User login
Blue Subcutaneous Nodules in a Young Service Member
DISCUSSION
A diagnosis of familial glomangiomatosis was made based on the clinical history and histopathologic findings from the punch biopsy. Glomus tumors are comprised of glomus cells, or undifferentiated smooth muscle cells responsible for thermoregulation.1 Glomus tumors are classified into 3 categories: solid (predominantly glomus cells), glomangiomas (predominantly blood vessels), and glomangiomyomas (predominantly smooth muscle cells).2 Glomangiomas, which comprise up to 20% of glomus tumors, typically present as bluish-purple, papular or nodular, hyperkeratotic lesions that are 2 to 10 mm in diameter.1 These lesions are tender to palpation and pain may worsen with exposure to cold. Glomangiomas are associated with a classic triad of symptoms that include hypersensitivity, intermittent pain, and pinpoint pain, but patients rarely present with all 3.3
Glomangiomas tend to occur in areas rich with glomus bodies—the distal extremities—specifically the palms, wrists, forearms, feet, and subungual regions; visceral organ involvement including the GI tract is very rare.1,4,5 About 38% to 68% of these lesions are hereditary or can be sporadic. If these lesions are hereditary, a patient is said to have familial glomangiomatosis. In familial glomangiomatosis, the glomulin gene is mutated in an autosomal dominant inheritance pattern with incomplete penetrance and variable expressivity. Inherited glomangiomas may present at birth or puberty similar to other vascular anomalies.4
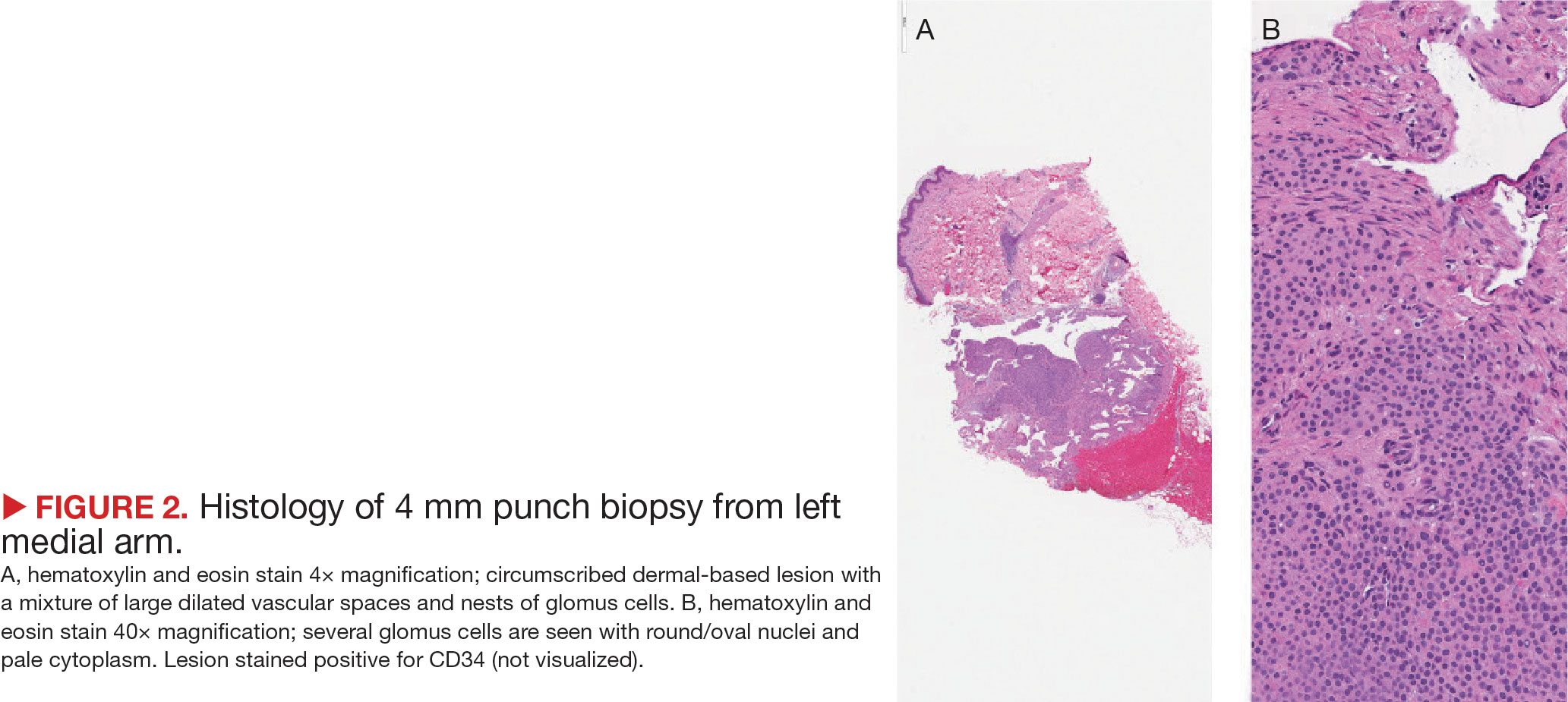
Histopathology of glomangiomas shows rows of glomus cells (modified smooth muscle cells) surrounding distorted venous channels.6,7 These lesions stain positive for CD34, vimentin, calponin, and α-smooth muscle actin, but are negative for desmin, S-100, and von Willebrand factor.1,8 Although the patient’s medical history and physical examination are important in establishing the diagnosis, histopathology is confirmatory.
While the punch biopsy results were pending, a complete blood count (CBC) and fecal occult blood test (FOBT) were ordered due to concerns for blue rubber bleb nevus syndrome (BRBNS), a rare disorder with about 200 reported cases. Patients present with multiple blue to violaceous compressible nodules that feel rubbery in consistency and may be painful with compression. Lesions may be up to 5 cm in diameter and with time, the GI tract may also become involved.9 In the GI tract, the small bowel is the most common site of involvement and patients may present with severe iron deficiency anemia due to hemorrhage.10 Histopathologic features are nonspecific and have features of venous malformations but may include large, tortuous, dilated vessels with a single endothelial lining with possible smooth muscle in vessel walls or calcifications.11 Due to concerns of BRBNS, laboratory studies (CBC and FOBT) were obtained but did not indicate the patient was experiencing a GI hemorrhage.
