User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
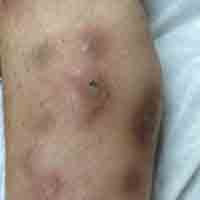
Hypopigmented Discoloration on the Thigh
The Diagnosis: Hypopigmented Mycosis Fungoides
The patient was started on clobetasol dipropionate cream 0.05% twice daily, which she did not tolerate due to a burning sensation on application. She then was started on narrowband UVB phototherapy 2 to 3 times weekly, and the hypopigmented areas began to improve. Narrowband UVB phototherapy was discontinued after 7 weeks due to the high cost to the patient, but the hypopigmented patches on the left thigh appeared to remit, and the patient did not return to the clinic for 6 months. She returned when the areas on the left thigh reappeared, along with new areas on the right buttock and right medial upper arm. Serial biopsies of the new patches also revealed a CD8+ atypical lymphocytic infiltrate consistent with hypopigmented patch-stage mycosis fungoides (MF). She was started on halobetasol ointment 0.05% twice daily to affected areas, which she tolerated well. Complete blood count and peripheral blood smear were unremarkable, and the patient continued to deny systemic symptoms. Over the next year, the patient's cutaneous findings continued to wax and wane with topical treatment, and she was referred to a regional cancer treatment center for a second opinion from a hematopathologist. Hematopathologic and dermatopathologic review of the case, including hematoxylin and eosin and immunohistochemical staining, was highly consistent with hypopigmented MF (Figures 1-3).
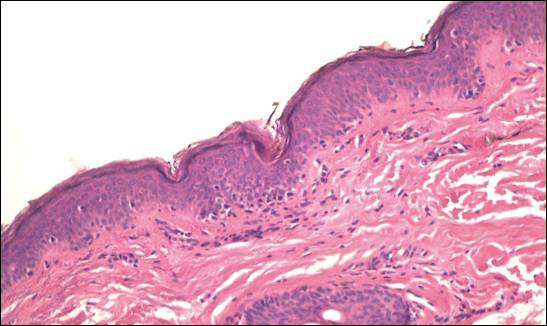
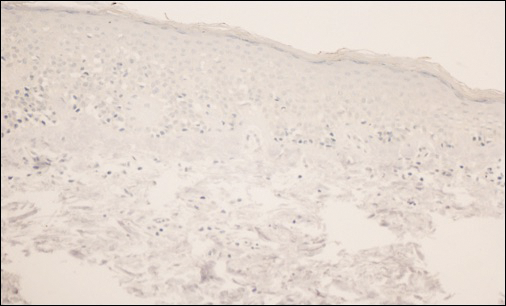
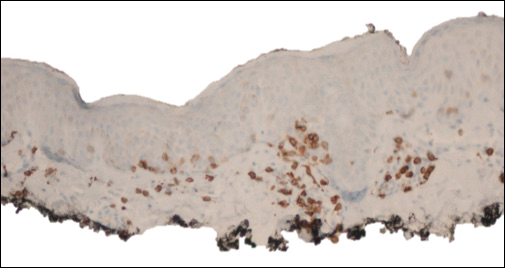
Mycosis fungoides is an uncommon disease characterized by atypical clonal T cells exhibiting epidermotropism. Most commonly, MF is characterized by a CD4+ lymphocytic infiltrate. Mycosis fungoides can be difficult to diagnose in its early stages, as it may resemble benign inflammatory conditions (eg, chronic atopic dermatitis, nummular eczema) and often requires biopsy and additional studies, such as immunohistochemistry, to secure a diagnosis. Hypopigmented MF is regarded as a subtype of MF, as it can exhibit different clinical and pathologic characteristics from classical MF. In particular, the lymphocytic phenotype in hypopigmented MF is more likely to be CD8+.
In general, the progression of MF is characterized as stage IA (patches or plaques involving less than 10% body surface area [BSA]), IB (patches or plaques involving ≥10% BSA without lymph node or visceral involvement), IIA (patches or plaques of any percentage of BSA with lymph node involvement), IIB (cutaneous tumors with or without lymph node involvement), III (erythroderma with low blood tumor burden), or IV (erythroderma with high blood tumor burden with or without visceral involvement). Hypopigmented MF generally presents in early patch stage and rarely progresses past stage IB, and thus generally has a favorable prognosis.1,2 Kim et al3 demonstrated that evolution from patch to plaque stage MF is accompanied by a shift in lymphocytes from the T helper 1 (Th1) to T helper 2 phenotype; therefore the Th1 phenotype, CD8+ T cells are associated with lower risk for disease progression. Other investigators also have hypothesized that predominance of Th1 phenotype, CD8+ T cells may have an immunoregulatory effect, thus preventing evolution of disease from patch to plaque stage and explaining why hypopigmented MF, with a predominantly CD8+ phenotype, confers better prognosis with less chance for disease progression than classical MF.4,5 The patch- or plaque-stage lesions of classical MF have a predilection for non-sun exposed areas (eg, buttocks, medial thighs, breasts),2 whereas hypopigmented MF tends to present with hypopigmented or depigmented lesions mainly distributed on the trunk, arms, and legs. These lesions may become more visible following sun exposure.1 The size of the hypopigmented lesions can vary, and patients may complain of pruritus with variable intensity.
Hypopigmented MF presents more commonly in younger populations, in contrast to classical MF.6-8 However, like classical MF, hypopigmented MF appears to more frequently affect individuals with darker Fitzpatrick skin types.1,9,10 Although it generally is accepted that hypopigmented MF does not favor either sex, some studies suggest that hypopigmented MF has a female predominance.6,10
Classical MF is characterized by an epidermotropic infiltrate of CD4+ T helper cells,10 whereas CD8+ epidermotropism is considered hallmark in hypopigmented MF.10-12 The other typical histopathologic features of hypopigmented MF generally are identical to those of classical MF, with solitary or small groups of atypical haloed lymphocytes within the basal layer, exocytosis of lymphocytes out of proportion to spongiosis, and papillary dermal fibrosis. Immunohistochemistry generally is helpful in distinguishing between classical MF and hypopigmented MF.
The clinical differential diagnosis for hypopigmented MF includes the early (inflammatory) stage of vitiligo, postinflammatory hypopigmentation, lichen sclerosus, pityriasis alba, and leprosy.
First-line treatment for hypopigmented MF consists of phototherapy/photochemotherapy and topical steroids.9,13 Narrowband UVB phototherapy has been used with good success in pediatric patients.14 However, narrowband UVB may not be as effective in darker-skinned individuals; it has been hypothesized that this lack of efficacy could be due to the protective effects of increased melanin in the skin.1 Other topical therapies may include topical carmustine and topical nitrogen mustard.
- Furlan FC, Sanches JA. Hypopigmented mycosis fungoides: a review of its clinical features and pathophysiology. An Bras Dermatol. 2013;88:954-960.
- Girardi M, Heald PW, Wilson LD. The pathogenesis of mycosis fungoides. N Engl J Med. 2004;350:1978-1988.
- Kim EJ, Hess S, Richardson SK, et al. Immunopathogenesis and therapy of cutaneous T cell lymphoma. J Clin Invest. 2005;115:798-812.
- Stone ML, Styles AR, Cockerell CJ, et al. Hypopigmented report of 7 cases and review of the literature. Cutis. 2001;67:133-138.
- Volkenandt M, Soyer HP, Cerroni L, et al. Molecular detection of clone-specific DNA in hypopigmented lesions of a patient with early evolving mycosis fungoides. Br J Dermatol. 1993;128:423-428.
- Furlan FC, Pereira BA, Sotto MN, et al. Hypopigmented mycosis fungoides versus mycosis fungoides with concomitant hypopigmented lesions: same disease or different variants of mycosis fungoides? Dermatology. 2014;229:271-274.
- Ardigó M, Borroni G, Muscardin L, et al. Hypopigmented mycosis fungoides in Caucasian patients: a clinicopathologic study of 7 cases. J Am Acad Dermatol. 2003;49:264-270.
- Boulos S, Vaid R, Aladily TN, et al. Clinical presentation, immunopathology, and treatment of juvenile-onset mycosis fungoides: a case series of 34 patients. J Am Acad Dermatol. 2014;71:1117-1126.
- Lambroza E, Cohen SR, Phelps R, et al. Hypopigmented variant of mycosis fungoides: demography, histopathology, and treatment of seven cases. J Am Acad Dermatol. 1995;32:987-993.
- El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, et al. Hypopigmented mycosis fungoides: Frequent expression of a CD8+ T-cell phenotype. Am J Surg Pathol. 2002;26:450-457.
- Furlan FC, de Paula Pereira BA, da Silva LF, et al. Loss of melanocytes in hypopigmented mycosis fungoides: a study of 18 patients. J Cutan Pathol. 2014;41:101-107.
- Tolkachjov SN, Comfere NI. Hypopigmented mycosis fungoides: a clinical mimicker of vitiligo. J Drugs Dermatol. 2015;14:193-194.
- Duarte I, Bedrikow, R, Aoki S. Mycosis fungoides: epidemiologic study of 17 cases and evaluation of PUVA photochemotherapy. An Bras Dermatol. 2006;81:40-45.
- Onsun N, Kural Y, Su O, et al. Hypopigmented mycosis fungoides associated with atopy in two children. Pediatr Dermatol. 2006;23:493-496.
The Diagnosis: Hypopigmented Mycosis Fungoides
The patient was started on clobetasol dipropionate cream 0.05% twice daily, which she did not tolerate due to a burning sensation on application. She then was started on narrowband UVB phototherapy 2 to 3 times weekly, and the hypopigmented areas began to improve. Narrowband UVB phototherapy was discontinued after 7 weeks due to the high cost to the patient, but the hypopigmented patches on the left thigh appeared to remit, and the patient did not return to the clinic for 6 months. She returned when the areas on the left thigh reappeared, along with new areas on the right buttock and right medial upper arm. Serial biopsies of the new patches also revealed a CD8+ atypical lymphocytic infiltrate consistent with hypopigmented patch-stage mycosis fungoides (MF). She was started on halobetasol ointment 0.05% twice daily to affected areas, which she tolerated well. Complete blood count and peripheral blood smear were unremarkable, and the patient continued to deny systemic symptoms. Over the next year, the patient's cutaneous findings continued to wax and wane with topical treatment, and she was referred to a regional cancer treatment center for a second opinion from a hematopathologist. Hematopathologic and dermatopathologic review of the case, including hematoxylin and eosin and immunohistochemical staining, was highly consistent with hypopigmented MF (Figures 1-3).
