User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Bullous Amyloidosis Masquerading as Pseudoporphyria
Cutaneous amyloidosis encompasses a variety of clinical presentations. Primary localized cutaneous amyloidosis comprises lichen amyloidosis, macular amyloidosis, and nodular amyloidosis.1 Macular and lichen amyloidosis result from keratin deposits, while nodular amyloidosis results from cutaneous infiltration of plasma cells.2 Primary systemic amyloidosis is due to a plasma cell dyscrasia, particularly multiple myeloma, while secondary systemic amyloidosis occurs in the setting of restrictive cardiomyopathy, congestive heart failure, renal dysfunction, or chronic inflammation, as seen with rheumatoid arthritis, tuberculosis, and various autoinflammatory disorders.2 Plasma cell proliferative disorders are associated with various skin disorders, which may result from aggregated misfolded monoclonal immunoglobulins, indicating light chain–related systemic amyloidosis. Mucocutaneous lesions can occur in 30% to 40% of cases of primary systemic amyloidosis and may present as purpura, ecchymoses, waxy thickening, plaques, subcutaneous nodules, and/or bullae.3,4 When blistering is present, the differential diagnosis is broad and includes autoimmune bullous disease, drug eruptions, enoxaparin-induced bullous hemorrhagic dermatosis, deposition diseases, allergic contact dermatitis, bullous cellulitis, bullous bite reactions, neutrophilic dermatosis, and bullous lichen sclerosus.5 Herein, we present a case of a woman with a bullous skin eruption who eventually was diagnosed with bullous amyloidosis subsequent to a diagnosis of multiple myeloma.
Case Report
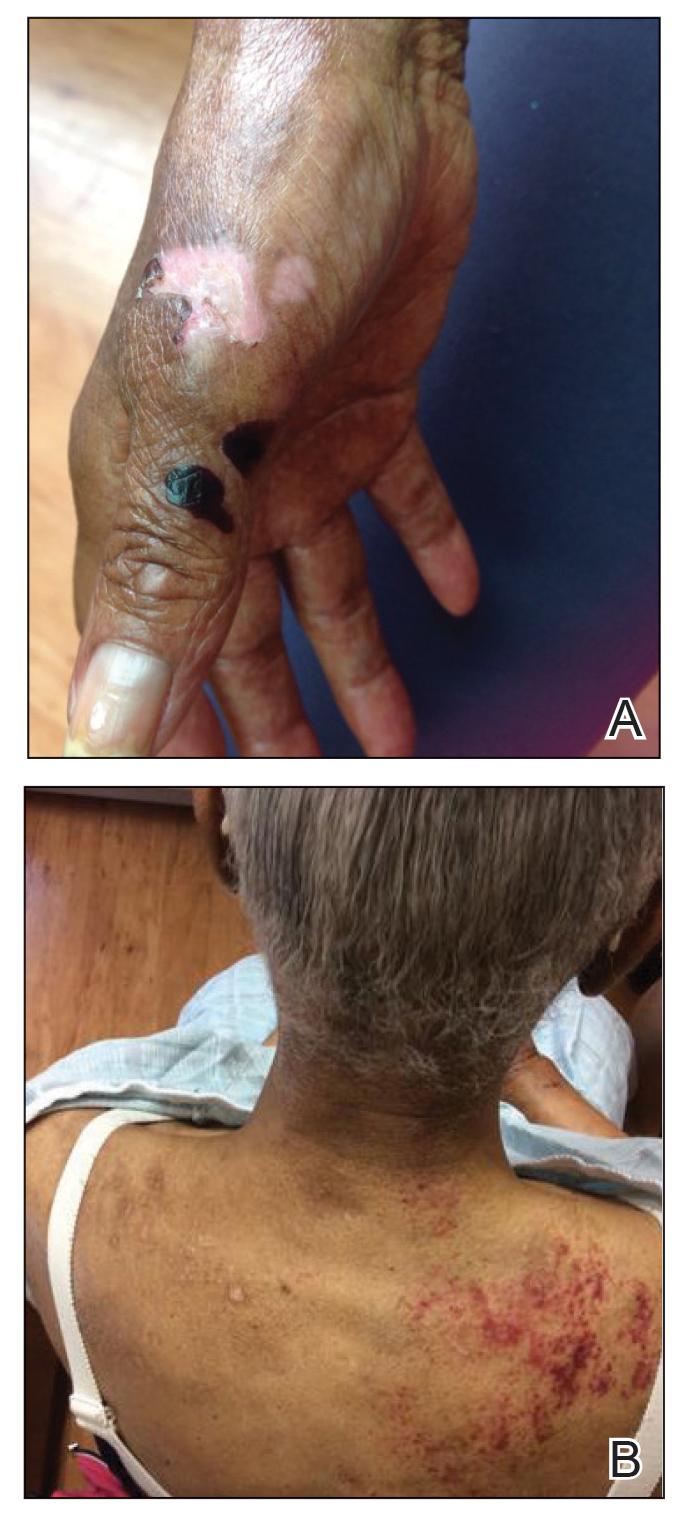
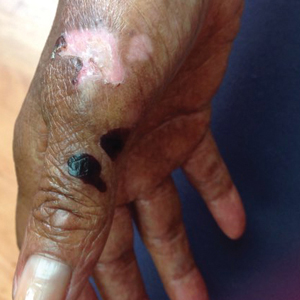
A 70-year-old woman presented to our dermatology clinic for evaluation of well-demarcated, hemorrhagic, flaccid vesicles and focal erosions with a rim of erythema on the distal forearms and hands. A shave biopsy from the right forearm showed cell-poor subepidermal vesicular dermatitis. Enzyme-linked immunosorbent assays for bullous pemphigoid antigens 1 and 2 as well as urinary porphyrins were negative. Direct immunofluorescence showed granular IgM at the basement membrane zone around vessels and cytoid bodies. At this time, a preliminary diagnosis of pseudoporphyria was suspected, though no classic medications (eg, nonsteroidal anti-inflammatory drugs, furosemide, antibiotics) or exogenous trigger factors (eg, UV light exposure, dialysis) were temporally related. Three months later, the patient presented with a large hemorrhagic bulla on the distal left forearm (Figure 1) and healing erosions on the dorsal fingers and upper back. Clobetasol ointment was initiated, as an autoimmune bullous dermatosis was suspected.

Approximately 1 year after she was first seen in our outpatient clinic, the patient was hospitalized for induction of chemotherapy—cyclophosphamide, bortezomib, and dexamethasone—for a new diagnosis of stage III multiple myeloma. A workup for back pain revealed multiple compression fractures and a plasma cell neoplasm with elevated λ light chains, which was confirmed with a bone marrow biopsy. During an inpatient dermatology consultation, we noted the development of intraoral hemorrhagic vesicles and worsening generalization of the hemorrhagic bullae, with healing erosions and intact hemorrhagic bullae on the dorsal hands, fingers (Figure 2), and upper back.
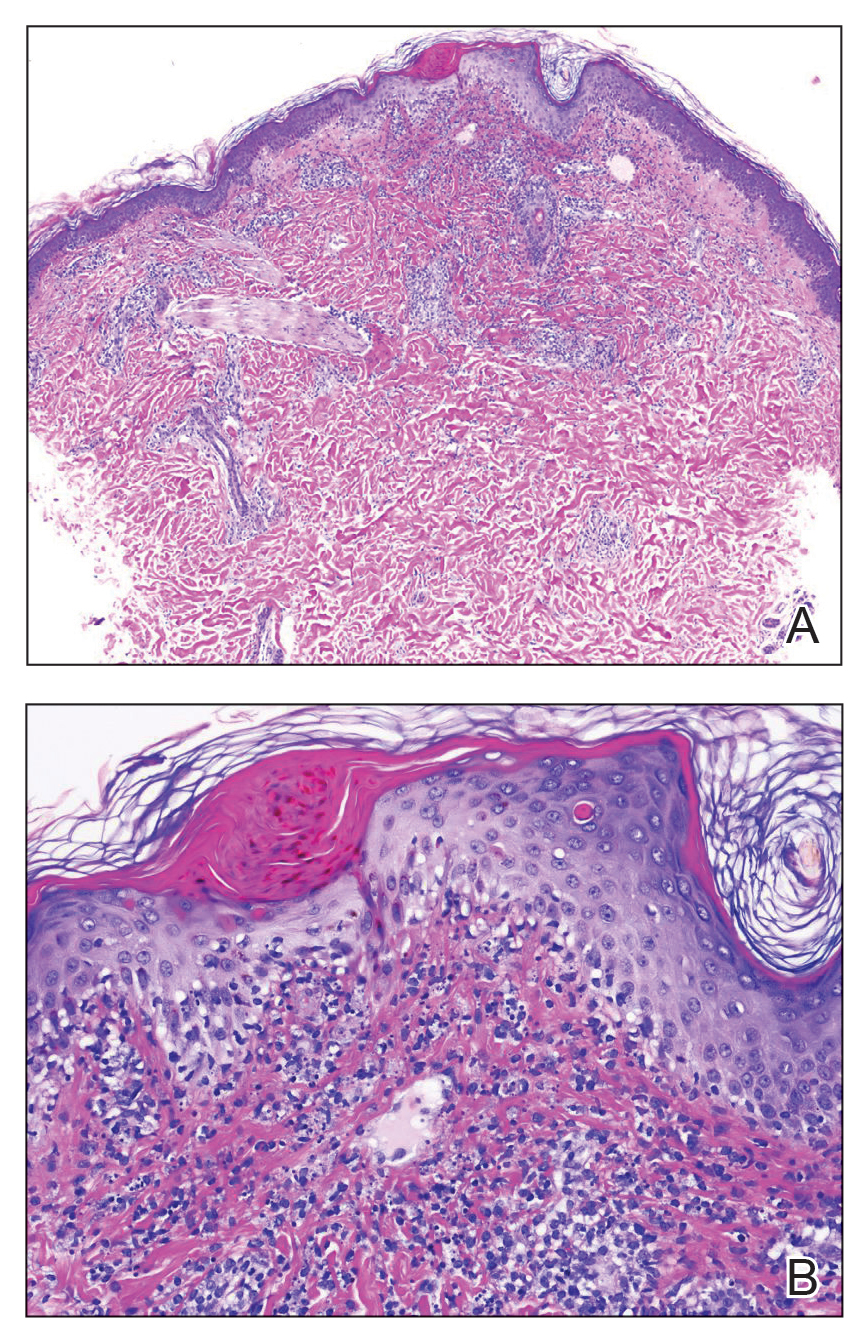
A repeat biopsy displayed bullous amyloidosis. Histopathologic examination revealed an ulcerated subepidermal blister with fibrin deposition at the ulcer base. A periadnexal, scant, eosinophilic deposition with extravasated red blood cells was appreciated. Amorphous eosinophilic deposits were found within the detached fragment of the epidermis and inflammatory infiltrate. A Congo red stain highlighted these areas with a salmon pink–colored material. Congo red staining showed a moderate amount of pale, apple green, birefringent deposit within these areas on polarized light examination.
A few months later, the patient was re-admitted, and the amount of skin detachment prompted the primary team to ask for another consultation. Although the extensive skin sloughing resembled toxic epidermal necrolysis, a repeat biopsy confirmed bullous amyloidosis.
Comment
Amyloidosis Histopathology—Amyloidoses represent a wide array of disorders with deposition of β-pleated sheets or amyloid fibrils, often with cutaneous manifestations.2,3 Primary systemic amyloidosis has been associated with underlying dyscrasia or multiple myeloma.6 In such cases, the skin lesions of multiple myeloma may result from a collection of misfolded monoclonal immunoglobulins or their fragments, as in light chain–related systemic amyloidosis.3 Histopathologically, both systemic and cutaneous amyloidosis appear similar and display deposition of amorphous, eosinophilic, fissured amyloid material in the dermis. Congo red stains the material orange-red and will display a characteristic apple green birefringence under polarized light.4 Although bullous amyloid lesions are rare, the cutaneous forms of these lesions can be an important sign of plasma cell dyscrasia.7
Presentation of Bullous Amyloidosis—Bullous manifestations rarely have been noted in the primary cutaneous forms of amyloidosis.5,8,9 Importantly, cutaneous blistering more often is linked to systemic forms of amyloidosis with multiorgan involvement, including primary systemic and myeloma-associated amyloidosis.5,10 However, patients with localized bullous cutaneous amyloidosis without systemic involvement also have been seen.10,11 Bullae may occur at any time, with contents that frequently are hemorrhagic due to capillary fragility.12,13 Bullous manifestations raise the differential diagnoses of bullous pemphigoid, epidermolysis bullosa acquisita, linear IgA disease, porphyria cutanea tarda, pseudoporphyria, bullous drug eruption, bullous eruption of renal dialysis, or bullous lupus erythematosus.5,13-17
In our patient, the acral distribution of bullae, presence of hemorrhage, chronicity of symptoms, and negative enzyme-linked immunosorbent assay initially suggested a diagnosis of pseudoporphyria. However, the presence of intraoral hemorrhagic vesicles and subsequent confirmatory pathology aided in differentiating bullous amyloidosis from pseudoporphyria. Nodular localized primary cutaneous amyloidosis, a rare form of skin-restricted amyloidoses, can coexist with bullous lesions. Of note, reported cases of nodular localized primary cutaneous amyloidosis did not result in development of multiple myeloma.5,10
Bullae are located either subepidermally or intradermally, and bullous lesions of cutaneous amyloidosis typically demonstrate subepidermal or superficial intradermal clefting on light microscopy.5,10,12 Histopathology of bullous amyloidosis shows intradermal or subepidermal blister formation and amorphous eosinophilic material showing apple green birefringence with Congo red staining deposited in the dermis and/or around the adipocytes and blood vessel walls.12,18-20 In prior cases, direct immunofluorescence of bullous amyloidosis revealed absent immunoglobulin (IgG, IgA, IgM) or complement (C3 and C9) deposits in the basement membrane zone or dermis.13,21,22 In these cases, electron microscopy was useful in diagnosis, as it showed the presence of amyloid deposits.21,22
Cause of Bullae—Various mechanisms are thought to trigger the blister formation in amyloidosis. Bullae created from trauma or friction often present as tense painful blisters that commonly are hemorrhagic.10,23 Amyloid deposits in the walls of blood vessels and the affinity of dermal amyloid in blood vessel walls to surrounding collagen likely leads to increased fragility of capillaries and the dermal matrix, hemorrhagic tendency, and infrapapillary blisters, thus creating hemorrhagic bullous eruptions.24,25 Specifically, close proximity of immunoglobulin-derived amyloid oligomers to epidermal keratinocytes may be toxic and therefore could trigger subepidermal bullous change.5 Additionally, alteration in the physicochemical properties of the amyloidal protein might explain bullous eruption.9 Trauma or rubbing of the hands and feet may precipitate the acral blister formation in bullous amyloidosis.5,11
Due to deposition of these amyloid fibrils, skin bleeding in these patients is called amyloid or pinch purpura. Vessel wall fragility and damage by amyloid are the principal causes of periorbital and gastrointestinal tract bleeding.26 Destruction of the lamina densa and widening of the intercellular space between keratinocytes by amyloid globules induce skin fragility.11
Although uncommon, various cases of bullous amyloidosis have been reported in the literature. Multiple myeloma patients represent the majority of those reported to have bullous amyloidosis.6,7,13,24,27-30 Plasmacytoma-associated bullous amyloid purpura and paraproteinemia also have been noted.25 Multiple myeloma with secondary AL amyloidosis has been seen with amyloid purpura and atraumatic ecchymoses of the face, highlighting the hemorrhage noted in these patients.26
Management of Amyloidosis—Various treatment options have been attempted for primary cutaneous amyloidosis, including oral retinoids, corticosteroids, cyclophosphamide, cyclosporine, amitriptyline, colchicine, cepharanthin, tacrolimus, dimethyl sulfoxide, vitamin D3 analogs, capsaicin, menthol, hydrocolloid dressings, surgical modalities, laser treatment, and phototherapy.1 There is no clear consensus for therapeutic modalities except for treating the underlying plasma cell dyscrasia in primary systemic amyloidosis.
Conclusion
We report the case of a patient displaying signs of pseudoporphyria that ultimately proved to be bullous amyloidosis, or what we termed pseudopseudoporphyria. Bullous amyloidosis should be considered in the differential diagnoses of hemorrhagic bullous skin eruptions. Particular attention should be given to a systemic workup for multiple myeloma when hemorrhagic vesicles/bullae are chronic and coexist with purpura, angina bullosa hemorrhagica, fatigue/weight loss, and/or macroglossia.
- Weidner T, Illing T, Elsner P. Primary localized cutaneous amyloidosis: a systematic treatment review. Am J Clin Dermatol. 2017;18:629-642.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Amyloidosis. Dermatology Essentials. Elsevier Saunders; 2014:341-345.
- Bhutani M, Shahid Z, Schnebelen A, et al. Cutaneous manifestations of multiple myeloma and other plasma cell proliferative disorders. Semin Oncol. 2016;43:395-400.
- Terushkin V, Boyd KP, Patel RR, et al. Primary localized cutaneous amyloidosis. Dermatol Online J. 2013;19:20711.
- LaChance A, Phelps A, Finch J, et al. Nodular localized primary cutaneous amyloidosis: a bullous variant. Clin Exp Dermatol. 2014;39:344-347.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
- Kanoh T. Bullous amyloidosis [in Japanese]. Rinsho Ketsueki. 1993;34:1050-1052.
- Johnson TM, Rapini RP, Hebert AA, et al. Bullous amyloidosis. Cutis. 1989;43:346-352.
- Houman MH, Smiti KM, Ben Ghorbel I, et al. Bullous amyloidosis. Ann Dermatol Venereol. 2002;129:299-302.
- Sanusi T, Li Y, Qian Y, et al. Primary localized cutaneous nodular amyloidosis with bullous lesions. Indian J Dermatol Venereol Leprol. 2015;81:400-402.
- Ochiai T, Morishima T, Hao T, et al. Bullous amyloidosis: the mechanism of blister formation revealed by electron microscopy. J Cutan Pathol. 2001;28:407-411.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Wang XD, Shen H, Liu ZH. Diffuse haemorrhagic bullous amyloidosis with multiple myeloma. Clin Exp Dermatol. 2008;33:94-96.
- Biswas P, Aggarwal I, Sen D, et al. Bullous pemphigoid clinically presenting as lichen amyloidosis. Indian J Dermatol Venereol Leprol. 2014;80:544-546.
- Bluhm JF 3rd. Bullous dermatosis vs amyloidosis. Arch Dermatol. 1981;117:252.
- Bluhm JF 3rd. Bullous amyloidosis vs epidermolysis bullosa acquisita. JAMA. 1981;245:32.
- Murphy GM, Wright J, Nicholls DS, et al. Sunbed-induced pseudoporphyria. Br J Dermatol. 1989;120:555-562.
- Pramatarov K, Lazarova A, Mateev G, et al. Bullous hemorrhagic primary systemic amyloidosis. Int J Dermatol. 1990;29:211-213.
- Bieber T, Ruzicka T, Linke RP, et al. Hemorrhagic bullous amyloidosis. a histologic, immunocytochemical, and ultrastructural study of two patients. Arch Dermatol. 1988;124:1683-1686.
- Khoo BP, Tay YK. Lichen amyloidosis: a bullous variant. Ann Acad Med Singapore. 2000;29:105-107.
- Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
- Schmutz JL, Barbaud A, Cuny JF, et al. Bullous amyloidosis [in French]. Ann Dermatol Venereol. 1988;115:295-301.
- Lachmann HJ, Hawkins PN. Amyloidosis of the skin. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. McGraw-Hill; 2012:1574-1583.
- Grundmann JU, Bonnekoh B, Gollnick H. Extensive haemorrhagic-bullous skin manifestation of systemic AA-amyloidosis associated with IgG lambda-myeloma. Eur J Dermatol. 2000;10:139-142.
- Hödl S, Turek TD, Kerl H. Plasmocytoma-associated bullous hemorrhagic amyloidosis of the skin [in German]. Hautarzt. 1982;33:556-558.
- Colucci G, Alberio L, Demarmels Biasiutti F, et al. Bilateral periorbital ecchymoses. an often missed sign of amyloid purpura. Hamostaseologie. 2014;34:249-252.
- Behera B, Pattnaik M, Sahu B, et al. Cutaneous manifestations of multiple myeloma. Indian J Dermatol. 2016;61:668-671.
- Fujita Y, Tsuji-Abe Y, Sato-Matsumura KC, et al. Nail dystrophy and blisters as sole manifestations in myeloma-associated amyloidosis. J Am Acad Dermatol. 2006;54:712-714.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Winzer M, Ruppert M, Baretton G, et al. Bullous poikilodermatitic amyloidosis of the skin with junctional bulla development in IgG light chain plasmacytoma of the lambda type. histology, immunohistology and electron microscopy [in German]. Hautarzt. 1992;43:199-204.
Cutaneous amyloidosis encompasses a variety of clinical presentations. Primary localized cutaneous amyloidosis comprises lichen amyloidosis, macular amyloidosis, and nodular amyloidosis.1 Macular and lichen amyloidosis result from keratin deposits, while nodular amyloidosis results from cutaneous infiltration of plasma cells.2 Primary systemic amyloidosis is due to a plasma cell dyscrasia, particularly multiple myeloma, while secondary systemic amyloidosis occurs in the setting of restrictive cardiomyopathy, congestive heart failure, renal dysfunction, or chronic inflammation, as seen with rheumatoid arthritis, tuberculosis, and various autoinflammatory disorders.2 Plasma cell proliferative disorders are associated with various skin disorders, which may result from aggregated misfolded monoclonal immunoglobulins, indicating light chain–related systemic amyloidosis. Mucocutaneous lesions can occur in 30% to 40% of cases of primary systemic amyloidosis and may present as purpura, ecchymoses, waxy thickening, plaques, subcutaneous nodules, and/or bullae.3,4 When blistering is present, the differential diagnosis is broad and includes autoimmune bullous disease, drug eruptions, enoxaparin-induced bullous hemorrhagic dermatosis, deposition diseases, allergic contact dermatitis, bullous cellulitis, bullous bite reactions, neutrophilic dermatosis, and bullous lichen sclerosus.5 Herein, we present a case of a woman with a bullous skin eruption who eventually was diagnosed with bullous amyloidosis subsequent to a diagnosis of multiple myeloma.
Case Report
A 70-year-old woman presented to our dermatology clinic for evaluation of well-demarcated, hemorrhagic, flaccid vesicles and focal erosions with a rim of erythema on the distal forearms and hands. A shave biopsy from the right forearm showed cell-poor subepidermal vesicular dermatitis. Enzyme-linked immunosorbent assays for bullous pemphigoid antigens 1 and 2 as well as urinary porphyrins were negative. Direct immunofluorescence showed granular IgM at the basement membrane zone around vessels and cytoid bodies. At this time, a preliminary diagnosis of pseudoporphyria was suspected, though no classic medications (eg, nonsteroidal anti-inflammatory drugs, furosemide, antibiotics) or exogenous trigger factors (eg, UV light exposure, dialysis) were temporally related. Three months later, the patient presented with a large hemorrhagic bulla on the distal left forearm (Figure 1) and healing erosions on the dorsal fingers and upper back. Clobetasol ointment was initiated, as an autoimmune bullous dermatosis was suspected.
Approximately 1 year after she was first seen in our outpatient clinic, the patient was hospitalized for induction of chemotherapy—cyclophosphamide, bortezomib, and dexamethasone—for a new diagnosis of stage III multiple myeloma. A workup for back pain revealed multiple compression fractures and a plasma cell neoplasm with elevated λ light chains, which was confirmed with a bone marrow biopsy. During an inpatient dermatology consultation, we noted the development of intraoral hemorrhagic vesicles and worsening generalization of the hemorrhagic bullae, with healing erosions and intact hemorrhagic bullae on the dorsal hands, fingers (Figure 2), and upper back.
A repeat biopsy displayed bullous amyloidosis. Histopathologic examination revealed an ulcerated subepidermal blister with fibrin deposition at the ulcer base. A periadnexal, scant, eosinophilic deposition with extravasated red blood cells was appreciated. Amorphous eosinophilic deposits were found within the detached fragment of the epidermis and inflammatory infiltrate. A Congo red stain highlighted these areas with a salmon pink–colored material. Congo red staining showed a moderate amount of pale, apple green, birefringent deposit within these areas on polarized light examination.
A few months later, the patient was re-admitted, and the amount of skin detachment prompted the primary team to ask for another consultation. Although the extensive skin sloughing resembled toxic epidermal necrolysis, a repeat biopsy confirmed bullous amyloidosis.
Comment
Amyloidosis Histopathology—Amyloidoses represent a wide array of disorders with deposition of β-pleated sheets or amyloid fibrils, often with cutaneous manifestations.2,3 Primary systemic amyloidosis has been associated with underlying dyscrasia or multiple myeloma.6 In such cases, the skin lesions of multiple myeloma may result from a collection of misfolded monoclonal immunoglobulins or their fragments, as in light chain–related systemic amyloidosis.3 Histopathologically, both systemic and cutaneous amyloidosis appear similar and display deposition of amorphous, eosinophilic, fissured amyloid material in the dermis. Congo red stains the material orange-red and will display a characteristic apple green birefringence under polarized light.4 Although bullous amyloid lesions are rare, the cutaneous forms of these lesions can be an important sign of plasma cell dyscrasia.7
Presentation of Bullous Amyloidosis—Bullous manifestations rarely have been noted in the primary cutaneous forms of amyloidosis.5,8,9 Importantly, cutaneous blistering more often is linked to systemic forms of amyloidosis with multiorgan involvement, including primary systemic and myeloma-associated amyloidosis.5,10 However, patients with localized bullous cutaneous amyloidosis without systemic involvement also have been seen.10,11 Bullae may occur at any time, with contents that frequently are hemorrhagic due to capillary fragility.12,13 Bullous manifestations raise the differential diagnoses of bullous pemphigoid, epidermolysis bullosa acquisita, linear IgA disease, porphyria cutanea tarda, pseudoporphyria, bullous drug eruption, bullous eruption of renal dialysis, or bullous lupus erythematosus.5,13-17
In our patient, the acral distribution of bullae, presence of hemorrhage, chronicity of symptoms, and negative enzyme-linked immunosorbent assay initially suggested a diagnosis of pseudoporphyria. However, the presence of intraoral hemorrhagic vesicles and subsequent confirmatory pathology aided in differentiating bullous amyloidosis from pseudoporphyria. Nodular localized primary cutaneous amyloidosis, a rare form of skin-restricted amyloidoses, can coexist with bullous lesions. Of note, reported cases of nodular localized primary cutaneous amyloidosis did not result in development of multiple myeloma.5,10
Bullae are located either subepidermally or intradermally, and bullous lesions of cutaneous amyloidosis typically demonstrate subepidermal or superficial intradermal clefting on light microscopy.5,10,12 Histopathology of bullous amyloidosis shows intradermal or subepidermal blister formation and amorphous eosinophilic material showing apple green birefringence with Congo red staining deposited in the dermis and/or around the adipocytes and blood vessel walls.12,18-20 In prior cases, direct immunofluorescence of bullous amyloidosis revealed absent immunoglobulin (IgG, IgA, IgM) or complement (C3 and C9) deposits in the basement membrane zone or dermis.13,21,22 In these cases, electron microscopy was useful in diagnosis, as it showed the presence of amyloid deposits.21,22
Cause of Bullae—Various mechanisms are thought to trigger the blister formation in amyloidosis. Bullae created from trauma or friction often present as tense painful blisters that commonly are hemorrhagic.10,23 Amyloid deposits in the walls of blood vessels and the affinity of dermal amyloid in blood vessel walls to surrounding collagen likely leads to increased fragility of capillaries and the dermal matrix, hemorrhagic tendency, and infrapapillary blisters, thus creating hemorrhagic bullous eruptions.24,25 Specifically, close proximity of immunoglobulin-derived amyloid oligomers to epidermal keratinocytes may be toxic and therefore could trigger subepidermal bullous change.5 Additionally, alteration in the physicochemical properties of the amyloidal protein might explain bullous eruption.9 Trauma or rubbing of the hands and feet may precipitate the acral blister formation in bullous amyloidosis.5,11
Due to deposition of these amyloid fibrils, skin bleeding in these patients is called amyloid or pinch purpura. Vessel wall fragility and damage by amyloid are the principal causes of periorbital and gastrointestinal tract bleeding.26 Destruction of the lamina densa and widening of the intercellular space between keratinocytes by amyloid globules induce skin fragility.11
Although uncommon, various cases of bullous amyloidosis have been reported in the literature. Multiple myeloma patients represent the majority of those reported to have bullous amyloidosis.6,7,13,24,27-30 Plasmacytoma-associated bullous amyloid purpura and paraproteinemia also have been noted.25 Multiple myeloma with secondary AL amyloidosis has been seen with amyloid purpura and atraumatic ecchymoses of the face, highlighting the hemorrhage noted in these patients.26
Management of Amyloidosis—Various treatment options have been attempted for primary cutaneous amyloidosis, including oral retinoids, corticosteroids, cyclophosphamide, cyclosporine, amitriptyline, colchicine, cepharanthin, tacrolimus, dimethyl sulfoxide, vitamin D3 analogs, capsaicin, menthol, hydrocolloid dressings, surgical modalities, laser treatment, and phototherapy.1 There is no clear consensus for therapeutic modalities except for treating the underlying plasma cell dyscrasia in primary systemic amyloidosis.
Conclusion
We report the case of a patient displaying signs of pseudoporphyria that ultimately proved to be bullous amyloidosis, or what we termed pseudopseudoporphyria. Bullous amyloidosis should be considered in the differential diagnoses of hemorrhagic bullous skin eruptions. Particular attention should be given to a systemic workup for multiple myeloma when hemorrhagic vesicles/bullae are chronic and coexist with purpura, angina bullosa hemorrhagica, fatigue/weight loss, and/or macroglossia.
Cutaneous amyloidosis encompasses a variety of clinical presentations. Primary localized cutaneous amyloidosis comprises lichen amyloidosis, macular amyloidosis, and nodular amyloidosis.1 Macular and lichen amyloidosis result from keratin deposits, while nodular amyloidosis results from cutaneous infiltration of plasma cells.2 Primary systemic amyloidosis is due to a plasma cell dyscrasia, particularly multiple myeloma, while secondary systemic amyloidosis occurs in the setting of restrictive cardiomyopathy, congestive heart failure, renal dysfunction, or chronic inflammation, as seen with rheumatoid arthritis, tuberculosis, and various autoinflammatory disorders.2 Plasma cell proliferative disorders are associated with various skin disorders, which may result from aggregated misfolded monoclonal immunoglobulins, indicating light chain–related systemic amyloidosis. Mucocutaneous lesions can occur in 30% to 40% of cases of primary systemic amyloidosis and may present as purpura, ecchymoses, waxy thickening, plaques, subcutaneous nodules, and/or bullae.3,4 When blistering is present, the differential diagnosis is broad and includes autoimmune bullous disease, drug eruptions, enoxaparin-induced bullous hemorrhagic dermatosis, deposition diseases, allergic contact dermatitis, bullous cellulitis, bullous bite reactions, neutrophilic dermatosis, and bullous lichen sclerosus.5 Herein, we present a case of a woman with a bullous skin eruption who eventually was diagnosed with bullous amyloidosis subsequent to a diagnosis of multiple myeloma.
Case Report
A 70-year-old woman presented to our dermatology clinic for evaluation of well-demarcated, hemorrhagic, flaccid vesicles and focal erosions with a rim of erythema on the distal forearms and hands. A shave biopsy from the right forearm showed cell-poor subepidermal vesicular dermatitis. Enzyme-linked immunosorbent assays for bullous pemphigoid antigens 1 and 2 as well as urinary porphyrins were negative. Direct immunofluorescence showed granular IgM at the basement membrane zone around vessels and cytoid bodies. At this time, a preliminary diagnosis of pseudoporphyria was suspected, though no classic medications (eg, nonsteroidal anti-inflammatory drugs, furosemide, antibiotics) or exogenous trigger factors (eg, UV light exposure, dialysis) were temporally related. Three months later, the patient presented with a large hemorrhagic bulla on the distal left forearm (Figure 1) and healing erosions on the dorsal fingers and upper back. Clobetasol ointment was initiated, as an autoimmune bullous dermatosis was suspected.
Approximately 1 year after she was first seen in our outpatient clinic, the patient was hospitalized for induction of chemotherapy—cyclophosphamide, bortezomib, and dexamethasone—for a new diagnosis of stage III multiple myeloma. A workup for back pain revealed multiple compression fractures and a plasma cell neoplasm with elevated λ light chains, which was confirmed with a bone marrow biopsy. During an inpatient dermatology consultation, we noted the development of intraoral hemorrhagic vesicles and worsening generalization of the hemorrhagic bullae, with healing erosions and intact hemorrhagic bullae on the dorsal hands, fingers (Figure 2), and upper back.
A repeat biopsy displayed bullous amyloidosis. Histopathologic examination revealed an ulcerated subepidermal blister with fibrin deposition at the ulcer base. A periadnexal, scant, eosinophilic deposition with extravasated red blood cells was appreciated. Amorphous eosinophilic deposits were found within the detached fragment of the epidermis and inflammatory infiltrate. A Congo red stain highlighted these areas with a salmon pink–colored material. Congo red staining showed a moderate amount of pale, apple green, birefringent deposit within these areas on polarized light examination.
A few months later, the patient was re-admitted, and the amount of skin detachment prompted the primary team to ask for another consultation. Although the extensive skin sloughing resembled toxic epidermal necrolysis, a repeat biopsy confirmed bullous amyloidosis.
Comment
Amyloidosis Histopathology—Amyloidoses represent a wide array of disorders with deposition of β-pleated sheets or amyloid fibrils, often with cutaneous manifestations.2,3 Primary systemic amyloidosis has been associated with underlying dyscrasia or multiple myeloma.6 In such cases, the skin lesions of multiple myeloma may result from a collection of misfolded monoclonal immunoglobulins or their fragments, as in light chain–related systemic amyloidosis.3 Histopathologically, both systemic and cutaneous amyloidosis appear similar and display deposition of amorphous, eosinophilic, fissured amyloid material in the dermis. Congo red stains the material orange-red and will display a characteristic apple green birefringence under polarized light.4 Although bullous amyloid lesions are rare, the cutaneous forms of these lesions can be an important sign of plasma cell dyscrasia.7
Presentation of Bullous Amyloidosis—Bullous manifestations rarely have been noted in the primary cutaneous forms of amyloidosis.5,8,9 Importantly, cutaneous blistering more often is linked to systemic forms of amyloidosis with multiorgan involvement, including primary systemic and myeloma-associated amyloidosis.5,10 However, patients with localized bullous cutaneous amyloidosis without systemic involvement also have been seen.10,11 Bullae may occur at any time, with contents that frequently are hemorrhagic due to capillary fragility.12,13 Bullous manifestations raise the differential diagnoses of bullous pemphigoid, epidermolysis bullosa acquisita, linear IgA disease, porphyria cutanea tarda, pseudoporphyria, bullous drug eruption, bullous eruption of renal dialysis, or bullous lupus erythematosus.5,13-17
In our patient, the acral distribution of bullae, presence of hemorrhage, chronicity of symptoms, and negative enzyme-linked immunosorbent assay initially suggested a diagnosis of pseudoporphyria. However, the presence of intraoral hemorrhagic vesicles and subsequent confirmatory pathology aided in differentiating bullous amyloidosis from pseudoporphyria. Nodular localized primary cutaneous amyloidosis, a rare form of skin-restricted amyloidoses, can coexist with bullous lesions. Of note, reported cases of nodular localized primary cutaneous amyloidosis did not result in development of multiple myeloma.5,10
Bullae are located either subepidermally or intradermally, and bullous lesions of cutaneous amyloidosis typically demonstrate subepidermal or superficial intradermal clefting on light microscopy.5,10,12 Histopathology of bullous amyloidosis shows intradermal or subepidermal blister formation and amorphous eosinophilic material showing apple green birefringence with Congo red staining deposited in the dermis and/or around the adipocytes and blood vessel walls.12,18-20 In prior cases, direct immunofluorescence of bullous amyloidosis revealed absent immunoglobulin (IgG, IgA, IgM) or complement (C3 and C9) deposits in the basement membrane zone or dermis.13,21,22 In these cases, electron microscopy was useful in diagnosis, as it showed the presence of amyloid deposits.21,22
Cause of Bullae—Various mechanisms are thought to trigger the blister formation in amyloidosis. Bullae created from trauma or friction often present as tense painful blisters that commonly are hemorrhagic.10,23 Amyloid deposits in the walls of blood vessels and the affinity of dermal amyloid in blood vessel walls to surrounding collagen likely leads to increased fragility of capillaries and the dermal matrix, hemorrhagic tendency, and infrapapillary blisters, thus creating hemorrhagic bullous eruptions.24,25 Specifically, close proximity of immunoglobulin-derived amyloid oligomers to epidermal keratinocytes may be toxic and therefore could trigger subepidermal bullous change.5 Additionally, alteration in the physicochemical properties of the amyloidal protein might explain bullous eruption.9 Trauma or rubbing of the hands and feet may precipitate the acral blister formation in bullous amyloidosis.5,11
Due to deposition of these amyloid fibrils, skin bleeding in these patients is called amyloid or pinch purpura. Vessel wall fragility and damage by amyloid are the principal causes of periorbital and gastrointestinal tract bleeding.26 Destruction of the lamina densa and widening of the intercellular space between keratinocytes by amyloid globules induce skin fragility.11
Although uncommon, various cases of bullous amyloidosis have been reported in the literature. Multiple myeloma patients represent the majority of those reported to have bullous amyloidosis.6,7,13,24,27-30 Plasmacytoma-associated bullous amyloid purpura and paraproteinemia also have been noted.25 Multiple myeloma with secondary AL amyloidosis has been seen with amyloid purpura and atraumatic ecchymoses of the face, highlighting the hemorrhage noted in these patients.26
Management of Amyloidosis—Various treatment options have been attempted for primary cutaneous amyloidosis, including oral retinoids, corticosteroids, cyclophosphamide, cyclosporine, amitriptyline, colchicine, cepharanthin, tacrolimus, dimethyl sulfoxide, vitamin D3 analogs, capsaicin, menthol, hydrocolloid dressings, surgical modalities, laser treatment, and phototherapy.1 There is no clear consensus for therapeutic modalities except for treating the underlying plasma cell dyscrasia in primary systemic amyloidosis.
Conclusion
We report the case of a patient displaying signs of pseudoporphyria that ultimately proved to be bullous amyloidosis, or what we termed pseudopseudoporphyria. Bullous amyloidosis should be considered in the differential diagnoses of hemorrhagic bullous skin eruptions. Particular attention should be given to a systemic workup for multiple myeloma when hemorrhagic vesicles/bullae are chronic and coexist with purpura, angina bullosa hemorrhagica, fatigue/weight loss, and/or macroglossia.
- Weidner T, Illing T, Elsner P. Primary localized cutaneous amyloidosis: a systematic treatment review. Am J Clin Dermatol. 2017;18:629-642.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Amyloidosis. Dermatology Essentials. Elsevier Saunders; 2014:341-345.
- Bhutani M, Shahid Z, Schnebelen A, et al. Cutaneous manifestations of multiple myeloma and other plasma cell proliferative disorders. Semin Oncol. 2016;43:395-400.
- Terushkin V, Boyd KP, Patel RR, et al. Primary localized cutaneous amyloidosis. Dermatol Online J. 2013;19:20711.
- LaChance A, Phelps A, Finch J, et al. Nodular localized primary cutaneous amyloidosis: a bullous variant. Clin Exp Dermatol. 2014;39:344-347.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
- Kanoh T. Bullous amyloidosis [in Japanese]. Rinsho Ketsueki. 1993;34:1050-1052.
- Johnson TM, Rapini RP, Hebert AA, et al. Bullous amyloidosis. Cutis. 1989;43:346-352.
- Houman MH, Smiti KM, Ben Ghorbel I, et al. Bullous amyloidosis. Ann Dermatol Venereol. 2002;129:299-302.
- Sanusi T, Li Y, Qian Y, et al. Primary localized cutaneous nodular amyloidosis with bullous lesions. Indian J Dermatol Venereol Leprol. 2015;81:400-402.
- Ochiai T, Morishima T, Hao T, et al. Bullous amyloidosis: the mechanism of blister formation revealed by electron microscopy. J Cutan Pathol. 2001;28:407-411.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Wang XD, Shen H, Liu ZH. Diffuse haemorrhagic bullous amyloidosis with multiple myeloma. Clin Exp Dermatol. 2008;33:94-96.
- Biswas P, Aggarwal I, Sen D, et al. Bullous pemphigoid clinically presenting as lichen amyloidosis. Indian J Dermatol Venereol Leprol. 2014;80:544-546.
- Bluhm JF 3rd. Bullous dermatosis vs amyloidosis. Arch Dermatol. 1981;117:252.
- Bluhm JF 3rd. Bullous amyloidosis vs epidermolysis bullosa acquisita. JAMA. 1981;245:32.
- Murphy GM, Wright J, Nicholls DS, et al. Sunbed-induced pseudoporphyria. Br J Dermatol. 1989;120:555-562.
- Pramatarov K, Lazarova A, Mateev G, et al. Bullous hemorrhagic primary systemic amyloidosis. Int J Dermatol. 1990;29:211-213.
- Bieber T, Ruzicka T, Linke RP, et al. Hemorrhagic bullous amyloidosis. a histologic, immunocytochemical, and ultrastructural study of two patients. Arch Dermatol. 1988;124:1683-1686.
- Khoo BP, Tay YK. Lichen amyloidosis: a bullous variant. Ann Acad Med Singapore. 2000;29:105-107.
- Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
- Schmutz JL, Barbaud A, Cuny JF, et al. Bullous amyloidosis [in French]. Ann Dermatol Venereol. 1988;115:295-301.
- Lachmann HJ, Hawkins PN. Amyloidosis of the skin. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. McGraw-Hill; 2012:1574-1583.
- Grundmann JU, Bonnekoh B, Gollnick H. Extensive haemorrhagic-bullous skin manifestation of systemic AA-amyloidosis associated with IgG lambda-myeloma. Eur J Dermatol. 2000;10:139-142.
- Hödl S, Turek TD, Kerl H. Plasmocytoma-associated bullous hemorrhagic amyloidosis of the skin [in German]. Hautarzt. 1982;33:556-558.
- Colucci G, Alberio L, Demarmels Biasiutti F, et al. Bilateral periorbital ecchymoses. an often missed sign of amyloid purpura. Hamostaseologie. 2014;34:249-252.
- Behera B, Pattnaik M, Sahu B, et al. Cutaneous manifestations of multiple myeloma. Indian J Dermatol. 2016;61:668-671.
- Fujita Y, Tsuji-Abe Y, Sato-Matsumura KC, et al. Nail dystrophy and blisters as sole manifestations in myeloma-associated amyloidosis. J Am Acad Dermatol. 2006;54:712-714.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Winzer M, Ruppert M, Baretton G, et al. Bullous poikilodermatitic amyloidosis of the skin with junctional bulla development in IgG light chain plasmacytoma of the lambda type. histology, immunohistology and electron microscopy [in German]. Hautarzt. 1992;43:199-204.
- Weidner T, Illing T, Elsner P. Primary localized cutaneous amyloidosis: a systematic treatment review. Am J Clin Dermatol. 2017;18:629-642.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Amyloidosis. Dermatology Essentials. Elsevier Saunders; 2014:341-345.
- Bhutani M, Shahid Z, Schnebelen A, et al. Cutaneous manifestations of multiple myeloma and other plasma cell proliferative disorders. Semin Oncol. 2016;43:395-400.
- Terushkin V, Boyd KP, Patel RR, et al. Primary localized cutaneous amyloidosis. Dermatol Online J. 2013;19:20711.
- LaChance A, Phelps A, Finch J, et al. Nodular localized primary cutaneous amyloidosis: a bullous variant. Clin Exp Dermatol. 2014;39:344-347.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
- Kanoh T. Bullous amyloidosis [in Japanese]. Rinsho Ketsueki. 1993;34:1050-1052.
- Johnson TM, Rapini RP, Hebert AA, et al. Bullous amyloidosis. Cutis. 1989;43:346-352.
- Houman MH, Smiti KM, Ben Ghorbel I, et al. Bullous amyloidosis. Ann Dermatol Venereol. 2002;129:299-302.
- Sanusi T, Li Y, Qian Y, et al. Primary localized cutaneous nodular amyloidosis with bullous lesions. Indian J Dermatol Venereol Leprol. 2015;81:400-402.
- Ochiai T, Morishima T, Hao T, et al. Bullous amyloidosis: the mechanism of blister formation revealed by electron microscopy. J Cutan Pathol. 2001;28:407-411.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Wang XD, Shen H, Liu ZH. Diffuse haemorrhagic bullous amyloidosis with multiple myeloma. Clin Exp Dermatol. 2008;33:94-96.
- Biswas P, Aggarwal I, Sen D, et al. Bullous pemphigoid clinically presenting as lichen amyloidosis. Indian J Dermatol Venereol Leprol. 2014;80:544-546.
- Bluhm JF 3rd. Bullous dermatosis vs amyloidosis. Arch Dermatol. 1981;117:252.
- Bluhm JF 3rd. Bullous amyloidosis vs epidermolysis bullosa acquisita. JAMA. 1981;245:32.
- Murphy GM, Wright J, Nicholls DS, et al. Sunbed-induced pseudoporphyria. Br J Dermatol. 1989;120:555-562.
- Pramatarov K, Lazarova A, Mateev G, et al. Bullous hemorrhagic primary systemic amyloidosis. Int J Dermatol. 1990;29:211-213.
- Bieber T, Ruzicka T, Linke RP, et al. Hemorrhagic bullous amyloidosis. a histologic, immunocytochemical, and ultrastructural study of two patients. Arch Dermatol. 1988;124:1683-1686.
- Khoo BP, Tay YK. Lichen amyloidosis: a bullous variant. Ann Acad Med Singapore. 2000;29:105-107.
- Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
- Schmutz JL, Barbaud A, Cuny JF, et al. Bullous amyloidosis [in French]. Ann Dermatol Venereol. 1988;115:295-301.
- Lachmann HJ, Hawkins PN. Amyloidosis of the skin. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. McGraw-Hill; 2012:1574-1583.
- Grundmann JU, Bonnekoh B, Gollnick H. Extensive haemorrhagic-bullous skin manifestation of systemic AA-amyloidosis associated with IgG lambda-myeloma. Eur J Dermatol. 2000;10:139-142.
- Hödl S, Turek TD, Kerl H. Plasmocytoma-associated bullous hemorrhagic amyloidosis of the skin [in German]. Hautarzt. 1982;33:556-558.
- Colucci G, Alberio L, Demarmels Biasiutti F, et al. Bilateral periorbital ecchymoses. an often missed sign of amyloid purpura. Hamostaseologie. 2014;34:249-252.
- Behera B, Pattnaik M, Sahu B, et al. Cutaneous manifestations of multiple myeloma. Indian J Dermatol. 2016;61:668-671.
- Fujita Y, Tsuji-Abe Y, Sato-Matsumura KC, et al. Nail dystrophy and blisters as sole manifestations in myeloma-associated amyloidosis. J Am Acad Dermatol. 2006;54:712-714.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Winzer M, Ruppert M, Baretton G, et al. Bullous poikilodermatitic amyloidosis of the skin with junctional bulla development in IgG light chain plasmacytoma of the lambda type. histology, immunohistology and electron microscopy [in German]. Hautarzt. 1992;43:199-204.
Practice Points
- Primary systemic amyloidosis, including the rare cutaneous bullous amyloidosis, often is difficult to diagnose and has been associated with underlying plasma cell dyscrasia or multiple myeloma.
- When evaluating patients with initially convincing signs of pseudoporphyria, it is imperative to consider the diagnosis of bullous amyloidosis, which additionally can present with intraoral hemorrhagic vesicles and have confirmatory histopathologic features.
- Further investigation for multiple myeloma is warranted when patients with a chronic hemorrhagic bullous condition also present with symptoms of purpura, angina bullosa hemorrhagica, fatigue, weight loss, and/or macroglossia. Accurate diagnosis of bullous amyloidosis and timely treatment of its underlying cause will contribute to better, more proactive patient care.
Kikuchi-Fujimoto Disease in an Adolescent Boy
To the Editor:
Kikuchi-Fujimoto Disease, also called histiocytic necrotizing lymphadenitis, was described in 1972 by both Kikuchi1 and Fujimoto et al.2 Most cases are reported in Asia, with limited reports in the United States.3-5 Kikuchi-Fujimoto disease is a rare, self-limiting condition consisting of benign lymphadenopathy and oftentimes fever and systemic symptoms. Lymph node involvement may mimic non-Hodgkin lymphoma or other reactive lymphadenopathy, rendering diagnostic accuracy challenging.5 Cutaneous manifestations are reported in only 16% to 40% of patients.6,7 Herein, we describe the clinical and pathologic features of a case of Kikuchi-Fujimoto disease with cutaneous involvement in an adolescent boy.
A 13-year-old adolescent boy with no notable medical history presented to the pediatric emergency department with cervical lymphadenopathy, weight loss, intermittent fever, and an evolving rash on the face, ears, arms, and thighs of 6 weeks’ duration. The illness began with enlarged lymph nodes and erythematous macules on the face and was diagnosed by his primary care physician as lymphadenitis that was unresponsive to clindamycin. Over the subsequent weeks, the rash worsened, and he developed intermittent fevers, night sweats, abdominal pain, and nausea with a 20-pound weight loss. He presented to the emergency department 3 weeks prior to the current admission and was noted to have elevated cytomegalovirus (CMV) IgM and IgG in addition to lymphopenia and anemia. He was discharged with outpatient follow-up. The rash progressed to involve the face, ears, arms, and thighs. One day prior to the current admission, the patient’s abdominal pain worsened acutely, and he experienced several episodes of emesis. He presented to the pediatric emergency department for further evaluation, and a dermatology consultation was requested at that time.
The patient’s rash was asymptomatic. In addition to the above symptoms, he also noted frequent nosebleeds, gingival bleeding, and diffuse myalgia that was most prominent on the hands and feet; he denied diarrhea, sick contacts, recent travel, or insect bites. His vital signs were normal, and he remained afebrile throughout the hospitalization. Physical examination revealed an ill-appearing patient with sunken eyes and dry lips. He had pink, oval, scaly plaques on the cheeks, ears, and arms (Figure 1). The thighs exhibited folliculocentric erythematous papules. The ocular conjunctivae were clear, but white exudative plaques were noted on the tongue. Tender, bilateral, cervical lymphadenopathy and diffuse abdominal tenderness with guarding and hepatosplenomegaly also were present. The fingers and toes were tender upon palpation.

