User login
Inhibitor may overcome ibrutinib resistance in MCL
Investigators have identified a mechanism of ibrutinib resistance in mantle cell lymphoma (MCL) and showed that a small molecule can overcome that resistance in vitro and in vivo.

The team found that ibrutinib-resistant MCL cells rely on oxidative phosphorylation (OXPHOS) and glutaminolysis to survive.
Targeting the OXPHOS pathway with a small molecule, IACS-010759, inhibited the proliferation of ibrutinib-resistant cells in vitro.
IACS-010759 also decreased tumor volume and improved survival in mouse models of ibrutinib-resistant MCL and double-hit B-cell lymphoma.
Now, IACS-10759 is being tested in phase 1 trials of lymphoma and solid tumors (NCT03291938) as well as acute myeloid leukemia (NCT02882321).
Liang Zhang, MD, PhD, of the University of Texas MD Anderson Cancer Center in Houston, and his colleagues conducted the preclinical research and described their findings in Science Translational Medicine.
The investigators sequenced samples from MCL patients with ibrutinib-sensitive and -resistant disease and found that “glutamine-fueled OXPHOS appears to be a prominent energy metabolism pathway in ibrutinib-resistant MCL cells.”
This finding prompted the team to test IACS-010759, an inhibitor of ETC complex I, in ibrutinib-resistant MCL. They theorized that the inhibitor would be effective because, during OXPHOS, electrons are transferred from electron donors to acceptors through the ETC in redox reactions that release energy to form ATP, and OXPHOS generates ATP to meet requirements for cell growth.
In experiments, IACS-010759 inhibited the proliferation of two ibrutinib-resistant MCL cell lines, Z-138 and Maver-1, in a dose-dependent manner.
The investigators also tested IACS-010759 in two mouse models of ibrutinib-resistant MCL. In both models, mice treated with IACS-010759 had a significant reduction in tumor volume, compared with controls. In one model, IACS-010759 extended survival by a median of 11 days.
Finally, the team tested IACS-010759 in a model of ibrutinib-resistant, double-hit (MYC and BCL-2) B-cell lymphoma with central nervous system involvement. Again, IACS-010759 significantly inhibited tumor growth. Compared to ibrutinib and vehicle control, IACS-010759 provided a median survival benefit of more than 20 days.
There were no toxicities associated with IACS-010759 treatment, according to the investigators.
This research was supported by the MD Anderson B Cell Lymphoma Moon Shot Project, Gary Rogers Foundation, Kinder Foundation, Cullen Foundation, Cancer Prevention Research Institute of Texas, and the National Institutes of Health. Most investigators reported having no competing interests, but two reported a patent (WO/2015/130790).
SOURCE: Zhang L et al. Sci Transl Med. 2019 May 8. doi: 10.1126/scitranslmed.aau1167.
Investigators have identified a mechanism of ibrutinib resistance in mantle cell lymphoma (MCL) and showed that a small molecule can overcome that resistance in vitro and in vivo.
The team found that ibrutinib-resistant MCL cells rely on oxidative phosphorylation (OXPHOS) and glutaminolysis to survive.
Targeting the OXPHOS pathway with a small molecule, IACS-010759, inhibited the proliferation of ibrutinib-resistant cells in vitro.
IACS-010759 also decreased tumor volume and improved survival in mouse models of ibrutinib-resistant MCL and double-hit B-cell lymphoma.
Now, IACS-10759 is being tested in phase 1 trials of lymphoma and solid tumors (NCT03291938) as well as acute myeloid leukemia (NCT02882321).
Liang Zhang, MD, PhD, of the University of Texas MD Anderson Cancer Center in Houston, and his colleagues conducted the preclinical research and described their findings in Science Translational Medicine.
The investigators sequenced samples from MCL patients with ibrutinib-sensitive and -resistant disease and found that “glutamine-fueled OXPHOS appears to be a prominent energy metabolism pathway in ibrutinib-resistant MCL cells.”
This finding prompted the team to test IACS-010759, an inhibitor of ETC complex I, in ibrutinib-resistant MCL. They theorized that the inhibitor would be effective because, during OXPHOS, electrons are transferred from electron donors to acceptors through the ETC in redox reactions that release energy to form ATP, and OXPHOS generates ATP to meet requirements for cell growth.
In experiments, IACS-010759 inhibited the proliferation of two ibrutinib-resistant MCL cell lines, Z-138 and Maver-1, in a dose-dependent manner.
The investigators also tested IACS-010759 in two mouse models of ibrutinib-resistant MCL. In both models, mice treated with IACS-010759 had a significant reduction in tumor volume, compared with controls. In one model, IACS-010759 extended survival by a median of 11 days.
Finally, the team tested IACS-010759 in a model of ibrutinib-resistant, double-hit (MYC and BCL-2) B-cell lymphoma with central nervous system involvement. Again, IACS-010759 significantly inhibited tumor growth. Compared to ibrutinib and vehicle control, IACS-010759 provided a median survival benefit of more than 20 days.
There were no toxicities associated with IACS-010759 treatment, according to the investigators.
This research was supported by the MD Anderson B Cell Lymphoma Moon Shot Project, Gary Rogers Foundation, Kinder Foundation, Cullen Foundation, Cancer Prevention Research Institute of Texas, and the National Institutes of Health. Most investigators reported having no competing interests, but two reported a patent (WO/2015/130790).
SOURCE: Zhang L et al. Sci Transl Med. 2019 May 8. doi: 10.1126/scitranslmed.aau1167.
Investigators have identified a mechanism of ibrutinib resistance in mantle cell lymphoma (MCL) and showed that a small molecule can overcome that resistance in vitro and in vivo.
The team found that ibrutinib-resistant MCL cells rely on oxidative phosphorylation (OXPHOS) and glutaminolysis to survive.
Targeting the OXPHOS pathway with a small molecule, IACS-010759, inhibited the proliferation of ibrutinib-resistant cells in vitro.
IACS-010759 also decreased tumor volume and improved survival in mouse models of ibrutinib-resistant MCL and double-hit B-cell lymphoma.
Now, IACS-10759 is being tested in phase 1 trials of lymphoma and solid tumors (NCT03291938) as well as acute myeloid leukemia (NCT02882321).
Liang Zhang, MD, PhD, of the University of Texas MD Anderson Cancer Center in Houston, and his colleagues conducted the preclinical research and described their findings in Science Translational Medicine.
The investigators sequenced samples from MCL patients with ibrutinib-sensitive and -resistant disease and found that “glutamine-fueled OXPHOS appears to be a prominent energy metabolism pathway in ibrutinib-resistant MCL cells.”
This finding prompted the team to test IACS-010759, an inhibitor of ETC complex I, in ibrutinib-resistant MCL. They theorized that the inhibitor would be effective because, during OXPHOS, electrons are transferred from electron donors to acceptors through the ETC in redox reactions that release energy to form ATP, and OXPHOS generates ATP to meet requirements for cell growth.
In experiments, IACS-010759 inhibited the proliferation of two ibrutinib-resistant MCL cell lines, Z-138 and Maver-1, in a dose-dependent manner.
The investigators also tested IACS-010759 in two mouse models of ibrutinib-resistant MCL. In both models, mice treated with IACS-010759 had a significant reduction in tumor volume, compared with controls. In one model, IACS-010759 extended survival by a median of 11 days.
Finally, the team tested IACS-010759 in a model of ibrutinib-resistant, double-hit (MYC and BCL-2) B-cell lymphoma with central nervous system involvement. Again, IACS-010759 significantly inhibited tumor growth. Compared to ibrutinib and vehicle control, IACS-010759 provided a median survival benefit of more than 20 days.
There were no toxicities associated with IACS-010759 treatment, according to the investigators.
This research was supported by the MD Anderson B Cell Lymphoma Moon Shot Project, Gary Rogers Foundation, Kinder Foundation, Cullen Foundation, Cancer Prevention Research Institute of Texas, and the National Institutes of Health. Most investigators reported having no competing interests, but two reported a patent (WO/2015/130790).
SOURCE: Zhang L et al. Sci Transl Med. 2019 May 8. doi: 10.1126/scitranslmed.aau1167.
FROM SCIENCE TRANSLATIONAL MEDICINE
Study finds lower quality of life for patients with hemophilia A
Real‐world data suggests that patients with hemophilia A with inhibitors had lower health-related quality of life (HRQOL) while receiving standard therapy, according to an international study.
“The objective of this analysis was to characterize disease‐specific HRQOL, overall health status and the effect of bleeding on health status,” wrote Johnny Mahlangu, MD, of the University of the Witwatersrand in Johannesburg, South Africa, and colleagues. The study was published in Haemophilia.
The researchers conducted a prospective, noninterventional study of 103 patients aged 12 years and older with hemophilia A who resided in several different countries, including Australia, Japan, South Africa, and the United States, among others.
The majority of participants (n = 75) received episodic treatment at study enrollment, while others (n = 28) received prophylactic-based therapy. Patients were treated with standard therapy, based on local institutional practice.
HRQOL outcome data were collected in adult and adolescent participants using the Haemophilia Quality of Life Questionnaire for Adults and the Haemophilia‐specific Quality of Life Questionnaire for Children Short Form. Other validated instruments were used to measure additional health‐related outcomes.
After analysis, the researchers found that HRQOL scores revealed impaired quality of life in adult and adolescent participants treated with both episodic and prophylactic regimens. The mean scores in the majority of HRQOL domains showed impairments occurring on average “sometimes” to “often,” the researchers reported.
Adults had highest scores, correlated with greatest impairments, in sports and leisure. Similarly, adolescents reported greatest impairment in the sports and school domain.
“These health‐related outcomes may result from a combination of poor bleed control and treatment burden,” the researchers wrote. “Compliance with prophylactic treatment was low, likely reflecting the high burden associated with standard therapies.”
The researchers acknowledged a key limitation of the study was participant dropout. As a result, some time-related data may be incomplete.
“These [data] demonstrate that patients with hemophilia A with inhibitors have impaired HRQOL, despite standard treatment, and that more effective treatment options are needed,” the researchers concluded.
The study was funded by F. Hoffmann-La Roche. The authors reported financial affiliations with Baxalta, Bayer, CSL Behring, Kaketsuken, Novo Nordisk, Pfizer, and several others.
SOURCE: Mahlangu J et al. Haemophilia. 2019 Apr 24. doi: 10.1111/hae.13731.
Real‐world data suggests that patients with hemophilia A with inhibitors had lower health-related quality of life (HRQOL) while receiving standard therapy, according to an international study.
“The objective of this analysis was to characterize disease‐specific HRQOL, overall health status and the effect of bleeding on health status,” wrote Johnny Mahlangu, MD, of the University of the Witwatersrand in Johannesburg, South Africa, and colleagues. The study was published in Haemophilia.
The researchers conducted a prospective, noninterventional study of 103 patients aged 12 years and older with hemophilia A who resided in several different countries, including Australia, Japan, South Africa, and the United States, among others.
The majority of participants (n = 75) received episodic treatment at study enrollment, while others (n = 28) received prophylactic-based therapy. Patients were treated with standard therapy, based on local institutional practice.
HRQOL outcome data were collected in adult and adolescent participants using the Haemophilia Quality of Life Questionnaire for Adults and the Haemophilia‐specific Quality of Life Questionnaire for Children Short Form. Other validated instruments were used to measure additional health‐related outcomes.
