcohol facilitates the inhibitory function of the GABAA receptor, allowing more GABA to traverse the receptor, and leading to alcohol’s intoxicating effects | Alcohol seems to inhibit the excitatory function of the NMDA glutamate receptor, believed to play a role in memory, learning, and generation of seizures | |
| During alcohol withdrawal, brain GABA concentrations fall below normal and GABAA receptor sensitivity may be reduced | Long-term alcohol exposure produces an adaptive increase in the function of NMDA receptors and results in development of glutamate-NMDA supersensitivity | |
| In the absence of alcohol, the resulting decrease in inhibitory function may contribute to symptoms of CNS hyperactivity associated with acute and protracted alcohol withdrawal | Acute alcohol withdrawal activates glutamate systems, leading to autonomic nervous system hyperactivity; alcohol withdrawal seizures are associated with increased NMDA receptor function | |
| GABA: gamma-aminobutyric acid; NMDA: N-methyl-D-aspartate | ||
| Source: Click here for a bibliography | ||
Table 3
Mechanisms of action of benzodiazepines vs 3 anticonvulsants
| Agent | Mechanism of action |
|---|---|
| Benzodiazepines | Activate GABAA chloride ionophore, increasing affinity of GABAA receptor for GABA and augmenting frequency of chloride channel openinga |
| Valproic acid | GABA modulation and possibly second messenger systems; may inhibit Na1+ and/or Ca2+ channel, thereby boosting GABA and glutamate actionb |
| Levetiracetam | Decreases high voltage activated Ca2+ channels; unique binding site (synaptic vesicle protein SV2A) is thought to be involved in calcium-dependent regulation of neurotransmitter vesicle exocytosisc |
| Gabapentin | GABA analog; unique binding site (Ca2+ channel subunit in brain) decreases calcium influx and inhibits release of excitatory amino acids and monoaminesd |
| GABA: gamma-aminobutyric acid | |
| Source: Click here for a bibliography | |
CASE REPORT 1: Valproic acid for alcohol overdose
After attempting suicide with an alcohol overdose, Ms. J, age 45, is transferred from the emergency room (ER) to our psychiatry consult service 10 hours after admission. Her symptoms include nausea, tremor, headaches, agitation, disorientation, and auditory hallucinations.
Medical history reveals 25 years of alcohol dependence, multiple hospitalizations for withdrawal, and many failed attempts to quit. Ms. J reports consuming an average of 16 drink equivalents (eg, 12 oz beers) daily but denies illicit drug use.
Lab values on admission include blood alcohol concentration (BAC) 290 mg/dL (0.29%), mean corpuscular volume (MCV) 96 fL, gamma-glutamyltransferase (GGT) 164 U/L, aspartate aminotransferase (AST) 43 U/L, alanine aminotransferase (ALT) 31 U/L, and alkaline phosphatase (ALP) 151 U/L. Urine drug screen, acetaminophen, salicylate, vitamin B1 (thiamine), B12 (cyanocobalamin), B9 (folate), and electrolytes (including magnesium) are normal.
We assess alcohol withdrawal severity using the CIWA-Ar (Click here to view/download a copy of this scale). Ms. J’s initial score is 17, indicating a risk of moderate alcohol withdrawal if untreated.
In the ER, Ms. J is placed on a symptom-triggered benzodiazepine detoxification protocol with lorazepam. We add IV valproic acid, 1,250 mg (based on 20 mg/kg body weight)13 divided into 2 doses over the first 24 hours, then maintain IV valproic acid at 500 mg twice daily (Table 4). Within 12 hours of starting combination therapy, Ms. J scores 7 on the CIWA-Ar—indicating mild withdrawal—with subsequent scores <5. She scores 0 with no residual withdrawal symptoms within 36 hours.
