User login
To the Editor:
A 7-day-old full-term infant presented to the neonatal intensive care unit with poor feeding and altered consciousness. She was born at 39 weeks and 3 days to a gravida 1 mother with a pregnancy history complicated by maternal chorioamnionitis and gestational diabetes. During labor, nonreassuring fetal heart tones and arrest of labor prompted an uncomplicated cesarean delivery with normal Apgar scores at birth. The infant’s family history revealed only beta thalassemia minor in her father. At 5 to 7 days of life, the mother noted difficulty with feeding and poor latch along with lethargy and depressed consciousness in the infant.
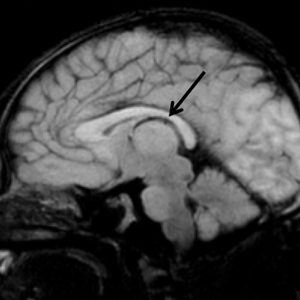
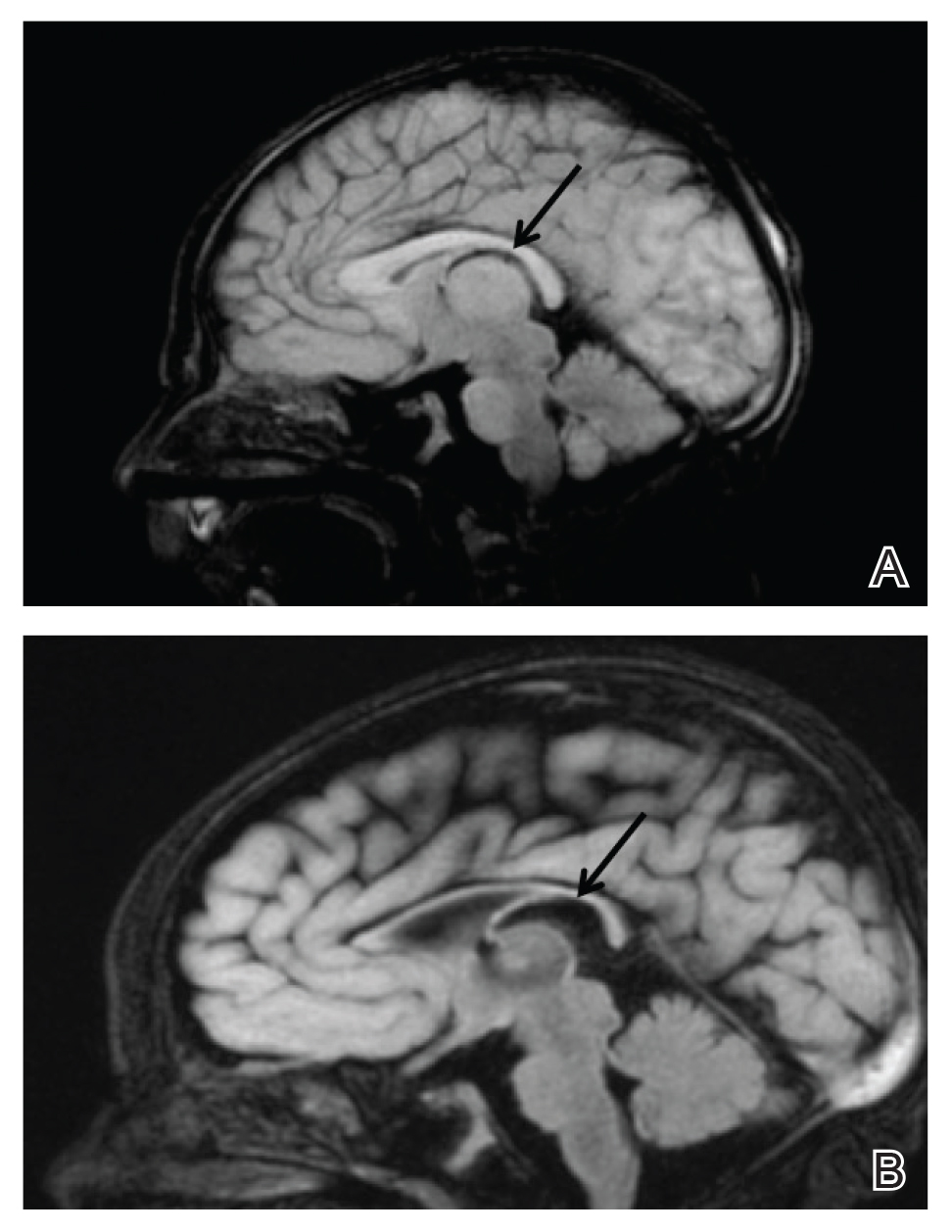
Upon arrival to the neonatal intensive care unit, the infant was noted to have rhythmic lip-smacking behavior, intermittent nystagmus, mild hypotonia, and clonic movements of the left upper extremity. An electroencephalogram was markedly abnormal, capturing multiple seizures in the bilateral cortical hemispheres. She was loaded with phenobarbital with no further seizure activity. Brain magnetic resonance imaging revealed innumerable punctate foci of restricted diffusion with corresponding punctate hemorrhage within the frontal and parietal white matter, as well as cortical diffusion restriction within the occipital lobe, inferior temporal lobe, bilateral thalami, and corpus callosum (Figure 1). An exhaustive infectious workup also was completed and was unremarkable, though she was treated with broad-spectrum antimicrobials, including intravenous acyclovir.