The differential diagnosis included Maffucci syndrome, also known as dyschondrodysplasia with hemangiomas, enchondromatosis with cavernous hemangiomas, or hemangiomatosis chondrodystrophic. Patients with Maffucci syndrome present with multiple enchondromas, soft tissue hemangiomas or lymphangiomas, and gliomas. These lesions tend to undergo malignant transformation from enchondromas to chondrosarcomas and hemangiomas to vascular sarcomas.12 This diagnosis was less likely in the patient in this case as there were no concerns of skeletal involvement upon history and physical examination.
Lastly, Klippel-Trénaunay syndrome can be associated with similar cutaneous vascular manifestations.13,14 This syndrome occurs due to somatic mutations altering angiogenesis during embryological development. This results in varicosities of superficial and deep venous systems, persistent embryonic veins, and valvular incompetence. However, these patients typically have capillary manifestations such as a flat, red, or purple port-wine stain present at birth and associated limb hypertrophy predominantly affecting a single lower limb.15,16 The patient reported not having the lesions present at birth and because bilateral upper/lower extremity and trunk involvement is rare in this syndrome, a Klippel-Trénaunay syndrome diagnosis was unlikely even in the absence of biopsy results.
Treatment
Based on pathology results, the patient was diagnosed with familial glomangiomatosis and a discussion of treatment options ensued. Asymptomatic lesions can be periodically managed. In addition, there are several treatments for symptomatic lesions. Symptomatic lesions may be tender to palpation and or hypersensitive to temperature change (cold). Though they exhibit slow growth, they can invade surrounding tissues including nerve sheaths which can worsen pain.
Surgical resection, sclerotherapy, laser therapy, and electron beam radiation have been used on patients with symptomatic lesions.8,17 Sclerotherapy involves introducing sterile solutions into a blood vessel’s lumen or into the vascular lesion itself to induce permanent endofibrosis and ablation.17 Hypertonic saline, sodium tetradecyl sulfate (STS), and absolute alcohol have been used to treat vascular anomalies as well as glomangiomas.17 Though case reports have noticed significant improvement in symptomatic lesions, sclerotherapy has been shown to be more effective in treating venous malformations than glomangiomas.18,19
A long-pulsed 1064-nm neodymium-doped yttrium aluminum garnet (Nd:YAG) laser has also been effective in treating larger glomangiomas that would otherwise be difficult to excise.20 The Nd:YAG laser has successfully treated lesions in patients with familial glomangiomatosis.21,22
Our patient opted for sclerotherapy with STS on symptomatic lesions of the bilateral upper extremities and trunk. The patient reported moderate improvement of some lesions at a 4-week follow-up appointment and sclerotherapy with STS was repeated.
It is important to note that if a glomangioma is fully excised, the prognosis is favorable; however, recurrence after surgical excision is seen in 10% to 33% of cases.23,24 Our patient had symptomatic lesions excised on the face, but they recurred. Glomangiomas confer a low risk of malignancy but some risk factors include lesions > 2 cm in size, deep lesions, muscle and/or bone invasion, and high mitotic activity.17,25 If left untreated, high-risk glomangiomas can potentially be life-threatening due to growth, bleeding, or vital organ obstruction.26
Primary Care Role
This patient was referred by his PCP assuming that these were symptomatic vascular lesions or telangiectasias (spider veins). Glomus cell tumors are classified as neurovascular neoplasms which may appear similar to vascular malformations or hemangiomas. 27 PCPs serve an important role in performing cutaneous biopsies to increase patient access to dermatologic care, increase patient awareness of skin conditions including skin cancer, and to potentially diagnose a malignant lesion.28 However, the PCP ultimately referred the patient to dermatology due to the number of growing, painful lesions. If the patient had a single lesion, it may have been appropriate to biopsy for diagnostic clarity.
A retrospective review found that the clinical diagnosis of glomus tumor showed concordance with histopathological diagnosis in 45.4% of cases. The most common alternate histopathological diagnoses were vascular tumors (25.9%) followed by other skin or soft tissue tumors like neuromas, leiomyomas, lipomas, or nevi.29 Even if the PCP performed an initial biopsy with high clinical suspicion of a vascular malformation, some glomus cell tumors may be vascular tumors and vice versa.
Though the patient’s history was consistent with the classic triad of glomangiomas including hypersensitivity, intermittent pain, and pinpoint pain, histopathology was necessary to confirm the diagnosis. Given that these appeared to be similar to telangiectasias to the PCP, a rare condition like BRBNS was likely not considered upon initial presentation. Furthermore, the patient had a negative review of systems to include GI symptoms like melena or hematochezia. The PCP had no concern of GI hemorrhage as these lesions can involve the GI tract. If the patient were to endorse additional symptoms, a CBC to evaluate for anemia as well as a GI referral would be warranted.
CONCLUSIONS
This case exhibits the importance of differentiating glomus cell tumors from other more common vascular anomalies via a patient’s history and histopathological findings. Diagnosis and treatment may be difficult depending on the extent of lesions.
- Brouillard P, Boon LM, Mulliken JB, et al. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (“glomangiomas”). Am J Hum Genet. 2002;70(4):866- 874. doi:10.1086/339492
- Chatterjee JS, Youssef AH, Brown RM, Nishikawa H. Congenital nodular multiple glomangioma: a case report. J Clin Pathol. 2005;58(1):102-103. doi:10.1136/jcp.2003.014324
- Larsen DK, Madsen PV. Ugeskr Laeger. 2018;180(30):V10170807.
- Boon LM, Brouillard P, Irrthum A, et al. A gene for inherited cutaneous venous anomalies (“glomangiomas”) localizes to chromosome 1p21-22. Am J Hum Genet. 1999;65(1):125-133. doi:10.1086/302450
- Tewattanarat N, Srinakarin J, Wongwiwatchai J, et al. Imaging of a glomus tumor of the liver in a child. Radiol Case Rep. 2020;15(4):311-315. doi:10.1016/j.radcr.2019.12.014
- Bolognia J, Schaffer JV, Cerroni L. Dermatology. 5th ed. Elsevier; 2024.