Ms. J requires lorazepam, 7 mg, during the 10 hours before valproic acid is added. She requires only 2 mg lorazepam over the next 3 days and reports no side effects related to IV valproic acid. At discharge, Ms. J begins extended-release oral valproic acid, 1,250 mg (based on 25 mg/kg body weight)13 once daily for 2 weeks, until she can obtain outpatient follow-up.
Table 4
Benzodiazepines and anticonvulsants for alcohol detoxification
| Benzodiazepines | Valproic acid | Levetiracetam | Gabapentin | |
|---|---|---|---|---|
| Loading dose | None | 20 mg/kg of body weight, divided into 2 doses for first 24 hours | 1,500 mg IV once daily | 400 mg PO qid |
| Maintenance dose | Day 1: 2 mg tid Day 2: 2 mg morning, 1 mg afternoon, 2 mg evening Day 3: 1 mg tid Day 4: 1 mg bid Day 5: 1 mg Day 6: none | 500 mg IV bid | Either 500 mg IV tid or 1,000 mg PO bid after 2 to 3 days of treatment | 1,200 mg PO tid |
| Side effects | Impaired consciousness, respiratory depression, hypotension | Dizziness, drowsiness, hair loss/thinning, nausea, tremor, weight gain | Somnolence, asthenia, dizziness, coordination difficulties | Somnolence, dizziness, ataxia, fatigue |
| Drug interactions | ↑ BZ: cimetidine, oral contraceptives, ethanol (acute), disulfiram, isoniazid, propranolol ↓ BZ: rifampin, ethanol (chronic) | ↑ VPA: aspirin, felbamate, fluoxetine, isoniazid ↓ VPA: carbamazepine, lamotrigine, phenobarbital, phenytoin, ritonavir | None | ↓ GBP 20%: antacids |
| BZ: benzodiazepine; GBP: gabapentin; PO: per os (by mouth); VPA: valproic acid | ||||
| Source: Click here for a bibliography | ||||
Less lorazepam needed
Adjunctive anticonvulsants can reduce the amount of lorazepam required during detoxification.14,15 Compared with benzodiazepine monotherapy, the advantages of combination therapy—particularly in outpatient alcohol withdrawal treatment and relapse prevention—include:
- minimal interaction with alcohol (avoiding increased psychomotor deficits, cognitive impairment, and intoxication)15
- lower abuse potential
- possible efficacy in mood stabilization before, during, and after withdrawal (Table 5).16
Given the risk of seizures during AWS, anticonvulsants seem to make empirical sense. One study reported a 1% incidence of withdrawal-related seizures in 545 alcohol-dependent inpatients treated with valproic acid.17 Another case series of 37 patients found no acute sequelae when valproic acid was used for AWS.18
Anticonvulsants such as valproic acid may reduce the frequency and severity of alcohol relapse, whereas benzodiazepines may increase relapse risk.19 During a 6-week trial, patients receiving valproic acid maintenance therapy had greater abstinence rates and improved drinking outcomes compared with detoxification-only groups.9
One disadvantage of valproic acid is potential hepatotoxicity, an important consideration in patients with liver damage. Fortunately, Ms. J’s AST and ALT values remained within normal limits during valproic acid treatment.
Table 5
Pharmacologic profiles of benzodiazepines vs 3 anticonvulsants
| Benzodiazepines | Valproic acid | Levetiracetam | Gabapentin | |
|---|---|---|---|---|
| Metabolism | CYP 2C19: diazepam CYP 3A3/4: alprazolam, clonazepam, diazepam, triazolam Phase II only: lorazepam, temazepam, oxazepam | >95% hepatic, of which <20% occurs via CYP isoenzymes | Not extensively metabolized; renal clearance; not involved with hepatic CYP isoenzymes | Not metabolized; secreted via kidneys as unchanged drug |
| Sedation | Mild to moderate | Mild to moderate | Mild to moderate | Moderate to severe |
| Synergistic effects with alcohol | Yes | No | No | No |
| Paradoxical disinhibition | Yes | No | No | No |
| Risk of addiction in outpatient therapy | Yes | No | No | No |
| CYP: cytochrome P450 | ||||
| Source: Click here for a bibliography | ||||
CASE REPORT 2: Levetiracetam for withdrawal seizures
Mr. H, age 42, presents to the ER after suffering a seizure. His medical history includes hypertension, alcohol dependence, and seizures during alcohol withdrawal. He denies a history of psychiatric illness, and his family history is unknown. He is noncompliant with hypertension treatment, which includes clonidine. Mr. H reports his usual alcohol consumption as a 6-pack of beer nightly during the week and a 12-pack nightly on weekends. He says his last drink was 4 days before admission.