After analysis, the researchers found that HRQOL scores revealed impaired quality of life in adult and adolescent participants treated with both episodic and prophylactic regimens. The mean scores in the majority of HRQOL domains showed impairments occurring on average “sometimes” to “often,” the researchers reported.
Adults had highest scores, correlated with greatest impairments, in sports and leisure. Similarly, adolescents reported greatest impairment in the sports and school domain.
“These health‐related outcomes may result from a combination of poor bleed control and treatment burden,” the researchers wrote. “Compliance with prophylactic treatment was low, likely reflecting the high burden associated with standard therapies.”
The researchers acknowledged a key limitation of the study was participant dropout. As a result, some time-related data may be incomplete.
“These [data] demonstrate that patients with hemophilia A with inhibitors have impaired HRQOL, despite standard treatment, and that more effective treatment options are needed,” the researchers concluded.
The study was funded by F. Hoffmann-La Roche. The authors reported financial affiliations with Baxalta, Bayer, CSL Behring, Kaketsuken, Novo Nordisk, Pfizer, and several others.
SOURCE: Mahlangu J et al. Haemophilia. 2019 Apr 24. doi: 10.1111/hae.13731.
Real‐world data suggests that patients with hemophilia A with inhibitors had lower health-related quality of life (HRQOL) while receiving standard therapy, according to an international study.
“The objective of this analysis was to characterize disease‐specific HRQOL, overall health status and the effect of bleeding on health status,” wrote Johnny Mahlangu, MD, of the University of the Witwatersrand in Johannesburg, South Africa, and colleagues. The study was published in Haemophilia.
The researchers conducted a prospective, noninterventional study of 103 patients aged 12 years and older with hemophilia A who resided in several different countries, including Australia, Japan, South Africa, and the United States, among others.
The majority of participants (n = 75) received episodic treatment at study enrollment, while others (n = 28) received prophylactic-based therapy. Patients were treated with standard therapy, based on local institutional practice.
HRQOL outcome data were collected in adult and adolescent participants using the Haemophilia Quality of Life Questionnaire for Adults and the Haemophilia‐specific Quality of Life Questionnaire for Children Short Form. Other validated instruments were used to measure additional health‐related outcomes.
After analysis, the researchers found that HRQOL scores revealed impaired quality of life in adult and adolescent participants treated with both episodic and prophylactic regimens. The mean scores in the majority of HRQOL domains showed impairments occurring on average “sometimes” to “often,” the researchers reported.
Adults had highest scores, correlated with greatest impairments, in sports and leisure. Similarly, adolescents reported greatest impairment in the sports and school domain.
“These health‐related outcomes may result from a combination of poor bleed control and treatment burden,” the researchers wrote. “Compliance with prophylactic treatment was low, likely reflecting the high burden associated with standard therapies.”
The researchers acknowledged a key limitation of the study was participant dropout. As a result, some time-related data may be incomplete.
“These [data] demonstrate that patients with hemophilia A with inhibitors have impaired HRQOL, despite standard treatment, and that more effective treatment options are needed,” the researchers concluded.
The study was funded by F. Hoffmann-La Roche. The authors reported financial affiliations with Baxalta, Bayer, CSL Behring, Kaketsuken, Novo Nordisk, Pfizer, and several others.
SOURCE: Mahlangu J et al. Haemophilia. 2019 Apr 24. doi: 10.1111/hae.13731.
FROM HAEMOPHILIA
New recommendations on immune tolerance induction in hemophilia A
New treatment recommendations, released by a panel of nine experts, offer consensus advice on the use of immune tolerance induction (ITI) therapy in patients with hemophilia A with inhibitors.
The recommendations from the Future of Immunotolerance Treatment (FIT) group were authored by a nine-member committee with expertise in the treatment of hemophilia. The authors attended three meetings from 2017-2018 to form a consensus on the use of nonfactor therapies with current inhibitor management strategies.
“The treatment of hemophilia A has evolved and a number of molecules that potentially can be used in the setting of patients with inhibitors have been developed, or are in various phases of development,” wrote Manuel Carcao, MD, of the University of Toronto and colleagues. The report is published in Haemophilia.
The current body of literature is lacking high-quality evidence on the concomitant use of nonfactor treatments, such as emicizumab, and current inhibitor therapies. The recommendations included the panel’s consensus opinions on treatment of inhibitors, with and without the use of nonreplacement therapies. The concurrent use of factor VIII replacement therapy and emicizumab could inhibit bleeding with lower dose immunotolerance strategies, according to the recommendations.
The group hypothesized that increased uptake of lower dose and lower frequency factor VIII ITI treatment strategies could reduce the likelihood of requiring central venous access while retaining a high probability of treatment success.
“In our new algorithm, we have indicated that one option might be to start patients on low-dose ITI with emicizumab regardless of their historical peak inhibitor titre. Patients could then escalate their ITI regimen should their response to low-dose ITI be deemed insufficient,” they wrote.
The experts provided a novel treatment algorithm for immune tolerance induction without emicizumab in addition to a new theoretical strategy with emicizumab.
Other recommendations included that patients with inhibitors should be offered at least one attempt at ITI and that monthly monitoring should be done and ITI dose and frequency should be adjusted based on changes in bleeding phenotype and inhibitor titer.
The authors acknowledged a current limitation is the lack of published evidence pertaining to the concurrent use of emicizumab and factor VIII replacement therapy.
“The FIT group sees the need for properly conducted prospective studies to evaluate the impact of adding emicizumab, and in the future, other nonfactor therapies, into the management of patients with inhibitors,” the experts wrote.
The manuscript was supported by Grifols. The authors reported financial disclosures related to Grifols and other companies.
SOURCE: Carcao M et al. Haemophilia. 2019 Apr 29. doi: 10.1111/hae.13762.
New treatment recommendations, released by a panel of nine experts, offer consensus advice on the use of immune tolerance induction (ITI) therapy in patients with hemophilia A with inhibitors.
The recommendations from the Future of Immunotolerance Treatment (FIT) group were authored by a nine-member committee with expertise in the treatment of hemophilia. The authors attended three meetings from 2017-2018 to form a consensus on the use of nonfactor therapies with current inhibitor management strategies.
“The treatment of hemophilia A has evolved and a number of molecules that potentially can be used in the setting of patients with inhibitors have been developed, or are in various phases of development,” wrote Manuel Carcao, MD, of the University of Toronto and colleagues. The report is published in Haemophilia.
The current body of literature is lacking high-quality evidence on the concomitant use of nonfactor treatments, such as emicizumab, and current inhibitor therapies. The recommendations included the panel’s consensus opinions on treatment of inhibitors, with and without the use of nonreplacement therapies. The concurrent use of factor VIII replacement therapy and emicizumab could inhibit bleeding with lower dose immunotolerance strategies, according to the recommendations.
The group hypothesized that increased uptake of lower dose and lower frequency factor VIII ITI treatment strategies could reduce the likelihood of requiring central venous access while retaining a high probability of treatment success.
“In our new algorithm, we have indicated that one option might be to start patients on low-dose ITI with emicizumab regardless of their historical peak inhibitor titre. Patients could then escalate their ITI regimen should their response to low-dose ITI be deemed insufficient,” they wrote.
The experts provided a novel treatment algorithm for immune tolerance induction without emicizumab in addition to a new theoretical strategy with emicizumab.
Other recommendations included that patients with inhibitors should be offered at least one attempt at ITI and that monthly monitoring should be done and ITI dose and frequency should be adjusted based on changes in bleeding phenotype and inhibitor titer.
The authors acknowledged a current limitation is the lack of published evidence pertaining to the concurrent use of emicizumab and factor VIII replacement therapy.
“The FIT group sees the need for properly conducted prospective studies to evaluate the impact of adding emicizumab, and in the future, other nonfactor therapies, into the management of patients with inhibitors,” the experts wrote.
The manuscript was supported by Grifols. The authors reported financial disclosures related to Grifols and other companies.
SOURCE: Carcao M et al. Haemophilia. 2019 Apr 29. doi: 10.1111/hae.13762.
New treatment recommendations, released by a panel of nine experts, offer consensus advice on the use of immune tolerance induction (ITI) therapy in patients with hemophilia A with inhibitors.
The recommendations from the Future of Immunotolerance Treatment (FIT) group were authored by a nine-member committee with expertise in the treatment of hemophilia. The authors attended three meetings from 2017-2018 to form a consensus on the use of nonfactor therapies with current inhibitor management strategies.
“The treatment of hemophilia A has evolved and a number of molecules that potentially can be used in the setting of patients with inhibitors have been developed, or are in various phases of development,” wrote Manuel Carcao, MD, of the University of Toronto and colleagues. The report is published in Haemophilia.
The current body of literature is lacking high-quality evidence on the concomitant use of nonfactor treatments, such as emicizumab, and current inhibitor therapies. The recommendations included the panel’s consensus opinions on treatment of inhibitors, with and without the use of nonreplacement therapies. The concurrent use of factor VIII replacement therapy and emicizumab could inhibit bleeding with lower dose immunotolerance strategies, according to the recommendations.
The group hypothesized that increased uptake of lower dose and lower frequency factor VIII ITI treatment strategies could reduce the likelihood of requiring central venous access while retaining a high probability of treatment success.
“In our new algorithm, we have indicated that one option might be to start patients on low-dose ITI with emicizumab regardless of their historical peak inhibitor titre. Patients could then escalate their ITI regimen should their response to low-dose ITI be deemed insufficient,” they wrote.
The experts provided a novel treatment algorithm for immune tolerance induction without emicizumab in addition to a new theoretical strategy with emicizumab.
Other recommendations included that patients with inhibitors should be offered at least one attempt at ITI and that monthly monitoring should be done and ITI dose and frequency should be adjusted based on changes in bleeding phenotype and inhibitor titer.
The authors acknowledged a current limitation is the lack of published evidence pertaining to the concurrent use of emicizumab and factor VIII replacement therapy.
“The FIT group sees the need for properly conducted prospective studies to evaluate the impact of adding emicizumab, and in the future, other nonfactor therapies, into the management of patients with inhibitors,” the experts wrote.
The manuscript was supported by Grifols. The authors reported financial disclosures related to Grifols and other companies.
SOURCE: Carcao M et al. Haemophilia. 2019 Apr 29. doi: 10.1111/hae.13762.
FROM HAEMOPHILIA
Study identifies GI bleed risk factors in von Willebrand disease
Researchers have identified several risk factors associated with gastrointestinal (GI) bleeding in adult patients with von Willebrand disease (VWD), based on findings from a retrospective analysis.
“[We] evaluated prevalence and risk factors of GIB among individuals with and without VWD, using a large national database,” wrote Anastasia Tsagianni, MD, of the University of Pittsburgh, and colleagues. The findings were published in Thrombosis Research.
The researchers retrospectively reviewed discharge data from the National Inpatient Sample database. The team analyzed correlates of GI bleeding among patients with VWD and estimated prevalence rates using measures in the database. Risk factors for GI bleeding in VWD were correlated via multivariable logistic regression.
Between Jan. 1, 2009, and Dec. 31, 2014, there were 16,640 admissions with VWD and 618 were admitted with GI bleeding, the researchers reported. After analysis, the researchers found that the prevalence of GI bleeding was 3.7% and 1.49% in VWD and non-VWD patients, respectively – a 2.5-fold greater rate in VWD patients.
“Comorbidities associated with greater [GI bleeding] risk in individuals with VWD include past surgery, hypertension, hyperlipidemia, smoking, renal disease, hepatitis C, thrombocytopenia, or liver disease,” the researchers wrote.
