User login
Artificial platelets increase survival in injured mice

Credit: Aaron Logan
New research suggests artificial platelets can aid natural platelets in the clotting process, thereby halting internal bleeding faster.
The artificial platelets, called hemostatic nanoparticles, dramatically increased survival rates in mouse models of blast trauma (hemorrhaging resulting from exposure to an explosion).
And the hemostatic nanoparticles did not appear to interfere with healing or cause any other complications for up to 3 weeks after injection.
Erin Lavik, ScD, of Case Western Reserve University in Cleveland, Ohio, and her colleagues recounted these results in Proceedings of the National Academy of Sciences.
Side effects and survival
The researchers developed a full-body-blast mouse model that replicates the injuries seen in people exposed to explosions. And they used these mice to compare the safety and efficacy of the hemostatic nanoparticles to no treatment, control nanoparticles, lactated Ringer’s solution, and rFVIIa.
The survival rate in mice treated with hemostatic nanoparticles increased to 95% at 1 hour post-trauma, compared to 60% for untreated mice. The odds ratio (OR) was 13.
When the researchers compared the hemostatic nanoparticles and lactated Ringer’s solution, the OR was 8.5. For hemostatic nanoparticles vs control nanoparticles, the OR was 7.1. And for hemostatic nanoparticles vs rFVIIa, the OR was 5.5.
Most mice were killed at the 1-hour time point, but the researchers maintained some mice in the nanoparticle groups so they could assess survival at 1 week and 3 weeks after the blast injury and examine the long-term effects of the nanoparticles.
The team said the vast majority of animals that survived at 1 hour lived to the end of the 3-week study. Differences in lethality at 1 week and 3 weeks were not statistically significant.
Furthermore, there were no unwanted side effects, such as accumulation of the nanoparticles, clot formation, or aberrant healing, during examinations at the 1-week and 3-week time points.
About the nanoparticles
The nanoparticles are made from short polymer chains already approved for other uses by the US Food and Drug Administration.
“They are incredibly simple . . . spheres with arms of peptides that react with activated blood platelets in damaged tissues to help clots form more quickly,” Dr Lavik said.
The dry particles remained viable after 2 weeks on a shelf. A medic in the field or an ambulance crew would add saline, shake and inject them, according to the researchers.
In earlier testing, rat models injected with the nanoparticles stopped bleeding faster than untreated models. Further research and testing are underway, and clinical trials are likely at least 5 years out, Dr Lavik said. ![]()
Credit: Aaron Logan
New research suggests artificial platelets can aid natural platelets in the clotting process, thereby halting internal bleeding faster.
The artificial platelets, called hemostatic nanoparticles, dramatically increased survival rates in mouse models of blast trauma (hemorrhaging resulting from exposure to an explosion).
And the hemostatic nanoparticles did not appear to interfere with healing or cause any other complications for up to 3 weeks after injection.
Erin Lavik, ScD, of Case Western Reserve University in Cleveland, Ohio, and her colleagues recounted these results in Proceedings of the National Academy of Sciences.
Side effects and survival
The researchers developed a full-body-blast mouse model that replicates the injuries seen in people exposed to explosions. And they used these mice to compare the safety and efficacy of the hemostatic nanoparticles to no treatment, control nanoparticles, lactated Ringer’s solution, and rFVIIa.
The survival rate in mice treated with hemostatic nanoparticles increased to 95% at 1 hour post-trauma, compared to 60% for untreated mice. The odds ratio (OR) was 13.
When the researchers compared the hemostatic nanoparticles and lactated Ringer’s solution, the OR was 8.5. For hemostatic nanoparticles vs control nanoparticles, the OR was 7.1. And for hemostatic nanoparticles vs rFVIIa, the OR was 5.5.
Most mice were killed at the 1-hour time point, but the researchers maintained some mice in the nanoparticle groups so they could assess survival at 1 week and 3 weeks after the blast injury and examine the long-term effects of the nanoparticles.
The team said the vast majority of animals that survived at 1 hour lived to the end of the 3-week study. Differences in lethality at 1 week and 3 weeks were not statistically significant.
Furthermore, there were no unwanted side effects, such as accumulation of the nanoparticles, clot formation, or aberrant healing, during examinations at the 1-week and 3-week time points.
About the nanoparticles
The nanoparticles are made from short polymer chains already approved for other uses by the US Food and Drug Administration.
“They are incredibly simple . . . spheres with arms of peptides that react with activated blood platelets in damaged tissues to help clots form more quickly,” Dr Lavik said.
The dry particles remained viable after 2 weeks on a shelf. A medic in the field or an ambulance crew would add saline, shake and inject them, according to the researchers.
In earlier testing, rat models injected with the nanoparticles stopped bleeding faster than untreated models. Further research and testing are underway, and clinical trials are likely at least 5 years out, Dr Lavik said. ![]()
Credit: Aaron Logan
New research suggests artificial platelets can aid natural platelets in the clotting process, thereby halting internal bleeding faster.
The artificial platelets, called hemostatic nanoparticles, dramatically increased survival rates in mouse models of blast trauma (hemorrhaging resulting from exposure to an explosion).
And the hemostatic nanoparticles did not appear to interfere with healing or cause any other complications for up to 3 weeks after injection.
Erin Lavik, ScD, of Case Western Reserve University in Cleveland, Ohio, and her colleagues recounted these results in Proceedings of the National Academy of Sciences.
Side effects and survival
The researchers developed a full-body-blast mouse model that replicates the injuries seen in people exposed to explosions. And they used these mice to compare the safety and efficacy of the hemostatic nanoparticles to no treatment, control nanoparticles, lactated Ringer’s solution, and rFVIIa.
The survival rate in mice treated with hemostatic nanoparticles increased to 95% at 1 hour post-trauma, compared to 60% for untreated mice. The odds ratio (OR) was 13.
When the researchers compared the hemostatic nanoparticles and lactated Ringer’s solution, the OR was 8.5. For hemostatic nanoparticles vs control nanoparticles, the OR was 7.1. And for hemostatic nanoparticles vs rFVIIa, the OR was 5.5.
Most mice were killed at the 1-hour time point, but the researchers maintained some mice in the nanoparticle groups so they could assess survival at 1 week and 3 weeks after the blast injury and examine the long-term effects of the nanoparticles.
The team said the vast majority of animals that survived at 1 hour lived to the end of the 3-week study. Differences in lethality at 1 week and 3 weeks were not statistically significant.
Furthermore, there were no unwanted side effects, such as accumulation of the nanoparticles, clot formation, or aberrant healing, during examinations at the 1-week and 3-week time points.
About the nanoparticles
The nanoparticles are made from short polymer chains already approved for other uses by the US Food and Drug Administration.
“They are incredibly simple . . . spheres with arms of peptides that react with activated blood platelets in damaged tissues to help clots form more quickly,” Dr Lavik said.
The dry particles remained viable after 2 weeks on a shelf. A medic in the field or an ambulance crew would add saline, shake and inject them, according to the researchers.
In earlier testing, rat models injected with the nanoparticles stopped bleeding faster than untreated models. Further research and testing are underway, and clinical trials are likely at least 5 years out, Dr Lavik said. ![]()
HIV+ cancer patients more likely to go untreated

lymphocyte; Credit: CDC
Cancer patients infected with HIV are less likely than their uninfected peers to receive cancer treatment, according to research published in the Journal of Clinical Oncology.
Results showed that HIV-positive patients were roughly twice as likely to go untreated for lymphomas and other cancers.
The researchers believe a lack of clinical trial data and treatment guidelines for HIV patients with cancer may contribute to this health disparity.
To assess the role of HIV status on cancer treatment, Gita Suneja, MD, of the University of Pennsylvania in Philadelphia, and her colleagues used data from the National Cancer Institute’s HIV/AIDS Cancer Match Study.
This included 3045 HIV-infected patients and 1,087,648 uninfected patients. The patients had been diagnosed with Hodgkin lymphoma, diffuse large B-cell lymphoma, or cervical, lung, anal, prostate, colorectal, or breast cancer from 1996 through 2010.
For each cancer type, the researchers assessed the relationship between HIV status and cancer treatment, adjusted for cancer stage, sex, age at cancer diagnosis, race/ethnicity, year of cancer diagnosis, and US state.
For all but 1 cancer type, there was a significantly higher proportion of HIV-infected patients who did not receive cancer treatment when compared with uninfected patients.
The adjusted odds ratios (aORs) were 1.67 for diffuse large B-cell lymphoma, 1.77 for Hodgkin lymphoma, 1.60 for cervical cancer, 1.79 for prostate cancer, 1.81 for breast cancer, 2.18 for lung cancer, and 2.27 for colorectal cancer.
Anal cancer was the only malignancy for which HIV status did not appear to impact treatment. The aOR was 1.01.