Laboratory workup at admission revealed the following: low white blood cell count, 2700/μL (reference range, 4500–11,000/μL); low hemoglobin, 9.6 g/dL (reference range, 14.0–17.5 g/dL); elevated aspartate aminotransferase, 91 U/L (reference range, 10–30 U/L); and elevated alanine aminotransferase, 118 U/L (reference range, 10–40 U/L). Lactate dehydrogenase (582 U/L [reference range, 100–200 U/L]), ferritin (1681 ng/mL [reference range, 15–200 ng/mL]), and C-reactive protein (6.0 mg/L [reference range, 0.08–3.1 mg/L]) also were elevated. A respiratory viral panel was unremarkable. Blood cultures were negative, and an HIV 1/2 assay was nonreactive. A chest radiograph demonstrated clear lung fields. Computed tomography of the abdomen and pelvis showed prominent mesenteric, ileocolic, and retroperitoneal lymph nodes.
The differential diagnoses at this time included acute connective tissue disease, a paraneoplastic phenomenon, cutaneous lymphoma, or an infectious etiology. A punch biopsy of the skin as well as tissue cultures were performed from a lesion on the right arm. Quantitative immunoglobulin (IgA, IgG, IgM) levels were checked, all of which were within reference range. An antinuclear antibody (ANA) assay and rheumatoid factor were normal.
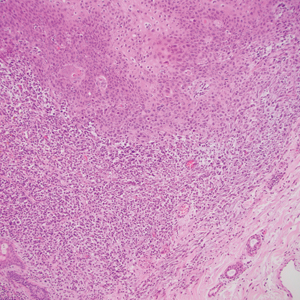
The tissue cultures were negative for bacteria, fungi, and mycobacteria. Microscopic examination of the skin biopsy revealed a moderate perivascular and interstitial infiltrate of predominantly histiocytes and lymphocytes with prominent karyorrhectic debris (nuclear dust) in the upper dermis as well as focal vacuolar interface changes with scattered necrotic keratinocytes in the epidermis (Figure 2). Based on these histopathologic findings, a diagnosis of Kikuchi-Fujimoto disease was considered. To confirm the diagnosis and to rule out the possibility of lymphoma, an excisional biopsy of the cervical lymph node was performed, which showed typical histopathologic features of histiocytic necrotizing lymphadenitis.
Given the patient’s clinical presentation with arthralgia, anorexia, lymphadenitis, and hepatosplenomegaly along with histopathologic findings from both the skin and lymph node biopsies, a diagnosis of Kikuchi-Fujimoto disease was made. The patient was conservatively managed with acetaminophen and was discharged with improvement in his appetite and systemic symptoms.
He was seen for follow-up 3 months later in the outpatient clinic. He denied any recurrence of systemic symptoms but endorsed a recent shedding of hair consistent with telogen effluvium. The rash had substantially improved, though residual asymptomatic erythematous plaques remained on the right forehead and right cheek (Figure 3). He was prescribed triamcinolone acetonide cream 0.1% to apply to the active area twice daily for the following 2 to 3 weeks.

