User login
A 55-year-old man with a 20-year history of type 2 diabetes mellitus was admitted to the hospital after presenting to the emergency department with an acute change in mental status. Three days earlier, he had begun to feel abdominal discomfort and dizziness, which gradually worsened.
On presentation, his Glasgow Coma Scale score was 13 out of 15 (eye-opening response 3 of 4, verbal response 4 of 5, motor response 6 of 6), his blood pressure was 221/114 mm Hg, and other vital signs were normal. Physical examination including a neurologic examination was normal. No gait abnormality or ataxia was noted.
When asked about current medications, he said that 2 years earlier he had missed an appointment with his primary care physician and so had never obtained refills of his diabetes medications.
Results of laboratory testing were as follows:
- Blood glucose 1,011 mg/dL (reference range 65–110)
- Hemoglobin A1c 17.8% (4%–5.6%)
- Sodium 126 mmol/L (135–145)
- Sodium corrected for serum glucose 141 mmol/L
- Potassium 3.2 mmol/L (3.5–5.0)
- Blood urea nitrogen 43.8 mg/dL (8–21)
- Calculated serum osmolality 324 mosm/kg (275–295).
Blood gas analysis showed no acidosis. Tests for urinary and serum ketones were negative. Computed tomography (CT) of the head without contrast was normal.
Based on the results of the evaluation, the patient’s condition was diagnosed as a hyperosmolar hyperglycemic state, presumably from dehydration and noncompliance with diabetes medications. His altered mental status was also attributed to this diagnosis. He was started on aggressive hydration and insulin infusion to correct the blood glucose level. Repeat laboratory testing 7 hours after admission revealed a blood glucose of 49 mg/dL, sodium 148 mmol/L, blood urea nitrogen 43 mg/dL, and calculated serum osmolality 290 mosm/kg.
The insulin infusion was suspended, and glucose infusion was started. With this treatment, his blood glucose level stabilized, but his Glasgow Coma Scale score was unchanged from the time of presentation. A neurologic examination at this time showed bilateral dysmetria. Cranial nerves were normal. Motor examination showed normal tone with a Medical Research Council score of 5 of 5 in all extremities. Sensory examination revealed a glove-and-stocking pattern of loss of vibratory sensation. Tendon reflexes were normal except for diminished bilateral knee-jerk and ankle-jerk responses.
.")
.")
OSMOTIC DEMYELINATION SYNDROME
.")
 were almost unchanged.")
Central pontine myelinolysis is a pivotal manifestation of the syndrome and is characterized by progressive lethargy, quadriparesis, dysarthria, ophthalmoplegia, dysphasia, ataxia, and reflex changes. Clinical symptoms of extrapontine myelinolysis are variable.4
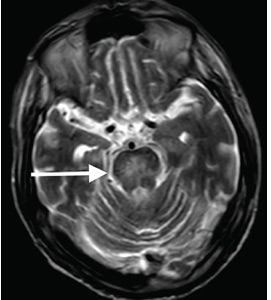
Although CT may underestimate osmotic demyelination syndrome, the typical radiologic findings on brain MRI are hyperintense lesions in the central pons or associated extrapontine structures on T2-weighted and fluid-attenuated inversion recovery sequences.4
A precise definition of hyperosmolar hyperglycemia does not exist. The Joint British Diabetes Societies for Inpatient Care suggested the following features: a measured osmolality of 320 mosm/kg or higher, a blood glucose level of 541 mg/dL or higher, severe dehydration, and feeling unwell.5
Our patient’s clinical course and high hemoglobin A1c suggested prolonged hyperglycemia and high serum osmolality before his admission. After his admission, aggressive hydration and insulin therapy corrected the hyperglycemia and serum osmolality too rapidly for his brain cells to adjust to the change. It was reasonable to suspect a hyperosmolar hyperglycemic state as one of the main causes of his mental status change and ataxia. This, along with lack of improvement in his impaired metal status and new-onset ataxia despite treatment, led to suspicion of osmotic demyelination syndrome. His diminished bilateral knee-jerk and ankle-jerk responses more likely represented diabetic neuropathy rather than osmotic demyelination syndrome.
