User login
Drug incompatible with certain devices, FDA warns
The US Food and Drug Administration (FDA) is warning healthcare professionals not to use Treanda (bendamustine hydrochloride) solution with closed-system transfer devices (CSTD), adapters, and syringes containing polycarbonate or acrylonitrile-butadiene-styrene (ABS).
Most marketed CSTDs contain either polycarbonate or ABS. And these materials dissolve when they come into contact with N, N-dimethylacetamide (DMA), an ingredient in Treanda solution.
This can lead to device failure, possible product contamination, and potential serious adverse health consequences, including skin reactions in healthcare professionals preparing and administering this product and the risk of small blood vessel blockage in patients.
Discovering the incompatibility
Treanda, which is manufactured by Teva, is used to treat patients with chronic lymphocytic leukemia and indolent B-cell non-Hodgkin lymphoma that has progressed during or within 6 months of treatment with rituximab or a rituximab-containing regimen.
Treanda is available as a solution—Treanda Injection (45 mg/0.5 mL or 180 mg/2 mL solution)—and a lyophilized powder—Treanda for Injection (25mg/vial or 100 mg/vial lyophilized powder).
The incompatibility of DMA with polycarbonate and ABS is only an issue with Treanda solution—not the lyophilized powder.
Since December 2014, Teva has received 40 complaints of the incompatibility issue, which was recently brought to the FDA’s attention. The agency also received a notification of device incompatibility with Treanda solution from a pharmacist.
These incompatibility issues included leaking of the CSTD, breaking or operational failure of the CSTD components, and a cloudy appearance or presence of particulate matter in the intravenous bag after dilution. To date, no adverse events have been reported related to the incompatibility.
FDA recommendations
The FDA has required label changes for both the solution and the powder formulations of Treanda to reflect the following safe preparation information.
The agency is recommending that healthcare professionals use Treanda solution only with polypropylene syringes containing a metal needle and a polypropylene hub. Polypropylene syringes are translucent in appearance.
Treanda solution should be inspected visually for particulate matter and discoloration prior to administration whenever the solution and container permit. The solution must be withdrawn and transferred for dilution in a biosafety cabinet or containment isolator.
If they aim to use a CSTD with Treanda solution, healthcare professionals should verify with the CSTD manufacturer or Teva US Medical Information (1-800-896-5855) that the CSTD is compatible with Treanda solution before preparing the drug.
Alternatively, healthcare professionals can use Treanda lyophilized powder with a CSTD. The solution and lyophilized powder formulations of Treanda should not be mixed.
For additional details on safe preparation of Treanda solution and lyophilized powder, see Teva’s Dear Health Care Provider letter.
Adverse events or quality problems associated with the use of Treanda products can be reported to the FDA’s MedWatch Adverse Event Reporting Program. ![]()
The US Food and Drug Administration (FDA) is warning healthcare professionals not to use Treanda (bendamustine hydrochloride) solution with closed-system transfer devices (CSTD), adapters, and syringes containing polycarbonate or acrylonitrile-butadiene-styrene (ABS).
Most marketed CSTDs contain either polycarbonate or ABS. And these materials dissolve when they come into contact with N, N-dimethylacetamide (DMA), an ingredient in Treanda solution.
This can lead to device failure, possible product contamination, and potential serious adverse health consequences, including skin reactions in healthcare professionals preparing and administering this product and the risk of small blood vessel blockage in patients.
Discovering the incompatibility
Treanda, which is manufactured by Teva, is used to treat patients with chronic lymphocytic leukemia and indolent B-cell non-Hodgkin lymphoma that has progressed during or within 6 months of treatment with rituximab or a rituximab-containing regimen.
Treanda is available as a solution—Treanda Injection (45 mg/0.5 mL or 180 mg/2 mL solution)—and a lyophilized powder—Treanda for Injection (25mg/vial or 100 mg/vial lyophilized powder).
The incompatibility of DMA with polycarbonate and ABS is only an issue with Treanda solution—not the lyophilized powder.
Since December 2014, Teva has received 40 complaints of the incompatibility issue, which was recently brought to the FDA’s attention. The agency also received a notification of device incompatibility with Treanda solution from a pharmacist.
These incompatibility issues included leaking of the CSTD, breaking or operational failure of the CSTD components, and a cloudy appearance or presence of particulate matter in the intravenous bag after dilution. To date, no adverse events have been reported related to the incompatibility.
FDA recommendations
The FDA has required label changes for both the solution and the powder formulations of Treanda to reflect the following safe preparation information.
The agency is recommending that healthcare professionals use Treanda solution only with polypropylene syringes containing a metal needle and a polypropylene hub. Polypropylene syringes are translucent in appearance.
Treanda solution should be inspected visually for particulate matter and discoloration prior to administration whenever the solution and container permit. The solution must be withdrawn and transferred for dilution in a biosafety cabinet or containment isolator.
If they aim to use a CSTD with Treanda solution, healthcare professionals should verify with the CSTD manufacturer or Teva US Medical Information (1-800-896-5855) that the CSTD is compatible with Treanda solution before preparing the drug.
Alternatively, healthcare professionals can use Treanda lyophilized powder with a CSTD. The solution and lyophilized powder formulations of Treanda should not be mixed.
For additional details on safe preparation of Treanda solution and lyophilized powder, see Teva’s Dear Health Care Provider letter.
Adverse events or quality problems associated with the use of Treanda products can be reported to the FDA’s MedWatch Adverse Event Reporting Program. ![]()
The US Food and Drug Administration (FDA) is warning healthcare professionals not to use Treanda (bendamustine hydrochloride) solution with closed-system transfer devices (CSTD), adapters, and syringes containing polycarbonate or acrylonitrile-butadiene-styrene (ABS).
Most marketed CSTDs contain either polycarbonate or ABS. And these materials dissolve when they come into contact with N, N-dimethylacetamide (DMA), an ingredient in Treanda solution.
This can lead to device failure, possible product contamination, and potential serious adverse health consequences, including skin reactions in healthcare professionals preparing and administering this product and the risk of small blood vessel blockage in patients.
Discovering the incompatibility
Treanda, which is manufactured by Teva, is used to treat patients with chronic lymphocytic leukemia and indolent B-cell non-Hodgkin lymphoma that has progressed during or within 6 months of treatment with rituximab or a rituximab-containing regimen.
Treanda is available as a solution—Treanda Injection (45 mg/0.5 mL or 180 mg/2 mL solution)—and a lyophilized powder—Treanda for Injection (25mg/vial or 100 mg/vial lyophilized powder).
The incompatibility of DMA with polycarbonate and ABS is only an issue with Treanda solution—not the lyophilized powder.
Since December 2014, Teva has received 40 complaints of the incompatibility issue, which was recently brought to the FDA’s attention. The agency also received a notification of device incompatibility with Treanda solution from a pharmacist.
These incompatibility issues included leaking of the CSTD, breaking or operational failure of the CSTD components, and a cloudy appearance or presence of particulate matter in the intravenous bag after dilution. To date, no adverse events have been reported related to the incompatibility.
FDA recommendations
The FDA has required label changes for both the solution and the powder formulations of Treanda to reflect the following safe preparation information.
The agency is recommending that healthcare professionals use Treanda solution only with polypropylene syringes containing a metal needle and a polypropylene hub. Polypropylene syringes are translucent in appearance.
Treanda solution should be inspected visually for particulate matter and discoloration prior to administration whenever the solution and container permit. The solution must be withdrawn and transferred for dilution in a biosafety cabinet or containment isolator.
If they aim to use a CSTD with Treanda solution, healthcare professionals should verify with the CSTD manufacturer or Teva US Medical Information (1-800-896-5855) that the CSTD is compatible with Treanda solution before preparing the drug.
Alternatively, healthcare professionals can use Treanda lyophilized powder with a CSTD. The solution and lyophilized powder formulations of Treanda should not be mixed.
For additional details on safe preparation of Treanda solution and lyophilized powder, see Teva’s Dear Health Care Provider letter.
Adverse events or quality problems associated with the use of Treanda products can be reported to the FDA’s MedWatch Adverse Event Reporting Program. ![]()
Clinical trial mandate not being followed

Photo by Esther Dyson
A study published in NEJM suggests that results from most trials on ClinicalTrials.gov are not posted on the site within a year of trial completion or termination, even though this violates the Food and Drug Administration Amendments Act (FDAAA) mandate.
Among all the trials analyzed, those funded by industry were the most likely to be publicly disclosed in a timely fashion, researchers found.
Even so, industry compliance with the FDAAA mandate was poor.
And compliance was worse for research funded by the National Institutes of Health (NIH) and other government and academic institutions.
Study authors said this lack of transparency has created a critical information gap about investigational drugs, devices, and biologic therapies that hampers progress and violates obligations to patients.
“Patients who participate in clinical research have the expectation that the risk of participation will be offset by the creation of generalizable knowledge and the advancement of science, and that is achieved through the availability of clinical trial results,” said study author Monique Anderson, MD, of the Duke Clinical Research Institute in Durham, North Carolina.
“Sponsors who lead clinical trials have an ethical and legal obligation to publically report their findings, whether the results are positive or negative.”
In 2000, the US Congress authorized the creation of the ClinicalTrials.gov registry to provide information about clinical trials. Seven years later, the FDAAA mandated that sponsors of most trials begin registering and reporting basic summary results on the registry so the American public could have access to the resulting data.
The requirement covers non-phase-1 trials of drugs, medical devices, or biologics that had at least 1 US research site. Trial results are to be reported by the sponsor within a year of completing data collection.
To gauge compliance with this mandate, Dr Anderson and her colleagues searched trials posted on ClinicalTrials.gov. They identified 13,327 trials that were likely to be subject to FDAAA provisions and were terminated or completed from January 1, 2008, through August 31, 2012.