Kikuchi-Fujimoto disease presents with a wide clinical spectrum, classically with benign lymphadenopathy and fever of unknown etiology.5,6 Lymphadenopathy most often is cervical (55%–99%)8 and unilateral,4,7 but patients can present with polyadenopathy (52%).7,8 Constitutional signs commonly include fever (35%–76%), weight loss, arthritis (5%–34%), and leukopenia (25%–74%).4,8,9
Cutaneous findings have been described in up to 40% of cases, of which clinical presentation is variable.6 Lesions may include blanchable, erythematous, painful, and/or indurated plaques, nodules, or maculopapules with confluence into patches, urticaria, morbilliform lesions, erythema multiforme, eyelid edema, leukocytoclastic vasculitis, papulopustules, ulcerated gingivae, and mucositis.6,7,10-13 Patients with skin lesions may be at an increased risk for developing systemic lupus erythematosus (SLE).8 Our patient presented with erythematous scaly plaques with a predominance of lesions in photodistributed locations, which clinically mimicked an underlying connective tissue disease process such as SLE.
Infectious agents such as CMV, parvovirus B19, human herpesvirus 6, human herpesvirus 8 and human T-cell lymphotropic virus 1, HIV, Yersinia enterocolitica, and Toxoplasma have all been implicated as possible causes of Kikuchi-Fujimoto disease, but studies have failed to provide convincing causal evidence.9,14,15 Our patient had positive IgM and IgG for CMV, which may have incited his disease.
Definitive diagnosis of Kikuchi-Fujimoto disease is made by lymph node excisional biopsy, which histologically exhibits a histiocytic cell proliferation with paracortical foci of necrosis and abundant karyorrhectic debris.5 Cutaneous histologic findings that support the diagnosis are variable and may include a dermal histiocytic infiltrate, epidermal change with necrotic keratinocytes, non-neutrophilic karyorrhectic debris, basal vacuolar change, papillary dermal edema, a nonspecific superficial and deep perivascular infiltrate, and a patchy infiltration of histiocytes and lymphocytes.6,13
Clinical and histopathological features of this disease can mimic other diseases, specifically SLE or lymphoma.7 An association with SLE has been suspected, though it is not well defined and more frequently is associated with cases from Asia than from Europe (28% and 9%, respectively).9 Patients presenting concomitantly with positive ANA, weight loss, arthralgia, and skin lesions are more likely to develop SLE.8 Furthermore, the cutaneous histologic finding of interface change suggests a link between the two diseases. As such, recommendations have been made for ANA screenings and follow-up of patients diagnosed with Kikuchi-Fujimoto disease for clinical evidence of autoimmune disease, particularly SLE.6 Although our patient did not have a positive ANA, his biopsy did demonstrate interface change, and he should be monitored for possible progression of disease in the future.
Kikuchi-Fujimoto disease differs from lymphoma, as it initially presents with rapid lymph node enlargement as opposed to the gradual enlargement seen in lymphoma. The lymph nodes in Kikuchi-Fujimoto disease often are firm and moveable compared to hard and immobile in lymphoma.3 Excisional lymph node biopsy is necessary for both confirming the diagnosis of Kikuchi-Fujimoto disease and ruling out lymphoma.5
Spontaneous resolution usually occurs in 1 to 4 months.3,6 As such, observation is the most common approach to management. When patients have symptoms that limit activities or cause undue distress such as fevers, joint pains, or abdominal pain, systemic treatment options may be desired. Symptomatic treatment can be managed with a short duration of oral corticosteroids,10,11 nonsteroidal anti-inflammatory drugs, antimalarials, and/or antipyretics.8-15 There are no guidelines regarding systemic steroid regimens, and various treatment schedules have been successful. Systemic therapy was considered for our patient for his weight loss and abdominal pain; however, by the time of discharge the patient was tolerating oral intake and his abdominal pain had improved.
- Kikuchi M. Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytosis. Nippon Ketsueki Gakkai Zasshi. 1972;35:379-380.
- Fujimoto Y, Kojima Y, Yamaguchi K. Cervical subacute necrotizing lymphadenitis: a new clinicopathological entity. Naika. 1972;30:920-927.
- Feder Jr HM, Liu J, Rezuke WN. Kikuchi disease in Connecticut. J Pediatr. 2014;164:196-200.
- Kang HM, Kim JY, Choi EH, et al. Clinical characteristics of severe histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto disease) in children. J Pediatr. 2016;171:208-212.
- Hutchinson CB, Wang E. Kikuchi-Fujimoto disease. Arch Pathol Lab Med. 2010;134:289-293.
- Atwater AR, Longly BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008;59:130-136.
- Yen H-R, Lin P-Y, Chuang W-Y, et al. Skin manifestations of Kikuchi-Fujimoto disease: case report and review. Eur J Pediatr. 2004;163:210-213.
- Dumas G, Prendki V, Haroche J, et al. Kikuchi-Fujimoto disease: retrospective study of 91 cases and review of literature. Medicine. 2014;93:372-382.
- Kuc ukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Yasukawa K, Matsumura T, Sato-Matsumura KC, et al. Kikuchi’s disease and the skin: case report and review of the literature. Br J Dermatol. 2001;144:885-889.
- Kaur S, Thami GP, Mohan H, et al. Kikuchi disease with facial rash and erythema multiforme. Pediatr Dermatol. 2001;18:403-405.
- Mauleón C, Valdivielso-Ramos M, Cabeza R, et al. Kikuchi disease with skin lesions mimicking lupus erythematosus. J Dermatol Case Rep. 2012;3:82-85.
- Obara K, Amoh Y. A case of Kikuchi’s disease (histiocytic necrotizing lymphoadenitis) with histiocytic cutaneous involvement. Rheumatol Int. 2015;35:1111-1113.
- Rosado FGN, Tang Y-W, Hasserjian RP, et al. Kikuchi-Fujimoto lymphadenitis: role of parvovirus B-19, Epstein-Barr virus, human herpesvirus 6, and human herpesvirus 8. Hum Pathol. 2013;44:255-259.
- Chiu CF, Chow KC, Lin TY, et al. Virus infection in patients with histiocytic necrotizing lymphadenitis in Taiwan. detection of Epstein-Barr virus, type I human T-cell lymphotropic virus, and parvovirus B19. Am J Clin Pathol. 2000;113:774-781.
To the Editor:
Kikuchi-Fujimoto Disease, also called histiocytic necrotizing lymphadenitis, was described in 1972 by both Kikuchi1 and Fujimoto et al.2 Most cases are reported in Asia, with limited reports in the United States.3-5 Kikuchi-Fujimoto disease is a rare, self-limiting condition consisting of benign lymphadenopathy and oftentimes fever and systemic symptoms. Lymph node involvement may mimic non-Hodgkin lymphoma or other reactive lymphadenopathy, rendering diagnostic accuracy challenging.5 Cutaneous manifestations are reported in only 16% to 40% of patients.6,7 Herein, we describe the clinical and pathologic features of a case of Kikuchi-Fujimoto disease with cutaneous involvement in an adolescent boy.
A 13-year-old adolescent boy with no notable medical history presented to the pediatric emergency department with cervical lymphadenopathy, weight loss, intermittent fever, and an evolving rash on the face, ears, arms, and thighs of 6 weeks’ duration. The illness began with enlarged lymph nodes and erythematous macules on the face and was diagnosed by his primary care physician as lymphadenitis that was unresponsive to clindamycin. Over the subsequent weeks, the rash worsened, and he developed intermittent fevers, night sweats, abdominal pain, and nausea with a 20-pound weight loss. He presented to the emergency department 3 weeks prior to the current admission and was noted to have elevated cytomegalovirus (CMV) IgM and IgG in addition to lymphopenia and anemia. He was discharged with outpatient follow-up. The rash progressed to involve the face, ears, arms, and thighs. One day prior to the current admission, the patient’s abdominal pain worsened acutely, and he experienced several episodes of emesis. He presented to the pediatric emergency department for further evaluation, and a dermatology consultation was requested at that time.
The patient’s rash was asymptomatic. In addition to the above symptoms, he also noted frequent nosebleeds, gingival bleeding, and diffuse myalgia that was most prominent on the hands and feet; he denied diarrhea, sick contacts, recent travel, or insect bites. His vital signs were normal, and he remained afebrile throughout the hospitalization. Physical examination revealed an ill-appearing patient with sunken eyes and dry lips. He had pink, oval, scaly plaques on the cheeks, ears, and arms (Figure 1). The thighs exhibited folliculocentric erythematous papules. The ocular conjunctivae were clear, but white exudative plaques were noted on the tongue. Tender, bilateral, cervical lymphadenopathy and diffuse abdominal tenderness with guarding and hepatosplenomegaly also were present. The fingers and toes were tender upon palpation.
Laboratory workup at admission revealed the following: low white blood cell count, 2700/μL (reference range, 4500–11,000/μL); low hemoglobin, 9.6 g/dL (reference range, 14.0–17.5 g/dL); elevated aspartate aminotransferase, 91 U/L (reference range, 10–30 U/L); and elevated alanine aminotransferase, 118 U/L (reference range, 10–40 U/L). Lactate dehydrogenase (582 U/L [reference range, 100–200 U/L]), ferritin (1681 ng/mL [reference range, 15–200 ng/mL]), and C-reactive protein (6.0 mg/L [reference range, 0.08–3.1 mg/L]) also were elevated. A respiratory viral panel was unremarkable. Blood cultures were negative, and an HIV 1/2 assay was nonreactive. A chest radiograph demonstrated clear lung fields. Computed tomography of the abdomen and pelvis showed prominent mesenteric, ileocolic, and retroperitoneal lymph nodes.
The differential diagnoses at this time included acute connective tissue disease, a paraneoplastic phenomenon, cutaneous lymphoma, or an infectious etiology. A punch biopsy of the skin as well as tissue cultures were performed from a lesion on the right arm. Quantitative immunoglobulin (IgA, IgG, IgM) levels were checked, all of which were within reference range. An antinuclear antibody (ANA) assay and rheumatoid factor were normal.
The tissue cultures were negative for bacteria, fungi, and mycobacteria. Microscopic examination of the skin biopsy revealed a moderate perivascular and interstitial infiltrate of predominantly histiocytes and lymphocytes with prominent karyorrhectic debris (nuclear dust) in the upper dermis as well as focal vacuolar interface changes with scattered necrotic keratinocytes in the epidermis (Figure 2). Based on these histopathologic findings, a diagnosis of Kikuchi-Fujimoto disease was considered. To confirm the diagnosis and to rule out the possibility of lymphoma, an excisional biopsy of the cervical lymph node was performed, which showed typical histopathologic features of histiocytic necrotizing lymphadenitis.
Given the patient’s clinical presentation with arthralgia, anorexia, lymphadenitis, and hepatosplenomegaly along with histopathologic findings from both the skin and lymph node biopsies, a diagnosis of Kikuchi-Fujimoto disease was made. The patient was conservatively managed with acetaminophen and was discharged with improvement in his appetite and systemic symptoms.
He was seen for follow-up 3 months later in the outpatient clinic. He denied any recurrence of systemic symptoms but endorsed a recent shedding of hair consistent with telogen effluvium. The rash had substantially improved, though residual asymptomatic erythematous plaques remained on the right forehead and right cheek (Figure 3). He was prescribed triamcinolone acetonide cream 0.1% to apply to the active area twice daily for the following 2 to 3 weeks.
Kikuchi-Fujimoto disease presents with a wide clinical spectrum, classically with benign lymphadenopathy and fever of unknown etiology.5,6 Lymphadenopathy most often is cervical (55%–99%)8 and unilateral,4,7 but patients can present with polyadenopathy (52%).7,8 Constitutional signs commonly include fever (35%–76%), weight loss, arthritis (5%–34%), and leukopenia (25%–74%).4,8,9
Cutaneous findings have been described in up to 40% of cases, of which clinical presentation is variable.6 Lesions may include blanchable, erythematous, painful, and/or indurated plaques, nodules, or maculopapules with confluence into patches, urticaria, morbilliform lesions, erythema multiforme, eyelid edema, leukocytoclastic vasculitis, papulopustules, ulcerated gingivae, and mucositis.6,7,10-13 Patients with skin lesions may be at an increased risk for developing systemic lupus erythematosus (SLE).8 Our patient presented with erythematous scaly plaques with a predominance of lesions in photodistributed locations, which clinically mimicked an underlying connective tissue disease process such as SLE.
Infectious agents such as CMV, parvovirus B19, human herpesvirus 6, human herpesvirus 8 and human T-cell lymphotropic virus 1, HIV, Yersinia enterocolitica, and Toxoplasma have all been implicated as possible causes of Kikuchi-Fujimoto disease, but studies have failed to provide convincing causal evidence.9,14,15 Our patient had positive IgM and IgG for CMV, which may have incited his disease.
Definitive diagnosis of Kikuchi-Fujimoto disease is made by lymph node excisional biopsy, which histologically exhibits a histiocytic cell proliferation with paracortical foci of necrosis and abundant karyorrhectic debris.5 Cutaneous histologic findings that support the diagnosis are variable and may include a dermal histiocytic infiltrate, epidermal change with necrotic keratinocytes, non-neutrophilic karyorrhectic debris, basal vacuolar change, papillary dermal edema, a nonspecific superficial and deep perivascular infiltrate, and a patchy infiltration of histiocytes and lymphocytes.6,13
Clinical and histopathological features of this disease can mimic other diseases, specifically SLE or lymphoma.7 An association with SLE has been suspected, though it is not well defined and more frequently is associated with cases from Asia than from Europe (28% and 9%, respectively).9 Patients presenting concomitantly with positive ANA, weight loss, arthralgia, and skin lesions are more likely to develop SLE.8 Furthermore, the cutaneous histologic finding of interface change suggests a link between the two diseases. As such, recommendations have been made for ANA screenings and follow-up of patients diagnosed with Kikuchi-Fujimoto disease for clinical evidence of autoimmune disease, particularly SLE.6 Although our patient did not have a positive ANA, his biopsy did demonstrate interface change, and he should be monitored for possible progression of disease in the future.
Kikuchi-Fujimoto disease differs from lymphoma, as it initially presents with rapid lymph node enlargement as opposed to the gradual enlargement seen in lymphoma. The lymph nodes in Kikuchi-Fujimoto disease often are firm and moveable compared to hard and immobile in lymphoma.3 Excisional lymph node biopsy is necessary for both confirming the diagnosis of Kikuchi-Fujimoto disease and ruling out lymphoma.5
Spontaneous resolution usually occurs in 1 to 4 months.3,6 As such, observation is the most common approach to management. When patients have symptoms that limit activities or cause undue distress such as fevers, joint pains, or abdominal pain, systemic treatment options may be desired. Symptomatic treatment can be managed with a short duration of oral corticosteroids,10,11 nonsteroidal anti-inflammatory drugs, antimalarials, and/or antipyretics.8-15 There are no guidelines regarding systemic steroid regimens, and various treatment schedules have been successful. Systemic therapy was considered for our patient for his weight loss and abdominal pain; however, by the time of discharge the patient was tolerating oral intake and his abdominal pain had improved.
To the Editor:
Kikuchi-Fujimoto Disease, also called histiocytic necrotizing lymphadenitis, was described in 1972 by both Kikuchi1 and Fujimoto et al.2 Most cases are reported in Asia, with limited reports in the United States.3-5 Kikuchi-Fujimoto disease is a rare, self-limiting condition consisting of benign lymphadenopathy and oftentimes fever and systemic symptoms. Lymph node involvement may mimic non-Hodgkin lymphoma or other reactive lymphadenopathy, rendering diagnostic accuracy challenging.5 Cutaneous manifestations are reported in only 16% to 40% of patients.6,7 Herein, we describe the clinical and pathologic features of a case of Kikuchi-Fujimoto disease with cutaneous involvement in an adolescent boy.
A 13-year-old adolescent boy with no notable medical history presented to the pediatric emergency department with cervical lymphadenopathy, weight loss, intermittent fever, and an evolving rash on the face, ears, arms, and thighs of 6 weeks’ duration. The illness began with enlarged lymph nodes and erythematous macules on the face and was diagnosed by his primary care physician as lymphadenitis that was unresponsive to clindamycin. Over the subsequent weeks, the rash worsened, and he developed intermittent fevers, night sweats, abdominal pain, and nausea with a 20-pound weight loss. He presented to the emergency department 3 weeks prior to the current admission and was noted to have elevated cytomegalovirus (CMV) IgM and IgG in addition to lymphopenia and anemia. He was discharged with outpatient follow-up. The rash progressed to involve the face, ears, arms, and thighs. One day prior to the current admission, the patient’s abdominal pain worsened acutely, and he experienced several episodes of emesis. He presented to the pediatric emergency department for further evaluation, and a dermatology consultation was requested at that time.
The patient’s rash was asymptomatic. In addition to the above symptoms, he also noted frequent nosebleeds, gingival bleeding, and diffuse myalgia that was most prominent on the hands and feet; he denied diarrhea, sick contacts, recent travel, or insect bites. His vital signs were normal, and he remained afebrile throughout the hospitalization. Physical examination revealed an ill-appearing patient with sunken eyes and dry lips. He had pink, oval, scaly plaques on the cheeks, ears, and arms (Figure 1). The thighs exhibited folliculocentric erythematous papules. The ocular conjunctivae were clear, but white exudative plaques were noted on the tongue. Tender, bilateral, cervical lymphadenopathy and diffuse abdominal tenderness with guarding and hepatosplenomegaly also were present. The fingers and toes were tender upon palpation.
Laboratory workup at admission revealed the following: low white blood cell count, 2700/μL (reference range, 4500–11,000/μL); low hemoglobin, 9.6 g/dL (reference range, 14.0–17.5 g/dL); elevated aspartate aminotransferase, 91 U/L (reference range, 10–30 U/L); and elevated alanine aminotransferase, 118 U/L (reference range, 10–40 U/L). Lactate dehydrogenase (582 U/L [reference range, 100–200 U/L]), ferritin (1681 ng/mL [reference range, 15–200 ng/mL]), and C-reactive protein (6.0 mg/L [reference range, 0.08–3.1 mg/L]) also were elevated. A respiratory viral panel was unremarkable. Blood cultures were negative, and an HIV 1/2 assay was nonreactive. A chest radiograph demonstrated clear lung fields. Computed tomography of the abdomen and pelvis showed prominent mesenteric, ileocolic, and retroperitoneal lymph nodes.
The differential diagnoses at this time included acute connective tissue disease, a paraneoplastic phenomenon, cutaneous lymphoma, or an infectious etiology. A punch biopsy of the skin as well as tissue cultures were performed from a lesion on the right arm. Quantitative immunoglobulin (IgA, IgG, IgM) levels were checked, all of which were within reference range. An antinuclear antibody (ANA) assay and rheumatoid factor were normal.
The tissue cultures were negative for bacteria, fungi, and mycobacteria. Microscopic examination of the skin biopsy revealed a moderate perivascular and interstitial infiltrate of predominantly histiocytes and lymphocytes with prominent karyorrhectic debris (nuclear dust) in the upper dermis as well as focal vacuolar interface changes with scattered necrotic keratinocytes in the epidermis (Figure 2). Based on these histopathologic findings, a diagnosis of Kikuchi-Fujimoto disease was considered. To confirm the diagnosis and to rule out the possibility of lymphoma, an excisional biopsy of the cervical lymph node was performed, which showed typical histopathologic features of histiocytic necrotizing lymphadenitis.
Given the patient’s clinical presentation with arthralgia, anorexia, lymphadenitis, and hepatosplenomegaly along with histopathologic findings from both the skin and lymph node biopsies, a diagnosis of Kikuchi-Fujimoto disease was made. The patient was conservatively managed with acetaminophen and was discharged with improvement in his appetite and systemic symptoms.
He was seen for follow-up 3 months later in the outpatient clinic. He denied any recurrence of systemic symptoms but endorsed a recent shedding of hair consistent with telogen effluvium. The rash had substantially improved, though residual asymptomatic erythematous plaques remained on the right forehead and right cheek (Figure 3). He was prescribed triamcinolone acetonide cream 0.1% to apply to the active area twice daily for the following 2 to 3 weeks.
Kikuchi-Fujimoto disease presents with a wide clinical spectrum, classically with benign lymphadenopathy and fever of unknown etiology.5,6 Lymphadenopathy most often is cervical (55%–99%)8 and unilateral,4,7 but patients can present with polyadenopathy (52%).7,8 Constitutional signs commonly include fever (35%–76%), weight loss, arthritis (5%–34%), and leukopenia (25%–74%).4,8,9
Cutaneous findings have been described in up to 40% of cases, of which clinical presentation is variable.6 Lesions may include blanchable, erythematous, painful, and/or indurated plaques, nodules, or maculopapules with confluence into patches, urticaria, morbilliform lesions, erythema multiforme, eyelid edema, leukocytoclastic vasculitis, papulopustules, ulcerated gingivae, and mucositis.6,7,10-13 Patients with skin lesions may be at an increased risk for developing systemic lupus erythematosus (SLE).8 Our patient presented with erythematous scaly plaques with a predominance of lesions in photodistributed locations, which clinically mimicked an underlying connective tissue disease process such as SLE.
Infectious agents such as CMV, parvovirus B19, human herpesvirus 6, human herpesvirus 8 and human T-cell lymphotropic virus 1, HIV, Yersinia enterocolitica, and Toxoplasma have all been implicated as possible causes of Kikuchi-Fujimoto disease, but studies have failed to provide convincing causal evidence.9,14,15 Our patient had positive IgM and IgG for CMV, which may have incited his disease.
Definitive diagnosis of Kikuchi-Fujimoto disease is made by lymph node excisional biopsy, which histologically exhibits a histiocytic cell proliferation with paracortical foci of necrosis and abundant karyorrhectic debris.5 Cutaneous histologic findings that support the diagnosis are variable and may include a dermal histiocytic infiltrate, epidermal change with necrotic keratinocytes, non-neutrophilic karyorrhectic debris, basal vacuolar change, papillary dermal edema, a nonspecific superficial and deep perivascular infiltrate, and a patchy infiltration of histiocytes and lymphocytes.6,13
Clinical and histopathological features of this disease can mimic other diseases, specifically SLE or lymphoma.7 An association with SLE has been suspected, though it is not well defined and more frequently is associated with cases from Asia than from Europe (28% and 9%, respectively).9 Patients presenting concomitantly with positive ANA, weight loss, arthralgia, and skin lesions are more likely to develop SLE.8 Furthermore, the cutaneous histologic finding of interface change suggests a link between the two diseases. As such, recommendations have been made for ANA screenings and follow-up of patients diagnosed with Kikuchi-Fujimoto disease for clinical evidence of autoimmune disease, particularly SLE.6 Although our patient did not have a positive ANA, his biopsy did demonstrate interface change, and he should be monitored for possible progression of disease in the future.
Kikuchi-Fujimoto disease differs from lymphoma, as it initially presents with rapid lymph node enlargement as opposed to the gradual enlargement seen in lymphoma. The lymph nodes in Kikuchi-Fujimoto disease often are firm and moveable compared to hard and immobile in lymphoma.3 Excisional lymph node biopsy is necessary for both confirming the diagnosis of Kikuchi-Fujimoto disease and ruling out lymphoma.5
Spontaneous resolution usually occurs in 1 to 4 months.3,6 As such, observation is the most common approach to management. When patients have symptoms that limit activities or cause undue distress such as fevers, joint pains, or abdominal pain, systemic treatment options may be desired. Symptomatic treatment can be managed with a short duration of oral corticosteroids,10,11 nonsteroidal anti-inflammatory drugs, antimalarials, and/or antipyretics.8-15 There are no guidelines regarding systemic steroid regimens, and various treatment schedules have been successful. Systemic therapy was considered for our patient for his weight loss and abdominal pain; however, by the time of discharge the patient was tolerating oral intake and his abdominal pain had improved.
- Kikuchi M. Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytosis. Nippon Ketsueki Gakkai Zasshi. 1972;35:379-380.
- Fujimoto Y, Kojima Y, Yamaguchi K. Cervical subacute necrotizing lymphadenitis: a new clinicopathological entity. Naika. 1972;30:920-927.
- Feder Jr HM, Liu J, Rezuke WN. Kikuchi disease in Connecticut. J Pediatr. 2014;164:196-200.
- Kang HM, Kim JY, Choi EH, et al. Clinical characteristics of severe histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto disease) in children. J Pediatr. 2016;171:208-212.
- Hutchinson CB, Wang E. Kikuchi-Fujimoto disease. Arch Pathol Lab Med. 2010;134:289-293.
- Atwater AR, Longly BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008;59:130-136.
- Yen H-R, Lin P-Y, Chuang W-Y, et al. Skin manifestations of Kikuchi-Fujimoto disease: case report and review. Eur J Pediatr. 2004;163:210-213.
- Dumas G, Prendki V, Haroche J, et al. Kikuchi-Fujimoto disease: retrospective study of 91 cases and review of literature. Medicine. 2014;93:372-382.
- Kuc ukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Yasukawa K, Matsumura T, Sato-Matsumura KC, et al. Kikuchi’s disease and the skin: case report and review of the literature. Br J Dermatol. 2001;144:885-889.
- Kaur S, Thami GP, Mohan H, et al. Kikuchi disease with facial rash and erythema multiforme. Pediatr Dermatol. 2001;18:403-405.
- Mauleón C, Valdivielso-Ramos M, Cabeza R, et al. Kikuchi disease with skin lesions mimicking lupus erythematosus. J Dermatol Case Rep. 2012;3:82-85.
- Obara K, Amoh Y. A case of Kikuchi’s disease (histiocytic necrotizing lymphoadenitis) with histiocytic cutaneous involvement. Rheumatol Int. 2015;35:1111-1113.
- Rosado FGN, Tang Y-W, Hasserjian RP, et al. Kikuchi-Fujimoto lymphadenitis: role of parvovirus B-19, Epstein-Barr virus, human herpesvirus 6, and human herpesvirus 8. Hum Pathol. 2013;44:255-259.
- Chiu CF, Chow KC, Lin TY, et al. Virus infection in patients with histiocytic necrotizing lymphadenitis in Taiwan. detection of Epstein-Barr virus, type I human T-cell lymphotropic virus, and parvovirus B19. Am J Clin Pathol. 2000;113:774-781.
- Kikuchi M. Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytosis. Nippon Ketsueki Gakkai Zasshi. 1972;35:379-380.
- Fujimoto Y, Kojima Y, Yamaguchi K. Cervical subacute necrotizing lymphadenitis: a new clinicopathological entity. Naika. 1972;30:920-927.
- Feder Jr HM, Liu J, Rezuke WN. Kikuchi disease in Connecticut. J Pediatr. 2014;164:196-200.
- Kang HM, Kim JY, Choi EH, et al. Clinical characteristics of severe histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto disease) in children. J Pediatr. 2016;171:208-212.
- Hutchinson CB, Wang E. Kikuchi-Fujimoto disease. Arch Pathol Lab Med. 2010;134:289-293.
- Atwater AR, Longly BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008;59:130-136.
- Yen H-R, Lin P-Y, Chuang W-Y, et al. Skin manifestations of Kikuchi-Fujimoto disease: case report and review. Eur J Pediatr. 2004;163:210-213.
- Dumas G, Prendki V, Haroche J, et al. Kikuchi-Fujimoto disease: retrospective study of 91 cases and review of literature. Medicine. 2014;93:372-382.
- Kuc ukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Yasukawa K, Matsumura T, Sato-Matsumura KC, et al. Kikuchi’s disease and the skin: case report and review of the literature. Br J Dermatol. 2001;144:885-889.
- Kaur S, Thami GP, Mohan H, et al. Kikuchi disease with facial rash and erythema multiforme. Pediatr Dermatol. 2001;18:403-405.
- Mauleón C, Valdivielso-Ramos M, Cabeza R, et al. Kikuchi disease with skin lesions mimicking lupus erythematosus. J Dermatol Case Rep. 2012;3:82-85.
- Obara K, Amoh Y. A case of Kikuchi’s disease (histiocytic necrotizing lymphoadenitis) with histiocytic cutaneous involvement. Rheumatol Int. 2015;35:1111-1113.
- Rosado FGN, Tang Y-W, Hasserjian RP, et al. Kikuchi-Fujimoto lymphadenitis: role of parvovirus B-19, Epstein-Barr virus, human herpesvirus 6, and human herpesvirus 8. Hum Pathol. 2013;44:255-259.
- Chiu CF, Chow KC, Lin TY, et al. Virus infection in patients with histiocytic necrotizing lymphadenitis in Taiwan. detection of Epstein-Barr virus, type I human T-cell lymphotropic virus, and parvovirus B19. Am J Clin Pathol. 2000;113:774-781.
Practice Points
- Kikuchi-Fujimoto disease is an uncommon, self-limited condition characterized by benign lymphadenopathy and variable systemic symptoms.
- Definitive diagnosis is made by excisional lymph node biopsy.
- Treatment options include oral corticosteroids, nonsteroidal anti-inflammatory drugs, antimalarials, and/or antipyretics.
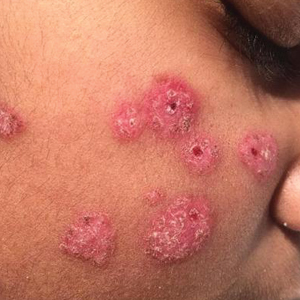
Chronic Hyperpigmented Patches on the Legs
The Diagnosis: Drug-Induced Hyperpigmentation
Additional history provided by the patient’s caretaker elucidated an extensive list of medications including chlorpromazine and minocycline, among several others. The caretaker revealed that the patient began treatment for acne vulgaris 2 years prior; despite the acne resolving, therapy was not discontinued. The blue-gray and brown pigmentation on our patient’s shins likely was attributed to a medication he was taking.
Both chlorpromazine and minocycline, among many other medications, are known to cause abnormal pigmentation of the skin.1 Minocycline is a tetracycline antibiotic prescribed for acne and other inflammatory cutaneous conditions. It is highly lipophilic, allowing it to reach high drug concentrations in the skin and nail unit.2 Patients taking minocycline long term and at high doses are at greatest risk for pigment deposition.3,4
Minocycline-induced hyperpigmentation is classified into 3 types. Type I describes blue-black deposition of pigment in acne scars and areas of inflammation, typically on facial skin.1,5 Histologically, type I stains positive for Perls Prussian blue, indicating an increased deposition of iron as hemosiderin,1 which likely occurs because minocycline is thought to play a role in defective clearance of hemosiderin from the dermis of injured tissue.5 Type II hyperpigmentation presents as bluegray pigment on the lower legs and occasionally the arms.6,7 Type II stains positive for both Perls Prussian blue and Fontana-Masson, demonstrating hemosiderin and melanin, respectively.6 The third form of hyperpigmentation results in diffuse, dark brown to gray pigmentation with a predilection for sun-exposed areas.8 Histology of type III shows increased pigment in the basal portion of the epidermis and brown-black pigment in macrophages of the dermis. Type III stains positive for Fontana-Masson and negative for Perls Prussian blue. The etiology of hyperpigmentation has been suspected to be caused by minocycline stimulating melanin production and/or deposition of minocycline-melanin complexes in dermal macrophages after a certain drug level; this largely is seen in patients receiving 100 to 200 mg daily as early as 1 year into treatment.8
Chlorpromazine is a typical antipsychotic that causes abnormal skin pigmentation in sun-exposed areas due to increased melanogenesis.9 Similar to type III minocyclineinduced hyperpigmentation, a histologic specimen may stain positive for Fontana-Masson yet negative for Perls Prussian blue. Lal et al10 demonstrated complete resolution of abnormal skin pigmentation within 5 years after stopping chlorpromazine. In contrast, minocyclineinduced hyperpigmentation may be permanent in some cases. There is substantial clinical and histologic overlap for drug-induced hyperpigmentation etiologies; it would behoove the clinician to focus on the most common locations affected and the generalized coloration.
Treatment of minocycline-induced hyperpigmentation includes the use of Q-switched lasers, specifically Q-switched ruby and Q-switched alexandrite.11 The use of the Q-switched Nd:YAG laser appears to be ineffective at clearing minocycline-induced pigmentation.7,11 In our patient, minocycline was discontinued immediately. Due to the patient’s critical condition, he deferred all other therapy. Erythema dyschromicum perstans, also referred to as ashy dermatosis, is an idiopathic form of hyperpigmentation.12 Lesions start as blue-gray to ashy gray macules, occasionally surrounded by a slightly erythematous, raised border.
Erythema dyschromicum perstans typically presents on the trunk, face, and arms of patients with Fitzpatrick skin types III and IV; it is considered a variant of lichen planus actinicus.12 Histologically, erythema dyschromicum perstans may mimic lichen planus pigmentosus (LPP); however, subtle differences exist to distinguish the 2 conditions. Erythema dyschromicum perstans demonstrates a mild lichenoid infiltrate, focal basal vacuolization at the dermoepidermal junction, and melanophage deposition.13 In contrast, LPP demonstrates pigmentary incontinence and a more severe inflammatory infiltrate. A perifollicular infiltrate and fibrosis also can be seen in LPP, which may explain the frontal fibrosing alopecia that often precedes LPP.13
Addison disease, also known as primary adrenal insufficiency, can cause diffuse hyperpigmentation in the skin, mucosae, and nail beds. The pigmentation is prominent in regions of naturally increased pigmentation, such as the flexural surfaces and intertriginous areas.14 Patients with adrenal insufficiency will have accompanying weight loss, hypotension, and fatigue, among other symptoms related to deficiency of cortisol and aldosterone. Skin biopsy shows acanthosis, hyperkeratosis, focal parakeratosis, spongiosis, superficial perivascular lymphocytic infiltrate, basal melanin deposition, and superficial dermal macrophages.15
Confluent and reticulated papillomatosis is an uncommon dermatosis that presents with multiple hyperpigmented macules and papules that coalesce to form patches and plaques centrally with reticulation in the periphery.16 Confluent and reticulated papillomatosis commonly presents on the upper trunk, axillae, and neck, though involvement can include flexural surfaces as well as the lower trunk and legs.16,17 Biopsy demonstrates undulating hyperkeratosis, papillomatosis, acanthosis, and negative fungal staining.16
Pretibial myxedema most commonly is associated with Graves disease and presents as well-defined thickening and induration with overlying pink or purple-brown papules in the pretibial region.18 An acral surface and mucin deposition within the entire dermis may be appreciated on histology with staining for colloidal iron or Alcian blue.
- Fenske NA, Millns JL, Greer KE. Minocycline-induced pigmentation at sites of cutaneous inflammation. JAMA. 1980;244:1103-1106. doi:10.1001/jama.1980.03310100021021
- Snodgrass A, Motaparthi K. Systemic antibacterial agents. In: Wolverton SE, Wu JJ, eds. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier; 2020:69-98.
- Eisen D, Hakim MD. Minocycline-induced pigmentation. incidence, prevention and management. Drug Saf. 1998;18:431-440. doi:10.2165/00002018-199818060-00004
- Goulden V, Glass D, Cunliffe WJ. Safety of long-term high-dose minocycline in the treatment of acne. Br J Dermatol. 1996;134:693-695. doi:10.1111/j.1365-2133.1996.tb06972.x
- Basler RS, Kohnen PW. Localized hemosiderosis as a sequela of acne. Arch Dermatol. 1978;114:1695-1697.
- Ridgway HA, Sonnex TS, Kennedy CT, et al. Hyperpigmentation associated with oral minocycline. Br J Dermatol. 1982;107:95-102. doi:10.1111/j.1365-2133.1982.tb00296.x
- Nisar MS, Iyer K, Brodell RT, et al. Minocycline-induced hyperpigmentation: comparison of 3 Q-switched lasers to reverse its effects. Clin Cosmet Investig Dermatol. 2013;6:159-162. doi:10.2147/CCID.S42166
- Simons JJ, Morales A. Minocycline and generalized cutaneous pigmentation. J Am Acad Dermatol. 1980;3:244-247. doi:10.1016/s0190 -9622(80)80186-1
- Perry TL, Culling CF, Berry K, et al. 7-Hydroxychlorpromazine: potential toxic drug metabolite in psychiatric patients. Science. 1964;146:81-83. doi:10.1126/science.146.3640.81
- Lal S, Bloom D, Silver B, et al. Replacement of chlorpromazine with other neuroleptics: effect on abnormal skin pigmentation and ocular changes. J Psychiatry Neurosci. 1993;18:173-177.
- Tsao H, Busam K, Barnhill RL, et al. Treatment of minocycline-induced hyperpigmentation with the Q-switched ruby laser. Arch Dermatol. 1996;132:1250-1251.
- Knox JM, Dodge BG, Freeman RG. Erythema dyschromicum perstans. Arch Dermatol. 1968;97:262-272. doi:10.1001 /archderm.1968.01610090034006
- Rutnin S, Udompanich S, Pratumchart N, et al. Ashy dermatosis and lichen planus pigmentosus: the histopathological differences. Biomed Res Int. 2019;2019:5829185. doi:10.1155/2019/5829185
- Montgomery H, O’Leary PA. Pigmentation of the skin in Addison’s disease, acanthosis nigricans and hemochromatosis. Arch Derm Syphilol. 1930;21:970-984. doi:10.1001 /archderm.1930.01440120072005
- Fernandez-Flores A, Cassarino DS. Histopathologic findings of cutaneous hyperpigmentation in Addison disease and immunostain of the melanocytic population. Am J Dermatopathol. 2017;39:924-927. doi:10.1097/DAD.0000000000000937
- Davis MD, Weenig RH, Camilleri MJ. Confluent and reticulate papillomatosis (Gougerot-Carteaud syndrome): a minocycline-responsive dermatosis without evidence for yeast in pathogenesis. a study of 39 patients and a proposal of diagnostic criteria. Br J Dermatol. 2006;154:287-293. doi:10.1111/j.1365-2133.2005.06955.x
- Jo S, Park HS, Cho S, et al. Updated diagnosis criteria for confluent and reticulated papillomatosis: a case report. Ann Dermatol. 2014; 26:409-410. doi:10.5021/ad.2014.26.3.409
- Lause M, Kamboj A, Fernandez Faith E. Dermatologic manifestations of endocrine disorders. Transl Pediatr. 2017;6:300-312. doi:10.21037 /tp.2017.09.08
The Diagnosis: Drug-Induced Hyperpigmentation
Additional history provided by the patient’s caretaker elucidated an extensive list of medications including chlorpromazine and minocycline, among several others. The caretaker revealed that the patient began treatment for acne vulgaris 2 years prior; despite the acne resolving, therapy was not discontinued. The blue-gray and brown pigmentation on our patient’s shins likely was attributed to a medication he was taking.
Both chlorpromazine and minocycline, among many other medications, are known to cause abnormal pigmentation of the skin.1 Minocycline is a tetracycline antibiotic prescribed for acne and other inflammatory cutaneous conditions. It is highly lipophilic, allowing it to reach high drug concentrations in the skin and nail unit.2 Patients taking minocycline long term and at high doses are at greatest risk for pigment deposition.3,4
