User login
Power lines don’t raise leukemia risk in kids
Living near overhead power lines in early life does not increase a child’s risk of developing leukemia, according to a study published in the British Journal of Cancer.
An earlier study using information on childhood leukemia diagnosed between 1962 and 1995 suggested there was an elevated risk for children born within 600 meters of overhead power lines.
But now, updated data indicate that children born after the 1980s don’t have an increased risk.
According to researchers, this strongly suggests there is no direct biological effect of power lines on leukemia risk.
They believe the previous findings could be explained by changes in the characteristics of people living near power lines. The results might also be a chance finding or have resulted from problems with the study design.
“It’s very encouraging to see that, in recent decades, there has been no increased risk of leukemia among children born near overhead power lines,” said lead study author Kathryn Bunch, of the University of Oxford.
“More research is needed to determine precisely why previous evidence suggested a risk prior to 1980, but parents can be reassured from the findings of this study that overhead power lines don’t increase their child’s risk of leukemia.”
Expanding on previous findings
Several years ago, Dr Bunch’s colleagues at the University of Oxford set out to determine if proximity to high-voltage power lines affected the risk of childhood cancers in England and Wales, using data spanning the period from 1962 to 1995.
The team found evidence to suggest a relationship between childhood leukemia risk and the proximity to power lines of the mother’s residence at the time of the child’s birth. This included all 400 kV and 275 kV power lines and a small fraction of 132 kV lines (Draper et al, BMJ 2005).
Dr Bunch and her colleagues decided to extend this study by including more recent data, as well as cases and control subjects from Scotland. The group evaluated 132 kV, 275 kV, and 400 kV power lines and looked at subjects living greater distances from the power lines than those included in the previous study.
The researchers analyzed 53,515 children enrolled in the National Registry of Childhood Tumours from 1962 to 2008 and a group of matched controls.
The team found that, for the entire study period, there was no evidence of an increased risk of leukemia among subjects living closer to power lines. The relative risk of leukemia for children living 0 m to 199 m from power lines, compared with those living 1000 m or more from power lines (for all voltages), was 1.12.
There did appear to be an increased risk of leukemia when the researchers analyzed data according to decade. However, this risk declined over time.
The relative risk of leukemia for children living 0 m to 199 m from power lines, compared with those living 1000 m or more from power lines, was 4.50 in the 1960s, 2.46 in the 1970s, 1.54 in the 1980s, 0.99 in the 1990s, and 0.71 in the 2000s.
The elevated risk in the 1980s was not statistically significant, the researchers noted. They also pointed out that, even in the decades when the risk appears to be present, there is no evidence that it extended beyond the 600 m limit of the original analysis.
The fact that the risk declined over time suggests the leukemia is unlikely to have arisen from any physical effect of the power lines, the researchers said. They believe it’s more likely the result of changing population characteristics. ![]()
Living near overhead power lines in early life does not increase a child’s risk of developing leukemia, according to a study published in the British Journal of Cancer.
An earlier study using information on childhood leukemia diagnosed between 1962 and 1995 suggested there was an elevated risk for children born within 600 meters of overhead power lines.
But now, updated data indicate that children born after the 1980s don’t have an increased risk.
According to researchers, this strongly suggests there is no direct biological effect of power lines on leukemia risk.
They believe the previous findings could be explained by changes in the characteristics of people living near power lines. The results might also be a chance finding or have resulted from problems with the study design.
“It’s very encouraging to see that, in recent decades, there has been no increased risk of leukemia among children born near overhead power lines,” said lead study author Kathryn Bunch, of the University of Oxford.
“More research is needed to determine precisely why previous evidence suggested a risk prior to 1980, but parents can be reassured from the findings of this study that overhead power lines don’t increase their child’s risk of leukemia.”
Expanding on previous findings
Several years ago, Dr Bunch’s colleagues at the University of Oxford set out to determine if proximity to high-voltage power lines affected the risk of childhood cancers in England and Wales, using data spanning the period from 1962 to 1995.
The team found evidence to suggest a relationship between childhood leukemia risk and the proximity to power lines of the mother’s residence at the time of the child’s birth. This included all 400 kV and 275 kV power lines and a small fraction of 132 kV lines (Draper et al, BMJ 2005).
Dr Bunch and her colleagues decided to extend this study by including more recent data, as well as cases and control subjects from Scotland. The group evaluated 132 kV, 275 kV, and 400 kV power lines and looked at subjects living greater distances from the power lines than those included in the previous study.
The researchers analyzed 53,515 children enrolled in the National Registry of Childhood Tumours from 1962 to 2008 and a group of matched controls.
The team found that, for the entire study period, there was no evidence of an increased risk of leukemia among subjects living closer to power lines. The relative risk of leukemia for children living 0 m to 199 m from power lines, compared with those living 1000 m or more from power lines (for all voltages), was 1.12.
There did appear to be an increased risk of leukemia when the researchers analyzed data according to decade. However, this risk declined over time.
The relative risk of leukemia for children living 0 m to 199 m from power lines, compared with those living 1000 m or more from power lines, was 4.50 in the 1960s, 2.46 in the 1970s, 1.54 in the 1980s, 0.99 in the 1990s, and 0.71 in the 2000s.
The elevated risk in the 1980s was not statistically significant, the researchers noted. They also pointed out that, even in the decades when the risk appears to be present, there is no evidence that it extended beyond the 600 m limit of the original analysis.
The fact that the risk declined over time suggests the leukemia is unlikely to have arisen from any physical effect of the power lines, the researchers said. They believe it’s more likely the result of changing population characteristics. ![]()
Living near overhead power lines in early life does not increase a child’s risk of developing leukemia, according to a study published in the British Journal of Cancer.
An earlier study using information on childhood leukemia diagnosed between 1962 and 1995 suggested there was an elevated risk for children born within 600 meters of overhead power lines.
But now, updated data indicate that children born after the 1980s don’t have an increased risk.
According to researchers, this strongly suggests there is no direct biological effect of power lines on leukemia risk.
They believe the previous findings could be explained by changes in the characteristics of people living near power lines. The results might also be a chance finding or have resulted from problems with the study design.
“It’s very encouraging to see that, in recent decades, there has been no increased risk of leukemia among children born near overhead power lines,” said lead study author Kathryn Bunch, of the University of Oxford.
“More research is needed to determine precisely why previous evidence suggested a risk prior to 1980, but parents can be reassured from the findings of this study that overhead power lines don’t increase their child’s risk of leukemia.”
Expanding on previous findings
Several years ago, Dr Bunch’s colleagues at the University of Oxford set out to determine if proximity to high-voltage power lines affected the risk of childhood cancers in England and Wales, using data spanning the period from 1962 to 1995.
The team found evidence to suggest a relationship between childhood leukemia risk and the proximity to power lines of the mother’s residence at the time of the child’s birth. This included all 400 kV and 275 kV power lines and a small fraction of 132 kV lines (Draper et al, BMJ 2005).
Dr Bunch and her colleagues decided to extend this study by including more recent data, as well as cases and control subjects from Scotland. The group evaluated 132 kV, 275 kV, and 400 kV power lines and looked at subjects living greater distances from the power lines than those included in the previous study.
The researchers analyzed 53,515 children enrolled in the National Registry of Childhood Tumours from 1962 to 2008 and a group of matched controls.
The team found that, for the entire study period, there was no evidence of an increased risk of leukemia among subjects living closer to power lines. The relative risk of leukemia for children living 0 m to 199 m from power lines, compared with those living 1000 m or more from power lines (for all voltages), was 1.12.
There did appear to be an increased risk of leukemia when the researchers analyzed data according to decade. However, this risk declined over time.
The relative risk of leukemia for children living 0 m to 199 m from power lines, compared with those living 1000 m or more from power lines, was 4.50 in the 1960s, 2.46 in the 1970s, 1.54 in the 1980s, 0.99 in the 1990s, and 0.71 in the 2000s.
The elevated risk in the 1980s was not statistically significant, the researchers noted. They also pointed out that, even in the decades when the risk appears to be present, there is no evidence that it extended beyond the 600 m limit of the original analysis.
The fact that the risk declined over time suggests the leukemia is unlikely to have arisen from any physical effect of the power lines, the researchers said. They believe it’s more likely the result of changing population characteristics. ![]()
Improving the efficacy of etoposide

after treatment with etoposide
Credit: CNIO
A compound that interferes with the cell cycle can increase the antineoplastic effects of etoposide, according to research published in Cell Reports.
Etoposide works by inhibiting topoisomerase II (TOP2), a protein needed for DNA repair during cell division.
Researchers discovered a relationship between TOP2 and Cdh1, a protein that (along with Cdc20) controls cell division by activating the anaphase-promoting complex/cyclosome (APC/C).
So the team hypothesized that combining etoposide with a compound that inhibits Cdh1 might improve etoposide’s antineoplastic effects. Experiments in cancer cell lines confirmed this theory.
Marcos Malumbres, PhD, of the Spanish National Cancer Research Centre (CNIO) in Madrid, and his colleagues began this research by investigating Cdh1 in vitro and in mouse models.
The team found that a decrease in Cdh1 activity increases cells’ TOP2 levels. So they decided to combine etoposide with a Cdh1 inhibitor and evaluate the effect on cancer cells, which divide more than normal cells and therefore have a greater dependency on TOP2 to maintain DNA integrity.
The researchers tested proTAME, a small molecule that targets APC/C-Cdh1 and APC/C-Cdc20, in combination with etoposide. And they found the drugs had a synergistic effect against cancer cells.
In experiments with a lung cancer cell line (A549) and 2 breast cancer cell lines (HeLa and MCF7), administering proTAME and etoposide together proved more effective than administering either compound alone.
The researchers believe these findings could apply to other malignancies as well. Etoposide has demonstrated activity against a number of cancers, including leukemias, lymphomas, and multiple myeloma.
