User login
To the Editor:
A 26-year-old man was referred to our clinic for evaluation of a persistent red rash to rule out cutaneous lupus erythematosus (LE). The patient was diagnosed at 12 years of age with autosomal-recessive chronic granulomatous disease (CGD)(nitroblue tetrazolium test, 5.0; low normal, 20.6), type p47phox mutation. At that time the patient had recurrent fevers, sinusitis, anemia, and noncaseating granulomatous liver lesions, but he lacked any cutaneous manifestations. The patient was then treated for approximately 2 years with interferon therapy but discontinued therapy given the absence of any signs or symptoms. He remained asymptomatic until approximately 16 years of age when he experienced the onset of an intermittently painful and pruritic rash on the face that slowly spread over the ensuing years to involve the trunk, arms, forearms, and hands. Although he reported that sunlight exacerbated the rash, the rash also persisted through the winter months when the majority of the sun-exposed areas of the trunk and arms were covered. He denied exposure to topical products and denied the use of any oral medications (prescription or over-the-counter).
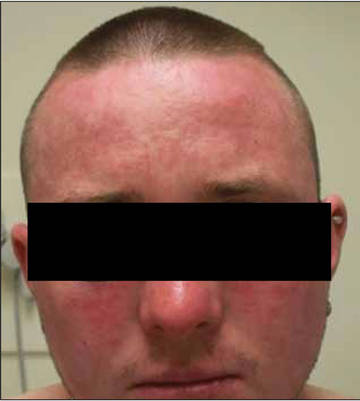
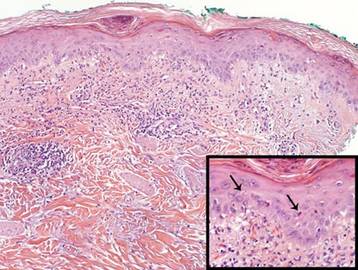
Review of systems was negative for fever, fatigue, malaise, headaches, joint pain, arthritis, oral ulcers, dyspnea, or dysuria. Physical examination revealed a well-defined exanthem comprised of erythematous, mildly indurated papules coalescing into larger plaques with white scale that were exclusively limited to the photodistributed areas of the face (Figure 1), neck, arms, forearms, hands, chest, and back. Laboratory test results included the following: minimally elevated erythrocyte sedimentation rate of 31 mm/h (reference range, 0–15 mm/h) and rheumatoid factor of 45 IU/mL (reference range, <20 IU/mL; negative antinuclear antibody screen, Sjögren syndrome antigens A and B, double-stranded DNA, anti–extractable nuclear antigen antibody test, and anti-Jo-1 antibody; complete blood cell count revealed no abnormalities; basic metabolic panel, C3 and C4, CH50, glucose-6-phosphate dehydrogenase activity, total plasma porphyrins, and testing for hepatitis B and C virus and human immunodeficiency virus serologies were negative. Skin biopsy from a lesion on the lateral arm showed features consistent with interface dermatitis (Figure 2). Additional skin biopsies for direct immunofluorescence showed linear deposition of IgG at the dermoepidermal junction, both from involved and uninvolved neck skin (more focally from the involved site). Extensive photopatch testing did not show any clinically relevant positive reactions.
|
Given the patient’s history of CGD and the extensive negative workup for rheumatologic, photoallergic, and phototoxic causes, the patient was diagnosed with a lupus-like rash of CGD. The rash failed to respond to rigorous sun avoidance and a 3-week on/1-week off regimen of high-potency class 1 topical steroids to the trunk, arms, forearms, and legs, and lower-potency class 4 topical steroid to the face, with disease flaring almost immediately on cessation of treatment during the rest weeks. Given the marked photodistribution resembling subacute cutaneous LE, oral hydroxychloroquine 200 mg (5.7 mg/kg) twice daily was initiated in addition to continued topical steroid therapy.
Four months after the addition of hydroxychloroquine, the patient showed considerable improvement of the rash. Seven months after initiation of hydroxychloroquine, the photodistributed rash was completely resolved and topical steroids were stopped. The rash remained in remission for an additional 24 months with hydroxychloroquine alone, at which time hydroxychloroquine was stopped; however, the rash flared 2 months later and hydroxychloroquine was restarted at 200 mg twice daily, resulting in clearance within 3 months. The patient was maintained on this dose of hydroxychloroquine.