Mycosis fungoides is an uncommon disease characterized by atypical clonal T cells exhibiting epidermotropism. Most commonly, MF is characterized by a CD4+ lymphocytic infiltrate. Mycosis fungoides can be difficult to diagnose in its early stages, as it may resemble benign inflammatory conditions (eg, chronic atopic dermatitis, nummular eczema) and often requires biopsy and additional studies, such as immunohistochemistry, to secure a diagnosis. Hypopigmented MF is regarded as a subtype of MF, as it can exhibit different clinical and pathologic characteristics from classical MF. In particular, the lymphocytic phenotype in hypopigmented MF is more likely to be CD8+.
In general, the progression of MF is characterized as stage IA (patches or plaques involving less than 10% body surface area [BSA]), IB (patches or plaques involving ≥10% BSA without lymph node or visceral involvement), IIA (patches or plaques of any percentage of BSA with lymph node involvement), IIB (cutaneous tumors with or without lymph node involvement), III (erythroderma with low blood tumor burden), or IV (erythroderma with high blood tumor burden with or without visceral involvement). Hypopigmented MF generally presents in early patch stage and rarely progresses past stage IB, and thus generally has a favorable prognosis.1,2 Kim et al3 demonstrated that evolution from patch to plaque stage MF is accompanied by a shift in lymphocytes from the T helper 1 (Th1) to T helper 2 phenotype; therefore the Th1 phenotype, CD8+ T cells are associated with lower risk for disease progression. Other investigators also have hypothesized that predominance of Th1 phenotype, CD8+ T cells may have an immunoregulatory effect, thus preventing evolution of disease from patch to plaque stage and explaining why hypopigmented MF, with a predominantly CD8+ phenotype, confers better prognosis with less chance for disease progression than classical MF.4,5 The patch- or plaque-stage lesions of classical MF have a predilection for non-sun exposed areas (eg, buttocks, medial thighs, breasts),2 whereas hypopigmented MF tends to present with hypopigmented or depigmented lesions mainly distributed on the trunk, arms, and legs. These lesions may become more visible following sun exposure.1 The size of the hypopigmented lesions can vary, and patients may complain of pruritus with variable intensity.
Hypopigmented MF presents more commonly in younger populations, in contrast to classical MF.6-8 However, like classical MF, hypopigmented MF appears to more frequently affect individuals with darker Fitzpatrick skin types.1,9,10 Although it generally is accepted that hypopigmented MF does not favor either sex, some studies suggest that hypopigmented MF has a female predominance.6,10
Classical MF is characterized by an epidermotropic infiltrate of CD4+ T helper cells,10 whereas CD8+ epidermotropism is considered hallmark in hypopigmented MF.10-12 The other typical histopathologic features of hypopigmented MF generally are identical to those of classical MF, with solitary or small groups of atypical haloed lymphocytes within the basal layer, exocytosis of lymphocytes out of proportion to spongiosis, and papillary dermal fibrosis. Immunohistochemistry generally is helpful in distinguishing between classical MF and hypopigmented MF.
The clinical differential diagnosis for hypopigmented MF includes the early (inflammatory) stage of vitiligo, postinflammatory hypopigmentation, lichen sclerosus, pityriasis alba, and leprosy.
First-line treatment for hypopigmented MF consists of phototherapy/photochemotherapy and topical steroids.9,13 Narrowband UVB phototherapy has been used with good success in pediatric patients.14 However, narrowband UVB may not be as effective in darker-skinned individuals; it has been hypothesized that this lack of efficacy could be due to the protective effects of increased melanin in the skin.1 Other topical therapies may include topical carmustine and topical nitrogen mustard.
The Diagnosis: Hypopigmented Mycosis Fungoides
The patient was started on clobetasol dipropionate cream 0.05% twice daily, which she did not tolerate due to a burning sensation on application. She then was started on narrowband UVB phototherapy 2 to 3 times weekly, and the hypopigmented areas began to improve. Narrowband UVB phototherapy was discontinued after 7 weeks due to the high cost to the patient, but the hypopigmented patches on the left thigh appeared to remit, and the patient did not return to the clinic for 6 months. She returned when the areas on the left thigh reappeared, along with new areas on the right buttock and right medial upper arm. Serial biopsies of the new patches also revealed a CD8+ atypical lymphocytic infiltrate consistent with hypopigmented patch-stage mycosis fungoides (MF). She was started on halobetasol ointment 0.05% twice daily to affected areas, which she tolerated well. Complete blood count and peripheral blood smear were unremarkable, and the patient continued to deny systemic symptoms. Over the next year, the patient's cutaneous findings continued to wax and wane with topical treatment, and she was referred to a regional cancer treatment center for a second opinion from a hematopathologist. Hematopathologic and dermatopathologic review of the case, including hematoxylin and eosin and immunohistochemical staining, was highly consistent with hypopigmented MF (Figures 1-3).
Mycosis fungoides is an uncommon disease characterized by atypical clonal T cells exhibiting epidermotropism. Most commonly, MF is characterized by a CD4+ lymphocytic infiltrate. Mycosis fungoides can be difficult to diagnose in its early stages, as it may resemble benign inflammatory conditions (eg, chronic atopic dermatitis, nummular eczema) and often requires biopsy and additional studies, such as immunohistochemistry, to secure a diagnosis. Hypopigmented MF is regarded as a subtype of MF, as it can exhibit different clinical and pathologic characteristics from classical MF. In particular, the lymphocytic phenotype in hypopigmented MF is more likely to be CD8+.
In general, the progression of MF is characterized as stage IA (patches or plaques involving less than 10% body surface area [BSA]), IB (patches or plaques involving ≥10% BSA without lymph node or visceral involvement), IIA (patches or plaques of any percentage of BSA with lymph node involvement), IIB (cutaneous tumors with or without lymph node involvement), III (erythroderma with low blood tumor burden), or IV (erythroderma with high blood tumor burden with or without visceral involvement). Hypopigmented MF generally presents in early patch stage and rarely progresses past stage IB, and thus generally has a favorable prognosis.1,2 Kim et al3 demonstrated that evolution from patch to plaque stage MF is accompanied by a shift in lymphocytes from the T helper 1 (Th1) to T helper 2 phenotype; therefore the Th1 phenotype, CD8+ T cells are associated with lower risk for disease progression. Other investigators also have hypothesized that predominance of Th1 phenotype, CD8+ T cells may have an immunoregulatory effect, thus preventing evolution of disease from patch to plaque stage and explaining why hypopigmented MF, with a predominantly CD8+ phenotype, confers better prognosis with less chance for disease progression than classical MF.4,5 The patch- or plaque-stage lesions of classical MF have a predilection for non-sun exposed areas (eg, buttocks, medial thighs, breasts),2 whereas hypopigmented MF tends to present with hypopigmented or depigmented lesions mainly distributed on the trunk, arms, and legs. These lesions may become more visible following sun exposure.1 The size of the hypopigmented lesions can vary, and patients may complain of pruritus with variable intensity.
Hypopigmented MF presents more commonly in younger populations, in contrast to classical MF.6-8 However, like classical MF, hypopigmented MF appears to more frequently affect individuals with darker Fitzpatrick skin types.1,9,10 Although it generally is accepted that hypopigmented MF does not favor either sex, some studies suggest that hypopigmented MF has a female predominance.6,10
Classical MF is characterized by an epidermotropic infiltrate of CD4+ T helper cells,10 whereas CD8+ epidermotropism is considered hallmark in hypopigmented MF.10-12 The other typical histopathologic features of hypopigmented MF generally are identical to those of classical MF, with solitary or small groups of atypical haloed lymphocytes within the basal layer, exocytosis of lymphocytes out of proportion to spongiosis, and papillary dermal fibrosis. Immunohistochemistry generally is helpful in distinguishing between classical MF and hypopigmented MF.
The clinical differential diagnosis for hypopigmented MF includes the early (inflammatory) stage of vitiligo, postinflammatory hypopigmentation, lichen sclerosus, pityriasis alba, and leprosy.
First-line treatment for hypopigmented MF consists of phototherapy/photochemotherapy and topical steroids.9,13 Narrowband UVB phototherapy has been used with good success in pediatric patients.14 However, narrowband UVB may not be as effective in darker-skinned individuals; it has been hypothesized that this lack of efficacy could be due to the protective effects of increased melanin in the skin.1 Other topical therapies may include topical carmustine and topical nitrogen mustard.
- Furlan FC, Sanches JA. Hypopigmented mycosis fungoides: a review of its clinical features and pathophysiology. An Bras Dermatol. 2013;88:954-960.
- Girardi M, Heald PW, Wilson LD. The pathogenesis of mycosis fungoides. N Engl J Med. 2004;350:1978-1988.
- Kim EJ, Hess S, Richardson SK, et al. Immunopathogenesis and therapy of cutaneous T cell lymphoma. J Clin Invest. 2005;115:798-812.
- Stone ML, Styles AR, Cockerell CJ, et al. Hypopigmented report of 7 cases and review of the literature. Cutis. 2001;67:133-138.
- Volkenandt M, Soyer HP, Cerroni L, et al. Molecular detection of clone-specific DNA in hypopigmented lesions of a patient with early evolving mycosis fungoides. Br J Dermatol. 1993;128:423-428.