The team said their next step is to study the etoposide-proTAME combination in patients and investigate the malignancies in which this therapeutic strategy would be most effective.
The researchers also noted that previous studies have shown Cdh1 is inactive in some patients due to various oncogenic mutations. So stratifying patients according to their tumor’s Cdh1 status could optimize treatment with etoposide. ![]()
after treatment with etoposide
Credit: CNIO
A compound that interferes with the cell cycle can increase the antineoplastic effects of etoposide, according to research published in Cell Reports.
Etoposide works by inhibiting topoisomerase II (TOP2), a protein needed for DNA repair during cell division.
Researchers discovered a relationship between TOP2 and Cdh1, a protein that (along with Cdc20) controls cell division by activating the anaphase-promoting complex/cyclosome (APC/C).
So the team hypothesized that combining etoposide with a compound that inhibits Cdh1 might improve etoposide’s antineoplastic effects. Experiments in cancer cell lines confirmed this theory.
Marcos Malumbres, PhD, of the Spanish National Cancer Research Centre (CNIO) in Madrid, and his colleagues began this research by investigating Cdh1 in vitro and in mouse models.
The team found that a decrease in Cdh1 activity increases cells’ TOP2 levels. So they decided to combine etoposide with a Cdh1 inhibitor and evaluate the effect on cancer cells, which divide more than normal cells and therefore have a greater dependency on TOP2 to maintain DNA integrity.
The researchers tested proTAME, a small molecule that targets APC/C-Cdh1 and APC/C-Cdc20, in combination with etoposide. And they found the drugs had a synergistic effect against cancer cells.
In experiments with a lung cancer cell line (A549) and 2 breast cancer cell lines (HeLa and MCF7), administering proTAME and etoposide together proved more effective than administering either compound alone.
The researchers believe these findings could apply to other malignancies as well. Etoposide has demonstrated activity against a number of cancers, including leukemias, lymphomas, and multiple myeloma.
The team said their next step is to study the etoposide-proTAME combination in patients and investigate the malignancies in which this therapeutic strategy would be most effective.
The researchers also noted that previous studies have shown Cdh1 is inactive in some patients due to various oncogenic mutations. So stratifying patients according to their tumor’s Cdh1 status could optimize treatment with etoposide. ![]()
after treatment with etoposide
Credit: CNIO
A compound that interferes with the cell cycle can increase the antineoplastic effects of etoposide, according to research published in Cell Reports.
Etoposide works by inhibiting topoisomerase II (TOP2), a protein needed for DNA repair during cell division.
Researchers discovered a relationship between TOP2 and Cdh1, a protein that (along with Cdc20) controls cell division by activating the anaphase-promoting complex/cyclosome (APC/C).
So the team hypothesized that combining etoposide with a compound that inhibits Cdh1 might improve etoposide’s antineoplastic effects. Experiments in cancer cell lines confirmed this theory.
Marcos Malumbres, PhD, of the Spanish National Cancer Research Centre (CNIO) in Madrid, and his colleagues began this research by investigating Cdh1 in vitro and in mouse models.
The team found that a decrease in Cdh1 activity increases cells’ TOP2 levels. So they decided to combine etoposide with a Cdh1 inhibitor and evaluate the effect on cancer cells, which divide more than normal cells and therefore have a greater dependency on TOP2 to maintain DNA integrity.
The researchers tested proTAME, a small molecule that targets APC/C-Cdh1 and APC/C-Cdc20, in combination with etoposide. And they found the drugs had a synergistic effect against cancer cells.
In experiments with a lung cancer cell line (A549) and 2 breast cancer cell lines (HeLa and MCF7), administering proTAME and etoposide together proved more effective than administering either compound alone.
The researchers believe these findings could apply to other malignancies as well. Etoposide has demonstrated activity against a number of cancers, including leukemias, lymphomas, and multiple myeloma.
The team said their next step is to study the etoposide-proTAME combination in patients and investigate the malignancies in which this therapeutic strategy would be most effective.
The researchers also noted that previous studies have shown Cdh1 is inactive in some patients due to various oncogenic mutations. So stratifying patients according to their tumor’s Cdh1 status could optimize treatment with etoposide. ![]()
National plan to lower HAIs shows signs of success, investigators find

Credit: CDC
New research suggests a federally sponsored plan to decrease the incidence of healthcare-acquired infections (HAIs) in the US was successful in addressing the challenges of prioritizing and coordinating strategies.
The plan has also been associated with reductions in the rates of HAIs, with progress made toward most targets where data are available.
Descriptions of the plan and its initial results appear in a series of articles published in a supplement to the February issue of Medical Care.
“Much progress has been made in raising awareness of and developing strategies for curbing the life-threatening infections that strike patients too often when they are receiving medical care,” said Katherine Kahn, MD, a leader of the project and professor at the Geffen School of Medicine at the University of California, Los Angeles.
“In order to make even more progress, we need to build our systems of care to be safer within and across hospitals, nursing homes, clinics, and community settings.”
In 2009, the US Department of Health and Humans Services released a national plan aimed at preventing HAIs, called “National Action Plan to Prevent Health Care-Associated Infections: Road Map to Elimination.”
Researchers performed an evaluation of the first few years of the plan, reviewing the structure of the effort, as well as the results thus far.
The plan focuses on evidence-based strategies, such as considering the benefits and risks when deciding about the use and duration of treatments like antibiotics.
Most of the prevention initiatives have focused on hospital settings, but the action plan has focused attention on efforts in other care settings, such as outpatient surgery centers, kidney dialysis centers, and long-term care facilities.
The investigators said these efforts have likely contributed to stakeholders’ reported perceptions of greater momentum in adopting strategies to prevent HAIs.
The national plan has generated clinical, political, and financial support for the complex efforts required to eliminate HAIs across federal, regional, state, and local settings.
Despite an influx of federal funding to support elimination of HAIs, the researchers said ongoing dedicated resources will be required to maintain momentum and sustain efforts made to date.
On the other hand, because future funding for efforts to further reduce HAIs is unclear, the investigators said it may be best to incorporate the efforts into the overall movement to improve patient safety. ![]()
Credit: CDC
New research suggests a federally sponsored plan to decrease the incidence of healthcare-acquired infections (HAIs) in the US was successful in addressing the challenges of prioritizing and coordinating strategies.
The plan has also been associated with reductions in the rates of HAIs, with progress made toward most targets where data are available.
Descriptions of the plan and its initial results appear in a series of articles published in a supplement to the February issue of Medical Care.
“Much progress has been made in raising awareness of and developing strategies for curbing the life-threatening infections that strike patients too often when they are receiving medical care,” said Katherine Kahn, MD, a leader of the project and professor at the Geffen School of Medicine at the University of California, Los Angeles.
“In order to make even more progress, we need to build our systems of care to be safer within and across hospitals, nursing homes, clinics, and community settings.”
In 2009, the US Department of Health and Humans Services released a national plan aimed at preventing HAIs, called “National Action Plan to Prevent Health Care-Associated Infections: Road Map to Elimination.”
Researchers performed an evaluation of the first few years of the plan, reviewing the structure of the effort, as well as the results thus far.
The plan focuses on evidence-based strategies, such as considering the benefits and risks when deciding about the use and duration of treatments like antibiotics.
Most of the prevention initiatives have focused on hospital settings, but the action plan has focused attention on efforts in other care settings, such as outpatient surgery centers, kidney dialysis centers, and long-term care facilities.
The investigators said these efforts have likely contributed to stakeholders’ reported perceptions of greater momentum in adopting strategies to prevent HAIs.
The national plan has generated clinical, political, and financial support for the complex efforts required to eliminate HAIs across federal, regional, state, and local settings.
Despite an influx of federal funding to support elimination of HAIs, the researchers said ongoing dedicated resources will be required to maintain momentum and sustain efforts made to date.
On the other hand, because future funding for efforts to further reduce HAIs is unclear, the investigators said it may be best to incorporate the efforts into the overall movement to improve patient safety. ![]()
Credit: CDC
New research suggests a federally sponsored plan to decrease the incidence of healthcare-acquired infections (HAIs) in the US was successful in addressing the challenges of prioritizing and coordinating strategies.
The plan has also been associated with reductions in the rates of HAIs, with progress made toward most targets where data are available.
Descriptions of the plan and its initial results appear in a series of articles published in a supplement to the February issue of Medical Care.
“Much progress has been made in raising awareness of and developing strategies for curbing the life-threatening infections that strike patients too often when they are receiving medical care,” said Katherine Kahn, MD, a leader of the project and professor at the Geffen School of Medicine at the University of California, Los Angeles.
“In order to make even more progress, we need to build our systems of care to be safer within and across hospitals, nursing homes, clinics, and community settings.”
In 2009, the US Department of Health and Humans Services released a national plan aimed at preventing HAIs, called “National Action Plan to Prevent Health Care-Associated Infections: Road Map to Elimination.”
Researchers performed an evaluation of the first few years of the plan, reviewing the structure of the effort, as well as the results thus far.
The plan focuses on evidence-based strategies, such as considering the benefits and risks when deciding about the use and duration of treatments like antibiotics.
Most of the prevention initiatives have focused on hospital settings, but the action plan has focused attention on efforts in other care settings, such as outpatient surgery centers, kidney dialysis centers, and long-term care facilities.
The investigators said these efforts have likely contributed to stakeholders’ reported perceptions of greater momentum in adopting strategies to prevent HAIs.
The national plan has generated clinical, political, and financial support for the complex efforts required to eliminate HAIs across federal, regional, state, and local settings.
Despite an influx of federal funding to support elimination of HAIs, the researchers said ongoing dedicated resources will be required to maintain momentum and sustain efforts made to date.
