User login
The Journal of Clinical Outcomes Management® is an independent, peer-reviewed journal offering evidence-based, practical information for improving the quality, safety, and value of health care.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
CDC: Masking no longer required in health care settings
It’s a “major departure” from the CDC’s previous recommendation of universal masking to fight the COVID-19 pandemic, The Hill says.
“Updates were made to reflect the high levels of vaccine-and infection-induced immunity and the availability of effective treatments and prevention tools,” the CDC’s new guidance says.
The agency now says that facilities in areas without high transmission can decide for themselves whether to require everyone – doctors, patients, and visitors – to wear masks.
Community transmission “is the metric currently recommended to guide select practices in healthcare settings to allow for earlier intervention, before there is strain on the health care system and to better protect the individuals seeking care in these settings,” the CDC said.
About 73% of the country is having “high” rates of transmission, The Hill said.
“Community transmission” is different from the “community level” metric that’s used for non–health care settings.
Community transmission refers to measures of the presence and spread of SARS-CoV-2, the CDC said. “Community levels place an emphasis on measures of the impact of COVID-19 in terms of hospitalizations and health care system strain, while accounting for transmission in the community.”
Just 7% of counties are considered high risk, while nearly 62 percent are low.
The new guidance applies wherever health care is delivered, including nursing homes and home health, the CDC said.
A version of this article first appeared on WebMD.com.
It’s a “major departure” from the CDC’s previous recommendation of universal masking to fight the COVID-19 pandemic, The Hill says.
“Updates were made to reflect the high levels of vaccine-and infection-induced immunity and the availability of effective treatments and prevention tools,” the CDC’s new guidance says.
The agency now says that facilities in areas without high transmission can decide for themselves whether to require everyone – doctors, patients, and visitors – to wear masks.
Community transmission “is the metric currently recommended to guide select practices in healthcare settings to allow for earlier intervention, before there is strain on the health care system and to better protect the individuals seeking care in these settings,” the CDC said.
About 73% of the country is having “high” rates of transmission, The Hill said.
“Community transmission” is different from the “community level” metric that’s used for non–health care settings.
Community transmission refers to measures of the presence and spread of SARS-CoV-2, the CDC said. “Community levels place an emphasis on measures of the impact of COVID-19 in terms of hospitalizations and health care system strain, while accounting for transmission in the community.”
Just 7% of counties are considered high risk, while nearly 62 percent are low.
The new guidance applies wherever health care is delivered, including nursing homes and home health, the CDC said.
A version of this article first appeared on WebMD.com.
It’s a “major departure” from the CDC’s previous recommendation of universal masking to fight the COVID-19 pandemic, The Hill says.
“Updates were made to reflect the high levels of vaccine-and infection-induced immunity and the availability of effective treatments and prevention tools,” the CDC’s new guidance says.
The agency now says that facilities in areas without high transmission can decide for themselves whether to require everyone – doctors, patients, and visitors – to wear masks.
Community transmission “is the metric currently recommended to guide select practices in healthcare settings to allow for earlier intervention, before there is strain on the health care system and to better protect the individuals seeking care in these settings,” the CDC said.
About 73% of the country is having “high” rates of transmission, The Hill said.
“Community transmission” is different from the “community level” metric that’s used for non–health care settings.
Community transmission refers to measures of the presence and spread of SARS-CoV-2, the CDC said. “Community levels place an emphasis on measures of the impact of COVID-19 in terms of hospitalizations and health care system strain, while accounting for transmission in the community.”
Just 7% of counties are considered high risk, while nearly 62 percent are low.
The new guidance applies wherever health care is delivered, including nursing homes and home health, the CDC said.
A version of this article first appeared on WebMD.com.
SMART-CHOICE 3-year results support dropping aspirin after PCI
Shortening the duration of dual-antiplatelet therapy (DAPT) and continuing with a P2Y12 inhibitor alone after percutaneous coronary intervention (PCI) was associated with a similar rate of ischemic events but with less bleeding than prolonged DAPT after 3 years of follow-up in the SMART-CHOICE trial.
“The current the investigators, with lead author Ki Hong Choi, MD, division of cardiology, department of medicine, Samsung Medical Center, Sungkyunkwan University, Seoul, South Korea, conclude.
The 3-year results from the study were published online in JAMA Cardiology.
The authors explain that although dual therapy with aspirin and a P2Y12 inhibitor after PCI with a drug-eluting stent (DES) is crucial to reduce the risk of ischemic events, it raises concerns about increased risk of bleeding, and the antiplatelet strategy after PCI is currently shifting to reduce the duration of DAPT.
Several recent randomized studies have consistently shown that a short duration of DAPT (1-3 months) followed by P2Y12 inhibitor monotherapy had ischemia protection effects comparable with that of DAPT of longer duration, and it was associated with a significantly reduced risk of bleeding events in patients who underwent PCI, they note. However, these studies have so far reported only 1-year outcomes, and long-term results are not yet available.
The SMART-CHOICE trial compared two antiplatelet strategies – 3 months of DAPT followed by long-term P2Y12 inhibitor monotherapy (mainly with clopidogrel) or prolonged DAPT for 12 months or longer – in 2,993 patients who had undergone PCI with a drug-eluting stent. Results at 12 months showed a similar rate of ischemic events with both strategies but a lower rate of bleeding in the group that received shortened DAPT.
The SMART-CHOICE investigators now report the 3-year results showing similar outcomes.
At 3 years, the primary endpoint, a composite of all-cause death, myocardial infarction, or stroke, had occurred in 6.3% of the shortened DAPT group and 6.1% in the prolonged DAPT group, giving a hazard ratio of 1.06 (95% confidence interval, 0.79-1.44).
But in the shortened DAPT group, the risk of bleeding was reduced. Bleeding Academic Research Consortium (BARC) types 2-5 bleeding had occurred in 3.2% of the shortened DAPT group and in 8.2% of the prolonged DAPT group (hazard ratio, 0.39; 95% CI, 0.28-0.55). Major bleeding, BARC types 3-5, occurred in 1.2% of the shortened DAPT group and in 2.4% of the prolonged DAPT group (HR, 0.56; 95% CI 0.31-0.99).
The landmark analyses between 3 months and 3 years and per-protocol analyses showed consistent results.
The researchers point out that this is the first trial to report on the long-term safety and efficacy of P2Y12-inhibitor monotherapy as long-term maintenance therapy for stable patients treated with PCI.
“Especially considering that extended DAPT significantly reduced the risks of ischemic events compared with aspirin monotherapy in a couple of trials, comparison between P2Y12-inhibitor monotherapy and prolonged DAPT for recurrent ischemic events over a longer period beyond 1 year is of great importance,” they say.
They cite two other trials – HOST-EXAM and GLOBAL LEADERS – which have shown P2Y12-inhibitor monotherapy to be superior to aspirin monotherapy in preventing both ischemic and bleeding events during the long-term maintenance period after PCI.
“Combining the results of the current study, HOST-EXAM trial, and landmark analysis of the GLOBAL LEADERS trial, long-term P2Y12-inhibitor monotherapy after a minimum period of DAPT might be the most reliable option from among aspirin monotherapy, P2Y12 monotherapy, and extended DAPT for maintenance therapy after stabilizing patients who have undergone PCI with a current-generation DES,” they conclude.
They note that the American College of Cardiology/American Heart Association/Society for Cardiovascular Angiography and Interventions guidelines for coronary artery revascularization newly recommends a shorter course of DAPT followed by P2Y12 monotherapy as a class IIa indication. The recommendation is based on results of five large, randomized clinical trials, including SMART-CHOICE, TWILIGHT, STOPDAPT-2, TICO, and GLOBAL LEADERS.
“The current results of extended follow-up from the SMART-CHOICE trial support evidence of aspirin-dropping strategy with indefinite use of P2Y12 inhibitor after minimum use of DAPT in patients who underwent PCI,” they say.
They point out that two further trials, A-CLOSE in high-risk patients and SMART-CHOICE III, will be helpful to confirm these findings.
P2Y12-inhibitor monotherapy ‘attractive concept’
In an accompanying editor’s note, Ajay Kirtane, MD, Columbia University Irving Medical Center/New York–Presbyterian Hospital, New York, and Roxana Mehran, MD, Icahn School of Medicine at Mount Sinai and the Cardiovascular Research Foundation, New York, note that current guidelines recommend 3-6 months of DAPT following PCI with current-generation drug-eluting stents in stable patients and 6-12 months or longer for those with acute coronary syndromes. For patients at higher risk of bleeding, even shorter DAPT durations can be considered on a case-by-case basis.
Historically, the component of DAPT subject to discontinuation decisions was the P2Y12 inhibitor (clopidogrel, prasugrel, or ticagrelor), but more recent trials have further explored whether discontinuation of the aspirin component of DAPT can mitigate bleeding while preserving anti-ischemic efficacy.
The editorialists explain that the concept of P2Y1-inhibitor monotherapy is attractive because it may optimize antiplatelet effects through a single agent that can avoid the gastrointestinal toxicity of aspirin as well as the increased bleeding that comes with combing multiple antithrombotic agents.
They suggest that the long-term results from the SMART-CHOICE trial “should lead clinicians to consider a strategy of monotherapy after a short period of DAPT as a viable one to mitigate bleeding risk,” although they also point out that SMART-CHOICE was underpowered to rigorously assess ischemic differences, so caution is warranted.
“For patients at greatest risk for recurrent ischemic events, the role of continued DAPT is always an option, but these data (and other consistent trials) give clinicians more options to pursue individualized treatment decisions,” they write.
“To some, the continually moving field of post-PCI antiplatelet therapy has provided too many choices, which can at times be dizzying. To us, every patient is different, and thoughtful evidence-based consideration is increasingly possible for many of our treatment decisions,” they conclude.
The SMART-CHOICE study was supported by unrestricted grants from the Korean Society of Interventional Cardiology, Abbott Vascular, Biotronik, and Boston Scientific.
A version of this article first appeared on Medscape.com.
Shortening the duration of dual-antiplatelet therapy (DAPT) and continuing with a P2Y12 inhibitor alone after percutaneous coronary intervention (PCI) was associated with a similar rate of ischemic events but with less bleeding than prolonged DAPT after 3 years of follow-up in the SMART-CHOICE trial.
“The current the investigators, with lead author Ki Hong Choi, MD, division of cardiology, department of medicine, Samsung Medical Center, Sungkyunkwan University, Seoul, South Korea, conclude.
The 3-year results from the study were published online in JAMA Cardiology.
The authors explain that although dual therapy with aspirin and a P2Y12 inhibitor after PCI with a drug-eluting stent (DES) is crucial to reduce the risk of ischemic events, it raises concerns about increased risk of bleeding, and the antiplatelet strategy after PCI is currently shifting to reduce the duration of DAPT.
Several recent randomized studies have consistently shown that a short duration of DAPT (1-3 months) followed by P2Y12 inhibitor monotherapy had ischemia protection effects comparable with that of DAPT of longer duration, and it was associated with a significantly reduced risk of bleeding events in patients who underwent PCI, they note. However, these studies have so far reported only 1-year outcomes, and long-term results are not yet available.
The SMART-CHOICE trial compared two antiplatelet strategies – 3 months of DAPT followed by long-term P2Y12 inhibitor monotherapy (mainly with clopidogrel) or prolonged DAPT for 12 months or longer – in 2,993 patients who had undergone PCI with a drug-eluting stent. Results at 12 months showed a similar rate of ischemic events with both strategies but a lower rate of bleeding in the group that received shortened DAPT.
The SMART-CHOICE investigators now report the 3-year results showing similar outcomes.
At 3 years, the primary endpoint, a composite of all-cause death, myocardial infarction, or stroke, had occurred in 6.3% of the shortened DAPT group and 6.1% in the prolonged DAPT group, giving a hazard ratio of 1.06 (95% confidence interval, 0.79-1.44).
But in the shortened DAPT group, the risk of bleeding was reduced. Bleeding Academic Research Consortium (BARC) types 2-5 bleeding had occurred in 3.2% of the shortened DAPT group and in 8.2% of the prolonged DAPT group (hazard ratio, 0.39; 95% CI, 0.28-0.55). Major bleeding, BARC types 3-5, occurred in 1.2% of the shortened DAPT group and in 2.4% of the prolonged DAPT group (HR, 0.56; 95% CI 0.31-0.99).
The landmark analyses between 3 months and 3 years and per-protocol analyses showed consistent results.
The researchers point out that this is the first trial to report on the long-term safety and efficacy of P2Y12-inhibitor monotherapy as long-term maintenance therapy for stable patients treated with PCI.
“Especially considering that extended DAPT significantly reduced the risks of ischemic events compared with aspirin monotherapy in a couple of trials, comparison between P2Y12-inhibitor monotherapy and prolonged DAPT for recurrent ischemic events over a longer period beyond 1 year is of great importance,” they say.
They cite two other trials – HOST-EXAM and GLOBAL LEADERS – which have shown P2Y12-inhibitor monotherapy to be superior to aspirin monotherapy in preventing both ischemic and bleeding events during the long-term maintenance period after PCI.
“Combining the results of the current study, HOST-EXAM trial, and landmark analysis of the GLOBAL LEADERS trial, long-term P2Y12-inhibitor monotherapy after a minimum period of DAPT might be the most reliable option from among aspirin monotherapy, P2Y12 monotherapy, and extended DAPT for maintenance therapy after stabilizing patients who have undergone PCI with a current-generation DES,” they conclude.
They note that the American College of Cardiology/American Heart Association/Society for Cardiovascular Angiography and Interventions guidelines for coronary artery revascularization newly recommends a shorter course of DAPT followed by P2Y12 monotherapy as a class IIa indication. The recommendation is based on results of five large, randomized clinical trials, including SMART-CHOICE, TWILIGHT, STOPDAPT-2, TICO, and GLOBAL LEADERS.
“The current results of extended follow-up from the SMART-CHOICE trial support evidence of aspirin-dropping strategy with indefinite use of P2Y12 inhibitor after minimum use of DAPT in patients who underwent PCI,” they say.
They point out that two further trials, A-CLOSE in high-risk patients and SMART-CHOICE III, will be helpful to confirm these findings.
P2Y12-inhibitor monotherapy ‘attractive concept’
In an accompanying editor’s note, Ajay Kirtane, MD, Columbia University Irving Medical Center/New York–Presbyterian Hospital, New York, and Roxana Mehran, MD, Icahn School of Medicine at Mount Sinai and the Cardiovascular Research Foundation, New York, note that current guidelines recommend 3-6 months of DAPT following PCI with current-generation drug-eluting stents in stable patients and 6-12 months or longer for those with acute coronary syndromes. For patients at higher risk of bleeding, even shorter DAPT durations can be considered on a case-by-case basis.
Historically, the component of DAPT subject to discontinuation decisions was the P2Y12 inhibitor (clopidogrel, prasugrel, or ticagrelor), but more recent trials have further explored whether discontinuation of the aspirin component of DAPT can mitigate bleeding while preserving anti-ischemic efficacy.
The editorialists explain that the concept of P2Y1-inhibitor monotherapy is attractive because it may optimize antiplatelet effects through a single agent that can avoid the gastrointestinal toxicity of aspirin as well as the increased bleeding that comes with combing multiple antithrombotic agents.
They suggest that the long-term results from the SMART-CHOICE trial “should lead clinicians to consider a strategy of monotherapy after a short period of DAPT as a viable one to mitigate bleeding risk,” although they also point out that SMART-CHOICE was underpowered to rigorously assess ischemic differences, so caution is warranted.
“For patients at greatest risk for recurrent ischemic events, the role of continued DAPT is always an option, but these data (and other consistent trials) give clinicians more options to pursue individualized treatment decisions,” they write.
“To some, the continually moving field of post-PCI antiplatelet therapy has provided too many choices, which can at times be dizzying. To us, every patient is different, and thoughtful evidence-based consideration is increasingly possible for many of our treatment decisions,” they conclude.
The SMART-CHOICE study was supported by unrestricted grants from the Korean Society of Interventional Cardiology, Abbott Vascular, Biotronik, and Boston Scientific.
A version of this article first appeared on Medscape.com.
Shortening the duration of dual-antiplatelet therapy (DAPT) and continuing with a P2Y12 inhibitor alone after percutaneous coronary intervention (PCI) was associated with a similar rate of ischemic events but with less bleeding than prolonged DAPT after 3 years of follow-up in the SMART-CHOICE trial.
“The current the investigators, with lead author Ki Hong Choi, MD, division of cardiology, department of medicine, Samsung Medical Center, Sungkyunkwan University, Seoul, South Korea, conclude.
The 3-year results from the study were published online in JAMA Cardiology.
The authors explain that although dual therapy with aspirin and a P2Y12 inhibitor after PCI with a drug-eluting stent (DES) is crucial to reduce the risk of ischemic events, it raises concerns about increased risk of bleeding, and the antiplatelet strategy after PCI is currently shifting to reduce the duration of DAPT.
Several recent randomized studies have consistently shown that a short duration of DAPT (1-3 months) followed by P2Y12 inhibitor monotherapy had ischemia protection effects comparable with that of DAPT of longer duration, and it was associated with a significantly reduced risk of bleeding events in patients who underwent PCI, they note. However, these studies have so far reported only 1-year outcomes, and long-term results are not yet available.
The SMART-CHOICE trial compared two antiplatelet strategies – 3 months of DAPT followed by long-term P2Y12 inhibitor monotherapy (mainly with clopidogrel) or prolonged DAPT for 12 months or longer – in 2,993 patients who had undergone PCI with a drug-eluting stent. Results at 12 months showed a similar rate of ischemic events with both strategies but a lower rate of bleeding in the group that received shortened DAPT.
The SMART-CHOICE investigators now report the 3-year results showing similar outcomes.
At 3 years, the primary endpoint, a composite of all-cause death, myocardial infarction, or stroke, had occurred in 6.3% of the shortened DAPT group and 6.1% in the prolonged DAPT group, giving a hazard ratio of 1.06 (95% confidence interval, 0.79-1.44).
But in the shortened DAPT group, the risk of bleeding was reduced. Bleeding Academic Research Consortium (BARC) types 2-5 bleeding had occurred in 3.2% of the shortened DAPT group and in 8.2% of the prolonged DAPT group (hazard ratio, 0.39; 95% CI, 0.28-0.55). Major bleeding, BARC types 3-5, occurred in 1.2% of the shortened DAPT group and in 2.4% of the prolonged DAPT group (HR, 0.56; 95% CI 0.31-0.99).
The landmark analyses between 3 months and 3 years and per-protocol analyses showed consistent results.