Most trials were industry-funded (65.6%), 14.2% were funded by the NIH, and the remaining 20.2% were funded by other government or academic institutions.
In all, 13.4% of trials had results posted on ClinicalTrials.gov within 12 months of trial completion/termination, and 6.1% of trials did not have results reported but had a legally acceptable delay because of a certification or an exemption request.
A higher percentage of trials—38.3%—had results posted on ClinicalTrials.gov at any time during the 5-year study period.
By September 27, 2013, sponsors for 15.8% of all the trials analyzed had submitted a certification or extension request to delay reporting to ClinicalTrials.gov. Of these, 23.0% had results reported during the study period.
At 12 months, 17% of industry-funded trials had results posted on ClinicalTrials.gov, as did 8.1% of trials funded by the NIH, and 5.7% of trials funded by other government or academic institutions. At 5 years, those percentages had increased to 41.5%, 38.9%, and 27.7%, respectively.
“The law requiring public disclosure was enacted amid concerns that sponsors and investigators were selectively publishing trials that favored sponsors’ interests,” Dr Anderson said. “Industry sponsors in particular were criticized for selective reporting.”
“Since the law’s enactment, many companies have developed disclosure policies and have actively pursued expanded public disclosure of data, but there may be a lack of knowledge about the law in academia, or a lack of resources to ensure timely reporting.”
Dr Anderson said penalties for failing to submit data within the 1-year reporting period could be as high as $10,000 a day and/or the loss of NIH funding, but enforcement has not occurred, pending a rule approval. ![]()
Photo by Esther Dyson
A study published in NEJM suggests that results from most trials on ClinicalTrials.gov are not posted on the site within a year of trial completion or termination, even though this violates the Food and Drug Administration Amendments Act (FDAAA) mandate.
Among all the trials analyzed, those funded by industry were the most likely to be publicly disclosed in a timely fashion, researchers found.
Even so, industry compliance with the FDAAA mandate was poor.
And compliance was worse for research funded by the National Institutes of Health (NIH) and other government and academic institutions.
Study authors said this lack of transparency has created a critical information gap about investigational drugs, devices, and biologic therapies that hampers progress and violates obligations to patients.
“Patients who participate in clinical research have the expectation that the risk of participation will be offset by the creation of generalizable knowledge and the advancement of science, and that is achieved through the availability of clinical trial results,” said study author Monique Anderson, MD, of the Duke Clinical Research Institute in Durham, North Carolina.
“Sponsors who lead clinical trials have an ethical and legal obligation to publically report their findings, whether the results are positive or negative.”
In 2000, the US Congress authorized the creation of the ClinicalTrials.gov registry to provide information about clinical trials. Seven years later, the FDAAA mandated that sponsors of most trials begin registering and reporting basic summary results on the registry so the American public could have access to the resulting data.
The requirement covers non-phase-1 trials of drugs, medical devices, or biologics that had at least 1 US research site. Trial results are to be reported by the sponsor within a year of completing data collection.
To gauge compliance with this mandate, Dr Anderson and her colleagues searched trials posted on ClinicalTrials.gov. They identified 13,327 trials that were likely to be subject to FDAAA provisions and were terminated or completed from January 1, 2008, through August 31, 2012.
Most trials were industry-funded (65.6%), 14.2% were funded by the NIH, and the remaining 20.2% were funded by other government or academic institutions.
In all, 13.4% of trials had results posted on ClinicalTrials.gov within 12 months of trial completion/termination, and 6.1% of trials did not have results reported but had a legally acceptable delay because of a certification or an exemption request.
A higher percentage of trials—38.3%—had results posted on ClinicalTrials.gov at any time during the 5-year study period.
By September 27, 2013, sponsors for 15.8% of all the trials analyzed had submitted a certification or extension request to delay reporting to ClinicalTrials.gov. Of these, 23.0% had results reported during the study period.
At 12 months, 17% of industry-funded trials had results posted on ClinicalTrials.gov, as did 8.1% of trials funded by the NIH, and 5.7% of trials funded by other government or academic institutions. At 5 years, those percentages had increased to 41.5%, 38.9%, and 27.7%, respectively.
“The law requiring public disclosure was enacted amid concerns that sponsors and investigators were selectively publishing trials that favored sponsors’ interests,” Dr Anderson said. “Industry sponsors in particular were criticized for selective reporting.”
“Since the law’s enactment, many companies have developed disclosure policies and have actively pursued expanded public disclosure of data, but there may be a lack of knowledge about the law in academia, or a lack of resources to ensure timely reporting.”
Dr Anderson said penalties for failing to submit data within the 1-year reporting period could be as high as $10,000 a day and/or the loss of NIH funding, but enforcement has not occurred, pending a rule approval. ![]()
Photo by Esther Dyson
A study published in NEJM suggests that results from most trials on ClinicalTrials.gov are not posted on the site within a year of trial completion or termination, even though this violates the Food and Drug Administration Amendments Act (FDAAA) mandate.
Among all the trials analyzed, those funded by industry were the most likely to be publicly disclosed in a timely fashion, researchers found.
Even so, industry compliance with the FDAAA mandate was poor.
And compliance was worse for research funded by the National Institutes of Health (NIH) and other government and academic institutions.
Study authors said this lack of transparency has created a critical information gap about investigational drugs, devices, and biologic therapies that hampers progress and violates obligations to patients.
“Patients who participate in clinical research have the expectation that the risk of participation will be offset by the creation of generalizable knowledge and the advancement of science, and that is achieved through the availability of clinical trial results,” said study author Monique Anderson, MD, of the Duke Clinical Research Institute in Durham, North Carolina.
“Sponsors who lead clinical trials have an ethical and legal obligation to publically report their findings, whether the results are positive or negative.”
In 2000, the US Congress authorized the creation of the ClinicalTrials.gov registry to provide information about clinical trials. Seven years later, the FDAAA mandated that sponsors of most trials begin registering and reporting basic summary results on the registry so the American public could have access to the resulting data.
The requirement covers non-phase-1 trials of drugs, medical devices, or biologics that had at least 1 US research site. Trial results are to be reported by the sponsor within a year of completing data collection.
To gauge compliance with this mandate, Dr Anderson and her colleagues searched trials posted on ClinicalTrials.gov. They identified 13,327 trials that were likely to be subject to FDAAA provisions and were terminated or completed from January 1, 2008, through August 31, 2012.
Most trials were industry-funded (65.6%), 14.2% were funded by the NIH, and the remaining 20.2% were funded by other government or academic institutions.
In all, 13.4% of trials had results posted on ClinicalTrials.gov within 12 months of trial completion/termination, and 6.1% of trials did not have results reported but had a legally acceptable delay because of a certification or an exemption request.
A higher percentage of trials—38.3%—had results posted on ClinicalTrials.gov at any time during the 5-year study period.
By September 27, 2013, sponsors for 15.8% of all the trials analyzed had submitted a certification or extension request to delay reporting to ClinicalTrials.gov. Of these, 23.0% had results reported during the study period.
At 12 months, 17% of industry-funded trials had results posted on ClinicalTrials.gov, as did 8.1% of trials funded by the NIH, and 5.7% of trials funded by other government or academic institutions. At 5 years, those percentages had increased to 41.5%, 38.9%, and 27.7%, respectively.
“The law requiring public disclosure was enacted amid concerns that sponsors and investigators were selectively publishing trials that favored sponsors’ interests,” Dr Anderson said. “Industry sponsors in particular were criticized for selective reporting.”
“Since the law’s enactment, many companies have developed disclosure policies and have actively pursued expanded public disclosure of data, but there may be a lack of knowledge about the law in academia, or a lack of resources to ensure timely reporting.”
Dr Anderson said penalties for failing to submit data within the 1-year reporting period could be as high as $10,000 a day and/or the loss of NIH funding, but enforcement has not occurred, pending a rule approval. ![]()
Plasma product can be stored longer, FDA says

Photo by Cristina Granados
The US Food and Drug Administration (FDA) has approved a revised label for the pooled plasma product Octaplas, increasing the product’s shelf life.
The new label says Octaplas can now be stored frozen, at or below -18°C (-0.4°F), for 3 years from the date of manufacture.
And thawed Octaplas should be used within 24 hours if refrigerated (between 1°C and 6°C/33.8°F to 42.8°F) or within 8 hours if stored at room temperature (between 20°C and 25°C/68°F to 77°F)
The previous product label said frozen Octaplas could be stored for 2 years, and thawed Octaplas should be used within 12 hours if stored between 2°C and 4°C (35.6°F to 39.2°F) or within 3 hours if stored between 20°C and 25°C (68°F to 77°F).
About Octaplas
Octaplas is a sterile, frozen solution of human plasma from several donors that has been treated with a solvent detergent process to minimize the risk of serious virus transmission. The plasma is collected from US donors who have been screened and tested for diseases transmitted by blood.
Octaplas gained FDA approval in January 2013. The product is indicated for the replacement of multiple coagulation factors in patients with acquired deficiencies due to liver disease or undergoing cardiac surgery or liver transplant. Octaplas can also be used for plasma exchange in patients with thrombotic thrombocytopenic purpura.
Octaplas is contraindicated in patients with immunoglobulin A deficiency, severe deficiency of protein S, history of hypersensitivity to fresh-frozen plasma or to plasma-derived products including any plasma protein, or a history of hypersensitivity reaction to Octaplas.
Serious adverse events observed in clinical trials of Octaplas were anaphylactic shock, citrate toxicity, and severe hypotension. The most common adverse events observed in 1% of patients or more included pruritus, urticaria, nausea, headache, and paresthesia.
