User login
- In low-risk patients, the physician should conduct a thorough history and physical examination. Routine laboratory testing may be useful, but further evaluation for underlying malignancy is unnecessary (B).
- Test for disorders associated with hypercoagulability under the following circumstances: when a thrombotic event occurs in a person younger than age 50; if a patient has a family history of venous thromboembolism; or if there are recurrent episodes of unexplained venous thromboembolism (C).
- When homocysteine levels are elevated in the presence of factor V Leiden or the prothrombin gene G20210A mutation, risk of recurrent thrombosis appears to be increased beyond the r isk associated with any one defect alone (B).
In which direction, and how aggressively, should the investigation proceed when common and obvious causes of venous thromboembolism—recent surgery, trauma, immobilization, or malignancy—are absent from a patient’s history?
Two causes of hypercoagulability warrant consideration: occult malignancy and coagulation disorders resulting in thrombophilia. This review provides guidance on diagnostic testing and extent of the work-up, summarized in an algorithm (Figure).
FIGURE
Evaluating idiopathic venous thromboembolism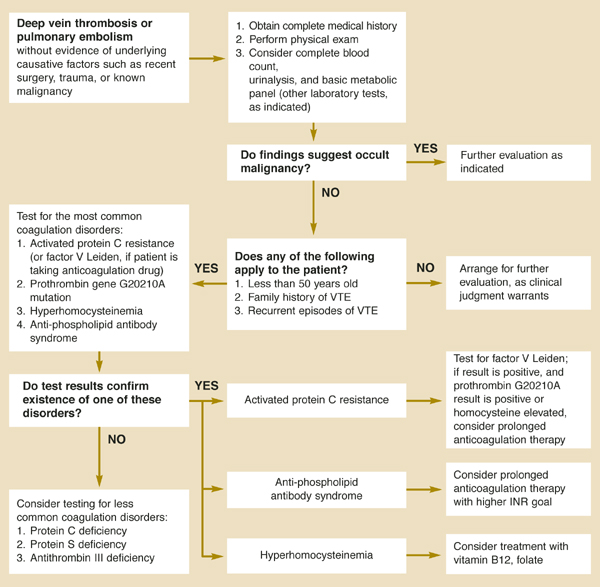
Malignancy and Venous Thromboembolism
Armand Trousseau first described the association between VTE and cancer nearly 150 years ago.5 For patients with known malignancy, a search for other possible causes of thrombosis is seldom needed (strength of recommendation [SOR]=B).
However, for individuals with idiopathic DVT or pulmonary embolism (PE), the clinician’s dilemma is in deciding how aggressively to look for occult malignancy. In a prospective study of 738 patients with objectively verified symptomatic deep vein thrombosis (DVT), cancer was the most common underlying cause and emerged as a major predictor of recurrent thrombotic events.6 Unfortunately, no laboratory test predicts occult cancer among patients with VTE.7
Risk of malignancy in per spective
Evidence of malignancy is usually discovered when taking a patient’s history and conducting a physical examination. Searching beyond the history and physical exam is seldom revealing.
In 1992, Prandoni and colleagues8 published a report of a study of 262 patients with symptomatic DVT, 250 of whom were followed for 2 years. One hundred seven patients had recognized nonmalignant risk factors for DVT and were not evaluated for cancer. Of the 155 patients with idiopathic venous thrombosis, 5 (3.3%) were discovered to have occult carcinoma. Malignancy was suggested in 4 of the 5 by history or physical examination.
In a similar study, Hettiarachchi et al9 evaluated 400 patients with confirmed DVT and found 70 (18%) had a diagnosis of cancer at the time of presentation. Of the remaining 326 patients (4 were lost to follow-up), 189 had recognized risk factors for DVT, 3 (1.6%) of whom were also found to have cancer; and 137 patients had unexplained DVT, 10 of whom (7.3%) were found to have occult carcinoma. As in the Prandoni study, most of the patients subsequently discovered to have cancer (10 of 13, 77%) had suggestive clinical findings in the history or physical examination.9
Venous thromboembolism and specific types of cancer
Two large, retrospective epidemiologic studies reviewed cases of thousands of patients in the Danish and Swedish National Patient Registries.10,11 Investigators for these studies found an approximately 30% increase in the diagnosis of cancer among patients with VTE compared with the general population. Because of their large size, both of these studies were able to demonstrate a significant association between thrombosis and pancreatic, liver, and ovarian cancers.
For liver and pancreatic cancers, consensus opinion suggests that early diagnosis does not change prognosis. Similarly, although some experts recommend ultrasound and Ca-125 testing to investigate possible ovarian cancer, no data support use of these tests in ovarian cancer screening.12
An evaluation of the Danish National Registry data has suggested that cancer diagnosed at the time of, or within a year of, the diagnosis of VTE is usually advanced and is associated with a poor prognosis.13 Indeed, the authors of the Danish study concluded that for patients with VTE, “[our] pragmatic recommendation [is] to use only simple methods of screening and to look for cancer in patients with signs and symptoms of cancer.”13
Recommended work-up
We conclude the literature8-13 does not support an aggressive search for hidden cancer in a patient with idiopathic VTE (SOR=B). Routine evaluation should include a careful history and physical examination. Because of their low cost, reliability, and ready availability, studies such as a complete blood count, basic chemistry panel, liver function tests, and urinalysis may be considered (SOR=B ). Examples of findings during the initial history, physical exam, and laboratory studies that should prompt further evaluation include anorexia, weight loss, cough, abdominal bloating, unexplained anemia, hyponatremia, hematuria, and abnormal liver enzymes.
It is estimated that more than 250,000 patients are hospitalized for venous thromboembolism (VTE) in the United Sates each year.1 The number of VTE cases annually in this country ranges from 600,000 to 2 million.2,3 The most common causes of VTE include surgery, trauma, hospital or nursing home confinement and malignant neoplasm.4
Unnecessary studies. Some authorities recommend a chest radiograph in the routine evaluation for occult malignancy,9 but its clinical utility for patients without pulmonary symptoms has not been clearly demonstrated. Because of their expense and low test yield, we do not advocate the use of more elaborate screens for occult malignancy, such as computerized tomography, magnetic resonance imaging, or serologic tumor markers.
Caveat. In the past, nearly all patients with pulmonary embolism or DVT were hospitalized to receive treatment with continuous intravenous heparin. Presumably these patients routinely received a careful history evaluation, physical examination, and standard blood work.
With increased use of low-molecular-weight heparins (given subcutaneously once or twice daily), many individuals with VTE are candidates for treatment partially or totally on an outpatient basis.14,15 Be sure that individuals receiving outpatient treatment for idiopathic VTE receive the same attention and routine work-up as hospitalized patients.
Coagulation disorders and Venous Thromboembolism
With advances in laboratory testing, more than half of idiopathic VTE cases can be attributed to a specific coagulation disorder. Data show that in a randomly selected group of patients with newly discovered DVT, 24% to 37% have an inherited predisposition to thrombosis.16-19
Deficiencies in protein C, protein S, and antithrombin were the first causes of inherited thrombophilia to be identified;20-22 however, these deficiencies are not particularly common. The most common causes of thrombophilic coagulation disorders are activated protein C resistance, the prothrombin gene G20210A mutation, and hyperhomocysteinemia.
When to look for prothrombotic defects
If, after ruling out occult malignancy, the cause of VTE remains uncertain, be judicious in deciding whether to run more tests for prothrombotic defects. We advocate pursuing further testing for patients likely to have an underlying hypercoagulable disorder, and for whom the identification of such a disorder would have management implications.
A decision to pursue testing with other patients should be made on a case-by-case basis. The yield in testing is low for many prothrombotic disorders, some tests are affected adversely by anticoagulation therapy, and the influence of a positive test result on patient management decisions remains unclear in many cases. Finally, serologic evaluation for thrombophilia would be costly if conducted for all patients with VTE, and the potential clinical benefit would be small.
Although large epidemiologic studies are lacking to help identify patients at increased risk of a hypercoagulable disorder, patients with clinically significant inherited thrombophilia tend to have VTE at a young age.23-25
In addition, advanced age alone is often regarded as an identifiable risk factor for DVT. A recent retrospective study demonstrated the risk for DVT rose rapidly during the 6th through 8th decades of life.26
We generally recommend testing for hypercoagulable disorders upon discovering one of the following findings:
- The first thrombotic event occurs when the patient is younger than 50 years
- A family history of VTE exists
- Episodes of unexplained VTE recur (SOR=C).
