User login
Amyotrophic lateral sclerosis (ALS) is probably best known to the majority of the US population as Lou Gehrig’s disease, named for the New York Yankee who died of ALS after a stellar 17-year baseball career in the 1920s and ’30s. Gradually, the general public is gaining familiarity with the characteristics of ALS, a disease with no single identifiable cause and no known cure.
About 5,600 people receive a diagnosis of ALS in the US each year (about 15 per day), making it the most common form of motor neuron disease.1 Most patients present with progressive muscle weakness in an extremity, which ultimately leads to respiratory failure and death.
Generally, patients with ALS have no cognitive disability and are aware of their physical decline. These patients are faced with important decisions as their condition worsens: Will they want nutritional support through a gastrostomy? For respiratory assistance, will their preference be noninvasive ventilation or mechanical ventilation via tracheostomy?
Not only do ALS patients undergo deterioration of motor function, but many experience muscle cramping, generalized pain, and depression. Only one medication is currently approved to treat ALS patients, with the benefit of prolonging life by a few months. In addition to symptom management, supportive measures, including physical, occupational, and speech therapy, exercise, nutrition, support groups, and counseling, are important tools to enhance quality of life for the patient with ALS.2
Primary care providers need to be aware of ALS, recognize its symptoms in their patients, and manage those affected on a case-by-case basis. Because of the challenges in diagnostic evaluation, the rapidly evolving nature of the disease, and the dire prognosis of ALS, any patient suspected of having ALS should be referred immediately to a neurologist. Providers need to educate patients and their caregivers regarding the disease process and ensure that patients receive appropriate care to meet their needs and preferences.
ALS DEFINED
ALS is a progressive neurodegenerative disease that affects both the upper and lower motor neurons. The disease is considered terminal. Although life may be prolonged by the one currently available pharmacologic agent, no treatment option is yet capable of stopping or reversing progression of the disease.3 While ALS was once believed to be a purely motor disorder, the accompanying degeneration of nonmotor brain regions, such as frontal and temporal cortical neurons, is considered by some to be part of the clinicopathologic spectrum of ALS.4
There are two forms of ALS: sporadic and familial. Sporadic ALS is by far the more common, accounting for 90% to 95% of cases. The remaining 5% to 10% of ALS cases are of the familial form, which can be autosomal-dominant or autosomal-recessive.5
The Degenerative Motor Neuron Diseases
Weakness and muscle wasting characterize several degenerative motor neuron diseases. In addition to ALS, these include primary lateral sclerosis, progressive muscular atrophy, progressive bulbar palsy, and pseudobulbar palsy.6
Primary lateral sclerosis involves upper motor neuron (UMN) dysfunction in the limbs.7Progressive muscular atrophy results from degeneration of the anterior horn cells in the spinal cord and is associated with lower motor neuron (LMN) deficits in the limbs.
Progressive bulbar palsy is a progressive UMN and LMN disorder of the cranial muscles. This condition may occasionally stay isolated in the bulbar segment, but more commonly, UMN and LMN signs and symptoms spread to involve other segments. This is then referred to as bulbar-onset ALS. There have been no reports of specific pathology in progressive bulbar palsy.
Pseudobulbar palsy results from an UMN lesion in the corticobulbar pathway in the pyramidal tract. It is characterized by difficulty chewing and swallowing, and slurred speech (manifestations that may also represent the initial presentation of ALS). Patients with pseudobulbar palsy may exhibit inappropriate, excessive yawning and emotional outbursts; these manifestations are referred to as emotional incontinence.8
EPIDEMIOLOGY
In North America, it is estimated that ALS affects 1.5 to 2.7 people per 100,000 between ages 20 and 80; most frequently, the disease presents between ages 55 and 65. ALS develops infrequently before age 30.3,9
While no gender difference is apparent in patients with familial ALS, sporadic ALS predominately affects males more than females (although the accepted ratio of 1.5:1 appears to be in decline9). After age 65, men and women are equally impacted.10
RISK FACTORS
Established risk factors for ALS include age and family history. Accumulating evidence suggests that military service and smoking may also contribute to the development of ALS.11-15 Children and siblings of ALS patients are at increased risk for ALS, while military personnel have 1.5 times the risk.11
Investigation of the precise link between military service and ALS is ongoing, but factors may include intense exertion, traumatic injuries, viral infections, and exposure to certain chemicals or metals.11 Research suggests the risk is independent of time period, years of service, or branch of service. Geographic location appears to be an independent factor, although there does seem to be a strong association between deployment during the 1991 Gulf War and the risk for ALS.12-14
Recently, smoking has been implicated as a potential risk factor in the disease.3,15 Certain environmental exposures have also been recognized as a possible risk factor. In Guam, an ALS-like syndrome has been identified among members of the Chamorro tribe. This syndrome has been linked to a neurotoxin in the seed of the cycad nut, a tropical plant endemic to the area, which was used in the 1950s and 1960s in the human food supply.3,16
ETIOLOGY
Familial ALS is a genetically transmitted degenerative disease. Twenty percent of cases involve the long arm of chromosome 21, which is responsible for coding of superoxide dismutase (SOD1). Mutations in SOD1, an RNA-processing protein called FUS, and the DNA-binding protein TDP-43, have all been identified in cases of familial ALS.17,18
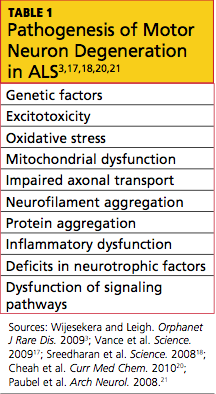
The exact etiology of sporadic ALS remains unknown, but gene mutations have also been implicated, including the ANG gene21 and TDP-43.18 Other theories include increased levels of glutamate (which have been detected in cerebrospinal fluid and serum of patients with ALS), mitochondrial dysfunction, free radical injury, programmed cell death, neurofilament defects, viral infections, and autoimmune dysfunction19,20 (see Table 13,17,18,20,21). It is possible that a combination of factors is involved in the development of sporadic ALS.
PATHOPHYSIOLOGY
The pathophysiology of ALS involves degeneration of the UMN and LMN axons, which leads to glial scarring and possible impairment of the glial cells’ ability to store excess glutamate.22 ALS affects the central nervous system, specifically the anterior horn cells in the spinal cord and the cranial nerve nuclei (X, XI, XII) of the LMNs, and the corticospinal tract and corticobulbar pathway of the UMNs. Bulbar and limb muscles innervated by LMNs are subject to atrophy, whereas cognition, coordination, sensation, the oculomotor system, and sphincters are typically spared.3
CLINICAL FEATURES
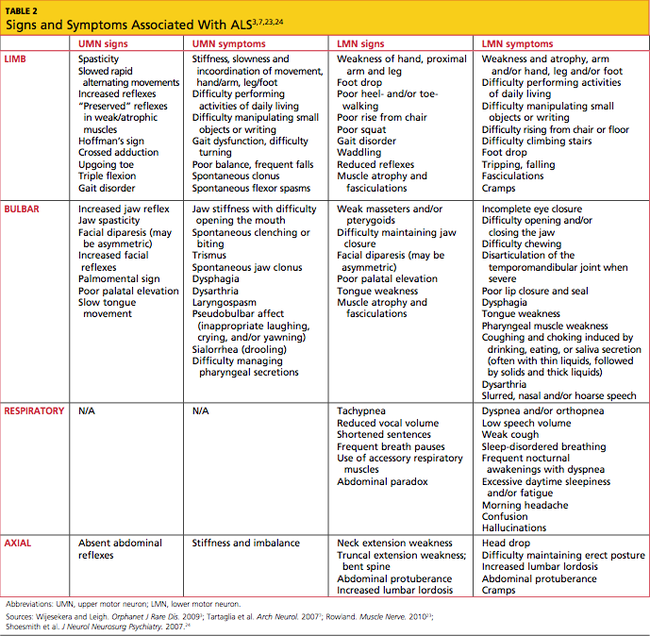
In most patients, ALS symptoms characterize either limb onset or bulbar onset (see Table 23,7,23,24), with limb onset being the more common (about 75% vs about 25% of cases, respectively).1 Typically, patients with limb-onset ALS complain of rapidly progressive, asymmetric weakness in an extremity, followed by focal muscle atrophy with cramping and fasciculations, and eventually, spasticity. Weakness generally begins in one hand, arm, foot, or leg. Patients may notice increased episodes of tripping, clumsiness when they run or walk, a “dropped foot” gait, and/or a decline in manual dexterity.25
The weakness often develops insidiously; patients may notice that symptoms are exacerbated by cold weather.3 Eventually, the bulbar muscles are affected, resulting in dysphagia, dysarthria, and dysphasia. Occasionally, patients encounter bladder dysfunction (urgent micturition), sensory symptoms, and cognitive symptoms (eg, dementia, parkinsonism).26 Multisystem involvement is possible. Ultimately, respiratory compromise or other pulmonary complications ensue, representing a primary cause of mortality in ALS patients.3,24
In bulbar-onset ALS, patients first notice symptoms of dysphasia and dysphagia. They may complain of slurred speech, nasal or low-volume speech, and/or inhibited tongue mobility. The risk for aspiration is increased. The majority of patients with bulbar-onset ALS experience sialorrhea (excessive drooling) because they have difficulty swallowing their saliva. In most patients, mild UMN-type bilateral facial weakness affects the lower half of the face.3 As is the case with limb-onset ALS, bulbar-onset ALS progresses to respiratory compromise.27
In less than 3% of patients with ALS, presentation begins with respiratory weakness and no significant limb or bulbar symptoms.24,28 Patients with respiratory-onset ALS experience symptoms associated with nocturnal hypoventilation, including daytime hypersomnolence, morning headaches, impaired concentration, irritability, anorexia, mood changes, dyspnea, orthopnea, and disturbed sleep; or they may experience type 2 respiratory failure.28,29
Patients with axial symptoms of ALS present with neck weakness and may complain of posterior neck pain or strain with a gradually worsening tendency of the head to tip forward. These patients often support the chin with one hand. Those with axial truncal weakness often complain of difficulty maintaining erect posture when standing and of stooping as they walk. Some patients support the trunk by placing their hands in their front pants pockets or on their upper thighs. They may report some relief when pushing a grocery cart.7,23,24
Symptoms of ALS can be present for weeks or months before a patient consults with a health care provider. The average time span from onset of initial symptoms to diagnosis of ALS is about one year.1 Due to the unpredictable pattern of progression and variability of symptoms among patients, it is difficult to approximate a time frame for symptom progression; for some patients, the disease progresses slowly, while others deteriorate rapidly.30
PHYSICAL EXAMINATION FINDINGS
At onset, the typical presentation of ALS includes muscle weakness in one limb as well as visible fasciculations. As the disease progresses, focal wasting of muscle groups occurs in all four extremities. Particularly involved are the muscles of the hands, forearms, or shoulders in the upper limbs; and of the proximal thigh or distal foot muscles in the lower limbs.31 Deep tendon reflexes are symmetrically brisk. Spasticity, evident in the upper limbs, may present as increased tone.32
In patients with bulbar dysfunction, dysarthria may arise from either LMN pathology or pseudobulbar palsy caused by a UMN disorder, leading to slow, slurred speech or speech with a nasal quality. Tongue fasciculations will be present, as will atrophy and diminished mobility of the tongue.33 The gag reflex remains intact, even brisk, but weakness may occur in the muscles of the soft palate.3 Facial weakness is sometimes seen late in the disease, as evidenced by difficulty sealing the lips or puffing out the cheeks. The jaw jerk will be brisk, indicating that cranial nerve V is intact.34
A pseudobulbar affect, which is best described as emotional lability, may be present. The patient may have a history of exaggerated expression of emotion, such as uncontrollable crying, laughing, or both.35 Cognition, coordination, sensation, the oculomotor system, and sphincters are generally spared. However, cases of frontotemporal dementias coexisting with ALS have been reported; affected patients exhibit cognitive impairment, compulsive behaviors, and personality changes, and they may experience shorter survival.4,24,36
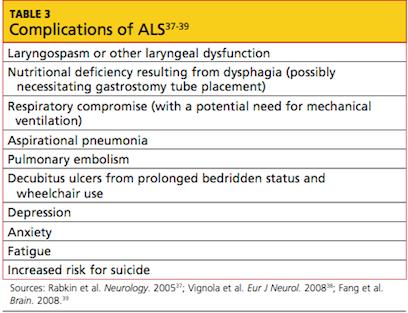
Evidence of known complications of ALS may also be noted during the physical examination; see Table 3.37-39
DIAGNOSIS OF ALS
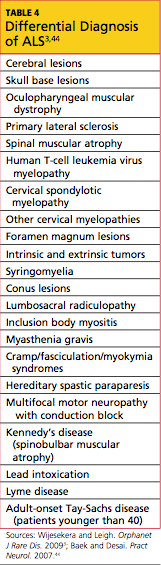
In current research, use of structural MRI, magnetic resonance spectroscopy, and diffusion tensor imaging is being examined to detect thinning in the primary motor cortex, fractional anisotropy in the corpus callosum, patterns of gray and white matter atrophy, and other proposed diagnostic markers for ALS.40-43 However, no single specific diagnostic test has yet been proven to identify ALS; rather, it remains a disease of exclusion (see Table 43,44). For a confirmed diagnosis of ALS to be made, the patient must display:
- Evidence of LMN degeneration as found through clinical, neuropathologic, or electrophysiological examination
- UMN degeneration detected by clinical examination, and
- Progressive spread of signs or symptoms within a region or to other regions.3,45-47
Other disease processes that might explain the signs of UMN and/or LMN degeneration must be excluded, such as cervical spinal disease, myasthenia gravis, multifocal motor neuropathy, lead intoxication, and Lyme disease.45
Imaging studies are not required in ALS cases that have clinically definite disease with bulbar or pseudobulbar onset.47 Otherwise, the essential role of neuroimaging is to exclude a treatable structural lesion that may mimic ALS by producing UMN and LMN signs in varying degrees.48
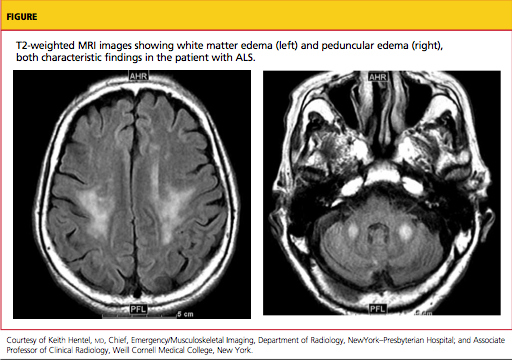
Typically, MRI of the head and spine is ordered in patients with suspected ALS (see figure); MRI can reveal lesions in the corticospinal tracts that occur in ALS. The most characteristic finding on T2-weighted MRI is hyperintensity of the corticospinal tracts, which is visualized best in the brain and brainstem, and to a lesser extent in the spinal cord.49 Decreased signal intensity in the motor cortex has been reported on MRI in cases of ALS.50
Additional diagnostic procedures that are useful in excluding other disease processes include blood and cerebrospinal fluid (CSF) samples,3,44 four-limb electromyography (EMG),44 nerve conduction studies, motor unit number estimations, and muscle biopsies.47
- Autopsy results of patients with ALS demonstrate:
- Neuron loss, especially in lumbar and cervical enlargements
- Nonexistent or atrophic neurons in the motor nuclei of the pons and medulla, and in the anterior horn cells of the spinal cord
- Degeneration of the lateral columns of the spinal cord, and
- Atrophy of the ventral roots.31,51
PROGNOSIS
The overall five-year survival rate for patients with ALS has been reported between 7% and 14%,52,53 and the mortality rate rises in patients older than 75 and in those with bulbar signs.30 The rate of disease progression varies greatly among ALS patients and may be hastened with advancing age, female gender, presence of bulbar features, and absence of a significant other.37,53,54
The average life span for a patient with limb-onset ALS is two to five years from diagnosis, whereas patients affected by the bulbar form usually succumb within six to 18 months.30,53 In patients who present with respiratory symptoms, Shoesmith et al24 have reported a mean time of 14.9 months from initial symptoms to need for full-time ventilation, and of 27.0 months from symptom onset to death.
Advance directives, end-of-life care, and respiratory and nutritional management become essential issues during late stages of ALS; thus, they should be discussed with patients and their relatives at the time of diagnosis or shortly thereafter.
TREATMENT
Pharmacologic Options
There are currently no treatments to halt the progression of ALS or to reverse the disease process. Riluzole, currently the only FDA-approved drug for treatment of ALS, has been shown to slow the loss of muscle strength and to prolong life by an average of two to three months.55 Riluzole targets and blocks glutamate transporters on the presynaptic neuron, decreasing glutamate release and reducing excitotoxicity20 (ie, overstimulation of the postsynaptic receptors). These effects support the theory that ALS may result from excess glutamate and help to explain the increased levels of glutamate found in the serum and CSF of ALS patients.19
Riluzole is typically dosed at 50 mg by mouth twice a day, although 200-mg/d doses have also been examined in clinical trials.20,55 Generally, the drug is well tolerated, with common adverse effects including asthenia, nausea, gastrointestinal upset, and abnormal liver test results. Liver function should be monitored regularly during riluzole therapy, with elevations in serum alanine transferase of particular concern.55
Additional agents have been used to reduce muscle spasticity, muscle cramping and fasciculations, and the associated pain some patients experience, attributable in part to lack of activity and/or inflammation.56 For spasticity, tizanidine or baclofen (orally, a maximum of 20 mg in divided doses; or lower doses administered intrathecally, for patients who experience sedation and fatigue with high oral doses56,57) are often used. Carbamazepine and phenytoin are most commonly used to relieve muscle cramps.56 For some patients, NSAIDs may be adequate to control moderate to severe pain, but others may require opioids.2,56,58
Depressive disorders must be identified accurately, using appropriate clinical tools, before SSRIs (eg, citalopram) or other medications (eg, amitriptyline) are prescribed3; these agents should not be used presumptively,37 as estimates of prevalence of depression among ALS patients range from 2% to 75%.2,37,38,54 Estimates of anxiety prevalence in these patients range from 0% to 30%, reflecting the importance of accurate diagnosis before lorazepam or other agents are prescribed.3,38
Promising results have been reported in the use of modafinil to manage fatigue in patients with ALS.59 For patients with pseudobulbar affect, dextromethorphan 20 mg/quinidine sulfate 10 mg is an FDA-approved treatment.35
Vitamins and other supplements, including creatine, vitamin E, coenzyme Q10, and acetylcysteine, have not been shown to improve survival in this patient population.3
Nonpharmacologic Interventions
Supportive measures for ALS include physical and occupational therapy. In speech and language therapy, breathing and relaxation patterns can be used to correct ineffective compensatory behaviors and help patients “economize” their speaking efforts.60
Nutritional support is also important in patients who have difficulty swallowing, although this can be alleviated somewhat by changes in posture (eg, lowering the chin before attempting to swallow) and by use of thickened fluids; those with immobility of the tongue may find swallowing easier with the head tilted back.60 Once oral alimentation is no longer possible, enteral tube feeding is an option that may prolong survival.60,61
As ALS progresses, dysphagia may be aggravated as the ability to cough, reflexively or voluntarily, is reduced.60 As breathing becomes increasingly difficult, patients may require respiratory support. Noninvasive ventilation, using either continuous or bilevel positive airway pressure, may be implemented early in patients with respiratory-onset ALS, and later in the disease process for other patients, to prevent apnea and hypoventilation.28,62 Mechanical ventilation via tracheostomy is the most invasive method to address respiratory dysfunction in patients with ALS; however, like noninvasive ventilation, nocturnal mechanical ventilation has been shown to extend survival in these patients.28,63,64
Diaphragm pacing stimulation (DPS) has emerged as a possible alternative to mechanical ventilation for ALS patients. The pacing system consists of a battery-operated external pulse generator with electrodes placed after laparoscopic mapping on the diaphragm.65 Natural respiration is mimicked as stimulation from the external pulse generator prompts the diaphragm to contract. Researchers have shown that the minimally invasive surgery (including use of general anesthesia) required to install the DPS system can be safely performed on patients with ALS, and its use can delay the need for a ventilator by 24 months.65,66 Use of the DPS device was granted FDA approval in 2011, under the Humanitarian Device Exemption program.67
Addressing Quality of Life
For the patient with ALS, quality of life is an important consideration throughout disease management. According to numerous research teams, quality of life for these patients depends less on physical function and strength and more on social relationships, existential issues, and spirituality.2,37,54 A high level of quality of life can be sustained in patients with ALS, despite the decline they experience in physical function.
Addressing depression and anxiety by nonpharmacologic means may be needed. Loss of physical strength and mobility and difficulties with speech, swallowing, and breathing can challenge even the strongest patient’s coping skills; even more difficult can be the increased dependence on caregivers, the loss of income, and the financial burdens incurred in health care–related expenses. In some patients, the severity of disease, the lack of effective treatments, and the loss of independence may trigger thoughts of suicide or the wish to “hasten death.”37,39 The importance of counseling, support groups, spirituality or religion, and palliative care, from early in the disease process, cannot be overstated.3,37
Some of these considerations may also be of benefit to spouses and other nonpaid caregivers of the patient with ALS, at least half of whom report feeling physically or psychologically unwell.37,68 Even when professional nursing services or hospice support are available, caregivers often devote 12 hours or more per day to nonprofessional patient care.69 Strategies that support the caregiver can reduce the patient’s perception of burden in that individual.37
CONCLUSION
The exact cause or causes of ALS remain unknown, making it difficult to predict who will present with a disease that appears impossible to prevent. Health care providers in any practice should be aware of the signs of ALS and familiar with its symptoms in order to provide optimal management for potentially affected patients.
Any cause for suspicion of ALS (ie, recent-onset limb weakness or atrophy; difficulty swallowing or speaking) warrants immediate patient referral to a neurologist. Evaluation should include a comprehensive history and physical examination, with emphasis on the musculoskeletal and neurologic exams; MRI of the spine and head, analysis of blood and CSF samples, EMG, and nerve conduction studies should be used to rule out treatable causes of limb weakness.
Potential complications of ALS, including nutritional deficiency, respiratory compromise, and depression, should be discussed early with patients and their caregivers. Management of the patient with ALS necessitates a multidisciplinary approach involving providers from several specialties to ensure that the many issues associated with this disease are being addressed. The priority of the health care provider should be to extend the patient’s survival while maintaining quality of life.
1. ALS Association. Facts you should know. www.alsa.org/about-als/facts-you-should-know.html. Accessed May 30, 2012.
2. Simmons Z. Management strategies for patients with amyotrophic lateral sclerosis from diagnosis through death. Neurologist. 2005;11(5):257-270.
3. Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet J Rare Dis. 2009;4:3.
4. Yoshida M. Amyotrophic lateral sclerosis with dementia: the clinicopathological spectrum. Neuropathology. 2004;24(1):87-102.
5. Byrne S, Walsh C, Lynch C, et al. Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2011;82(6):623-627.
6. National Institute of Neurological Disorders and Stroke, NIH. Motor neuron diseases fact sheet. www.ninds.nih.gov/disorders/motor_neuron_diseases/detail_motor_neuron_diseases.htm. Accessed May 30, 2012.
7. Tartaglia MC, Rowe A, Findlater K. Differentiation between primary lateral sclerosis and amyotrophic lateral sclerosis: examination of symptoms and signs at disease onset and during follow-up. Arch Neurol. 2007;64(2):232-236.
8. Strowd RE, Cartwright MS, Okun MS, et al. Pseudobulbar affect: prevalence and quality of life impact in movement disorders. J Neurol. 2010;257(8):1382-1387.
9. Beghi E, Logroscino G, Chiò A, et al; EURALS Consortium. The epidemiology of ALS and the role of population-based registries. Biochim Biophys Acta. 2006;1762(11-12):1150-1157.
10. McCombe PA, Henderson RD. Effects of gender in amyotrophic lateral sclerosis. Gend Med. 2010;7(6):557-570.
11. Weisskopf MG, Morozova N, O’Reilly EJ, et al. Prospective study of chemical exposures and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2009;80(5):558-561.
12. ALS Association. ALS in the military: unexpected consequences of military service (2011). www.alsa.org/assets/pdfs/advocacy/als_military_paper.pdf. Accessed May 22, 2012.
13. Horner RD, Kamins KG, Feussner JR, et al. Occurrence of amyotrophic lateral sclerosis among Gulf War veterans. Neurology. 2003;61(6):742-749.