Transfusion reactions can occur with ABO blood group mismatches. High infusion rates can induce hypervolemia with consequent pulmonary edema or cardiac failure. Excessive bleeding due to hyperfibrinolysis can occur due to low levels of alpha2-antiplasmin.
Thrombosis can occur due to low levels of protein S. Citrate toxicity can occur with transfusion rates exceeding 1 mL/kg/min of Octaplas. As Octaplas is made from human plasma, it may carry a risk of transmitting infectious agents, such as viruses, the variant Creutzfeldt-Jakob disease agent, and, theoretically, the Creutzfeldt-Jakob disease agent.
For more details on Octaplas, see the complete prescribing information. ![]()
Photo by Cristina Granados
The US Food and Drug Administration (FDA) has approved a revised label for the pooled plasma product Octaplas, increasing the product’s shelf life.
The new label says Octaplas can now be stored frozen, at or below -18°C (-0.4°F), for 3 years from the date of manufacture.
And thawed Octaplas should be used within 24 hours if refrigerated (between 1°C and 6°C/33.8°F to 42.8°F) or within 8 hours if stored at room temperature (between 20°C and 25°C/68°F to 77°F)
The previous product label said frozen Octaplas could be stored for 2 years, and thawed Octaplas should be used within 12 hours if stored between 2°C and 4°C (35.6°F to 39.2°F) or within 3 hours if stored between 20°C and 25°C (68°F to 77°F).
About Octaplas
Octaplas is a sterile, frozen solution of human plasma from several donors that has been treated with a solvent detergent process to minimize the risk of serious virus transmission. The plasma is collected from US donors who have been screened and tested for diseases transmitted by blood.
Octaplas gained FDA approval in January 2013. The product is indicated for the replacement of multiple coagulation factors in patients with acquired deficiencies due to liver disease or undergoing cardiac surgery or liver transplant. Octaplas can also be used for plasma exchange in patients with thrombotic thrombocytopenic purpura.
Octaplas is contraindicated in patients with immunoglobulin A deficiency, severe deficiency of protein S, history of hypersensitivity to fresh-frozen plasma or to plasma-derived products including any plasma protein, or a history of hypersensitivity reaction to Octaplas.
Serious adverse events observed in clinical trials of Octaplas were anaphylactic shock, citrate toxicity, and severe hypotension. The most common adverse events observed in 1% of patients or more included pruritus, urticaria, nausea, headache, and paresthesia.
Transfusion reactions can occur with ABO blood group mismatches. High infusion rates can induce hypervolemia with consequent pulmonary edema or cardiac failure. Excessive bleeding due to hyperfibrinolysis can occur due to low levels of alpha2-antiplasmin.
Thrombosis can occur due to low levels of protein S. Citrate toxicity can occur with transfusion rates exceeding 1 mL/kg/min of Octaplas. As Octaplas is made from human plasma, it may carry a risk of transmitting infectious agents, such as viruses, the variant Creutzfeldt-Jakob disease agent, and, theoretically, the Creutzfeldt-Jakob disease agent.
For more details on Octaplas, see the complete prescribing information. ![]()
Photo by Cristina Granados
The US Food and Drug Administration (FDA) has approved a revised label for the pooled plasma product Octaplas, increasing the product’s shelf life.
The new label says Octaplas can now be stored frozen, at or below -18°C (-0.4°F), for 3 years from the date of manufacture.
And thawed Octaplas should be used within 24 hours if refrigerated (between 1°C and 6°C/33.8°F to 42.8°F) or within 8 hours if stored at room temperature (between 20°C and 25°C/68°F to 77°F)
The previous product label said frozen Octaplas could be stored for 2 years, and thawed Octaplas should be used within 12 hours if stored between 2°C and 4°C (35.6°F to 39.2°F) or within 3 hours if stored between 20°C and 25°C (68°F to 77°F).
About Octaplas
Octaplas is a sterile, frozen solution of human plasma from several donors that has been treated with a solvent detergent process to minimize the risk of serious virus transmission. The plasma is collected from US donors who have been screened and tested for diseases transmitted by blood.
Octaplas gained FDA approval in January 2013. The product is indicated for the replacement of multiple coagulation factors in patients with acquired deficiencies due to liver disease or undergoing cardiac surgery or liver transplant. Octaplas can also be used for plasma exchange in patients with thrombotic thrombocytopenic purpura.
Octaplas is contraindicated in patients with immunoglobulin A deficiency, severe deficiency of protein S, history of hypersensitivity to fresh-frozen plasma or to plasma-derived products including any plasma protein, or a history of hypersensitivity reaction to Octaplas.
Serious adverse events observed in clinical trials of Octaplas were anaphylactic shock, citrate toxicity, and severe hypotension. The most common adverse events observed in 1% of patients or more included pruritus, urticaria, nausea, headache, and paresthesia.
Transfusion reactions can occur with ABO blood group mismatches. High infusion rates can induce hypervolemia with consequent pulmonary edema or cardiac failure. Excessive bleeding due to hyperfibrinolysis can occur due to low levels of alpha2-antiplasmin.
Thrombosis can occur due to low levels of protein S. Citrate toxicity can occur with transfusion rates exceeding 1 mL/kg/min of Octaplas. As Octaplas is made from human plasma, it may carry a risk of transmitting infectious agents, such as viruses, the variant Creutzfeldt-Jakob disease agent, and, theoretically, the Creutzfeldt-Jakob disease agent.
For more details on Octaplas, see the complete prescribing information. ![]()
Polymer can stop lethal bleeding in vivo
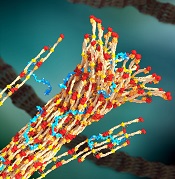
a blood clot, with PolySTAT
(blue) binding strands together
Image by William Walker–
University of Washington
Preclinical research suggests an injectable polymer known as PolySTAT may one day be able to halt life-threatening bleeding in soldiers and trauma patients.
Once injected, this hemostatic polymer circulates in the blood, homes to sites of vascular injury, and promotes the formation of blood clots.
In experiments with rats, 100% of animals injected with PolySTAT survived a typically lethal injury to the femoral artery. In comparison, 0% to 40% of controls survived.
“Most of the patients who die from bleeding die quickly,” said Nathan White, MD, of the University of Washington in Seattle.
“[PolySTAT] is something you could potentially put in a syringe inside a backpack and give right away to reduce blood loss and keep people alive long enough to make it to medical care.”
Dr White and his colleagues described their work with PolySTAT in Science Translational Medicine. A related Focus article addressed the promises and challenges of advancing PolySTAT and other clotting approaches from proof-of-principle to clinical development.
PolySTAT induces hemostasis by cross-linking the fibrin matrix within blood clots, just as factor XIII does. But the researchers said PolySTAT offers greater protection against natural enzymes that dissolve blood clots.
That’s because PolySTAT binds to fibrin monomers and is uniformly integrated into fibrin fibers during polymerization. This produces a fortified, hybrid polymer network that can resist enzymatic degradation.
In vitro experiments showed that PolySTAT accelerated clotting kinetics, increased the strength of blood clots, and delayed clot breakdown.
The researchers also assessed how PolySTAT affected rats following a femoral artery injury, comparing results with PolySTAT to those with volume control (0.9% saline), a nonbinding scrambled control polymer (PolySCRAM), rat albumin, and human FXIIIa.
The team found that PolySTAT conferred superior survival by reducing blood loss and fluid resuscitation requirements.
All of the rats treated with PolySTAT (5/5) survived to the end of the experiment, compared to none of the rats that received albumin, 20% that received PolySCRAM or FXIIIa, and 40% that received volume control.
The researchers said PolySTAT’s initial safety profile looks promising, but they are still planning to test the polymer on larger animals and conduct additional screening to find out if PolySTAT binds to any other unintended substances.
The team also plans to investigate PolySTAT’s potential for treating hemophilia and for integration into bandages. ![]()
a blood clot, with PolySTAT
(blue) binding strands together
Image by William Walker–
University of Washington
Preclinical research suggests an injectable polymer known as PolySTAT may one day be able to halt life-threatening bleeding in soldiers and trauma patients.
Once injected, this hemostatic polymer circulates in the blood, homes to sites of vascular injury, and promotes the formation of blood clots.
In experiments with rats, 100% of animals injected with PolySTAT survived a typically lethal injury to the femoral artery. In comparison, 0% to 40% of controls survived.
“Most of the patients who die from bleeding die quickly,” said Nathan White, MD, of the University of Washington in Seattle.
“[PolySTAT] is something you could potentially put in a syringe inside a backpack and give right away to reduce blood loss and keep people alive long enough to make it to medical care.”
Dr White and his colleagues described their work with PolySTAT in Science Translational Medicine. A related Focus article addressed the promises and challenges of advancing PolySTAT and other clotting approaches from proof-of-principle to clinical development.
PolySTAT induces hemostasis by cross-linking the fibrin matrix within blood clots, just as factor XIII does. But the researchers said PolySTAT offers greater protection against natural enzymes that dissolve blood clots.
That’s because PolySTAT binds to fibrin monomers and is uniformly integrated into fibrin fibers during polymerization. This produces a fortified, hybrid polymer network that can resist enzymatic degradation.
In vitro experiments showed that PolySTAT accelerated clotting kinetics, increased the strength of blood clots, and delayed clot breakdown.
The researchers also assessed how PolySTAT affected rats following a femoral artery injury, comparing results with PolySTAT to those with volume control (0.9% saline), a nonbinding scrambled control polymer (PolySCRAM), rat albumin, and human FXIIIa.
The team found that PolySTAT conferred superior survival by reducing blood loss and fluid resuscitation requirements.