On the other hand, because future funding for efforts to further reduce HAIs is unclear, the investigators said it may be best to incorporate the efforts into the overall movement to improve patient safety. ![]()
Unique protein found in MM patients

Credit: Rhoda Baer
Researchers say they have discovered a bacterial protein that attaches to virtually any antibody and prevents it from binding to its target.
It appears that this molecule, called mycoplasma protein (or protein M), helps some bacteria evade the immune response and establish long-term infections.
The researchers discovered protein M in samples from multiple myeloma (MM) patients.
And the team believes the protein could be engineered to target cancerous B cells in patients with MM and other B-cell malignancies.
Protein M might also become a target for new antibacterial drugs, and it could prove useful for preparing highly pure antibodies for research and drug manufacturing.
Richard A. Lerner, MD, of The Scripps Research Institute (TSRI) in La Jolla, California, and his colleagues described their discovery of protein M in Science.
The discovery originated from an effort to understand the origin of MM. Clonal B-cell proliferation, as well as MM and lymphomas, can result from chronic infections by organisms such as Escherichia coli, Helicobacter pylori, and hepatitis C virus.
To better understand this process, Dr Lerner and his colleagues investigated mycoplasma, a parasite that infects people chronically and is largely confined to the cell surface.
In a search for factors associated with long-term mycoplasma infection, the team tested samples of antibodies from MM patients’ blood against a variety of mycoplasma species. One of the proteins recognized by the antibodies was from Mycoplasma genitalium, which causes sexually transmitted infections.
To the researchers’ surprise, every antibody sample tested showed reactivity to this protein. But further tests made it clear that these antibody reactions were not in response to mass infection with M genitalium.
Instead, the M genitalium protein, which the team named protein M, appeared to have evolved simply to bind to any antibody it encounters.
“It binds to every antibody generically—capable of hijacking the entire diversity of antibody repertoire—but, at the same time, it blocks the specific interaction between that antibody and its intended biomolecular target,” said Rajesh Grover, PhD, of TSRI.
To better understand how protein M works, the researchers took a structural biology approach. Using X-ray crystallography and other techniques, the team determined the protein’s 3D atomic structure while it was bound to various human antibodies.
Compared to thousands of known structures in the Protein Data Bank, the worldwide structure database, protein M appeared to be unique. The data also revealed that protein M binds to a small, conserved region at the outer tip of every antibody’s antigen-binding arm.
“It likely extends the other end of itself, like a tail, over the antibody’s main antigen-binding region,” said Xueyong Zhu, PhD, of TSRI.
The team is now studying protein M’s function during M genitalium infections. It seems likely that the protein evolved to help M genitalium cope with the immune response, as it has one of the smallest bacterial genomes in nature.
“It appears to represent an elegant evolutionary solution to the special problem that mycoplasma have in evading the adaptive immune system,” Dr Grover said. “The smallest parasitic [bacterium] on planet Earth seems to have evolved the most sophisticated invading molecular machine.”
If protein M is confirmed as a universal “decoy” for antibodies, it will become a target for new drugs, the researchers said. This could make it easier to treat chronic, sometimes silent, infections by M genitalium and any other microbes that have evolved a similar antibody-thwarting defense.
In principle, protein M also could be engineered to target specific groups of B cells, delivering cytotoxic agents to cancerous B cells in patients with MM and lymphomas.
But the most immediate use of protein M, according to the researchers, is likely to be as a tool for grabbing antibodies in test tubes and cell cultures. This would allow the preparation of highly pure antibodies for research and drug manufacturing. Other generic antibody-binding proteins have been put to use in this way, but, so far, it appears that none does the job quite as well as protein M.
“It may be the most useful antibody purification device ever found,” said Dr Lerner, who is now working to commercialize the protein. ![]()
Credit: Rhoda Baer
Researchers say they have discovered a bacterial protein that attaches to virtually any antibody and prevents it from binding to its target.
It appears that this molecule, called mycoplasma protein (or protein M), helps some bacteria evade the immune response and establish long-term infections.
The researchers discovered protein M in samples from multiple myeloma (MM) patients.
And the team believes the protein could be engineered to target cancerous B cells in patients with MM and other B-cell malignancies.
Protein M might also become a target for new antibacterial drugs, and it could prove useful for preparing highly pure antibodies for research and drug manufacturing.
Richard A. Lerner, MD, of The Scripps Research Institute (TSRI) in La Jolla, California, and his colleagues described their discovery of protein M in Science.
The discovery originated from an effort to understand the origin of MM. Clonal B-cell proliferation, as well as MM and lymphomas, can result from chronic infections by organisms such as Escherichia coli, Helicobacter pylori, and hepatitis C virus.
To better understand this process, Dr Lerner and his colleagues investigated mycoplasma, a parasite that infects people chronically and is largely confined to the cell surface.
In a search for factors associated with long-term mycoplasma infection, the team tested samples of antibodies from MM patients’ blood against a variety of mycoplasma species. One of the proteins recognized by the antibodies was from Mycoplasma genitalium, which causes sexually transmitted infections.
To the researchers’ surprise, every antibody sample tested showed reactivity to this protein. But further tests made it clear that these antibody reactions were not in response to mass infection with M genitalium.
Instead, the M genitalium protein, which the team named protein M, appeared to have evolved simply to bind to any antibody it encounters.
“It binds to every antibody generically—capable of hijacking the entire diversity of antibody repertoire—but, at the same time, it blocks the specific interaction between that antibody and its intended biomolecular target,” said Rajesh Grover, PhD, of TSRI.
To better understand how protein M works, the researchers took a structural biology approach. Using X-ray crystallography and other techniques, the team determined the protein’s 3D atomic structure while it was bound to various human antibodies.
Compared to thousands of known structures in the Protein Data Bank, the worldwide structure database, protein M appeared to be unique. The data also revealed that protein M binds to a small, conserved region at the outer tip of every antibody’s antigen-binding arm.
“It likely extends the other end of itself, like a tail, over the antibody’s main antigen-binding region,” said Xueyong Zhu, PhD, of TSRI.
The team is now studying protein M’s function during M genitalium infections. It seems likely that the protein evolved to help M genitalium cope with the immune response, as it has one of the smallest bacterial genomes in nature.
“It appears to represent an elegant evolutionary solution to the special problem that mycoplasma have in evading the adaptive immune system,” Dr Grover said. “The smallest parasitic [bacterium] on planet Earth seems to have evolved the most sophisticated invading molecular machine.”
If protein M is confirmed as a universal “decoy” for antibodies, it will become a target for new drugs, the researchers said. This could make it easier to treat chronic, sometimes silent, infections by M genitalium and any other microbes that have evolved a similar antibody-thwarting defense.
In principle, protein M also could be engineered to target specific groups of B cells, delivering cytotoxic agents to cancerous B cells in patients with MM and lymphomas.
But the most immediate use of protein M, according to the researchers, is likely to be as a tool for grabbing antibodies in test tubes and cell cultures. This would allow the preparation of highly pure antibodies for research and drug manufacturing. Other generic antibody-binding proteins have been put to use in this way, but, so far, it appears that none does the job quite as well as protein M.
“It may be the most useful antibody purification device ever found,” said Dr Lerner, who is now working to commercialize the protein. ![]()
Credit: Rhoda Baer
Researchers say they have discovered a bacterial protein that attaches to virtually any antibody and prevents it from binding to its target.
It appears that this molecule, called mycoplasma protein (or protein M), helps some bacteria evade the immune response and establish long-term infections.
The researchers discovered protein M in samples from multiple myeloma (MM) patients.
And the team believes the protein could be engineered to target cancerous B cells in patients with MM and other B-cell malignancies.
Protein M might also become a target for new antibacterial drugs, and it could prove useful for preparing highly pure antibodies for research and drug manufacturing.
Richard A. Lerner, MD, of The Scripps Research Institute (TSRI) in La Jolla, California, and his colleagues described their discovery of protein M in Science.
The discovery originated from an effort to understand the origin of MM. Clonal B-cell proliferation, as well as MM and lymphomas, can result from chronic infections by organisms such as Escherichia coli, Helicobacter pylori, and hepatitis C virus.
To better understand this process, Dr Lerner and his colleagues investigated mycoplasma, a parasite that infects people chronically and is largely confined to the cell surface.
In a search for factors associated with long-term mycoplasma infection, the team tested samples of antibodies from MM patients’ blood against a variety of mycoplasma species. One of the proteins recognized by the antibodies was from Mycoplasma genitalium, which causes sexually transmitted infections.
To the researchers’ surprise, every antibody sample tested showed reactivity to this protein. But further tests made it clear that these antibody reactions were not in response to mass infection with M genitalium.
Instead, the M genitalium protein, which the team named protein M, appeared to have evolved simply to bind to any antibody it encounters.
“It binds to every antibody generically—capable of hijacking the entire diversity of antibody repertoire—but, at the same time, it blocks the specific interaction between that antibody and its intended biomolecular target,” said Rajesh Grover, PhD, of TSRI.
To better understand how protein M works, the researchers took a structural biology approach. Using X-ray crystallography and other techniques, the team determined the protein’s 3D atomic structure while it was bound to various human antibodies.
Compared to thousands of known structures in the Protein Data Bank, the worldwide structure database, protein M appeared to be unique. The data also revealed that protein M binds to a small, conserved region at the outer tip of every antibody’s antigen-binding arm.
“It likely extends the other end of itself, like a tail, over the antibody’s main antigen-binding region,” said Xueyong Zhu, PhD, of TSRI.
The team is now studying protein M’s function during M genitalium infections. It seems likely that the protein evolved to help M genitalium cope with the immune response, as it has one of the smallest bacterial genomes in nature.
“It appears to represent an elegant evolutionary solution to the special problem that mycoplasma have in evading the adaptive immune system,” Dr Grover said. “The smallest parasitic [bacterium] on planet Earth seems to have evolved the most sophisticated invading molecular machine.”
