User login
Before the introduction of insulin, there were few reported cases of pregnancy complicated by diabetes because women with the disease too often did not live to childbearing age, and when they did, they were often counseled to terminate their pregnancies. Perinatal and maternal mortality in the limited number of reported pregnancies were 70% and 40%, respectively,1 making the risks of continuing the pregnancy quite high.
After insulin became available, maternal mortality dropped dramatically, down to a few percent. Perinatal mortality also declined, but it took several decades to achieve a similar magnitude of reduction.2 Today, with insulin therapy and tight glucose control as well as improved perinatal care, almost all women with diabetes can contemplate pregnancy with greater hope for normal outcomes.

Problems persist, however. Maternal diabetes continues to cause a variety of adverse outcomes, including infants large for gestational age, prematurity, and structural birth defects. Birth defects and prematurity, in fact, are the top causes of the unacceptably high infant mortality rate in the United States – a rate that is about 70% higher than the average in comparable developed countries.3
Infant mortality is considered an indicator of population health and of the development of a country; to reduce its rate, we must address these two areas.
Women with type 1 and type 2 diabetes are five times more likely to have a child with birth defects than are nondiabetic women.4 Up to 10% of women with preexisting diabetes will have fetuses with a major congenital malformation.5
Over the years we have been striving in our Center for Birth Defects Research to understand the pathomechanisms and the molecular and epigenetic alterations behind the high rates of birth defects in the offspring of women with preexisting diabetes. We have focused on heart defects and neural tube defects (particularly the latter), which together cause significant mortality, morbidity, disability, and human suffering.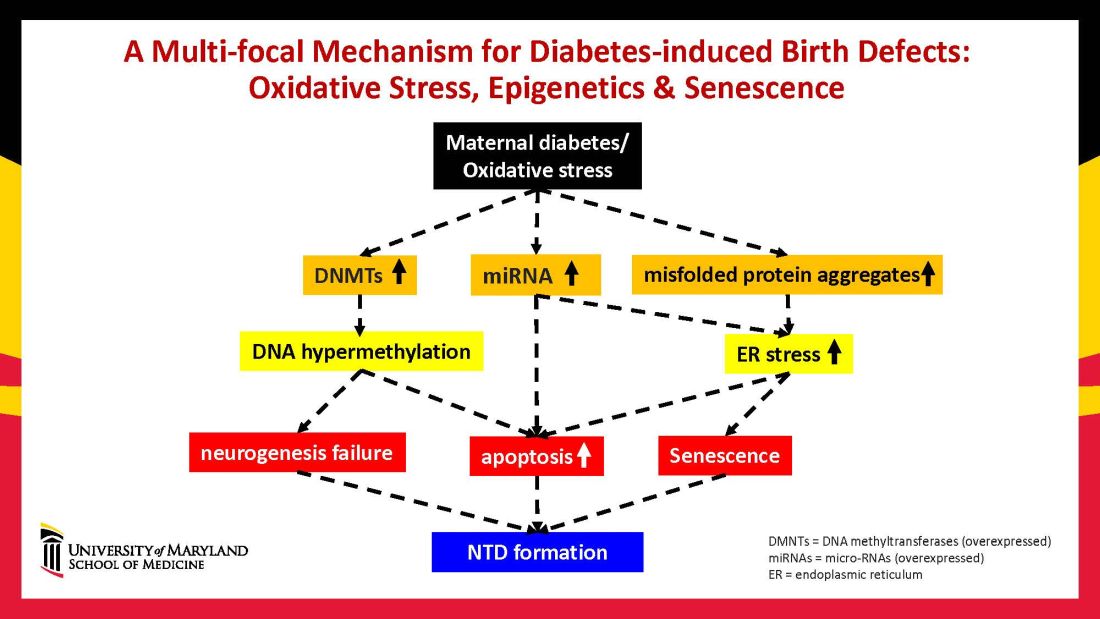
Using animal models that mimic human diabetic pregnancy, we have made significant strides in our understanding of the mechanisms, uncovering molecular pathways involving oxidative stress, senescence/premature cellular aging, and epigenetic modifications (Figure 1). Understanding these pathways is providing us, in turn, with potential therapeutic targets and approaches that may be used in the future to prevent birth defects in women who enter pregnancy with type 1 or type 2 diabetes.