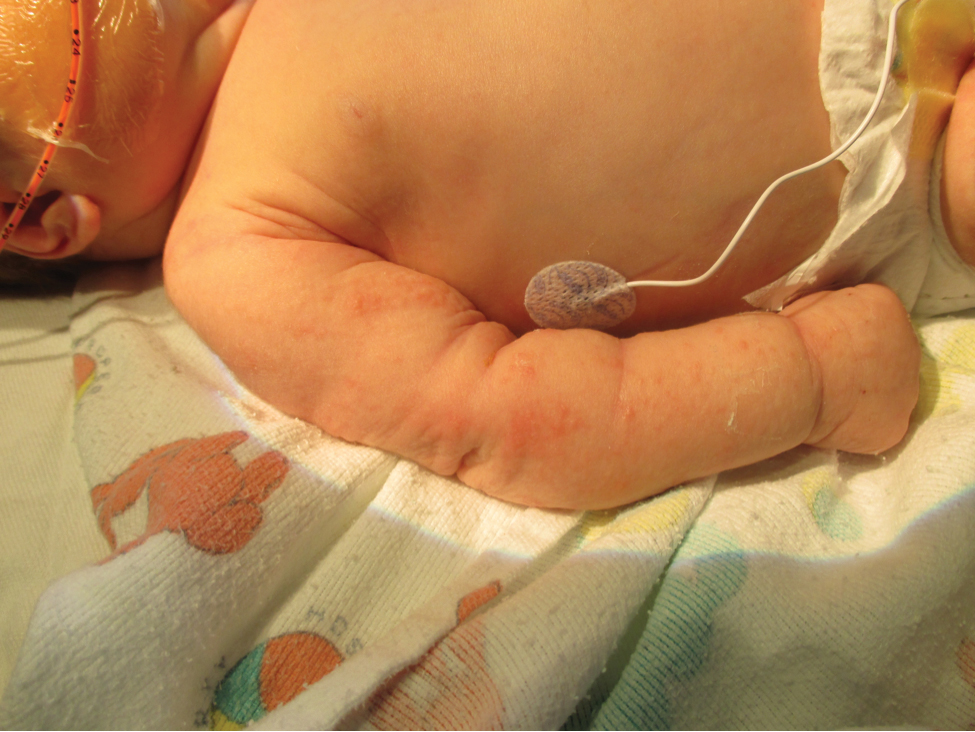
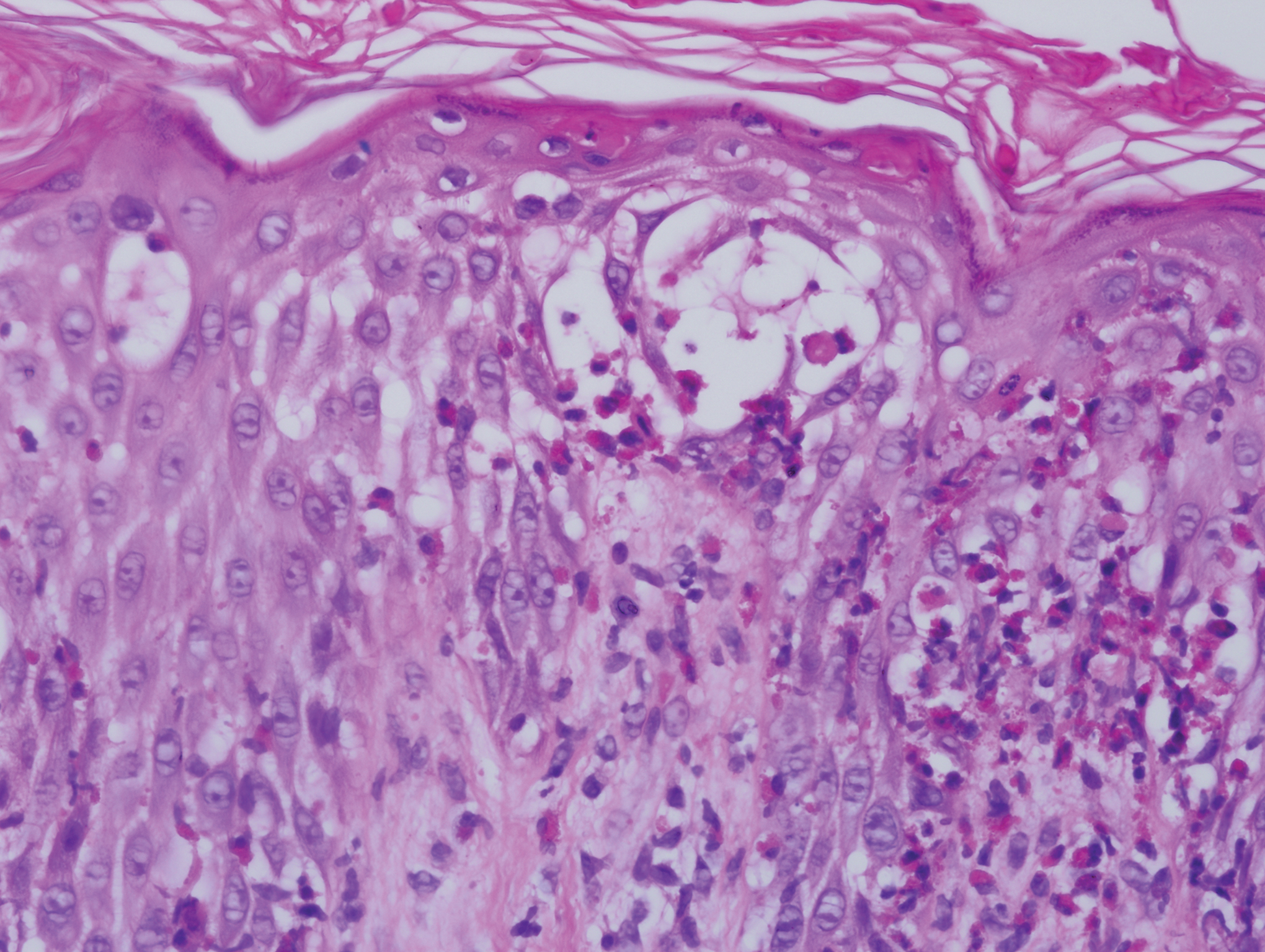
Five days after being hospitalized (day 10 of life), a vesicular rash was noted on the arms and legs (Figure 2). Discussion with the patient’s mother revealed that the first signs of unusual skin lesions occurred as early as several days prior. There were no oral mucosal lesions or gross ocular abnormalities. No nail changes were appreciated. A bedside Tzanck preparation was negative for viral cytopathic changes. A skin biopsy was performed that demonstrated eosinophilic spongiosis with necrotic keratinocytes, typical of the vesicular stage of incontinentia pigmenti (IP)(Figure 3). An ophthalmology examination showed an arteriovenous malformation of the right eye with subtle neovascularization at the infratemporal periphery, consistent with known ocular manifestations of IP. The infant’s mother reported no history of notable dental abnormalities, hair loss, skin rashes, or nail changes. Genetic testing demonstrated the common IKBKG (inhibitor of κ light polypeptide gene enhancer in B cells, kinase gamma [formerly known as NEMO]) gene deletion on the X chromosome, consistent with IP.
She successfully underwent retinal laser ablative therapy for the ocular manifestations without further evidence of neovascularization. She developed a mild cataract that was not visually significant and required no intervention. Her brain abnormalities were thought to represent foci of necrosis with superimposed hemorrhagic transformation due to spontaneous degeneration of brain cells in which the mutated X chromosome was activated. No further treatment was indicated beyond suppression of the consequent seizures. There was no notable cortical edema or other medical indication for systemic glucocorticoid therapy. Phenobarbital was continued without further seizure events.
Several months after the initial presentation, a follow-up electroencephalogram was normal. Phenobarbital was slowly weaned and finally discontinued approximately 6 months after the initial event with no other reported seizures. She currently is achieving normal developmental milestones with the exception of slight motor delay and expected residual hypotonia.