The researchers point out that this is the first trial to report on the long-term safety and efficacy of P2Y12-inhibitor monotherapy as long-term maintenance therapy for stable patients treated with PCI.
“Especially considering that extended DAPT significantly reduced the risks of ischemic events compared with aspirin monotherapy in a couple of trials, comparison between P2Y12-inhibitor monotherapy and prolonged DAPT for recurrent ischemic events over a longer period beyond 1 year is of great importance,” they say.
They cite two other trials – HOST-EXAM and GLOBAL LEADERS – which have shown P2Y12-inhibitor monotherapy to be superior to aspirin monotherapy in preventing both ischemic and bleeding events during the long-term maintenance period after PCI.
“Combining the results of the current study, HOST-EXAM trial, and landmark analysis of the GLOBAL LEADERS trial, long-term P2Y12-inhibitor monotherapy after a minimum period of DAPT might be the most reliable option from among aspirin monotherapy, P2Y12 monotherapy, and extended DAPT for maintenance therapy after stabilizing patients who have undergone PCI with a current-generation DES,” they conclude.
They note that the American College of Cardiology/American Heart Association/Society for Cardiovascular Angiography and Interventions guidelines for coronary artery revascularization newly recommends a shorter course of DAPT followed by P2Y12 monotherapy as a class IIa indication. The recommendation is based on results of five large, randomized clinical trials, including SMART-CHOICE, TWILIGHT, STOPDAPT-2, TICO, and GLOBAL LEADERS.
“The current results of extended follow-up from the SMART-CHOICE trial support evidence of aspirin-dropping strategy with indefinite use of P2Y12 inhibitor after minimum use of DAPT in patients who underwent PCI,” they say.
They point out that two further trials, A-CLOSE in high-risk patients and SMART-CHOICE III, will be helpful to confirm these findings.
P2Y12-inhibitor monotherapy ‘attractive concept’
In an accompanying editor’s note, Ajay Kirtane, MD, Columbia University Irving Medical Center/New York–Presbyterian Hospital, New York, and Roxana Mehran, MD, Icahn School of Medicine at Mount Sinai and the Cardiovascular Research Foundation, New York, note that current guidelines recommend 3-6 months of DAPT following PCI with current-generation drug-eluting stents in stable patients and 6-12 months or longer for those with acute coronary syndromes. For patients at higher risk of bleeding, even shorter DAPT durations can be considered on a case-by-case basis.
Historically, the component of DAPT subject to discontinuation decisions was the P2Y12 inhibitor (clopidogrel, prasugrel, or ticagrelor), but more recent trials have further explored whether discontinuation of the aspirin component of DAPT can mitigate bleeding while preserving anti-ischemic efficacy.
The editorialists explain that the concept of P2Y1-inhibitor monotherapy is attractive because it may optimize antiplatelet effects through a single agent that can avoid the gastrointestinal toxicity of aspirin as well as the increased bleeding that comes with combing multiple antithrombotic agents.
They suggest that the long-term results from the SMART-CHOICE trial “should lead clinicians to consider a strategy of monotherapy after a short period of DAPT as a viable one to mitigate bleeding risk,” although they also point out that SMART-CHOICE was underpowered to rigorously assess ischemic differences, so caution is warranted.
“For patients at greatest risk for recurrent ischemic events, the role of continued DAPT is always an option, but these data (and other consistent trials) give clinicians more options to pursue individualized treatment decisions,” they write.
“To some, the continually moving field of post-PCI antiplatelet therapy has provided too many choices, which can at times be dizzying. To us, every patient is different, and thoughtful evidence-based consideration is increasingly possible for many of our treatment decisions,” they conclude.
The SMART-CHOICE study was supported by unrestricted grants from the Korean Society of Interventional Cardiology, Abbott Vascular, Biotronik, and Boston Scientific.
A version of this article first appeared on Medscape.com.
FROM JAMA CARDIOLOGY
How to improve diagnosis of HFpEF, common in diabetes
STOCKHOLM – Recent study results confirm that two agents from the sodium-glucose cotransporter 2 (SGLT2) inhibitor class can significantly cut the incidence of adverse cardiovascular events in patients with heart failure with reduced ejection fraction (HFpEF), a disease especially common in people with type 2 diabetes, obesity, or both.
And findings from secondary analyses of the studies – including one reported at the annual meeting of the European Association for the Study of Diabetes – show that these SGLT2 inhibitors work as well for cutting incident adverse events (cardiovascular death or worsening heart failure) in patients with HFpEF and diabetes as they do for people with normal blood glucose levels.
But delivering treatment with these proven agents, dapagliflozin (Farxiga) and empagliflozin (Jardiance), first requires diagnosis of HFpEF, a task that clinicians have historically fallen short in accomplishing.
When in 2021, results from the EMPEROR-Preserved trial with empagliflozin and when in September 2022 results from the DELIVER trial with dapagliflozin established the efficacy of these two SGLT2 inhibitors as the first treatments proven to benefit patients with HFpEF, they also raised the stakes for clinicians to be much more diligent and systematic in evaluating people at high risk for developing HFpEF because of having type 2 diabetes or obesity, two of the most potent risk factors for this form of heart failure.
‘Vigilance ... needs to increase’
“Vigilance for HFpEF needs to increase because we can now help these patients,” declared Lars H. Lund, MD, PhD, speaking at the meeting. “Type 2 diabetes dramatically increases the incidence of HFpEF,” and the mechanisms by which it does this are “especially amenable to treatment with SGLT2 inhibitors,” said Dr. Lund, a cardiologist and heart failure specialist at the Karolinska Institute, Stockholm.
HFpEF has a history of going undetected in people with type 2 diabetes, an ironic situation given its high incidence as well as the elevated rate of adverse cardiovascular events when heart failure occurs in patients with type 2 diabetes compared with patients who do not have diabetes.
The key, say experts, is for clinicians to maintain a high index of suspicion for signs and symptoms of heart failure in people with type 2 diabetes and to regularly assess them, starting with just a few simple questions that probe for the presence of dyspnea, exertional fatigue, or both, an approach not widely employed up to now.
Clinicians who care for people with type 2 diabetes must become “alert to thinking about heart failure and alert to asking questions about signs and symptoms” that flag the presence of HFpEF, advised Naveed Sattar, MBChB, PhD, a professor of metabolic medicine at the University of Glasgow.
Soon, medical groups will issue guidelines for appropriate assessment for the presence of HFpEF in people with type 2 diabetes, Dr. Sattar predicted in an interview.
A need to probe
“You can’t simply ask patients with type 2 diabetes whether they have shortness of breath or exertional fatigue and stop there,” because often their first response will be no.
“Commonly, patients will initially say they have no dyspnea, but when you probe further, you find symptoms,” noted Mikhail N. Kosiborod, MD, codirector of Saint Luke’s Cardiometabolic Center of Excellence in Kansas City, Mo.
These people are often sedentary, so they frequently don’t experience shortness of breath at baseline, Dr. Kosiborod said in an interview. In some cases, they may limit their activity because of their exertional intolerance.
Once a person’s suggestive symptoms become known, the next step is to measure the serum level of N-terminal pro-B-type natriuretic peptide (NT-proBNP), a biomarker considered to be a generally reliable signal of existing heart failure when elevated.
Any value above 125 pg/mL is suggestive of prevalent heart failure and should lead to the next diagnostic step of echocardiography, Dr. Sattar said.
Elevated NT-proBNP has such good positive predictive value for identifying heart failure that it is tempting to use it broadly in people with type 2 diabetes. A 2022 consensus report from the American Diabetes Association says that “measurement of a natriuretic peptide [such as NT-proBNP] or high-sensitivity cardiac troponin is recommended on at least a yearly basis to identify the earliest HF [heart failure] stages and implement strategies to prevent transition to symptomatic HF.”
Test costs require targeting
But because of the relatively high current price for an NT-proBNP test, the cost-benefit ratio for widespread annual testing of all people with type 2 diabetes would be poor, some experts caution.
“Screening everyone may not be the right answer. Hundreds of millions of people worldwide” have type 2 diabetes. “You first need to target evaluation to people with symptoms,” advised Dr. Kosiborod.
He also warned that a low NT-proBNP level does not always rule out HFpEF, especially among people with type 2 diabetes who also have overweight or obesity, because NT-proBNP levels can be “artificially low” in people with obesity.
Other potential aids to diagnosis are assessment scores that researchers have developed, such as the H2FPEF score, which relies on variables that include age, obesity, and the presence of atrial fibrillation and hypertension.
However, this score also requires an echocardiography examination, another test that would have a questionable cost-benefit ratio if performed widely for patients with type 2 diabetes without targeting, Dr. Kosiborod said.
SGLT2 inhibitors benefit HFpEF regardless of glucose levels
A prespecified analysis of the DELIVER results that divided the study cohort on the basis of their glycemic status proved the efficacy of the SGLT2 inhibitor dapagliflozin for patients with HFpEF regardless of whether or not they had type 2 diabetes, prediabetes, or were normoglycemic at entry into the study, Silvio E. Inzucchi, MD, reported at the EASD meeting.

Treatment with dapagliflozin cut the incidence of the trial’s primary outcome of cardiovascular death or worsening heart failure by a significant 18% relative to placebo among all enrolled patients.
The new analysis reported by Dr. Inzucchi showed that treatment was associated with a 23% relative risk reduction among those with normoglycemia, a 13% reduction among those with prediabetes, and a 19% reduction among those with type 2 diabetes, with no signal of a significant difference among the three subgroups.
“There was no statistical interaction between categorical glycemic subgrouping and dapagliflozin’s treatment effect,” concluded Dr. Inzucchi, director of the Yale Medicine Diabetes Center, New Haven, Conn.
He also reported that, among the 6,259 people in the trial with HFpEF, 50% had diabetes, 31% had prediabetes, and a scant 19% had normoglycemia. The finding highlights once again the high prevalence of dysglycemia among people with HFpEF.
Previously, a prespecified secondary analysis of data from the EMPEROR-Preserved trial yielded similar findings for empagliflozin that showed the agent’s efficacy for people with HFpEF across the range of glucose levels.
The DELIVER trial was funded by AstraZeneca, the company that markets dapagliflozin (Farxiga). The EMPEROR-Preserved trial was sponsored by Boehringer Ingelheim and Eli Lilly, the companies that jointly market empagliflozin (Jardiance). Dr. Lund has been a consultant to AstraZeneca and Boehringer Ingelheim and to numerous other companies, and he is a stockholder in AnaCardio. Dr. Sattar has been a consultant to and has received research support from AstraZeneca and Boehringer Ingelheim, and he has been a consultant with numerous companies. Dr. Kosiborod has been a consultant to and has received research funding from AstraZeneca and Boehringer Ingelheim and has been a consultant to Eli Lilly and numerous other companies. Dr. Inzucchi has been a consultant to, given talks on behalf of, or served on trial committees for Abbott, AstraZeneca, Boehringer Ingelheim, Esperion, Lexicon, Merck, Novo Nordisk, Pfizer, and vTv Therapetics.
A version of this article first appeared on Medscape.com.
STOCKHOLM – Recent study results confirm that two agents from the sodium-glucose cotransporter 2 (SGLT2) inhibitor class can significantly cut the incidence of adverse cardiovascular events in patients with heart failure with reduced ejection fraction (HFpEF), a disease especially common in people with type 2 diabetes, obesity, or both.
And findings from secondary analyses of the studies – including one reported at the annual meeting of the European Association for the Study of Diabetes – show that these SGLT2 inhibitors work as well for cutting incident adverse events (cardiovascular death or worsening heart failure) in patients with HFpEF and diabetes as they do for people with normal blood glucose levels.
But delivering treatment with these proven agents, dapagliflozin (Farxiga) and empagliflozin (Jardiance), first requires diagnosis of HFpEF, a task that clinicians have historically fallen short in accomplishing.
When in 2021, results from the EMPEROR-Preserved trial with empagliflozin and when in September 2022 results from the DELIVER trial with dapagliflozin established the efficacy of these two SGLT2 inhibitors as the first treatments proven to benefit patients with HFpEF, they also raised the stakes for clinicians to be much more diligent and systematic in evaluating people at high risk for developing HFpEF because of having type 2 diabetes or obesity, two of the most potent risk factors for this form of heart failure.
‘Vigilance ... needs to increase’
“Vigilance for HFpEF needs to increase because we can now help these patients,” declared Lars H. Lund, MD, PhD, speaking at the meeting. “Type 2 diabetes dramatically increases the incidence of HFpEF,” and the mechanisms by which it does this are “especially amenable to treatment with SGLT2 inhibitors,” said Dr. Lund, a cardiologist and heart failure specialist at the Karolinska Institute, Stockholm.
HFpEF has a history of going undetected in people with type 2 diabetes, an ironic situation given its high incidence as well as the elevated rate of adverse cardiovascular events when heart failure occurs in patients with type 2 diabetes compared with patients who do not have diabetes.
The key, say experts, is for clinicians to maintain a high index of suspicion for signs and symptoms of heart failure in people with type 2 diabetes and to regularly assess them, starting with just a few simple questions that probe for the presence of dyspnea, exertional fatigue, or both, an approach not widely employed up to now.
Clinicians who care for people with type 2 diabetes must become “alert to thinking about heart failure and alert to asking questions about signs and symptoms” that flag the presence of HFpEF, advised Naveed Sattar, MBChB, PhD, a professor of metabolic medicine at the University of Glasgow.
Soon, medical groups will issue guidelines for appropriate assessment for the presence of HFpEF in people with type 2 diabetes, Dr. Sattar predicted in an interview.
A need to probe
“You can’t simply ask patients with type 2 diabetes whether they have shortness of breath or exertional fatigue and stop there,” because often their first response will be no.
“Commonly, patients will initially say they have no dyspnea, but when you probe further, you find symptoms,” noted Mikhail N. Kosiborod, MD, codirector of Saint Luke’s Cardiometabolic Center of Excellence in Kansas City, Mo.
These people are often sedentary, so they frequently don’t experience shortness of breath at baseline, Dr. Kosiborod said in an interview. In some cases, they may limit their activity because of their exertional intolerance.
Once a person’s suggestive symptoms become known, the next step is to measure the serum level of N-terminal pro-B-type natriuretic peptide (NT-proBNP), a biomarker considered to be a generally reliable signal of existing heart failure when elevated.
Any value above 125 pg/mL is suggestive of prevalent heart failure and should lead to the next diagnostic step of echocardiography, Dr. Sattar said.
Elevated NT-proBNP has such good positive predictive value for identifying heart failure that it is tempting to use it broadly in people with type 2 diabetes. A 2022 consensus report from the American Diabetes Association says that “measurement of a natriuretic peptide [such as NT-proBNP] or high-sensitivity cardiac troponin is recommended on at least a yearly basis to identify the earliest HF [heart failure] stages and implement strategies to prevent transition to symptomatic HF.”
Test costs require targeting
But because of the relatively high current price for an NT-proBNP test, the cost-benefit ratio for widespread annual testing of all people with type 2 diabetes would be poor, some experts caution.
“Screening everyone may not be the right answer. Hundreds of millions of people worldwide” have type 2 diabetes. “You first need to target evaluation to people with symptoms,” advised Dr. Kosiborod.
He also warned that a low NT-proBNP level does not always rule out HFpEF, especially among people with type 2 diabetes who also have overweight or obesity, because NT-proBNP levels can be “artificially low” in people with obesity.
Other potential aids to diagnosis are assessment scores that researchers have developed, such as the H2FPEF score, which relies on variables that include age, obesity, and the presence of atrial fibrillation and hypertension.
However, this score also requires an echocardiography examination, another test that would have a questionable cost-benefit ratio if performed widely for patients with type 2 diabetes without targeting, Dr. Kosiborod said.
SGLT2 inhibitors benefit HFpEF regardless of glucose levels
A prespecified analysis of the DELIVER results that divided the study cohort on the basis of their glycemic status proved the efficacy of the SGLT2 inhibitor dapagliflozin for patients with HFpEF regardless of whether or not they had type 2 diabetes, prediabetes, or were normoglycemic at entry into the study, Silvio E. Inzucchi, MD, reported at the EASD meeting.
Treatment with dapagliflozin cut the incidence of the trial’s primary outcome of cardiovascular death or worsening heart failure by a significant 18% relative to placebo among all enrolled patients.
The new analysis reported by Dr. Inzucchi showed that treatment was associated with a 23% relative risk reduction among those with normoglycemia, a 13% reduction among those with prediabetes, and a 19% reduction among those with type 2 diabetes, with no signal of a significant difference among the three subgroups.
“There was no statistical interaction between categorical glycemic subgrouping and dapagliflozin’s treatment effect,” concluded Dr. Inzucchi, director of the Yale Medicine Diabetes Center, New Haven, Conn.
He also reported that, among the 6,259 people in the trial with HFpEF, 50% had diabetes, 31% had prediabetes, and a scant 19% had normoglycemia. The finding highlights once again the high prevalence of dysglycemia among people with HFpEF.
Previously, a prespecified secondary analysis of data from the EMPEROR-Preserved trial yielded similar findings for empagliflozin that showed the agent’s efficacy for people with HFpEF across the range of glucose levels.
The DELIVER trial was funded by AstraZeneca, the company that markets dapagliflozin (Farxiga). The EMPEROR-Preserved trial was sponsored by Boehringer Ingelheim and Eli Lilly, the companies that jointly market empagliflozin (Jardiance). Dr. Lund has been a consultant to AstraZeneca and Boehringer Ingelheim and to numerous other companies, and he is a stockholder in AnaCardio. Dr. Sattar has been a consultant to and has received research support from AstraZeneca and Boehringer Ingelheim, and he has been a consultant with numerous companies. Dr. Kosiborod has been a consultant to and has received research funding from AstraZeneca and Boehringer Ingelheim and has been a consultant to Eli Lilly and numerous other companies. Dr. Inzucchi has been a consultant to, given talks on behalf of, or served on trial committees for Abbott, AstraZeneca, Boehringer Ingelheim, Esperion, Lexicon, Merck, Novo Nordisk, Pfizer, and vTv Therapetics.
A version of this article first appeared on Medscape.com.
STOCKHOLM – Recent study results confirm that two agents from the sodium-glucose cotransporter 2 (SGLT2) inhibitor class can significantly cut the incidence of adverse cardiovascular events in patients with heart failure with reduced ejection fraction (HFpEF), a disease especially common in people with type 2 diabetes, obesity, or both.