Unraveling the role of oxidative stress
Our mouse models accurately reflect the human conditions of diabetes in pregnancy and diabetic embryopathy. Offspring of mice with type 1 and type 2 diabetes have a similarly higher rate of neural tube defects and congenital heart disease, compared to mice without diabetes. We observe a similar incidence of anencephaly and spina bifida, and of cardiac septation defects in the mouse embryo hearts, for instance.

A primary mechanism and causal event of diabetic embryopathy is hyperglycemia-induced apoptosis in embryonic cells. Excessive cell death in the neural epithelium or in the developing heart leads to abnormal organogenesis and dysfunctional developmental events that cause birth defects. We have identified pathways leading to apoptosis, and have found that many of these pathways crosstalk with each other.
Hyperglycemia induces oxidative stress – one of these pathways – by causing sustained generation of reactive oxygen species. The cells’ mitochondrial function is significantly impaired by the hyperglycemia response, and this diabetes-induced mitochondrial dysfunction further increases the production of reactive oxygen species and a weakening of the endogenous cellular antioxidant systems, both of which then exacerbate oxidative stress.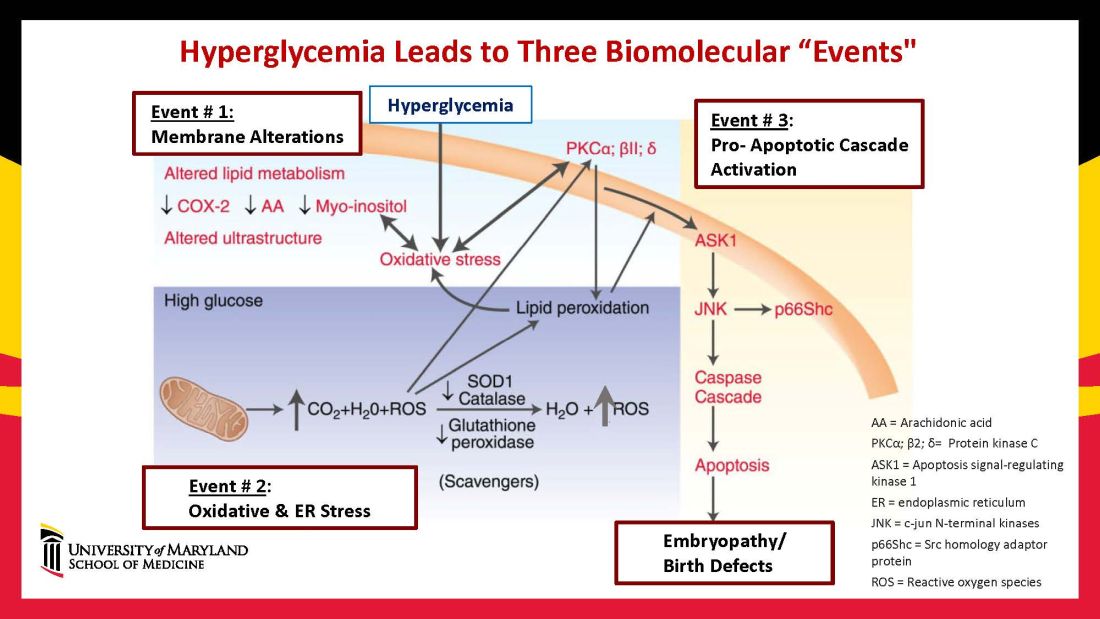
Our research has detailed what happens downstream. We’ve learned that oxidative stress in embryos exposed to maternal diabetes activates a cascade of proapoptotic kinase signaling molecules – for example, protein kinase C isoforms such as PKCalpha; apoptosis signal-regulating kinase 1; and c-Jun-N-terminal kinases – that ultimately lead to abnormal cell death in the neuroepithelium before neural tube closure (Figure 2).5
Hyperglycemia also alters membrane biochemistry in the developing embryo, suppressing lipids including arachidonic acid and myoinositol, and induces the elevation of other molecules that cause newly synthesized proteins to be misfolded. A build-up of misfolded/unfolded proteins triggers or exacerbates endoplasmic reticulum stress, which, like oxidative stress, plays a role in the activation of proapoptotic kinase signaling and apoptosis.6
When we’ve deleted genes for some of the proapoptotic kinase–signaling intermediates, or otherwise inhibited oxidative and endoplasmic reticulum stresses, we’ve been able to ameliorate neural cell apoptosis and the formation of neural tube defects. Studying the processes both forward and backward gives us confidence that the pathways are real and important, and that altering the pathways can alter the outcomes.