During treatment, the patient had an episode of extensive furunculosis caused by Staphylococcus aureus that was successfully treated with a 14-day course of oral doxycycline 100 mg twice daily. He has since been maintained on prophylactic intranasal mupirocin ointment 2% for the first several days of each month and daily benzoyl peroxide wash 10% without further episodes. He also developed a single lesion of alopecia areata that was successfully treated with intralesional steroid injections.
Chronic granulomatous disease can either be X-linked or, less commonly, autosomal recessive, resulting from a defect in components of the nicotinamide adenine dinucleotide phosphate oxidase complex that is necessary to generate reactive oxygen intermediates for killing phagocytosed microbes. Cutaneous manifestations are relatively common in CGD (60%–70% of cases)1 and include infectious lesions (eg, recurrent mucous membrane infections, impetigo, carbuncles, otitis externa, suppurative lymphadenopathy) as well as the less common chronic inflammatory conditions such as lupus-like eruption, aphthous stomatitis, Raynaud phenomenon, arcuate dermal erythema, and Jessner lymphocytic infiltrate.2 The pathognomonic clinical feature of CGD is the presence of characteristic multinucleated giant cell granulomas distributed in multiple organ systems such as the gastrointestinal system, causing pyloric and/or small bowel obstruction, and the genitourinary system, causing ureter and/or bladder outlet obstruction.3
Importantly, CGD patients also demonstrate immune-related inflammatory disorders, most commonly inflammatory bowel disease, IgA nephropathy, sarcoidosis, and juvenile idiopathic arthritis.3 In addition, both CGD patients and female carriers of X-linked CGD have been reported to demonstrate lupus-like rashes that share overlapping clinical and histologic features with the rashes seen in true discoid LE and tumid LE patients without CGD.4-6 This lupus-like rash is more commonly observed in adulthood and in carriers, possibly secondary to the high childhood mortality rate of CGD patients.4,6
De Ravin et al3 proposed that autoimmune conditions arising in CGD patients who have met established criteria for a particular autoimmune disease should be treated for that condition rather than consider it as a part of the CGD spectrum. This theory has important therapeutic implications, including initiating paradoxical corticosteroid and/or steroid-sparing immunosuppressive agents in this otherwise immunocompromised patient population. They reported a 21-year-old man with cutaneous LE lesions and negative lupus serologies whose lesions were refractory to topical steroids but responded to systemic prednisone, requiring a low-dose alternate-day maintenance regimen.3 Beyond the development of a true autoimmune disease associated with CGD, systemic medications, specifically voriconazole, have been implicated as an alternative etiology for this rash in CGD.7 While important to consider, our patient’s rash presented in the absence of any systemic medications, supporting the former etiology over the latter.
Our case demonstrates the utility of hydroxychloroquine to treat the lupus-like rash of CGD. Similarly, the lupus-like symptoms of female carriers of X-linked CGD, predominantly with negative lupus serologies, also have been reported to respond to hydroxychloroquine and mepacrine.4,5,8-10 Interestingly, the utility of monotherapy with hydroxychloroquine may extend beyond treating cutaneous lupus-like lesions, as this regimen also was reported to successfully treat gastric granulomatous involvement in a CGD patient.11
Chronic granulomatous disease often is fatal in early childhood or adolescence due to sequelae from infections or chronic granulomatous infiltration of internal organs. Residual reactive oxygen intermediate production was shown to be a predictor of overall survival, and CGD patients with 1% of normal reactive oxygen intermediate production by neutrophils had a greater likelihood of survival.12 In this regard, the otherwise good health of our patient at the time of presentation was consistent with his initial nitroblue tetrazolium test showing some residual oxidative activity, emphasizing the phenotypic variability of this rare genetic disorder and the importance of considering CGD in the diagnosis of seronegative cutaneous lupus-like reactions.
1. Dohil M, Prendiville JS, Crawford RI, et al. Cutaneous manifestations of chronic granulomatous disease. a report of four cases and review of the literature. J Am Acad Dermatol. 1997;36(6, pt 1):899-907.
2. Chowdhury MM, Anstey A, Matthews CN. The dermatosis of chronic granulomatous disease. Clin Exp Dermatol. 2000;25:190-194.