Incontinentia pigmenti, also known as Bloch-Sulzberger syndrome, is a rare multisystem neuroectodermal disorder, primarily affecting the skin, central nervous system (CNS), and retinas. The disorder can be inherited in an X-linked dominant fashion and appears almost exclusively in women with typical in utero lethality seen in males. Most affected individuals have a sporadic, or de novo, mutation, which was likely the case in our patient given that her mother demonstrated no signs or symptoms.1 The pathogenesis of disease is a defect at chromosome Xq28 that is a region encoding the nuclear factor–κB essential modulator, IKBKG. Absence or mutation of IKBKG in IP results in failure to activate nuclear factor–κB and leaves cells vulnerable to cytokine-mediated apoptosis, especially after exposure to tumor necrosis factor α.2
Clinical manifestations of IP are present at or soon after birth. The cutaneous findings of this disorder are classically described as a step-wise progression through 4 distinct stages: (1) a linear and/or whorled vesicular eruption predominantly on the extremities at birth or within the first few weeks of life; (2) thickened linear or whorled verrucous plaques; (3) hyperpigmented streaks and whorls that may or may not correspond with prior affected areas that may resolve by adolescence; and (4) hypopigmented, possibly atrophic plaques on the extremities that may persist lifelong. Importantly, not every patient will experience each of these stages. Overlap can occur, and the time course of each stage is highly variable. Other ectodermal manifestations include dental abnormalities such as small, misshaped, or missing teeth; alopecia; and nail abnormalities. Ocular abnormalities associated with IP primarily occur in the retina, including vascular occlusion, neovascularization, hemorrhages, foveal abnormalities, as well as exudative and tractional detachments.3,4
It is crucial to recognize CNS anomalies in association with the cutaneous findings of IP, as CNS pathology can be severe with profound developmental implications. Central nervous system findings have been noted to correlate with the appearance of the vesicular stage of IP. A high index of suspicion is needed, as the disease can demonstrate progression within a short time.5-8 The most frequent anomalies include seizures, motor impairment, intellectual disability, and microcephaly.9,10 Some of the most commonly identified CNS lesions on imaging include necrosis or brain infarcts, atrophy, and lesions of the corpus callosum.7
The pathogenesis of observed CNS changes in IP is not well understood. There have been numerous proposals of a vascular mechanism, and a microangiopathic process appears to be most plausible. Mutations in IKBKG may result in interruption of signaling via vascular endothelial growth factor receptor 3 with a consequent impact on angiogenesis, supporting a vascular mechanism. Additionally, mutations in IKBKG lead to activation of eotaxin, an eosinophil-selective chemokine.9 Eotaxin activation results in eosinophilic degranulation that mediates the classic eosinophilic infiltrate seen in the classic skin histology of IP. Additionally, it has been shown that eotaxin is strongly expressed by endothelial cells in IP, and more abundant eosinophil degranulation may play a role in mediating vaso-occlusion.7 Other studies have found that the highest expression level of the IKBKG gene is in the CNS, potentially explaining the extensive imaging findings of hemorrhage and diffusion restriction in our patient. These features likely are attributable to apoptosis of cells possessing the mutated IKBKG gene.9-11
- Ehrenreich M, Tarlow MM, Godlewska-Janusz E, et al. Incontinentia pigmenti (Bloch-Sulzberger syndrome): a systemic disorder. Cutis. 2007;79:355-362.
- Smahi A, Courtois G, Rabia SH, et al. The NF-kappaB signaling pathway in human diseases: from incontinentia pigmenti to ectodermal dysplasias and immune-deficiency syndromes. Hum Mol Genet. 2002;11:2371-2375.
- O’Doherty M, McCreery K, Green AJ, et al. Incontinentia pigmenti—ophthalmological observation of a series of cases and review of the literature. Br J Ophthalmol. 2011;95:11-16.
- Swinney CC, Han DP, Karth PA. Incontinentia pigmenti: a comprehensive review and update. Ophthalmic Surg Lasers Imaging Retina. 2015;46:650-657.
- Hennel SJ, Ekert PG, Volpe JJ, et al. Insights into the pathogenesis of cerebral lesions in incontinentia pigmenti. Pediatr Neurol. 2003;29:148-150.
- Maingay-de Groof F, Lequin MH, Roofthooft DW, et al. Extensive cerebral infarction in the newborn due to incontinentia pigmenti. Eur J Paediatr Neurol. 2008;12:284-289.
- Minic´ S, Trpinac D, Obradovic´ M. Systematic review of central nervous system anomalies in incontinentia pigmenti. Orphanet J Rare Dis. 2013;8:25-35.