Activated protein C resistance (Factor V Leiden mutation)
Prevalence. Approximately 90% of cases of activated protein C resistance are due to a substitution of glutamine for arginine at position 506 on the factor V gene, the so-called Leiden mutation.27
Factor V Leiden mutation varies greatly by race. In North America, this mutation is found in the heterozygous form in approximately 5% of Caucasians, 2% of Hispanics, 1% of African Americans, and less than 0.5% of Asians.28 Persons heterozygous for factor V Leiden have approximately a 7-fold increase in the relative risk;29 persons homozygous for the mutation have approximately an 80-fold increase in the relative risk.30
Detection. Activated protein C resistance is typically assessed by mixing patient plasma with factor V-deficient plasma. This clotting assay is not affected by oral anticoagulation medication but is affected by heparin. A positive test result for activated protein C resistance typically warrants a polymerase chain reaction assay to distinguish the factor V Leiden mutation from other causes of activated protein C resistance.
Testing considerations. Although factor V Leiden mutation is the cause of activated protein C resistance in most cases, we recommend that activated protein C resistance testing be done first, as it is a less expensive and more widely available test than the DNA-based factor V Leiden mutation test. However, if a patient is taking heparin at the time of evaluation for thrombophilia, it may be necessary to defer activated protein C resistance testing or proceed directly to factor V Leiden DNA-based testing.
Prothrombin gene G2 0210A mutation
Prevalence. Like the factor V Leiden mutation, the prothrombin gene G20210A mutation is more common among Caucasians than among those of African or Asian descent.31 Prothrombin mutation is estimated to cause a 2.5-fold relative risk of first DVT.32
Testing considerations. If cost or availability of the polymerase chain reaction assay are issues, a reasonable course of action would be to reserve the assay for patients with factor V Leiden mutation. Data suggest that patients with both factor V and prothrombin gene G20210A mutations are at significantly higher risk for recurrent VTE; consider prolonged anticoagulation.33
Hyperhomocysteinemia
Evidence accumulating over the past decade has shown an increased risk of VTE with elevated homocysteine levels. A case-control study by den Heijer et al34 in 1996 demonstrated a relative risk for first thrombosis of 2.5 in those with a homocysteine level above the 95th percentile of the control group’s levels (which in that study corresponded to a homocysteine level of 18.5 micromoles per liter or above). This predisposition to thrombosis has also been demonstrated in subsequent meta-analyses.35,36
When homocysteine levels are elevated in the presence of factor V Leiden or the prothrombin gene G20210A mutation, risk of thrombosis appears to increase beyond that associated with any one defect alone, perhaps as high as 50-fold risk.37-39
Management Implications of Test Results
If testing for activated protein C resistance is possible and the result is positive, confirmatory testing for factor V Leiden is indicated. If the patient is homozygous for factor V Leiden (rare), or is heterozygous for factor V Leiden and has the prothrombin gene G20210A defect, consider a prolonged course of anticoagulation, perhaps even for the rest of the patient’s life.
We base these suggestions on the high rate of VTE detected among individuals who are homozygous for factor V Leiden30 and on a recent study that showed the relative risk of recurrent thrombosis to be 2.6 for those with factor V Leiden and prothrombin gene G20210A mutations, versus for those heterozygous for factor V Leiden alone.33 This recent study also showed that the relative risk of recurrent thrombosis was not increased in those heterozygous for the Factor V Leiden mutation alone compared to those without this mutation.33
If either the factor V Leiden or the prothrombin gene G20210A mutation is present, consider prolonging the planned course of anticoagulation (although the data to support this decision are less compelling). Data are limited and conflicting regarding the appropriate length and intensity of anticoagulation therapy for patients with inherited prothrombotic defects.33,40-42 When prolonging anticoagulation for these patients, try to balance the risk of a recurrent thrombotic event with the risk of a bleeding complication from chronic anticoagulation.
Prolonged anticoagulation may also be indicated if the homocysteine level is elevated and either the factor V Leiden or the prothrombin gene G20210A mutation is present. In addition, therapy with folate, pyridoxine, and vitamin B12 should be initiated in cases of elevated homocyteine levels. We recommend the use of these vitamins based on a meta-analysis which demonstrated that folate supplementation at a dose of 0.5 mg to 5 mg/day lowered homocysteine levels by approximately 25% and vitamin B12 supplementation at a dose of 0.5 mg/day led to a further reduction of approximately 7%. In this same meta-analysis, pyridoxine at a mean dose of 16.5 mg daily did not demonstrate further lowering of the homocysteine level; however, as it is safe, inexpensive and well-tolerated, we still recommend its use.43
Finally, while less common than the hypercoagulable states discussed above, the antiphospholipid antibody syndrome should be targeted as part of first-tier testing because of its clear impact on management. If the antiphospholipid antibody testing (such as the lupus anticoagulant or anticardiolipin antibody) results are positive, lifelong anticoagulation44 and perhaps a higher target international normalized ratio are considerations.
Second-tier testing: Pursuing less common defects
If the preceding evaluation is unrevealing for a patient with high-risk characteristics, consider pursuing “second-tier” testing. Such testing may include functional and immunologic assays for protein C, protein S, and anti-thrombin III deficiencies, all of which are heterozygous abnormalities caused by multiple mutations and, thus, not detectable with genetic assays.
Patients will likely be taking anticoagulation medication when these tests are administered; thus, test results must be interpreted with caution. Testing for these deficiencies is most reliable if conducted at least 2 weeks after anticoagulation has been discontinued. Again, the incidence of protein C, protein S, and antithrombin III deficiencies is low, and the yield of testing therefore is also likely to be low.
We would not recommend discontinuing anticoagulation to conduct testing for these uncommon defects once it has been initiated following an acute thrombotic event. Rather, testing should be pursued once the planned course of anticoagulation with warfarin has been completed (for example, 6 to 9 months).
Other inherited thrombophilic disorders include heparin cofactor II deficiency, plasminogen deficiency, dysfibrinogenemia, factor XII deficiency, and increased factor VIII coagulant activity. These disorders are rare and their clinical importance remains unclear. Testing for these disorders, if desired, is best conducted in consultation with a hematologist or coagulation specialist.
Corresponding author
Charles F.S. Locke, MD, Johns Hopkins Community Physicians, 2360 W. Joppa Rd., Baltimore, MD 21093. E-mail: clocke@jhmi.edu.
1. Goldhaber SZ. Pulmonary embolism. N Engl J Med 1998;339:93-104.
2. Deitcher SR. Overview of enoxaparin in the treatment of deep vein thrombosis. Am J Manag Care 2000;6(suppl):S1026-S1033.
3. Hirsh J, Hoak J. Management of deep vein thrombosis and pulmonary embolism. A statement for healthcare professionals. Council on Thrombosis (in consultation with the Council on Cardiovascular Radiology), American Heart Association. Circulation. 1996;93:2212-2245.
4. Heit JA, Silverstein MD, Mohr DN, et al. Risk factors for deep vein thrombosis and pulmonary embolism. Arch Int Med 2000;160:809-815.
5. Trousseau A. Phlegmasia alba dolens. In: Clinique Medicale de l’Hotel-Dieu de Paris. Vol. 3. 2nd ed. Paris: JB Bailliere; 1865;654-712.
6. Hansson PO, Sorbo J, Eriksson H. Recurrent venous thromboembolism after deep vein thrombosis: incidence and risk factors. Arch Intern Med 2000;160:769-774.
7. Naschitz JE, Yeshurun D, Lev LM. Thromboembolism in cancer. Cancer 1993;71:1384-1390.
8. Prandoni P, Lensing AW, Buller HR, et al. Deep-vein thrombosis and the incidence of subsequent symptomatic cancer. N Engl J Med 1992;327:1128-1133.
9. Hettiarachchi RJ, Lok J, Prins MH, et al. Undiagnosed malignancy in patients with deep vein thrombosis: incidence, risk indicators, and diagnosis. Cancer 1998;83:180-185.
10. Baron JA, Gridley G, Weiderpass E, et al. Venous thromboembolism and cancer. Lancet 1998;351:1077-1080.
11. Sorenson HT, Mellemkjaer L, Steffensen FH, et al. The risk of a diagnosis of cancer after primary deep venous thrombosis or pulmonary embolism. N Engl J Med 1998;338:1169-1173.
12. Jacobs I, Davies AP, Bridges J, et al. Prevalence screening for ovarian cancer in postmenopausal women by CA 125 measurement and ultrasonography. BMJ 1993;306:1030-1034.