All of the rats treated with PolySTAT (5/5) survived to the end of the experiment, compared to none of the rats that received albumin, 20% that received PolySCRAM or FXIIIa, and 40% that received volume control.
The researchers said PolySTAT’s initial safety profile looks promising, but they are still planning to test the polymer on larger animals and conduct additional screening to find out if PolySTAT binds to any other unintended substances.
The team also plans to investigate PolySTAT’s potential for treating hemophilia and for integration into bandages. ![]()
a blood clot, with PolySTAT
(blue) binding strands together
Image by William Walker–
University of Washington
Preclinical research suggests an injectable polymer known as PolySTAT may one day be able to halt life-threatening bleeding in soldiers and trauma patients.
Once injected, this hemostatic polymer circulates in the blood, homes to sites of vascular injury, and promotes the formation of blood clots.
In experiments with rats, 100% of animals injected with PolySTAT survived a typically lethal injury to the femoral artery. In comparison, 0% to 40% of controls survived.
“Most of the patients who die from bleeding die quickly,” said Nathan White, MD, of the University of Washington in Seattle.
“[PolySTAT] is something you could potentially put in a syringe inside a backpack and give right away to reduce blood loss and keep people alive long enough to make it to medical care.”
Dr White and his colleagues described their work with PolySTAT in Science Translational Medicine. A related Focus article addressed the promises and challenges of advancing PolySTAT and other clotting approaches from proof-of-principle to clinical development.
PolySTAT induces hemostasis by cross-linking the fibrin matrix within blood clots, just as factor XIII does. But the researchers said PolySTAT offers greater protection against natural enzymes that dissolve blood clots.
That’s because PolySTAT binds to fibrin monomers and is uniformly integrated into fibrin fibers during polymerization. This produces a fortified, hybrid polymer network that can resist enzymatic degradation.
In vitro experiments showed that PolySTAT accelerated clotting kinetics, increased the strength of blood clots, and delayed clot breakdown.
The researchers also assessed how PolySTAT affected rats following a femoral artery injury, comparing results with PolySTAT to those with volume control (0.9% saline), a nonbinding scrambled control polymer (PolySCRAM), rat albumin, and human FXIIIa.
The team found that PolySTAT conferred superior survival by reducing blood loss and fluid resuscitation requirements.
All of the rats treated with PolySTAT (5/5) survived to the end of the experiment, compared to none of the rats that received albumin, 20% that received PolySCRAM or FXIIIa, and 40% that received volume control.
The researchers said PolySTAT’s initial safety profile looks promising, but they are still planning to test the polymer on larger animals and conduct additional screening to find out if PolySTAT binds to any other unintended substances.
The team also plans to investigate PolySTAT’s potential for treating hemophilia and for integration into bandages. ![]()
Rehospitalization after severe sepsis often avoidable

A new analysis indicates that patients hospitalized for severe sepsis are often readmitted within 90 days, and many of these readmissions may be preventable.
About 43% of the patients studied were readmitted to the hospital within 90 days of their sepsis hospitalization.
And 42% of these hospitalizations were due to conditions that could potentially be prevented or treated early to avoid hospitalization, according to researchers.
Hallie C. Prescott, MD, of the University of Michigan, Ann Arbor, and her colleagues reported these findings in JAMA.
The researchers analyzed participants in the nationally representative US Health and Retirement Study, a sample of households with adults 50 years of age or older that is linked to Medicare claims (1998-2010).
The team examined the most common readmission diagnoses among patients who were hospitalized for severe sepsis, the extent to which readmissions might have been preventable, and whether the pattern of readmission diagnoses differed compared with that of other acute medical conditions.
To gauge what proportion of rehospitalizations might have been preventable, the researchers looked at ambulatory-care-sensitive conditions (ACSCs) identified by the Agency for Healthcare Research and Quality. They also expanded the definition of ACSCs to include conditions that aren’t common among the general population but arise more often in sepsis survivors.
So their potentially preventable readmission diagnoses included pneumonia, hypertension, dehydration, asthma, urinary tract infection, chronic obstructive pulmonary disease exacerbation, perforated appendix, diabetes, angina, congestive heart failure, sepsis, acute renal failure, skin or soft tissue infection, and aspiration pneumonitis.
Dr Prescott and her colleagues identified 2617 hospitalizations for severe sepsis that could be matched to hospitalizations for other acute medical conditions. And they found that 1115 of the severe sepsis survivors (42.6%) were rehospitalized within 90 days.
The 10 most common readmission diagnoses following severe sepsis were sepsis (6.4%), congestive heart failure (5.5%), pneumonia (3.5%), acute renal failure (3.3%), rehabilitation (2.8%), respiratory failure (2.5%), complication related to a device, implant, or graft (2%), exacerbation of chronic obstructive pulmonary disorder (1.9%), aspiration pneumonitis (1.8%), and urinary tract infection (1.7%).
Readmissions for a primary diagnosis of infection (sepsis, pneumonia, urinary tract, and skin or soft tissue infection) occurred in 11.9% of severe sepsis survivors and 8.0% of patients with acute medical conditions (P<0.001).
Likewise, readmissions for ACSCs were more common after severe sepsis than for patients with acute conditions—21.6% and 19.1%, respectively (P=0.02)—and accounted for a greater proportion of all 90-day readmissions—41.6% and 37%.1, respectively (P=0.009).
“Many of these conditions can be managed if the patient can get in to see a doctor at the start of the illness, meaning that we potentially avoid hospitalization,” Dr Prescott said. “We need to assess their vulnerability and design a better landing pad for patients when they leave the hospital, and avoid the second hit that derails recovery.” ![]()
A new analysis indicates that patients hospitalized for severe sepsis are often readmitted within 90 days, and many of these readmissions may be preventable.
About 43% of the patients studied were readmitted to the hospital within 90 days of their sepsis hospitalization.
And 42% of these hospitalizations were due to conditions that could potentially be prevented or treated early to avoid hospitalization, according to researchers.
Hallie C. Prescott, MD, of the University of Michigan, Ann Arbor, and her colleagues reported these findings in JAMA.
The researchers analyzed participants in the nationally representative US Health and Retirement Study, a sample of households with adults 50 years of age or older that is linked to Medicare claims (1998-2010).
The team examined the most common readmission diagnoses among patients who were hospitalized for severe sepsis, the extent to which readmissions might have been preventable, and whether the pattern of readmission diagnoses differed compared with that of other acute medical conditions.
To gauge what proportion of rehospitalizations might have been preventable, the researchers looked at ambulatory-care-sensitive conditions (ACSCs) identified by the Agency for Healthcare Research and Quality. They also expanded the definition of ACSCs to include conditions that aren’t common among the general population but arise more often in sepsis survivors.
So their potentially preventable readmission diagnoses included pneumonia, hypertension, dehydration, asthma, urinary tract infection, chronic obstructive pulmonary disease exacerbation, perforated appendix, diabetes, angina, congestive heart failure, sepsis, acute renal failure, skin or soft tissue infection, and aspiration pneumonitis.
Dr Prescott and her colleagues identified 2617 hospitalizations for severe sepsis that could be matched to hospitalizations for other acute medical conditions. And they found that 1115 of the severe sepsis survivors (42.6%) were rehospitalized within 90 days.
The 10 most common readmission diagnoses following severe sepsis were sepsis (6.4%), congestive heart failure (5.5%), pneumonia (3.5%), acute renal failure (3.3%), rehabilitation (2.8%), respiratory failure (2.5%), complication related to a device, implant, or graft (2%), exacerbation of chronic obstructive pulmonary disorder (1.9%), aspiration pneumonitis (1.8%), and urinary tract infection (1.7%).
Readmissions for a primary diagnosis of infection (sepsis, pneumonia, urinary tract, and skin or soft tissue infection) occurred in 11.9% of severe sepsis survivors and 8.0% of patients with acute medical conditions (P<0.001).
Likewise, readmissions for ACSCs were more common after severe sepsis than for patients with acute conditions—21.6% and 19.1%, respectively (P=0.02)—and accounted for a greater proportion of all 90-day readmissions—41.6% and 37%.1, respectively (P=0.009).
“Many of these conditions can be managed if the patient can get in to see a doctor at the start of the illness, meaning that we potentially avoid hospitalization,” Dr Prescott said. “We need to assess their vulnerability and design a better landing pad for patients when they leave the hospital, and avoid the second hit that derails recovery.” ![]()
A new analysis indicates that patients hospitalized for severe sepsis are often readmitted within 90 days, and many of these readmissions may be preventable.
About 43% of the patients studied were readmitted to the hospital within 90 days of their sepsis hospitalization.
And 42% of these hospitalizations were due to conditions that could potentially be prevented or treated early to avoid hospitalization, according to researchers.
Hallie C. Prescott, MD, of the University of Michigan, Ann Arbor, and her colleagues reported these findings in JAMA.
The researchers analyzed participants in the nationally representative US Health and Retirement Study, a sample of households with adults 50 years of age or older that is linked to Medicare claims (1998-2010).
The team examined the most common readmission diagnoses among patients who were hospitalized for severe sepsis, the extent to which readmissions might have been preventable, and whether the pattern of readmission diagnoses differed compared with that of other acute medical conditions.
To gauge what proportion of rehospitalizations might have been preventable, the researchers looked at ambulatory-care-sensitive conditions (ACSCs) identified by the Agency for Healthcare Research and Quality. They also expanded the definition of ACSCs to include conditions that aren’t common among the general population but arise more often in sepsis survivors.
So their potentially preventable readmission diagnoses included pneumonia, hypertension, dehydration, asthma, urinary tract infection, chronic obstructive pulmonary disease exacerbation, perforated appendix, diabetes, angina, congestive heart failure, sepsis, acute renal failure, skin or soft tissue infection, and aspiration pneumonitis.
