User login
Mitchel is a reporter for MDedge based in the Philadelphia area. He started with the company in 1992, when it was International Medical News Group (IMNG), and has since covered a range of medical specialties. Mitchel trained as a virologist at Roswell Park Memorial Institute in Buffalo, and then worked briefly as a researcher at Boston Children's Hospital before pivoting to journalism as a AAAS Mass Media Fellow in 1980. His first reporting job was with Science Digest magazine, and from the mid-1980s to early-1990s he was a reporter with Medical World News. @mitchelzoler
Insulin Degludec Edges Glargine on Hypoglycemia Rate
BERLIN – Insulin degludec, an investigational insulin with a prolonged half-life and a reportedly flatter pharmacokinetic profile with fewer high and low levels, showed a modest hypoglycemic edge over insulin glargine in a 52-week head-to-head comparison in patients with type 2 diabetes.
While insulin degludec (IDeg) ran statistically even with insulin glargine for total hypoglycemic episodes and for severe hyperglycemic episodes, the IDeg formulation led to significantly fewer nocturnal hypoglycemic episodes, with a relative risk reduction of 36%. However, because all hypoglycemic episodes were infrequent, the results showed an absolute reduction average of about 0.3 nocturnal episodes per patient on glargine to about 0.2 episodes per patient on degludec, Dr. Bernard Zinman reported at the meeting.

"You need to treat four patients for a year to prevent one nocturnal episode. To me, that sounds like a good deal," said Dr. Zinman, director of the Centre for Diabetes at Mount Sinai Hospital in Toronto. In contrast, the study results also showed that 50 patients with type 2 diabetes would need IDeg treatment for a year to prevent one severe hypoglycemic episode, compared with glargine, he added.
"We want to encourage using insulin earlier in patients with type 2 diabetes, but many physicians are afraid of insulin because of [the associated risk of] hypoglycemia. I think that physicians will use degludec preferentially in patients at high risk for hypoglycemia," Dr. Zinman said in an interview. He included in this risk group younger patients with type 1 diabetes, patients with type 1 diabetes who take insulin as multiple-dose injections, patients maintained on a tight-control regimen, and patients with a history of hypoglycemia. The low numbers of hypoglycemia episodes seen in his results reflected selection of low-risk patients, he added. The results "underestimate the clinical value [of preventing hypoglycemia] because in routine clinical practice hypoglycemia is more of an issue" than it was in this trial.
An application to approve IDeg for treating patients with either type 1 or type 2 diabetes is pending before the Food and Drug Administration, and last July Novo Nordisk, the company developing the drug, said that it expected a FDA advisory committee to consider the application in November 2012.
The trial enrolled 1,030 patients with established type 2 diabetes inadequately controlled by metformin and possibly other oral drugs and no history of insulin treatment. The researchers randomized 773 patients to once-daily treatment with IDeg and 257 to a once daily regimen of insulin glargine, with the dosage titrated to achieve pre-breakfast plasma glucose of 3.9-4.9 mmol/L. The patients average 59 years old, their average body mass index was about 31 kg/m2, their average hemoglobin A1c was 8.2%, and they had type 2 diabetes for an average of about 9 years.
Throughout the 52-week study, average HbA1c levels in the two treatment arms tracked together and, at 1 year, the two groups had virtually identical average levels of about 7.3%. Fasting plasma glucose also tracked at similar rates in both arms during the study but, at 1 year, the average level was 0.43 mmol/L lower in the degludec group, a statistically significant difference. The average daily insulin dosage received by patients in each of the two arms was also virtually identical.
The rate of all confirmed hypoglycemia episodes was 18% lower among the IDeg-treated patients, compared with the controls, but this difference did not reach statistical significance. Severe hypoglycemia episodes – defined as events where the patient required help – showed an 86% relative risk reduction, but because the event rates in both arms was small this difference was also not statistically significant.
An additional analysis also looked at the incidence of hypoglycemic episodes during the maintenance phase of treatment in the study, after the first 16 weeks. During maintenance, the two arms failed to show a significant difference in any measure of hypoglycemic episodes except for nocturnal events, which degludec treatment cut by 49% relative to the glargine group, a statistically significant difference.
Other than hypoglycemia the two insulin analogues showed similar safety profiles.
The study was sponsored by Novo Nordisk, the company developing IDeg. Dr. Zinman said that he has been an advisor to Novo Nordisk, Merck, Sanofi-Aventis, and Boehringer Ingelheim, and research funding from Novo Nordisk, Merck, and Boehringer Ingelheim.
The value shown by insulin degludec, compared with insulin glargine, in this study boils down to whether or not the incidence of hypoglycemia episodes among patients treated with glargine is too high. In the results reported by Dr. Zinman, the hypoglycemic event rates were lower than in other studies. The results of his study show that existing insulin analogues, such as glargine, perform very well and are very safe.

|
|
Will further reduction in the rate of nocturnal hypoglycemia events, from a rate that is already low, motivate physicians to prescribe insulin degludec? That remains to be seen. New insulin analogues will have difficulty penetrating the market unless they show substantial differences compared with existing analogues, or unless they are not priced above existing analogues.
Jack L. Leahy, M.D., is professor and director of endocrinology at the University of Vermont in Burlington. He said that he has been an adviser to and has received research funding from Novo Nordisk, Merck, Sanofi-Aventis, and other companies. He made these comments in an interview.
The value shown by insulin degludec, compared with insulin glargine, in this study boils down to whether or not the incidence of hypoglycemia episodes among patients treated with glargine is too high. In the results reported by Dr. Zinman, the hypoglycemic event rates were lower than in other studies. The results of his study show that existing insulin analogues, such as glargine, perform very well and are very safe.
|
|
|
Will further reduction in the rate of nocturnal hypoglycemia events, from a rate that is already low, motivate physicians to prescribe insulin degludec? That remains to be seen. New insulin analogues will have difficulty penetrating the market unless they show substantial differences compared with existing analogues, or unless they are not priced above existing analogues.
Jack L. Leahy, M.D., is professor and director of endocrinology at the University of Vermont in Burlington. He said that he has been an adviser to and has received research funding from Novo Nordisk, Merck, Sanofi-Aventis, and other companies. He made these comments in an interview.
The value shown by insulin degludec, compared with insulin glargine, in this study boils down to whether or not the incidence of hypoglycemia episodes among patients treated with glargine is too high. In the results reported by Dr. Zinman, the hypoglycemic event rates were lower than in other studies. The results of his study show that existing insulin analogues, such as glargine, perform very well and are very safe.
|
|
|
Will further reduction in the rate of nocturnal hypoglycemia events, from a rate that is already low, motivate physicians to prescribe insulin degludec? That remains to be seen. New insulin analogues will have difficulty penetrating the market unless they show substantial differences compared with existing analogues, or unless they are not priced above existing analogues.
Jack L. Leahy, M.D., is professor and director of endocrinology at the University of Vermont in Burlington. He said that he has been an adviser to and has received research funding from Novo Nordisk, Merck, Sanofi-Aventis, and other companies. He made these comments in an interview.
BERLIN – Insulin degludec, an investigational insulin with a prolonged half-life and a reportedly flatter pharmacokinetic profile with fewer high and low levels, showed a modest hypoglycemic edge over insulin glargine in a 52-week head-to-head comparison in patients with type 2 diabetes.
While insulin degludec (IDeg) ran statistically even with insulin glargine for total hypoglycemic episodes and for severe hyperglycemic episodes, the IDeg formulation led to significantly fewer nocturnal hypoglycemic episodes, with a relative risk reduction of 36%. However, because all hypoglycemic episodes were infrequent, the results showed an absolute reduction average of about 0.3 nocturnal episodes per patient on glargine to about 0.2 episodes per patient on degludec, Dr. Bernard Zinman reported at the meeting.
"You need to treat four patients for a year to prevent one nocturnal episode. To me, that sounds like a good deal," said Dr. Zinman, director of the Centre for Diabetes at Mount Sinai Hospital in Toronto. In contrast, the study results also showed that 50 patients with type 2 diabetes would need IDeg treatment for a year to prevent one severe hypoglycemic episode, compared with glargine, he added.
"We want to encourage using insulin earlier in patients with type 2 diabetes, but many physicians are afraid of insulin because of [the associated risk of] hypoglycemia. I think that physicians will use degludec preferentially in patients at high risk for hypoglycemia," Dr. Zinman said in an interview. He included in this risk group younger patients with type 1 diabetes, patients with type 1 diabetes who take insulin as multiple-dose injections, patients maintained on a tight-control regimen, and patients with a history of hypoglycemia. The low numbers of hypoglycemia episodes seen in his results reflected selection of low-risk patients, he added. The results "underestimate the clinical value [of preventing hypoglycemia] because in routine clinical practice hypoglycemia is more of an issue" than it was in this trial.
An application to approve IDeg for treating patients with either type 1 or type 2 diabetes is pending before the Food and Drug Administration, and last July Novo Nordisk, the company developing the drug, said that it expected a FDA advisory committee to consider the application in November 2012.
The trial enrolled 1,030 patients with established type 2 diabetes inadequately controlled by metformin and possibly other oral drugs and no history of insulin treatment. The researchers randomized 773 patients to once-daily treatment with IDeg and 257 to a once daily regimen of insulin glargine, with the dosage titrated to achieve pre-breakfast plasma glucose of 3.9-4.9 mmol/L. The patients average 59 years old, their average body mass index was about 31 kg/m2, their average hemoglobin A1c was 8.2%, and they had type 2 diabetes for an average of about 9 years.
Throughout the 52-week study, average HbA1c levels in the two treatment arms tracked together and, at 1 year, the two groups had virtually identical average levels of about 7.3%. Fasting plasma glucose also tracked at similar rates in both arms during the study but, at 1 year, the average level was 0.43 mmol/L lower in the degludec group, a statistically significant difference. The average daily insulin dosage received by patients in each of the two arms was also virtually identical.
The rate of all confirmed hypoglycemia episodes was 18% lower among the IDeg-treated patients, compared with the controls, but this difference did not reach statistical significance. Severe hypoglycemia episodes – defined as events where the patient required help – showed an 86% relative risk reduction, but because the event rates in both arms was small this difference was also not statistically significant.
An additional analysis also looked at the incidence of hypoglycemic episodes during the maintenance phase of treatment in the study, after the first 16 weeks. During maintenance, the two arms failed to show a significant difference in any measure of hypoglycemic episodes except for nocturnal events, which degludec treatment cut by 49% relative to the glargine group, a statistically significant difference.
Other than hypoglycemia the two insulin analogues showed similar safety profiles.
The study was sponsored by Novo Nordisk, the company developing IDeg. Dr. Zinman said that he has been an advisor to Novo Nordisk, Merck, Sanofi-Aventis, and Boehringer Ingelheim, and research funding from Novo Nordisk, Merck, and Boehringer Ingelheim.
BERLIN – Insulin degludec, an investigational insulin with a prolonged half-life and a reportedly flatter pharmacokinetic profile with fewer high and low levels, showed a modest hypoglycemic edge over insulin glargine in a 52-week head-to-head comparison in patients with type 2 diabetes.
While insulin degludec (IDeg) ran statistically even with insulin glargine for total hypoglycemic episodes and for severe hyperglycemic episodes, the IDeg formulation led to significantly fewer nocturnal hypoglycemic episodes, with a relative risk reduction of 36%. However, because all hypoglycemic episodes were infrequent, the results showed an absolute reduction average of about 0.3 nocturnal episodes per patient on glargine to about 0.2 episodes per patient on degludec, Dr. Bernard Zinman reported at the meeting.
"You need to treat four patients for a year to prevent one nocturnal episode. To me, that sounds like a good deal," said Dr. Zinman, director of the Centre for Diabetes at Mount Sinai Hospital in Toronto. In contrast, the study results also showed that 50 patients with type 2 diabetes would need IDeg treatment for a year to prevent one severe hypoglycemic episode, compared with glargine, he added.
"We want to encourage using insulin earlier in patients with type 2 diabetes, but many physicians are afraid of insulin because of [the associated risk of] hypoglycemia. I think that physicians will use degludec preferentially in patients at high risk for hypoglycemia," Dr. Zinman said in an interview. He included in this risk group younger patients with type 1 diabetes, patients with type 1 diabetes who take insulin as multiple-dose injections, patients maintained on a tight-control regimen, and patients with a history of hypoglycemia. The low numbers of hypoglycemia episodes seen in his results reflected selection of low-risk patients, he added. The results "underestimate the clinical value [of preventing hypoglycemia] because in routine clinical practice hypoglycemia is more of an issue" than it was in this trial.
An application to approve IDeg for treating patients with either type 1 or type 2 diabetes is pending before the Food and Drug Administration, and last July Novo Nordisk, the company developing the drug, said that it expected a FDA advisory committee to consider the application in November 2012.
The trial enrolled 1,030 patients with established type 2 diabetes inadequately controlled by metformin and possibly other oral drugs and no history of insulin treatment. The researchers randomized 773 patients to once-daily treatment with IDeg and 257 to a once daily regimen of insulin glargine, with the dosage titrated to achieve pre-breakfast plasma glucose of 3.9-4.9 mmol/L. The patients average 59 years old, their average body mass index was about 31 kg/m2, their average hemoglobin A1c was 8.2%, and they had type 2 diabetes for an average of about 9 years.
Throughout the 52-week study, average HbA1c levels in the two treatment arms tracked together and, at 1 year, the two groups had virtually identical average levels of about 7.3%. Fasting plasma glucose also tracked at similar rates in both arms during the study but, at 1 year, the average level was 0.43 mmol/L lower in the degludec group, a statistically significant difference. The average daily insulin dosage received by patients in each of the two arms was also virtually identical.
The rate of all confirmed hypoglycemia episodes was 18% lower among the IDeg-treated patients, compared with the controls, but this difference did not reach statistical significance. Severe hypoglycemia episodes – defined as events where the patient required help – showed an 86% relative risk reduction, but because the event rates in both arms was small this difference was also not statistically significant.
An additional analysis also looked at the incidence of hypoglycemic episodes during the maintenance phase of treatment in the study, after the first 16 weeks. During maintenance, the two arms failed to show a significant difference in any measure of hypoglycemic episodes except for nocturnal events, which degludec treatment cut by 49% relative to the glargine group, a statistically significant difference.
Other than hypoglycemia the two insulin analogues showed similar safety profiles.
The study was sponsored by Novo Nordisk, the company developing IDeg. Dr. Zinman said that he has been an advisor to Novo Nordisk, Merck, Sanofi-Aventis, and Boehringer Ingelheim, and research funding from Novo Nordisk, Merck, and Boehringer Ingelheim.
AT THE ANNUAL MEETING OF THE EUROPEAN ASSOCIATION FOR THE STUDY OF DIABETES
Major Finding: Insulin degludec produced a statistically significant 36% relative cut in nocturnal hypoglycemic episodes, compared with glargine during 1 year.
Data Source: Results were taken from a randomized, multicenter trial that enrolled 1,030 patients with type 2 diabetes.
Disclosures: The study was sponsored by Novo Nordisk, the company developing insulin degludec. Dr. Zinman said that he has been an adviser to Novo Nordisk, Merck, Sanofi-Aventis, and Boehringer Ingelheim, and research funding from Novo Nordisk, Merck, and Boehringer Ingelheim

Apixaban Cuts Bleeding Risk in Renal Dysfunction Patients
MUNICH - Patients with renal dysfunction as well as atrial fibrillation who received apixaban to prevent strokes and systemic embolism had the biggest drop in major bleeding events compared with control patients on warfarin in a prespecified substudy of the ARISTOTLE trial.
"Our findings suggest that apixaban may be particularly suited to address the unmet need for more effective and safe stroke prevention in patients with atrial fibrillation and renal dysfunction," Dr. Stefan H. Hohnloser said at the annual congress of the European Society of Cardiology.