14. Horner RD, Grambow SC, Coffman DJ, et al. Amyotrophic lateral sclerosis among 1991 Gulf War veterans: evidence for a time-limited outbreak. Neuroepidemiology. 2008;31(1):28-32.
15. Wang H, O’Reilly EJ, Weisskopf MG, et al. Smoking and risk of amyotrophic lateral sclerosis: a pooled analysis of 5 prospective cohorts. Arch Neurol. 2011;68(2):207-213.
16. Steele JC, McGeer PL. The ALS/PDC syndrome of Guam and the cycad hypothesis. Neurology. 2008;70(21):1984-1990.
17. Vance C, Rogelj B, Hortobágyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208-1211.
18. Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):1668-1672.
19. Pioro EP, Majors AAW, Mitsumoto H, et al. 1H-MRS evidence of neurodegeneration and excess glutamate + glutamine in ALS medulla. Neurology. 1999;53(1):71-79.
20. Cheah BC, Vucic S, Krishnan AV, Kiernan MC. Riluzole, neuroprotection and amyotrophic lateral sclerosis. Curr Med Chem. 2010;17(18):1942-1959.
21. Paubel A, Violette J, Amy M, et al. Mutations of the ANG gene in French patients with sporadic amyotrophic lateral sclerosis. Arch Neurol. 2008;65(10):1333-1336.
22. Neusch C, Bähr M, Schneider-Gold C. Glia cells in amyotrophic lateral sclerosis: new clues to understanding an old disease? Muscle Nerve. 2007;35(6):712-724.
23. Rowland LP. Progressive muscular atrophy and other lower motor neuron syndromes of adults. Muscle Nerve. 2010;41(12):161-165.
24. Shoesmith CL, Findlater K, Rowe A, Strong MJ. Progression of amyotrophic lateral sclerosis with respiratory onset. J Neurol Neurosurg Psychiatry. 2007;78(6):629-631.
25. Traynor BJ, Codd MB, Corr B, et al. Clinical features of amyotrophic lateral sclerosis according to the El Escorial and Airlie House diagnostic criteria: a population-based study. Arch Neurol. 2000;57(8):1171-1176.
26. Phukan J, Pender NP, Hardiman O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol. 2007;6(11):994-1003.
27. Chen R, Grand’Maison F, Strong MJ, et al. Motor neuron disease presenting as acute respiratory failure: a clinical and pathological study. J Neurol Neurosurg Psychiatry. 1996;60(4):455-458.
28. Gautier G, Verscheuren A, Monnier A, et al. ALS with respiratory onset: clinical features and effects of non-invasive ventilation on the prognosis. Amyotroph Lateral Scler. 2010;11(4):379-382.
29. Hasan A, Saxena AB, Ahmed SM, Swamy TLN. Amyotrophic lateral sclerosis presenting with orthopnea in a patient with COPD and obstructive sleep apnea. J Med All Sci. 2011;1(1):46-49.
30. Zoccolella S, Beghi E, Palagano G, et al. Analysis of survival and prognostic factors in amyotrophic lateral sclerosis: a population based study. J Neurol Neurosurg Psychiatry. 2008;79(1):33-37.
31. Brown RH Jr. Amyotrophic lateral sclerosis and other motor neuron diseases. In: Fauci AS, Braunwald E, Kasper DL, ed al, eds. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw Hill Medical; 2008:2572-2576.
32. Ince PG, Lowe J, Shaw PJ. Amyotrophic lateral sclerosis: current issues in classification, pathogenesis and molecular pathology. Neuropathol Appl Neurobiol. 1998;24(2):104-117.
33. Atsumi T, Miyatake T. Morphometry of the degenerative process in the hypoglossal nerves in amyotrophic lateral sclerosis. Acta Neuropathol. 1987;73(1):25-31.
34. Shimizu T, Komori T, Kato S, et al. Masseter inhibitory reflex in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001;2(4):189-195.
35. Rosen H. Dextromethorphan/quinidine sulfate (Zenvia) for pseudobulbar affect. Drugs Today (Barc). 2008;44(9):661-668.
36. Rippon GA, Scarmeas N, Gordon PH, et al. An observational study of cognitive impairment in amyotrophic lateral sclerosis. Arch Neurol. 2006;63(3):345-352.
37. Rabkin JG, Albert SM, Del Bene ML, et al. Prevalence of depressive disorders and change over time in late-stage ALS. Neurology. 2005;65(1):62-67.
38. Vignola A, Guzzo A, Calvo A, et al. Anxiety undermines quality of life in ALS patients and caregivers. Eur J Neurol. 2008;15(11):1231-1236.
39. Fang F, Valdimarsdóttir U, Fürst CJ, et al. Suicide among patients with amyotrophic lateral sclerosis. Brain. 2008;131(pt 10):2729-2733.
40. Verstraete E, Veldink JH, Hendrikse J, et al. Structural MRI reveals cortical thinning in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2012;83(4):383-388.
41. Turner MR, Modo M. Advances in the application of MRI to amyotrophic lateral sclerosis. Expert Opin Med Diagn. 2010;4(6):483-496.
42. Cirillo M, Esposito F, Tedeschi G, et al. Widespread microstructural white matter involvement in amyotrophic lateral sclerosis: a whole-brain DTI study. AJNR Am J Neuroradiol. 2012 Feb 2. [Epub ahead of print]
43. Canu E, Agosta F, Riva N, et al. The topography of brain microstructural damage in amyotrophic lateral sclerosis assessed using diffusion tensor MR imaging. AJNR Am J Neuroradiol. 2011;32(7):1307-1314.
44. Baek WS, Desai NP. ALS: pitfalls in the diagnosis. Pract Neurol. 2007;7(2):74-81.
45. ALS Association. Criteria for the diagnosis of ALS. www.alsa.org/als-care/resources/publica tions-videos/factsheets/criteria-for-diagnosis.html. Accessed May 31, 2012.
46. de Carvalho M, Dengler R, Eisen A, et al. Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol. 2008;119(3):497-503.
47. Brooks BR, Miller RG, Swash M, Munsat TL; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293-299.
48. Waragai M. MRI and clinical features of amyotrophic lateral sclerosis. Neuroradiology. 1997;39(12):847-851.
49. Thorpe JW, Moseley IF, Hawkes CH, et al. Brain and spinal cord MRI in motor neuron disease. J Neurol Neurosurg Psychiatry. 1996; 61(3):314-317.
50. Oba H, Araki T, Ohtomo K, et al. Amyotrophic lateral sclerosis: T2 shortening in motor cortex at MR imaging. Radiology. 1993;189 (3):843-846.
51. Piao YS, Wakabayashi K, Kakita A, et al. Neuropathology with clinical correlations of sporadic amyotrophic lateral sclerosis: 102 autopsy cases examined between 1962 and 2000. Brain Pathol. 2003;13(1):10-22.
52. Mateen FJ, Carone M, Sorenson EJ. Patients who survive 5 years or more with ALS in Olmsted County, 1925-2004. J Neurol Neurosurg Psychiatry. 2010;81(10):1144-1146.
53. del Aguila MA, Longstreth WT Jr, McGuire V, et al. Prognosis in amyotrophic lateral sclerosis: a population-based study. Neurology. 2003;60(5):813-819.
54. Matuz T, Birbaumer N, Hautzinger M, Kübler A. Coping with amyotrophic lateral sclerosis: an integrative view. J Neurol Neurosurg Psychiatry. 2010;81(8):893-898.
55. Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2012 Mar 14;3:CD001447.
56. Handy CR, Krudy C, Boulis N, Federici T. Pain in amyotrophic lateral sclerosis: a neglected aspect of disease. Neurol Res Int. 2011; 2011:403808. Epub 2011 May 3.
57. McClelland S 3rd, Bethoux FA, Boulis NM, et al. Intrathecal baclofen for spasticity-related pain in amyotrophic lateral sclerosis: efficacy and factors associated with pain relief. Muscle Nerve. 2008;37(3):396-398.
58. Andersen PM, Borasio GD, Dengler R, et al; EALSC Working Group. Good practice in the management of amyotrophic lateral sclerosis: clinical guidelines. Amyotroph Lateral Scler. 2007;8(4):195-213.
59. Rabkin JG, Gordon PH, McElhiney M, et al. Modafinil treatment of fatigue in patients with ALS: a placebo-controlled study. Muscle Nerve. 2009;39(3):297-303.
60. Kühnlein P, Gdynia HJ, Sperfeld AD, et al. Diagnosis and treatment of bulbar symptoms in amyotrophic lateral sclerosis. Nat Clin Pract Neurol. 2008;4(7):366-374.
61. Katzberg HD, Benatar M. Enteral tube feeding for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev. 2011 Jan 19;(1):CD004030.
62. Lechtzin N, Scott Y, Busse AM, et al. Early use of non-invasive ventilation prolongs survival in subjects with ALS. Amyotroph Lateral Scler. 2007;8(3):185-188.
63. Annane D, Orlikowski D, Chevret S, et al. Nocturnal mechanical ventilation for chronic hypoventilation in patients with neuromuscular and chest wall disorders. Cochrane Database Syst Rev. 2007 Oct 17;(4):CD001941.
64. Radunovic A, Annane D, Jewitt K, Mustfa N. Mechanical ventilation for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev. 2009 Oct 7;(4):CD004427.
65. Onders RP, Elmo MJ, Khansarinia S, et al. Complete worldwide operative experience in laparoscopic diaphragm pacing: results and differences in spinal cord injured patients and amyotrophic lateral sclerosis patients. Surg Endosc. 2009;23(7):1433-1440.
66. Onders RP, Carlin AM, Elmo M, et al. Amyotrophic lateral sclerosis: the Midwestern surgical experience with the diaphragm pacing stimulation system shows that general anesthesia can be safely performed. Am J Surg. 2009;197(3):386-390.
67. US Food and Drug Administration. Medical devices: NeuRx Diaphragm Pacing System™—H100006. www.fda.gov/MedicalDevices/Prod uctsandMedicalProcedures/DeviceApprovalsand Clearances/Recently-ApprovedDevices/ucm278684.htm. Accessed May 31, 2012.
68. Rabkin JG, Wagner GJ, Del Bene M. Resilience and distress among amyotrophic lateral sclerosis patients and caregivers. Psychosom Med. 2000; 62(2):271-279.
69. Chiò A, Gauthier A, Vignola A, et al. Caregiver time use in ALS. Neurology. 2006;67(5):902-904.
Amyotrophic lateral sclerosis (ALS) is probably best known to the majority of the US population as Lou Gehrig’s disease, named for the New York Yankee who died of ALS after a stellar 17-year baseball career in the 1920s and ’30s. Gradually, the general public is gaining familiarity with the characteristics of ALS, a disease with no single identifiable cause and no known cure.
About 5,600 people receive a diagnosis of ALS in the US each year (about 15 per day), making it the most common form of motor neuron disease.1 Most patients present with progressive muscle weakness in an extremity, which ultimately leads to respiratory failure and death.
Generally, patients with ALS have no cognitive disability and are aware of their physical decline. These patients are faced with important decisions as their condition worsens: Will they want nutritional support through a gastrostomy? For respiratory assistance, will their preference be noninvasive ventilation or mechanical ventilation via tracheostomy?
Not only do ALS patients undergo deterioration of motor function, but many experience muscle cramping, generalized pain, and depression. Only one medication is currently approved to treat ALS patients, with the benefit of prolonging life by a few months. In addition to symptom management, supportive measures, including physical, occupational, and speech therapy, exercise, nutrition, support groups, and counseling, are important tools to enhance quality of life for the patient with ALS.2
Primary care providers need to be aware of ALS, recognize its symptoms in their patients, and manage those affected on a case-by-case basis. Because of the challenges in diagnostic evaluation, the rapidly evolving nature of the disease, and the dire prognosis of ALS, any patient suspected of having ALS should be referred immediately to a neurologist. Providers need to educate patients and their caregivers regarding the disease process and ensure that patients receive appropriate care to meet their needs and preferences.