Osmotic demyelination syndrome has seldom been reported as a complication of hyperosmolar hyperglycemia.6–13 And extrapontine myelinolysis with hyperosmolar hyperglycemia is extremely rare, with only 2 reports to date to the best of our knowledge.6,10
There is no specific treatment for osmotic demyelination syndrome except for supportive care and treatment of coexisting conditions. Once an osmotic derangement is identified, we recommend correcting chronically elevated serum glucose values gradually to avoid overtreatment, just as we would do with elevated serum sodium levels. Changes in neurologic findings, serum blood glucose level, and serum osmolality should be followed closely. A review showed that a favorable recovery from osmotic demyelination syndrome is possible even with severe neurologic deficits.4
TAKE-AWAY POINTS
- Osmotic demyelination syndrome is a rare but severe complication of a hyperosmolar hyperglycemic state.
- Physicians should be aware not only of changes in serum sodium, but also of changes in serum osmolality and serum glucose.
- When a new-onset neurologic deficit is found during treatment of a hyperosmolar hyperglycemic state, suspect osmotic demyelination syndrome, monitor changes in serum osmolality, and consider brain MRI.
- Brown WD. Osmotic demyelination disorders: central pontine and extrapontine myelinolysis. Curr Opin Neurol 2000; 13(6):691–697. pmid:11148672
- Laureno R, Karp BI. Myelinolysis after correction of hyponatraemia. Ann Intern Med 1997; 126(1):57–62. pmid:8992924
- Adams RD, Victor M, Mancall EL. Central pontine myelinolysis: a hitherto undescribed disease occurring in alcoholic and malnourished patients. AMA Arch Neurol Psychiatry 1959; 81(2):154–172. pmid:13616772
- Singh TD, Fugate JE, Rabinstein AA. Central pontine and extrapontine myelinolysis: a systematic review. Eur J Neurol 2014; 21(12):1443–1450. doi:10.1111/ene.12571
- Scott AR; Joint British Diabetes Societies (JBDS) for Inpatient Care; JBDS Hyperosmolar Hyperglycaemic Guidelines Group. Management of hyperosmolar hyperglycaemic state in adults with diabetes. Diabet Med 2015; 32(6):714–724. doi:10.1111/dme.12757
- McComb RD, Pfeiffer RF, Casey JH, Wolcott G, Till DJ. Lateral pontine and extrapontine myelinolysis associated with hypernatremia and hyperglycemia. Clin Neuropathol 1989; 8(6):284–288. pmid:2695277
- O’Malley G, Moran C, Draman MS, et al. Central pontine myelinolysis complicating treatment of the hyperglycaemic hyperosmolar state. Ann Clin Biochem 2008; 45(pt 4):440–443. doi:10.1258/acb.2008.007171
- Burns JD, Kosa SC, Wijdicks EF. Central pontine myelinolysis in a patient with hyperosmolar hyperglycemia and consistently normal serum sodium. Neurocrit Care 2009; 11(2):251–254. doi:10.1007/s12028-009-9241-9
- Mao S, Liu Z, Ding M. Central pontine myelinolysis in a patient with epilepsia partialis continua and hyperglycaemic hyperosmolar state. Ann Clin Biochem 2011; 48(pt 1):79–82. doi:10.1258/acb.2010.010152. Epub 2010 Nov 23.
- Guerrero WR, Dababneh H, Nadeau SE. Hemiparesis, encephalopathy, and extrapontine osmotic myelinolysis in the setting of hyperosmolar hyperglycemia. J Clin Neurosci 2013; 20(6):894–896. doi:10.1016/j.jocn.2012.05.045
- Hegazi MO, Mashankar A. Central pontine myelinolysis in the hyperosmolar hyperglycaemic state. Med Princ Pract 2013; 22(1):96–99. doi:10.1159/000341718
- Rodríguez-Velver KV, Soto-Garcia AJ, Zapata-Rivera MA, Montes-Villarreal J, Villarreal-Pérez JZ, Rodríguez-Gutiérrez R. Osmotic demyelination syndrome as the initial manifestation of a hyperosmolar hyperglycemic state. Case Rep Neurol Med 2014; 2014:652523. doi:10.1155/2014/652523
- Chang YM. Central pontine myelinolysis associated with diabetic hyperglycemia. JSM Clin Case Rep 2014; 2(6):1059.