13. Sorensen HT, Mellemkjaer L, Olsen JH, et al. Prognosis of cancers associated with venous thromboembolism. N Engl J Med 2000;343:1846-1850.
14. Levine M, Gent M, Hirsh J, et al. A comparison of low-molecular-weight heparin administered primarily at home with unfractionated heparin administered in the hospital for proximal deep-vein thrombosis. N Engl J Med 1996;334:677-681.
15. Koopman M, Prandoni P, Piovella F, et al. for the Tasman Study Group. Treatment of venous thrombosis with intravenous unfractionated heparin administered in the hospital as compared with subcutaneous low-molecular-weight heparin administered at home. N Engl J Med 1996;334:682-687.
16. Mateo J, Oliver A, Borrell M, Sala N, Fontcuberta J. Laboratory evaluation and clinical characteristics of 2,132 consecutive unselected patients with venous thromboembolism—results of the Spanish Multicentric Study on Thrombophilia (EMET-Study). Thromb Haemost 1997;77:444-451.
17. Ben-Tal O, Zivelin A, Seligsohn U. The relative frequency of hereditary thrombotic disorders among 107 patients with thrombophilia in Israel. Thromb Haemost 1989;61:50-54.
18. Ridker PM, Hennekens CH, Lindpaintner K, et al. Mutation in the gene coding for coagulation factor V and the risk of myocardial infarction, stroke, and venous thrombosis in apparently healthy men. N Engl J Med 1995;332:912-917.
19. Margaglione M, Brancaccio V, Giuliani N, et al. Increased risk for venous thrombosis in carriers of the prothrombin G—>A20210 gene variant. Ann Intern Med 1998;129:89-93.
20. Egeberg O. Inherited antithrombin deficiency causing thrombophilia. Thromb Diath Haemorrh 1965;13:516.-
21. Griffith JH, Evatt B, Zimmerman TS, et al. Deficiency of protein C in congenital thrombotic disease. J Clin Invest 1981;68:1370-1373.
22. Comp PC, Nixon RR, Cooper MR, et al. Familial protein S deficiency is associated with recurrent thrombosis. J Clin Invest 1984;74:2082-2088.
23. Hillarp A, Dahlback B, Zoller B. Activated protein C resistance: from phenotype to genotype and clinical practice. Blood Rev 1995;9:201-212.
24. Zoller B, Berntsdotter A, Garcia de Frutos P, et al. Resistance to activated protein C as an additional genetic risk factor in hereditary deficiency of protein S. Blood 1995;85:3518-3523.
25. Van Boven HH, Reitsma PH, Rosendaal FR, et al. Factor V Leiden (FV R506Q) in families with inherited antithrombin deficiency. Thromb Haemost 1996;75:417-421.
26. Heit JA, Silverstein MD, Mohr DN, et al. Predictors of recurrence after deep vein thrombosis and pulmonary embolism. Arch Intern Med 2000;160:809-815.
27. Bertina RM, Koeleman BPC, Koster T, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994;369:64-67.
28. Ridker PM, Miletich JP, Hennekens CH, Buring JE. Ethnic distribution of factor V Leiden in 4047 men and women. Implications for venous thromboembolism screening. JAMA 1997;277:1305-1307.
29. Koster T, Rosendaal FR, de Ronde H, Briet E, Vandenbroucke JP, Bertina RM. Venous thrombosis due to poor anticoagulant response to activated protein C: Leiden thrombophilia study. Lancet 1993;342:1503-1506.
30. Rosendaal FR, Koster T, Vandenbroucke JP, Reitsma PH. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance). Blood 1995;85:1504-1508.
31. Rosendaal FR, Doggen CJ, Zivelin A, et al. Geographic distribution of the 20210 G to A prothrombin variant. Thromb Haemost 1998;79:706-708.
32. Poort SR, Rosendaal FR, Reitsma PH, Bertina RM. A common genetic variation in the 3´ untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood 1996;88:3698-3703.
33. De Stefano V, Martinelli I, Mannucci PM, et al. The risk of recurrent deep venous thrombosis among heterozygous carriers of both factor V Leiden and the G20210A prothrombin mutation. N Engl J Med 1999;341:801-806.
34. den Heijer M, Koster T, Blom HJ, et al. Hyperhomocysteinemia as a risk factor for deep-vein thrombosis. N Engl J Med 1996;334:759-762.
35. Ray JG. Meta-analysis of hyperhomocysteinemia as a risk factor for venous thromboembolic disease. Arch Intern Med 1998;158:2101-2106.
36. den Heijer M, Rosendaal FR, Blom HJ, Gerrits WB, Bos GM. Hyperhomocysteinemia and venous thrombosis: a meta-analysis. Thromb Haemost 1998;80:874-877.
37. Ridker PM, Hennekens CH, Selhub J, Miletich JP, Malinow MR, Stampfer MJ. Interrelation of hyperhomocyst(e)inemia, factor V Leiden, and risk of future venous thromboembolism. Circulation 1997;95:1777-1782.
38. Kluijtmans LA, den Heijer M, Reitsma PH, Heil SG, Blom HJ, Rosendaal FR. Thermolabile methylenetetrahydrofolate reductase and factor V Leiden in the risk of deep-vein thrombosis. Thromb Haemost 1998;79:254-258.
39. DeStefano V, Zappacosta B, Persichilli S, et al. Prevalence of mild hyperhomocysteinaemia and association with thrombophilic genotypes (factor V Leiden and prothrombin G20210A) in Italian patients with venous thromboembolic disease. Br J Haematol 1999;106:564-568.
40. Kearon C, Gent M, Hirsh J, et al. A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 1999;340:901-907.
41. Simioni P, Prandoni P, Lensing AWA, et al. The risk of recurrent venous thromboembolism in patients with an Arg506—>Gln mutation in the gene for factor V (factor V Leiden). N Engl J Med 1997;336:399-403.
42. Ridker PM, Miletich JP, Stampfer MJ, Goldhaber SZ, Lindpaintner K, Hennekens CH. Factor V Leiden and risks of recurrent idiopathic venous thromboembolism. Circulation 1995;92:2800-2802.
43. Lowering blood homocysteine with folic acid based supplements: meta-analysis of randomised trials. Homocysteine Lowering Trialists’ Collaboration. BMJ 1998;316:894-898.
44. McCrae KR. Antiphospholipid antibody associated thrombosis: a consensus for treatment? Lupus 1996;5:560-570.
- In low-risk patients, the physician should conduct a thorough history and physical examination. Routine laboratory testing may be useful, but further evaluation for underlying malignancy is unnecessary (B).
- Test for disorders associated with hypercoagulability under the following circumstances: when a thrombotic event occurs in a person younger than age 50; if a patient has a family history of venous thromboembolism; or if there are recurrent episodes of unexplained venous thromboembolism (C).
- When homocysteine levels are elevated in the presence of factor V Leiden or the prothrombin gene G20210A mutation, risk of recurrent thrombosis appears to be increased beyond the r isk associated with any one defect alone (B).
In which direction, and how aggressively, should the investigation proceed when common and obvious causes of venous thromboembolism—recent surgery, trauma, immobilization, or malignancy—are absent from a patient’s history?
Two causes of hypercoagulability warrant consideration: occult malignancy and coagulation disorders resulting in thrombophilia. This review provides guidance on diagnostic testing and extent of the work-up, summarized in an algorithm (Figure).
FIGURE
Evaluating idiopathic venous thromboembolism
Malignancy and Venous Thromboembolism
Armand Trousseau first described the association between VTE and cancer nearly 150 years ago.5 For patients with known malignancy, a search for other possible causes of thrombosis is seldom needed (strength of recommendation [SOR]=B).
However, for individuals with idiopathic DVT or pulmonary embolism (PE), the clinician’s dilemma is in deciding how aggressively to look for occult malignancy. In a prospective study of 738 patients with objectively verified symptomatic deep vein thrombosis (DVT), cancer was the most common underlying cause and emerged as a major predictor of recurrent thrombotic events.6 Unfortunately, no laboratory test predicts occult cancer among patients with VTE.7
Risk of malignancy in per spective
Evidence of malignancy is usually discovered when taking a patient’s history and conducting a physical examination. Searching beyond the history and physical exam is seldom revealing.
In 1992, Prandoni and colleagues8 published a report of a study of 262 patients with symptomatic DVT, 250 of whom were followed for 2 years. One hundred seven patients had recognized nonmalignant risk factors for DVT and were not evaluated for cancer. Of the 155 patients with idiopathic venous thrombosis, 5 (3.3%) were discovered to have occult carcinoma. Malignancy was suggested in 4 of the 5 by history or physical examination.