Reduced autophagy and induction of cellular senescence
Just as mitochondria are negatively affected by hyperglycemic conditions, so are autophagosomes – organelles that play a key role in removing abnormal or damaged stem cells and cellular components (including unfolded protein aggregates) and in maintaining cellular homeostasis. A high level of autophagy is essential for neural tube closure as well as cardiac morphogenesis.
In our models, maternal diabetes significantly suppressed the process of autophagy in neuroepithelial cells. We have identified responsible molecular intermediates and a key regulating gene for autophagy impairment and have found that deletion of the gene restores autophagy and reduces the development of neural tube defects.4 Administration of a naturally occurring compound, trehalose, which reactivates autophagy, had a similar effect.7Exposure to hyperglycemia not only causes cell death and suppresses autophagy, it also impairs other aspects of cellular function. More recently, we have shown that cells in the neuroepithelium become quiescent and cease proliferating. The quiescent cells, those cells with premature aging markers, also produce cytokines that influence the functioning and development of neighboring cells, causing additional cell death.
All told, premature senescence in the neuroepithelium adversely affects the neurulation process, leading to neural tube defects. In our mouse model, the senomorphic agent rapamycin suppressed cellular senescence, reduced the number of apoptotic neuroepithelial cells, and reduced the formation of neural tube defects.8
The role of epigenetics, future interventions
Epigenetics – the process by which gene expression and function can be modified by environmental conditions without modification of the DNA sequence – has become an additional area of focus in diabetic embryopathy. Our lab has studied the overexpression of both DNA methyltransferases (DNMTs) that cause DNA hypermethylation, and of microRNAs (miRNAs) that can suppress gene expression at the posttranscriptional level. Both are considered to be primary epigenetic mechanisms involved in human diseases and it appears that they are influential in the incidence of birth defects in diabetic mothers.
In our mouse models, maternal diabetes induces DNA hypermethylation via the increase of DNMTs, leading to the silencing of genes essential for neural tube closure and formation of the developing heart. MiRNAs also play a role; in addition to finding altered DNMT activity in the neural epithelium and other tissues of diabetes-exposed embryos, we also found altered miRNA expression. By deleting miRNA genes or by inhibiting DNMT activity through treatment with antioxidants, we saw significant reductions in birth defects.