A 55-year-old man with a 20-year history of type 2 diabetes mellitus was admitted to the hospital after presenting to the emergency department with an acute change in mental status. Three days earlier, he had begun to feel abdominal discomfort and dizziness, which gradually worsened.
On presentation, his Glasgow Coma Scale score was 13 out of 15 (eye-opening response 3 of 4, verbal response 4 of 5, motor response 6 of 6), his blood pressure was 221/114 mm Hg, and other vital signs were normal. Physical examination including a neurologic examination was normal. No gait abnormality or ataxia was noted.
When asked about current medications, he said that 2 years earlier he had missed an appointment with his primary care physician and so had never obtained refills of his diabetes medications.
Results of laboratory testing were as follows:
- Blood glucose 1,011 mg/dL (reference range 65–110)
- Hemoglobin A1c 17.8% (4%–5.6%)
- Sodium 126 mmol/L (135–145)
- Sodium corrected for serum glucose 141 mmol/L
- Potassium 3.2 mmol/L (3.5–5.0)
- Blood urea nitrogen 43.8 mg/dL (8–21)
- Calculated serum osmolality 324 mosm/kg (275–295).
Blood gas analysis showed no acidosis. Tests for urinary and serum ketones were negative. Computed tomography (CT) of the head without contrast was normal.
Based on the results of the evaluation, the patient’s condition was diagnosed as a hyperosmolar hyperglycemic state, presumably from dehydration and noncompliance with diabetes medications. His altered mental status was also attributed to this diagnosis. He was started on aggressive hydration and insulin infusion to correct the blood glucose level. Repeat laboratory testing 7 hours after admission revealed a blood glucose of 49 mg/dL, sodium 148 mmol/L, blood urea nitrogen 43 mg/dL, and calculated serum osmolality 290 mosm/kg.
The insulin infusion was suspended, and glucose infusion was started. With this treatment, his blood glucose level stabilized, but his Glasgow Coma Scale score was unchanged from the time of presentation. A neurologic examination at this time showed bilateral dysmetria. Cranial nerves were normal. Motor examination showed normal tone with a Medical Research Council score of 5 of 5 in all extremities. Sensory examination revealed a glove-and-stocking pattern of loss of vibratory sensation. Tendon reflexes were normal except for diminished bilateral knee-jerk and ankle-jerk responses.
OSMOTIC DEMYELINATION SYNDROME
Central pontine myelinolysis is a pivotal manifestation of the syndrome and is characterized by progressive lethargy, quadriparesis, dysarthria, ophthalmoplegia, dysphasia, ataxia, and reflex changes. Clinical symptoms of extrapontine myelinolysis are variable.4
Although CT may underestimate osmotic demyelination syndrome, the typical radiologic findings on brain MRI are hyperintense lesions in the central pons or associated extrapontine structures on T2-weighted and fluid-attenuated inversion recovery sequences.4
A precise definition of hyperosmolar hyperglycemia does not exist. The Joint British Diabetes Societies for Inpatient Care suggested the following features: a measured osmolality of 320 mosm/kg or higher, a blood glucose level of 541 mg/dL or higher, severe dehydration, and feeling unwell.5
Our patient’s clinical course and high hemoglobin A1c suggested prolonged hyperglycemia and high serum osmolality before his admission. After his admission, aggressive hydration and insulin therapy corrected the hyperglycemia and serum osmolality too rapidly for his brain cells to adjust to the change. It was reasonable to suspect a hyperosmolar hyperglycemic state as one of the main causes of his mental status change and ataxia. This, along with lack of improvement in his impaired metal status and new-onset ataxia despite treatment, led to suspicion of osmotic demyelination syndrome. His diminished bilateral knee-jerk and ankle-jerk responses more likely represented diabetic neuropathy rather than osmotic demyelination syndrome.