- Wolf NI, Kramer N, Harting I, et al. Diffuse cortical necrosis in a neonate with incontinentia pigmenti and an encephalitis-like presentation. AJNR Am J Neuroradiol. 2005;26:1580-1582.
- Phan TA, Wargon O, Turner AM. Incontinentia pigmenti case series: clinical spectrum of incontinentia pigmenti in 53 female patients and their relatives. Clin Exp Dermatol. 2005;30:474-480.
- Volpe J. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res. 2001;50:553-562.
- Pascual-Castroviejo I, Pascual-Pascual SI, Velazquez-Fragua R, et al. Incontinentia pigmenti: clinical and neuroimaging findings in a series of 12 patients. Neurologia. 2006;21:239-248.
To the Editor:
A 7-day-old full-term infant presented to the neonatal intensive care unit with poor feeding and altered consciousness. She was born at 39 weeks and 3 days to a gravida 1 mother with a pregnancy history complicated by maternal chorioamnionitis and gestational diabetes. During labor, nonreassuring fetal heart tones and arrest of labor prompted an uncomplicated cesarean delivery with normal Apgar scores at birth. The infant’s family history revealed only beta thalassemia minor in her father. At 5 to 7 days of life, the mother noted difficulty with feeding and poor latch along with lethargy and depressed consciousness in the infant.
Upon arrival to the neonatal intensive care unit, the infant was noted to have rhythmic lip-smacking behavior, intermittent nystagmus, mild hypotonia, and clonic movements of the left upper extremity. An electroencephalogram was markedly abnormal, capturing multiple seizures in the bilateral cortical hemispheres. She was loaded with phenobarbital with no further seizure activity. Brain magnetic resonance imaging revealed innumerable punctate foci of restricted diffusion with corresponding punctate hemorrhage within the frontal and parietal white matter, as well as cortical diffusion restriction within the occipital lobe, inferior temporal lobe, bilateral thalami, and corpus callosum (Figure 1). An exhaustive infectious workup also was completed and was unremarkable, though she was treated with broad-spectrum antimicrobials, including intravenous acyclovir.
Five days after being hospitalized (day 10 of life), a vesicular rash was noted on the arms and legs (Figure 2). Discussion with the patient’s mother revealed that the first signs of unusual skin lesions occurred as early as several days prior. There were no oral mucosal lesions or gross ocular abnormalities. No nail changes were appreciated. A bedside Tzanck preparation was negative for viral cytopathic changes. A skin biopsy was performed that demonstrated eosinophilic spongiosis with necrotic keratinocytes, typical of the vesicular stage of incontinentia pigmenti (IP)(Figure 3). An ophthalmology examination showed an arteriovenous malformation of the right eye with subtle neovascularization at the infratemporal periphery, consistent with known ocular manifestations of IP. The infant’s mother reported no history of notable dental abnormalities, hair loss, skin rashes, or nail changes. Genetic testing demonstrated the common IKBKG (inhibitor of κ light polypeptide gene enhancer in B cells, kinase gamma [formerly known as NEMO]) gene deletion on the X chromosome, consistent with IP.
She successfully underwent retinal laser ablative therapy for the ocular manifestations without further evidence of neovascularization. She developed a mild cataract that was not visually significant and required no intervention. Her brain abnormalities were thought to represent foci of necrosis with superimposed hemorrhagic transformation due to spontaneous degeneration of brain cells in which the mutated X chromosome was activated. No further treatment was indicated beyond suppression of the consequent seizures. There was no notable cortical edema or other medical indication for systemic glucocorticoid therapy. Phenobarbital was continued without further seizure events.
Several months after the initial presentation, a follow-up electroencephalogram was normal. Phenobarbital was slowly weaned and finally discontinued approximately 6 months after the initial event with no other reported seizures. She currently is achieving normal developmental milestones with the exception of slight motor delay and expected residual hypotonia.