In a similar study, Hettiarachchi et al9 evaluated 400 patients with confirmed DVT and found 70 (18%) had a diagnosis of cancer at the time of presentation. Of the remaining 326 patients (4 were lost to follow-up), 189 had recognized risk factors for DVT, 3 (1.6%) of whom were also found to have cancer; and 137 patients had unexplained DVT, 10 of whom (7.3%) were found to have occult carcinoma. As in the Prandoni study, most of the patients subsequently discovered to have cancer (10 of 13, 77%) had suggestive clinical findings in the history or physical examination.9
Venous thromboembolism and specific types of cancer
Two large, retrospective epidemiologic studies reviewed cases of thousands of patients in the Danish and Swedish National Patient Registries.10,11 Investigators for these studies found an approximately 30% increase in the diagnosis of cancer among patients with VTE compared with the general population. Because of their large size, both of these studies were able to demonstrate a significant association between thrombosis and pancreatic, liver, and ovarian cancers.
For liver and pancreatic cancers, consensus opinion suggests that early diagnosis does not change prognosis. Similarly, although some experts recommend ultrasound and Ca-125 testing to investigate possible ovarian cancer, no data support use of these tests in ovarian cancer screening.12
An evaluation of the Danish National Registry data has suggested that cancer diagnosed at the time of, or within a year of, the diagnosis of VTE is usually advanced and is associated with a poor prognosis.13 Indeed, the authors of the Danish study concluded that for patients with VTE, “[our] pragmatic recommendation [is] to use only simple methods of screening and to look for cancer in patients with signs and symptoms of cancer.”13
Recommended work-up
We conclude the literature8-13 does not support an aggressive search for hidden cancer in a patient with idiopathic VTE (SOR=B). Routine evaluation should include a careful history and physical examination. Because of their low cost, reliability, and ready availability, studies such as a complete blood count, basic chemistry panel, liver function tests, and urinalysis may be considered (SOR=B ). Examples of findings during the initial history, physical exam, and laboratory studies that should prompt further evaluation include anorexia, weight loss, cough, abdominal bloating, unexplained anemia, hyponatremia, hematuria, and abnormal liver enzymes.
It is estimated that more than 250,000 patients are hospitalized for venous thromboembolism (VTE) in the United Sates each year.1 The number of VTE cases annually in this country ranges from 600,000 to 2 million.2,3 The most common causes of VTE include surgery, trauma, hospital or nursing home confinement and malignant neoplasm.4
Unnecessary studies. Some authorities recommend a chest radiograph in the routine evaluation for occult malignancy,9 but its clinical utility for patients without pulmonary symptoms has not been clearly demonstrated. Because of their expense and low test yield, we do not advocate the use of more elaborate screens for occult malignancy, such as computerized tomography, magnetic resonance imaging, or serologic tumor markers.
Caveat. In the past, nearly all patients with pulmonary embolism or DVT were hospitalized to receive treatment with continuous intravenous heparin. Presumably these patients routinely received a careful history evaluation, physical examination, and standard blood work.
With increased use of low-molecular-weight heparins (given subcutaneously once or twice daily), many individuals with VTE are candidates for treatment partially or totally on an outpatient basis.14,15 Be sure that individuals receiving outpatient treatment for idiopathic VTE receive the same attention and routine work-up as hospitalized patients.
Coagulation disorders and Venous Thromboembolism
With advances in laboratory testing, more than half of idiopathic VTE cases can be attributed to a specific coagulation disorder. Data show that in a randomly selected group of patients with newly discovered DVT, 24% to 37% have an inherited predisposition to thrombosis.16-19
Deficiencies in protein C, protein S, and antithrombin were the first causes of inherited thrombophilia to be identified;20-22 however, these deficiencies are not particularly common. The most common causes of thrombophilic coagulation disorders are activated protein C resistance, the prothrombin gene G20210A mutation, and hyperhomocysteinemia.
When to look for prothrombotic defects
If, after ruling out occult malignancy, the cause of VTE remains uncertain, be judicious in deciding whether to run more tests for prothrombotic defects. We advocate pursuing further testing for patients likely to have an underlying hypercoagulable disorder, and for whom the identification of such a disorder would have management implications.
A decision to pursue testing with other patients should be made on a case-by-case basis. The yield in testing is low for many prothrombotic disorders, some tests are affected adversely by anticoagulation therapy, and the influence of a positive test result on patient management decisions remains unclear in many cases. Finally, serologic evaluation for thrombophilia would be costly if conducted for all patients with VTE, and the potential clinical benefit would be small.
Although large epidemiologic studies are lacking to help identify patients at increased risk of a hypercoagulable disorder, patients with clinically significant inherited thrombophilia tend to have VTE at a young age.23-25
In addition, advanced age alone is often regarded as an identifiable risk factor for DVT. A recent retrospective study demonstrated the risk for DVT rose rapidly during the 6th through 8th decades of life.26
We generally recommend testing for hypercoagulable disorders upon discovering one of the following findings:
- The first thrombotic event occurs when the patient is younger than 50 years
- A family history of VTE exists
- Episodes of unexplained VTE recur (SOR=C).
Activated protein C resistance (Factor V Leiden mutation)
Prevalence. Approximately 90% of cases of activated protein C resistance are due to a substitution of glutamine for arginine at position 506 on the factor V gene, the so-called Leiden mutation.27
Factor V Leiden mutation varies greatly by race. In North America, this mutation is found in the heterozygous form in approximately 5% of Caucasians, 2% of Hispanics, 1% of African Americans, and less than 0.5% of Asians.28 Persons heterozygous for factor V Leiden have approximately a 7-fold increase in the relative risk;29 persons homozygous for the mutation have approximately an 80-fold increase in the relative risk.30
Detection. Activated protein C resistance is typically assessed by mixing patient plasma with factor V-deficient plasma. This clotting assay is not affected by oral anticoagulation medication but is affected by heparin. A positive test result for activated protein C resistance typically warrants a polymerase chain reaction assay to distinguish the factor V Leiden mutation from other causes of activated protein C resistance.
Testing considerations. Although factor V Leiden mutation is the cause of activated protein C resistance in most cases, we recommend that activated protein C resistance testing be done first, as it is a less expensive and more widely available test than the DNA-based factor V Leiden mutation test. However, if a patient is taking heparin at the time of evaluation for thrombophilia, it may be necessary to defer activated protein C resistance testing or proceed directly to factor V Leiden DNA-based testing.
Prothrombin gene G2 0210A mutation
Prevalence. Like the factor V Leiden mutation, the prothrombin gene G20210A mutation is more common among Caucasians than among those of African or Asian descent.31 Prothrombin mutation is estimated to cause a 2.5-fold relative risk of first DVT.32
Testing considerations. If cost or availability of the polymerase chain reaction assay are issues, a reasonable course of action would be to reserve the assay for patients with factor V Leiden mutation. Data suggest that patients with both factor V and prothrombin gene G20210A mutations are at significantly higher risk for recurrent VTE; consider prolonged anticoagulation.33
Hyperhomocysteinemia
Evidence accumulating over the past decade has shown an increased risk of VTE with elevated homocysteine levels. A case-control study by den Heijer et al34 in 1996 demonstrated a relative risk for first thrombosis of 2.5 in those with a homocysteine level above the 95th percentile of the control group’s levels (which in that study corresponded to a homocysteine level of 18.5 micromoles per liter or above). This predisposition to thrombosis has also been demonstrated in subsequent meta-analyses.35,36
When homocysteine levels are elevated in the presence of factor V Leiden or the prothrombin gene G20210A mutation, risk of thrombosis appears to increase beyond that associated with any one defect alone, perhaps as high as 50-fold risk.37-39
Management Implications of Test Results
If testing for activated protein C resistance is possible and the result is positive, confirmatory testing for factor V Leiden is indicated. If the patient is homozygous for factor V Leiden (rare), or is heterozygous for factor V Leiden and has the prothrombin gene G20210A defect, consider a prolonged course of anticoagulation, perhaps even for the rest of the patient’s life.
