User login
Why EBV-positive lymphomas resist IFN therapy

Credit: Ed Uthman
New research has revealed how Epstein Barr virus (EBV) and other herpes viruses outwit the body’s immune response.
It seems these viruses carry microRNAs (miRNAs) that block the interferon (IFN) response—when immune cells release IFN to prevent viral replication, which often kills or slows the growth of infected host cells.
This appears to explain why patients with EBV-positive lymphomas and other viral cancers may resist treatment with IFN.
Jennifer Cox, a graduate student at the University of Texas Austin, and her colleagues recounted these findings in PNAS.
The team noted that many viruses, including EBV, carry miRNAs they use to hijack natural processes in a host’s cells during an infection.
Viral miRNAs are known to prevent host cell death, promote host cell growth, and dampen the host cell’s viral defenses. However, scientists don’t yet know which viral miRNAs perform which functions.
To gain some insight, Cox and her colleagues screened a library of more than 70 human viral miRNAs. This revealed 3 unrelated miRNAs from distantly related herpes viruses that significantly inhibited IFN signaling.
The 5’ and 3’ derivatives from EBV-encoded miR-BART-18 precursor miRNA and the orthologous precursor miRNA from Rhesus lymphocryptovirus all reduced expression of the cyclic AMP-responsive element-binding protein (CBP), which, as part of the p300-CBP complex, mediates IFN signaling.
When the researchers restored miR-BART-18 to cells infected with an EBV miRNA mutant, they observed a cellular growth advantage upon IFN treatment. And they found that miRNAs from other herpes viruses were able to complement this activity.
The team also showed that blocking miR-BART-18 function in an EBV-positive tumor cell line rendered cells more susceptible to IFN-mediated effects.
“[These findings] could explain the variability seen in the success of previous interferon-based cancer treatments,” Cox said. “While this work does not immediately identify new drugs, the fact that such different tumor viruses have converged on the same strategy makes this an exciting pursuit for future therapies against viral cancers.” ![]()
Credit: Ed Uthman
New research has revealed how Epstein Barr virus (EBV) and other herpes viruses outwit the body’s immune response.
It seems these viruses carry microRNAs (miRNAs) that block the interferon (IFN) response—when immune cells release IFN to prevent viral replication, which often kills or slows the growth of infected host cells.
This appears to explain why patients with EBV-positive lymphomas and other viral cancers may resist treatment with IFN.
Jennifer Cox, a graduate student at the University of Texas Austin, and her colleagues recounted these findings in PNAS.
The team noted that many viruses, including EBV, carry miRNAs they use to hijack natural processes in a host’s cells during an infection.
Viral miRNAs are known to prevent host cell death, promote host cell growth, and dampen the host cell’s viral defenses. However, scientists don’t yet know which viral miRNAs perform which functions.
To gain some insight, Cox and her colleagues screened a library of more than 70 human viral miRNAs. This revealed 3 unrelated miRNAs from distantly related herpes viruses that significantly inhibited IFN signaling.
The 5’ and 3’ derivatives from EBV-encoded miR-BART-18 precursor miRNA and the orthologous precursor miRNA from Rhesus lymphocryptovirus all reduced expression of the cyclic AMP-responsive element-binding protein (CBP), which, as part of the p300-CBP complex, mediates IFN signaling.
When the researchers restored miR-BART-18 to cells infected with an EBV miRNA mutant, they observed a cellular growth advantage upon IFN treatment. And they found that miRNAs from other herpes viruses were able to complement this activity.
The team also showed that blocking miR-BART-18 function in an EBV-positive tumor cell line rendered cells more susceptible to IFN-mediated effects.
“[These findings] could explain the variability seen in the success of previous interferon-based cancer treatments,” Cox said. “While this work does not immediately identify new drugs, the fact that such different tumor viruses have converged on the same strategy makes this an exciting pursuit for future therapies against viral cancers.” ![]()
Credit: Ed Uthman
New research has revealed how Epstein Barr virus (EBV) and other herpes viruses outwit the body’s immune response.
It seems these viruses carry microRNAs (miRNAs) that block the interferon (IFN) response—when immune cells release IFN to prevent viral replication, which often kills or slows the growth of infected host cells.
This appears to explain why patients with EBV-positive lymphomas and other viral cancers may resist treatment with IFN.
Jennifer Cox, a graduate student at the University of Texas Austin, and her colleagues recounted these findings in PNAS.
The team noted that many viruses, including EBV, carry miRNAs they use to hijack natural processes in a host’s cells during an infection.
Viral miRNAs are known to prevent host cell death, promote host cell growth, and dampen the host cell’s viral defenses. However, scientists don’t yet know which viral miRNAs perform which functions.
To gain some insight, Cox and her colleagues screened a library of more than 70 human viral miRNAs. This revealed 3 unrelated miRNAs from distantly related herpes viruses that significantly inhibited IFN signaling.
The 5’ and 3’ derivatives from EBV-encoded miR-BART-18 precursor miRNA and the orthologous precursor miRNA from Rhesus lymphocryptovirus all reduced expression of the cyclic AMP-responsive element-binding protein (CBP), which, as part of the p300-CBP complex, mediates IFN signaling.
When the researchers restored miR-BART-18 to cells infected with an EBV miRNA mutant, they observed a cellular growth advantage upon IFN treatment. And they found that miRNAs from other herpes viruses were able to complement this activity.
The team also showed that blocking miR-BART-18 function in an EBV-positive tumor cell line rendered cells more susceptible to IFN-mediated effects.
“[These findings] could explain the variability seen in the success of previous interferon-based cancer treatments,” Cox said. “While this work does not immediately identify new drugs, the fact that such different tumor viruses have converged on the same strategy makes this an exciting pursuit for future therapies against viral cancers.” ![]()
Drug on the fast track to treat HAE

within pancreatic tissue
Credit: Louisa Howard
The US Food and Drug Administration (FDA) has granted fast track designation for BCX4161, an oral inhibitor of plasma kallikrein intended to treat hereditary angioedema (HAE).
Uncontrolled activation of plasma kallikrein, caused by deficiency of its physiological inhibitor (C1 inhibitor) in HAE, results in acute systemic edema.
By inhibiting plasma kallikrein, BCX4161 suppresses the production of bradykinin, the mediator of acute swelling attacks in HAE patients.
HAE is a severely debilitating and potentially fatal condition that occurs in approximately 1 in 50,000 people. Symptoms include recurrent episodes of edema in various locations, as well as bouts of excruciating abdominal pain, nausea, and vomiting that are caused by swelling in the intestinal walls.
HAE patients have a defect in the gene that controls C1 inhibitor, and this results in the production of inadequate or non-functioning C1 inhibitor protein.
Normal C1 inhibitor helps regulate the biochemical interactions of blood-based systems involved in disease-fighting, inflammatory response, and coagulation.
Because defective C1 inhibitor does not adequately perform its regulatory function, a biochemical imbalance can occur and produce unwanted peptides that induce the capillaries to release fluids into surrounding tissue, causing edema.
BCX4161 trials
In May 2014, BioCryst Pharmaceuticals, the company developing BCX4161, announced results from the phase 2a OPuS-1 trial.
OPuS-1 investigators evaluated 400 mg of BCX4161 administered 3 times a day for 28 days in HAE patients with a high angioedema attack frequency (≥ 1 per week), in a randomized, placebo-controlled, 2-period cross-over design.
BCX4161 demonstrated a significant reduction in mean attack rate compared to placebo. The mean attack rate per patient-week was 0.82 on BCX4161 treatment and 1.27 on placebo (P<0.001).
The mean number of attack-free days during each treatment period improved from 19 for placebo to 22 for BCX4161 (P=0.008). Three subjects were attack-free during the BCX4161 period, compared to none during the placebo period.
BCX4161 was generally well-tolerated, BioCryst reported, with an adverse event profile similar to that observed for placebo. There was one serious adverse event reported, an abdominal HAE attack during the placebo period.
In December, the first patient was dosed in the OPuS-2 trial, a double-blind, randomized, placebo- controlled trial conducted in the US and European Union.
Study investigators will evaluate the efficacy and safety of BCX4161 treatment for 12 weeks in patients with HAE. BioCryst expects to report results from OPuS-2 by the end of 2015.
About fast track designation
The FDA’s fast track process is designed to facilitate the development and expedite the review and approval of drugs intended to treat serious or life-threatening conditions that also address unmet medical needs.
A drug that receives fast track designation is usually eligible for more frequent written communication and meetings with the FDA to discuss the drug’s development plan and the collection of appropriate data supporting drug approval.
Priority review and rolling review may be granted if relevant criteria are met. Rolling review allows a drug company to submit completed sections of its new drug application on an ongoing basis, rather than wait until the entire application is complete. ![]()
within pancreatic tissue
Credit: Louisa Howard
The US Food and Drug Administration (FDA) has granted fast track designation for BCX4161, an oral inhibitor of plasma kallikrein intended to treat hereditary angioedema (HAE).
Uncontrolled activation of plasma kallikrein, caused by deficiency of its physiological inhibitor (C1 inhibitor) in HAE, results in acute systemic edema.
By inhibiting plasma kallikrein, BCX4161 suppresses the production of bradykinin, the mediator of acute swelling attacks in HAE patients.
HAE is a severely debilitating and potentially fatal condition that occurs in approximately 1 in 50,000 people. Symptoms include recurrent episodes of edema in various locations, as well as bouts of excruciating abdominal pain, nausea, and vomiting that are caused by swelling in the intestinal walls.
HAE patients have a defect in the gene that controls C1 inhibitor, and this results in the production of inadequate or non-functioning C1 inhibitor protein.
Normal C1 inhibitor helps regulate the biochemical interactions of blood-based systems involved in disease-fighting, inflammatory response, and coagulation.
Because defective C1 inhibitor does not adequately perform its regulatory function, a biochemical imbalance can occur and produce unwanted peptides that induce the capillaries to release fluids into surrounding tissue, causing edema.
BCX4161 trials
In May 2014, BioCryst Pharmaceuticals, the company developing BCX4161, announced results from the phase 2a OPuS-1 trial.
OPuS-1 investigators evaluated 400 mg of BCX4161 administered 3 times a day for 28 days in HAE patients with a high angioedema attack frequency (≥ 1 per week), in a randomized, placebo-controlled, 2-period cross-over design.