- Elston D, Ferringer T, Ko CJ, Peckham S, High WA, DiCaudo DJ. Dermatopathology. 3rd ed. Elsevier; 2018.
- Leger M, Patel U, Mandal R, et al. Glomangioma. Dermatol Online J. 2010;16(11):11.
- Jin XL, Wang ZH, Xiao XB, Huang LS, Zhao XY. Blue rub ber bleb nevus syndrome: a case report and literature review. World J Gastroenterol. 2014;20(45):17254-17259. doi:10.3748/wjg.v20.i45.17254
- Aravindan U, Ganesan R, Thamarai Kannan M. Surgery for blue rubber bleb nevus syndrome-a case report. Indian J Surg. 2018;80(3):272-274. doi:10.1007/s12262-017-1715-y
- Dobru D, Seuchea N, Dorin M, Careianu V. Blue rubber bleb nevus syndrome: case report and literature review. Rom J Gastroenterol. 2004;13(3):237-240.
- Prokopchuk O, Andres S, Becker K, Holzapfel K, Hartmann D, Friess H. Maffucci syndrome and neoplasms: a case report and review of the literature. BMC Res Notes. 2016;9:126. doi:10.1186/s13104-016-1913-x
- Wang SK, Drucker NA, Gupta AK, Marshalleck FE, Dalsing MC. Diagnosis and management of the venous malformations of Klippel-Trénaunay syndrome. J Vasc Surg Venous Lymphat Disord. 2017;5(4):587-595. doi:10.1016/j.jvsv.2016.10.084
- Yamaki T, Konoeda H, Fujisawa D, et al. Prevalence of various congenital vascular malformations in patients with Klippel- Trenaunay syndrome. J Vasc Surg Venous Lymphat Disord. 2013;1(2):187-193. doi:10.1016/j.jvsv.2012.07.010
- Alwalid O, Makamure J, Cheng QG, et al. Radiological aspect of Klippel-Trénaunay Syndrome: a case series with review of literature. Curr Med Sci. 2018;38(5):925-931. doi:10.1007/s11596-018-1964-4
- Sung HM, Chung HY, Lee SJ, et al. Clinical experience of the Klippel-Trenaunay Syndrome. Arch Plast Surg. Sep 2015;42(5):552-558. doi:10.5999/aps.2015.42.5.552
- Jha A, Khunger N, Malarvizhi K, Ramesh V, Singh A. Familial disseminated cutaneous glomuvenous malformation: treatment with polidocanol sclerotherapy. J Cutan Aesthet Surg. 2016;9(4):266-269. doi:10.4103/0974-2077.197083
- Enjolras O, Ciabrini D, Mazoyer E, Laurian C, Herbreteau D. Extensive pure venous malformations in the upper or lower limb: a review of 27 cases. J Am Acad Dermatol. 1997;36(2 Pt 1):219-225. doi:10.1016/s0190-9622(97)70284-6
- Berenguer B, Burrows PE, Zurakowski D, Mulliken JB. Sclerotherapy of craniofacial venous malformations: complications and results. Plast Reconstr Surg. 1999;104(1):1-15.
- Rivers JK, Rivers CA, Li MK, Martinka M. Laser therapy for an acquired glomuvenous malformation (glomus tumour): a nonsurgical approach. J Cutan Med Surg. 2016;20(1):80-183. doi:10.1177/1203475415596121
- Phillips CB, Guerrero C, Theos A. Nd:YAG laser offers promising treatment option for familial glomuvenous malformation. Dermatol Online J. 2015;21(4).
- Jha A, Ramesh V, Singh A. Disseminated cutaneous glomuvenous malformation. Indian J Dermatol Venereol Leprol. 2014;80(6):556-558. doi:10.4103/0378-6323.144200
- Gonçalves R, Lopes A, Júlio C, Durão C, de Mello RA. Knee glomangioma: a rare location for a glomus tumor. Rare Tumors. 2014;6(4):5588. doi:10.4081/rt.2014.5588
- Cabral CR, Oliveira Filho J, Matsumoto JL, Cignachi S, Tebet AC, Nasser KaR. Type 2 segmental glomangioma- -Case report. An Bras Dermatol. 2015;90(3 Suppl 1):97-100. doi:10.1590/abd1806-4841.20152483
- Tony G, Hauxwell S, Nair N, Harrison DA, Richards PJ. Large plaque-like glomangioma in a patient with multiple glomus tumours: review of imaging and histology. Clin Exp Dermatol. 2013;38(7):693-700. doi:10.1111/ced.12122
- Boon LM, Mulliken JB, Enjolras O, Vikkula M. Glomuvenous malformation (glomangioma) and venous malformation: distinct clinicopathologic and genetic entities. Arch Dermatol. 2004;140(8):971-976. doi:10.1001/archderm.140.8.971
- Honsawek S, Kitidumrongsook P, Luangjarmekorn P, Pataradool K, Thanakit V, Patradul A. Glomus tumors of the fingers: Expression of vascular endothelial growth factor. World J Orthop. 2016;7(12):843-846. doi:10.5312/wjo.v7.i12.843
- Jones TP, Boiko PE, Piepkorn MW. Skin biopsy indications in primary care practice: a population-based study. J Am Board Fam Pract. 1996;9(6):397-404.