And findings from secondary analyses of the studies – including one reported at the annual meeting of the European Association for the Study of Diabetes – show that these SGLT2 inhibitors work as well for cutting incident adverse events (cardiovascular death or worsening heart failure) in patients with HFpEF and diabetes as they do for people with normal blood glucose levels.
But delivering treatment with these proven agents, dapagliflozin (Farxiga) and empagliflozin (Jardiance), first requires diagnosis of HFpEF, a task that clinicians have historically fallen short in accomplishing.
When in 2021, results from the EMPEROR-Preserved trial with empagliflozin and when in September 2022 results from the DELIVER trial with dapagliflozin established the efficacy of these two SGLT2 inhibitors as the first treatments proven to benefit patients with HFpEF, they also raised the stakes for clinicians to be much more diligent and systematic in evaluating people at high risk for developing HFpEF because of having type 2 diabetes or obesity, two of the most potent risk factors for this form of heart failure.
‘Vigilance ... needs to increase’
“Vigilance for HFpEF needs to increase because we can now help these patients,” declared Lars H. Lund, MD, PhD, speaking at the meeting. “Type 2 diabetes dramatically increases the incidence of HFpEF,” and the mechanisms by which it does this are “especially amenable to treatment with SGLT2 inhibitors,” said Dr. Lund, a cardiologist and heart failure specialist at the Karolinska Institute, Stockholm.
HFpEF has a history of going undetected in people with type 2 diabetes, an ironic situation given its high incidence as well as the elevated rate of adverse cardiovascular events when heart failure occurs in patients with type 2 diabetes compared with patients who do not have diabetes.
The key, say experts, is for clinicians to maintain a high index of suspicion for signs and symptoms of heart failure in people with type 2 diabetes and to regularly assess them, starting with just a few simple questions that probe for the presence of dyspnea, exertional fatigue, or both, an approach not widely employed up to now.
Clinicians who care for people with type 2 diabetes must become “alert to thinking about heart failure and alert to asking questions about signs and symptoms” that flag the presence of HFpEF, advised Naveed Sattar, MBChB, PhD, a professor of metabolic medicine at the University of Glasgow.
Soon, medical groups will issue guidelines for appropriate assessment for the presence of HFpEF in people with type 2 diabetes, Dr. Sattar predicted in an interview.
A need to probe
“You can’t simply ask patients with type 2 diabetes whether they have shortness of breath or exertional fatigue and stop there,” because often their first response will be no.
“Commonly, patients will initially say they have no dyspnea, but when you probe further, you find symptoms,” noted Mikhail N. Kosiborod, MD, codirector of Saint Luke’s Cardiometabolic Center of Excellence in Kansas City, Mo.
These people are often sedentary, so they frequently don’t experience shortness of breath at baseline, Dr. Kosiborod said in an interview. In some cases, they may limit their activity because of their exertional intolerance.
Once a person’s suggestive symptoms become known, the next step is to measure the serum level of N-terminal pro-B-type natriuretic peptide (NT-proBNP), a biomarker considered to be a generally reliable signal of existing heart failure when elevated.
Any value above 125 pg/mL is suggestive of prevalent heart failure and should lead to the next diagnostic step of echocardiography, Dr. Sattar said.
Elevated NT-proBNP has such good positive predictive value for identifying heart failure that it is tempting to use it broadly in people with type 2 diabetes. A 2022 consensus report from the American Diabetes Association says that “measurement of a natriuretic peptide [such as NT-proBNP] or high-sensitivity cardiac troponin is recommended on at least a yearly basis to identify the earliest HF [heart failure] stages and implement strategies to prevent transition to symptomatic HF.”
Test costs require targeting
But because of the relatively high current price for an NT-proBNP test, the cost-benefit ratio for widespread annual testing of all people with type 2 diabetes would be poor, some experts caution.
“Screening everyone may not be the right answer. Hundreds of millions of people worldwide” have type 2 diabetes. “You first need to target evaluation to people with symptoms,” advised Dr. Kosiborod.
He also warned that a low NT-proBNP level does not always rule out HFpEF, especially among people with type 2 diabetes who also have overweight or obesity, because NT-proBNP levels can be “artificially low” in people with obesity.
Other potential aids to diagnosis are assessment scores that researchers have developed, such as the H2FPEF score, which relies on variables that include age, obesity, and the presence of atrial fibrillation and hypertension.
However, this score also requires an echocardiography examination, another test that would have a questionable cost-benefit ratio if performed widely for patients with type 2 diabetes without targeting, Dr. Kosiborod said.
SGLT2 inhibitors benefit HFpEF regardless of glucose levels
A prespecified analysis of the DELIVER results that divided the study cohort on the basis of their glycemic status proved the efficacy of the SGLT2 inhibitor dapagliflozin for patients with HFpEF regardless of whether or not they had type 2 diabetes, prediabetes, or were normoglycemic at entry into the study, Silvio E. Inzucchi, MD, reported at the EASD meeting.
Treatment with dapagliflozin cut the incidence of the trial’s primary outcome of cardiovascular death or worsening heart failure by a significant 18% relative to placebo among all enrolled patients.
The new analysis reported by Dr. Inzucchi showed that treatment was associated with a 23% relative risk reduction among those with normoglycemia, a 13% reduction among those with prediabetes, and a 19% reduction among those with type 2 diabetes, with no signal of a significant difference among the three subgroups.
“There was no statistical interaction between categorical glycemic subgrouping and dapagliflozin’s treatment effect,” concluded Dr. Inzucchi, director of the Yale Medicine Diabetes Center, New Haven, Conn.
He also reported that, among the 6,259 people in the trial with HFpEF, 50% had diabetes, 31% had prediabetes, and a scant 19% had normoglycemia. The finding highlights once again the high prevalence of dysglycemia among people with HFpEF.
Previously, a prespecified secondary analysis of data from the EMPEROR-Preserved trial yielded similar findings for empagliflozin that showed the agent’s efficacy for people with HFpEF across the range of glucose levels.
The DELIVER trial was funded by AstraZeneca, the company that markets dapagliflozin (Farxiga). The EMPEROR-Preserved trial was sponsored by Boehringer Ingelheim and Eli Lilly, the companies that jointly market empagliflozin (Jardiance). Dr. Lund has been a consultant to AstraZeneca and Boehringer Ingelheim and to numerous other companies, and he is a stockholder in AnaCardio. Dr. Sattar has been a consultant to and has received research support from AstraZeneca and Boehringer Ingelheim, and he has been a consultant with numerous companies. Dr. Kosiborod has been a consultant to and has received research funding from AstraZeneca and Boehringer Ingelheim and has been a consultant to Eli Lilly and numerous other companies. Dr. Inzucchi has been a consultant to, given talks on behalf of, or served on trial committees for Abbott, AstraZeneca, Boehringer Ingelheim, Esperion, Lexicon, Merck, Novo Nordisk, Pfizer, and vTv Therapetics.
A version of this article first appeared on Medscape.com.
AT EASD 2022
Risk-adapted screening strategy could reduce colonoscopy use
The Asia-Pacific Colorectal Screening (APCS) scoring system, combined with a stool DNA test, could improve the detection of advanced colorectal neoplasms and limit colonoscopy use, according to a new study published in Clinical Gastroenterology and Hepatology.
Although a colonoscopy can detect both colorectal cancer and precancerous lesions, using it as the primary screening tool can cause barriers due to high costs, limited resources, and low compliance, wrote Junfeng Xu of the gastroenterology department at the First Medical Center of Chinese PLA General Hospital, Beijing, and colleagues.
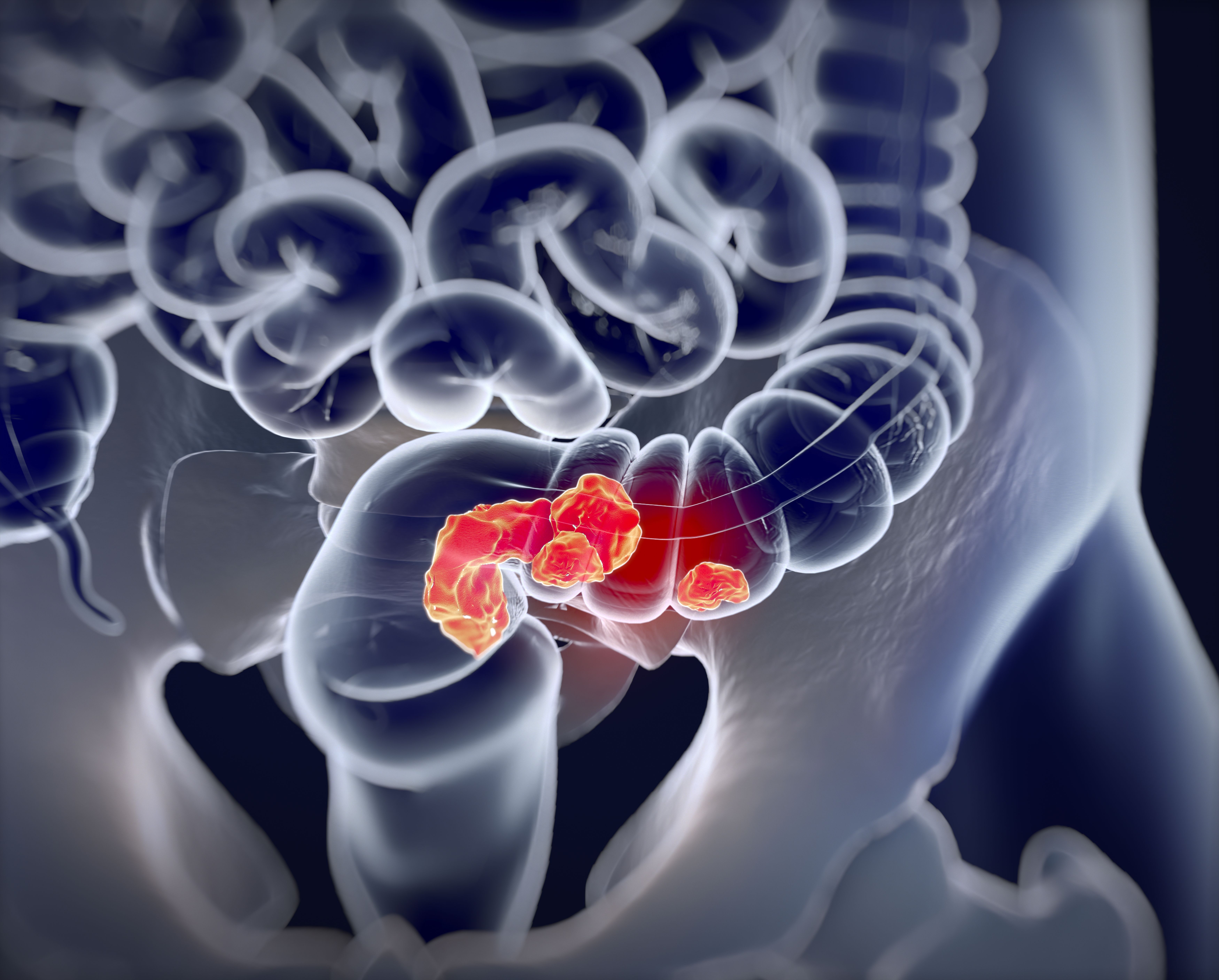
“Therefore, a more efficient risk-adapted screening approach for selection of colonoscopy is recommended,” the authors wrote. “This APCS-based algorithm for triaging subjects can significantly reduce the colonoscopy workload by half.”
Colorectal cancer is the third most common cancer worldwide, and in China, it was the third most diagnosed cancer and fourth most common cause of cancer-related death in 2016. In countries with limited health care resources, risk-adapted screening strategies could be more cost-effective and accessible than traditional screening strategies, the authors wrote.
Developed by the Asia-Pacific Working Group on CRC, the APCS score is based on age, sex, smoking status, and family history of colorectal cancer. Those with a score of 0-1 are considered low-risk, while scores of 2-3 are considered moderate-risk, and scores of 4-7 are considered high-risk.
In a cross-sectional, multicenter observational study, the investigators calculated APCS scores for 2,439 participants who visited eight outpatient clinics or cancer screening centers across four provinces in China between August 2017 and April 2019. Colonoscopy appointments were scheduled for all participants.
Participants provided test results for a stool DNA test (SDC2 and SFRP2 tests) and fecal immunochemical test (FIT), with colonoscopy outcomes used as the gold standard. The researchers used the manufacturer’s recommended hemoglobin threshold of 20 μg/g of dry stool for a positive result on FIT, in addition to a threshold of 4.4 μg/g to match the specificity of the stool DNA test.
Among all participants, 42 patients (1.9%) had colorectal cancer, 302 patients (13.5%) had advanced adenoma, and 551 patients (24.6%) had nonadvanced adenoma on colonoscopy.
Based on the APCS score, 946 participants (38.8%) were categorized as high risk, and they had a 1.8-fold increase in risk for advanced neoplasms (95% confidence interval, 1.4-2.3), as compared with the low- and moderate-risk groups.
Compared with direct colonoscopy, the combination of APCS score and stool DNA test detected 95.2% of invasive cancers (among 40 out of 42 patients) and 73.5% of advanced neoplasms (among 253 of 344 patients). The colonoscopy workload was 47.1% with this strategy. The risk-adapted screening approach required significantly fewer colonoscopies for detecting one advanced neoplasm than a direct colonoscopy screening, at 4.17 versus 6.51 (P < .001).
“Our findings provide critical references for designing effective population-based CRC screening strategies in the future, especially in resource-constrained countries and regions,” the authors concluded.
Avoiding complications
“Colonoscopy is expensive and time consuming for both the patient and the health care system. Colonoscopy is also not without risk since bowel perforation, post-procedural bleeding, and sedation-related complications all occur at low but measurable rates,” said Reid Ness, MD, associate professor of medicine and gastroenterologist at Vanderbilt University Medical Center, Nashville, Tenn.

“The primary strength of the study was that all patients eventually received colonoscopy, allowing for the best estimate of strategy sensitivity for advanced colorectal neoplasia,” he said.
At the same time, the cross-sectional study was unable to estimate the benefit of using this type of screening strategy over time, Dr. Ness said. With limited endoscopic resources available in many countries, however, clinicians need better modalities and strategies for noninvasive identification of advanced colorectal neoplasia, he added.
“Since less than 5% of the population will eventually develop colorectal cancer, the overwhelming majority can only be discomfited and possibly injured through colonoscopy screening,” he said. “For these reasons, the use of a CRC screening strategy that minimizes the use of colonoscopy without compromising the identification rate for advanced colorectal neoplasia is best for both the patient and the health care system.”
The study was supported by grants from the Beijing Municipal Science and Technology Commission. The authors declared no conflicts of interest. Dr. Ness reported no relevant disclosures.
This article was updated Oct. 4, 2022.
The Asia-Pacific Colorectal Screening (APCS) scoring system, combined with a stool DNA test, could improve the detection of advanced colorectal neoplasms and limit colonoscopy use, according to a new study published in Clinical Gastroenterology and Hepatology.
Although a colonoscopy can detect both colorectal cancer and precancerous lesions, using it as the primary screening tool can cause barriers due to high costs, limited resources, and low compliance, wrote Junfeng Xu of the gastroenterology department at the First Medical Center of Chinese PLA General Hospital, Beijing, and colleagues.
“Therefore, a more efficient risk-adapted screening approach for selection of colonoscopy is recommended,” the authors wrote. “This APCS-based algorithm for triaging subjects can significantly reduce the colonoscopy workload by half.”
Colorectal cancer is the third most common cancer worldwide, and in China, it was the third most diagnosed cancer and fourth most common cause of cancer-related death in 2016. In countries with limited health care resources, risk-adapted screening strategies could be more cost-effective and accessible than traditional screening strategies, the authors wrote.
Developed by the Asia-Pacific Working Group on CRC, the APCS score is based on age, sex, smoking status, and family history of colorectal cancer. Those with a score of 0-1 are considered low-risk, while scores of 2-3 are considered moderate-risk, and scores of 4-7 are considered high-risk.
In a cross-sectional, multicenter observational study, the investigators calculated APCS scores for 2,439 participants who visited eight outpatient clinics or cancer screening centers across four provinces in China between August 2017 and April 2019. Colonoscopy appointments were scheduled for all participants.
Participants provided test results for a stool DNA test (SDC2 and SFRP2 tests) and fecal immunochemical test (FIT), with colonoscopy outcomes used as the gold standard. The researchers used the manufacturer’s recommended hemoglobin threshold of 20 μg/g of dry stool for a positive result on FIT, in addition to a threshold of 4.4 μg/g to match the specificity of the stool DNA test.
Among all participants, 42 patients (1.9%) had colorectal cancer, 302 patients (13.5%) had advanced adenoma, and 551 patients (24.6%) had nonadvanced adenoma on colonoscopy.
Based on the APCS score, 946 participants (38.8%) were categorized as high risk, and they had a 1.8-fold increase in risk for advanced neoplasms (95% confidence interval, 1.4-2.3), as compared with the low- and moderate-risk groups.
Compared with direct colonoscopy, the combination of APCS score and stool DNA test detected 95.2% of invasive cancers (among 40 out of 42 patients) and 73.5% of advanced neoplasms (among 253 of 344 patients). The colonoscopy workload was 47.1% with this strategy. The risk-adapted screening approach required significantly fewer colonoscopies for detecting one advanced neoplasm than a direct colonoscopy screening, at 4.17 versus 6.51 (P < .001).
“Our findings provide critical references for designing effective population-based CRC screening strategies in the future, especially in resource-constrained countries and regions,” the authors concluded.
Avoiding complications
“Colonoscopy is expensive and time consuming for both the patient and the health care system. Colonoscopy is also not without risk since bowel perforation, post-procedural bleeding, and sedation-related complications all occur at low but measurable rates,” said Reid Ness, MD, associate professor of medicine and gastroenterologist at Vanderbilt University Medical Center, Nashville, Tenn.
“The primary strength of the study was that all patients eventually received colonoscopy, allowing for the best estimate of strategy sensitivity for advanced colorectal neoplasia,” he said.
At the same time, the cross-sectional study was unable to estimate the benefit of using this type of screening strategy over time, Dr. Ness said. With limited endoscopic resources available in many countries, however, clinicians need better modalities and strategies for noninvasive identification of advanced colorectal neoplasia, he added.
“Since less than 5% of the population will eventually develop colorectal cancer, the overwhelming majority can only be discomfited and possibly injured through colonoscopy screening,” he said. “For these reasons, the use of a CRC screening strategy that minimizes the use of colonoscopy without compromising the identification rate for advanced colorectal neoplasia is best for both the patient and the health care system.”