Among HIV-infected individuals, factors independently associated with a lack of cancer treatment included low CD4 count, male sex with injection drug use as mode of HIV exposure, age 45 to 64 years, black race, and distant or unknown cancer stage.
“In my clinical experience, I have seen uncertainty surrounding treatment of HIV-infected cancer patients,” Dr Suneja said. “Patients with HIV have typically been excluded from clinical trials, and, therefore, oncologists do not know if the best available treatments are equally safe and effective in those with HIV.”
“Many oncologists rely on guidelines based on such trials for treatment decision-making, and, in the absence of guidance, they may elect not to treat HIV-infected cancer patients due to concerns about adverse side effects or poor survival. This could help explain, in part, why many HIV-positive cancer patients are not receiving appropriate cancer care.”
Therefore, Dr Suneja and her colleagues recommend that cancer trials begin enrolling HIV-infected patients and cancer management guidelines incorporate recommendations for HIV-infected patients. ![]()
lymphocyte; Credit: CDC
Cancer patients infected with HIV are less likely than their uninfected peers to receive cancer treatment, according to research published in the Journal of Clinical Oncology.
Results showed that HIV-positive patients were roughly twice as likely to go untreated for lymphomas and other cancers.
The researchers believe a lack of clinical trial data and treatment guidelines for HIV patients with cancer may contribute to this health disparity.
To assess the role of HIV status on cancer treatment, Gita Suneja, MD, of the University of Pennsylvania in Philadelphia, and her colleagues used data from the National Cancer Institute’s HIV/AIDS Cancer Match Study.
This included 3045 HIV-infected patients and 1,087,648 uninfected patients. The patients had been diagnosed with Hodgkin lymphoma, diffuse large B-cell lymphoma, or cervical, lung, anal, prostate, colorectal, or breast cancer from 1996 through 2010.
For each cancer type, the researchers assessed the relationship between HIV status and cancer treatment, adjusted for cancer stage, sex, age at cancer diagnosis, race/ethnicity, year of cancer diagnosis, and US state.
For all but 1 cancer type, there was a significantly higher proportion of HIV-infected patients who did not receive cancer treatment when compared with uninfected patients.
The adjusted odds ratios (aORs) were 1.67 for diffuse large B-cell lymphoma, 1.77 for Hodgkin lymphoma, 1.60 for cervical cancer, 1.79 for prostate cancer, 1.81 for breast cancer, 2.18 for lung cancer, and 2.27 for colorectal cancer.
Anal cancer was the only malignancy for which HIV status did not appear to impact treatment. The aOR was 1.01.
Among HIV-infected individuals, factors independently associated with a lack of cancer treatment included low CD4 count, male sex with injection drug use as mode of HIV exposure, age 45 to 64 years, black race, and distant or unknown cancer stage.
“In my clinical experience, I have seen uncertainty surrounding treatment of HIV-infected cancer patients,” Dr Suneja said. “Patients with HIV have typically been excluded from clinical trials, and, therefore, oncologists do not know if the best available treatments are equally safe and effective in those with HIV.”
“Many oncologists rely on guidelines based on such trials for treatment decision-making, and, in the absence of guidance, they may elect not to treat HIV-infected cancer patients due to concerns about adverse side effects or poor survival. This could help explain, in part, why many HIV-positive cancer patients are not receiving appropriate cancer care.”
Therefore, Dr Suneja and her colleagues recommend that cancer trials begin enrolling HIV-infected patients and cancer management guidelines incorporate recommendations for HIV-infected patients. ![]()
lymphocyte; Credit: CDC
Cancer patients infected with HIV are less likely than their uninfected peers to receive cancer treatment, according to research published in the Journal of Clinical Oncology.
Results showed that HIV-positive patients were roughly twice as likely to go untreated for lymphomas and other cancers.
The researchers believe a lack of clinical trial data and treatment guidelines for HIV patients with cancer may contribute to this health disparity.
To assess the role of HIV status on cancer treatment, Gita Suneja, MD, of the University of Pennsylvania in Philadelphia, and her colleagues used data from the National Cancer Institute’s HIV/AIDS Cancer Match Study.
This included 3045 HIV-infected patients and 1,087,648 uninfected patients. The patients had been diagnosed with Hodgkin lymphoma, diffuse large B-cell lymphoma, or cervical, lung, anal, prostate, colorectal, or breast cancer from 1996 through 2010.
For each cancer type, the researchers assessed the relationship between HIV status and cancer treatment, adjusted for cancer stage, sex, age at cancer diagnosis, race/ethnicity, year of cancer diagnosis, and US state.
For all but 1 cancer type, there was a significantly higher proportion of HIV-infected patients who did not receive cancer treatment when compared with uninfected patients.
The adjusted odds ratios (aORs) were 1.67 for diffuse large B-cell lymphoma, 1.77 for Hodgkin lymphoma, 1.60 for cervical cancer, 1.79 for prostate cancer, 1.81 for breast cancer, 2.18 for lung cancer, and 2.27 for colorectal cancer.
Anal cancer was the only malignancy for which HIV status did not appear to impact treatment. The aOR was 1.01.
Among HIV-infected individuals, factors independently associated with a lack of cancer treatment included low CD4 count, male sex with injection drug use as mode of HIV exposure, age 45 to 64 years, black race, and distant or unknown cancer stage.
“In my clinical experience, I have seen uncertainty surrounding treatment of HIV-infected cancer patients,” Dr Suneja said. “Patients with HIV have typically been excluded from clinical trials, and, therefore, oncologists do not know if the best available treatments are equally safe and effective in those with HIV.”
“Many oncologists rely on guidelines based on such trials for treatment decision-making, and, in the absence of guidance, they may elect not to treat HIV-infected cancer patients due to concerns about adverse side effects or poor survival. This could help explain, in part, why many HIV-positive cancer patients are not receiving appropriate cancer care.”
Therefore, Dr Suneja and her colleagues recommend that cancer trials begin enrolling HIV-infected patients and cancer management guidelines incorporate recommendations for HIV-infected patients. ![]()
Group engineers versatile RBCs

Researchers say they’ve modified red blood cells (RBCs) to carry a range of payloads—from drugs to vaccines to imaging agents.
“We wanted to create high-value red cells that do more than simply carry oxygen,” said study author Harvey Lodish, PhD, of the Whitehead Institute in Cambridge, Massachusetts.
“Here, we’ve laid out the technology to make mouse and human red blood cells in culture that can express what we want and potentially be used for therapeutic or diagnostic purposes.”
Dr Lodish and his colleagues detailed the technology in Proceedings of the National Academy of Sciences.
The team noted that RBCs are an attractive vehicle for a variety of reasons, including their abundance and their long lifespan.
Perhaps most importantly, during RBC production, progenitor cells jettison their nuclei and all DNA therein. Without a nucleus, a mature RBC lacks any genetic material or any signs of earlier genetic manipulation that could result in tumor formation or other adverse effects.
Exploiting this characteristic, Dr Lodish and his colleagues introduced into early stage RBC progenitors genes coding for specific, slightly modified, normal red cell surface proteins.
As the RBCs approach maturity and enucleate, the proteins remain on the cell surface, where they are modified by a protein-labeling technique known as sortagging.
The technique relies on the bacterial enzyme sortase A to establish a strong chemical bond between the surface protein and a substance of choice, be it a small-molecule therapeutic or an antibody capable of binding a toxin. The modifications leave the cells and their surfaces unharmed.
“Because the modified human red blood cells can circulate in the body for up to 4 months, one could envision a scenario in which the cells are used to introduce antibodies that neutralize a toxin,” said Hidde L. Ploegh, PhD, also of the Whitehead Institute. “The result would be long-lasting reserves of antitoxin antibodies.”
The approach has captured the attention of the US military and its Defense Advanced Research Projects Agency, which is supporting the research in the interest of developing treatments or vaccines effective against biological weapons.
Dr Lodish believes the applications are potentially vast and may include RBCs modified to bind and remove bad cholesterol from the bloodstream, carry clot-busting proteins to treat ischemic strokes or deep-vein thrombosis, or deliver anti-inflammatory antibodies to alleviate chronic inflammation.
Dr Ploegh said there is evidence to suggest that modified RBCs could be used to suppress the unwanted immune response that often accompanies treatment with protein-based therapies.
He is exploring whether these RBCs could be used to prime the immune system to allow patients to better tolerate treatment with such therapies. ![]()
Researchers say they’ve modified red blood cells (RBCs) to carry a range of payloads—from drugs to vaccines to imaging agents.
“We wanted to create high-value red cells that do more than simply carry oxygen,” said study author Harvey Lodish, PhD, of the Whitehead Institute in Cambridge, Massachusetts.