If protein M is confirmed as a universal “decoy” for antibodies, it will become a target for new drugs, the researchers said. This could make it easier to treat chronic, sometimes silent, infections by M genitalium and any other microbes that have evolved a similar antibody-thwarting defense.
In principle, protein M also could be engineered to target specific groups of B cells, delivering cytotoxic agents to cancerous B cells in patients with MM and lymphomas.
But the most immediate use of protein M, according to the researchers, is likely to be as a tool for grabbing antibodies in test tubes and cell cultures. This would allow the preparation of highly pure antibodies for research and drug manufacturing. Other generic antibody-binding proteins have been put to use in this way, but, so far, it appears that none does the job quite as well as protein M.
“It may be the most useful antibody purification device ever found,” said Dr Lerner, who is now working to commercialize the protein. ![]()
Sequencing reveals therapeutic target for leukemias

of General Medical Sciences
By analyzing the whole genomes of 3-year-old twin sisters—one healthy and one with multi-lineage leukemia (MLL)—researchers have identified a possible therapeutic target for leukemias.
Their research pointed to a molecular pathway involving the gene SETD2, which can mutate in blood cells as DNA is being transcribed and replicated.
The team confirmed the importance of this pathway via follow-up experiments using samples from leukemia patients and mouse models of the disease.
“We reasoned that monozygotic twins discordant for human leukemia would have identical inherited genetic backgrounds and well-matched tissue-specific events,” said Gang Huang, PhD, of Cincinnati Children’s Hospital Medical Center in Ohio.
“This provided a strong basis for comparison and analysis. We identified a gene mutation involving SETD2 that contributes to the initiation and progression of leukemia by promoting the self-renewal potential of leukemia stem cells.”
Dr Huang and his colleagues recounted this discovery in Nature Genetics.
The team showed that the onset of aggressive and acute leukemia is fueled by a spiraling cascade of multiple genetic mutations and chromosomal translocations.
In comparing data from the twins, the researchers identified a chromosomal translocation in the MLL-NRIP3 fusion gene.
When they activated MLL-NRIP3 in mouse models, the animals developed MLL, but it took a long period of time. This suggests that additional epigenetic and molecular events must be involved to induce full-blown leukemia.
The researchers went on to show that activation of MLL-NRIP3 cooperated with the molecular cascade (including mutations in SETD2) to cause leukemia.
The initial clue came when the team was looking for additional genomic alterations in the leukemic cells of the sick twin. They discovered that activation of MLL-NRIP3 started the molecular cascade that led to bi-allelic mutations in SETD2, a tumor suppressor that regulates the histone modification protein H3K36me3.
During transcriptional elongation, SETD2 and H3K36me3 normally mark the zone for accurate gene transcription along the DNA. In the case of the sick twin, the mutations and molecular cascade disrupted the H3K36me3 mark, leading to abnormal transcription and MLL.
To confirm the importance of these findings, the researchers analyzed blood samples from 241 patients—134 with acute myeloid leukemia and 107 with acute lymphoblastic leukemia. This revealed 19 somatic SETD2 mutations in 15 patients (6.2%).
SETD2 mutations were more common in patients with MLL rearrangements than those without—22.6% (6/27) and 4.6% (8/173), respectively. And patients with SETD2 mutations had decreased levels of global H3K36me3.
In follow-up tests on cell cultures of pre-leukemic cells and mouse models, the researchers saw the same progression of genetic mutations and related molecular events fuel the growth of leukemic cells.
The team also noticed that SETD2 mutation activated 2 genes—mTOR and JAK-STAT—that are known to contribute to leukemia and other cancers. So the researchers decided to test 2 mTOR inhibitors—Torin1 and rapamycin—on pre-leukemic cells generated by SETD2 mutations.
That treatment prompted a marked decrease in cell growth, indicating that SETD2 mutations activate numerous molecular pathways to generate leukemia.
Dr Huang said the tests also suggest there are multiple opportunities to find new molecular targets for developing more effective drugs—in particular, those that would target the MLL fusion-SETD2-H3K36me3 pathway to treat acute and aggressive leukemias.
The researchers are following up their current study by identifying additional pathways activated by mutations of SETD2. They also are looking for possible new molecular targets and therapeutic strategies to block disruptions in the SETD2-H3K36me3 pathway. ![]()
of General Medical Sciences
By analyzing the whole genomes of 3-year-old twin sisters—one healthy and one with multi-lineage leukemia (MLL)—researchers have identified a possible therapeutic target for leukemias.
Their research pointed to a molecular pathway involving the gene SETD2, which can mutate in blood cells as DNA is being transcribed and replicated.
The team confirmed the importance of this pathway via follow-up experiments using samples from leukemia patients and mouse models of the disease.
“We reasoned that monozygotic twins discordant for human leukemia would have identical inherited genetic backgrounds and well-matched tissue-specific events,” said Gang Huang, PhD, of Cincinnati Children’s Hospital Medical Center in Ohio.
“This provided a strong basis for comparison and analysis. We identified a gene mutation involving SETD2 that contributes to the initiation and progression of leukemia by promoting the self-renewal potential of leukemia stem cells.”
Dr Huang and his colleagues recounted this discovery in Nature Genetics.
The team showed that the onset of aggressive and acute leukemia is fueled by a spiraling cascade of multiple genetic mutations and chromosomal translocations.
In comparing data from the twins, the researchers identified a chromosomal translocation in the MLL-NRIP3 fusion gene.
When they activated MLL-NRIP3 in mouse models, the animals developed MLL, but it took a long period of time. This suggests that additional epigenetic and molecular events must be involved to induce full-blown leukemia.
The researchers went on to show that activation of MLL-NRIP3 cooperated with the molecular cascade (including mutations in SETD2) to cause leukemia.
The initial clue came when the team was looking for additional genomic alterations in the leukemic cells of the sick twin. They discovered that activation of MLL-NRIP3 started the molecular cascade that led to bi-allelic mutations in SETD2, a tumor suppressor that regulates the histone modification protein H3K36me3.
During transcriptional elongation, SETD2 and H3K36me3 normally mark the zone for accurate gene transcription along the DNA. In the case of the sick twin, the mutations and molecular cascade disrupted the H3K36me3 mark, leading to abnormal transcription and MLL.
To confirm the importance of these findings, the researchers analyzed blood samples from 241 patients—134 with acute myeloid leukemia and 107 with acute lymphoblastic leukemia. This revealed 19 somatic SETD2 mutations in 15 patients (6.2%).
SETD2 mutations were more common in patients with MLL rearrangements than those without—22.6% (6/27) and 4.6% (8/173), respectively. And patients with SETD2 mutations had decreased levels of global H3K36me3.
In follow-up tests on cell cultures of pre-leukemic cells and mouse models, the researchers saw the same progression of genetic mutations and related molecular events fuel the growth of leukemic cells.
The team also noticed that SETD2 mutation activated 2 genes—mTOR and JAK-STAT—that are known to contribute to leukemia and other cancers. So the researchers decided to test 2 mTOR inhibitors—Torin1 and rapamycin—on pre-leukemic cells generated by SETD2 mutations.
That treatment prompted a marked decrease in cell growth, indicating that SETD2 mutations activate numerous molecular pathways to generate leukemia.
Dr Huang said the tests also suggest there are multiple opportunities to find new molecular targets for developing more effective drugs—in particular, those that would target the MLL fusion-SETD2-H3K36me3 pathway to treat acute and aggressive leukemias.
The researchers are following up their current study by identifying additional pathways activated by mutations of SETD2. They also are looking for possible new molecular targets and therapeutic strategies to block disruptions in the SETD2-H3K36me3 pathway. ![]()
of General Medical Sciences
By analyzing the whole genomes of 3-year-old twin sisters—one healthy and one with multi-lineage leukemia (MLL)—researchers have identified a possible therapeutic target for leukemias.
Their research pointed to a molecular pathway involving the gene SETD2, which can mutate in blood cells as DNA is being transcribed and replicated.
The team confirmed the importance of this pathway via follow-up experiments using samples from leukemia patients and mouse models of the disease.
“We reasoned that monozygotic twins discordant for human leukemia would have identical inherited genetic backgrounds and well-matched tissue-specific events,” said Gang Huang, PhD, of Cincinnati Children’s Hospital Medical Center in Ohio.
“This provided a strong basis for comparison and analysis. We identified a gene mutation involving SETD2 that contributes to the initiation and progression of leukemia by promoting the self-renewal potential of leukemia stem cells.”
Dr Huang and his colleagues recounted this discovery in Nature Genetics.
The team showed that the onset of aggressive and acute leukemia is fueled by a spiraling cascade of multiple genetic mutations and chromosomal translocations.
In comparing data from the twins, the researchers identified a chromosomal translocation in the MLL-NRIP3 fusion gene.
When they activated MLL-NRIP3 in mouse models, the animals developed MLL, but it took a long period of time. This suggests that additional epigenetic and molecular events must be involved to induce full-blown leukemia.
The researchers went on to show that activation of MLL-NRIP3 cooperated with the molecular cascade (including mutations in SETD2) to cause leukemia.
The initial clue came when the team was looking for additional genomic alterations in the leukemic cells of the sick twin. They discovered that activation of MLL-NRIP3 started the molecular cascade that led to bi-allelic mutations in SETD2, a tumor suppressor that regulates the histone modification protein H3K36me3.
During transcriptional elongation, SETD2 and H3K36me3 normally mark the zone for accurate gene transcription along the DNA. In the case of the sick twin, the mutations and molecular cascade disrupted the H3K36me3 mark, leading to abnormal transcription and MLL.
To confirm the importance of these findings, the researchers analyzed blood samples from 241 patients—134 with acute myeloid leukemia and 107 with acute lymphoblastic leukemia. This revealed 19 somatic SETD2 mutations in 15 patients (6.2%).