Dr Prescott and her colleagues identified 2617 hospitalizations for severe sepsis that could be matched to hospitalizations for other acute medical conditions. And they found that 1115 of the severe sepsis survivors (42.6%) were rehospitalized within 90 days.
The 10 most common readmission diagnoses following severe sepsis were sepsis (6.4%), congestive heart failure (5.5%), pneumonia (3.5%), acute renal failure (3.3%), rehabilitation (2.8%), respiratory failure (2.5%), complication related to a device, implant, or graft (2%), exacerbation of chronic obstructive pulmonary disorder (1.9%), aspiration pneumonitis (1.8%), and urinary tract infection (1.7%).
Readmissions for a primary diagnosis of infection (sepsis, pneumonia, urinary tract, and skin or soft tissue infection) occurred in 11.9% of severe sepsis survivors and 8.0% of patients with acute medical conditions (P<0.001).
Likewise, readmissions for ACSCs were more common after severe sepsis than for patients with acute conditions—21.6% and 19.1%, respectively (P=0.02)—and accounted for a greater proportion of all 90-day readmissions—41.6% and 37%.1, respectively (P=0.009).
“Many of these conditions can be managed if the patient can get in to see a doctor at the start of the illness, meaning that we potentially avoid hospitalization,” Dr Prescott said. “We need to assess their vulnerability and design a better landing pad for patients when they leave the hospital, and avoid the second hit that derails recovery.” ![]()
Researchers generate RBCs to treat SCD
Image by Ying Wang–
Johns Hopkins Medicine
Researchers say they have devised a technique for generating normal, mature red blood cells (RBCs) from patients with sickle cell disease (SCD).
The team hopes that, ultimately, the RBCs could be transfused back into the patients from which they are derived and eliminate the need for donor transfusion in SCD.
Linzhao Cheng, PhD, of the Johns Hopkins School of Medicine in Baltimore, Maryland, and his colleagues described the technique in Stem Cells.
Dr Cheng noted that SCD patients often require RBC transfusions, but over time, their bodies may begin to mount an immune response against the foreign blood.
“Their bodies quickly kill off the blood cells,” Dr Cheng said. “So they have to get transfusions more and more frequently.”
A solution, Dr Cheng and his colleagues thought, would be to generate RBCs for transfusion using a patient’s own cells.
To do this, the researchers first took hematopoietic cells from an SCD patient and generated induced pluripotent stem cells (iPSCs).
Then, the team used the gene-editing technique CRISPR/Cas9 to target the homozygous SCD mutation (nt. 69A>T) in the HBB gene and ensure the RBCs they generated would not be sickled.
Finally, they coaxed the iPSCs into mature RBCs that expressed the corrected HBB gene. The edited iPSCs generated RBCs just as efficiently as iPSCs that hadn’t been subjected to CRISPR/Cas9.
And the level of HBB protein expression in the RBCs derived from edited iPSCs was similar to that of RBCs generated from unedited iPSCs.
Dr Cheng noted that, to become medically useful, this method will have to be made more efficient and scaled up significantly. And the cells would need to be tested for safety.
“[Nevertheless,] this study shows it may be possible in the not-too-distant future to provide patients with sickle cell disease with an exciting new treatment option,” Dr Cheng said.
He and his colleagues believe this method of RBC generation may also be applicable for other blood disorders. And they think it might be possible to edit cells from healthy individuals so they can resist malaria and other infectious agents.
Another research group has reported the ability to correct the SCD mutation using zinc-finger nucleases. ![]()
Image by Ying Wang–
Johns Hopkins Medicine
Researchers say they have devised a technique for generating normal, mature red blood cells (RBCs) from patients with sickle cell disease (SCD).
The team hopes that, ultimately, the RBCs could be transfused back into the patients from which they are derived and eliminate the need for donor transfusion in SCD.
Linzhao Cheng, PhD, of the Johns Hopkins School of Medicine in Baltimore, Maryland, and his colleagues described the technique in Stem Cells.
Dr Cheng noted that SCD patients often require RBC transfusions, but over time, their bodies may begin to mount an immune response against the foreign blood.
“Their bodies quickly kill off the blood cells,” Dr Cheng said. “So they have to get transfusions more and more frequently.”
A solution, Dr Cheng and his colleagues thought, would be to generate RBCs for transfusion using a patient’s own cells.
To do this, the researchers first took hematopoietic cells from an SCD patient and generated induced pluripotent stem cells (iPSCs).
Then, the team used the gene-editing technique CRISPR/Cas9 to target the homozygous SCD mutation (nt. 69A>T) in the HBB gene and ensure the RBCs they generated would not be sickled.
Finally, they coaxed the iPSCs into mature RBCs that expressed the corrected HBB gene. The edited iPSCs generated RBCs just as efficiently as iPSCs that hadn’t been subjected to CRISPR/Cas9.
And the level of HBB protein expression in the RBCs derived from edited iPSCs was similar to that of RBCs generated from unedited iPSCs.
Dr Cheng noted that, to become medically useful, this method will have to be made more efficient and scaled up significantly. And the cells would need to be tested for safety.
“[Nevertheless,] this study shows it may be possible in the not-too-distant future to provide patients with sickle cell disease with an exciting new treatment option,” Dr Cheng said.
He and his colleagues believe this method of RBC generation may also be applicable for other blood disorders. And they think it might be possible to edit cells from healthy individuals so they can resist malaria and other infectious agents.
Another research group has reported the ability to correct the SCD mutation using zinc-finger nucleases. ![]()
Image by Ying Wang–
Johns Hopkins Medicine
Researchers say they have devised a technique for generating normal, mature red blood cells (RBCs) from patients with sickle cell disease (SCD).
The team hopes that, ultimately, the RBCs could be transfused back into the patients from which they are derived and eliminate the need for donor transfusion in SCD.
Linzhao Cheng, PhD, of the Johns Hopkins School of Medicine in Baltimore, Maryland, and his colleagues described the technique in Stem Cells.
Dr Cheng noted that SCD patients often require RBC transfusions, but over time, their bodies may begin to mount an immune response against the foreign blood.
“Their bodies quickly kill off the blood cells,” Dr Cheng said. “So they have to get transfusions more and more frequently.”
A solution, Dr Cheng and his colleagues thought, would be to generate RBCs for transfusion using a patient’s own cells.
To do this, the researchers first took hematopoietic cells from an SCD patient and generated induced pluripotent stem cells (iPSCs).
Then, the team used the gene-editing technique CRISPR/Cas9 to target the homozygous SCD mutation (nt. 69A>T) in the HBB gene and ensure the RBCs they generated would not be sickled.
Finally, they coaxed the iPSCs into mature RBCs that expressed the corrected HBB gene. The edited iPSCs generated RBCs just as efficiently as iPSCs that hadn’t been subjected to CRISPR/Cas9.
And the level of HBB protein expression in the RBCs derived from edited iPSCs was similar to that of RBCs generated from unedited iPSCs.
Dr Cheng noted that, to become medically useful, this method will have to be made more efficient and scaled up significantly. And the cells would need to be tested for safety.
“[Nevertheless,] this study shows it may be possible in the not-too-distant future to provide patients with sickle cell disease with an exciting new treatment option,” Dr Cheng said.
He and his colleagues believe this method of RBC generation may also be applicable for other blood disorders. And they think it might be possible to edit cells from healthy individuals so they can resist malaria and other infectious agents.
Another research group has reported the ability to correct the SCD mutation using zinc-finger nucleases. ![]()
Group identifies new subtype of ALL

Photo by Steven Harbour
Researchers say they have discovered a new subtype of acute lymphoblastic leukemia (ALL) that is sensitive to drugs already approved to treat other hematologic malignancies.
The team uncovered cases of ALL that were dependent upon tonic pre-B-cell-receptor (BCR) signaling and therefore sensitive to drugs that inhibit tyrosine kinases downstream of the pre-BCR.
The group also developed a test that can identify patients with this subtype of ALL.
“We hope patients in this newly identified subset can be treated with these targeted drugs, . . . which are powerfully effective in the mouse experiments we have conducted on ALL,” said Markus Müschen, MD, PhD, of the University of California, San Francisco.
Dr Müschen and his colleagues described this work in Cancer Cell.
The researchers studied samples from 830 patients enrolled in 4 ongoing ALL trials and found tonic pre-BCR signaling in 112 patients (13.5%). Virtually all of the bone marrow slices from these patients showed “beautiful staining” of BCL6 expression, Dr Müschen said. (Two of the patients had weak staining.)
On the other hand, no BCL6 staining was observed in patients lacking pre-BCR signaling. These results suggest that BCL6 is a biomarker for pre-BCR signaling. And by testing patients for BCL6, we may be able to identify those who will respond to treatment with pre-BCR signaling inhibitors, the researchers said.
The team tested a range of pre-BCR signaling inhibitors in vitro. And they found a few compounds that were effective against pre-BCR+ ALL—the SYK inhibitor PRT062607, the BTK inhibitor ibrutinib, the SRC inhibitor dasatinib, and the PIK3δ inhibitor idelalisib.
Subsequent experiments revealed that dasatinib had the strongest antileukemic effect, so the researchers tested the drug in mouse models of pre-BCR+ ALL. Dasatinib significantly delayed leukemic expansion and prolonged overall survival in some mice, while completely eradicating the disease in other mice.
Dr Müschen said that dasatinib and other pre-BCR signaling inhibitors may be able to reduce the amount of conventional chemotherapy given to patients with pre-BCR+ ALL, or even replace chemotherapy altogether.