“Here, we’ve laid out the technology to make mouse and human red blood cells in culture that can express what we want and potentially be used for therapeutic or diagnostic purposes.”
Dr Lodish and his colleagues detailed the technology in Proceedings of the National Academy of Sciences.
The team noted that RBCs are an attractive vehicle for a variety of reasons, including their abundance and their long lifespan.
Perhaps most importantly, during RBC production, progenitor cells jettison their nuclei and all DNA therein. Without a nucleus, a mature RBC lacks any genetic material or any signs of earlier genetic manipulation that could result in tumor formation or other adverse effects.
Exploiting this characteristic, Dr Lodish and his colleagues introduced into early stage RBC progenitors genes coding for specific, slightly modified, normal red cell surface proteins.
As the RBCs approach maturity and enucleate, the proteins remain on the cell surface, where they are modified by a protein-labeling technique known as sortagging.
The technique relies on the bacterial enzyme sortase A to establish a strong chemical bond between the surface protein and a substance of choice, be it a small-molecule therapeutic or an antibody capable of binding a toxin. The modifications leave the cells and their surfaces unharmed.
“Because the modified human red blood cells can circulate in the body for up to 4 months, one could envision a scenario in which the cells are used to introduce antibodies that neutralize a toxin,” said Hidde L. Ploegh, PhD, also of the Whitehead Institute. “The result would be long-lasting reserves of antitoxin antibodies.”
The approach has captured the attention of the US military and its Defense Advanced Research Projects Agency, which is supporting the research in the interest of developing treatments or vaccines effective against biological weapons.
Dr Lodish believes the applications are potentially vast and may include RBCs modified to bind and remove bad cholesterol from the bloodstream, carry clot-busting proteins to treat ischemic strokes or deep-vein thrombosis, or deliver anti-inflammatory antibodies to alleviate chronic inflammation.
Dr Ploegh said there is evidence to suggest that modified RBCs could be used to suppress the unwanted immune response that often accompanies treatment with protein-based therapies.
He is exploring whether these RBCs could be used to prime the immune system to allow patients to better tolerate treatment with such therapies. ![]()
Researchers say they’ve modified red blood cells (RBCs) to carry a range of payloads—from drugs to vaccines to imaging agents.
“We wanted to create high-value red cells that do more than simply carry oxygen,” said study author Harvey Lodish, PhD, of the Whitehead Institute in Cambridge, Massachusetts.
“Here, we’ve laid out the technology to make mouse and human red blood cells in culture that can express what we want and potentially be used for therapeutic or diagnostic purposes.”
Dr Lodish and his colleagues detailed the technology in Proceedings of the National Academy of Sciences.
The team noted that RBCs are an attractive vehicle for a variety of reasons, including their abundance and their long lifespan.
Perhaps most importantly, during RBC production, progenitor cells jettison their nuclei and all DNA therein. Without a nucleus, a mature RBC lacks any genetic material or any signs of earlier genetic manipulation that could result in tumor formation or other adverse effects.
Exploiting this characteristic, Dr Lodish and his colleagues introduced into early stage RBC progenitors genes coding for specific, slightly modified, normal red cell surface proteins.
As the RBCs approach maturity and enucleate, the proteins remain on the cell surface, where they are modified by a protein-labeling technique known as sortagging.
The technique relies on the bacterial enzyme sortase A to establish a strong chemical bond between the surface protein and a substance of choice, be it a small-molecule therapeutic or an antibody capable of binding a toxin. The modifications leave the cells and their surfaces unharmed.
“Because the modified human red blood cells can circulate in the body for up to 4 months, one could envision a scenario in which the cells are used to introduce antibodies that neutralize a toxin,” said Hidde L. Ploegh, PhD, also of the Whitehead Institute. “The result would be long-lasting reserves of antitoxin antibodies.”
The approach has captured the attention of the US military and its Defense Advanced Research Projects Agency, which is supporting the research in the interest of developing treatments or vaccines effective against biological weapons.
Dr Lodish believes the applications are potentially vast and may include RBCs modified to bind and remove bad cholesterol from the bloodstream, carry clot-busting proteins to treat ischemic strokes or deep-vein thrombosis, or deliver anti-inflammatory antibodies to alleviate chronic inflammation.
Dr Ploegh said there is evidence to suggest that modified RBCs could be used to suppress the unwanted immune response that often accompanies treatment with protein-based therapies.
He is exploring whether these RBCs could be used to prime the immune system to allow patients to better tolerate treatment with such therapies. ![]()
Enhancing gene delivery to HSCs
![]()
Credit: Chad McNeeley
Scientists say they’ve overcome a major hurdle to developing gene therapies for blood disorders.
They found the drug rapamycin could help them bypass the natural defenses of hematopoietic stem cells (HSCs) and deliver therapeutic doses of disease-fighting genes, without compromising HSC function.
The team believes this discovery could lead to more effective and affordable long-term treatments for disorders such as leukemia and sickle cell anemia.
Bruce Torbett, PhD, of The Scripps Research Institute in La Jolla, California, and his colleagues reported their findings in Blood.
Past research showed that HIV vectors can deliver genes to HSCs. However, when scientists extract HSCs from the body for gene therapy, HIV vectors are usually able to deliver genes to about 30% to 40% of the cells.
For leukemia, leukodystrophy, or genetic diseases where treatment requires a reasonable number of healthy cells derived from stem cells, this number may be too low for therapeutic purposes.
This limitation prompted Dr Torbett and his colleagues to test whether rapamycin could improve delivery of a gene to HSCs. Rapamycin was selected based on its ability to control virus entry and slow cell growth.
The researchers began by isolating stem cells from cord blood samples. They exposed the HSCs to rapamycin and HIV vectors engineered to deliver a gene for a green florescent protein. This fluorescence provided a visual marker that helped the team track gene delivery.
They saw a big difference in both mouse and human stem cells treated with rapamycin, where therapeutic genes were inserted into up to 80% of cells. This property had never been connected to rapamycin before.
The researchers also found that rapamycin can keep HSCs from differentiating as quickly when taken out of the body for gene therapy.
“We wanted to make sure the conditions we will use preserve stem cells, so if we transplant them back into our animal models, they act just like the original stem cells,” Dr Torbett said. “We showed that, in 2 sets of animal models, stem cells remain and produce gene-modified cells.”
The scientists hope these methods could someday be useful in the clinic.
“Our methods could reduce costs and the amount of preparation that goes into modifying blood stem cells using viral vector gene therapy,” said Cathy Wang, also of The Scripps Research Institute. “It would make gene therapy accessible to a lot more patients.”
She said the team’s next steps are to carry out preclinical studies using rapamycin with stem cells in other animal models and then test the method in humans. The researchers are also working to delineate the dual pathways of rapamycin’s mechanism of action in HSCs. ![]()
![]()
Credit: Chad McNeeley
Scientists say they’ve overcome a major hurdle to developing gene therapies for blood disorders.
They found the drug rapamycin could help them bypass the natural defenses of hematopoietic stem cells (HSCs) and deliver therapeutic doses of disease-fighting genes, without compromising HSC function.
The team believes this discovery could lead to more effective and affordable long-term treatments for disorders such as leukemia and sickle cell anemia.
Bruce Torbett, PhD, of The Scripps Research Institute in La Jolla, California, and his colleagues reported their findings in Blood.
Past research showed that HIV vectors can deliver genes to HSCs. However, when scientists extract HSCs from the body for gene therapy, HIV vectors are usually able to deliver genes to about 30% to 40% of the cells.
For leukemia, leukodystrophy, or genetic diseases where treatment requires a reasonable number of healthy cells derived from stem cells, this number may be too low for therapeutic purposes.
This limitation prompted Dr Torbett and his colleagues to test whether rapamycin could improve delivery of a gene to HSCs. Rapamycin was selected based on its ability to control virus entry and slow cell growth.
The researchers began by isolating stem cells from cord blood samples. They exposed the HSCs to rapamycin and HIV vectors engineered to deliver a gene for a green florescent protein. This fluorescence provided a visual marker that helped the team track gene delivery.
They saw a big difference in both mouse and human stem cells treated with rapamycin, where therapeutic genes were inserted into up to 80% of cells. This property had never been connected to rapamycin before.
The researchers also found that rapamycin can keep HSCs from differentiating as quickly when taken out of the body for gene therapy.
“We wanted to make sure the conditions we will use preserve stem cells, so if we transplant them back into our animal models, they act just like the original stem cells,” Dr Torbett said. “We showed that, in 2 sets of animal models, stem cells remain and produce gene-modified cells.”
The scientists hope these methods could someday be useful in the clinic.
“Our methods could reduce costs and the amount of preparation that goes into modifying blood stem cells using viral vector gene therapy,” said Cathy Wang, also of The Scripps Research Institute. “It would make gene therapy accessible to a lot more patients.”