- Furlan FC, Pereira BA, Sotto MN, et al. Hypopigmented mycosis fungoides versus mycosis fungoides with concomitant hypopigmented lesions: same disease or different variants of mycosis fungoides? Dermatology. 2014;229:271-274.
- Ardigó M, Borroni G, Muscardin L, et al. Hypopigmented mycosis fungoides in Caucasian patients: a clinicopathologic study of 7 cases. J Am Acad Dermatol. 2003;49:264-270.
- Boulos S, Vaid R, Aladily TN, et al. Clinical presentation, immunopathology, and treatment of juvenile-onset mycosis fungoides: a case series of 34 patients. J Am Acad Dermatol. 2014;71:1117-1126.
- Lambroza E, Cohen SR, Phelps R, et al. Hypopigmented variant of mycosis fungoides: demography, histopathology, and treatment of seven cases. J Am Acad Dermatol. 1995;32:987-993.
- El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, et al. Hypopigmented mycosis fungoides: Frequent expression of a CD8+ T-cell phenotype. Am J Surg Pathol. 2002;26:450-457.
- Furlan FC, de Paula Pereira BA, da Silva LF, et al. Loss of melanocytes in hypopigmented mycosis fungoides: a study of 18 patients. J Cutan Pathol. 2014;41:101-107.
- Tolkachjov SN, Comfere NI. Hypopigmented mycosis fungoides: a clinical mimicker of vitiligo. J Drugs Dermatol. 2015;14:193-194.
- Duarte I, Bedrikow, R, Aoki S. Mycosis fungoides: epidemiologic study of 17 cases and evaluation of PUVA photochemotherapy. An Bras Dermatol. 2006;81:40-45.
- Onsun N, Kural Y, Su O, et al. Hypopigmented mycosis fungoides associated with atopy in two children. Pediatr Dermatol. 2006;23:493-496.
- Furlan FC, Sanches JA. Hypopigmented mycosis fungoides: a review of its clinical features and pathophysiology. An Bras Dermatol. 2013;88:954-960.
- Girardi M, Heald PW, Wilson LD. The pathogenesis of mycosis fungoides. N Engl J Med. 2004;350:1978-1988.
- Kim EJ, Hess S, Richardson SK, et al. Immunopathogenesis and therapy of cutaneous T cell lymphoma. J Clin Invest. 2005;115:798-812.
- Stone ML, Styles AR, Cockerell CJ, et al. Hypopigmented report of 7 cases and review of the literature. Cutis. 2001;67:133-138.
- Volkenandt M, Soyer HP, Cerroni L, et al. Molecular detection of clone-specific DNA in hypopigmented lesions of a patient with early evolving mycosis fungoides. Br J Dermatol. 1993;128:423-428.
- Furlan FC, Pereira BA, Sotto MN, et al. Hypopigmented mycosis fungoides versus mycosis fungoides with concomitant hypopigmented lesions: same disease or different variants of mycosis fungoides? Dermatology. 2014;229:271-274.
- Ardigó M, Borroni G, Muscardin L, et al. Hypopigmented mycosis fungoides in Caucasian patients: a clinicopathologic study of 7 cases. J Am Acad Dermatol. 2003;49:264-270.
- Boulos S, Vaid R, Aladily TN, et al. Clinical presentation, immunopathology, and treatment of juvenile-onset mycosis fungoides: a case series of 34 patients. J Am Acad Dermatol. 2014;71:1117-1126.
- Lambroza E, Cohen SR, Phelps R, et al. Hypopigmented variant of mycosis fungoides: demography, histopathology, and treatment of seven cases. J Am Acad Dermatol. 1995;32:987-993.
- El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, et al. Hypopigmented mycosis fungoides: Frequent expression of a CD8+ T-cell phenotype. Am J Surg Pathol. 2002;26:450-457.
- Furlan FC, de Paula Pereira BA, da Silva LF, et al. Loss of melanocytes in hypopigmented mycosis fungoides: a study of 18 patients. J Cutan Pathol. 2014;41:101-107.
- Tolkachjov SN, Comfere NI. Hypopigmented mycosis fungoides: a clinical mimicker of vitiligo. J Drugs Dermatol. 2015;14:193-194.
- Duarte I, Bedrikow, R, Aoki S. Mycosis fungoides: epidemiologic study of 17 cases and evaluation of PUVA photochemotherapy. An Bras Dermatol. 2006;81:40-45.
- Onsun N, Kural Y, Su O, et al. Hypopigmented mycosis fungoides associated with atopy in two children. Pediatr Dermatol. 2006;23:493-496.
A 39-year-old woman presented with 2 areas of hypopigmented discoloration on the left thigh of 6 months' duration. The hypopigmentation was more visible following sun exposure because the areas did not tan. The patient had not sought prior treatment for the discoloration and denied any previous rash or trauma to the area. Her medical history was remarkable for hypothyroidism associated with mild and transient alopecia, acne, and xerosis. Her daily medications included oral contraceptive pills (norgestimate/ethinyl estradiol), oral levothyroxine/liothyronine, and sulfacetamide lotion 10%. She denied any allergies, and the remainder of her medical, surgical, social, and family history was unremarkable. A review of systems was negative for enlarged lymph nodes, fever, night sweats, and fatigue. Physical examination revealed 2 subtle hypopigmented patches with fine, atrophic, cigarette paper-like wrinkling distributed on the left medial and posterior upper thigh. Initial biopsy of the hypopigmented patches revealed a CD8+ lymphocytic infiltrate with an atypical interface.
National Health Council to Host Workshop for Patient Organizations
The National Health Council will host a free workshop in Washington, DC, for patient advocacy organizations on Feb. 15, 2018 on engaging in quality measurement and research. More.
The National Health Council will host a free workshop in Washington, DC, for patient advocacy organizations on Feb. 15, 2018 on engaging in quality measurement and research. More.
The National Health Council will host a free workshop in Washington, DC, for patient advocacy organizations on Feb. 15, 2018 on engaging in quality measurement and research. More.
NORD Call for Abstracts Reopened
Grants are available through the NORD Research Program for the study of cat eye syndrome, malonic aciduria, and post-orgasmic illness syndrome. View the RFPs here.
Grants are available through the NORD Research Program for the study of cat eye syndrome, malonic aciduria, and post-orgasmic illness syndrome. View the RFPs here.
Grants are available through the NORD Research Program for the study of cat eye syndrome, malonic aciduria, and post-orgasmic illness syndrome. View the RFPs here.
NORD Submits Comments to the Centers for Medicare and Medicaid Services
NORD has been, and will continue to be, engaged in a number of activities to protect access to quality health care coverage for Medicaid beneficiaries. Most recently, NORD has submitted comments to the Centers for Medicare and Medicaid Services (CMS) regarding Kansas’ effort to implement work requirements and a lifetime limit in its Medicaid program. Read the comments.
NORD has been, and will continue to be, engaged in a number of activities to protect access to quality health care coverage for Medicaid beneficiaries. Most recently, NORD has submitted comments to the Centers for Medicare and Medicaid Services (CMS) regarding Kansas’ effort to implement work requirements and a lifetime limit in its Medicaid program. Read the comments.
NORD has been, and will continue to be, engaged in a number of activities to protect access to quality health care coverage for Medicaid beneficiaries. Most recently, NORD has submitted comments to the Centers for Medicare and Medicaid Services (CMS) regarding Kansas’ effort to implement work requirements and a lifetime limit in its Medicaid program. Read the comments.
NORD Advocates for Iowa Co-pay Choice Bill
NORD has been advocating for the Iowa Co-pay Choice Bill (SSB 3004). This legislation would help with rising out-of-pocket costs. Specifically, SSB 3004 gives choice and co-pay predictability to Iowa families by requiring that insurance providers offer a minimum number of plans that provide a traditional co-pay option, as opposed to co-insurance. NORD will continue to advocate for this legislation throughout February.
NORD has been advocating for the Iowa Co-pay Choice Bill (SSB 3004). This legislation would help with rising out-of-pocket costs. Specifically, SSB 3004 gives choice and co-pay predictability to Iowa families by requiring that insurance providers offer a minimum number of plans that provide a traditional co-pay option, as opposed to co-insurance. NORD will continue to advocate for this legislation throughout February.
NORD has been advocating for the Iowa Co-pay Choice Bill (SSB 3004). This legislation would help with rising out-of-pocket costs. Specifically, SSB 3004 gives choice and co-pay predictability to Iowa families by requiring that insurance providers offer a minimum number of plans that provide a traditional co-pay option, as opposed to co-insurance. NORD will continue to advocate for this legislation throughout February.
Letter to Congress: NORD and Others Oppose “Right to Try”
NORD and 40 other patient organizations and professional societies have sent a letter to the leadership of the US House of Representatives explaining why they oppose “Right to Try” legislation currently being considered by Congress. Signers of the letter include the American Society of Clinical Oncology, Leukemia & Lymphoma Society, American Lung Association, and National Health Council. The organizations say they support patients being given access to unapproved therapies but believe the bills under consideration will not increase patient access.
NORD and 40 other patient organizations and professional societies have sent a letter to the leadership of the US House of Representatives explaining why they oppose “Right to Try” legislation currently being considered by Congress. Signers of the letter include the American Society of Clinical Oncology, Leukemia & Lymphoma Society, American Lung Association, and National Health Council. The organizations say they support patients being given access to unapproved therapies but believe the bills under consideration will not increase patient access.
NORD and 40 other patient organizations and professional societies have sent a letter to the leadership of the US House of Representatives explaining why they oppose “Right to Try” legislation currently being considered by Congress. Signers of the letter include the American Society of Clinical Oncology, Leukemia & Lymphoma Society, American Lung Association, and National Health Council. The organizations say they support patients being given access to unapproved therapies but believe the bills under consideration will not increase patient access.