“In our experiments with mice, both combination therapy with low-dose chemotherapy and single-agent targeted therapy each worked very well,” he said. ![]()
Photo by Steven Harbour
Researchers say they have discovered a new subtype of acute lymphoblastic leukemia (ALL) that is sensitive to drugs already approved to treat other hematologic malignancies.
The team uncovered cases of ALL that were dependent upon tonic pre-B-cell-receptor (BCR) signaling and therefore sensitive to drugs that inhibit tyrosine kinases downstream of the pre-BCR.
The group also developed a test that can identify patients with this subtype of ALL.
“We hope patients in this newly identified subset can be treated with these targeted drugs, . . . which are powerfully effective in the mouse experiments we have conducted on ALL,” said Markus Müschen, MD, PhD, of the University of California, San Francisco.
Dr Müschen and his colleagues described this work in Cancer Cell.
The researchers studied samples from 830 patients enrolled in 4 ongoing ALL trials and found tonic pre-BCR signaling in 112 patients (13.5%). Virtually all of the bone marrow slices from these patients showed “beautiful staining” of BCL6 expression, Dr Müschen said. (Two of the patients had weak staining.)
On the other hand, no BCL6 staining was observed in patients lacking pre-BCR signaling. These results suggest that BCL6 is a biomarker for pre-BCR signaling. And by testing patients for BCL6, we may be able to identify those who will respond to treatment with pre-BCR signaling inhibitors, the researchers said.
The team tested a range of pre-BCR signaling inhibitors in vitro. And they found a few compounds that were effective against pre-BCR+ ALL—the SYK inhibitor PRT062607, the BTK inhibitor ibrutinib, the SRC inhibitor dasatinib, and the PIK3δ inhibitor idelalisib.
Subsequent experiments revealed that dasatinib had the strongest antileukemic effect, so the researchers tested the drug in mouse models of pre-BCR+ ALL. Dasatinib significantly delayed leukemic expansion and prolonged overall survival in some mice, while completely eradicating the disease in other mice.
Dr Müschen said that dasatinib and other pre-BCR signaling inhibitors may be able to reduce the amount of conventional chemotherapy given to patients with pre-BCR+ ALL, or even replace chemotherapy altogether.
“In our experiments with mice, both combination therapy with low-dose chemotherapy and single-agent targeted therapy each worked very well,” he said. ![]()
Photo by Steven Harbour
Researchers say they have discovered a new subtype of acute lymphoblastic leukemia (ALL) that is sensitive to drugs already approved to treat other hematologic malignancies.
The team uncovered cases of ALL that were dependent upon tonic pre-B-cell-receptor (BCR) signaling and therefore sensitive to drugs that inhibit tyrosine kinases downstream of the pre-BCR.
The group also developed a test that can identify patients with this subtype of ALL.
“We hope patients in this newly identified subset can be treated with these targeted drugs, . . . which are powerfully effective in the mouse experiments we have conducted on ALL,” said Markus Müschen, MD, PhD, of the University of California, San Francisco.
Dr Müschen and his colleagues described this work in Cancer Cell.
The researchers studied samples from 830 patients enrolled in 4 ongoing ALL trials and found tonic pre-BCR signaling in 112 patients (13.5%). Virtually all of the bone marrow slices from these patients showed “beautiful staining” of BCL6 expression, Dr Müschen said. (Two of the patients had weak staining.)
On the other hand, no BCL6 staining was observed in patients lacking pre-BCR signaling. These results suggest that BCL6 is a biomarker for pre-BCR signaling. And by testing patients for BCL6, we may be able to identify those who will respond to treatment with pre-BCR signaling inhibitors, the researchers said.
The team tested a range of pre-BCR signaling inhibitors in vitro. And they found a few compounds that were effective against pre-BCR+ ALL—the SYK inhibitor PRT062607, the BTK inhibitor ibrutinib, the SRC inhibitor dasatinib, and the PIK3δ inhibitor idelalisib.
Subsequent experiments revealed that dasatinib had the strongest antileukemic effect, so the researchers tested the drug in mouse models of pre-BCR+ ALL. Dasatinib significantly delayed leukemic expansion and prolonged overall survival in some mice, while completely eradicating the disease in other mice.
Dr Müschen said that dasatinib and other pre-BCR signaling inhibitors may be able to reduce the amount of conventional chemotherapy given to patients with pre-BCR+ ALL, or even replace chemotherapy altogether.
“In our experiments with mice, both combination therapy with low-dose chemotherapy and single-agent targeted therapy each worked very well,” he said.
Inhibitor outperforms BAT in myelofibrosis trial

Image by Peter Anderson
Results of a phase 3 trial suggest the JAK2/FLT3 inhibitor pacritinib may be more effective for patients with myelofibrosis (MF) than best available therapy (BAT), excluding JAK inhibitors.
Patients who received pacritinib were more likely than those who received BAT to see a reduction in spleen volume and to become transfusion-independent,
regardless of their platelet counts.
In fact, pacritinib proved particularly effective among patients with severe thrombocytopenia.
CTI Biopharma and Baxter International, Inc., the companies developing pacritinib, announced these results from the PERSIST-1 trial yesterday.
“Despite the introduction of JAK2 inhibitors as effective therapies for patients with myelofibrosis, there remains a treatment gap for patients with disease-related or treatment-emergent thrombocytopenia,” said study investigator Claire Harrison, MD, of Guy’s Hospital in London, UK.
“The currently approved drug [ruxolitinib] may require dose titration to less effective doses in this patient population, thus limiting our ability to effectively treat them. Results from the PERSIST-1 randomized trial demonstrate that pacritinib could address this unmet medical need.”
PERSIST-1 is a randomized (2:1), phase 3 trial comparing the safety and efficacy of pacritinib to BAT, other than JAK inhibitors. Investigators enrolled 327 patients with primary and secondary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF.
The study’s primary endpoint was the proportion of patients achieving a 35% or greater reduction in spleen volume from baseline to week 24, as measured by MRI or CT, when compared with BAT. Once patients completed 24 weeks of treatment, or if their disease progressed, they could cross over from the BAT arm to the pacritinib arm.
Investigators found that PERSIST-1 met its primary endpoint in the intent-to-treat population. Pacritinib produced a significantly better rate of spleen volume reduction (P=0.0003) when compared to BAT.
The same was true among patients with platelet counts of less than 100,000/μL and less than 50,000/μL, both subgroups that were stratified at randomization.
The magnitude of treatment effect was consistent with previously reported phase 2 results, with the greatest reduction observed among the sickest patients (platelet counts <50,000/μL).
Fifty patients were transfusion-dependent at study entry (receiving ≥ 6 units of red blood cells over 90 days pre-entry). And, compared to BAT, pacritinib resulted in a clinically meaningful percentage of patients becoming transfusion-independent.
Ultimately, 79% of patients in the BAT arm crossed over to the pacritinib arm.
Investigators said the safety profile in the PERSIST-1 trial was consistent with prior phase 2 trials, as the most common treatment-emergent adverse events were diarrhea, nausea, and vomiting. However, the incidence of grade 3 events was lower than previously observed in phase 2 trials. And no grade 4 gastrointestinal adverse events were reported.
Three patients discontinued treatment with pacritinib, and 9 required dose reductions for diarrhea. A preliminary analysis suggested that few patients discontinued pacritinib or required a dose reduction due to treatment-related anemia or thrombocytopenia.
Additional data from ongoing analyses will be submitted for presentation at an upcoming scientific meeting, according to Baxter and CTI Biopharma.
Image by Peter Anderson
Results of a phase 3 trial suggest the JAK2/FLT3 inhibitor pacritinib may be more effective for patients with myelofibrosis (MF) than best available therapy (BAT), excluding JAK inhibitors.
Patients who received pacritinib were more likely than those who received BAT to see a reduction in spleen volume and to become transfusion-independent,
regardless of their platelet counts.
In fact, pacritinib proved particularly effective among patients with severe thrombocytopenia.
CTI Biopharma and Baxter International, Inc., the companies developing pacritinib, announced these results from the PERSIST-1 trial yesterday.
“Despite the introduction of JAK2 inhibitors as effective therapies for patients with myelofibrosis, there remains a treatment gap for patients with disease-related or treatment-emergent thrombocytopenia,” said study investigator Claire Harrison, MD, of Guy’s Hospital in London, UK.
“The currently approved drug [ruxolitinib] may require dose titration to less effective doses in this patient population, thus limiting our ability to effectively treat them. Results from the PERSIST-1 randomized trial demonstrate that pacritinib could address this unmet medical need.”
PERSIST-1 is a randomized (2:1), phase 3 trial comparing the safety and efficacy of pacritinib to BAT, other than JAK inhibitors. Investigators enrolled 327 patients with primary and secondary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF.
The study’s primary endpoint was the proportion of patients achieving a 35% or greater reduction in spleen volume from baseline to week 24, as measured by MRI or CT, when compared with BAT. Once patients completed 24 weeks of treatment, or if their disease progressed, they could cross over from the BAT arm to the pacritinib arm.
Investigators found that PERSIST-1 met its primary endpoint in the intent-to-treat population. Pacritinib produced a significantly better rate of spleen volume reduction (P=0.0003) when compared to BAT.
The same was true among patients with platelet counts of less than 100,000/μL and less than 50,000/μL, both subgroups that were stratified at randomization.
The magnitude of treatment effect was consistent with previously reported phase 2 results, with the greatest reduction observed among the sickest patients (platelet counts <50,000/μL).
Fifty patients were transfusion-dependent at study entry (receiving ≥ 6 units of red blood cells over 90 days pre-entry). And, compared to BAT, pacritinib resulted in a clinically meaningful percentage of patients becoming transfusion-independent.
Ultimately, 79% of patients in the BAT arm crossed over to the pacritinib arm.