The finding is especially relevant to practice because of the high coprevalence of atrial fibrillation and chronic kidney disease. A recent U.S. study of patients with chronic kidney disease found an 18% prevalence of atrial fibrillation – two- to threefold higher than in the general population – and a greater than 25% prevalence of atrial fibrillation among patients with renal dysfunction who were at least 70 year old (Am. Heart J. 2010;159:1102-7).
But while apixiban triggered significantly fewer major bleeds compared with warfarin among patients with renal dysfunction enrolled in the pivotal Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE) trial, apixaban’s efficacy for stroke and embolism protection in atrial fibrillation patients with renal dysfunction was just as good as it was in patients with normal renal activity.
"When compared with warfarin, apixaban treatment reduced the rate of stroke, death, and major bleeding, regardless of renal function," Dr. Hohnloser and his associates wrote in a published report on their findings that appeared online concurrent with his report at the meeting (Eur. Heart J. 2012 [doi:10.1093/eurheartj/ehs274]).
"We know that patients with atrial fibrillation and impaired renal function not only have a high stroke rate, but also have a significantly higher bleeding rate with respect to patients with normal renal function. The message from the new analysis is that, in patients with impaired renal function, apixaban is superior to warfarin with respect to the risk of major bleeding," said Dr. Gerhard Hindricks, professor and director of the department of electrophysiology at the Leipzig (Germany) University Heart Center.
The new findings also drew a sharp contrast between apixaban and another new oral anticoagulant, dabigatran, noted Dr. Hindricks and Dr. Jan Steffel of University Hospital Zurich in an editorial they wrote that appeared online concurrent with the main report (Eur. Heart J. 2012 [doi:10.1093/eurheartj/ehs267]).
"Dabigatran, which is 80% renally cleared, has a significant potential for severe bleeding in patients with reduced renal function," they wrote.
In addition, they added, "major bleeding events are the single most prevalent reason why proper anticoagulation is withheld in patients with atrial fibrillation, especially those with impaired renal function. ... Chronic kidney disease certainly appears to be the ‘Achilles heel’ of dabigatran, as accumulation is likely to occur due to mainly renal elimination. Hence, in our view of the available data, apixaban would probably be the preferred agent over dabigatran in these patients."
Dr. Hindricks and Dr. Steffel concluded that the new findings from ARISTOTLE provide "solid evidence for the superiority of apixaban in patients with atrial fibrillation and chronic kidney disease. In the light of these data, apixaban appears to be a very appealing option for these individuals, potentially leading to a substantial increase in the numbers of appropriately anticoagulated patients."
The substudy analysis calculated estimated glomerular filtration rates (eGFR) for patients enrolled in ARISTOTLE by three difference methods: the Cockcroft-Gault formula, the Chronic Kidney Disease-Epidemiology Collaboration formula, and based on cystatin C measurement. It focused on 18,122 of the patients enrolled in ARISTOTLE who had data available that allowed calculation of their eGFR. All three methods found that about 15% of the enrolled patients had an eGFR of 50 mL/min per 1.73 m2 or less.
The new analysis showed that patients with an eGFR of 50 mL/min or less had the greatest reduction in major bleeding episodes during apixaban treatment, compared with warfarin treatment. Among patients with impaired renal function, the bleeding risk on apixaban dropped by 35%-52%, depending on which formula the researchers used to identify patients with an eGFR that low, reported Dr. Hohnloser, professor and director of the department of clinical electrophysiology at J.W. Goethe University in Frankfurt, Germany.
In contrast, the primary efficacy end point of stroke or systemic embolism occurred consistently less often among patients treated with apixaban than in those on warfarin regardless of their eGFR.
The ARISTOTLE trial was sponsored by Bristol-Myers Squibb and Pfizer, the companies that market apixaban (Eliquis). Dr. Hohnloser said that he has received consulting and lecture fees from Bristol-Myers Squibb, Pfizer, and other drug companies and research grants from Sanofi-Aventis and St. Jude. Dr. Hindricks said that he has received honoraria from and has been a consultant to Biosense, Stereotaxis, St. Jude, and Biotronik.
Renal dysfunction is highly prevalent in patients with atrial fibrillation and is associated with both stroke and bleeding risk. In ARISTOTLE, the overall findings of the trial are consistent with the outcomes seen in patients with moderate renal dysfunction, with an estimated glomerular filtration rate (eGFR) of less than 50 mL/min per 1.73 m2.
The ARISTOTLE results showed that the rate of ischemic stroke was twofold higher, the rate of major bleeds was threefold higher, and the mortality rate was threefold higher among patients with an eGFR of 50 mL/min per 1.73 m2 or less compared with patients with an eGFR of greater than 80 mL/min per 1.73 m2.
The benefit over warfarin of reduced bleeding appears to be more marked in patients with moderate renal dysfunction. These patients received a reduced apixaban dosage, 2.5 mg b.i.d. instead of the full dosage of 5 mg b.i.d., but results of a sensitivity analysis by the researchers suggested that this reduced dosage was not the key factor producing the reduced rate of major bleeds.
In my view, apixaban provides a treatment option and advantages over warfarin in atrial fibrillation patients with moderate renal dysfunction, a group of patients who currently receive suboptimal management.
Keith A.A. Fox, M.D., is professor of cardiology at the University of Edinburgh. He said that he has received grant funding and honoraria from Sanofi, Lilly, Bayer/Johnson & Johnson, Astra Zeneca, and Boehringer Ingelheim. He made these comments as designated discussant for the report by Dr. Hohnloser.
Renal dysfunction is highly prevalent in patients with atrial fibrillation and is associated with both stroke and bleeding risk. In ARISTOTLE, the overall findings of the trial are consistent with the outcomes seen in patients with moderate renal dysfunction, with an estimated glomerular filtration rate (eGFR) of less than 50 mL/min per 1.73 m2.
The ARISTOTLE results showed that the rate of ischemic stroke was twofold higher, the rate of major bleeds was threefold higher, and the mortality rate was threefold higher among patients with an eGFR of 50 mL/min per 1.73 m2 or less compared with patients with an eGFR of greater than 80 mL/min per 1.73 m2.
The benefit over warfarin of reduced bleeding appears to be more marked in patients with moderate renal dysfunction. These patients received a reduced apixaban dosage, 2.5 mg b.i.d. instead of the full dosage of 5 mg b.i.d., but results of a sensitivity analysis by the researchers suggested that this reduced dosage was not the key factor producing the reduced rate of major bleeds.
In my view, apixaban provides a treatment option and advantages over warfarin in atrial fibrillation patients with moderate renal dysfunction, a group of patients who currently receive suboptimal management.
Keith A.A. Fox, M.D., is professor of cardiology at the University of Edinburgh. He said that he has received grant funding and honoraria from Sanofi, Lilly, Bayer/Johnson & Johnson, Astra Zeneca, and Boehringer Ingelheim. He made these comments as designated discussant for the report by Dr. Hohnloser.
Renal dysfunction is highly prevalent in patients with atrial fibrillation and is associated with both stroke and bleeding risk. In ARISTOTLE, the overall findings of the trial are consistent with the outcomes seen in patients with moderate renal dysfunction, with an estimated glomerular filtration rate (eGFR) of less than 50 mL/min per 1.73 m2.
The ARISTOTLE results showed that the rate of ischemic stroke was twofold higher, the rate of major bleeds was threefold higher, and the mortality rate was threefold higher among patients with an eGFR of 50 mL/min per 1.73 m2 or less compared with patients with an eGFR of greater than 80 mL/min per 1.73 m2.
The benefit over warfarin of reduced bleeding appears to be more marked in patients with moderate renal dysfunction. These patients received a reduced apixaban dosage, 2.5 mg b.i.d. instead of the full dosage of 5 mg b.i.d., but results of a sensitivity analysis by the researchers suggested that this reduced dosage was not the key factor producing the reduced rate of major bleeds.
In my view, apixaban provides a treatment option and advantages over warfarin in atrial fibrillation patients with moderate renal dysfunction, a group of patients who currently receive suboptimal management.
Keith A.A. Fox, M.D., is professor of cardiology at the University of Edinburgh. He said that he has received grant funding and honoraria from Sanofi, Lilly, Bayer/Johnson & Johnson, Astra Zeneca, and Boehringer Ingelheim. He made these comments as designated discussant for the report by Dr. Hohnloser.
MUNICH - Patients with renal dysfunction as well as atrial fibrillation who received apixaban to prevent strokes and systemic embolism had the biggest drop in major bleeding events compared with control patients on warfarin in a prespecified substudy of the ARISTOTLE trial.
"Our findings suggest that apixaban may be particularly suited to address the unmet need for more effective and safe stroke prevention in patients with atrial fibrillation and renal dysfunction," Dr. Stefan H. Hohnloser said at the annual congress of the European Society of Cardiology.
The finding is especially relevant to practice because of the high coprevalence of atrial fibrillation and chronic kidney disease. A recent U.S. study of patients with chronic kidney disease found an 18% prevalence of atrial fibrillation – two- to threefold higher than in the general population – and a greater than 25% prevalence of atrial fibrillation among patients with renal dysfunction who were at least 70 year old (Am. Heart J. 2010;159:1102-7).
But while apixiban triggered significantly fewer major bleeds compared with warfarin among patients with renal dysfunction enrolled in the pivotal Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE) trial, apixaban’s efficacy for stroke and embolism protection in atrial fibrillation patients with renal dysfunction was just as good as it was in patients with normal renal activity.
"When compared with warfarin, apixaban treatment reduced the rate of stroke, death, and major bleeding, regardless of renal function," Dr. Hohnloser and his associates wrote in a published report on their findings that appeared online concurrent with his report at the meeting (Eur. Heart J. 2012 [doi:10.1093/eurheartj/ehs274]).
"We know that patients with atrial fibrillation and impaired renal function not only have a high stroke rate, but also have a significantly higher bleeding rate with respect to patients with normal renal function. The message from the new analysis is that, in patients with impaired renal function, apixaban is superior to warfarin with respect to the risk of major bleeding," said Dr. Gerhard Hindricks, professor and director of the department of electrophysiology at the Leipzig (Germany) University Heart Center.
The new findings also drew a sharp contrast between apixaban and another new oral anticoagulant, dabigatran, noted Dr. Hindricks and Dr. Jan Steffel of University Hospital Zurich in an editorial they wrote that appeared online concurrent with the main report (Eur. Heart J. 2012 [doi:10.1093/eurheartj/ehs267]).
"Dabigatran, which is 80% renally cleared, has a significant potential for severe bleeding in patients with reduced renal function," they wrote.
In addition, they added, "major bleeding events are the single most prevalent reason why proper anticoagulation is withheld in patients with atrial fibrillation, especially those with impaired renal function. ... Chronic kidney disease certainly appears to be the ‘Achilles heel’ of dabigatran, as accumulation is likely to occur due to mainly renal elimination. Hence, in our view of the available data, apixaban would probably be the preferred agent over dabigatran in these patients."
Dr. Hindricks and Dr. Steffel concluded that the new findings from ARISTOTLE provide "solid evidence for the superiority of apixaban in patients with atrial fibrillation and chronic kidney disease. In the light of these data, apixaban appears to be a very appealing option for these individuals, potentially leading to a substantial increase in the numbers of appropriately anticoagulated patients."
The substudy analysis calculated estimated glomerular filtration rates (eGFR) for patients enrolled in ARISTOTLE by three difference methods: the Cockcroft-Gault formula, the Chronic Kidney Disease-Epidemiology Collaboration formula, and based on cystatin C measurement. It focused on 18,122 of the patients enrolled in ARISTOTLE who had data available that allowed calculation of their eGFR. All three methods found that about 15% of the enrolled patients had an eGFR of 50 mL/min per 1.73 m2 or less.
The new analysis showed that patients with an eGFR of 50 mL/min or less had the greatest reduction in major bleeding episodes during apixaban treatment, compared with warfarin treatment. Among patients with impaired renal function, the bleeding risk on apixaban dropped by 35%-52%, depending on which formula the researchers used to identify patients with an eGFR that low, reported Dr. Hohnloser, professor and director of the department of clinical electrophysiology at J.W. Goethe University in Frankfurt, Germany.
In contrast, the primary efficacy end point of stroke or systemic embolism occurred consistently less often among patients treated with apixaban than in those on warfarin regardless of their eGFR.
The ARISTOTLE trial was sponsored by Bristol-Myers Squibb and Pfizer, the companies that market apixaban (Eliquis). Dr. Hohnloser said that he has received consulting and lecture fees from Bristol-Myers Squibb, Pfizer, and other drug companies and research grants from Sanofi-Aventis and St. Jude. Dr. Hindricks said that he has received honoraria from and has been a consultant to Biosense, Stereotaxis, St. Jude, and Biotronik.
MUNICH - Patients with renal dysfunction as well as atrial fibrillation who received apixaban to prevent strokes and systemic embolism had the biggest drop in major bleeding events compared with control patients on warfarin in a prespecified substudy of the ARISTOTLE trial.
"Our findings suggest that apixaban may be particularly suited to address the unmet need for more effective and safe stroke prevention in patients with atrial fibrillation and renal dysfunction," Dr. Stefan H. Hohnloser said at the annual congress of the European Society of Cardiology.
The finding is especially relevant to practice because of the high coprevalence of atrial fibrillation and chronic kidney disease. A recent U.S. study of patients with chronic kidney disease found an 18% prevalence of atrial fibrillation – two- to threefold higher than in the general population – and a greater than 25% prevalence of atrial fibrillation among patients with renal dysfunction who were at least 70 year old (Am. Heart J. 2010;159:1102-7).
But while apixiban triggered significantly fewer major bleeds compared with warfarin among patients with renal dysfunction enrolled in the pivotal Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE) trial, apixaban’s efficacy for stroke and embolism protection in atrial fibrillation patients with renal dysfunction was just as good as it was in patients with normal renal activity.
"When compared with warfarin, apixaban treatment reduced the rate of stroke, death, and major bleeding, regardless of renal function," Dr. Hohnloser and his associates wrote in a published report on their findings that appeared online concurrent with his report at the meeting (Eur. Heart J. 2012 [doi:10.1093/eurheartj/ehs274]).
"We know that patients with atrial fibrillation and impaired renal function not only have a high stroke rate, but also have a significantly higher bleeding rate with respect to patients with normal renal function. The message from the new analysis is that, in patients with impaired renal function, apixaban is superior to warfarin with respect to the risk of major bleeding," said Dr. Gerhard Hindricks, professor and director of the department of electrophysiology at the Leipzig (Germany) University Heart Center.
The new findings also drew a sharp contrast between apixaban and another new oral anticoagulant, dabigatran, noted Dr. Hindricks and Dr. Jan Steffel of University Hospital Zurich in an editorial they wrote that appeared online concurrent with the main report (Eur. Heart J. 2012 [doi:10.1093/eurheartj/ehs267]).
"Dabigatran, which is 80% renally cleared, has a significant potential for severe bleeding in patients with reduced renal function," they wrote.
In addition, they added, "major bleeding events are the single most prevalent reason why proper anticoagulation is withheld in patients with atrial fibrillation, especially those with impaired renal function. ... Chronic kidney disease certainly appears to be the ‘Achilles heel’ of dabigatran, as accumulation is likely to occur due to mainly renal elimination. Hence, in our view of the available data, apixaban would probably be the preferred agent over dabigatran in these patients."
Dr. Hindricks and Dr. Steffel concluded that the new findings from ARISTOTLE provide "solid evidence for the superiority of apixaban in patients with atrial fibrillation and chronic kidney disease. In the light of these data, apixaban appears to be a very appealing option for these individuals, potentially leading to a substantial increase in the numbers of appropriately anticoagulated patients."
The substudy analysis calculated estimated glomerular filtration rates (eGFR) for patients enrolled in ARISTOTLE by three difference methods: the Cockcroft-Gault formula, the Chronic Kidney Disease-Epidemiology Collaboration formula, and based on cystatin C measurement. It focused on 18,122 of the patients enrolled in ARISTOTLE who had data available that allowed calculation of their eGFR. All three methods found that about 15% of the enrolled patients had an eGFR of 50 mL/min per 1.73 m2 or less.
The new analysis showed that patients with an eGFR of 50 mL/min or less had the greatest reduction in major bleeding episodes during apixaban treatment, compared with warfarin treatment. Among patients with impaired renal function, the bleeding risk on apixaban dropped by 35%-52%, depending on which formula the researchers used to identify patients with an eGFR that low, reported Dr. Hohnloser, professor and director of the department of clinical electrophysiology at J.W. Goethe University in Frankfurt, Germany.
In contrast, the primary efficacy end point of stroke or systemic embolism occurred consistently less often among patients treated with apixaban than in those on warfarin regardless of their eGFR.
The ARISTOTLE trial was sponsored by Bristol-Myers Squibb and Pfizer, the companies that market apixaban (Eliquis). Dr. Hohnloser said that he has received consulting and lecture fees from Bristol-Myers Squibb, Pfizer, and other drug companies and research grants from Sanofi-Aventis and St. Jude. Dr. Hindricks said that he has received honoraria from and has been a consultant to Biosense, Stereotaxis, St. Jude, and Biotronik.
AT THE ANNUAL CONGRESS OF THE EUROPEAN SOCIETY OF CARDIOLOGY
Major Finding: Among atrial fibrillation patients with renal dysfunction, apixiban produced 35%-52% fewer major bleeding episodes compared with warfarin.
Data Source: A prespecified substudy of ARISTOTLE, a multicenter, randomized trial of 18,201 patients with atrial fibrillation.
Disclosures: The ARISTOTLE trial was sponsored by Bristol-Myers Squibb and Pfizer, the companies that market apixaban (Eliquis). Dr. Hohnloser said that he has received consulting and lecture fees from Bristol-Myers Squibb, Pfizer, and other drug companies and research grants from sanofi-aventis and St. Jude. Dr. Hindricks said that he has received honoraria from and has been a consultant to Biosense, Stereotaxis, St. Jude, and Biotronik.