- Mravic M, LaChaud G, Nguyen A, Scott MA, Dry SM, James AW. Clinical and histopathological diagnosis of glomus tumor: an institutional experience of 138 cases. Int J Surg Pathol. 2015;23(3):181-188. doi:10.1177/1066896914567330
DISCUSSION
A diagnosis of familial glomangiomatosis was made based on the clinical history and histopathologic findings from the punch biopsy. Glomus tumors are comprised of glomus cells, or undifferentiated smooth muscle cells responsible for thermoregulation.1 Glomus tumors are classified into 3 categories: solid (predominantly glomus cells), glomangiomas (predominantly blood vessels), and glomangiomyomas (predominantly smooth muscle cells).2 Glomangiomas, which comprise up to 20% of glomus tumors, typically present as bluish-purple, papular or nodular, hyperkeratotic lesions that are 2 to 10 mm in diameter.1 These lesions are tender to palpation and pain may worsen with exposure to cold. Glomangiomas are associated with a classic triad of symptoms that include hypersensitivity, intermittent pain, and pinpoint pain, but patients rarely present with all 3.3
Glomangiomas tend to occur in areas rich with glomus bodies—the distal extremities—specifically the palms, wrists, forearms, feet, and subungual regions; visceral organ involvement including the GI tract is very rare.1,4,5 About 38% to 68% of these lesions are hereditary or can be sporadic. If these lesions are hereditary, a patient is said to have familial glomangiomatosis. In familial glomangiomatosis, the glomulin gene is mutated in an autosomal dominant inheritance pattern with incomplete penetrance and variable expressivity. Inherited glomangiomas may present at birth or puberty similar to other vascular anomalies.4
Histopathology of glomangiomas shows rows of glomus cells (modified smooth muscle cells) surrounding distorted venous channels.6,7 These lesions stain positive for CD34, vimentin, calponin, and α-smooth muscle actin, but are negative for desmin, S-100, and von Willebrand factor.1,8 Although the patient’s medical history and physical examination are important in establishing the diagnosis, histopathology is confirmatory.
While the punch biopsy results were pending, a complete blood count (CBC) and fecal occult blood test (FOBT) were ordered due to concerns for blue rubber bleb nevus syndrome (BRBNS), a rare disorder with about 200 reported cases. Patients present with multiple blue to violaceous compressible nodules that feel rubbery in consistency and may be painful with compression. Lesions may be up to 5 cm in diameter and with time, the GI tract may also become involved.9 In the GI tract, the small bowel is the most common site of involvement and patients may present with severe iron deficiency anemia due to hemorrhage.10 Histopathologic features are nonspecific and have features of venous malformations but may include large, tortuous, dilated vessels with a single endothelial lining with possible smooth muscle in vessel walls or calcifications.11 Due to concerns of BRBNS, laboratory studies (CBC and FOBT) were obtained but did not indicate the patient was experiencing a GI hemorrhage.
The differential diagnosis included Maffucci syndrome, also known as dyschondrodysplasia with hemangiomas, enchondromatosis with cavernous hemangiomas, or hemangiomatosis chondrodystrophic. Patients with Maffucci syndrome present with multiple enchondromas, soft tissue hemangiomas or lymphangiomas, and gliomas. These lesions tend to undergo malignant transformation from enchondromas to chondrosarcomas and hemangiomas to vascular sarcomas.12 This diagnosis was less likely in the patient in this case as there were no concerns of skeletal involvement upon history and physical examination.
Lastly, Klippel-Trénaunay syndrome can be associated with similar cutaneous vascular manifestations.13,14 This syndrome occurs due to somatic mutations altering angiogenesis during embryological development. This results in varicosities of superficial and deep venous systems, persistent embryonic veins, and valvular incompetence. However, these patients typically have capillary manifestations such as a flat, red, or purple port-wine stain present at birth and associated limb hypertrophy predominantly affecting a single lower limb.15,16 The patient reported not having the lesions present at birth and because bilateral upper/lower extremity and trunk involvement is rare in this syndrome, a Klippel-Trénaunay syndrome diagnosis was unlikely even in the absence of biopsy results.
Treatment
Based on pathology results, the patient was diagnosed with familial glomangiomatosis and a discussion of treatment options ensued. Asymptomatic lesions can be periodically managed. In addition, there are several treatments for symptomatic lesions. Symptomatic lesions may be tender to palpation and or hypersensitive to temperature change (cold). Though they exhibit slow growth, they can invade surrounding tissues including nerve sheaths which can worsen pain.
Surgical resection, sclerotherapy, laser therapy, and electron beam radiation have been used on patients with symptomatic lesions.8,17 Sclerotherapy involves introducing sterile solutions into a blood vessel’s lumen or into the vascular lesion itself to induce permanent endofibrosis and ablation.17 Hypertonic saline, sodium tetradecyl sulfate (STS), and absolute alcohol have been used to treat vascular anomalies as well as glomangiomas.17 Though case reports have noticed significant improvement in symptomatic lesions, sclerotherapy has been shown to be more effective in treating venous malformations than glomangiomas.18,19
A long-pulsed 1064-nm neodymium-doped yttrium aluminum garnet (Nd:YAG) laser has also been effective in treating larger glomangiomas that would otherwise be difficult to excise.20 The Nd:YAG laser has successfully treated lesions in patients with familial glomangiomatosis.21,22
Our patient opted for sclerotherapy with STS on symptomatic lesions of the bilateral upper extremities and trunk. The patient reported moderate improvement of some lesions at a 4-week follow-up appointment and sclerotherapy with STS was repeated.
It is important to note that if a glomangioma is fully excised, the prognosis is favorable; however, recurrence after surgical excision is seen in 10% to 33% of cases.23,24 Our patient had symptomatic lesions excised on the face, but they recurred. Glomangiomas confer a low risk of malignancy but some risk factors include lesions > 2 cm in size, deep lesions, muscle and/or bone invasion, and high mitotic activity.17,25 If left untreated, high-risk glomangiomas can potentially be life-threatening due to growth, bleeding, or vital organ obstruction.26
Primary Care Role
This patient was referred by his PCP assuming that these were symptomatic vascular lesions or telangiectasias (spider veins). Glomus cell tumors are classified as neurovascular neoplasms which may appear similar to vascular malformations or hemangiomas. 27 PCPs serve an important role in performing cutaneous biopsies to increase patient access to dermatologic care, increase patient awareness of skin conditions including skin cancer, and to potentially diagnose a malignant lesion.28 However, the PCP ultimately referred the patient to dermatology due to the number of growing, painful lesions. If the patient had a single lesion, it may have been appropriate to biopsy for diagnostic clarity.
A retrospective review found that the clinical diagnosis of glomus tumor showed concordance with histopathological diagnosis in 45.4% of cases. The most common alternate histopathological diagnoses were vascular tumors (25.9%) followed by other skin or soft tissue tumors like neuromas, leiomyomas, lipomas, or nevi.29 Even if the PCP performed an initial biopsy with high clinical suspicion of a vascular malformation, some glomus cell tumors may be vascular tumors and vice versa.