Osmotic demyelination syndrome has seldom been reported as a complication of hyperosmolar hyperglycemia.6–13 And extrapontine myelinolysis with hyperosmolar hyperglycemia is extremely rare, with only 2 reports to date to the best of our knowledge.6,10
There is no specific treatment for osmotic demyelination syndrome except for supportive care and treatment of coexisting conditions. Once an osmotic derangement is identified, we recommend correcting chronically elevated serum glucose values gradually to avoid overtreatment, just as we would do with elevated serum sodium levels. Changes in neurologic findings, serum blood glucose level, and serum osmolality should be followed closely. A review showed that a favorable recovery from osmotic demyelination syndrome is possible even with severe neurologic deficits.4
TAKE-AWAY POINTS
- Osmotic demyelination syndrome is a rare but severe complication of a hyperosmolar hyperglycemic state.
- Physicians should be aware not only of changes in serum sodium, but also of changes in serum osmolality and serum glucose.
- When a new-onset neurologic deficit is found during treatment of a hyperosmolar hyperglycemic state, suspect osmotic demyelination syndrome, monitor changes in serum osmolality, and consider brain MRI.
A 55-year-old man with a 20-year history of type 2 diabetes mellitus was admitted to the hospital after presenting to the emergency department with an acute change in mental status. Three days earlier, he had begun to feel abdominal discomfort and dizziness, which gradually worsened.
On presentation, his Glasgow Coma Scale score was 13 out of 15 (eye-opening response 3 of 4, verbal response 4 of 5, motor response 6 of 6), his blood pressure was 221/114 mm Hg, and other vital signs were normal. Physical examination including a neurologic examination was normal. No gait abnormality or ataxia was noted.
When asked about current medications, he said that 2 years earlier he had missed an appointment with his primary care physician and so had never obtained refills of his diabetes medications.
Results of laboratory testing were as follows:
- Blood glucose 1,011 mg/dL (reference range 65–110)
- Hemoglobin A1c 17.8% (4%–5.6%)
- Sodium 126 mmol/L (135–145)
- Sodium corrected for serum glucose 141 mmol/L
- Potassium 3.2 mmol/L (3.5–5.0)
- Blood urea nitrogen 43.8 mg/dL (8–21)
- Calculated serum osmolality 324 mosm/kg (275–295).
Blood gas analysis showed no acidosis. Tests for urinary and serum ketones were negative. Computed tomography (CT) of the head without contrast was normal.
Based on the results of the evaluation, the patient’s condition was diagnosed as a hyperosmolar hyperglycemic state, presumably from dehydration and noncompliance with diabetes medications. His altered mental status was also attributed to this diagnosis. He was started on aggressive hydration and insulin infusion to correct the blood glucose level. Repeat laboratory testing 7 hours after admission revealed a blood glucose of 49 mg/dL, sodium 148 mmol/L, blood urea nitrogen 43 mg/dL, and calculated serum osmolality 290 mosm/kg.
The insulin infusion was suspended, and glucose infusion was started. With this treatment, his blood glucose level stabilized, but his Glasgow Coma Scale score was unchanged from the time of presentation. A neurologic examination at this time showed bilateral dysmetria. Cranial nerves were normal. Motor examination showed normal tone with a Medical Research Council score of 5 of 5 in all extremities. Sensory examination revealed a glove-and-stocking pattern of loss of vibratory sensation. Tendon reflexes were normal except for diminished bilateral knee-jerk and ankle-jerk responses.
OSMOTIC DEMYELINATION SYNDROME
Central pontine myelinolysis is a pivotal manifestation of the syndrome and is characterized by progressive lethargy, quadriparesis, dysarthria, ophthalmoplegia, dysphasia, ataxia, and reflex changes. Clinical symptoms of extrapontine myelinolysis are variable.4
Although CT may underestimate osmotic demyelination syndrome, the typical radiologic findings on brain MRI are hyperintense lesions in the central pons or associated extrapontine structures on T2-weighted and fluid-attenuated inversion recovery sequences.4
A precise definition of hyperosmolar hyperglycemia does not exist. The Joint British Diabetes Societies for Inpatient Care suggested the following features: a measured osmolality of 320 mosm/kg or higher, a blood glucose level of 541 mg/dL or higher, severe dehydration, and feeling unwell.5
Our patient’s clinical course and high hemoglobin A1c suggested prolonged hyperglycemia and high serum osmolality before his admission. After his admission, aggressive hydration and insulin therapy corrected the hyperglycemia and serum osmolality too rapidly for his brain cells to adjust to the change. It was reasonable to suspect a hyperosmolar hyperglycemic state as one of the main causes of his mental status change and ataxia. This, along with lack of improvement in his impaired metal status and new-onset ataxia despite treatment, led to suspicion of osmotic demyelination syndrome. His diminished bilateral knee-jerk and ankle-jerk responses more likely represented diabetic neuropathy rather than osmotic demyelination syndrome.