BCX4161 demonstrated a significant reduction in mean attack rate compared to placebo. The mean attack rate per patient-week was 0.82 on BCX4161 treatment and 1.27 on placebo (P<0.001).
The mean number of attack-free days during each treatment period improved from 19 for placebo to 22 for BCX4161 (P=0.008). Three subjects were attack-free during the BCX4161 period, compared to none during the placebo period.
BCX4161 was generally well-tolerated, BioCryst reported, with an adverse event profile similar to that observed for placebo. There was one serious adverse event reported, an abdominal HAE attack during the placebo period.
In December, the first patient was dosed in the OPuS-2 trial, a double-blind, randomized, placebo- controlled trial conducted in the US and European Union.
Study investigators will evaluate the efficacy and safety of BCX4161 treatment for 12 weeks in patients with HAE. BioCryst expects to report results from OPuS-2 by the end of 2015.
About fast track designation
The FDA’s fast track process is designed to facilitate the development and expedite the review and approval of drugs intended to treat serious or life-threatening conditions that also address unmet medical needs.
A drug that receives fast track designation is usually eligible for more frequent written communication and meetings with the FDA to discuss the drug’s development plan and the collection of appropriate data supporting drug approval.
Priority review and rolling review may be granted if relevant criteria are met. Rolling review allows a drug company to submit completed sections of its new drug application on an ongoing basis, rather than wait until the entire application is complete. ![]()
within pancreatic tissue
Credit: Louisa Howard
The US Food and Drug Administration (FDA) has granted fast track designation for BCX4161, an oral inhibitor of plasma kallikrein intended to treat hereditary angioedema (HAE).
Uncontrolled activation of plasma kallikrein, caused by deficiency of its physiological inhibitor (C1 inhibitor) in HAE, results in acute systemic edema.
By inhibiting plasma kallikrein, BCX4161 suppresses the production of bradykinin, the mediator of acute swelling attacks in HAE patients.
HAE is a severely debilitating and potentially fatal condition that occurs in approximately 1 in 50,000 people. Symptoms include recurrent episodes of edema in various locations, as well as bouts of excruciating abdominal pain, nausea, and vomiting that are caused by swelling in the intestinal walls.
HAE patients have a defect in the gene that controls C1 inhibitor, and this results in the production of inadequate or non-functioning C1 inhibitor protein.
Normal C1 inhibitor helps regulate the biochemical interactions of blood-based systems involved in disease-fighting, inflammatory response, and coagulation.
Because defective C1 inhibitor does not adequately perform its regulatory function, a biochemical imbalance can occur and produce unwanted peptides that induce the capillaries to release fluids into surrounding tissue, causing edema.
BCX4161 trials
In May 2014, BioCryst Pharmaceuticals, the company developing BCX4161, announced results from the phase 2a OPuS-1 trial.
OPuS-1 investigators evaluated 400 mg of BCX4161 administered 3 times a day for 28 days in HAE patients with a high angioedema attack frequency (≥ 1 per week), in a randomized, placebo-controlled, 2-period cross-over design.
BCX4161 demonstrated a significant reduction in mean attack rate compared to placebo. The mean attack rate per patient-week was 0.82 on BCX4161 treatment and 1.27 on placebo (P<0.001).
The mean number of attack-free days during each treatment period improved from 19 for placebo to 22 for BCX4161 (P=0.008). Three subjects were attack-free during the BCX4161 period, compared to none during the placebo period.
BCX4161 was generally well-tolerated, BioCryst reported, with an adverse event profile similar to that observed for placebo. There was one serious adverse event reported, an abdominal HAE attack during the placebo period.
In December, the first patient was dosed in the OPuS-2 trial, a double-blind, randomized, placebo- controlled trial conducted in the US and European Union.
Study investigators will evaluate the efficacy and safety of BCX4161 treatment for 12 weeks in patients with HAE. BioCryst expects to report results from OPuS-2 by the end of 2015.
About fast track designation
The FDA’s fast track process is designed to facilitate the development and expedite the review and approval of drugs intended to treat serious or life-threatening conditions that also address unmet medical needs.
A drug that receives fast track designation is usually eligible for more frequent written communication and meetings with the FDA to discuss the drug’s development plan and the collection of appropriate data supporting drug approval.
Priority review and rolling review may be granted if relevant criteria are met. Rolling review allows a drug company to submit completed sections of its new drug application on an ongoing basis, rather than wait until the entire application is complete. ![]()
Team discovers new mechanisms of platelet formation
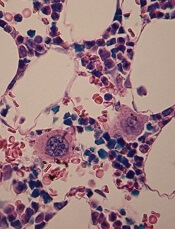
in the bone marrow
Researchers have found that megakaryocytes can produce platelets via different polyploidization mechanisms.
Experiments suggested that endomitosis is the major megakaryocyte polyploidization mechanism, but megakaryocytes can generate platelets even in the absence of endomitosis—via endocycles and re-replication.
These findings may have implications for treating cancers as well as thrombocytopenia, the researchers said.
They described their discoveries in Developmental Cell.
Marcos Malumbres, PhD, of Centro Nacional de Investigaciones Oncologicas (CNIO) in Madrid, Spain, and his colleagues performed a genetic analysis of endomitosis, in which megakaryocytes become polyploid by entering mitosis but aborting anaphase.
The team studied mice lacking key proteins involved in mitotic entry (Cdk1 and Cdk2), mitotic progression (Aurora B), or mitotic exit (the anaphase-promoting complex [APC/C] cofactor Cdc20) during megakaryocyte maturation.
They found that Aurora B is not required for megakaryocyte maturation, but Cdc20 is. In mice lacking Cdc20, the researchers observed mitotic arrest and severe thrombocytopenia. The team said this suggests endomitosis is the major megakaryocyte polyploidization mechanism in vivo.
So they were surprised to discover that deleting Cdk1 prevents endomitosis but does not hinder platelet formation.
“Whilst the elimination of the main proteins that regulate megakaryocyte growth generates, as we thought, a reduction in the production of platelets, this didn’t happen when we removed Cdk1,” Dr Malumbres said. “[Even] when Cdk1 was absent, the megakaryocytes were able to grow in size in a similar way to normal cells.”
“[Further] analysis revealed that cells deficient in Cdk1 underwent cellular reprogramming towards a process known as endocycles, which can also be seen in other types of cells, such as certain cells in the placenta,” said Marianna Trakala, also of CNIO.
In endocycles, cells successively replicate their genomes without segregating chromosomes during mitosis.
The researchers uncovered an additional mechanism for megakaryocyte polyploidy when they studied mice lacking both Cdk1 and Cdk2. The deletion of both Cdks resulted in aberrant re-replication, in which the genome is replicated more than once per cell cycle, but platelet levels were not affected.
In fact, the team found that the loss of Cdk1 alone or Cdk1 and Cdk2 together can significantly rescue proplatelet formation and increase platelet levels in Cdc20-null mice.
“We immediately asked ourselves if, by reprogramming the cell cycle towards endocycles, we could correct the thrombocytopenia induced in other models,” Dr Malumbres said.
And when the researchers eliminated Cdk1 in Cdc20-null mice with severe thrombocytopenia, they observed an increase in platelet production. Results were similar when the team ablated Cdk1 and Cdk2 in Cdc20-null mice.
In addition to their implications for treating thrombocytopenia, these results could aid the design of cancer treatments, the researchers said. The findings reveal the different requirements that normal or tumor cells have toward cell-cycle regulators.
Specifically, the research suggests that deleting Cdk1 or other cell-cycle proteins is lethal for tumor cells but does not affect megakaryocytes. So drugs that inhibit these proteins could be used to treat malignancies such as pro-megakaryocytic leukemias. ![]()
in the bone marrow
Researchers have found that megakaryocytes can produce platelets via different polyploidization mechanisms.
Experiments suggested that endomitosis is the major megakaryocyte polyploidization mechanism, but megakaryocytes can generate platelets even in the absence of endomitosis—via endocycles and re-replication.
These findings may have implications for treating cancers as well as thrombocytopenia, the researchers said.
They described their discoveries in Developmental Cell.
Marcos Malumbres, PhD, of Centro Nacional de Investigaciones Oncologicas (CNIO) in Madrid, Spain, and his colleagues performed a genetic analysis of endomitosis, in which megakaryocytes become polyploid by entering mitosis but aborting anaphase.
The team studied mice lacking key proteins involved in mitotic entry (Cdk1 and Cdk2), mitotic progression (Aurora B), or mitotic exit (the anaphase-promoting complex [APC/C] cofactor Cdc20) during megakaryocyte maturation.
They found that Aurora B is not required for megakaryocyte maturation, but Cdc20 is. In mice lacking Cdc20, the researchers observed mitotic arrest and severe thrombocytopenia. The team said this suggests endomitosis is the major megakaryocyte polyploidization mechanism in vivo.
So they were surprised to discover that deleting Cdk1 prevents endomitosis but does not hinder platelet formation.
“Whilst the elimination of the main proteins that regulate megakaryocyte growth generates, as we thought, a reduction in the production of platelets, this didn’t happen when we removed Cdk1,” Dr Malumbres said. “[Even] when Cdk1 was absent, the megakaryocytes were able to grow in size in a similar way to normal cells.”
“[Further] analysis revealed that cells deficient in Cdk1 underwent cellular reprogramming towards a process known as endocycles, which can also be seen in other types of cells, such as certain cells in the placenta,” said Marianna Trakala, also of CNIO.
In endocycles, cells successively replicate their genomes without segregating chromosomes during mitosis.
The researchers uncovered an additional mechanism for megakaryocyte polyploidy when they studied mice lacking both Cdk1 and Cdk2. The deletion of both Cdks resulted in aberrant re-replication, in which the genome is replicated more than once per cell cycle, but platelet levels were not affected.
In fact, the team found that the loss of Cdk1 alone or Cdk1 and Cdk2 together can significantly rescue proplatelet formation and increase platelet levels in Cdc20-null mice.
“We immediately asked ourselves if, by reprogramming the cell cycle towards endocycles, we could correct the thrombocytopenia induced in other models,” Dr Malumbres said.
And when the researchers eliminated Cdk1 in Cdc20-null mice with severe thrombocytopenia, they observed an increase in platelet production. Results were similar when the team ablated Cdk1 and Cdk2 in Cdc20-null mice.
In addition to their implications for treating thrombocytopenia, these results could aid the design of cancer treatments, the researchers said. The findings reveal the different requirements that normal or tumor cells have toward cell-cycle regulators.