Though the patient’s history was consistent with the classic triad of glomangiomas including hypersensitivity, intermittent pain, and pinpoint pain, histopathology was necessary to confirm the diagnosis. Given that these appeared to be similar to telangiectasias to the PCP, a rare condition like BRBNS was likely not considered upon initial presentation. Furthermore, the patient had a negative review of systems to include GI symptoms like melena or hematochezia. The PCP had no concern of GI hemorrhage as these lesions can involve the GI tract. If the patient were to endorse additional symptoms, a CBC to evaluate for anemia as well as a GI referral would be warranted.
CONCLUSIONS
This case exhibits the importance of differentiating glomus cell tumors from other more common vascular anomalies via a patient’s history and histopathological findings. Diagnosis and treatment may be difficult depending on the extent of lesions.
DISCUSSION
A diagnosis of familial glomangiomatosis was made based on the clinical history and histopathologic findings from the punch biopsy. Glomus tumors are comprised of glomus cells, or undifferentiated smooth muscle cells responsible for thermoregulation.1 Glomus tumors are classified into 3 categories: solid (predominantly glomus cells), glomangiomas (predominantly blood vessels), and glomangiomyomas (predominantly smooth muscle cells).2 Glomangiomas, which comprise up to 20% of glomus tumors, typically present as bluish-purple, papular or nodular, hyperkeratotic lesions that are 2 to 10 mm in diameter.1 These lesions are tender to palpation and pain may worsen with exposure to cold. Glomangiomas are associated with a classic triad of symptoms that include hypersensitivity, intermittent pain, and pinpoint pain, but patients rarely present with all 3.3
Glomangiomas tend to occur in areas rich with glomus bodies—the distal extremities—specifically the palms, wrists, forearms, feet, and subungual regions; visceral organ involvement including the GI tract is very rare.1,4,5 About 38% to 68% of these lesions are hereditary or can be sporadic. If these lesions are hereditary, a patient is said to have familial glomangiomatosis. In familial glomangiomatosis, the glomulin gene is mutated in an autosomal dominant inheritance pattern with incomplete penetrance and variable expressivity. Inherited glomangiomas may present at birth or puberty similar to other vascular anomalies.4
Histopathology of glomangiomas shows rows of glomus cells (modified smooth muscle cells) surrounding distorted venous channels.6,7 These lesions stain positive for CD34, vimentin, calponin, and α-smooth muscle actin, but are negative for desmin, S-100, and von Willebrand factor.1,8 Although the patient’s medical history and physical examination are important in establishing the diagnosis, histopathology is confirmatory.
While the punch biopsy results were pending, a complete blood count (CBC) and fecal occult blood test (FOBT) were ordered due to concerns for blue rubber bleb nevus syndrome (BRBNS), a rare disorder with about 200 reported cases. Patients present with multiple blue to violaceous compressible nodules that feel rubbery in consistency and may be painful with compression. Lesions may be up to 5 cm in diameter and with time, the GI tract may also become involved.9 In the GI tract, the small bowel is the most common site of involvement and patients may present with severe iron deficiency anemia due to hemorrhage.10 Histopathologic features are nonspecific and have features of venous malformations but may include large, tortuous, dilated vessels with a single endothelial lining with possible smooth muscle in vessel walls or calcifications.11 Due to concerns of BRBNS, laboratory studies (CBC and FOBT) were obtained but did not indicate the patient was experiencing a GI hemorrhage.
The differential diagnosis included Maffucci syndrome, also known as dyschondrodysplasia with hemangiomas, enchondromatosis with cavernous hemangiomas, or hemangiomatosis chondrodystrophic. Patients with Maffucci syndrome present with multiple enchondromas, soft tissue hemangiomas or lymphangiomas, and gliomas. These lesions tend to undergo malignant transformation from enchondromas to chondrosarcomas and hemangiomas to vascular sarcomas.12 This diagnosis was less likely in the patient in this case as there were no concerns of skeletal involvement upon history and physical examination.
Lastly, Klippel-Trénaunay syndrome can be associated with similar cutaneous vascular manifestations.13,14 This syndrome occurs due to somatic mutations altering angiogenesis during embryological development. This results in varicosities of superficial and deep venous systems, persistent embryonic veins, and valvular incompetence. However, these patients typically have capillary manifestations such as a flat, red, or purple port-wine stain present at birth and associated limb hypertrophy predominantly affecting a single lower limb.15,16 The patient reported not having the lesions present at birth and because bilateral upper/lower extremity and trunk involvement is rare in this syndrome, a Klippel-Trénaunay syndrome diagnosis was unlikely even in the absence of biopsy results.
Treatment
Based on pathology results, the patient was diagnosed with familial glomangiomatosis and a discussion of treatment options ensued. Asymptomatic lesions can be periodically managed. In addition, there are several treatments for symptomatic lesions. Symptomatic lesions may be tender to palpation and or hypersensitive to temperature change (cold). Though they exhibit slow growth, they can invade surrounding tissues including nerve sheaths which can worsen pain.
Surgical resection, sclerotherapy, laser therapy, and electron beam radiation have been used on patients with symptomatic lesions.8,17 Sclerotherapy involves introducing sterile solutions into a blood vessel’s lumen or into the vascular lesion itself to induce permanent endofibrosis and ablation.17 Hypertonic saline, sodium tetradecyl sulfate (STS), and absolute alcohol have been used to treat vascular anomalies as well as glomangiomas.17 Though case reports have noticed significant improvement in symptomatic lesions, sclerotherapy has been shown to be more effective in treating venous malformations than glomangiomas.18,19
A long-pulsed 1064-nm neodymium-doped yttrium aluminum garnet (Nd:YAG) laser has also been effective in treating larger glomangiomas that would otherwise be difficult to excise.20 The Nd:YAG laser has successfully treated lesions in patients with familial glomangiomatosis.21,22
Our patient opted for sclerotherapy with STS on symptomatic lesions of the bilateral upper extremities and trunk. The patient reported moderate improvement of some lesions at a 4-week follow-up appointment and sclerotherapy with STS was repeated.