She said the team’s next steps are to carry out preclinical studies using rapamycin with stem cells in other animal models and then test the method in humans. The researchers are also working to delineate the dual pathways of rapamycin’s mechanism of action in HSCs. ![]()
![]()
Credit: Chad McNeeley
Scientists say they’ve overcome a major hurdle to developing gene therapies for blood disorders.
They found the drug rapamycin could help them bypass the natural defenses of hematopoietic stem cells (HSCs) and deliver therapeutic doses of disease-fighting genes, without compromising HSC function.
The team believes this discovery could lead to more effective and affordable long-term treatments for disorders such as leukemia and sickle cell anemia.
Bruce Torbett, PhD, of The Scripps Research Institute in La Jolla, California, and his colleagues reported their findings in Blood.
Past research showed that HIV vectors can deliver genes to HSCs. However, when scientists extract HSCs from the body for gene therapy, HIV vectors are usually able to deliver genes to about 30% to 40% of the cells.
For leukemia, leukodystrophy, or genetic diseases where treatment requires a reasonable number of healthy cells derived from stem cells, this number may be too low for therapeutic purposes.
This limitation prompted Dr Torbett and his colleagues to test whether rapamycin could improve delivery of a gene to HSCs. Rapamycin was selected based on its ability to control virus entry and slow cell growth.
The researchers began by isolating stem cells from cord blood samples. They exposed the HSCs to rapamycin and HIV vectors engineered to deliver a gene for a green florescent protein. This fluorescence provided a visual marker that helped the team track gene delivery.
They saw a big difference in both mouse and human stem cells treated with rapamycin, where therapeutic genes were inserted into up to 80% of cells. This property had never been connected to rapamycin before.
The researchers also found that rapamycin can keep HSCs from differentiating as quickly when taken out of the body for gene therapy.
“We wanted to make sure the conditions we will use preserve stem cells, so if we transplant them back into our animal models, they act just like the original stem cells,” Dr Torbett said. “We showed that, in 2 sets of animal models, stem cells remain and produce gene-modified cells.”
The scientists hope these methods could someday be useful in the clinic.
“Our methods could reduce costs and the amount of preparation that goes into modifying blood stem cells using viral vector gene therapy,” said Cathy Wang, also of The Scripps Research Institute. “It would make gene therapy accessible to a lot more patients.”
She said the team’s next steps are to carry out preclinical studies using rapamycin with stem cells in other animal models and then test the method in humans. The researchers are also working to delineate the dual pathways of rapamycin’s mechanism of action in HSCs. ![]()
Technology lowers stress among pediatric patients
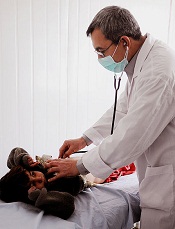
Credit: Logan Tuttle
A new study suggests videoconferencing with family and friends can lower stress for pediatric patients who are hospitalized for an extended period.
UC Davis Children’s Hospital provides these patients with laptops, webcams, and secure Internet connections for videoconferencing.
And anecdotal accounts have suggested the service, called Family-Link, benefits patients. But researchers wanted more concrete evidence that Family-Link can reduce anxiety.
To that end, James Marcin, MD, and his colleagues studied 367 children who were hospitalized at UC Davis for at least 4 days.
Two hundred and thirty-two patients took advantage of the videoconferencing service, and 135 did not. The researchers used the Parent-Guardian Stress Survey to assess the children’s anxiety levels, both at admission and discharge.
The survey included 4 question groups centered on each child’s behavior and emotions, staff communication, sights and sounds, and the child’s appearance. Parents/guardians were asked whether the child exhibited a variety of behaviors, such as being demanding, frightened, angry, or confused.
The survey also included questions about the impact of monitoring equipment on stress levels and the staff’s ability to communicate important details about the child’s care.
Overall, children who used Family-Link experienced a greater reduction in stress than children who did not use the service.
The researchers were surprised to find this effect was even more pronounced for children who lived closer to the hospital and had shorter hospitalizations. This group experienced a 37% stress reduction when using Family-Link.
“This study shows that we have another tool to help children during their hospital stays,” said Nikki Yang, first author on the study. “The improvement in stress scores shows that Family-Link is really helping many children and might possibly be improving outcomes.”
Yang and her colleagues reported these findings in Pediatrics. ![]()
Credit: Logan Tuttle
A new study suggests videoconferencing with family and friends can lower stress for pediatric patients who are hospitalized for an extended period.
UC Davis Children’s Hospital provides these patients with laptops, webcams, and secure Internet connections for videoconferencing.
And anecdotal accounts have suggested the service, called Family-Link, benefits patients. But researchers wanted more concrete evidence that Family-Link can reduce anxiety.
To that end, James Marcin, MD, and his colleagues studied 367 children who were hospitalized at UC Davis for at least 4 days.
Two hundred and thirty-two patients took advantage of the videoconferencing service, and 135 did not. The researchers used the Parent-Guardian Stress Survey to assess the children’s anxiety levels, both at admission and discharge.
The survey included 4 question groups centered on each child’s behavior and emotions, staff communication, sights and sounds, and the child’s appearance. Parents/guardians were asked whether the child exhibited a variety of behaviors, such as being demanding, frightened, angry, or confused.
The survey also included questions about the impact of monitoring equipment on stress levels and the staff’s ability to communicate important details about the child’s care.
Overall, children who used Family-Link experienced a greater reduction in stress than children who did not use the service.
The researchers were surprised to find this effect was even more pronounced for children who lived closer to the hospital and had shorter hospitalizations. This group experienced a 37% stress reduction when using Family-Link.
“This study shows that we have another tool to help children during their hospital stays,” said Nikki Yang, first author on the study. “The improvement in stress scores shows that Family-Link is really helping many children and might possibly be improving outcomes.”
Yang and her colleagues reported these findings in Pediatrics. ![]()
Credit: Logan Tuttle
A new study suggests videoconferencing with family and friends can lower stress for pediatric patients who are hospitalized for an extended period.
UC Davis Children’s Hospital provides these patients with laptops, webcams, and secure Internet connections for videoconferencing.
And anecdotal accounts have suggested the service, called Family-Link, benefits patients. But researchers wanted more concrete evidence that Family-Link can reduce anxiety.
To that end, James Marcin, MD, and his colleagues studied 367 children who were hospitalized at UC Davis for at least 4 days.
Two hundred and thirty-two patients took advantage of the videoconferencing service, and 135 did not. The researchers used the Parent-Guardian Stress Survey to assess the children’s anxiety levels, both at admission and discharge.
The survey included 4 question groups centered on each child’s behavior and emotions, staff communication, sights and sounds, and the child’s appearance. Parents/guardians were asked whether the child exhibited a variety of behaviors, such as being demanding, frightened, angry, or confused.
The survey also included questions about the impact of monitoring equipment on stress levels and the staff’s ability to communicate important details about the child’s care.
Overall, children who used Family-Link experienced a greater reduction in stress than children who did not use the service.
The researchers were surprised to find this effect was even more pronounced for children who lived closer to the hospital and had shorter hospitalizations. This group experienced a 37% stress reduction when using Family-Link.
“This study shows that we have another tool to help children during their hospital stays,” said Nikki Yang, first author on the study. “The improvement in stress scores shows that Family-Link is really helping many children and might possibly be improving outcomes.”
Yang and her colleagues reported these findings in Pediatrics. ![]()
Ofatumumab falls short in CLL, DLBCL

Credit: Linda Bartlett
The anti-CD20 antibody ofatumumab (Arzerra) has failed to meet the primary endpoint in two phase 3 trials, according to the companies developing the drug.
Ofatumumab failed to improve progression-free survival (PFS) when compared to physicians’ choice in patients with bulky, fludarabine-refractory chronic lymphocytic leukemia (CLL).
Likewise, ofatumumab plus chemotherapy failed to improve PFS when compared to rituximab plus chemotherapy in patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL).
GlaxoSmithKline (GSK) and Genmab recently announced these headline results but said the full analyses of safety and efficacy data are underway and will be completed in the coming months.
However, based on these early results, the companies said they are unlikely to pursue regulatory filings for ofatumumab in either indication.
CLL trial
In the OMB114242 study, researchers enrolled 122 patients with bulky, fludarabine-refractory CLL. Patients were randomized to receive ofatumumab or physicians’ choice (2:1).
Patients randomized to ofatumumab received an initial dose of 300 mg, followed 1 week later by 2000 mg once weekly for 7 weeks, followed 4 weeks later by 1 infusion of 2000 mg every 4 weeks, for a total treatment duration of 6 to 12 months. Patients in the physicians’ choice arm received a treatment regimen chosen by a physician for up to 6 months.