Investigators said the safety profile in the PERSIST-1 trial was consistent with prior phase 2 trials, as the most common treatment-emergent adverse events were diarrhea, nausea, and vomiting. However, the incidence of grade 3 events was lower than previously observed in phase 2 trials. And no grade 4 gastrointestinal adverse events were reported.
Three patients discontinued treatment with pacritinib, and 9 required dose reductions for diarrhea. A preliminary analysis suggested that few patients discontinued pacritinib or required a dose reduction due to treatment-related anemia or thrombocytopenia.
Additional data from ongoing analyses will be submitted for presentation at an upcoming scientific meeting, according to Baxter and CTI Biopharma.
Image by Peter Anderson
Results of a phase 3 trial suggest the JAK2/FLT3 inhibitor pacritinib may be more effective for patients with myelofibrosis (MF) than best available therapy (BAT), excluding JAK inhibitors.
Patients who received pacritinib were more likely than those who received BAT to see a reduction in spleen volume and to become transfusion-independent,
regardless of their platelet counts.
In fact, pacritinib proved particularly effective among patients with severe thrombocytopenia.
CTI Biopharma and Baxter International, Inc., the companies developing pacritinib, announced these results from the PERSIST-1 trial yesterday.
“Despite the introduction of JAK2 inhibitors as effective therapies for patients with myelofibrosis, there remains a treatment gap for patients with disease-related or treatment-emergent thrombocytopenia,” said study investigator Claire Harrison, MD, of Guy’s Hospital in London, UK.
“The currently approved drug [ruxolitinib] may require dose titration to less effective doses in this patient population, thus limiting our ability to effectively treat them. Results from the PERSIST-1 randomized trial demonstrate that pacritinib could address this unmet medical need.”
PERSIST-1 is a randomized (2:1), phase 3 trial comparing the safety and efficacy of pacritinib to BAT, other than JAK inhibitors. Investigators enrolled 327 patients with primary and secondary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF.
The study’s primary endpoint was the proportion of patients achieving a 35% or greater reduction in spleen volume from baseline to week 24, as measured by MRI or CT, when compared with BAT. Once patients completed 24 weeks of treatment, or if their disease progressed, they could cross over from the BAT arm to the pacritinib arm.
Investigators found that PERSIST-1 met its primary endpoint in the intent-to-treat population. Pacritinib produced a significantly better rate of spleen volume reduction (P=0.0003) when compared to BAT.
The same was true among patients with platelet counts of less than 100,000/μL and less than 50,000/μL, both subgroups that were stratified at randomization.
The magnitude of treatment effect was consistent with previously reported phase 2 results, with the greatest reduction observed among the sickest patients (platelet counts <50,000/μL).
Fifty patients were transfusion-dependent at study entry (receiving ≥ 6 units of red blood cells over 90 days pre-entry). And, compared to BAT, pacritinib resulted in a clinically meaningful percentage of patients becoming transfusion-independent.
Ultimately, 79% of patients in the BAT arm crossed over to the pacritinib arm.
Investigators said the safety profile in the PERSIST-1 trial was consistent with prior phase 2 trials, as the most common treatment-emergent adverse events were diarrhea, nausea, and vomiting. However, the incidence of grade 3 events was lower than previously observed in phase 2 trials. And no grade 4 gastrointestinal adverse events were reported.
Three patients discontinued treatment with pacritinib, and 9 required dose reductions for diarrhea. A preliminary analysis suggested that few patients discontinued pacritinib or required a dose reduction due to treatment-related anemia or thrombocytopenia.
Additional data from ongoing analyses will be submitted for presentation at an upcoming scientific meeting, according to Baxter and CTI Biopharma.
ASH advocates use of systems-based hematologists

Photo courtesy of CDC
The American Society of Hematology (ASH) has released a report proposing a new role for hematologists specializing in non-malignant blood disorders.
ASH partnered with the healthcare consulting firm The Lewin Group to identify emerging career opportunities for health system- and hospital-based hematologists and to provide guidance on pursuing those opportunities.
The resulting report, published in Blood, outlines a few models for a systems-based clinical hematologist.
The report’s authors noted that demand for hematology expertise remains high nationwide. However, ASH and its members are concerned that changes to academic training will hinder both the recruitment of new talent to the field and the retention of seasoned experts.
The authors said that today’s hematology trainees are unlikely to receive the same non-malignant training as many “classic” hematologists trained in prior decades. And training shortfalls are further compounded by the fact that primary care physicians do not have the expertise to manage common blood disorders, which increases referrals to hematologists.
This results in higher demand for a smaller pool of hematologists entering the field with adequate training to effectively and efficiently manage non-malignant disorders.
“Given the rapid evolution and complexity of the field, the time is appropriate to identify career pathways that attract and enable physicians to practice non-malignant hematology in a sustainable manner,” said author Janis L. Abkowitz, MD, of the University of Washington in Seattle.
She and her colleagues noted that, in response to these challenges, US hematologists are defining new paths and assuming more centralized positions in large and small healthcare systems.
These systems-based hematologists are specialty-trained physicians—employed by a hospital, medical center, or health system—who optimize individual patient care as well as the overall system of healthcare delivery for patients with blood disorders.
For example, a systems-based hematologist could work closely with surgeons to minimize perioperative bleeding and could manage care pathways for patients with chronic blood diseases.
The report offered 4 examples where the involvement of a systems-based hematologist would lead to cost-effective decision-making. These were based upon interviews with 14 early adoptors of the systems-based approach to hematology.
The first example was heparin-induced thrombocytopenia (HIT). A systems-based hematologist could implement care pathways that focus on HIT by working to reduce unnecessary heparin exposure, optimizing laboratory testing for suspected HIT, and reducing unnecessary procedures in patients.
The second example was thrombotic thrombocytopenic purpura (TTP). A systems-based hematologist could optimize testing for TTP, which may reduce system-wide plasma use.
The third example was a medical director for hemostasis and thrombosis. A systems-based hematologist could foster appropriate and safe practices, including the implementation of and adherence to preventive care for thrombotic events and the optimal use of anticoagulant medications.
The fourth example was non-malignant hematology consultation in an accountable care organization (ACO) environment. The authors noted that ACOs have enabled more patients to be served by a health system, but there are fewer incentives for physicians to manage common hematology-related issues. A funded systems-based hematologist could ensure that patients have more timely access to hematology consultations.
“A systems-based hematologist position presents a unique opportunity for hematologists to design new models for care delivery and demonstrate their ability to improve clinical outcomes while maintaining or reducing costs,” Dr Abkowitz said. “Just as blood must flow throughout the body, the expertise of hematology must flow throughout the healthcare system.”
As a next step, ASH has invited its members to share practice models they have developed and examples of how they have collaborated with others to improve healthcare outcomes, reduce complications, and eliminate unnecessary spending.
Photo courtesy of CDC
The American Society of Hematology (ASH) has released a report proposing a new role for hematologists specializing in non-malignant blood disorders.
ASH partnered with the healthcare consulting firm The Lewin Group to identify emerging career opportunities for health system- and hospital-based hematologists and to provide guidance on pursuing those opportunities.
The resulting report, published in Blood, outlines a few models for a systems-based clinical hematologist.
The report’s authors noted that demand for hematology expertise remains high nationwide. However, ASH and its members are concerned that changes to academic training will hinder both the recruitment of new talent to the field and the retention of seasoned experts.
The authors said that today’s hematology trainees are unlikely to receive the same non-malignant training as many “classic” hematologists trained in prior decades. And training shortfalls are further compounded by the fact that primary care physicians do not have the expertise to manage common blood disorders, which increases referrals to hematologists.
This results in higher demand for a smaller pool of hematologists entering the field with adequate training to effectively and efficiently manage non-malignant disorders.
“Given the rapid evolution and complexity of the field, the time is appropriate to identify career pathways that attract and enable physicians to practice non-malignant hematology in a sustainable manner,” said author Janis L. Abkowitz, MD, of the University of Washington in Seattle.
She and her colleagues noted that, in response to these challenges, US hematologists are defining new paths and assuming more centralized positions in large and small healthcare systems.
These systems-based hematologists are specialty-trained physicians—employed by a hospital, medical center, or health system—who optimize individual patient care as well as the overall system of healthcare delivery for patients with blood disorders.
For example, a systems-based hematologist could work closely with surgeons to minimize perioperative bleeding and could manage care pathways for patients with chronic blood diseases.
The report offered 4 examples where the involvement of a systems-based hematologist would lead to cost-effective decision-making. These were based upon interviews with 14 early adoptors of the systems-based approach to hematology.
The first example was heparin-induced thrombocytopenia (HIT). A systems-based hematologist could implement care pathways that focus on HIT by working to reduce unnecessary heparin exposure, optimizing laboratory testing for suspected HIT, and reducing unnecessary procedures in patients.
The second example was thrombotic thrombocytopenic purpura (TTP). A systems-based hematologist could optimize testing for TTP, which may reduce system-wide plasma use.
The third example was a medical director for hemostasis and thrombosis. A systems-based hematologist could foster appropriate and safe practices, including the implementation of and adherence to preventive care for thrombotic events and the optimal use of anticoagulant medications.
The fourth example was non-malignant hematology consultation in an accountable care organization (ACO) environment. The authors noted that ACOs have enabled more patients to be served by a health system, but there are fewer incentives for physicians to manage common hematology-related issues. A funded systems-based hematologist could ensure that patients have more timely access to hematology consultations.
“A systems-based hematologist position presents a unique opportunity for hematologists to design new models for care delivery and demonstrate their ability to improve clinical outcomes while maintaining or reducing costs,” Dr Abkowitz said. “Just as blood must flow throughout the body, the expertise of hematology must flow throughout the healthcare system.”