CRT's Mild Heart Failure Benefits Persist 5 Years
MUNICH – Cardiac resynchronization in patients with mild heart failure produced durable benefits during an average 4.5 years of follow-up, with evidence for ongoing cardiac remodeling and reduced disease progression in a series of 419 patients.
"The benefits of CRT [cardiac-resynchronization therapy] persisted, indicating that CRT attenuates disease progression in mildly symptomatic heart failure patients with wide QRS," Dr. Cecilia Linde said at the annual congress of the European Society of Cardiology.
"The results clearly show that the reverse remodeling produced by CRT was sustained over 5 years, and that’s good news for the treatment of patients with heart failure and an indication for cardiac resynchronization," commented Dr. Gerhard Hindricks, professor and director of the department of electrophysiology at the Leipzig (Germany) University Heart Center.
The findings "confirm the results from RAFT [Resynchronization-Defibrillation for Ambulatory Heart Failure Trial (N. Engl. J. Med. 2010;363:2385-95)] with respect to the long-term outcome of CRT in patients with NYHA [New York Heart Association] class II heart failure for survival and heart failure hospitalization, and provide novel, long-term information on reverse remodeling" from CRT in these patients, commented Dr. Angelo Auricchio, director of the cardiac electrophysiology program at the Ticino Cardiac Center in Lugano, Switzerland.
The data analyzed by Dr. Linde and her associates came from 419 patients originally enrolled in the "CRT on" arm of the REVERSE (Resynchronization Reverses Remodeling in Systolic Left Ventricular Dysfunction) trial, which enrolled patients with NYHA class I or II heart failure and a QRS duration or at least 120 msec. The primary end point of the study was the composite of death or heart failure hospitalization during the first 12 months of active CRT use compared with patients who received a CRT device that had not been turned on (J. Am. Coll. Cardiol. 2008;52:1834-43). The new analysis reported by Dr. Linde focused on 419 patients initially randomized to the CRT-on arm who continued on active CRT use for up to 5 years, with an average follow-up of 4.5 years. The researchers collected complete follow-up data for 95% of these patients through 3 years, for 89% through 4 years, and for 85% through 5 years.
At baseline, the patients had an average age of 63 years; 78% were men, and 82% had NYHA class I heart failure, with a average left ventricular ejection fraction of 27%. Their average QRS duration at entry was 153 msec, and 56% of the patients had an ischemic etiology for their heart failure.
Annual follow-up findings showed a consistent, linear rate of death and first heart failure hospitalization throughout follow-up, with an annual mortality rate of 3% and an annual rate of death or first heart-failure hospitalization of 6%, said Dr. Linde, a cardiologist at the Karolinska Institute in Stockholm.
The results also showed a stable pattern of left-ventricular reverse remodeling following the first year on CRT. After 12 months, patients had an average cut in the left ventricular end diastolic volume index of 19 mL/m2; 25 mL/m2 after 24 months; and 23 mL/m2 after 5 years. The average drop in left ventricular end systolic volume index was 18 mL/m2 after 12 months; 24 mL/m2 after 24 months; and 23 mL/m2 at 5 years. Other measures of heart failure severity – the 6-minute walk, the Minnesota Living With Heart Failure score, and the Kansas City Cardiomyopathy Questionnaire score all improved significantly during the first 12 months on CRT therapy, and then held fairly steady during the subsequent 4 years. At 12 months, about 43% of patients had NYHA class I disease, about 53% had class II disease, and the rest had class III disease (which compared with rates of 18% with class I and 82% with class II at baseline). After 5 years, roughly 34% of patients had NYHA class I heart failure, about 56% had class II, about 9% had class III, and about 1% had class IV disease.
The majority of left-ventricular lead complications – the main adverse event of treatment – occurred during the first 12 months, with a cumulative incidence of 9% after 1 year. At 5 years, 13% of patients had experienced a lead complication.
The REVERSE trial was sponsored by Medtronic. Dr. Linde said that she has been a consultant to Medtronic and St. Jude and received research grants from Medtronic. Dr. Hindricks said that he received honoraria from and served on advisory boards for Biosense, Stereotaxis, St. Jude, and Biotronik. Dr. Auricchio said that he was a speaker for, consultant to, and received honoraria from Medtronic, St. Jude, and several other device and drug companies.
MUNICH – Cardiac resynchronization in patients with mild heart failure produced durable benefits during an average 4.5 years of follow-up, with evidence for ongoing cardiac remodeling and reduced disease progression in a series of 419 patients.
"The benefits of CRT [cardiac-resynchronization therapy] persisted, indicating that CRT attenuates disease progression in mildly symptomatic heart failure patients with wide QRS," Dr. Cecilia Linde said at the annual congress of the European Society of Cardiology.
"The results clearly show that the reverse remodeling produced by CRT was sustained over 5 years, and that’s good news for the treatment of patients with heart failure and an indication for cardiac resynchronization," commented Dr. Gerhard Hindricks, professor and director of the department of electrophysiology at the Leipzig (Germany) University Heart Center.
The findings "confirm the results from RAFT [Resynchronization-Defibrillation for Ambulatory Heart Failure Trial (N. Engl. J. Med. 2010;363:2385-95)] with respect to the long-term outcome of CRT in patients with NYHA [New York Heart Association] class II heart failure for survival and heart failure hospitalization, and provide novel, long-term information on reverse remodeling" from CRT in these patients, commented Dr. Angelo Auricchio, director of the cardiac electrophysiology program at the Ticino Cardiac Center in Lugano, Switzerland.
The data analyzed by Dr. Linde and her associates came from 419 patients originally enrolled in the "CRT on" arm of the REVERSE (Resynchronization Reverses Remodeling in Systolic Left Ventricular Dysfunction) trial, which enrolled patients with NYHA class I or II heart failure and a QRS duration or at least 120 msec. The primary end point of the study was the composite of death or heart failure hospitalization during the first 12 months of active CRT use compared with patients who received a CRT device that had not been turned on (J. Am. Coll. Cardiol. 2008;52:1834-43). The new analysis reported by Dr. Linde focused on 419 patients initially randomized to the CRT-on arm who continued on active CRT use for up to 5 years, with an average follow-up of 4.5 years. The researchers collected complete follow-up data for 95% of these patients through 3 years, for 89% through 4 years, and for 85% through 5 years.
At baseline, the patients had an average age of 63 years; 78% were men, and 82% had NYHA class I heart failure, with a average left ventricular ejection fraction of 27%. Their average QRS duration at entry was 153 msec, and 56% of the patients had an ischemic etiology for their heart failure.
Annual follow-up findings showed a consistent, linear rate of death and first heart failure hospitalization throughout follow-up, with an annual mortality rate of 3% and an annual rate of death or first heart-failure hospitalization of 6%, said Dr. Linde, a cardiologist at the Karolinska Institute in Stockholm.
The results also showed a stable pattern of left-ventricular reverse remodeling following the first year on CRT. After 12 months, patients had an average cut in the left ventricular end diastolic volume index of 19 mL/m2; 25 mL/m2 after 24 months; and 23 mL/m2 after 5 years. The average drop in left ventricular end systolic volume index was 18 mL/m2 after 12 months; 24 mL/m2 after 24 months; and 23 mL/m2 at 5 years. Other measures of heart failure severity – the 6-minute walk, the Minnesota Living With Heart Failure score, and the Kansas City Cardiomyopathy Questionnaire score all improved significantly during the first 12 months on CRT therapy, and then held fairly steady during the subsequent 4 years. At 12 months, about 43% of patients had NYHA class I disease, about 53% had class II disease, and the rest had class III disease (which compared with rates of 18% with class I and 82% with class II at baseline). After 5 years, roughly 34% of patients had NYHA class I heart failure, about 56% had class II, about 9% had class III, and about 1% had class IV disease.
The majority of left-ventricular lead complications – the main adverse event of treatment – occurred during the first 12 months, with a cumulative incidence of 9% after 1 year. At 5 years, 13% of patients had experienced a lead complication.
The REVERSE trial was sponsored by Medtronic. Dr. Linde said that she has been a consultant to Medtronic and St. Jude and received research grants from Medtronic. Dr. Hindricks said that he received honoraria from and served on advisory boards for Biosense, Stereotaxis, St. Jude, and Biotronik. Dr. Auricchio said that he was a speaker for, consultant to, and received honoraria from Medtronic, St. Jude, and several other device and drug companies.
MUNICH – Cardiac resynchronization in patients with mild heart failure produced durable benefits during an average 4.5 years of follow-up, with evidence for ongoing cardiac remodeling and reduced disease progression in a series of 419 patients.
"The benefits of CRT [cardiac-resynchronization therapy] persisted, indicating that CRT attenuates disease progression in mildly symptomatic heart failure patients with wide QRS," Dr. Cecilia Linde said at the annual congress of the European Society of Cardiology.
"The results clearly show that the reverse remodeling produced by CRT was sustained over 5 years, and that’s good news for the treatment of patients with heart failure and an indication for cardiac resynchronization," commented Dr. Gerhard Hindricks, professor and director of the department of electrophysiology at the Leipzig (Germany) University Heart Center.
The findings "confirm the results from RAFT [Resynchronization-Defibrillation for Ambulatory Heart Failure Trial (N. Engl. J. Med. 2010;363:2385-95)] with respect to the long-term outcome of CRT in patients with NYHA [New York Heart Association] class II heart failure for survival and heart failure hospitalization, and provide novel, long-term information on reverse remodeling" from CRT in these patients, commented Dr. Angelo Auricchio, director of the cardiac electrophysiology program at the Ticino Cardiac Center in Lugano, Switzerland.
The data analyzed by Dr. Linde and her associates came from 419 patients originally enrolled in the "CRT on" arm of the REVERSE (Resynchronization Reverses Remodeling in Systolic Left Ventricular Dysfunction) trial, which enrolled patients with NYHA class I or II heart failure and a QRS duration or at least 120 msec. The primary end point of the study was the composite of death or heart failure hospitalization during the first 12 months of active CRT use compared with patients who received a CRT device that had not been turned on (J. Am. Coll. Cardiol. 2008;52:1834-43). The new analysis reported by Dr. Linde focused on 419 patients initially randomized to the CRT-on arm who continued on active CRT use for up to 5 years, with an average follow-up of 4.5 years. The researchers collected complete follow-up data for 95% of these patients through 3 years, for 89% through 4 years, and for 85% through 5 years.
At baseline, the patients had an average age of 63 years; 78% were men, and 82% had NYHA class I heart failure, with a average left ventricular ejection fraction of 27%. Their average QRS duration at entry was 153 msec, and 56% of the patients had an ischemic etiology for their heart failure.
Annual follow-up findings showed a consistent, linear rate of death and first heart failure hospitalization throughout follow-up, with an annual mortality rate of 3% and an annual rate of death or first heart-failure hospitalization of 6%, said Dr. Linde, a cardiologist at the Karolinska Institute in Stockholm.
The results also showed a stable pattern of left-ventricular reverse remodeling following the first year on CRT. After 12 months, patients had an average cut in the left ventricular end diastolic volume index of 19 mL/m2; 25 mL/m2 after 24 months; and 23 mL/m2 after 5 years. The average drop in left ventricular end systolic volume index was 18 mL/m2 after 12 months; 24 mL/m2 after 24 months; and 23 mL/m2 at 5 years. Other measures of heart failure severity – the 6-minute walk, the Minnesota Living With Heart Failure score, and the Kansas City Cardiomyopathy Questionnaire score all improved significantly during the first 12 months on CRT therapy, and then held fairly steady during the subsequent 4 years. At 12 months, about 43% of patients had NYHA class I disease, about 53% had class II disease, and the rest had class III disease (which compared with rates of 18% with class I and 82% with class II at baseline). After 5 years, roughly 34% of patients had NYHA class I heart failure, about 56% had class II, about 9% had class III, and about 1% had class IV disease.
The majority of left-ventricular lead complications – the main adverse event of treatment – occurred during the first 12 months, with a cumulative incidence of 9% after 1 year. At 5 years, 13% of patients had experienced a lead complication.
The REVERSE trial was sponsored by Medtronic. Dr. Linde said that she has been a consultant to Medtronic and St. Jude and received research grants from Medtronic. Dr. Hindricks said that he received honoraria from and served on advisory boards for Biosense, Stereotaxis, St. Jude, and Biotronik. Dr. Auricchio said that he was a speaker for, consultant to, and received honoraria from Medtronic, St. Jude, and several other device and drug companies.
AT THE ANNUAL CONGRESS OF THE EUROPEAN SOCIETY OF CARDIOLOGY
Major Finding: Five years of CRT cut average left-ventricular end-systolic volume index by 23 mL/m2 compared with 18 mL/m2 after 12 months.
Data Source: Data came from follow-up of 419 patients with mild heart failure randomized to the CRT-on arm of the REVERSE trial.
Disclosures: The REVERSE trial was sponsored by Medtronic. Dr. Linde said that she has been a consultant to Medtronic and St. Jude and received research grants from Medtronic. Dr. Hindricks said that he received honoraria from and served on advisory boards for Biosense, Stereotaxis, St. Jude, and Biotronik. Dr. Auricchio said that he was a speaker for, consultant to, and received honoraria from Medtronic, St. Jude, and several other device and drug companies.
Severe Psoriasis Linked to Doubled Diabetes Risk
MUNICH – Patients with severe psoriasis have a twofold increased risk of developing new-onset diabetes based on a review of more than 4 million Danish children and adults, the first nationwide cohort to be evaluated for a link between the two diseases.
"Our results underline the importance of considering psoriatic patients as a high-risk population in terms of diabetes and cardiovascular risk," Mr. Usman Khalid said at the Annual Congress of the European Society of Cardiology. "Screening for diabetes and cardiovascular risk factors in patients with psoriasis is warranted," and follows existing guidelines for managing patients with psoriasis, said Mr. Khalid, a researcher in the cardiovascular research unit of Gentofte Hospital in Copenhagen.

The likely mechanism underlying the association is the inflammatory state of patients with psoriasis, he added.
"These observations are new, interesting, and important," commented Dr. Lars Rydén, professor of cardiology at the Karolinska Institute in Stockholm. Physicians should add "carefully looking for diabetes" to their existing routine screening in psoriasis patients, he said.
The study used Danish national population and patient records for more than 4.6 million Danish citizens aged 10 years or older from 1997 through the end of 2009. The researchers excluded the 97,000 people who had diabetes, psoriasis, or both at entry into the database, which left just more than 4.5 million people, of whom 52,613 developed new-onset psoriasis during follow-up, and 4,465,643 people without psoriasis who served as the reference population. The researchers defined severe psoriasis as a case that required hospitalization at least three times, or patients diagnosed with psoriatic arthritis. The cohort of children and adults with incidence psoriasis included 45,829 mild cases, and 6,784 severe cases.
The researchers tallied the number of people who developed new-onset diabetes, both among those who never had psoriasis during the study period, and among those who developed psoriasis. They identified new diabetes cases based on initiation of treatment with one or more glucose-lowering drugs. During follow-up, the number of new cases of diabetes was 3.67/1,000 person-years among those with no psoriasis, 6.93/1,000 patient-years among patients with mild psoriasis, and 9.65/1,000 patient-years among patients with severe psoriasis. The vast majority of the diabetes that developed was type 2.
Using adjustments that controlled for potential confounders at baseline, including age, sex, comorbidities, medications, and socioeconomic status, the researchers found that, compared with the people without psoriasis, those with mild psoriasis had a statistically significant 47% increased incidence of diabetes, and severe psoriasis linked with a statistically significant twofold increased risk for diabetes, Mr. Khalid reported. The median time from onset of psoriasis to the first treatment for diabetes was about 6 years.
The analysis notably focused on patients who developed diabetes following initial development of psoriasis, which provided insight into the sequence of the two diseases that had not been available in previously-reported studies, noted Dr. Ole Ahlehoff, a cardiology researcher at Gentofte Hospital and collaborator on the study.
"I suggest screening patients with psoriasis once a year for cardiovascular risk factors, including hypertension, dyslipidemia, lifestyle factors, and diabetes based on their glucose level," said Dr. Ahlehoff, who spoke about the research at a press conference. Medical groups have released guidelines that recommend annual risk assessment for patients with severe psoriasis, such as psoriatic arthritis, including the European League Against Arthritis (Ann. Rheum. Dis. 2010;69:325-31), and the Scottish Intercollegiate Guidelines Network.
Mr. Khalid and Dr. Ahlehoff said that they had no disclosures. Dr. Rydén had no relevant disclosures.
MUNICH – Patients with severe psoriasis have a twofold increased risk of developing new-onset diabetes based on a review of more than 4 million Danish children and adults, the first nationwide cohort to be evaluated for a link between the two diseases.
"Our results underline the importance of considering psoriatic patients as a high-risk population in terms of diabetes and cardiovascular risk," Mr. Usman Khalid said at the Annual Congress of the European Society of Cardiology. "Screening for diabetes and cardiovascular risk factors in patients with psoriasis is warranted," and follows existing guidelines for managing patients with psoriasis, said Mr. Khalid, a researcher in the cardiovascular research unit of Gentofte Hospital in Copenhagen.
The likely mechanism underlying the association is the inflammatory state of patients with psoriasis, he added.
"These observations are new, interesting, and important," commented Dr. Lars Rydén, professor of cardiology at the Karolinska Institute in Stockholm. Physicians should add "carefully looking for diabetes" to their existing routine screening in psoriasis patients, he said.
The study used Danish national population and patient records for more than 4.6 million Danish citizens aged 10 years or older from 1997 through the end of 2009. The researchers excluded the 97,000 people who had diabetes, psoriasis, or both at entry into the database, which left just more than 4.5 million people, of whom 52,613 developed new-onset psoriasis during follow-up, and 4,465,643 people without psoriasis who served as the reference population. The researchers defined severe psoriasis as a case that required hospitalization at least three times, or patients diagnosed with psoriatic arthritis. The cohort of children and adults with incidence psoriasis included 45,829 mild cases, and 6,784 severe cases.
The researchers tallied the number of people who developed new-onset diabetes, both among those who never had psoriasis during the study period, and among those who developed psoriasis. They identified new diabetes cases based on initiation of treatment with one or more glucose-lowering drugs. During follow-up, the number of new cases of diabetes was 3.67/1,000 person-years among those with no psoriasis, 6.93/1,000 patient-years among patients with mild psoriasis, and 9.65/1,000 patient-years among patients with severe psoriasis. The vast majority of the diabetes that developed was type 2.
Using adjustments that controlled for potential confounders at baseline, including age, sex, comorbidities, medications, and socioeconomic status, the researchers found that, compared with the people without psoriasis, those with mild psoriasis had a statistically significant 47% increased incidence of diabetes, and severe psoriasis linked with a statistically significant twofold increased risk for diabetes, Mr. Khalid reported. The median time from onset of psoriasis to the first treatment for diabetes was about 6 years.
The analysis notably focused on patients who developed diabetes following initial development of psoriasis, which provided insight into the sequence of the two diseases that had not been available in previously-reported studies, noted Dr. Ole Ahlehoff, a cardiology researcher at Gentofte Hospital and collaborator on the study.
"I suggest screening patients with psoriasis once a year for cardiovascular risk factors, including hypertension, dyslipidemia, lifestyle factors, and diabetes based on their glucose level," said Dr. Ahlehoff, who spoke about the research at a press conference. Medical groups have released guidelines that recommend annual risk assessment for patients with severe psoriasis, such as psoriatic arthritis, including the European League Against Arthritis (Ann. Rheum. Dis. 2010;69:325-31), and the Scottish Intercollegiate Guidelines Network.
Mr. Khalid and Dr. Ahlehoff said that they had no disclosures. Dr. Rydén had no relevant disclosures.
MUNICH – Patients with severe psoriasis have a twofold increased risk of developing new-onset diabetes based on a review of more than 4 million Danish children and adults, the first nationwide cohort to be evaluated for a link between the two diseases.
"Our results underline the importance of considering psoriatic patients as a high-risk population in terms of diabetes and cardiovascular risk," Mr. Usman Khalid said at the Annual Congress of the European Society of Cardiology. "Screening for diabetes and cardiovascular risk factors in patients with psoriasis is warranted," and follows existing guidelines for managing patients with psoriasis, said Mr. Khalid, a researcher in the cardiovascular research unit of Gentofte Hospital in Copenhagen.
The likely mechanism underlying the association is the inflammatory state of patients with psoriasis, he added.
"These observations are new, interesting, and important," commented Dr. Lars Rydén, professor of cardiology at the Karolinska Institute in Stockholm. Physicians should add "carefully looking for diabetes" to their existing routine screening in psoriasis patients, he said.
The study used Danish national population and patient records for more than 4.6 million Danish citizens aged 10 years or older from 1997 through the end of 2009. The researchers excluded the 97,000 people who had diabetes, psoriasis, or both at entry into the database, which left just more than 4.5 million people, of whom 52,613 developed new-onset psoriasis during follow-up, and 4,465,643 people without psoriasis who served as the reference population. The researchers defined severe psoriasis as a case that required hospitalization at least three times, or patients diagnosed with psoriatic arthritis. The cohort of children and adults with incidence psoriasis included 45,829 mild cases, and 6,784 severe cases.
The researchers tallied the number of people who developed new-onset diabetes, both among those who never had psoriasis during the study period, and among those who developed psoriasis. They identified new diabetes cases based on initiation of treatment with one or more glucose-lowering drugs. During follow-up, the number of new cases of diabetes was 3.67/1,000 person-years among those with no psoriasis, 6.93/1,000 patient-years among patients with mild psoriasis, and 9.65/1,000 patient-years among patients with severe psoriasis. The vast majority of the diabetes that developed was type 2.
Using adjustments that controlled for potential confounders at baseline, including age, sex, comorbidities, medications, and socioeconomic status, the researchers found that, compared with the people without psoriasis, those with mild psoriasis had a statistically significant 47% increased incidence of diabetes, and severe psoriasis linked with a statistically significant twofold increased risk for diabetes, Mr. Khalid reported. The median time from onset of psoriasis to the first treatment for diabetes was about 6 years.
The analysis notably focused on patients who developed diabetes following initial development of psoriasis, which provided insight into the sequence of the two diseases that had not been available in previously-reported studies, noted Dr. Ole Ahlehoff, a cardiology researcher at Gentofte Hospital and collaborator on the study.
"I suggest screening patients with psoriasis once a year for cardiovascular risk factors, including hypertension, dyslipidemia, lifestyle factors, and diabetes based on their glucose level," said Dr. Ahlehoff, who spoke about the research at a press conference. Medical groups have released guidelines that recommend annual risk assessment for patients with severe psoriasis, such as psoriatic arthritis, including the European League Against Arthritis (Ann. Rheum. Dis. 2010;69:325-31), and the Scottish Intercollegiate Guidelines Network.
Mr. Khalid and Dr. Ahlehoff said that they had no disclosures. Dr. Rydén had no relevant disclosures.
AT THE ANNUAL CONGRESS OF THE EUROPEAN SOCIETY OF CARDIOLOGY
Major Finding: New-onset, severe psoriasis doubled the risk for incident diabetes during a median of 6 years compared with people without psoriasis.
Data Source: Data came from an analysis of 4.5 million Danish children and adults followed during 1997-2009.
Disclosures: Mr. Khalid and Dr. Ahlehoff said that they had no disclosures. Dr. Rydén had no relevant disclosures.