In the multivariable analysis, the factors associated with GI bleeding were smoking status (odds ratio, 1.40), African American race (OR, 1.80), male gender (OR, 1.61), angiodysplasia (OR, 104.06), colonic diverticulitis (OR, 16.66), and hepatitis C (OR 2.17). These variables were similar to risk factors identified in the non-VWD group, the researchers noted.
“Although significant, age did not appear to be a strong risk factor for either group,” they wrote. “Steroids were not associated with increased risk for [GI bleeding] in either group.”
A key limitation of the study was the use of discharge diagnosis codes as an inclusion method, the researchers noted. As a result, misclassification bias could be present, they added.
The Pennsylvania Department of Health and the Health Resources and Services Administration funded the study. The authors reported having no conflicts of interest.
SOURCE: Tsagianni A et al. Thromb Res. 2019 Apr 17. doi: 10.1016/j.thromres.2019.04.017.
Researchers have identified several risk factors associated with gastrointestinal (GI) bleeding in adult patients with von Willebrand disease (VWD), based on findings from a retrospective analysis.
“[We] evaluated prevalence and risk factors of GIB among individuals with and without VWD, using a large national database,” wrote Anastasia Tsagianni, MD, of the University of Pittsburgh, and colleagues. The findings were published in Thrombosis Research.
The researchers retrospectively reviewed discharge data from the National Inpatient Sample database. The team analyzed correlates of GI bleeding among patients with VWD and estimated prevalence rates using measures in the database. Risk factors for GI bleeding in VWD were correlated via multivariable logistic regression.
Between Jan. 1, 2009, and Dec. 31, 2014, there were 16,640 admissions with VWD and 618 were admitted with GI bleeding, the researchers reported. After analysis, the researchers found that the prevalence of GI bleeding was 3.7% and 1.49% in VWD and non-VWD patients, respectively – a 2.5-fold greater rate in VWD patients.
“Comorbidities associated with greater [GI bleeding] risk in individuals with VWD include past surgery, hypertension, hyperlipidemia, smoking, renal disease, hepatitis C, thrombocytopenia, or liver disease,” the researchers wrote.
In the multivariable analysis, the factors associated with GI bleeding were smoking status (odds ratio, 1.40), African American race (OR, 1.80), male gender (OR, 1.61), angiodysplasia (OR, 104.06), colonic diverticulitis (OR, 16.66), and hepatitis C (OR 2.17). These variables were similar to risk factors identified in the non-VWD group, the researchers noted.
“Although significant, age did not appear to be a strong risk factor for either group,” they wrote. “Steroids were not associated with increased risk for [GI bleeding] in either group.”
A key limitation of the study was the use of discharge diagnosis codes as an inclusion method, the researchers noted. As a result, misclassification bias could be present, they added.
The Pennsylvania Department of Health and the Health Resources and Services Administration funded the study. The authors reported having no conflicts of interest.
SOURCE: Tsagianni A et al. Thromb Res. 2019 Apr 17. doi: 10.1016/j.thromres.2019.04.017.
Researchers have identified several risk factors associated with gastrointestinal (GI) bleeding in adult patients with von Willebrand disease (VWD), based on findings from a retrospective analysis.
“[We] evaluated prevalence and risk factors of GIB among individuals with and without VWD, using a large national database,” wrote Anastasia Tsagianni, MD, of the University of Pittsburgh, and colleagues. The findings were published in Thrombosis Research.
The researchers retrospectively reviewed discharge data from the National Inpatient Sample database. The team analyzed correlates of GI bleeding among patients with VWD and estimated prevalence rates using measures in the database. Risk factors for GI bleeding in VWD were correlated via multivariable logistic regression.
Between Jan. 1, 2009, and Dec. 31, 2014, there were 16,640 admissions with VWD and 618 were admitted with GI bleeding, the researchers reported. After analysis, the researchers found that the prevalence of GI bleeding was 3.7% and 1.49% in VWD and non-VWD patients, respectively – a 2.5-fold greater rate in VWD patients.
“Comorbidities associated with greater [GI bleeding] risk in individuals with VWD include past surgery, hypertension, hyperlipidemia, smoking, renal disease, hepatitis C, thrombocytopenia, or liver disease,” the researchers wrote.
In the multivariable analysis, the factors associated with GI bleeding were smoking status (odds ratio, 1.40), African American race (OR, 1.80), male gender (OR, 1.61), angiodysplasia (OR, 104.06), colonic diverticulitis (OR, 16.66), and hepatitis C (OR 2.17). These variables were similar to risk factors identified in the non-VWD group, the researchers noted.
“Although significant, age did not appear to be a strong risk factor for either group,” they wrote. “Steroids were not associated with increased risk for [GI bleeding] in either group.”
A key limitation of the study was the use of discharge diagnosis codes as an inclusion method, the researchers noted. As a result, misclassification bias could be present, they added.
The Pennsylvania Department of Health and the Health Resources and Services Administration funded the study. The authors reported having no conflicts of interest.
SOURCE: Tsagianni A et al. Thromb Res. 2019 Apr 17. doi: 10.1016/j.thromres.2019.04.017.
FROM THROMBOSIS RESEARCH
Key clinical point: Researchers identified several risk factors associated for gastrointestinal bleeding in adult patients with von Willebrand disease.
Major finding: In a multivariate analysis, the most significant factors associated with gastrointestinal bleeding in von Willebrand disease were smoking status, African American race, male gender, angiodysplasia, diverticulitis, and hepatitis C.
Study details: A retrospective analysis of 16,640 patients with the disease.
Disclosures: The Pennsylvania Department of Health and the Health Resources and Services Administration funded the study. The authors reported having no conflicts of interest.
Source: Tsagianni A et al. Thromb Res. 2019 Apr 17. doi: 10.1016/j.thromres.2019.04.017.
Researchers propose new risk groups for NK-AML
NEWPORT BEACH, CALIF. – New research suggests patients with normal karyotype acute myeloid leukemia (NK-AML) can be divided into four risk groups associated with overall survival.
Investigators used machine learning algorithms to study the association between mutations and overall survival in 1,352 patients with NK-AML. The analysis revealed combinations of mutations that could be used to classify NK-AML patients into favorable, intermediate-1, intermediate-2, and unfavorable risk groups.
For example, patients who had NPM1 mutations but wild-type FLT3-ITD and DNMT3A, had a median overall survival of 99.1 months and could be classified as favorable risk. Conversely, patients who had NPM1, FLT3-ITD, and DNMT3A mutations, had a median overall survival of 13.4 months and could be classified as unfavorable risk.
Aziz Nazha, MD, of the Cleveland Clinic, and his colleagues conducted this research and presented the findings at the Acute Leukemia Forum of Hemedicus.
The investigators looked at genomic and clinical data from 1,352 patients with NK-AML. The patients were a median age of 55 years and had a median white blood cell count of 21.3 x 109/L, a median hemoglobin of 9.1 g/dL, and a median platelet count of 61 x 109/L. More than half of patients (57.3%) were male.
The patients were screened for 35 genes that are commonly mutated in AML and other myeloid malignancies. The investigators used machine learning algorithms, including random survival forest and recommender system algorithms, to study the association between mutations and overall survival in an “unbiased” way.
Dr. Nazha said there were a median of three mutations per patient sample, and “there are some competing interests between those mutations to impact the prognosis of the patient.”
The investigators used the mutations and their associations with overall survival to classify patients into the risk groups outlined in the table below.
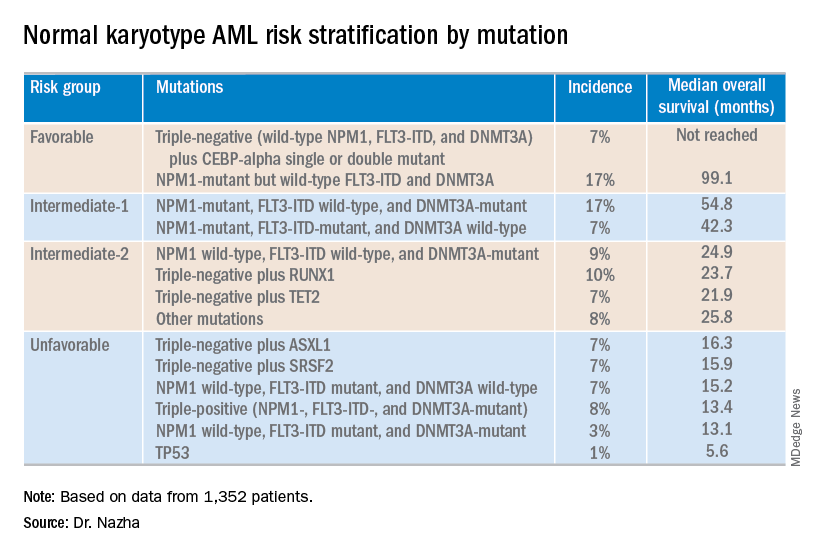
These findings can improve the risk stratification of NK-AML and may aid physicians in making treatment decisions, according to Dr. Nazha and his colleagues. To move this work forward, the investigators are attempting to develop a personalized model that can make predictions specific to an individual patient based on that patient’s mutation information.
Dr. Nazha reported having no financial disclosures relevant to this research. Other investigators reported relationships with the Munich Leukemia Laboratory.
The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
NEWPORT BEACH, CALIF. – New research suggests patients with normal karyotype acute myeloid leukemia (NK-AML) can be divided into four risk groups associated with overall survival.
Investigators used machine learning algorithms to study the association between mutations and overall survival in 1,352 patients with NK-AML. The analysis revealed combinations of mutations that could be used to classify NK-AML patients into favorable, intermediate-1, intermediate-2, and unfavorable risk groups.
For example, patients who had NPM1 mutations but wild-type FLT3-ITD and DNMT3A, had a median overall survival of 99.1 months and could be classified as favorable risk. Conversely, patients who had NPM1, FLT3-ITD, and DNMT3A mutations, had a median overall survival of 13.4 months and could be classified as unfavorable risk.
Aziz Nazha, MD, of the Cleveland Clinic, and his colleagues conducted this research and presented the findings at the Acute Leukemia Forum of Hemedicus.
The investigators looked at genomic and clinical data from 1,352 patients with NK-AML. The patients were a median age of 55 years and had a median white blood cell count of 21.3 x 109/L, a median hemoglobin of 9.1 g/dL, and a median platelet count of 61 x 109/L. More than half of patients (57.3%) were male.
The patients were screened for 35 genes that are commonly mutated in AML and other myeloid malignancies. The investigators used machine learning algorithms, including random survival forest and recommender system algorithms, to study the association between mutations and overall survival in an “unbiased” way.
Dr. Nazha said there were a median of three mutations per patient sample, and “there are some competing interests between those mutations to impact the prognosis of the patient.”
The investigators used the mutations and their associations with overall survival to classify patients into the risk groups outlined in the table below.
These findings can improve the risk stratification of NK-AML and may aid physicians in making treatment decisions, according to Dr. Nazha and his colleagues. To move this work forward, the investigators are attempting to develop a personalized model that can make predictions specific to an individual patient based on that patient’s mutation information.
Dr. Nazha reported having no financial disclosures relevant to this research. Other investigators reported relationships with the Munich Leukemia Laboratory.
The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
NEWPORT BEACH, CALIF. – New research suggests patients with normal karyotype acute myeloid leukemia (NK-AML) can be divided into four risk groups associated with overall survival.