SETD2 mutations were more common in patients with MLL rearrangements than those without—22.6% (6/27) and 4.6% (8/173), respectively. And patients with SETD2 mutations had decreased levels of global H3K36me3.
In follow-up tests on cell cultures of pre-leukemic cells and mouse models, the researchers saw the same progression of genetic mutations and related molecular events fuel the growth of leukemic cells.
The team also noticed that SETD2 mutation activated 2 genes—mTOR and JAK-STAT—that are known to contribute to leukemia and other cancers. So the researchers decided to test 2 mTOR inhibitors—Torin1 and rapamycin—on pre-leukemic cells generated by SETD2 mutations.
That treatment prompted a marked decrease in cell growth, indicating that SETD2 mutations activate numerous molecular pathways to generate leukemia.
Dr Huang said the tests also suggest there are multiple opportunities to find new molecular targets for developing more effective drugs—in particular, those that would target the MLL fusion-SETD2-H3K36me3 pathway to treat acute and aggressive leukemias.
The researchers are following up their current study by identifying additional pathways activated by mutations of SETD2. They also are looking for possible new molecular targets and therapeutic strategies to block disruptions in the SETD2-H3K36me3 pathway. ![]()
Team discovers global regulator of RNA editing
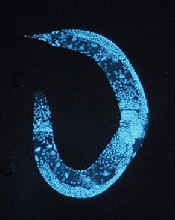
Credit: UC San Diego
School of Medicine
Scientists say they have identified a protein that broadly regulates how genetic information transcribed from DNA to messenger RNA (mRNA) is processed and ultimately translated into the myriad proteins necessary for life.
The group’s work shows that the protein ADR-1 binds to mRNA and then enhances RNA editing.
This process allows a gene to be present as multiple mRNAs that can each affect gene expression differently.
The research appears in Cell Reports.
“Problems with RNA editing show up in many human diseases, including those of neurodegeneration, cancer, and blood disorders,” said study author Gene Yeo, PhD, of the University of California, San Diego.
“This is the first time that a single protein has been identified that broadly regulates RNA editing. There are probably hundreds more. Our approach provides
a method to screen for them and opens up new ways to study human biology and disease.”
Using the model organism Caenorhabditis elegans, Dr Yeo and his colleagues identified more than 400 new mRNA editing sites. As the majority of them were regulated by ADR-1, the team declared the protein the first global regulator of RNA editing.
“What we’ve determined is that this protein’s ability to alter editing of mRNAs is not specific to just a few genes, but, instead, its ability to bind to mRNAs is required for proper RNA editing of most mRNAs,” said study author Michael C. Washburn, of Indiana University in Bloomington.
The group found that the region of ADR-1 protein that binds to target mRNAs in C elegans is required for regulating editing. This region is present in many human proteins, and a protein similar to ADR-1 is specifically expressed in human neurons.
“So it is likely that a similar mechanism exists to regulate editing in humans,” said study author Heather A. Hundley, PhD, also of Indiana University.
“Further work in our lab will be aimed at understanding the detailed mechanism of how these proteins regulate editing, in turn providing an inroad to developing therapeutics that modulate editing for the treatment of human diseases.”
C elegans, like humans, highly expresses a family of proteins in the nervous system called adenosine deaminases acting on RNA (ADARs), a family that includes ADR-1.
ADARs change specific nucleotides in RNA in a process called adenosine-to-inosine editing (A-to-I editing) that diversifies genetic information to specify different amino acids, splice sites, and structures.
Scientists currently estimate there are between 400,000 and 1 million A-to-I editing events in noncoding regions of the human transcriptome.
Newly synthesized RNA encodes the exact information found in DNA. But it’s when later RNA editing occurs that RNA gets altered, and this change is most often catalyzed by ADARs.
“One thing we also know is that ADAR protein levels are not altered in disease, implying that other mechanisms are also at work regulating ADAR-mediated RNA editing,” Dr Hundley said. “By identifying this major regulator of noncoding editing in C elegans, we can now focus on dissecting the regulatory mechanism and determining the conservation of this regulatory protein in human cells.” ![]()
Credit: UC San Diego
School of Medicine
Scientists say they have identified a protein that broadly regulates how genetic information transcribed from DNA to messenger RNA (mRNA) is processed and ultimately translated into the myriad proteins necessary for life.
The group’s work shows that the protein ADR-1 binds to mRNA and then enhances RNA editing.
This process allows a gene to be present as multiple mRNAs that can each affect gene expression differently.
The research appears in Cell Reports.
“Problems with RNA editing show up in many human diseases, including those of neurodegeneration, cancer, and blood disorders,” said study author Gene Yeo, PhD, of the University of California, San Diego.
“This is the first time that a single protein has been identified that broadly regulates RNA editing. There are probably hundreds more. Our approach provides
a method to screen for them and opens up new ways to study human biology and disease.”
Using the model organism Caenorhabditis elegans, Dr Yeo and his colleagues identified more than 400 new mRNA editing sites. As the majority of them were regulated by ADR-1, the team declared the protein the first global regulator of RNA editing.
“What we’ve determined is that this protein’s ability to alter editing of mRNAs is not specific to just a few genes, but, instead, its ability to bind to mRNAs is required for proper RNA editing of most mRNAs,” said study author Michael C. Washburn, of Indiana University in Bloomington.
The group found that the region of ADR-1 protein that binds to target mRNAs in C elegans is required for regulating editing. This region is present in many human proteins, and a protein similar to ADR-1 is specifically expressed in human neurons.
“So it is likely that a similar mechanism exists to regulate editing in humans,” said study author Heather A. Hundley, PhD, also of Indiana University.
“Further work in our lab will be aimed at understanding the detailed mechanism of how these proteins regulate editing, in turn providing an inroad to developing therapeutics that modulate editing for the treatment of human diseases.”
C elegans, like humans, highly expresses a family of proteins in the nervous system called adenosine deaminases acting on RNA (ADARs), a family that includes ADR-1.
ADARs change specific nucleotides in RNA in a process called adenosine-to-inosine editing (A-to-I editing) that diversifies genetic information to specify different amino acids, splice sites, and structures.
Scientists currently estimate there are between 400,000 and 1 million A-to-I editing events in noncoding regions of the human transcriptome.
Newly synthesized RNA encodes the exact information found in DNA. But it’s when later RNA editing occurs that RNA gets altered, and this change is most often catalyzed by ADARs.
“One thing we also know is that ADAR protein levels are not altered in disease, implying that other mechanisms are also at work regulating ADAR-mediated RNA editing,” Dr Hundley said. “By identifying this major regulator of noncoding editing in C elegans, we can now focus on dissecting the regulatory mechanism and determining the conservation of this regulatory protein in human cells.” ![]()
Credit: UC San Diego
School of Medicine
Scientists say they have identified a protein that broadly regulates how genetic information transcribed from DNA to messenger RNA (mRNA) is processed and ultimately translated into the myriad proteins necessary for life.
The group’s work shows that the protein ADR-1 binds to mRNA and then enhances RNA editing.
This process allows a gene to be present as multiple mRNAs that can each affect gene expression differently.
The research appears in Cell Reports.
“Problems with RNA editing show up in many human diseases, including those of neurodegeneration, cancer, and blood disorders,” said study author Gene Yeo, PhD, of the University of California, San Diego.
“This is the first time that a single protein has been identified that broadly regulates RNA editing. There are probably hundreds more. Our approach provides
a method to screen for them and opens up new ways to study human biology and disease.”
Using the model organism Caenorhabditis elegans, Dr Yeo and his colleagues identified more than 400 new mRNA editing sites. As the majority of them were regulated by ADR-1, the team declared the protein the first global regulator of RNA editing.
“What we’ve determined is that this protein’s ability to alter editing of mRNAs is not specific to just a few genes, but, instead, its ability to bind to mRNAs is required for proper RNA editing of most mRNAs,” said study author Michael C. Washburn, of Indiana University in Bloomington.
The group found that the region of ADR-1 protein that binds to target mRNAs in C elegans is required for regulating editing. This region is present in many human proteins, and a protein similar to ADR-1 is specifically expressed in human neurons.
“So it is likely that a similar mechanism exists to regulate editing in humans,” said study author Heather A. Hundley, PhD, also of Indiana University.
“Further work in our lab will be aimed at understanding the detailed mechanism of how these proteins regulate editing, in turn providing an inroad to developing therapeutics that modulate editing for the treatment of human diseases.”
C elegans, like humans, highly expresses a family of proteins in the nervous system called adenosine deaminases acting on RNA (ADARs), a family that includes ADR-1.
ADARs change specific nucleotides in RNA in a process called adenosine-to-inosine editing (A-to-I editing) that diversifies genetic information to specify different amino acids, splice sites, and structures.
Scientists currently estimate there are between 400,000 and 1 million A-to-I editing events in noncoding regions of the human transcriptome.
Newly synthesized RNA encodes the exact information found in DNA. But it’s when later RNA editing occurs that RNA gets altered, and this change is most often catalyzed by ADARs.
“One thing we also know is that ADAR protein levels are not altered in disease, implying that other mechanisms are also at work regulating ADAR-mediated RNA editing,” Dr Hundley said. “By identifying this major regulator of noncoding editing in C elegans, we can now focus on dissecting the regulatory mechanism and determining the conservation of this regulatory protein in human cells.” ![]()
Leaders agree on SGR bill but funding still a question

Credit: Lawrence Jackson
Leaders from the US Senate and House of Representatives have agreed on a plan to replace the Medicare sustainable growth rate (SGR) formula, but they must still agree on how to pay for it.
The group’s bill, called the SGR Repeal and Medicare Provider Payment Modernization Act of 2014, would remove the threat of a 23.7% cut in Medicare payments that is set to take effect on April 1.