Incontinentia pigmenti, also known as Bloch-Sulzberger syndrome, is a rare multisystem neuroectodermal disorder, primarily affecting the skin, central nervous system (CNS), and retinas. The disorder can be inherited in an X-linked dominant fashion and appears almost exclusively in women with typical in utero lethality seen in males. Most affected individuals have a sporadic, or de novo, mutation, which was likely the case in our patient given that her mother demonstrated no signs or symptoms.1 The pathogenesis of disease is a defect at chromosome Xq28 that is a region encoding the nuclear factor–κB essential modulator, IKBKG. Absence or mutation of IKBKG in IP results in failure to activate nuclear factor–κB and leaves cells vulnerable to cytokine-mediated apoptosis, especially after exposure to tumor necrosis factor α.2
Clinical manifestations of IP are present at or soon after birth. The cutaneous findings of this disorder are classically described as a step-wise progression through 4 distinct stages: (1) a linear and/or whorled vesicular eruption predominantly on the extremities at birth or within the first few weeks of life; (2) thickened linear or whorled verrucous plaques; (3) hyperpigmented streaks and whorls that may or may not correspond with prior affected areas that may resolve by adolescence; and (4) hypopigmented, possibly atrophic plaques on the extremities that may persist lifelong. Importantly, not every patient will experience each of these stages. Overlap can occur, and the time course of each stage is highly variable. Other ectodermal manifestations include dental abnormalities such as small, misshaped, or missing teeth; alopecia; and nail abnormalities. Ocular abnormalities associated with IP primarily occur in the retina, including vascular occlusion, neovascularization, hemorrhages, foveal abnormalities, as well as exudative and tractional detachments.3,4
It is crucial to recognize CNS anomalies in association with the cutaneous findings of IP, as CNS pathology can be severe with profound developmental implications. Central nervous system findings have been noted to correlate with the appearance of the vesicular stage of IP. A high index of suspicion is needed, as the disease can demonstrate progression within a short time.5-8 The most frequent anomalies include seizures, motor impairment, intellectual disability, and microcephaly.9,10 Some of the most commonly identified CNS lesions on imaging include necrosis or brain infarcts, atrophy, and lesions of the corpus callosum.7
The pathogenesis of observed CNS changes in IP is not well understood. There have been numerous proposals of a vascular mechanism, and a microangiopathic process appears to be most plausible. Mutations in IKBKG may result in interruption of signaling via vascular endothelial growth factor receptor 3 with a consequent impact on angiogenesis, supporting a vascular mechanism. Additionally, mutations in IKBKG lead to activation of eotaxin, an eosinophil-selective chemokine.9 Eotaxin activation results in eosinophilic degranulation that mediates the classic eosinophilic infiltrate seen in the classic skin histology of IP. Additionally, it has been shown that eotaxin is strongly expressed by endothelial cells in IP, and more abundant eosinophil degranulation may play a role in mediating vaso-occlusion.7 Other studies have found that the highest expression level of the IKBKG gene is in the CNS, potentially explaining the extensive imaging findings of hemorrhage and diffusion restriction in our patient. These features likely are attributable to apoptosis of cells possessing the mutated IKBKG gene.9-11
To the Editor:
A 7-day-old full-term infant presented to the neonatal intensive care unit with poor feeding and altered consciousness. She was born at 39 weeks and 3 days to a gravida 1 mother with a pregnancy history complicated by maternal chorioamnionitis and gestational diabetes. During labor, nonreassuring fetal heart tones and arrest of labor prompted an uncomplicated cesarean delivery with normal Apgar scores at birth. The infant’s family history revealed only beta thalassemia minor in her father. At 5 to 7 days of life, the mother noted difficulty with feeding and poor latch along with lethargy and depressed consciousness in the infant.
Upon arrival to the neonatal intensive care unit, the infant was noted to have rhythmic lip-smacking behavior, intermittent nystagmus, mild hypotonia, and clonic movements of the left upper extremity. An electroencephalogram was markedly abnormal, capturing multiple seizures in the bilateral cortical hemispheres. She was loaded with phenobarbital with no further seizure activity. Brain magnetic resonance imaging revealed innumerable punctate foci of restricted diffusion with corresponding punctate hemorrhage within the frontal and parietal white matter, as well as cortical diffusion restriction within the occipital lobe, inferior temporal lobe, bilateral thalami, and corpus callosum (Figure 1). An exhaustive infectious workup also was completed and was unremarkable, though she was treated with broad-spectrum antimicrobials, including intravenous acyclovir.
Five days after being hospitalized (day 10 of life), a vesicular rash was noted on the arms and legs (Figure 2). Discussion with the patient’s mother revealed that the first signs of unusual skin lesions occurred as early as several days prior. There were no oral mucosal lesions or gross ocular abnormalities. No nail changes were appreciated. A bedside Tzanck preparation was negative for viral cytopathic changes. A skin biopsy was performed that demonstrated eosinophilic spongiosis with necrotic keratinocytes, typical of the vesicular stage of incontinentia pigmenti (IP)(Figure 3). An ophthalmology examination showed an arteriovenous malformation of the right eye with subtle neovascularization at the infratemporal periphery, consistent with known ocular manifestations of IP. The infant’s mother reported no history of notable dental abnormalities, hair loss, skin rashes, or nail changes. Genetic testing demonstrated the common IKBKG (inhibitor of κ light polypeptide gene enhancer in B cells, kinase gamma [formerly known as NEMO]) gene deletion on the X chromosome, consistent with IP.