The study was supported by grants from the Beijing Municipal Science and Technology Commission. The authors declared no conflicts of interest. Dr. Ness reported no relevant disclosures.
This article was updated Oct. 4, 2022.
The Asia-Pacific Colorectal Screening (APCS) scoring system, combined with a stool DNA test, could improve the detection of advanced colorectal neoplasms and limit colonoscopy use, according to a new study published in Clinical Gastroenterology and Hepatology.
Although a colonoscopy can detect both colorectal cancer and precancerous lesions, using it as the primary screening tool can cause barriers due to high costs, limited resources, and low compliance, wrote Junfeng Xu of the gastroenterology department at the First Medical Center of Chinese PLA General Hospital, Beijing, and colleagues.
“Therefore, a more efficient risk-adapted screening approach for selection of colonoscopy is recommended,” the authors wrote. “This APCS-based algorithm for triaging subjects can significantly reduce the colonoscopy workload by half.”
Colorectal cancer is the third most common cancer worldwide, and in China, it was the third most diagnosed cancer and fourth most common cause of cancer-related death in 2016. In countries with limited health care resources, risk-adapted screening strategies could be more cost-effective and accessible than traditional screening strategies, the authors wrote.
Developed by the Asia-Pacific Working Group on CRC, the APCS score is based on age, sex, smoking status, and family history of colorectal cancer. Those with a score of 0-1 are considered low-risk, while scores of 2-3 are considered moderate-risk, and scores of 4-7 are considered high-risk.
In a cross-sectional, multicenter observational study, the investigators calculated APCS scores for 2,439 participants who visited eight outpatient clinics or cancer screening centers across four provinces in China between August 2017 and April 2019. Colonoscopy appointments were scheduled for all participants.
Participants provided test results for a stool DNA test (SDC2 and SFRP2 tests) and fecal immunochemical test (FIT), with colonoscopy outcomes used as the gold standard. The researchers used the manufacturer’s recommended hemoglobin threshold of 20 μg/g of dry stool for a positive result on FIT, in addition to a threshold of 4.4 μg/g to match the specificity of the stool DNA test.
Among all participants, 42 patients (1.9%) had colorectal cancer, 302 patients (13.5%) had advanced adenoma, and 551 patients (24.6%) had nonadvanced adenoma on colonoscopy.
Based on the APCS score, 946 participants (38.8%) were categorized as high risk, and they had a 1.8-fold increase in risk for advanced neoplasms (95% confidence interval, 1.4-2.3), as compared with the low- and moderate-risk groups.
Compared with direct colonoscopy, the combination of APCS score and stool DNA test detected 95.2% of invasive cancers (among 40 out of 42 patients) and 73.5% of advanced neoplasms (among 253 of 344 patients). The colonoscopy workload was 47.1% with this strategy. The risk-adapted screening approach required significantly fewer colonoscopies for detecting one advanced neoplasm than a direct colonoscopy screening, at 4.17 versus 6.51 (P < .001).
“Our findings provide critical references for designing effective population-based CRC screening strategies in the future, especially in resource-constrained countries and regions,” the authors concluded.
Avoiding complications
“Colonoscopy is expensive and time consuming for both the patient and the health care system. Colonoscopy is also not without risk since bowel perforation, post-procedural bleeding, and sedation-related complications all occur at low but measurable rates,” said Reid Ness, MD, associate professor of medicine and gastroenterologist at Vanderbilt University Medical Center, Nashville, Tenn.
“The primary strength of the study was that all patients eventually received colonoscopy, allowing for the best estimate of strategy sensitivity for advanced colorectal neoplasia,” he said.
At the same time, the cross-sectional study was unable to estimate the benefit of using this type of screening strategy over time, Dr. Ness said. With limited endoscopic resources available in many countries, however, clinicians need better modalities and strategies for noninvasive identification of advanced colorectal neoplasia, he added.
“Since less than 5% of the population will eventually develop colorectal cancer, the overwhelming majority can only be discomfited and possibly injured through colonoscopy screening,” he said. “For these reasons, the use of a CRC screening strategy that minimizes the use of colonoscopy without compromising the identification rate for advanced colorectal neoplasia is best for both the patient and the health care system.”
The study was supported by grants from the Beijing Municipal Science and Technology Commission. The authors declared no conflicts of interest. Dr. Ness reported no relevant disclosures.
This article was updated Oct. 4, 2022.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Medical record documentation: What to do, and what to avoid
Medical record documentation serves as a reminder of previous discussions with patients and what happened during their visits, a reimbursement justification for services, a communication tool to coordinate care with current and future clinicians, and a basis for defense in legal or regulatory matters.1,2 Documentation should be thorough, accurate, timely, and objective, with the ultimate goal of communicating our thoughts in an easily understood manner to other clinicians or attorneys.2 If we fail to achieve this goal, we may inadvertently give the impression that our care was hurried, incomplete, or thoughtless.2
Although not an exhaustive list, this article outlines strategies to employ and practices to avoid in our documentation efforts so we may enhance our defense in case of litigation and ensure the smooth transition of care for our patients.
Strategies to employ
Proper and accurate documentation details the course of patient care, and we should describe our thoughts in a clear and logical manner. Doing so minimizes the risk of misinterpretation by other clinicians or attorneys. Make sure the documentation of each appointment details the reason(s) for the patient’s visit, the effectiveness of treatment, possible treatment nonadherence, our clinical assessment, treatment consent, changes to the patient’s treatment plan, follow-up plans, reasons for not pursuing certain actions (eg, hospitalization), and a suicide risk assessment (and/or a violence risk assessment, if clinically indicated).2 Document missed or rescheduled appointments, and telephone and electronic contact with patients. Also be sure to use only commonly approved abbreviations.2 Document these items sooner rather than later because doing so improves the credibility of your charting.1 If you are handwriting notes, add the date and time to each encounter and make sure your handwriting is legible. Describe the behaviors of patients in objective and nonjudgmental terms.3 Documenting quotes from patients can convey crucial information about what was considered when making clinical decisions.1
Practices to avoid
If there is a need to make changes to previous entries, ensure these corrections are not mistaken for alterations. Each health care institution has its own policy for making corrections and addenda to medical records. Corrections to a patient’s medical record are acceptable, provided they are done appropriately, as I outlined in a previous Pearls article.4 Minimize or eliminate the copying and pasting of information; doing so can improve the efficiency of our documentation, but the practice can undermine the quality of the medical record, increase the risk of outdated and repetitive information being included, lead to clinical errors, and lead to overbilling of services.5 Finally, be sure to avoid speculation, personal commentary about patients and their family members, and language with negative connotations (unless such language is a direct quote from the patient).2,3
1. Mossman D. Tips to make documentation easier, faster, and more satisfying. Current Psychiatry. 2008;7(2):80,84-86.
2. Staus C. Documentation: your very best defense. Psychiatric News. 2022;57(4):7,19.
3. Nelson KJ. How to use patient-centered language in documentation. Current Psychiatry. 2011;10(10):70.
4. Joshi KG. Metadata, malpractice claims, and making changes to the EHR. Current Psychiatry. 2021;20(3):e1-e3. doi:10.12788/cp.0106
5. Neal D. Do’s and don’ts of electronic documentation. Psychiatric News. 2021;56(8):7.
Medical record documentation serves as a reminder of previous discussions with patients and what happened during their visits, a reimbursement justification for services, a communication tool to coordinate care with current and future clinicians, and a basis for defense in legal or regulatory matters.1,2 Documentation should be thorough, accurate, timely, and objective, with the ultimate goal of communicating our thoughts in an easily understood manner to other clinicians or attorneys.2 If we fail to achieve this goal, we may inadvertently give the impression that our care was hurried, incomplete, or thoughtless.2
Although not an exhaustive list, this article outlines strategies to employ and practices to avoid in our documentation efforts so we may enhance our defense in case of litigation and ensure the smooth transition of care for our patients.
Strategies to employ
Proper and accurate documentation details the course of patient care, and we should describe our thoughts in a clear and logical manner. Doing so minimizes the risk of misinterpretation by other clinicians or attorneys. Make sure the documentation of each appointment details the reason(s) for the patient’s visit, the effectiveness of treatment, possible treatment nonadherence, our clinical assessment, treatment consent, changes to the patient’s treatment plan, follow-up plans, reasons for not pursuing certain actions (eg, hospitalization), and a suicide risk assessment (and/or a violence risk assessment, if clinically indicated).2 Document missed or rescheduled appointments, and telephone and electronic contact with patients. Also be sure to use only commonly approved abbreviations.2 Document these items sooner rather than later because doing so improves the credibility of your charting.1 If you are handwriting notes, add the date and time to each encounter and make sure your handwriting is legible. Describe the behaviors of patients in objective and nonjudgmental terms.3 Documenting quotes from patients can convey crucial information about what was considered when making clinical decisions.1
Practices to avoid
If there is a need to make changes to previous entries, ensure these corrections are not mistaken for alterations. Each health care institution has its own policy for making corrections and addenda to medical records. Corrections to a patient’s medical record are acceptable, provided they are done appropriately, as I outlined in a previous Pearls article.4 Minimize or eliminate the copying and pasting of information; doing so can improve the efficiency of our documentation, but the practice can undermine the quality of the medical record, increase the risk of outdated and repetitive information being included, lead to clinical errors, and lead to overbilling of services.5 Finally, be sure to avoid speculation, personal commentary about patients and their family members, and language with negative connotations (unless such language is a direct quote from the patient).2,3
Medical record documentation serves as a reminder of previous discussions with patients and what happened during their visits, a reimbursement justification for services, a communication tool to coordinate care with current and future clinicians, and a basis for defense in legal or regulatory matters.1,2 Documentation should be thorough, accurate, timely, and objective, with the ultimate goal of communicating our thoughts in an easily understood manner to other clinicians or attorneys.2 If we fail to achieve this goal, we may inadvertently give the impression that our care was hurried, incomplete, or thoughtless.2
Although not an exhaustive list, this article outlines strategies to employ and practices to avoid in our documentation efforts so we may enhance our defense in case of litigation and ensure the smooth transition of care for our patients.
Strategies to employ
Proper and accurate documentation details the course of patient care, and we should describe our thoughts in a clear and logical manner. Doing so minimizes the risk of misinterpretation by other clinicians or attorneys. Make sure the documentation of each appointment details the reason(s) for the patient’s visit, the effectiveness of treatment, possible treatment nonadherence, our clinical assessment, treatment consent, changes to the patient’s treatment plan, follow-up plans, reasons for not pursuing certain actions (eg, hospitalization), and a suicide risk assessment (and/or a violence risk assessment, if clinically indicated).2 Document missed or rescheduled appointments, and telephone and electronic contact with patients. Also be sure to use only commonly approved abbreviations.2 Document these items sooner rather than later because doing so improves the credibility of your charting.1 If you are handwriting notes, add the date and time to each encounter and make sure your handwriting is legible. Describe the behaviors of patients in objective and nonjudgmental terms.3 Documenting quotes from patients can convey crucial information about what was considered when making clinical decisions.1
Practices to avoid
If there is a need to make changes to previous entries, ensure these corrections are not mistaken for alterations. Each health care institution has its own policy for making corrections and addenda to medical records. Corrections to a patient’s medical record are acceptable, provided they are done appropriately, as I outlined in a previous Pearls article.4 Minimize or eliminate the copying and pasting of information; doing so can improve the efficiency of our documentation, but the practice can undermine the quality of the medical record, increase the risk of outdated and repetitive information being included, lead to clinical errors, and lead to overbilling of services.5 Finally, be sure to avoid speculation, personal commentary about patients and their family members, and language with negative connotations (unless such language is a direct quote from the patient).2,3
1. Mossman D. Tips to make documentation easier, faster, and more satisfying. Current Psychiatry. 2008;7(2):80,84-86.
2. Staus C. Documentation: your very best defense. Psychiatric News. 2022;57(4):7,19.
3. Nelson KJ. How to use patient-centered language in documentation. Current Psychiatry. 2011;10(10):70.
4. Joshi KG. Metadata, malpractice claims, and making changes to the EHR. Current Psychiatry. 2021;20(3):e1-e3. doi:10.12788/cp.0106
5. Neal D. Do’s and don’ts of electronic documentation. Psychiatric News. 2021;56(8):7.
1. Mossman D. Tips to make documentation easier, faster, and more satisfying. Current Psychiatry. 2008;7(2):80,84-86.
2. Staus C. Documentation: your very best defense. Psychiatric News. 2022;57(4):7,19.
3. Nelson KJ. How to use patient-centered language in documentation. Current Psychiatry. 2011;10(10):70.
4. Joshi KG. Metadata, malpractice claims, and making changes to the EHR. Current Psychiatry. 2021;20(3):e1-e3. doi:10.12788/cp.0106
5. Neal D. Do’s and don’ts of electronic documentation. Psychiatric News. 2021;56(8):7.

Sex differences seen in inflammatory arthritis health care use
Women with inflammatory arthritis (IA) are more likely to use healthcare services than men, a Canadian study found. The results suggest there are biological differences in disease course and sociocultural differences in health care access and patient behavior among the sexes, Sanjana Tarannum said in a presentation at the Lancet Summit on Sex and Gender in Rheumatology.
Ms. Tarannum and colleagues also recently published the study in Annals of the Rheumatic Diseases.
Effectively managing IA patients calls for timely access to and appropriate use of health care resources, said Ms. Tarannum, of the Women’s College Research Institute in Toronto.
Sex and gender are often used interchangeably but they refer to different things. “Sex is the biological characteristic of being male or female. It relates to disease inheritance patterns, pain processing mechanisms, and immune dysregulation in the context of inflammatory arthritis,” Ms. Tarannum said during her presentation.
Gender is a sociocultural construct associated with masculine or feminine traits. In the context of IA, gender relates to coping strategies, pain perception and reporting, and health care–seeking behavior of patients and interaction with care providers.
A patient’s sex relates to healthcare encounters, time to diagnosis, and prescription patterns. These all affect disease outcomes. Previous studies have yielded inconsistent results and mainly focused on rheumatoid arthritis rather than other IA types such as ankylosing spondylitis (AS).
Ms. Tarannum and colleagues sought to compare health care usage between male and female patients for musculoskeletal-related issues before and after IA diagnosis. They used Ontario administrative health data to create three cohorts of patients with RA, AS, and psoriatic arthritis (PsA), the three most common types of IA. The patients were diagnosed during 2010-2017, and outcomes were assessed in each year for 3 years before and after diagnosis.
Health care use indicators included visits to physicians, musculoskeletal imaging, laboratory tests, and dispensation of drugs. Regression models adjusting for sociodemographic factors and comorbidities were used to compare male and female patients.
Sex-related differences emerge in all IA groups
The investigators assessed 41,277 patients with RA (69% female), 8,150 patients with AS (51% female), and 6,446 patients with PsA (54% female). Male patients had more cardiovascular disease, whereas female patients had higher incidences of depression and osteoporosis.
Similar trends of sex-related differences emerged in all three cohorts. Before diagnosis, female patients were more likely to visit rheumatologists or family physicians for musculoskeletal reasons or use musculoskeletal imaging and laboratory tests. Women were also more likely to remain in rheumatology care after diagnosis.
Men were more likely to visit the ED for musculoskeletal reasons immediately before diagnosis.
No sex- or gender-related differences were observed in medication use, although older females with RA or AS were more likely to get prescriptions for NSAIDs and opioids and conventional disease-modifying antirheumatic drugs, respectively.
The findings show that overall musculoskeletal health care use was higher in female patients with IA. “Sex differences were more pronounced the earlier the encounter was from the time of diagnosis and tended to diminish with time,” Ms. Tarannum observed. Sex differences were also more prominent in the RA and AS cohorts.
Women seek out care, do repeat visits
Several reasons may explain why utilization was higher in females. Women with IA have a higher overall risk of musculoskeletal conditions such as osteoarthritis, which could have driven the health care encounters. Numerous studies have also reported that female patients have a lower threshold for pain as well as a greater tendency to seek out health care.
Additionally, female patients often present with pain and fatigue, which are often misdiagnosed as fibromyalgia or depression. Therefore, they often require repeated health care encounters to arrive at an IA diagnosis, Ms. Tarannum said.
An early prodromal phase in females could have triggered a health care encounter as well.
Men, by comparison, are more likely to have acute-onset or severe disease. Objective signs and radiologic features can facilitate diagnosis in men, she said. Male patients also show more reluctance in seeking care, have a higher threshold for pain, and are less likely to have a usual source of care such as a family physician.
Higher confidence in hospital-based emergency services also could have resulted in more ED visits and lower health care use in men. Better response to treatments could also have resulted in fewer episodes of rheumatology care after diagnosis.
The results aren’t surprising, said Scott Zashin, MD, a rheumatologist in Dallas who wasn’t a part of the study.
“At least in terms of musculoskeletal disorders, my clinical experience suggests that women are more compliant with their follow-up than male patients. Especially with gout, a common type of arthritis in men, male patients may wait until their symptoms are severe before seeking medical attention,” Dr. Zashin said.
The Enid Walker Graduate Student Award for Research in Women’s Health provided funding for this study.
A version of this article first appeared on Medscape.com.
Women with inflammatory arthritis (IA) are more likely to use healthcare services than men, a Canadian study found. The results suggest there are biological differences in disease course and sociocultural differences in health care access and patient behavior among the sexes, Sanjana Tarannum said in a presentation at the Lancet Summit on Sex and Gender in Rheumatology.
Ms. Tarannum and colleagues also recently published the study in Annals of the Rheumatic Diseases.
Effectively managing IA patients calls for timely access to and appropriate use of health care resources, said Ms. Tarannum, of the Women’s College Research Institute in Toronto.
Sex and gender are often used interchangeably but they refer to different things. “Sex is the biological characteristic of being male or female. It relates to disease inheritance patterns, pain processing mechanisms, and immune dysregulation in the context of inflammatory arthritis,” Ms. Tarannum said during her presentation.
Gender is a sociocultural construct associated with masculine or feminine traits. In the context of IA, gender relates to coping strategies, pain perception and reporting, and health care–seeking behavior of patients and interaction with care providers.
A patient’s sex relates to healthcare encounters, time to diagnosis, and prescription patterns. These all affect disease outcomes. Previous studies have yielded inconsistent results and mainly focused on rheumatoid arthritis rather than other IA types such as ankylosing spondylitis (AS).
Ms. Tarannum and colleagues sought to compare health care usage between male and female patients for musculoskeletal-related issues before and after IA diagnosis. They used Ontario administrative health data to create three cohorts of patients with RA, AS, and psoriatic arthritis (PsA), the three most common types of IA. The patients were diagnosed during 2010-2017, and outcomes were assessed in each year for 3 years before and after diagnosis.