Specifically, the research suggests that deleting Cdk1 or other cell-cycle proteins is lethal for tumor cells but does not affect megakaryocytes. So drugs that inhibit these proteins could be used to treat malignancies such as pro-megakaryocytic leukemias. ![]()
in the bone marrow
Researchers have found that megakaryocytes can produce platelets via different polyploidization mechanisms.
Experiments suggested that endomitosis is the major megakaryocyte polyploidization mechanism, but megakaryocytes can generate platelets even in the absence of endomitosis—via endocycles and re-replication.
These findings may have implications for treating cancers as well as thrombocytopenia, the researchers said.
They described their discoveries in Developmental Cell.
Marcos Malumbres, PhD, of Centro Nacional de Investigaciones Oncologicas (CNIO) in Madrid, Spain, and his colleagues performed a genetic analysis of endomitosis, in which megakaryocytes become polyploid by entering mitosis but aborting anaphase.
The team studied mice lacking key proteins involved in mitotic entry (Cdk1 and Cdk2), mitotic progression (Aurora B), or mitotic exit (the anaphase-promoting complex [APC/C] cofactor Cdc20) during megakaryocyte maturation.
They found that Aurora B is not required for megakaryocyte maturation, but Cdc20 is. In mice lacking Cdc20, the researchers observed mitotic arrest and severe thrombocytopenia. The team said this suggests endomitosis is the major megakaryocyte polyploidization mechanism in vivo.
So they were surprised to discover that deleting Cdk1 prevents endomitosis but does not hinder platelet formation.
“Whilst the elimination of the main proteins that regulate megakaryocyte growth generates, as we thought, a reduction in the production of platelets, this didn’t happen when we removed Cdk1,” Dr Malumbres said. “[Even] when Cdk1 was absent, the megakaryocytes were able to grow in size in a similar way to normal cells.”
“[Further] analysis revealed that cells deficient in Cdk1 underwent cellular reprogramming towards a process known as endocycles, which can also be seen in other types of cells, such as certain cells in the placenta,” said Marianna Trakala, also of CNIO.
In endocycles, cells successively replicate their genomes without segregating chromosomes during mitosis.
The researchers uncovered an additional mechanism for megakaryocyte polyploidy when they studied mice lacking both Cdk1 and Cdk2. The deletion of both Cdks resulted in aberrant re-replication, in which the genome is replicated more than once per cell cycle, but platelet levels were not affected.
In fact, the team found that the loss of Cdk1 alone or Cdk1 and Cdk2 together can significantly rescue proplatelet formation and increase platelet levels in Cdc20-null mice.
“We immediately asked ourselves if, by reprogramming the cell cycle towards endocycles, we could correct the thrombocytopenia induced in other models,” Dr Malumbres said.
And when the researchers eliminated Cdk1 in Cdc20-null mice with severe thrombocytopenia, they observed an increase in platelet production. Results were similar when the team ablated Cdk1 and Cdk2 in Cdc20-null mice.
In addition to their implications for treating thrombocytopenia, these results could aid the design of cancer treatments, the researchers said. The findings reveal the different requirements that normal or tumor cells have toward cell-cycle regulators.
Specifically, the research suggests that deleting Cdk1 or other cell-cycle proteins is lethal for tumor cells but does not affect megakaryocytes. So drugs that inhibit these proteins could be used to treat malignancies such as pro-megakaryocytic leukemias. ![]()
Fusion protein fights resistant ALL

In a preclinical study, a fusion protein targeted treatment-resistant leukemia, demonstrating superiority over both chemotherapy and radiation.
The protein, CD19L-sTRAIL, induced apoptosis in radiation-resistant cells from patients with B-precursor acute lymphoblastic leukemia (ALL) and in
mouse models of the disease.
Additionally, mice that received CD19L-sTRAIL had significantly longer event-free survival than mice that received chemotherapy.
An account of this study appears in The Journal of Clinical Investigation.
Study investigators knew that TNF-related apoptosis-inducing ligand (TRAIL) can cause apoptosis in cancer cells by binding to TRAIL-receptor 1 and TRAIL-receptor 2.
“TRAIL is a naturally occurring part of the body’s immune system that kills cancer cells without toxicity to normal cells,” said Faith Uckun, MD, PhD, of Children’s Hospital Los Angeles in California.
“However, earlier clinical trials using TRAIL as a potential anticancer medicine candidate have not been successful, largely because of its propensity to bind, not only to cancer cells, but also to ‘decoy’ receptors.”
But Dr Uckun and her colleagues had discovered CD19-ligand (CD19L), a natural ligand of human CD19 that is expressed by almost all ALL cells. And they hypothesized that fusing CD19L to the portion of TRAIL that can kill cancer cells (known as sTRAIL) would produce a powerful weapon against leukemia cells.
The resulting fusion protein, CD19L-sTRAIL, would seek out, bind, and destroy only leukemia cells carrying CD19 as the target docking site.
In experiments, the investigators showed that their engineering converted sTRAIL into a much more potent “membrane-anchored” form that is capable of triggering apoptosis, even in the most aggressive and therapy-resistant ALL cells.
“Due to its ability to anchor to the surface of cancer cells via CD19, CD19L-sTRAIL was 100,000-fold more potent than sTRAIL and consistently killed more than 99% of aggressive leukemia cells taken directly from children with ALL—not only in the test tube but also in mice,” Dr Uckun said.
At a 2.1 pM concentration, CD19L-sTRAIL caused 84.0±4.7% apoptosis in leukemia cells from patients with B-precursor ALL, whereas radiation with 2 Gy γ-rays caused 45.0±9.0% apoptosis (P<0.0001). Higher concentrations of CD19L-sTRAIL prompted an apoptosis rate of more than 90%.
In cells from mouse models of B-precursor ALL, 2.1 pM of CD19L-sTRAIL caused 91.4±5.4% apoptosis, compared to 16.0±4.6% with 2 Gy γ-rays (P<0.0001).
In addition, administering 2 or 3 doses of CD19L-sTRAIL significantly improved event-free survival in mice challenged with an otherwise fatal dose of leukemia cells.
The median event-free survival was 17 days in control mice; 20 days in mice treated with either vincristine, dexamethasone, and peg-asparaginase or vincristine, doxorubicin, and peg-asparaginase; and 58 days in mice that received 2- to 3-day treatment with CD19L-sTRAIL (P<0.0001 vs controls; P=0.0002 vs chemo).
The investigators said these results support the clinical potential of CD19L-sTRAIL as a new agent against B-precursor ALL.
“We are hopeful that the knowledge gained from this study will open a new range of effective treatment opportunities for children with recurrent leukemia,” Dr Uckun said. ![]()
In a preclinical study, a fusion protein targeted treatment-resistant leukemia, demonstrating superiority over both chemotherapy and radiation.
The protein, CD19L-sTRAIL, induced apoptosis in radiation-resistant cells from patients with B-precursor acute lymphoblastic leukemia (ALL) and in
mouse models of the disease.
Additionally, mice that received CD19L-sTRAIL had significantly longer event-free survival than mice that received chemotherapy.
An account of this study appears in The Journal of Clinical Investigation.
Study investigators knew that TNF-related apoptosis-inducing ligand (TRAIL) can cause apoptosis in cancer cells by binding to TRAIL-receptor 1 and TRAIL-receptor 2.
“TRAIL is a naturally occurring part of the body’s immune system that kills cancer cells without toxicity to normal cells,” said Faith Uckun, MD, PhD, of Children’s Hospital Los Angeles in California.
“However, earlier clinical trials using TRAIL as a potential anticancer medicine candidate have not been successful, largely because of its propensity to bind, not only to cancer cells, but also to ‘decoy’ receptors.”
But Dr Uckun and her colleagues had discovered CD19-ligand (CD19L), a natural ligand of human CD19 that is expressed by almost all ALL cells. And they hypothesized that fusing CD19L to the portion of TRAIL that can kill cancer cells (known as sTRAIL) would produce a powerful weapon against leukemia cells.
The resulting fusion protein, CD19L-sTRAIL, would seek out, bind, and destroy only leukemia cells carrying CD19 as the target docking site.
In experiments, the investigators showed that their engineering converted sTRAIL into a much more potent “membrane-anchored” form that is capable of triggering apoptosis, even in the most aggressive and therapy-resistant ALL cells.
“Due to its ability to anchor to the surface of cancer cells via CD19, CD19L-sTRAIL was 100,000-fold more potent than sTRAIL and consistently killed more than 99% of aggressive leukemia cells taken directly from children with ALL—not only in the test tube but also in mice,” Dr Uckun said.
At a 2.1 pM concentration, CD19L-sTRAIL caused 84.0±4.7% apoptosis in leukemia cells from patients with B-precursor ALL, whereas radiation with 2 Gy γ-rays caused 45.0±9.0% apoptosis (P<0.0001). Higher concentrations of CD19L-sTRAIL prompted an apoptosis rate of more than 90%.
In cells from mouse models of B-precursor ALL, 2.1 pM of CD19L-sTRAIL caused 91.4±5.4% apoptosis, compared to 16.0±4.6% with 2 Gy γ-rays (P<0.0001).
In addition, administering 2 or 3 doses of CD19L-sTRAIL significantly improved event-free survival in mice challenged with an otherwise fatal dose of leukemia cells.
The median event-free survival was 17 days in control mice; 20 days in mice treated with either vincristine, dexamethasone, and peg-asparaginase or vincristine, doxorubicin, and peg-asparaginase; and 58 days in mice that received 2- to 3-day treatment with CD19L-sTRAIL (P<0.0001 vs controls; P=0.0002 vs chemo).
The investigators said these results support the clinical potential of CD19L-sTRAIL as a new agent against B-precursor ALL.
“We are hopeful that the knowledge gained from this study will open a new range of effective treatment opportunities for children with recurrent leukemia,” Dr Uckun said. ![]()
In a preclinical study, a fusion protein targeted treatment-resistant leukemia, demonstrating superiority over both chemotherapy and radiation.
The protein, CD19L-sTRAIL, induced apoptosis in radiation-resistant cells from patients with B-precursor acute lymphoblastic leukemia (ALL) and in
mouse models of the disease.
Additionally, mice that received CD19L-sTRAIL had significantly longer event-free survival than mice that received chemotherapy.