The primary endpoint was PFS, and secondary objectives were to evaluate response, overall survival, safety, tolerability, and health-related quality of life.
The median PFS, as assessed by an independent review committee, was 5.36 months for ofatumumab and 3.61 months for physicians’ choice (hazard ratio 0.79, P=0.267).
“It was our priority to share this result with the scientific community as soon it became available,” said Rafael Amado, MD, Head of Oncology R&D at GSK. “We will now work to further analyze the data and to better understand the totality of the efficacy and safety findings.”
This study was conducted to meet the requirements from the European Commission for the conditional approval of ofatumumab for the treatment of CLL in patients who are refractory to fludarabine and alemtuzumab. The current indications in the European Union and the United States do not include bulky, fludarabine-refractory CLL patients.
“Although ofatumumab performed broadly in line with previous data, today’s result is disappointing,” said Jan van de Winkel, PhD, Chief Executive Officer of Genmab. “Based on this result, we do not anticipate applying for a label expansion for ofatumumab in this specific refractory CLL population.”
DLBCL trial
The ORCHARRD study included 447 patients who were refractory to, or had relapsed following, first-line treatment with rituximab in combination with a chemotherapy regimen containing anthracycline or anthracenedione. Patients were also eligible for autologous stem cell transplant.
Patients were randomized 1:1 to receive 3 cycles of either ofatumumab or rituximab in combination with DHAP (dexamethasone, cytarabine, and cisplatin) salvage chemotherapy. After the third treatment cycle, patients who obtained a complete or partial response received high-dose chemotherapy followed by transplant.
The primary endpoint was PFS. But GSK and Genmab reported no statistically significant difference in PFS between the treatment arms.
There were no differences in adverse events (AEs) leading to treatment discontinuation, grade 3 or higher AEs, or severe AEs between the treatment arms. However, there were more dose interruptions and delays due to infusion reactions and increased serum creatinine in the ofatumumab-plus-chemotherapy arm, which require further analysis.
“We plan to submit detailed data from the ofatumumab ORCHARRD study in DLBCL for presentation at a medical conference later this year, which we hope will provide further clarity on today’s headline results,” Dr van de Winkel said. “Based on [these early] results, we are unlikely to move forward with a regulatory filing.” ![]()
Credit: Linda Bartlett
The anti-CD20 antibody ofatumumab (Arzerra) has failed to meet the primary endpoint in two phase 3 trials, according to the companies developing the drug.
Ofatumumab failed to improve progression-free survival (PFS) when compared to physicians’ choice in patients with bulky, fludarabine-refractory chronic lymphocytic leukemia (CLL).
Likewise, ofatumumab plus chemotherapy failed to improve PFS when compared to rituximab plus chemotherapy in patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL).
GlaxoSmithKline (GSK) and Genmab recently announced these headline results but said the full analyses of safety and efficacy data are underway and will be completed in the coming months.
However, based on these early results, the companies said they are unlikely to pursue regulatory filings for ofatumumab in either indication.
CLL trial
In the OMB114242 study, researchers enrolled 122 patients with bulky, fludarabine-refractory CLL. Patients were randomized to receive ofatumumab or physicians’ choice (2:1).
Patients randomized to ofatumumab received an initial dose of 300 mg, followed 1 week later by 2000 mg once weekly for 7 weeks, followed 4 weeks later by 1 infusion of 2000 mg every 4 weeks, for a total treatment duration of 6 to 12 months. Patients in the physicians’ choice arm received a treatment regimen chosen by a physician for up to 6 months.
The primary endpoint was PFS, and secondary objectives were to evaluate response, overall survival, safety, tolerability, and health-related quality of life.
The median PFS, as assessed by an independent review committee, was 5.36 months for ofatumumab and 3.61 months for physicians’ choice (hazard ratio 0.79, P=0.267).
“It was our priority to share this result with the scientific community as soon it became available,” said Rafael Amado, MD, Head of Oncology R&D at GSK. “We will now work to further analyze the data and to better understand the totality of the efficacy and safety findings.”
This study was conducted to meet the requirements from the European Commission for the conditional approval of ofatumumab for the treatment of CLL in patients who are refractory to fludarabine and alemtuzumab. The current indications in the European Union and the United States do not include bulky, fludarabine-refractory CLL patients.
“Although ofatumumab performed broadly in line with previous data, today’s result is disappointing,” said Jan van de Winkel, PhD, Chief Executive Officer of Genmab. “Based on this result, we do not anticipate applying for a label expansion for ofatumumab in this specific refractory CLL population.”
DLBCL trial
The ORCHARRD study included 447 patients who were refractory to, or had relapsed following, first-line treatment with rituximab in combination with a chemotherapy regimen containing anthracycline or anthracenedione. Patients were also eligible for autologous stem cell transplant.
Patients were randomized 1:1 to receive 3 cycles of either ofatumumab or rituximab in combination with DHAP (dexamethasone, cytarabine, and cisplatin) salvage chemotherapy. After the third treatment cycle, patients who obtained a complete or partial response received high-dose chemotherapy followed by transplant.
The primary endpoint was PFS. But GSK and Genmab reported no statistically significant difference in PFS between the treatment arms.
There were no differences in adverse events (AEs) leading to treatment discontinuation, grade 3 or higher AEs, or severe AEs between the treatment arms. However, there were more dose interruptions and delays due to infusion reactions and increased serum creatinine in the ofatumumab-plus-chemotherapy arm, which require further analysis.
“We plan to submit detailed data from the ofatumumab ORCHARRD study in DLBCL for presentation at a medical conference later this year, which we hope will provide further clarity on today’s headline results,” Dr van de Winkel said. “Based on [these early] results, we are unlikely to move forward with a regulatory filing.” ![]()
Credit: Linda Bartlett
The anti-CD20 antibody ofatumumab (Arzerra) has failed to meet the primary endpoint in two phase 3 trials, according to the companies developing the drug.
Ofatumumab failed to improve progression-free survival (PFS) when compared to physicians’ choice in patients with bulky, fludarabine-refractory chronic lymphocytic leukemia (CLL).
Likewise, ofatumumab plus chemotherapy failed to improve PFS when compared to rituximab plus chemotherapy in patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL).
GlaxoSmithKline (GSK) and Genmab recently announced these headline results but said the full analyses of safety and efficacy data are underway and will be completed in the coming months.
However, based on these early results, the companies said they are unlikely to pursue regulatory filings for ofatumumab in either indication.
CLL trial
In the OMB114242 study, researchers enrolled 122 patients with bulky, fludarabine-refractory CLL. Patients were randomized to receive ofatumumab or physicians’ choice (2:1).
Patients randomized to ofatumumab received an initial dose of 300 mg, followed 1 week later by 2000 mg once weekly for 7 weeks, followed 4 weeks later by 1 infusion of 2000 mg every 4 weeks, for a total treatment duration of 6 to 12 months. Patients in the physicians’ choice arm received a treatment regimen chosen by a physician for up to 6 months.
The primary endpoint was PFS, and secondary objectives were to evaluate response, overall survival, safety, tolerability, and health-related quality of life.
The median PFS, as assessed by an independent review committee, was 5.36 months for ofatumumab and 3.61 months for physicians’ choice (hazard ratio 0.79, P=0.267).
“It was our priority to share this result with the scientific community as soon it became available,” said Rafael Amado, MD, Head of Oncology R&D at GSK. “We will now work to further analyze the data and to better understand the totality of the efficacy and safety findings.”
This study was conducted to meet the requirements from the European Commission for the conditional approval of ofatumumab for the treatment of CLL in patients who are refractory to fludarabine and alemtuzumab. The current indications in the European Union and the United States do not include bulky, fludarabine-refractory CLL patients.
“Although ofatumumab performed broadly in line with previous data, today’s result is disappointing,” said Jan van de Winkel, PhD, Chief Executive Officer of Genmab. “Based on this result, we do not anticipate applying for a label expansion for ofatumumab in this specific refractory CLL population.”
DLBCL trial
The ORCHARRD study included 447 patients who were refractory to, or had relapsed following, first-line treatment with rituximab in combination with a chemotherapy regimen containing anthracycline or anthracenedione. Patients were also eligible for autologous stem cell transplant.
Patients were randomized 1:1 to receive 3 cycles of either ofatumumab or rituximab in combination with DHAP (dexamethasone, cytarabine, and cisplatin) salvage chemotherapy. After the third treatment cycle, patients who obtained a complete or partial response received high-dose chemotherapy followed by transplant.
The primary endpoint was PFS. But GSK and Genmab reported no statistically significant difference in PFS between the treatment arms.
There were no differences in adverse events (AEs) leading to treatment discontinuation, grade 3 or higher AEs, or severe AEs between the treatment arms. However, there were more dose interruptions and delays due to infusion reactions and increased serum creatinine in the ofatumumab-plus-chemotherapy arm, which require further analysis.