In one study of the green tea polyphenol epigallocatechin gallate (EGCG), we demonstrated inhibition of diabetes-elevated DNMT expression and activity and suppression of DNA hypermethylation. The expression of genes essential for neural tube closure was restored, with a subsequent reduction in neural tube defects from 29.5% to 2% in embryos treated with EGCG.9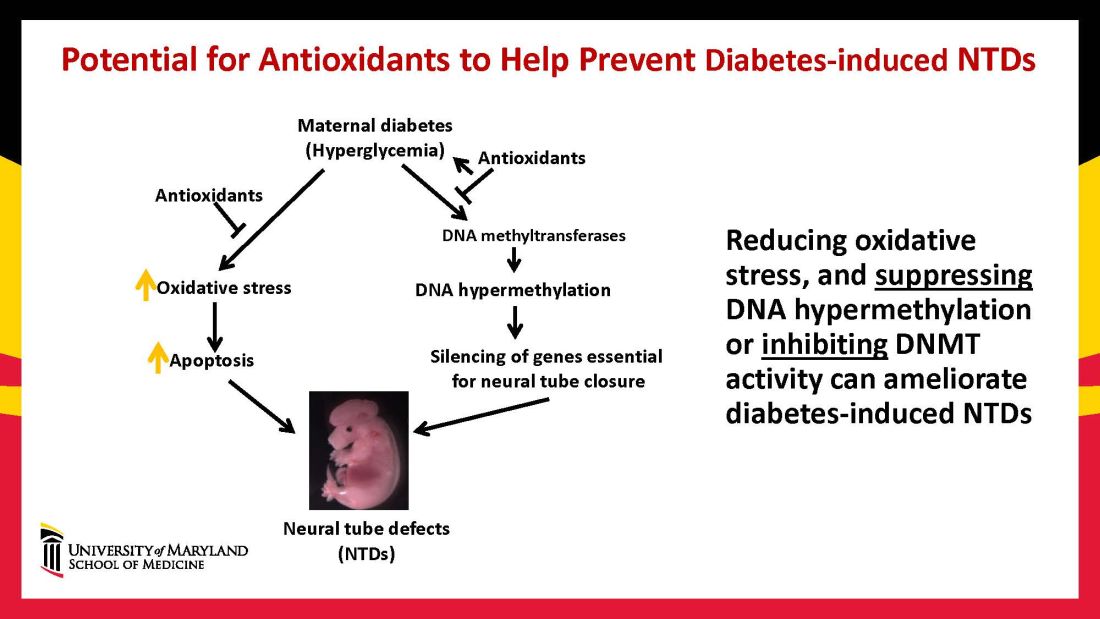
Our interventions to reverse or alter the mechanisms and pathways leading to birth defects have not only helped prove causation, but have given us hope for the future. Antioxidants are among the compounds that could be used as dietary supplements during pregnancy to prevent structural birth defects (Figure 3). Other compounds could activate the process of autophagy (for example, trehalose) and antisenescence compounds similar to rapamycin could be used to reduce numbers of senescent cells in the neuroepithelium or the developing heart.
Dr. Reece and Dr. Yang reported no relevant disclosures.
Dr. Reece, a maternal-fetal medicine specialist, is dean emeritus of the University of Maryland School of Medicine, former university executive vice president, endowed professor and director of CARTI, and codirector of the Center for Birth Defects.
*This story was updated on Nov. 3, 2022
References
1. Z Zhiyong and Reece EA. Clin Lab Med. 2013;33(2)207-33.
2. Reece EA and Coustan DR. Diabetes and obesity in women. Wolters Kluwer: 2019. 4th ed. (https://www.amazon.com/Diabetes-Obesity-Women-Albert-Reece/dp/1496390547).
3. The Peterson-KFF Health System Tracker. www.healthsystemtracker.org.
4. Wang F et al. Nat. Commun. 2017;8:15182.
5. Yang P et al. Am J Obstet Gynecol. 2015;212(5):569-79.
6. Li X et al. Diabetes. 2013 Feb;62(2):599-608.
7. Xu C et al. Am J Physiol Endocrinol Metab. 2013 Sep 1;305(5):E667-78.
8. Xu C et al. Sci Adv. 2021;7(27):eabf5089.
9. Zhong J et al. Am J Obstet Gynecol. 2016 Sep;215(3):368.e1-10.
Before the introduction of insulin, there were few reported cases of pregnancy complicated by diabetes because women with the disease too often did not live to childbearing age, and when they did, they were often counseled to terminate their pregnancies. Perinatal and maternal mortality in the limited number of reported pregnancies were 70% and 40%, respectively,1 making the risks of continuing the pregnancy quite high.
After insulin became available, maternal mortality dropped dramatically, down to a few percent. Perinatal mortality also declined, but it took several decades to achieve a similar magnitude of reduction.2 Today, with insulin therapy and tight glucose control as well as improved perinatal care, almost all women with diabetes can contemplate pregnancy with greater hope for normal outcomes.