Rare Disease Day Events Planned for Campuses, Hospitals, and State Legislatures
On February 28, 2018, Rare Disease Day will be observed around the world. This annual observance is intended to promote awareness of the 7,000 diseases considered rare in the US, the need for research and clinical guidelines, and the challenges and opportunities for improving the quality of life for patients. Educational events are planned at numerous hospitals, universities, schools, and community centers across the US. NORD will be hosting “State House Events” with patient advocates in more than 30 states to educate state legislators on issues related to state policy. Visit the national website hosted by NORD. Visit the international website.
On February 28, 2018, Rare Disease Day will be observed around the world. This annual observance is intended to promote awareness of the 7,000 diseases considered rare in the US, the need for research and clinical guidelines, and the challenges and opportunities for improving the quality of life for patients. Educational events are planned at numerous hospitals, universities, schools, and community centers across the US. NORD will be hosting “State House Events” with patient advocates in more than 30 states to educate state legislators on issues related to state policy. Visit the national website hosted by NORD. Visit the international website.
On February 28, 2018, Rare Disease Day will be observed around the world. This annual observance is intended to promote awareness of the 7,000 diseases considered rare in the US, the need for research and clinical guidelines, and the challenges and opportunities for improving the quality of life for patients. Educational events are planned at numerous hospitals, universities, schools, and community centers across the US. NORD will be hosting “State House Events” with patient advocates in more than 30 states to educate state legislators on issues related to state policy. Visit the national website hosted by NORD. Visit the international website.
NORD to Moderate Panel at NIH Rare Disease Day Event
NORD Board Chair Marshall Summar, MD, of Children’s National Health System, will moderate a panel on the importance of collaborating with patient organizations in rare disease research at the annual National Institutes of Health (NIH) Rare Disease Day event on March 1, 2018. The event is open to the public and will be webcast. Other speakers will include NIH Director Francis Collins, MD, PhD; representatives of the Rare Disease Congressional Caucus; NIH researchers; and leaders of patient organizations. Topics will include gene therapy, gene editing, and engaging the next generation of the rare diseases community. For information or to register, visit the NIH web page.
NORD Board Chair Marshall Summar, MD, of Children’s National Health System, will moderate a panel on the importance of collaborating with patient organizations in rare disease research at the annual National Institutes of Health (NIH) Rare Disease Day event on March 1, 2018. The event is open to the public and will be webcast. Other speakers will include NIH Director Francis Collins, MD, PhD; representatives of the Rare Disease Congressional Caucus; NIH researchers; and leaders of patient organizations. Topics will include gene therapy, gene editing, and engaging the next generation of the rare diseases community. For information or to register, visit the NIH web page.
NORD Board Chair Marshall Summar, MD, of Children’s National Health System, will moderate a panel on the importance of collaborating with patient organizations in rare disease research at the annual National Institutes of Health (NIH) Rare Disease Day event on March 1, 2018. The event is open to the public and will be webcast. Other speakers will include NIH Director Francis Collins, MD, PhD; representatives of the Rare Disease Congressional Caucus; NIH researchers; and leaders of patient organizations. Topics will include gene therapy, gene editing, and engaging the next generation of the rare diseases community. For information or to register, visit the NIH web page.
A Peek at Our February 2018 Issue
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Panniculitis, Pancreatitis, and Polyarthritis: A Rare Clinical Syndrome
Pancreatic panniculitis is a rare disease contributing to widespread fat necrosis in patients with underlying pancreatic disorders. This entity was first described in 1883,1 but it was not until 1947 that it was reported in the English-language literature.2 Patients with pancreatitis infrequently develop extrapancreatic manifestations. It has been estimated that only 2% to 3% of patients worldwide with an underlying pancreatic disease develop cutaneous lesions.3 Patients who develop pancreatic panniculitis typically present with tender, edematous, erythematous to brown, subcutaneous nodules on the lower legs with the tendency for spontaneous ulceration. Lesions tend to exude a viscous, yellow-brown, oily substance that represents liquefactive necrosis of enzymatic fat in subcutaneous tissue. Cutaneous lesions may precede, occur simultaneously, or follow the development of an underlying pancreatic disorder. Rarely, patients may develop inflammatory arthritis secondary to intraosseous fat necrosis, completing the triad of findings diagnostic for panniculitis, pancreatitis, and polyarthritis (PPP) syndrome. Although the underlying pancreatic pathology may vary, roughly 80% of cases worldwide have acute/chronic pancreatitis or pancreatic carcinoma, most commonly acinar cell carcinoma.4-6 Less common pancreatic disorders include pancreatic pseudocyst, pancreatic divisum, and vascular pancreatic fistulas.7 Narváez et al8 found that of the 25 cases of PPP syndrome reported in the literature, 68% (17/25) were men, 32% (8/25) were women, 56% (14/25) were younger than 50 years, and 64% (16/25) had a history of prior or current alcohol abuse.
Case Report
A 68-year-old man with a history of hypertension, gastroesophageal reflux disease, chronic pancreatitis of unknown etiology, and arthritis presented to our clinic for evaluation of painful skin nodules on the lower legs of 8 months’ duration, in addition to joint pain and swelling of the metacarpophalangeal (MCP), metatarsophalangeal, and ankle joints. He had a history of numerous hospital admissions over the last 2 years for pancreatitis and was being managed by the rheumatology department for arthritic symptoms.
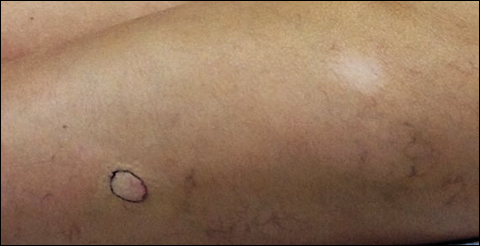
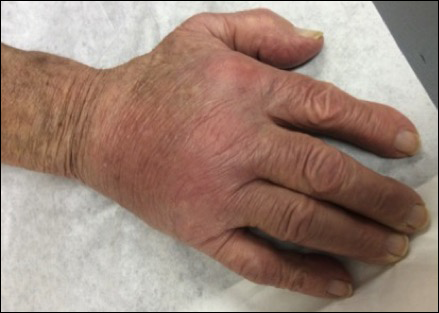
Physical examination revealed multiple 1- to 4-cm, ill-defined, erythematous to brown, subcutaneous nodules on the bilateral lower legs (Figure 1) and right inferomedial thigh that were tender to palpation. Marked erythema and edema of the MCP and metatarsophalangeal joints (Figure 2) and bilateral ankles were observed. Diffuse 2+ pitting edema was present in the bilateral lower extremities, along with areas of hyperpigmentation overlying resolving lesions.

Laboratory data revealed an elevated lipase level (>16,000 U/L [reference range, 31–186 U/L]), amylase level (>4700 U/L [reference range, 27–131 U/L]), erythrocyte sedimentation rate (94 mm/h [reference range, 0–20 mm/h]), and C-reactive protein level (93.5 mg/L [0.08–3.1 mg/L]). The patient had more than 6 episodes of recurrent idiopathic pancreatitis over the last 2 years, though symptoms of abdominal pain were minimal to nonexistent. Liver function tests and alcohol, calcium, and triglyceride levels all were within reference range. Rheumatoid factor and antinuclear antibodies were negative.
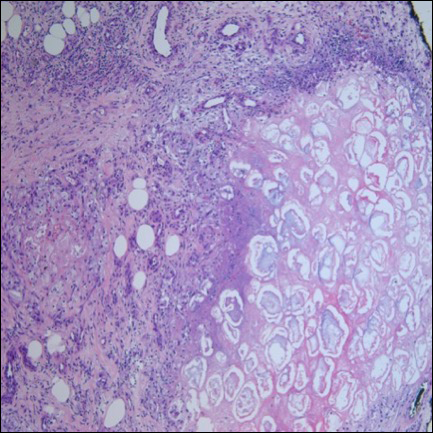
Ultrasonography showed no evidence of cholelithiasis. Computed tomography of the abdomen and pelvis demonstrated a 1.8×1.4-cm hypodense lesion within the pancreatic head with calcifications and mild proximal pancreatic ductal dilatation (Figure 3). However, multiple magnetic resonance cholangiopancreatography examinations and endoscopic ultrasounds with fine-needle aspiration specimens were performed, all negative for malignancy. Computed tomography of the left ankle demonstrated evidence of bony cortical destruction in the lateral aspect of the posterior calcaneus. Bone biopsy specimens demonstrated mild chronic inflammation with no evidence of osteomyelitis. A serum uric acid level was found to be 4.4 mg/dL (reference range, 4.0–8.0 mg/dL) and a joint aspirate demonstrated turbid fluid with lipoid material and no evidence of crystals or organisms on culture. Furthermore, a 4-mm punch biopsy of a nodule on the right leg revealed extensive lobular and septal liquefactive adipocyte necrosis with scattered neutrophils and lymphocytes (Figure 4). Aggregates of fine granular basophilic material were observed with prominent adipocyte degeneration and calcification.

Symptomatic treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) along with intralesional, topical, and oral corticosteroids had proven ineffective in the management of this patient. He was subsequently referred to the surgery department for a pancreaticoduodenectomy (Whipple procedure) with notable improvement in pancreatic enzyme levels, lower leg subcutaneous nodules, and arthritis weeks after surgery.
Comment
A triad of pancreatic panniculitis, pancreatitis, and polyarthritis characterizes a rare entity known as PPP syndrome. Pancreatic panniculitis is a rare form of subcutaneous lobular fat necrosis associated with various underlying pancreatic disorders. Approximately 0.3% to 3.0% of patients with an underlying pancreatic disorder are affected with pancreatic panniculitis.9 Pancreatic panniculitis has been found in roughly 2% to 3% of patients with acute or chronic pancreatitis and pancreatic carcinoma, most commonly the acinar cell type.10 Narváez et al8 reported that nearly two-thirds of patients diagnosed with PPP syndrome have minimal to absent abdominal symptoms that often lead to misdiagnosis and affect the overall prognosis of patients with pancreatic disease. Any delay in the diagnosis of PPP syndrome leads to a worse prognosis, with a mortality rate reported to be approximately 24%.8 Potts et al5 provided a review of 27 patients with pancreatic panniculitis in which all 8 patients with pancreatic carcinoma and 42% (8/19) of patients with pancreatitis died.