Mr. H scores 19 on the CIWA-Ar, placing him at risk for moderate withdrawal. Head CT shows diffuse atrophy, without evidence of an acute intracranial process. BAC is zero on admission, and urine drug screen is negative. Amylase, lipase, and lactate dehydrogenase (LDH) levels suggest acute pancreatitis. AST is elevated to 131 U/L, ALT is elevated to 42 U/L, but MCV is within normal limits.
The psychiatric service is consulted on day 2 of admission, and we prescribe levetiracetam, 500 mg IV every 8 hours.20 IV lorazepam also is available as needed: 1 mg every 8 hours for the first 2 days, then 1 mg every 12 hours for 2 days, then 1 mg every 24 hours. The patient’s CIWA-Ar score is 9 on days 2 and 3 of admission, followed by scores consistently between 2 and 3 after scheduled levetiracetam administration. Mr. H requires 3 mg of lorazepam the remainder of his hospitalization. He is discharged on day 7 with a CIWA-Ar score of 2, and reports no adverse effects related to levetiracetam. He leaves the hospital with a 2-week prescription for oral levetiracetam, 500 mg tid.
Advantages of levetiracetam
Levetiracetam is FDA-approved for adjunctive treatment of adults with partial-onset seizures.21 Successful AWS treatment with adjunctive levetiracetam has been supported by few but promising studies.10,20 Potential advantages of levetiracetam in detoxification include:
- a lack of GABAergic properties, which limits the risk of intoxication or respiratory insufficiency when combined with alcohol21
- low drug-drug interaction risk because of nonhepatic metabolism and primary renal excretion.22,23
We selected levetiracetam for Mr. H because of his history of alcohol withdrawal seizures and acute pancreatitis. Anticonvulsants may be more effective than lorazepam in reducing the risk of alcohol withdrawal seizures,24 and we felt valproic acid might not be safe for him because of its low but real risk of pancreatitis.13 We based our levetiracetam dosing on a small open-label trial20 and product information for treating adults with partial-onset seizures.25
Studies also demonstrate levetiracetam’s potential for relapse prevention during outpatient therapy. In a 10-week trial, levetiracetam decreased the number of standard drinks in alcohol-dependent patients from 5.3 to 1.7 per day.10 This was a small open trial, however, and large controlled trials support the usefulness of other, FDA-approved medications—including disulfiram, naltrexone, and acamprosate—for alcohol relapse prevention.
CASE REPORT 3: Gabapentin for acute withdrawal
Mr. B, age 38, presents to the ER after a 13-day alcohol binge. He has been drinking increasing amounts of alcohol over 6 weeks. Three months earlier, Mr. B was admitted for alcohol withdrawal treatment and received 49 mg of lorazepam over 3 days. This resulted in his transfer from the step-down unit to the intensive care unit for increased agitation, possibly caused by paradoxical disinhibition from excessive lorazepam use.26
Mr. B’s medical history is significant for alcohol-induced seizures, DTs, traumatic brain injury related to craniotomy, and right arm amputation. Mr. B drinks approximately 24 beers per day. He denies tobacco use but admits to past use of cocaine, marijuana, and heroin.