Minocycline-induced hyperpigmentation is classified into 3 types. Type I describes blue-black deposition of pigment in acne scars and areas of inflammation, typically on facial skin.1,5 Histologically, type I stains positive for Perls Prussian blue, indicating an increased deposition of iron as hemosiderin,1 which likely occurs because minocycline is thought to play a role in defective clearance of hemosiderin from the dermis of injured tissue.5 Type II hyperpigmentation presents as bluegray pigment on the lower legs and occasionally the arms.6,7 Type II stains positive for both Perls Prussian blue and Fontana-Masson, demonstrating hemosiderin and melanin, respectively.6 The third form of hyperpigmentation results in diffuse, dark brown to gray pigmentation with a predilection for sun-exposed areas.8 Histology of type III shows increased pigment in the basal portion of the epidermis and brown-black pigment in macrophages of the dermis. Type III stains positive for Fontana-Masson and negative for Perls Prussian blue. The etiology of hyperpigmentation has been suspected to be caused by minocycline stimulating melanin production and/or deposition of minocycline-melanin complexes in dermal macrophages after a certain drug level; this largely is seen in patients receiving 100 to 200 mg daily as early as 1 year into treatment.8
Chlorpromazine is a typical antipsychotic that causes abnormal skin pigmentation in sun-exposed areas due to increased melanogenesis.9 Similar to type III minocyclineinduced hyperpigmentation, a histologic specimen may stain positive for Fontana-Masson yet negative for Perls Prussian blue. Lal et al10 demonstrated complete resolution of abnormal skin pigmentation within 5 years after stopping chlorpromazine. In contrast, minocyclineinduced hyperpigmentation may be permanent in some cases. There is substantial clinical and histologic overlap for drug-induced hyperpigmentation etiologies; it would behoove the clinician to focus on the most common locations affected and the generalized coloration.
Treatment of minocycline-induced hyperpigmentation includes the use of Q-switched lasers, specifically Q-switched ruby and Q-switched alexandrite.11 The use of the Q-switched Nd:YAG laser appears to be ineffective at clearing minocycline-induced pigmentation.7,11 In our patient, minocycline was discontinued immediately. Due to the patient’s critical condition, he deferred all other therapy. Erythema dyschromicum perstans, also referred to as ashy dermatosis, is an idiopathic form of hyperpigmentation.12 Lesions start as blue-gray to ashy gray macules, occasionally surrounded by a slightly erythematous, raised border.
Erythema dyschromicum perstans typically presents on the trunk, face, and arms of patients with Fitzpatrick skin types III and IV; it is considered a variant of lichen planus actinicus.12 Histologically, erythema dyschromicum perstans may mimic lichen planus pigmentosus (LPP); however, subtle differences exist to distinguish the 2 conditions. Erythema dyschromicum perstans demonstrates a mild lichenoid infiltrate, focal basal vacuolization at the dermoepidermal junction, and melanophage deposition.13 In contrast, LPP demonstrates pigmentary incontinence and a more severe inflammatory infiltrate. A perifollicular infiltrate and fibrosis also can be seen in LPP, which may explain the frontal fibrosing alopecia that often precedes LPP.13
Addison disease, also known as primary adrenal insufficiency, can cause diffuse hyperpigmentation in the skin, mucosae, and nail beds. The pigmentation is prominent in regions of naturally increased pigmentation, such as the flexural surfaces and intertriginous areas.14 Patients with adrenal insufficiency will have accompanying weight loss, hypotension, and fatigue, among other symptoms related to deficiency of cortisol and aldosterone. Skin biopsy shows acanthosis, hyperkeratosis, focal parakeratosis, spongiosis, superficial perivascular lymphocytic infiltrate, basal melanin deposition, and superficial dermal macrophages.15
Confluent and reticulated papillomatosis is an uncommon dermatosis that presents with multiple hyperpigmented macules and papules that coalesce to form patches and plaques centrally with reticulation in the periphery.16 Confluent and reticulated papillomatosis commonly presents on the upper trunk, axillae, and neck, though involvement can include flexural surfaces as well as the lower trunk and legs.16,17 Biopsy demonstrates undulating hyperkeratosis, papillomatosis, acanthosis, and negative fungal staining.16
Pretibial myxedema most commonly is associated with Graves disease and presents as well-defined thickening and induration with overlying pink or purple-brown papules in the pretibial region.18 An acral surface and mucin deposition within the entire dermis may be appreciated on histology with staining for colloidal iron or Alcian blue.
The Diagnosis: Drug-Induced Hyperpigmentation
Additional history provided by the patient’s caretaker elucidated an extensive list of medications including chlorpromazine and minocycline, among several others. The caretaker revealed that the patient began treatment for acne vulgaris 2 years prior; despite the acne resolving, therapy was not discontinued. The blue-gray and brown pigmentation on our patient’s shins likely was attributed to a medication he was taking.
Both chlorpromazine and minocycline, among many other medications, are known to cause abnormal pigmentation of the skin.1 Minocycline is a tetracycline antibiotic prescribed for acne and other inflammatory cutaneous conditions. It is highly lipophilic, allowing it to reach high drug concentrations in the skin and nail unit.2 Patients taking minocycline long term and at high doses are at greatest risk for pigment deposition.3,4
Minocycline-induced hyperpigmentation is classified into 3 types. Type I describes blue-black deposition of pigment in acne scars and areas of inflammation, typically on facial skin.1,5 Histologically, type I stains positive for Perls Prussian blue, indicating an increased deposition of iron as hemosiderin,1 which likely occurs because minocycline is thought to play a role in defective clearance of hemosiderin from the dermis of injured tissue.5 Type II hyperpigmentation presents as bluegray pigment on the lower legs and occasionally the arms.6,7 Type II stains positive for both Perls Prussian blue and Fontana-Masson, demonstrating hemosiderin and melanin, respectively.6 The third form of hyperpigmentation results in diffuse, dark brown to gray pigmentation with a predilection for sun-exposed areas.8 Histology of type III shows increased pigment in the basal portion of the epidermis and brown-black pigment in macrophages of the dermis. Type III stains positive for Fontana-Masson and negative for Perls Prussian blue. The etiology of hyperpigmentation has been suspected to be caused by minocycline stimulating melanin production and/or deposition of minocycline-melanin complexes in dermal macrophages after a certain drug level; this largely is seen in patients receiving 100 to 200 mg daily as early as 1 year into treatment.8
Chlorpromazine is a typical antipsychotic that causes abnormal skin pigmentation in sun-exposed areas due to increased melanogenesis.9 Similar to type III minocyclineinduced hyperpigmentation, a histologic specimen may stain positive for Fontana-Masson yet negative for Perls Prussian blue. Lal et al10 demonstrated complete resolution of abnormal skin pigmentation within 5 years after stopping chlorpromazine. In contrast, minocyclineinduced hyperpigmentation may be permanent in some cases. There is substantial clinical and histologic overlap for drug-induced hyperpigmentation etiologies; it would behoove the clinician to focus on the most common locations affected and the generalized coloration.
Treatment of minocycline-induced hyperpigmentation includes the use of Q-switched lasers, specifically Q-switched ruby and Q-switched alexandrite.11 The use of the Q-switched Nd:YAG laser appears to be ineffective at clearing minocycline-induced pigmentation.7,11 In our patient, minocycline was discontinued immediately. Due to the patient’s critical condition, he deferred all other therapy. Erythema dyschromicum perstans, also referred to as ashy dermatosis, is an idiopathic form of hyperpigmentation.12 Lesions start as blue-gray to ashy gray macules, occasionally surrounded by a slightly erythematous, raised border.
Erythema dyschromicum perstans typically presents on the trunk, face, and arms of patients with Fitzpatrick skin types III and IV; it is considered a variant of lichen planus actinicus.12 Histologically, erythema dyschromicum perstans may mimic lichen planus pigmentosus (LPP); however, subtle differences exist to distinguish the 2 conditions. Erythema dyschromicum perstans demonstrates a mild lichenoid infiltrate, focal basal vacuolization at the dermoepidermal junction, and melanophage deposition.13 In contrast, LPP demonstrates pigmentary incontinence and a more severe inflammatory infiltrate. A perifollicular infiltrate and fibrosis also can be seen in LPP, which may explain the frontal fibrosing alopecia that often precedes LPP.13
Addison disease, also known as primary adrenal insufficiency, can cause diffuse hyperpigmentation in the skin, mucosae, and nail beds. The pigmentation is prominent in regions of naturally increased pigmentation, such as the flexural surfaces and intertriginous areas.14 Patients with adrenal insufficiency will have accompanying weight loss, hypotension, and fatigue, among other symptoms related to deficiency of cortisol and aldosterone. Skin biopsy shows acanthosis, hyperkeratosis, focal parakeratosis, spongiosis, superficial perivascular lymphocytic infiltrate, basal melanin deposition, and superficial dermal macrophages.15
Confluent and reticulated papillomatosis is an uncommon dermatosis that presents with multiple hyperpigmented macules and papules that coalesce to form patches and plaques centrally with reticulation in the periphery.16 Confluent and reticulated papillomatosis commonly presents on the upper trunk, axillae, and neck, though involvement can include flexural surfaces as well as the lower trunk and legs.16,17 Biopsy demonstrates undulating hyperkeratosis, papillomatosis, acanthosis, and negative fungal staining.16
Pretibial myxedema most commonly is associated with Graves disease and presents as well-defined thickening and induration with overlying pink or purple-brown papules in the pretibial region.18 An acral surface and mucin deposition within the entire dermis may be appreciated on histology with staining for colloidal iron or Alcian blue.
- Fenske NA, Millns JL, Greer KE. Minocycline-induced pigmentation at sites of cutaneous inflammation. JAMA. 1980;244:1103-1106. doi:10.1001/jama.1980.03310100021021
- Snodgrass A, Motaparthi K. Systemic antibacterial agents. In: Wolverton SE, Wu JJ, eds. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier; 2020:69-98.
- Eisen D, Hakim MD. Minocycline-induced pigmentation. incidence, prevention and management. Drug Saf. 1998;18:431-440. doi:10.2165/00002018-199818060-00004
- Goulden V, Glass D, Cunliffe WJ. Safety of long-term high-dose minocycline in the treatment of acne. Br J Dermatol. 1996;134:693-695. doi:10.1111/j.1365-2133.1996.tb06972.x
- Basler RS, Kohnen PW. Localized hemosiderosis as a sequela of acne. Arch Dermatol. 1978;114:1695-1697.
- Ridgway HA, Sonnex TS, Kennedy CT, et al. Hyperpigmentation associated with oral minocycline. Br J Dermatol. 1982;107:95-102. doi:10.1111/j.1365-2133.1982.tb00296.x
- Nisar MS, Iyer K, Brodell RT, et al. Minocycline-induced hyperpigmentation: comparison of 3 Q-switched lasers to reverse its effects. Clin Cosmet Investig Dermatol. 2013;6:159-162. doi:10.2147/CCID.S42166
- Simons JJ, Morales A. Minocycline and generalized cutaneous pigmentation. J Am Acad Dermatol. 1980;3:244-247. doi:10.1016/s0190 -9622(80)80186-1
- Perry TL, Culling CF, Berry K, et al. 7-Hydroxychlorpromazine: potential toxic drug metabolite in psychiatric patients. Science. 1964;146:81-83. doi:10.1126/science.146.3640.81
- Lal S, Bloom D, Silver B, et al. Replacement of chlorpromazine with other neuroleptics: effect on abnormal skin pigmentation and ocular changes. J Psychiatry Neurosci. 1993;18:173-177.
- Tsao H, Busam K, Barnhill RL, et al. Treatment of minocycline-induced hyperpigmentation with the Q-switched ruby laser. Arch Dermatol. 1996;132:1250-1251.
- Knox JM, Dodge BG, Freeman RG. Erythema dyschromicum perstans. Arch Dermatol. 1968;97:262-272. doi:10.1001 /archderm.1968.01610090034006
- Rutnin S, Udompanich S, Pratumchart N, et al. Ashy dermatosis and lichen planus pigmentosus: the histopathological differences. Biomed Res Int. 2019;2019:5829185. doi:10.1155/2019/5829185
- Montgomery H, O’Leary PA. Pigmentation of the skin in Addison’s disease, acanthosis nigricans and hemochromatosis. Arch Derm Syphilol. 1930;21:970-984. doi:10.1001 /archderm.1930.01440120072005
- Fernandez-Flores A, Cassarino DS. Histopathologic findings of cutaneous hyperpigmentation in Addison disease and immunostain of the melanocytic population. Am J Dermatopathol. 2017;39:924-927. doi:10.1097/DAD.0000000000000937
- Davis MD, Weenig RH, Camilleri MJ. Confluent and reticulate papillomatosis (Gougerot-Carteaud syndrome): a minocycline-responsive dermatosis without evidence for yeast in pathogenesis. a study of 39 patients and a proposal of diagnostic criteria. Br J Dermatol. 2006;154:287-293. doi:10.1111/j.1365-2133.2005.06955.x
- Jo S, Park HS, Cho S, et al. Updated diagnosis criteria for confluent and reticulated papillomatosis: a case report. Ann Dermatol. 2014; 26:409-410. doi:10.5021/ad.2014.26.3.409
- Lause M, Kamboj A, Fernandez Faith E. Dermatologic manifestations of endocrine disorders. Transl Pediatr. 2017;6:300-312. doi:10.21037 /tp.2017.09.08
- Fenske NA, Millns JL, Greer KE. Minocycline-induced pigmentation at sites of cutaneous inflammation. JAMA. 1980;244:1103-1106. doi:10.1001/jama.1980.03310100021021
- Snodgrass A, Motaparthi K. Systemic antibacterial agents. In: Wolverton SE, Wu JJ, eds. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier; 2020:69-98.
- Eisen D, Hakim MD. Minocycline-induced pigmentation. incidence, prevention and management. Drug Saf. 1998;18:431-440. doi:10.2165/00002018-199818060-00004
- Goulden V, Glass D, Cunliffe WJ. Safety of long-term high-dose minocycline in the treatment of acne. Br J Dermatol. 1996;134:693-695. doi:10.1111/j.1365-2133.1996.tb06972.x
- Basler RS, Kohnen PW. Localized hemosiderosis as a sequela of acne. Arch Dermatol. 1978;114:1695-1697.
- Ridgway HA, Sonnex TS, Kennedy CT, et al. Hyperpigmentation associated with oral minocycline. Br J Dermatol. 1982;107:95-102. doi:10.1111/j.1365-2133.1982.tb00296.x
- Nisar MS, Iyer K, Brodell RT, et al. Minocycline-induced hyperpigmentation: comparison of 3 Q-switched lasers to reverse its effects. Clin Cosmet Investig Dermatol. 2013;6:159-162. doi:10.2147/CCID.S42166
- Simons JJ, Morales A. Minocycline and generalized cutaneous pigmentation. J Am Acad Dermatol. 1980;3:244-247. doi:10.1016/s0190 -9622(80)80186-1
- Perry TL, Culling CF, Berry K, et al. 7-Hydroxychlorpromazine: potential toxic drug metabolite in psychiatric patients. Science. 1964;146:81-83. doi:10.1126/science.146.3640.81
- Lal S, Bloom D, Silver B, et al. Replacement of chlorpromazine with other neuroleptics: effect on abnormal skin pigmentation and ocular changes. J Psychiatry Neurosci. 1993;18:173-177.
- Tsao H, Busam K, Barnhill RL, et al. Treatment of minocycline-induced hyperpigmentation with the Q-switched ruby laser. Arch Dermatol. 1996;132:1250-1251.
- Knox JM, Dodge BG, Freeman RG. Erythema dyschromicum perstans. Arch Dermatol. 1968;97:262-272. doi:10.1001 /archderm.1968.01610090034006
- Rutnin S, Udompanich S, Pratumchart N, et al. Ashy dermatosis and lichen planus pigmentosus: the histopathological differences. Biomed Res Int. 2019;2019:5829185. doi:10.1155/2019/5829185
- Montgomery H, O’Leary PA. Pigmentation of the skin in Addison’s disease, acanthosis nigricans and hemochromatosis. Arch Derm Syphilol. 1930;21:970-984. doi:10.1001 /archderm.1930.01440120072005
- Fernandez-Flores A, Cassarino DS. Histopathologic findings of cutaneous hyperpigmentation in Addison disease and immunostain of the melanocytic population. Am J Dermatopathol. 2017;39:924-927. doi:10.1097/DAD.0000000000000937
- Davis MD, Weenig RH, Camilleri MJ. Confluent and reticulate papillomatosis (Gougerot-Carteaud syndrome): a minocycline-responsive dermatosis without evidence for yeast in pathogenesis. a study of 39 patients and a proposal of diagnostic criteria. Br J Dermatol. 2006;154:287-293. doi:10.1111/j.1365-2133.2005.06955.x
- Jo S, Park HS, Cho S, et al. Updated diagnosis criteria for confluent and reticulated papillomatosis: a case report. Ann Dermatol. 2014; 26:409-410. doi:10.5021/ad.2014.26.3.409
- Lause M, Kamboj A, Fernandez Faith E. Dermatologic manifestations of endocrine disorders. Transl Pediatr. 2017;6:300-312. doi:10.21037 /tp.2017.09.08
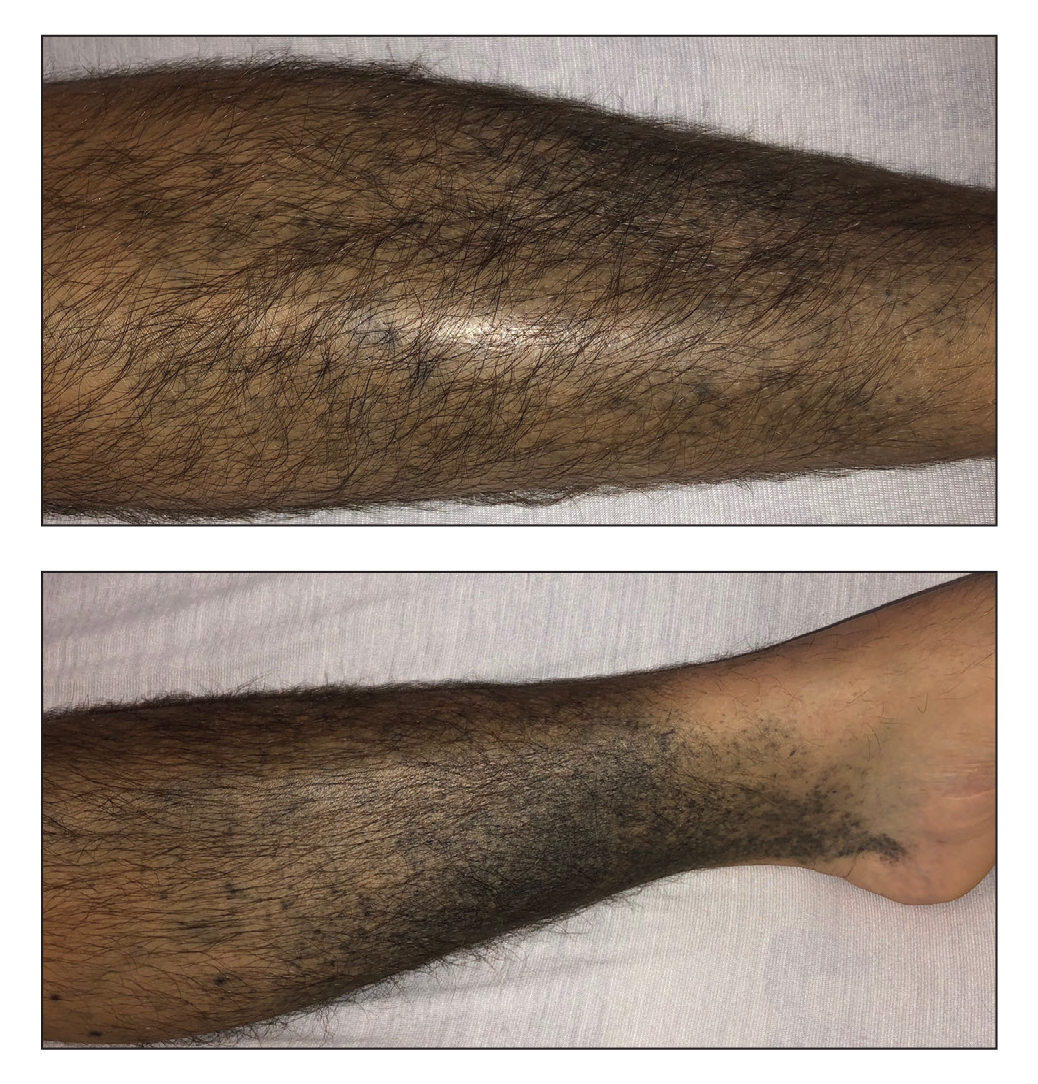
A 37-year-old man with a history of cerebral palsy, bipolar disorder, and impulse control disorder presented to the emergency department with breathing difficulty and worsening malaise. The patient subsequently was intubated due to hypoxic respiratory failure and was found to be positive for SARS-CoV-2. He was admitted to the intensive care unit, and dermatology was consulted due to concern that the cutaneous findings were demonstrative of a vasculitic process. Physical examination revealed diffuse, symmetric, dark brown to blue-gray macules coalescing into patches on the anterior tibia (top) and covering the entire lower leg (bottom). The patches were mottled and did not blanch with pressure. According to the patient’s caretaker, the leg hyperpigmentation had been present for 2 years.
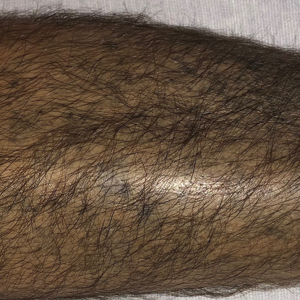
Pedunculated Tumor on the Posterior Neck
The Diagnosis: Nodular Hidradenoma
A biopsy of the nodule showed a large, fungating, well-circumscribed, multilobulated neoplasm composed of primarily monotonous eosinophilic cells in a background of keloidal stroma (Figure). There was a minority population of small, monotonous, clear cells within the lobules, and no glandular structures were noted. Neither cytological nor architectural atypia were evident. MART-1/Melan-A and S-100 stains were negative, consistent with a diagnosis of benign nodular hidradenoma.
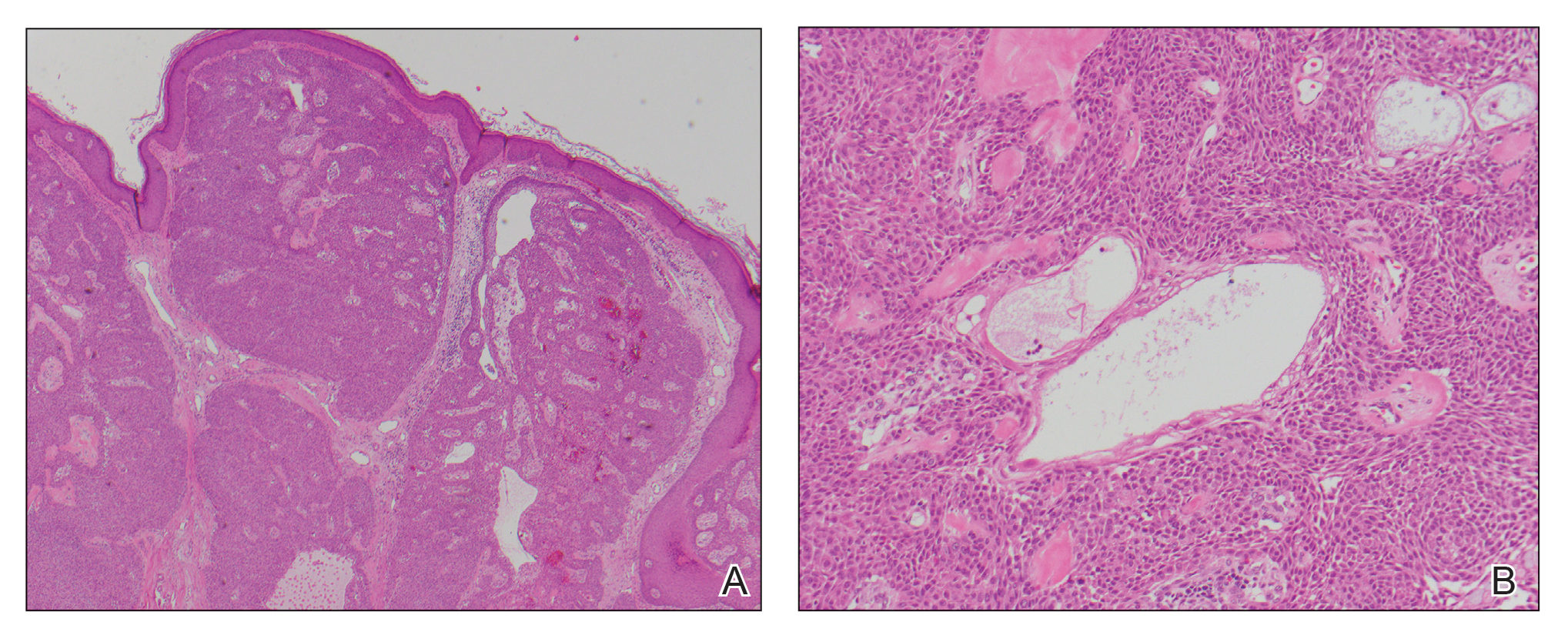
Nodular hidradenoma (also known as acrospiroma, solid-cystic hidradenoma, clear cell hidradenoma, and eccrine sweat gland adenoma) is a benign adnexal tumor of the apocrine or eccrine glands.1,2 Nodular hidradenoma can arise at any cutaneous site but most commonly arises on the head and anterior portion of the trunk and rarely on the extremities.2 It presents as a solitary nodular, cystic, or pedunculated mass that can reach up to several centimeters in diameter.2,3 Nodular hidradenoma more commonly affects women compared to men with a ratio of 1.7 to 1 and commonly presents between the third and fifth decades of life, with an average age at presentation of 37.2 years.2,4 There can be associated skin changes, including smoothening, thickening, ulceration, and bluish discoloration. Dermoscopy commonly shows a pinkish homogenous area that extends throughout the entire lesion. This homogenous area less commonly can be bluish, brownish, or pink-blue. Most nodular hidradenomas also can exhibit vascularization, with arborizing telangiectases, polymorphous atypical vessels, and linear irregular vessels being most common; however, this is not specific to nodular hidradenoma.3 Occasionally, tumors can drain serous or hemorrhagic fluid. Nodular hidradenoma commonly is a slow-growing tumor.5 Rapid increase in tumor size can be indicative of malignant transformation, hemorrhage into the tumor, or trauma to the area.2
Histologically, nodular hidradenoma consists of a circumscribed, nonencapsulated, multilobular tumor commonly found in the dermis and sometimes extending into the subcutaneous tissue. There usually is no epidermal attachment, and the overlying epidermis largely is normal. The tumor consists of large multilobulated areas of epithelial cells, tubular lamina, and large cystic areas filled with homogenous eosinophilic material.1 It notably is composed of 2 epithelial cell types: (1) fusiform cells with elongated vesicular nuclei and basophilic cytoplasm, and (2) large polygonal cells with round eccentric nuclei and eosinophilic, periodic acid–Schiff–positive cytoplasm that washes away during fixation, giving the appearance of clear cells.5 Both types of cells are small, monotonous, and void of mitosis or dyskeratosis. Although there can be ducts with apocrine secretion present within the lobulated tumor, they are not consistently found. Due to the varying features that are neither mandatory nor consistent to arrive at this diagnosis, some dermatopathologists view the term hidradenoma as a catch-all term that includes several different types of benign sweat gland tumors. Some authors divide the terminology into apocrine hidradenoma and eccrine hidradenoma based on whether the tumor is composed of solely clear mucinous cells, or poroid and cuticular cells, respectively.
Although nodular hidradenoma classically is a benign tumor, total surgical excision is recommended due to the rare risk for malignant transformation. Rarely, longstanding hidradenomas can metastasize to lymph nodes, bone, or viscera; in these instances, metastatic hidradenoma has a 5-year survival rate of 30%. Recurrence may occur in tumors that are inadequately excised, and the rate of recurrence is estimated to be approximately 10% of surgically excised tumors.5 However, utilization of Mohs micrographic surgery for excision of nodular hidradenoma is associated with a reduced recurrence rate.6
Keloids present as painful, sometimes pruritic, raised scars that extend beyond the boundary of the initial injury, commonly arising on the shoulder, upper arm, and chest. Histopathology reveals nodules of thick hyalinized collagen bundles, keloidal collagen with mucinous ground substance, and few fibroblasts.7
Metastatic renal cell carcinoma to the skin most commonly presents on the face and scalp as a nodular, rapidly growing, round to oval lesion that is flesh colored to reddish purple in a patient with history of renal cell carcinoma.8 Histopathology shows clusters of atypical, nucleated clear cells surrounded by chicken wire vasculature.8,9
Verruca vulgaris is caused by human papillomavirus and most commonly occurs on the hands and feet. It presents as a pink to white, sessile lesion with a verrucous surface and exophytic growths. Histopathology shows acanthosis; hypergranulosis; exophytic projections with a fibrovascular core; inward cupping of the rete ridges; and koilocytes, which are cells with an eccentric, raisinlike nucleus and vacuolated cytoplasm in the granular layer of the epidermis.10
Similar to nodular hidradenoma, nodular melanoma most commonly presents on the head and neck as a symmetric, elevated, amelanotic nodule, but in contrast to nodular hidradenoma, it typically is confined to a smaller diameter.11 Histologically, it is characterized by sheets of atypical, commonly epithelioid melanocytes with a lack of maturation and brisk mitotic activity extending through the epidermis and dermis with lateral extension limited to less than 3 rete ridges.12
- Patterson JW, Weedon D. Tumors of cutaneous appendages. In: Patterson JW, Weedon D. Weedon’s Skin Pathology. 5th ed. Elsevier; 2020:951-1016.
- Ngo N, Susa M, Nakagawa T, et al. Malignant transformation of nodular hidradenoma in the lower leg. Case Rep Oncol. 2018;11:298-304. doi:10.1159/000489255
- Zaballos P, Gómez-Martín I, Martin JM, et al. Dermoscopy of adnexal tumors. Dermatol Clin. 2018;36:397-412. doi:10.1016/j .det.2018.05.007
- Hernández-Pérez E, Cestoni-Parducci R. Nodular hidradenoma and hidradenocarcinoma: a 10-year review. J Am Acad Dermatol. 1985; 12:15-20. doi:10.1016/s0190-9622(85)70002-3
- Stratigos AJ, Olbricht S, Kwan TH, et al. Nodular hidradenoma. Dermatol Surg. 1998;24:387-391. doi:10.1111/j.1524-4725.1998.tb04173.x
- Yavel R, Hinshaw M, Rao V, et al. Hidradenomas and a hidradenocarcinoma of the scalp managed using Mohs micrographic surgery and a multidisciplinary approach. Dermatol Surg. 2009;35:273-281. doi:10.1111/j.1524-4725.2008.34424.x
- Lee JY-Y, Yang C-C, Chao S-C, et al. Histopathological differential diagnosis of keloid and hypertrophic scar. Am J Dermatopathol. 2004;26:379-384. doi:10.1097/00000372-200410000-00006
- Ferhatoglu MF, Senol K, Filiz AI. Skin metastasis of renal cell carcinoma: a case report. Cureus. 2018;10:E3614. doi:10.7759/cureus.3614
- Jaitly V, Jahan-Tigh R, Belousova T, et al. Case report and literature review of nodular hidradenoma, a rare adnexal tumor that mimics breast carcinoma, in a 20-year-old woman. Lab Med. 2019;50:320-325. doi:10.1093/labmed/lmy084
- Betz SJ. HPV-related papillary lesions of the oral mucosa: a review. Head Neck Pathol. 2019;13:80-90. doi:10.1007/s12105-019-01003-7
- Kalkhoran S, Milne O, Zalaudek I, et al. Historical, clinical, and dermoscopic characteristics of thin nodular melanoma. Arch Dermatol. 2010;146:311-318. doi:10.1001/archdermatol.2009.369
- Smoller BR. Histologic criteria for diagnosing primary cutaneous malignant melanoma. Mod Pathol. 2006;19(suppl 2):S34-S40. doi:10.1038 /modpathol.3800508
The Diagnosis: Nodular Hidradenoma
A biopsy of the nodule showed a large, fungating, well-circumscribed, multilobulated neoplasm composed of primarily monotonous eosinophilic cells in a background of keloidal stroma (Figure). There was a minority population of small, monotonous, clear cells within the lobules, and no glandular structures were noted. Neither cytological nor architectural atypia were evident. MART-1/Melan-A and S-100 stains were negative, consistent with a diagnosis of benign nodular hidradenoma.
Nodular hidradenoma (also known as acrospiroma, solid-cystic hidradenoma, clear cell hidradenoma, and eccrine sweat gland adenoma) is a benign adnexal tumor of the apocrine or eccrine glands.1,2 Nodular hidradenoma can arise at any cutaneous site but most commonly arises on the head and anterior portion of the trunk and rarely on the extremities.2 It presents as a solitary nodular, cystic, or pedunculated mass that can reach up to several centimeters in diameter.2,3 Nodular hidradenoma more commonly affects women compared to men with a ratio of 1.7 to 1 and commonly presents between the third and fifth decades of life, with an average age at presentation of 37.2 years.2,4 There can be associated skin changes, including smoothening, thickening, ulceration, and bluish discoloration. Dermoscopy commonly shows a pinkish homogenous area that extends throughout the entire lesion. This homogenous area less commonly can be bluish, brownish, or pink-blue. Most nodular hidradenomas also can exhibit vascularization, with arborizing telangiectases, polymorphous atypical vessels, and linear irregular vessels being most common; however, this is not specific to nodular hidradenoma.3 Occasionally, tumors can drain serous or hemorrhagic fluid. Nodular hidradenoma commonly is a slow-growing tumor.5 Rapid increase in tumor size can be indicative of malignant transformation, hemorrhage into the tumor, or trauma to the area.2
Histologically, nodular hidradenoma consists of a circumscribed, nonencapsulated, multilobular tumor commonly found in the dermis and sometimes extending into the subcutaneous tissue. There usually is no epidermal attachment, and the overlying epidermis largely is normal. The tumor consists of large multilobulated areas of epithelial cells, tubular lamina, and large cystic areas filled with homogenous eosinophilic material.1 It notably is composed of 2 epithelial cell types: (1) fusiform cells with elongated vesicular nuclei and basophilic cytoplasm, and (2) large polygonal cells with round eccentric nuclei and eosinophilic, periodic acid–Schiff–positive cytoplasm that washes away during fixation, giving the appearance of clear cells.5 Both types of cells are small, monotonous, and void of mitosis or dyskeratosis. Although there can be ducts with apocrine secretion present within the lobulated tumor, they are not consistently found. Due to the varying features that are neither mandatory nor consistent to arrive at this diagnosis, some dermatopathologists view the term hidradenoma as a catch-all term that includes several different types of benign sweat gland tumors. Some authors divide the terminology into apocrine hidradenoma and eccrine hidradenoma based on whether the tumor is composed of solely clear mucinous cells, or poroid and cuticular cells, respectively.
Although nodular hidradenoma classically is a benign tumor, total surgical excision is recommended due to the rare risk for malignant transformation. Rarely, longstanding hidradenomas can metastasize to lymph nodes, bone, or viscera; in these instances, metastatic hidradenoma has a 5-year survival rate of 30%. Recurrence may occur in tumors that are inadequately excised, and the rate of recurrence is estimated to be approximately 10% of surgically excised tumors.5 However, utilization of Mohs micrographic surgery for excision of nodular hidradenoma is associated with a reduced recurrence rate.6
Keloids present as painful, sometimes pruritic, raised scars that extend beyond the boundary of the initial injury, commonly arising on the shoulder, upper arm, and chest. Histopathology reveals nodules of thick hyalinized collagen bundles, keloidal collagen with mucinous ground substance, and few fibroblasts.7
Metastatic renal cell carcinoma to the skin most commonly presents on the face and scalp as a nodular, rapidly growing, round to oval lesion that is flesh colored to reddish purple in a patient with history of renal cell carcinoma.8 Histopathology shows clusters of atypical, nucleated clear cells surrounded by chicken wire vasculature.8,9
Verruca vulgaris is caused by human papillomavirus and most commonly occurs on the hands and feet. It presents as a pink to white, sessile lesion with a verrucous surface and exophytic growths. Histopathology shows acanthosis; hypergranulosis; exophytic projections with a fibrovascular core; inward cupping of the rete ridges; and koilocytes, which are cells with an eccentric, raisinlike nucleus and vacuolated cytoplasm in the granular layer of the epidermis.10
Similar to nodular hidradenoma, nodular melanoma most commonly presents on the head and neck as a symmetric, elevated, amelanotic nodule, but in contrast to nodular hidradenoma, it typically is confined to a smaller diameter.11 Histologically, it is characterized by sheets of atypical, commonly epithelioid melanocytes with a lack of maturation and brisk mitotic activity extending through the epidermis and dermis with lateral extension limited to less than 3 rete ridges.12
The Diagnosis: Nodular Hidradenoma
A biopsy of the nodule showed a large, fungating, well-circumscribed, multilobulated neoplasm composed of primarily monotonous eosinophilic cells in a background of keloidal stroma (Figure). There was a minority population of small, monotonous, clear cells within the lobules, and no glandular structures were noted. Neither cytological nor architectural atypia were evident. MART-1/Melan-A and S-100 stains were negative, consistent with a diagnosis of benign nodular hidradenoma.
Nodular hidradenoma (also known as acrospiroma, solid-cystic hidradenoma, clear cell hidradenoma, and eccrine sweat gland adenoma) is a benign adnexal tumor of the apocrine or eccrine glands.1,2 Nodular hidradenoma can arise at any cutaneous site but most commonly arises on the head and anterior portion of the trunk and rarely on the extremities.2 It presents as a solitary nodular, cystic, or pedunculated mass that can reach up to several centimeters in diameter.2,3 Nodular hidradenoma more commonly affects women compared to men with a ratio of 1.7 to 1 and commonly presents between the third and fifth decades of life, with an average age at presentation of 37.2 years.2,4 There can be associated skin changes, including smoothening, thickening, ulceration, and bluish discoloration. Dermoscopy commonly shows a pinkish homogenous area that extends throughout the entire lesion. This homogenous area less commonly can be bluish, brownish, or pink-blue. Most nodular hidradenomas also can exhibit vascularization, with arborizing telangiectases, polymorphous atypical vessels, and linear irregular vessels being most common; however, this is not specific to nodular hidradenoma.3 Occasionally, tumors can drain serous or hemorrhagic fluid. Nodular hidradenoma commonly is a slow-growing tumor.5 Rapid increase in tumor size can be indicative of malignant transformation, hemorrhage into the tumor, or trauma to the area.2
Histologically, nodular hidradenoma consists of a circumscribed, nonencapsulated, multilobular tumor commonly found in the dermis and sometimes extending into the subcutaneous tissue. There usually is no epidermal attachment, and the overlying epidermis largely is normal. The tumor consists of large multilobulated areas of epithelial cells, tubular lamina, and large cystic areas filled with homogenous eosinophilic material.1 It notably is composed of 2 epithelial cell types: (1) fusiform cells with elongated vesicular nuclei and basophilic cytoplasm, and (2) large polygonal cells with round eccentric nuclei and eosinophilic, periodic acid–Schiff–positive cytoplasm that washes away during fixation, giving the appearance of clear cells.5 Both types of cells are small, monotonous, and void of mitosis or dyskeratosis. Although there can be ducts with apocrine secretion present within the lobulated tumor, they are not consistently found. Due to the varying features that are neither mandatory nor consistent to arrive at this diagnosis, some dermatopathologists view the term hidradenoma as a catch-all term that includes several different types of benign sweat gland tumors. Some authors divide the terminology into apocrine hidradenoma and eccrine hidradenoma based on whether the tumor is composed of solely clear mucinous cells, or poroid and cuticular cells, respectively.
Although nodular hidradenoma classically is a benign tumor, total surgical excision is recommended due to the rare risk for malignant transformation. Rarely, longstanding hidradenomas can metastasize to lymph nodes, bone, or viscera; in these instances, metastatic hidradenoma has a 5-year survival rate of 30%. Recurrence may occur in tumors that are inadequately excised, and the rate of recurrence is estimated to be approximately 10% of surgically excised tumors.5 However, utilization of Mohs micrographic surgery for excision of nodular hidradenoma is associated with a reduced recurrence rate.6
Keloids present as painful, sometimes pruritic, raised scars that extend beyond the boundary of the initial injury, commonly arising on the shoulder, upper arm, and chest. Histopathology reveals nodules of thick hyalinized collagen bundles, keloidal collagen with mucinous ground substance, and few fibroblasts.7
Metastatic renal cell carcinoma to the skin most commonly presents on the face and scalp as a nodular, rapidly growing, round to oval lesion that is flesh colored to reddish purple in a patient with history of renal cell carcinoma.8 Histopathology shows clusters of atypical, nucleated clear cells surrounded by chicken wire vasculature.8,9
Verruca vulgaris is caused by human papillomavirus and most commonly occurs on the hands and feet. It presents as a pink to white, sessile lesion with a verrucous surface and exophytic growths. Histopathology shows acanthosis; hypergranulosis; exophytic projections with a fibrovascular core; inward cupping of the rete ridges; and koilocytes, which are cells with an eccentric, raisinlike nucleus and vacuolated cytoplasm in the granular layer of the epidermis.10
Similar to nodular hidradenoma, nodular melanoma most commonly presents on the head and neck as a symmetric, elevated, amelanotic nodule, but in contrast to nodular hidradenoma, it typically is confined to a smaller diameter.11 Histologically, it is characterized by sheets of atypical, commonly epithelioid melanocytes with a lack of maturation and brisk mitotic activity extending through the epidermis and dermis with lateral extension limited to less than 3 rete ridges.12
- Patterson JW, Weedon D. Tumors of cutaneous appendages. In: Patterson JW, Weedon D. Weedon’s Skin Pathology. 5th ed. Elsevier; 2020:951-1016.
- Ngo N, Susa M, Nakagawa T, et al. Malignant transformation of nodular hidradenoma in the lower leg. Case Rep Oncol. 2018;11:298-304. doi:10.1159/000489255
- Zaballos P, Gómez-Martín I, Martin JM, et al. Dermoscopy of adnexal tumors. Dermatol Clin. 2018;36:397-412. doi:10.1016/j .det.2018.05.007
- Hernández-Pérez E, Cestoni-Parducci R. Nodular hidradenoma and hidradenocarcinoma: a 10-year review. J Am Acad Dermatol. 1985; 12:15-20. doi:10.1016/s0190-9622(85)70002-3
- Stratigos AJ, Olbricht S, Kwan TH, et al. Nodular hidradenoma. Dermatol Surg. 1998;24:387-391. doi:10.1111/j.1524-4725.1998.tb04173.x
- Yavel R, Hinshaw M, Rao V, et al. Hidradenomas and a hidradenocarcinoma of the scalp managed using Mohs micrographic surgery and a multidisciplinary approach. Dermatol Surg. 2009;35:273-281. doi:10.1111/j.1524-4725.2008.34424.x
- Lee JY-Y, Yang C-C, Chao S-C, et al. Histopathological differential diagnosis of keloid and hypertrophic scar. Am J Dermatopathol. 2004;26:379-384. doi:10.1097/00000372-200410000-00006
- Ferhatoglu MF, Senol K, Filiz AI. Skin metastasis of renal cell carcinoma: a case report. Cureus. 2018;10:E3614. doi:10.7759/cureus.3614
- Jaitly V, Jahan-Tigh R, Belousova T, et al. Case report and literature review of nodular hidradenoma, a rare adnexal tumor that mimics breast carcinoma, in a 20-year-old woman. Lab Med. 2019;50:320-325. doi:10.1093/labmed/lmy084
- Betz SJ. HPV-related papillary lesions of the oral mucosa: a review. Head Neck Pathol. 2019;13:80-90. doi:10.1007/s12105-019-01003-7
- Kalkhoran S, Milne O, Zalaudek I, et al. Historical, clinical, and dermoscopic characteristics of thin nodular melanoma. Arch Dermatol. 2010;146:311-318. doi:10.1001/archdermatol.2009.369
- Smoller BR. Histologic criteria for diagnosing primary cutaneous malignant melanoma. Mod Pathol. 2006;19(suppl 2):S34-S40. doi:10.1038 /modpathol.3800508
- Patterson JW, Weedon D. Tumors of cutaneous appendages. In: Patterson JW, Weedon D. Weedon’s Skin Pathology. 5th ed. Elsevier; 2020:951-1016.
- Ngo N, Susa M, Nakagawa T, et al. Malignant transformation of nodular hidradenoma in the lower leg. Case Rep Oncol. 2018;11:298-304. doi:10.1159/000489255
- Zaballos P, Gómez-Martín I, Martin JM, et al. Dermoscopy of adnexal tumors. Dermatol Clin. 2018;36:397-412. doi:10.1016/j .det.2018.05.007
- Hernández-Pérez E, Cestoni-Parducci R. Nodular hidradenoma and hidradenocarcinoma: a 10-year review. J Am Acad Dermatol. 1985; 12:15-20. doi:10.1016/s0190-9622(85)70002-3
- Stratigos AJ, Olbricht S, Kwan TH, et al. Nodular hidradenoma. Dermatol Surg. 1998;24:387-391. doi:10.1111/j.1524-4725.1998.tb04173.x
- Yavel R, Hinshaw M, Rao V, et al. Hidradenomas and a hidradenocarcinoma of the scalp managed using Mohs micrographic surgery and a multidisciplinary approach. Dermatol Surg. 2009;35:273-281. doi:10.1111/j.1524-4725.2008.34424.x
- Lee JY-Y, Yang C-C, Chao S-C, et al. Histopathological differential diagnosis of keloid and hypertrophic scar. Am J Dermatopathol. 2004;26:379-384. doi:10.1097/00000372-200410000-00006
- Ferhatoglu MF, Senol K, Filiz AI. Skin metastasis of renal cell carcinoma: a case report. Cureus. 2018;10:E3614. doi:10.7759/cureus.3614
- Jaitly V, Jahan-Tigh R, Belousova T, et al. Case report and literature review of nodular hidradenoma, a rare adnexal tumor that mimics breast carcinoma, in a 20-year-old woman. Lab Med. 2019;50:320-325. doi:10.1093/labmed/lmy084
- Betz SJ. HPV-related papillary lesions of the oral mucosa: a review. Head Neck Pathol. 2019;13:80-90. doi:10.1007/s12105-019-01003-7
- Kalkhoran S, Milne O, Zalaudek I, et al. Historical, clinical, and dermoscopic characteristics of thin nodular melanoma. Arch Dermatol. 2010;146:311-318. doi:10.1001/archdermatol.2009.369
- Smoller BR. Histologic criteria for diagnosing primary cutaneous malignant melanoma. Mod Pathol. 2006;19(suppl 2):S34-S40. doi:10.1038 /modpathol.3800508