Quinine Doesn't Mix Well With Heart Failure, Ischemic Disease
MUNICH – Treatment with quinine increased the mortality of heart failure patients who also had ischemic heart disease and received a beta-blocker by a statistically significant 16% in a study of more than 136,000 Danish heart failure patients during 1997-2010.
Although experts suggest that such use of quinine is not commonplace in the United States, the analysis of Danish patient and population registries showed that quinine was used in the treatment of 10% of HF patients. Quinine, an antimalarial drug, is often prescribed off-label to treat leg cramps, a common problem in patients with HF, Dr. Charlotte Andersson said at the annual congress of the European Society of Cardiology.

"Clinicians should attempt to find a treatment for leg cramps other than prescribing quinine" in patients with chronic HF, especially those with ischemic heart disease, said Dr. Andersson, a cardiologist at Gentofte Hospital in Copenhagen. She also expressed surprise that the dangerous interaction was greatest among patients who also received a beta-blocker drug.
"I would have expected patients on beta-blockers to be protected, but they weren’t, so probably conduction blocks [caused by quinine] are the problem," she said. "We found the highest risk of death during the first week of quinine treatment in patients with ischemic heart disease and on a beta-blocker, but if quinine was used chronically it still increased the risk."
"This is very interesting and important information," commented Dr. Marco Metra, a cardiologist at the University of Brescia, Italy. "The question is, what is the mechanism of the interaction of quinine and beta-blockers? We think that beta-blockers are protective against arrhythmias caused by QT prolongation. In this case, [the problem may be] bradycardia" caused by a beta-blocker that interacts with an effect from quinine, he suggested.
Dr. Andersson and her associates reviewed Danish national patient and population registry records collected during 1997-2010, which included 136,427 patients discharged from a hospital with a diagnosis of heart failure and who were alive at least 30 days following discharge. Their average age was 74 years, 47% were women, and 65% died during a median follow-up of 2.8 years.
The records showed that 14,306 patients (10%) had received at least one course of treatment with quinine, at a dosage of 100 or 200 mg/day. Overall, patients who received quinine had a 3% increased risk of death after adjustment for age and HF severity, which was estimated based on the dosage of loop diuretics they received. This increased risk fell just short of statistical significance.
However, further analysis showed that specific subgroups of HF patients faced a higher mortality threat from quinine treatment. Patients with ischemic heart disease (37% of the HF population) who received treatment with a beta-blocker (60% of the HF patients with ischemic heart disease) had the highest mortality when they received quinine, 16% higher than for similar patients who did not get quinine, after adjustment for age and HF severity. This meant that for every 38 patients with this clinical profile treated with quinine, 1 died.
Among HF patients with ischemic heart disease who did not receive a beta-blocker, the mortality rate was elevated by a statistically significant 8% among quinine users compared with quinine nonusers; 1 death occurred for every 46 patients in this category. And among HF patients who received a beta-blocker but who did not have ischemic heart disease, quinine treatment raised mortality by a statistically significant 9%, or 1 death for every 97 patients treated with quinine who fit this clinical profile.
Dr. Andersson said that she and her associates had no disclosures.
MUNICH – Treatment with quinine increased the mortality of heart failure patients who also had ischemic heart disease and received a beta-blocker by a statistically significant 16% in a study of more than 136,000 Danish heart failure patients during 1997-2010.
Although experts suggest that such use of quinine is not commonplace in the United States, the analysis of Danish patient and population registries showed that quinine was used in the treatment of 10% of HF patients. Quinine, an antimalarial drug, is often prescribed off-label to treat leg cramps, a common problem in patients with HF, Dr. Charlotte Andersson said at the annual congress of the European Society of Cardiology.
"Clinicians should attempt to find a treatment for leg cramps other than prescribing quinine" in patients with chronic HF, especially those with ischemic heart disease, said Dr. Andersson, a cardiologist at Gentofte Hospital in Copenhagen. She also expressed surprise that the dangerous interaction was greatest among patients who also received a beta-blocker drug.
"I would have expected patients on beta-blockers to be protected, but they weren’t, so probably conduction blocks [caused by quinine] are the problem," she said. "We found the highest risk of death during the first week of quinine treatment in patients with ischemic heart disease and on a beta-blocker, but if quinine was used chronically it still increased the risk."
"This is very interesting and important information," commented Dr. Marco Metra, a cardiologist at the University of Brescia, Italy. "The question is, what is the mechanism of the interaction of quinine and beta-blockers? We think that beta-blockers are protective against arrhythmias caused by QT prolongation. In this case, [the problem may be] bradycardia" caused by a beta-blocker that interacts with an effect from quinine, he suggested.
Dr. Andersson and her associates reviewed Danish national patient and population registry records collected during 1997-2010, which included 136,427 patients discharged from a hospital with a diagnosis of heart failure and who were alive at least 30 days following discharge. Their average age was 74 years, 47% were women, and 65% died during a median follow-up of 2.8 years.
The records showed that 14,306 patients (10%) had received at least one course of treatment with quinine, at a dosage of 100 or 200 mg/day. Overall, patients who received quinine had a 3% increased risk of death after adjustment for age and HF severity, which was estimated based on the dosage of loop diuretics they received. This increased risk fell just short of statistical significance.
However, further analysis showed that specific subgroups of HF patients faced a higher mortality threat from quinine treatment. Patients with ischemic heart disease (37% of the HF population) who received treatment with a beta-blocker (60% of the HF patients with ischemic heart disease) had the highest mortality when they received quinine, 16% higher than for similar patients who did not get quinine, after adjustment for age and HF severity. This meant that for every 38 patients with this clinical profile treated with quinine, 1 died.
Among HF patients with ischemic heart disease who did not receive a beta-blocker, the mortality rate was elevated by a statistically significant 8% among quinine users compared with quinine nonusers; 1 death occurred for every 46 patients in this category. And among HF patients who received a beta-blocker but who did not have ischemic heart disease, quinine treatment raised mortality by a statistically significant 9%, or 1 death for every 97 patients treated with quinine who fit this clinical profile.
Dr. Andersson said that she and her associates had no disclosures.
MUNICH – Treatment with quinine increased the mortality of heart failure patients who also had ischemic heart disease and received a beta-blocker by a statistically significant 16% in a study of more than 136,000 Danish heart failure patients during 1997-2010.
Although experts suggest that such use of quinine is not commonplace in the United States, the analysis of Danish patient and population registries showed that quinine was used in the treatment of 10% of HF patients. Quinine, an antimalarial drug, is often prescribed off-label to treat leg cramps, a common problem in patients with HF, Dr. Charlotte Andersson said at the annual congress of the European Society of Cardiology.
"Clinicians should attempt to find a treatment for leg cramps other than prescribing quinine" in patients with chronic HF, especially those with ischemic heart disease, said Dr. Andersson, a cardiologist at Gentofte Hospital in Copenhagen. She also expressed surprise that the dangerous interaction was greatest among patients who also received a beta-blocker drug.
"I would have expected patients on beta-blockers to be protected, but they weren’t, so probably conduction blocks [caused by quinine] are the problem," she said. "We found the highest risk of death during the first week of quinine treatment in patients with ischemic heart disease and on a beta-blocker, but if quinine was used chronically it still increased the risk."
"This is very interesting and important information," commented Dr. Marco Metra, a cardiologist at the University of Brescia, Italy. "The question is, what is the mechanism of the interaction of quinine and beta-blockers? We think that beta-blockers are protective against arrhythmias caused by QT prolongation. In this case, [the problem may be] bradycardia" caused by a beta-blocker that interacts with an effect from quinine, he suggested.
Dr. Andersson and her associates reviewed Danish national patient and population registry records collected during 1997-2010, which included 136,427 patients discharged from a hospital with a diagnosis of heart failure and who were alive at least 30 days following discharge. Their average age was 74 years, 47% were women, and 65% died during a median follow-up of 2.8 years.
The records showed that 14,306 patients (10%) had received at least one course of treatment with quinine, at a dosage of 100 or 200 mg/day. Overall, patients who received quinine had a 3% increased risk of death after adjustment for age and HF severity, which was estimated based on the dosage of loop diuretics they received. This increased risk fell just short of statistical significance.
However, further analysis showed that specific subgroups of HF patients faced a higher mortality threat from quinine treatment. Patients with ischemic heart disease (37% of the HF population) who received treatment with a beta-blocker (60% of the HF patients with ischemic heart disease) had the highest mortality when they received quinine, 16% higher than for similar patients who did not get quinine, after adjustment for age and HF severity. This meant that for every 38 patients with this clinical profile treated with quinine, 1 died.
Among HF patients with ischemic heart disease who did not receive a beta-blocker, the mortality rate was elevated by a statistically significant 8% among quinine users compared with quinine nonusers; 1 death occurred for every 46 patients in this category. And among HF patients who received a beta-blocker but who did not have ischemic heart disease, quinine treatment raised mortality by a statistically significant 9%, or 1 death for every 97 patients treated with quinine who fit this clinical profile.
Dr. Andersson said that she and her associates had no disclosures.
AT THE ANNUAL CONGRESS OF THE EUROPEAN SOCIETY OF CARDIOLOGY
Major Finding: Among patients with heart failure and ischemic heart disease on beta-blocker treatment, quinine dosing boosted mortality by 16%.
Data Source: This was a review of Danish patient and population records that included 136,427 patients discharged with a diagnosis of heart failure during 1997-2010.
Disclosures: Dr. Andersson said that she and her associates had no disclosures.

Adding Clopidogrel to Anticoagulant Is Plenty for PCI
MUNICH – Patients who require chronic oral anticoagulant treatment and received a coronary stent had significantly fewer bleeding events and better outcomes if they received only added clopidogrel than if they were treated with clopidogrel plus aspirin on top of their anticoagulant in the WOEST trial.
As the first study to prospectively test the best combination of antiplatelet drugs to use on top of anticoagulant treatment in patients following coronary stenting, experts hailed the finding as important evidence to address a common and vexing clinical situation.

"We propose that a strategy of oral anticoagulants plus clopidogrel, but without aspirin could be applied in this group of high-risk patients who are on an oral anticoagulant and undergo percutaneous coronary intervention," said Dr. Willem Dewilde, lead investigator for the study, at the Annual Congress of the European Society of Cardiology.
"We are faced with a huge clinical problem, with an increasing number of patients with atrial fibrillation who undergo stent procedures, so this finding has major implications," commented Dr. David R. Holmes Jr., professor of medicine and a cardiologist at the Mayo Clinic in Rochester, Minn. "Bleeding is such a huge issue, and we now have scientific data that says [withholding aspirin] is safe and produces less bleeding."
"The taboo against discontinuing or omitting aspirin has been broken," commented Dr. Marco Valgimigli, director of the coronary catheterization laboratory at the University Hospital of Ferrara (Italy).
The WOEST (What is the Optimal Antiplatelet and Anticoagulant Therapy in Patients with Oral Anticoagulation and Coronary Stenting) trial enrolled 573 patients who required chronic oral anticoagulant treatment and were scheduled for coronary stenting at 15 centers in the Netherlands and Belgium. The patients averaged about 70 years of age, and about 80% were men. A total of 70% received daily, oral treatment with an anticoagulant for atrial fibrillation or flutter, about 10% for a mechanical valve, and the remaining 20% had other reasons for their regimen, including treatment of a pulmonary embolus. All patients received warfarin or another oral anticoagulant such as acenocoumarol. About two-thirds of the patients received a drug-eluting coronary stent, with the remainder receiving a bare-metal stent.
Following stenting, the researchers randomized patients to a regimen of their oral anticoagulant plus 75 mg clopidogrel daily (dual therapy), or their oral anticoagulant, clopidogrel, and 80 mg aspirin daily (triple therapy). The antiplatelet part of the regimen continued for at least 1 month in patients who received a bare-metal stent and for a year in those who got a drug-eluting stent.