We base these suggestions on the high rate of VTE detected among individuals who are homozygous for factor V Leiden30 and on a recent study that showed the relative risk of recurrent thrombosis to be 2.6 for those with factor V Leiden and prothrombin gene G20210A mutations, versus for those heterozygous for factor V Leiden alone.33 This recent study also showed that the relative risk of recurrent thrombosis was not increased in those heterozygous for the Factor V Leiden mutation alone compared to those without this mutation.33
If either the factor V Leiden or the prothrombin gene G20210A mutation is present, consider prolonging the planned course of anticoagulation (although the data to support this decision are less compelling). Data are limited and conflicting regarding the appropriate length and intensity of anticoagulation therapy for patients with inherited prothrombotic defects.33,40-42 When prolonging anticoagulation for these patients, try to balance the risk of a recurrent thrombotic event with the risk of a bleeding complication from chronic anticoagulation.
Prolonged anticoagulation may also be indicated if the homocysteine level is elevated and either the factor V Leiden or the prothrombin gene G20210A mutation is present. In addition, therapy with folate, pyridoxine, and vitamin B12 should be initiated in cases of elevated homocyteine levels. We recommend the use of these vitamins based on a meta-analysis which demonstrated that folate supplementation at a dose of 0.5 mg to 5 mg/day lowered homocysteine levels by approximately 25% and vitamin B12 supplementation at a dose of 0.5 mg/day led to a further reduction of approximately 7%. In this same meta-analysis, pyridoxine at a mean dose of 16.5 mg daily did not demonstrate further lowering of the homocysteine level; however, as it is safe, inexpensive and well-tolerated, we still recommend its use.43
Finally, while less common than the hypercoagulable states discussed above, the antiphospholipid antibody syndrome should be targeted as part of first-tier testing because of its clear impact on management. If the antiphospholipid antibody testing (such as the lupus anticoagulant or anticardiolipin antibody) results are positive, lifelong anticoagulation44 and perhaps a higher target international normalized ratio are considerations.
Second-tier testing: Pursuing less common defects
If the preceding evaluation is unrevealing for a patient with high-risk characteristics, consider pursuing “second-tier” testing. Such testing may include functional and immunologic assays for protein C, protein S, and anti-thrombin III deficiencies, all of which are heterozygous abnormalities caused by multiple mutations and, thus, not detectable with genetic assays.
Patients will likely be taking anticoagulation medication when these tests are administered; thus, test results must be interpreted with caution. Testing for these deficiencies is most reliable if conducted at least 2 weeks after anticoagulation has been discontinued. Again, the incidence of protein C, protein S, and antithrombin III deficiencies is low, and the yield of testing therefore is also likely to be low.
We would not recommend discontinuing anticoagulation to conduct testing for these uncommon defects once it has been initiated following an acute thrombotic event. Rather, testing should be pursued once the planned course of anticoagulation with warfarin has been completed (for example, 6 to 9 months).
Other inherited thrombophilic disorders include heparin cofactor II deficiency, plasminogen deficiency, dysfibrinogenemia, factor XII deficiency, and increased factor VIII coagulant activity. These disorders are rare and their clinical importance remains unclear. Testing for these disorders, if desired, is best conducted in consultation with a hematologist or coagulation specialist.
Corresponding author
Charles F.S. Locke, MD, Johns Hopkins Community Physicians, 2360 W. Joppa Rd., Baltimore, MD 21093. E-mail: clocke@jhmi.edu.
- In low-risk patients, the physician should conduct a thorough history and physical examination. Routine laboratory testing may be useful, but further evaluation for underlying malignancy is unnecessary (B).
- Test for disorders associated with hypercoagulability under the following circumstances: when a thrombotic event occurs in a person younger than age 50; if a patient has a family history of venous thromboembolism; or if there are recurrent episodes of unexplained venous thromboembolism (C).
- When homocysteine levels are elevated in the presence of factor V Leiden or the prothrombin gene G20210A mutation, risk of recurrent thrombosis appears to be increased beyond the r isk associated with any one defect alone (B).
In which direction, and how aggressively, should the investigation proceed when common and obvious causes of venous thromboembolism—recent surgery, trauma, immobilization, or malignancy—are absent from a patient’s history?
Two causes of hypercoagulability warrant consideration: occult malignancy and coagulation disorders resulting in thrombophilia. This review provides guidance on diagnostic testing and extent of the work-up, summarized in an algorithm (Figure).
FIGURE
Evaluating idiopathic venous thromboembolism
Malignancy and Venous Thromboembolism
Armand Trousseau first described the association between VTE and cancer nearly 150 years ago.5 For patients with known malignancy, a search for other possible causes of thrombosis is seldom needed (strength of recommendation [SOR]=B).
However, for individuals with idiopathic DVT or pulmonary embolism (PE), the clinician’s dilemma is in deciding how aggressively to look for occult malignancy. In a prospective study of 738 patients with objectively verified symptomatic deep vein thrombosis (DVT), cancer was the most common underlying cause and emerged as a major predictor of recurrent thrombotic events.6 Unfortunately, no laboratory test predicts occult cancer among patients with VTE.7
Risk of malignancy in per spective
Evidence of malignancy is usually discovered when taking a patient’s history and conducting a physical examination. Searching beyond the history and physical exam is seldom revealing.
In 1992, Prandoni and colleagues8 published a report of a study of 262 patients with symptomatic DVT, 250 of whom were followed for 2 years. One hundred seven patients had recognized nonmalignant risk factors for DVT and were not evaluated for cancer. Of the 155 patients with idiopathic venous thrombosis, 5 (3.3%) were discovered to have occult carcinoma. Malignancy was suggested in 4 of the 5 by history or physical examination.
In a similar study, Hettiarachchi et al9 evaluated 400 patients with confirmed DVT and found 70 (18%) had a diagnosis of cancer at the time of presentation. Of the remaining 326 patients (4 were lost to follow-up), 189 had recognized risk factors for DVT, 3 (1.6%) of whom were also found to have cancer; and 137 patients had unexplained DVT, 10 of whom (7.3%) were found to have occult carcinoma. As in the Prandoni study, most of the patients subsequently discovered to have cancer (10 of 13, 77%) had suggestive clinical findings in the history or physical examination.9
Venous thromboembolism and specific types of cancer
Two large, retrospective epidemiologic studies reviewed cases of thousands of patients in the Danish and Swedish National Patient Registries.10,11 Investigators for these studies found an approximately 30% increase in the diagnosis of cancer among patients with VTE compared with the general population. Because of their large size, both of these studies were able to demonstrate a significant association between thrombosis and pancreatic, liver, and ovarian cancers.
For liver and pancreatic cancers, consensus opinion suggests that early diagnosis does not change prognosis. Similarly, although some experts recommend ultrasound and Ca-125 testing to investigate possible ovarian cancer, no data support use of these tests in ovarian cancer screening.12
An evaluation of the Danish National Registry data has suggested that cancer diagnosed at the time of, or within a year of, the diagnosis of VTE is usually advanced and is associated with a poor prognosis.13 Indeed, the authors of the Danish study concluded that for patients with VTE, “[our] pragmatic recommendation [is] to use only simple methods of screening and to look for cancer in patients with signs and symptoms of cancer.”13
Recommended work-up
We conclude the literature8-13 does not support an aggressive search for hidden cancer in a patient with idiopathic VTE (SOR=B). Routine evaluation should include a careful history and physical examination. Because of their low cost, reliability, and ready availability, studies such as a complete blood count, basic chemistry panel, liver function tests, and urinalysis may be considered (SOR=B ). Examples of findings during the initial history, physical exam, and laboratory studies that should prompt further evaluation include anorexia, weight loss, cough, abdominal bloating, unexplained anemia, hyponatremia, hematuria, and abnormal liver enzymes.
It is estimated that more than 250,000 patients are hospitalized for venous thromboembolism (VTE) in the United Sates each year.1 The number of VTE cases annually in this country ranges from 600,000 to 2 million.2,3 The most common causes of VTE include surgery, trauma, hospital or nursing home confinement and malignant neoplasm.4
Unnecessary studies. Some authorities recommend a chest radiograph in the routine evaluation for occult malignancy,9 but its clinical utility for patients without pulmonary symptoms has not been clearly demonstrated. Because of their expense and low test yield, we do not advocate the use of more elaborate screens for occult malignancy, such as computerized tomography, magnetic resonance imaging, or serologic tumor markers.
Caveat. In the past, nearly all patients with pulmonary embolism or DVT were hospitalized to receive treatment with continuous intravenous heparin. Presumably these patients routinely received a careful history evaluation, physical examination, and standard blood work.