And it would provide annual payment updates of 0.5% for the next 5 years, as Medicare transitions to a payment system designed to reward physicians based on care quality, not quantity.
“This is a significant step forward in our long-standing effort to replace the flawed physician update formula with a 21st-century system focused on quality and value rather than the quantity of services,” said House Ways and Means Committee Ranking Member Sander Levin (D-Mich.).
“For the first time in many years, we have agreement among the bipartisan leadership of the 3 committees that oversee Medicare. Now, we have to turn to the thorny issue of offsets.”
The Congressional Budget Office has estimated that the new plan would increase direct spending by $150.4 billion from 2014 to 2023, although other sources have said the cost would be about $126 billion for the same time period.
Repealing the SGR
The SGR calls for annual, automatic cuts in Medicare payments to physicians. It was established in 1997 to control physician spending, but, over the years, the necessary cuts have not occurred.
Since 2003, Congress has spent about $150 billion to provide short-term fixes to spare physicians from the cuts. Without another short-term fix or legislation to permanently eliminate the SGR, physicians would see a 23.7% cut in Medicare payments starting April 1.
For years, Congress has been trying to eliminate the SGR, but agreeing on a plan to do so—and a way to pay for it—has proven difficult. Now, Congress says it has brought “renewed commitment to repealing and replacing the flawed SGR update mechanism.”
Provisions of the bill
The SGR Repeal and Medicare Provider Payment Modernization Act of 2014 would institute a 0.5% annual payment increase for physicians from 2014 through 2018. According to Congress, this would help the transition to a merit-based incentive payment system (MIPS), beginning in 2018.
The MIPS consolidates the 3 existing incentive programs—the Physician Quality Reporting System, the Value-Based Payment Modifier, and the Meaningful Use of Electronic Health Records—into a single program that would reward providers who meet performance thresholds.
The bill would also give a 5% bonus to providers who receive a significant portion of their revenue from an alternative payment model (APM) or patient-centered medical home (PCMH). Participants would need to receive at least 25% of their Medicare revenue through an APM in 2018-2019, and this threshold would increase over time.
The bill would provide incentives for participation in private-payer APMs as well. And it would establish a technical advisory committee to review and recommend physician-developed APMs based on criteria developed through an open-comment process.
Finally, the bill would expand the use of Medicare data. Quality and utilization data would be posted on the Physician Compare website with the goal of helping patients to make more informed decisions about their care.
Furthermore, “qualified entities” would be allowed to provide analyses and underlying data to providers, although this provision is subject to privacy and security laws. And qualified clinical data registries would be allowed to purchase claims data.
To read the bill in full, visit Congress.gov. ![]()
Credit: Lawrence Jackson
Leaders from the US Senate and House of Representatives have agreed on a plan to replace the Medicare sustainable growth rate (SGR) formula, but they must still agree on how to pay for it.
The group’s bill, called the SGR Repeal and Medicare Provider Payment Modernization Act of 2014, would remove the threat of a 23.7% cut in Medicare payments that is set to take effect on April 1.
And it would provide annual payment updates of 0.5% for the next 5 years, as Medicare transitions to a payment system designed to reward physicians based on care quality, not quantity.
“This is a significant step forward in our long-standing effort to replace the flawed physician update formula with a 21st-century system focused on quality and value rather than the quantity of services,” said House Ways and Means Committee Ranking Member Sander Levin (D-Mich.).
“For the first time in many years, we have agreement among the bipartisan leadership of the 3 committees that oversee Medicare. Now, we have to turn to the thorny issue of offsets.”
The Congressional Budget Office has estimated that the new plan would increase direct spending by $150.4 billion from 2014 to 2023, although other sources have said the cost would be about $126 billion for the same time period.
Repealing the SGR
The SGR calls for annual, automatic cuts in Medicare payments to physicians. It was established in 1997 to control physician spending, but, over the years, the necessary cuts have not occurred.
Since 2003, Congress has spent about $150 billion to provide short-term fixes to spare physicians from the cuts. Without another short-term fix or legislation to permanently eliminate the SGR, physicians would see a 23.7% cut in Medicare payments starting April 1.
For years, Congress has been trying to eliminate the SGR, but agreeing on a plan to do so—and a way to pay for it—has proven difficult. Now, Congress says it has brought “renewed commitment to repealing and replacing the flawed SGR update mechanism.”
Provisions of the bill
The SGR Repeal and Medicare Provider Payment Modernization Act of 2014 would institute a 0.5% annual payment increase for physicians from 2014 through 2018. According to Congress, this would help the transition to a merit-based incentive payment system (MIPS), beginning in 2018.
The MIPS consolidates the 3 existing incentive programs—the Physician Quality Reporting System, the Value-Based Payment Modifier, and the Meaningful Use of Electronic Health Records—into a single program that would reward providers who meet performance thresholds.
The bill would also give a 5% bonus to providers who receive a significant portion of their revenue from an alternative payment model (APM) or patient-centered medical home (PCMH). Participants would need to receive at least 25% of their Medicare revenue through an APM in 2018-2019, and this threshold would increase over time.
The bill would provide incentives for participation in private-payer APMs as well. And it would establish a technical advisory committee to review and recommend physician-developed APMs based on criteria developed through an open-comment process.
Finally, the bill would expand the use of Medicare data. Quality and utilization data would be posted on the Physician Compare website with the goal of helping patients to make more informed decisions about their care.
Furthermore, “qualified entities” would be allowed to provide analyses and underlying data to providers, although this provision is subject to privacy and security laws. And qualified clinical data registries would be allowed to purchase claims data.
To read the bill in full, visit Congress.gov. ![]()
Credit: Lawrence Jackson
Leaders from the US Senate and House of Representatives have agreed on a plan to replace the Medicare sustainable growth rate (SGR) formula, but they must still agree on how to pay for it.
The group’s bill, called the SGR Repeal and Medicare Provider Payment Modernization Act of 2014, would remove the threat of a 23.7% cut in Medicare payments that is set to take effect on April 1.
And it would provide annual payment updates of 0.5% for the next 5 years, as Medicare transitions to a payment system designed to reward physicians based on care quality, not quantity.
“This is a significant step forward in our long-standing effort to replace the flawed physician update formula with a 21st-century system focused on quality and value rather than the quantity of services,” said House Ways and Means Committee Ranking Member Sander Levin (D-Mich.).
“For the first time in many years, we have agreement among the bipartisan leadership of the 3 committees that oversee Medicare. Now, we have to turn to the thorny issue of offsets.”
The Congressional Budget Office has estimated that the new plan would increase direct spending by $150.4 billion from 2014 to 2023, although other sources have said the cost would be about $126 billion for the same time period.
Repealing the SGR
The SGR calls for annual, automatic cuts in Medicare payments to physicians. It was established in 1997 to control physician spending, but, over the years, the necessary cuts have not occurred.
Since 2003, Congress has spent about $150 billion to provide short-term fixes to spare physicians from the cuts. Without another short-term fix or legislation to permanently eliminate the SGR, physicians would see a 23.7% cut in Medicare payments starting April 1.
For years, Congress has been trying to eliminate the SGR, but agreeing on a plan to do so—and a way to pay for it—has proven difficult. Now, Congress says it has brought “renewed commitment to repealing and replacing the flawed SGR update mechanism.”
Provisions of the bill
The SGR Repeal and Medicare Provider Payment Modernization Act of 2014 would institute a 0.5% annual payment increase for physicians from 2014 through 2018. According to Congress, this would help the transition to a merit-based incentive payment system (MIPS), beginning in 2018.
The MIPS consolidates the 3 existing incentive programs—the Physician Quality Reporting System, the Value-Based Payment Modifier, and the Meaningful Use of Electronic Health Records—into a single program that would reward providers who meet performance thresholds.
The bill would also give a 5% bonus to providers who receive a significant portion of their revenue from an alternative payment model (APM) or patient-centered medical home (PCMH). Participants would need to receive at least 25% of their Medicare revenue through an APM in 2018-2019, and this threshold would increase over time.
The bill would provide incentives for participation in private-payer APMs as well. And it would establish a technical advisory committee to review and recommend physician-developed APMs based on criteria developed through an open-comment process.
Finally, the bill would expand the use of Medicare data. Quality and utilization data would be posted on the Physician Compare website with the goal of helping patients to make more informed decisions about their care.
Furthermore, “qualified entities” would be allowed to provide analyses and underlying data to providers, although this provision is subject to privacy and security laws. And qualified clinical data registries would be allowed to purchase claims data.
To read the bill in full, visit Congress.gov.
Defect causes bone marrow failure, group finds
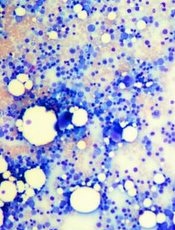
Credit: Daniel E. Sabath
Researchers say they’ve discovered a distinct bone-marrow-failure syndrome and the genetic defect that causes it.
In its natural form, the gene ERCC6L2 plays a role in DNA repair and mitochondrial function.
But investigators found evidence to suggest that mutations in ERCC6L2, and the subsequent DNA damage, were the underlying cause of tri-lineage bone marrow failure in a pair of patients with neurological dysfunction.
“New DNA sequencing technology has enabled us to identify and define a new gene defect which causes a particular type of bone marrow failure,” said Inderjeet Dokal, MD, of Queen Mary University of London in the UK.
“Clinicians treating patients with bone marrow failure should now include analysis for this gene in their investigation.”
Dr Dokal and his colleagues described this research in The American Journal of Human Genetics.
The team performed exome sequencing in 3 patients with genetically uncharacterized, tri-lineage bone marrow failure.
The patients came from consanguineous families (their parents were first-degree cousins), they had developmental delays characterized by learning disabilities, and 2 of the patients had microcephaly.
