User login
Diagnosis: Lymphomatoid papulosis
Lymphomatoid papulosis is a rare, chronic, and benign papulonodular or papulonecrotic skin disorder. LyP affects people of all ages, and its peak incidence occurs in the 5th decade. It generally has no predilection for a particular sex; however, some have reported a slight predominance in males. Patients of all races may be diagnosed with LyP, although it is less common in black patients. In addition, 10% of LyP cases are associated with anaplastic large cell lymphoma, cutaneous T-cell lymphoma (mycosis fungoides), or Hodgkin’s lymphoma.
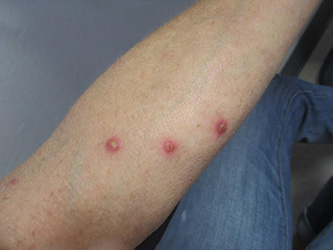
Patients typically present with multiple erythematous papules that evolve into red-brown papulopustular or papulovesicular lesions. The papules may be mildly pruritic or asymptomatic and can be few in number to hundreds at presentation. The lesions usually appear in crops that resolve within 2-8 weeks with or without scarring, and can continue this cyclic process for months to years. The arms, legs, and trunk are most commonly affected, although LyP can present anywhere on the body.
The diagnosis of LyP is classically based upon histopathologic examination. Hematoxylin and eosin staining reveals a dense dermal infiltrate of atypical lymphocytes along with numerous eosinophils and neutrophils; lymphocytes are CD30+. Vessels in the dermis also appear with fibrin deposition, endothelial edema, and red blood cell extravasation. In addition, LyP can be classified as type A, type B, type C, and/or type D. These subtypes are determined by the size of atypical lymphocytes, type of atypical cells, location and amount of infiltrate, and CD30 and CD8 staining.
The differential diagnosis of LyP includes anaplastic large cell lymphoma, cutaneous T-cell lymphoma, folliculitis, insect bites, Langerhans cell histiocytosis, leukemia cutis, milia, miliaria, and scabies.
The etiology of LyP is unknown. It is unclear whether the proliferation of T cells is a benign and chronic disorder initiated by a stimulus or an indolent T-cell malignancy that the immune system monitors and only allows for localized, cutaneous involvement.
Mild forms of LyP can often be managed with topical corticosteroids. However, other therapies such as intralesional corticosteroids, phototherapy (UVB or PUVA), tetracycline antibiotics, and methotrexate are effective in treating cases of more persistent and widespread disease.
Our patient’s biopsy showed an irregular epidermis with scale, focal ulceration, scattered eosinophils, and dermal lymphocytes and histiocytes present in a perivascular pattern. Many of the lymphoid cells were enlarged, hyperchromatic, and irregular. Immunohistochemical staining was CD30+. These histologic changes were most consistent with lymphomatoid papulosis.
Dr. Bilu Martin is in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit your case for possible publication, send an email to dermnews@frontlinemedcom.com.
Diagnosis: Lymphomatoid papulosis
Lymphomatoid papulosis is a rare, chronic, and benign papulonodular or papulonecrotic skin disorder. LyP affects people of all ages, and its peak incidence occurs in the 5th decade. It generally has no predilection for a particular sex; however, some have reported a slight predominance in males. Patients of all races may be diagnosed with LyP, although it is less common in black patients. In addition, 10% of LyP cases are associated with anaplastic large cell lymphoma, cutaneous T-cell lymphoma (mycosis fungoides), or Hodgkin’s lymphoma.
Patients typically present with multiple erythematous papules that evolve into red-brown papulopustular or papulovesicular lesions. The papules may be mildly pruritic or asymptomatic and can be few in number to hundreds at presentation. The lesions usually appear in crops that resolve within 2-8 weeks with or without scarring, and can continue this cyclic process for months to years. The arms, legs, and trunk are most commonly affected, although LyP can present anywhere on the body.
The diagnosis of LyP is classically based upon histopathologic examination. Hematoxylin and eosin staining reveals a dense dermal infiltrate of atypical lymphocytes along with numerous eosinophils and neutrophils; lymphocytes are CD30+. Vessels in the dermis also appear with fibrin deposition, endothelial edema, and red blood cell extravasation. In addition, LyP can be classified as type A, type B, type C, and/or type D. These subtypes are determined by the size of atypical lymphocytes, type of atypical cells, location and amount of infiltrate, and CD30 and CD8 staining.