Pancreatic panniculitis in the setting of PPP syndrome commonly presents with erythematous to brown, exquisitely tender, edematous, subcutaneous nodules on the lower legs. Lesions can range in size from several millimeters to 5 cm. The subcutaneous nodules may spontaneously ulcerate and exude oily viscous material from the liquefactive necrosis of adipocytes. In approximately 40% of patients, skin lesions are the presenting feature.11 Lesions typically resolve only after the pancreatic inflammation regresses, leaving behind atrophic hyperpigmented scars.3 Other presenting symptoms may include joint pain, pitting edema, and subcutaneous nodules, which can precede the diagnosis by up to 9 months.
The exact pathogenesis of PPP syndrome remains unclear. The most widely recognized hypothesis suggests that pancreatic enzymes (eg, trypsin, amylase, lipase, phospholipase A) released from the damaged pancreas are transported through the bloodstream to distant visceral and soft tissue sites, leading to lipolysis and inflammation to the surrounding subcutis and bone marrow.3 Ferrari et al12 reported this effect as a product of the accumulation of high levels of free fatty acids within the joint space by the action of lipolytic pancreatic enzymes on adipose cell membranes, resulting in acute arthritis.
Histopathologic findings of pancreatic panniculitis vary based on the acuity of the disease. Acute lesions typically demonstrate lobular and septal panniculitis. Szymanski and Bluefarb13 described the pathognomonic histologic findings of focal liquefactive necrosis and anucleate necrotic adipocytes surrounded by a shadowy and thickened cell membrane signifying the characteristic ghost cells. Fine basophilic material also may be seen intermixed with the necrotic adipocytes, representing saponified calcium. A brisk inflammatory infiltrate involving lymphocytes, macrophages, and neutrophils tends to surround the areas of necrotic adipocytes. Chronic lesions often demonstrate a paucity of fat necrosis and ghost cells and more granulomatous infiltrate. Langerhans giant cells, macrophages, and lymphocytes predominate in the subcutaneous fat.
Laboratory findings associated with pancreatic panniculitis may include elevated serum amylase, lipase, and/or trypsin levels. Not all the enzymes have to be elevated simultaneously. On occasion, one enzyme may be within reference range while the others are elevated. Rarely, patients may have an elevated lipase level with no signs of underlying pancreatic disease, which demonstrates that panniculitis does not correlate with the enzyme levels. In all cases of suspected pancreatic panniculitis, a complete laboratory workup is recommended including lipase, amylase, and trypsin serum levels. Eosinophilia may be a prominent finding in patients with pancreatic panniculitis and tends to occur in association with an underlying pancreatic carcinoma. Patients with pancreatic panniculitis associated with pancreatic carcinoma tend to have more severe, diffuse, and persistent subcutaneous nodules that often are refractory to treatment with frequent recurrence. A rare constellation of findings known as Schmid triad is comprised of panniculitis, polyarthritis, and eosinophilia and typically portends a poor prognosis secondary to an underlying pancreatic tumor.14 Cutaneous nodules may predate the diagnosis of pancreatic carcinoma by several months, thus signifying the need for a high index of suspicion in patients with lower leg subcutaneous nodules.
Joint disease most commonly involves the ankles, knees, wrists, and MCP joints.5,6,11 It has been suggested that arthritic symptoms are from periarticular fat necrosis or a direct extension from the necrotic subcutaneous tissue to the adjacent joint space.15 Dahl et al3 reported the composition of joint effusion fluid in 3 patients with PPP syndrome. The aspirate in all 3 patients contained viscous yellow material similar to the necrotic adipose tissue seen draining from subcutaneous nodules. Joint aspirate analysis demonstrated increased concentration of free fatty acids in the joint fluid consistent with severe lipolysis.3
The PPP syndrome acronym may be misleading to physicians, as arthritis is not always polyarticular. Dahl et al3 reported that monoarticular or oligoarticular arthritic symptoms were present in 56% of patients studied. In rare cases, the arthritic symptoms antedated the diagnosis of clinically asymptomatic pancreatic disease. Arthritis can be either symmetric or asymmetric and infrequently follows a chronic course, leading to radiographic lytic lesions and symptoms that often are unresponsive to conventional therapy.16
Treatment of PPP syndrome is largely supportive, with a focus on correcting the underlying pancreatic disease. It is imperative to identify any complicating factors contributing to high levels of circulating pancreatic enzymes. Pseudocysts must be addressed if discovered in these patients, as they often perpetuate the substantial release of pancreatic enzymes into the serum, leading to characteristic subcutaneous fat necrosis and arthritis. Sepsis also is a concern, likely secondary to bacterial colonization of the ulcerated subcutaneous nodules and compromised skin barrier. Nonsteroidal anti-inflammatory drugs and corticosteroids have been used for symptomatic relief but usually are ineffective and have not been shown to reduce the duration of the disease.12,16 Octreotide has been utilized and may potentially reduce pancreatic enzyme secretion leading to improvement in cutaneous and musculoskeletal lesions.17 Plasmapheresis has been used as an adjuvant treatment in patients with persistent hyperamylasemia and hyperlipasemia, but reports are anecdotal. Often reserved for severe disease, cholecystectomy, pancreatic duct removal, and pancreaticoduodenectomy have demonstrated success in the management of chronic pancreatitis and panniculitis. Dahl et al3 reported 2 cases in which cholecystectomy was performed with complete resolution of the skin and pancreatic disease. Our patient was initially treated symptomatically with NSAIDs and corticosteroids but there was no clinical response. The patient eventually underwent a pancreaticoduodenectomy 9 months after the onset of symptoms with complete resolution of joint pain and swelling, greater than 50% resolution of his lower leg subcutaneous nodules, and remarkable reduction in amylase and lipase levels on 1-month follow-up.
Conclusion
Panniculitis, pancreatitis, and polyarthritis syndrome is a rare diagnosis characterized by a triad of pancreatic panniculitis, pancreatitis, and polyarthritis. Adjuvant therapies for PPP syndrome, such as NSAIDs, corticosteroids, plasmapheresis, and octreotide, have been used with equivocal results, but definitive treatment requires correction of the primary pancreatic disorder. More importantly, many pancreatic diseases can cause pancreatic panniculitis, but extensive, refractory, or ulcerated cases could be an early indicator of an occult pancreatic malignancy and should prompt early evaluation with a multidisciplinary approach. This approach should incorporate management from dermatology, internal medicine, rheumatology, gastroenterology, surgery, and primary care.
- Chiari H. Uber die Sogenannte Fettnekrose. Prag Med Wochenschr. 1883;8:285-286, 299-301.
- Blauvelt H. Case of acute pancreatitis with subcutaneous fat necrosis. Br J Surg. 1946;34:207-208.
- Dahl PR, Su D, Cullimore KC, et al. Pancreatic panniculitis. J Am Acad Dermatol. 1995;33:413-417.
- Mullen GT, Caperton EM Jr, Crespin SR, et al. Arthritis and skin lesions resembling erythema nodosum in pancreatic disease. Ann Intern Med. 1968;68:75-87.
- Potts DE, Mass MF, Iseman MD. Syndrome and pancreatic disease, subcutaneous fat necrosis and polyserositis: case report and review of literature. Am J Med. 1975;58:417-423.
- Sorensen EV. Subcutaneous fat necrosis in pancreatic disease: a review and two new case reports. J Clin Gastroenterol. 1988;10:71-75.
- García-Romero D, Vanaclocha F. Pancreatic panniculitis. Dermatol Clin. 2008;26:465-470.
- Narváez J, Bianchi M, Santo P, et al. Pancreatitis, panniculitis, and polyarthritis. Semin Arthritis Rheum. 2010;39:417-423.
- Rongioetti F, Caputo V. Pancreatic panniculitis. G Ital Dermatol Venereol. 2013;148:419-425.
- Poelman SM, Nguyen K. Pancreatic panniculitis associated with acinar cell pancreatic carcinoma. J Cutan Med Surg. 2008;12:38-42.
- Hughes SH, Apisarnthanarax P, Mullins F. Subcutaneous fat necrosis associated with pancreatic disease. Arch Dermatol. 1975:111:506-510.
- Ferrari R, Wendelboe M, Ford PM, et al. Pancreatitis arthritis with periarticular fat necrosis. J Rheumatol. 1993;20:1436-1437.
- Szymanski FJ, Bluefarb SM. Nodular fat necrosis and pancreatic diseases. Arch Dermatol. 1961;83:224-229.
- Beltraminelly HS, Buechner SA, Hausermann P. Pancreatic panniculitis in a patient with an acinar cell cystadenocarcinoma of the pancreas. Dermatology. 2004;208:265-267.
- Burns WA, Matthews MJ, Hamosh M, et al. Lipase-secreting acinar cell carcinoma of the pancreas with polyarthropathy: a light and electron microscopic, histochemical, and biochemical study. Cancer. 1974;33:1002-1009.
- Baron M, Paltiel H, Lander P. Aseptic necrosis of the talus and calcaneal insufficiency fractures in a patient with pancreatitis, subcutaneous fat necrosis, and arthritis. Arthritis Rheum. 1984;27:1309-1313.
- Zundler S, Erber R, Agaimy A, et al. Pancreatic panniculitis in a patient with pancreatic-type acinar cell carcinoma of the liver—case report and review of literature. BMC Cancer. 2016;16:130.