“We plan to submit detailed data from the ofatumumab ORCHARRD study in DLBCL for presentation at a medical conference later this year, which we hope will provide further clarity on today’s headline results,” Dr van de Winkel said. “Based on [these early] results, we are unlikely to move forward with a regulatory filing.” ![]()
CHMP recommends apixaban for VTE

Credit: Andre E.X. Brown
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended that apixaban (Eliquis) be granted marketing authorization for the treatment and prevention of venous thromboembolism (VTE) in adults.
The European Commission will take the CHMP’s opinion into account when deciding whether to approve the drug for this indication in all 28 European Union member states, plus Iceland and Norway.
The CHMP’s recommendation of apixaban was based on results from the AMPLIFY and AMPLIFY-EXTENSION studies.
The phase 3 AMPLIFY trial enrolled 5395 patients with confirmed, symptomatic deep vein thrombosis (DVT) or pulmonary embolism (PE) requiring treatment for 6 months.
About half of patients (n=2691) were randomized to apixaban, and the other half (n=2704) were randomized to standard of care, which was initial enoxaparin treatment overlapped by warfarin therapy.
The primary efficacy outcome was the composite endpoint of recurrent, symptomatic VTE or VTE-related death. This occurred in 59 patients in the apixaban arm (2.3%) and 71 patients (2.7%) in the standard-therapy arm (P<0.0001 for noninferiority).
The composite endpoint of major bleeding and clinically relevant, nonmajor bleeding occurred in 4.3% of patients in the apixaban arm and 9.7% of patients in the standard-therapy arm (P<0.001).
The phase 3 AMPLIFY-EXTENSION trial included 2486 patients with prior VTE who had completed 6 to 12 months of anticoagulation treatment for DVT or PE.
Eight hundred and forty-two patients were randomized to apixaban at 2.5 mg, 815 were randomized to apixaban at 5 mg, and 829 were randomized to placebo.
The study’s primary efficacy endpoint was recurrent VTE or all-cause death. During the 12-month active study period, these events occurred in 32 patients (3.8%) in the 2.5-mg arm, 34 patients (4.2%) in the 5-mg arm, and 96 patients (11.6%) in the placebo arm. Both apixaban doses were significantly superior to placebo (P<0.001).
The primary safety endpoint was the incidence of major bleeding, which occurred in 2 patients (0.2%) in the 2.5-mg arm, 1 patient (0.1%) in the 5-mg arm, and 4 patients (0.5%) in the placebo arm. ![]()
Credit: Andre E.X. Brown
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended that apixaban (Eliquis) be granted marketing authorization for the treatment and prevention of venous thromboembolism (VTE) in adults.
The European Commission will take the CHMP’s opinion into account when deciding whether to approve the drug for this indication in all 28 European Union member states, plus Iceland and Norway.
The CHMP’s recommendation of apixaban was based on results from the AMPLIFY and AMPLIFY-EXTENSION studies.
The phase 3 AMPLIFY trial enrolled 5395 patients with confirmed, symptomatic deep vein thrombosis (DVT) or pulmonary embolism (PE) requiring treatment for 6 months.
About half of patients (n=2691) were randomized to apixaban, and the other half (n=2704) were randomized to standard of care, which was initial enoxaparin treatment overlapped by warfarin therapy.
The primary efficacy outcome was the composite endpoint of recurrent, symptomatic VTE or VTE-related death. This occurred in 59 patients in the apixaban arm (2.3%) and 71 patients (2.7%) in the standard-therapy arm (P<0.0001 for noninferiority).
The composite endpoint of major bleeding and clinically relevant, nonmajor bleeding occurred in 4.3% of patients in the apixaban arm and 9.7% of patients in the standard-therapy arm (P<0.001).
The phase 3 AMPLIFY-EXTENSION trial included 2486 patients with prior VTE who had completed 6 to 12 months of anticoagulation treatment for DVT or PE.
Eight hundred and forty-two patients were randomized to apixaban at 2.5 mg, 815 were randomized to apixaban at 5 mg, and 829 were randomized to placebo.
The study’s primary efficacy endpoint was recurrent VTE or all-cause death. During the 12-month active study period, these events occurred in 32 patients (3.8%) in the 2.5-mg arm, 34 patients (4.2%) in the 5-mg arm, and 96 patients (11.6%) in the placebo arm. Both apixaban doses were significantly superior to placebo (P<0.001).
The primary safety endpoint was the incidence of major bleeding, which occurred in 2 patients (0.2%) in the 2.5-mg arm, 1 patient (0.1%) in the 5-mg arm, and 4 patients (0.5%) in the placebo arm. ![]()
Credit: Andre E.X. Brown
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended that apixaban (Eliquis) be granted marketing authorization for the treatment and prevention of venous thromboembolism (VTE) in adults.
The European Commission will take the CHMP’s opinion into account when deciding whether to approve the drug for this indication in all 28 European Union member states, plus Iceland and Norway.
The CHMP’s recommendation of apixaban was based on results from the AMPLIFY and AMPLIFY-EXTENSION studies.
The phase 3 AMPLIFY trial enrolled 5395 patients with confirmed, symptomatic deep vein thrombosis (DVT) or pulmonary embolism (PE) requiring treatment for 6 months.
About half of patients (n=2691) were randomized to apixaban, and the other half (n=2704) were randomized to standard of care, which was initial enoxaparin treatment overlapped by warfarin therapy.
The primary efficacy outcome was the composite endpoint of recurrent, symptomatic VTE or VTE-related death. This occurred in 59 patients in the apixaban arm (2.3%) and 71 patients (2.7%) in the standard-therapy arm (P<0.0001 for noninferiority).
The composite endpoint of major bleeding and clinically relevant, nonmajor bleeding occurred in 4.3% of patients in the apixaban arm and 9.7% of patients in the standard-therapy arm (P<0.001).
The phase 3 AMPLIFY-EXTENSION trial included 2486 patients with prior VTE who had completed 6 to 12 months of anticoagulation treatment for DVT or PE.
Eight hundred and forty-two patients were randomized to apixaban at 2.5 mg, 815 were randomized to apixaban at 5 mg, and 829 were randomized to placebo.
The study’s primary efficacy endpoint was recurrent VTE or all-cause death. During the 12-month active study period, these events occurred in 32 patients (3.8%) in the 2.5-mg arm, 34 patients (4.2%) in the 5-mg arm, and 96 patients (11.6%) in the placebo arm. Both apixaban doses were significantly superior to placebo (P<0.001).
The primary safety endpoint was the incidence of major bleeding, which occurred in 2 patients (0.2%) in the 2.5-mg arm, 1 patient (0.1%) in the 5-mg arm, and 4 patients (0.5%) in the placebo arm. ![]()
VSTs can target up to 5 viruses

for Cell and Gene Therapy
at Baylor College of Medicine
New virus-specific T-cells (VSTs) can effectively target multiple viruses, thereby clearing and preventing infections in transplant recipients, researchers have reported in Science Translational Medicine.
VSTs have proven effective in previous studies, but they are typically costly, can take months to produce, and often target a single virus.
The new VSTs took about 10 days to produce, are more cost-effective than standard therapy, and can target up to 5 viruses, according to the researchers.
Ann Leen, PhD, of the Baylor College of Medicine in Houston, and her colleagues developed the technique to produce the VSTs, which can target Epstein-Barr virus (EBV), adenovirus (ADV), cytomegalovirus (CMV), BK virus (BKV), and human herpesvirus 6 (HHV6).
“Unlike conventional antiviral drugs, our therapy improves virus-specific T-cell immunity—the root cause of post-transplant viral infections—providing both an effective and safe strategy to treat viruses,” Dr Leen said.
“Additionally, we can readily produce both individualized products and T-cell banks for third-party use, facilitating the extension of T-cell therapy to a standard of care for transplant recipients.”
The researchers generated 48 VST cell lines by stimulating peripheral blood mononuclear cells from allogeneic donors with overlapping peptide libraries that incorporate EBV, CMV, ADV, BKV, and HHV6 antigens.
The team then tested these VSTs in 11 patients who had received allogeneic transplants to treat leukemia, lymphoma, sickle cell disease, and other hematologic and immunodeficient disorders.
Eight of the patients had up to 4 active infections with the targeted viruses. Three patients received the VSTs to prevent infection.
The VSTs produced an overall 94% virological and clinical response that was sustained long-term. Of the 3 patients who received VSTs prophylactically, all remained free of infection for more than 3 months after infusion.
There were no immediate infusion-related toxicities. One patient developed de novo graft-vs-host disease of the skin that improved with topical steroids.
Two patients who received VSTs as prophylaxis developed transplant-associated microangiopathy, but the researchers considered this to be unrelated to VST infusion.