Problems persist, however. Maternal diabetes continues to cause a variety of adverse outcomes, including infants large for gestational age, prematurity, and structural birth defects. Birth defects and prematurity, in fact, are the top causes of the unacceptably high infant mortality rate in the United States – a rate that is about 70% higher than the average in comparable developed countries.3
Infant mortality is considered an indicator of population health and of the development of a country; to reduce its rate, we must address these two areas.
Women with type 1 and type 2 diabetes are five times more likely to have a child with birth defects than are nondiabetic women.4 Up to 10% of women with preexisting diabetes will have fetuses with a major congenital malformation.5
Over the years we have been striving in our Center for Birth Defects Research to understand the pathomechanisms and the molecular and epigenetic alterations behind the high rates of birth defects in the offspring of women with preexisting diabetes. We have focused on heart defects and neural tube defects (particularly the latter), which together cause significant mortality, morbidity, disability, and human suffering.
Using animal models that mimic human diabetic pregnancy, we have made significant strides in our understanding of the mechanisms, uncovering molecular pathways involving oxidative stress, senescence/premature cellular aging, and epigenetic modifications (Figure 1). Understanding these pathways is providing us, in turn, with potential therapeutic targets and approaches that may be used in the future to prevent birth defects in women who enter pregnancy with type 1 or type 2 diabetes.
Unraveling the role of oxidative stress
Our mouse models accurately reflect the human conditions of diabetes in pregnancy and diabetic embryopathy. Offspring of mice with type 1 and type 2 diabetes have a similarly higher rate of neural tube defects and congenital heart disease, compared to mice without diabetes. We observe a similar incidence of anencephaly and spina bifida, and of cardiac septation defects in the mouse embryo hearts, for instance.
A primary mechanism and causal event of diabetic embryopathy is hyperglycemia-induced apoptosis in embryonic cells. Excessive cell death in the neural epithelium or in the developing heart leads to abnormal organogenesis and dysfunctional developmental events that cause birth defects. We have identified pathways leading to apoptosis, and have found that many of these pathways crosstalk with each other.
Hyperglycemia induces oxidative stress – one of these pathways – by causing sustained generation of reactive oxygen species. The cells’ mitochondrial function is significantly impaired by the hyperglycemia response, and this diabetes-induced mitochondrial dysfunction further increases the production of reactive oxygen species and a weakening of the endogenous cellular antioxidant systems, both of which then exacerbate oxidative stress.
Our research has detailed what happens downstream. We’ve learned that oxidative stress in embryos exposed to maternal diabetes activates a cascade of proapoptotic kinase signaling molecules – for example, protein kinase C isoforms such as PKCalpha; apoptosis signal-regulating kinase 1; and c-Jun-N-terminal kinases – that ultimately lead to abnormal cell death in the neuroepithelium before neural tube closure (Figure 2).5
Hyperglycemia also alters membrane biochemistry in the developing embryo, suppressing lipids including arachidonic acid and myoinositol, and induces the elevation of other molecules that cause newly synthesized proteins to be misfolded. A build-up of misfolded/unfolded proteins triggers or exacerbates endoplasmic reticulum stress, which, like oxidative stress, plays a role in the activation of proapoptotic kinase signaling and apoptosis.6
When we’ve deleted genes for some of the proapoptotic kinase–signaling intermediates, or otherwise inhibited oxidative and endoplasmic reticulum stresses, we’ve been able to ameliorate neural cell apoptosis and the formation of neural tube defects. Studying the processes both forward and backward gives us confidence that the pathways are real and important, and that altering the pathways can alter the outcomes.
Reduced autophagy and induction of cellular senescence
Just as mitochondria are negatively affected by hyperglycemic conditions, so are autophagosomes – organelles that play a key role in removing abnormal or damaged stem cells and cellular components (including unfolded protein aggregates) and in maintaining cellular homeostasis. A high level of autophagy is essential for neural tube closure as well as cardiac morphogenesis.