After a year of follow-up, the incidence of the primary end point – total bleeding events – occurred in 20% of the 279 evaluable patients who received dual therapy and in 45% of 284 evaluable patients on triple therapy, a statistically significant difference, reported Dr. Dewilde, a cardiologist at TweeSteden Hospital in Tilberg, the Netherlands. The study’s secondary end point – the combined rate of death, myocardial infarction, target vessel revascularization, stroke, or stent thrombosis – occurred in 11% of patients on double therapy and 18% of those on triple therapy, a statistically significant difference.
Patients on dual therapy had a significantly lower rate of all-cause death, compared with the triple-therapy patients, 3% compared with 6%, and numerically lower levels of stent thrombosis, stroke, and myocardial infarction, although the differences for these individual end points were not statistically significant.
The dual-therapy patients also had significantly lower rates of minimal or minor bleeds, measured by the TIMI (Thrombolysis in Myocardial Infarction) criteria, and the rate of TIMI major bleeds was also lower on dual therapy, but not significantly. The dual-therapy patients had strikingly lower numbers of bleeds at gastrointestinal sites and skin sites, compared with the triple-therapy patients.
"TIMI minimal and minor bleeds are anything but minor from a clinical standpoint," noted Dr. Valgimigli. "Double therapy with clopidogrel and warfarin seems a very reasonable therapy based on WOEST, especially in DES-treated patients."
Dr. Dewilde and Dr. Holmes said that they had no disclosures. Dr. Valgimigli said that he has served on the speakers bureau for AstraZeneca, Eli Lilly, Iroko, and other companies that market vascular devices. He has also been on advisory boards for Eli Lilly, and several companies that make vascular devices. Dr. Smith said that he had no disclosures.
I have participated on committees that made recommendations on managing patients following percutaneous coronary interventions, and for secondary prevention of coronary disease, and in both cases, we realized that there was a major need for a randomized, controlled trial that examined exactly the question addressed by WOEST: What is the best and safest antiplatelet regimen to use on patients who have just received a coronary stent and who require oral anticoagulant therapy because they also have atrial fibrillation, a mechanical valve, or some other indication? Until now, the recommendations we came up with could only be based on expert opinion.
Because the WOEST trial is the first to address this important issue, it will need careful review so that we can decide how it should affect practice. This review will have to closely examine several important issues: Was the WOEST study adequately powered to legitimately address the issues of safety and efficacy in these patients, including the rate of stent thrombosis? Also, we will need to know the international normalized ratio (INR) that these patients maintained throughout the 1-year follow-up of the study. Were there differences in the INRs that could explain the bleeding differences? Another issue is the patient’s age.

|
|
Having data from a good, prospective, controlled trial that looked at the question of how to manage patients who received a coronary stent while on oral anticoagulant therapy is a significant step forward, but it would be premature to make changes in the recommended treatment of patients like these until all elements of the study’s design and findings undergo careful analysis.
Sidney C. Smith, M.D., is a professor of medicine and an interventional cardiologist at the Center for Heart & Vascular Care of the University of North Carolina in Chapel Hill. He chaired the committee of the American Heart Association and American College of Cardiology that issued recommendations for the secondary prevention of coronary disease in 2011 (Circulation 2011;124:2458-73). He said that he had no disclosures. Dr. Smith made these comments in an interview.
What is the Optimal Antiplatelet and Anticoagulant Therapy in Patients with Oral Anticoagulation and Coronary Stenting trial,
I have participated on committees that made recommendations on managing patients following percutaneous coronary interventions, and for secondary prevention of coronary disease, and in both cases, we realized that there was a major need for a randomized, controlled trial that examined exactly the question addressed by WOEST: What is the best and safest antiplatelet regimen to use on patients who have just received a coronary stent and who require oral anticoagulant therapy because they also have atrial fibrillation, a mechanical valve, or some other indication? Until now, the recommendations we came up with could only be based on expert opinion.
Because the WOEST trial is the first to address this important issue, it will need careful review so that we can decide how it should affect practice. This review will have to closely examine several important issues: Was the WOEST study adequately powered to legitimately address the issues of safety and efficacy in these patients, including the rate of stent thrombosis? Also, we will need to know the international normalized ratio (INR) that these patients maintained throughout the 1-year follow-up of the study. Were there differences in the INRs that could explain the bleeding differences? Another issue is the patient’s age.
|
|
|
Having data from a good, prospective, controlled trial that looked at the question of how to manage patients who received a coronary stent while on oral anticoagulant therapy is a significant step forward, but it would be premature to make changes in the recommended treatment of patients like these until all elements of the study’s design and findings undergo careful analysis.
Sidney C. Smith, M.D., is a professor of medicine and an interventional cardiologist at the Center for Heart & Vascular Care of the University of North Carolina in Chapel Hill. He chaired the committee of the American Heart Association and American College of Cardiology that issued recommendations for the secondary prevention of coronary disease in 2011 (Circulation 2011;124:2458-73). He said that he had no disclosures. Dr. Smith made these comments in an interview.
I have participated on committees that made recommendations on managing patients following percutaneous coronary interventions, and for secondary prevention of coronary disease, and in both cases, we realized that there was a major need for a randomized, controlled trial that examined exactly the question addressed by WOEST: What is the best and safest antiplatelet regimen to use on patients who have just received a coronary stent and who require oral anticoagulant therapy because they also have atrial fibrillation, a mechanical valve, or some other indication? Until now, the recommendations we came up with could only be based on expert opinion.
Because the WOEST trial is the first to address this important issue, it will need careful review so that we can decide how it should affect practice. This review will have to closely examine several important issues: Was the WOEST study adequately powered to legitimately address the issues of safety and efficacy in these patients, including the rate of stent thrombosis? Also, we will need to know the international normalized ratio (INR) that these patients maintained throughout the 1-year follow-up of the study. Were there differences in the INRs that could explain the bleeding differences? Another issue is the patient’s age.
|
|
|
Having data from a good, prospective, controlled trial that looked at the question of how to manage patients who received a coronary stent while on oral anticoagulant therapy is a significant step forward, but it would be premature to make changes in the recommended treatment of patients like these until all elements of the study’s design and findings undergo careful analysis.
Sidney C. Smith, M.D., is a professor of medicine and an interventional cardiologist at the Center for Heart & Vascular Care of the University of North Carolina in Chapel Hill. He chaired the committee of the American Heart Association and American College of Cardiology that issued recommendations for the secondary prevention of coronary disease in 2011 (Circulation 2011;124:2458-73). He said that he had no disclosures. Dr. Smith made these comments in an interview.
MUNICH – Patients who require chronic oral anticoagulant treatment and received a coronary stent had significantly fewer bleeding events and better outcomes if they received only added clopidogrel than if they were treated with clopidogrel plus aspirin on top of their anticoagulant in the WOEST trial.
As the first study to prospectively test the best combination of antiplatelet drugs to use on top of anticoagulant treatment in patients following coronary stenting, experts hailed the finding as important evidence to address a common and vexing clinical situation.
"We propose that a strategy of oral anticoagulants plus clopidogrel, but without aspirin could be applied in this group of high-risk patients who are on an oral anticoagulant and undergo percutaneous coronary intervention," said Dr. Willem Dewilde, lead investigator for the study, at the Annual Congress of the European Society of Cardiology.
"We are faced with a huge clinical problem, with an increasing number of patients with atrial fibrillation who undergo stent procedures, so this finding has major implications," commented Dr. David R. Holmes Jr., professor of medicine and a cardiologist at the Mayo Clinic in Rochester, Minn. "Bleeding is such a huge issue, and we now have scientific data that says [withholding aspirin] is safe and produces less bleeding."
"The taboo against discontinuing or omitting aspirin has been broken," commented Dr. Marco Valgimigli, director of the coronary catheterization laboratory at the University Hospital of Ferrara (Italy).
The WOEST (What is the Optimal Antiplatelet and Anticoagulant Therapy in Patients with Oral Anticoagulation and Coronary Stenting) trial enrolled 573 patients who required chronic oral anticoagulant treatment and were scheduled for coronary stenting at 15 centers in the Netherlands and Belgium. The patients averaged about 70 years of age, and about 80% were men. A total of 70% received daily, oral treatment with an anticoagulant for atrial fibrillation or flutter, about 10% for a mechanical valve, and the remaining 20% had other reasons for their regimen, including treatment of a pulmonary embolus. All patients received warfarin or another oral anticoagulant such as acenocoumarol. About two-thirds of the patients received a drug-eluting coronary stent, with the remainder receiving a bare-metal stent.
Following stenting, the researchers randomized patients to a regimen of their oral anticoagulant plus 75 mg clopidogrel daily (dual therapy), or their oral anticoagulant, clopidogrel, and 80 mg aspirin daily (triple therapy). The antiplatelet part of the regimen continued for at least 1 month in patients who received a bare-metal stent and for a year in those who got a drug-eluting stent.
After a year of follow-up, the incidence of the primary end point – total bleeding events – occurred in 20% of the 279 evaluable patients who received dual therapy and in 45% of 284 evaluable patients on triple therapy, a statistically significant difference, reported Dr. Dewilde, a cardiologist at TweeSteden Hospital in Tilberg, the Netherlands. The study’s secondary end point – the combined rate of death, myocardial infarction, target vessel revascularization, stroke, or stent thrombosis – occurred in 11% of patients on double therapy and 18% of those on triple therapy, a statistically significant difference.
Patients on dual therapy had a significantly lower rate of all-cause death, compared with the triple-therapy patients, 3% compared with 6%, and numerically lower levels of stent thrombosis, stroke, and myocardial infarction, although the differences for these individual end points were not statistically significant.
The dual-therapy patients also had significantly lower rates of minimal or minor bleeds, measured by the TIMI (Thrombolysis in Myocardial Infarction) criteria, and the rate of TIMI major bleeds was also lower on dual therapy, but not significantly. The dual-therapy patients had strikingly lower numbers of bleeds at gastrointestinal sites and skin sites, compared with the triple-therapy patients.
"TIMI minimal and minor bleeds are anything but minor from a clinical standpoint," noted Dr. Valgimigli. "Double therapy with clopidogrel and warfarin seems a very reasonable therapy based on WOEST, especially in DES-treated patients."
Dr. Dewilde and Dr. Holmes said that they had no disclosures. Dr. Valgimigli said that he has served on the speakers bureau for AstraZeneca, Eli Lilly, Iroko, and other companies that market vascular devices. He has also been on advisory boards for Eli Lilly, and several companies that make vascular devices. Dr. Smith said that he had no disclosures.
MUNICH – Patients who require chronic oral anticoagulant treatment and received a coronary stent had significantly fewer bleeding events and better outcomes if they received only added clopidogrel than if they were treated with clopidogrel plus aspirin on top of their anticoagulant in the WOEST trial.
As the first study to prospectively test the best combination of antiplatelet drugs to use on top of anticoagulant treatment in patients following coronary stenting, experts hailed the finding as important evidence to address a common and vexing clinical situation.
"We propose that a strategy of oral anticoagulants plus clopidogrel, but without aspirin could be applied in this group of high-risk patients who are on an oral anticoagulant and undergo percutaneous coronary intervention," said Dr. Willem Dewilde, lead investigator for the study, at the Annual Congress of the European Society of Cardiology.
"We are faced with a huge clinical problem, with an increasing number of patients with atrial fibrillation who undergo stent procedures, so this finding has major implications," commented Dr. David R. Holmes Jr., professor of medicine and a cardiologist at the Mayo Clinic in Rochester, Minn. "Bleeding is such a huge issue, and we now have scientific data that says [withholding aspirin] is safe and produces less bleeding."
"The taboo against discontinuing or omitting aspirin has been broken," commented Dr. Marco Valgimigli, director of the coronary catheterization laboratory at the University Hospital of Ferrara (Italy).
The WOEST (What is the Optimal Antiplatelet and Anticoagulant Therapy in Patients with Oral Anticoagulation and Coronary Stenting) trial enrolled 573 patients who required chronic oral anticoagulant treatment and were scheduled for coronary stenting at 15 centers in the Netherlands and Belgium. The patients averaged about 70 years of age, and about 80% were men. A total of 70% received daily, oral treatment with an anticoagulant for atrial fibrillation or flutter, about 10% for a mechanical valve, and the remaining 20% had other reasons for their regimen, including treatment of a pulmonary embolus. All patients received warfarin or another oral anticoagulant such as acenocoumarol. About two-thirds of the patients received a drug-eluting coronary stent, with the remainder receiving a bare-metal stent.
Following stenting, the researchers randomized patients to a regimen of their oral anticoagulant plus 75 mg clopidogrel daily (dual therapy), or their oral anticoagulant, clopidogrel, and 80 mg aspirin daily (triple therapy). The antiplatelet part of the regimen continued for at least 1 month in patients who received a bare-metal stent and for a year in those who got a drug-eluting stent.
After a year of follow-up, the incidence of the primary end point – total bleeding events – occurred in 20% of the 279 evaluable patients who received dual therapy and in 45% of 284 evaluable patients on triple therapy, a statistically significant difference, reported Dr. Dewilde, a cardiologist at TweeSteden Hospital in Tilberg, the Netherlands. The study’s secondary end point – the combined rate of death, myocardial infarction, target vessel revascularization, stroke, or stent thrombosis – occurred in 11% of patients on double therapy and 18% of those on triple therapy, a statistically significant difference.
Patients on dual therapy had a significantly lower rate of all-cause death, compared with the triple-therapy patients, 3% compared with 6%, and numerically lower levels of stent thrombosis, stroke, and myocardial infarction, although the differences for these individual end points were not statistically significant.
The dual-therapy patients also had significantly lower rates of minimal or minor bleeds, measured by the TIMI (Thrombolysis in Myocardial Infarction) criteria, and the rate of TIMI major bleeds was also lower on dual therapy, but not significantly. The dual-therapy patients had strikingly lower numbers of bleeds at gastrointestinal sites and skin sites, compared with the triple-therapy patients.
"TIMI minimal and minor bleeds are anything but minor from a clinical standpoint," noted Dr. Valgimigli. "Double therapy with clopidogrel and warfarin seems a very reasonable therapy based on WOEST, especially in DES-treated patients."
Dr. Dewilde and Dr. Holmes said that they had no disclosures. Dr. Valgimigli said that he has served on the speakers bureau for AstraZeneca, Eli Lilly, Iroko, and other companies that market vascular devices. He has also been on advisory boards for Eli Lilly, and several companies that make vascular devices. Dr. Smith said that he had no disclosures.
What is the Optimal Antiplatelet and Anticoagulant Therapy in Patients with Oral Anticoagulation and Coronary Stenting trial,
What is the Optimal Antiplatelet and Anticoagulant Therapy in Patients with Oral Anticoagulation and Coronary Stenting trial,
AT THE ANNUAL CONGRESS OF THE EUROPEAN SOCIETY OF CARDIOLOGY
Major Finding: In coronary-stented patients on oral anticoagulation, bleeds occurred in 20% on clopidogrel only and 45% on clopidogrel and aspirin.
Data Source: Data came from the WOEST study, which randomized 573 patients at 15 centers in the Netherlands and Belgium and followed them for 1 year.
Disclosures: Dr. Dewilde said that he and his associates on the WOEST study had no disclosures. Dr. Holmes said that he had no disclosures. Dr. Dr. Valgimigli said that he has served on the speakers bureau for AstraZeneca, Eli Lilly, Iroko, and other companies that market vascular devices. He has also been on advisory boards for Eli Lilly and several companies that make vascular devices

Anti-TNF Use Linked to Cardiovascular-Disease Drop in RA
Evidence continues to accumulate that the potent anti-inflammatory effect of drugs that block tumor necrosis factor can significantly dampen cardiovascular-disease risk in patients with rheumatoid arthritis, even though definitive proof from a prospective trial is still lacking.
Two recent pieces of suggestive evidence came from a meta-analysis of four placebo-controlled trials of adalimumab (Humira) that together included nearly 2,500 patients with rheumatoid arthritis, and from two prospective cohort studies of 828 RA patients that compared the outcomes of those treated with either adalimumab or etanercept (Enbrel) to the outcomes of similar patients who did not receive an anti–tumor necrosis factor drug.
In both studies, treatment with an anti-TNF agent was linked to a statistically significant cut in cardiovascular (CV) events of about 50%.
These results support another recent, similar finding reported in June at the Annual European Congress of Rheumatology in London. In that study, analysis of medical records from more than 109,000 U.S. patients with RA showed that every 6 months of treatment with an anti-TNF drug reduced the rate of CV events by 13%, compared with RA patients who did not receive a TNF blocker.
The meta-analysis of four trials included data collected in the ARMADA (Arthritis Rheum. 2003;48:35-45), DEO19 (Arthritis Rheum. 2004;50:1400-11), PREMIER (Arthritis Rheum. 2006;54:26-37), and OPTIMA (Ann. Rheum. Dis. 2012 May [doi: 10.1136/annrheumdis-2011-201247]) trials. Collectively, these four studies included 1,411 RA patients treated with both adalimumab and methotrexate, and 1,036 patients who received methotrexate but no anti-TNF drug. At baseline, patients in these two treatment groups had similar demographic and CV disease risk profiles.