On admission, Mr. B’s BAC is 360 mg/dL (0.36%), AST is elevated at 72 U/L, ALT at 42 U/L, and LDH significantly elevated at 384 U/L. Urine drug screen is negative, and his CIWA-Ar score is 23. His score of –1 on the Richmond Agitation and Sedation Scale (RASS)27 correlates with very mild sedation.
Guided by Bonnet et al28 and clinical experience, we start Mr. B on gabapentin, 1,200 mg tid, and IV lorazepam, 2 mg every 8 hours as needed for breakthrough withdrawal. We decrease lorazepam by 50% every other day until Mr. B is discharged. On days 2, 3, and 4, Mr. B’s CIWA-Ar scores are 6, 9, and 2, respectively. His RASS score drops from –1 on days 1 and 2 to 0 until discharge, indicating an alert and calm state.
Mr. B requires a total of 2 mg of lorazepam throughout hospitalization. He finishes alcohol detoxification on day 4 and is discharged with a prescription for gabapentin, 1,200 mg tid. Two weeks later, when he is admitted to a 28-day inpatient alcohol rehabilitation unit, Mr. B has not relapsed.
More abstinent days
Gabapentin is FDA-approved as adjunctive therapy for partial seizures. Off-label, it has been generally efficacious as an adjunct in alcohol detoxification.29-32 We chose adjunctive anticonvulsant therapy for Mr. B because of his history of alcohol-induced seizures. We chose gabapentin instead of valproic acid because of Mr. B’s liver damage and gabapentin’s lack of hepatic metabolism.
Gabapentin may reduce alcohol consumption and craving in alcohol-dependent patients. By increasing the number of abstinent days, gabapentin may help patients maintain abstinence.33 Gabapentin does not appear to interact clinically with alcohol, causing neither sedation nor synergistic effects.34 Its relative lack of abuse potential may be valuable in outpatient alcohol withdrawal treatment and in maintaining alcohol abstinence after detoxification.
Related resource
- Asplund CA, Aaronson JW, Aaronson HE. 3 regimens for alcohol withdrawal and detoxification. J Fam Pract. 2004;53(7):545-554. www.jfponline.com/Pages.asp?AID=1730.
Drug brand names
- Acamprosate • Campral
- Alprazolam • Xanax
- Carbamazepine • Carbatrol
- Cimetidine • Tagamet
- Clonazepam • Klonopin
- Clonidine • Catapres
- Diazepam • Valium
- Disulfiram • Antabuse
- Felbamate • Felbatol
- Fluoxetine • Prozac
- Gabapentin • Neurontin
- Isoniazid • Nydrazid
- Lamotrigine • Lamictal
- Levetiracetam • Keppra
- Lorazepam • Ativan
- Naltrexone • ReVia, Vivitrol
- Oxazepam • Serax
- Phenobarbital • Luminal
- Phenytoin • Dilantin
- Propranolol • Inderal
- Rifampin • Rifadin
- Ritonavir • Norvir
- Temazepam • Restoril
- Triazolam • Halcion
- Valproic acid • Depakote, Depakene
Disclosures
Dr. Spiegel is a speaker for Pfizer Inc. and GlaxoSmithKline.
Dr. Radac reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
The authors thank Rishi Laroia, MD, Robert Swanson, MD, and Adam W. Coe, MD for their contributions to this article.
1. Mayo-Smith MF, Cushman P, Hill AJ, et al. Pharmacological management of alcohol withdrawal. JAMA. 1997;278:144-151.
2. Myrick H, Brady KT, Malcom R. Divalproex in the treatment of alcohol withdrawal—statistical data included. Am J Drug Alcohol Abuse. 2000;26(1):155-160.
3. Johnson BA, Swift RM, Addolorato G, et al. Safety and efficacy of GABAergic medications for treating alcoholism. Alcohol Clin Exp Res. 2005;29(2):248-254.
4. Lum E, Gorman SK, Slavik RS. Valproic acid management of acute alcohol withdrawal. Ann Pharmacother. 2006;40(3):441-448.