3. De Ravin SS, Naumann N, Cowen EW, et al. Chronic granulomatous disease as a risk factor for autoimmune disease [published online ahead of print September 26, 2008]. J Allergy Clin Immunol. 2008;122:1097-1103.
4. Kragballe K, Borregaard N, Brandrup F, et al. Relation of monocyte and neutrophil oxidative metabolism to skin and oral lesions in carriers of chronic granulomatous disease. Clin Exp Immunol. 1981;43:390-398.
5. Barton LL, Johnson CR. Discoid lupus erythematosus and X-linked chronic granulomatous disease. Pediatr Dermatol. 1986;3:376-379.
6. Córdoba-Guijarro S, Feal C, Daudén E, et al. Lupus erythematosus-like lesions in a carrier of X-linked chronic granulomatous disease. J Eur Acad Dermatol Venereol. 2000;14:409-411.
7. Gomez-Moyano E, Vera-Casaño A, Moreno-Perez D, et al. Lupus erythematosus-like lesions by voriconazole in an infant with chronic granulomatous disease. Pediatr Dermatol. 2010;27:105-106.
8. Brandrup F, Koch C, Petri M, et al. Discoid lupus erythematosus-like lesions and stomatitis in female carriers of X-linked chronic granulomatous disease. Br J Dermatol. 1981;104:495-505.
9. Cale CM, Morton L, Goldblatt D. Cutaneous and other lupus-like symptoms in carriers of X-linked chronic granulomatous disease: incidence and autoimmune serology. Clin Exp Immunol. 2007;148:79-84.
10. Levinsky RJ, Harvey BA, Roberton DM, et al. A polymorph bactericidal defect and a lupus-like syndrome. Arch Dis Child. 1981;56:382-385.
11. Arlet JB, Aouba A, Suarez F, et al. Efficiency of hydroxychloroquine in the treatment of granulomatous complications in chronic granulomatous disease. Eur J Gastroenterol Hepatol. 2008;20:142-144.
12. Kuhns DB, Alvord WG, Heller T, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363:2600-2610.
To the Editor:
A 26-year-old man was referred to our clinic for evaluation of a persistent red rash to rule out cutaneous lupus erythematosus (LE). The patient was diagnosed at 12 years of age with autosomal-recessive chronic granulomatous disease (CGD)(nitroblue tetrazolium test, 5.0; low normal, 20.6), type p47phox mutation. At that time the patient had recurrent fevers, sinusitis, anemia, and noncaseating granulomatous liver lesions, but he lacked any cutaneous manifestations. The patient was then treated for approximately 2 years with interferon therapy but discontinued therapy given the absence of any signs or symptoms. He remained asymptomatic until approximately 16 years of age when he experienced the onset of an intermittently painful and pruritic rash on the face that slowly spread over the ensuing years to involve the trunk, arms, forearms, and hands. Although he reported that sunlight exacerbated the rash, the rash also persisted through the winter months when the majority of the sun-exposed areas of the trunk and arms were covered. He denied exposure to topical products and denied the use of any oral medications (prescription or over-the-counter).
Review of systems was negative for fever, fatigue, malaise, headaches, joint pain, arthritis, oral ulcers, dyspnea, or dysuria. Physical examination revealed a well-defined exanthem comprised of erythematous, mildly indurated papules coalescing into larger plaques with white scale that were exclusively limited to the photodistributed areas of the face (Figure 1), neck, arms, forearms, hands, chest, and back. Laboratory test results included the following: minimally elevated erythrocyte sedimentation rate of 31 mm/h (reference range, 0–15 mm/h) and rheumatoid factor of 45 IU/mL (reference range, <20 IU/mL; negative antinuclear antibody screen, Sjögren syndrome antigens A and B, double-stranded DNA, anti–extractable nuclear antigen antibody test, and anti-Jo-1 antibody; complete blood cell count revealed no abnormalities; basic metabolic panel, C3 and C4, CH50, glucose-6-phosphate dehydrogenase activity, total plasma porphyrins, and testing for hepatitis B and C virus and human immunodeficiency virus serologies were negative. Skin biopsy from a lesion on the lateral arm showed features consistent with interface dermatitis (Figure 2). Additional skin biopsies for direct immunofluorescence showed linear deposition of IgG at the dermoepidermal junction, both from involved and uninvolved neck skin (more focally from the involved site). Extensive photopatch testing did not show any clinically relevant positive reactions.