Pancreatic panniculitis is a rare disease contributing to widespread fat necrosis in patients with underlying pancreatic disorders. This entity was first described in 1883,1 but it was not until 1947 that it was reported in the English-language literature.2 Patients with pancreatitis infrequently develop extrapancreatic manifestations. It has been estimated that only 2% to 3% of patients worldwide with an underlying pancreatic disease develop cutaneous lesions.3 Patients who develop pancreatic panniculitis typically present with tender, edematous, erythematous to brown, subcutaneous nodules on the lower legs with the tendency for spontaneous ulceration. Lesions tend to exude a viscous, yellow-brown, oily substance that represents liquefactive necrosis of enzymatic fat in subcutaneous tissue. Cutaneous lesions may precede, occur simultaneously, or follow the development of an underlying pancreatic disorder. Rarely, patients may develop inflammatory arthritis secondary to intraosseous fat necrosis, completing the triad of findings diagnostic for panniculitis, pancreatitis, and polyarthritis (PPP) syndrome. Although the underlying pancreatic pathology may vary, roughly 80% of cases worldwide have acute/chronic pancreatitis or pancreatic carcinoma, most commonly acinar cell carcinoma.4-6 Less common pancreatic disorders include pancreatic pseudocyst, pancreatic divisum, and vascular pancreatic fistulas.7 Narváez et al8 found that of the 25 cases of PPP syndrome reported in the literature, 68% (17/25) were men, 32% (8/25) were women, 56% (14/25) were younger than 50 years, and 64% (16/25) had a history of prior or current alcohol abuse.
Case Report
A 68-year-old man with a history of hypertension, gastroesophageal reflux disease, chronic pancreatitis of unknown etiology, and arthritis presented to our clinic for evaluation of painful skin nodules on the lower legs of 8 months’ duration, in addition to joint pain and swelling of the metacarpophalangeal (MCP), metatarsophalangeal, and ankle joints. He had a history of numerous hospital admissions over the last 2 years for pancreatitis and was being managed by the rheumatology department for arthritic symptoms.
Physical examination revealed multiple 1- to 4-cm, ill-defined, erythematous to brown, subcutaneous nodules on the bilateral lower legs (Figure 1) and right inferomedial thigh that were tender to palpation. Marked erythema and edema of the MCP and metatarsophalangeal joints (Figure 2) and bilateral ankles were observed. Diffuse 2+ pitting edema was present in the bilateral lower extremities, along with areas of hyperpigmentation overlying resolving lesions.
Laboratory data revealed an elevated lipase level (>16,000 U/L [reference range, 31–186 U/L]), amylase level (>4700 U/L [reference range, 27–131 U/L]), erythrocyte sedimentation rate (94 mm/h [reference range, 0–20 mm/h]), and C-reactive protein level (93.5 mg/L [0.08–3.1 mg/L]). The patient had more than 6 episodes of recurrent idiopathic pancreatitis over the last 2 years, though symptoms of abdominal pain were minimal to nonexistent. Liver function tests and alcohol, calcium, and triglyceride levels all were within reference range. Rheumatoid factor and antinuclear antibodies were negative.
Ultrasonography showed no evidence of cholelithiasis. Computed tomography of the abdomen and pelvis demonstrated a 1.8×1.4-cm hypodense lesion within the pancreatic head with calcifications and mild proximal pancreatic ductal dilatation (Figure 3). However, multiple magnetic resonance cholangiopancreatography examinations and endoscopic ultrasounds with fine-needle aspiration specimens were performed, all negative for malignancy. Computed tomography of the left ankle demonstrated evidence of bony cortical destruction in the lateral aspect of the posterior calcaneus. Bone biopsy specimens demonstrated mild chronic inflammation with no evidence of osteomyelitis. A serum uric acid level was found to be 4.4 mg/dL (reference range, 4.0–8.0 mg/dL) and a joint aspirate demonstrated turbid fluid with lipoid material and no evidence of crystals or organisms on culture. Furthermore, a 4-mm punch biopsy of a nodule on the right leg revealed extensive lobular and septal liquefactive adipocyte necrosis with scattered neutrophils and lymphocytes (Figure 4). Aggregates of fine granular basophilic material were observed with prominent adipocyte degeneration and calcification.
Symptomatic treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) along with intralesional, topical, and oral corticosteroids had proven ineffective in the management of this patient. He was subsequently referred to the surgery department for a pancreaticoduodenectomy (Whipple procedure) with notable improvement in pancreatic enzyme levels, lower leg subcutaneous nodules, and arthritis weeks after surgery.
Comment
A triad of pancreatic panniculitis, pancreatitis, and polyarthritis characterizes a rare entity known as PPP syndrome. Pancreatic panniculitis is a rare form of subcutaneous lobular fat necrosis associated with various underlying pancreatic disorders. Approximately 0.3% to 3.0% of patients with an underlying pancreatic disorder are affected with pancreatic panniculitis.9 Pancreatic panniculitis has been found in roughly 2% to 3% of patients with acute or chronic pancreatitis and pancreatic carcinoma, most commonly the acinar cell type.10 Narváez et al8 reported that nearly two-thirds of patients diagnosed with PPP syndrome have minimal to absent abdominal symptoms that often lead to misdiagnosis and affect the overall prognosis of patients with pancreatic disease. Any delay in the diagnosis of PPP syndrome leads to a worse prognosis, with a mortality rate reported to be approximately 24%.8 Potts et al5 provided a review of 27 patients with pancreatic panniculitis in which all 8 patients with pancreatic carcinoma and 42% (8/19) of patients with pancreatitis died.
Pancreatic panniculitis in the setting of PPP syndrome commonly presents with erythematous to brown, exquisitely tender, edematous, subcutaneous nodules on the lower legs. Lesions can range in size from several millimeters to 5 cm. The subcutaneous nodules may spontaneously ulcerate and exude oily viscous material from the liquefactive necrosis of adipocytes. In approximately 40% of patients, skin lesions are the presenting feature.11 Lesions typically resolve only after the pancreatic inflammation regresses, leaving behind atrophic hyperpigmented scars.3 Other presenting symptoms may include joint pain, pitting edema, and subcutaneous nodules, which can precede the diagnosis by up to 9 months.
The exact pathogenesis of PPP syndrome remains unclear. The most widely recognized hypothesis suggests that pancreatic enzymes (eg, trypsin, amylase, lipase, phospholipase A) released from the damaged pancreas are transported through the bloodstream to distant visceral and soft tissue sites, leading to lipolysis and inflammation to the surrounding subcutis and bone marrow.3 Ferrari et al12 reported this effect as a product of the accumulation of high levels of free fatty acids within the joint space by the action of lipolytic pancreatic enzymes on adipose cell membranes, resulting in acute arthritis.
Histopathologic findings of pancreatic panniculitis vary based on the acuity of the disease. Acute lesions typically demonstrate lobular and septal panniculitis. Szymanski and Bluefarb13 described the pathognomonic histologic findings of focal liquefactive necrosis and anucleate necrotic adipocytes surrounded by a shadowy and thickened cell membrane signifying the characteristic ghost cells. Fine basophilic material also may be seen intermixed with the necrotic adipocytes, representing saponified calcium. A brisk inflammatory infiltrate involving lymphocytes, macrophages, and neutrophils tends to surround the areas of necrotic adipocytes. Chronic lesions often demonstrate a paucity of fat necrosis and ghost cells and more granulomatous infiltrate. Langerhans giant cells, macrophages, and lymphocytes predominate in the subcutaneous fat.
Laboratory findings associated with pancreatic panniculitis may include elevated serum amylase, lipase, and/or trypsin levels. Not all the enzymes have to be elevated simultaneously. On occasion, one enzyme may be within reference range while the others are elevated. Rarely, patients may have an elevated lipase level with no signs of underlying pancreatic disease, which demonstrates that panniculitis does not correlate with the enzyme levels. In all cases of suspected pancreatic panniculitis, a complete laboratory workup is recommended including lipase, amylase, and trypsin serum levels. Eosinophilia may be a prominent finding in patients with pancreatic panniculitis and tends to occur in association with an underlying pancreatic carcinoma. Patients with pancreatic panniculitis associated with pancreatic carcinoma tend to have more severe, diffuse, and persistent subcutaneous nodules that often are refractory to treatment with frequent recurrence. A rare constellation of findings known as Schmid triad is comprised of panniculitis, polyarthritis, and eosinophilia and typically portends a poor prognosis secondary to an underlying pancreatic tumor.14 Cutaneous nodules may predate the diagnosis of pancreatic carcinoma by several months, thus signifying the need for a high index of suspicion in patients with lower leg subcutaneous nodules.
Joint disease most commonly involves the ankles, knees, wrists, and MCP joints.5,6,11 It has been suggested that arthritic symptoms are from periarticular fat necrosis or a direct extension from the necrotic subcutaneous tissue to the adjacent joint space.15 Dahl et al3 reported the composition of joint effusion fluid in 3 patients with PPP syndrome. The aspirate in all 3 patients contained viscous yellow material similar to the necrotic adipose tissue seen draining from subcutaneous nodules. Joint aspirate analysis demonstrated increased concentration of free fatty acids in the joint fluid consistent with severe lipolysis.3
The PPP syndrome acronym may be misleading to physicians, as arthritis is not always polyarticular. Dahl et al3 reported that monoarticular or oligoarticular arthritic symptoms were present in 56% of patients studied. In rare cases, the arthritic symptoms antedated the diagnosis of clinically asymptomatic pancreatic disease. Arthritis can be either symmetric or asymmetric and infrequently follows a chronic course, leading to radiographic lytic lesions and symptoms that often are unresponsive to conventional therapy.16
Treatment of PPP syndrome is largely supportive, with a focus on correcting the underlying pancreatic disease. It is imperative to identify any complicating factors contributing to high levels of circulating pancreatic enzymes. Pseudocysts must be addressed if discovered in these patients, as they often perpetuate the substantial release of pancreatic enzymes into the serum, leading to characteristic subcutaneous fat necrosis and arthritis. Sepsis also is a concern, likely secondary to bacterial colonization of the ulcerated subcutaneous nodules and compromised skin barrier. Nonsteroidal anti-inflammatory drugs and corticosteroids have been used for symptomatic relief but usually are ineffective and have not been shown to reduce the duration of the disease.12,16 Octreotide has been utilized and may potentially reduce pancreatic enzyme secretion leading to improvement in cutaneous and musculoskeletal lesions.17 Plasmapheresis has been used as an adjuvant treatment in patients with persistent hyperamylasemia and hyperlipasemia, but reports are anecdotal. Often reserved for severe disease, cholecystectomy, pancreatic duct removal, and pancreaticoduodenectomy have demonstrated success in the management of chronic pancreatitis and panniculitis. Dahl et al3 reported 2 cases in which cholecystectomy was performed with complete resolution of the skin and pancreatic disease. Our patient was initially treated symptomatically with NSAIDs and corticosteroids but there was no clinical response. The patient eventually underwent a pancreaticoduodenectomy 9 months after the onset of symptoms with complete resolution of joint pain and swelling, greater than 50% resolution of his lower leg subcutaneous nodules, and remarkable reduction in amylase and lipase levels on 1-month follow-up.