Health care use indicators included visits to physicians, musculoskeletal imaging, laboratory tests, and dispensation of drugs. Regression models adjusting for sociodemographic factors and comorbidities were used to compare male and female patients.
Sex-related differences emerge in all IA groups
The investigators assessed 41,277 patients with RA (69% female), 8,150 patients with AS (51% female), and 6,446 patients with PsA (54% female). Male patients had more cardiovascular disease, whereas female patients had higher incidences of depression and osteoporosis.
Similar trends of sex-related differences emerged in all three cohorts. Before diagnosis, female patients were more likely to visit rheumatologists or family physicians for musculoskeletal reasons or use musculoskeletal imaging and laboratory tests. Women were also more likely to remain in rheumatology care after diagnosis.
Men were more likely to visit the ED for musculoskeletal reasons immediately before diagnosis.
No sex- or gender-related differences were observed in medication use, although older females with RA or AS were more likely to get prescriptions for NSAIDs and opioids and conventional disease-modifying antirheumatic drugs, respectively.
The findings show that overall musculoskeletal health care use was higher in female patients with IA. “Sex differences were more pronounced the earlier the encounter was from the time of diagnosis and tended to diminish with time,” Ms. Tarannum observed. Sex differences were also more prominent in the RA and AS cohorts.
Women seek out care, do repeat visits
Several reasons may explain why utilization was higher in females. Women with IA have a higher overall risk of musculoskeletal conditions such as osteoarthritis, which could have driven the health care encounters. Numerous studies have also reported that female patients have a lower threshold for pain as well as a greater tendency to seek out health care.
Additionally, female patients often present with pain and fatigue, which are often misdiagnosed as fibromyalgia or depression. Therefore, they often require repeated health care encounters to arrive at an IA diagnosis, Ms. Tarannum said.
An early prodromal phase in females could have triggered a health care encounter as well.
Men, by comparison, are more likely to have acute-onset or severe disease. Objective signs and radiologic features can facilitate diagnosis in men, she said. Male patients also show more reluctance in seeking care, have a higher threshold for pain, and are less likely to have a usual source of care such as a family physician.
Higher confidence in hospital-based emergency services also could have resulted in more ED visits and lower health care use in men. Better response to treatments could also have resulted in fewer episodes of rheumatology care after diagnosis.
The results aren’t surprising, said Scott Zashin, MD, a rheumatologist in Dallas who wasn’t a part of the study.
“At least in terms of musculoskeletal disorders, my clinical experience suggests that women are more compliant with their follow-up than male patients. Especially with gout, a common type of arthritis in men, male patients may wait until their symptoms are severe before seeking medical attention,” Dr. Zashin said.
The Enid Walker Graduate Student Award for Research in Women’s Health provided funding for this study.
A version of this article first appeared on Medscape.com.
Women with inflammatory arthritis (IA) are more likely to use healthcare services than men, a Canadian study found. The results suggest there are biological differences in disease course and sociocultural differences in health care access and patient behavior among the sexes, Sanjana Tarannum said in a presentation at the Lancet Summit on Sex and Gender in Rheumatology.
Ms. Tarannum and colleagues also recently published the study in Annals of the Rheumatic Diseases.
Effectively managing IA patients calls for timely access to and appropriate use of health care resources, said Ms. Tarannum, of the Women’s College Research Institute in Toronto.
Sex and gender are often used interchangeably but they refer to different things. “Sex is the biological characteristic of being male or female. It relates to disease inheritance patterns, pain processing mechanisms, and immune dysregulation in the context of inflammatory arthritis,” Ms. Tarannum said during her presentation.
Gender is a sociocultural construct associated with masculine or feminine traits. In the context of IA, gender relates to coping strategies, pain perception and reporting, and health care–seeking behavior of patients and interaction with care providers.
A patient’s sex relates to healthcare encounters, time to diagnosis, and prescription patterns. These all affect disease outcomes. Previous studies have yielded inconsistent results and mainly focused on rheumatoid arthritis rather than other IA types such as ankylosing spondylitis (AS).
Ms. Tarannum and colleagues sought to compare health care usage between male and female patients for musculoskeletal-related issues before and after IA diagnosis. They used Ontario administrative health data to create three cohorts of patients with RA, AS, and psoriatic arthritis (PsA), the three most common types of IA. The patients were diagnosed during 2010-2017, and outcomes were assessed in each year for 3 years before and after diagnosis.
Health care use indicators included visits to physicians, musculoskeletal imaging, laboratory tests, and dispensation of drugs. Regression models adjusting for sociodemographic factors and comorbidities were used to compare male and female patients.
Sex-related differences emerge in all IA groups
The investigators assessed 41,277 patients with RA (69% female), 8,150 patients with AS (51% female), and 6,446 patients with PsA (54% female). Male patients had more cardiovascular disease, whereas female patients had higher incidences of depression and osteoporosis.
Similar trends of sex-related differences emerged in all three cohorts. Before diagnosis, female patients were more likely to visit rheumatologists or family physicians for musculoskeletal reasons or use musculoskeletal imaging and laboratory tests. Women were also more likely to remain in rheumatology care after diagnosis.
Men were more likely to visit the ED for musculoskeletal reasons immediately before diagnosis.
No sex- or gender-related differences were observed in medication use, although older females with RA or AS were more likely to get prescriptions for NSAIDs and opioids and conventional disease-modifying antirheumatic drugs, respectively.
The findings show that overall musculoskeletal health care use was higher in female patients with IA. “Sex differences were more pronounced the earlier the encounter was from the time of diagnosis and tended to diminish with time,” Ms. Tarannum observed. Sex differences were also more prominent in the RA and AS cohorts.
Women seek out care, do repeat visits
Several reasons may explain why utilization was higher in females. Women with IA have a higher overall risk of musculoskeletal conditions such as osteoarthritis, which could have driven the health care encounters. Numerous studies have also reported that female patients have a lower threshold for pain as well as a greater tendency to seek out health care.
Additionally, female patients often present with pain and fatigue, which are often misdiagnosed as fibromyalgia or depression. Therefore, they often require repeated health care encounters to arrive at an IA diagnosis, Ms. Tarannum said.
An early prodromal phase in females could have triggered a health care encounter as well.
Men, by comparison, are more likely to have acute-onset or severe disease. Objective signs and radiologic features can facilitate diagnosis in men, she said. Male patients also show more reluctance in seeking care, have a higher threshold for pain, and are less likely to have a usual source of care such as a family physician.
Higher confidence in hospital-based emergency services also could have resulted in more ED visits and lower health care use in men. Better response to treatments could also have resulted in fewer episodes of rheumatology care after diagnosis.
The results aren’t surprising, said Scott Zashin, MD, a rheumatologist in Dallas who wasn’t a part of the study.
“At least in terms of musculoskeletal disorders, my clinical experience suggests that women are more compliant with their follow-up than male patients. Especially with gout, a common type of arthritis in men, male patients may wait until their symptoms are severe before seeking medical attention,” Dr. Zashin said.
The Enid Walker Graduate Student Award for Research in Women’s Health provided funding for this study.
A version of this article first appeared on Medscape.com.
FROM THE LANCET SUMMIT ON SEX AND GENDER IN RHEUMATOLOGY
Why can’t U.K. immunocompromised patients get Evusheld?
This transcript has been edited for clarity.
I’m David Kerr, professor of cancer medicine at Oxford. As I’m gearing up to have my autumnal COVID-19 booster vaccine,
This was developed by AstraZeneca. It’s a combination of two relatively long-acting antibodies (tixagevimab and cilgavimab) that bind to the spike protein on the outside of the SARS-CoV-2 virus, the virus that causes COVID-19. The antibody binds to the spike protein and prevents it from binding to and infecting or damaging cells, so it’s what’s called preexposure prophylaxis.
Although vaccination is still the best approach to protecting against and, one would hope, conferring a degree of herd protection to our population as a whole, there are some people who cannot mount an appropriate immune response and we have to take care of these folks. Because the vaccines don’t work very well for them, the vaccine itself is not sufficient to protect them.
Evusheld, in trials that have been done hitherto, can protect people who can’t mount an immune response from being infected. Between 75% and 80% of patients treated with Evusheld didn’t get COVID-19. The duration of effect seemed to be at least for 6 months, possibly longer, so it’s a really good result. This caused our medicines regulatory authority in the United Kingdom to approve the drug in March of this year. Although the drug has been approved, it’s not yet funded and not yet available for vulnerable patients.
These are patients who, for reasons of inborn genetic diseases, cannot mount an immune response; patients who are pharmacologically immune depleted; patients who have had transplants and are on immunosuppressive drugs; and some of our cancer patients, particularly those with blood or hematologic malignancies, who can receive very heavy treatment that can pound the immune system to bits.
These are, in the population as a whole, relatively small numbers, but an important number of people who are still vulnerable to developing COVID-19 despite vaccination.
Why isn’t the drug available? We have a two-stage process in the United Kingdom. We have the scientists and regulatory authorities looking at the evidence and data and saying, “Yes, it stacks up. This drug is effective and safe to some extent.”
The second phase is a health technology assessment undertaken by NICE, our National Institute for Health and Care Excellence – something that I’ve talked about a number of times before and the sometimes seemingly arbitrary decisions that they make. NICE hasn’t evaluated the drug yet, and the British government has held out because they are arguing that we don’t have enough data.
The trials with Evusheld were done before the Omicron variant dominated, as it does now; therefore, they are looking to try to work with AstraZeneca to generate more real-world data to show that Evusheld would prevent infection from the Omicron variant of the virus. Equally as important, how long does that protection last? Is it as protective against Omicron, and what’s the duration of that protection? Those bits of work are going on now.
Some real-world data are starting to emerge, showing that Evusheld will offer some degree of protection against Omicron, but there are still question marks about duration and the proportion of the population that would benefit.
NICE aren’t due to report on this – although the drug was approved in March of this year – until next year some time. That’s what’s caused a degree of consternation in the community of patients that we serve. Some of my clinical colleagues are beating the drum, saying, “We must have this drug now.” We’re still waiting on NICE to announce.
One obvious way to go around this is the government, which has bent over backwards in the United Kingdom to do as much as it can to protect the population from COVID-19. There was fantastic vaccine rollout and an extraordinary economic package to support individuals during lockdown to maintain the workforce, to support families and people at home. They’ve done a fantastic job.
Wanting to damp down this controversy, perhaps the sensible thing would be to ask NICE to evaluate the data that they have just now, to allow AstraZeneca to present whatever real-world evidence they have, and although it may not be perfect, it may be sufficient – we don’t know – to pass the NICE health technology assessment.
Watch this space. Let’s see what happens. If I were government, that’s what I would do. I would ask NICE to bring their appraisal forward. I would ask them to work with AstraZeneca to go over every ounce and iota of data that they have to see if this drug will be sufficiently effective and sufficiently cost-effective to be used before winter comes. I think the whole world is holding its breath, expecting another COVID-19 winter surge. Now would be the time to act.
What do you think? Here we are in the United Kingdom discussing yet another quasi–”health-rationing” problem. It’s not. This is about collecting more data and being as rational as possible. Can we accelerate that process? Perhaps.
Thanks for listening. I’d be very grateful for any comments that you might choose to make.
David J. Kerr, MD, DSc, is a professor of cancer medicine at the University of Oxford (England). He disclosed financial relationships with Oxford Cancer Biomarkers, Afrox, GlaxoSmithKline, Bayer HealthCare Pharmaceuticals, Genomic Health, Merck Serono, Roche, and Celleron Therapeutics.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
I’m David Kerr, professor of cancer medicine at Oxford. As I’m gearing up to have my autumnal COVID-19 booster vaccine,
This was developed by AstraZeneca. It’s a combination of two relatively long-acting antibodies (tixagevimab and cilgavimab) that bind to the spike protein on the outside of the SARS-CoV-2 virus, the virus that causes COVID-19. The antibody binds to the spike protein and prevents it from binding to and infecting or damaging cells, so it’s what’s called preexposure prophylaxis.
Although vaccination is still the best approach to protecting against and, one would hope, conferring a degree of herd protection to our population as a whole, there are some people who cannot mount an appropriate immune response and we have to take care of these folks. Because the vaccines don’t work very well for them, the vaccine itself is not sufficient to protect them.
Evusheld, in trials that have been done hitherto, can protect people who can’t mount an immune response from being infected. Between 75% and 80% of patients treated with Evusheld didn’t get COVID-19. The duration of effect seemed to be at least for 6 months, possibly longer, so it’s a really good result. This caused our medicines regulatory authority in the United Kingdom to approve the drug in March of this year. Although the drug has been approved, it’s not yet funded and not yet available for vulnerable patients.
These are patients who, for reasons of inborn genetic diseases, cannot mount an immune response; patients who are pharmacologically immune depleted; patients who have had transplants and are on immunosuppressive drugs; and some of our cancer patients, particularly those with blood or hematologic malignancies, who can receive very heavy treatment that can pound the immune system to bits.
These are, in the population as a whole, relatively small numbers, but an important number of people who are still vulnerable to developing COVID-19 despite vaccination.
Why isn’t the drug available? We have a two-stage process in the United Kingdom. We have the scientists and regulatory authorities looking at the evidence and data and saying, “Yes, it stacks up. This drug is effective and safe to some extent.”
The second phase is a health technology assessment undertaken by NICE, our National Institute for Health and Care Excellence – something that I’ve talked about a number of times before and the sometimes seemingly arbitrary decisions that they make. NICE hasn’t evaluated the drug yet, and the British government has held out because they are arguing that we don’t have enough data.
The trials with Evusheld were done before the Omicron variant dominated, as it does now; therefore, they are looking to try to work with AstraZeneca to generate more real-world data to show that Evusheld would prevent infection from the Omicron variant of the virus. Equally as important, how long does that protection last? Is it as protective against Omicron, and what’s the duration of that protection? Those bits of work are going on now.
Some real-world data are starting to emerge, showing that Evusheld will offer some degree of protection against Omicron, but there are still question marks about duration and the proportion of the population that would benefit.
NICE aren’t due to report on this – although the drug was approved in March of this year – until next year some time. That’s what’s caused a degree of consternation in the community of patients that we serve. Some of my clinical colleagues are beating the drum, saying, “We must have this drug now.” We’re still waiting on NICE to announce.
One obvious way to go around this is the government, which has bent over backwards in the United Kingdom to do as much as it can to protect the population from COVID-19. There was fantastic vaccine rollout and an extraordinary economic package to support individuals during lockdown to maintain the workforce, to support families and people at home. They’ve done a fantastic job.
Wanting to damp down this controversy, perhaps the sensible thing would be to ask NICE to evaluate the data that they have just now, to allow AstraZeneca to present whatever real-world evidence they have, and although it may not be perfect, it may be sufficient – we don’t know – to pass the NICE health technology assessment.
Watch this space. Let’s see what happens. If I were government, that’s what I would do. I would ask NICE to bring their appraisal forward. I would ask them to work with AstraZeneca to go over every ounce and iota of data that they have to see if this drug will be sufficiently effective and sufficiently cost-effective to be used before winter comes. I think the whole world is holding its breath, expecting another COVID-19 winter surge. Now would be the time to act.
What do you think? Here we are in the United Kingdom discussing yet another quasi–”health-rationing” problem. It’s not. This is about collecting more data and being as rational as possible. Can we accelerate that process? Perhaps.
Thanks for listening. I’d be very grateful for any comments that you might choose to make.
David J. Kerr, MD, DSc, is a professor of cancer medicine at the University of Oxford (England). He disclosed financial relationships with Oxford Cancer Biomarkers, Afrox, GlaxoSmithKline, Bayer HealthCare Pharmaceuticals, Genomic Health, Merck Serono, Roche, and Celleron Therapeutics.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
I’m David Kerr, professor of cancer medicine at Oxford. As I’m gearing up to have my autumnal COVID-19 booster vaccine,
This was developed by AstraZeneca. It’s a combination of two relatively long-acting antibodies (tixagevimab and cilgavimab) that bind to the spike protein on the outside of the SARS-CoV-2 virus, the virus that causes COVID-19. The antibody binds to the spike protein and prevents it from binding to and infecting or damaging cells, so it’s what’s called preexposure prophylaxis.
Although vaccination is still the best approach to protecting against and, one would hope, conferring a degree of herd protection to our population as a whole, there are some people who cannot mount an appropriate immune response and we have to take care of these folks. Because the vaccines don’t work very well for them, the vaccine itself is not sufficient to protect them.
Evusheld, in trials that have been done hitherto, can protect people who can’t mount an immune response from being infected. Between 75% and 80% of patients treated with Evusheld didn’t get COVID-19. The duration of effect seemed to be at least for 6 months, possibly longer, so it’s a really good result. This caused our medicines regulatory authority in the United Kingdom to approve the drug in March of this year. Although the drug has been approved, it’s not yet funded and not yet available for vulnerable patients.
These are patients who, for reasons of inborn genetic diseases, cannot mount an immune response; patients who are pharmacologically immune depleted; patients who have had transplants and are on immunosuppressive drugs; and some of our cancer patients, particularly those with blood or hematologic malignancies, who can receive very heavy treatment that can pound the immune system to bits.
These are, in the population as a whole, relatively small numbers, but an important number of people who are still vulnerable to developing COVID-19 despite vaccination.
Why isn’t the drug available? We have a two-stage process in the United Kingdom. We have the scientists and regulatory authorities looking at the evidence and data and saying, “Yes, it stacks up. This drug is effective and safe to some extent.”
The second phase is a health technology assessment undertaken by NICE, our National Institute for Health and Care Excellence – something that I’ve talked about a number of times before and the sometimes seemingly arbitrary decisions that they make. NICE hasn’t evaluated the drug yet, and the British government has held out because they are arguing that we don’t have enough data.
The trials with Evusheld were done before the Omicron variant dominated, as it does now; therefore, they are looking to try to work with AstraZeneca to generate more real-world data to show that Evusheld would prevent infection from the Omicron variant of the virus. Equally as important, how long does that protection last? Is it as protective against Omicron, and what’s the duration of that protection? Those bits of work are going on now.
Some real-world data are starting to emerge, showing that Evusheld will offer some degree of protection against Omicron, but there are still question marks about duration and the proportion of the population that would benefit.
NICE aren’t due to report on this – although the drug was approved in March of this year – until next year some time. That’s what’s caused a degree of consternation in the community of patients that we serve. Some of my clinical colleagues are beating the drum, saying, “We must have this drug now.” We’re still waiting on NICE to announce.