The sequencing did not uncover variations in any of the known genes associated with bone marrow failure. And the researchers could not find any obvious disease-causing variants in 1 of the patients.
However, the other 2 patients shared homozygous truncating mutations in ERCC6L2—c.1963C>T (p.Arg655*) and c.1236_1239delAACA (p.Thr413Cysfs*2). The c.1963C>T variant had already been identified, but, to the researchers’ knowledge, the other variant had not.
Additional experiments suggested that these mutations affect the subcellular localization and stability of ERCC6L2.
The investigators then speculated that ERCC6L2 plays a role in the DNA-damage response. To test that theory, they mimicked the truncating mutations by knocking down ERCC6L2 expression in human A549 cells.
This significantly reduced cell viability when the cells were exposed to the DNA-damaging agents mitomycin C and irofulven.
To further confirm their theory, the researchers looked at another marker of DNA damage. Previous research had suggested that Snf2 protein complexes are involved in the recruitment of γH2AX, a phosphorylated form of histone 2A, to sites of DNA damage.
So the team performed immunostaining with a γH2AX-specific antibody. And they found that ERCC6L2-knockdown cells displayed H2AX phosphorylation, an effect that increased upon genotoxic stress (treatment with irofulven).
These results indicate that ERCC6L2 plays a role in the DNA-damage-response pathway, and knockdown of this gene sensitizes cells to genotoxic agents.
Additional experiments showed that ERCC6L2 translocated to the mitochondria and the nucleus in response to DNA damage. And ERCC6L2 knockdown induced intracellular reactive oxygen species (ROS).
But introducing the ROS scavenger N-acetyl cysteine diminished the cytotoxicity induced by irofulven, and it halted ERCCGL2 traffic to the mitochondria and nucleus.
The investigators said these results point to a distinct bone-marrow-failure syndrome resulting from mutations in ERCC6L2.
“This is a promising finding which we hope, one day, could lead to finding an effective treatment for this type of gene defect,” Dr Dokal said. “Now [that] we know this research technique works, we plan to carry out further studies to shed more light on the genetic basis of many other cases of bone marrow failure.”
Credit: Daniel E. Sabath
Researchers say they’ve discovered a distinct bone-marrow-failure syndrome and the genetic defect that causes it.
In its natural form, the gene ERCC6L2 plays a role in DNA repair and mitochondrial function.
But investigators found evidence to suggest that mutations in ERCC6L2, and the subsequent DNA damage, were the underlying cause of tri-lineage bone marrow failure in a pair of patients with neurological dysfunction.
“New DNA sequencing technology has enabled us to identify and define a new gene defect which causes a particular type of bone marrow failure,” said Inderjeet Dokal, MD, of Queen Mary University of London in the UK.
“Clinicians treating patients with bone marrow failure should now include analysis for this gene in their investigation.”
Dr Dokal and his colleagues described this research in The American Journal of Human Genetics.
The team performed exome sequencing in 3 patients with genetically uncharacterized, tri-lineage bone marrow failure.
The patients came from consanguineous families (their parents were first-degree cousins), they had developmental delays characterized by learning disabilities, and 2 of the patients had microcephaly.
The sequencing did not uncover variations in any of the known genes associated with bone marrow failure. And the researchers could not find any obvious disease-causing variants in 1 of the patients.
However, the other 2 patients shared homozygous truncating mutations in ERCC6L2—c.1963C>T (p.Arg655*) and c.1236_1239delAACA (p.Thr413Cysfs*2). The c.1963C>T variant had already been identified, but, to the researchers’ knowledge, the other variant had not.
Additional experiments suggested that these mutations affect the subcellular localization and stability of ERCC6L2.
The investigators then speculated that ERCC6L2 plays a role in the DNA-damage response. To test that theory, they mimicked the truncating mutations by knocking down ERCC6L2 expression in human A549 cells.
This significantly reduced cell viability when the cells were exposed to the DNA-damaging agents mitomycin C and irofulven.
To further confirm their theory, the researchers looked at another marker of DNA damage. Previous research had suggested that Snf2 protein complexes are involved in the recruitment of γH2AX, a phosphorylated form of histone 2A, to sites of DNA damage.
So the team performed immunostaining with a γH2AX-specific antibody. And they found that ERCC6L2-knockdown cells displayed H2AX phosphorylation, an effect that increased upon genotoxic stress (treatment with irofulven).
These results indicate that ERCC6L2 plays a role in the DNA-damage-response pathway, and knockdown of this gene sensitizes cells to genotoxic agents.
Additional experiments showed that ERCC6L2 translocated to the mitochondria and the nucleus in response to DNA damage. And ERCC6L2 knockdown induced intracellular reactive oxygen species (ROS).
But introducing the ROS scavenger N-acetyl cysteine diminished the cytotoxicity induced by irofulven, and it halted ERCCGL2 traffic to the mitochondria and nucleus.
The investigators said these results point to a distinct bone-marrow-failure syndrome resulting from mutations in ERCC6L2.
“This is a promising finding which we hope, one day, could lead to finding an effective treatment for this type of gene defect,” Dr Dokal said. “Now [that] we know this research technique works, we plan to carry out further studies to shed more light on the genetic basis of many other cases of bone marrow failure.”
Credit: Daniel E. Sabath
Researchers say they’ve discovered a distinct bone-marrow-failure syndrome and the genetic defect that causes it.
In its natural form, the gene ERCC6L2 plays a role in DNA repair and mitochondrial function.
But investigators found evidence to suggest that mutations in ERCC6L2, and the subsequent DNA damage, were the underlying cause of tri-lineage bone marrow failure in a pair of patients with neurological dysfunction.
“New DNA sequencing technology has enabled us to identify and define a new gene defect which causes a particular type of bone marrow failure,” said Inderjeet Dokal, MD, of Queen Mary University of London in the UK.
“Clinicians treating patients with bone marrow failure should now include analysis for this gene in their investigation.”
Dr Dokal and his colleagues described this research in The American Journal of Human Genetics.
The team performed exome sequencing in 3 patients with genetically uncharacterized, tri-lineage bone marrow failure.
The patients came from consanguineous families (their parents were first-degree cousins), they had developmental delays characterized by learning disabilities, and 2 of the patients had microcephaly.
The sequencing did not uncover variations in any of the known genes associated with bone marrow failure. And the researchers could not find any obvious disease-causing variants in 1 of the patients.
However, the other 2 patients shared homozygous truncating mutations in ERCC6L2—c.1963C>T (p.Arg655*) and c.1236_1239delAACA (p.Thr413Cysfs*2). The c.1963C>T variant had already been identified, but, to the researchers’ knowledge, the other variant had not.
Additional experiments suggested that these mutations affect the subcellular localization and stability of ERCC6L2.
The investigators then speculated that ERCC6L2 plays a role in the DNA-damage response. To test that theory, they mimicked the truncating mutations by knocking down ERCC6L2 expression in human A549 cells.
This significantly reduced cell viability when the cells were exposed to the DNA-damaging agents mitomycin C and irofulven.
To further confirm their theory, the researchers looked at another marker of DNA damage. Previous research had suggested that Snf2 protein complexes are involved in the recruitment of γH2AX, a phosphorylated form of histone 2A, to sites of DNA damage.
So the team performed immunostaining with a γH2AX-specific antibody. And they found that ERCC6L2-knockdown cells displayed H2AX phosphorylation, an effect that increased upon genotoxic stress (treatment with irofulven).
These results indicate that ERCC6L2 plays a role in the DNA-damage-response pathway, and knockdown of this gene sensitizes cells to genotoxic agents.
Additional experiments showed that ERCC6L2 translocated to the mitochondria and the nucleus in response to DNA damage. And ERCC6L2 knockdown induced intracellular reactive oxygen species (ROS).
But introducing the ROS scavenger N-acetyl cysteine diminished the cytotoxicity induced by irofulven, and it halted ERCCGL2 traffic to the mitochondria and nucleus.
The investigators said these results point to a distinct bone-marrow-failure syndrome resulting from mutations in ERCC6L2.
“This is a promising finding which we hope, one day, could lead to finding an effective treatment for this type of gene defect,” Dr Dokal said. “Now [that] we know this research technique works, we plan to carry out further studies to shed more light on the genetic basis of many other cases of bone marrow failure.”
Climate change may increase malaria incidence
Credit: James Gathany
New research indicates that, toward the end of the century, climate change may make tropical highland regions suitable breeding grounds for malaria. And this could greatly increase the incidence of the disease.
Scientists compared the latest predictions for global warming with a range of statistical models commonly used to predict the spread of malaria.
And the models suggested that the changing climate will allow malaria to move into higher altitudes during warmer seasons and become permanently resident in larger areas.
This would mainly affect Africa but would also have an impact in Asia and South America.
In eastern Africa, the change could result in an additional 100 million people being exposed to malaria by the end of the 2080s, the researchers calculated.
However, they noted that the size of the impact is highly variable, as it can be affected by a number of factors.
“[W]e expect increased urbanization in these areas over the next 70 years,” said study author Cyril Caminade, PhD, of the University of Liverpool in the UK.
“Other developments, such as land use changes, population movements, and economic growth, will also have to be accounted for in future studies. What is clear is that diseases such as malaria are going to be moving, and this is a crucial element of how we prepare for the effects of climate change.”
The comparison of these models has not been carried out before, and by doing so, the researchers were able to find that malaria spreading to tropical highland areas was the one area in which the models agreed.
There was distinct variation in other parts of the world. Two of the models predicted that malaria would move northward, eventually spreading into Europe, Russia, and North America. Others showed that malaria would move north, but only as far as North Africa, where it was eliminated in the 20th century.
Credit: James Gathany
New research indicates that, toward the end of the century, climate change may make tropical highland regions suitable breeding grounds for malaria. And this could greatly increase the incidence of the disease.