In our models, maternal diabetes significantly suppressed the process of autophagy in neuroepithelial cells. We have identified responsible molecular intermediates and a key regulating gene for autophagy impairment and have found that deletion of the gene restores autophagy and reduces the development of neural tube defects.4 Administration of a naturally occurring compound, trehalose, which reactivates autophagy, had a similar effect.7Exposure to hyperglycemia not only causes cell death and suppresses autophagy, it also impairs other aspects of cellular function. More recently, we have shown that cells in the neuroepithelium become quiescent and cease proliferating. The quiescent cells, those cells with premature aging markers, also produce cytokines that influence the functioning and development of neighboring cells, causing additional cell death.
All told, premature senescence in the neuroepithelium adversely affects the neurulation process, leading to neural tube defects. In our mouse model, the senomorphic agent rapamycin suppressed cellular senescence, reduced the number of apoptotic neuroepithelial cells, and reduced the formation of neural tube defects.8
The role of epigenetics, future interventions
Epigenetics – the process by which gene expression and function can be modified by environmental conditions without modification of the DNA sequence – has become an additional area of focus in diabetic embryopathy. Our lab has studied the overexpression of both DNA methyltransferases (DNMTs) that cause DNA hypermethylation, and of microRNAs (miRNAs) that can suppress gene expression at the posttranscriptional level. Both are considered to be primary epigenetic mechanisms involved in human diseases and it appears that they are influential in the incidence of birth defects in diabetic mothers.
In our mouse models, maternal diabetes induces DNA hypermethylation via the increase of DNMTs, leading to the silencing of genes essential for neural tube closure and formation of the developing heart. MiRNAs also play a role; in addition to finding altered DNMT activity in the neural epithelium and other tissues of diabetes-exposed embryos, we also found altered miRNA expression. By deleting miRNA genes or by inhibiting DNMT activity through treatment with antioxidants, we saw significant reductions in birth defects.
In one study of the green tea polyphenol epigallocatechin gallate (EGCG), we demonstrated inhibition of diabetes-elevated DNMT expression and activity and suppression of DNA hypermethylation. The expression of genes essential for neural tube closure was restored, with a subsequent reduction in neural tube defects from 29.5% to 2% in embryos treated with EGCG.9
Our interventions to reverse or alter the mechanisms and pathways leading to birth defects have not only helped prove causation, but have given us hope for the future. Antioxidants are among the compounds that could be used as dietary supplements during pregnancy to prevent structural birth defects (Figure 3). Other compounds could activate the process of autophagy (for example, trehalose) and antisenescence compounds similar to rapamycin could be used to reduce numbers of senescent cells in the neuroepithelium or the developing heart.
Dr. Reece and Dr. Yang reported no relevant disclosures.
Dr. Reece, a maternal-fetal medicine specialist, is dean emeritus of the University of Maryland School of Medicine, former university executive vice president, endowed professor and director of CARTI, and codirector of the Center for Birth Defects.
*This story was updated on Nov. 3, 2022
References
1. Z Zhiyong and Reece EA. Clin Lab Med. 2013;33(2)207-33.
2. Reece EA and Coustan DR. Diabetes and obesity in women. Wolters Kluwer: 2019. 4th ed. (https://www.amazon.com/Diabetes-Obesity-Women-Albert-Reece/dp/1496390547).
3. The Peterson-KFF Health System Tracker. www.healthsystemtracker.org.
4. Wang F et al. Nat. Commun. 2017;8:15182.
5. Yang P et al. Am J Obstet Gynecol. 2015;212(5):569-79.
6. Li X et al. Diabetes. 2013 Feb;62(2):599-608.
7. Xu C et al. Am J Physiol Endocrinol Metab. 2013 Sep 1;305(5):E667-78.
8. Xu C et al. Sci Adv. 2021;7(27):eabf5089.
9. Zhong J et al. Am J Obstet Gynecol. 2016 Sep;215(3):368.e1-10.
Before the introduction of insulin, there were few reported cases of pregnancy complicated by diabetes because women with the disease too often did not live to childbearing age, and when they did, they were often counseled to terminate their pregnancies. Perinatal and maternal mortality in the limited number of reported pregnancies were 70% and 40%, respectively,1 making the risks of continuing the pregnancy quite high.