An account of this study appears in The Journal of Clinical Investigation.
Study investigators knew that TNF-related apoptosis-inducing ligand (TRAIL) can cause apoptosis in cancer cells by binding to TRAIL-receptor 1 and TRAIL-receptor 2.
“TRAIL is a naturally occurring part of the body’s immune system that kills cancer cells without toxicity to normal cells,” said Faith Uckun, MD, PhD, of Children’s Hospital Los Angeles in California.
“However, earlier clinical trials using TRAIL as a potential anticancer medicine candidate have not been successful, largely because of its propensity to bind, not only to cancer cells, but also to ‘decoy’ receptors.”
But Dr Uckun and her colleagues had discovered CD19-ligand (CD19L), a natural ligand of human CD19 that is expressed by almost all ALL cells. And they hypothesized that fusing CD19L to the portion of TRAIL that can kill cancer cells (known as sTRAIL) would produce a powerful weapon against leukemia cells.
The resulting fusion protein, CD19L-sTRAIL, would seek out, bind, and destroy only leukemia cells carrying CD19 as the target docking site.
In experiments, the investigators showed that their engineering converted sTRAIL into a much more potent “membrane-anchored” form that is capable of triggering apoptosis, even in the most aggressive and therapy-resistant ALL cells.
“Due to its ability to anchor to the surface of cancer cells via CD19, CD19L-sTRAIL was 100,000-fold more potent than sTRAIL and consistently killed more than 99% of aggressive leukemia cells taken directly from children with ALL—not only in the test tube but also in mice,” Dr Uckun said.
At a 2.1 pM concentration, CD19L-sTRAIL caused 84.0±4.7% apoptosis in leukemia cells from patients with B-precursor ALL, whereas radiation with 2 Gy γ-rays caused 45.0±9.0% apoptosis (P<0.0001). Higher concentrations of CD19L-sTRAIL prompted an apoptosis rate of more than 90%.
In cells from mouse models of B-precursor ALL, 2.1 pM of CD19L-sTRAIL caused 91.4±5.4% apoptosis, compared to 16.0±4.6% with 2 Gy γ-rays (P<0.0001).
In addition, administering 2 or 3 doses of CD19L-sTRAIL significantly improved event-free survival in mice challenged with an otherwise fatal dose of leukemia cells.
The median event-free survival was 17 days in control mice; 20 days in mice treated with either vincristine, dexamethasone, and peg-asparaginase or vincristine, doxorubicin, and peg-asparaginase; and 58 days in mice that received 2- to 3-day treatment with CD19L-sTRAIL (P<0.0001 vs controls; P=0.0002 vs chemo).
The investigators said these results support the clinical potential of CD19L-sTRAIL as a new agent against B-precursor ALL.
“We are hopeful that the knowledge gained from this study will open a new range of effective treatment opportunities for children with recurrent leukemia,” Dr Uckun said. ![]()
Product granted orphan designation for GVHD

Credit: PLOS ONE
The European Medicines Agency (EMA) has granted orphan drug status to ApoCell for the prevention of graft-vs-host disease (GVHD).
ApoCell consists of matched-donor, mononuclear-enriched, early apoptotic cells. It has been shown to immunomodulate macrophages and dendritic cells, both of which are involved in GVHD pathogenesis.
ApoCell showed early promise in a phase 1/2a study, and a phase 2b/3 trial of the product is set to begin this year.
ApoCell, which is under development by Enlivex Therapeutics, received orphan drug designation in the US in 2013.
In the European Union (EU), orphan designation is granted to products intended for the treatment, prevention, or diagnosis of a life-threatening or chronically debilitating disease with a prevalence of no more than 5 in 10,000, with no satisfactory method of diagnosis, prevention, or treatment authorized by the EMA.
Orphan designation in the EU comes with a number of benefits, including protocol assistance and 10-year market exclusivity following regulatory approval.
Phase 1/2a trial of ApoCell
For this study, researchers tested an infusion of ApoCell, in addition to cyclosporine and methotrexate, as prophylaxis for acute GVHD.
The trial enrolled 13 leukemia patients who had a median age of 37 (range, 20-59). All of the patients received an HLA-matched, myeloablative, allogeneic hematopoietic stem cell transplant (HSCT) from a related donor.
All patients engrafted. The median time to neutrophil recovery was 13 days after HSCT (range, 11 to 19), and the median time to platelet recovery was 15 days (range, 11 to 59).
At 100 days post-HSCT, the cumulative incidence of acute grade 2-4 GVHD was 23.1%, and the incidence of acute grade 3-4 GVHD was 15.4%. None of the patients developed acute GVHD beyond day 100.
Among patients who received the 2 higher doses of ApoCell (n=6), the rate of acute grade 2-4 GVHD was 0%.
Ten patients experienced serious adverse events, but 7 were considered unrelated to ApoCell, and 3 were likely unrelated to the treatment.
The nonrelapse mortality rate was 7.7% at both 100 and 180 days after HSCT. The overall survival rate was 92% at 100 days and 85% at 180 days.
The researchers said these results suggest a single infusion of ApoCell is safe and may effectively prevent acute GVHD. ![]()
Credit: PLOS ONE
The European Medicines Agency (EMA) has granted orphan drug status to ApoCell for the prevention of graft-vs-host disease (GVHD).
ApoCell consists of matched-donor, mononuclear-enriched, early apoptotic cells. It has been shown to immunomodulate macrophages and dendritic cells, both of which are involved in GVHD pathogenesis.
ApoCell showed early promise in a phase 1/2a study, and a phase 2b/3 trial of the product is set to begin this year.
ApoCell, which is under development by Enlivex Therapeutics, received orphan drug designation in the US in 2013.
In the European Union (EU), orphan designation is granted to products intended for the treatment, prevention, or diagnosis of a life-threatening or chronically debilitating disease with a prevalence of no more than 5 in 10,000, with no satisfactory method of diagnosis, prevention, or treatment authorized by the EMA.
Orphan designation in the EU comes with a number of benefits, including protocol assistance and 10-year market exclusivity following regulatory approval.
Phase 1/2a trial of ApoCell
For this study, researchers tested an infusion of ApoCell, in addition to cyclosporine and methotrexate, as prophylaxis for acute GVHD.
The trial enrolled 13 leukemia patients who had a median age of 37 (range, 20-59). All of the patients received an HLA-matched, myeloablative, allogeneic hematopoietic stem cell transplant (HSCT) from a related donor.
All patients engrafted. The median time to neutrophil recovery was 13 days after HSCT (range, 11 to 19), and the median time to platelet recovery was 15 days (range, 11 to 59).
At 100 days post-HSCT, the cumulative incidence of acute grade 2-4 GVHD was 23.1%, and the incidence of acute grade 3-4 GVHD was 15.4%. None of the patients developed acute GVHD beyond day 100.
Among patients who received the 2 higher doses of ApoCell (n=6), the rate of acute grade 2-4 GVHD was 0%.
Ten patients experienced serious adverse events, but 7 were considered unrelated to ApoCell, and 3 were likely unrelated to the treatment.
The nonrelapse mortality rate was 7.7% at both 100 and 180 days after HSCT. The overall survival rate was 92% at 100 days and 85% at 180 days.
The researchers said these results suggest a single infusion of ApoCell is safe and may effectively prevent acute GVHD. ![]()
Credit: PLOS ONE
The European Medicines Agency (EMA) has granted orphan drug status to ApoCell for the prevention of graft-vs-host disease (GVHD).
ApoCell consists of matched-donor, mononuclear-enriched, early apoptotic cells. It has been shown to immunomodulate macrophages and dendritic cells, both of which are involved in GVHD pathogenesis.
ApoCell showed early promise in a phase 1/2a study, and a phase 2b/3 trial of the product is set to begin this year.
ApoCell, which is under development by Enlivex Therapeutics, received orphan drug designation in the US in 2013.
In the European Union (EU), orphan designation is granted to products intended for the treatment, prevention, or diagnosis of a life-threatening or chronically debilitating disease with a prevalence of no more than 5 in 10,000, with no satisfactory method of diagnosis, prevention, or treatment authorized by the EMA.
Orphan designation in the EU comes with a number of benefits, including protocol assistance and 10-year market exclusivity following regulatory approval.
Phase 1/2a trial of ApoCell
For this study, researchers tested an infusion of ApoCell, in addition to cyclosporine and methotrexate, as prophylaxis for acute GVHD.
The trial enrolled 13 leukemia patients who had a median age of 37 (range, 20-59). All of the patients received an HLA-matched, myeloablative, allogeneic hematopoietic stem cell transplant (HSCT) from a related donor.
All patients engrafted. The median time to neutrophil recovery was 13 days after HSCT (range, 11 to 19), and the median time to platelet recovery was 15 days (range, 11 to 59).
At 100 days post-HSCT, the cumulative incidence of acute grade 2-4 GVHD was 23.1%, and the incidence of acute grade 3-4 GVHD was 15.4%. None of the patients developed acute GVHD beyond day 100.
Among patients who received the 2 higher doses of ApoCell (n=6), the rate of acute grade 2-4 GVHD was 0%.
Ten patients experienced serious adverse events, but 7 were considered unrelated to ApoCell, and 3 were likely unrelated to the treatment.
The nonrelapse mortality rate was 7.7% at both 100 and 180 days after HSCT. The overall survival rate was 92% at 100 days and 85% at 180 days.
The researchers said these results suggest a single infusion of ApoCell is safe and may effectively prevent acute GVHD. ![]()
NICE recommends rivaroxaban for ACS
Credit: NHS
In its final draft guidance, the UK’s National Institute for Health and Care Excellence (NICE) is recommending rivaroxaban (Xarelto) as an option for preventing adverse outcomes in patients with acute coronary syndromes (ACS).
The agency said it supports use of the factor Xa inhibitor in combination with aspirin and clopidogrel or aspirin alone to prevent atherothrombotic events in ACS patients with elevated cardiac biomarkers.
This includes patients who have had ST-segment-elevation myocardial infarctions (STEMIs) or non-ST-segment myocardial infarctions (NSTEMIs) but not unstable angina. In unstable angina, damage to the heart is not severe enough to result in the release of biomarkers into the blood, so this condition is not included in the guidance.
“Because rivaroxaban is associated with a higher risk of causing bleeding than clopidogrel in combination with aspirin or aspirin alone, the draft guidance recommends that, before starting treatment, doctors should carry out a careful assessment of a person’s bleeding risk,” said Carole Longson, NICE Health Technology Evaluation Centre Director.