During study periods that ranged from 16 to 104 weeks, the incidence of major adverse CV events was 1.3% in patients who received adalimumab, and 2% in those who didn’t. This difference represents a statistically significant, 2/3 drop in the risk for a major CV event in a proportional-hazard model, said Dr. Gerd Burmester, lead investigator for the analysis and a rheumatologist and professor of medicine at Charité Hospital in Berlin. His analysis also showed a statistically significant risk reduction with adalimumab in the rate of nonfatal myocardial infarction, but no significant effect of adalimumab on the rates of nonfatal myocardial infarction or nonfatal stroke compared with patients not receiving adalimumab.
A limitation of the analysis was that none of the four trials was powered to assess CV outcomes, Dr. Burmester said.
The second study used data collected from two cohorts: the CARRÉ (Cardiovascular Research and Rheumatoid Arthritis) study, a prospective Dutch cohort study of 309 randomly selected RA patients who were not treated with a TNF blocker; and the Biologics cohort, which involves 519 Dutch RA patients who have been followed since they began treatment with an anti-TNF drug for the first time, either adalimumab or etanercept.
During follow-up, there were 8 CV events per 1,000 patient-years in the cohort receiving an anti-TNF drug, compared with 23 events per 1,000 patient-years in patients not on an anti-TNF agent, a statistically significant reduction. In a proportional hazards model that adjusted for baseline differences in age and gender, treatment with an anti-TNF agent reduced the rate of CV events by about half, a statistically significant effect, said Dr. Alper M. van Sijl, a researcher at the Reade Centre for Rehabilitation and Rheumatology at the VU Medical Center in Amsterdam.
"Our observations confirm the association between strong suppression of inflammation [with an anti-TNF drug] and curbing the cardiovascular risk in RA," said Dr. van Sijl and his colleagues. But they cautioned that because of the design of the study it remains unclear whether this was a real effect of the anti-TNF drugs, or a bias to treat patients with lower CV disease risk with a TNF blocker.
Dr. Burmester said that he has been a consultant to, served as a speaker for, and received research support from Abbott, the company that markets adalimumab. Dr. van Silj said that he had no disclosures.
Evidence continues to accumulate that the potent anti-inflammatory effect of drugs that block tumor necrosis factor can significantly dampen cardiovascular-disease risk in patients with rheumatoid arthritis, even though definitive proof from a prospective trial is still lacking.
Two recent pieces of suggestive evidence came from a meta-analysis of four placebo-controlled trials of adalimumab (Humira) that together included nearly 2,500 patients with rheumatoid arthritis, and from two prospective cohort studies of 828 RA patients that compared the outcomes of those treated with either adalimumab or etanercept (Enbrel) to the outcomes of similar patients who did not receive an anti–tumor necrosis factor drug.
In both studies, treatment with an anti-TNF agent was linked to a statistically significant cut in cardiovascular (CV) events of about 50%.
These results support another recent, similar finding reported in June at the Annual European Congress of Rheumatology in London. In that study, analysis of medical records from more than 109,000 U.S. patients with RA showed that every 6 months of treatment with an anti-TNF drug reduced the rate of CV events by 13%, compared with RA patients who did not receive a TNF blocker.
The meta-analysis of four trials included data collected in the ARMADA (Arthritis Rheum. 2003;48:35-45), DEO19 (Arthritis Rheum. 2004;50:1400-11), PREMIER (Arthritis Rheum. 2006;54:26-37), and OPTIMA (Ann. Rheum. Dis. 2012 May [doi: 10.1136/annrheumdis-2011-201247]) trials. Collectively, these four studies included 1,411 RA patients treated with both adalimumab and methotrexate, and 1,036 patients who received methotrexate but no anti-TNF drug. At baseline, patients in these two treatment groups had similar demographic and CV disease risk profiles.
During study periods that ranged from 16 to 104 weeks, the incidence of major adverse CV events was 1.3% in patients who received adalimumab, and 2% in those who didn’t. This difference represents a statistically significant, 2/3 drop in the risk for a major CV event in a proportional-hazard model, said Dr. Gerd Burmester, lead investigator for the analysis and a rheumatologist and professor of medicine at Charité Hospital in Berlin. His analysis also showed a statistically significant risk reduction with adalimumab in the rate of nonfatal myocardial infarction, but no significant effect of adalimumab on the rates of nonfatal myocardial infarction or nonfatal stroke compared with patients not receiving adalimumab.
A limitation of the analysis was that none of the four trials was powered to assess CV outcomes, Dr. Burmester said.
The second study used data collected from two cohorts: the CARRÉ (Cardiovascular Research and Rheumatoid Arthritis) study, a prospective Dutch cohort study of 309 randomly selected RA patients who were not treated with a TNF blocker; and the Biologics cohort, which involves 519 Dutch RA patients who have been followed since they began treatment with an anti-TNF drug for the first time, either adalimumab or etanercept.
During follow-up, there were 8 CV events per 1,000 patient-years in the cohort receiving an anti-TNF drug, compared with 23 events per 1,000 patient-years in patients not on an anti-TNF agent, a statistically significant reduction. In a proportional hazards model that adjusted for baseline differences in age and gender, treatment with an anti-TNF agent reduced the rate of CV events by about half, a statistically significant effect, said Dr. Alper M. van Sijl, a researcher at the Reade Centre for Rehabilitation and Rheumatology at the VU Medical Center in Amsterdam.
"Our observations confirm the association between strong suppression of inflammation [with an anti-TNF drug] and curbing the cardiovascular risk in RA," said Dr. van Sijl and his colleagues. But they cautioned that because of the design of the study it remains unclear whether this was a real effect of the anti-TNF drugs, or a bias to treat patients with lower CV disease risk with a TNF blocker.
Dr. Burmester said that he has been a consultant to, served as a speaker for, and received research support from Abbott, the company that markets adalimumab. Dr. van Silj said that he had no disclosures.
Evidence continues to accumulate that the potent anti-inflammatory effect of drugs that block tumor necrosis factor can significantly dampen cardiovascular-disease risk in patients with rheumatoid arthritis, even though definitive proof from a prospective trial is still lacking.
Two recent pieces of suggestive evidence came from a meta-analysis of four placebo-controlled trials of adalimumab (Humira) that together included nearly 2,500 patients with rheumatoid arthritis, and from two prospective cohort studies of 828 RA patients that compared the outcomes of those treated with either adalimumab or etanercept (Enbrel) to the outcomes of similar patients who did not receive an anti–tumor necrosis factor drug.
In both studies, treatment with an anti-TNF agent was linked to a statistically significant cut in cardiovascular (CV) events of about 50%.
These results support another recent, similar finding reported in June at the Annual European Congress of Rheumatology in London. In that study, analysis of medical records from more than 109,000 U.S. patients with RA showed that every 6 months of treatment with an anti-TNF drug reduced the rate of CV events by 13%, compared with RA patients who did not receive a TNF blocker.
The meta-analysis of four trials included data collected in the ARMADA (Arthritis Rheum. 2003;48:35-45), DEO19 (Arthritis Rheum. 2004;50:1400-11), PREMIER (Arthritis Rheum. 2006;54:26-37), and OPTIMA (Ann. Rheum. Dis. 2012 May [doi: 10.1136/annrheumdis-2011-201247]) trials. Collectively, these four studies included 1,411 RA patients treated with both adalimumab and methotrexate, and 1,036 patients who received methotrexate but no anti-TNF drug. At baseline, patients in these two treatment groups had similar demographic and CV disease risk profiles.
During study periods that ranged from 16 to 104 weeks, the incidence of major adverse CV events was 1.3% in patients who received adalimumab, and 2% in those who didn’t. This difference represents a statistically significant, 2/3 drop in the risk for a major CV event in a proportional-hazard model, said Dr. Gerd Burmester, lead investigator for the analysis and a rheumatologist and professor of medicine at Charité Hospital in Berlin. His analysis also showed a statistically significant risk reduction with adalimumab in the rate of nonfatal myocardial infarction, but no significant effect of adalimumab on the rates of nonfatal myocardial infarction or nonfatal stroke compared with patients not receiving adalimumab.
A limitation of the analysis was that none of the four trials was powered to assess CV outcomes, Dr. Burmester said.
The second study used data collected from two cohorts: the CARRÉ (Cardiovascular Research and Rheumatoid Arthritis) study, a prospective Dutch cohort study of 309 randomly selected RA patients who were not treated with a TNF blocker; and the Biologics cohort, which involves 519 Dutch RA patients who have been followed since they began treatment with an anti-TNF drug for the first time, either adalimumab or etanercept.
During follow-up, there were 8 CV events per 1,000 patient-years in the cohort receiving an anti-TNF drug, compared with 23 events per 1,000 patient-years in patients not on an anti-TNF agent, a statistically significant reduction. In a proportional hazards model that adjusted for baseline differences in age and gender, treatment with an anti-TNF agent reduced the rate of CV events by about half, a statistically significant effect, said Dr. Alper M. van Sijl, a researcher at the Reade Centre for Rehabilitation and Rheumatology at the VU Medical Center in Amsterdam.
"Our observations confirm the association between strong suppression of inflammation [with an anti-TNF drug] and curbing the cardiovascular risk in RA," said Dr. van Sijl and his colleagues. But they cautioned that because of the design of the study it remains unclear whether this was a real effect of the anti-TNF drugs, or a bias to treat patients with lower CV disease risk with a TNF blocker.
Dr. Burmester said that he has been a consultant to, served as a speaker for, and received research support from Abbott, the company that markets adalimumab. Dr. van Silj said that he had no disclosures.

A-Fib Ablation Shown Safe During Cardiac Surgery
MUNICH – Left atrial ablative treatment for atrial fibrillation was safe when administered during open heart surgery in a multicenter, randomized trial of 224 patients who all had atrial fibrillation and required heart surgery for another reason.
The treatment was also effective, resulting in a significantly higher rate of sinus rhythm among the patients treated with ablation compared with the control patients who underwent open-heart surgery alone, Dr. Petr Widimsky reported at the annual congress of the European Society of Cardiology.

Some experts who heard the findings expressed some skepticism about the pattern of anti-arrhythmic effects and other atrial fibrillation–related outcomes during follow-up.
"The rhythm outcomes had some surprises. All the benefit was in patients with long-standing, persistent atrial fibrillation; that’s very surprising because it contrasts with previous findings from both catheter ablation and surgical ablation," said Dr. Gerhard Hindricks, a professor of medicine and director of the department of electrophysiology at the Heart Center of Leipzig (Germany) University.
"The secondary outcomes were also a surprise, because the atrial fibrillation response did not translate into a change in treatment" in the use of anti-arrhythmic drugs, and the two groups showed no differences in the 1-year rates of stroke, major bleeding, and all-cause mortality despite the reported different rates of continued AF. In an editorial that accompanied the published paper, Dr. Hindricks cited the inadequacy of assessing atrial arrhythmia at 1-year follow-up with 24-hour Holter ECG monitoring. "This follow-up regimen is by far not enough to generate reliable and solid results," Dr. Hindricks and his coauthor wrote (Eur. Heart J. 2012;33 [doi: 10.1093/eurheartj/ehs294]).
Despite these unexpected findings, "surgical ablation of atrial fibrillation should be cautiously indicated in asymptomatic patients scheduled for cardiac surgery," Dr. Hindricks said in comments he made at the meeting.
"This was the largest" of several randomized, prospective studies that have assessed ablation of atrial fibrillation during cardiac surgery, said Dr. Riccardo Cappato, director of the Center of Clinical Arrhythmia and Electrophysiology at the Policlinico San Donato in Milan. "The increased probability of maintaining sinus rhythm in patients with permanent atrial fibrillation does not seem to have been at the expense of higher perioperative risk exposure."

The PRAGUE-12 study enrolled 224 patients with AF who required cardiac surgery for coronary artery bypass, valve repair or replacement, or both at three centers in the Czech Republic and Slovakia during 2007-2011. The investigators randomized 117 patients to undergo atrial ablation during their surgery, and 107 who received no ablation with their surgery and served as controls. The surgeons could use whichever energy source for ablation they preferred, but 97% used cryoablation. The ablation-lesion set was the same for all patients in that treatment arm.
The patients averaged about 70 years old, and about 58% were men. Roughly half the patients had long-standing persistent AF, about a quarter had persistent AF, and the remaining quarter had paroxysmal AF.
All patients underwent open surgery using a median sternotomy with cardiopulmonary bypass and cardioplegic heart arrest. Adding ablation to the procedures increased the total surgical time by an average of 20 minutes, and increased the period of cardiopulmonary bypass and the cross-clamp time by an average of 28 minutes.
The primary safety end point was the combined rate of death, stroke, myocardial ischemia, or renal failure requiring dialysis at 30 days after surgery; this occurred in 10% of the patients who had ablation and 15% of the control patients, a nonsignificant difference, reported Dr. Widimsky, professor and head of the Cardiocenter of Charles University in Prague. Each component of the combined adverse event was similar between the two study arms. At 1 year after surgery, the combined adverse event rate was 41% in the patients treated with AF ablation and 40% in the controls.
The study’s primary efficacy end point was the prevalence of sinus rhythm in patients as measured by 24-hour Holter ECG monitoring, which occurred in 60% of the patients who underwent ablation and in 36% of the controls, a significant difference. This difference was driven by the outcome difference among the subgroup of patients who entered the study with long-standing persistent AF. In this subgroup, the proportion of patients in sinus rhythm when assessed after 1 year was 53% in the ablated group and 14% in the controls, a significant difference. In contrast, the prevalence of sinus rhythm at 1 year did not differ significantly between the two treatment arms in patients who entered the study with paroxysmal AF, or in those who entered with persistent AF.
Concurrent with the presentation of PRAGUE-12, the results also appeared in an article published online (Eur. Heart J. 2012;33 [doi: 10.1093/eurheartj/ehs290]).
Dr. Widimsky and his associates said that they had no disclosures.
When Dr. James L. Cox introduced the Cox-Maze III procedure for the surgical disruption of atrial fibrillation, the technique involved prolonged cutting and sewing, and the added time on cardiopulmonary bypass and cross-clamping this produced led to a lot of morbidity, including neurologic complications and renal failure.
The current study used a modified Maze procedure that primarily used cryoablation. Because no cutting and sewing were involved, the surgery was much easier, and it produced fewer complications. Surgeons are interested in an atrial fibrillation procedure that is safe and increases the likelihood of leading to sinus rhythm.