5. Trevisan LA, Boutros N, Petrakis IL, et al. Complications of alcohol withdrawal: pathophysiological insights. Alcohol Health Res World. 1998;22(1):61-66.
6. Esel E. Neurobiology of alcohol withdrawal inhibitory and excitatory neurotransmitters. Turk Psikiyatri Derg. 2006;7(2):129-138.
7. Myrick H, Brady KT. The use of divalproex in the treatment of addictive disorders. Psychopharmacol Bull. 2003;37(suppl 2):89-97.
8. Brady KT, Myrick H, Henderson S, et al. The use of divalproex in alcohol relapse prevention: a pilot study. Drug Alcohol Depend. 2002;67(3):323-330.
9. Longo L, Campbell T, Hubatch S. Divalproex sodium (Depakote) for alcohol withdrawal and relapse prevention. J Addict Dis. 2002;21(2):55-64.
10. Sarid-Segal O, Piechniczek-Buczek J, Knapp C, et al. The effects of levetiracetam on alcohol consumption in alcohol-dependent subjects: an open label study. Am J Drug Alcohol Abuse. 2008;34(4):441-447.
11. American Psychiatric Association. Treatments of psychiatric disorders: a task force report of the American Psychiatric Association. Washington, DC: American Psychiatric Association Press; 1989:187.
12. Mason BJ, Light JM, Williams LD, et al. Proof-of-concept human laboratory study for protracted abstinence in alcohol dependence: effects of gabapentin. Addict Biol. 2009;14(1):73-83.
13. Physicians’ Desk Reference 2009. 63rd ed. Montvale, NJ: Physicians’ Desk Reference; 2008:423-431.
14. Malcolm R, Ballenger JC, Sturgis ET, et al. Double-blind controlled trial comparing carbamazepine to oxazepam treatment of alcohol withdrawal. Am J Psychiatry. 1989;146:617-621.
15. Myrick H, Anton R, Voronin K, et al. A double-blind evaluation of gabapentin on alcohol effects and drinking in a clinical laboratory paradigm. Alcohol Clin Exp Res. 2007;31(2):221-227.
16. Malcolm R, Myrick H, Brady KT, et al. Update on anticonvulsants for the treatment of alcohol withdrawal. Am J Addict. 2001;10(suppl):16-23.
17. Davis LL, Ryan W, Adinoff B, et al. Comprehensive review of the psychiatric uses of valproate. J Clin Psychopharm. 2000;20(1 suppl 1):1S-17S.
18. Rosenthal RN, Perkel C, Singh P, et al. A pilot open randomized trial of valproate and phenobarbital in the treatment of acute alcohol withdrawal. Am J Addict. 1998;7:189-197.
19. Book SW, Myrick H. Novel anticonvulsants in the treatment of alcoholism. Expert Opin Investig Drugs. 2005;14(4):371-376.
20. Krebs M, Leopold K, Richter C, et al. Levetiracetam for the treatment of alcohol withdrawal syndrome: an open-label pilot trial. J Clin Psychopharmacol. 2006;26(3):347-349.
21. LaRoche SM, Helmers SL. The new antiepileptic drugs: scientific review. JAMA. 2004;291:605-614.
22. Chabolla DR, Harnois DM, Meschia JF. Levetiracetam monotherapy for liver transplant patients with seizures. Transplant Proc. 2003;35:1480-1481.
23. Paul F, Meencke HJ. Levetiracetam in focal epilepsy and hepatic porphyria: a case report. Epilepsia. 2004;45:559-560.
24. Ntais C, Pakos E, Kyzas P, et al. Benzodiazepines for alcohol withdrawal. Cochrane Database Syst Rev. 2005;(3):CD005063.-
25. Physicians’ Desk Reference 2009. 63rd ed. Montvale, NJ: Physicians’ Desk Reference; 2008:3131-3143.
26. Saias T, Gallarda T. Paradoxical aggressive reactions to benzodiazepine use: a review. Encephale. 2008;34(4):330-336.