ALS DEFINED
ALS is a progressive neurodegenerative disease that affects both the upper and lower motor neurons. The disease is considered terminal. Although life may be prolonged by the one currently available pharmacologic agent, no treatment option is yet capable of stopping or reversing progression of the disease.3 While ALS was once believed to be a purely motor disorder, the accompanying degeneration of nonmotor brain regions, such as frontal and temporal cortical neurons, is considered by some to be part of the clinicopathologic spectrum of ALS.4
There are two forms of ALS: sporadic and familial. Sporadic ALS is by far the more common, accounting for 90% to 95% of cases. The remaining 5% to 10% of ALS cases are of the familial form, which can be autosomal-dominant or autosomal-recessive.5
The Degenerative Motor Neuron Diseases
Weakness and muscle wasting characterize several degenerative motor neuron diseases. In addition to ALS, these include primary lateral sclerosis, progressive muscular atrophy, progressive bulbar palsy, and pseudobulbar palsy.6
Primary lateral sclerosis involves upper motor neuron (UMN) dysfunction in the limbs.7Progressive muscular atrophy results from degeneration of the anterior horn cells in the spinal cord and is associated with lower motor neuron (LMN) deficits in the limbs.
Progressive bulbar palsy is a progressive UMN and LMN disorder of the cranial muscles. This condition may occasionally stay isolated in the bulbar segment, but more commonly, UMN and LMN signs and symptoms spread to involve other segments. This is then referred to as bulbar-onset ALS. There have been no reports of specific pathology in progressive bulbar palsy.
Pseudobulbar palsy results from an UMN lesion in the corticobulbar pathway in the pyramidal tract. It is characterized by difficulty chewing and swallowing, and slurred speech (manifestations that may also represent the initial presentation of ALS). Patients with pseudobulbar palsy may exhibit inappropriate, excessive yawning and emotional outbursts; these manifestations are referred to as emotional incontinence.8
EPIDEMIOLOGY
In North America, it is estimated that ALS affects 1.5 to 2.7 people per 100,000 between ages 20 and 80; most frequently, the disease presents between ages 55 and 65. ALS develops infrequently before age 30.3,9
While no gender difference is apparent in patients with familial ALS, sporadic ALS predominately affects males more than females (although the accepted ratio of 1.5:1 appears to be in decline9). After age 65, men and women are equally impacted.10
RISK FACTORS
Established risk factors for ALS include age and family history. Accumulating evidence suggests that military service and smoking may also contribute to the development of ALS.11-15 Children and siblings of ALS patients are at increased risk for ALS, while military personnel have 1.5 times the risk.11
Investigation of the precise link between military service and ALS is ongoing, but factors may include intense exertion, traumatic injuries, viral infections, and exposure to certain chemicals or metals.11 Research suggests the risk is independent of time period, years of service, or branch of service. Geographic location appears to be an independent factor, although there does seem to be a strong association between deployment during the 1991 Gulf War and the risk for ALS.12-14
Recently, smoking has been implicated as a potential risk factor in the disease.3,15 Certain environmental exposures have also been recognized as a possible risk factor. In Guam, an ALS-like syndrome has been identified among members of the Chamorro tribe. This syndrome has been linked to a neurotoxin in the seed of the cycad nut, a tropical plant endemic to the area, which was used in the 1950s and 1960s in the human food supply.3,16
ETIOLOGY
Familial ALS is a genetically transmitted degenerative disease. Twenty percent of cases involve the long arm of chromosome 21, which is responsible for coding of superoxide dismutase (SOD1). Mutations in SOD1, an RNA-processing protein called FUS, and the DNA-binding protein TDP-43, have all been identified in cases of familial ALS.17,18
The exact etiology of sporadic ALS remains unknown, but gene mutations have also been implicated, including the ANG gene21 and TDP-43.18 Other theories include increased levels of glutamate (which have been detected in cerebrospinal fluid and serum of patients with ALS), mitochondrial dysfunction, free radical injury, programmed cell death, neurofilament defects, viral infections, and autoimmune dysfunction19,20 (see Table 13,17,18,20,21). It is possible that a combination of factors is involved in the development of sporadic ALS.
PATHOPHYSIOLOGY
The pathophysiology of ALS involves degeneration of the UMN and LMN axons, which leads to glial scarring and possible impairment of the glial cells’ ability to store excess glutamate.22 ALS affects the central nervous system, specifically the anterior horn cells in the spinal cord and the cranial nerve nuclei (X, XI, XII) of the LMNs, and the corticospinal tract and corticobulbar pathway of the UMNs. Bulbar and limb muscles innervated by LMNs are subject to atrophy, whereas cognition, coordination, sensation, the oculomotor system, and sphincters are typically spared.3
CLINICAL FEATURES
In most patients, ALS symptoms characterize either limb onset or bulbar onset (see Table 23,7,23,24), with limb onset being the more common (about 75% vs about 25% of cases, respectively).1 Typically, patients with limb-onset ALS complain of rapidly progressive, asymmetric weakness in an extremity, followed by focal muscle atrophy with cramping and fasciculations, and eventually, spasticity. Weakness generally begins in one hand, arm, foot, or leg. Patients may notice increased episodes of tripping, clumsiness when they run or walk, a “dropped foot” gait, and/or a decline in manual dexterity.25
The weakness often develops insidiously; patients may notice that symptoms are exacerbated by cold weather.3 Eventually, the bulbar muscles are affected, resulting in dysphagia, dysarthria, and dysphasia. Occasionally, patients encounter bladder dysfunction (urgent micturition), sensory symptoms, and cognitive symptoms (eg, dementia, parkinsonism).26 Multisystem involvement is possible. Ultimately, respiratory compromise or other pulmonary complications ensue, representing a primary cause of mortality in ALS patients.3,24
In bulbar-onset ALS, patients first notice symptoms of dysphasia and dysphagia. They may complain of slurred speech, nasal or low-volume speech, and/or inhibited tongue mobility. The risk for aspiration is increased. The majority of patients with bulbar-onset ALS experience sialorrhea (excessive drooling) because they have difficulty swallowing their saliva. In most patients, mild UMN-type bilateral facial weakness affects the lower half of the face.3 As is the case with limb-onset ALS, bulbar-onset ALS progresses to respiratory compromise.27
In less than 3% of patients with ALS, presentation begins with respiratory weakness and no significant limb or bulbar symptoms.24,28 Patients with respiratory-onset ALS experience symptoms associated with nocturnal hypoventilation, including daytime hypersomnolence, morning headaches, impaired concentration, irritability, anorexia, mood changes, dyspnea, orthopnea, and disturbed sleep; or they may experience type 2 respiratory failure.28,29
Patients with axial symptoms of ALS present with neck weakness and may complain of posterior neck pain or strain with a gradually worsening tendency of the head to tip forward. These patients often support the chin with one hand. Those with axial truncal weakness often complain of difficulty maintaining erect posture when standing and of stooping as they walk. Some patients support the trunk by placing their hands in their front pants pockets or on their upper thighs. They may report some relief when pushing a grocery cart.7,23,24
Symptoms of ALS can be present for weeks or months before a patient consults with a health care provider. The average time span from onset of initial symptoms to diagnosis of ALS is about one year.1 Due to the unpredictable pattern of progression and variability of symptoms among patients, it is difficult to approximate a time frame for symptom progression; for some patients, the disease progresses slowly, while others deteriorate rapidly.30
PHYSICAL EXAMINATION FINDINGS
At onset, the typical presentation of ALS includes muscle weakness in one limb as well as visible fasciculations. As the disease progresses, focal wasting of muscle groups occurs in all four extremities. Particularly involved are the muscles of the hands, forearms, or shoulders in the upper limbs; and of the proximal thigh or distal foot muscles in the lower limbs.31 Deep tendon reflexes are symmetrically brisk. Spasticity, evident in the upper limbs, may present as increased tone.32
In patients with bulbar dysfunction, dysarthria may arise from either LMN pathology or pseudobulbar palsy caused by a UMN disorder, leading to slow, slurred speech or speech with a nasal quality. Tongue fasciculations will be present, as will atrophy and diminished mobility of the tongue.33 The gag reflex remains intact, even brisk, but weakness may occur in the muscles of the soft palate.3 Facial weakness is sometimes seen late in the disease, as evidenced by difficulty sealing the lips or puffing out the cheeks. The jaw jerk will be brisk, indicating that cranial nerve V is intact.34
A pseudobulbar affect, which is best described as emotional lability, may be present. The patient may have a history of exaggerated expression of emotion, such as uncontrollable crying, laughing, or both.35 Cognition, coordination, sensation, the oculomotor system, and sphincters are generally spared. However, cases of frontotemporal dementias coexisting with ALS have been reported; affected patients exhibit cognitive impairment, compulsive behaviors, and personality changes, and they may experience shorter survival.4,24,36
Evidence of known complications of ALS may also be noted during the physical examination; see Table 3.37-39
DIAGNOSIS OF ALS
In current research, use of structural MRI, magnetic resonance spectroscopy, and diffusion tensor imaging is being examined to detect thinning in the primary motor cortex, fractional anisotropy in the corpus callosum, patterns of gray and white matter atrophy, and other proposed diagnostic markers for ALS.40-43 However, no single specific diagnostic test has yet been proven to identify ALS; rather, it remains a disease of exclusion (see Table 43,44). For a confirmed diagnosis of ALS to be made, the patient must display:
- Evidence of LMN degeneration as found through clinical, neuropathologic, or electrophysiological examination
- UMN degeneration detected by clinical examination, and
- Progressive spread of signs or symptoms within a region or to other regions.3,45-47
Other disease processes that might explain the signs of UMN and/or LMN degeneration must be excluded, such as cervical spinal disease, myasthenia gravis, multifocal motor neuropathy, lead intoxication, and Lyme disease.45
Imaging studies are not required in ALS cases that have clinically definite disease with bulbar or pseudobulbar onset.47 Otherwise, the essential role of neuroimaging is to exclude a treatable structural lesion that may mimic ALS by producing UMN and LMN signs in varying degrees.48
Typically, MRI of the head and spine is ordered in patients with suspected ALS (see figure); MRI can reveal lesions in the corticospinal tracts that occur in ALS. The most characteristic finding on T2-weighted MRI is hyperintensity of the corticospinal tracts, which is visualized best in the brain and brainstem, and to a lesser extent in the spinal cord.49 Decreased signal intensity in the motor cortex has been reported on MRI in cases of ALS.50
Additional diagnostic procedures that are useful in excluding other disease processes include blood and cerebrospinal fluid (CSF) samples,3,44 four-limb electromyography (EMG),44 nerve conduction studies, motor unit number estimations, and muscle biopsies.47
- Autopsy results of patients with ALS demonstrate:
- Neuron loss, especially in lumbar and cervical enlargements
- Nonexistent or atrophic neurons in the motor nuclei of the pons and medulla, and in the anterior horn cells of the spinal cord
- Degeneration of the lateral columns of the spinal cord, and
- Atrophy of the ventral roots.31,51
PROGNOSIS
The overall five-year survival rate for patients with ALS has been reported between 7% and 14%,52,53 and the mortality rate rises in patients older than 75 and in those with bulbar signs.30 The rate of disease progression varies greatly among ALS patients and may be hastened with advancing age, female gender, presence of bulbar features, and absence of a significant other.37,53,54
The average life span for a patient with limb-onset ALS is two to five years from diagnosis, whereas patients affected by the bulbar form usually succumb within six to 18 months.30,53 In patients who present with respiratory symptoms, Shoesmith et al24 have reported a mean time of 14.9 months from initial symptoms to need for full-time ventilation, and of 27.0 months from symptom onset to death.
Advance directives, end-of-life care, and respiratory and nutritional management become essential issues during late stages of ALS; thus, they should be discussed with patients and their relatives at the time of diagnosis or shortly thereafter.