“The decision to start treatment should be made after an informed discussion between the doctor and patient about the benefits and risks of rivaroxaban. Also, because there is limited experience of treatment with rivaroxaban up to 24 months, the draft guidance recommends careful consideration should be given to whether treatment is continued beyond 12 months.”
The final draft guidance is now with consultees, who have the opportunity to appeal against it. Once NICE issues its final guidance on a technology, it replaces local recommendations.
Cost and clinical effectiveness
An appraisal committee advising NICE concluded that rivaroxaban given at 2.5 mg twice daily in combination with aspirin plus clopidogrel or with aspirin alone was more effective than aspirin plus clopidogrel or aspirin alone for preventing further cardiovascular deaths and myocardial infarction in patients with ACS and raised cardiac biomarkers.
The committee also found rivaroxaban to be cost-effective. According to Bayer Healthcare (the company co-developing rivaroxaban with Janssen Research & Development, LLC), the base-case incremental cost-effectiveness ratio (ICER) was £6203 per quality-adjusted life-year (QALY) gained. The evidence review group’s preferred base-case estimate was £5622 per QALY gained.
The committee said there is uncertainty about the validity of these results, which were based on the ATLAS-ACS 2-TIMI 51 trial, because of the risk of bias resulting from missing trial data and informative censoring.
However, the committee considered that the ICERs presented were all within the range that could be considered cost-effective, and adjusting for the various types of bias that might have occurred was unlikely to increase the ICER to the extent that it would become unacceptable.
The list price of rivaroxaban is £58.88 per 2.5 mg, 56-capsule pack (excluding value-added tax). The license dose is 2.5 mg twice daily, which equates to a price of £2.10 per day.
Assuming a treatment duration of 12 months, total acquisition costs are £766.50. Costs may vary in different settings because of negotiated procurement discounts. ![]()
Credit: NHS
In its final draft guidance, the UK’s National Institute for Health and Care Excellence (NICE) is recommending rivaroxaban (Xarelto) as an option for preventing adverse outcomes in patients with acute coronary syndromes (ACS).
The agency said it supports use of the factor Xa inhibitor in combination with aspirin and clopidogrel or aspirin alone to prevent atherothrombotic events in ACS patients with elevated cardiac biomarkers.
This includes patients who have had ST-segment-elevation myocardial infarctions (STEMIs) or non-ST-segment myocardial infarctions (NSTEMIs) but not unstable angina. In unstable angina, damage to the heart is not severe enough to result in the release of biomarkers into the blood, so this condition is not included in the guidance.
“Because rivaroxaban is associated with a higher risk of causing bleeding than clopidogrel in combination with aspirin or aspirin alone, the draft guidance recommends that, before starting treatment, doctors should carry out a careful assessment of a person’s bleeding risk,” said Carole Longson, NICE Health Technology Evaluation Centre Director.
“The decision to start treatment should be made after an informed discussion between the doctor and patient about the benefits and risks of rivaroxaban. Also, because there is limited experience of treatment with rivaroxaban up to 24 months, the draft guidance recommends careful consideration should be given to whether treatment is continued beyond 12 months.”
The final draft guidance is now with consultees, who have the opportunity to appeal against it. Once NICE issues its final guidance on a technology, it replaces local recommendations.
Cost and clinical effectiveness
An appraisal committee advising NICE concluded that rivaroxaban given at 2.5 mg twice daily in combination with aspirin plus clopidogrel or with aspirin alone was more effective than aspirin plus clopidogrel or aspirin alone for preventing further cardiovascular deaths and myocardial infarction in patients with ACS and raised cardiac biomarkers.
The committee also found rivaroxaban to be cost-effective. According to Bayer Healthcare (the company co-developing rivaroxaban with Janssen Research & Development, LLC), the base-case incremental cost-effectiveness ratio (ICER) was £6203 per quality-adjusted life-year (QALY) gained. The evidence review group’s preferred base-case estimate was £5622 per QALY gained.
The committee said there is uncertainty about the validity of these results, which were based on the ATLAS-ACS 2-TIMI 51 trial, because of the risk of bias resulting from missing trial data and informative censoring.
However, the committee considered that the ICERs presented were all within the range that could be considered cost-effective, and adjusting for the various types of bias that might have occurred was unlikely to increase the ICER to the extent that it would become unacceptable.
The list price of rivaroxaban is £58.88 per 2.5 mg, 56-capsule pack (excluding value-added tax). The license dose is 2.5 mg twice daily, which equates to a price of £2.10 per day.
Assuming a treatment duration of 12 months, total acquisition costs are £766.50. Costs may vary in different settings because of negotiated procurement discounts. ![]()
Credit: NHS
In its final draft guidance, the UK’s National Institute for Health and Care Excellence (NICE) is recommending rivaroxaban (Xarelto) as an option for preventing adverse outcomes in patients with acute coronary syndromes (ACS).
The agency said it supports use of the factor Xa inhibitor in combination with aspirin and clopidogrel or aspirin alone to prevent atherothrombotic events in ACS patients with elevated cardiac biomarkers.
This includes patients who have had ST-segment-elevation myocardial infarctions (STEMIs) or non-ST-segment myocardial infarctions (NSTEMIs) but not unstable angina. In unstable angina, damage to the heart is not severe enough to result in the release of biomarkers into the blood, so this condition is not included in the guidance.
“Because rivaroxaban is associated with a higher risk of causing bleeding than clopidogrel in combination with aspirin or aspirin alone, the draft guidance recommends that, before starting treatment, doctors should carry out a careful assessment of a person’s bleeding risk,” said Carole Longson, NICE Health Technology Evaluation Centre Director.
“The decision to start treatment should be made after an informed discussion between the doctor and patient about the benefits and risks of rivaroxaban. Also, because there is limited experience of treatment with rivaroxaban up to 24 months, the draft guidance recommends careful consideration should be given to whether treatment is continued beyond 12 months.”
The final draft guidance is now with consultees, who have the opportunity to appeal against it. Once NICE issues its final guidance on a technology, it replaces local recommendations.
Cost and clinical effectiveness
An appraisal committee advising NICE concluded that rivaroxaban given at 2.5 mg twice daily in combination with aspirin plus clopidogrel or with aspirin alone was more effective than aspirin plus clopidogrel or aspirin alone for preventing further cardiovascular deaths and myocardial infarction in patients with ACS and raised cardiac biomarkers.
The committee also found rivaroxaban to be cost-effective. According to Bayer Healthcare (the company co-developing rivaroxaban with Janssen Research & Development, LLC), the base-case incremental cost-effectiveness ratio (ICER) was £6203 per quality-adjusted life-year (QALY) gained. The evidence review group’s preferred base-case estimate was £5622 per QALY gained.
The committee said there is uncertainty about the validity of these results, which were based on the ATLAS-ACS 2-TIMI 51 trial, because of the risk of bias resulting from missing trial data and informative censoring.
However, the committee considered that the ICERs presented were all within the range that could be considered cost-effective, and adjusting for the various types of bias that might have occurred was unlikely to increase the ICER to the extent that it would become unacceptable.
The list price of rivaroxaban is £58.88 per 2.5 mg, 56-capsule pack (excluding value-added tax). The license dose is 2.5 mg twice daily, which equates to a price of £2.10 per day.
Assuming a treatment duration of 12 months, total acquisition costs are £766.50. Costs may vary in different settings because of negotiated procurement discounts. ![]()
Deaths in young leukemia patients on decline in UK

Credit: Bill Branson
Cancer deaths in children, adolescents, and young adults in the UK have fallen by 58% in the past 40 years, according to new figures from Cancer Research UK.*
Among cancer patients age 24 and under, the number of deaths each year dropped from about 1300 in the mid-1970s (1975-1977) to around 550 in more recent years (2010-2012).
And patients with leukemia have seen the steepest decline in mortality.
This is key because, according to Cancer Research UK, leukemia is the most commonly diagnosed cancer in children aged 14 and under and the sixth most commonly diagnosed cancer among 15- to 24-year-olds.
“These figures are testament to the real progress we’re making in treating children and young people with cancer,” said Pam Kearns, PhD, director of the Cancer Research UK Clinical Trials Unit in Birmingham.
“But hundreds of young people are dying from cancer each year in the UK, which means there’s still much more we need to do.”
Around 1600 children and 2200 adolescents and young adults are diagnosed with cancer every year in the UK.
And cancer remains the biggest killer of children and young people in the UK. It is the most common cause of death in children and young women aged 15 to 24 and the fourth most common cause of death in young men aged 15 to 24.
Still, the latest figures from Cancer Research UK show progress.
The mortality rate for all cancer patients ages 0 to 24 fell from 63.7 per million in 1975-1977 to 36.7 per million in 2001-2003 and then to 27.9 per million in 2010-2012. So the mortality rate has fallen 56% since 1975 and 24% in the last decade.
The average number of cancer deaths among children age 0 to 24 fell from 1325 in 1975-1977 to 681 in 2001-2003 and then to 550 in 2010-2012. That translates to a 58% decrease in deaths from 1975 and a 19% decrease in the last decade.
For leukemia patients ages 0 to 24, the mortality rate fell from 9.6 per million in 2001-2003 to 5.6 per million in 2010-2012, which translates to a 42% decrease. The annual average number of deaths dropped from 179 in 2001-2003 to 110 in 2010-2012, which translates to a 39% decrease.
“Cancer causes more deaths among children and young people than any other disease in the UK, so it’s hugely encouraging to see that death toll now falling steadily,” said Harpal Kumar, DSc, Cancer Research UK’s chief executive.
“But as the largest funder of research into children’s cancers in the UK, we will keep going until no young lives are lost to cancer.”
To that end, Cancer Research UK has launched “Cancer Research UK Kids & Teens,” an ongoing campaign to fund more research to find better treatments for young cancer patients.
“Money raised by ‘Cancer Research UK Kids & Teens’ will be restricted to research into cancers affecting children, teenagers, and young adults, enabling us to better understand these cancers and find better and kinder treatments and cures,” Dr Kumar said. “In the next 5 to 10 years, Cancer Research UK hopes to double the amount it spends on these cancers.” ![]()
*Some of the information in this article is not available on the Cancer Research UK website (and was sent to HematologyTimes upon request), but similar data and additional details are available on the organization’s Cancer Stats page.