|
Mitchel L. Zoler/IMNG Medical Media
|
This study is important because it shows that this method can be used safely and it improves the end point of resting sinus rhythm. It is paradoxical that the main rhythm benefit was in patients with long-standing persistent atrial fibrillation, because usually that is harder to treat. But this may have been a methodological issue, because they measured the 1-year rhythm outcome without doing prolonged, continuous ECG monitoring. It’s hard to define success in patients with paroxysmal atrial fibrillation unless you monitor patients continuously for a week.
The main message from this study was that the ablation procedure was safe during cardiac surgery and did not add much time or complexity to the surgery. I think these results will increase the use of ablation during cardiac surgery for other reasons. It is a major contribution.
Miguel Sousa Uva, M.D., a cardiac surgeon at Hospital da Cruz Vermelha in Lisbon, made these comments in an interview. He said that he had no disclosures.
When Dr. James L. Cox introduced the Cox-Maze III procedure for the surgical disruption of atrial fibrillation, the technique involved prolonged cutting and sewing, and the added time on cardiopulmonary bypass and cross-clamping this produced led to a lot of morbidity, including neurologic complications and renal failure.
The current study used a modified Maze procedure that primarily used cryoablation. Because no cutting and sewing were involved, the surgery was much easier, and it produced fewer complications. Surgeons are interested in an atrial fibrillation procedure that is safe and increases the likelihood of leading to sinus rhythm.
|
|
Mitchel L. Zoler/IMNG Medical Media
|
This study is important because it shows that this method can be used safely and it improves the end point of resting sinus rhythm. It is paradoxical that the main rhythm benefit was in patients with long-standing persistent atrial fibrillation, because usually that is harder to treat. But this may have been a methodological issue, because they measured the 1-year rhythm outcome without doing prolonged, continuous ECG monitoring. It’s hard to define success in patients with paroxysmal atrial fibrillation unless you monitor patients continuously for a week.
The main message from this study was that the ablation procedure was safe during cardiac surgery and did not add much time or complexity to the surgery. I think these results will increase the use of ablation during cardiac surgery for other reasons. It is a major contribution.
Miguel Sousa Uva, M.D., a cardiac surgeon at Hospital da Cruz Vermelha in Lisbon, made these comments in an interview. He said that he had no disclosures.
When Dr. James L. Cox introduced the Cox-Maze III procedure for the surgical disruption of atrial fibrillation, the technique involved prolonged cutting and sewing, and the added time on cardiopulmonary bypass and cross-clamping this produced led to a lot of morbidity, including neurologic complications and renal failure.
The current study used a modified Maze procedure that primarily used cryoablation. Because no cutting and sewing were involved, the surgery was much easier, and it produced fewer complications. Surgeons are interested in an atrial fibrillation procedure that is safe and increases the likelihood of leading to sinus rhythm.
|
|
Mitchel L. Zoler/IMNG Medical Media
|
This study is important because it shows that this method can be used safely and it improves the end point of resting sinus rhythm. It is paradoxical that the main rhythm benefit was in patients with long-standing persistent atrial fibrillation, because usually that is harder to treat. But this may have been a methodological issue, because they measured the 1-year rhythm outcome without doing prolonged, continuous ECG monitoring. It’s hard to define success in patients with paroxysmal atrial fibrillation unless you monitor patients continuously for a week.
The main message from this study was that the ablation procedure was safe during cardiac surgery and did not add much time or complexity to the surgery. I think these results will increase the use of ablation during cardiac surgery for other reasons. It is a major contribution.
Miguel Sousa Uva, M.D., a cardiac surgeon at Hospital da Cruz Vermelha in Lisbon, made these comments in an interview. He said that he had no disclosures.
MUNICH – Left atrial ablative treatment for atrial fibrillation was safe when administered during open heart surgery in a multicenter, randomized trial of 224 patients who all had atrial fibrillation and required heart surgery for another reason.
The treatment was also effective, resulting in a significantly higher rate of sinus rhythm among the patients treated with ablation compared with the control patients who underwent open-heart surgery alone, Dr. Petr Widimsky reported at the annual congress of the European Society of Cardiology.
Some experts who heard the findings expressed some skepticism about the pattern of anti-arrhythmic effects and other atrial fibrillation–related outcomes during follow-up.
"The rhythm outcomes had some surprises. All the benefit was in patients with long-standing, persistent atrial fibrillation; that’s very surprising because it contrasts with previous findings from both catheter ablation and surgical ablation," said Dr. Gerhard Hindricks, a professor of medicine and director of the department of electrophysiology at the Heart Center of Leipzig (Germany) University.
"The secondary outcomes were also a surprise, because the atrial fibrillation response did not translate into a change in treatment" in the use of anti-arrhythmic drugs, and the two groups showed no differences in the 1-year rates of stroke, major bleeding, and all-cause mortality despite the reported different rates of continued AF. In an editorial that accompanied the published paper, Dr. Hindricks cited the inadequacy of assessing atrial arrhythmia at 1-year follow-up with 24-hour Holter ECG monitoring. "This follow-up regimen is by far not enough to generate reliable and solid results," Dr. Hindricks and his coauthor wrote (Eur. Heart J. 2012;33 [doi: 10.1093/eurheartj/ehs294]).
Despite these unexpected findings, "surgical ablation of atrial fibrillation should be cautiously indicated in asymptomatic patients scheduled for cardiac surgery," Dr. Hindricks said in comments he made at the meeting.
"This was the largest" of several randomized, prospective studies that have assessed ablation of atrial fibrillation during cardiac surgery, said Dr. Riccardo Cappato, director of the Center of Clinical Arrhythmia and Electrophysiology at the Policlinico San Donato in Milan. "The increased probability of maintaining sinus rhythm in patients with permanent atrial fibrillation does not seem to have been at the expense of higher perioperative risk exposure."
The PRAGUE-12 study enrolled 224 patients with AF who required cardiac surgery for coronary artery bypass, valve repair or replacement, or both at three centers in the Czech Republic and Slovakia during 2007-2011. The investigators randomized 117 patients to undergo atrial ablation during their surgery, and 107 who received no ablation with their surgery and served as controls. The surgeons could use whichever energy source for ablation they preferred, but 97% used cryoablation. The ablation-lesion set was the same for all patients in that treatment arm.
The patients averaged about 70 years old, and about 58% were men. Roughly half the patients had long-standing persistent AF, about a quarter had persistent AF, and the remaining quarter had paroxysmal AF.
All patients underwent open surgery using a median sternotomy with cardiopulmonary bypass and cardioplegic heart arrest. Adding ablation to the procedures increased the total surgical time by an average of 20 minutes, and increased the period of cardiopulmonary bypass and the cross-clamp time by an average of 28 minutes.
The primary safety end point was the combined rate of death, stroke, myocardial ischemia, or renal failure requiring dialysis at 30 days after surgery; this occurred in 10% of the patients who had ablation and 15% of the control patients, a nonsignificant difference, reported Dr. Widimsky, professor and head of the Cardiocenter of Charles University in Prague. Each component of the combined adverse event was similar between the two study arms. At 1 year after surgery, the combined adverse event rate was 41% in the patients treated with AF ablation and 40% in the controls.
The study’s primary efficacy end point was the prevalence of sinus rhythm in patients as measured by 24-hour Holter ECG monitoring, which occurred in 60% of the patients who underwent ablation and in 36% of the controls, a significant difference. This difference was driven by the outcome difference among the subgroup of patients who entered the study with long-standing persistent AF. In this subgroup, the proportion of patients in sinus rhythm when assessed after 1 year was 53% in the ablated group and 14% in the controls, a significant difference. In contrast, the prevalence of sinus rhythm at 1 year did not differ significantly between the two treatment arms in patients who entered the study with paroxysmal AF, or in those who entered with persistent AF.
Concurrent with the presentation of PRAGUE-12, the results also appeared in an article published online (Eur. Heart J. 2012;33 [doi: 10.1093/eurheartj/ehs290]).
Dr. Widimsky and his associates said that they had no disclosures.
MUNICH – Left atrial ablative treatment for atrial fibrillation was safe when administered during open heart surgery in a multicenter, randomized trial of 224 patients who all had atrial fibrillation and required heart surgery for another reason.
The treatment was also effective, resulting in a significantly higher rate of sinus rhythm among the patients treated with ablation compared with the control patients who underwent open-heart surgery alone, Dr. Petr Widimsky reported at the annual congress of the European Society of Cardiology.
Some experts who heard the findings expressed some skepticism about the pattern of anti-arrhythmic effects and other atrial fibrillation–related outcomes during follow-up.
"The rhythm outcomes had some surprises. All the benefit was in patients with long-standing, persistent atrial fibrillation; that’s very surprising because it contrasts with previous findings from both catheter ablation and surgical ablation," said Dr. Gerhard Hindricks, a professor of medicine and director of the department of electrophysiology at the Heart Center of Leipzig (Germany) University.
"The secondary outcomes were also a surprise, because the atrial fibrillation response did not translate into a change in treatment" in the use of anti-arrhythmic drugs, and the two groups showed no differences in the 1-year rates of stroke, major bleeding, and all-cause mortality despite the reported different rates of continued AF. In an editorial that accompanied the published paper, Dr. Hindricks cited the inadequacy of assessing atrial arrhythmia at 1-year follow-up with 24-hour Holter ECG monitoring. "This follow-up regimen is by far not enough to generate reliable and solid results," Dr. Hindricks and his coauthor wrote (Eur. Heart J. 2012;33 [doi: 10.1093/eurheartj/ehs294]).
Despite these unexpected findings, "surgical ablation of atrial fibrillation should be cautiously indicated in asymptomatic patients scheduled for cardiac surgery," Dr. Hindricks said in comments he made at the meeting.
"This was the largest" of several randomized, prospective studies that have assessed ablation of atrial fibrillation during cardiac surgery, said Dr. Riccardo Cappato, director of the Center of Clinical Arrhythmia and Electrophysiology at the Policlinico San Donato in Milan. "The increased probability of maintaining sinus rhythm in patients with permanent atrial fibrillation does not seem to have been at the expense of higher perioperative risk exposure."
The PRAGUE-12 study enrolled 224 patients with AF who required cardiac surgery for coronary artery bypass, valve repair or replacement, or both at three centers in the Czech Republic and Slovakia during 2007-2011. The investigators randomized 117 patients to undergo atrial ablation during their surgery, and 107 who received no ablation with their surgery and served as controls. The surgeons could use whichever energy source for ablation they preferred, but 97% used cryoablation. The ablation-lesion set was the same for all patients in that treatment arm.
The patients averaged about 70 years old, and about 58% were men. Roughly half the patients had long-standing persistent AF, about a quarter had persistent AF, and the remaining quarter had paroxysmal AF.
All patients underwent open surgery using a median sternotomy with cardiopulmonary bypass and cardioplegic heart arrest. Adding ablation to the procedures increased the total surgical time by an average of 20 minutes, and increased the period of cardiopulmonary bypass and the cross-clamp time by an average of 28 minutes.
The primary safety end point was the combined rate of death, stroke, myocardial ischemia, or renal failure requiring dialysis at 30 days after surgery; this occurred in 10% of the patients who had ablation and 15% of the control patients, a nonsignificant difference, reported Dr. Widimsky, professor and head of the Cardiocenter of Charles University in Prague. Each component of the combined adverse event was similar between the two study arms. At 1 year after surgery, the combined adverse event rate was 41% in the patients treated with AF ablation and 40% in the controls.
The study’s primary efficacy end point was the prevalence of sinus rhythm in patients as measured by 24-hour Holter ECG monitoring, which occurred in 60% of the patients who underwent ablation and in 36% of the controls, a significant difference. This difference was driven by the outcome difference among the subgroup of patients who entered the study with long-standing persistent AF. In this subgroup, the proportion of patients in sinus rhythm when assessed after 1 year was 53% in the ablated group and 14% in the controls, a significant difference. In contrast, the prevalence of sinus rhythm at 1 year did not differ significantly between the two treatment arms in patients who entered the study with paroxysmal AF, or in those who entered with persistent AF.
Concurrent with the presentation of PRAGUE-12, the results also appeared in an article published online (Eur. Heart J. 2012;33 [doi: 10.1093/eurheartj/ehs290]).
Dr. Widimsky and his associates said that they had no disclosures.
AT THE ANNUAL CONGRESS OF THE EUROPEAN SOCIETY OF CARDIOLOGY
Major Finding: After 30 days, the adverse event rate was 10% in cardiac surgery patients also undergoing ablation and 15% in patients without ablation, a difference that was not significant.
Data Source: PRAGUE-12 was a randomized trial that enrolled 224 patients with atrial fibrillation who also required cardiac surgery for another reason at three centers in the Czech Republic and Slovakia.
Disclosures: Dr. Widimsky and his associates said that they had no disclosures.

TRILOGY: Prasugrel, Clopidogrel Look Similar in ACS Patients
MUNICH – Prasugrel had no overall efficacy advantage over clopidogrel when treating patients with non–ST-elevation acute coronary syndrome in a prospective, randomized trial with more than 9,000 patients.
Results from the Targeted Platelet Inhibition to Clarify the Optimal Strategy to Medically Manage Acute Coronary Syndrome (TRILOGY ACS) did provide reassuring, new safety data for prasugrel compared with clopidogrel in the enrolled patient population.