She successfully underwent retinal laser ablative therapy for the ocular manifestations without further evidence of neovascularization. She developed a mild cataract that was not visually significant and required no intervention. Her brain abnormalities were thought to represent foci of necrosis with superimposed hemorrhagic transformation due to spontaneous degeneration of brain cells in which the mutated X chromosome was activated. No further treatment was indicated beyond suppression of the consequent seizures. There was no notable cortical edema or other medical indication for systemic glucocorticoid therapy. Phenobarbital was continued without further seizure events.
Several months after the initial presentation, a follow-up electroencephalogram was normal. Phenobarbital was slowly weaned and finally discontinued approximately 6 months after the initial event with no other reported seizures. She currently is achieving normal developmental milestones with the exception of slight motor delay and expected residual hypotonia.
Incontinentia pigmenti, also known as Bloch-Sulzberger syndrome, is a rare multisystem neuroectodermal disorder, primarily affecting the skin, central nervous system (CNS), and retinas. The disorder can be inherited in an X-linked dominant fashion and appears almost exclusively in women with typical in utero lethality seen in males. Most affected individuals have a sporadic, or de novo, mutation, which was likely the case in our patient given that her mother demonstrated no signs or symptoms.1 The pathogenesis of disease is a defect at chromosome Xq28 that is a region encoding the nuclear factor–κB essential modulator, IKBKG. Absence or mutation of IKBKG in IP results in failure to activate nuclear factor–κB and leaves cells vulnerable to cytokine-mediated apoptosis, especially after exposure to tumor necrosis factor α.2
Clinical manifestations of IP are present at or soon after birth. The cutaneous findings of this disorder are classically described as a step-wise progression through 4 distinct stages: (1) a linear and/or whorled vesicular eruption predominantly on the extremities at birth or within the first few weeks of life; (2) thickened linear or whorled verrucous plaques; (3) hyperpigmented streaks and whorls that may or may not correspond with prior affected areas that may resolve by adolescence; and (4) hypopigmented, possibly atrophic plaques on the extremities that may persist lifelong. Importantly, not every patient will experience each of these stages. Overlap can occur, and the time course of each stage is highly variable. Other ectodermal manifestations include dental abnormalities such as small, misshaped, or missing teeth; alopecia; and nail abnormalities. Ocular abnormalities associated with IP primarily occur in the retina, including vascular occlusion, neovascularization, hemorrhages, foveal abnormalities, as well as exudative and tractional detachments.3,4
It is crucial to recognize CNS anomalies in association with the cutaneous findings of IP, as CNS pathology can be severe with profound developmental implications. Central nervous system findings have been noted to correlate with the appearance of the vesicular stage of IP. A high index of suspicion is needed, as the disease can demonstrate progression within a short time.5-8 The most frequent anomalies include seizures, motor impairment, intellectual disability, and microcephaly.9,10 Some of the most commonly identified CNS lesions on imaging include necrosis or brain infarcts, atrophy, and lesions of the corpus callosum.7
The pathogenesis of observed CNS changes in IP is not well understood. There have been numerous proposals of a vascular mechanism, and a microangiopathic process appears to be most plausible. Mutations in IKBKG may result in interruption of signaling via vascular endothelial growth factor receptor 3 with a consequent impact on angiogenesis, supporting a vascular mechanism. Additionally, mutations in IKBKG lead to activation of eotaxin, an eosinophil-selective chemokine.9 Eotaxin activation results in eosinophilic degranulation that mediates the classic eosinophilic infiltrate seen in the classic skin histology of IP. Additionally, it has been shown that eotaxin is strongly expressed by endothelial cells in IP, and more abundant eosinophil degranulation may play a role in mediating vaso-occlusion.7 Other studies have found that the highest expression level of the IKBKG gene is in the CNS, potentially explaining the extensive imaging findings of hemorrhage and diffusion restriction in our patient. These features likely are attributable to apoptosis of cells possessing the mutated IKBKG gene.9-11
- Ehrenreich M, Tarlow MM, Godlewska-Janusz E, et al. Incontinentia pigmenti (Bloch-Sulzberger syndrome): a systemic disorder. Cutis. 2007;79:355-362.