As a next step, ASH has invited its members to share practice models they have developed and examples of how they have collaborated with others to improve healthcare outcomes, reduce complications, and eliminate unnecessary spending.
Photo courtesy of CDC
The American Society of Hematology (ASH) has released a report proposing a new role for hematologists specializing in non-malignant blood disorders.
ASH partnered with the healthcare consulting firm The Lewin Group to identify emerging career opportunities for health system- and hospital-based hematologists and to provide guidance on pursuing those opportunities.
The resulting report, published in Blood, outlines a few models for a systems-based clinical hematologist.
The report’s authors noted that demand for hematology expertise remains high nationwide. However, ASH and its members are concerned that changes to academic training will hinder both the recruitment of new talent to the field and the retention of seasoned experts.
The authors said that today’s hematology trainees are unlikely to receive the same non-malignant training as many “classic” hematologists trained in prior decades. And training shortfalls are further compounded by the fact that primary care physicians do not have the expertise to manage common blood disorders, which increases referrals to hematologists.
This results in higher demand for a smaller pool of hematologists entering the field with adequate training to effectively and efficiently manage non-malignant disorders.
“Given the rapid evolution and complexity of the field, the time is appropriate to identify career pathways that attract and enable physicians to practice non-malignant hematology in a sustainable manner,” said author Janis L. Abkowitz, MD, of the University of Washington in Seattle.
She and her colleagues noted that, in response to these challenges, US hematologists are defining new paths and assuming more centralized positions in large and small healthcare systems.
These systems-based hematologists are specialty-trained physicians—employed by a hospital, medical center, or health system—who optimize individual patient care as well as the overall system of healthcare delivery for patients with blood disorders.
For example, a systems-based hematologist could work closely with surgeons to minimize perioperative bleeding and could manage care pathways for patients with chronic blood diseases.
The report offered 4 examples where the involvement of a systems-based hematologist would lead to cost-effective decision-making. These were based upon interviews with 14 early adoptors of the systems-based approach to hematology.
The first example was heparin-induced thrombocytopenia (HIT). A systems-based hematologist could implement care pathways that focus on HIT by working to reduce unnecessary heparin exposure, optimizing laboratory testing for suspected HIT, and reducing unnecessary procedures in patients.
The second example was thrombotic thrombocytopenic purpura (TTP). A systems-based hematologist could optimize testing for TTP, which may reduce system-wide plasma use.
The third example was a medical director for hemostasis and thrombosis. A systems-based hematologist could foster appropriate and safe practices, including the implementation of and adherence to preventive care for thrombotic events and the optimal use of anticoagulant medications.
The fourth example was non-malignant hematology consultation in an accountable care organization (ACO) environment. The authors noted that ACOs have enabled more patients to be served by a health system, but there are fewer incentives for physicians to manage common hematology-related issues. A funded systems-based hematologist could ensure that patients have more timely access to hematology consultations.
“A systems-based hematologist position presents a unique opportunity for hematologists to design new models for care delivery and demonstrate their ability to improve clinical outcomes while maintaining or reducing costs,” Dr Abkowitz said. “Just as blood must flow throughout the body, the expertise of hematology must flow throughout the healthcare system.”
As a next step, ASH has invited its members to share practice models they have developed and examples of how they have collaborated with others to improve healthcare outcomes, reduce complications, and eliminate unnecessary spending.
New test can better predict cytokine storm, team says

Photo by Juan D. Alfonso
Scientists have developed a test that uses cells from a single donor’s blood to predict whether a new drug will cause a cytokine storm.
The group says this is an improvement over current tests, which use endothelial cells and peripheral blood mononuclear cells (PBMCs) from two separate donors and can therefore produce inaccurate results.
Furthermore, current tests cannot differentiate drugs that induce a mild cytokine storm from those that induce a severe one, but the new test can.
Jane Mitchell, PhD, of the National Heart and Lung Institute at Imperial College London in the UK, and her colleagues described the new test in The FASEB Journal.
Current tests for cytokine storm reactions use endothelial cells taken from the vessels of one donor and PBMCs from a different donor because endothelial cells are normally only grown from tissue removed in surgery or post-mortem, or from umbilical vessels after birth.
When cells from two different donors are used, one may have an immune reaction to the other. And this can result in the test falsely showing a severe immune reaction to a drug that is safe.
Dr Mitchell and her colleagues say they have solved this problem by isolating stem cells from the blood of a volunteer and using them to grow endothelial cells in a dish. The team then added PBMCs to the donor’s own endothelial cells to recreate the unique conditions found in their blood vessels.
When the scientists added the immunomodulatory drug TGN1412, the mixture of cells released a cytokine storm, as would happen inside the human body.
Responses to other drugs were consistent with those observed in humans as well. There was a modest response to alemtuzumab (Campath) and no response to the control antibodies trastuzumab (Herceptin), bevacizumab (Avastin), and ofatumumab (Arzerra).
“As biological therapies become more mainstream, it’s more likely that drugs being tested on humans for the first time will have unexpected and potentially catastrophic effects,” Dr Mitchell said.
“We’ve used adult stem cell technology to develop a laboratory test that could prevent another disaster like the TGN1412 trial [in which 6 healthy young men developed multi-organ failure]. Drug companies have the technical capacity to start using this test now, but we’re working on developing an off-the-shelf kit, which will make it easy to use on a large scale.”
The team has collaborated with the National Institute for Biological Standards and Control to validate the test and are now working with the clinical trials company Quintiles to develop the technology further.
Photo by Juan D. Alfonso
Scientists have developed a test that uses cells from a single donor’s blood to predict whether a new drug will cause a cytokine storm.
The group says this is an improvement over current tests, which use endothelial cells and peripheral blood mononuclear cells (PBMCs) from two separate donors and can therefore produce inaccurate results.
Furthermore, current tests cannot differentiate drugs that induce a mild cytokine storm from those that induce a severe one, but the new test can.
Jane Mitchell, PhD, of the National Heart and Lung Institute at Imperial College London in the UK, and her colleagues described the new test in The FASEB Journal.
Current tests for cytokine storm reactions use endothelial cells taken from the vessels of one donor and PBMCs from a different donor because endothelial cells are normally only grown from tissue removed in surgery or post-mortem, or from umbilical vessels after birth.
When cells from two different donors are used, one may have an immune reaction to the other. And this can result in the test falsely showing a severe immune reaction to a drug that is safe.
Dr Mitchell and her colleagues say they have solved this problem by isolating stem cells from the blood of a volunteer and using them to grow endothelial cells in a dish. The team then added PBMCs to the donor’s own endothelial cells to recreate the unique conditions found in their blood vessels.
When the scientists added the immunomodulatory drug TGN1412, the mixture of cells released a cytokine storm, as would happen inside the human body.
Responses to other drugs were consistent with those observed in humans as well. There was a modest response to alemtuzumab (Campath) and no response to the control antibodies trastuzumab (Herceptin), bevacizumab (Avastin), and ofatumumab (Arzerra).
“As biological therapies become more mainstream, it’s more likely that drugs being tested on humans for the first time will have unexpected and potentially catastrophic effects,” Dr Mitchell said.
“We’ve used adult stem cell technology to develop a laboratory test that could prevent another disaster like the TGN1412 trial [in which 6 healthy young men developed multi-organ failure]. Drug companies have the technical capacity to start using this test now, but we’re working on developing an off-the-shelf kit, which will make it easy to use on a large scale.”
The team has collaborated with the National Institute for Biological Standards and Control to validate the test and are now working with the clinical trials company Quintiles to develop the technology further.
Photo by Juan D. Alfonso
Scientists have developed a test that uses cells from a single donor’s blood to predict whether a new drug will cause a cytokine storm.
The group says this is an improvement over current tests, which use endothelial cells and peripheral blood mononuclear cells (PBMCs) from two separate donors and can therefore produce inaccurate results.
Furthermore, current tests cannot differentiate drugs that induce a mild cytokine storm from those that induce a severe one, but the new test can.
Jane Mitchell, PhD, of the National Heart and Lung Institute at Imperial College London in the UK, and her colleagues described the new test in The FASEB Journal.
Current tests for cytokine storm reactions use endothelial cells taken from the vessels of one donor and PBMCs from a different donor because endothelial cells are normally only grown from tissue removed in surgery or post-mortem, or from umbilical vessels after birth.
When cells from two different donors are used, one may have an immune reaction to the other. And this can result in the test falsely showing a severe immune reaction to a drug that is safe.
Dr Mitchell and her colleagues say they have solved this problem by isolating stem cells from the blood of a volunteer and using them to grow endothelial cells in a dish. The team then added PBMCs to the donor’s own endothelial cells to recreate the unique conditions found in their blood vessels.
When the scientists added the immunomodulatory drug TGN1412, the mixture of cells released a cytokine storm, as would happen inside the human body.
Responses to other drugs were consistent with those observed in humans as well. There was a modest response to alemtuzumab (Campath) and no response to the control antibodies trastuzumab (Herceptin), bevacizumab (Avastin), and ofatumumab (Arzerra).
“As biological therapies become more mainstream, it’s more likely that drugs being tested on humans for the first time will have unexpected and potentially catastrophic effects,” Dr Mitchell said.
“We’ve used adult stem cell technology to develop a laboratory test that could prevent another disaster like the TGN1412 trial [in which 6 healthy young men developed multi-organ failure]. Drug companies have the technical capacity to start using this test now, but we’re working on developing an off-the-shelf kit, which will make it easy to use on a large scale.”
The team has collaborated with the National Institute for Biological Standards and Control to validate the test and are now working with the clinical trials company Quintiles to develop the technology further.