They noted that this is the first time BKV and HHV6 reactivations have been controlled using VSTs. Dr Leen’s team had previously reported promising results with VSTs targeting EBV, CMV, and ADV.
“This study translated improved manufacturing techniques developed in Dr Leen’s laboratory to the clinic and showed that virus-specific T cells produced with the new method could target new viruses and be ready for clinical use after 10 days,” said Helen Heslop, MD, also of Baylor College of Medicine.
“These advances mean that this therapy could be available for more patients to treat viral infections and provide long-lasting protection.” ![]()
for Cell and Gene Therapy
at Baylor College of Medicine
New virus-specific T-cells (VSTs) can effectively target multiple viruses, thereby clearing and preventing infections in transplant recipients, researchers have reported in Science Translational Medicine.
VSTs have proven effective in previous studies, but they are typically costly, can take months to produce, and often target a single virus.
The new VSTs took about 10 days to produce, are more cost-effective than standard therapy, and can target up to 5 viruses, according to the researchers.
Ann Leen, PhD, of the Baylor College of Medicine in Houston, and her colleagues developed the technique to produce the VSTs, which can target Epstein-Barr virus (EBV), adenovirus (ADV), cytomegalovirus (CMV), BK virus (BKV), and human herpesvirus 6 (HHV6).
“Unlike conventional antiviral drugs, our therapy improves virus-specific T-cell immunity—the root cause of post-transplant viral infections—providing both an effective and safe strategy to treat viruses,” Dr Leen said.
“Additionally, we can readily produce both individualized products and T-cell banks for third-party use, facilitating the extension of T-cell therapy to a standard of care for transplant recipients.”
The researchers generated 48 VST cell lines by stimulating peripheral blood mononuclear cells from allogeneic donors with overlapping peptide libraries that incorporate EBV, CMV, ADV, BKV, and HHV6 antigens.
The team then tested these VSTs in 11 patients who had received allogeneic transplants to treat leukemia, lymphoma, sickle cell disease, and other hematologic and immunodeficient disorders.
Eight of the patients had up to 4 active infections with the targeted viruses. Three patients received the VSTs to prevent infection.
The VSTs produced an overall 94% virological and clinical response that was sustained long-term. Of the 3 patients who received VSTs prophylactically, all remained free of infection for more than 3 months after infusion.
There were no immediate infusion-related toxicities. One patient developed de novo graft-vs-host disease of the skin that improved with topical steroids.
Two patients who received VSTs as prophylaxis developed transplant-associated microangiopathy, but the researchers considered this to be unrelated to VST infusion.
They noted that this is the first time BKV and HHV6 reactivations have been controlled using VSTs. Dr Leen’s team had previously reported promising results with VSTs targeting EBV, CMV, and ADV.
“This study translated improved manufacturing techniques developed in Dr Leen’s laboratory to the clinic and showed that virus-specific T cells produced with the new method could target new viruses and be ready for clinical use after 10 days,” said Helen Heslop, MD, also of Baylor College of Medicine.
“These advances mean that this therapy could be available for more patients to treat viral infections and provide long-lasting protection.” ![]()
for Cell and Gene Therapy
at Baylor College of Medicine
New virus-specific T-cells (VSTs) can effectively target multiple viruses, thereby clearing and preventing infections in transplant recipients, researchers have reported in Science Translational Medicine.
VSTs have proven effective in previous studies, but they are typically costly, can take months to produce, and often target a single virus.
The new VSTs took about 10 days to produce, are more cost-effective than standard therapy, and can target up to 5 viruses, according to the researchers.
Ann Leen, PhD, of the Baylor College of Medicine in Houston, and her colleagues developed the technique to produce the VSTs, which can target Epstein-Barr virus (EBV), adenovirus (ADV), cytomegalovirus (CMV), BK virus (BKV), and human herpesvirus 6 (HHV6).
“Unlike conventional antiviral drugs, our therapy improves virus-specific T-cell immunity—the root cause of post-transplant viral infections—providing both an effective and safe strategy to treat viruses,” Dr Leen said.
“Additionally, we can readily produce both individualized products and T-cell banks for third-party use, facilitating the extension of T-cell therapy to a standard of care for transplant recipients.”
The researchers generated 48 VST cell lines by stimulating peripheral blood mononuclear cells from allogeneic donors with overlapping peptide libraries that incorporate EBV, CMV, ADV, BKV, and HHV6 antigens.
The team then tested these VSTs in 11 patients who had received allogeneic transplants to treat leukemia, lymphoma, sickle cell disease, and other hematologic and immunodeficient disorders.
Eight of the patients had up to 4 active infections with the targeted viruses. Three patients received the VSTs to prevent infection.
The VSTs produced an overall 94% virological and clinical response that was sustained long-term. Of the 3 patients who received VSTs prophylactically, all remained free of infection for more than 3 months after infusion.
There were no immediate infusion-related toxicities. One patient developed de novo graft-vs-host disease of the skin that improved with topical steroids.
Two patients who received VSTs as prophylaxis developed transplant-associated microangiopathy, but the researchers considered this to be unrelated to VST infusion.
They noted that this is the first time BKV and HHV6 reactivations have been controlled using VSTs. Dr Leen’s team had previously reported promising results with VSTs targeting EBV, CMV, and ADV.
“This study translated improved manufacturing techniques developed in Dr Leen’s laboratory to the clinic and showed that virus-specific T cells produced with the new method could target new viruses and be ready for clinical use after 10 days,” said Helen Heslop, MD, also of Baylor College of Medicine.
“These advances mean that this therapy could be available for more patients to treat viral infections and provide long-lasting protection.” ![]()
System can attenuate thrombin generation

Credit: Kevin MacKenzie
Results of a phase 1 trial suggest the REG2 anticoagulation system can attenuate thrombin generation and then restore thrombin following reversal.
The REG2 system consists of pegnivacogin, a subcutaneously administered aptamer factor IXa inhibitor, and its intravenous active control agent, anivamersen.
REG2 is formulated to provide a controllable level of anticoagulation for up to 2 weeks for sub-acute uses, especially in cases where a patient may be unable to swallow an oral anticoagulant.
Results observed with REG2 were published in the Journal of Thrombosis and Thrombolysis. The study was sponsored by Regado Biosciences, Inc, the company developing the REG2 system.
The study included 32 healthy volunteers who were enrolled sequentially into 4 cohorts. Patients in cohorts 1-3 were randomized (3:1) to ascending doses of pegnivacogin—from 0.5 mg/kg to 3 mg/kg—or placebo, with no anivamersen administered.
In cohort 4, all patients received 2 mg/kg of pegnivacogin and were randomized (1:1) to reversal by 1 dose or multiple doses over time of 1 mg/kg of anivamersen.
The researchers drew blood samples in the presence or absence of corn trypsin inhibitor at specified times within each dosing cohort. They initiated thrombin generation with tissue factor and measured thrombin generation kinetics using the calibrated automated thrombogram (CAT).
The team found that REG2 attenuated thrombin generation in a dose-dependent manner. All parameters of the CAT assay, except for lag time, showed a dose- and concentration-dependent response to pegnivacogin.
Specifically, there was a decrease in velocity index (VIx) from baseline to 24 hours after pegnivacogin administration. The largest percent change in VIx was in cohort 3 (3 mg/kg). There was a 90% decrease at 24 hours post-dose. This remained stable up to 48 hours in cohort 1 and up to 96 hours in cohorts 2 and 3.
Similarly, mean peak thrombin generation (PTG) values decreased as pegnivacogin dose and plasma concentrations increased. Cohort 3 had an 80% decrease in PTG at 24 hours post-dose. The percent change in mean PTG was consistent and preserved for at least 24 hours for all pegnivacogin doses. In cohorts 2 and 3, it was stable for at least 96 hours.
Endogenous thrombin potential (ETP) levels also decreased after pegnivacogin administration. Again, the largest decrease occurred in cohort 3, which exhibited a 65% decrease at 24 hours. Beyond that, the percent change in ETP was stable within each dose group, up to the last recorded measurement at 96 hours.
Peak time (PTm) was positively correlated with pegnivacogin concentration and relative activated partial thromboplastin time values. The correlation between PTm and quartile of pegnivacogin concentration was statistically significant (P=0.04) but weaker than that of ETP (P=0.001), PTG (P<0.001), or VIx (P<0.001). The changes in PTm persisted for at least 96 hours after pegnivacogin administration.
Finally, the researchers found that anivamersen reversed the effects of pegnivacogin, restoring thrombin generation without rebound effect. ![]()
Credit: Kevin MacKenzie
Results of a phase 1 trial suggest the REG2 anticoagulation system can attenuate thrombin generation and then restore thrombin following reversal.