It is important to note that if a glomangioma is fully excised, the prognosis is favorable; however, recurrence after surgical excision is seen in 10% to 33% of cases.23,24 Our patient had symptomatic lesions excised on the face, but they recurred. Glomangiomas confer a low risk of malignancy but some risk factors include lesions > 2 cm in size, deep lesions, muscle and/or bone invasion, and high mitotic activity.17,25 If left untreated, high-risk glomangiomas can potentially be life-threatening due to growth, bleeding, or vital organ obstruction.26
Primary Care Role
This patient was referred by his PCP assuming that these were symptomatic vascular lesions or telangiectasias (spider veins). Glomus cell tumors are classified as neurovascular neoplasms which may appear similar to vascular malformations or hemangiomas. 27 PCPs serve an important role in performing cutaneous biopsies to increase patient access to dermatologic care, increase patient awareness of skin conditions including skin cancer, and to potentially diagnose a malignant lesion.28 However, the PCP ultimately referred the patient to dermatology due to the number of growing, painful lesions. If the patient had a single lesion, it may have been appropriate to biopsy for diagnostic clarity.
A retrospective review found that the clinical diagnosis of glomus tumor showed concordance with histopathological diagnosis in 45.4% of cases. The most common alternate histopathological diagnoses were vascular tumors (25.9%) followed by other skin or soft tissue tumors like neuromas, leiomyomas, lipomas, or nevi.29 Even if the PCP performed an initial biopsy with high clinical suspicion of a vascular malformation, some glomus cell tumors may be vascular tumors and vice versa.
Though the patient’s history was consistent with the classic triad of glomangiomas including hypersensitivity, intermittent pain, and pinpoint pain, histopathology was necessary to confirm the diagnosis. Given that these appeared to be similar to telangiectasias to the PCP, a rare condition like BRBNS was likely not considered upon initial presentation. Furthermore, the patient had a negative review of systems to include GI symptoms like melena or hematochezia. The PCP had no concern of GI hemorrhage as these lesions can involve the GI tract. If the patient were to endorse additional symptoms, a CBC to evaluate for anemia as well as a GI referral would be warranted.
CONCLUSIONS
This case exhibits the importance of differentiating glomus cell tumors from other more common vascular anomalies via a patient’s history and histopathological findings. Diagnosis and treatment may be difficult depending on the extent of lesions.
- Brouillard P, Boon LM, Mulliken JB, et al. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (“glomangiomas”). Am J Hum Genet. 2002;70(4):866- 874. doi:10.1086/339492
- Chatterjee JS, Youssef AH, Brown RM, Nishikawa H. Congenital nodular multiple glomangioma: a case report. J Clin Pathol. 2005;58(1):102-103. doi:10.1136/jcp.2003.014324
- Larsen DK, Madsen PV. Ugeskr Laeger. 2018;180(30):V10170807.
- Boon LM, Brouillard P, Irrthum A, et al. A gene for inherited cutaneous venous anomalies (“glomangiomas”) localizes to chromosome 1p21-22. Am J Hum Genet. 1999;65(1):125-133. doi:10.1086/302450
- Tewattanarat N, Srinakarin J, Wongwiwatchai J, et al. Imaging of a glomus tumor of the liver in a child. Radiol Case Rep. 2020;15(4):311-315. doi:10.1016/j.radcr.2019.12.014
- Bolognia J, Schaffer JV, Cerroni L. Dermatology. 5th ed. Elsevier; 2024.
- Elston D, Ferringer T, Ko CJ, Peckham S, High WA, DiCaudo DJ. Dermatopathology. 3rd ed. Elsevier; 2018.
- Leger M, Patel U, Mandal R, et al. Glomangioma. Dermatol Online J. 2010;16(11):11.
- Jin XL, Wang ZH, Xiao XB, Huang LS, Zhao XY. Blue rub ber bleb nevus syndrome: a case report and literature review. World J Gastroenterol. 2014;20(45):17254-17259. doi:10.3748/wjg.v20.i45.17254
- Aravindan U, Ganesan R, Thamarai Kannan M. Surgery for blue rubber bleb nevus syndrome-a case report. Indian J Surg. 2018;80(3):272-274. doi:10.1007/s12262-017-1715-y
- Dobru D, Seuchea N, Dorin M, Careianu V. Blue rubber bleb nevus syndrome: case report and literature review. Rom J Gastroenterol. 2004;13(3):237-240.
- Prokopchuk O, Andres S, Becker K, Holzapfel K, Hartmann D, Friess H. Maffucci syndrome and neoplasms: a case report and review of the literature. BMC Res Notes. 2016;9:126. doi:10.1186/s13104-016-1913-x
- Wang SK, Drucker NA, Gupta AK, Marshalleck FE, Dalsing MC. Diagnosis and management of the venous malformations of Klippel-Trénaunay syndrome. J Vasc Surg Venous Lymphat Disord. 2017;5(4):587-595. doi:10.1016/j.jvsv.2016.10.084
- Yamaki T, Konoeda H, Fujisawa D, et al. Prevalence of various congenital vascular malformations in patients with Klippel- Trenaunay syndrome. J Vasc Surg Venous Lymphat Disord. 2013;1(2):187-193. doi:10.1016/j.jvsv.2012.07.010
- Alwalid O, Makamure J, Cheng QG, et al. Radiological aspect of Klippel-Trénaunay Syndrome: a case series with review of literature. Curr Med Sci. 2018;38(5):925-931. doi:10.1007/s11596-018-1964-4
- Sung HM, Chung HY, Lee SJ, et al. Clinical experience of the Klippel-Trenaunay Syndrome. Arch Plast Surg. Sep 2015;42(5):552-558. doi:10.5999/aps.2015.42.5.552
- Jha A, Khunger N, Malarvizhi K, Ramesh V, Singh A. Familial disseminated cutaneous glomuvenous malformation: treatment with polidocanol sclerotherapy. J Cutan Aesthet Surg. 2016;9(4):266-269. doi:10.4103/0974-2077.197083
- Enjolras O, Ciabrini D, Mazoyer E, Laurian C, Herbreteau D. Extensive pure venous malformations in the upper or lower limb: a review of 27 cases. J Am Acad Dermatol. 1997;36(2 Pt 1):219-225. doi:10.1016/s0190-9622(97)70284-6
- Berenguer B, Burrows PE, Zurakowski D, Mulliken JB. Sclerotherapy of craniofacial venous malformations: complications and results. Plast Reconstr Surg. 1999;104(1):1-15.