Credit: Bill Branson
Cancer deaths in children, adolescents, and young adults in the UK have fallen by 58% in the past 40 years, according to new figures from Cancer Research UK.*
Among cancer patients age 24 and under, the number of deaths each year dropped from about 1300 in the mid-1970s (1975-1977) to around 550 in more recent years (2010-2012).
And patients with leukemia have seen the steepest decline in mortality.
This is key because, according to Cancer Research UK, leukemia is the most commonly diagnosed cancer in children aged 14 and under and the sixth most commonly diagnosed cancer among 15- to 24-year-olds.
“These figures are testament to the real progress we’re making in treating children and young people with cancer,” said Pam Kearns, PhD, director of the Cancer Research UK Clinical Trials Unit in Birmingham.
“But hundreds of young people are dying from cancer each year in the UK, which means there’s still much more we need to do.”
Around 1600 children and 2200 adolescents and young adults are diagnosed with cancer every year in the UK.
And cancer remains the biggest killer of children and young people in the UK. It is the most common cause of death in children and young women aged 15 to 24 and the fourth most common cause of death in young men aged 15 to 24.
Still, the latest figures from Cancer Research UK show progress.
The mortality rate for all cancer patients ages 0 to 24 fell from 63.7 per million in 1975-1977 to 36.7 per million in 2001-2003 and then to 27.9 per million in 2010-2012. So the mortality rate has fallen 56% since 1975 and 24% in the last decade.
The average number of cancer deaths among children age 0 to 24 fell from 1325 in 1975-1977 to 681 in 2001-2003 and then to 550 in 2010-2012. That translates to a 58% decrease in deaths from 1975 and a 19% decrease in the last decade.
For leukemia patients ages 0 to 24, the mortality rate fell from 9.6 per million in 2001-2003 to 5.6 per million in 2010-2012, which translates to a 42% decrease. The annual average number of deaths dropped from 179 in 2001-2003 to 110 in 2010-2012, which translates to a 39% decrease.
“Cancer causes more deaths among children and young people than any other disease in the UK, so it’s hugely encouraging to see that death toll now falling steadily,” said Harpal Kumar, DSc, Cancer Research UK’s chief executive.
“But as the largest funder of research into children’s cancers in the UK, we will keep going until no young lives are lost to cancer.”
To that end, Cancer Research UK has launched “Cancer Research UK Kids & Teens,” an ongoing campaign to fund more research to find better treatments for young cancer patients.
“Money raised by ‘Cancer Research UK Kids & Teens’ will be restricted to research into cancers affecting children, teenagers, and young adults, enabling us to better understand these cancers and find better and kinder treatments and cures,” Dr Kumar said. “In the next 5 to 10 years, Cancer Research UK hopes to double the amount it spends on these cancers.” ![]()
*Some of the information in this article is not available on the Cancer Research UK website (and was sent to HematologyTimes upon request), but similar data and additional details are available on the organization’s Cancer Stats page.
Credit: Bill Branson
Cancer deaths in children, adolescents, and young adults in the UK have fallen by 58% in the past 40 years, according to new figures from Cancer Research UK.*
Among cancer patients age 24 and under, the number of deaths each year dropped from about 1300 in the mid-1970s (1975-1977) to around 550 in more recent years (2010-2012).
And patients with leukemia have seen the steepest decline in mortality.
This is key because, according to Cancer Research UK, leukemia is the most commonly diagnosed cancer in children aged 14 and under and the sixth most commonly diagnosed cancer among 15- to 24-year-olds.
“These figures are testament to the real progress we’re making in treating children and young people with cancer,” said Pam Kearns, PhD, director of the Cancer Research UK Clinical Trials Unit in Birmingham.
“But hundreds of young people are dying from cancer each year in the UK, which means there’s still much more we need to do.”
Around 1600 children and 2200 adolescents and young adults are diagnosed with cancer every year in the UK.
And cancer remains the biggest killer of children and young people in the UK. It is the most common cause of death in children and young women aged 15 to 24 and the fourth most common cause of death in young men aged 15 to 24.
Still, the latest figures from Cancer Research UK show progress.
The mortality rate for all cancer patients ages 0 to 24 fell from 63.7 per million in 1975-1977 to 36.7 per million in 2001-2003 and then to 27.9 per million in 2010-2012. So the mortality rate has fallen 56% since 1975 and 24% in the last decade.
The average number of cancer deaths among children age 0 to 24 fell from 1325 in 1975-1977 to 681 in 2001-2003 and then to 550 in 2010-2012. That translates to a 58% decrease in deaths from 1975 and a 19% decrease in the last decade.
For leukemia patients ages 0 to 24, the mortality rate fell from 9.6 per million in 2001-2003 to 5.6 per million in 2010-2012, which translates to a 42% decrease. The annual average number of deaths dropped from 179 in 2001-2003 to 110 in 2010-2012, which translates to a 39% decrease.
“Cancer causes more deaths among children and young people than any other disease in the UK, so it’s hugely encouraging to see that death toll now falling steadily,” said Harpal Kumar, DSc, Cancer Research UK’s chief executive.
“But as the largest funder of research into children’s cancers in the UK, we will keep going until no young lives are lost to cancer.”
To that end, Cancer Research UK has launched “Cancer Research UK Kids & Teens,” an ongoing campaign to fund more research to find better treatments for young cancer patients.
“Money raised by ‘Cancer Research UK Kids & Teens’ will be restricted to research into cancers affecting children, teenagers, and young adults, enabling us to better understand these cancers and find better and kinder treatments and cures,” Dr Kumar said. “In the next 5 to 10 years, Cancer Research UK hopes to double the amount it spends on these cancers.”
*Some of the information in this article is not available on the Cancer Research UK website (and was sent to HematologyTimes upon request), but similar data and additional details are available on the organization’s Cancer Stats page.
EMA wants to suspend drugs due to data manipulation

Credit: Steven Harbour
The European Medicines Agency (EMA) has recommended suspending a number of drugs that were approved in the European Union (EU) based on clinical studies conducted at GVK Biosciences in Hyderabad, India.
An inspection of the site suggested GVK Bio employees may have manipulated data from trials that took place there.
So the EMA compiled a list of drugs—which includes clopidogrel, dexamethasone, and tacrolimus, among others—that should be suspended.
However, the EMA said there is no evidence of harm or lack of effectiveness linked to the conduct of studies by GVK Bio, and patients should continue taking their medicines as prescribed.
The EMA’s opinion has been forwarded to the European Commission (EC), which will adopt a legally binding decision.
It was at the request of the EC that the EMA’s Committee for Medicinal Products for Human Use (CHMP) reviewed the drugs set to be suspended.
An inspection by the French medicines agency, Agence nationale de sécurité du médicament et des produits de santé (ANSM), in May 2014 uncovered “non-compliance with good clinical practice” at the GVK Bio site in Hyderabad.
The ANSM inspector analyzed 9 studies conducted there from 2008 to 2013 and found evidence suggesting that data from electrocardiograms (ECGs) had been manipulated. It appeared that GVK Bio employees were taking multiple ECGs of one volunteer and presenting them as ECGs of other volunteers.
The EMA said the systematic nature of these data manipulations, the extended period of time during which they took place, and the number of staff members involved casts doubt on the integrity of the way trials were performed at the Hyderabad facility and on the reliability of data generated there.
With this in mind, the CHMP looked at more than 1000 pharmaceutical forms and strengths of medicines studied at the site. For more than 300 of the medications, there was sufficient data from other sources to support the drugs’ authorization, so these will remain on the market in the EU.
For drugs that lack data from other studies, the CHMP recommended suspension unless they are of critical importance for patients because alternatives will not be able to meet patients’ needs.
Whether a medicine is critical for patients will be determined by the national authorities of EU member states, depending on the situation in their country. For drugs that are considered critical, companies developing those drugs will be given 12 months to submit additional data.
The CHMP’s recommendation will be sent to the EC for a legally binding decision that will apply to all EU member states whether or not they have taken interim measures to suspend medicines.
GVK Bio responds
GVK Bio said an internal audit conducted following the ANSM inspection suggested that standard operating procedures were followed for the 9 trials analyzed. The organization also sought the opinion of 4 independent cardiologists, who said it was difficult to determine if the ECGs belong to the same volunteer or more than one person.
GVK Bio presented this information to the ANSM, but the agency concluded that the studies did not meet good clinical practice guidelines and should be rejected.
Following subsequent meetings and analyses, the EMA came to a similar conclusion—that the overall findings cast doubt on the results of trials conducted at the Hyderabad facility from 2008 to 2014.
GVK Bio said it respects the EMA’s decision, but their recommended suspension of drugs is “unprecedented and highly disproportional” for a few reasons.
First, the ANSM said the ECGs in question were not essential to demonstrate the bioequivalence of the drugs tested, and the agency’s recommendation to reject the 9 studies was a “precautionary” measure.
The ANSM also said the observations made during the inspection should not be extrapolated to other trial-related activities at the Hyderabad site or GVK Bio’s other site in Ahmedabad.
And finally, the EMA itself said there is no evidence proving that the drugs in question are ineffective or pose a risk to human health.
Credit: Steven Harbour
The European Medicines Agency (EMA) has recommended suspending a number of drugs that were approved in the European Union (EU) based on clinical studies conducted at GVK Biosciences in Hyderabad, India.
An inspection of the site suggested GVK Bio employees may have manipulated data from trials that took place there.
So the EMA compiled a list of drugs—which includes clopidogrel, dexamethasone, and tacrolimus, among others—that should be suspended.
However, the EMA said there is no evidence of harm or lack of effectiveness linked to the conduct of studies by GVK Bio, and patients should continue taking their medicines as prescribed.
The EMA’s opinion has been forwarded to the European Commission (EC), which will adopt a legally binding decision.
It was at the request of the EC that the EMA’s Committee for Medicinal Products for Human Use (CHMP) reviewed the drugs set to be suspended.
An inspection by the French medicines agency, Agence nationale de sécurité du médicament et des produits de santé (ANSM), in May 2014 uncovered “non-compliance with good clinical practice” at the GVK Bio site in Hyderabad.
The ANSM inspector analyzed 9 studies conducted there from 2008 to 2013 and found evidence suggesting that data from electrocardiograms (ECGs) had been manipulated. It appeared that GVK Bio employees were taking multiple ECGs of one volunteer and presenting them as ECGs of other volunteers.