Conclusion
Panniculitis, pancreatitis, and polyarthritis syndrome is a rare diagnosis characterized by a triad of pancreatic panniculitis, pancreatitis, and polyarthritis. Adjuvant therapies for PPP syndrome, such as NSAIDs, corticosteroids, plasmapheresis, and octreotide, have been used with equivocal results, but definitive treatment requires correction of the primary pancreatic disorder. More importantly, many pancreatic diseases can cause pancreatic panniculitis, but extensive, refractory, or ulcerated cases could be an early indicator of an occult pancreatic malignancy and should prompt early evaluation with a multidisciplinary approach. This approach should incorporate management from dermatology, internal medicine, rheumatology, gastroenterology, surgery, and primary care.
Pancreatic panniculitis is a rare disease contributing to widespread fat necrosis in patients with underlying pancreatic disorders. This entity was first described in 1883,1 but it was not until 1947 that it was reported in the English-language literature.2 Patients with pancreatitis infrequently develop extrapancreatic manifestations. It has been estimated that only 2% to 3% of patients worldwide with an underlying pancreatic disease develop cutaneous lesions.3 Patients who develop pancreatic panniculitis typically present with tender, edematous, erythematous to brown, subcutaneous nodules on the lower legs with the tendency for spontaneous ulceration. Lesions tend to exude a viscous, yellow-brown, oily substance that represents liquefactive necrosis of enzymatic fat in subcutaneous tissue. Cutaneous lesions may precede, occur simultaneously, or follow the development of an underlying pancreatic disorder. Rarely, patients may develop inflammatory arthritis secondary to intraosseous fat necrosis, completing the triad of findings diagnostic for panniculitis, pancreatitis, and polyarthritis (PPP) syndrome. Although the underlying pancreatic pathology may vary, roughly 80% of cases worldwide have acute/chronic pancreatitis or pancreatic carcinoma, most commonly acinar cell carcinoma.4-6 Less common pancreatic disorders include pancreatic pseudocyst, pancreatic divisum, and vascular pancreatic fistulas.7 Narváez et al8 found that of the 25 cases of PPP syndrome reported in the literature, 68% (17/25) were men, 32% (8/25) were women, 56% (14/25) were younger than 50 years, and 64% (16/25) had a history of prior or current alcohol abuse.
Case Report
A 68-year-old man with a history of hypertension, gastroesophageal reflux disease, chronic pancreatitis of unknown etiology, and arthritis presented to our clinic for evaluation of painful skin nodules on the lower legs of 8 months’ duration, in addition to joint pain and swelling of the metacarpophalangeal (MCP), metatarsophalangeal, and ankle joints. He had a history of numerous hospital admissions over the last 2 years for pancreatitis and was being managed by the rheumatology department for arthritic symptoms.
Physical examination revealed multiple 1- to 4-cm, ill-defined, erythematous to brown, subcutaneous nodules on the bilateral lower legs (Figure 1) and right inferomedial thigh that were tender to palpation. Marked erythema and edema of the MCP and metatarsophalangeal joints (Figure 2) and bilateral ankles were observed. Diffuse 2+ pitting edema was present in the bilateral lower extremities, along with areas of hyperpigmentation overlying resolving lesions.
Laboratory data revealed an elevated lipase level (>16,000 U/L [reference range, 31–186 U/L]), amylase level (>4700 U/L [reference range, 27–131 U/L]), erythrocyte sedimentation rate (94 mm/h [reference range, 0–20 mm/h]), and C-reactive protein level (93.5 mg/L [0.08–3.1 mg/L]). The patient had more than 6 episodes of recurrent idiopathic pancreatitis over the last 2 years, though symptoms of abdominal pain were minimal to nonexistent. Liver function tests and alcohol, calcium, and triglyceride levels all were within reference range. Rheumatoid factor and antinuclear antibodies were negative.
Ultrasonography showed no evidence of cholelithiasis. Computed tomography of the abdomen and pelvis demonstrated a 1.8×1.4-cm hypodense lesion within the pancreatic head with calcifications and mild proximal pancreatic ductal dilatation (Figure 3). However, multiple magnetic resonance cholangiopancreatography examinations and endoscopic ultrasounds with fine-needle aspiration specimens were performed, all negative for malignancy. Computed tomography of the left ankle demonstrated evidence of bony cortical destruction in the lateral aspect of the posterior calcaneus. Bone biopsy specimens demonstrated mild chronic inflammation with no evidence of osteomyelitis. A serum uric acid level was found to be 4.4 mg/dL (reference range, 4.0–8.0 mg/dL) and a joint aspirate demonstrated turbid fluid with lipoid material and no evidence of crystals or organisms on culture. Furthermore, a 4-mm punch biopsy of a nodule on the right leg revealed extensive lobular and septal liquefactive adipocyte necrosis with scattered neutrophils and lymphocytes (Figure 4). Aggregates of fine granular basophilic material were observed with prominent adipocyte degeneration and calcification.
Symptomatic treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) along with intralesional, topical, and oral corticosteroids had proven ineffective in the management of this patient. He was subsequently referred to the surgery department for a pancreaticoduodenectomy (Whipple procedure) with notable improvement in pancreatic enzyme levels, lower leg subcutaneous nodules, and arthritis weeks after surgery.
Comment
A triad of pancreatic panniculitis, pancreatitis, and polyarthritis characterizes a rare entity known as PPP syndrome. Pancreatic panniculitis is a rare form of subcutaneous lobular fat necrosis associated with various underlying pancreatic disorders. Approximately 0.3% to 3.0% of patients with an underlying pancreatic disorder are affected with pancreatic panniculitis.9 Pancreatic panniculitis has been found in roughly 2% to 3% of patients with acute or chronic pancreatitis and pancreatic carcinoma, most commonly the acinar cell type.10 Narváez et al8 reported that nearly two-thirds of patients diagnosed with PPP syndrome have minimal to absent abdominal symptoms that often lead to misdiagnosis and affect the overall prognosis of patients with pancreatic disease. Any delay in the diagnosis of PPP syndrome leads to a worse prognosis, with a mortality rate reported to be approximately 24%.8 Potts et al5 provided a review of 27 patients with pancreatic panniculitis in which all 8 patients with pancreatic carcinoma and 42% (8/19) of patients with pancreatitis died.
Pancreatic panniculitis in the setting of PPP syndrome commonly presents with erythematous to brown, exquisitely tender, edematous, subcutaneous nodules on the lower legs. Lesions can range in size from several millimeters to 5 cm. The subcutaneous nodules may spontaneously ulcerate and exude oily viscous material from the liquefactive necrosis of adipocytes. In approximately 40% of patients, skin lesions are the presenting feature.11 Lesions typically resolve only after the pancreatic inflammation regresses, leaving behind atrophic hyperpigmented scars.3 Other presenting symptoms may include joint pain, pitting edema, and subcutaneous nodules, which can precede the diagnosis by up to 9 months.
The exact pathogenesis of PPP syndrome remains unclear. The most widely recognized hypothesis suggests that pancreatic enzymes (eg, trypsin, amylase, lipase, phospholipase A) released from the damaged pancreas are transported through the bloodstream to distant visceral and soft tissue sites, leading to lipolysis and inflammation to the surrounding subcutis and bone marrow.3 Ferrari et al12 reported this effect as a product of the accumulation of high levels of free fatty acids within the joint space by the action of lipolytic pancreatic enzymes on adipose cell membranes, resulting in acute arthritis.
Histopathologic findings of pancreatic panniculitis vary based on the acuity of the disease. Acute lesions typically demonstrate lobular and septal panniculitis. Szymanski and Bluefarb13 described the pathognomonic histologic findings of focal liquefactive necrosis and anucleate necrotic adipocytes surrounded by a shadowy and thickened cell membrane signifying the characteristic ghost cells. Fine basophilic material also may be seen intermixed with the necrotic adipocytes, representing saponified calcium. A brisk inflammatory infiltrate involving lymphocytes, macrophages, and neutrophils tends to surround the areas of necrotic adipocytes. Chronic lesions often demonstrate a paucity of fat necrosis and ghost cells and more granulomatous infiltrate. Langerhans giant cells, macrophages, and lymphocytes predominate in the subcutaneous fat.
Laboratory findings associated with pancreatic panniculitis may include elevated serum amylase, lipase, and/or trypsin levels. Not all the enzymes have to be elevated simultaneously. On occasion, one enzyme may be within reference range while the others are elevated. Rarely, patients may have an elevated lipase level with no signs of underlying pancreatic disease, which demonstrates that panniculitis does not correlate with the enzyme levels. In all cases of suspected pancreatic panniculitis, a complete laboratory workup is recommended including lipase, amylase, and trypsin serum levels. Eosinophilia may be a prominent finding in patients with pancreatic panniculitis and tends to occur in association with an underlying pancreatic carcinoma. Patients with pancreatic panniculitis associated with pancreatic carcinoma tend to have more severe, diffuse, and persistent subcutaneous nodules that often are refractory to treatment with frequent recurrence. A rare constellation of findings known as Schmid triad is comprised of panniculitis, polyarthritis, and eosinophilia and typically portends a poor prognosis secondary to an underlying pancreatic tumor.14 Cutaneous nodules may predate the diagnosis of pancreatic carcinoma by several months, thus signifying the need for a high index of suspicion in patients with lower leg subcutaneous nodules.