- Smahi A, Courtois G, Rabia SH, et al. The NF-kappaB signaling pathway in human diseases: from incontinentia pigmenti to ectodermal dysplasias and immune-deficiency syndromes. Hum Mol Genet. 2002;11:2371-2375.
- O’Doherty M, McCreery K, Green AJ, et al. Incontinentia pigmenti—ophthalmological observation of a series of cases and review of the literature. Br J Ophthalmol. 2011;95:11-16.
- Swinney CC, Han DP, Karth PA. Incontinentia pigmenti: a comprehensive review and update. Ophthalmic Surg Lasers Imaging Retina. 2015;46:650-657.
- Hennel SJ, Ekert PG, Volpe JJ, et al. Insights into the pathogenesis of cerebral lesions in incontinentia pigmenti. Pediatr Neurol. 2003;29:148-150.
- Maingay-de Groof F, Lequin MH, Roofthooft DW, et al. Extensive cerebral infarction in the newborn due to incontinentia pigmenti. Eur J Paediatr Neurol. 2008;12:284-289.
- Minic´ S, Trpinac D, Obradovic´ M. Systematic review of central nervous system anomalies in incontinentia pigmenti. Orphanet J Rare Dis. 2013;8:25-35.
- Wolf NI, Kramer N, Harting I, et al. Diffuse cortical necrosis in a neonate with incontinentia pigmenti and an encephalitis-like presentation. AJNR Am J Neuroradiol. 2005;26:1580-1582.
- Phan TA, Wargon O, Turner AM. Incontinentia pigmenti case series: clinical spectrum of incontinentia pigmenti in 53 female patients and their relatives. Clin Exp Dermatol. 2005;30:474-480.
- Volpe J. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res. 2001;50:553-562.
- Pascual-Castroviejo I, Pascual-Pascual SI, Velazquez-Fragua R, et al. Incontinentia pigmenti: clinical and neuroimaging findings in a series of 12 patients. Neurologia. 2006;21:239-248.
- Ehrenreich M, Tarlow MM, Godlewska-Janusz E, et al. Incontinentia pigmenti (Bloch-Sulzberger syndrome): a systemic disorder. Cutis. 2007;79:355-362.
- Smahi A, Courtois G, Rabia SH, et al. The NF-kappaB signaling pathway in human diseases: from incontinentia pigmenti to ectodermal dysplasias and immune-deficiency syndromes. Hum Mol Genet. 2002;11:2371-2375.
- O’Doherty M, McCreery K, Green AJ, et al. Incontinentia pigmenti—ophthalmological observation of a series of cases and review of the literature. Br J Ophthalmol. 2011;95:11-16.
- Swinney CC, Han DP, Karth PA. Incontinentia pigmenti: a comprehensive review and update. Ophthalmic Surg Lasers Imaging Retina. 2015;46:650-657.
- Hennel SJ, Ekert PG, Volpe JJ, et al. Insights into the pathogenesis of cerebral lesions in incontinentia pigmenti. Pediatr Neurol. 2003;29:148-150.
- Maingay-de Groof F, Lequin MH, Roofthooft DW, et al. Extensive cerebral infarction in the newborn due to incontinentia pigmenti. Eur J Paediatr Neurol. 2008;12:284-289.
- Minic´ S, Trpinac D, Obradovic´ M. Systematic review of central nervous system anomalies in incontinentia pigmenti. Orphanet J Rare Dis. 2013;8:25-35.
- Wolf NI, Kramer N, Harting I, et al. Diffuse cortical necrosis in a neonate with incontinentia pigmenti and an encephalitis-like presentation. AJNR Am J Neuroradiol. 2005;26:1580-1582.
- Phan TA, Wargon O, Turner AM. Incontinentia pigmenti case series: clinical spectrum of incontinentia pigmenti in 53 female patients and their relatives. Clin Exp Dermatol. 2005;30:474-480.
- Volpe J. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res. 2001;50:553-562.
- Pascual-Castroviejo I, Pascual-Pascual SI, Velazquez-Fragua R, et al. Incontinentia pigmenti: clinical and neuroimaging findings in a series of 12 patients. Neurologia. 2006;21:239-248.
Practice Points
- Central nervous system involvement in incontinentia pigmenti (IP) may be profound and can present prior to the classic cutaneous findings.
- A high index of suspicion for IP should be maintained in neonatal vesicular eruptions of unclear etiology, especially in the setting of unexplained seizures and/or abnormal brain imaging.