|
Given the patient’s history of CGD and the extensive negative workup for rheumatologic, photoallergic, and phototoxic causes, the patient was diagnosed with a lupus-like rash of CGD. The rash failed to respond to rigorous sun avoidance and a 3-week on/1-week off regimen of high-potency class 1 topical steroids to the trunk, arms, forearms, and legs, and lower-potency class 4 topical steroid to the face, with disease flaring almost immediately on cessation of treatment during the rest weeks. Given the marked photodistribution resembling subacute cutaneous LE, oral hydroxychloroquine 200 mg (5.7 mg/kg) twice daily was initiated in addition to continued topical steroid therapy.
Four months after the addition of hydroxychloroquine, the patient showed considerable improvement of the rash. Seven months after initiation of hydroxychloroquine, the photodistributed rash was completely resolved and topical steroids were stopped. The rash remained in remission for an additional 24 months with hydroxychloroquine alone, at which time hydroxychloroquine was stopped; however, the rash flared 2 months later and hydroxychloroquine was restarted at 200 mg twice daily, resulting in clearance within 3 months. The patient was maintained on this dose of hydroxychloroquine.
During treatment, the patient had an episode of extensive furunculosis caused by Staphylococcus aureus that was successfully treated with a 14-day course of oral doxycycline 100 mg twice daily. He has since been maintained on prophylactic intranasal mupirocin ointment 2% for the first several days of each month and daily benzoyl peroxide wash 10% without further episodes. He also developed a single lesion of alopecia areata that was successfully treated with intralesional steroid injections.
Chronic granulomatous disease can either be X-linked or, less commonly, autosomal recessive, resulting from a defect in components of the nicotinamide adenine dinucleotide phosphate oxidase complex that is necessary to generate reactive oxygen intermediates for killing phagocytosed microbes. Cutaneous manifestations are relatively common in CGD (60%–70% of cases)1 and include infectious lesions (eg, recurrent mucous membrane infections, impetigo, carbuncles, otitis externa, suppurative lymphadenopathy) as well as the less common chronic inflammatory conditions such as lupus-like eruption, aphthous stomatitis, Raynaud phenomenon, arcuate dermal erythema, and Jessner lymphocytic infiltrate.2 The pathognomonic clinical feature of CGD is the presence of characteristic multinucleated giant cell granulomas distributed in multiple organ systems such as the gastrointestinal system, causing pyloric and/or small bowel obstruction, and the genitourinary system, causing ureter and/or bladder outlet obstruction.3
Importantly, CGD patients also demonstrate immune-related inflammatory disorders, most commonly inflammatory bowel disease, IgA nephropathy, sarcoidosis, and juvenile idiopathic arthritis.3 In addition, both CGD patients and female carriers of X-linked CGD have been reported to demonstrate lupus-like rashes that share overlapping clinical and histologic features with the rashes seen in true discoid LE and tumid LE patients without CGD.4-6 This lupus-like rash is more commonly observed in adulthood and in carriers, possibly secondary to the high childhood mortality rate of CGD patients.4,6
De Ravin et al3 proposed that autoimmune conditions arising in CGD patients who have met established criteria for a particular autoimmune disease should be treated for that condition rather than consider it as a part of the CGD spectrum. This theory has important therapeutic implications, including initiating paradoxical corticosteroid and/or steroid-sparing immunosuppressive agents in this otherwise immunocompromised patient population. They reported a 21-year-old man with cutaneous LE lesions and negative lupus serologies whose lesions were refractory to topical steroids but responded to systemic prednisone, requiring a low-dose alternate-day maintenance regimen.3 Beyond the development of a true autoimmune disease associated with CGD, systemic medications, specifically voriconazole, have been implicated as an alternative etiology for this rash in CGD.7 While important to consider, our patient’s rash presented in the absence of any systemic medications, supporting the former etiology over the latter.