Investigators used machine learning algorithms to study the association between mutations and overall survival in 1,352 patients with NK-AML. The analysis revealed combinations of mutations that could be used to classify NK-AML patients into favorable, intermediate-1, intermediate-2, and unfavorable risk groups.
For example, patients who had NPM1 mutations but wild-type FLT3-ITD and DNMT3A, had a median overall survival of 99.1 months and could be classified as favorable risk. Conversely, patients who had NPM1, FLT3-ITD, and DNMT3A mutations, had a median overall survival of 13.4 months and could be classified as unfavorable risk.
Aziz Nazha, MD, of the Cleveland Clinic, and his colleagues conducted this research and presented the findings at the Acute Leukemia Forum of Hemedicus.
The investigators looked at genomic and clinical data from 1,352 patients with NK-AML. The patients were a median age of 55 years and had a median white blood cell count of 21.3 x 109/L, a median hemoglobin of 9.1 g/dL, and a median platelet count of 61 x 109/L. More than half of patients (57.3%) were male.
The patients were screened for 35 genes that are commonly mutated in AML and other myeloid malignancies. The investigators used machine learning algorithms, including random survival forest and recommender system algorithms, to study the association between mutations and overall survival in an “unbiased” way.
Dr. Nazha said there were a median of three mutations per patient sample, and “there are some competing interests between those mutations to impact the prognosis of the patient.”
The investigators used the mutations and their associations with overall survival to classify patients into the risk groups outlined in the table below.
These findings can improve the risk stratification of NK-AML and may aid physicians in making treatment decisions, according to Dr. Nazha and his colleagues. To move this work forward, the investigators are attempting to develop a personalized model that can make predictions specific to an individual patient based on that patient’s mutation information.
Dr. Nazha reported having no financial disclosures relevant to this research. Other investigators reported relationships with the Munich Leukemia Laboratory.
The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
REPORTING FROM ALF 2019
Gabapentin falls short in treating sickle cell pain
NEW ORLEANS – Adding gabapentin to standard therapy did not significantly reduce vaso-occlusive pain in most patients with sickle cell disease enrolled in a phase 2 trial.

In the entire cohort, there were no significant differences in pain response between patients who received gabapentin and those who received placebo. However, patients with the HbSS genotype had a significantly greater decrease in pain score from baseline to discharge if they received gabapentin rather than placebo.
Additional studies are needed to confirm these findings because this trial was limited by a small sample size, according to study investigator Latika Puri, MD, of St. Jude Children’s Research Hospital in Memphis. Dr. Puri presented the trial at the annual meeting of the American Society of Pediatric Hematology/Oncology.
The trial included 86 evaluable patients who had vaso-occlusive pain and a pain score of at least 4. All patients received standard therapy for vaso-occlusive pain and were randomized to receive placebo (n = 44) or a single oral dose of gabapentin at 15 mg/kg (n = 42).
Baseline characteristics were similar between the treatment arms. For the entire cohort, the mean age was 11.8 years (range, 1-21 years), and 51% of patients were male. Forty-four patients had the HbSS genotype, 25 had the HbSC genotype, 8 had HbS/beta0-thalassemia, and 9 had other genotypes.
The mean pain score at baseline was 7.8 for the entire cohort, 8.0 for the gabapentin arm, and 7.7 for the placebo arm.
For the entire cohort, there was no significant difference in pain response between the gabapentin and placebo arms.
The proportion of patients who experienced a greater than 33% decrease in pain from baseline to 3 hours posttreatment was 67% in the gabapentin arm and 59% in the placebo arm (P = .23). The proportion of patients who experienced a greater than 33% decrease from baseline to discharge from the acute care clinic was 75% and 61%, respectively (P = .18).
In the entire cohort, decreases in pain scores from baseline to 3 hours posttreatment were not significantly different between the gabapentin and placebo arms, at 1.3 and 0.7, respectively (P = .74). Likewise, decreases in pain scores from baseline to discharge were not significantly different, at 1.6 and 0.8 (P = .38).
Among patients who had the HbSS genotype, there was a significantly greater decrease in pain score from baseline to discharge in the gabapentin arm than in the placebo arm, 5.9 versus 3.6 (P = .03). However, there were no other significant differences in pain response for the HbSS subgroup.
There were no significant differences in opioid consumption or hospitalization for the HbSS subgroup or the entire cohort. For the entire cohort, the mean morphine equivalent dose from baseline to 3 hours posttreatment was 0.16 mg/kg in the gabapentin arm and 0.17 mg/kg in the placebo arm (P = .89). For the HbSS subgroup, the mean dose was 0.16 mg/kg and 0.15 mg/kg, respectively (P = .93).
In the entire cohort, 24% of patients in the gabapentin arm and 27% of those in the placebo arm were hospitalized (P = .71). In the HbSS subgroup, hospitalizations occurred in 11% and 35% (P = .15).
Dr. Puri pointed out several challenges that led to limitations in this study. Specifically, the investigators had to obtain patient consent while delivering standard treatment, while patients were in pain and distress, and from patients who had already received opioids and were sleepy. Additionally, gabapentin had to be delivered within 1 hour of opioid administration, and a lack of after-hours staff limited enrollment.
“These challenges led to one of our biggest limitations, which was a small sample size, leading to a limited power to observe real differences,” Dr. Puri said. “We also defined a very short time period of evaluation for the primary outcomes; that was 3 hours from the gabapentin dose or placebo dose. This limited our capability to see real differences if they existed.”
Dr. Puri said additional studies with larger sample sizes are needed to confirm these findings. She added that efforts to better characterize pain in sickle cell disease could reveal patients who may benefit from gabapentin because they have a neuropathic component to their pain.
The trial was sponsored by St. Jude Children’s Research Hospital in collaboration with Scan|Design Foundation. Dr. Puri did not provide disclosure information at the meeting.
SOURCE: Puri L et al. ASPHO 2019, Abstract 2011.
NEW ORLEANS – Adding gabapentin to standard therapy did not significantly reduce vaso-occlusive pain in most patients with sickle cell disease enrolled in a phase 2 trial.
In the entire cohort, there were no significant differences in pain response between patients who received gabapentin and those who received placebo. However, patients with the HbSS genotype had a significantly greater decrease in pain score from baseline to discharge if they received gabapentin rather than placebo.
Additional studies are needed to confirm these findings because this trial was limited by a small sample size, according to study investigator Latika Puri, MD, of St. Jude Children’s Research Hospital in Memphis. Dr. Puri presented the trial at the annual meeting of the American Society of Pediatric Hematology/Oncology.
The trial included 86 evaluable patients who had vaso-occlusive pain and a pain score of at least 4. All patients received standard therapy for vaso-occlusive pain and were randomized to receive placebo (n = 44) or a single oral dose of gabapentin at 15 mg/kg (n = 42).
Baseline characteristics were similar between the treatment arms. For the entire cohort, the mean age was 11.8 years (range, 1-21 years), and 51% of patients were male. Forty-four patients had the HbSS genotype, 25 had the HbSC genotype, 8 had HbS/beta0-thalassemia, and 9 had other genotypes.
The mean pain score at baseline was 7.8 for the entire cohort, 8.0 for the gabapentin arm, and 7.7 for the placebo arm.
For the entire cohort, there was no significant difference in pain response between the gabapentin and placebo arms.
The proportion of patients who experienced a greater than 33% decrease in pain from baseline to 3 hours posttreatment was 67% in the gabapentin arm and 59% in the placebo arm (P = .23). The proportion of patients who experienced a greater than 33% decrease from baseline to discharge from the acute care clinic was 75% and 61%, respectively (P = .18).
In the entire cohort, decreases in pain scores from baseline to 3 hours posttreatment were not significantly different between the gabapentin and placebo arms, at 1.3 and 0.7, respectively (P = .74). Likewise, decreases in pain scores from baseline to discharge were not significantly different, at 1.6 and 0.8 (P = .38).
Among patients who had the HbSS genotype, there was a significantly greater decrease in pain score from baseline to discharge in the gabapentin arm than in the placebo arm, 5.9 versus 3.6 (P = .03). However, there were no other significant differences in pain response for the HbSS subgroup.
There were no significant differences in opioid consumption or hospitalization for the HbSS subgroup or the entire cohort. For the entire cohort, the mean morphine equivalent dose from baseline to 3 hours posttreatment was 0.16 mg/kg in the gabapentin arm and 0.17 mg/kg in the placebo arm (P = .89). For the HbSS subgroup, the mean dose was 0.16 mg/kg and 0.15 mg/kg, respectively (P = .93).
In the entire cohort, 24% of patients in the gabapentin arm and 27% of those in the placebo arm were hospitalized (P = .71). In the HbSS subgroup, hospitalizations occurred in 11% and 35% (P = .15).
Dr. Puri pointed out several challenges that led to limitations in this study. Specifically, the investigators had to obtain patient consent while delivering standard treatment, while patients were in pain and distress, and from patients who had already received opioids and were sleepy. Additionally, gabapentin had to be delivered within 1 hour of opioid administration, and a lack of after-hours staff limited enrollment.
“These challenges led to one of our biggest limitations, which was a small sample size, leading to a limited power to observe real differences,” Dr. Puri said. “We also defined a very short time period of evaluation for the primary outcomes; that was 3 hours from the gabapentin dose or placebo dose. This limited our capability to see real differences if they existed.”
Dr. Puri said additional studies with larger sample sizes are needed to confirm these findings. She added that efforts to better characterize pain in sickle cell disease could reveal patients who may benefit from gabapentin because they have a neuropathic component to their pain.
The trial was sponsored by St. Jude Children’s Research Hospital in collaboration with Scan|Design Foundation. Dr. Puri did not provide disclosure information at the meeting.
SOURCE: Puri L et al. ASPHO 2019, Abstract 2011.
NEW ORLEANS – Adding gabapentin to standard therapy did not significantly reduce vaso-occlusive pain in most patients with sickle cell disease enrolled in a phase 2 trial.
In the entire cohort, there were no significant differences in pain response between patients who received gabapentin and those who received placebo. However, patients with the HbSS genotype had a significantly greater decrease in pain score from baseline to discharge if they received gabapentin rather than placebo.
Additional studies are needed to confirm these findings because this trial was limited by a small sample size, according to study investigator Latika Puri, MD, of St. Jude Children’s Research Hospital in Memphis. Dr. Puri presented the trial at the annual meeting of the American Society of Pediatric Hematology/Oncology.
The trial included 86 evaluable patients who had vaso-occlusive pain and a pain score of at least 4. All patients received standard therapy for vaso-occlusive pain and were randomized to receive placebo (n = 44) or a single oral dose of gabapentin at 15 mg/kg (n = 42).
Baseline characteristics were similar between the treatment arms. For the entire cohort, the mean age was 11.8 years (range, 1-21 years), and 51% of patients were male. Forty-four patients had the HbSS genotype, 25 had the HbSC genotype, 8 had HbS/beta0-thalassemia, and 9 had other genotypes.
The mean pain score at baseline was 7.8 for the entire cohort, 8.0 for the gabapentin arm, and 7.7 for the placebo arm.
For the entire cohort, there was no significant difference in pain response between the gabapentin and placebo arms.
The proportion of patients who experienced a greater than 33% decrease in pain from baseline to 3 hours posttreatment was 67% in the gabapentin arm and 59% in the placebo arm (P = .23). The proportion of patients who experienced a greater than 33% decrease from baseline to discharge from the acute care clinic was 75% and 61%, respectively (P = .18).