The REG2 system consists of pegnivacogin, a subcutaneously administered aptamer factor IXa inhibitor, and its intravenous active control agent, anivamersen.
REG2 is formulated to provide a controllable level of anticoagulation for up to 2 weeks for sub-acute uses, especially in cases where a patient may be unable to swallow an oral anticoagulant.
Results observed with REG2 were published in the Journal of Thrombosis and Thrombolysis. The study was sponsored by Regado Biosciences, Inc, the company developing the REG2 system.
The study included 32 healthy volunteers who were enrolled sequentially into 4 cohorts. Patients in cohorts 1-3 were randomized (3:1) to ascending doses of pegnivacogin—from 0.5 mg/kg to 3 mg/kg—or placebo, with no anivamersen administered.
In cohort 4, all patients received 2 mg/kg of pegnivacogin and were randomized (1:1) to reversal by 1 dose or multiple doses over time of 1 mg/kg of anivamersen.
The researchers drew blood samples in the presence or absence of corn trypsin inhibitor at specified times within each dosing cohort. They initiated thrombin generation with tissue factor and measured thrombin generation kinetics using the calibrated automated thrombogram (CAT).
The team found that REG2 attenuated thrombin generation in a dose-dependent manner. All parameters of the CAT assay, except for lag time, showed a dose- and concentration-dependent response to pegnivacogin.
Specifically, there was a decrease in velocity index (VIx) from baseline to 24 hours after pegnivacogin administration. The largest percent change in VIx was in cohort 3 (3 mg/kg). There was a 90% decrease at 24 hours post-dose. This remained stable up to 48 hours in cohort 1 and up to 96 hours in cohorts 2 and 3.
Similarly, mean peak thrombin generation (PTG) values decreased as pegnivacogin dose and plasma concentrations increased. Cohort 3 had an 80% decrease in PTG at 24 hours post-dose. The percent change in mean PTG was consistent and preserved for at least 24 hours for all pegnivacogin doses. In cohorts 2 and 3, it was stable for at least 96 hours.
Endogenous thrombin potential (ETP) levels also decreased after pegnivacogin administration. Again, the largest decrease occurred in cohort 3, which exhibited a 65% decrease at 24 hours. Beyond that, the percent change in ETP was stable within each dose group, up to the last recorded measurement at 96 hours.
Peak time (PTm) was positively correlated with pegnivacogin concentration and relative activated partial thromboplastin time values. The correlation between PTm and quartile of pegnivacogin concentration was statistically significant (P=0.04) but weaker than that of ETP (P=0.001), PTG (P<0.001), or VIx (P<0.001). The changes in PTm persisted for at least 96 hours after pegnivacogin administration.
Finally, the researchers found that anivamersen reversed the effects of pegnivacogin, restoring thrombin generation without rebound effect. ![]()
Credit: Kevin MacKenzie
Results of a phase 1 trial suggest the REG2 anticoagulation system can attenuate thrombin generation and then restore thrombin following reversal.
The REG2 system consists of pegnivacogin, a subcutaneously administered aptamer factor IXa inhibitor, and its intravenous active control agent, anivamersen.
REG2 is formulated to provide a controllable level of anticoagulation for up to 2 weeks for sub-acute uses, especially in cases where a patient may be unable to swallow an oral anticoagulant.
Results observed with REG2 were published in the Journal of Thrombosis and Thrombolysis. The study was sponsored by Regado Biosciences, Inc, the company developing the REG2 system.
The study included 32 healthy volunteers who were enrolled sequentially into 4 cohorts. Patients in cohorts 1-3 were randomized (3:1) to ascending doses of pegnivacogin—from 0.5 mg/kg to 3 mg/kg—or placebo, with no anivamersen administered.
In cohort 4, all patients received 2 mg/kg of pegnivacogin and were randomized (1:1) to reversal by 1 dose or multiple doses over time of 1 mg/kg of anivamersen.
The researchers drew blood samples in the presence or absence of corn trypsin inhibitor at specified times within each dosing cohort. They initiated thrombin generation with tissue factor and measured thrombin generation kinetics using the calibrated automated thrombogram (CAT).
The team found that REG2 attenuated thrombin generation in a dose-dependent manner. All parameters of the CAT assay, except for lag time, showed a dose- and concentration-dependent response to pegnivacogin.
Specifically, there was a decrease in velocity index (VIx) from baseline to 24 hours after pegnivacogin administration. The largest percent change in VIx was in cohort 3 (3 mg/kg). There was a 90% decrease at 24 hours post-dose. This remained stable up to 48 hours in cohort 1 and up to 96 hours in cohorts 2 and 3.
Similarly, mean peak thrombin generation (PTG) values decreased as pegnivacogin dose and plasma concentrations increased. Cohort 3 had an 80% decrease in PTG at 24 hours post-dose. The percent change in mean PTG was consistent and preserved for at least 24 hours for all pegnivacogin doses. In cohorts 2 and 3, it was stable for at least 96 hours.
Endogenous thrombin potential (ETP) levels also decreased after pegnivacogin administration. Again, the largest decrease occurred in cohort 3, which exhibited a 65% decrease at 24 hours. Beyond that, the percent change in ETP was stable within each dose group, up to the last recorded measurement at 96 hours.
Peak time (PTm) was positively correlated with pegnivacogin concentration and relative activated partial thromboplastin time values. The correlation between PTm and quartile of pegnivacogin concentration was statistically significant (P=0.04) but weaker than that of ETP (P=0.001), PTG (P<0.001), or VIx (P<0.001). The changes in PTm persisted for at least 96 hours after pegnivacogin administration.
Finally, the researchers found that anivamersen reversed the effects of pegnivacogin, restoring thrombin generation without rebound effect.
Study reveals new risk factors for bloodstream infections

Current guidelines to help prevent bloodstream infections during intravenous feeding may need revisions to strengthen protection for patients, a new study suggests.
These guidelines restrict how long a single bag of parenteral nutrition containing lipids can be used, due to the ability of lipids to encourage the growth of microorganisms.
But researchers found the guidelines do not account for other independent factors that can affect the growth of potentially deadly microorganisms.
Their findings appear in the Journal of Parenteral and Enteral Nutrition.
Peter David Austin, of the University of Southampton in the UK, and his colleagues looked at the growth of Escherichia coli and Enterococcus durans in parenteral nutrition to identify factors that can affect microbial growth.
They assessed the growth of E coli and E durans in quadruplicate in 12 different patients who received parenteral nutrition, with and without lipid, in varying glucose concentrations.
Results showed that glucose concentration, the proportion of glucose to lipid, and osmolarity all affected microbial growth, in addition to the presence of lipids.
The researchers therefore recommend these additional factors be considered when making clinical and policy decisions to limit the potential growth of microorganisms in parenteral nutrition.
Current guidelines to help prevent bloodstream infections during intravenous feeding may need revisions to strengthen protection for patients, a new study suggests.
These guidelines restrict how long a single bag of parenteral nutrition containing lipids can be used, due to the ability of lipids to encourage the growth of microorganisms.
But researchers found the guidelines do not account for other independent factors that can affect the growth of potentially deadly microorganisms.
Their findings appear in the Journal of Parenteral and Enteral Nutrition.
Peter David Austin, of the University of Southampton in the UK, and his colleagues looked at the growth of Escherichia coli and Enterococcus durans in parenteral nutrition to identify factors that can affect microbial growth.
They assessed the growth of E coli and E durans in quadruplicate in 12 different patients who received parenteral nutrition, with and without lipid, in varying glucose concentrations.
Results showed that glucose concentration, the proportion of glucose to lipid, and osmolarity all affected microbial growth, in addition to the presence of lipids.
The researchers therefore recommend these additional factors be considered when making clinical and policy decisions to limit the potential growth of microorganisms in parenteral nutrition.
Current guidelines to help prevent bloodstream infections during intravenous feeding may need revisions to strengthen protection for patients, a new study suggests.
These guidelines restrict how long a single bag of parenteral nutrition containing lipids can be used, due to the ability of lipids to encourage the growth of microorganisms.
But researchers found the guidelines do not account for other independent factors that can affect the growth of potentially deadly microorganisms.
Their findings appear in the Journal of Parenteral and Enteral Nutrition.
Peter David Austin, of the University of Southampton in the UK, and his colleagues looked at the growth of Escherichia coli and Enterococcus durans in parenteral nutrition to identify factors that can affect microbial growth.
They assessed the growth of E coli and E durans in quadruplicate in 12 different patients who received parenteral nutrition, with and without lipid, in varying glucose concentrations.
Results showed that glucose concentration, the proportion of glucose to lipid, and osmolarity all affected microbial growth, in addition to the presence of lipids.
The researchers therefore recommend these additional factors be considered when making clinical and policy decisions to limit the potential growth of microorganisms in parenteral nutrition.