With increased use of low-molecular-weight heparins (given subcutaneously once or twice daily), many individuals with VTE are candidates for treatment partially or totally on an outpatient basis.14,15 Be sure that individuals receiving outpatient treatment for idiopathic VTE receive the same attention and routine work-up as hospitalized patients.
Coagulation disorders and Venous Thromboembolism
With advances in laboratory testing, more than half of idiopathic VTE cases can be attributed to a specific coagulation disorder. Data show that in a randomly selected group of patients with newly discovered DVT, 24% to 37% have an inherited predisposition to thrombosis.16-19
Deficiencies in protein C, protein S, and antithrombin were the first causes of inherited thrombophilia to be identified;20-22 however, these deficiencies are not particularly common. The most common causes of thrombophilic coagulation disorders are activated protein C resistance, the prothrombin gene G20210A mutation, and hyperhomocysteinemia.
When to look for prothrombotic defects
If, after ruling out occult malignancy, the cause of VTE remains uncertain, be judicious in deciding whether to run more tests for prothrombotic defects. We advocate pursuing further testing for patients likely to have an underlying hypercoagulable disorder, and for whom the identification of such a disorder would have management implications.
A decision to pursue testing with other patients should be made on a case-by-case basis. The yield in testing is low for many prothrombotic disorders, some tests are affected adversely by anticoagulation therapy, and the influence of a positive test result on patient management decisions remains unclear in many cases. Finally, serologic evaluation for thrombophilia would be costly if conducted for all patients with VTE, and the potential clinical benefit would be small.
Although large epidemiologic studies are lacking to help identify patients at increased risk of a hypercoagulable disorder, patients with clinically significant inherited thrombophilia tend to have VTE at a young age.23-25
In addition, advanced age alone is often regarded as an identifiable risk factor for DVT. A recent retrospective study demonstrated the risk for DVT rose rapidly during the 6th through 8th decades of life.26
We generally recommend testing for hypercoagulable disorders upon discovering one of the following findings:
- The first thrombotic event occurs when the patient is younger than 50 years
- A family history of VTE exists
- Episodes of unexplained VTE recur (SOR=C).
Activated protein C resistance (Factor V Leiden mutation)
Prevalence. Approximately 90% of cases of activated protein C resistance are due to a substitution of glutamine for arginine at position 506 on the factor V gene, the so-called Leiden mutation.27
Factor V Leiden mutation varies greatly by race. In North America, this mutation is found in the heterozygous form in approximately 5% of Caucasians, 2% of Hispanics, 1% of African Americans, and less than 0.5% of Asians.28 Persons heterozygous for factor V Leiden have approximately a 7-fold increase in the relative risk;29 persons homozygous for the mutation have approximately an 80-fold increase in the relative risk.30
Detection. Activated protein C resistance is typically assessed by mixing patient plasma with factor V-deficient plasma. This clotting assay is not affected by oral anticoagulation medication but is affected by heparin. A positive test result for activated protein C resistance typically warrants a polymerase chain reaction assay to distinguish the factor V Leiden mutation from other causes of activated protein C resistance.
Testing considerations. Although factor V Leiden mutation is the cause of activated protein C resistance in most cases, we recommend that activated protein C resistance testing be done first, as it is a less expensive and more widely available test than the DNA-based factor V Leiden mutation test. However, if a patient is taking heparin at the time of evaluation for thrombophilia, it may be necessary to defer activated protein C resistance testing or proceed directly to factor V Leiden DNA-based testing.
Prothrombin gene G2 0210A mutation
Prevalence. Like the factor V Leiden mutation, the prothrombin gene G20210A mutation is more common among Caucasians than among those of African or Asian descent.31 Prothrombin mutation is estimated to cause a 2.5-fold relative risk of first DVT.32
Testing considerations. If cost or availability of the polymerase chain reaction assay are issues, a reasonable course of action would be to reserve the assay for patients with factor V Leiden mutation. Data suggest that patients with both factor V and prothrombin gene G20210A mutations are at significantly higher risk for recurrent VTE; consider prolonged anticoagulation.33
Hyperhomocysteinemia
Evidence accumulating over the past decade has shown an increased risk of VTE with elevated homocysteine levels. A case-control study by den Heijer et al34 in 1996 demonstrated a relative risk for first thrombosis of 2.5 in those with a homocysteine level above the 95th percentile of the control group’s levels (which in that study corresponded to a homocysteine level of 18.5 micromoles per liter or above). This predisposition to thrombosis has also been demonstrated in subsequent meta-analyses.35,36
When homocysteine levels are elevated in the presence of factor V Leiden or the prothrombin gene G20210A mutation, risk of thrombosis appears to increase beyond that associated with any one defect alone, perhaps as high as 50-fold risk.37-39
Management Implications of Test Results
If testing for activated protein C resistance is possible and the result is positive, confirmatory testing for factor V Leiden is indicated. If the patient is homozygous for factor V Leiden (rare), or is heterozygous for factor V Leiden and has the prothrombin gene G20210A defect, consider a prolonged course of anticoagulation, perhaps even for the rest of the patient’s life.
We base these suggestions on the high rate of VTE detected among individuals who are homozygous for factor V Leiden30 and on a recent study that showed the relative risk of recurrent thrombosis to be 2.6 for those with factor V Leiden and prothrombin gene G20210A mutations, versus for those heterozygous for factor V Leiden alone.33 This recent study also showed that the relative risk of recurrent thrombosis was not increased in those heterozygous for the Factor V Leiden mutation alone compared to those without this mutation.33
If either the factor V Leiden or the prothrombin gene G20210A mutation is present, consider prolonging the planned course of anticoagulation (although the data to support this decision are less compelling). Data are limited and conflicting regarding the appropriate length and intensity of anticoagulation therapy for patients with inherited prothrombotic defects.33,40-42 When prolonging anticoagulation for these patients, try to balance the risk of a recurrent thrombotic event with the risk of a bleeding complication from chronic anticoagulation.
Prolonged anticoagulation may also be indicated if the homocysteine level is elevated and either the factor V Leiden or the prothrombin gene G20210A mutation is present. In addition, therapy with folate, pyridoxine, and vitamin B12 should be initiated in cases of elevated homocyteine levels. We recommend the use of these vitamins based on a meta-analysis which demonstrated that folate supplementation at a dose of 0.5 mg to 5 mg/day lowered homocysteine levels by approximately 25% and vitamin B12 supplementation at a dose of 0.5 mg/day led to a further reduction of approximately 7%. In this same meta-analysis, pyridoxine at a mean dose of 16.5 mg daily did not demonstrate further lowering of the homocysteine level; however, as it is safe, inexpensive and well-tolerated, we still recommend its use.43
Finally, while less common than the hypercoagulable states discussed above, the antiphospholipid antibody syndrome should be targeted as part of first-tier testing because of its clear impact on management. If the antiphospholipid antibody testing (such as the lupus anticoagulant or anticardiolipin antibody) results are positive, lifelong anticoagulation44 and perhaps a higher target international normalized ratio are considerations.
Second-tier testing: Pursuing less common defects
If the preceding evaluation is unrevealing for a patient with high-risk characteristics, consider pursuing “second-tier” testing. Such testing may include functional and immunologic assays for protein C, protein S, and anti-thrombin III deficiencies, all of which are heterozygous abnormalities caused by multiple mutations and, thus, not detectable with genetic assays.
Patients will likely be taking anticoagulation medication when these tests are administered; thus, test results must be interpreted with caution. Testing for these deficiencies is most reliable if conducted at least 2 weeks after anticoagulation has been discontinued. Again, the incidence of protein C, protein S, and antithrombin III deficiencies is low, and the yield of testing therefore is also likely to be low.
We would not recommend discontinuing anticoagulation to conduct testing for these uncommon defects once it has been initiated following an acute thrombotic event. Rather, testing should be pursued once the planned course of anticoagulation with warfarin has been completed (for example, 6 to 9 months).
Other inherited thrombophilic disorders include heparin cofactor II deficiency, plasminogen deficiency, dysfibrinogenemia, factor XII deficiency, and increased factor VIII coagulant activity. These disorders are rare and their clinical importance remains unclear. Testing for these disorders, if desired, is best conducted in consultation with a hematologist or coagulation specialist.
Corresponding author
Charles F.S. Locke, MD, Johns Hopkins Community Physicians, 2360 W. Joppa Rd., Baltimore, MD 21093. E-mail: clocke@jhmi.edu.
1. Goldhaber SZ. Pulmonary embolism. N Engl J Med 1998;339:93-104.
2. Deitcher SR. Overview of enoxaparin in the treatment of deep vein thrombosis. Am J Manag Care 2000;6(suppl):S1026-S1033.