In the entire cohort, decreases in pain scores from baseline to 3 hours posttreatment were not significantly different between the gabapentin and placebo arms, at 1.3 and 0.7, respectively (P = .74). Likewise, decreases in pain scores from baseline to discharge were not significantly different, at 1.6 and 0.8 (P = .38).
Among patients who had the HbSS genotype, there was a significantly greater decrease in pain score from baseline to discharge in the gabapentin arm than in the placebo arm, 5.9 versus 3.6 (P = .03). However, there were no other significant differences in pain response for the HbSS subgroup.
There were no significant differences in opioid consumption or hospitalization for the HbSS subgroup or the entire cohort. For the entire cohort, the mean morphine equivalent dose from baseline to 3 hours posttreatment was 0.16 mg/kg in the gabapentin arm and 0.17 mg/kg in the placebo arm (P = .89). For the HbSS subgroup, the mean dose was 0.16 mg/kg and 0.15 mg/kg, respectively (P = .93).
In the entire cohort, 24% of patients in the gabapentin arm and 27% of those in the placebo arm were hospitalized (P = .71). In the HbSS subgroup, hospitalizations occurred in 11% and 35% (P = .15).
Dr. Puri pointed out several challenges that led to limitations in this study. Specifically, the investigators had to obtain patient consent while delivering standard treatment, while patients were in pain and distress, and from patients who had already received opioids and were sleepy. Additionally, gabapentin had to be delivered within 1 hour of opioid administration, and a lack of after-hours staff limited enrollment.
“These challenges led to one of our biggest limitations, which was a small sample size, leading to a limited power to observe real differences,” Dr. Puri said. “We also defined a very short time period of evaluation for the primary outcomes; that was 3 hours from the gabapentin dose or placebo dose. This limited our capability to see real differences if they existed.”
Dr. Puri said additional studies with larger sample sizes are needed to confirm these findings. She added that efforts to better characterize pain in sickle cell disease could reveal patients who may benefit from gabapentin because they have a neuropathic component to their pain.
The trial was sponsored by St. Jude Children’s Research Hospital in collaboration with Scan|Design Foundation. Dr. Puri did not provide disclosure information at the meeting.
SOURCE: Puri L et al. ASPHO 2019, Abstract 2011.
REPORTING FROM THE 2019 ASPHO CONFERENCE
Key clinical point:
Major finding: The proportion of patients who experienced a greater than 33% decrease in pain from baseline to 3 hours posttreatment was 67% in the gabapentin arm and 59% in the placebo arm (P = .23).
Study details: A phase 2 trial of 86 evaluable patients.
Disclosures: The trial was sponsored by St. Jude Children’s Research Hospital in collaboration with Scan|Design Foundation. The speaker did not provide disclosure information at the meeting.
Source: Puri L et al. 2019 ASPHO Conference, Abstract 2011.
Combo proves most effective in HMA-naive, higher-risk MDS
NEWPORT BEACH, CALIF. – The combination of oral rigosertib and azacitidine is proceeding to a phase 3 trial in patients with myelodysplastic syndromes (MDS), but it isn’t clear if the combination will continue to be developed for acute myeloid leukemia (AML).
In a phase 1/2 trial, oral rigosertib plus azacitidine produced a 90% response rate in higher-risk MDS patients who were naive to hypomethylating agents (HMAs), a 54% response rate in higher-risk MDS patients who had failed HMA therapy, and a 50% response rate in patients with AML.
Genitourinary toxicities were initially a concern in this trial, but researchers found ways to mitigate the risk of these toxicities, according to Richard Woodman, MD, chief medical officer and senior vice president of research and development at Onconova Therapeutics, the company developing rigosertib.
Dr. Woodman and his colleagues presented results from the phase 1/2 trial in two posters at the Acute Leukemia Forum of Hemedicus.
Results in AML
The researchers reported phase 1 results in 17 patients with AML. Eleven patients had AML, according to investigator assessment, and six patients had refractory anemia with excess blasts in transformation, according to French American British criteria, as well as least 20% excess blasts at baseline.
The median age of the patients was 73 years, and 53% were men. Two patients had received no prior therapies, six patients had relapsed disease, and nine were refractory to their last therapy.
Patients received oral rigosertib at escalating doses twice daily on days 1-21 of a 28-day cycle. The recommended phase 2 dose was 840 mg daily (560 mg in the morning and 280 mg in the afternoon), but there were two expansion cohorts in which patients received 1,120 mg daily (560 mg twice a day or 840 mg in the morning and 280 mg in the afternoon). The patients also received azacitidine at 75 mg/m2 per day subcutaneously or intravenously for 7 days starting on day 8.
Patients received a median of three treatment cycles. Fifteen of the 17 patients (88%) discontinued treatment, most because of progressive disease (n = 5), toxicity (n = 4), or death (n = 3).
Twelve patients were evaluable for response, and six (50%) responded. One patient achieved a morphologic complete remission (CR), three achieved a morphologic leukemia-free state, and two had a partial response.
The most common treatment-emergent adverse events (TEAEs) were fatigue (53%), diarrhea (53%), nausea (53%), constipation (47%), back pain (41%), pyrexia (41%), and pneumonia (35%). Grade 3 or higher TEAEs included pneumonia (35%) and anemia (24%).
These results haven’t provided a clear way forward for oral rigosertib and azacitidine in AML. Dr. Woodman said the researchers will have to review past studies and evaluate how AML patients (with at least 20% blasts) have responded to intravenous rigosertib, consult experts in the field, and then decide how they will move forward with oral rigosertib and azacitidine in AML.
Results in MDS
Dr. Woodman and his colleagues presented data on 74 patients with higher-risk MDS. The median age was 69 years, and 59% were men. Most patients were high risk (n = 23) or very high risk (n = 33), according to the Revised International Prognostic Scoring System.
The patients received oral rigosertib at a dose of 840 mg/day or higher on days 1-21 of a 28-day cycle. They also received azacitidine at 75 mg/m2 per day subcutaneously or intravenously for 7 days starting on day 8.
The median duration of treatment was 7.8 months in patients who were HMA naive and 4.9 months in patients who failed HMA therapy. The most common reasons for treatment discontinuation in the HMA-naive patients were toxicity (n = 8), progression (n = 7), and patient request (n = 7). The most common reasons for discontinuation in patients who had failed HMA therapy were progression (n = 12), toxicity (n = 5), and investigator decision (n = 4).
In total, 55 patients were evaluable for response, 26 who had failed HMA therapy and 29 who were HMA naive.
“The best responses, not surprisingly, were in patients that were HMA naive,” Dr. Woodman said.
In the HMA-naive patients, the overall response rate was 90%. Ten patients had a CR, five had a marrow CR with hematologic improvement, three had hematologic improvement alone, eight had a marrow CR alone, and three patients had stable disease. None of the patients progressed.
In the patients who had failed HMA therapy, the overall response rate was 54%. One patient achieved a CR, one had a partial response, five had a marrow CR with hematologic improvement, two had hematologic improvement alone, five had a marrow CR alone, seven had stable disease, and five progressed.
The median duration of response was 10.8 months in patients who failed HMA therapy and 12.2 months in the HMA-naive patients.
The most common TEAEs in the entire MDS cohort were hematuria (45%), constipation (43%), diarrhea (42%), fatigue (42%), dysuria (38%), pyrexia (36%), nausea (35%), neutropenia (31%), and thrombocytopenia (30%).
Grade 3 or higher TEAEs were neutropenia (27%), thrombocytopenia (26%), hematuria (9%), dysuria (9%), diarrhea (5%), fatigue (4%), and pyrexia (1%).
Dr. Woodman said patients who were most likely to be at risk for genitourinary toxicities (hematuria and dysuria) were those who weren’t well hydrated, took rigosertib at night, and didn’t void their bladders before bedtime. He said the researchers’ hypothesis is that there is some local bladder irritation in that setting.
However, the researchers found ways to mitigate the risk of genitourinary toxicities, including:
- Requiring the second dose of rigosertib to be taken in the afternoon rather than evening (about 3 p.m.).
- Asking patients to consume at least 2 liters of fluid per day.
- Having patients empty their bladders before bedtime.
- Assessing urine pH roughly 2 hours after the morning dose of rigosertib and prescribing sodium bicarbonate if the pH is less than 7.5.
Dr. Woodman said the phase 2 results in MDS patients have prompted the development of a phase 3 trial in which researchers will compare oral rigosertib plus azacitidine to azacitidine plus placebo.
Dr. Woodman is employed by Onconova Therapeutics, which sponsored the phase 1/2 trial. The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
NEWPORT BEACH, CALIF. – The combination of oral rigosertib and azacitidine is proceeding to a phase 3 trial in patients with myelodysplastic syndromes (MDS), but it isn’t clear if the combination will continue to be developed for acute myeloid leukemia (AML).
In a phase 1/2 trial, oral rigosertib plus azacitidine produced a 90% response rate in higher-risk MDS patients who were naive to hypomethylating agents (HMAs), a 54% response rate in higher-risk MDS patients who had failed HMA therapy, and a 50% response rate in patients with AML.
Genitourinary toxicities were initially a concern in this trial, but researchers found ways to mitigate the risk of these toxicities, according to Richard Woodman, MD, chief medical officer and senior vice president of research and development at Onconova Therapeutics, the company developing rigosertib.
Dr. Woodman and his colleagues presented results from the phase 1/2 trial in two posters at the Acute Leukemia Forum of Hemedicus.
Results in AML
The researchers reported phase 1 results in 17 patients with AML. Eleven patients had AML, according to investigator assessment, and six patients had refractory anemia with excess blasts in transformation, according to French American British criteria, as well as least 20% excess blasts at baseline.
The median age of the patients was 73 years, and 53% were men. Two patients had received no prior therapies, six patients had relapsed disease, and nine were refractory to their last therapy.
Patients received oral rigosertib at escalating doses twice daily on days 1-21 of a 28-day cycle. The recommended phase 2 dose was 840 mg daily (560 mg in the morning and 280 mg in the afternoon), but there were two expansion cohorts in which patients received 1,120 mg daily (560 mg twice a day or 840 mg in the morning and 280 mg in the afternoon). The patients also received azacitidine at 75 mg/m2 per day subcutaneously or intravenously for 7 days starting on day 8.
Patients received a median of three treatment cycles. Fifteen of the 17 patients (88%) discontinued treatment, most because of progressive disease (n = 5), toxicity (n = 4), or death (n = 3).
Twelve patients were evaluable for response, and six (50%) responded. One patient achieved a morphologic complete remission (CR), three achieved a morphologic leukemia-free state, and two had a partial response.
The most common treatment-emergent adverse events (TEAEs) were fatigue (53%), diarrhea (53%), nausea (53%), constipation (47%), back pain (41%), pyrexia (41%), and pneumonia (35%). Grade 3 or higher TEAEs included pneumonia (35%) and anemia (24%).
These results haven’t provided a clear way forward for oral rigosertib and azacitidine in AML. Dr. Woodman said the researchers will have to review past studies and evaluate how AML patients (with at least 20% blasts) have responded to intravenous rigosertib, consult experts in the field, and then decide how they will move forward with oral rigosertib and azacitidine in AML.