A 56-year-old man presented with a progressively enlarging lesion on the posterior neck of 8 months’ duration. He reported localized pruritus of the lesion that improved with triamcinolone cream 0.05% and oral hydroxyzine as well as occasional irritation of the mass with oozing of clear fluid and blood. He denied associated pain and constitutional symptoms. Physical examination revealed a 2.5-cm, nodular, pedunculated, rubbery mass with foci of crusting on the central posterior neck. The mass was flesh colored to pink, and no lymphadenopathy was noted on physical examination.
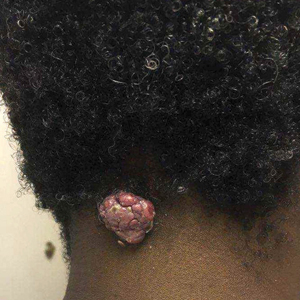
Botulinum Toxin for the Treatment of Intractable Raynaud Phenomenon
To the Editor:
Raynaud phenomenon (RP) is an episodic vasospasm of the digits that can lead to ulceration, gangrene, and autoamputation with prolonged ischemia. OnabotulinumtoxinA has been implemented as a treatment of intractable RP by paralyzing the muscles of the digital arteries. We report a case of a woman with severe RP secondary to systemic lupus erythematosus (SLE) who was treated with onabotulinumtoxinA injections after multiple treatment modalities failed to improve her condition. We describe the dosage and injection technique used to produce clinical improvement in our patient and compare it to prior reports in the literature.
A 33-year-old woman presented to the emergency department for worsening foot pain of 5 days' duration with dusky purple color changes concerning for impending Raynaud crisis related to RP. The patient had a history of antiphospholipid antibody syndrome (APS) and SLE with overlapping symptoms of polymyositis and scleroderma. She had been hospitalized for RP multiple times prior to the current admission. She was medically managed with nifedipine, sildenafil, losartan potassium, aspirin, alprostadil, and prostaglandin infusions, and was surgically managed with a right-hand sympathectomy and right ulnar artery bypass graft that had subsequently thrombosed. At the current presentation, she had painful dusky toes on both feet though more pronounced on the left foot. She endorsed foot pain while walking and tenderness to palpation of the fingers, which were minimally improved with intravenous prostaglandins.
Physical examination revealed blanching of the digits in both hands with pits in the right fourth and left first digits. Dusky patches overlaid all the toes as well as the superior plantar aspects of the feet (Figure 1). Given the history of APS, a punch biopsy was performed on the left medial plantar foot and results showed no histologic evidence of vasculitis or vasculopathy. Necrotic foci were present on the left and right second metatarsal bones, which were not reperfusable (Figure 2). The clinical findings and punch biopsy results favored RP as opposed to vasculopathy from APS.
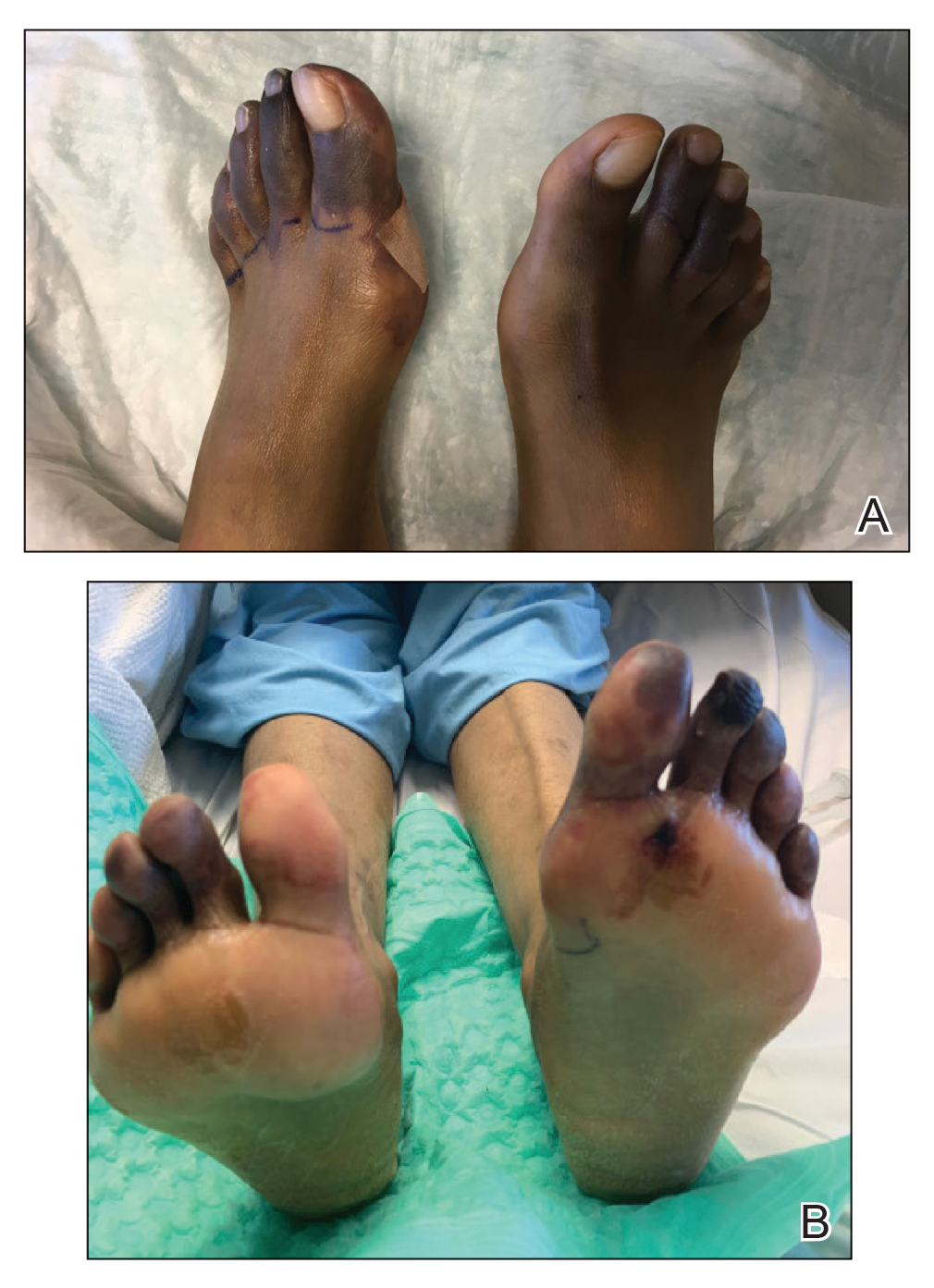
Several interventions were attempted, and after 4 days with no response, the patient agreed to receive treatment with onabotulinumtoxinA. OnabotulinumtoxinA (5 U) was injected into the subcutaneous tissue of the medial and lateral aspects of each of the first and second toes near the proximal phalanges (40 U total). However, treatment could not be completed due to severe pain caused by the injections despite preprocedure regional nerve blocks to both lower extremities, preinjection icing, and lorazepam. Two days later, the patient tolerated onabotulinumtoxinA injections of all remaining digits of both feet (60 U total). She noted slight clinical improvement soon thereafter. One week after treatment of all 10 toes, she reported decreased pain and reduced duskiness of both feet (Figure 3).
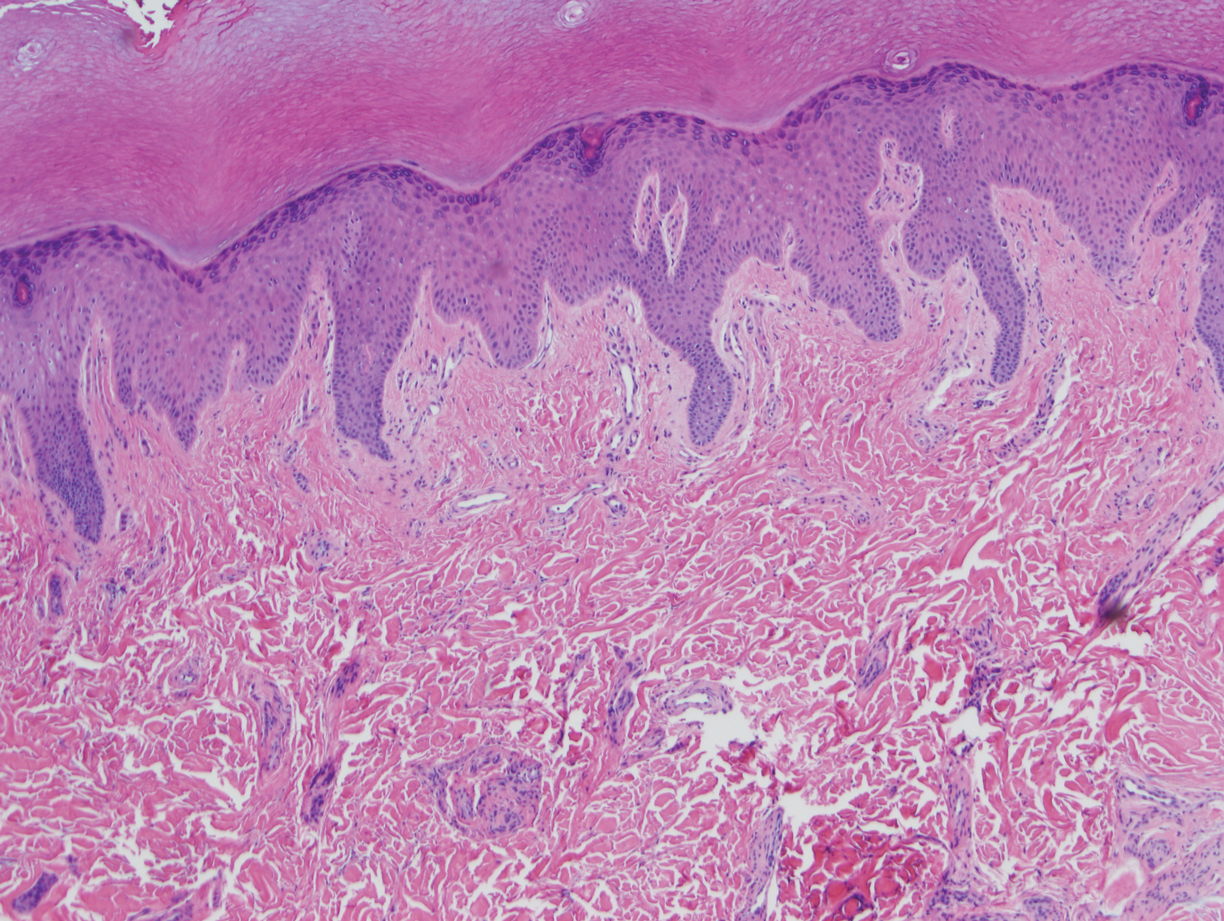
One month later, the patient endorsed recurring pain in the hands and feet. Physical examination revealed reticular cyanosis and increased violaceous patches of the hands; the feet were overall unchanged from the prior hospitalization. At 4-month follow-up, there was gangrene on the left second, third, and fifth toe in addition to areas of induration noted on the fingers. She was repeatedly hospitalized over the next 6 months for pain management and gangrene of the toes, and finally underwent an amputation of the left and right second toe at the proximal and middle phalanx, respectively. She currently is continuing extensive medical management for pain and gangrene of the digits; she has not received additional onabotulinumtoxinA injections.
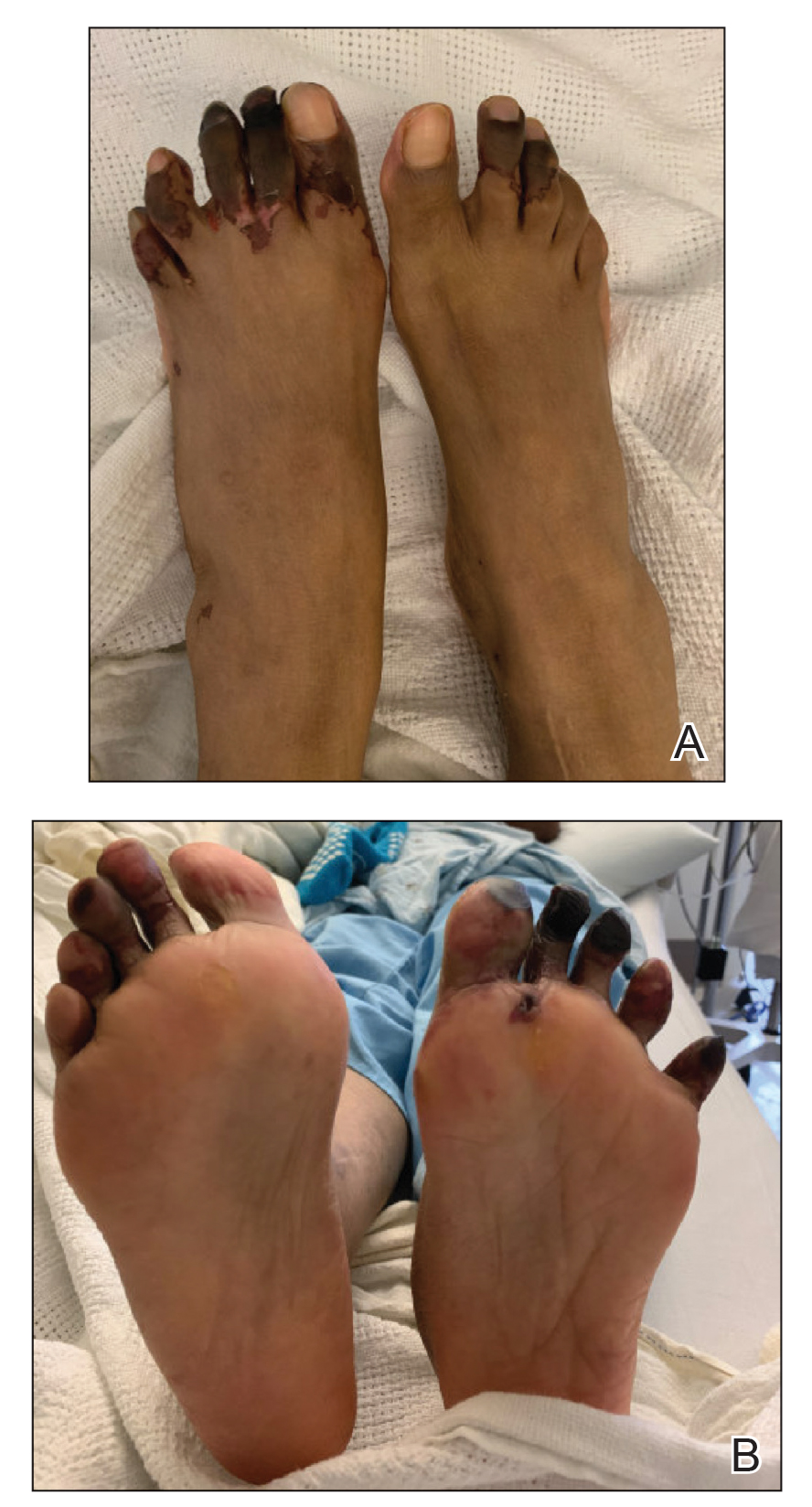
Raynaud phenomenon is a vascular disorder characterized by intermittent arteriolar vasospasm of the digits, often due to cold temperature or stress. Approximately 90% of RP cases are primarily idiopathic, with the remaining cases secondary to other diseases, typically systemic sclerosis, SLE, or mixed connective tissue disease.1 Symptoms present with characteristic changing of hands from white (ischemia) to blue (hypoxia) to red (reperfusion). Episodic attacks of vasospasm and ischemia can be painful and lead to digital ulcerations and necrosis of the digits or hands. Other complications including digital tuft pits, pterygium inversum unguis, or torturous nail fold capillaries with capillary dropout also may be seen.2
Although the etiology is multifactorial, the pathophysiology primarily is due to an imbalance of vasodilation and vasoconstriction. Perturbed levels of vasodilatory mediators include nitric oxide, prostacyclin, and calcitonin gene-related peptide.3 Meanwhile, abnormal neural sympathetic control of α-adrenergic receptors located on smooth muscle vasculature and subsequent endothelial hyperproliferation may contribute to inappropriate vasoconstriction.4
The first-line therapy for mild to moderate disease refractory to conservative management includes monotherapy with dihydropyridine calcium channel blockers. For severe disease, combination therapy involves addition of other classes of medications including phosphodiesterase 5 inhibitors, topical nitrates, angiotensin receptor blockers, or selective serotonin reuptake inhibitors. Intravenous prostacyclin, endothelin receptor blockers, and onabotulinumtoxinA injections may be added as third-line therapy. Finally, surgical management including sympathectomy with continued pharmacologic therapy may be needed for disease recalcitrant to the aforementioned options.2
OnabotulinumtoxinA is a neurotoxin produced by the bacterium Clostridium botulinum. The toxin’s mechanism of action involves inhibition of the release of presynaptic acetylcholine-containing vesicles at the neuromuscular junction through cleavage of sensory nerve action potential receptor proteins. In addition, it inhibits smooth muscle vasoconstriction and pain by blocking α2-adrenergic receptors on blood vessels and chronic pain-transmitting C fibers in nerves, respectively.3,5
Only recently has onabotulinumtoxinA been used for treatment of RP. Botulinum toxin is approved for the treatment of spastic and dystonic diseases such as blepharospasm, headaches in patients with chronic migraines, upper limb spasticity, cervical dystonia, torticollis, ocular strabismus, and hyperhidrosis.3 However, the versatility of its therapeutic effects is evident in its broad off-label clinical applications, including achalasia; carpal tunnel syndrome; and spasticity relating to stroke, paraplegia, and cerebral palsy, among many others.5
Few studies have analyzed the use of onabotulinumtoxinA for the treatment of RP.3,6 There is no consensus yet regarding dose, dilution, or injection sites. One vial of onabotulinumtoxinA contains 100 U and is reconstituted in 20 mL of normal saline to produce 5 U/mL. The simplest technique involves the injection of 5 U into the medial and lateral aspects of each finger at its base, at the level of or just proximal to the A1 pulley, for a total of 50 U per hand.7 In the foot, injection can be made at the base of each toe near the proximal phalanges. A regimen of 50 to 100 U per hand was used by Neumeister et al5 on 19 patients, who subsequently standardized it to 10 U on each neurovascular bundle in a follow-up study,7 giving a total volume of 2 mL per injection. Associated pain or a burning sensation initially may be experienced, which may be mitigated by a lidocaine hydrochloride wrist block prior to injection.7 This technique produced immediate and lasting pain relief, increased tissue perfusion, and resolved digital ulcers in 28 of 33 patients. Most patients reported immediate relief, and a few noted gradual reduction in pain and resolution of chronic ulcers within 2 months. Of the 33 patients, 7 (21.2%) required repeat injections for recurrent pain, but the majority were pain free up to 6 years later with a single injection schedule.7
Injection into the palmar region, wrists, and/or fingers also may be performed. Effects of using different injection sites (eg, neurovascular bundle, distal palm, proximal hand) have been explored and were not notably different between these locations.8 Lastly, the frequency of injections may be attenuated according to the spectrum and severity of the patient’s symptoms. In a report of 11 patients who received a total of 100 U of onabotulinumtoxinA per hand, 5 required repeat injections within 3 to 8 months.9
Studies have reported onabotulinumtoxinA to be a promising option for the treatment of intractable symptoms. Likewise, our patient had a notable reduction in pain with signs of clinical improvement within 24 to 48 hours after injection. The need for amputation 6 months later likely was because the patient’s toes were already necrosing prior to treatment with onabotulinumtoxinA. Thus, the timing of intervention may play a critical role in response to onabotulinumtoxinA injections, particularly because the severity of our patient’s presentation was comparable to other cases reported in the literature. Even in reports using a smaller dose—2 U injected into each toe as opposed to 10 U per toe, as in our case—follow-up showed favorable results.10 In other reports, response can be perceived within days to a week, with remarkable improvement of numbness, pain, digit color, and wound resolution, in addition to decreased frequency and severity of attacks. Moreover, greater vasodilation and subsequent tissue perfusion have been evidenced by objective measures including digital transcutaneous oxygen saturation and Doppler sonography.7,8 Side effects, which are minimal and temporary, include local pain triggering a vasospastic attack and intrinsic muscle weakness; more rarely, dysesthesia and thenar eminence atrophy have been reported.11
Available studies have shown onabotulinumtoxinA to produce favorable results in the treatment of vasospastic disease. We suspect that an earlier intervention for our patient—before necrosis of the toes developed—would have led to a more positive outcome, consistent with other reports. Treatment with onabotulinumtoxinA is an approach to consider when the standard-of-care treatments for RP have been exhausted, as timely intervention may prevent the need for surgery. The indications and appropriate dosing protocol remain to be defined, in addition to more thorough evaluation of its efficacy relative to other medical and surgical options.
- Neumeister MW. The role of botulinum toxin in vasospastic disorders of the hand. Hand Clin. 2015;31:23-37. doi:10.1016/j.hcl.2014.09.003
- Bakst R, Merola JF, Franks AG, et al. Raynaud’s phenomenon: pathogenesis and management. J Am Acad Dermatol. 2008;59:633-653. doi:10.1016/j.jaad.2008.06.004
- Iorio ML, Masden DL, Higgins JP. Botulinum toxin a treatment of Raynaud’s phenomenon: a review. Semin Arthritis Rheum. 2012;41:599-603. doi:10.1016/j.semarthrit.2011.07.006
- Wigley FM, Flavahan NA. Raynaud’s phenomenon. N Engl J Med. 2016;375:556-565. doi:10.1056/NEJMra1507638
- Neumeister MW, Chambers CB, Herron MS, et al. Botox therapy for ischemic digits. Plast Reconstr Surg. 2009;124:191-200. doi:10.1097/PRS.0b013e3181a80576
- Sycha T, Graninger M, Auff E, et al. Botulinum toxin in the treatment of Raynaud’s phenomenon: a pilot study. Eur J Clin Invest. 2004;34:312-313. doi:10.1016/j.jaad.2013.06.029
- Neumeister MW. Botulinum toxin type A in the treatment of Raynaud’s phenomenon. J Hand Surg Am. 2010;35:2085-2092. doi:10.1016/j.jhsa.2010.09.019
- Fregene A, Ditmars D, Siddiqui A. Botulinum toxin type A: a treatment option for digital ischemia in patients with Raynaud’s phenomenon. J Hand Surg Am. 2009;34:446-452. doi:10.1016/j.jhsa.2008.11.026
- Van Beek AL, Lim PK, Gear AJL, et al. Management of vasospastic disorders with botulinum toxin A. Plast Reconstr Surg. 2007;119:217-226. doi:10.1097/01.prs.0000244860.00674.57
- Dhaliwal K, Griffin M, Denton CP, et al. The novel use of botulinum toxin A for the treatment of Raynaud’s phenomenon in the toes. BMJ Case Rep. 2018;2018:2017-2019. doi:10.1136/bcr-2017-219348
- Eickhoff JC, Smith JK, Landau ME, et al. Iatrogenic thenar eminence atrophy after Botox A injection for secondary Raynaud phenomenon. J Clin Rheumatol. 2016;22:395-396. doi:10.1097/RHU.0000000000000450
To the Editor:
Raynaud phenomenon (RP) is an episodic vasospasm of the digits that can lead to ulceration, gangrene, and autoamputation with prolonged ischemia. OnabotulinumtoxinA has been implemented as a treatment of intractable RP by paralyzing the muscles of the digital arteries. We report a case of a woman with severe RP secondary to systemic lupus erythematosus (SLE) who was treated with onabotulinumtoxinA injections after multiple treatment modalities failed to improve her condition. We describe the dosage and injection technique used to produce clinical improvement in our patient and compare it to prior reports in the literature.
A 33-year-old woman presented to the emergency department for worsening foot pain of 5 days' duration with dusky purple color changes concerning for impending Raynaud crisis related to RP. The patient had a history of antiphospholipid antibody syndrome (APS) and SLE with overlapping symptoms of polymyositis and scleroderma. She had been hospitalized for RP multiple times prior to the current admission. She was medically managed with nifedipine, sildenafil, losartan potassium, aspirin, alprostadil, and prostaglandin infusions, and was surgically managed with a right-hand sympathectomy and right ulnar artery bypass graft that had subsequently thrombosed. At the current presentation, she had painful dusky toes on both feet though more pronounced on the left foot. She endorsed foot pain while walking and tenderness to palpation of the fingers, which were minimally improved with intravenous prostaglandins.
Physical examination revealed blanching of the digits in both hands with pits in the right fourth and left first digits. Dusky patches overlaid all the toes as well as the superior plantar aspects of the feet (Figure 1). Given the history of APS, a punch biopsy was performed on the left medial plantar foot and results showed no histologic evidence of vasculitis or vasculopathy. Necrotic foci were present on the left and right second metatarsal bones, which were not reperfusable (Figure 2). The clinical findings and punch biopsy results favored RP as opposed to vasculopathy from APS.
Several interventions were attempted, and after 4 days with no response, the patient agreed to receive treatment with onabotulinumtoxinA. OnabotulinumtoxinA (5 U) was injected into the subcutaneous tissue of the medial and lateral aspects of each of the first and second toes near the proximal phalanges (40 U total). However, treatment could not be completed due to severe pain caused by the injections despite preprocedure regional nerve blocks to both lower extremities, preinjection icing, and lorazepam. Two days later, the patient tolerated onabotulinumtoxinA injections of all remaining digits of both feet (60 U total). She noted slight clinical improvement soon thereafter. One week after treatment of all 10 toes, she reported decreased pain and reduced duskiness of both feet (Figure 3).
One month later, the patient endorsed recurring pain in the hands and feet. Physical examination revealed reticular cyanosis and increased violaceous patches of the hands; the feet were overall unchanged from the prior hospitalization. At 4-month follow-up, there was gangrene on the left second, third, and fifth toe in addition to areas of induration noted on the fingers. She was repeatedly hospitalized over the next 6 months for pain management and gangrene of the toes, and finally underwent an amputation of the left and right second toe at the proximal and middle phalanx, respectively. She currently is continuing extensive medical management for pain and gangrene of the digits; she has not received additional onabotulinumtoxinA injections.
Raynaud phenomenon is a vascular disorder characterized by intermittent arteriolar vasospasm of the digits, often due to cold temperature or stress. Approximately 90% of RP cases are primarily idiopathic, with the remaining cases secondary to other diseases, typically systemic sclerosis, SLE, or mixed connective tissue disease.1 Symptoms present with characteristic changing of hands from white (ischemia) to blue (hypoxia) to red (reperfusion). Episodic attacks of vasospasm and ischemia can be painful and lead to digital ulcerations and necrosis of the digits or hands. Other complications including digital tuft pits, pterygium inversum unguis, or torturous nail fold capillaries with capillary dropout also may be seen.2
Although the etiology is multifactorial, the pathophysiology primarily is due to an imbalance of vasodilation and vasoconstriction. Perturbed levels of vasodilatory mediators include nitric oxide, prostacyclin, and calcitonin gene-related peptide.3 Meanwhile, abnormal neural sympathetic control of α-adrenergic receptors located on smooth muscle vasculature and subsequent endothelial hyperproliferation may contribute to inappropriate vasoconstriction.4
The first-line therapy for mild to moderate disease refractory to conservative management includes monotherapy with dihydropyridine calcium channel blockers. For severe disease, combination therapy involves addition of other classes of medications including phosphodiesterase 5 inhibitors, topical nitrates, angiotensin receptor blockers, or selective serotonin reuptake inhibitors. Intravenous prostacyclin, endothelin receptor blockers, and onabotulinumtoxinA injections may be added as third-line therapy. Finally, surgical management including sympathectomy with continued pharmacologic therapy may be needed for disease recalcitrant to the aforementioned options.2
OnabotulinumtoxinA is a neurotoxin produced by the bacterium Clostridium botulinum. The toxin’s mechanism of action involves inhibition of the release of presynaptic acetylcholine-containing vesicles at the neuromuscular junction through cleavage of sensory nerve action potential receptor proteins. In addition, it inhibits smooth muscle vasoconstriction and pain by blocking α2-adrenergic receptors on blood vessels and chronic pain-transmitting C fibers in nerves, respectively.3,5
Only recently has onabotulinumtoxinA been used for treatment of RP. Botulinum toxin is approved for the treatment of spastic and dystonic diseases such as blepharospasm, headaches in patients with chronic migraines, upper limb spasticity, cervical dystonia, torticollis, ocular strabismus, and hyperhidrosis.3 However, the versatility of its therapeutic effects is evident in its broad off-label clinical applications, including achalasia; carpal tunnel syndrome; and spasticity relating to stroke, paraplegia, and cerebral palsy, among many others.5
Few studies have analyzed the use of onabotulinumtoxinA for the treatment of RP.3,6 There is no consensus yet regarding dose, dilution, or injection sites. One vial of onabotulinumtoxinA contains 100 U and is reconstituted in 20 mL of normal saline to produce 5 U/mL. The simplest technique involves the injection of 5 U into the medial and lateral aspects of each finger at its base, at the level of or just proximal to the A1 pulley, for a total of 50 U per hand.7 In the foot, injection can be made at the base of each toe near the proximal phalanges. A regimen of 50 to 100 U per hand was used by Neumeister et al5 on 19 patients, who subsequently standardized it to 10 U on each neurovascular bundle in a follow-up study,7 giving a total volume of 2 mL per injection. Associated pain or a burning sensation initially may be experienced, which may be mitigated by a lidocaine hydrochloride wrist block prior to injection.7 This technique produced immediate and lasting pain relief, increased tissue perfusion, and resolved digital ulcers in 28 of 33 patients. Most patients reported immediate relief, and a few noted gradual reduction in pain and resolution of chronic ulcers within 2 months. Of the 33 patients, 7 (21.2%) required repeat injections for recurrent pain, but the majority were pain free up to 6 years later with a single injection schedule.7
Injection into the palmar region, wrists, and/or fingers also may be performed. Effects of using different injection sites (eg, neurovascular bundle, distal palm, proximal hand) have been explored and were not notably different between these locations.8 Lastly, the frequency of injections may be attenuated according to the spectrum and severity of the patient’s symptoms. In a report of 11 patients who received a total of 100 U of onabotulinumtoxinA per hand, 5 required repeat injections within 3 to 8 months.9
Studies have reported onabotulinumtoxinA to be a promising option for the treatment of intractable symptoms. Likewise, our patient had a notable reduction in pain with signs of clinical improvement within 24 to 48 hours after injection. The need for amputation 6 months later likely was because the patient’s toes were already necrosing prior to treatment with onabotulinumtoxinA. Thus, the timing of intervention may play a critical role in response to onabotulinumtoxinA injections, particularly because the severity of our patient’s presentation was comparable to other cases reported in the literature. Even in reports using a smaller dose—2 U injected into each toe as opposed to 10 U per toe, as in our case—follow-up showed favorable results.10 In other reports, response can be perceived within days to a week, with remarkable improvement of numbness, pain, digit color, and wound resolution, in addition to decreased frequency and severity of attacks. Moreover, greater vasodilation and subsequent tissue perfusion have been evidenced by objective measures including digital transcutaneous oxygen saturation and Doppler sonography.7,8 Side effects, which are minimal and temporary, include local pain triggering a vasospastic attack and intrinsic muscle weakness; more rarely, dysesthesia and thenar eminence atrophy have been reported.11
Available studies have shown onabotulinumtoxinA to produce favorable results in the treatment of vasospastic disease. We suspect that an earlier intervention for our patient—before necrosis of the toes developed—would have led to a more positive outcome, consistent with other reports. Treatment with onabotulinumtoxinA is an approach to consider when the standard-of-care treatments for RP have been exhausted, as timely intervention may prevent the need for surgery. The indications and appropriate dosing protocol remain to be defined, in addition to more thorough evaluation of its efficacy relative to other medical and surgical options.
To the Editor:
Raynaud phenomenon (RP) is an episodic vasospasm of the digits that can lead to ulceration, gangrene, and autoamputation with prolonged ischemia. OnabotulinumtoxinA has been implemented as a treatment of intractable RP by paralyzing the muscles of the digital arteries. We report a case of a woman with severe RP secondary to systemic lupus erythematosus (SLE) who was treated with onabotulinumtoxinA injections after multiple treatment modalities failed to improve her condition. We describe the dosage and injection technique used to produce clinical improvement in our patient and compare it to prior reports in the literature.
A 33-year-old woman presented to the emergency department for worsening foot pain of 5 days' duration with dusky purple color changes concerning for impending Raynaud crisis related to RP. The patient had a history of antiphospholipid antibody syndrome (APS) and SLE with overlapping symptoms of polymyositis and scleroderma. She had been hospitalized for RP multiple times prior to the current admission. She was medically managed with nifedipine, sildenafil, losartan potassium, aspirin, alprostadil, and prostaglandin infusions, and was surgically managed with a right-hand sympathectomy and right ulnar artery bypass graft that had subsequently thrombosed. At the current presentation, she had painful dusky toes on both feet though more pronounced on the left foot. She endorsed foot pain while walking and tenderness to palpation of the fingers, which were minimally improved with intravenous prostaglandins.
Physical examination revealed blanching of the digits in both hands with pits in the right fourth and left first digits. Dusky patches overlaid all the toes as well as the superior plantar aspects of the feet (Figure 1). Given the history of APS, a punch biopsy was performed on the left medial plantar foot and results showed no histologic evidence of vasculitis or vasculopathy. Necrotic foci were present on the left and right second metatarsal bones, which were not reperfusable (Figure 2). The clinical findings and punch biopsy results favored RP as opposed to vasculopathy from APS.
Several interventions were attempted, and after 4 days with no response, the patient agreed to receive treatment with onabotulinumtoxinA. OnabotulinumtoxinA (5 U) was injected into the subcutaneous tissue of the medial and lateral aspects of each of the first and second toes near the proximal phalanges (40 U total). However, treatment could not be completed due to severe pain caused by the injections despite preprocedure regional nerve blocks to both lower extremities, preinjection icing, and lorazepam. Two days later, the patient tolerated onabotulinumtoxinA injections of all remaining digits of both feet (60 U total). She noted slight clinical improvement soon thereafter. One week after treatment of all 10 toes, she reported decreased pain and reduced duskiness of both feet (Figure 3).
One month later, the patient endorsed recurring pain in the hands and feet. Physical examination revealed reticular cyanosis and increased violaceous patches of the hands; the feet were overall unchanged from the prior hospitalization. At 4-month follow-up, there was gangrene on the left second, third, and fifth toe in addition to areas of induration noted on the fingers. She was repeatedly hospitalized over the next 6 months for pain management and gangrene of the toes, and finally underwent an amputation of the left and right second toe at the proximal and middle phalanx, respectively. She currently is continuing extensive medical management for pain and gangrene of the digits; she has not received additional onabotulinumtoxinA injections.
Raynaud phenomenon is a vascular disorder characterized by intermittent arteriolar vasospasm of the digits, often due to cold temperature or stress. Approximately 90% of RP cases are primarily idiopathic, with the remaining cases secondary to other diseases, typically systemic sclerosis, SLE, or mixed connective tissue disease.1 Symptoms present with characteristic changing of hands from white (ischemia) to blue (hypoxia) to red (reperfusion). Episodic attacks of vasospasm and ischemia can be painful and lead to digital ulcerations and necrosis of the digits or hands. Other complications including digital tuft pits, pterygium inversum unguis, or torturous nail fold capillaries with capillary dropout also may be seen.2
Although the etiology is multifactorial, the pathophysiology primarily is due to an imbalance of vasodilation and vasoconstriction. Perturbed levels of vasodilatory mediators include nitric oxide, prostacyclin, and calcitonin gene-related peptide.3 Meanwhile, abnormal neural sympathetic control of α-adrenergic receptors located on smooth muscle vasculature and subsequent endothelial hyperproliferation may contribute to inappropriate vasoconstriction.4
The first-line therapy for mild to moderate disease refractory to conservative management includes monotherapy with dihydropyridine calcium channel blockers. For severe disease, combination therapy involves addition of other classes of medications including phosphodiesterase 5 inhibitors, topical nitrates, angiotensin receptor blockers, or selective serotonin reuptake inhibitors. Intravenous prostacyclin, endothelin receptor blockers, and onabotulinumtoxinA injections may be added as third-line therapy. Finally, surgical management including sympathectomy with continued pharmacologic therapy may be needed for disease recalcitrant to the aforementioned options.2
OnabotulinumtoxinA is a neurotoxin produced by the bacterium Clostridium botulinum. The toxin’s mechanism of action involves inhibition of the release of presynaptic acetylcholine-containing vesicles at the neuromuscular junction through cleavage of sensory nerve action potential receptor proteins. In addition, it inhibits smooth muscle vasoconstriction and pain by blocking α2-adrenergic receptors on blood vessels and chronic pain-transmitting C fibers in nerves, respectively.3,5
Only recently has onabotulinumtoxinA been used for treatment of RP. Botulinum toxin is approved for the treatment of spastic and dystonic diseases such as blepharospasm, headaches in patients with chronic migraines, upper limb spasticity, cervical dystonia, torticollis, ocular strabismus, and hyperhidrosis.3 However, the versatility of its therapeutic effects is evident in its broad off-label clinical applications, including achalasia; carpal tunnel syndrome; and spasticity relating to stroke, paraplegia, and cerebral palsy, among many others.5
Few studies have analyzed the use of onabotulinumtoxinA for the treatment of RP.3,6 There is no consensus yet regarding dose, dilution, or injection sites. One vial of onabotulinumtoxinA contains 100 U and is reconstituted in 20 mL of normal saline to produce 5 U/mL. The simplest technique involves the injection of 5 U into the medial and lateral aspects of each finger at its base, at the level of or just proximal to the A1 pulley, for a total of 50 U per hand.7 In the foot, injection can be made at the base of each toe near the proximal phalanges. A regimen of 50 to 100 U per hand was used by Neumeister et al5 on 19 patients, who subsequently standardized it to 10 U on each neurovascular bundle in a follow-up study,7 giving a total volume of 2 mL per injection. Associated pain or a burning sensation initially may be experienced, which may be mitigated by a lidocaine hydrochloride wrist block prior to injection.7 This technique produced immediate and lasting pain relief, increased tissue perfusion, and resolved digital ulcers in 28 of 33 patients. Most patients reported immediate relief, and a few noted gradual reduction in pain and resolution of chronic ulcers within 2 months. Of the 33 patients, 7 (21.2%) required repeat injections for recurrent pain, but the majority were pain free up to 6 years later with a single injection schedule.7
Injection into the palmar region, wrists, and/or fingers also may be performed. Effects of using different injection sites (eg, neurovascular bundle, distal palm, proximal hand) have been explored and were not notably different between these locations.8 Lastly, the frequency of injections may be attenuated according to the spectrum and severity of the patient’s symptoms. In a report of 11 patients who received a total of 100 U of onabotulinumtoxinA per hand, 5 required repeat injections within 3 to 8 months.9
Studies have reported onabotulinumtoxinA to be a promising option for the treatment of intractable symptoms. Likewise, our patient had a notable reduction in pain with signs of clinical improvement within 24 to 48 hours after injection. The need for amputation 6 months later likely was because the patient’s toes were already necrosing prior to treatment with onabotulinumtoxinA. Thus, the timing of intervention may play a critical role in response to onabotulinumtoxinA injections, particularly because the severity of our patient’s presentation was comparable to other cases reported in the literature. Even in reports using a smaller dose—2 U injected into each toe as opposed to 10 U per toe, as in our case—follow-up showed favorable results.10 In other reports, response can be perceived within days to a week, with remarkable improvement of numbness, pain, digit color, and wound resolution, in addition to decreased frequency and severity of attacks. Moreover, greater vasodilation and subsequent tissue perfusion have been evidenced by objective measures including digital transcutaneous oxygen saturation and Doppler sonography.7,8 Side effects, which are minimal and temporary, include local pain triggering a vasospastic attack and intrinsic muscle weakness; more rarely, dysesthesia and thenar eminence atrophy have been reported.11
Available studies have shown onabotulinumtoxinA to produce favorable results in the treatment of vasospastic disease. We suspect that an earlier intervention for our patient—before necrosis of the toes developed—would have led to a more positive outcome, consistent with other reports. Treatment with onabotulinumtoxinA is an approach to consider when the standard-of-care treatments for RP have been exhausted, as timely intervention may prevent the need for surgery. The indications and appropriate dosing protocol remain to be defined, in addition to more thorough evaluation of its efficacy relative to other medical and surgical options.
- Neumeister MW. The role of botulinum toxin in vasospastic disorders of the hand. Hand Clin. 2015;31:23-37. doi:10.1016/j.hcl.2014.09.003
- Bakst R, Merola JF, Franks AG, et al. Raynaud’s phenomenon: pathogenesis and management. J Am Acad Dermatol. 2008;59:633-653. doi:10.1016/j.jaad.2008.06.004
- Iorio ML, Masden DL, Higgins JP. Botulinum toxin a treatment of Raynaud’s phenomenon: a review. Semin Arthritis Rheum. 2012;41:599-603. doi:10.1016/j.semarthrit.2011.07.006
- Wigley FM, Flavahan NA. Raynaud’s phenomenon. N Engl J Med. 2016;375:556-565. doi:10.1056/NEJMra1507638
- Neumeister MW, Chambers CB, Herron MS, et al. Botox therapy for ischemic digits. Plast Reconstr Surg. 2009;124:191-200. doi:10.1097/PRS.0b013e3181a80576
- Sycha T, Graninger M, Auff E, et al. Botulinum toxin in the treatment of Raynaud’s phenomenon: a pilot study. Eur J Clin Invest. 2004;34:312-313. doi:10.1016/j.jaad.2013.06.029
- Neumeister MW. Botulinum toxin type A in the treatment of Raynaud’s phenomenon. J Hand Surg Am. 2010;35:2085-2092. doi:10.1016/j.jhsa.2010.09.019
- Fregene A, Ditmars D, Siddiqui A. Botulinum toxin type A: a treatment option for digital ischemia in patients with Raynaud’s phenomenon. J Hand Surg Am. 2009;34:446-452. doi:10.1016/j.jhsa.2008.11.026
- Van Beek AL, Lim PK, Gear AJL, et al. Management of vasospastic disorders with botulinum toxin A. Plast Reconstr Surg. 2007;119:217-226. doi:10.1097/01.prs.0000244860.00674.57
- Dhaliwal K, Griffin M, Denton CP, et al. The novel use of botulinum toxin A for the treatment of Raynaud’s phenomenon in the toes. BMJ Case Rep. 2018;2018:2017-2019. doi:10.1136/bcr-2017-219348
- Eickhoff JC, Smith JK, Landau ME, et al. Iatrogenic thenar eminence atrophy after Botox A injection for secondary Raynaud phenomenon. J Clin Rheumatol. 2016;22:395-396. doi:10.1097/RHU.0000000000000450
- Neumeister MW. The role of botulinum toxin in vasospastic disorders of the hand. Hand Clin. 2015;31:23-37. doi:10.1016/j.hcl.2014.09.003
- Bakst R, Merola JF, Franks AG, et al. Raynaud’s phenomenon: pathogenesis and management. J Am Acad Dermatol. 2008;59:633-653. doi:10.1016/j.jaad.2008.06.004
- Iorio ML, Masden DL, Higgins JP. Botulinum toxin a treatment of Raynaud’s phenomenon: a review. Semin Arthritis Rheum. 2012;41:599-603. doi:10.1016/j.semarthrit.2011.07.006
- Wigley FM, Flavahan NA. Raynaud’s phenomenon. N Engl J Med. 2016;375:556-565. doi:10.1056/NEJMra1507638
- Neumeister MW, Chambers CB, Herron MS, et al. Botox therapy for ischemic digits. Plast Reconstr Surg. 2009;124:191-200. doi:10.1097/PRS.0b013e3181a80576
- Sycha T, Graninger M, Auff E, et al. Botulinum toxin in the treatment of Raynaud’s phenomenon: a pilot study. Eur J Clin Invest. 2004;34:312-313. doi:10.1016/j.jaad.2013.06.029
- Neumeister MW. Botulinum toxin type A in the treatment of Raynaud’s phenomenon. J Hand Surg Am. 2010;35:2085-2092. doi:10.1016/j.jhsa.2010.09.019
- Fregene A, Ditmars D, Siddiqui A. Botulinum toxin type A: a treatment option for digital ischemia in patients with Raynaud’s phenomenon. J Hand Surg Am. 2009;34:446-452. doi:10.1016/j.jhsa.2008.11.026
- Van Beek AL, Lim PK, Gear AJL, et al. Management of vasospastic disorders with botulinum toxin A. Plast Reconstr Surg. 2007;119:217-226. doi:10.1097/01.prs.0000244860.00674.57
- Dhaliwal K, Griffin M, Denton CP, et al. The novel use of botulinum toxin A for the treatment of Raynaud’s phenomenon in the toes. BMJ Case Rep. 2018;2018:2017-2019. doi:10.1136/bcr-2017-219348
- Eickhoff JC, Smith JK, Landau ME, et al. Iatrogenic thenar eminence atrophy after Botox A injection for secondary Raynaud phenomenon. J Clin Rheumatol. 2016;22:395-396. doi:10.1097/RHU.0000000000000450
Practice Points
- Raynaud phenomenon (RP) is a vascular disorder characterized by episodic vasospasms of the digits often due to cold temperature or stress.
- OnabotulinumtoxinA has been implemented as a treatment of intractable RP after failure with traditional treatments, such as calcium channel blockers, angiotensin receptor blockers, prostaglandins, endothelin receptor blockers, and phosphodiesterase 5 inhibitors.
- A standard technique of delivery of onabotulinumtoxinA involves injection of 5 U/mL into the medial and lateral aspects of each finger at its base (near the metacarpal head) for a total of 50 U per hand or foot.
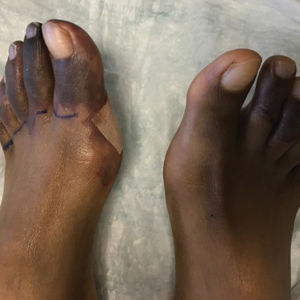
Flagellate Shiitake Mushroom Reaction With Histologic Features of Acute Generalized Exanthematous Pustulosis
To the Editor:
A 59-year-old man presented with a severely pruritic rash on the legs, arms, abdomen, groin, and buttocks of 3 days’ duration. He reported subjective fever and chills. Prior to the appearance of the rash, the patient and his family had eaten shiitake mushrooms daily for 3 days. He denied any new medications in the last several months or any recent upper respiratory or gastrointestinal tract illnesses. His medical history included type 2 diabetes mellitus and diabetes-induced end-stage renal disease requiring home peritoneal dialysis. His long-term medications for diabetes mellitus, hypertension, benign prostatic hyperplasia, hyperlipidemia, and insomnia included amlodipine, atorvastatin, finasteride, gabapentin, insulin glargine, linagliptin, metoprolol, and mirtazapine.
Physical examination revealed an afebrile man with medium brown skin tone and diffuse, bright red, erythematous patches on the lower legs, axillae, medial forearms, lateral trunk, lower abdomen, and groin. There were distinct flagellate, linear, red patches on the lower legs (Figure 1). In addition, small clusters of 1- to 2-mm superficial pustules were present on the right upper medial thigh and left forearm with micropapules grouped in the skin folds.