The EMA said the systematic nature of these data manipulations, the extended period of time during which they took place, and the number of staff members involved casts doubt on the integrity of the way trials were performed at the Hyderabad facility and on the reliability of data generated there.
With this in mind, the CHMP looked at more than 1000 pharmaceutical forms and strengths of medicines studied at the site. For more than 300 of the medications, there was sufficient data from other sources to support the drugs’ authorization, so these will remain on the market in the EU.
For drugs that lack data from other studies, the CHMP recommended suspension unless they are of critical importance for patients because alternatives will not be able to meet patients’ needs.
Whether a medicine is critical for patients will be determined by the national authorities of EU member states, depending on the situation in their country. For drugs that are considered critical, companies developing those drugs will be given 12 months to submit additional data.
The CHMP’s recommendation will be sent to the EC for a legally binding decision that will apply to all EU member states whether or not they have taken interim measures to suspend medicines.
GVK Bio responds
GVK Bio said an internal audit conducted following the ANSM inspection suggested that standard operating procedures were followed for the 9 trials analyzed. The organization also sought the opinion of 4 independent cardiologists, who said it was difficult to determine if the ECGs belong to the same volunteer or more than one person.
GVK Bio presented this information to the ANSM, but the agency concluded that the studies did not meet good clinical practice guidelines and should be rejected.
Following subsequent meetings and analyses, the EMA came to a similar conclusion—that the overall findings cast doubt on the results of trials conducted at the Hyderabad facility from 2008 to 2014.
GVK Bio said it respects the EMA’s decision, but their recommended suspension of drugs is “unprecedented and highly disproportional” for a few reasons.
First, the ANSM said the ECGs in question were not essential to demonstrate the bioequivalence of the drugs tested, and the agency’s recommendation to reject the 9 studies was a “precautionary” measure.
The ANSM also said the observations made during the inspection should not be extrapolated to other trial-related activities at the Hyderabad site or GVK Bio’s other site in Ahmedabad.
And finally, the EMA itself said there is no evidence proving that the drugs in question are ineffective or pose a risk to human health.
Credit: Steven Harbour
The European Medicines Agency (EMA) has recommended suspending a number of drugs that were approved in the European Union (EU) based on clinical studies conducted at GVK Biosciences in Hyderabad, India.
An inspection of the site suggested GVK Bio employees may have manipulated data from trials that took place there.
So the EMA compiled a list of drugs—which includes clopidogrel, dexamethasone, and tacrolimus, among others—that should be suspended.
However, the EMA said there is no evidence of harm or lack of effectiveness linked to the conduct of studies by GVK Bio, and patients should continue taking their medicines as prescribed.
The EMA’s opinion has been forwarded to the European Commission (EC), which will adopt a legally binding decision.
It was at the request of the EC that the EMA’s Committee for Medicinal Products for Human Use (CHMP) reviewed the drugs set to be suspended.
An inspection by the French medicines agency, Agence nationale de sécurité du médicament et des produits de santé (ANSM), in May 2014 uncovered “non-compliance with good clinical practice” at the GVK Bio site in Hyderabad.
The ANSM inspector analyzed 9 studies conducted there from 2008 to 2013 and found evidence suggesting that data from electrocardiograms (ECGs) had been manipulated. It appeared that GVK Bio employees were taking multiple ECGs of one volunteer and presenting them as ECGs of other volunteers.
The EMA said the systematic nature of these data manipulations, the extended period of time during which they took place, and the number of staff members involved casts doubt on the integrity of the way trials were performed at the Hyderabad facility and on the reliability of data generated there.
With this in mind, the CHMP looked at more than 1000 pharmaceutical forms and strengths of medicines studied at the site. For more than 300 of the medications, there was sufficient data from other sources to support the drugs’ authorization, so these will remain on the market in the EU.
For drugs that lack data from other studies, the CHMP recommended suspension unless they are of critical importance for patients because alternatives will not be able to meet patients’ needs.
Whether a medicine is critical for patients will be determined by the national authorities of EU member states, depending on the situation in their country. For drugs that are considered critical, companies developing those drugs will be given 12 months to submit additional data.
The CHMP’s recommendation will be sent to the EC for a legally binding decision that will apply to all EU member states whether or not they have taken interim measures to suspend medicines.
GVK Bio responds
GVK Bio said an internal audit conducted following the ANSM inspection suggested that standard operating procedures were followed for the 9 trials analyzed. The organization also sought the opinion of 4 independent cardiologists, who said it was difficult to determine if the ECGs belong to the same volunteer or more than one person.
GVK Bio presented this information to the ANSM, but the agency concluded that the studies did not meet good clinical practice guidelines and should be rejected.
Following subsequent meetings and analyses, the EMA came to a similar conclusion—that the overall findings cast doubt on the results of trials conducted at the Hyderabad facility from 2008 to 2014.
GVK Bio said it respects the EMA’s decision, but their recommended suspension of drugs is “unprecedented and highly disproportional” for a few reasons.
First, the ANSM said the ECGs in question were not essential to demonstrate the bioequivalence of the drugs tested, and the agency’s recommendation to reject the 9 studies was a “precautionary” measure.
The ANSM also said the observations made during the inspection should not be extrapolated to other trial-related activities at the Hyderabad site or GVK Bio’s other site in Ahmedabad.
And finally, the EMA itself said there is no evidence proving that the drugs in question are ineffective or pose a risk to human health.
CHMP recommends products for thrombosis, hemostasis
Credit: Piotr Bodzek
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) is recommending approval for the antiplatelet agent cangrelor (Kengrexal) and a hemostatic sealant powder to be marketed as Raplixa (formerly known as Fibrocaps).
Cangrelor is an intravenous antiplatelet agent that provides immediate and reversible P2Y12 inhibition. It is intended to prevent thrombosis in the acute care setting.
The sealant powder is a ready-to-use, biologically active, powdered fibrin sealant that provides hemostasis in a range of bleeding settings. It is intended for use in patients undergoing surgery.
Neither product has been approved for commercial use in any market, and both are under review by the US Food and Drug Administration as well as the European Medicines Agency/European Commission.
The European Commission generally follows CHMP recommendations for marketing authorizations and delivers its final decision within 3 months of the CHMP recommendation. Decisions are applicable to all 28 member states of the European Union, plus Iceland, Norway, and Liechtenstein.
About cangrelor
Cangrelor is intended to prevent platelet activation and aggregation that leads to thrombosis in the acute care setting, particularly in patients undergoing percutaneous coronary intervention (PCI).
The CHMP is recommending cangrelor for European marketing authorization based on results from the CHAMPION PHOENIX trial, which was funded by the drug’s developer, The Medicines Company.
This phase 3, randomized, double-blind trial was a comparison of cangrelor and oral clopidogrel in 11,145 patients undergoing PCI.
In the initial analysis of the trial data, researchers found that cangrelor reduced the overall odds of complications from stenting procedures—including death, myocardial infarction, ischemia-driven revascularization, and stent thrombosis—compared to clopidogrel.
However, cangrelor also increased the risk of major and minor bleeding, as well as transient dyspnea.
Results of a second analysis suggested cangrelor can reduce the risk of stent thrombosis alone in patients undergoing PCI, when compared to clopidogrel. Cangrelor was an independent predictor of freedom from stent thrombosis at 30 days after PCI.
About Raplixa
This sealant powder is a mixture of 2 essential blood clotting proteins, fibrinogen and thrombin, formulated as a dry powder for topical use. It is intended to aid hemostasis during surgery.
The CHMP is recommending Raplixa (formerly Fibrocaps) for European marketing authorization based on results of the phase 3 FINISH-3 trial, which was funded by ProFibrix, Inc., the company that was developing the product at the time. (The Medicines Company recently purchased all the outstanding equity of ProFibrix.)
In this randomized, single-blind, controlled trial, researchers compared Raplixa administered with a gelatin sponge to the gelatin sponge alone as a hemostat for surgical bleeding in 4 indications: spinal, hepatic, vascular, and soft tissue dissection.
Raplixa significantly reduced the median time to hemostasis and the restricted mean time to hemostasis, compared with the gelatin sponge alone, for all 4 indications. Raplixa also significantly increased the probability of hemostasis at 3- and 5-minute time points.
The incidence of adverse events was generally similar between the treatment arms. However, non-neutralizing, anti-thrombin antibodies were slightly more common in the sponge-alone arm.
Credit: Piotr Bodzek
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) is recommending approval for the antiplatelet agent cangrelor (Kengrexal) and a hemostatic sealant powder to be marketed as Raplixa (formerly known as Fibrocaps).
Cangrelor is an intravenous antiplatelet agent that provides immediate and reversible P2Y12 inhibition. It is intended to prevent thrombosis in the acute care setting.
The sealant powder is a ready-to-use, biologically active, powdered fibrin sealant that provides hemostasis in a range of bleeding settings. It is intended for use in patients undergoing surgery.
Neither product has been approved for commercial use in any market, and both are under review by the US Food and Drug Administration as well as the European Medicines Agency/European Commission.
The European Commission generally follows CHMP recommendations for marketing authorizations and delivers its final decision within 3 months of the CHMP recommendation. Decisions are applicable to all 28 member states of the European Union, plus Iceland, Norway, and Liechtenstein.
About cangrelor
Cangrelor is intended to prevent platelet activation and aggregation that leads to thrombosis in the acute care setting, particularly in patients undergoing percutaneous coronary intervention (PCI).
The CHMP is recommending cangrelor for European marketing authorization based on results from the CHAMPION PHOENIX trial, which was funded by the drug’s developer, The Medicines Company.
This phase 3, randomized, double-blind trial was a comparison of cangrelor and oral clopidogrel in 11,145 patients undergoing PCI.
In the initial analysis of the trial data, researchers found that cangrelor reduced the overall odds of complications from stenting procedures—including death, myocardial infarction, ischemia-driven revascularization, and stent thrombosis—compared to clopidogrel.
However, cangrelor also increased the risk of major and minor bleeding, as well as transient dyspnea.
Results of a second analysis suggested cangrelor can reduce the risk of stent thrombosis alone in patients undergoing PCI, when compared to clopidogrel. Cangrelor was an independent predictor of freedom from stent thrombosis at 30 days after PCI.
About Raplixa
This sealant powder is a mixture of 2 essential blood clotting proteins, fibrinogen and thrombin, formulated as a dry powder for topical use. It is intended to aid hemostasis during surgery.