The differential diagnosis of LyP includes anaplastic large cell lymphoma, cutaneous T-cell lymphoma, folliculitis, insect bites, Langerhans cell histiocytosis, leukemia cutis, milia, miliaria, and scabies.
The etiology of LyP is unknown. It is unclear whether the proliferation of T cells is a benign and chronic disorder initiated by a stimulus or an indolent T-cell malignancy that the immune system monitors and only allows for localized, cutaneous involvement.
Mild forms of LyP can often be managed with topical corticosteroids. However, other therapies such as intralesional corticosteroids, phototherapy (UVB or PUVA), tetracycline antibiotics, and methotrexate are effective in treating cases of more persistent and widespread disease.
Our patient’s biopsy showed an irregular epidermis with scale, focal ulceration, scattered eosinophils, and dermal lymphocytes and histiocytes present in a perivascular pattern. Many of the lymphoid cells were enlarged, hyperchromatic, and irregular. Immunohistochemical staining was CD30+. These histologic changes were most consistent with lymphomatoid papulosis.
Dr. Bilu Martin is in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit your case for possible publication, send an email to dermnews@frontlinemedcom.com.
Diagnosis: Lymphomatoid papulosis
Lymphomatoid papulosis is a rare, chronic, and benign papulonodular or papulonecrotic skin disorder. LyP affects people of all ages, and its peak incidence occurs in the 5th decade. It generally has no predilection for a particular sex; however, some have reported a slight predominance in males. Patients of all races may be diagnosed with LyP, although it is less common in black patients. In addition, 10% of LyP cases are associated with anaplastic large cell lymphoma, cutaneous T-cell lymphoma (mycosis fungoides), or Hodgkin’s lymphoma.
Patients typically present with multiple erythematous papules that evolve into red-brown papulopustular or papulovesicular lesions. The papules may be mildly pruritic or asymptomatic and can be few in number to hundreds at presentation. The lesions usually appear in crops that resolve within 2-8 weeks with or without scarring, and can continue this cyclic process for months to years. The arms, legs, and trunk are most commonly affected, although LyP can present anywhere on the body.
The diagnosis of LyP is classically based upon histopathologic examination. Hematoxylin and eosin staining reveals a dense dermal infiltrate of atypical lymphocytes along with numerous eosinophils and neutrophils; lymphocytes are CD30+. Vessels in the dermis also appear with fibrin deposition, endothelial edema, and red blood cell extravasation. In addition, LyP can be classified as type A, type B, type C, and/or type D. These subtypes are determined by the size of atypical lymphocytes, type of atypical cells, location and amount of infiltrate, and CD30 and CD8 staining.
The differential diagnosis of LyP includes anaplastic large cell lymphoma, cutaneous T-cell lymphoma, folliculitis, insect bites, Langerhans cell histiocytosis, leukemia cutis, milia, miliaria, and scabies.
The etiology of LyP is unknown. It is unclear whether the proliferation of T cells is a benign and chronic disorder initiated by a stimulus or an indolent T-cell malignancy that the immune system monitors and only allows for localized, cutaneous involvement.
Mild forms of LyP can often be managed with topical corticosteroids. However, other therapies such as intralesional corticosteroids, phototherapy (UVB or PUVA), tetracycline antibiotics, and methotrexate are effective in treating cases of more persistent and widespread disease.
Our patient’s biopsy showed an irregular epidermis with scale, focal ulceration, scattered eosinophils, and dermal lymphocytes and histiocytes present in a perivascular pattern. Many of the lymphoid cells were enlarged, hyperchromatic, and irregular. Immunohistochemical staining was CD30+. These histologic changes were most consistent with lymphomatoid papulosis.
Dr. Bilu Martin is in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit your case for possible publication, send an email to dermnews@frontlinemedcom.com.
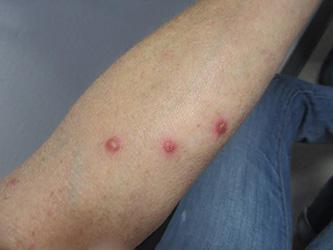
A 61-year-old woman with no significant past medical history presented with a rash on her arms. The lesions were pruritic and showed mild improvement after she took an oral antihistamine. The patient stated that she had similar outbreaks in the past that were treated with minocycline by an outside dermatologist. At that time, one lesion was cultured, showing no evidence of bacteria or herpes. She denied any history of gardening. On physical exam, she had erythematous papules and pustules in a sporotrichoid pattern on the arms, axilla, upper back, and antecubital fossa. A biopsy was performed.