One obvious way to go around this is the government, which has bent over backwards in the United Kingdom to do as much as it can to protect the population from COVID-19. There was fantastic vaccine rollout and an extraordinary economic package to support individuals during lockdown to maintain the workforce, to support families and people at home. They’ve done a fantastic job.
Wanting to damp down this controversy, perhaps the sensible thing would be to ask NICE to evaluate the data that they have just now, to allow AstraZeneca to present whatever real-world evidence they have, and although it may not be perfect, it may be sufficient – we don’t know – to pass the NICE health technology assessment.
Watch this space. Let’s see what happens. If I were government, that’s what I would do. I would ask NICE to bring their appraisal forward. I would ask them to work with AstraZeneca to go over every ounce and iota of data that they have to see if this drug will be sufficiently effective and sufficiently cost-effective to be used before winter comes. I think the whole world is holding its breath, expecting another COVID-19 winter surge. Now would be the time to act.
What do you think? Here we are in the United Kingdom discussing yet another quasi–”health-rationing” problem. It’s not. This is about collecting more data and being as rational as possible. Can we accelerate that process? Perhaps.
Thanks for listening. I’d be very grateful for any comments that you might choose to make.
David J. Kerr, MD, DSc, is a professor of cancer medicine at the University of Oxford (England). He disclosed financial relationships with Oxford Cancer Biomarkers, Afrox, GlaxoSmithKline, Bayer HealthCare Pharmaceuticals, Genomic Health, Merck Serono, Roche, and Celleron Therapeutics.
A version of this article first appeared on Medscape.com.
Cost paramount when choosing metastatic breast cancer treatment
While efficacy and quality of life outcomes are similar across commonly used treatments for endocrine-refractory or triple-negative metastatic breast cancer, the costs of these agents vary widely, a recent analysis reveals.
Notably, the authors found that
Given “razor thin” differences in outcomes, cost should become a major consideration, the researchers concluded.
“As a society, we urgently need more strategies to reduce cancer drug costs without compromising outcomes, and our analysis provides quantifiable evidence to help providers choose lower priced, but equally effective sequences of drugs,” first author Stephanie B. Wheeler, PhD, from the University of North Carolina at Chapel Hill, explained in a press release.
Although the drugs Dr. Wheeler and colleagues studied are reimbursed in the metastatic breast cancer setting, “the optimal sequencing of them has been unclear, which has led to considerable variation in physician preference and practice,” Dr. Wheeler said.
In the study, published in the Journal of Clinical Oncology, Dr. Wheeler and colleagues estimated the cost-effectiveness of different therapeutic options from the first- to third-line setting for this patient population.
The researchers used three dynamic microsimulation computer models to predict how hypothetical sets of 10,000 patients with specific types of metastatic breast cancer would respond to various therapy types and sequences. The cohorts were grouped according to prior chemotherapy exposure: cohort 1 had no taxane or anthracycline exposure, cohort 2 had taxane and anthracycline exposure, and cohort 3 had taxane exposure but was anthracycline naive.
On the basis of feedback from oncologists, the investigators focused on different agents in the three cohorts: paclitaxel, capecitabine, or pegylated liposomal doxorubicin for cohort 1; eribulin, capecitabine, or carboplatin for cohort 2; and pegylated liposomal doxorubicin, capecitabine, or eribulin for cohort 3.
Overall, the models showed “nearly indistinguishable differences” in quality of life. In fact, the “razor-thin incremental differences in quality-adjusted survival” across the treatment sequences often amounted to differences of only a few days or weeks, the authors noted, adding that, even in the most extreme of cases, 3 weeks separated the best and worst options for quality-adjusted life-years.
But the models did show considerable differences in costs.
The authors found that, for cohort 1, treatment with paclitaxel followed by capecitabine and then pegylated liposomal doxorubicin corresponded to the highest expected quality-adjusted life-year gain and the lowest costs – $686 per month versus the highest cost option of $1,765.
For cohort 2, treatment with carboplatin followed by capecitabine and then eribulin corresponded to the highest expected quality-adjusted life-year gain and lowest costs.
For cohort 3, treatment sequences beginning with capecitabine or pegylated liposomal doxorubicin followed by eribulin was most cost effective.
Notably, the authors found that eribulin – the most expensive treatment with a high expected adverse event burden – performed particularly poorly in the two cohorts in which it was evaluated, “suggesting it should be used last in a sequence, on the basis of cost-effectiveness alone.”
In other words, “more spending on cancer care does not necessarily confer greater health benefits,” said Dr. Wheeler, also a professor of health policy.
“I hope our study will help expand the framework that we use to make these decisions from one where we just think about the biologic action of the drug to one where we also consider the bigger picture of what the treatment experience is like for the patient, including their financial burden, investment of time, and side effects,” study coauthor Katherine E. Reeder-Hayes, MD, section chief of breast oncology at UNC, said in the press release.
The results demonstrate that therapeutic decisions in the endocrine-refractory or triple-negative metastatic setting “may prioritize costs without affecting clinical outcomes” and highlight the direct impact that a “high-quality, transparent, and accessible economic analysis” can have on patient care, Scott D. Ramsey, MD, PhD, of Fred Hutchinson Cancer Research Center, Seattle, and colleagues wrote in an accompanying editorial.
Following the treatment sequences outlined in this study would “reduce patient financial burden and save our health system hundreds of millions of dollars annually,” the editorialists wrote.
As for next steps, Dr. Wheeler and colleagues have developed a financial navigation program to help patients manage their out-of-pocket cancer care costs and are currently scaling up the intervention in nine rural and nonrural oncology practices across North Carolina.
The study was supported by the Center for Disease Control and Prevention through the Prevention Research Centers Program. Dr. Wheeler has received research funding and payment for travel, accommodations, and expenses from Pfizer. Dr. Ramsey has had consulting or advisory roles and has received research funding and/or payment for travel, accommodations, and expenses from Bayer, Genentech, AstraZeneca, Merck, GRAIL, Seattle Genetics, Biovica, and/or Flatiron Health. Because of their editorial roles at the journal, the Journal of Clinical Oncology recused Dr. Wheeler and Dr. Ramsey from having any role in the peer review of their respective manuscripts.
A version of this article first appeared on Medscape.com.
While efficacy and quality of life outcomes are similar across commonly used treatments for endocrine-refractory or triple-negative metastatic breast cancer, the costs of these agents vary widely, a recent analysis reveals.
Notably, the authors found that
Given “razor thin” differences in outcomes, cost should become a major consideration, the researchers concluded.
“As a society, we urgently need more strategies to reduce cancer drug costs without compromising outcomes, and our analysis provides quantifiable evidence to help providers choose lower priced, but equally effective sequences of drugs,” first author Stephanie B. Wheeler, PhD, from the University of North Carolina at Chapel Hill, explained in a press release.
Although the drugs Dr. Wheeler and colleagues studied are reimbursed in the metastatic breast cancer setting, “the optimal sequencing of them has been unclear, which has led to considerable variation in physician preference and practice,” Dr. Wheeler said.
In the study, published in the Journal of Clinical Oncology, Dr. Wheeler and colleagues estimated the cost-effectiveness of different therapeutic options from the first- to third-line setting for this patient population.
The researchers used three dynamic microsimulation computer models to predict how hypothetical sets of 10,000 patients with specific types of metastatic breast cancer would respond to various therapy types and sequences. The cohorts were grouped according to prior chemotherapy exposure: cohort 1 had no taxane or anthracycline exposure, cohort 2 had taxane and anthracycline exposure, and cohort 3 had taxane exposure but was anthracycline naive.
On the basis of feedback from oncologists, the investigators focused on different agents in the three cohorts: paclitaxel, capecitabine, or pegylated liposomal doxorubicin for cohort 1; eribulin, capecitabine, or carboplatin for cohort 2; and pegylated liposomal doxorubicin, capecitabine, or eribulin for cohort 3.
Overall, the models showed “nearly indistinguishable differences” in quality of life. In fact, the “razor-thin incremental differences in quality-adjusted survival” across the treatment sequences often amounted to differences of only a few days or weeks, the authors noted, adding that, even in the most extreme of cases, 3 weeks separated the best and worst options for quality-adjusted life-years.
But the models did show considerable differences in costs.
The authors found that, for cohort 1, treatment with paclitaxel followed by capecitabine and then pegylated liposomal doxorubicin corresponded to the highest expected quality-adjusted life-year gain and the lowest costs – $686 per month versus the highest cost option of $1,765.
For cohort 2, treatment with carboplatin followed by capecitabine and then eribulin corresponded to the highest expected quality-adjusted life-year gain and lowest costs.
For cohort 3, treatment sequences beginning with capecitabine or pegylated liposomal doxorubicin followed by eribulin was most cost effective.
Notably, the authors found that eribulin – the most expensive treatment with a high expected adverse event burden – performed particularly poorly in the two cohorts in which it was evaluated, “suggesting it should be used last in a sequence, on the basis of cost-effectiveness alone.”
In other words, “more spending on cancer care does not necessarily confer greater health benefits,” said Dr. Wheeler, also a professor of health policy.
“I hope our study will help expand the framework that we use to make these decisions from one where we just think about the biologic action of the drug to one where we also consider the bigger picture of what the treatment experience is like for the patient, including their financial burden, investment of time, and side effects,” study coauthor Katherine E. Reeder-Hayes, MD, section chief of breast oncology at UNC, said in the press release.
The results demonstrate that therapeutic decisions in the endocrine-refractory or triple-negative metastatic setting “may prioritize costs without affecting clinical outcomes” and highlight the direct impact that a “high-quality, transparent, and accessible economic analysis” can have on patient care, Scott D. Ramsey, MD, PhD, of Fred Hutchinson Cancer Research Center, Seattle, and colleagues wrote in an accompanying editorial.
Following the treatment sequences outlined in this study would “reduce patient financial burden and save our health system hundreds of millions of dollars annually,” the editorialists wrote.
As for next steps, Dr. Wheeler and colleagues have developed a financial navigation program to help patients manage their out-of-pocket cancer care costs and are currently scaling up the intervention in nine rural and nonrural oncology practices across North Carolina.
The study was supported by the Center for Disease Control and Prevention through the Prevention Research Centers Program. Dr. Wheeler has received research funding and payment for travel, accommodations, and expenses from Pfizer. Dr. Ramsey has had consulting or advisory roles and has received research funding and/or payment for travel, accommodations, and expenses from Bayer, Genentech, AstraZeneca, Merck, GRAIL, Seattle Genetics, Biovica, and/or Flatiron Health. Because of their editorial roles at the journal, the Journal of Clinical Oncology recused Dr. Wheeler and Dr. Ramsey from having any role in the peer review of their respective manuscripts.
A version of this article first appeared on Medscape.com.
While efficacy and quality of life outcomes are similar across commonly used treatments for endocrine-refractory or triple-negative metastatic breast cancer, the costs of these agents vary widely, a recent analysis reveals.
Notably, the authors found that
Given “razor thin” differences in outcomes, cost should become a major consideration, the researchers concluded.
“As a society, we urgently need more strategies to reduce cancer drug costs without compromising outcomes, and our analysis provides quantifiable evidence to help providers choose lower priced, but equally effective sequences of drugs,” first author Stephanie B. Wheeler, PhD, from the University of North Carolina at Chapel Hill, explained in a press release.
Although the drugs Dr. Wheeler and colleagues studied are reimbursed in the metastatic breast cancer setting, “the optimal sequencing of them has been unclear, which has led to considerable variation in physician preference and practice,” Dr. Wheeler said.
In the study, published in the Journal of Clinical Oncology, Dr. Wheeler and colleagues estimated the cost-effectiveness of different therapeutic options from the first- to third-line setting for this patient population.
The researchers used three dynamic microsimulation computer models to predict how hypothetical sets of 10,000 patients with specific types of metastatic breast cancer would respond to various therapy types and sequences. The cohorts were grouped according to prior chemotherapy exposure: cohort 1 had no taxane or anthracycline exposure, cohort 2 had taxane and anthracycline exposure, and cohort 3 had taxane exposure but was anthracycline naive.
On the basis of feedback from oncologists, the investigators focused on different agents in the three cohorts: paclitaxel, capecitabine, or pegylated liposomal doxorubicin for cohort 1; eribulin, capecitabine, or carboplatin for cohort 2; and pegylated liposomal doxorubicin, capecitabine, or eribulin for cohort 3.
Overall, the models showed “nearly indistinguishable differences” in quality of life. In fact, the “razor-thin incremental differences in quality-adjusted survival” across the treatment sequences often amounted to differences of only a few days or weeks, the authors noted, adding that, even in the most extreme of cases, 3 weeks separated the best and worst options for quality-adjusted life-years.
But the models did show considerable differences in costs.
The authors found that, for cohort 1, treatment with paclitaxel followed by capecitabine and then pegylated liposomal doxorubicin corresponded to the highest expected quality-adjusted life-year gain and the lowest costs – $686 per month versus the highest cost option of $1,765.
For cohort 2, treatment with carboplatin followed by capecitabine and then eribulin corresponded to the highest expected quality-adjusted life-year gain and lowest costs.
For cohort 3, treatment sequences beginning with capecitabine or pegylated liposomal doxorubicin followed by eribulin was most cost effective.
Notably, the authors found that eribulin – the most expensive treatment with a high expected adverse event burden – performed particularly poorly in the two cohorts in which it was evaluated, “suggesting it should be used last in a sequence, on the basis of cost-effectiveness alone.”
In other words, “more spending on cancer care does not necessarily confer greater health benefits,” said Dr. Wheeler, also a professor of health policy.
“I hope our study will help expand the framework that we use to make these decisions from one where we just think about the biologic action of the drug to one where we also consider the bigger picture of what the treatment experience is like for the patient, including their financial burden, investment of time, and side effects,” study coauthor Katherine E. Reeder-Hayes, MD, section chief of breast oncology at UNC, said in the press release.
The results demonstrate that therapeutic decisions in the endocrine-refractory or triple-negative metastatic setting “may prioritize costs without affecting clinical outcomes” and highlight the direct impact that a “high-quality, transparent, and accessible economic analysis” can have on patient care, Scott D. Ramsey, MD, PhD, of Fred Hutchinson Cancer Research Center, Seattle, and colleagues wrote in an accompanying editorial.
Following the treatment sequences outlined in this study would “reduce patient financial burden and save our health system hundreds of millions of dollars annually,” the editorialists wrote.
As for next steps, Dr. Wheeler and colleagues have developed a financial navigation program to help patients manage their out-of-pocket cancer care costs and are currently scaling up the intervention in nine rural and nonrural oncology practices across North Carolina.
The study was supported by the Center for Disease Control and Prevention through the Prevention Research Centers Program. Dr. Wheeler has received research funding and payment for travel, accommodations, and expenses from Pfizer. Dr. Ramsey has had consulting or advisory roles and has received research funding and/or payment for travel, accommodations, and expenses from Bayer, Genentech, AstraZeneca, Merck, GRAIL, Seattle Genetics, Biovica, and/or Flatiron Health. Because of their editorial roles at the journal, the Journal of Clinical Oncology recused Dr. Wheeler and Dr. Ramsey from having any role in the peer review of their respective manuscripts.
A version of this article first appeared on Medscape.com.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Aspirin primary prevention benefit in those with raised Lp(a)?
Aspirin may be of specific benefit for the primary prevention of cardiovascular disease in individuals with raised Lp(a) levels, a new study has suggested.
The study analyzed data from the ASPREE (ASPirin in Reducing Events in the Elderly) trial, which randomized 19,000 individuals aged 70 years or older without a history of cardiovascular disease to aspirin (100 mg/day) or placebo. While the main results, reported previously, showed no net benefit of aspirin in the overall population, the current analysis suggests there may be a benefit in individuals with raised Lp(a) levels.
The current analysis was published online in the Journal of the American College of Cardiology.
“Our study provides evidence that aspirin may specifically benefit older individuals with genotypes for elevated plasma Lp(a) in the setting of high-risk primary prevention of cardiovascular events and that overall benefit may outweigh harm related to major bleeding,” the authors, led by Paul Lacaze, PhD, Monash University, Melbourne, conclude.
They also point out that similar observations have been previously seen in another large aspirin primary prevention study conducted in younger women, the Women’s Health Study, and the current analysis provides validation of those findings.
“Our results provide new evidence to support the potential use of aspirin to target individuals with elevated Lp(a) for the primary prevention of cardiovascular events,” the researchers say.
They acknowledge that these results would be strengthened by the use of directly measured plasma Lp(a) levels, in addition to Lp(a) genotypes.
But they add: “Nonetheless, given the lack of any currently approved therapies for targeting elevated Lp(a), our findings may have widespread clinical implications, adding evidence to the rationale that aspirin may be a viable option for reducing Lp(a)-mediated cardiovascular risk.”
Dr. Lacaze and colleagues explain that elevated plasma Lp(a) levels confer up to fourfold increased risk of cardiovascular disease, with around 20%-30% of the general population affected. Despite the high burden and prevalence of elevated plasma Lp(a), there are currently no approved pharmacologic therapies targeting this lipoprotein. Although promising candidates are in development for the secondary prevention of Lp(a)-mediated cardiovascular disease, it will be many years before these candidates are assessed for primary prevention.
For the current study, researchers analyzed data from 12,815 ASPREE participants who had undergone genotyping and compared outcomes with aspirin versus placebo in those with and without genotypes associated with elevated Lp(a) levels.
Results showed that individuals with elevated Lp(a)-associated genotypes, defined in two different ways, showed a reduction in ischemic events with aspirin versus placebo, and this benefit was not outweighed by an increased bleeding risk.
Specifically, in the placebo group, individuals who carried the rs3798220-C allele, which is known to be associated with raised Lp(a) levels, making up 3.2% of the genotyped population in the study, had an almost twofold increased risk of major adverse cardiovascular events than those not carrying this genotype. However, the risk was attenuated in the aspirin group, with carriers of the rs3798220-C allele actually having a lower rate of cardiovascular events than noncarriers.
In addition, rs3798220-C carrier status was not significantly associated with increased risk of clinically significant bleeding events in the aspirin group.
Similar results were seen with the second way of identifying patients with a high risk of elevated Lp(a) levels using a 43-variant genetic risk score (LPA-GRS).
In the whole study population, aspirin reduced major adverse cardiovascular events by 1.7 events per 1,000 person-years and increased clinically significant bleeding events by 1.7 events per 1,000 person-years, suggesting parity between overall benefit versus harm.