Results in MDS
Dr. Woodman and his colleagues presented data on 74 patients with higher-risk MDS. The median age was 69 years, and 59% were men. Most patients were high risk (n = 23) or very high risk (n = 33), according to the Revised International Prognostic Scoring System.
The patients received oral rigosertib at a dose of 840 mg/day or higher on days 1-21 of a 28-day cycle. They also received azacitidine at 75 mg/m2 per day subcutaneously or intravenously for 7 days starting on day 8.
The median duration of treatment was 7.8 months in patients who were HMA naive and 4.9 months in patients who failed HMA therapy. The most common reasons for treatment discontinuation in the HMA-naive patients were toxicity (n = 8), progression (n = 7), and patient request (n = 7). The most common reasons for discontinuation in patients who had failed HMA therapy were progression (n = 12), toxicity (n = 5), and investigator decision (n = 4).
In total, 55 patients were evaluable for response, 26 who had failed HMA therapy and 29 who were HMA naive.
“The best responses, not surprisingly, were in patients that were HMA naive,” Dr. Woodman said.
In the HMA-naive patients, the overall response rate was 90%. Ten patients had a CR, five had a marrow CR with hematologic improvement, three had hematologic improvement alone, eight had a marrow CR alone, and three patients had stable disease. None of the patients progressed.
In the patients who had failed HMA therapy, the overall response rate was 54%. One patient achieved a CR, one had a partial response, five had a marrow CR with hematologic improvement, two had hematologic improvement alone, five had a marrow CR alone, seven had stable disease, and five progressed.
The median duration of response was 10.8 months in patients who failed HMA therapy and 12.2 months in the HMA-naive patients.
The most common TEAEs in the entire MDS cohort were hematuria (45%), constipation (43%), diarrhea (42%), fatigue (42%), dysuria (38%), pyrexia (36%), nausea (35%), neutropenia (31%), and thrombocytopenia (30%).
Grade 3 or higher TEAEs were neutropenia (27%), thrombocytopenia (26%), hematuria (9%), dysuria (9%), diarrhea (5%), fatigue (4%), and pyrexia (1%).
Dr. Woodman said patients who were most likely to be at risk for genitourinary toxicities (hematuria and dysuria) were those who weren’t well hydrated, took rigosertib at night, and didn’t void their bladders before bedtime. He said the researchers’ hypothesis is that there is some local bladder irritation in that setting.
However, the researchers found ways to mitigate the risk of genitourinary toxicities, including:
- Requiring the second dose of rigosertib to be taken in the afternoon rather than evening (about 3 p.m.).
- Asking patients to consume at least 2 liters of fluid per day.
- Having patients empty their bladders before bedtime.
- Assessing urine pH roughly 2 hours after the morning dose of rigosertib and prescribing sodium bicarbonate if the pH is less than 7.5.
Dr. Woodman said the phase 2 results in MDS patients have prompted the development of a phase 3 trial in which researchers will compare oral rigosertib plus azacitidine to azacitidine plus placebo.
Dr. Woodman is employed by Onconova Therapeutics, which sponsored the phase 1/2 trial. The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
NEWPORT BEACH, CALIF. – The combination of oral rigosertib and azacitidine is proceeding to a phase 3 trial in patients with myelodysplastic syndromes (MDS), but it isn’t clear if the combination will continue to be developed for acute myeloid leukemia (AML).
In a phase 1/2 trial, oral rigosertib plus azacitidine produced a 90% response rate in higher-risk MDS patients who were naive to hypomethylating agents (HMAs), a 54% response rate in higher-risk MDS patients who had failed HMA therapy, and a 50% response rate in patients with AML.
Genitourinary toxicities were initially a concern in this trial, but researchers found ways to mitigate the risk of these toxicities, according to Richard Woodman, MD, chief medical officer and senior vice president of research and development at Onconova Therapeutics, the company developing rigosertib.
Dr. Woodman and his colleagues presented results from the phase 1/2 trial in two posters at the Acute Leukemia Forum of Hemedicus.
Results in AML
The researchers reported phase 1 results in 17 patients with AML. Eleven patients had AML, according to investigator assessment, and six patients had refractory anemia with excess blasts in transformation, according to French American British criteria, as well as least 20% excess blasts at baseline.
The median age of the patients was 73 years, and 53% were men. Two patients had received no prior therapies, six patients had relapsed disease, and nine were refractory to their last therapy.
Patients received oral rigosertib at escalating doses twice daily on days 1-21 of a 28-day cycle. The recommended phase 2 dose was 840 mg daily (560 mg in the morning and 280 mg in the afternoon), but there were two expansion cohorts in which patients received 1,120 mg daily (560 mg twice a day or 840 mg in the morning and 280 mg in the afternoon). The patients also received azacitidine at 75 mg/m2 per day subcutaneously or intravenously for 7 days starting on day 8.
Patients received a median of three treatment cycles. Fifteen of the 17 patients (88%) discontinued treatment, most because of progressive disease (n = 5), toxicity (n = 4), or death (n = 3).
Twelve patients were evaluable for response, and six (50%) responded. One patient achieved a morphologic complete remission (CR), three achieved a morphologic leukemia-free state, and two had a partial response.
The most common treatment-emergent adverse events (TEAEs) were fatigue (53%), diarrhea (53%), nausea (53%), constipation (47%), back pain (41%), pyrexia (41%), and pneumonia (35%). Grade 3 or higher TEAEs included pneumonia (35%) and anemia (24%).
These results haven’t provided a clear way forward for oral rigosertib and azacitidine in AML. Dr. Woodman said the researchers will have to review past studies and evaluate how AML patients (with at least 20% blasts) have responded to intravenous rigosertib, consult experts in the field, and then decide how they will move forward with oral rigosertib and azacitidine in AML.
Results in MDS
Dr. Woodman and his colleagues presented data on 74 patients with higher-risk MDS. The median age was 69 years, and 59% were men. Most patients were high risk (n = 23) or very high risk (n = 33), according to the Revised International Prognostic Scoring System.
The patients received oral rigosertib at a dose of 840 mg/day or higher on days 1-21 of a 28-day cycle. They also received azacitidine at 75 mg/m2 per day subcutaneously or intravenously for 7 days starting on day 8.
The median duration of treatment was 7.8 months in patients who were HMA naive and 4.9 months in patients who failed HMA therapy. The most common reasons for treatment discontinuation in the HMA-naive patients were toxicity (n = 8), progression (n = 7), and patient request (n = 7). The most common reasons for discontinuation in patients who had failed HMA therapy were progression (n = 12), toxicity (n = 5), and investigator decision (n = 4).
In total, 55 patients were evaluable for response, 26 who had failed HMA therapy and 29 who were HMA naive.
“The best responses, not surprisingly, were in patients that were HMA naive,” Dr. Woodman said.
In the HMA-naive patients, the overall response rate was 90%. Ten patients had a CR, five had a marrow CR with hematologic improvement, three had hematologic improvement alone, eight had a marrow CR alone, and three patients had stable disease. None of the patients progressed.
In the patients who had failed HMA therapy, the overall response rate was 54%. One patient achieved a CR, one had a partial response, five had a marrow CR with hematologic improvement, two had hematologic improvement alone, five had a marrow CR alone, seven had stable disease, and five progressed.
The median duration of response was 10.8 months in patients who failed HMA therapy and 12.2 months in the HMA-naive patients.
The most common TEAEs in the entire MDS cohort were hematuria (45%), constipation (43%), diarrhea (42%), fatigue (42%), dysuria (38%), pyrexia (36%), nausea (35%), neutropenia (31%), and thrombocytopenia (30%).
Grade 3 or higher TEAEs were neutropenia (27%), thrombocytopenia (26%), hematuria (9%), dysuria (9%), diarrhea (5%), fatigue (4%), and pyrexia (1%).
Dr. Woodman said patients who were most likely to be at risk for genitourinary toxicities (hematuria and dysuria) were those who weren’t well hydrated, took rigosertib at night, and didn’t void their bladders before bedtime. He said the researchers’ hypothesis is that there is some local bladder irritation in that setting.
However, the researchers found ways to mitigate the risk of genitourinary toxicities, including:
- Requiring the second dose of rigosertib to be taken in the afternoon rather than evening (about 3 p.m.).
- Asking patients to consume at least 2 liters of fluid per day.
- Having patients empty their bladders before bedtime.
- Assessing urine pH roughly 2 hours after the morning dose of rigosertib and prescribing sodium bicarbonate if the pH is less than 7.5.
Dr. Woodman said the phase 2 results in MDS patients have prompted the development of a phase 3 trial in which researchers will compare oral rigosertib plus azacitidine to azacitidine plus placebo.
Dr. Woodman is employed by Onconova Therapeutics, which sponsored the phase 1/2 trial. The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
REPORTING FROM ALF 2019

More abnormal cells linked to poorer ASCT outcomes in MDS
NEWPORT BEACH, CALIF. – Researchers say they’ve found an association between the percentage of cytogenetically abnormal cells at allogeneic stem cell transplant (ASCT) and posttransplant outcomes in patients with myelodysplastic syndromes (MDS).
Patients who had more than 60% cytogenetically abnormal cells at ASCT had significantly inferior overall survival (OS) and relapse-free survival (RFS), compared to patients with fewer abnormal cells.
Dipenkumar Modi, MD, of Barbara Ann Karmanos Cancer Institute at Wayne State University in Detroit, and his colleagues conducted this research and presented the results at the Acute Leukemia Forum of Hemedicus.
The researchers studied 109 adult MDS patients who underwent ASCT from January 2000 through December 2016. The patients were divided into three groups based on the percentage of cytogenetically abnormal cells at ASCT:
- Group 1 had less than 30% (n = 22)
- Group 2 had 30%-60% (n = 23)
- Group 3 had greater than 60% (n = 64).
Baseline characteristics were largely similar between the groups. However, patients in group 3 were significantly more likely than those in groups 1 and 2 to have del(5q) and monosomy 5+7 (P = .048).
Patients in group 1 had a significantly higher percentage of bone marrow transplants (as opposed to peripheral blood stem cell transplants) than patients in groups 2 and 3 (P = .039). And patients in group 1 had significantly fewer blasts at ASCT than patients in groups 2 and 3 (P = .011).
The researchers found no significant between-group differences in relapse and nonrelapse mortality, but there were significant differences in OS and RFS.
Patients in group 3 had inferior RFS compared to patients in group 1, which was the reference group. The hazard ratio (HR) was 2.503 (P = .013) in a univariable analysis and 2.196 (P = .049) in a multivariable analysis.
Group 3 also had inferior OS compared to group 1. The hazard ratio was 2.589 (P = .021) in a univariable analysis and 2.478 (P = .040) in a multivariable analysis.
There was no significant difference in RFS or OS between groups 1 and 2. The HR for RFS in group 2 was 1.879 (P = .148) in a univariable analysis and 1.365 (P = .506) in a multivariable analysis. The HR for OS was 1.997 (P = .155) and 1.413 (P = .511), respectively.
Dr. Modi said these results suggest patients with greater than 60% cytogenetically abnormal cells at ASCT should be monitored more closely after transplant, and their immunosuppressive medication should be tapered as soon as possible.
Dr. Modi and his colleagues reported having no conflicts of interest relevant to this research.
The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
NEWPORT BEACH, CALIF. – Researchers say they’ve found an association between the percentage of cytogenetically abnormal cells at allogeneic stem cell transplant (ASCT) and posttransplant outcomes in patients with myelodysplastic syndromes (MDS).