Our case demonstrates the utility of hydroxychloroquine to treat the lupus-like rash of CGD. Similarly, the lupus-like symptoms of female carriers of X-linked CGD, predominantly with negative lupus serologies, also have been reported to respond to hydroxychloroquine and mepacrine.4,5,8-10 Interestingly, the utility of monotherapy with hydroxychloroquine may extend beyond treating cutaneous lupus-like lesions, as this regimen also was reported to successfully treat gastric granulomatous involvement in a CGD patient.11
Chronic granulomatous disease often is fatal in early childhood or adolescence due to sequelae from infections or chronic granulomatous infiltration of internal organs. Residual reactive oxygen intermediate production was shown to be a predictor of overall survival, and CGD patients with 1% of normal reactive oxygen intermediate production by neutrophils had a greater likelihood of survival.12 In this regard, the otherwise good health of our patient at the time of presentation was consistent with his initial nitroblue tetrazolium test showing some residual oxidative activity, emphasizing the phenotypic variability of this rare genetic disorder and the importance of considering CGD in the diagnosis of seronegative cutaneous lupus-like reactions.
To the Editor:
A 26-year-old man was referred to our clinic for evaluation of a persistent red rash to rule out cutaneous lupus erythematosus (LE). The patient was diagnosed at 12 years of age with autosomal-recessive chronic granulomatous disease (CGD)(nitroblue tetrazolium test, 5.0; low normal, 20.6), type p47phox mutation. At that time the patient had recurrent fevers, sinusitis, anemia, and noncaseating granulomatous liver lesions, but he lacked any cutaneous manifestations. The patient was then treated for approximately 2 years with interferon therapy but discontinued therapy given the absence of any signs or symptoms. He remained asymptomatic until approximately 16 years of age when he experienced the onset of an intermittently painful and pruritic rash on the face that slowly spread over the ensuing years to involve the trunk, arms, forearms, and hands. Although he reported that sunlight exacerbated the rash, the rash also persisted through the winter months when the majority of the sun-exposed areas of the trunk and arms were covered. He denied exposure to topical products and denied the use of any oral medications (prescription or over-the-counter).
Review of systems was negative for fever, fatigue, malaise, headaches, joint pain, arthritis, oral ulcers, dyspnea, or dysuria. Physical examination revealed a well-defined exanthem comprised of erythematous, mildly indurated papules coalescing into larger plaques with white scale that were exclusively limited to the photodistributed areas of the face (Figure 1), neck, arms, forearms, hands, chest, and back. Laboratory test results included the following: minimally elevated erythrocyte sedimentation rate of 31 mm/h (reference range, 0–15 mm/h) and rheumatoid factor of 45 IU/mL (reference range, <20 IU/mL; negative antinuclear antibody screen, Sjögren syndrome antigens A and B, double-stranded DNA, anti–extractable nuclear antigen antibody test, and anti-Jo-1 antibody; complete blood cell count revealed no abnormalities; basic metabolic panel, C3 and C4, CH50, glucose-6-phosphate dehydrogenase activity, total plasma porphyrins, and testing for hepatitis B and C virus and human immunodeficiency virus serologies were negative. Skin biopsy from a lesion on the lateral arm showed features consistent with interface dermatitis (Figure 2). Additional skin biopsies for direct immunofluorescence showed linear deposition of IgG at the dermoepidermal junction, both from involved and uninvolved neck skin (more focally from the involved site). Extensive photopatch testing did not show any clinically relevant positive reactions.
|
Given the patient’s history of CGD and the extensive negative workup for rheumatologic, photoallergic, and phototoxic causes, the patient was diagnosed with a lupus-like rash of CGD. The rash failed to respond to rigorous sun avoidance and a 3-week on/1-week off regimen of high-potency class 1 topical steroids to the trunk, arms, forearms, and legs, and lower-potency class 4 topical steroid to the face, with disease flaring almost immediately on cessation of treatment during the rest weeks. Given the marked photodistribution resembling subacute cutaneous LE, oral hydroxychloroquine 200 mg (5.7 mg/kg) twice daily was initiated in addition to continued topical steroid therapy.
Four months after the addition of hydroxychloroquine, the patient showed considerable improvement of the rash. Seven months after initiation of hydroxychloroquine, the photodistributed rash was completely resolved and topical steroids were stopped. The rash remained in remission for an additional 24 months with hydroxychloroquine alone, at which time hydroxychloroquine was stopped; however, the rash flared 2 months later and hydroxychloroquine was restarted at 200 mg twice daily, resulting in clearance within 3 months. The patient was maintained on this dose of hydroxychloroquine.