However, in the rs3798220-C subgroup, aspirin reduced major adverse cardiovascular events by 11.4 events per 1,000 person-years (a more than sixfold higher magnitude of cardiovascular disease risk reduction than in the overall cohort), with a bleeding risk of 3.3 events per 1,000 person-years, the researchers report.
“Hence in rs3798220-C carriers, aspirin appeared to have a net benefit of 8.1 events per 1,000 person-years,” they state.
In the highest LPA-GRS quintile, aspirin reduced major adverse cardiovascular events by 3.3 events per 1,000 person-years (approximately twofold higher magnitude of risk reduction, compared with the overall cohort), with an increase in bleeding risk of 1.6 events per 1,000 person-years (almost identical bleeding risk to the overall cohort). This shifted the benefit versus harm balance in the highest LPA-GRS quintile to a net benefit of 1.7 events per 1,000 person-years.
Similar findings in the Women’s Health Study
Dr. Lacaze and colleagues point out that similar results have also been seen in another large aspirin primary prevention study – the Women’s Health Study (WHS).
The WHS compared aspirin 100 mg every other day with placebo in initially healthy younger women. Previously reported results showed that women carrying the rs3798220-C variant, associated with highly elevated Lp(a) levels, had a twofold higher risk of cardiovascular events than noncarrier women in the placebo group, but this risk was reduced in the aspirin group. And there was no increased risk of bleeding in women with elevated Lp(a).
“These results, in the absence of any other randomized controlled trial evidence or approved therapy for treating Lp(a)-associated risk, have been used by some physicians as justification for prescribing aspirin in patients with elevated Lp(a),” Dr. Lacaze and colleagues note.
“In the present study of the ASPREE trial population, our results were consistent with the WHS analysis, despite randomizing older individuals (both men and women),” they add.
They say this validation of the WHS result provides evidence that a very high-risk subgroup of individuals with highly elevated Lp(a) – those carrying the rs3798220-C allele – may benefit from low-dose aspirin for the primary prevention of cardiovascular events. Further, the benefits in this subgroup specifically may outweigh any bleeding risk.
But they point out that rs3798220-C carriers comprise only a small portion of all individuals with elevated Lp(a) in the general population, while the polygenic LPA-GRS explains about 60% of the variation in directly measured plasma Lp(a) levels and has the potential advantage of being able to identify a larger group of individuals at increased risk.
The researchers note, however, that it is not clear to what extent the LPA-GRS results add further evidence to suggest that individuals with elevated Lp(a), beyond rs3798220-C carriers, may be more likely to benefit from aspirin.
“If the benefit of aspirin extends beyond very high-risk rs3798220-C carriers alone, to the broader 20%-30% of individuals with elevated Lp(a), the potential utility of aspirin for the primary prevention of cardiovascular events would increase substantially,” they say.
‘Very high clinical relevance’
In an accompanying editorial, Ana Devesa, MD, Borja Ibanez, MD, PhD, and Valentin Fuster, MD, PhD, The National Center for Cardiovascular Research, Madrid, say that: “[Dr.] Lacaze et al. are to be congratulated for a study of very high clinical relevance that represents a first indication for primary prevention for patients at high cardiovascular risk.”
They explain that the pathogenic mechanism of Lp(a) is believed to be a combination of prothrombotic and proatherogenic effects, and the current findings support the hypothesis that the prothrombotic mechanism of Lp(a) is mediated by platelet aggregation.
This would explain the occurrence of thrombotic events in the presence of atherosclerosis in that elevated Lp(a) levels may induce platelet adhesion and aggregation to the activated atherosclerotic plaque, thus enhancing the atherothrombotic process. Moreover, activated platelets release several mediators that result in cell adhesion and attraction of chemokines and proinflammatory cytokines, driving an inflammatory response and mediating atherosclerosis progression, they add.
The editorialists highlight the limitations of the study already acknowledged by the authors: The analysis used genotypes rather than elevated Lp(a) levels and included only those of European ancestry, meaning the results are difficult to extrapolate to other populations.
“The next steps in clinical practice should be defined, and there are still questions to be answered,” they conclude. “Will every patient benefit from antithrombotic therapies? Should all patients who have elevated Lp(a) levels be treated with aspirin?”
The ASPREE Biobank is supported by grants from the Commonwealth Scientific and Industrial Research Organisation, Monash University, Menzies Research Institute, Australian National University, University of Melbourne, National Institutes of Health, National Health and Medical Research Council of Australia, and the Victorian Cancer Agency. Dr. Lacaze is supported by a National Heart Foundation Future Leader Fellowship.
A version of this article first appeared on Medscape.com.
Aspirin may be of specific benefit for the primary prevention of cardiovascular disease in individuals with raised Lp(a) levels, a new study has suggested.
The study analyzed data from the ASPREE (ASPirin in Reducing Events in the Elderly) trial, which randomized 19,000 individuals aged 70 years or older without a history of cardiovascular disease to aspirin (100 mg/day) or placebo. While the main results, reported previously, showed no net benefit of aspirin in the overall population, the current analysis suggests there may be a benefit in individuals with raised Lp(a) levels.
The current analysis was published online in the Journal of the American College of Cardiology.
“Our study provides evidence that aspirin may specifically benefit older individuals with genotypes for elevated plasma Lp(a) in the setting of high-risk primary prevention of cardiovascular events and that overall benefit may outweigh harm related to major bleeding,” the authors, led by Paul Lacaze, PhD, Monash University, Melbourne, conclude.
They also point out that similar observations have been previously seen in another large aspirin primary prevention study conducted in younger women, the Women’s Health Study, and the current analysis provides validation of those findings.
“Our results provide new evidence to support the potential use of aspirin to target individuals with elevated Lp(a) for the primary prevention of cardiovascular events,” the researchers say.
They acknowledge that these results would be strengthened by the use of directly measured plasma Lp(a) levels, in addition to Lp(a) genotypes.
But they add: “Nonetheless, given the lack of any currently approved therapies for targeting elevated Lp(a), our findings may have widespread clinical implications, adding evidence to the rationale that aspirin may be a viable option for reducing Lp(a)-mediated cardiovascular risk.”
Dr. Lacaze and colleagues explain that elevated plasma Lp(a) levels confer up to fourfold increased risk of cardiovascular disease, with around 20%-30% of the general population affected. Despite the high burden and prevalence of elevated plasma Lp(a), there are currently no approved pharmacologic therapies targeting this lipoprotein. Although promising candidates are in development for the secondary prevention of Lp(a)-mediated cardiovascular disease, it will be many years before these candidates are assessed for primary prevention.
For the current study, researchers analyzed data from 12,815 ASPREE participants who had undergone genotyping and compared outcomes with aspirin versus placebo in those with and without genotypes associated with elevated Lp(a) levels.
Results showed that individuals with elevated Lp(a)-associated genotypes, defined in two different ways, showed a reduction in ischemic events with aspirin versus placebo, and this benefit was not outweighed by an increased bleeding risk.
Specifically, in the placebo group, individuals who carried the rs3798220-C allele, which is known to be associated with raised Lp(a) levels, making up 3.2% of the genotyped population in the study, had an almost twofold increased risk of major adverse cardiovascular events than those not carrying this genotype. However, the risk was attenuated in the aspirin group, with carriers of the rs3798220-C allele actually having a lower rate of cardiovascular events than noncarriers.
In addition, rs3798220-C carrier status was not significantly associated with increased risk of clinically significant bleeding events in the aspirin group.
Similar results were seen with the second way of identifying patients with a high risk of elevated Lp(a) levels using a 43-variant genetic risk score (LPA-GRS).
In the whole study population, aspirin reduced major adverse cardiovascular events by 1.7 events per 1,000 person-years and increased clinically significant bleeding events by 1.7 events per 1,000 person-years, suggesting parity between overall benefit versus harm.
However, in the rs3798220-C subgroup, aspirin reduced major adverse cardiovascular events by 11.4 events per 1,000 person-years (a more than sixfold higher magnitude of cardiovascular disease risk reduction than in the overall cohort), with a bleeding risk of 3.3 events per 1,000 person-years, the researchers report.
“Hence in rs3798220-C carriers, aspirin appeared to have a net benefit of 8.1 events per 1,000 person-years,” they state.
In the highest LPA-GRS quintile, aspirin reduced major adverse cardiovascular events by 3.3 events per 1,000 person-years (approximately twofold higher magnitude of risk reduction, compared with the overall cohort), with an increase in bleeding risk of 1.6 events per 1,000 person-years (almost identical bleeding risk to the overall cohort). This shifted the benefit versus harm balance in the highest LPA-GRS quintile to a net benefit of 1.7 events per 1,000 person-years.
Similar findings in the Women’s Health Study
Dr. Lacaze and colleagues point out that similar results have also been seen in another large aspirin primary prevention study – the Women’s Health Study (WHS).
The WHS compared aspirin 100 mg every other day with placebo in initially healthy younger women. Previously reported results showed that women carrying the rs3798220-C variant, associated with highly elevated Lp(a) levels, had a twofold higher risk of cardiovascular events than noncarrier women in the placebo group, but this risk was reduced in the aspirin group. And there was no increased risk of bleeding in women with elevated Lp(a).
“These results, in the absence of any other randomized controlled trial evidence or approved therapy for treating Lp(a)-associated risk, have been used by some physicians as justification for prescribing aspirin in patients with elevated Lp(a),” Dr. Lacaze and colleagues note.
“In the present study of the ASPREE trial population, our results were consistent with the WHS analysis, despite randomizing older individuals (both men and women),” they add.
They say this validation of the WHS result provides evidence that a very high-risk subgroup of individuals with highly elevated Lp(a) – those carrying the rs3798220-C allele – may benefit from low-dose aspirin for the primary prevention of cardiovascular events. Further, the benefits in this subgroup specifically may outweigh any bleeding risk.
But they point out that rs3798220-C carriers comprise only a small portion of all individuals with elevated Lp(a) in the general population, while the polygenic LPA-GRS explains about 60% of the variation in directly measured plasma Lp(a) levels and has the potential advantage of being able to identify a larger group of individuals at increased risk.
The researchers note, however, that it is not clear to what extent the LPA-GRS results add further evidence to suggest that individuals with elevated Lp(a), beyond rs3798220-C carriers, may be more likely to benefit from aspirin.
“If the benefit of aspirin extends beyond very high-risk rs3798220-C carriers alone, to the broader 20%-30% of individuals with elevated Lp(a), the potential utility of aspirin for the primary prevention of cardiovascular events would increase substantially,” they say.
‘Very high clinical relevance’
In an accompanying editorial, Ana Devesa, MD, Borja Ibanez, MD, PhD, and Valentin Fuster, MD, PhD, The National Center for Cardiovascular Research, Madrid, say that: “[Dr.] Lacaze et al. are to be congratulated for a study of very high clinical relevance that represents a first indication for primary prevention for patients at high cardiovascular risk.”
They explain that the pathogenic mechanism of Lp(a) is believed to be a combination of prothrombotic and proatherogenic effects, and the current findings support the hypothesis that the prothrombotic mechanism of Lp(a) is mediated by platelet aggregation.
This would explain the occurrence of thrombotic events in the presence of atherosclerosis in that elevated Lp(a) levels may induce platelet adhesion and aggregation to the activated atherosclerotic plaque, thus enhancing the atherothrombotic process. Moreover, activated platelets release several mediators that result in cell adhesion and attraction of chemokines and proinflammatory cytokines, driving an inflammatory response and mediating atherosclerosis progression, they add.
The editorialists highlight the limitations of the study already acknowledged by the authors: The analysis used genotypes rather than elevated Lp(a) levels and included only those of European ancestry, meaning the results are difficult to extrapolate to other populations.
“The next steps in clinical practice should be defined, and there are still questions to be answered,” they conclude. “Will every patient benefit from antithrombotic therapies? Should all patients who have elevated Lp(a) levels be treated with aspirin?”
The ASPREE Biobank is supported by grants from the Commonwealth Scientific and Industrial Research Organisation, Monash University, Menzies Research Institute, Australian National University, University of Melbourne, National Institutes of Health, National Health and Medical Research Council of Australia, and the Victorian Cancer Agency. Dr. Lacaze is supported by a National Heart Foundation Future Leader Fellowship.
A version of this article first appeared on Medscape.com.
Aspirin may be of specific benefit for the primary prevention of cardiovascular disease in individuals with raised Lp(a) levels, a new study has suggested.
The study analyzed data from the ASPREE (ASPirin in Reducing Events in the Elderly) trial, which randomized 19,000 individuals aged 70 years or older without a history of cardiovascular disease to aspirin (100 mg/day) or placebo. While the main results, reported previously, showed no net benefit of aspirin in the overall population, the current analysis suggests there may be a benefit in individuals with raised Lp(a) levels.
The current analysis was published online in the Journal of the American College of Cardiology.
“Our study provides evidence that aspirin may specifically benefit older individuals with genotypes for elevated plasma Lp(a) in the setting of high-risk primary prevention of cardiovascular events and that overall benefit may outweigh harm related to major bleeding,” the authors, led by Paul Lacaze, PhD, Monash University, Melbourne, conclude.
They also point out that similar observations have been previously seen in another large aspirin primary prevention study conducted in younger women, the Women’s Health Study, and the current analysis provides validation of those findings.
“Our results provide new evidence to support the potential use of aspirin to target individuals with elevated Lp(a) for the primary prevention of cardiovascular events,” the researchers say.
They acknowledge that these results would be strengthened by the use of directly measured plasma Lp(a) levels, in addition to Lp(a) genotypes.
But they add: “Nonetheless, given the lack of any currently approved therapies for targeting elevated Lp(a), our findings may have widespread clinical implications, adding evidence to the rationale that aspirin may be a viable option for reducing Lp(a)-mediated cardiovascular risk.”
Dr. Lacaze and colleagues explain that elevated plasma Lp(a) levels confer up to fourfold increased risk of cardiovascular disease, with around 20%-30% of the general population affected. Despite the high burden and prevalence of elevated plasma Lp(a), there are currently no approved pharmacologic therapies targeting this lipoprotein. Although promising candidates are in development for the secondary prevention of Lp(a)-mediated cardiovascular disease, it will be many years before these candidates are assessed for primary prevention.
For the current study, researchers analyzed data from 12,815 ASPREE participants who had undergone genotyping and compared outcomes with aspirin versus placebo in those with and without genotypes associated with elevated Lp(a) levels.
Results showed that individuals with elevated Lp(a)-associated genotypes, defined in two different ways, showed a reduction in ischemic events with aspirin versus placebo, and this benefit was not outweighed by an increased bleeding risk.
Specifically, in the placebo group, individuals who carried the rs3798220-C allele, which is known to be associated with raised Lp(a) levels, making up 3.2% of the genotyped population in the study, had an almost twofold increased risk of major adverse cardiovascular events than those not carrying this genotype. However, the risk was attenuated in the aspirin group, with carriers of the rs3798220-C allele actually having a lower rate of cardiovascular events than noncarriers.
In addition, rs3798220-C carrier status was not significantly associated with increased risk of clinically significant bleeding events in the aspirin group.
Similar results were seen with the second way of identifying patients with a high risk of elevated Lp(a) levels using a 43-variant genetic risk score (LPA-GRS).
In the whole study population, aspirin reduced major adverse cardiovascular events by 1.7 events per 1,000 person-years and increased clinically significant bleeding events by 1.7 events per 1,000 person-years, suggesting parity between overall benefit versus harm.
However, in the rs3798220-C subgroup, aspirin reduced major adverse cardiovascular events by 11.4 events per 1,000 person-years (a more than sixfold higher magnitude of cardiovascular disease risk reduction than in the overall cohort), with a bleeding risk of 3.3 events per 1,000 person-years, the researchers report.
“Hence in rs3798220-C carriers, aspirin appeared to have a net benefit of 8.1 events per 1,000 person-years,” they state.
In the highest LPA-GRS quintile, aspirin reduced major adverse cardiovascular events by 3.3 events per 1,000 person-years (approximately twofold higher magnitude of risk reduction, compared with the overall cohort), with an increase in bleeding risk of 1.6 events per 1,000 person-years (almost identical bleeding risk to the overall cohort). This shifted the benefit versus harm balance in the highest LPA-GRS quintile to a net benefit of 1.7 events per 1,000 person-years.
Similar findings in the Women’s Health Study
Dr. Lacaze and colleagues point out that similar results have also been seen in another large aspirin primary prevention study – the Women’s Health Study (WHS).
The WHS compared aspirin 100 mg every other day with placebo in initially healthy younger women. Previously reported results showed that women carrying the rs3798220-C variant, associated with highly elevated Lp(a) levels, had a twofold higher risk of cardiovascular events than noncarrier women in the placebo group, but this risk was reduced in the aspirin group. And there was no increased risk of bleeding in women with elevated Lp(a).
“These results, in the absence of any other randomized controlled trial evidence or approved therapy for treating Lp(a)-associated risk, have been used by some physicians as justification for prescribing aspirin in patients with elevated Lp(a),” Dr. Lacaze and colleagues note.
“In the present study of the ASPREE trial population, our results were consistent with the WHS analysis, despite randomizing older individuals (both men and women),” they add.
They say this validation of the WHS result provides evidence that a very high-risk subgroup of individuals with highly elevated Lp(a) – those carrying the rs3798220-C allele – may benefit from low-dose aspirin for the primary prevention of cardiovascular events. Further, the benefits in this subgroup specifically may outweigh any bleeding risk.
But they point out that rs3798220-C carriers comprise only a small portion of all individuals with elevated Lp(a) in the general population, while the polygenic LPA-GRS explains about 60% of the variation in directly measured plasma Lp(a) levels and has the potential advantage of being able to identify a larger group of individuals at increased risk.
The researchers note, however, that it is not clear to what extent the LPA-GRS results add further evidence to suggest that individuals with elevated Lp(a), beyond rs3798220-C carriers, may be more likely to benefit from aspirin.
“If the benefit of aspirin extends beyond very high-risk rs3798220-C carriers alone, to the broader 20%-30% of individuals with elevated Lp(a), the potential utility of aspirin for the primary prevention of cardiovascular events would increase substantially,” they say.
‘Very high clinical relevance’
In an accompanying editorial, Ana Devesa, MD, Borja Ibanez, MD, PhD, and Valentin Fuster, MD, PhD, The National Center for Cardiovascular Research, Madrid, say that: “[Dr.] Lacaze et al. are to be congratulated for a study of very high clinical relevance that represents a first indication for primary prevention for patients at high cardiovascular risk.”
They explain that the pathogenic mechanism of Lp(a) is believed to be a combination of prothrombotic and proatherogenic effects, and the current findings support the hypothesis that the prothrombotic mechanism of Lp(a) is mediated by platelet aggregation.