After insulin became available, maternal mortality dropped dramatically, down to a few percent. Perinatal mortality also declined, but it took several decades to achieve a similar magnitude of reduction.2 Today, with insulin therapy and tight glucose control as well as improved perinatal care, almost all women with diabetes can contemplate pregnancy with greater hope for normal outcomes.
Problems persist, however. Maternal diabetes continues to cause a variety of adverse outcomes, including infants large for gestational age, prematurity, and structural birth defects. Birth defects and prematurity, in fact, are the top causes of the unacceptably high infant mortality rate in the United States – a rate that is about 70% higher than the average in comparable developed countries.3
Infant mortality is considered an indicator of population health and of the development of a country; to reduce its rate, we must address these two areas.
Women with type 1 and type 2 diabetes are five times more likely to have a child with birth defects than are nondiabetic women.4 Up to 10% of women with preexisting diabetes will have fetuses with a major congenital malformation.5
Over the years we have been striving in our Center for Birth Defects Research to understand the pathomechanisms and the molecular and epigenetic alterations behind the high rates of birth defects in the offspring of women with preexisting diabetes. We have focused on heart defects and neural tube defects (particularly the latter), which together cause significant mortality, morbidity, disability, and human suffering.
Using animal models that mimic human diabetic pregnancy, we have made significant strides in our understanding of the mechanisms, uncovering molecular pathways involving oxidative stress, senescence/premature cellular aging, and epigenetic modifications (Figure 1). Understanding these pathways is providing us, in turn, with potential therapeutic targets and approaches that may be used in the future to prevent birth defects in women who enter pregnancy with type 1 or type 2 diabetes.
Unraveling the role of oxidative stress
Our mouse models accurately reflect the human conditions of diabetes in pregnancy and diabetic embryopathy. Offspring of mice with type 1 and type 2 diabetes have a similarly higher rate of neural tube defects and congenital heart disease, compared to mice without diabetes. We observe a similar incidence of anencephaly and spina bifida, and of cardiac septation defects in the mouse embryo hearts, for instance.
A primary mechanism and causal event of diabetic embryopathy is hyperglycemia-induced apoptosis in embryonic cells. Excessive cell death in the neural epithelium or in the developing heart leads to abnormal organogenesis and dysfunctional developmental events that cause birth defects. We have identified pathways leading to apoptosis, and have found that many of these pathways crosstalk with each other.
Hyperglycemia induces oxidative stress – one of these pathways – by causing sustained generation of reactive oxygen species. The cells’ mitochondrial function is significantly impaired by the hyperglycemia response, and this diabetes-induced mitochondrial dysfunction further increases the production of reactive oxygen species and a weakening of the endogenous cellular antioxidant systems, both of which then exacerbate oxidative stress.
Our research has detailed what happens downstream. We’ve learned that oxidative stress in embryos exposed to maternal diabetes activates a cascade of proapoptotic kinase signaling molecules – for example, protein kinase C isoforms such as PKCalpha; apoptosis signal-regulating kinase 1; and c-Jun-N-terminal kinases – that ultimately lead to abnormal cell death in the neuroepithelium before neural tube closure (Figure 2).5
Hyperglycemia also alters membrane biochemistry in the developing embryo, suppressing lipids including arachidonic acid and myoinositol, and induces the elevation of other molecules that cause newly synthesized proteins to be misfolded. A build-up of misfolded/unfolded proteins triggers or exacerbates endoplasmic reticulum stress, which, like oxidative stress, plays a role in the activation of proapoptotic kinase signaling and apoptosis.6
When we’ve deleted genes for some of the proapoptotic kinase–signaling intermediates, or otherwise inhibited oxidative and endoplasmic reticulum stresses, we’ve been able to ameliorate neural cell apoptosis and the formation of neural tube defects. Studying the processes both forward and backward gives us confidence that the pathways are real and important, and that altering the pathways can alter the outcomes.
Reduced autophagy and induction of cellular senescence
Just as mitochondria are negatively affected by hyperglycemic conditions, so are autophagosomes – organelles that play a key role in removing abnormal or damaged stem cells and cellular components (including unfolded protein aggregates) and in maintaining cellular homeostasis. A high level of autophagy is essential for neural tube closure as well as cardiac morphogenesis.