27. Sessler CN, Gosnell MS, Grap MJ, et al. The Richmond Agitation–Sedation Scale: validity and reliability in adult intensive care unit patients. Am J Respir Crit Care Med. 2002;166:1338-1344.
28. Bonnet U, Banger M, Leweke FM, et al. Treatment of acute alcohol withdrawal with gabapentin: results from a controlled two-center trial. J Clin Psychopharmacol. 2003;23(5):514-519.
29. Myrick H, Malcolm R, Brady KT. Gabapentin treatment of alcohol withdrawal. Am J Psychiatry. 1998;155:1626.-
30. Bonnet U, Banger M, Leweke FM, et al. Treatment of alcohol withdrawal syndrome with gabapentin. Pharmacopsychiatry. 1999;32:107-109.
31. Mariani JJ, Rosenthal RN, Tross S, et al. A randomized, open-label, controlled trial of gabapentin and phenobarbital in the treatment of alcohol withdrawal. Am J Addict. 2006;15(1):76-84.
32. Voris J, Smith NL, Rao SM, et al. Gabapentin for the treatment of ethanol withdrawal. Subst Abus. 2003;24(2):129-132.
33. Furieri FA, Nakamura-Palacios EM. Gabapentin reduces alcohol consumption and craving: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2007;68(11):1691-1700.
34. Bisaga A, Evans SM. The acute effects of gabapentin in combination with alcohol in heavy drinkers. Drug Alcohol Depend. 2006;83(1):25-32.
1. Mayo-Smith MF, Cushman P, Hill AJ, et al. Pharmacological management of alcohol withdrawal. JAMA. 1997;278:144-151.
2. Myrick H, Brady KT, Malcom R. Divalproex in the treatment of alcohol withdrawal—statistical data included. Am J Drug Alcohol Abuse. 2000;26(1):155-160.
3. Johnson BA, Swift RM, Addolorato G, et al. Safety and efficacy of GABAergic medications for treating alcoholism. Alcohol Clin Exp Res. 2005;29(2):248-254.
4. Lum E, Gorman SK, Slavik RS. Valproic acid management of acute alcohol withdrawal. Ann Pharmacother. 2006;40(3):441-448.
5. Trevisan LA, Boutros N, Petrakis IL, et al. Complications of alcohol withdrawal: pathophysiological insights. Alcohol Health Res World. 1998;22(1):61-66.
6. Esel E. Neurobiology of alcohol withdrawal inhibitory and excitatory neurotransmitters. Turk Psikiyatri Derg. 2006;7(2):129-138.
7. Myrick H, Brady KT. The use of divalproex in the treatment of addictive disorders. Psychopharmacol Bull. 2003;37(suppl 2):89-97.
8. Brady KT, Myrick H, Henderson S, et al. The use of divalproex in alcohol relapse prevention: a pilot study. Drug Alcohol Depend. 2002;67(3):323-330.
9. Longo L, Campbell T, Hubatch S. Divalproex sodium (Depakote) for alcohol withdrawal and relapse prevention. J Addict Dis. 2002;21(2):55-64.
10. Sarid-Segal O, Piechniczek-Buczek J, Knapp C, et al. The effects of levetiracetam on alcohol consumption in alcohol-dependent subjects: an open label study. Am J Drug Alcohol Abuse. 2008;34(4):441-447.
11. American Psychiatric Association. Treatments of psychiatric disorders: a task force report of the American Psychiatric Association. Washington, DC: American Psychiatric Association Press; 1989:187.
12. Mason BJ, Light JM, Williams LD, et al. Proof-of-concept human laboratory study for protracted abstinence in alcohol dependence: effects of gabapentin. Addict Biol. 2009;14(1):73-83.