This would explain the occurrence of thrombotic events in the presence of atherosclerosis in that elevated Lp(a) levels may induce platelet adhesion and aggregation to the activated atherosclerotic plaque, thus enhancing the atherothrombotic process. Moreover, activated platelets release several mediators that result in cell adhesion and attraction of chemokines and proinflammatory cytokines, driving an inflammatory response and mediating atherosclerosis progression, they add.
The editorialists highlight the limitations of the study already acknowledged by the authors: The analysis used genotypes rather than elevated Lp(a) levels and included only those of European ancestry, meaning the results are difficult to extrapolate to other populations.
“The next steps in clinical practice should be defined, and there are still questions to be answered,” they conclude. “Will every patient benefit from antithrombotic therapies? Should all patients who have elevated Lp(a) levels be treated with aspirin?”
The ASPREE Biobank is supported by grants from the Commonwealth Scientific and Industrial Research Organisation, Monash University, Menzies Research Institute, Australian National University, University of Melbourne, National Institutes of Health, National Health and Medical Research Council of Australia, and the Victorian Cancer Agency. Dr. Lacaze is supported by a National Heart Foundation Future Leader Fellowship.
A version of this article first appeared on Medscape.com.
ALS drug gets FDA panel thumbs-up after rare second look
In a rare second review of a new drug application,
By a vote of 7-2, the FDA Peripheral and Central Nervous System Drugs Advisory Committee reversed course on AMX0035 (Amylyx Pharmaceuticals), a combination of sodium phenylbutyrate and taurursodiol.
The panel previously voted 6-4 to reject the drug, ruling that data provided by Amylyx had failed to demonstrate that the survival benefit reported in the only clinical trial of AMX0035 so far was a direct result of the drug.
This time, two panelists who previously voted no were swayed by the drug maker’s new analysis of previously presented research, more than 1,300 public comments in support of the drug, supportive testimony from ALS patients and clinicians, and assurances from company executives that Amylyx would pull the drug from the market if results of an ongoing phase 3 clinical trial show the drug doesn’t work.
“As in March, today we have to have an internal dialogue between our scientific scrutiny and clinical compassion,” said Liana G. Apostolova, MD, from Indiana University, Indianapolis, who originally voted against the application.
“Today I also saw additional confirmatory evidence that was not unequivocally persuasive but was nonetheless reassuring,” Dr. Apostolova said. “Because of that I am voting in support of AMX0035.”
A rare second chance
ALS (Lou Gehrig’s disease) is a progressive, fatal neurodegenerative disease affecting nerve cells in the brain and spinal cord that causes loss of motor control. It is rare, affecting about 30,000 people in the United States with another 5,000 new cases diagnosed each year. Most people with the disease die within 2 years of diagnosis.
The FDA has approved two therapies for ALS, but both have limited efficacy.
Typically, FDA approval requires two large studies or one study with a “very persuasive” effect on survival.
Amylyx’s application is based on a single study, the multicenter, two-phase CENTAUR trial. In that trial, 137 people with ALS received AMX0035 or placebo for 24 weeks.
Researchers found that patients receiving AMX0035 had a 25% slower decline in function, compared with the those taking placebo. A change of 20% or more is considered clinically meaningful.
The investigators also found a statistically significant median difference of 4.8 months in time to death, first hospitalization, or tracheostomy/permanent assisted ventilation in the group originally assigned to receive AMX0035 compared with the group originally assigned to receive placebo (hazard ratio, 0.62; P = .023).
In the panel’s previous vote against the drug application, members cited several issues with the study, concluding that it did not offer persuasive or robust evidence of efficacy. They also cited missing data assumptions in the primary analysis, issues of randomization and imbalances in concomitant use of riluzole and edaravone, the two FDA-approved drugs for ALS.
The FDA later requested additional information from Amylyx, delayed its final ruling on the new drug application to Sept. 29, and called for a second review meeting – a virtually unheard-of move.
An FDA review posted in advance of the meeting Sept. 29 had hinted at a different outcome. In that report, regulators said new data from Amylyx were not “sufficiently independent or persuasive” to establish effectiveness.
However, FDA officials in the meeting stressed the importance of considering unmet medical need in ALS in the panel’s decision-making process.
“Recognizing the substantial unmet medical need in ALS, we feel that it is important that the committee is afforded the opportunity to consider this new information, along with the information presented at the prior meeting, in that context,” Billy Dunn, MD, director of the FDA Office of Neuroscience, said during the meeting.
Panelists heard additional data that Amylyx claims confirms the results of the CENTAUR study, including new analyses of the previously submitted survival data and new data from that study and an open-label extension.
They also provided new information on a biomarker data from a phase 2 study of AMX0035 to treat Alzheimer’s disease.
“I think we note the limitations of the analyses, but we still haven’t taken it off the table that they could be considered as confirmatory evidence and that’s why we’re here today,” said Teresa Buracchio, MD, director of the division of neurology for the FDA.
Two members of the panel who voted no in March stuck with that position at the Sept. 29 meeting.
“Unfortunately, I don’t believe the new evidence we’ve reviewed, while promising, combined with that prior evidence, constitutes substantial evidence of effectiveness,” said panelist Caleb Alexander, MD, a professor of epidemiology and medicine at the Johns Hopkins University Center for Drug Safety and Effectiveness, Baltimore.
Dr. Alexander, who also voted no in March, said that post hoc data presented at the meeting were not enough to assuage concerns that led him and others to reject the drug in March.
A challenging situation
Amylyx is currently leading the 48-week international, phase 3, placebo-controlled PHOENIX clinical trial of AMX0035. The study has enrolled about half of its 600-patient target.
“Undoubtedly, the results of the phase 3 study would be highly informative for a regulatory decision on the current ... review for AMX0035,” said Emily Freilich, MD, of the FDA.
However, results aren’t expected until late 2023 or early 2024, which “places the agency in a challenging situation of potentially making a regulatory decision that may not be subsequently confirmed by the results of the ongoing study.”
In June, Amylyx received conditional approval in Canada for the drug, but final approval depends on the outcome of the PHOENIX trial. The FDA does not offer a conditional approval track.
“If AMX0035 is not approved now, the FDA anticipated decision will likely happen in 2025, underscoring the critical importance of today’s outcome,” said Tammy Sarnelli, MPAHC, global head of Regulatory Affairs for Amylyx Pharmaceuticals.
If the FDA were to approve AMX0035 and results from the PHOENIX trial ultimately fail to prove efficacy, Justin Klee, co-CEO and cofounder of Amylyx Pharmaceuticals, said the company would withdraw the drug.
“To be clear, if PHOENIX is not successful, we will do what is right for patients, which includes voluntarily removing the product from the market,” Mr. Klee said.
Regardless of the company’s decision, FDA officials noted that the agency does have the ability to recall a drug from the market if studies show that it no longer meets requirements for approval.
“The FDA, with all due respect, significantly understates the complexity and likelihood of their pulling a product from the market,” Dr. Alexander said. “Whether or not they can ultimately pull a product from the market is no substitute for the evidentiary thresholds that are required for market access.”
A version of this article first appeared on Medscape.com.
In a rare second review of a new drug application,
By a vote of 7-2, the FDA Peripheral and Central Nervous System Drugs Advisory Committee reversed course on AMX0035 (Amylyx Pharmaceuticals), a combination of sodium phenylbutyrate and taurursodiol.
The panel previously voted 6-4 to reject the drug, ruling that data provided by Amylyx had failed to demonstrate that the survival benefit reported in the only clinical trial of AMX0035 so far was a direct result of the drug.
This time, two panelists who previously voted no were swayed by the drug maker’s new analysis of previously presented research, more than 1,300 public comments in support of the drug, supportive testimony from ALS patients and clinicians, and assurances from company executives that Amylyx would pull the drug from the market if results of an ongoing phase 3 clinical trial show the drug doesn’t work.
“As in March, today we have to have an internal dialogue between our scientific scrutiny and clinical compassion,” said Liana G. Apostolova, MD, from Indiana University, Indianapolis, who originally voted against the application.
“Today I also saw additional confirmatory evidence that was not unequivocally persuasive but was nonetheless reassuring,” Dr. Apostolova said. “Because of that I am voting in support of AMX0035.”
A rare second chance
ALS (Lou Gehrig’s disease) is a progressive, fatal neurodegenerative disease affecting nerve cells in the brain and spinal cord that causes loss of motor control. It is rare, affecting about 30,000 people in the United States with another 5,000 new cases diagnosed each year. Most people with the disease die within 2 years of diagnosis.
The FDA has approved two therapies for ALS, but both have limited efficacy.
Typically, FDA approval requires two large studies or one study with a “very persuasive” effect on survival.
Amylyx’s application is based on a single study, the multicenter, two-phase CENTAUR trial. In that trial, 137 people with ALS received AMX0035 or placebo for 24 weeks.
Researchers found that patients receiving AMX0035 had a 25% slower decline in function, compared with the those taking placebo. A change of 20% or more is considered clinically meaningful.
The investigators also found a statistically significant median difference of 4.8 months in time to death, first hospitalization, or tracheostomy/permanent assisted ventilation in the group originally assigned to receive AMX0035 compared with the group originally assigned to receive placebo (hazard ratio, 0.62; P = .023).
In the panel’s previous vote against the drug application, members cited several issues with the study, concluding that it did not offer persuasive or robust evidence of efficacy. They also cited missing data assumptions in the primary analysis, issues of randomization and imbalances in concomitant use of riluzole and edaravone, the two FDA-approved drugs for ALS.
The FDA later requested additional information from Amylyx, delayed its final ruling on the new drug application to Sept. 29, and called for a second review meeting – a virtually unheard-of move.
An FDA review posted in advance of the meeting Sept. 29 had hinted at a different outcome. In that report, regulators said new data from Amylyx were not “sufficiently independent or persuasive” to establish effectiveness.
However, FDA officials in the meeting stressed the importance of considering unmet medical need in ALS in the panel’s decision-making process.
“Recognizing the substantial unmet medical need in ALS, we feel that it is important that the committee is afforded the opportunity to consider this new information, along with the information presented at the prior meeting, in that context,” Billy Dunn, MD, director of the FDA Office of Neuroscience, said during the meeting.
Panelists heard additional data that Amylyx claims confirms the results of the CENTAUR study, including new analyses of the previously submitted survival data and new data from that study and an open-label extension.
They also provided new information on a biomarker data from a phase 2 study of AMX0035 to treat Alzheimer’s disease.
“I think we note the limitations of the analyses, but we still haven’t taken it off the table that they could be considered as confirmatory evidence and that’s why we’re here today,” said Teresa Buracchio, MD, director of the division of neurology for the FDA.
Two members of the panel who voted no in March stuck with that position at the Sept. 29 meeting.
“Unfortunately, I don’t believe the new evidence we’ve reviewed, while promising, combined with that prior evidence, constitutes substantial evidence of effectiveness,” said panelist Caleb Alexander, MD, a professor of epidemiology and medicine at the Johns Hopkins University Center for Drug Safety and Effectiveness, Baltimore.
Dr. Alexander, who also voted no in March, said that post hoc data presented at the meeting were not enough to assuage concerns that led him and others to reject the drug in March.
A challenging situation
Amylyx is currently leading the 48-week international, phase 3, placebo-controlled PHOENIX clinical trial of AMX0035. The study has enrolled about half of its 600-patient target.
“Undoubtedly, the results of the phase 3 study would be highly informative for a regulatory decision on the current ... review for AMX0035,” said Emily Freilich, MD, of the FDA.
However, results aren’t expected until late 2023 or early 2024, which “places the agency in a challenging situation of potentially making a regulatory decision that may not be subsequently confirmed by the results of the ongoing study.”
In June, Amylyx received conditional approval in Canada for the drug, but final approval depends on the outcome of the PHOENIX trial. The FDA does not offer a conditional approval track.
“If AMX0035 is not approved now, the FDA anticipated decision will likely happen in 2025, underscoring the critical importance of today’s outcome,” said Tammy Sarnelli, MPAHC, global head of Regulatory Affairs for Amylyx Pharmaceuticals.
If the FDA were to approve AMX0035 and results from the PHOENIX trial ultimately fail to prove efficacy, Justin Klee, co-CEO and cofounder of Amylyx Pharmaceuticals, said the company would withdraw the drug.
“To be clear, if PHOENIX is not successful, we will do what is right for patients, which includes voluntarily removing the product from the market,” Mr. Klee said.
Regardless of the company’s decision, FDA officials noted that the agency does have the ability to recall a drug from the market if studies show that it no longer meets requirements for approval.
“The FDA, with all due respect, significantly understates the complexity and likelihood of their pulling a product from the market,” Dr. Alexander said. “Whether or not they can ultimately pull a product from the market is no substitute for the evidentiary thresholds that are required for market access.”
A version of this article first appeared on Medscape.com.
In a rare second review of a new drug application,
By a vote of 7-2, the FDA Peripheral and Central Nervous System Drugs Advisory Committee reversed course on AMX0035 (Amylyx Pharmaceuticals), a combination of sodium phenylbutyrate and taurursodiol.
The panel previously voted 6-4 to reject the drug, ruling that data provided by Amylyx had failed to demonstrate that the survival benefit reported in the only clinical trial of AMX0035 so far was a direct result of the drug.
This time, two panelists who previously voted no were swayed by the drug maker’s new analysis of previously presented research, more than 1,300 public comments in support of the drug, supportive testimony from ALS patients and clinicians, and assurances from company executives that Amylyx would pull the drug from the market if results of an ongoing phase 3 clinical trial show the drug doesn’t work.
“As in March, today we have to have an internal dialogue between our scientific scrutiny and clinical compassion,” said Liana G. Apostolova, MD, from Indiana University, Indianapolis, who originally voted against the application.
“Today I also saw additional confirmatory evidence that was not unequivocally persuasive but was nonetheless reassuring,” Dr. Apostolova said. “Because of that I am voting in support of AMX0035.”
A rare second chance
ALS (Lou Gehrig’s disease) is a progressive, fatal neurodegenerative disease affecting nerve cells in the brain and spinal cord that causes loss of motor control. It is rare, affecting about 30,000 people in the United States with another 5,000 new cases diagnosed each year. Most people with the disease die within 2 years of diagnosis.
The FDA has approved two therapies for ALS, but both have limited efficacy.
Typically, FDA approval requires two large studies or one study with a “very persuasive” effect on survival.
Amylyx’s application is based on a single study, the multicenter, two-phase CENTAUR trial. In that trial, 137 people with ALS received AMX0035 or placebo for 24 weeks.
Researchers found that patients receiving AMX0035 had a 25% slower decline in function, compared with the those taking placebo. A change of 20% or more is considered clinically meaningful.
The investigators also found a statistically significant median difference of 4.8 months in time to death, first hospitalization, or tracheostomy/permanent assisted ventilation in the group originally assigned to receive AMX0035 compared with the group originally assigned to receive placebo (hazard ratio, 0.62; P = .023).
In the panel’s previous vote against the drug application, members cited several issues with the study, concluding that it did not offer persuasive or robust evidence of efficacy. They also cited missing data assumptions in the primary analysis, issues of randomization and imbalances in concomitant use of riluzole and edaravone, the two FDA-approved drugs for ALS.
The FDA later requested additional information from Amylyx, delayed its final ruling on the new drug application to Sept. 29, and called for a second review meeting – a virtually unheard-of move.
An FDA review posted in advance of the meeting Sept. 29 had hinted at a different outcome. In that report, regulators said new data from Amylyx were not “sufficiently independent or persuasive” to establish effectiveness.
However, FDA officials in the meeting stressed the importance of considering unmet medical need in ALS in the panel’s decision-making process.
“Recognizing the substantial unmet medical need in ALS, we feel that it is important that the committee is afforded the opportunity to consider this new information, along with the information presented at the prior meeting, in that context,” Billy Dunn, MD, director of the FDA Office of Neuroscience, said during the meeting.
Panelists heard additional data that Amylyx claims confirms the results of the CENTAUR study, including new analyses of the previously submitted survival data and new data from that study and an open-label extension.
They also provided new information on a biomarker data from a phase 2 study of AMX0035 to treat Alzheimer’s disease.
“I think we note the limitations of the analyses, but we still haven’t taken it off the table that they could be considered as confirmatory evidence and that’s why we’re here today,” said Teresa Buracchio, MD, director of the division of neurology for the FDA.
Two members of the panel who voted no in March stuck with that position at the Sept. 29 meeting.
“Unfortunately, I don’t believe the new evidence we’ve reviewed, while promising, combined with that prior evidence, constitutes substantial evidence of effectiveness,” said panelist Caleb Alexander, MD, a professor of epidemiology and medicine at the Johns Hopkins University Center for Drug Safety and Effectiveness, Baltimore.
Dr. Alexander, who also voted no in March, said that post hoc data presented at the meeting were not enough to assuage concerns that led him and others to reject the drug in March.
A challenging situation
Amylyx is currently leading the 48-week international, phase 3, placebo-controlled PHOENIX clinical trial of AMX0035. The study has enrolled about half of its 600-patient target.
“Undoubtedly, the results of the phase 3 study would be highly informative for a regulatory decision on the current ... review for AMX0035,” said Emily Freilich, MD, of the FDA.
However, results aren’t expected until late 2023 or early 2024, which “places the agency in a challenging situation of potentially making a regulatory decision that may not be subsequently confirmed by the results of the ongoing study.”
In June, Amylyx received conditional approval in Canada for the drug, but final approval depends on the outcome of the PHOENIX trial. The FDA does not offer a conditional approval track.
“If AMX0035 is not approved now, the FDA anticipated decision will likely happen in 2025, underscoring the critical importance of today’s outcome,” said Tammy Sarnelli, MPAHC, global head of Regulatory Affairs for Amylyx Pharmaceuticals.
If the FDA were to approve AMX0035 and results from the PHOENIX trial ultimately fail to prove efficacy, Justin Klee, co-CEO and cofounder of Amylyx Pharmaceuticals, said the company would withdraw the drug.
“To be clear, if PHOENIX is not successful, we will do what is right for patients, which includes voluntarily removing the product from the market,” Mr. Klee said.
Regardless of the company’s decision, FDA officials noted that the agency does have the ability to recall a drug from the market if studies show that it no longer meets requirements for approval.
“The FDA, with all due respect, significantly understates the complexity and likelihood of their pulling a product from the market,” Dr. Alexander said. “Whether or not they can ultimately pull a product from the market is no substitute for the evidentiary thresholds that are required for market access.”
A version of this article first appeared on Medscape.com.