In our models, maternal diabetes significantly suppressed the process of autophagy in neuroepithelial cells. We have identified responsible molecular intermediates and a key regulating gene for autophagy impairment and have found that deletion of the gene restores autophagy and reduces the development of neural tube defects.4 Administration of a naturally occurring compound, trehalose, which reactivates autophagy, had a similar effect.7Exposure to hyperglycemia not only causes cell death and suppresses autophagy, it also impairs other aspects of cellular function. More recently, we have shown that cells in the neuroepithelium become quiescent and cease proliferating. The quiescent cells, those cells with premature aging markers, also produce cytokines that influence the functioning and development of neighboring cells, causing additional cell death.
All told, premature senescence in the neuroepithelium adversely affects the neurulation process, leading to neural tube defects. In our mouse model, the senomorphic agent rapamycin suppressed cellular senescence, reduced the number of apoptotic neuroepithelial cells, and reduced the formation of neural tube defects.8
The role of epigenetics, future interventions
Epigenetics – the process by which gene expression and function can be modified by environmental conditions without modification of the DNA sequence – has become an additional area of focus in diabetic embryopathy. Our lab has studied the overexpression of both DNA methyltransferases (DNMTs) that cause DNA hypermethylation, and of microRNAs (miRNAs) that can suppress gene expression at the posttranscriptional level. Both are considered to be primary epigenetic mechanisms involved in human diseases and it appears that they are influential in the incidence of birth defects in diabetic mothers.
In our mouse models, maternal diabetes induces DNA hypermethylation via the increase of DNMTs, leading to the silencing of genes essential for neural tube closure and formation of the developing heart. MiRNAs also play a role; in addition to finding altered DNMT activity in the neural epithelium and other tissues of diabetes-exposed embryos, we also found altered miRNA expression. By deleting miRNA genes or by inhibiting DNMT activity through treatment with antioxidants, we saw significant reductions in birth defects.
In one study of the green tea polyphenol epigallocatechin gallate (EGCG), we demonstrated inhibition of diabetes-elevated DNMT expression and activity and suppression of DNA hypermethylation. The expression of genes essential for neural tube closure was restored, with a subsequent reduction in neural tube defects from 29.5% to 2% in embryos treated with EGCG.9
Our interventions to reverse or alter the mechanisms and pathways leading to birth defects have not only helped prove causation, but have given us hope for the future. Antioxidants are among the compounds that could be used as dietary supplements during pregnancy to prevent structural birth defects (Figure 3). Other compounds could activate the process of autophagy (for example, trehalose) and antisenescence compounds similar to rapamycin could be used to reduce numbers of senescent cells in the neuroepithelium or the developing heart.
Dr. Reece and Dr. Yang reported no relevant disclosures.
Dr. Reece, a maternal-fetal medicine specialist, is dean emeritus of the University of Maryland School of Medicine, former university executive vice president, endowed professor and director of CARTI, and codirector of the Center for Birth Defects.
*This story was updated on Nov. 3, 2022
References
1. Z Zhiyong and Reece EA. Clin Lab Med. 2013;33(2)207-33.
2. Reece EA and Coustan DR. Diabetes and obesity in women. Wolters Kluwer: 2019. 4th ed. (https://www.amazon.com/Diabetes-Obesity-Women-Albert-Reece/dp/1496390547).
3. The Peterson-KFF Health System Tracker. www.healthsystemtracker.org.
4. Wang F et al. Nat. Commun. 2017;8:15182.
5. Yang P et al. Am J Obstet Gynecol. 2015;212(5):569-79.
6. Li X et al. Diabetes. 2013 Feb;62(2):599-608.
7. Xu C et al. Am J Physiol Endocrinol Metab. 2013 Sep 1;305(5):E667-78.
8. Xu C et al. Sci Adv. 2021;7(27):eabf5089.
9. Zhong J et al. Am J Obstet Gynecol. 2016 Sep;215(3):368.e1-10.