Joint disease most commonly involves the ankles, knees, wrists, and MCP joints.5,6,11 It has been suggested that arthritic symptoms are from periarticular fat necrosis or a direct extension from the necrotic subcutaneous tissue to the adjacent joint space.15 Dahl et al3 reported the composition of joint effusion fluid in 3 patients with PPP syndrome. The aspirate in all 3 patients contained viscous yellow material similar to the necrotic adipose tissue seen draining from subcutaneous nodules. Joint aspirate analysis demonstrated increased concentration of free fatty acids in the joint fluid consistent with severe lipolysis.3
The PPP syndrome acronym may be misleading to physicians, as arthritis is not always polyarticular. Dahl et al3 reported that monoarticular or oligoarticular arthritic symptoms were present in 56% of patients studied. In rare cases, the arthritic symptoms antedated the diagnosis of clinically asymptomatic pancreatic disease. Arthritis can be either symmetric or asymmetric and infrequently follows a chronic course, leading to radiographic lytic lesions and symptoms that often are unresponsive to conventional therapy.16
Treatment of PPP syndrome is largely supportive, with a focus on correcting the underlying pancreatic disease. It is imperative to identify any complicating factors contributing to high levels of circulating pancreatic enzymes. Pseudocysts must be addressed if discovered in these patients, as they often perpetuate the substantial release of pancreatic enzymes into the serum, leading to characteristic subcutaneous fat necrosis and arthritis. Sepsis also is a concern, likely secondary to bacterial colonization of the ulcerated subcutaneous nodules and compromised skin barrier. Nonsteroidal anti-inflammatory drugs and corticosteroids have been used for symptomatic relief but usually are ineffective and have not been shown to reduce the duration of the disease.12,16 Octreotide has been utilized and may potentially reduce pancreatic enzyme secretion leading to improvement in cutaneous and musculoskeletal lesions.17 Plasmapheresis has been used as an adjuvant treatment in patients with persistent hyperamylasemia and hyperlipasemia, but reports are anecdotal. Often reserved for severe disease, cholecystectomy, pancreatic duct removal, and pancreaticoduodenectomy have demonstrated success in the management of chronic pancreatitis and panniculitis. Dahl et al3 reported 2 cases in which cholecystectomy was performed with complete resolution of the skin and pancreatic disease. Our patient was initially treated symptomatically with NSAIDs and corticosteroids but there was no clinical response. The patient eventually underwent a pancreaticoduodenectomy 9 months after the onset of symptoms with complete resolution of joint pain and swelling, greater than 50% resolution of his lower leg subcutaneous nodules, and remarkable reduction in amylase and lipase levels on 1-month follow-up.
Conclusion
Panniculitis, pancreatitis, and polyarthritis syndrome is a rare diagnosis characterized by a triad of pancreatic panniculitis, pancreatitis, and polyarthritis. Adjuvant therapies for PPP syndrome, such as NSAIDs, corticosteroids, plasmapheresis, and octreotide, have been used with equivocal results, but definitive treatment requires correction of the primary pancreatic disorder. More importantly, many pancreatic diseases can cause pancreatic panniculitis, but extensive, refractory, or ulcerated cases could be an early indicator of an occult pancreatic malignancy and should prompt early evaluation with a multidisciplinary approach. This approach should incorporate management from dermatology, internal medicine, rheumatology, gastroenterology, surgery, and primary care.
- Chiari H. Uber die Sogenannte Fettnekrose. Prag Med Wochenschr. 1883;8:285-286, 299-301.
- Blauvelt H. Case of acute pancreatitis with subcutaneous fat necrosis. Br J Surg. 1946;34:207-208.
- Dahl PR, Su D, Cullimore KC, et al. Pancreatic panniculitis. J Am Acad Dermatol. 1995;33:413-417.
- Mullen GT, Caperton EM Jr, Crespin SR, et al. Arthritis and skin lesions resembling erythema nodosum in pancreatic disease. Ann Intern Med. 1968;68:75-87.
- Potts DE, Mass MF, Iseman MD. Syndrome and pancreatic disease, subcutaneous fat necrosis and polyserositis: case report and review of literature. Am J Med. 1975;58:417-423.
- Sorensen EV. Subcutaneous fat necrosis in pancreatic disease: a review and two new case reports. J Clin Gastroenterol. 1988;10:71-75.
- García-Romero D, Vanaclocha F. Pancreatic panniculitis. Dermatol Clin. 2008;26:465-470.
- Narváez J, Bianchi M, Santo P, et al. Pancreatitis, panniculitis, and polyarthritis. Semin Arthritis Rheum. 2010;39:417-423.
- Rongioetti F, Caputo V. Pancreatic panniculitis. G Ital Dermatol Venereol. 2013;148:419-425.
- Poelman SM, Nguyen K. Pancreatic panniculitis associated with acinar cell pancreatic carcinoma. J Cutan Med Surg. 2008;12:38-42.
- Hughes SH, Apisarnthanarax P, Mullins F. Subcutaneous fat necrosis associated with pancreatic disease. Arch Dermatol. 1975:111:506-510.
- Ferrari R, Wendelboe M, Ford PM, et al. Pancreatitis arthritis with periarticular fat necrosis. J Rheumatol. 1993;20:1436-1437.
- Szymanski FJ, Bluefarb SM. Nodular fat necrosis and pancreatic diseases. Arch Dermatol. 1961;83:224-229.
- Beltraminelly HS, Buechner SA, Hausermann P. Pancreatic panniculitis in a patient with an acinar cell cystadenocarcinoma of the pancreas. Dermatology. 2004;208:265-267.
- Burns WA, Matthews MJ, Hamosh M, et al. Lipase-secreting acinar cell carcinoma of the pancreas with polyarthropathy: a light and electron microscopic, histochemical, and biochemical study. Cancer. 1974;33:1002-1009.
- Baron M, Paltiel H, Lander P. Aseptic necrosis of the talus and calcaneal insufficiency fractures in a patient with pancreatitis, subcutaneous fat necrosis, and arthritis. Arthritis Rheum. 1984;27:1309-1313.
- Zundler S, Erber R, Agaimy A, et al. Pancreatic panniculitis in a patient with pancreatic-type acinar cell carcinoma of the liver—case report and review of literature. BMC Cancer. 2016;16:130.
- Chiari H. Uber die Sogenannte Fettnekrose. Prag Med Wochenschr. 1883;8:285-286, 299-301.
- Blauvelt H. Case of acute pancreatitis with subcutaneous fat necrosis. Br J Surg. 1946;34:207-208.
- Dahl PR, Su D, Cullimore KC, et al. Pancreatic panniculitis. J Am Acad Dermatol. 1995;33:413-417.
- Mullen GT, Caperton EM Jr, Crespin SR, et al. Arthritis and skin lesions resembling erythema nodosum in pancreatic disease. Ann Intern Med. 1968;68:75-87.
- Potts DE, Mass MF, Iseman MD. Syndrome and pancreatic disease, subcutaneous fat necrosis and polyserositis: case report and review of literature. Am J Med. 1975;58:417-423.
- Sorensen EV. Subcutaneous fat necrosis in pancreatic disease: a review and two new case reports. J Clin Gastroenterol. 1988;10:71-75.
- García-Romero D, Vanaclocha F. Pancreatic panniculitis. Dermatol Clin. 2008;26:465-470.
- Narváez J, Bianchi M, Santo P, et al. Pancreatitis, panniculitis, and polyarthritis. Semin Arthritis Rheum. 2010;39:417-423.
- Rongioetti F, Caputo V. Pancreatic panniculitis. G Ital Dermatol Venereol. 2013;148:419-425.
- Poelman SM, Nguyen K. Pancreatic panniculitis associated with acinar cell pancreatic carcinoma. J Cutan Med Surg. 2008;12:38-42.
- Hughes SH, Apisarnthanarax P, Mullins F. Subcutaneous fat necrosis associated with pancreatic disease. Arch Dermatol. 1975:111:506-510.
- Ferrari R, Wendelboe M, Ford PM, et al. Pancreatitis arthritis with periarticular fat necrosis. J Rheumatol. 1993;20:1436-1437.
- Szymanski FJ, Bluefarb SM. Nodular fat necrosis and pancreatic diseases. Arch Dermatol. 1961;83:224-229.
- Beltraminelly HS, Buechner SA, Hausermann P. Pancreatic panniculitis in a patient with an acinar cell cystadenocarcinoma of the pancreas. Dermatology. 2004;208:265-267.
- Burns WA, Matthews MJ, Hamosh M, et al. Lipase-secreting acinar cell carcinoma of the pancreas with polyarthropathy: a light and electron microscopic, histochemical, and biochemical study. Cancer. 1974;33:1002-1009.
- Baron M, Paltiel H, Lander P. Aseptic necrosis of the talus and calcaneal insufficiency fractures in a patient with pancreatitis, subcutaneous fat necrosis, and arthritis. Arthritis Rheum. 1984;27:1309-1313.
- Zundler S, Erber R, Agaimy A, et al. Pancreatic panniculitis in a patient with pancreatic-type acinar cell carcinoma of the liver—case report and review of literature. BMC Cancer. 2016;16:130.
Practice Points
- Recognition of skin lesions in a patient with a history of pancreatitis may represent a rare entity known as pancreatic panniculitis.
- Panniculitis, pancreatitis, and polyarthritis (PPP) syndrome is a rare diagnosis characterized by a triad of pancreatic panniculitis, pancreatitis, and polyarthritis.
- A rare constellation of findings known as Schmid triad is comprised of panniculitis, polyarthritis, and eosinophilia and typically portends a poor prognosis secondary to an underlying pancreatic tumor.
- These findings should prompt early evaluation with a multidisciplinary approach.