Osmotic demyelination syndrome has seldom been reported as a complication of hyperosmolar hyperglycemia.6–13 And extrapontine myelinolysis with hyperosmolar hyperglycemia is extremely rare, with only 2 reports to date to the best of our knowledge.6,10
There is no specific treatment for osmotic demyelination syndrome except for supportive care and treatment of coexisting conditions. Once an osmotic derangement is identified, we recommend correcting chronically elevated serum glucose values gradually to avoid overtreatment, just as we would do with elevated serum sodium levels. Changes in neurologic findings, serum blood glucose level, and serum osmolality should be followed closely. A review showed that a favorable recovery from osmotic demyelination syndrome is possible even with severe neurologic deficits.4
TAKE-AWAY POINTS
- Osmotic demyelination syndrome is a rare but severe complication of a hyperosmolar hyperglycemic state.
- Physicians should be aware not only of changes in serum sodium, but also of changes in serum osmolality and serum glucose.
- When a new-onset neurologic deficit is found during treatment of a hyperosmolar hyperglycemic state, suspect osmotic demyelination syndrome, monitor changes in serum osmolality, and consider brain MRI.
- Brown WD. Osmotic demyelination disorders: central pontine and extrapontine myelinolysis. Curr Opin Neurol 2000; 13(6):691–697. pmid:11148672
- Laureno R, Karp BI. Myelinolysis after correction of hyponatraemia. Ann Intern Med 1997; 126(1):57–62. pmid:8992924
- Adams RD, Victor M, Mancall EL. Central pontine myelinolysis: a hitherto undescribed disease occurring in alcoholic and malnourished patients. AMA Arch Neurol Psychiatry 1959; 81(2):154–172. pmid:13616772
- Singh TD, Fugate JE, Rabinstein AA. Central pontine and extrapontine myelinolysis: a systematic review. Eur J Neurol 2014; 21(12):1443–1450. doi:10.1111/ene.12571
- Scott AR; Joint British Diabetes Societies (JBDS) for Inpatient Care; JBDS Hyperosmolar Hyperglycaemic Guidelines Group. Management of hyperosmolar hyperglycaemic state in adults with diabetes. Diabet Med 2015; 32(6):714–724. doi:10.1111/dme.12757
- McComb RD, Pfeiffer RF, Casey JH, Wolcott G, Till DJ. Lateral pontine and extrapontine myelinolysis associated with hypernatremia and hyperglycemia. Clin Neuropathol 1989; 8(6):284–288. pmid:2695277
- O’Malley G, Moran C, Draman MS, et al. Central pontine myelinolysis complicating treatment of the hyperglycaemic hyperosmolar state. Ann Clin Biochem 2008; 45(pt 4):440–443. doi:10.1258/acb.2008.007171
- Burns JD, Kosa SC, Wijdicks EF. Central pontine myelinolysis in a patient with hyperosmolar hyperglycemia and consistently normal serum sodium. Neurocrit Care 2009; 11(2):251–254. doi:10.1007/s12028-009-9241-9
- Mao S, Liu Z, Ding M. Central pontine myelinolysis in a patient with epilepsia partialis continua and hyperglycaemic hyperosmolar state. Ann Clin Biochem 2011; 48(pt 1):79–82. doi:10.1258/acb.2010.010152. Epub 2010 Nov 23.