A shave biopsy specimen from a pustule on the right upper medial thigh revealed spongiotic dermatitis with neutrophilic subcorneal pustule formation and frequent eosinophils (Figure 2). The dermis contained scattered mixed inflammatory cells including neutrophils, eosinophils, lymphocytes, and histiocytes (Figure 3). These histologic findings were consistent with acute generalized exanthematous pustulosis (AGEP). No biopsy was performed on the flagellate patches due to its clinically distinct presentation and well-established association with shiitake mushroom ingestion.

The patient was treated with triamcinolone ointment and systemic corticosteroids to reduce pruritus and quickly clear the lesions due to his comorbidities. He recovered completely within 1 week and had no evidence of postinflammatory hyperpigmentation from the flagellate dermatitis.
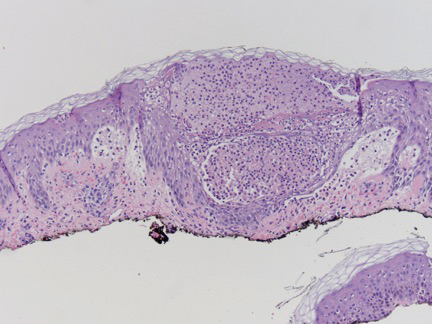
Flagellate dermatitis is an intensely pruritic dermatitis characterized by 1-mm, disseminated, erythematous papules in a linear grouped arrangement secondary to koebnerization due to the patient scratching. It was first described in 1977 by Nakamura.1 Although it rarely is seen outside of China and Japan, there are well-established associations of flagellate dermatitis with bleomycin and shiitake mushroom (Lentinula edodes) ingestion. One key clinical difference between the two causes is that postinflammatory hyperpigmentation changes usually are seen with bleomycin-induced flagellate dermatitis and typically are not present with shiitake mushroom–induced flagellate dermatitis.2 Following ingestion of shiitake mushrooms, the median time of onset of presentation typically is 24 hours but ranges from 12 hours to 5 days. Most patients completely recover by 3 weeks, with or without treatment.3 Although the pathogenesis of shiitake mushroom–induced flagellate dermatitis is not clear, the most common theory is a toxic reaction to lentinan, a polysaccharide isolated from shiitake mushrooms. However, type I and IV allergic hypersensitivities also have been supported by the time of onset, clearance, severe pruritus, benefit from steroids and antihistamines, and lack of grouped outbreaks in people exposed to shared meals containing shiitake mushrooms.3,4 Furthermore, there is a case of patch test–confirmed allergic contact dermatitis to shiitake mushrooms, demonstrating a 1+ reaction at 96 hours to the cap of a shiitake mushroom but a negative pin-prick test at 20 minutes, suggesting type IV hypersensitivity.5 An additional case revealed a positive skin-prick test with formation of a 4-mm wheal and subsequent pruritic papules and vesicles appearing 48 to 72 hours later at the prick site.6 Subsequent cases have been reported in association with consumption of raw shiitake mushrooms, but cases have been reported after consumption of fully cooked mushrooms, which does not support a toxin-mediated theory, as cooking the mushroom before consumption likely would denature or change the structure of the suspected toxin.2
Acute generalized exanthematous pustulosis is a rare eruption that occurs due to ingestion of a causative agent, usually an antibiotic, and is characterized by the presence of fever and disseminated, erythematous, pinpoint, sterile pustules on the skin and mucous membranes. It affects 1 to 5 persons per million per year, with more than 90% of cases attributed to drug ingestion.7 Spontaneous resolution can be expected within 15 days of its onset; however, there is a mortality rate of up to 5% that occurs most often in those with severe comorbidities or in older patients, for whom systemic corticosteroid therapy may be justified.7,8 A multinational case-control study conducted to evaluate the risk of AGEP associated with certain drugs revealed macrolides (namely pristinamycin); β-lactam antibiotics including penicillin, aminopenicillin, and cephalosporin; quinolones; hydroxychloroquine; anti-infective sulfonamides; terbinafine; and diltiazem as the most strongly associated culprits.9 Our patient’s flagellate dermatitis was unique in that it also showed histologic features of AGEP. The pathogenesis of drug-induced AGEP has been partially elucidated and involves activation of drug-specific CD4+ and CD8+ T cells that migrate to the skin and participate in apoptotic signaling of keratinocytes and recruitment of neutrophils and eosinophils, which form subcorneal sterile pustules.7 In a study of severe cutaneous adverse drug reactions, 50% (7/14) of patients with AGEP had positive patch tests to the causative agent.10 This T cell–dependent response explains why the condition responds to systemic corticosteroids. Additionally, our case report of shiitake mushroom–induced flagellate dermatitis with histologic features of AGEP suggests that the pathogenesis of flagellate dermatitis may be a T cell–mediated type IV hypersensitivity reaction. The time of onset, lack of grouped outbreaks in those sharing shiitake mushroom–containing meals, severe pruritus, lack of cases demonstrating an anaphylactic or wheal and flare response, benefit of steroids, and a case with histologic features of AGEP all lend support to this theory.
We report a case of shiitake mushroom–induced flagellate dermatitis with histologic features of AGEP. The time course, histologic features of AGEP, absence of new medications, and resolution with discontinuation of shiitake mushrooms lends support of the hypothesis that the pathogenesis of shiitake mushroom–induced flagellate dermatitis is similar to AGEP’s type IV hypersensitivity reaction. To further elucidate its pathogenesis, skin prick testing and patch testing with shiitake mushrooms in patients exhibiting shiitake mushroom–induced flagellate dermatitis may prove to be beneficial.
- Nakamura T. Toxicoderma caused by shiitake (Lentinus edodes)[in Japanese]. Jpn J Clin Dermatol. 1977;31:65-68.
- Chu EY, Anand D, Dawn A, et al. Shiitake dermatitis: a report of 3 cases and review of the literature. Cutis. 2013;91:287-290.
- Boels D, Landreau A, Bruneau C, et al. Shiitake dermatitis recorded by French Poison Control Centers—new case series with clinical observations. Clin Toxicol (Phila). 2014;52:625-628.
- Nakamura T. Shiitake (Lentinus edodes) dermatitis. Contact Dermatitis. 1992;27:65-70.
- Curnow P, Tam M. Contact dermatitis to shiitake mushroom. Australas J Dermatol. 2003;44:155-157.
- Lippert U, Martin V, Schwertfeger C, et al. Shiitake dermatitis. Br J Dermatol. 2003;148:178-179.
- Fernando SL. Acute generalised exanthematous pustulosis. Australas J Dermatol. 2012;53:87-92.
- Sidoroff A, Halevy S, Bavinck JN, et al. Acute generalized exanthematous pustulosis (AGEP)—a clinical reaction pattern. J Cutan Pathol. 2001;28:113-119.
- Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)—results of a multinational case-control study (EuroSCAR). Br J Dermatol. 2007;157:989-996.
- Wolkenstein P, Chosidow O, Flechet ML, et al. Patch testing in severe cutaneous adverse drug reactions, including Stevens-Johnson syndrome and toxic epidermal necrolysis. Contact Dermatitis. 1996;35:234-236.
To the Editor:
A 59-year-old man presented with a severely pruritic rash on the legs, arms, abdomen, groin, and buttocks of 3 days’ duration. He reported subjective fever and chills. Prior to the appearance of the rash, the patient and his family had eaten shiitake mushrooms daily for 3 days. He denied any new medications in the last several months or any recent upper respiratory or gastrointestinal tract illnesses. His medical history included type 2 diabetes mellitus and diabetes-induced end-stage renal disease requiring home peritoneal dialysis. His long-term medications for diabetes mellitus, hypertension, benign prostatic hyperplasia, hyperlipidemia, and insomnia included amlodipine, atorvastatin, finasteride, gabapentin, insulin glargine, linagliptin, metoprolol, and mirtazapine.
Physical examination revealed an afebrile man with medium brown skin tone and diffuse, bright red, erythematous patches on the lower legs, axillae, medial forearms, lateral trunk, lower abdomen, and groin. There were distinct flagellate, linear, red patches on the lower legs (Figure 1). In addition, small clusters of 1- to 2-mm superficial pustules were present on the right upper medial thigh and left forearm with micropapules grouped in the skin folds.
A shave biopsy specimen from a pustule on the right upper medial thigh revealed spongiotic dermatitis with neutrophilic subcorneal pustule formation and frequent eosinophils (Figure 2). The dermis contained scattered mixed inflammatory cells including neutrophils, eosinophils, lymphocytes, and histiocytes (Figure 3). These histologic findings were consistent with acute generalized exanthematous pustulosis (AGEP). No biopsy was performed on the flagellate patches due to its clinically distinct presentation and well-established association with shiitake mushroom ingestion.
The patient was treated with triamcinolone ointment and systemic corticosteroids to reduce pruritus and quickly clear the lesions due to his comorbidities. He recovered completely within 1 week and had no evidence of postinflammatory hyperpigmentation from the flagellate dermatitis.
Flagellate dermatitis is an intensely pruritic dermatitis characterized by 1-mm, disseminated, erythematous papules in a linear grouped arrangement secondary to koebnerization due to the patient scratching. It was first described in 1977 by Nakamura.1 Although it rarely is seen outside of China and Japan, there are well-established associations of flagellate dermatitis with bleomycin and shiitake mushroom (Lentinula edodes) ingestion. One key clinical difference between the two causes is that postinflammatory hyperpigmentation changes usually are seen with bleomycin-induced flagellate dermatitis and typically are not present with shiitake mushroom–induced flagellate dermatitis.2 Following ingestion of shiitake mushrooms, the median time of onset of presentation typically is 24 hours but ranges from 12 hours to 5 days. Most patients completely recover by 3 weeks, with or without treatment.3 Although the pathogenesis of shiitake mushroom–induced flagellate dermatitis is not clear, the most common theory is a toxic reaction to lentinan, a polysaccharide isolated from shiitake mushrooms. However, type I and IV allergic hypersensitivities also have been supported by the time of onset, clearance, severe pruritus, benefit from steroids and antihistamines, and lack of grouped outbreaks in people exposed to shared meals containing shiitake mushrooms.3,4 Furthermore, there is a case of patch test–confirmed allergic contact dermatitis to shiitake mushrooms, demonstrating a 1+ reaction at 96 hours to the cap of a shiitake mushroom but a negative pin-prick test at 20 minutes, suggesting type IV hypersensitivity.5 An additional case revealed a positive skin-prick test with formation of a 4-mm wheal and subsequent pruritic papules and vesicles appearing 48 to 72 hours later at the prick site.6 Subsequent cases have been reported in association with consumption of raw shiitake mushrooms, but cases have been reported after consumption of fully cooked mushrooms, which does not support a toxin-mediated theory, as cooking the mushroom before consumption likely would denature or change the structure of the suspected toxin.2
Acute generalized exanthematous pustulosis is a rare eruption that occurs due to ingestion of a causative agent, usually an antibiotic, and is characterized by the presence of fever and disseminated, erythematous, pinpoint, sterile pustules on the skin and mucous membranes. It affects 1 to 5 persons per million per year, with more than 90% of cases attributed to drug ingestion.7 Spontaneous resolution can be expected within 15 days of its onset; however, there is a mortality rate of up to 5% that occurs most often in those with severe comorbidities or in older patients, for whom systemic corticosteroid therapy may be justified.7,8 A multinational case-control study conducted to evaluate the risk of AGEP associated with certain drugs revealed macrolides (namely pristinamycin); β-lactam antibiotics including penicillin, aminopenicillin, and cephalosporin; quinolones; hydroxychloroquine; anti-infective sulfonamides; terbinafine; and diltiazem as the most strongly associated culprits.9 Our patient’s flagellate dermatitis was unique in that it also showed histologic features of AGEP. The pathogenesis of drug-induced AGEP has been partially elucidated and involves activation of drug-specific CD4+ and CD8+ T cells that migrate to the skin and participate in apoptotic signaling of keratinocytes and recruitment of neutrophils and eosinophils, which form subcorneal sterile pustules.7 In a study of severe cutaneous adverse drug reactions, 50% (7/14) of patients with AGEP had positive patch tests to the causative agent.10 This T cell–dependent response explains why the condition responds to systemic corticosteroids. Additionally, our case report of shiitake mushroom–induced flagellate dermatitis with histologic features of AGEP suggests that the pathogenesis of flagellate dermatitis may be a T cell–mediated type IV hypersensitivity reaction. The time of onset, lack of grouped outbreaks in those sharing shiitake mushroom–containing meals, severe pruritus, lack of cases demonstrating an anaphylactic or wheal and flare response, benefit of steroids, and a case with histologic features of AGEP all lend support to this theory.
We report a case of shiitake mushroom–induced flagellate dermatitis with histologic features of AGEP. The time course, histologic features of AGEP, absence of new medications, and resolution with discontinuation of shiitake mushrooms lends support of the hypothesis that the pathogenesis of shiitake mushroom–induced flagellate dermatitis is similar to AGEP’s type IV hypersensitivity reaction. To further elucidate its pathogenesis, skin prick testing and patch testing with shiitake mushrooms in patients exhibiting shiitake mushroom–induced flagellate dermatitis may prove to be beneficial.
To the Editor:
A 59-year-old man presented with a severely pruritic rash on the legs, arms, abdomen, groin, and buttocks of 3 days’ duration. He reported subjective fever and chills. Prior to the appearance of the rash, the patient and his family had eaten shiitake mushrooms daily for 3 days. He denied any new medications in the last several months or any recent upper respiratory or gastrointestinal tract illnesses. His medical history included type 2 diabetes mellitus and diabetes-induced end-stage renal disease requiring home peritoneal dialysis. His long-term medications for diabetes mellitus, hypertension, benign prostatic hyperplasia, hyperlipidemia, and insomnia included amlodipine, atorvastatin, finasteride, gabapentin, insulin glargine, linagliptin, metoprolol, and mirtazapine.
Physical examination revealed an afebrile man with medium brown skin tone and diffuse, bright red, erythematous patches on the lower legs, axillae, medial forearms, lateral trunk, lower abdomen, and groin. There were distinct flagellate, linear, red patches on the lower legs (Figure 1). In addition, small clusters of 1- to 2-mm superficial pustules were present on the right upper medial thigh and left forearm with micropapules grouped in the skin folds.
A shave biopsy specimen from a pustule on the right upper medial thigh revealed spongiotic dermatitis with neutrophilic subcorneal pustule formation and frequent eosinophils (Figure 2). The dermis contained scattered mixed inflammatory cells including neutrophils, eosinophils, lymphocytes, and histiocytes (Figure 3). These histologic findings were consistent with acute generalized exanthematous pustulosis (AGEP). No biopsy was performed on the flagellate patches due to its clinically distinct presentation and well-established association with shiitake mushroom ingestion.
The patient was treated with triamcinolone ointment and systemic corticosteroids to reduce pruritus and quickly clear the lesions due to his comorbidities. He recovered completely within 1 week and had no evidence of postinflammatory hyperpigmentation from the flagellate dermatitis.
Flagellate dermatitis is an intensely pruritic dermatitis characterized by 1-mm, disseminated, erythematous papules in a linear grouped arrangement secondary to koebnerization due to the patient scratching. It was first described in 1977 by Nakamura.1 Although it rarely is seen outside of China and Japan, there are well-established associations of flagellate dermatitis with bleomycin and shiitake mushroom (Lentinula edodes) ingestion. One key clinical difference between the two causes is that postinflammatory hyperpigmentation changes usually are seen with bleomycin-induced flagellate dermatitis and typically are not present with shiitake mushroom–induced flagellate dermatitis.2 Following ingestion of shiitake mushrooms, the median time of onset of presentation typically is 24 hours but ranges from 12 hours to 5 days. Most patients completely recover by 3 weeks, with or without treatment.3 Although the pathogenesis of shiitake mushroom–induced flagellate dermatitis is not clear, the most common theory is a toxic reaction to lentinan, a polysaccharide isolated from shiitake mushrooms. However, type I and IV allergic hypersensitivities also have been supported by the time of onset, clearance, severe pruritus, benefit from steroids and antihistamines, and lack of grouped outbreaks in people exposed to shared meals containing shiitake mushrooms.3,4 Furthermore, there is a case of patch test–confirmed allergic contact dermatitis to shiitake mushrooms, demonstrating a 1+ reaction at 96 hours to the cap of a shiitake mushroom but a negative pin-prick test at 20 minutes, suggesting type IV hypersensitivity.5 An additional case revealed a positive skin-prick test with formation of a 4-mm wheal and subsequent pruritic papules and vesicles appearing 48 to 72 hours later at the prick site.6 Subsequent cases have been reported in association with consumption of raw shiitake mushrooms, but cases have been reported after consumption of fully cooked mushrooms, which does not support a toxin-mediated theory, as cooking the mushroom before consumption likely would denature or change the structure of the suspected toxin.2
Acute generalized exanthematous pustulosis is a rare eruption that occurs due to ingestion of a causative agent, usually an antibiotic, and is characterized by the presence of fever and disseminated, erythematous, pinpoint, sterile pustules on the skin and mucous membranes. It affects 1 to 5 persons per million per year, with more than 90% of cases attributed to drug ingestion.7 Spontaneous resolution can be expected within 15 days of its onset; however, there is a mortality rate of up to 5% that occurs most often in those with severe comorbidities or in older patients, for whom systemic corticosteroid therapy may be justified.7,8 A multinational case-control study conducted to evaluate the risk of AGEP associated with certain drugs revealed macrolides (namely pristinamycin); β-lactam antibiotics including penicillin, aminopenicillin, and cephalosporin; quinolones; hydroxychloroquine; anti-infective sulfonamides; terbinafine; and diltiazem as the most strongly associated culprits.9 Our patient’s flagellate dermatitis was unique in that it also showed histologic features of AGEP. The pathogenesis of drug-induced AGEP has been partially elucidated and involves activation of drug-specific CD4+ and CD8+ T cells that migrate to the skin and participate in apoptotic signaling of keratinocytes and recruitment of neutrophils and eosinophils, which form subcorneal sterile pustules.7 In a study of severe cutaneous adverse drug reactions, 50% (7/14) of patients with AGEP had positive patch tests to the causative agent.10 This T cell–dependent response explains why the condition responds to systemic corticosteroids. Additionally, our case report of shiitake mushroom–induced flagellate dermatitis with histologic features of AGEP suggests that the pathogenesis of flagellate dermatitis may be a T cell–mediated type IV hypersensitivity reaction. The time of onset, lack of grouped outbreaks in those sharing shiitake mushroom–containing meals, severe pruritus, lack of cases demonstrating an anaphylactic or wheal and flare response, benefit of steroids, and a case with histologic features of AGEP all lend support to this theory.
We report a case of shiitake mushroom–induced flagellate dermatitis with histologic features of AGEP. The time course, histologic features of AGEP, absence of new medications, and resolution with discontinuation of shiitake mushrooms lends support of the hypothesis that the pathogenesis of shiitake mushroom–induced flagellate dermatitis is similar to AGEP’s type IV hypersensitivity reaction. To further elucidate its pathogenesis, skin prick testing and patch testing with shiitake mushrooms in patients exhibiting shiitake mushroom–induced flagellate dermatitis may prove to be beneficial.
- Nakamura T. Toxicoderma caused by shiitake (Lentinus edodes)[in Japanese]. Jpn J Clin Dermatol. 1977;31:65-68.
- Chu EY, Anand D, Dawn A, et al. Shiitake dermatitis: a report of 3 cases and review of the literature. Cutis. 2013;91:287-290.
- Boels D, Landreau A, Bruneau C, et al. Shiitake dermatitis recorded by French Poison Control Centers—new case series with clinical observations. Clin Toxicol (Phila). 2014;52:625-628.
- Nakamura T. Shiitake (Lentinus edodes) dermatitis. Contact Dermatitis. 1992;27:65-70.
- Curnow P, Tam M. Contact dermatitis to shiitake mushroom. Australas J Dermatol. 2003;44:155-157.
- Lippert U, Martin V, Schwertfeger C, et al. Shiitake dermatitis. Br J Dermatol. 2003;148:178-179.
- Fernando SL. Acute generalised exanthematous pustulosis. Australas J Dermatol. 2012;53:87-92.
- Sidoroff A, Halevy S, Bavinck JN, et al. Acute generalized exanthematous pustulosis (AGEP)—a clinical reaction pattern. J Cutan Pathol. 2001;28:113-119.
- Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)—results of a multinational case-control study (EuroSCAR). Br J Dermatol. 2007;157:989-996.
- Wolkenstein P, Chosidow O, Flechet ML, et al. Patch testing in severe cutaneous adverse drug reactions, including Stevens-Johnson syndrome and toxic epidermal necrolysis. Contact Dermatitis. 1996;35:234-236.
- Nakamura T. Toxicoderma caused by shiitake (Lentinus edodes)[in Japanese]. Jpn J Clin Dermatol. 1977;31:65-68.
- Chu EY, Anand D, Dawn A, et al. Shiitake dermatitis: a report of 3 cases and review of the literature. Cutis. 2013;91:287-290.
- Boels D, Landreau A, Bruneau C, et al. Shiitake dermatitis recorded by French Poison Control Centers—new case series with clinical observations. Clin Toxicol (Phila). 2014;52:625-628.
- Nakamura T. Shiitake (Lentinus edodes) dermatitis. Contact Dermatitis. 1992;27:65-70.
- Curnow P, Tam M. Contact dermatitis to shiitake mushroom. Australas J Dermatol. 2003;44:155-157.
- Lippert U, Martin V, Schwertfeger C, et al. Shiitake dermatitis. Br J Dermatol. 2003;148:178-179.
- Fernando SL. Acute generalised exanthematous pustulosis. Australas J Dermatol. 2012;53:87-92.
- Sidoroff A, Halevy S, Bavinck JN, et al. Acute generalized exanthematous pustulosis (AGEP)—a clinical reaction pattern. J Cutan Pathol. 2001;28:113-119.
- Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)—results of a multinational case-control study (EuroSCAR). Br J Dermatol. 2007;157:989-996.
- Wolkenstein P, Chosidow O, Flechet ML, et al. Patch testing in severe cutaneous adverse drug reactions, including Stevens-Johnson syndrome and toxic epidermal necrolysis. Contact Dermatitis. 1996;35:234-236.
Practice Points
- Ingestion of shiitake mushrooms and bleomycin is associated with flagellate dermatitis.
- Acute generalized exanthematous pustulosis (AGEP) is a rare condition associated with certain drug ingestion.
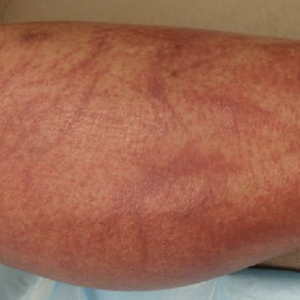
Overlapping Phenotypic Features of PTEN Hamartoma Tumor Syndrome and Birt-Hogg-Dubé Syndrome
To the Editor:
PTEN hamartoma tumor syndrome (PHTS) encompasses a spectrum of disorders that most commonly are caused by autosomal-dominant germline mutations in the phosphatase and tensin homolog, PTEN, tumor suppressor gene on chromosome 10q23. We describe a patient who presented with clinical features of PHTS and Birt-Hogg-Dubé syndrome (BHDS). Because the genetic mutations associated with both PHTS and BHDS result in altered mammalian target of rapamycin (mTOR) signaling, patients may have overlapping phenotypic features.
A 51-year-old man with a history of multiple carcinomas presented for evaluation of flesh-colored papules on the cheeks, nose, tongue, and hands, in addition to numerous skin tags on the neck, axillae, and lower abdomen bilaterally. His medical history was notable for several nasal and gastrointestinal tract polyps, chromophobe renal cell carcinoma, cutaneous lipomas, atypical carcinoid syndrome of the right lung, and a multinodular thyroid. His family history was notable for small cell lung cancer in his father, breast cancer and pancreatic cancer in his maternal aunt, esophageal cancer in his maternal grandfather, and celiac disease in his daughter.

Clinical examination revealed flesh-colored, dome-shaped papules measuring 1 to 2 mm in diameter on the nose and cheeks (Figure 1). He had hyperkeratotic papules on the dorsal fingers, consistent with acral keratoses. Additionally, multiple flesh-colored papules with a cobblestonelike appearance were noted on the oral mucosa (Figure 2). Other findings included pedunculated papules on the neck, axillae, and lower abdomen bilaterally, consistent with fibroepithelial polyps, as well as hyperpigmented velvety plaques on the axillae, characteristic of acanthosis nigricans (Figure 3). A shave biopsy of a papule on the right cheek revealed a proliferation of plump stellate fibroblasts, small blood vessels, and thick collagen bundles, characteristic of a fibrous papule (Figure 4).

Differential diagnoses for our patient included BHDS and Cowden syndrome (CS). Due to the combination of extensive family history of multiorgan cancers as well as the clinical findings, he was referred to a geneticist for further evaluation. Genetic analysis was positive for a heterozygous mutation variant of uncertain significance in the PTEN gene.

The PHTS disorders include CS, Bannayan-Riley-Ruvalcaba syndrome, Lhermitte-Duclos disease, Proteus syndrome, and Proteus-like syndrome (Table).1-9 Our patient’s clinical findings were indicative of CS, a rare genodermatosis characterized by multiple hamartomas and neoplasms of ectodermal, mesodermal, and endodermal origin.1 Most CS patients develop trichilemmomas of the central face, mucocutaneous papillomatous papules, and acral and plantar keratoses by the third decade of life.1 Importantly, CS patients have an increased risk for breast, thyroid, renal, endometrial, and colorectal cancers, as well as melanoma, with estimated lifetime risks of 85%, 35%, 33%, 28%, 9%, and 6%, respectively.2,10

Regarding the pathophysiology of PHTS disorders, PTEN encodes a phosphatase that inhibits phosphoinositide 3-kinase/Akt and mTOR signaling pathways, thereby controlling cell proliferation, cell-cycle progression, and apoptosis.2,3 Loss of PTEN function, as seen in CS patients, results in an increased risk for cancer.2 Other genetic diseases, including juvenile polyposis syndrome, Proteus syndrome, tuberous sclerosis, and Peutz-Jeghers syndrome, have phenotypic similarities to PHTS.3 Specifically, loss-of-function mutations of TSC1 and TSC2, tumor suppressor genes associated with tuberous sclerosis, similarly result in dysregulation of mTOR signaling.

Our patient also had some clinical features characteristic of BHDS, such as flesh-colored facial papules, acrochordonlike lesions, and chromophobe renal cell carcinoma.11 Birt-Hogg-Dubé syndrome most often is caused by an autosomal-dominant germline mutation in FLCN, a tumor suppressor gene.11 Interestingly, FLCN interacts with AMP-activated protein kinase to help regulate mTOR signaling, which may explain phenotypic similarities seen in CS and BHDS.12
Because the PHTS disorders and BHDS result in similar functional consequences on the mTOR signaling pathway, patients can present with overlapping clinical features that may be diagnostically challenging. Management includes patient education regarding cancer risk, surveillance for early detection of malignancy, and genetic counseling for family members.2 It is important for clinicians to appreciate phenotypic similarities between PHTS and other disorders affecting mTOR signaling to prevent delays in diagnosis.
- Nosé V. Genodermatosis affecting the skin and mucosa of the head and neck: clinicopathologic, genetic, and molecular aspect—PTEN-hamartoma tumor syndrome/Cowden syndrome. Head Neck Pathol. 2016;10:131-138.
- Porto A, Roider E, Ruzicka T. Cowden syndrome: report of a case and brief review of literature. An Bras Dermatol. 2013;88(6 suppl 1):S52-S55.
- Leslie N, Longy M. Inherited PTEN mutations and the prediction of phenotype. Semin Cell Dev Biol. 2016;52:30-38.
- The National Comprehensive Cancer Network (NCCN) clinical practice guidelines in oncology. genetic/familial high-risk assessment: breast and ovarian (version 1.2017). Published September 19, 2016. Accessed August 11, 2021. https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf
- Laury AR, Bongiovanni M, Tille J, et al. Thyroid pathology in PTEN-hamartoma tumor syndrome: characteristic findings of a distinct entity. Thyroid. 2011;21:135-144.
- Eng C. PTEN hamartoma tumor syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. University of Washington; 2001.
- Golden N, Tjokorda MGB, Sri M, et al. Management of unusual dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease) in a developing country: case report and review of the literature. Asian J Neurosurg. 2016;11:170.
- Biesecker LG, Happle R, Mulliken JB, et al. Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999;84:389-395.
- Busa T, Milh M, Degardin N, et al. Clinical presentation of PTEN mutations in childhood in the absence of family history of Cowden syndrome. Eur J Paediatr Neurol. 2015;19:188-192.
- Tan MH, Mester JL, Ngeow J, et al. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18:400-407.
- Ponti G, Pellacani G, Seidenari S, et al. Cancer-associated genodermatoses: skin neoplasms as clues to hereditary tumor syndromes. Crit Rev Oncol Hematol. 2013;85:239-256.
- Baba M, Hong S, Sharma N, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A. 2006;103:15552-15557.
To the Editor:
PTEN hamartoma tumor syndrome (PHTS) encompasses a spectrum of disorders that most commonly are caused by autosomal-dominant germline mutations in the phosphatase and tensin homolog, PTEN, tumor suppressor gene on chromosome 10q23. We describe a patient who presented with clinical features of PHTS and Birt-Hogg-Dubé syndrome (BHDS). Because the genetic mutations associated with both PHTS and BHDS result in altered mammalian target of rapamycin (mTOR) signaling, patients may have overlapping phenotypic features.
A 51-year-old man with a history of multiple carcinomas presented for evaluation of flesh-colored papules on the cheeks, nose, tongue, and hands, in addition to numerous skin tags on the neck, axillae, and lower abdomen bilaterally. His medical history was notable for several nasal and gastrointestinal tract polyps, chromophobe renal cell carcinoma, cutaneous lipomas, atypical carcinoid syndrome of the right lung, and a multinodular thyroid. His family history was notable for small cell lung cancer in his father, breast cancer and pancreatic cancer in his maternal aunt, esophageal cancer in his maternal grandfather, and celiac disease in his daughter.
Clinical examination revealed flesh-colored, dome-shaped papules measuring 1 to 2 mm in diameter on the nose and cheeks (Figure 1). He had hyperkeratotic papules on the dorsal fingers, consistent with acral keratoses. Additionally, multiple flesh-colored papules with a cobblestonelike appearance were noted on the oral mucosa (Figure 2). Other findings included pedunculated papules on the neck, axillae, and lower abdomen bilaterally, consistent with fibroepithelial polyps, as well as hyperpigmented velvety plaques on the axillae, characteristic of acanthosis nigricans (Figure 3). A shave biopsy of a papule on the right cheek revealed a proliferation of plump stellate fibroblasts, small blood vessels, and thick collagen bundles, characteristic of a fibrous papule (Figure 4).
Differential diagnoses for our patient included BHDS and Cowden syndrome (CS). Due to the combination of extensive family history of multiorgan cancers as well as the clinical findings, he was referred to a geneticist for further evaluation. Genetic analysis was positive for a heterozygous mutation variant of uncertain significance in the PTEN gene.
The PHTS disorders include CS, Bannayan-Riley-Ruvalcaba syndrome, Lhermitte-Duclos disease, Proteus syndrome, and Proteus-like syndrome (Table).1-9 Our patient’s clinical findings were indicative of CS, a rare genodermatosis characterized by multiple hamartomas and neoplasms of ectodermal, mesodermal, and endodermal origin.1 Most CS patients develop trichilemmomas of the central face, mucocutaneous papillomatous papules, and acral and plantar keratoses by the third decade of life.1 Importantly, CS patients have an increased risk for breast, thyroid, renal, endometrial, and colorectal cancers, as well as melanoma, with estimated lifetime risks of 85%, 35%, 33%, 28%, 9%, and 6%, respectively.2,10
Regarding the pathophysiology of PHTS disorders, PTEN encodes a phosphatase that inhibits phosphoinositide 3-kinase/Akt and mTOR signaling pathways, thereby controlling cell proliferation, cell-cycle progression, and apoptosis.2,3 Loss of PTEN function, as seen in CS patients, results in an increased risk for cancer.2 Other genetic diseases, including juvenile polyposis syndrome, Proteus syndrome, tuberous sclerosis, and Peutz-Jeghers syndrome, have phenotypic similarities to PHTS.3 Specifically, loss-of-function mutations of TSC1 and TSC2, tumor suppressor genes associated with tuberous sclerosis, similarly result in dysregulation of mTOR signaling.
Our patient also had some clinical features characteristic of BHDS, such as flesh-colored facial papules, acrochordonlike lesions, and chromophobe renal cell carcinoma.11 Birt-Hogg-Dubé syndrome most often is caused by an autosomal-dominant germline mutation in FLCN, a tumor suppressor gene.11 Interestingly, FLCN interacts with AMP-activated protein kinase to help regulate mTOR signaling, which may explain phenotypic similarities seen in CS and BHDS.12
Because the PHTS disorders and BHDS result in similar functional consequences on the mTOR signaling pathway, patients can present with overlapping clinical features that may be diagnostically challenging. Management includes patient education regarding cancer risk, surveillance for early detection of malignancy, and genetic counseling for family members.2 It is important for clinicians to appreciate phenotypic similarities between PHTS and other disorders affecting mTOR signaling to prevent delays in diagnosis.
To the Editor:
PTEN hamartoma tumor syndrome (PHTS) encompasses a spectrum of disorders that most commonly are caused by autosomal-dominant germline mutations in the phosphatase and tensin homolog, PTEN, tumor suppressor gene on chromosome 10q23. We describe a patient who presented with clinical features of PHTS and Birt-Hogg-Dubé syndrome (BHDS). Because the genetic mutations associated with both PHTS and BHDS result in altered mammalian target of rapamycin (mTOR) signaling, patients may have overlapping phenotypic features.
A 51-year-old man with a history of multiple carcinomas presented for evaluation of flesh-colored papules on the cheeks, nose, tongue, and hands, in addition to numerous skin tags on the neck, axillae, and lower abdomen bilaterally. His medical history was notable for several nasal and gastrointestinal tract polyps, chromophobe renal cell carcinoma, cutaneous lipomas, atypical carcinoid syndrome of the right lung, and a multinodular thyroid. His family history was notable for small cell lung cancer in his father, breast cancer and pancreatic cancer in his maternal aunt, esophageal cancer in his maternal grandfather, and celiac disease in his daughter.
Clinical examination revealed flesh-colored, dome-shaped papules measuring 1 to 2 mm in diameter on the nose and cheeks (Figure 1). He had hyperkeratotic papules on the dorsal fingers, consistent with acral keratoses. Additionally, multiple flesh-colored papules with a cobblestonelike appearance were noted on the oral mucosa (Figure 2). Other findings included pedunculated papules on the neck, axillae, and lower abdomen bilaterally, consistent with fibroepithelial polyps, as well as hyperpigmented velvety plaques on the axillae, characteristic of acanthosis nigricans (Figure 3). A shave biopsy of a papule on the right cheek revealed a proliferation of plump stellate fibroblasts, small blood vessels, and thick collagen bundles, characteristic of a fibrous papule (Figure 4).
Differential diagnoses for our patient included BHDS and Cowden syndrome (CS). Due to the combination of extensive family history of multiorgan cancers as well as the clinical findings, he was referred to a geneticist for further evaluation. Genetic analysis was positive for a heterozygous mutation variant of uncertain significance in the PTEN gene.
The PHTS disorders include CS, Bannayan-Riley-Ruvalcaba syndrome, Lhermitte-Duclos disease, Proteus syndrome, and Proteus-like syndrome (Table).1-9 Our patient’s clinical findings were indicative of CS, a rare genodermatosis characterized by multiple hamartomas and neoplasms of ectodermal, mesodermal, and endodermal origin.1 Most CS patients develop trichilemmomas of the central face, mucocutaneous papillomatous papules, and acral and plantar keratoses by the third decade of life.1 Importantly, CS patients have an increased risk for breast, thyroid, renal, endometrial, and colorectal cancers, as well as melanoma, with estimated lifetime risks of 85%, 35%, 33%, 28%, 9%, and 6%, respectively.2,10
Regarding the pathophysiology of PHTS disorders, PTEN encodes a phosphatase that inhibits phosphoinositide 3-kinase/Akt and mTOR signaling pathways, thereby controlling cell proliferation, cell-cycle progression, and apoptosis.2,3 Loss of PTEN function, as seen in CS patients, results in an increased risk for cancer.2 Other genetic diseases, including juvenile polyposis syndrome, Proteus syndrome, tuberous sclerosis, and Peutz-Jeghers syndrome, have phenotypic similarities to PHTS.3 Specifically, loss-of-function mutations of TSC1 and TSC2, tumor suppressor genes associated with tuberous sclerosis, similarly result in dysregulation of mTOR signaling.
Our patient also had some clinical features characteristic of BHDS, such as flesh-colored facial papules, acrochordonlike lesions, and chromophobe renal cell carcinoma.11 Birt-Hogg-Dubé syndrome most often is caused by an autosomal-dominant germline mutation in FLCN, a tumor suppressor gene.11 Interestingly, FLCN interacts with AMP-activated protein kinase to help regulate mTOR signaling, which may explain phenotypic similarities seen in CS and BHDS.12
Because the PHTS disorders and BHDS result in similar functional consequences on the mTOR signaling pathway, patients can present with overlapping clinical features that may be diagnostically challenging. Management includes patient education regarding cancer risk, surveillance for early detection of malignancy, and genetic counseling for family members.2 It is important for clinicians to appreciate phenotypic similarities between PHTS and other disorders affecting mTOR signaling to prevent delays in diagnosis.
- Nosé V. Genodermatosis affecting the skin and mucosa of the head and neck: clinicopathologic, genetic, and molecular aspect—PTEN-hamartoma tumor syndrome/Cowden syndrome. Head Neck Pathol. 2016;10:131-138.
- Porto A, Roider E, Ruzicka T. Cowden syndrome: report of a case and brief review of literature. An Bras Dermatol. 2013;88(6 suppl 1):S52-S55.
- Leslie N, Longy M. Inherited PTEN mutations and the prediction of phenotype. Semin Cell Dev Biol. 2016;52:30-38.
- The National Comprehensive Cancer Network (NCCN) clinical practice guidelines in oncology. genetic/familial high-risk assessment: breast and ovarian (version 1.2017). Published September 19, 2016. Accessed August 11, 2021. https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf
- Laury AR, Bongiovanni M, Tille J, et al. Thyroid pathology in PTEN-hamartoma tumor syndrome: characteristic findings of a distinct entity. Thyroid. 2011;21:135-144.
- Eng C. PTEN hamartoma tumor syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. University of Washington; 2001.
- Golden N, Tjokorda MGB, Sri M, et al. Management of unusual dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease) in a developing country: case report and review of the literature. Asian J Neurosurg. 2016;11:170.
- Biesecker LG, Happle R, Mulliken JB, et al. Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999;84:389-395.
- Busa T, Milh M, Degardin N, et al. Clinical presentation of PTEN mutations in childhood in the absence of family history of Cowden syndrome. Eur J Paediatr Neurol. 2015;19:188-192.
- Tan MH, Mester JL, Ngeow J, et al. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18:400-407.
- Ponti G, Pellacani G, Seidenari S, et al. Cancer-associated genodermatoses: skin neoplasms as clues to hereditary tumor syndromes. Crit Rev Oncol Hematol. 2013;85:239-256.
- Baba M, Hong S, Sharma N, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A. 2006;103:15552-15557.
- Nosé V. Genodermatosis affecting the skin and mucosa of the head and neck: clinicopathologic, genetic, and molecular aspect—PTEN-hamartoma tumor syndrome/Cowden syndrome. Head Neck Pathol. 2016;10:131-138.
- Porto A, Roider E, Ruzicka T. Cowden syndrome: report of a case and brief review of literature. An Bras Dermatol. 2013;88(6 suppl 1):S52-S55.
- Leslie N, Longy M. Inherited PTEN mutations and the prediction of phenotype. Semin Cell Dev Biol. 2016;52:30-38.
- The National Comprehensive Cancer Network (NCCN) clinical practice guidelines in oncology. genetic/familial high-risk assessment: breast and ovarian (version 1.2017). Published September 19, 2016. Accessed August 11, 2021. https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf
- Laury AR, Bongiovanni M, Tille J, et al. Thyroid pathology in PTEN-hamartoma tumor syndrome: characteristic findings of a distinct entity. Thyroid. 2011;21:135-144.
- Eng C. PTEN hamartoma tumor syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. University of Washington; 2001.
- Golden N, Tjokorda MGB, Sri M, et al. Management of unusual dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease) in a developing country: case report and review of the literature. Asian J Neurosurg. 2016;11:170.
- Biesecker LG, Happle R, Mulliken JB, et al. Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999;84:389-395.
- Busa T, Milh M, Degardin N, et al. Clinical presentation of PTEN mutations in childhood in the absence of family history of Cowden syndrome. Eur J Paediatr Neurol. 2015;19:188-192.
- Tan MH, Mester JL, Ngeow J, et al. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18:400-407.
- Ponti G, Pellacani G, Seidenari S, et al. Cancer-associated genodermatoses: skin neoplasms as clues to hereditary tumor syndromes. Crit Rev Oncol Hematol. 2013;85:239-256.
- Baba M, Hong S, Sharma N, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A. 2006;103:15552-15557.
PRACTICE POINTS
- PTEN hamartoma tumor syndrome (PHTS) represents a spectrum of disorders caused by autosomal-dominant germline mutations in PTEN.
- Our patient presented with phenotypic features of PHTS and Birt-Hogg-Dubé syndrome. Given that both syndromes cause alterations in mammalian target of rapamycin signaling, overlapping phenotypic features may be seen.
- Recognizing overlapping phenotypic features of these syndromes will allow for timely diagnosis and surveillance for malignancy.
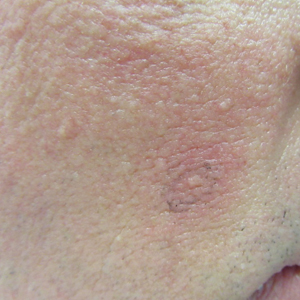
Velvety Plaques on the Abdomen and Extremities
The Diagnosis: Dermatitis Neglecta
A punch biopsy of the abdomen revealed hyperkeratosis and mild papillomatosis (Figure), which can be seen in dermatitis neglecta (DN) and acanthosis nigricans (AN) as well as confluent and reticulated papillomatosis (CARP). Due to the patient’s history of mood and psychotic disorders, collateral information was obtained from the patient’s family, who reported that the patient had a depressed mood in the last few months and was not showering or caring for herself during this period. There was no additional personal or family history of skin disease. Clinical and histopathologic findings led to a diagnosis of DN. Following recommendations for daily cleansing with soap and water along with topical ammonium lactate, near-complete resolution of the rash was achieved in 3 weeks.
Dermatitis neglecta, or unwashed dermatosis, is a skin condition that occurs secondary to poor hygiene, which was first reported in 1995 by Poskitt et al.1 Avoidance of washing in affected areas can be due to physical disability, pain after injury, neurological deficit, or psychologically induced fear or neglect. Sebum, sweat, corneocytes, and bacteria combine into compact adherent crusts of dirt, which appear as hyperkeratotic plaques with cornflakelike scale.2,3 Despite its innate simplicity, DN is a diagnostic challenge, as it clinically and histologically mimics other dermatoses including AN, terra firmaforme dermatosis, and CARP.2,4 Ultimately, the diagnosis of DN can be made when a history of poor hygiene is probable or elicited, and lesions can be removed with soap and water. Treatment of DN includes daily cleansing with soap and water; however, resistant lesions or extensive disease may require keratolytic agents, as in our patient.2-4 In contrast, terra firma-forme dermatosis, which may look similar, is not due to poor hygiene, and the lesions typically are resistant to soap and water, classically requiring isopropyl alcohol for removal. Overall, maintained awareness of DN is imperative, as early diagnosis can avoid unnecessary biopsies and more complex treatment measures as well as facilitate coordination of care when additional medical or psychiatric concerns are present.