TREATMENT
Pharmacologic Options
There are currently no treatments to halt the progression of ALS or to reverse the disease process. Riluzole, currently the only FDA-approved drug for treatment of ALS, has been shown to slow the loss of muscle strength and to prolong life by an average of two to three months.55 Riluzole targets and blocks glutamate transporters on the presynaptic neuron, decreasing glutamate release and reducing excitotoxicity20 (ie, overstimulation of the postsynaptic receptors). These effects support the theory that ALS may result from excess glutamate and help to explain the increased levels of glutamate found in the serum and CSF of ALS patients.19
Riluzole is typically dosed at 50 mg by mouth twice a day, although 200-mg/d doses have also been examined in clinical trials.20,55 Generally, the drug is well tolerated, with common adverse effects including asthenia, nausea, gastrointestinal upset, and abnormal liver test results. Liver function should be monitored regularly during riluzole therapy, with elevations in serum alanine transferase of particular concern.55
Additional agents have been used to reduce muscle spasticity, muscle cramping and fasciculations, and the associated pain some patients experience, attributable in part to lack of activity and/or inflammation.56 For spasticity, tizanidine or baclofen (orally, a maximum of 20 mg in divided doses; or lower doses administered intrathecally, for patients who experience sedation and fatigue with high oral doses56,57) are often used. Carbamazepine and phenytoin are most commonly used to relieve muscle cramps.56 For some patients, NSAIDs may be adequate to control moderate to severe pain, but others may require opioids.2,56,58
Depressive disorders must be identified accurately, using appropriate clinical tools, before SSRIs (eg, citalopram) or other medications (eg, amitriptyline) are prescribed3; these agents should not be used presumptively,37 as estimates of prevalence of depression among ALS patients range from 2% to 75%.2,37,38,54 Estimates of anxiety prevalence in these patients range from 0% to 30%, reflecting the importance of accurate diagnosis before lorazepam or other agents are prescribed.3,38
Promising results have been reported in the use of modafinil to manage fatigue in patients with ALS.59 For patients with pseudobulbar affect, dextromethorphan 20 mg/quinidine sulfate 10 mg is an FDA-approved treatment.35
Vitamins and other supplements, including creatine, vitamin E, coenzyme Q10, and acetylcysteine, have not been shown to improve survival in this patient population.3
Nonpharmacologic Interventions
Supportive measures for ALS include physical and occupational therapy. In speech and language therapy, breathing and relaxation patterns can be used to correct ineffective compensatory behaviors and help patients “economize” their speaking efforts.60
Nutritional support is also important in patients who have difficulty swallowing, although this can be alleviated somewhat by changes in posture (eg, lowering the chin before attempting to swallow) and by use of thickened fluids; those with immobility of the tongue may find swallowing easier with the head tilted back.60 Once oral alimentation is no longer possible, enteral tube feeding is an option that may prolong survival.60,61
As ALS progresses, dysphagia may be aggravated as the ability to cough, reflexively or voluntarily, is reduced.60 As breathing becomes increasingly difficult, patients may require respiratory support. Noninvasive ventilation, using either continuous or bilevel positive airway pressure, may be implemented early in patients with respiratory-onset ALS, and later in the disease process for other patients, to prevent apnea and hypoventilation.28,62 Mechanical ventilation via tracheostomy is the most invasive method to address respiratory dysfunction in patients with ALS; however, like noninvasive ventilation, nocturnal mechanical ventilation has been shown to extend survival in these patients.28,63,64
Diaphragm pacing stimulation (DPS) has emerged as a possible alternative to mechanical ventilation for ALS patients. The pacing system consists of a battery-operated external pulse generator with electrodes placed after laparoscopic mapping on the diaphragm.65 Natural respiration is mimicked as stimulation from the external pulse generator prompts the diaphragm to contract. Researchers have shown that the minimally invasive surgery (including use of general anesthesia) required to install the DPS system can be safely performed on patients with ALS, and its use can delay the need for a ventilator by 24 months.65,66 Use of the DPS device was granted FDA approval in 2011, under the Humanitarian Device Exemption program.67
Addressing Quality of Life
For the patient with ALS, quality of life is an important consideration throughout disease management. According to numerous research teams, quality of life for these patients depends less on physical function and strength and more on social relationships, existential issues, and spirituality.2,37,54 A high level of quality of life can be sustained in patients with ALS, despite the decline they experience in physical function.
Addressing depression and anxiety by nonpharmacologic means may be needed. Loss of physical strength and mobility and difficulties with speech, swallowing, and breathing can challenge even the strongest patient’s coping skills; even more difficult can be the increased dependence on caregivers, the loss of income, and the financial burdens incurred in health care–related expenses. In some patients, the severity of disease, the lack of effective treatments, and the loss of independence may trigger thoughts of suicide or the wish to “hasten death.”37,39 The importance of counseling, support groups, spirituality or religion, and palliative care, from early in the disease process, cannot be overstated.3,37
Some of these considerations may also be of benefit to spouses and other nonpaid caregivers of the patient with ALS, at least half of whom report feeling physically or psychologically unwell.37,68 Even when professional nursing services or hospice support are available, caregivers often devote 12 hours or more per day to nonprofessional patient care.69 Strategies that support the caregiver can reduce the patient’s perception of burden in that individual.37
CONCLUSION
The exact cause or causes of ALS remain unknown, making it difficult to predict who will present with a disease that appears impossible to prevent. Health care providers in any practice should be aware of the signs of ALS and familiar with its symptoms in order to provide optimal management for potentially affected patients.
Any cause for suspicion of ALS (ie, recent-onset limb weakness or atrophy; difficulty swallowing or speaking) warrants immediate patient referral to a neurologist. Evaluation should include a comprehensive history and physical examination, with emphasis on the musculoskeletal and neurologic exams; MRI of the spine and head, analysis of blood and CSF samples, EMG, and nerve conduction studies should be used to rule out treatable causes of limb weakness.
Potential complications of ALS, including nutritional deficiency, respiratory compromise, and depression, should be discussed early with patients and their caregivers. Management of the patient with ALS necessitates a multidisciplinary approach involving providers from several specialties to ensure that the many issues associated with this disease are being addressed. The priority of the health care provider should be to extend the patient’s survival while maintaining quality of life.
Amyotrophic lateral sclerosis (ALS) is probably best known to the majority of the US population as Lou Gehrig’s disease, named for the New York Yankee who died of ALS after a stellar 17-year baseball career in the 1920s and ’30s. Gradually, the general public is gaining familiarity with the characteristics of ALS, a disease with no single identifiable cause and no known cure.
About 5,600 people receive a diagnosis of ALS in the US each year (about 15 per day), making it the most common form of motor neuron disease.1 Most patients present with progressive muscle weakness in an extremity, which ultimately leads to respiratory failure and death.
Generally, patients with ALS have no cognitive disability and are aware of their physical decline. These patients are faced with important decisions as their condition worsens: Will they want nutritional support through a gastrostomy? For respiratory assistance, will their preference be noninvasive ventilation or mechanical ventilation via tracheostomy?
Not only do ALS patients undergo deterioration of motor function, but many experience muscle cramping, generalized pain, and depression. Only one medication is currently approved to treat ALS patients, with the benefit of prolonging life by a few months. In addition to symptom management, supportive measures, including physical, occupational, and speech therapy, exercise, nutrition, support groups, and counseling, are important tools to enhance quality of life for the patient with ALS.2
Primary care providers need to be aware of ALS, recognize its symptoms in their patients, and manage those affected on a case-by-case basis. Because of the challenges in diagnostic evaluation, the rapidly evolving nature of the disease, and the dire prognosis of ALS, any patient suspected of having ALS should be referred immediately to a neurologist. Providers need to educate patients and their caregivers regarding the disease process and ensure that patients receive appropriate care to meet their needs and preferences.
ALS DEFINED
ALS is a progressive neurodegenerative disease that affects both the upper and lower motor neurons. The disease is considered terminal. Although life may be prolonged by the one currently available pharmacologic agent, no treatment option is yet capable of stopping or reversing progression of the disease.3 While ALS was once believed to be a purely motor disorder, the accompanying degeneration of nonmotor brain regions, such as frontal and temporal cortical neurons, is considered by some to be part of the clinicopathologic spectrum of ALS.4
There are two forms of ALS: sporadic and familial. Sporadic ALS is by far the more common, accounting for 90% to 95% of cases. The remaining 5% to 10% of ALS cases are of the familial form, which can be autosomal-dominant or autosomal-recessive.5
The Degenerative Motor Neuron Diseases
Weakness and muscle wasting characterize several degenerative motor neuron diseases. In addition to ALS, these include primary lateral sclerosis, progressive muscular atrophy, progressive bulbar palsy, and pseudobulbar palsy.6
Primary lateral sclerosis involves upper motor neuron (UMN) dysfunction in the limbs.7Progressive muscular atrophy results from degeneration of the anterior horn cells in the spinal cord and is associated with lower motor neuron (LMN) deficits in the limbs.
Progressive bulbar palsy is a progressive UMN and LMN disorder of the cranial muscles. This condition may occasionally stay isolated in the bulbar segment, but more commonly, UMN and LMN signs and symptoms spread to involve other segments. This is then referred to as bulbar-onset ALS. There have been no reports of specific pathology in progressive bulbar palsy.
Pseudobulbar palsy results from an UMN lesion in the corticobulbar pathway in the pyramidal tract. It is characterized by difficulty chewing and swallowing, and slurred speech (manifestations that may also represent the initial presentation of ALS). Patients with pseudobulbar palsy may exhibit inappropriate, excessive yawning and emotional outbursts; these manifestations are referred to as emotional incontinence.8
EPIDEMIOLOGY
In North America, it is estimated that ALS affects 1.5 to 2.7 people per 100,000 between ages 20 and 80; most frequently, the disease presents between ages 55 and 65. ALS develops infrequently before age 30.3,9
While no gender difference is apparent in patients with familial ALS, sporadic ALS predominately affects males more than females (although the accepted ratio of 1.5:1 appears to be in decline9). After age 65, men and women are equally impacted.10
RISK FACTORS
Established risk factors for ALS include age and family history. Accumulating evidence suggests that military service and smoking may also contribute to the development of ALS.11-15 Children and siblings of ALS patients are at increased risk for ALS, while military personnel have 1.5 times the risk.11
Investigation of the precise link between military service and ALS is ongoing, but factors may include intense exertion, traumatic injuries, viral infections, and exposure to certain chemicals or metals.11 Research suggests the risk is independent of time period, years of service, or branch of service. Geographic location appears to be an independent factor, although there does seem to be a strong association between deployment during the 1991 Gulf War and the risk for ALS.12-14
Recently, smoking has been implicated as a potential risk factor in the disease.3,15 Certain environmental exposures have also been recognized as a possible risk factor. In Guam, an ALS-like syndrome has been identified among members of the Chamorro tribe. This syndrome has been linked to a neurotoxin in the seed of the cycad nut, a tropical plant endemic to the area, which was used in the 1950s and 1960s in the human food supply.3,16
ETIOLOGY
Familial ALS is a genetically transmitted degenerative disease. Twenty percent of cases involve the long arm of chromosome 21, which is responsible for coding of superoxide dismutase (SOD1). Mutations in SOD1, an RNA-processing protein called FUS, and the DNA-binding protein TDP-43, have all been identified in cases of familial ALS.17,18
The exact etiology of sporadic ALS remains unknown, but gene mutations have also been implicated, including the ANG gene21 and TDP-43.18 Other theories include increased levels of glutamate (which have been detected in cerebrospinal fluid and serum of patients with ALS), mitochondrial dysfunction, free radical injury, programmed cell death, neurofilament defects, viral infections, and autoimmune dysfunction19,20 (see Table 13,17,18,20,21). It is possible that a combination of factors is involved in the development of sporadic ALS.