- Rivers JK, Rivers CA, Li MK, Martinka M. Laser therapy for an acquired glomuvenous malformation (glomus tumour): a nonsurgical approach. J Cutan Med Surg. 2016;20(1):80-183. doi:10.1177/1203475415596121
- Phillips CB, Guerrero C, Theos A. Nd:YAG laser offers promising treatment option for familial glomuvenous malformation. Dermatol Online J. 2015;21(4).
- Jha A, Ramesh V, Singh A. Disseminated cutaneous glomuvenous malformation. Indian J Dermatol Venereol Leprol. 2014;80(6):556-558. doi:10.4103/0378-6323.144200
- Gonçalves R, Lopes A, Júlio C, Durão C, de Mello RA. Knee glomangioma: a rare location for a glomus tumor. Rare Tumors. 2014;6(4):5588. doi:10.4081/rt.2014.5588
- Cabral CR, Oliveira Filho J, Matsumoto JL, Cignachi S, Tebet AC, Nasser KaR. Type 2 segmental glomangioma- -Case report. An Bras Dermatol. 2015;90(3 Suppl 1):97-100. doi:10.1590/abd1806-4841.20152483
- Tony G, Hauxwell S, Nair N, Harrison DA, Richards PJ. Large plaque-like glomangioma in a patient with multiple glomus tumours: review of imaging and histology. Clin Exp Dermatol. 2013;38(7):693-700. doi:10.1111/ced.12122
- Boon LM, Mulliken JB, Enjolras O, Vikkula M. Glomuvenous malformation (glomangioma) and venous malformation: distinct clinicopathologic and genetic entities. Arch Dermatol. 2004;140(8):971-976. doi:10.1001/archderm.140.8.971
- Honsawek S, Kitidumrongsook P, Luangjarmekorn P, Pataradool K, Thanakit V, Patradul A. Glomus tumors of the fingers: Expression of vascular endothelial growth factor. World J Orthop. 2016;7(12):843-846. doi:10.5312/wjo.v7.i12.843
- Jones TP, Boiko PE, Piepkorn MW. Skin biopsy indications in primary care practice: a population-based study. J Am Board Fam Pract. 1996;9(6):397-404.
- Mravic M, LaChaud G, Nguyen A, Scott MA, Dry SM, James AW. Clinical and histopathological diagnosis of glomus tumor: an institutional experience of 138 cases. Int J Surg Pathol. 2015;23(3):181-188. doi:10.1177/1066896914567330
- Brouillard P, Boon LM, Mulliken JB, et al. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (“glomangiomas”). Am J Hum Genet. 2002;70(4):866- 874. doi:10.1086/339492
- Chatterjee JS, Youssef AH, Brown RM, Nishikawa H. Congenital nodular multiple glomangioma: a case report. J Clin Pathol. 2005;58(1):102-103. doi:10.1136/jcp.2003.014324
- Larsen DK, Madsen PV. Ugeskr Laeger. 2018;180(30):V10170807.
- Boon LM, Brouillard P, Irrthum A, et al. A gene for inherited cutaneous venous anomalies (“glomangiomas”) localizes to chromosome 1p21-22. Am J Hum Genet. 1999;65(1):125-133. doi:10.1086/302450
- Tewattanarat N, Srinakarin J, Wongwiwatchai J, et al. Imaging of a glomus tumor of the liver in a child. Radiol Case Rep. 2020;15(4):311-315. doi:10.1016/j.radcr.2019.12.014
- Bolognia J, Schaffer JV, Cerroni L. Dermatology. 5th ed. Elsevier; 2024.
- Elston D, Ferringer T, Ko CJ, Peckham S, High WA, DiCaudo DJ. Dermatopathology. 3rd ed. Elsevier; 2018.
- Leger M, Patel U, Mandal R, et al. Glomangioma. Dermatol Online J. 2010;16(11):11.
- Jin XL, Wang ZH, Xiao XB, Huang LS, Zhao XY. Blue rub ber bleb nevus syndrome: a case report and literature review. World J Gastroenterol. 2014;20(45):17254-17259. doi:10.3748/wjg.v20.i45.17254
- Aravindan U, Ganesan R, Thamarai Kannan M. Surgery for blue rubber bleb nevus syndrome-a case report. Indian J Surg. 2018;80(3):272-274. doi:10.1007/s12262-017-1715-y
- Dobru D, Seuchea N, Dorin M, Careianu V. Blue rubber bleb nevus syndrome: case report and literature review. Rom J Gastroenterol. 2004;13(3):237-240.
- Prokopchuk O, Andres S, Becker K, Holzapfel K, Hartmann D, Friess H. Maffucci syndrome and neoplasms: a case report and review of the literature. BMC Res Notes. 2016;9:126. doi:10.1186/s13104-016-1913-x
- Wang SK, Drucker NA, Gupta AK, Marshalleck FE, Dalsing MC. Diagnosis and management of the venous malformations of Klippel-Trénaunay syndrome. J Vasc Surg Venous Lymphat Disord. 2017;5(4):587-595. doi:10.1016/j.jvsv.2016.10.084
- Yamaki T, Konoeda H, Fujisawa D, et al. Prevalence of various congenital vascular malformations in patients with Klippel- Trenaunay syndrome. J Vasc Surg Venous Lymphat Disord. 2013;1(2):187-193. doi:10.1016/j.jvsv.2012.07.010
- Alwalid O, Makamure J, Cheng QG, et al. Radiological aspect of Klippel-Trénaunay Syndrome: a case series with review of literature. Curr Med Sci. 2018;38(5):925-931. doi:10.1007/s11596-018-1964-4
- Sung HM, Chung HY, Lee SJ, et al. Clinical experience of the Klippel-Trenaunay Syndrome. Arch Plast Surg. Sep 2015;42(5):552-558. doi:10.5999/aps.2015.42.5.552
- Jha A, Khunger N, Malarvizhi K, Ramesh V, Singh A. Familial disseminated cutaneous glomuvenous malformation: treatment with polidocanol sclerotherapy. J Cutan Aesthet Surg. 2016;9(4):266-269. doi:10.4103/0974-2077.197083
- Enjolras O, Ciabrini D, Mazoyer E, Laurian C, Herbreteau D. Extensive pure venous malformations in the upper or lower limb: a review of 27 cases. J Am Acad Dermatol. 1997;36(2 Pt 1):219-225. doi:10.1016/s0190-9622(97)70284-6
- Berenguer B, Burrows PE, Zurakowski D, Mulliken JB. Sclerotherapy of craniofacial venous malformations: complications and results. Plast Reconstr Surg. 1999;104(1):1-15.