Although the diagnoses of DN and terra firma-forme dermatosis can be distinguished based on the patient’s clinical history and response to simple cleansing measures alone, the alternate diagnoses can be excluded based on different clinical distributions and response to other treatment modalities but sometimes may require clinicopathologic correlation for definitive diagnosis. Our patient had a biopsy diagnosis of psoriasiform dermatitis from an outside provider, but neither her clinical disease nor repeated histopathologic findings supported a diagnosis of psoriasis or other classic psoriasiform dermatoses such as contact dermatitis, dermatophyte/ candidal infection, seborrheic dermatitis, pityriasis rubra pilaris, pityriasis rosea, scabies, or syphilis.
It is imperative to exclude alternative diagnoses because they can have systemic implications and can misguide treatment, as was done initially with our patient. Psoriasis vulgaris in its classic form is a chronic inflammatory skin disease that manifests as sharply demarcated, erythematous plaques with overlying thick silvery scale; it has the additional histologic findings of neutrophilic spongiform pustules in the epidermis, tortuous blood vessels in the papillary dermis, and neutrophils and parakeratosis in the stratum corneum. In its benign form, AN is associated with endocrinopathies, most commonly obesity and insulin-resistant diabetes mellitus, and presents as hyperkeratotic, velvety, hyperpigmented plaques typically limited to the neck and axillae. Malignant AN spontaneously arises in association with systemic malignancy and can be extensive and generalized.5 Treatment of AN primarily focuses on resolution of the underlying systemic disease; however, cosmetic treatment with topical or oral retinoids may hasten resolution of cutaneous disease.6 Confluent and reticulated papillomatosis is characterized by reticulated hyperkeratotic plaques with a common distribution over the central and upper trunk. Unlike DN and AN, which may occur at any age, CARP typically is seen in adolescents and young adults.7 There is no evidence-based gold standard for the management of CARP; however, the successful use of various antibiotics, antifungals, and retinoids—alone or in combination—has been reported.8 Overall, compared to the other entities in the differential diagnosis, DN easily can be prevented with consistent use of soap and water and may be underreported given the asymptomatic nature of the disease and the typical patient population.
- Poskitt L, Wayte J, Wojnarowska F, et al. ‘Dermatitis neglecta’: unwashed dermatosis. Br J Dermatol. 1995;132:827-829.
- Perez-Rodriguez IM, Munoz-Garza FZ, Ocampo-Candiani J. An unusually severe case of dermatosis neglecta: a diagnostic challenge. Case Rep Dermatol. 2014;6:194-199.
- Park JM, Roh MR, Kwon JE, et al. A case of generalized dermatitis neglecta mimicking psoriasis vulgaris. Arch Dermatol. 2010;146:1050-1051.
- Lopes S, Vide J, Antunes I, et al. Dermatitis neglecta: a challenging diagnosis in psychodermatology. Acta Dermatovenerol Alp Pannonica Adriat. 2018;27:109-110.
- Shah KR, Boland CR, Patel M, et al. Cutaneous manifestations of gastrointestinal disease: part I. J Am Acad Dermatol. 2013;68:189. e1-21; quiz 210.
- Patel NU, Roach C, Alinia H, et al. Current treatment options for acanthosis nigricans. Clin Cosmet Investig Dermatol. 2018; 11:407-413.
- Kurtyka DJ, Burke KT, DeKlotz CMC. Use of topical sirolimus (rapamycin) for treating confluent and reticulated papillomatosis. JAMA Dermatol. 2021;157:121-123.
- Mufti A, Sachdeva M, Maliyar K, et al. Treatment outcomes in confluent and reticulated papillomatosis: a systematic review. J Am Acad Dermatol. 2021;84:825-829.
The Diagnosis: Dermatitis Neglecta
A punch biopsy of the abdomen revealed hyperkeratosis and mild papillomatosis (Figure), which can be seen in dermatitis neglecta (DN) and acanthosis nigricans (AN) as well as confluent and reticulated papillomatosis (CARP). Due to the patient’s history of mood and psychotic disorders, collateral information was obtained from the patient’s family, who reported that the patient had a depressed mood in the last few months and was not showering or caring for herself during this period. There was no additional personal or family history of skin disease. Clinical and histopathologic findings led to a diagnosis of DN. Following recommendations for daily cleansing with soap and water along with topical ammonium lactate, near-complete resolution of the rash was achieved in 3 weeks.
Dermatitis neglecta, or unwashed dermatosis, is a skin condition that occurs secondary to poor hygiene, which was first reported in 1995 by Poskitt et al.1 Avoidance of washing in affected areas can be due to physical disability, pain after injury, neurological deficit, or psychologically induced fear or neglect. Sebum, sweat, corneocytes, and bacteria combine into compact adherent crusts of dirt, which appear as hyperkeratotic plaques with cornflakelike scale.2,3 Despite its innate simplicity, DN is a diagnostic challenge, as it clinically and histologically mimics other dermatoses including AN, terra firmaforme dermatosis, and CARP.2,4 Ultimately, the diagnosis of DN can be made when a history of poor hygiene is probable or elicited, and lesions can be removed with soap and water. Treatment of DN includes daily cleansing with soap and water; however, resistant lesions or extensive disease may require keratolytic agents, as in our patient.2-4 In contrast, terra firma-forme dermatosis, which may look similar, is not due to poor hygiene, and the lesions typically are resistant to soap and water, classically requiring isopropyl alcohol for removal. Overall, maintained awareness of DN is imperative, as early diagnosis can avoid unnecessary biopsies and more complex treatment measures as well as facilitate coordination of care when additional medical or psychiatric concerns are present.
Although the diagnoses of DN and terra firma-forme dermatosis can be distinguished based on the patient’s clinical history and response to simple cleansing measures alone, the alternate diagnoses can be excluded based on different clinical distributions and response to other treatment modalities but sometimes may require clinicopathologic correlation for definitive diagnosis. Our patient had a biopsy diagnosis of psoriasiform dermatitis from an outside provider, but neither her clinical disease nor repeated histopathologic findings supported a diagnosis of psoriasis or other classic psoriasiform dermatoses such as contact dermatitis, dermatophyte/ candidal infection, seborrheic dermatitis, pityriasis rubra pilaris, pityriasis rosea, scabies, or syphilis.
It is imperative to exclude alternative diagnoses because they can have systemic implications and can misguide treatment, as was done initially with our patient. Psoriasis vulgaris in its classic form is a chronic inflammatory skin disease that manifests as sharply demarcated, erythematous plaques with overlying thick silvery scale; it has the additional histologic findings of neutrophilic spongiform pustules in the epidermis, tortuous blood vessels in the papillary dermis, and neutrophils and parakeratosis in the stratum corneum. In its benign form, AN is associated with endocrinopathies, most commonly obesity and insulin-resistant diabetes mellitus, and presents as hyperkeratotic, velvety, hyperpigmented plaques typically limited to the neck and axillae. Malignant AN spontaneously arises in association with systemic malignancy and can be extensive and generalized.5 Treatment of AN primarily focuses on resolution of the underlying systemic disease; however, cosmetic treatment with topical or oral retinoids may hasten resolution of cutaneous disease.6 Confluent and reticulated papillomatosis is characterized by reticulated hyperkeratotic plaques with a common distribution over the central and upper trunk. Unlike DN and AN, which may occur at any age, CARP typically is seen in adolescents and young adults.7 There is no evidence-based gold standard for the management of CARP; however, the successful use of various antibiotics, antifungals, and retinoids—alone or in combination—has been reported.8 Overall, compared to the other entities in the differential diagnosis, DN easily can be prevented with consistent use of soap and water and may be underreported given the asymptomatic nature of the disease and the typical patient population.
The Diagnosis: Dermatitis Neglecta
A punch biopsy of the abdomen revealed hyperkeratosis and mild papillomatosis (Figure), which can be seen in dermatitis neglecta (DN) and acanthosis nigricans (AN) as well as confluent and reticulated papillomatosis (CARP). Due to the patient’s history of mood and psychotic disorders, collateral information was obtained from the patient’s family, who reported that the patient had a depressed mood in the last few months and was not showering or caring for herself during this period. There was no additional personal or family history of skin disease. Clinical and histopathologic findings led to a diagnosis of DN. Following recommendations for daily cleansing with soap and water along with topical ammonium lactate, near-complete resolution of the rash was achieved in 3 weeks.
Dermatitis neglecta, or unwashed dermatosis, is a skin condition that occurs secondary to poor hygiene, which was first reported in 1995 by Poskitt et al.1 Avoidance of washing in affected areas can be due to physical disability, pain after injury, neurological deficit, or psychologically induced fear or neglect. Sebum, sweat, corneocytes, and bacteria combine into compact adherent crusts of dirt, which appear as hyperkeratotic plaques with cornflakelike scale.2,3 Despite its innate simplicity, DN is a diagnostic challenge, as it clinically and histologically mimics other dermatoses including AN, terra firmaforme dermatosis, and CARP.2,4 Ultimately, the diagnosis of DN can be made when a history of poor hygiene is probable or elicited, and lesions can be removed with soap and water. Treatment of DN includes daily cleansing with soap and water; however, resistant lesions or extensive disease may require keratolytic agents, as in our patient.2-4 In contrast, terra firma-forme dermatosis, which may look similar, is not due to poor hygiene, and the lesions typically are resistant to soap and water, classically requiring isopropyl alcohol for removal. Overall, maintained awareness of DN is imperative, as early diagnosis can avoid unnecessary biopsies and more complex treatment measures as well as facilitate coordination of care when additional medical or psychiatric concerns are present.
Although the diagnoses of DN and terra firma-forme dermatosis can be distinguished based on the patient’s clinical history and response to simple cleansing measures alone, the alternate diagnoses can be excluded based on different clinical distributions and response to other treatment modalities but sometimes may require clinicopathologic correlation for definitive diagnosis. Our patient had a biopsy diagnosis of psoriasiform dermatitis from an outside provider, but neither her clinical disease nor repeated histopathologic findings supported a diagnosis of psoriasis or other classic psoriasiform dermatoses such as contact dermatitis, dermatophyte/ candidal infection, seborrheic dermatitis, pityriasis rubra pilaris, pityriasis rosea, scabies, or syphilis.
It is imperative to exclude alternative diagnoses because they can have systemic implications and can misguide treatment, as was done initially with our patient. Psoriasis vulgaris in its classic form is a chronic inflammatory skin disease that manifests as sharply demarcated, erythematous plaques with overlying thick silvery scale; it has the additional histologic findings of neutrophilic spongiform pustules in the epidermis, tortuous blood vessels in the papillary dermis, and neutrophils and parakeratosis in the stratum corneum. In its benign form, AN is associated with endocrinopathies, most commonly obesity and insulin-resistant diabetes mellitus, and presents as hyperkeratotic, velvety, hyperpigmented plaques typically limited to the neck and axillae. Malignant AN spontaneously arises in association with systemic malignancy and can be extensive and generalized.5 Treatment of AN primarily focuses on resolution of the underlying systemic disease; however, cosmetic treatment with topical or oral retinoids may hasten resolution of cutaneous disease.6 Confluent and reticulated papillomatosis is characterized by reticulated hyperkeratotic plaques with a common distribution over the central and upper trunk. Unlike DN and AN, which may occur at any age, CARP typically is seen in adolescents and young adults.7 There is no evidence-based gold standard for the management of CARP; however, the successful use of various antibiotics, antifungals, and retinoids—alone or in combination—has been reported.8 Overall, compared to the other entities in the differential diagnosis, DN easily can be prevented with consistent use of soap and water and may be underreported given the asymptomatic nature of the disease and the typical patient population.
- Poskitt L, Wayte J, Wojnarowska F, et al. ‘Dermatitis neglecta’: unwashed dermatosis. Br J Dermatol. 1995;132:827-829.
- Perez-Rodriguez IM, Munoz-Garza FZ, Ocampo-Candiani J. An unusually severe case of dermatosis neglecta: a diagnostic challenge. Case Rep Dermatol. 2014;6:194-199.
- Park JM, Roh MR, Kwon JE, et al. A case of generalized dermatitis neglecta mimicking psoriasis vulgaris. Arch Dermatol. 2010;146:1050-1051.
- Lopes S, Vide J, Antunes I, et al. Dermatitis neglecta: a challenging diagnosis in psychodermatology. Acta Dermatovenerol Alp Pannonica Adriat. 2018;27:109-110.
- Shah KR, Boland CR, Patel M, et al. Cutaneous manifestations of gastrointestinal disease: part I. J Am Acad Dermatol. 2013;68:189. e1-21; quiz 210.
- Patel NU, Roach C, Alinia H, et al. Current treatment options for acanthosis nigricans. Clin Cosmet Investig Dermatol. 2018; 11:407-413.
- Kurtyka DJ, Burke KT, DeKlotz CMC. Use of topical sirolimus (rapamycin) for treating confluent and reticulated papillomatosis. JAMA Dermatol. 2021;157:121-123.
- Mufti A, Sachdeva M, Maliyar K, et al. Treatment outcomes in confluent and reticulated papillomatosis: a systematic review. J Am Acad Dermatol. 2021;84:825-829.
- Poskitt L, Wayte J, Wojnarowska F, et al. ‘Dermatitis neglecta’: unwashed dermatosis. Br J Dermatol. 1995;132:827-829.
- Perez-Rodriguez IM, Munoz-Garza FZ, Ocampo-Candiani J. An unusually severe case of dermatosis neglecta: a diagnostic challenge. Case Rep Dermatol. 2014;6:194-199.
- Park JM, Roh MR, Kwon JE, et al. A case of generalized dermatitis neglecta mimicking psoriasis vulgaris. Arch Dermatol. 2010;146:1050-1051.
- Lopes S, Vide J, Antunes I, et al. Dermatitis neglecta: a challenging diagnosis in psychodermatology. Acta Dermatovenerol Alp Pannonica Adriat. 2018;27:109-110.
- Shah KR, Boland CR, Patel M, et al. Cutaneous manifestations of gastrointestinal disease: part I. J Am Acad Dermatol. 2013;68:189. e1-21; quiz 210.
- Patel NU, Roach C, Alinia H, et al. Current treatment options for acanthosis nigricans. Clin Cosmet Investig Dermatol. 2018; 11:407-413.
- Kurtyka DJ, Burke KT, DeKlotz CMC. Use of topical sirolimus (rapamycin) for treating confluent and reticulated papillomatosis. JAMA Dermatol. 2021;157:121-123.
- Mufti A, Sachdeva M, Maliyar K, et al. Treatment outcomes in confluent and reticulated papillomatosis: a systematic review. J Am Acad Dermatol. 2021;84:825-829.

A 28-year-old woman was admitted to the medicine service with bilateral pedal numbness and ataxia, as well as an asymptomatic rash on the neck, chest, abdomen, and extremities of a few months’ duration. The patient was seen by an outside dermatologist for the same rash 1 month prior, at which time a punch biopsy of the right forearm was suggestive of psoriasiform dermatitis; however, the rash failed to improve with topical ammonium lactate and corticosteroids. During the current admission, the patient was found to have low methylmalonic acid and vitamin B1 levels; however, vitamin B12, thyroid studies, rapid plasma reagin test, and inflammatory markers, as well as central and peripheral imaging and nerve conduction studies were normal.
Dermatology was consulted. Physical examination revealed retention hyperkeratosis on the neck that was wipeable with 70% isopropyl alcohol, as well as nonwipeable, thin, reticulated plaques on the mid chest and thick velvety plaques on the abdomen and bilateral extremities. There was notable sparing of areas with natural occlusion such as the back and body folds. A punch biopsy of the abdomen was performed.
Acne Vulgaris

THE COMPARISON
A A 27-year-old Hispanic woman with comedonal and inflammatory acne. Erythema is prominent around the inflammatory lesions. Note the pustule on the cheek surrounded by pink color.
B A teenaged Black boy with acne papules and pustules on the face. There are comedones, hyperpigmented macules, and pustules on the cheek.
C A teenaged Black girl with pomade acne. The patient used various hair care products, which obstructed the pilosebaceous units on the forehead.
Epidemiology
Acne is a leading dermatologic condition in individuals with skin of color in the United States.1
Key clinical features in people with darker skin tones include:
- erythematous or hyperpigmented papules or comedones
- hyperpigmented macules and postinflammatory hyperpigmentation (PIH)
- increased risk for keloidal scars.2
Worth noting
- Patients with darker skin tones may be more concerned with the dark marks (also referred to as scars or manchas in Spanish) than the acne itself. This PIH may be viewed by patients as the major problem.
- Acne medications such as azelaic acid and some retinoids (when applied appropriately) can treat both acne and PIH.3
- Irritation from topical acne medications, including retinoid dermatitis, may lead to more PIH. Using noncomedogenic moisturizers and applying medication appropriately (ie, a pea-sized amount of topical retinoid per application) may help limit irritation.4,5
- One type of acne seen more commonly, although not exclusively, in Black patients is pomade acne, which principally appears on the forehead and is associated with use of hair care and styling products (Figure, C).
Health disparity highlight
Disparities in access to health care exist for those with dermatologic concerns. According to one study, African American (28.5%) and Hispanic patients (23.9%) were less likely to be seen by a dermatologist solely for the diagnosis of a dermatologic condition compared to Asian and Pacific Islander patients (36.7%) or White patients (43.2%).1
Noting that isotretinoin is the most potent systemic therapy for severe cystic acne vulgaris, Bell et al6 reported that Black patients had lower odds of receiving isotretinoin compared to White patients. Hispanic patients had lower odds of receiving a topical retinoid, tretinoin, than non-Hispanic patients.6
- Davis SA, Narahari S, Feldman SR, et al. Top dermatologic conditions in patients of color: an analysis of nationally representative data. J Drugs Dermatol. 2012;11:466-473.
- Alexis AF, Woolery-Lloyd H, Williams K, et al. Racial/ethnic variations in acne: implications for treatment and skin care recommendations for acne patients with skin of color. J Drugs Dermatol. 2021;20:716-725.
- Woolery-Lloyd HC, Keri J, Doig S. Retinoids and azelaic acid to treat acne and hyperpigmentation in skin of color. J Drugs Dermatol. 2013;12:434-437.
- Grayson C, Heath C. Tips for addressing common conditions affecting pediatric and adolescent patients with skin of color [published online March 2, 2021]. Pediatr Dermatol. doi:10.1111/pde.14525
- Alexis AD, Harper JC, Stein Gold L, et al. Treating acne in patients with skin of color. Semin Cutan Med Surg. 2018;37(suppl 3):S71-S73.
- Bell MA, Whang KA, Thomas J, et al. Racial and ethnic disparities in access to emerging and frontline therapies in common dermatological conditions: a cross-sectional study. J Natl Med Assoc. 2020;112:650-653.
THE COMPARISON
A A 27-year-old Hispanic woman with comedonal and inflammatory acne. Erythema is prominent around the inflammatory lesions. Note the pustule on the cheek surrounded by pink color.
B A teenaged Black boy with acne papules and pustules on the face. There are comedones, hyperpigmented macules, and pustules on the cheek.
C A teenaged Black girl with pomade acne. The patient used various hair care products, which obstructed the pilosebaceous units on the forehead.
Epidemiology
Acne is a leading dermatologic condition in individuals with skin of color in the United States.1
Key clinical features in people with darker skin tones include:
- erythematous or hyperpigmented papules or comedones
- hyperpigmented macules and postinflammatory hyperpigmentation (PIH)
- increased risk for keloidal scars.2
Worth noting
- Patients with darker skin tones may be more concerned with the dark marks (also referred to as scars or manchas in Spanish) than the acne itself. This PIH may be viewed by patients as the major problem.
- Acne medications such as azelaic acid and some retinoids (when applied appropriately) can treat both acne and PIH.3
- Irritation from topical acne medications, including retinoid dermatitis, may lead to more PIH. Using noncomedogenic moisturizers and applying medication appropriately (ie, a pea-sized amount of topical retinoid per application) may help limit irritation.4,5
- One type of acne seen more commonly, although not exclusively, in Black patients is pomade acne, which principally appears on the forehead and is associated with use of hair care and styling products (Figure, C).
Health disparity highlight
Disparities in access to health care exist for those with dermatologic concerns. According to one study, African American (28.5%) and Hispanic patients (23.9%) were less likely to be seen by a dermatologist solely for the diagnosis of a dermatologic condition compared to Asian and Pacific Islander patients (36.7%) or White patients (43.2%).1
Noting that isotretinoin is the most potent systemic therapy for severe cystic acne vulgaris, Bell et al6 reported that Black patients had lower odds of receiving isotretinoin compared to White patients. Hispanic patients had lower odds of receiving a topical retinoid, tretinoin, than non-Hispanic patients.6
THE COMPARISON
A A 27-year-old Hispanic woman with comedonal and inflammatory acne. Erythema is prominent around the inflammatory lesions. Note the pustule on the cheek surrounded by pink color.
B A teenaged Black boy with acne papules and pustules on the face. There are comedones, hyperpigmented macules, and pustules on the cheek.
C A teenaged Black girl with pomade acne. The patient used various hair care products, which obstructed the pilosebaceous units on the forehead.
Epidemiology
Acne is a leading dermatologic condition in individuals with skin of color in the United States.1
Key clinical features in people with darker skin tones include:
- erythematous or hyperpigmented papules or comedones
- hyperpigmented macules and postinflammatory hyperpigmentation (PIH)
- increased risk for keloidal scars.2
Worth noting
- Patients with darker skin tones may be more concerned with the dark marks (also referred to as scars or manchas in Spanish) than the acne itself. This PIH may be viewed by patients as the major problem.
- Acne medications such as azelaic acid and some retinoids (when applied appropriately) can treat both acne and PIH.3
- Irritation from topical acne medications, including retinoid dermatitis, may lead to more PIH. Using noncomedogenic moisturizers and applying medication appropriately (ie, a pea-sized amount of topical retinoid per application) may help limit irritation.4,5
- One type of acne seen more commonly, although not exclusively, in Black patients is pomade acne, which principally appears on the forehead and is associated with use of hair care and styling products (Figure, C).
Health disparity highlight
Disparities in access to health care exist for those with dermatologic concerns. According to one study, African American (28.5%) and Hispanic patients (23.9%) were less likely to be seen by a dermatologist solely for the diagnosis of a dermatologic condition compared to Asian and Pacific Islander patients (36.7%) or White patients (43.2%).1
Noting that isotretinoin is the most potent systemic therapy for severe cystic acne vulgaris, Bell et al6 reported that Black patients had lower odds of receiving isotretinoin compared to White patients. Hispanic patients had lower odds of receiving a topical retinoid, tretinoin, than non-Hispanic patients.6
- Davis SA, Narahari S, Feldman SR, et al. Top dermatologic conditions in patients of color: an analysis of nationally representative data. J Drugs Dermatol. 2012;11:466-473.
- Alexis AF, Woolery-Lloyd H, Williams K, et al. Racial/ethnic variations in acne: implications for treatment and skin care recommendations for acne patients with skin of color. J Drugs Dermatol. 2021;20:716-725.
- Woolery-Lloyd HC, Keri J, Doig S. Retinoids and azelaic acid to treat acne and hyperpigmentation in skin of color. J Drugs Dermatol. 2013;12:434-437.
- Grayson C, Heath C. Tips for addressing common conditions affecting pediatric and adolescent patients with skin of color [published online March 2, 2021]. Pediatr Dermatol. doi:10.1111/pde.14525
- Alexis AD, Harper JC, Stein Gold L, et al. Treating acne in patients with skin of color. Semin Cutan Med Surg. 2018;37(suppl 3):S71-S73.
- Bell MA, Whang KA, Thomas J, et al. Racial and ethnic disparities in access to emerging and frontline therapies in common dermatological conditions: a cross-sectional study. J Natl Med Assoc. 2020;112:650-653.
- Davis SA, Narahari S, Feldman SR, et al. Top dermatologic conditions in patients of color: an analysis of nationally representative data. J Drugs Dermatol. 2012;11:466-473.
- Alexis AF, Woolery-Lloyd H, Williams K, et al. Racial/ethnic variations in acne: implications for treatment and skin care recommendations for acne patients with skin of color. J Drugs Dermatol. 2021;20:716-725.
- Woolery-Lloyd HC, Keri J, Doig S. Retinoids and azelaic acid to treat acne and hyperpigmentation in skin of color. J Drugs Dermatol. 2013;12:434-437.
- Grayson C, Heath C. Tips for addressing common conditions affecting pediatric and adolescent patients with skin of color [published online March 2, 2021]. Pediatr Dermatol. doi:10.1111/pde.14525
- Alexis AD, Harper JC, Stein Gold L, et al. Treating acne in patients with skin of color. Semin Cutan Med Surg. 2018;37(suppl 3):S71-S73.
- Bell MA, Whang KA, Thomas J, et al. Racial and ethnic disparities in access to emerging and frontline therapies in common dermatological conditions: a cross-sectional study. J Natl Med Assoc. 2020;112:650-653.