PATHOPHYSIOLOGY
The pathophysiology of ALS involves degeneration of the UMN and LMN axons, which leads to glial scarring and possible impairment of the glial cells’ ability to store excess glutamate.22 ALS affects the central nervous system, specifically the anterior horn cells in the spinal cord and the cranial nerve nuclei (X, XI, XII) of the LMNs, and the corticospinal tract and corticobulbar pathway of the UMNs. Bulbar and limb muscles innervated by LMNs are subject to atrophy, whereas cognition, coordination, sensation, the oculomotor system, and sphincters are typically spared.3
CLINICAL FEATURES
In most patients, ALS symptoms characterize either limb onset or bulbar onset (see Table 23,7,23,24), with limb onset being the more common (about 75% vs about 25% of cases, respectively).1 Typically, patients with limb-onset ALS complain of rapidly progressive, asymmetric weakness in an extremity, followed by focal muscle atrophy with cramping and fasciculations, and eventually, spasticity. Weakness generally begins in one hand, arm, foot, or leg. Patients may notice increased episodes of tripping, clumsiness when they run or walk, a “dropped foot” gait, and/or a decline in manual dexterity.25
The weakness often develops insidiously; patients may notice that symptoms are exacerbated by cold weather.3 Eventually, the bulbar muscles are affected, resulting in dysphagia, dysarthria, and dysphasia. Occasionally, patients encounter bladder dysfunction (urgent micturition), sensory symptoms, and cognitive symptoms (eg, dementia, parkinsonism).26 Multisystem involvement is possible. Ultimately, respiratory compromise or other pulmonary complications ensue, representing a primary cause of mortality in ALS patients.3,24
In bulbar-onset ALS, patients first notice symptoms of dysphasia and dysphagia. They may complain of slurred speech, nasal or low-volume speech, and/or inhibited tongue mobility. The risk for aspiration is increased. The majority of patients with bulbar-onset ALS experience sialorrhea (excessive drooling) because they have difficulty swallowing their saliva. In most patients, mild UMN-type bilateral facial weakness affects the lower half of the face.3 As is the case with limb-onset ALS, bulbar-onset ALS progresses to respiratory compromise.27
In less than 3% of patients with ALS, presentation begins with respiratory weakness and no significant limb or bulbar symptoms.24,28 Patients with respiratory-onset ALS experience symptoms associated with nocturnal hypoventilation, including daytime hypersomnolence, morning headaches, impaired concentration, irritability, anorexia, mood changes, dyspnea, orthopnea, and disturbed sleep; or they may experience type 2 respiratory failure.28,29
Patients with axial symptoms of ALS present with neck weakness and may complain of posterior neck pain or strain with a gradually worsening tendency of the head to tip forward. These patients often support the chin with one hand. Those with axial truncal weakness often complain of difficulty maintaining erect posture when standing and of stooping as they walk. Some patients support the trunk by placing their hands in their front pants pockets or on their upper thighs. They may report some relief when pushing a grocery cart.7,23,24
Symptoms of ALS can be present for weeks or months before a patient consults with a health care provider. The average time span from onset of initial symptoms to diagnosis of ALS is about one year.1 Due to the unpredictable pattern of progression and variability of symptoms among patients, it is difficult to approximate a time frame for symptom progression; for some patients, the disease progresses slowly, while others deteriorate rapidly.30
PHYSICAL EXAMINATION FINDINGS
At onset, the typical presentation of ALS includes muscle weakness in one limb as well as visible fasciculations. As the disease progresses, focal wasting of muscle groups occurs in all four extremities. Particularly involved are the muscles of the hands, forearms, or shoulders in the upper limbs; and of the proximal thigh or distal foot muscles in the lower limbs.31 Deep tendon reflexes are symmetrically brisk. Spasticity, evident in the upper limbs, may present as increased tone.32
In patients with bulbar dysfunction, dysarthria may arise from either LMN pathology or pseudobulbar palsy caused by a UMN disorder, leading to slow, slurred speech or speech with a nasal quality. Tongue fasciculations will be present, as will atrophy and diminished mobility of the tongue.33 The gag reflex remains intact, even brisk, but weakness may occur in the muscles of the soft palate.3 Facial weakness is sometimes seen late in the disease, as evidenced by difficulty sealing the lips or puffing out the cheeks. The jaw jerk will be brisk, indicating that cranial nerve V is intact.34
A pseudobulbar affect, which is best described as emotional lability, may be present. The patient may have a history of exaggerated expression of emotion, such as uncontrollable crying, laughing, or both.35 Cognition, coordination, sensation, the oculomotor system, and sphincters are generally spared. However, cases of frontotemporal dementias coexisting with ALS have been reported; affected patients exhibit cognitive impairment, compulsive behaviors, and personality changes, and they may experience shorter survival.4,24,36
Evidence of known complications of ALS may also be noted during the physical examination; see Table 3.37-39
DIAGNOSIS OF ALS
In current research, use of structural MRI, magnetic resonance spectroscopy, and diffusion tensor imaging is being examined to detect thinning in the primary motor cortex, fractional anisotropy in the corpus callosum, patterns of gray and white matter atrophy, and other proposed diagnostic markers for ALS.40-43 However, no single specific diagnostic test has yet been proven to identify ALS; rather, it remains a disease of exclusion (see Table 43,44). For a confirmed diagnosis of ALS to be made, the patient must display:
- Evidence of LMN degeneration as found through clinical, neuropathologic, or electrophysiological examination
- UMN degeneration detected by clinical examination, and
- Progressive spread of signs or symptoms within a region or to other regions.3,45-47
Other disease processes that might explain the signs of UMN and/or LMN degeneration must be excluded, such as cervical spinal disease, myasthenia gravis, multifocal motor neuropathy, lead intoxication, and Lyme disease.45
Imaging studies are not required in ALS cases that have clinically definite disease with bulbar or pseudobulbar onset.47 Otherwise, the essential role of neuroimaging is to exclude a treatable structural lesion that may mimic ALS by producing UMN and LMN signs in varying degrees.48
Typically, MRI of the head and spine is ordered in patients with suspected ALS (see figure); MRI can reveal lesions in the corticospinal tracts that occur in ALS. The most characteristic finding on T2-weighted MRI is hyperintensity of the corticospinal tracts, which is visualized best in the brain and brainstem, and to a lesser extent in the spinal cord.49 Decreased signal intensity in the motor cortex has been reported on MRI in cases of ALS.50
Additional diagnostic procedures that are useful in excluding other disease processes include blood and cerebrospinal fluid (CSF) samples,3,44 four-limb electromyography (EMG),44 nerve conduction studies, motor unit number estimations, and muscle biopsies.47
- Autopsy results of patients with ALS demonstrate:
- Neuron loss, especially in lumbar and cervical enlargements
- Nonexistent or atrophic neurons in the motor nuclei of the pons and medulla, and in the anterior horn cells of the spinal cord
- Degeneration of the lateral columns of the spinal cord, and
- Atrophy of the ventral roots.31,51
PROGNOSIS
The overall five-year survival rate for patients with ALS has been reported between 7% and 14%,52,53 and the mortality rate rises in patients older than 75 and in those with bulbar signs.30 The rate of disease progression varies greatly among ALS patients and may be hastened with advancing age, female gender, presence of bulbar features, and absence of a significant other.37,53,54
The average life span for a patient with limb-onset ALS is two to five years from diagnosis, whereas patients affected by the bulbar form usually succumb within six to 18 months.30,53 In patients who present with respiratory symptoms, Shoesmith et al24 have reported a mean time of 14.9 months from initial symptoms to need for full-time ventilation, and of 27.0 months from symptom onset to death.
Advance directives, end-of-life care, and respiratory and nutritional management become essential issues during late stages of ALS; thus, they should be discussed with patients and their relatives at the time of diagnosis or shortly thereafter.
TREATMENT
Pharmacologic Options
There are currently no treatments to halt the progression of ALS or to reverse the disease process. Riluzole, currently the only FDA-approved drug for treatment of ALS, has been shown to slow the loss of muscle strength and to prolong life by an average of two to three months.55 Riluzole targets and blocks glutamate transporters on the presynaptic neuron, decreasing glutamate release and reducing excitotoxicity20 (ie, overstimulation of the postsynaptic receptors). These effects support the theory that ALS may result from excess glutamate and help to explain the increased levels of glutamate found in the serum and CSF of ALS patients.19
Riluzole is typically dosed at 50 mg by mouth twice a day, although 200-mg/d doses have also been examined in clinical trials.20,55 Generally, the drug is well tolerated, with common adverse effects including asthenia, nausea, gastrointestinal upset, and abnormal liver test results. Liver function should be monitored regularly during riluzole therapy, with elevations in serum alanine transferase of particular concern.55
Additional agents have been used to reduce muscle spasticity, muscle cramping and fasciculations, and the associated pain some patients experience, attributable in part to lack of activity and/or inflammation.56 For spasticity, tizanidine or baclofen (orally, a maximum of 20 mg in divided doses; or lower doses administered intrathecally, for patients who experience sedation and fatigue with high oral doses56,57) are often used. Carbamazepine and phenytoin are most commonly used to relieve muscle cramps.56 For some patients, NSAIDs may be adequate to control moderate to severe pain, but others may require opioids.2,56,58
Depressive disorders must be identified accurately, using appropriate clinical tools, before SSRIs (eg, citalopram) or other medications (eg, amitriptyline) are prescribed3; these agents should not be used presumptively,37 as estimates of prevalence of depression among ALS patients range from 2% to 75%.2,37,38,54 Estimates of anxiety prevalence in these patients range from 0% to 30%, reflecting the importance of accurate diagnosis before lorazepam or other agents are prescribed.3,38
Promising results have been reported in the use of modafinil to manage fatigue in patients with ALS.59 For patients with pseudobulbar affect, dextromethorphan 20 mg/quinidine sulfate 10 mg is an FDA-approved treatment.35
Vitamins and other supplements, including creatine, vitamin E, coenzyme Q10, and acetylcysteine, have not been shown to improve survival in this patient population.3
Nonpharmacologic Interventions
Supportive measures for ALS include physical and occupational therapy. In speech and language therapy, breathing and relaxation patterns can be used to correct ineffective compensatory behaviors and help patients “economize” their speaking efforts.60
Nutritional support is also important in patients who have difficulty swallowing, although this can be alleviated somewhat by changes in posture (eg, lowering the chin before attempting to swallow) and by use of thickened fluids; those with immobility of the tongue may find swallowing easier with the head tilted back.60 Once oral alimentation is no longer possible, enteral tube feeding is an option that may prolong survival.60,61
As ALS progresses, dysphagia may be aggravated as the ability to cough, reflexively or voluntarily, is reduced.60 As breathing becomes increasingly difficult, patients may require respiratory support. Noninvasive ventilation, using either continuous or bilevel positive airway pressure, may be implemented early in patients with respiratory-onset ALS, and later in the disease process for other patients, to prevent apnea and hypoventilation.28,62 Mechanical ventilation via tracheostomy is the most invasive method to address respiratory dysfunction in patients with ALS; however, like noninvasive ventilation, nocturnal mechanical ventilation has been shown to extend survival in these patients.28,63,64
Diaphragm pacing stimulation (DPS) has emerged as a possible alternative to mechanical ventilation for ALS patients. The pacing system consists of a battery-operated external pulse generator with electrodes placed after laparoscopic mapping on the diaphragm.65 Natural respiration is mimicked as stimulation from the external pulse generator prompts the diaphragm to contract. Researchers have shown that the minimally invasive surgery (including use of general anesthesia) required to install the DPS system can be safely performed on patients with ALS, and its use can delay the need for a ventilator by 24 months.65,66 Use of the DPS device was granted FDA approval in 2011, under the Humanitarian Device Exemption program.67
Addressing Quality of Life
For the patient with ALS, quality of life is an important consideration throughout disease management. According to numerous research teams, quality of life for these patients depends less on physical function and strength and more on social relationships, existential issues, and spirituality.2,37,54 A high level of quality of life can be sustained in patients with ALS, despite the decline they experience in physical function.
Addressing depression and anxiety by nonpharmacologic means may be needed. Loss of physical strength and mobility and difficulties with speech, swallowing, and breathing can challenge even the strongest patient’s coping skills; even more difficult can be the increased dependence on caregivers, the loss of income, and the financial burdens incurred in health care–related expenses. In some patients, the severity of disease, the lack of effective treatments, and the loss of independence may trigger thoughts of suicide or the wish to “hasten death.”37,39 The importance of counseling, support groups, spirituality or religion, and palliative care, from early in the disease process, cannot be overstated.3,37
Some of these considerations may also be of benefit to spouses and other nonpaid caregivers of the patient with ALS, at least half of whom report feeling physically or psychologically unwell.37,68 Even when professional nursing services or hospice support are available, caregivers often devote 12 hours or more per day to nonprofessional patient care.69 Strategies that support the caregiver can reduce the patient’s perception of burden in that individual.37
CONCLUSION
The exact cause or causes of ALS remain unknown, making it difficult to predict who will present with a disease that appears impossible to prevent. Health care providers in any practice should be aware of the signs of ALS and familiar with its symptoms in order to provide optimal management for potentially affected patients.
Any cause for suspicion of ALS (ie, recent-onset limb weakness or atrophy; difficulty swallowing or speaking) warrants immediate patient referral to a neurologist. Evaluation should include a comprehensive history and physical examination, with emphasis on the musculoskeletal and neurologic exams; MRI of the spine and head, analysis of blood and CSF samples, EMG, and nerve conduction studies should be used to rule out treatable causes of limb weakness.
Potential complications of ALS, including nutritional deficiency, respiratory compromise, and depression, should be discussed early with patients and their caregivers. Management of the patient with ALS necessitates a multidisciplinary approach involving providers from several specialties to ensure that the many issues associated with this disease are being addressed. The priority of the health care provider should be to extend the patient’s survival while maintaining quality of life.
1. ALS Association. Facts you should know. www.alsa.org/about-als/facts-you-should-know.html. Accessed May 30, 2012.
2. Simmons Z. Management strategies for patients with amyotrophic lateral sclerosis from diagnosis through death. Neurologist. 2005;11(5):257-270.
3. Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet J Rare Dis. 2009;4:3.
4. Yoshida M. Amyotrophic lateral sclerosis with dementia: the clinicopathological spectrum. Neuropathology. 2004;24(1):87-102.
5. Byrne S, Walsh C, Lynch C, et al. Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2011;82(6):623-627.
6. National Institute of Neurological Disorders and Stroke, NIH. Motor neuron diseases fact sheet. www.ninds.nih.gov/disorders/motor_neuron_diseases/detail_motor_neuron_diseases.htm. Accessed May 30, 2012.
7. Tartaglia MC, Rowe A, Findlater K. Differentiation between primary lateral sclerosis and amyotrophic lateral sclerosis: examination of symptoms and signs at disease onset and during follow-up. Arch Neurol. 2007;64(2):232-236.
8. Strowd RE, Cartwright MS, Okun MS, et al. Pseudobulbar affect: prevalence and quality of life impact in movement disorders. J Neurol. 2010;257(8):1382-1387.
9. Beghi E, Logroscino G, Chiò A, et al; EURALS Consortium. The epidemiology of ALS and the role of population-based registries. Biochim Biophys Acta. 2006;1762(11-12):1150-1157.
10. McCombe PA, Henderson RD. Effects of gender in amyotrophic lateral sclerosis. Gend Med. 2010;7(6):557-570.
11. Weisskopf MG, Morozova N, O’Reilly EJ, et al. Prospective study of chemical exposures and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2009;80(5):558-561.
12. ALS Association. ALS in the military: unexpected consequences of military service (2011). www.alsa.org/assets/pdfs/advocacy/als_military_paper.pdf. Accessed May 22, 2012.
13. Horner RD, Kamins KG, Feussner JR, et al. Occurrence of amyotrophic lateral sclerosis among Gulf War veterans. Neurology. 2003;61(6):742-749.
14. Horner RD, Grambow SC, Coffman DJ, et al. Amyotrophic lateral sclerosis among 1991 Gulf War veterans: evidence for a time-limited outbreak. Neuroepidemiology. 2008;31(1):28-32.
15. Wang H, O’Reilly EJ, Weisskopf MG, et al. Smoking and risk of amyotrophic lateral sclerosis: a pooled analysis of 5 prospective cohorts. Arch Neurol. 2011;68(2):207-213.
16. Steele JC, McGeer PL. The ALS/PDC syndrome of Guam and the cycad hypothesis. Neurology. 2008;70(21):1984-1990.
17. Vance C, Rogelj B, Hortobágyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208-1211.
18. Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):1668-1672.
19. Pioro EP, Majors AAW, Mitsumoto H, et al. 1H-MRS evidence of neurodegeneration and excess glutamate + glutamine in ALS medulla. Neurology. 1999;53(1):71-79.
20. Cheah BC, Vucic S, Krishnan AV, Kiernan MC. Riluzole, neuroprotection and amyotrophic lateral sclerosis. Curr Med Chem. 2010;17(18):1942-1959.
21. Paubel A, Violette J, Amy M, et al. Mutations of the ANG gene in French patients with sporadic amyotrophic lateral sclerosis. Arch Neurol. 2008;65(10):1333-1336.
22. Neusch C, Bähr M, Schneider-Gold C. Glia cells in amyotrophic lateral sclerosis: new clues to understanding an old disease? Muscle Nerve. 2007;35(6):712-724.
23. Rowland LP. Progressive muscular atrophy and other lower motor neuron syndromes of adults. Muscle Nerve. 2010;41(12):161-165.
24. Shoesmith CL, Findlater K, Rowe A, Strong MJ. Progression of amyotrophic lateral sclerosis with respiratory onset. J Neurol Neurosurg Psychiatry. 2007;78(6):629-631.
25. Traynor BJ, Codd MB, Corr B, et al. Clinical features of amyotrophic lateral sclerosis according to the El Escorial and Airlie House diagnostic criteria: a population-based study. Arch Neurol. 2000;57(8):1171-1176.
26. Phukan J, Pender NP, Hardiman O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol. 2007;6(11):994-1003.
27. Chen R, Grand’Maison F, Strong MJ, et al. Motor neuron disease presenting as acute respiratory failure: a clinical and pathological study. J Neurol Neurosurg Psychiatry. 1996;60(4):455-458.
28. Gautier G, Verscheuren A, Monnier A, et al. ALS with respiratory onset: clinical features and effects of non-invasive ventilation on the prognosis. Amyotroph Lateral Scler. 2010;11(4):379-382.
29. Hasan A, Saxena AB, Ahmed SM, Swamy TLN. Amyotrophic lateral sclerosis presenting with orthopnea in a patient with COPD and obstructive sleep apnea. J Med All Sci. 2011;1(1):46-49.
30. Zoccolella S, Beghi E, Palagano G, et al. Analysis of survival and prognostic factors in amyotrophic lateral sclerosis: a population based study. J Neurol Neurosurg Psychiatry. 2008;79(1):33-37.
31. Brown RH Jr. Amyotrophic lateral sclerosis and other motor neuron diseases. In: Fauci AS, Braunwald E, Kasper DL, ed al, eds. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw Hill Medical; 2008:2572-2576.
32. Ince PG, Lowe J, Shaw PJ. Amyotrophic lateral sclerosis: current issues in classification, pathogenesis and molecular pathology. Neuropathol Appl Neurobiol. 1998;24(2):104-117.
33. Atsumi T, Miyatake T. Morphometry of the degenerative process in the hypoglossal nerves in amyotrophic lateral sclerosis. Acta Neuropathol. 1987;73(1):25-31.
34. Shimizu T, Komori T, Kato S, et al. Masseter inhibitory reflex in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001;2(4):189-195.
35. Rosen H. Dextromethorphan/quinidine sulfate (Zenvia) for pseudobulbar affect. Drugs Today (Barc). 2008;44(9):661-668.
36. Rippon GA, Scarmeas N, Gordon PH, et al. An observational study of cognitive impairment in amyotrophic lateral sclerosis. Arch Neurol. 2006;63(3):345-352.
37. Rabkin JG, Albert SM, Del Bene ML, et al. Prevalence of depressive disorders and change over time in late-stage ALS. Neurology. 2005;65(1):62-67.
38. Vignola A, Guzzo A, Calvo A, et al. Anxiety undermines quality of life in ALS patients and caregivers. Eur J Neurol. 2008;15(11):1231-1236.
39. Fang F, Valdimarsdóttir U, Fürst CJ, et al. Suicide among patients with amyotrophic lateral sclerosis. Brain. 2008;131(pt 10):2729-2733.
40. Verstraete E, Veldink JH, Hendrikse J, et al. Structural MRI reveals cortical thinning in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2012;83(4):383-388.
41. Turner MR, Modo M. Advances in the application of MRI to amyotrophic lateral sclerosis. Expert Opin Med Diagn. 2010;4(6):483-496.
42. Cirillo M, Esposito F, Tedeschi G, et al. Widespread microstructural white matter involvement in amyotrophic lateral sclerosis: a whole-brain DTI study. AJNR Am J Neuroradiol. 2012 Feb 2. [Epub ahead of print]
43. Canu E, Agosta F, Riva N, et al. The topography of brain microstructural damage in amyotrophic lateral sclerosis assessed using diffusion tensor MR imaging. AJNR Am J Neuroradiol. 2011;32(7):1307-1314.
44. Baek WS, Desai NP. ALS: pitfalls in the diagnosis. Pract Neurol. 2007;7(2):74-81.
45. ALS Association. Criteria for the diagnosis of ALS. www.alsa.org/als-care/resources/publica tions-videos/factsheets/criteria-for-diagnosis.html. Accessed May 31, 2012.
46. de Carvalho M, Dengler R, Eisen A, et al. Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol. 2008;119(3):497-503.
47. Brooks BR, Miller RG, Swash M, Munsat TL; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293-299.
48. Waragai M. MRI and clinical features of amyotrophic lateral sclerosis. Neuroradiology. 1997;39(12):847-851.
49. Thorpe JW, Moseley IF, Hawkes CH, et al. Brain and spinal cord MRI in motor neuron disease. J Neurol Neurosurg Psychiatry. 1996; 61(3):314-317.
50. Oba H, Araki T, Ohtomo K, et al. Amyotrophic lateral sclerosis: T2 shortening in motor cortex at MR imaging. Radiology. 1993;189 (3):843-846.
51. Piao YS, Wakabayashi K, Kakita A, et al. Neuropathology with clinical correlations of sporadic amyotrophic lateral sclerosis: 102 autopsy cases examined between 1962 and 2000. Brain Pathol. 2003;13(1):10-22.
52. Mateen FJ, Carone M, Sorenson EJ. Patients who survive 5 years or more with ALS in Olmsted County, 1925-2004. J Neurol Neurosurg Psychiatry. 2010;81(10):1144-1146.
53. del Aguila MA, Longstreth WT Jr, McGuire V, et al. Prognosis in amyotrophic lateral sclerosis: a population-based study. Neurology. 2003;60(5):813-819.
54. Matuz T, Birbaumer N, Hautzinger M, Kübler A. Coping with amyotrophic lateral sclerosis: an integrative view. J Neurol Neurosurg Psychiatry. 2010;81(8):893-898.
55. Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2012 Mar 14;3:CD001447.
56. Handy CR, Krudy C, Boulis N, Federici T. Pain in amyotrophic lateral sclerosis: a neglected aspect of disease. Neurol Res Int. 2011; 2011:403808. Epub 2011 May 3.
57. McClelland S 3rd, Bethoux FA, Boulis NM, et al. Intrathecal baclofen for spasticity-related pain in amyotrophic lateral sclerosis: efficacy and factors associated with pain relief. Muscle Nerve. 2008;37(3):396-398.
58. Andersen PM, Borasio GD, Dengler R, et al; EALSC Working Group. Good practice in the management of amyotrophic lateral sclerosis: clinical guidelines. Amyotroph Lateral Scler. 2007;8(4):195-213.
59. Rabkin JG, Gordon PH, McElhiney M, et al. Modafinil treatment of fatigue in patients with ALS: a placebo-controlled study. Muscle Nerve. 2009;39(3):297-303.
60. Kühnlein P, Gdynia HJ, Sperfeld AD, et al. Diagnosis and treatment of bulbar symptoms in amyotrophic lateral sclerosis. Nat Clin Pract Neurol. 2008;4(7):366-374.
61. Katzberg HD, Benatar M. Enteral tube feeding for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev. 2011 Jan 19;(1):CD004030.
62. Lechtzin N, Scott Y, Busse AM, et al. Early use of non-invasive ventilation prolongs survival in subjects with ALS. Amyotroph Lateral Scler. 2007;8(3):185-188.
63. Annane D, Orlikowski D, Chevret S, et al. Nocturnal mechanical ventilation for chronic hypoventilation in patients with neuromuscular and chest wall disorders. Cochrane Database Syst Rev. 2007 Oct 17;(4):CD001941.
64. Radunovic A, Annane D, Jewitt K, Mustfa N. Mechanical ventilation for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev. 2009 Oct 7;(4):CD004427.
65. Onders RP, Elmo MJ, Khansarinia S, et al. Complete worldwide operative experience in laparoscopic diaphragm pacing: results and differences in spinal cord injured patients and amyotrophic lateral sclerosis patients. Surg Endosc. 2009;23(7):1433-1440.
66. Onders RP, Carlin AM, Elmo M, et al. Amyotrophic lateral sclerosis: the Midwestern surgical experience with the diaphragm pacing stimulation system shows that general anesthesia can be safely performed. Am J Surg. 2009;197(3):386-390.
67. US Food and Drug Administration. Medical devices: NeuRx Diaphragm Pacing System™—H100006. www.fda.gov/MedicalDevices/Prod uctsandMedicalProcedures/DeviceApprovalsand Clearances/Recently-ApprovedDevices/ucm278684.htm. Accessed May 31, 2012.
68. Rabkin JG, Wagner GJ, Del Bene M. Resilience and distress among amyotrophic lateral sclerosis patients and caregivers. Psychosom Med. 2000; 62(2):271-279.
69. Chiò A, Gauthier A, Vignola A, et al. Caregiver time use in ALS. Neurology. 2006;67(5):902-904.
1. ALS Association. Facts you should know. www.alsa.org/about-als/facts-you-should-know.html. Accessed May 30, 2012.
2. Simmons Z. Management strategies for patients with amyotrophic lateral sclerosis from diagnosis through death. Neurologist. 2005;11(5):257-270.
3. Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet J Rare Dis. 2009;4:3.
4. Yoshida M. Amyotrophic lateral sclerosis with dementia: the clinicopathological spectrum. Neuropathology. 2004;24(1):87-102.
5. Byrne S, Walsh C, Lynch C, et al. Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2011;82(6):623-627.
6. National Institute of Neurological Disorders and Stroke, NIH. Motor neuron diseases fact sheet. www.ninds.nih.gov/disorders/motor_neuron_diseases/detail_motor_neuron_diseases.htm. Accessed May 30, 2012.
7. Tartaglia MC, Rowe A, Findlater K. Differentiation between primary lateral sclerosis and amyotrophic lateral sclerosis: examination of symptoms and signs at disease onset and during follow-up. Arch Neurol. 2007;64(2):232-236.
8. Strowd RE, Cartwright MS, Okun MS, et al. Pseudobulbar affect: prevalence and quality of life impact in movement disorders. J Neurol. 2010;257(8):1382-1387.
9. Beghi E, Logroscino G, Chiò A, et al; EURALS Consortium. The epidemiology of ALS and the role of population-based registries. Biochim Biophys Acta. 2006;1762(11-12):1150-1157.
10. McCombe PA, Henderson RD. Effects of gender in amyotrophic lateral sclerosis. Gend Med. 2010;7(6):557-570.
11. Weisskopf MG, Morozova N, O’Reilly EJ, et al. Prospective study of chemical exposures and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2009;80(5):558-561.
12. ALS Association. ALS in the military: unexpected consequences of military service (2011). www.alsa.org/assets/pdfs/advocacy/als_military_paper.pdf. Accessed May 22, 2012.
13. Horner RD, Kamins KG, Feussner JR, et al. Occurrence of amyotrophic lateral sclerosis among Gulf War veterans. Neurology. 2003;61(6):742-749.
14. Horner RD, Grambow SC, Coffman DJ, et al. Amyotrophic lateral sclerosis among 1991 Gulf War veterans: evidence for a time-limited outbreak. Neuroepidemiology. 2008;31(1):28-32.
15. Wang H, O’Reilly EJ, Weisskopf MG, et al. Smoking and risk of amyotrophic lateral sclerosis: a pooled analysis of 5 prospective cohorts. Arch Neurol. 2011;68(2):207-213.
16. Steele JC, McGeer PL. The ALS/PDC syndrome of Guam and the cycad hypothesis. Neurology. 2008;70(21):1984-1990.
17. Vance C, Rogelj B, Hortobágyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208-1211.
18. Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):1668-1672.
19. Pioro EP, Majors AAW, Mitsumoto H, et al. 1H-MRS evidence of neurodegeneration and excess glutamate + glutamine in ALS medulla. Neurology. 1999;53(1):71-79.
20. Cheah BC, Vucic S, Krishnan AV, Kiernan MC. Riluzole, neuroprotection and amyotrophic lateral sclerosis. Curr Med Chem. 2010;17(18):1942-1959.
21. Paubel A, Violette J, Amy M, et al. Mutations of the ANG gene in French patients with sporadic amyotrophic lateral sclerosis. Arch Neurol. 2008;65(10):1333-1336.
22. Neusch C, Bähr M, Schneider-Gold C. Glia cells in amyotrophic lateral sclerosis: new clues to understanding an old disease? Muscle Nerve. 2007;35(6):712-724.
23. Rowland LP. Progressive muscular atrophy and other lower motor neuron syndromes of adults. Muscle Nerve. 2010;41(12):161-165.
24. Shoesmith CL, Findlater K, Rowe A, Strong MJ. Progression of amyotrophic lateral sclerosis with respiratory onset. J Neurol Neurosurg Psychiatry. 2007;78(6):629-631.
25. Traynor BJ, Codd MB, Corr B, et al. Clinical features of amyotrophic lateral sclerosis according to the El Escorial and Airlie House diagnostic criteria: a population-based study. Arch Neurol. 2000;57(8):1171-1176.
26. Phukan J, Pender NP, Hardiman O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol. 2007;6(11):994-1003.
27. Chen R, Grand’Maison F, Strong MJ, et al. Motor neuron disease presenting as acute respiratory failure: a clinical and pathological study. J Neurol Neurosurg Psychiatry. 1996;60(4):455-458.
28. Gautier G, Verscheuren A, Monnier A, et al. ALS with respiratory onset: clinical features and effects of non-invasive ventilation on the prognosis. Amyotroph Lateral Scler. 2010;11(4):379-382.
29. Hasan A, Saxena AB, Ahmed SM, Swamy TLN. Amyotrophic lateral sclerosis presenting with orthopnea in a patient with COPD and obstructive sleep apnea. J Med All Sci. 2011;1(1):46-49.
30. Zoccolella S, Beghi E, Palagano G, et al. Analysis of survival and prognostic factors in amyotrophic lateral sclerosis: a population based study. J Neurol Neurosurg Psychiatry. 2008;79(1):33-37.
31. Brown RH Jr. Amyotrophic lateral sclerosis and other motor neuron diseases. In: Fauci AS, Braunwald E, Kasper DL, ed al, eds. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw Hill Medical; 2008:2572-2576.
32. Ince PG, Lowe J, Shaw PJ. Amyotrophic lateral sclerosis: current issues in classification, pathogenesis and molecular pathology. Neuropathol Appl Neurobiol. 1998;24(2):104-117.
33. Atsumi T, Miyatake T. Morphometry of the degenerative process in the hypoglossal nerves in amyotrophic lateral sclerosis. Acta Neuropathol. 1987;73(1):25-31.
34. Shimizu T, Komori T, Kato S, et al. Masseter inhibitory reflex in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001;2(4):189-195.
35. Rosen H. Dextromethorphan/quinidine sulfate (Zenvia) for pseudobulbar affect. Drugs Today (Barc). 2008;44(9):661-668.
36. Rippon GA, Scarmeas N, Gordon PH, et al. An observational study of cognitive impairment in amyotrophic lateral sclerosis. Arch Neurol. 2006;63(3):345-352.
37. Rabkin JG, Albert SM, Del Bene ML, et al. Prevalence of depressive disorders and change over time in late-stage ALS. Neurology. 2005;65(1):62-67.
38. Vignola A, Guzzo A, Calvo A, et al. Anxiety undermines quality of life in ALS patients and caregivers. Eur J Neurol. 2008;15(11):1231-1236.
39. Fang F, Valdimarsdóttir U, Fürst CJ, et al. Suicide among patients with amyotrophic lateral sclerosis. Brain. 2008;131(pt 10):2729-2733.
40. Verstraete E, Veldink JH, Hendrikse J, et al. Structural MRI reveals cortical thinning in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2012;83(4):383-388.
41. Turner MR, Modo M. Advances in the application of MRI to amyotrophic lateral sclerosis. Expert Opin Med Diagn. 2010;4(6):483-496.
42. Cirillo M, Esposito F, Tedeschi G, et al. Widespread microstructural white matter involvement in amyotrophic lateral sclerosis: a whole-brain DTI study. AJNR Am J Neuroradiol. 2012 Feb 2. [Epub ahead of print]
43. Canu E, Agosta F, Riva N, et al. The topography of brain microstructural damage in amyotrophic lateral sclerosis assessed using diffusion tensor MR imaging. AJNR Am J Neuroradiol. 2011;32(7):1307-1314.
44. Baek WS, Desai NP. ALS: pitfalls in the diagnosis. Pract Neurol. 2007;7(2):74-81.
45. ALS Association. Criteria for the diagnosis of ALS. www.alsa.org/als-care/resources/publica tions-videos/factsheets/criteria-for-diagnosis.html. Accessed May 31, 2012.
46. de Carvalho M, Dengler R, Eisen A, et al. Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol. 2008;119(3):497-503.
47. Brooks BR, Miller RG, Swash M, Munsat TL; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293-299.
48. Waragai M. MRI and clinical features of amyotrophic lateral sclerosis. Neuroradiology. 1997;39(12):847-851.
49. Thorpe JW, Moseley IF, Hawkes CH, et al. Brain and spinal cord MRI in motor neuron disease. J Neurol Neurosurg Psychiatry. 1996; 61(3):314-317.
50. Oba H, Araki T, Ohtomo K, et al. Amyotrophic lateral sclerosis: T2 shortening in motor cortex at MR imaging. Radiology. 1993;189 (3):843-846.
51. Piao YS, Wakabayashi K, Kakita A, et al. Neuropathology with clinical correlations of sporadic amyotrophic lateral sclerosis: 102 autopsy cases examined between 1962 and 2000. Brain Pathol. 2003;13(1):10-22.
52. Mateen FJ, Carone M, Sorenson EJ. Patients who survive 5 years or more with ALS in Olmsted County, 1925-2004. J Neurol Neurosurg Psychiatry. 2010;81(10):1144-1146.
53. del Aguila MA, Longstreth WT Jr, McGuire V, et al. Prognosis in amyotrophic lateral sclerosis: a population-based study. Neurology. 2003;60(5):813-819.
54. Matuz T, Birbaumer N, Hautzinger M, Kübler A. Coping with amyotrophic lateral sclerosis: an integrative view. J Neurol Neurosurg Psychiatry. 2010;81(8):893-898.
55. Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2012 Mar 14;3:CD001447.
56. Handy CR, Krudy C, Boulis N, Federici T. Pain in amyotrophic lateral sclerosis: a neglected aspect of disease. Neurol Res Int. 2011; 2011:403808. Epub 2011 May 3.
57. McClelland S 3rd, Bethoux FA, Boulis NM, et al. Intrathecal baclofen for spasticity-related pain in amyotrophic lateral sclerosis: efficacy and factors associated with pain relief. Muscle Nerve. 2008;37(3):396-398.
58. Andersen PM, Borasio GD, Dengler R, et al; EALSC Working Group. Good practice in the management of amyotrophic lateral sclerosis: clinical guidelines. Amyotroph Lateral Scler. 2007;8(4):195-213.
59. Rabkin JG, Gordon PH, McElhiney M, et al. Modafinil treatment of fatigue in patients with ALS: a placebo-controlled study. Muscle Nerve. 2009;39(3):297-303.
60. Kühnlein P, Gdynia HJ, Sperfeld AD, et al. Diagnosis and treatment of bulbar symptoms in amyotrophic lateral sclerosis. Nat Clin Pract Neurol. 2008;4(7):366-374.
61. Katzberg HD, Benatar M. Enteral tube feeding for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev. 2011 Jan 19;(1):CD004030.
62. Lechtzin N, Scott Y, Busse AM, et al. Early use of non-invasive ventilation prolongs survival in subjects with ALS. Amyotroph Lateral Scler. 2007;8(3):185-188.
63. Annane D, Orlikowski D, Chevret S, et al. Nocturnal mechanical ventilation for chronic hypoventilation in patients with neuromuscular and chest wall disorders. Cochrane Database Syst Rev. 2007 Oct 17;(4):CD001941.
64. Radunovic A, Annane D, Jewitt K, Mustfa N. Mechanical ventilation for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev. 2009 Oct 7;(4):CD004427.
65. Onders RP, Elmo MJ, Khansarinia S, et al. Complete worldwide operative experience in laparoscopic diaphragm pacing: results and differences in spinal cord injured patients and amyotrophic lateral sclerosis patients. Surg Endosc. 2009;23(7):1433-1440.
66. Onders RP, Carlin AM, Elmo M, et al. Amyotrophic lateral sclerosis: the Midwestern surgical experience with the diaphragm pacing stimulation system shows that general anesthesia can be safely performed. Am J Surg. 2009;197(3):386-390.
67. US Food and Drug Administration. Medical devices: NeuRx Diaphragm Pacing System™—H100006. www.fda.gov/MedicalDevices/Prod uctsandMedicalProcedures/DeviceApprovalsand Clearances/Recently-ApprovedDevices/ucm278684.htm. Accessed May 31, 2012.
68. Rabkin JG, Wagner GJ, Del Bene M. Resilience and distress among amyotrophic lateral sclerosis patients and caregivers. Psychosom Med. 2000; 62(2):271-279.
69. Chiò A, Gauthier A, Vignola A, et al. Caregiver time use in ALS. Neurology. 2006;67(5):902-904.