3. Hirsh J, Hoak J. Management of deep vein thrombosis and pulmonary embolism. A statement for healthcare professionals. Council on Thrombosis (in consultation with the Council on Cardiovascular Radiology), American Heart Association. Circulation. 1996;93:2212-2245.
4. Heit JA, Silverstein MD, Mohr DN, et al. Risk factors for deep vein thrombosis and pulmonary embolism. Arch Int Med 2000;160:809-815.
5. Trousseau A. Phlegmasia alba dolens. In: Clinique Medicale de l’Hotel-Dieu de Paris. Vol. 3. 2nd ed. Paris: JB Bailliere; 1865;654-712.
6. Hansson PO, Sorbo J, Eriksson H. Recurrent venous thromboembolism after deep vein thrombosis: incidence and risk factors. Arch Intern Med 2000;160:769-774.
7. Naschitz JE, Yeshurun D, Lev LM. Thromboembolism in cancer. Cancer 1993;71:1384-1390.
8. Prandoni P, Lensing AW, Buller HR, et al. Deep-vein thrombosis and the incidence of subsequent symptomatic cancer. N Engl J Med 1992;327:1128-1133.
9. Hettiarachchi RJ, Lok J, Prins MH, et al. Undiagnosed malignancy in patients with deep vein thrombosis: incidence, risk indicators, and diagnosis. Cancer 1998;83:180-185.
10. Baron JA, Gridley G, Weiderpass E, et al. Venous thromboembolism and cancer. Lancet 1998;351:1077-1080.
11. Sorenson HT, Mellemkjaer L, Steffensen FH, et al. The risk of a diagnosis of cancer after primary deep venous thrombosis or pulmonary embolism. N Engl J Med 1998;338:1169-1173.
12. Jacobs I, Davies AP, Bridges J, et al. Prevalence screening for ovarian cancer in postmenopausal women by CA 125 measurement and ultrasonography. BMJ 1993;306:1030-1034.
13. Sorensen HT, Mellemkjaer L, Olsen JH, et al. Prognosis of cancers associated with venous thromboembolism. N Engl J Med 2000;343:1846-1850.
14. Levine M, Gent M, Hirsh J, et al. A comparison of low-molecular-weight heparin administered primarily at home with unfractionated heparin administered in the hospital for proximal deep-vein thrombosis. N Engl J Med 1996;334:677-681.
15. Koopman M, Prandoni P, Piovella F, et al. for the Tasman Study Group. Treatment of venous thrombosis with intravenous unfractionated heparin administered in the hospital as compared with subcutaneous low-molecular-weight heparin administered at home. N Engl J Med 1996;334:682-687.
16. Mateo J, Oliver A, Borrell M, Sala N, Fontcuberta J. Laboratory evaluation and clinical characteristics of 2,132 consecutive unselected patients with venous thromboembolism—results of the Spanish Multicentric Study on Thrombophilia (EMET-Study). Thromb Haemost 1997;77:444-451.
17. Ben-Tal O, Zivelin A, Seligsohn U. The relative frequency of hereditary thrombotic disorders among 107 patients with thrombophilia in Israel. Thromb Haemost 1989;61:50-54.
18. Ridker PM, Hennekens CH, Lindpaintner K, et al. Mutation in the gene coding for coagulation factor V and the risk of myocardial infarction, stroke, and venous thrombosis in apparently healthy men. N Engl J Med 1995;332:912-917.
19. Margaglione M, Brancaccio V, Giuliani N, et al. Increased risk for venous thrombosis in carriers of the prothrombin G—>A20210 gene variant. Ann Intern Med 1998;129:89-93.
20. Egeberg O. Inherited antithrombin deficiency causing thrombophilia. Thromb Diath Haemorrh 1965;13:516.-
21. Griffith JH, Evatt B, Zimmerman TS, et al. Deficiency of protein C in congenital thrombotic disease. J Clin Invest 1981;68:1370-1373.
22. Comp PC, Nixon RR, Cooper MR, et al. Familial protein S deficiency is associated with recurrent thrombosis. J Clin Invest 1984;74:2082-2088.
23. Hillarp A, Dahlback B, Zoller B. Activated protein C resistance: from phenotype to genotype and clinical practice. Blood Rev 1995;9:201-212.
24. Zoller B, Berntsdotter A, Garcia de Frutos P, et al. Resistance to activated protein C as an additional genetic risk factor in hereditary deficiency of protein S. Blood 1995;85:3518-3523.
25. Van Boven HH, Reitsma PH, Rosendaal FR, et al. Factor V Leiden (FV R506Q) in families with inherited antithrombin deficiency. Thromb Haemost 1996;75:417-421.
26. Heit JA, Silverstein MD, Mohr DN, et al. Predictors of recurrence after deep vein thrombosis and pulmonary embolism. Arch Intern Med 2000;160:809-815.
27. Bertina RM, Koeleman BPC, Koster T, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994;369:64-67.
28. Ridker PM, Miletich JP, Hennekens CH, Buring JE. Ethnic distribution of factor V Leiden in 4047 men and women. Implications for venous thromboembolism screening. JAMA 1997;277:1305-1307.
29. Koster T, Rosendaal FR, de Ronde H, Briet E, Vandenbroucke JP, Bertina RM. Venous thrombosis due to poor anticoagulant response to activated protein C: Leiden thrombophilia study. Lancet 1993;342:1503-1506.
30. Rosendaal FR, Koster T, Vandenbroucke JP, Reitsma PH. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance). Blood 1995;85:1504-1508.
31. Rosendaal FR, Doggen CJ, Zivelin A, et al. Geographic distribution of the 20210 G to A prothrombin variant. Thromb Haemost 1998;79:706-708.
32. Poort SR, Rosendaal FR, Reitsma PH, Bertina RM. A common genetic variation in the 3´ untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood 1996;88:3698-3703.
33. De Stefano V, Martinelli I, Mannucci PM, et al. The risk of recurrent deep venous thrombosis among heterozygous carriers of both factor V Leiden and the G20210A prothrombin mutation. N Engl J Med 1999;341:801-806.
34. den Heijer M, Koster T, Blom HJ, et al. Hyperhomocysteinemia as a risk factor for deep-vein thrombosis. N Engl J Med 1996;334:759-762.
35. Ray JG. Meta-analysis of hyperhomocysteinemia as a risk factor for venous thromboembolic disease. Arch Intern Med 1998;158:2101-2106.
36. den Heijer M, Rosendaal FR, Blom HJ, Gerrits WB, Bos GM. Hyperhomocysteinemia and venous thrombosis: a meta-analysis. Thromb Haemost 1998;80:874-877.
37. Ridker PM, Hennekens CH, Selhub J, Miletich JP, Malinow MR, Stampfer MJ. Interrelation of hyperhomocyst(e)inemia, factor V Leiden, and risk of future venous thromboembolism. Circulation 1997;95:1777-1782.
38. Kluijtmans LA, den Heijer M, Reitsma PH, Heil SG, Blom HJ, Rosendaal FR. Thermolabile methylenetetrahydrofolate reductase and factor V Leiden in the risk of deep-vein thrombosis. Thromb Haemost 1998;79:254-258.
39. DeStefano V, Zappacosta B, Persichilli S, et al. Prevalence of mild hyperhomocysteinaemia and association with thrombophilic genotypes (factor V Leiden and prothrombin G20210A) in Italian patients with venous thromboembolic disease. Br J Haematol 1999;106:564-568.
40. Kearon C, Gent M, Hirsh J, et al. A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 1999;340:901-907.
41. Simioni P, Prandoni P, Lensing AWA, et al. The risk of recurrent venous thromboembolism in patients with an Arg506—>Gln mutation in the gene for factor V (factor V Leiden). N Engl J Med 1997;336:399-403.
42. Ridker PM, Miletich JP, Stampfer MJ, Goldhaber SZ, Lindpaintner K, Hennekens CH. Factor V Leiden and risks of recurrent idiopathic venous thromboembolism. Circulation 1995;92:2800-2802.
43. Lowering blood homocysteine with folic acid based supplements: meta-analysis of randomised trials. Homocysteine Lowering Trialists’ Collaboration. BMJ 1998;316:894-898.
44. McCrae KR. Antiphospholipid antibody associated thrombosis: a consensus for treatment? Lupus 1996;5:560-570.
1. Goldhaber SZ. Pulmonary embolism. N Engl J Med 1998;339:93-104.
2. Deitcher SR. Overview of enoxaparin in the treatment of deep vein thrombosis. Am J Manag Care 2000;6(suppl):S1026-S1033.