- Rivers JK, Rivers CA, Li MK, Martinka M. Laser therapy for an acquired glomuvenous malformation (glomus tumour): a nonsurgical approach. J Cutan Med Surg. 2016;20(1):80-183. doi:10.1177/1203475415596121
- Phillips CB, Guerrero C, Theos A. Nd:YAG laser offers promising treatment option for familial glomuvenous malformation. Dermatol Online J. 2015;21(4).
- Jha A, Ramesh V, Singh A. Disseminated cutaneous glomuvenous malformation. Indian J Dermatol Venereol Leprol. 2014;80(6):556-558. doi:10.4103/0378-6323.144200
- Gonçalves R, Lopes A, Júlio C, Durão C, de Mello RA. Knee glomangioma: a rare location for a glomus tumor. Rare Tumors. 2014;6(4):5588. doi:10.4081/rt.2014.5588
- Cabral CR, Oliveira Filho J, Matsumoto JL, Cignachi S, Tebet AC, Nasser KaR. Type 2 segmental glomangioma- -Case report. An Bras Dermatol. 2015;90(3 Suppl 1):97-100. doi:10.1590/abd1806-4841.20152483
- Tony G, Hauxwell S, Nair N, Harrison DA, Richards PJ. Large plaque-like glomangioma in a patient with multiple glomus tumours: review of imaging and histology. Clin Exp Dermatol. 2013;38(7):693-700. doi:10.1111/ced.12122
- Boon LM, Mulliken JB, Enjolras O, Vikkula M. Glomuvenous malformation (glomangioma) and venous malformation: distinct clinicopathologic and genetic entities. Arch Dermatol. 2004;140(8):971-976. doi:10.1001/archderm.140.8.971
- Honsawek S, Kitidumrongsook P, Luangjarmekorn P, Pataradool K, Thanakit V, Patradul A. Glomus tumors of the fingers: Expression of vascular endothelial growth factor. World J Orthop. 2016;7(12):843-846. doi:10.5312/wjo.v7.i12.843
- Jones TP, Boiko PE, Piepkorn MW. Skin biopsy indications in primary care practice: a population-based study. J Am Board Fam Pract. 1996;9(6):397-404.
- Mravic M, LaChaud G, Nguyen A, Scott MA, Dry SM, James AW. Clinical and histopathological diagnosis of glomus tumor: an institutional experience of 138 cases. Int J Surg Pathol. 2015;23(3):181-188. doi:10.1177/1066896914567330
Blue Subcutaneous Nodules in a Young Service Member
Blue Subcutaneous Nodules in a Young Service Member
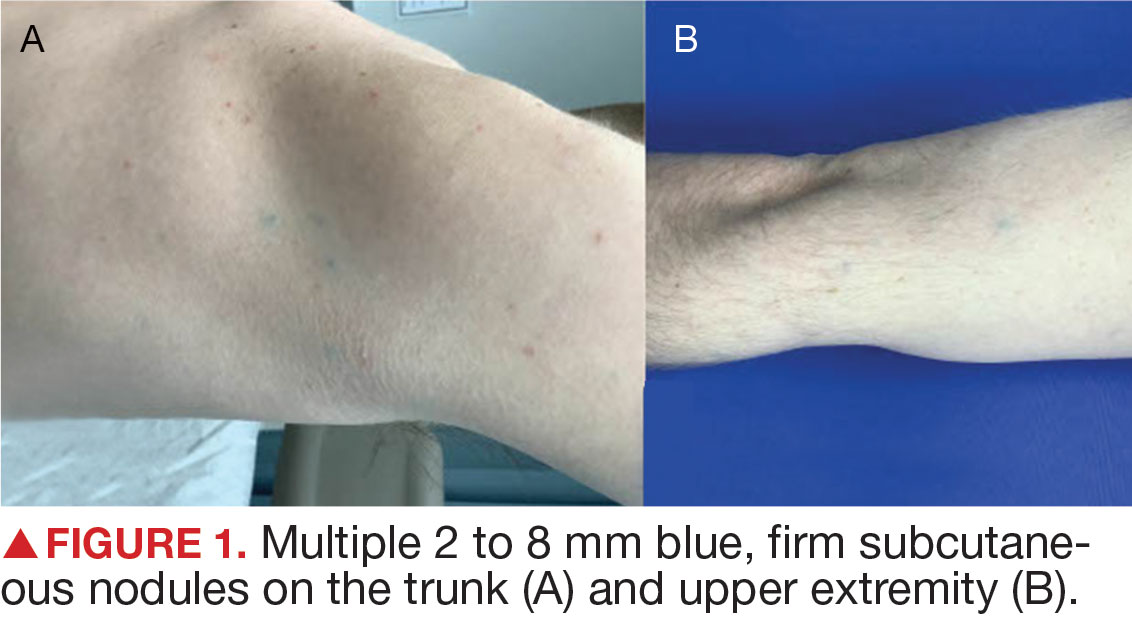
A 26-year-old male with Fitzpatrick skin type II presented for evaluation in the dermatology clinic after being referred by his primary care practitioner (PCP) with a complaint of spider veins. The patient reported a lifelong history of blue subcutaneous nodules that initially appeared on his face during childhood but have since involved his trunk and upper and lower extremities. The patient reported that some of the nodules were painful and increased in size with exercise. His medical history was unremarkable with no other chronic conditions or daily medication use. The patient reported no gastrointestinal (GI) symptoms, melena, or hematochezia. The patient’s mother had similar nodules but his 7 siblings did not.
Upon physical examination, numerous blue subcutaneous nodules, 2 to 8 mm in size, were scattered across his trunk, and proximal and distal extremities were present (Figure 1). The physical examination was otherwise unremarkable. Upon discussing differential diagnosis of these lesions with the patient, he was amenable to a punch biopsy for further diagnostic clarity (Figure 2).