13. Physicians’ Desk Reference 2009. 63rd ed. Montvale, NJ: Physicians’ Desk Reference; 2008:423-431.
14. Malcolm R, Ballenger JC, Sturgis ET, et al. Double-blind controlled trial comparing carbamazepine to oxazepam treatment of alcohol withdrawal. Am J Psychiatry. 1989;146:617-621.
15. Myrick H, Anton R, Voronin K, et al. A double-blind evaluation of gabapentin on alcohol effects and drinking in a clinical laboratory paradigm. Alcohol Clin Exp Res. 2007;31(2):221-227.
16. Malcolm R, Myrick H, Brady KT, et al. Update on anticonvulsants for the treatment of alcohol withdrawal. Am J Addict. 2001;10(suppl):16-23.
17. Davis LL, Ryan W, Adinoff B, et al. Comprehensive review of the psychiatric uses of valproate. J Clin Psychopharm. 2000;20(1 suppl 1):1S-17S.
18. Rosenthal RN, Perkel C, Singh P, et al. A pilot open randomized trial of valproate and phenobarbital in the treatment of acute alcohol withdrawal. Am J Addict. 1998;7:189-197.
19. Book SW, Myrick H. Novel anticonvulsants in the treatment of alcoholism. Expert Opin Investig Drugs. 2005;14(4):371-376.
20. Krebs M, Leopold K, Richter C, et al. Levetiracetam for the treatment of alcohol withdrawal syndrome: an open-label pilot trial. J Clin Psychopharmacol. 2006;26(3):347-349.
21. LaRoche SM, Helmers SL. The new antiepileptic drugs: scientific review. JAMA. 2004;291:605-614.
22. Chabolla DR, Harnois DM, Meschia JF. Levetiracetam monotherapy for liver transplant patients with seizures. Transplant Proc. 2003;35:1480-1481.
23. Paul F, Meencke HJ. Levetiracetam in focal epilepsy and hepatic porphyria: a case report. Epilepsia. 2004;45:559-560.
24. Ntais C, Pakos E, Kyzas P, et al. Benzodiazepines for alcohol withdrawal. Cochrane Database Syst Rev. 2005;(3):CD005063.-
25. Physicians’ Desk Reference 2009. 63rd ed. Montvale, NJ: Physicians’ Desk Reference; 2008:3131-3143.
26. Saias T, Gallarda T. Paradoxical aggressive reactions to benzodiazepine use: a review. Encephale. 2008;34(4):330-336.
27. Sessler CN, Gosnell MS, Grap MJ, et al. The Richmond Agitation–Sedation Scale: validity and reliability in adult intensive care unit patients. Am J Respir Crit Care Med. 2002;166:1338-1344.
28. Bonnet U, Banger M, Leweke FM, et al. Treatment of acute alcohol withdrawal with gabapentin: results from a controlled two-center trial. J Clin Psychopharmacol. 2003;23(5):514-519.
29. Myrick H, Malcolm R, Brady KT. Gabapentin treatment of alcohol withdrawal. Am J Psychiatry. 1998;155:1626.-
30. Bonnet U, Banger M, Leweke FM, et al. Treatment of alcohol withdrawal syndrome with gabapentin. Pharmacopsychiatry. 1999;32:107-109.
31. Mariani JJ, Rosenthal RN, Tross S, et al. A randomized, open-label, controlled trial of gabapentin and phenobarbital in the treatment of alcohol withdrawal. Am J Addict. 2006;15(1):76-84.
32. Voris J, Smith NL, Rao SM, et al. Gabapentin for the treatment of ethanol withdrawal. Subst Abus. 2003;24(2):129-132.
33. Furieri FA, Nakamura-Palacios EM. Gabapentin reduces alcohol consumption and craving: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2007;68(11):1691-1700.
34. Bisaga A, Evans SM. The acute effects of gabapentin in combination with alcohol in heavy drinkers. Drug Alcohol Depend. 2006;83(1):25-32.