Scientists compared the latest predictions for global warming with a range of statistical models commonly used to predict the spread of malaria.
And the models suggested that the changing climate will allow malaria to move into higher altitudes during warmer seasons and become permanently resident in larger areas.
This would mainly affect Africa but would also have an impact in Asia and South America.
In eastern Africa, the change could result in an additional 100 million people being exposed to malaria by the end of the 2080s, the researchers calculated.
However, they noted that the size of the impact is highly variable, as it can be affected by a number of factors.
“[W]e expect increased urbanization in these areas over the next 70 years,” said study author Cyril Caminade, PhD, of the University of Liverpool in the UK.
“Other developments, such as land use changes, population movements, and economic growth, will also have to be accounted for in future studies. What is clear is that diseases such as malaria are going to be moving, and this is a crucial element of how we prepare for the effects of climate change.”
The comparison of these models has not been carried out before, and by doing so, the researchers were able to find that malaria spreading to tropical highland areas was the one area in which the models agreed.
There was distinct variation in other parts of the world. Two of the models predicted that malaria would move northward, eventually spreading into Europe, Russia, and North America. Others showed that malaria would move north, but only as far as North Africa, where it was eliminated in the 20th century.
Credit: James Gathany
New research indicates that, toward the end of the century, climate change may make tropical highland regions suitable breeding grounds for malaria. And this could greatly increase the incidence of the disease.
Scientists compared the latest predictions for global warming with a range of statistical models commonly used to predict the spread of malaria.
And the models suggested that the changing climate will allow malaria to move into higher altitudes during warmer seasons and become permanently resident in larger areas.
This would mainly affect Africa but would also have an impact in Asia and South America.
In eastern Africa, the change could result in an additional 100 million people being exposed to malaria by the end of the 2080s, the researchers calculated.
However, they noted that the size of the impact is highly variable, as it can be affected by a number of factors.
“[W]e expect increased urbanization in these areas over the next 70 years,” said study author Cyril Caminade, PhD, of the University of Liverpool in the UK.
“Other developments, such as land use changes, population movements, and economic growth, will also have to be accounted for in future studies. What is clear is that diseases such as malaria are going to be moving, and this is a crucial element of how we prepare for the effects of climate change.”
The comparison of these models has not been carried out before, and by doing so, the researchers were able to find that malaria spreading to tropical highland areas was the one area in which the models agreed.
There was distinct variation in other parts of the world. Two of the models predicted that malaria would move northward, eventually spreading into Europe, Russia, and North America. Others showed that malaria would move north, but only as far as North Africa, where it was eliminated in the 20th century.
Method captures images of multiple cell components

(green), mitochondria (purple),
Golgi apparatus (red), and
peroxisomes (yellow) in a cell
Credit: Maier Avendano
A new microscopy method allows scientists to image many cellular components at once, according to a paper published in Nature Methods.
Such images could shed light on complex cellular pathways and might lead to new ways to diagnose disease, track its progress, or monitor treatment effectiveness at a cellular level, the researchers said.
They noted that today’s imaging methods typically spot, at most, 3 or 4 types of biomolecules simultaneously.
But to truly understand complex cellular functions, it’s important to be able to visualize most or all of the molecules at once, said study author Peng Yin, PhD, of the Wyss Institute at Harvard Medical School in Boston.
“If you can see only a few things at a time, you are missing the big picture,” Dr Yin said.
So he and his colleagues sought a way to take aerial views of cells that could show dozens of types of biomolecules. They decided to build upon a method called DNA-PAINT, which can create snapshots of up to 3 molecules at once by labeling them with different colored dyes.
The team modified DNA-PAINT to create a method called Exchange-PAINT. Exchange-PAINT relies on the fact that DNA strands with the correct sequence of nucleotides bind specifically to partner strands with complementary sequences.
The researchers label a biomolecule they want to visualize with a short DNA tag. Then, they add to the solution a partner strand carrying a fluorescent dye that lights up only when the 2 strands pair up.
When that partner strand binds the tagged biomolecule, it lights up, then lets go, causing the biomolecule to “blink” at a precise rate the researchers can control. The team uses this blinking to obtain ultra-sharp images.
They repeat the process to visualize a second target, a third, and so on. Then, they overlay the resulting images to create a composite image in which each biomolecule is assigned a different color.
This allows them to create false-color images that simultaneously show many types of biomolecules—far more than they could simultaneously visualize by labeling them with different colored dyes. And these false-color images allow them to spot enough biomolecules at once to capture the entire scene.
To test Exchange-PAINT, the researchers created 10 unique pieces of folded DNA that resembled the numerals 0 through 9. These numerals could be resolved with less than 10 nanometers resolution, or 1/20th of the diffraction limit.
The team was able to use Exchange-PAINT to capture clear images of the 10 different DNA origami structures in one image. They also used the method to capture images of fixed human cells, with each color tagging a different cellular component—microtubules, mitochondria, Golgi apparatus, or peroxisomes.
Dr Yin expects that, with further development, this method could be used to visualize dozens of cellular components at once.
(green), mitochondria (purple),
Golgi apparatus (red), and
peroxisomes (yellow) in a cell
Credit: Maier Avendano
A new microscopy method allows scientists to image many cellular components at once, according to a paper published in Nature Methods.
Such images could shed light on complex cellular pathways and might lead to new ways to diagnose disease, track its progress, or monitor treatment effectiveness at a cellular level, the researchers said.
They noted that today’s imaging methods typically spot, at most, 3 or 4 types of biomolecules simultaneously.
But to truly understand complex cellular functions, it’s important to be able to visualize most or all of the molecules at once, said study author Peng Yin, PhD, of the Wyss Institute at Harvard Medical School in Boston.
“If you can see only a few things at a time, you are missing the big picture,” Dr Yin said.
So he and his colleagues sought a way to take aerial views of cells that could show dozens of types of biomolecules. They decided to build upon a method called DNA-PAINT, which can create snapshots of up to 3 molecules at once by labeling them with different colored dyes.
The team modified DNA-PAINT to create a method called Exchange-PAINT. Exchange-PAINT relies on the fact that DNA strands with the correct sequence of nucleotides bind specifically to partner strands with complementary sequences.
The researchers label a biomolecule they want to visualize with a short DNA tag. Then, they add to the solution a partner strand carrying a fluorescent dye that lights up only when the 2 strands pair up.
When that partner strand binds the tagged biomolecule, it lights up, then lets go, causing the biomolecule to “blink” at a precise rate the researchers can control. The team uses this blinking to obtain ultra-sharp images.
They repeat the process to visualize a second target, a third, and so on. Then, they overlay the resulting images to create a composite image in which each biomolecule is assigned a different color.
This allows them to create false-color images that simultaneously show many types of biomolecules—far more than they could simultaneously visualize by labeling them with different colored dyes. And these false-color images allow them to spot enough biomolecules at once to capture the entire scene.
To test Exchange-PAINT, the researchers created 10 unique pieces of folded DNA that resembled the numerals 0 through 9. These numerals could be resolved with less than 10 nanometers resolution, or 1/20th of the diffraction limit.
The team was able to use Exchange-PAINT to capture clear images of the 10 different DNA origami structures in one image. They also used the method to capture images of fixed human cells, with each color tagging a different cellular component—microtubules, mitochondria, Golgi apparatus, or peroxisomes.
Dr Yin expects that, with further development, this method could be used to visualize dozens of cellular components at once.
(green), mitochondria (purple),
Golgi apparatus (red), and
peroxisomes (yellow) in a cell
Credit: Maier Avendano
A new microscopy method allows scientists to image many cellular components at once, according to a paper published in Nature Methods.
Such images could shed light on complex cellular pathways and might lead to new ways to diagnose disease, track its progress, or monitor treatment effectiveness at a cellular level, the researchers said.
They noted that today’s imaging methods typically spot, at most, 3 or 4 types of biomolecules simultaneously.
But to truly understand complex cellular functions, it’s important to be able to visualize most or all of the molecules at once, said study author Peng Yin, PhD, of the Wyss Institute at Harvard Medical School in Boston.
“If you can see only a few things at a time, you are missing the big picture,” Dr Yin said.
So he and his colleagues sought a way to take aerial views of cells that could show dozens of types of biomolecules. They decided to build upon a method called DNA-PAINT, which can create snapshots of up to 3 molecules at once by labeling them with different colored dyes.
The team modified DNA-PAINT to create a method called Exchange-PAINT. Exchange-PAINT relies on the fact that DNA strands with the correct sequence of nucleotides bind specifically to partner strands with complementary sequences.
The researchers label a biomolecule they want to visualize with a short DNA tag. Then, they add to the solution a partner strand carrying a fluorescent dye that lights up only when the 2 strands pair up.
When that partner strand binds the tagged biomolecule, it lights up, then lets go, causing the biomolecule to “blink” at a precise rate the researchers can control. The team uses this blinking to obtain ultra-sharp images.
They repeat the process to visualize a second target, a third, and so on. Then, they overlay the resulting images to create a composite image in which each biomolecule is assigned a different color.
This allows them to create false-color images that simultaneously show many types of biomolecules—far more than they could simultaneously visualize by labeling them with different colored dyes. And these false-color images allow them to spot enough biomolecules at once to capture the entire scene.
To test Exchange-PAINT, the researchers created 10 unique pieces of folded DNA that resembled the numerals 0 through 9. These numerals could be resolved with less than 10 nanometers resolution, or 1/20th of the diffraction limit.
The team was able to use Exchange-PAINT to capture clear images of the 10 different DNA origami structures in one image. They also used the method to capture images of fixed human cells, with each color tagging a different cellular component—microtubules, mitochondria, Golgi apparatus, or peroxisomes.
Dr Yin expects that, with further development, this method could be used to visualize dozens of cellular components at once.