- Guerrero WR, Dababneh H, Nadeau SE. Hemiparesis, encephalopathy, and extrapontine osmotic myelinolysis in the setting of hyperosmolar hyperglycemia. J Clin Neurosci 2013; 20(6):894–896. doi:10.1016/j.jocn.2012.05.045
- Hegazi MO, Mashankar A. Central pontine myelinolysis in the hyperosmolar hyperglycaemic state. Med Princ Pract 2013; 22(1):96–99. doi:10.1159/000341718
- Rodríguez-Velver KV, Soto-Garcia AJ, Zapata-Rivera MA, Montes-Villarreal J, Villarreal-Pérez JZ, Rodríguez-Gutiérrez R. Osmotic demyelination syndrome as the initial manifestation of a hyperosmolar hyperglycemic state. Case Rep Neurol Med 2014; 2014:652523. doi:10.1155/2014/652523
- Chang YM. Central pontine myelinolysis associated with diabetic hyperglycemia. JSM Clin Case Rep 2014; 2(6):1059.
- Brown WD. Osmotic demyelination disorders: central pontine and extrapontine myelinolysis. Curr Opin Neurol 2000; 13(6):691–697. pmid:11148672
- Laureno R, Karp BI. Myelinolysis after correction of hyponatraemia. Ann Intern Med 1997; 126(1):57–62. pmid:8992924
- Adams RD, Victor M, Mancall EL. Central pontine myelinolysis: a hitherto undescribed disease occurring in alcoholic and malnourished patients. AMA Arch Neurol Psychiatry 1959; 81(2):154–172. pmid:13616772
- Singh TD, Fugate JE, Rabinstein AA. Central pontine and extrapontine myelinolysis: a systematic review. Eur J Neurol 2014; 21(12):1443–1450. doi:10.1111/ene.12571
- Scott AR; Joint British Diabetes Societies (JBDS) for Inpatient Care; JBDS Hyperosmolar Hyperglycaemic Guidelines Group. Management of hyperosmolar hyperglycaemic state in adults with diabetes. Diabet Med 2015; 32(6):714–724. doi:10.1111/dme.12757
- McComb RD, Pfeiffer RF, Casey JH, Wolcott G, Till DJ. Lateral pontine and extrapontine myelinolysis associated with hypernatremia and hyperglycemia. Clin Neuropathol 1989; 8(6):284–288. pmid:2695277
- O’Malley G, Moran C, Draman MS, et al. Central pontine myelinolysis complicating treatment of the hyperglycaemic hyperosmolar state. Ann Clin Biochem 2008; 45(pt 4):440–443. doi:10.1258/acb.2008.007171
- Burns JD, Kosa SC, Wijdicks EF. Central pontine myelinolysis in a patient with hyperosmolar hyperglycemia and consistently normal serum sodium. Neurocrit Care 2009; 11(2):251–254. doi:10.1007/s12028-009-9241-9
- Mao S, Liu Z, Ding M. Central pontine myelinolysis in a patient with epilepsia partialis continua and hyperglycaemic hyperosmolar state. Ann Clin Biochem 2011; 48(pt 1):79–82. doi:10.1258/acb.2010.010152. Epub 2010 Nov 23.
- Guerrero WR, Dababneh H, Nadeau SE. Hemiparesis, encephalopathy, and extrapontine osmotic myelinolysis in the setting of hyperosmolar hyperglycemia. J Clin Neurosci 2013; 20(6):894–896. doi:10.1016/j.jocn.2012.05.045
- Hegazi MO, Mashankar A. Central pontine myelinolysis in the hyperosmolar hyperglycaemic state. Med Princ Pract 2013; 22(1):96–99. doi:10.1159/000341718
- Rodríguez-Velver KV, Soto-Garcia AJ, Zapata-Rivera MA, Montes-Villarreal J, Villarreal-Pérez JZ, Rodríguez-Gutiérrez R. Osmotic demyelination syndrome as the initial manifestation of a hyperosmolar hyperglycemic state. Case Rep Neurol Med 2014; 2014:652523. doi:10.1155/2014/652523
- Chang YM. Central pontine myelinolysis associated with diabetic hyperglycemia. JSM Clin Case Rep 2014; 2(6):1059.