Erythematous and Ulcerated Plaque on the Left Temple
The Diagnosis: Primary Cutaneous Carcinosarcoma
The immunohistochemical findings supported an epithelial component consistent with moderately differentiated squamous cell carcinoma (SCC) and a mesenchymal component with features consistent with a sarcoma. Consequently, the lesion was diagnosed as a primary cutaneous carcinosarcoma (PCCS).
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm consisting of malignant epithelial (carcinoma) and mesenchymal (sarcoma) components.1 Primary cutaneous carcinosarcomas are uncommon, poorly understood, primary cutaneous tumors.2,3 Characteristic of this tumor, cytokeratins highlight the epithelial component while vimentin highlights the mesenchymal component.4 Histologically, the sarcomatous components of PCCS often are highly variable, with an absence of transitional areas within the epithelial component, which frequently resembles basal cell carcinoma and/ or SCC.5-7 Primary cutaneous carcinosarcoma favors areas of chronic UV radiation exposure, particularly on the head and neck. Most tumors present with a slowly growing, polypoid, flesh-colored to erythematous nodule due to the infiltrative mesenchymal component.7 Primary cutaneous carcinosarcoma primarily is diagnosed in elderly patients, with the majority of cases diagnosed in the eighth or ninth decades of life (range, 32–98 years).1,8 Men appear to be twice as likely to be diagnosed with a PCCS compared to women.1 Primary cutaneous carcinosarcomas are recognized as aggressive tumors with a high propensity to metastasize and recur locally, necessitating early diagnosis and treatment.4 Accurate diagnosis of PCCSs can be challenging due to the biphasic nature of the neoplasm as well as poor differentiation or unequal proportions of the epithelial and mesenchymal components.5 Additionally, overlapping diagnostic criteria coupled with vague demarcation between soft-tissue sarcomas and distinct carcinomas also may contribute to a delay in diagnosis.9 Treatment is achieved surgically by complete wide resection, with no evidence to support the use of adjuvant or neoadjuvant external beam radiation therapy. Due to the small number of reported cases, no treatment recommendations currently exist.1
Surgical management with wide local excision has been disappointing, with recurrence rates reported as high as 33%.6 Primary cutaneous carcinosarcoma has an estimated overall recurrence rate of 19% and a 5-year disease-free rate of 50%.10 Risk factors associated with poorer prognosis include tumors with adnexal subtype, age less than 65 years, rapid tumor growth, a tumor greater than 20 mm at presentation, and a long-standing tumor lasting up to 30 years.2,4 Although wide local excision and Mohs micrographic surgery (MMS) both have been utilized successfully, MMS has been shown to result in a cure rate of greater than 98%.6
Atypical fibroxanthoma (AFX) is a cutaneous tumor of fibrohistiocytic mesenchymal origin that typically manifests on sun-damaged skin in elderly individuals. Clinically, it presents as a rapidly growing neoplasm that often ulcerates and bleeds. These heterogenous neoplasms have several distinct characteristics, including dense cellularity with disorganized, large, pleomorphic, and atypical-appearing spindle-shaped cells arising in the upper layers of the dermis, often disseminating into the reticular dermis and occasionally into the subcutaneous fat (Figure 1). The neoplastic cells often exhibit hyperchromic and irregular nuclei, multinucleated giant cells, and atypical mitotic figures. In most cases, negative immunohistochemical staining with SOX-10, S-100, cytokeratins, desmin, and caldesmon will allow pathologists to differentiate between AFX and other common tumors on the differential diagnosis, such as SCC, melanoma, and leiomyosarcoma. CD10 and procollagen type 1 are positive antigenic markers in AFX, but they are not specific. The standard treatment of AFX includes wide local excision or MMS for superior margin control.11
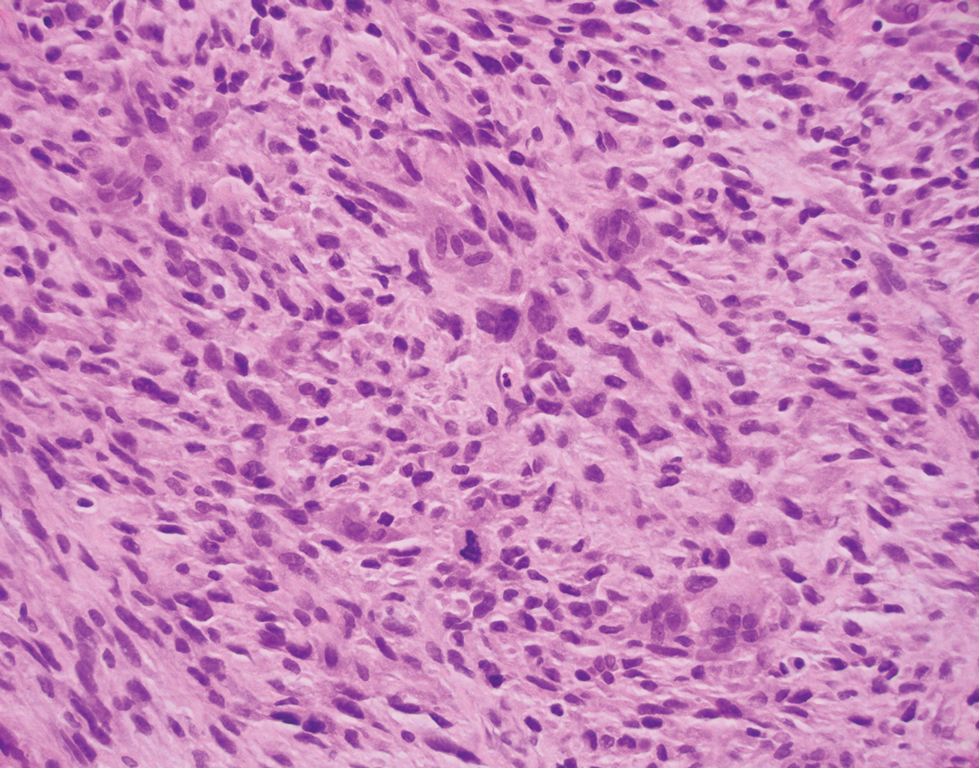
Spindle cell SCC presents as a raised or exophytic nodule, often with spontaneous bleeding and central ulceration. It usually presents on sun-damaged skin or in individuals with a history of ionizing radiation. Histologically, it is characterized by atypical spindleshaped keratinocytes in the dermis existing as single cells or cohesive nests along with keratin pearls (Figure 2). The atypical spindle cells may comprise the entire tumor or only a small portion. The use of immunohistochemical markers often is required to establish a definitive diagnosis. Spindle cell SCC stains positively, albeit frequently focally, for p63, p40, and high-molecular-weight cytokeratins such as cytokeratin 5/6, while S-100 protein, SOX-10, MART-1/Melan-A, and muscle-specific actin stains typically are negative. Wide local excision or MMS is recommended for treatment of these lesions.12
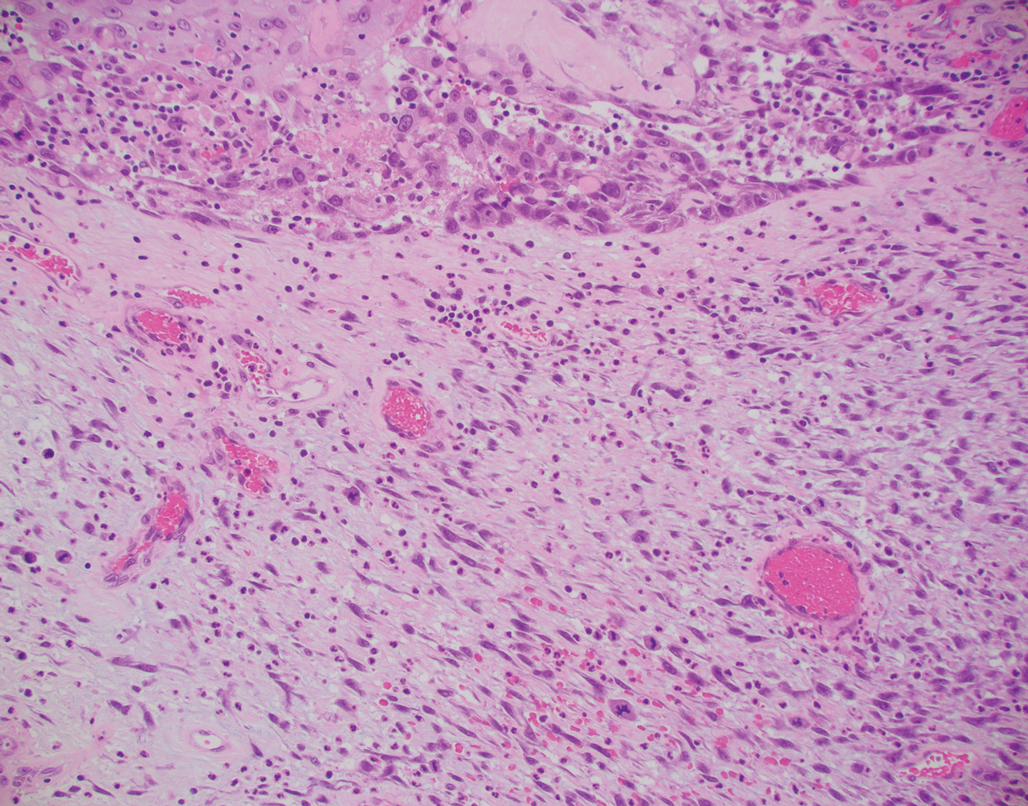
Primary cutaneous myoepithelial carcinomas are uncommon neoplasms of myoepithelial differentiation. Clinically, they often arise as soft nodular lesions on the head, neck, and lower extremities with a bimodal age distribution (50 years). Histologically cutaneous myoepithelial tumors are well-differentiated, dermal-based nodules without connection to the overlying epidermis (Figure 3). The myoepithelial cells can exhibit spindled, epithelioid, plasmacytoid, or clear cell morphologic features and show variability in cell growth patterns. One of the most common growth patterns is oval to round cells forming cords and chains in a chondromyxoid stroma. Most cases display an immunophenotyped co-expression of an epithelial cytokeratin and S-100 protein. Myoepithelial markers also may be present, including keratins, smooth muscle actin, calponin, glial fibrillary acidic protein, p63, and desmin. Surgical removal with wide local excision or MMS is essential.13
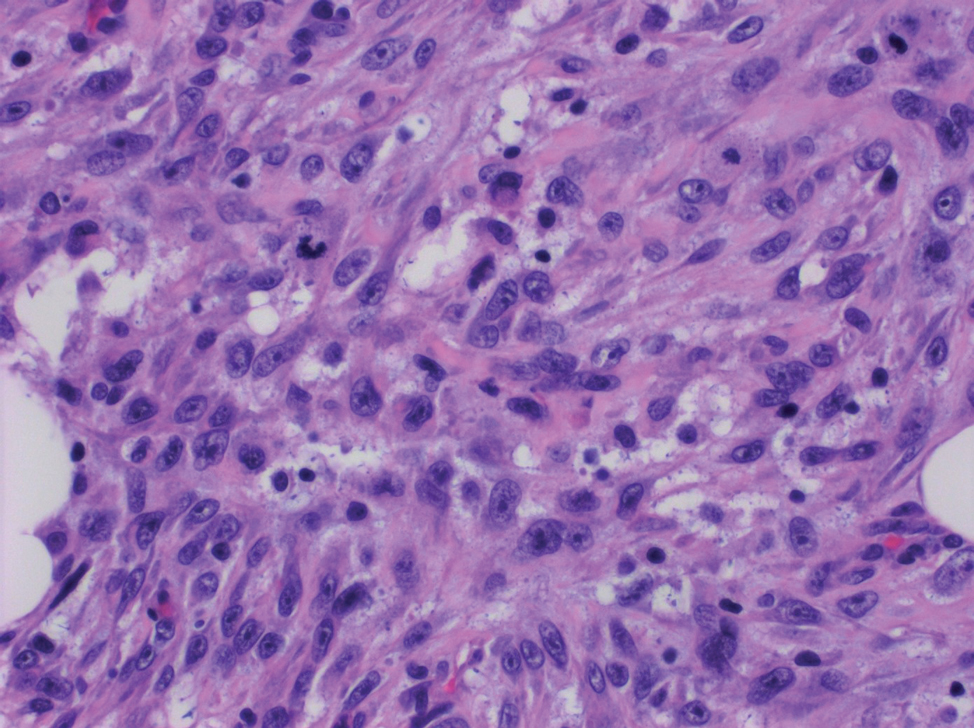
Leiomyosarcoma (LMS) is a tumor that originates from smooth muscle and rarely develops in the dermis.14 Pleomorphic LMS is a morphologic variant of LMS that has a low propensity to metastasize but commonly exhibits local recurrence.15 Leiomyosarcoma can present in any age group but most commonly manifests in individuals aged 50 to 70 years. Clinically, LMS presents as a firm solitary nodule with a smooth pink surface or a more exophytic tumor with a reddish or brown color on the extensor surface of the lower limbs; it is less common on the scalp and face.14 Histologically, most cases of pleomorphic LMS show small foci of fascicles consisting of smooth muscle tumor cells in addition to cellular pleomorphism (Figure 4).15 Many of these cells demonstrate a clear perinuclear vacuole that generally is appreciated in neoplastic smooth muscle cells.14 Pleomorphic LMS typically stains positively for at least one smooth muscle marker including desmin, h-caldesmon, muscle-specific actin, α-smooth muscle actin, or smooth muscle myosin in the leiomyosarcomatous fascicular areas.16 Complete surgical excision is the treatment of choice, and the best results are obtained with MMS.14
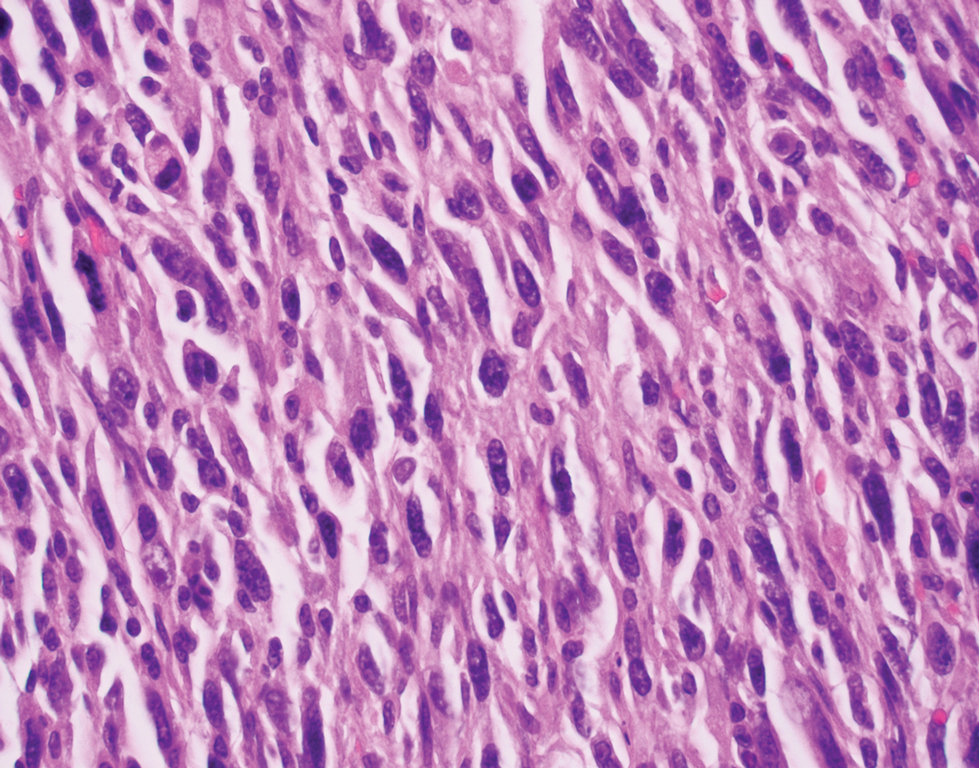
- Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
- Bourgeault E, Alain J, Gagne E. Primary cutaneous carcinosarcoma of the basal cell subtype should be treated as a high-risk basal cell carcinoma. J Cutan Med Surg. 2015;19:407-411.
- West L, Srivastava D. Cutaneous carcinosarcoma of the medial canthus discovered on Mohs debulk analysis. Dermatol Surg. 2019;45:1700-1702.
- Kwan JM, Satter EK. Carcinosarcoma: a primary cutaneous tumor with biphasic differentiation. Cutis. 2013;92:247-249.
- Suh KY, Lacouture M, Gerami P. p63 in primary cutaneous carcinosarcoma. Am J Dermatopathol. 2007;29:374‐377.
- Ruiz-Villaverde R, Aneiros-Fernandez J. Primary cutaneous carcinosarcoma: a cutaneous neoplasm with an exceptional presentation. Sultan Qaboos Univ Med J. 2018;18:E114-E115.
- Smart CN, Pucci RA, Binder SW, et al. Cutaneous carcinosarcoma with myoepithelial differentiation: immunohistochemical and cytogenetic analysis of a case presenting in an unusual location. Am J Dermatopathol. 2009;31:715‐717.
- Clark JJ, Bowen AR, Bowen GM, et al. Cutaneous carcinosarcoma: a series of six cases and a review of the literature. J Cutan Pathol. 2017;44:34‐44.
- Müller CS, Pföhler C, Schiekofer C, et al. Primary cutaneous carcinosarcomas: a morphological histogenetic concept revisited. Am J Dermatopathol. 2014;36:328‐339.
- Bellew S, Del Rosso JQ, Mobini N. Primary carcinosarcoma of the ear: case report and review of the literature. J Clin Aesthet Dermatol. 2009;2:33‐35.
- Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
- Soleymani T, Aasi SZ, Novoa R, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma: updates on classification and management. Dermatol Clin. 2019;37:253-259.
- Parekh V, Seykora JT. Cutaneous squamous cell carcinoma. Clin Lab Med. 2017;37:503-525.
- Johnson GE, Stevens K, Morrison AO, et al. Cutaneous myoepithelial carcinoma with disseminated metastases. Cutis. 2017;99:E19-E26.
- Llombart B, Serra-Guillén C, Requena C, et al. Leiomyosarcoma and pleomorphic dermal sarcoma: guidelines for diagnosis and treatment. Actas Dermosifiliogr. 2019;110:4-11.
- Oda Y, Miyajima K, Kawaguchi K, et al. Pleomorphic leiomyosarcoma: clinicopathologic and immunohistochemical study with special emphasis on its distinction from ordinary leiomyosarcoma and malignant fibrous histiocytoma. Am J Surg Pathol. 2001;25:1030-1038.
The Diagnosis: Primary Cutaneous Carcinosarcoma
The immunohistochemical findings supported an epithelial component consistent with moderately differentiated squamous cell carcinoma (SCC) and a mesenchymal component with features consistent with a sarcoma. Consequently, the lesion was diagnosed as a primary cutaneous carcinosarcoma (PCCS).
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm consisting of malignant epithelial (carcinoma) and mesenchymal (sarcoma) components.1 Primary cutaneous carcinosarcomas are uncommon, poorly understood, primary cutaneous tumors.2,3 Characteristic of this tumor, cytokeratins highlight the epithelial component while vimentin highlights the mesenchymal component.4 Histologically, the sarcomatous components of PCCS often are highly variable, with an absence of transitional areas within the epithelial component, which frequently resembles basal cell carcinoma and/ or SCC.5-7 Primary cutaneous carcinosarcoma favors areas of chronic UV radiation exposure, particularly on the head and neck. Most tumors present with a slowly growing, polypoid, flesh-colored to erythematous nodule due to the infiltrative mesenchymal component.7 Primary cutaneous carcinosarcoma primarily is diagnosed in elderly patients, with the majority of cases diagnosed in the eighth or ninth decades of life (range, 32–98 years).1,8 Men appear to be twice as likely to be diagnosed with a PCCS compared to women.1 Primary cutaneous carcinosarcomas are recognized as aggressive tumors with a high propensity to metastasize and recur locally, necessitating early diagnosis and treatment.4 Accurate diagnosis of PCCSs can be challenging due to the biphasic nature of the neoplasm as well as poor differentiation or unequal proportions of the epithelial and mesenchymal components.5 Additionally, overlapping diagnostic criteria coupled with vague demarcation between soft-tissue sarcomas and distinct carcinomas also may contribute to a delay in diagnosis.9 Treatment is achieved surgically by complete wide resection, with no evidence to support the use of adjuvant or neoadjuvant external beam radiation therapy. Due to the small number of reported cases, no treatment recommendations currently exist.1
Surgical management with wide local excision has been disappointing, with recurrence rates reported as high as 33%.6 Primary cutaneous carcinosarcoma has an estimated overall recurrence rate of 19% and a 5-year disease-free rate of 50%.10 Risk factors associated with poorer prognosis include tumors with adnexal subtype, age less than 65 years, rapid tumor growth, a tumor greater than 20 mm at presentation, and a long-standing tumor lasting up to 30 years.2,4 Although wide local excision and Mohs micrographic surgery (MMS) both have been utilized successfully, MMS has been shown to result in a cure rate of greater than 98%.6
Atypical fibroxanthoma (AFX) is a cutaneous tumor of fibrohistiocytic mesenchymal origin that typically manifests on sun-damaged skin in elderly individuals. Clinically, it presents as a rapidly growing neoplasm that often ulcerates and bleeds. These heterogenous neoplasms have several distinct characteristics, including dense cellularity with disorganized, large, pleomorphic, and atypical-appearing spindle-shaped cells arising in the upper layers of the dermis, often disseminating into the reticular dermis and occasionally into the subcutaneous fat (Figure 1). The neoplastic cells often exhibit hyperchromic and irregular nuclei, multinucleated giant cells, and atypical mitotic figures. In most cases, negative immunohistochemical staining with SOX-10, S-100, cytokeratins, desmin, and caldesmon will allow pathologists to differentiate between AFX and other common tumors on the differential diagnosis, such as SCC, melanoma, and leiomyosarcoma. CD10 and procollagen type 1 are positive antigenic markers in AFX, but they are not specific. The standard treatment of AFX includes wide local excision or MMS for superior margin control.11
Spindle cell SCC presents as a raised or exophytic nodule, often with spontaneous bleeding and central ulceration. It usually presents on sun-damaged skin or in individuals with a history of ionizing radiation. Histologically, it is characterized by atypical spindleshaped keratinocytes in the dermis existing as single cells or cohesive nests along with keratin pearls (Figure 2). The atypical spindle cells may comprise the entire tumor or only a small portion. The use of immunohistochemical markers often is required to establish a definitive diagnosis. Spindle cell SCC stains positively, albeit frequently focally, for p63, p40, and high-molecular-weight cytokeratins such as cytokeratin 5/6, while S-100 protein, SOX-10, MART-1/Melan-A, and muscle-specific actin stains typically are negative. Wide local excision or MMS is recommended for treatment of these lesions.12
Primary cutaneous myoepithelial carcinomas are uncommon neoplasms of myoepithelial differentiation. Clinically, they often arise as soft nodular lesions on the head, neck, and lower extremities with a bimodal age distribution (50 years). Histologically cutaneous myoepithelial tumors are well-differentiated, dermal-based nodules without connection to the overlying epidermis (Figure 3). The myoepithelial cells can exhibit spindled, epithelioid, plasmacytoid, or clear cell morphologic features and show variability in cell growth patterns. One of the most common growth patterns is oval to round cells forming cords and chains in a chondromyxoid stroma. Most cases display an immunophenotyped co-expression of an epithelial cytokeratin and S-100 protein. Myoepithelial markers also may be present, including keratins, smooth muscle actin, calponin, glial fibrillary acidic protein, p63, and desmin. Surgical removal with wide local excision or MMS is essential.13
Leiomyosarcoma (LMS) is a tumor that originates from smooth muscle and rarely develops in the dermis.14 Pleomorphic LMS is a morphologic variant of LMS that has a low propensity to metastasize but commonly exhibits local recurrence.15 Leiomyosarcoma can present in any age group but most commonly manifests in individuals aged 50 to 70 years. Clinically, LMS presents as a firm solitary nodule with a smooth pink surface or a more exophytic tumor with a reddish or brown color on the extensor surface of the lower limbs; it is less common on the scalp and face.14 Histologically, most cases of pleomorphic LMS show small foci of fascicles consisting of smooth muscle tumor cells in addition to cellular pleomorphism (Figure 4).15 Many of these cells demonstrate a clear perinuclear vacuole that generally is appreciated in neoplastic smooth muscle cells.14 Pleomorphic LMS typically stains positively for at least one smooth muscle marker including desmin, h-caldesmon, muscle-specific actin, α-smooth muscle actin, or smooth muscle myosin in the leiomyosarcomatous fascicular areas.16 Complete surgical excision is the treatment of choice, and the best results are obtained with MMS.14
The Diagnosis: Primary Cutaneous Carcinosarcoma
The immunohistochemical findings supported an epithelial component consistent with moderately differentiated squamous cell carcinoma (SCC) and a mesenchymal component with features consistent with a sarcoma. Consequently, the lesion was diagnosed as a primary cutaneous carcinosarcoma (PCCS).
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm consisting of malignant epithelial (carcinoma) and mesenchymal (sarcoma) components.1 Primary cutaneous carcinosarcomas are uncommon, poorly understood, primary cutaneous tumors.2,3 Characteristic of this tumor, cytokeratins highlight the epithelial component while vimentin highlights the mesenchymal component.4 Histologically, the sarcomatous components of PCCS often are highly variable, with an absence of transitional areas within the epithelial component, which frequently resembles basal cell carcinoma and/ or SCC.5-7 Primary cutaneous carcinosarcoma favors areas of chronic UV radiation exposure, particularly on the head and neck. Most tumors present with a slowly growing, polypoid, flesh-colored to erythematous nodule due to the infiltrative mesenchymal component.7 Primary cutaneous carcinosarcoma primarily is diagnosed in elderly patients, with the majority of cases diagnosed in the eighth or ninth decades of life (range, 32–98 years).1,8 Men appear to be twice as likely to be diagnosed with a PCCS compared to women.1 Primary cutaneous carcinosarcomas are recognized as aggressive tumors with a high propensity to metastasize and recur locally, necessitating early diagnosis and treatment.4 Accurate diagnosis of PCCSs can be challenging due to the biphasic nature of the neoplasm as well as poor differentiation or unequal proportions of the epithelial and mesenchymal components.5 Additionally, overlapping diagnostic criteria coupled with vague demarcation between soft-tissue sarcomas and distinct carcinomas also may contribute to a delay in diagnosis.9 Treatment is achieved surgically by complete wide resection, with no evidence to support the use of adjuvant or neoadjuvant external beam radiation therapy. Due to the small number of reported cases, no treatment recommendations currently exist.1
Surgical management with wide local excision has been disappointing, with recurrence rates reported as high as 33%.6 Primary cutaneous carcinosarcoma has an estimated overall recurrence rate of 19% and a 5-year disease-free rate of 50%.10 Risk factors associated with poorer prognosis include tumors with adnexal subtype, age less than 65 years, rapid tumor growth, a tumor greater than 20 mm at presentation, and a long-standing tumor lasting up to 30 years.2,4 Although wide local excision and Mohs micrographic surgery (MMS) both have been utilized successfully, MMS has been shown to result in a cure rate of greater than 98%.6
Atypical fibroxanthoma (AFX) is a cutaneous tumor of fibrohistiocytic mesenchymal origin that typically manifests on sun-damaged skin in elderly individuals. Clinically, it presents as a rapidly growing neoplasm that often ulcerates and bleeds. These heterogenous neoplasms have several distinct characteristics, including dense cellularity with disorganized, large, pleomorphic, and atypical-appearing spindle-shaped cells arising in the upper layers of the dermis, often disseminating into the reticular dermis and occasionally into the subcutaneous fat (Figure 1). The neoplastic cells often exhibit hyperchromic and irregular nuclei, multinucleated giant cells, and atypical mitotic figures. In most cases, negative immunohistochemical staining with SOX-10, S-100, cytokeratins, desmin, and caldesmon will allow pathologists to differentiate between AFX and other common tumors on the differential diagnosis, such as SCC, melanoma, and leiomyosarcoma. CD10 and procollagen type 1 are positive antigenic markers in AFX, but they are not specific. The standard treatment of AFX includes wide local excision or MMS for superior margin control.11
Spindle cell SCC presents as a raised or exophytic nodule, often with spontaneous bleeding and central ulceration. It usually presents on sun-damaged skin or in individuals with a history of ionizing radiation. Histologically, it is characterized by atypical spindleshaped keratinocytes in the dermis existing as single cells or cohesive nests along with keratin pearls (Figure 2). The atypical spindle cells may comprise the entire tumor or only a small portion. The use of immunohistochemical markers often is required to establish a definitive diagnosis. Spindle cell SCC stains positively, albeit frequently focally, for p63, p40, and high-molecular-weight cytokeratins such as cytokeratin 5/6, while S-100 protein, SOX-10, MART-1/Melan-A, and muscle-specific actin stains typically are negative. Wide local excision or MMS is recommended for treatment of these lesions.12
Primary cutaneous myoepithelial carcinomas are uncommon neoplasms of myoepithelial differentiation. Clinically, they often arise as soft nodular lesions on the head, neck, and lower extremities with a bimodal age distribution (50 years). Histologically cutaneous myoepithelial tumors are well-differentiated, dermal-based nodules without connection to the overlying epidermis (Figure 3). The myoepithelial cells can exhibit spindled, epithelioid, plasmacytoid, or clear cell morphologic features and show variability in cell growth patterns. One of the most common growth patterns is oval to round cells forming cords and chains in a chondromyxoid stroma. Most cases display an immunophenotyped co-expression of an epithelial cytokeratin and S-100 protein. Myoepithelial markers also may be present, including keratins, smooth muscle actin, calponin, glial fibrillary acidic protein, p63, and desmin. Surgical removal with wide local excision or MMS is essential.13
Leiomyosarcoma (LMS) is a tumor that originates from smooth muscle and rarely develops in the dermis.14 Pleomorphic LMS is a morphologic variant of LMS that has a low propensity to metastasize but commonly exhibits local recurrence.15 Leiomyosarcoma can present in any age group but most commonly manifests in individuals aged 50 to 70 years. Clinically, LMS presents as a firm solitary nodule with a smooth pink surface or a more exophytic tumor with a reddish or brown color on the extensor surface of the lower limbs; it is less common on the scalp and face.14 Histologically, most cases of pleomorphic LMS show small foci of fascicles consisting of smooth muscle tumor cells in addition to cellular pleomorphism (Figure 4).15 Many of these cells demonstrate a clear perinuclear vacuole that generally is appreciated in neoplastic smooth muscle cells.14 Pleomorphic LMS typically stains positively for at least one smooth muscle marker including desmin, h-caldesmon, muscle-specific actin, α-smooth muscle actin, or smooth muscle myosin in the leiomyosarcomatous fascicular areas.16 Complete surgical excision is the treatment of choice, and the best results are obtained with MMS.14
- Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
- Bourgeault E, Alain J, Gagne E. Primary cutaneous carcinosarcoma of the basal cell subtype should be treated as a high-risk basal cell carcinoma. J Cutan Med Surg. 2015;19:407-411.
- West L, Srivastava D. Cutaneous carcinosarcoma of the medial canthus discovered on Mohs debulk analysis. Dermatol Surg. 2019;45:1700-1702.
- Kwan JM, Satter EK. Carcinosarcoma: a primary cutaneous tumor with biphasic differentiation. Cutis. 2013;92:247-249.
- Suh KY, Lacouture M, Gerami P. p63 in primary cutaneous carcinosarcoma. Am J Dermatopathol. 2007;29:374‐377.
- Ruiz-Villaverde R, Aneiros-Fernandez J. Primary cutaneous carcinosarcoma: a cutaneous neoplasm with an exceptional presentation. Sultan Qaboos Univ Med J. 2018;18:E114-E115.
- Smart CN, Pucci RA, Binder SW, et al. Cutaneous carcinosarcoma with myoepithelial differentiation: immunohistochemical and cytogenetic analysis of a case presenting in an unusual location. Am J Dermatopathol. 2009;31:715‐717.
- Clark JJ, Bowen AR, Bowen GM, et al. Cutaneous carcinosarcoma: a series of six cases and a review of the literature. J Cutan Pathol. 2017;44:34‐44.
- Müller CS, Pföhler C, Schiekofer C, et al. Primary cutaneous carcinosarcomas: a morphological histogenetic concept revisited. Am J Dermatopathol. 2014;36:328‐339.
- Bellew S, Del Rosso JQ, Mobini N. Primary carcinosarcoma of the ear: case report and review of the literature. J Clin Aesthet Dermatol. 2009;2:33‐35.
- Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
- Soleymani T, Aasi SZ, Novoa R, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma: updates on classification and management. Dermatol Clin. 2019;37:253-259.
- Parekh V, Seykora JT. Cutaneous squamous cell carcinoma. Clin Lab Med. 2017;37:503-525.
- Johnson GE, Stevens K, Morrison AO, et al. Cutaneous myoepithelial carcinoma with disseminated metastases. Cutis. 2017;99:E19-E26.
- Llombart B, Serra-Guillén C, Requena C, et al. Leiomyosarcoma and pleomorphic dermal sarcoma: guidelines for diagnosis and treatment. Actas Dermosifiliogr. 2019;110:4-11.
- Oda Y, Miyajima K, Kawaguchi K, et al. Pleomorphic leiomyosarcoma: clinicopathologic and immunohistochemical study with special emphasis on its distinction from ordinary leiomyosarcoma and malignant fibrous histiocytoma. Am J Surg Pathol. 2001;25:1030-1038.
- Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
- Bourgeault E, Alain J, Gagne E. Primary cutaneous carcinosarcoma of the basal cell subtype should be treated as a high-risk basal cell carcinoma. J Cutan Med Surg. 2015;19:407-411.
- West L, Srivastava D. Cutaneous carcinosarcoma of the medial canthus discovered on Mohs debulk analysis. Dermatol Surg. 2019;45:1700-1702.
- Kwan JM, Satter EK. Carcinosarcoma: a primary cutaneous tumor with biphasic differentiation. Cutis. 2013;92:247-249.
- Suh KY, Lacouture M, Gerami P. p63 in primary cutaneous carcinosarcoma. Am J Dermatopathol. 2007;29:374‐377.
- Ruiz-Villaverde R, Aneiros-Fernandez J. Primary cutaneous carcinosarcoma: a cutaneous neoplasm with an exceptional presentation. Sultan Qaboos Univ Med J. 2018;18:E114-E115.
- Smart CN, Pucci RA, Binder SW, et al. Cutaneous carcinosarcoma with myoepithelial differentiation: immunohistochemical and cytogenetic analysis of a case presenting in an unusual location. Am J Dermatopathol. 2009;31:715‐717.
- Clark JJ, Bowen AR, Bowen GM, et al. Cutaneous carcinosarcoma: a series of six cases and a review of the literature. J Cutan Pathol. 2017;44:34‐44.
- Müller CS, Pföhler C, Schiekofer C, et al. Primary cutaneous carcinosarcomas: a morphological histogenetic concept revisited. Am J Dermatopathol. 2014;36:328‐339.
- Bellew S, Del Rosso JQ, Mobini N. Primary carcinosarcoma of the ear: case report and review of the literature. J Clin Aesthet Dermatol. 2009;2:33‐35.
- Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
- Soleymani T, Aasi SZ, Novoa R, et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma: updates on classification and management. Dermatol Clin. 2019;37:253-259.
- Parekh V, Seykora JT. Cutaneous squamous cell carcinoma. Clin Lab Med. 2017;37:503-525.
- Johnson GE, Stevens K, Morrison AO, et al. Cutaneous myoepithelial carcinoma with disseminated metastases. Cutis. 2017;99:E19-E26.
- Llombart B, Serra-Guillén C, Requena C, et al. Leiomyosarcoma and pleomorphic dermal sarcoma: guidelines for diagnosis and treatment. Actas Dermosifiliogr. 2019;110:4-11.
- Oda Y, Miyajima K, Kawaguchi K, et al. Pleomorphic leiomyosarcoma: clinicopathologic and immunohistochemical study with special emphasis on its distinction from ordinary leiomyosarcoma and malignant fibrous histiocytoma. Am J Surg Pathol. 2001;25:1030-1038.
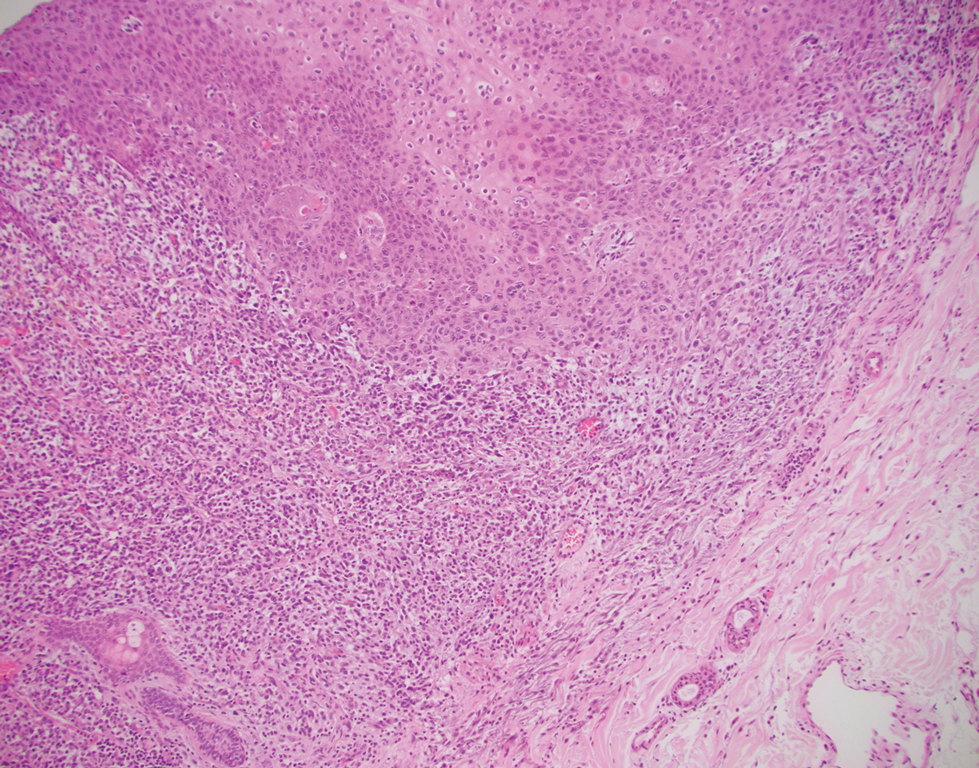
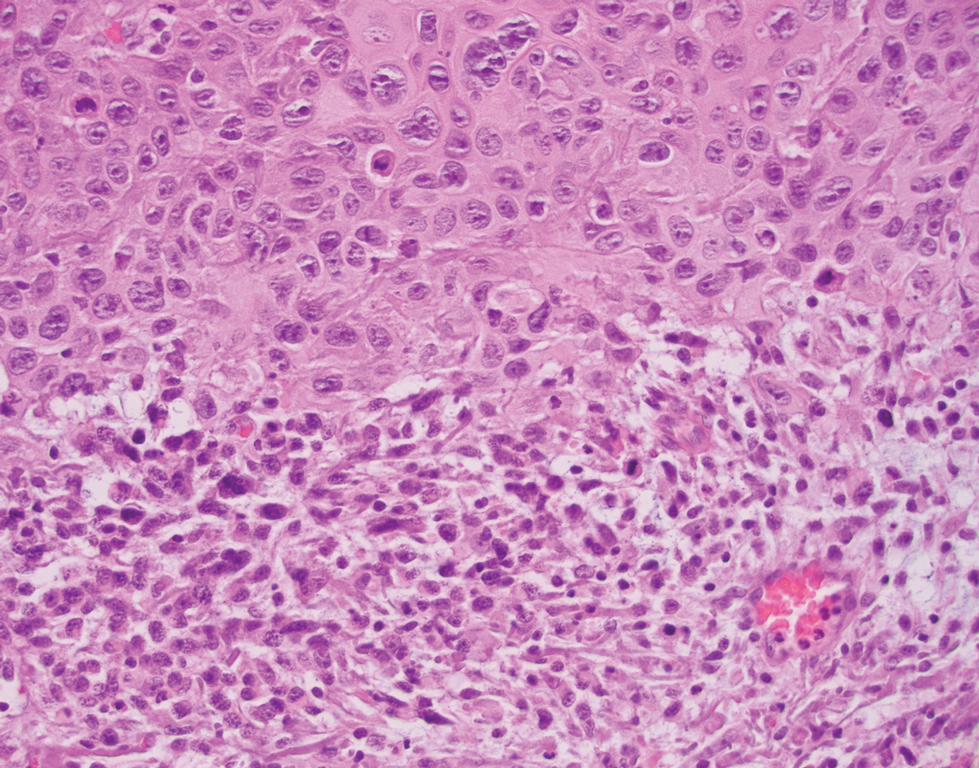
A 72-year-old man with a history of nonmelanoma skin cancer and lung transplant maintained on stable doses of prednisone and tacrolimus presented with a 1.3×1.8-cm, slow-growing, well-demarcated, ulcerated, erythematous plaque with overlying serous crust on the left temple of 6 months’ duration. No cervical or axillary lymphadenopathy was appreciated on physical examination. A biopsy was performed followed by Mohs micrographic surgery. Microscopic examination of the debulking specimen revealed atypical spindle cells in the papillary and reticular dermis radiating from a central focus of a moderately differentiated squamous cell carcinoma. The squamous cells stained positive for cytokeratin 5/6, pankeratin, and p40, while the spindle cells stained positive only for vimentin.