During treatment, the patient had an episode of extensive furunculosis caused by Staphylococcus aureus that was successfully treated with a 14-day course of oral doxycycline 100 mg twice daily. He has since been maintained on prophylactic intranasal mupirocin ointment 2% for the first several days of each month and daily benzoyl peroxide wash 10% without further episodes. He also developed a single lesion of alopecia areata that was successfully treated with intralesional steroid injections.
Chronic granulomatous disease can either be X-linked or, less commonly, autosomal recessive, resulting from a defect in components of the nicotinamide adenine dinucleotide phosphate oxidase complex that is necessary to generate reactive oxygen intermediates for killing phagocytosed microbes. Cutaneous manifestations are relatively common in CGD (60%–70% of cases)1 and include infectious lesions (eg, recurrent mucous membrane infections, impetigo, carbuncles, otitis externa, suppurative lymphadenopathy) as well as the less common chronic inflammatory conditions such as lupus-like eruption, aphthous stomatitis, Raynaud phenomenon, arcuate dermal erythema, and Jessner lymphocytic infiltrate.2 The pathognomonic clinical feature of CGD is the presence of characteristic multinucleated giant cell granulomas distributed in multiple organ systems such as the gastrointestinal system, causing pyloric and/or small bowel obstruction, and the genitourinary system, causing ureter and/or bladder outlet obstruction.3
Importantly, CGD patients also demonstrate immune-related inflammatory disorders, most commonly inflammatory bowel disease, IgA nephropathy, sarcoidosis, and juvenile idiopathic arthritis.3 In addition, both CGD patients and female carriers of X-linked CGD have been reported to demonstrate lupus-like rashes that share overlapping clinical and histologic features with the rashes seen in true discoid LE and tumid LE patients without CGD.4-6 This lupus-like rash is more commonly observed in adulthood and in carriers, possibly secondary to the high childhood mortality rate of CGD patients.4,6
De Ravin et al3 proposed that autoimmune conditions arising in CGD patients who have met established criteria for a particular autoimmune disease should be treated for that condition rather than consider it as a part of the CGD spectrum. This theory has important therapeutic implications, including initiating paradoxical corticosteroid and/or steroid-sparing immunosuppressive agents in this otherwise immunocompromised patient population. They reported a 21-year-old man with cutaneous LE lesions and negative lupus serologies whose lesions were refractory to topical steroids but responded to systemic prednisone, requiring a low-dose alternate-day maintenance regimen.3 Beyond the development of a true autoimmune disease associated with CGD, systemic medications, specifically voriconazole, have been implicated as an alternative etiology for this rash in CGD.7 While important to consider, our patient’s rash presented in the absence of any systemic medications, supporting the former etiology over the latter.
Our case demonstrates the utility of hydroxychloroquine to treat the lupus-like rash of CGD. Similarly, the lupus-like symptoms of female carriers of X-linked CGD, predominantly with negative lupus serologies, also have been reported to respond to hydroxychloroquine and mepacrine.4,5,8-10 Interestingly, the utility of monotherapy with hydroxychloroquine may extend beyond treating cutaneous lupus-like lesions, as this regimen also was reported to successfully treat gastric granulomatous involvement in a CGD patient.11
Chronic granulomatous disease often is fatal in early childhood or adolescence due to sequelae from infections or chronic granulomatous infiltration of internal organs. Residual reactive oxygen intermediate production was shown to be a predictor of overall survival, and CGD patients with 1% of normal reactive oxygen intermediate production by neutrophils had a greater likelihood of survival.12 In this regard, the otherwise good health of our patient at the time of presentation was consistent with his initial nitroblue tetrazolium test showing some residual oxidative activity, emphasizing the phenotypic variability of this rare genetic disorder and the importance of considering CGD in the diagnosis of seronegative cutaneous lupus-like reactions.
1. Dohil M, Prendiville JS, Crawford RI, et al. Cutaneous manifestations of chronic granulomatous disease. a report of four cases and review of the literature. J Am Acad Dermatol. 1997;36(6, pt 1):899-907.
2. Chowdhury MM, Anstey A, Matthews CN. The dermatosis of chronic granulomatous disease. Clin Exp Dermatol. 2000;25:190-194.
3. De Ravin SS, Naumann N, Cowen EW, et al. Chronic granulomatous disease as a risk factor for autoimmune disease [published online ahead of print September 26, 2008]. J Allergy Clin Immunol. 2008;122:1097-1103.