Patients who had more than 60% cytogenetically abnormal cells at ASCT had significantly inferior overall survival (OS) and relapse-free survival (RFS), compared to patients with fewer abnormal cells.
Dipenkumar Modi, MD, of Barbara Ann Karmanos Cancer Institute at Wayne State University in Detroit, and his colleagues conducted this research and presented the results at the Acute Leukemia Forum of Hemedicus.
The researchers studied 109 adult MDS patients who underwent ASCT from January 2000 through December 2016. The patients were divided into three groups based on the percentage of cytogenetically abnormal cells at ASCT:
- Group 1 had less than 30% (n = 22)
- Group 2 had 30%-60% (n = 23)
- Group 3 had greater than 60% (n = 64).
Baseline characteristics were largely similar between the groups. However, patients in group 3 were significantly more likely than those in groups 1 and 2 to have del(5q) and monosomy 5+7 (P = .048).
Patients in group 1 had a significantly higher percentage of bone marrow transplants (as opposed to peripheral blood stem cell transplants) than patients in groups 2 and 3 (P = .039). And patients in group 1 had significantly fewer blasts at ASCT than patients in groups 2 and 3 (P = .011).
The researchers found no significant between-group differences in relapse and nonrelapse mortality, but there were significant differences in OS and RFS.
Patients in group 3 had inferior RFS compared to patients in group 1, which was the reference group. The hazard ratio (HR) was 2.503 (P = .013) in a univariable analysis and 2.196 (P = .049) in a multivariable analysis.
Group 3 also had inferior OS compared to group 1. The hazard ratio was 2.589 (P = .021) in a univariable analysis and 2.478 (P = .040) in a multivariable analysis.
There was no significant difference in RFS or OS between groups 1 and 2. The HR for RFS in group 2 was 1.879 (P = .148) in a univariable analysis and 1.365 (P = .506) in a multivariable analysis. The HR for OS was 1.997 (P = .155) and 1.413 (P = .511), respectively.
Dr. Modi said these results suggest patients with greater than 60% cytogenetically abnormal cells at ASCT should be monitored more closely after transplant, and their immunosuppressive medication should be tapered as soon as possible.
Dr. Modi and his colleagues reported having no conflicts of interest relevant to this research.
The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
NEWPORT BEACH, CALIF. – Researchers say they’ve found an association between the percentage of cytogenetically abnormal cells at allogeneic stem cell transplant (ASCT) and posttransplant outcomes in patients with myelodysplastic syndromes (MDS).
Patients who had more than 60% cytogenetically abnormal cells at ASCT had significantly inferior overall survival (OS) and relapse-free survival (RFS), compared to patients with fewer abnormal cells.
Dipenkumar Modi, MD, of Barbara Ann Karmanos Cancer Institute at Wayne State University in Detroit, and his colleagues conducted this research and presented the results at the Acute Leukemia Forum of Hemedicus.
The researchers studied 109 adult MDS patients who underwent ASCT from January 2000 through December 2016. The patients were divided into three groups based on the percentage of cytogenetically abnormal cells at ASCT:
- Group 1 had less than 30% (n = 22)
- Group 2 had 30%-60% (n = 23)
- Group 3 had greater than 60% (n = 64).
Baseline characteristics were largely similar between the groups. However, patients in group 3 were significantly more likely than those in groups 1 and 2 to have del(5q) and monosomy 5+7 (P = .048).
Patients in group 1 had a significantly higher percentage of bone marrow transplants (as opposed to peripheral blood stem cell transplants) than patients in groups 2 and 3 (P = .039). And patients in group 1 had significantly fewer blasts at ASCT than patients in groups 2 and 3 (P = .011).
The researchers found no significant between-group differences in relapse and nonrelapse mortality, but there were significant differences in OS and RFS.
Patients in group 3 had inferior RFS compared to patients in group 1, which was the reference group. The hazard ratio (HR) was 2.503 (P = .013) in a univariable analysis and 2.196 (P = .049) in a multivariable analysis.
Group 3 also had inferior OS compared to group 1. The hazard ratio was 2.589 (P = .021) in a univariable analysis and 2.478 (P = .040) in a multivariable analysis.
There was no significant difference in RFS or OS between groups 1 and 2. The HR for RFS in group 2 was 1.879 (P = .148) in a univariable analysis and 1.365 (P = .506) in a multivariable analysis. The HR for OS was 1.997 (P = .155) and 1.413 (P = .511), respectively.
Dr. Modi said these results suggest patients with greater than 60% cytogenetically abnormal cells at ASCT should be monitored more closely after transplant, and their immunosuppressive medication should be tapered as soon as possible.
Dr. Modi and his colleagues reported having no conflicts of interest relevant to this research.
The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
REPORTING FROM ALF 2019

FDA approves ivosidenib frontline for certain AML patients
The Food and Drug Administration has approved ivosidenib (Tibsovo) for newly diagnosed acute myeloid leukemia (AML) with a susceptible IDH1 mutation in patients who are at least 75 years old or have comorbidities preventing the use of intensive induction chemotherapy.

In July 2018, the FDA approved ivosidenib for adults with relapsed or refractory AML with a susceptible IDH1 mutation.
The latest approval was based on results from an open-label, single-arm, multicenter trial of patients with newly diagnosed AML with an IDH1 mutation. Patients were treated with 500 mg ivosidenib daily until disease progression, development of unacceptable toxicity, or hematopoietic stem cell transplantation; the median age of the 28 patients treated with ivosidenib was 77 years.
Of the 28 patients treated, 12 achieved complete remission or complete remission with partial hematologic recovery; 7 of the 17 transfusion-dependent patients achieved transfusion independence for at least 8 weeks.
The most common adverse events were diarrhea, fatigue, edema, decreased appetite, leukocytosis, nausea, arthralgia, abdominal pain, dyspnea, differentiation syndrome, and myalgia. The drug’s prescribing information includes a boxed warning on the risk of differentiation syndrome.
“The recommended ivosidenib dose is 500 mg orally once daily with or without food until disease progression or unacceptable toxicity. For patients without disease progression or unacceptable toxicity, treatment is recommended for a minimum of 6 months to allow time for clinical response,” the FDA noted.
Find the full press release on the FDA website.
The Food and Drug Administration has approved ivosidenib (Tibsovo) for newly diagnosed acute myeloid leukemia (AML) with a susceptible IDH1 mutation in patients who are at least 75 years old or have comorbidities preventing the use of intensive induction chemotherapy.
In July 2018, the FDA approved ivosidenib for adults with relapsed or refractory AML with a susceptible IDH1 mutation.
The latest approval was based on results from an open-label, single-arm, multicenter trial of patients with newly diagnosed AML with an IDH1 mutation. Patients were treated with 500 mg ivosidenib daily until disease progression, development of unacceptable toxicity, or hematopoietic stem cell transplantation; the median age of the 28 patients treated with ivosidenib was 77 years.
Of the 28 patients treated, 12 achieved complete remission or complete remission with partial hematologic recovery; 7 of the 17 transfusion-dependent patients achieved transfusion independence for at least 8 weeks.
The most common adverse events were diarrhea, fatigue, edema, decreased appetite, leukocytosis, nausea, arthralgia, abdominal pain, dyspnea, differentiation syndrome, and myalgia. The drug’s prescribing information includes a boxed warning on the risk of differentiation syndrome.
“The recommended ivosidenib dose is 500 mg orally once daily with or without food until disease progression or unacceptable toxicity. For patients without disease progression or unacceptable toxicity, treatment is recommended for a minimum of 6 months to allow time for clinical response,” the FDA noted.
Find the full press release on the FDA website.
The Food and Drug Administration has approved ivosidenib (Tibsovo) for newly diagnosed acute myeloid leukemia (AML) with a susceptible IDH1 mutation in patients who are at least 75 years old or have comorbidities preventing the use of intensive induction chemotherapy.
In July 2018, the FDA approved ivosidenib for adults with relapsed or refractory AML with a susceptible IDH1 mutation.
The latest approval was based on results from an open-label, single-arm, multicenter trial of patients with newly diagnosed AML with an IDH1 mutation. Patients were treated with 500 mg ivosidenib daily until disease progression, development of unacceptable toxicity, or hematopoietic stem cell transplantation; the median age of the 28 patients treated with ivosidenib was 77 years.
Of the 28 patients treated, 12 achieved complete remission or complete remission with partial hematologic recovery; 7 of the 17 transfusion-dependent patients achieved transfusion independence for at least 8 weeks.
The most common adverse events were diarrhea, fatigue, edema, decreased appetite, leukocytosis, nausea, arthralgia, abdominal pain, dyspnea, differentiation syndrome, and myalgia. The drug’s prescribing information includes a boxed warning on the risk of differentiation syndrome.
“The recommended ivosidenib dose is 500 mg orally once daily with or without food until disease progression or unacceptable toxicity. For patients without disease progression or unacceptable toxicity, treatment is recommended for a minimum of 6 months to allow time for clinical response,” the FDA noted.
Find the full press release on the FDA website.
LentiGlobin reduces transfusion dependence in young thalassemia patients
NEW ORLEANS – The gene therapy LentiGlobin can reduce transfusion dependence in children and young adults with non-beta0/beta0 thalassemia, according to two trials.

In a phase 1/2 trial, 8 of 10 of patients achieved transfusion independence at a median follow-up of 36.0 months. In a phase 3 trial, transfusion independence was achieved by 2 of 3 patients with follow-up of at least 12 months.
Timothy S. Olson, MD, PhD, of Children’s Hospital of Philadelphia, presented results from the phase 1/2 HGB-204 trial and the phase 3 HGB-207 trial at the annual meeting of the American Society of Pediatric Hematology/Oncology.
Treatment
In both trials, patients received granulocyte colony-stimulating factor and plerixafor for hematopoietic stem cell mobilization. Their cells were collected via apheresis and transduced with the betibeglogene darolentivec (BB305) lentiviral vector. The patients received busulfan (for an average of 4 days) as conditioning and were infused with the transduced cells.
The manufacturing process for LentiGlobin was refined in the HGB-207 trial, which translated to a product with a higher vector copy number and higher proportion of CD34+ cells transduced, Dr. Olson said.
The median vector copy number was 3.1 in the HGB-207 trial and 0.7 in the HGB-204 trial. The median proportion of CD34+ cells transfused was 81% and 29%, respectively. The median cell dose was 7.7 x 106 CD34+ cells/kg and 7.1 x 106 CD34+ cells/kg, respectively.
HGB-204 patients and efficacy
The HGB-204 trial included 10 patients with non-beta0/beta0 genotypes – 6 with betaE/beta0, 1 with beta+/beta0, 2 with beta+/beta+, and 1 with an “other” genotype.
The patients’ median age at consent was 19.5 years (range, 16-34). The annualized median prestudy red blood cell (RBC) transfusion volume was 151 mL/kg per year.
At a median follow-up of 36 months, 8 of the 10 patients achieved transfusion independence. The median duration of transfusion independence was 38 months. The median weighted average hemoglobin during transfusion independence was 10.2 g/dL.
“Two patients did not achieve transfusion independence, and both patients were on the lower end of the spectrum both in terms of vector copy number per cell and the percentage of CD34+ cells that were successfully transduced,” Dr. Olson said. “Both patients actually experienced a reduction in the annualized transfusion volume requirements of between 43% and 77%.”