The CHMP is recommending Raplixa (formerly Fibrocaps) for European marketing authorization based on results of the phase 3 FINISH-3 trial, which was funded by ProFibrix, Inc., the company that was developing the product at the time. (The Medicines Company recently purchased all the outstanding equity of ProFibrix.)
In this randomized, single-blind, controlled trial, researchers compared Raplixa administered with a gelatin sponge to the gelatin sponge alone as a hemostat for surgical bleeding in 4 indications: spinal, hepatic, vascular, and soft tissue dissection.
Raplixa significantly reduced the median time to hemostasis and the restricted mean time to hemostasis, compared with the gelatin sponge alone, for all 4 indications. Raplixa also significantly increased the probability of hemostasis at 3- and 5-minute time points.
The incidence of adverse events was generally similar between the treatment arms. However, non-neutralizing, anti-thrombin antibodies were slightly more common in the sponge-alone arm.
Credit: Piotr Bodzek
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) is recommending approval for the antiplatelet agent cangrelor (Kengrexal) and a hemostatic sealant powder to be marketed as Raplixa (formerly known as Fibrocaps).
Cangrelor is an intravenous antiplatelet agent that provides immediate and reversible P2Y12 inhibition. It is intended to prevent thrombosis in the acute care setting.
The sealant powder is a ready-to-use, biologically active, powdered fibrin sealant that provides hemostasis in a range of bleeding settings. It is intended for use in patients undergoing surgery.
Neither product has been approved for commercial use in any market, and both are under review by the US Food and Drug Administration as well as the European Medicines Agency/European Commission.
The European Commission generally follows CHMP recommendations for marketing authorizations and delivers its final decision within 3 months of the CHMP recommendation. Decisions are applicable to all 28 member states of the European Union, plus Iceland, Norway, and Liechtenstein.
About cangrelor
Cangrelor is intended to prevent platelet activation and aggregation that leads to thrombosis in the acute care setting, particularly in patients undergoing percutaneous coronary intervention (PCI).
The CHMP is recommending cangrelor for European marketing authorization based on results from the CHAMPION PHOENIX trial, which was funded by the drug’s developer, The Medicines Company.
This phase 3, randomized, double-blind trial was a comparison of cangrelor and oral clopidogrel in 11,145 patients undergoing PCI.
In the initial analysis of the trial data, researchers found that cangrelor reduced the overall odds of complications from stenting procedures—including death, myocardial infarction, ischemia-driven revascularization, and stent thrombosis—compared to clopidogrel.
However, cangrelor also increased the risk of major and minor bleeding, as well as transient dyspnea.
Results of a second analysis suggested cangrelor can reduce the risk of stent thrombosis alone in patients undergoing PCI, when compared to clopidogrel. Cangrelor was an independent predictor of freedom from stent thrombosis at 30 days after PCI.
About Raplixa
This sealant powder is a mixture of 2 essential blood clotting proteins, fibrinogen and thrombin, formulated as a dry powder for topical use. It is intended to aid hemostasis during surgery.
The CHMP is recommending Raplixa (formerly Fibrocaps) for European marketing authorization based on results of the phase 3 FINISH-3 trial, which was funded by ProFibrix, Inc., the company that was developing the product at the time. (The Medicines Company recently purchased all the outstanding equity of ProFibrix.)
In this randomized, single-blind, controlled trial, researchers compared Raplixa administered with a gelatin sponge to the gelatin sponge alone as a hemostat for surgical bleeding in 4 indications: spinal, hepatic, vascular, and soft tissue dissection.
Raplixa significantly reduced the median time to hemostasis and the restricted mean time to hemostasis, compared with the gelatin sponge alone, for all 4 indications. Raplixa also significantly increased the probability of hemostasis at 3- and 5-minute time points.
The incidence of adverse events was generally similar between the treatment arms. However, non-neutralizing, anti-thrombin antibodies were slightly more common in the sponge-alone arm.
CHMP recommends ruxolitinib for PV

Credit: Zak Hubbard
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) is recommending the approval of ruxolitinib (Jakavi) to
treat adults with polycythemia vera (PV) who are resistant to or cannot tolerate hydroxyurea.
The European Commission generally follows the CHMP’s advice and delivers its final decision within 3 months of the CHMP recommendation.
If approved, ruxolitinib would be the first targeted treatment option for PV patients in Europe.
The European Commission’s decision will be applicable to all 28 member states of the European Union, plus Iceland, Norway, and Liechtenstein.
About ruxolitinib
Ruxolitinib is an oral inhibitor of the JAK 1 and JAK 2 tyrosine kinases. It was approved by the European Commission in August 2012 to treat adults with primary myelofibrosis (MF), post-PV MF, or post-essential thrombocythemia MF. The drug is approved to treat MF in more than 70 countries.
Novartis licensed ruxolitinib from Incyte Corporation (which markets the drug as Jakafi in the US) for development and commercialization outside the US. Ruxolitinib is approved in the US to treat patients with MF and those with PV.
Ruxolitinib in PV: The RESPONSE trial
The CHMP’s recommendation to approve ruxolitinib for PV was based on results from the phase 3 RESPONSE trial, which was funded by Incyte.
The trial included 222 patients who had PV for at least 24 weeks. All patients had an inadequate response to or could not tolerate hydroxyurea, had undergone a phlebotomy, and had an enlarged spleen.
They were randomized to receive ruxolitinib starting at 10 mg twice daily or best available therapy (BAT) as determined by the investigator. The ruxolitinib dose was adjusted as needed.
The study was designed to measure the reduced need for phlebotomy beginning at week 8 and continuing through week 32, in addition to at least a 35% reduction in spleen volume at week 32.
Twenty-one percent of ruxolitinib-treated patients met this endpoint, compared to 1% of patients who received BAT.
Ruxolitinib was generally well-tolerated, but 3.6% of patients discontinued treatment due to adverse events, compared to 1.8% of patients on BAT.
The most common hematologic adverse events associated with ruxolitinib were anemia and thrombocytopenia. The most common non-hematologic events were headache, diarrhea, fatigue, dizziness, constipation, and shingles.
Credit: Zak Hubbard
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) is recommending the approval of ruxolitinib (Jakavi) to
treat adults with polycythemia vera (PV) who are resistant to or cannot tolerate hydroxyurea.
The European Commission generally follows the CHMP’s advice and delivers its final decision within 3 months of the CHMP recommendation.
If approved, ruxolitinib would be the first targeted treatment option for PV patients in Europe.
The European Commission’s decision will be applicable to all 28 member states of the European Union, plus Iceland, Norway, and Liechtenstein.
About ruxolitinib
Ruxolitinib is an oral inhibitor of the JAK 1 and JAK 2 tyrosine kinases. It was approved by the European Commission in August 2012 to treat adults with primary myelofibrosis (MF), post-PV MF, or post-essential thrombocythemia MF. The drug is approved to treat MF in more than 70 countries.
Novartis licensed ruxolitinib from Incyte Corporation (which markets the drug as Jakafi in the US) for development and commercialization outside the US. Ruxolitinib is approved in the US to treat patients with MF and those with PV.
Ruxolitinib in PV: The RESPONSE trial
The CHMP’s recommendation to approve ruxolitinib for PV was based on results from the phase 3 RESPONSE trial, which was funded by Incyte.
The trial included 222 patients who had PV for at least 24 weeks. All patients had an inadequate response to or could not tolerate hydroxyurea, had undergone a phlebotomy, and had an enlarged spleen.
They were randomized to receive ruxolitinib starting at 10 mg twice daily or best available therapy (BAT) as determined by the investigator. The ruxolitinib dose was adjusted as needed.
The study was designed to measure the reduced need for phlebotomy beginning at week 8 and continuing through week 32, in addition to at least a 35% reduction in spleen volume at week 32.
Twenty-one percent of ruxolitinib-treated patients met this endpoint, compared to 1% of patients who received BAT.
Ruxolitinib was generally well-tolerated, but 3.6% of patients discontinued treatment due to adverse events, compared to 1.8% of patients on BAT.
The most common hematologic adverse events associated with ruxolitinib were anemia and thrombocytopenia. The most common non-hematologic events were headache, diarrhea, fatigue, dizziness, constipation, and shingles.
Credit: Zak Hubbard
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) is recommending the approval of ruxolitinib (Jakavi) to
treat adults with polycythemia vera (PV) who are resistant to or cannot tolerate hydroxyurea.
The European Commission generally follows the CHMP’s advice and delivers its final decision within 3 months of the CHMP recommendation.
If approved, ruxolitinib would be the first targeted treatment option for PV patients in Europe.
The European Commission’s decision will be applicable to all 28 member states of the European Union, plus Iceland, Norway, and Liechtenstein.
About ruxolitinib
Ruxolitinib is an oral inhibitor of the JAK 1 and JAK 2 tyrosine kinases. It was approved by the European Commission in August 2012 to treat adults with primary myelofibrosis (MF), post-PV MF, or post-essential thrombocythemia MF. The drug is approved to treat MF in more than 70 countries.
Novartis licensed ruxolitinib from Incyte Corporation (which markets the drug as Jakafi in the US) for development and commercialization outside the US. Ruxolitinib is approved in the US to treat patients with MF and those with PV.
Ruxolitinib in PV: The RESPONSE trial
The CHMP’s recommendation to approve ruxolitinib for PV was based on results from the phase 3 RESPONSE trial, which was funded by Incyte.
The trial included 222 patients who had PV for at least 24 weeks. All patients had an inadequate response to or could not tolerate hydroxyurea, had undergone a phlebotomy, and had an enlarged spleen.
They were randomized to receive ruxolitinib starting at 10 mg twice daily or best available therapy (BAT) as determined by the investigator. The ruxolitinib dose was adjusted as needed.
The study was designed to measure the reduced need for phlebotomy beginning at week 8 and continuing through week 32, in addition to at least a 35% reduction in spleen volume at week 32.
Twenty-one percent of ruxolitinib-treated patients met this endpoint, compared to 1% of patients who received BAT.
Ruxolitinib was generally well-tolerated, but 3.6% of patients discontinued treatment due to adverse events, compared to 1.8% of patients on BAT.
The most common hematologic adverse events associated with ruxolitinib were anemia and thrombocytopenia. The most common non-hematologic events were headache, diarrhea, fatigue, dizziness, constipation, and shingles.