3. Hirsh J, Hoak J. Management of deep vein thrombosis and pulmonary embolism. A statement for healthcare professionals. Council on Thrombosis (in consultation with the Council on Cardiovascular Radiology), American Heart Association. Circulation. 1996;93:2212-2245.
4. Heit JA, Silverstein MD, Mohr DN, et al. Risk factors for deep vein thrombosis and pulmonary embolism. Arch Int Med 2000;160:809-815.
5. Trousseau A. Phlegmasia alba dolens. In: Clinique Medicale de l’Hotel-Dieu de Paris. Vol. 3. 2nd ed. Paris: JB Bailliere; 1865;654-712.
6. Hansson PO, Sorbo J, Eriksson H. Recurrent venous thromboembolism after deep vein thrombosis: incidence and risk factors. Arch Intern Med 2000;160:769-774.
7. Naschitz JE, Yeshurun D, Lev LM. Thromboembolism in cancer. Cancer 1993;71:1384-1390.
8. Prandoni P, Lensing AW, Buller HR, et al. Deep-vein thrombosis and the incidence of subsequent symptomatic cancer. N Engl J Med 1992;327:1128-1133.
9. Hettiarachchi RJ, Lok J, Prins MH, et al. Undiagnosed malignancy in patients with deep vein thrombosis: incidence, risk indicators, and diagnosis. Cancer 1998;83:180-185.
10. Baron JA, Gridley G, Weiderpass E, et al. Venous thromboembolism and cancer. Lancet 1998;351:1077-1080.
11. Sorenson HT, Mellemkjaer L, Steffensen FH, et al. The risk of a diagnosis of cancer after primary deep venous thrombosis or pulmonary embolism. N Engl J Med 1998;338:1169-1173.
12. Jacobs I, Davies AP, Bridges J, et al. Prevalence screening for ovarian cancer in postmenopausal women by CA 125 measurement and ultrasonography. BMJ 1993;306:1030-1034.
13. Sorensen HT, Mellemkjaer L, Olsen JH, et al. Prognosis of cancers associated with venous thromboembolism. N Engl J Med 2000;343:1846-1850.
14. Levine M, Gent M, Hirsh J, et al. A comparison of low-molecular-weight heparin administered primarily at home with unfractionated heparin administered in the hospital for proximal deep-vein thrombosis. N Engl J Med 1996;334:677-681.
15. Koopman M, Prandoni P, Piovella F, et al. for the Tasman Study Group. Treatment of venous thrombosis with intravenous unfractionated heparin administered in the hospital as compared with subcutaneous low-molecular-weight heparin administered at home. N Engl J Med 1996;334:682-687.
16. Mateo J, Oliver A, Borrell M, Sala N, Fontcuberta J. Laboratory evaluation and clinical characteristics of 2,132 consecutive unselected patients with venous thromboembolism—results of the Spanish Multicentric Study on Thrombophilia (EMET-Study). Thromb Haemost 1997;77:444-451.
17. Ben-Tal O, Zivelin A, Seligsohn U. The relative frequency of hereditary thrombotic disorders among 107 patients with thrombophilia in Israel. Thromb Haemost 1989;61:50-54.
18. Ridker PM, Hennekens CH, Lindpaintner K, et al. Mutation in the gene coding for coagulation factor V and the risk of myocardial infarction, stroke, and venous thrombosis in apparently healthy men. N Engl J Med 1995;332:912-917.
19. Margaglione M, Brancaccio V, Giuliani N, et al. Increased risk for venous thrombosis in carriers of the prothrombin G—>A20210 gene variant. Ann Intern Med 1998;129:89-93.
20. Egeberg O. Inherited antithrombin deficiency causing thrombophilia. Thromb Diath Haemorrh 1965;13:516.-
21. Griffith JH, Evatt B, Zimmerman TS, et al. Deficiency of protein C in congenital thrombotic disease. J Clin Invest 1981;68:1370-1373.
22. Comp PC, Nixon RR, Cooper MR, et al. Familial protein S deficiency is associated with recurrent thrombosis. J Clin Invest 1984;74:2082-2088.
23. Hillarp A, Dahlback B, Zoller B. Activated protein C resistance: from phenotype to genotype and clinical practice. Blood Rev 1995;9:201-212.
24. Zoller B, Berntsdotter A, Garcia de Frutos P, et al. Resistance to activated protein C as an additional genetic risk factor in hereditary deficiency of protein S. Blood 1995;85:3518-3523.
25. Van Boven HH, Reitsma PH, Rosendaal FR, et al. Factor V Leiden (FV R506Q) in families with inherited antithrombin deficiency. Thromb Haemost 1996;75:417-421.
26. Heit JA, Silverstein MD, Mohr DN, et al. Predictors of recurrence after deep vein thrombosis and pulmonary embolism. Arch Intern Med 2000;160:809-815.
27. Bertina RM, Koeleman BPC, Koster T, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994;369:64-67.
28. Ridker PM, Miletich JP, Hennekens CH, Buring JE. Ethnic distribution of factor V Leiden in 4047 men and women. Implications for venous thromboembolism screening. JAMA 1997;277:1305-1307.
29. Koster T, Rosendaal FR, de Ronde H, Briet E, Vandenbroucke JP, Bertina RM. Venous thrombosis due to poor anticoagulant response to activated protein C: Leiden thrombophilia study. Lancet 1993;342:1503-1506.
30. Rosendaal FR, Koster T, Vandenbroucke JP, Reitsma PH. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance). Blood 1995;85:1504-1508.
31. Rosendaal FR, Doggen CJ, Zivelin A, et al. Geographic distribution of the 20210 G to A prothrombin variant. Thromb Haemost 1998;79:706-708.
32. Poort SR, Rosendaal FR, Reitsma PH, Bertina RM. A common genetic variation in the 3´ untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood 1996;88:3698-3703.
33. De Stefano V, Martinelli I, Mannucci PM, et al. The risk of recurrent deep venous thrombosis among heterozygous carriers of both factor V Leiden and the G20210A prothrombin mutation. N Engl J Med 1999;341:801-806.
34. den Heijer M, Koster T, Blom HJ, et al. Hyperhomocysteinemia as a risk factor for deep-vein thrombosis. N Engl J Med 1996;334:759-762.
35. Ray JG. Meta-analysis of hyperhomocysteinemia as a risk factor for venous thromboembolic disease. Arch Intern Med 1998;158:2101-2106.
36. den Heijer M, Rosendaal FR, Blom HJ, Gerrits WB, Bos GM. Hyperhomocysteinemia and venous thrombosis: a meta-analysis. Thromb Haemost 1998;80:874-877.
37. Ridker PM, Hennekens CH, Selhub J, Miletich JP, Malinow MR, Stampfer MJ. Interrelation of hyperhomocyst(e)inemia, factor V Leiden, and risk of future venous thromboembolism. Circulation 1997;95:1777-1782.
38. Kluijtmans LA, den Heijer M, Reitsma PH, Heil SG, Blom HJ, Rosendaal FR. Thermolabile methylenetetrahydrofolate reductase and factor V Leiden in the risk of deep-vein thrombosis. Thromb Haemost 1998;79:254-258.
39. DeStefano V, Zappacosta B, Persichilli S, et al. Prevalence of mild hyperhomocysteinaemia and association with thrombophilic genotypes (factor V Leiden and prothrombin G20210A) in Italian patients with venous thromboembolic disease. Br J Haematol 1999;106:564-568.
40. Kearon C, Gent M, Hirsh J, et al. A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 1999;340:901-907.
41. Simioni P, Prandoni P, Lensing AWA, et al. The risk of recurrent venous thromboembolism in patients with an Arg506—>Gln mutation in the gene for factor V (factor V Leiden). N Engl J Med 1997;336:399-403.
42. Ridker PM, Miletich JP, Stampfer MJ, Goldhaber SZ, Lindpaintner K, Hennekens CH. Factor V Leiden and risks of recurrent idiopathic venous thromboembolism. Circulation 1995;92:2800-2802.
43. Lowering blood homocysteine with folic acid based supplements: meta-analysis of randomised trials. Homocysteine Lowering Trialists’ Collaboration. BMJ 1998;316:894-898.
44. McCrae KR. Antiphospholipid antibody associated thrombosis: a consensus for treatment? Lupus 1996;5:560-570.