HGB-207 patients and efficacy
The HGB-207 trial included 16 patients with non-beta0/beta0 genotypes – 6 with betaE/beta0, 7 with beta+/beta0, and 3 with the beta+/beta+ genotype.
The patients’ median age at consent was 19 years . The annualized median prestudy RBC transfusion volume was 192 mL/kg per year.
The median follow-up in this trial is 9.3 months. Ten of 11 patients with at least 3 months of follow-up are transfusion-free with hemoglobin levels greater than 11 g/dL.
Two patients have achieved transfusion independence according to the protocol definition, which is weighted average hemoglobin of 9 g/dL or greater without any RBC transfusions for at least 12 months.
“In the one patient in this study who did not achieve transfusion independence, the vector-derived hemoglobin was quite low, and this correlated with a very low vector copy number seen in circulating peripheral blood mononuclear cells,” Dr. Olson said.
It isn’t clear why this occurred, however, as the vector copy number wasn’t especially low in the LentiGlobin product the patient received. Therefore, the researchers are still investigating why this patient failed to achieve transfusion independence.
Safety in both trials
“Very importantly, there were no deaths, there were no engraftment failures, there was no evidence of vector-mediated replication-competent lentivirus, and integration site analysis revealed no evidence of clonal dominance,” Dr. Olson said.
He added that most of the grade 3 or greater adverse events seen in both trials were directly attributable to busulfan-based myeloablative conditioning, including four episodes of veno-occlusive disease.
Nonhematologic grade 3 or higher adverse events in HGB-204 included stomatitis (n = 8), febrile neutropenia (n = 6), irregular menstruation (n = 3), pharyngeal inflammation (n = 2), and veno-occlusive liver disease (n = 1).
Nonhematologic grade 3 or higher adverse events in HGB-207 included stomatitis (n = 9), febrile neutropenia (n = 4), pharyngeal inflammation (n = 2), epistaxis (n = 3), pyrexia (n = 3), veno-occlusive liver disease (n = 3), ALT increase (n = 2), bilirubin increase (n = 2), and hypoxia (n = 2).
One patient in HGB-207 had grade 3 thrombocytopenia considered possibly related to LentiGlobin.
Dr. Olson reported advisory board engagement with bluebird bio, which sponsored both trials.
SOURCE: Olson TS et al. ASPHO 2019. Abstract 2002.
NEW ORLEANS – The gene therapy LentiGlobin can reduce transfusion dependence in children and young adults with non-beta0/beta0 thalassemia, according to two trials.
In a phase 1/2 trial, 8 of 10 of patients achieved transfusion independence at a median follow-up of 36.0 months. In a phase 3 trial, transfusion independence was achieved by 2 of 3 patients with follow-up of at least 12 months.
Timothy S. Olson, MD, PhD, of Children’s Hospital of Philadelphia, presented results from the phase 1/2 HGB-204 trial and the phase 3 HGB-207 trial at the annual meeting of the American Society of Pediatric Hematology/Oncology.
Treatment
In both trials, patients received granulocyte colony-stimulating factor and plerixafor for hematopoietic stem cell mobilization. Their cells were collected via apheresis and transduced with the betibeglogene darolentivec (BB305) lentiviral vector. The patients received busulfan (for an average of 4 days) as conditioning and were infused with the transduced cells.
The manufacturing process for LentiGlobin was refined in the HGB-207 trial, which translated to a product with a higher vector copy number and higher proportion of CD34+ cells transduced, Dr. Olson said.
The median vector copy number was 3.1 in the HGB-207 trial and 0.7 in the HGB-204 trial. The median proportion of CD34+ cells transfused was 81% and 29%, respectively. The median cell dose was 7.7 x 106 CD34+ cells/kg and 7.1 x 106 CD34+ cells/kg, respectively.
HGB-204 patients and efficacy
The HGB-204 trial included 10 patients with non-beta0/beta0 genotypes – 6 with betaE/beta0, 1 with beta+/beta0, 2 with beta+/beta+, and 1 with an “other” genotype.
The patients’ median age at consent was 19.5 years (range, 16-34). The annualized median prestudy red blood cell (RBC) transfusion volume was 151 mL/kg per year.
At a median follow-up of 36 months, 8 of the 10 patients achieved transfusion independence. The median duration of transfusion independence was 38 months. The median weighted average hemoglobin during transfusion independence was 10.2 g/dL.
“Two patients did not achieve transfusion independence, and both patients were on the lower end of the spectrum both in terms of vector copy number per cell and the percentage of CD34+ cells that were successfully transduced,” Dr. Olson said. “Both patients actually experienced a reduction in the annualized transfusion volume requirements of between 43% and 77%.”
HGB-207 patients and efficacy
The HGB-207 trial included 16 patients with non-beta0/beta0 genotypes – 6 with betaE/beta0, 7 with beta+/beta0, and 3 with the beta+/beta+ genotype.
The patients’ median age at consent was 19 years . The annualized median prestudy RBC transfusion volume was 192 mL/kg per year.
The median follow-up in this trial is 9.3 months. Ten of 11 patients with at least 3 months of follow-up are transfusion-free with hemoglobin levels greater than 11 g/dL.
Two patients have achieved transfusion independence according to the protocol definition, which is weighted average hemoglobin of 9 g/dL or greater without any RBC transfusions for at least 12 months.
“In the one patient in this study who did not achieve transfusion independence, the vector-derived hemoglobin was quite low, and this correlated with a very low vector copy number seen in circulating peripheral blood mononuclear cells,” Dr. Olson said.
It isn’t clear why this occurred, however, as the vector copy number wasn’t especially low in the LentiGlobin product the patient received. Therefore, the researchers are still investigating why this patient failed to achieve transfusion independence.
Safety in both trials
“Very importantly, there were no deaths, there were no engraftment failures, there was no evidence of vector-mediated replication-competent lentivirus, and integration site analysis revealed no evidence of clonal dominance,” Dr. Olson said.
He added that most of the grade 3 or greater adverse events seen in both trials were directly attributable to busulfan-based myeloablative conditioning, including four episodes of veno-occlusive disease.
Nonhematologic grade 3 or higher adverse events in HGB-204 included stomatitis (n = 8), febrile neutropenia (n = 6), irregular menstruation (n = 3), pharyngeal inflammation (n = 2), and veno-occlusive liver disease (n = 1).
Nonhematologic grade 3 or higher adverse events in HGB-207 included stomatitis (n = 9), febrile neutropenia (n = 4), pharyngeal inflammation (n = 2), epistaxis (n = 3), pyrexia (n = 3), veno-occlusive liver disease (n = 3), ALT increase (n = 2), bilirubin increase (n = 2), and hypoxia (n = 2).
One patient in HGB-207 had grade 3 thrombocytopenia considered possibly related to LentiGlobin.
Dr. Olson reported advisory board engagement with bluebird bio, which sponsored both trials.
SOURCE: Olson TS et al. ASPHO 2019. Abstract 2002.
NEW ORLEANS – The gene therapy LentiGlobin can reduce transfusion dependence in children and young adults with non-beta0/beta0 thalassemia, according to two trials.
In a phase 1/2 trial, 8 of 10 of patients achieved transfusion independence at a median follow-up of 36.0 months. In a phase 3 trial, transfusion independence was achieved by 2 of 3 patients with follow-up of at least 12 months.
Timothy S. Olson, MD, PhD, of Children’s Hospital of Philadelphia, presented results from the phase 1/2 HGB-204 trial and the phase 3 HGB-207 trial at the annual meeting of the American Society of Pediatric Hematology/Oncology.
Treatment
In both trials, patients received granulocyte colony-stimulating factor and plerixafor for hematopoietic stem cell mobilization. Their cells were collected via apheresis and transduced with the betibeglogene darolentivec (BB305) lentiviral vector. The patients received busulfan (for an average of 4 days) as conditioning and were infused with the transduced cells.
The manufacturing process for LentiGlobin was refined in the HGB-207 trial, which translated to a product with a higher vector copy number and higher proportion of CD34+ cells transduced, Dr. Olson said.
The median vector copy number was 3.1 in the HGB-207 trial and 0.7 in the HGB-204 trial. The median proportion of CD34+ cells transfused was 81% and 29%, respectively. The median cell dose was 7.7 x 106 CD34+ cells/kg and 7.1 x 106 CD34+ cells/kg, respectively.
HGB-204 patients and efficacy
The HGB-204 trial included 10 patients with non-beta0/beta0 genotypes – 6 with betaE/beta0, 1 with beta+/beta0, 2 with beta+/beta+, and 1 with an “other” genotype.
The patients’ median age at consent was 19.5 years (range, 16-34). The annualized median prestudy red blood cell (RBC) transfusion volume was 151 mL/kg per year.
At a median follow-up of 36 months, 8 of the 10 patients achieved transfusion independence. The median duration of transfusion independence was 38 months. The median weighted average hemoglobin during transfusion independence was 10.2 g/dL.
“Two patients did not achieve transfusion independence, and both patients were on the lower end of the spectrum both in terms of vector copy number per cell and the percentage of CD34+ cells that were successfully transduced,” Dr. Olson said. “Both patients actually experienced a reduction in the annualized transfusion volume requirements of between 43% and 77%.”
HGB-207 patients and efficacy
The HGB-207 trial included 16 patients with non-beta0/beta0 genotypes – 6 with betaE/beta0, 7 with beta+/beta0, and 3 with the beta+/beta+ genotype.
The patients’ median age at consent was 19 years . The annualized median prestudy RBC transfusion volume was 192 mL/kg per year.
The median follow-up in this trial is 9.3 months. Ten of 11 patients with at least 3 months of follow-up are transfusion-free with hemoglobin levels greater than 11 g/dL.
Two patients have achieved transfusion independence according to the protocol definition, which is weighted average hemoglobin of 9 g/dL or greater without any RBC transfusions for at least 12 months.
“In the one patient in this study who did not achieve transfusion independence, the vector-derived hemoglobin was quite low, and this correlated with a very low vector copy number seen in circulating peripheral blood mononuclear cells,” Dr. Olson said.
It isn’t clear why this occurred, however, as the vector copy number wasn’t especially low in the LentiGlobin product the patient received. Therefore, the researchers are still investigating why this patient failed to achieve transfusion independence.
Safety in both trials
“Very importantly, there were no deaths, there were no engraftment failures, there was no evidence of vector-mediated replication-competent lentivirus, and integration site analysis revealed no evidence of clonal dominance,” Dr. Olson said.
He added that most of the grade 3 or greater adverse events seen in both trials were directly attributable to busulfan-based myeloablative conditioning, including four episodes of veno-occlusive disease.
Nonhematologic grade 3 or higher adverse events in HGB-204 included stomatitis (n = 8), febrile neutropenia (n = 6), irregular menstruation (n = 3), pharyngeal inflammation (n = 2), and veno-occlusive liver disease (n = 1).
Nonhematologic grade 3 or higher adverse events in HGB-207 included stomatitis (n = 9), febrile neutropenia (n = 4), pharyngeal inflammation (n = 2), epistaxis (n = 3), pyrexia (n = 3), veno-occlusive liver disease (n = 3), ALT increase (n = 2), bilirubin increase (n = 2), and hypoxia (n = 2).
One patient in HGB-207 had grade 3 thrombocytopenia considered possibly related to LentiGlobin.
Dr. Olson reported advisory board engagement with bluebird bio, which sponsored both trials.
SOURCE: Olson TS et al. ASPHO 2019. Abstract 2002.
REPORTING FROM 2019 ASPHO CONFERENCE