4. Kragballe K, Borregaard N, Brandrup F, et al. Relation of monocyte and neutrophil oxidative metabolism to skin and oral lesions in carriers of chronic granulomatous disease. Clin Exp Immunol. 1981;43:390-398.
5. Barton LL, Johnson CR. Discoid lupus erythematosus and X-linked chronic granulomatous disease. Pediatr Dermatol. 1986;3:376-379.
6. Córdoba-Guijarro S, Feal C, Daudén E, et al. Lupus erythematosus-like lesions in a carrier of X-linked chronic granulomatous disease. J Eur Acad Dermatol Venereol. 2000;14:409-411.
7. Gomez-Moyano E, Vera-Casaño A, Moreno-Perez D, et al. Lupus erythematosus-like lesions by voriconazole in an infant with chronic granulomatous disease. Pediatr Dermatol. 2010;27:105-106.
8. Brandrup F, Koch C, Petri M, et al. Discoid lupus erythematosus-like lesions and stomatitis in female carriers of X-linked chronic granulomatous disease. Br J Dermatol. 1981;104:495-505.
9. Cale CM, Morton L, Goldblatt D. Cutaneous and other lupus-like symptoms in carriers of X-linked chronic granulomatous disease: incidence and autoimmune serology. Clin Exp Immunol. 2007;148:79-84.
10. Levinsky RJ, Harvey BA, Roberton DM, et al. A polymorph bactericidal defect and a lupus-like syndrome. Arch Dis Child. 1981;56:382-385.
11. Arlet JB, Aouba A, Suarez F, et al. Efficiency of hydroxychloroquine in the treatment of granulomatous complications in chronic granulomatous disease. Eur J Gastroenterol Hepatol. 2008;20:142-144.
12. Kuhns DB, Alvord WG, Heller T, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363:2600-2610.
1. Dohil M, Prendiville JS, Crawford RI, et al. Cutaneous manifestations of chronic granulomatous disease. a report of four cases and review of the literature. J Am Acad Dermatol. 1997;36(6, pt 1):899-907.
2. Chowdhury MM, Anstey A, Matthews CN. The dermatosis of chronic granulomatous disease. Clin Exp Dermatol. 2000;25:190-194.
3. De Ravin SS, Naumann N, Cowen EW, et al. Chronic granulomatous disease as a risk factor for autoimmune disease [published online ahead of print September 26, 2008]. J Allergy Clin Immunol. 2008;122:1097-1103.
4. Kragballe K, Borregaard N, Brandrup F, et al. Relation of monocyte and neutrophil oxidative metabolism to skin and oral lesions in carriers of chronic granulomatous disease. Clin Exp Immunol. 1981;43:390-398.
5. Barton LL, Johnson CR. Discoid lupus erythematosus and X-linked chronic granulomatous disease. Pediatr Dermatol. 1986;3:376-379.
6. Córdoba-Guijarro S, Feal C, Daudén E, et al. Lupus erythematosus-like lesions in a carrier of X-linked chronic granulomatous disease. J Eur Acad Dermatol Venereol. 2000;14:409-411.
7. Gomez-Moyano E, Vera-Casaño A, Moreno-Perez D, et al. Lupus erythematosus-like lesions by voriconazole in an infant with chronic granulomatous disease. Pediatr Dermatol. 2010;27:105-106.
8. Brandrup F, Koch C, Petri M, et al. Discoid lupus erythematosus-like lesions and stomatitis in female carriers of X-linked chronic granulomatous disease. Br J Dermatol. 1981;104:495-505.
9. Cale CM, Morton L, Goldblatt D. Cutaneous and other lupus-like symptoms in carriers of X-linked chronic granulomatous disease: incidence and autoimmune serology. Clin Exp Immunol. 2007;148:79-84.
10. Levinsky RJ, Harvey BA, Roberton DM, et al. A polymorph bactericidal defect and a lupus-like syndrome. Arch Dis Child. 1981;56:382-385.
11. Arlet JB, Aouba A, Suarez F, et al. Efficiency of hydroxychloroquine in the treatment of granulomatous complications in chronic granulomatous disease. Eur J Gastroenterol Hepatol. 2008;20:142-144.
12. Kuhns DB, Alvord WG, Heller T, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363:2600-2610.