The study outcomes also supplied very suggestive evidence for certain efficacy advantages of prasugrel compared with clopidogrel in the study’s target patients. But prasugrel’s inability to beat clopidogrel for the study’s primary efficacy end point produced an inescapable argument against springing for the more expensive prasugrel rather than the cheaper clopidogrel in most non–ST-elevation ACS patients who initially undergo medical management rather than revascularization therapy.
"In the largest trial to date of ACS patients managed medically without revascularization, prasugrel was not statistically different from clopidogrel during 2.5 years of follow-up among patients less than 75 years of age," Dr. Matthew T. Roe said at the meeting. Concurrent with his presentation, the results appeared online (N.Engl.J.Med.2012;367:doi:10.1056/NEJMoa1205512).
Current European Society of Cardiology guidelines for managing patients with ACS without ST-segment elevation say that prasugrel is indicated for patients with known coronary anatomy who are proceeding to PCI (Eur. Heart J. 2011;32:2999-3054). "I believe these recommendations should not be changed," commented Dr. Raffaele De Caterina, professor and director of the division of cardiology at G. d’Annunzio University in Chieti, Italy. In contrast, the same guidelines say that another potent antiplatelet drug, ticagrelor, is recommended for all patients at moderate-to-high risk of ischemic events regardless of their initial treatment strategy. The ticagrelor recommendation was largely based on the results of a prespecified sub-analysis from the Platelet Inhibition and Patient Outcomes (PLATO) trial published last year (BMJ 2011;342:d3527), Dr. De Caterina said.
The new safety findings came from the TRILOGY ACS study design, which called for cutting the standard 10 mg daily dosage of prasugrel in half for patients aged 75 or older, and for patients who weighed less than 60 kg. The consequence was that the rates of severe or intracranial bleeds in patients in the prasugrel arm were not significantly different from patients in the clopidogrel arm, a marked contrast to the results from an earlier major trial that compared prasugrel and clopidogrel, the Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel-Thrombolysis in Myocardial Infarction (TRITON-TIMI) 38 trial (N. Engl. J. Med. 2007;357:2001-15).
"A reasonable solution for the problem of excess bleeding in the elderly appears to have been successfully arrived at by modifying the dose," commented Dr. Elliott M. Antman, director of the cardiac unit at Brigham and Women’s Hospital in Boston and lead investigator for the TRITON-TIMI 38 trial.
"The safety here was very comforting, because this trial had the longest follow-up of any antiplatelet treatment agent. The results point out clearly that one drug dose does not fit all," said Dr. E. Magnus Ohman, professor of medicine at Duke University in Durham, N.C., and co-lead investigator for the TRILOGY ACS trial along with Dr. Roe.
The trial’s main efficacy end point focused on the 7,243 enrolled patients who were younger than 75 years old. During a median follow-up of 17 months and as long as 30 months, patients treated with prasugrel had a 13.9% rate of the primary, combined end point of cardiovascular death, myocardial infarction, or stroke, compared with a 16% rate among patients randomized to clopidogrel. This difference did not reach statistical significance, reported Dr. Roe, a Duke cardiologist.
The trial included a prespecified, secondary analysis that focused on recurrent cardiovascular events (not just the incidence of initial events used for the primary analysis). Total recurrent events occurred in 459 patients treated with prasugrel and 530 patients treated with clopidogrel, a 15% relative hazard reduction in favor of prasugrel that reached statistical significance.
In addition, the rate of the primary efficacy end point in the two treatment arms was virtually identical during the first 12 months of follow-up. Subsequently the two arms showed a clear split, with the prasugrel-treated patients having a significant, 28% reduced rate of primary outcomes after the study’s first 12 months, a statistically significant difference in a post-hoc analysis.
The delayed benefit from prasugrel compared with clopidogrel in this trial contrasted with the TRITON-TIMI 38 results, which showed an immediate benefit from prasugrel in patients who underwent percutaneous coronary interventions. A likely explanation is that the highly thrombogenic stents that the TRITON-TIMI 38 patients received posed an acute thrombotic risk, while thrombotic events occurred more slowly in the medically managed patients enrolled in TRILOGY ASC, Dr. Antman suggested in an interview.
The TRILOGY ACS trial was sponsored by Eli Lilly and Daiichi Sankyo, the companies that market prasugrel (Effient). Dr. Roe said that he has received consulting fees from Eli Lilly and Daiichi Sankyo and other drug companies, research grants from Eli Lilly and other drug companies, and lecture fees from AstraZeneca and Janssen. Dr. De Caterina said that he has been a speaker for and received honoraria from Lilly, Daiichi Sankyo, AstraZeneca, and Bayer. Dr. Antman said that he served as lead investigator for the TRITON-TIMI 38 trial. Dr. Ohman said that he received grant support and travel expenses from Eli Lilly and Daiichi Sankyo, and consulting and lecture fees from other drug companies.
The results from TRILOGY ACS clearly showed no overall advantage for treating acute coronary syndrome patients who are managed medically with prasugrel compared with clopidogrel for the study’s primary outcome. The efficacy results showed two very interesting advantages to prasugrel treatment: a statistically significant reduction in recurrent ischemic coronary events, and a statistically significant reduction in events starting a year after the onset of treatment. The recurrent-event analysis was prespecified, and is a very clinically relevant endpoint; the 1-year landmark analysis was done post hoc. Because the primary end point showed no significant benefit, both of these other outcomes must be considered just hypothesis generating.
The safety results for prasugrel were very reassuring. They indicated that barring patients with a history of stroke from the trial and using a reduced, 5 mg/day dosage for patients aged 75 or older and for patients who weighed less than 60 kg eliminated the excess risk for major bleeding events compared with clopidogrel, which were seen in the prior comparison of prasugrel and clopidogrel, the TRITON-TIMI 38 study.
In the United States clopidogrel is now available as a generic drug and is substantially cheaper than prasugrel. If cost were not an issue, then I might be inclined to use prasugrel over clopidogrel in the types of patients enrolled in TRILOGY ACS because of the signals of possible superior efficacy and no added safety risk. But cost is an issue and will remain so for several more years until prasugrel goes off patent. Until then, the possible advantages of prasugrel over clopidogrel do not tip the balance compared with prasugrel’s excess expense.
But the issue of which drug to choose in these patients doesn’t end there, because there is a third option, ticagrelor. A prespecified sub-analysis of the PLATO trial, which compared ticagrelor with clopidogrel in acute coronary syndrome patients, looked at the comparison between the two drugs specifically in patients with non–ST-segment elevation acute coronary syndrome who did not initially undergo invasive management. This is a population of patients very similar to those enrolled into TRILOGY ACS. The subanalysis showed that ticagrelor had a significant efficacy advantage over clopidogrel that was similar to what was seen in the entire PLATO trial, and that provides a compelling reason to consider ticagrelor over clopidogrel in these patients.
But the overall PLATO results as well as this subanalysis also showed a statistically significant excess risk for major bleeding events for ticagrelor, compared with clopidogrel. And ticagrelor, like prasugrel, is substantially more expensive than clopidogrel.
A further complication is that ticagrelor is a twice-daily drug, while clopidogrel and prasugrel are administered once daily.
If I were treating the TRILOGY ACS type of patients, cost were no object, the patients seemed likely to be compliant with their drug regimen, and their bleeding risk was relatively low, then I might want to go with ticagrelor to take advantage of its efficacy edge over clopidogrel. But if cost were a significant consideration, or I had doubts about regular patient compliance with a b.i.d. regimen, or the patient had a high risk for bleeding complications, then clopidogrel might seem like the most attractive option.
DEEPAK L. BHATT, M.D., is chief of cardiology at the VA Boston Healthcare System. Dr. Bhatt served as a coinvestigator and a member of the steering committee for TRILOGY ACS. He said that he has received grant support from several drug companies, but not from the companies that sponsored TRILOGY ACS. Dr. Bhatt made these comments in an interview.
The results from TRILOGY ACS clearly showed no overall advantage for treating acute coronary syndrome patients who are managed medically with prasugrel compared with clopidogrel for the study’s primary outcome. The efficacy results showed two very interesting advantages to prasugrel treatment: a statistically significant reduction in recurrent ischemic coronary events, and a statistically significant reduction in events starting a year after the onset of treatment. The recurrent-event analysis was prespecified, and is a very clinically relevant endpoint; the 1-year landmark analysis was done post hoc. Because the primary end point showed no significant benefit, both of these other outcomes must be considered just hypothesis generating.
The safety results for prasugrel were very reassuring. They indicated that barring patients with a history of stroke from the trial and using a reduced, 5 mg/day dosage for patients aged 75 or older and for patients who weighed less than 60 kg eliminated the excess risk for major bleeding events compared with clopidogrel, which were seen in the prior comparison of prasugrel and clopidogrel, the TRITON-TIMI 38 study.
In the United States clopidogrel is now available as a generic drug and is substantially cheaper than prasugrel. If cost were not an issue, then I might be inclined to use prasugrel over clopidogrel in the types of patients enrolled in TRILOGY ACS because of the signals of possible superior efficacy and no added safety risk. But cost is an issue and will remain so for several more years until prasugrel goes off patent. Until then, the possible advantages of prasugrel over clopidogrel do not tip the balance compared with prasugrel’s excess expense.
But the issue of which drug to choose in these patients doesn’t end there, because there is a third option, ticagrelor. A prespecified sub-analysis of the PLATO trial, which compared ticagrelor with clopidogrel in acute coronary syndrome patients, looked at the comparison between the two drugs specifically in patients with non–ST-segment elevation acute coronary syndrome who did not initially undergo invasive management. This is a population of patients very similar to those enrolled into TRILOGY ACS. The subanalysis showed that ticagrelor had a significant efficacy advantage over clopidogrel that was similar to what was seen in the entire PLATO trial, and that provides a compelling reason to consider ticagrelor over clopidogrel in these patients.
But the overall PLATO results as well as this subanalysis also showed a statistically significant excess risk for major bleeding events for ticagrelor, compared with clopidogrel. And ticagrelor, like prasugrel, is substantially more expensive than clopidogrel.
A further complication is that ticagrelor is a twice-daily drug, while clopidogrel and prasugrel are administered once daily.
If I were treating the TRILOGY ACS type of patients, cost were no object, the patients seemed likely to be compliant with their drug regimen, and their bleeding risk was relatively low, then I might want to go with ticagrelor to take advantage of its efficacy edge over clopidogrel. But if cost were a significant consideration, or I had doubts about regular patient compliance with a b.i.d. regimen, or the patient had a high risk for bleeding complications, then clopidogrel might seem like the most attractive option.
DEEPAK L. BHATT, M.D., is chief of cardiology at the VA Boston Healthcare System. Dr. Bhatt served as a coinvestigator and a member of the steering committee for TRILOGY ACS. He said that he has received grant support from several drug companies, but not from the companies that sponsored TRILOGY ACS. Dr. Bhatt made these comments in an interview.
The results from TRILOGY ACS clearly showed no overall advantage for treating acute coronary syndrome patients who are managed medically with prasugrel compared with clopidogrel for the study’s primary outcome. The efficacy results showed two very interesting advantages to prasugrel treatment: a statistically significant reduction in recurrent ischemic coronary events, and a statistically significant reduction in events starting a year after the onset of treatment. The recurrent-event analysis was prespecified, and is a very clinically relevant endpoint; the 1-year landmark analysis was done post hoc. Because the primary end point showed no significant benefit, both of these other outcomes must be considered just hypothesis generating.
The safety results for prasugrel were very reassuring. They indicated that barring patients with a history of stroke from the trial and using a reduced, 5 mg/day dosage for patients aged 75 or older and for patients who weighed less than 60 kg eliminated the excess risk for major bleeding events compared with clopidogrel, which were seen in the prior comparison of prasugrel and clopidogrel, the TRITON-TIMI 38 study.
In the United States clopidogrel is now available as a generic drug and is substantially cheaper than prasugrel. If cost were not an issue, then I might be inclined to use prasugrel over clopidogrel in the types of patients enrolled in TRILOGY ACS because of the signals of possible superior efficacy and no added safety risk. But cost is an issue and will remain so for several more years until prasugrel goes off patent. Until then, the possible advantages of prasugrel over clopidogrel do not tip the balance compared with prasugrel’s excess expense.
But the issue of which drug to choose in these patients doesn’t end there, because there is a third option, ticagrelor. A prespecified sub-analysis of the PLATO trial, which compared ticagrelor with clopidogrel in acute coronary syndrome patients, looked at the comparison between the two drugs specifically in patients with non–ST-segment elevation acute coronary syndrome who did not initially undergo invasive management. This is a population of patients very similar to those enrolled into TRILOGY ACS. The subanalysis showed that ticagrelor had a significant efficacy advantage over clopidogrel that was similar to what was seen in the entire PLATO trial, and that provides a compelling reason to consider ticagrelor over clopidogrel in these patients.
But the overall PLATO results as well as this subanalysis also showed a statistically significant excess risk for major bleeding events for ticagrelor, compared with clopidogrel. And ticagrelor, like prasugrel, is substantially more expensive than clopidogrel.
A further complication is that ticagrelor is a twice-daily drug, while clopidogrel and prasugrel are administered once daily.
If I were treating the TRILOGY ACS type of patients, cost were no object, the patients seemed likely to be compliant with their drug regimen, and their bleeding risk was relatively low, then I might want to go with ticagrelor to take advantage of its efficacy edge over clopidogrel. But if cost were a significant consideration, or I had doubts about regular patient compliance with a b.i.d. regimen, or the patient had a high risk for bleeding complications, then clopidogrel might seem like the most attractive option.
DEEPAK L. BHATT, M.D., is chief of cardiology at the VA Boston Healthcare System. Dr. Bhatt served as a coinvestigator and a member of the steering committee for TRILOGY ACS. He said that he has received grant support from several drug companies, but not from the companies that sponsored TRILOGY ACS. Dr. Bhatt made these comments in an interview.
MUNICH – Prasugrel had no overall efficacy advantage over clopidogrel when treating patients with non–ST-elevation acute coronary syndrome in a prospective, randomized trial with more than 9,000 patients.
Results from the Targeted Platelet Inhibition to Clarify the Optimal Strategy to Medically Manage Acute Coronary Syndrome (TRILOGY ACS) did provide reassuring, new safety data for prasugrel compared with clopidogrel in the enrolled patient population.
The study outcomes also supplied very suggestive evidence for certain efficacy advantages of prasugrel compared with clopidogrel in the study’s target patients. But prasugrel’s inability to beat clopidogrel for the study’s primary efficacy end point produced an inescapable argument against springing for the more expensive prasugrel rather than the cheaper clopidogrel in most non–ST-elevation ACS patients who initially undergo medical management rather than revascularization therapy.
"In the largest trial to date of ACS patients managed medically without revascularization, prasugrel was not statistically different from clopidogrel during 2.5 years of follow-up among patients less than 75 years of age," Dr. Matthew T. Roe said at the meeting. Concurrent with his presentation, the results appeared online (N.Engl.J.Med.2012;367:doi:10.1056/NEJMoa1205512).
Current European Society of Cardiology guidelines for managing patients with ACS without ST-segment elevation say that prasugrel is indicated for patients with known coronary anatomy who are proceeding to PCI (Eur. Heart J. 2011;32:2999-3054). "I believe these recommendations should not be changed," commented Dr. Raffaele De Caterina, professor and director of the division of cardiology at G. d’Annunzio University in Chieti, Italy. In contrast, the same guidelines say that another potent antiplatelet drug, ticagrelor, is recommended for all patients at moderate-to-high risk of ischemic events regardless of their initial treatment strategy. The ticagrelor recommendation was largely based on the results of a prespecified sub-analysis from the Platelet Inhibition and Patient Outcomes (PLATO) trial published last year (BMJ 2011;342:d3527), Dr. De Caterina said.
The new safety findings came from the TRILOGY ACS study design, which called for cutting the standard 10 mg daily dosage of prasugrel in half for patients aged 75 or older, and for patients who weighed less than 60 kg. The consequence was that the rates of severe or intracranial bleeds in patients in the prasugrel arm were not significantly different from patients in the clopidogrel arm, a marked contrast to the results from an earlier major trial that compared prasugrel and clopidogrel, the Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel-Thrombolysis in Myocardial Infarction (TRITON-TIMI) 38 trial (N. Engl. J. Med. 2007;357:2001-15).
"A reasonable solution for the problem of excess bleeding in the elderly appears to have been successfully arrived at by modifying the dose," commented Dr. Elliott M. Antman, director of the cardiac unit at Brigham and Women’s Hospital in Boston and lead investigator for the TRITON-TIMI 38 trial.
"The safety here was very comforting, because this trial had the longest follow-up of any antiplatelet treatment agent. The results point out clearly that one drug dose does not fit all," said Dr. E. Magnus Ohman, professor of medicine at Duke University in Durham, N.C., and co-lead investigator for the TRILOGY ACS trial along with Dr. Roe.
The trial’s main efficacy end point focused on the 7,243 enrolled patients who were younger than 75 years old. During a median follow-up of 17 months and as long as 30 months, patients treated with prasugrel had a 13.9% rate of the primary, combined end point of cardiovascular death, myocardial infarction, or stroke, compared with a 16% rate among patients randomized to clopidogrel. This difference did not reach statistical significance, reported Dr. Roe, a Duke cardiologist.
The trial included a prespecified, secondary analysis that focused on recurrent cardiovascular events (not just the incidence of initial events used for the primary analysis). Total recurrent events occurred in 459 patients treated with prasugrel and 530 patients treated with clopidogrel, a 15% relative hazard reduction in favor of prasugrel that reached statistical significance.
In addition, the rate of the primary efficacy end point in the two treatment arms was virtually identical during the first 12 months of follow-up. Subsequently the two arms showed a clear split, with the prasugrel-treated patients having a significant, 28% reduced rate of primary outcomes after the study’s first 12 months, a statistically significant difference in a post-hoc analysis.
The delayed benefit from prasugrel compared with clopidogrel in this trial contrasted with the TRITON-TIMI 38 results, which showed an immediate benefit from prasugrel in patients who underwent percutaneous coronary interventions. A likely explanation is that the highly thrombogenic stents that the TRITON-TIMI 38 patients received posed an acute thrombotic risk, while thrombotic events occurred more slowly in the medically managed patients enrolled in TRILOGY ASC, Dr. Antman suggested in an interview.
The TRILOGY ACS trial was sponsored by Eli Lilly and Daiichi Sankyo, the companies that market prasugrel (Effient). Dr. Roe said that he has received consulting fees from Eli Lilly and Daiichi Sankyo and other drug companies, research grants from Eli Lilly and other drug companies, and lecture fees from AstraZeneca and Janssen. Dr. De Caterina said that he has been a speaker for and received honoraria from Lilly, Daiichi Sankyo, AstraZeneca, and Bayer. Dr. Antman said that he served as lead investigator for the TRITON-TIMI 38 trial. Dr. Ohman said that he received grant support and travel expenses from Eli Lilly and Daiichi Sankyo, and consulting and lecture fees from other drug companies.
MUNICH – Prasugrel had no overall efficacy advantage over clopidogrel when treating patients with non–ST-elevation acute coronary syndrome in a prospective, randomized trial with more than 9,000 patients.
Results from the Targeted Platelet Inhibition to Clarify the Optimal Strategy to Medically Manage Acute Coronary Syndrome (TRILOGY ACS) did provide reassuring, new safety data for prasugrel compared with clopidogrel in the enrolled patient population.
The study outcomes also supplied very suggestive evidence for certain efficacy advantages of prasugrel compared with clopidogrel in the study’s target patients. But prasugrel’s inability to beat clopidogrel for the study’s primary efficacy end point produced an inescapable argument against springing for the more expensive prasugrel rather than the cheaper clopidogrel in most non–ST-elevation ACS patients who initially undergo medical management rather than revascularization therapy.
"In the largest trial to date of ACS patients managed medically without revascularization, prasugrel was not statistically different from clopidogrel during 2.5 years of follow-up among patients less than 75 years of age," Dr. Matthew T. Roe said at the meeting. Concurrent with his presentation, the results appeared online (N.Engl.J.Med.2012;367:doi:10.1056/NEJMoa1205512).
Current European Society of Cardiology guidelines for managing patients with ACS without ST-segment elevation say that prasugrel is indicated for patients with known coronary anatomy who are proceeding to PCI (Eur. Heart J. 2011;32:2999-3054). "I believe these recommendations should not be changed," commented Dr. Raffaele De Caterina, professor and director of the division of cardiology at G. d’Annunzio University in Chieti, Italy. In contrast, the same guidelines say that another potent antiplatelet drug, ticagrelor, is recommended for all patients at moderate-to-high risk of ischemic events regardless of their initial treatment strategy. The ticagrelor recommendation was largely based on the results of a prespecified sub-analysis from the Platelet Inhibition and Patient Outcomes (PLATO) trial published last year (BMJ 2011;342:d3527), Dr. De Caterina said.
The new safety findings came from the TRILOGY ACS study design, which called for cutting the standard 10 mg daily dosage of prasugrel in half for patients aged 75 or older, and for patients who weighed less than 60 kg. The consequence was that the rates of severe or intracranial bleeds in patients in the prasugrel arm were not significantly different from patients in the clopidogrel arm, a marked contrast to the results from an earlier major trial that compared prasugrel and clopidogrel, the Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel-Thrombolysis in Myocardial Infarction (TRITON-TIMI) 38 trial (N. Engl. J. Med. 2007;357:2001-15).
"A reasonable solution for the problem of excess bleeding in the elderly appears to have been successfully arrived at by modifying the dose," commented Dr. Elliott M. Antman, director of the cardiac unit at Brigham and Women’s Hospital in Boston and lead investigator for the TRITON-TIMI 38 trial.
"The safety here was very comforting, because this trial had the longest follow-up of any antiplatelet treatment agent. The results point out clearly that one drug dose does not fit all," said Dr. E. Magnus Ohman, professor of medicine at Duke University in Durham, N.C., and co-lead investigator for the TRILOGY ACS trial along with Dr. Roe.
The trial’s main efficacy end point focused on the 7,243 enrolled patients who were younger than 75 years old. During a median follow-up of 17 months and as long as 30 months, patients treated with prasugrel had a 13.9% rate of the primary, combined end point of cardiovascular death, myocardial infarction, or stroke, compared with a 16% rate among patients randomized to clopidogrel. This difference did not reach statistical significance, reported Dr. Roe, a Duke cardiologist.
The trial included a prespecified, secondary analysis that focused on recurrent cardiovascular events (not just the incidence of initial events used for the primary analysis). Total recurrent events occurred in 459 patients treated with prasugrel and 530 patients treated with clopidogrel, a 15% relative hazard reduction in favor of prasugrel that reached statistical significance.
In addition, the rate of the primary efficacy end point in the two treatment arms was virtually identical during the first 12 months of follow-up. Subsequently the two arms showed a clear split, with the prasugrel-treated patients having a significant, 28% reduced rate of primary outcomes after the study’s first 12 months, a statistically significant difference in a post-hoc analysis.
The delayed benefit from prasugrel compared with clopidogrel in this trial contrasted with the TRITON-TIMI 38 results, which showed an immediate benefit from prasugrel in patients who underwent percutaneous coronary interventions. A likely explanation is that the highly thrombogenic stents that the TRITON-TIMI 38 patients received posed an acute thrombotic risk, while thrombotic events occurred more slowly in the medically managed patients enrolled in TRILOGY ASC, Dr. Antman suggested in an interview.
The TRILOGY ACS trial was sponsored by Eli Lilly and Daiichi Sankyo, the companies that market prasugrel (Effient). Dr. Roe said that he has received consulting fees from Eli Lilly and Daiichi Sankyo and other drug companies, research grants from Eli Lilly and other drug companies, and lecture fees from AstraZeneca and Janssen. Dr. De Caterina said that he has been a speaker for and received honoraria from Lilly, Daiichi Sankyo, AstraZeneca, and Bayer. Dr. Antman said that he served as lead investigator for the TRITON-TIMI 38 trial. Dr. Ohman said that he received grant support and travel expenses from Eli Lilly and Daiichi Sankyo, and consulting and lecture fees from other drug companies.
AT THE ANNUAL CONGRESS OF THE EUROPEAN SOCIETY OF CARDIOLOGY
Major Finding: Prasugrel showed no overall efficacy edge over clopidogrel in medically managed patients with non–ST-segment elevation acute coronary syndrome.
Data Source: The TRILOGY ACS trial randomized 9,326 patients with non–ST-elevation ACS who did not undergo initial revascularization therapy.
Disclosures: TRILOGY ACS was sponsored by Eli Lilly and Daiichi Sankyo, the companies that market prasugrel (Effient).

LVADs for Severe Heart Failure Gradually Take Hold
A sea change in management of severe heart failure began two and a half years ago, in January 2010, when the Food and Drug Administration approved U.S. marketing of the HeartMate II continuous-flow, left ventricular assist device for destination therapy. Patients, cardiologists, and cardiothoracic surgeons at last had a reasonably durable, effective, and relatively safe alternative to heart transplant to offer patients at the end stage of deteriorating heart function.
The Heart Mate II quickly supplanted the prior-generation, pulsatile-flow devices, and 2010 also saw a 10-fold spike in the number of left ventricular assist devices (LVADs) placed in U.S. patients as destination therapy.
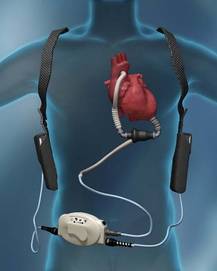
During the first half of 2011, 6-month survival among U.S. patients with a continuous-flow LVAD (which means the HeartMate II, the only continuous-flow device on the U.S. market) was 89%, and 12-month survival during 2010 was 81% (J. Heart Lung Transplant 2012;31:117-26, putting the continuous-flow LVAD in at least the same ballpark as heart transplant, which has a 5-year survival of about 80% and a median survival of about 10 years in U.S. patients.
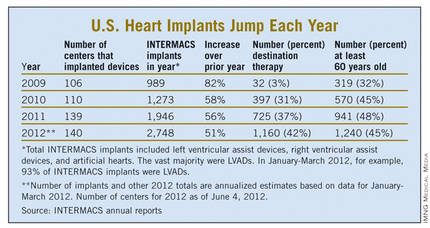
But despite estimates that many thousands, if not tens of thousands, of Americans meet the clinical criteria to qualify for placement of an LVAD, the reality is that in the 30 months since the HeartMate II became available for routine destination therapy through mid-2012, only about 4,600 were placed in U.S. patients. In addition, just slightly more than a third of those patients, roughly 1,700, received their LVAD explicitly for destination therapy.
Even in 2012, a majority of patients who received an LVAD got their device with the understanding that it was as a bridge to transplant, an LVAD placed with temporary intent to help patients survive and improve clinically until their LVAD could be swapped out and replaced by a transplanted heart.
LVAD Numbers Up, but Still Low
If the availability of the HeartMate II for destination therapy in routine practice was a revolution for the management of many patients with New York Heart Association class IV heart failure – which is how many experts in this field view the device – it has so far been a revolution played out in slow motion. In large part that’s because the field started small, and stayed small until recently. At the end of 2008, fewer than 1,000 patients had ever received a ventricular assist device or a total artificial heart. Even though the number nearly doubled in 2009, by decade’s end the cumulative total still remained under 2,000.

Despite the low numbers compared with the estimated need, many physicians and surgeons who specialize in advanced heart failure say they are satisfied with the pace at which continuous-flow LVADs have entered patients over the past 2 years. Though they acknowledge that the number of potential U.S. candidates for an LVAD undoubtedly is far larger than the nearly 4,600 who received one since early 2010, they note that the implantation rate grew in a steady and robust way during the past 30 months, as has the number of centers performing the surgery. With a better-than-50% year-over-year growth spurt in both 2010 and 2011, and with that pace continuing into the first months of 2012, the 140 U.S. centers that now place LVADs are on track to do nearly 3,000 this year (although only 42% were for destination therapy during January-March 2012).
"You probably don’t want it to increase too rapidly, because it is still high risk and expensive," said Dr. David E. Lanfear, a cardiologist specializing in advanced heart failure and transplantation at Henry Ford Hospital in Detroit. LVAD placements "have not reached the maximum number of patients who they could help, but you need to be cautious because it requires specialized expertise to do it properly. I wouldn’t expect that [during 2 years] it would immediately reach maximum use. It’s still in the growth phase. There are a lot of patients out there, so it will continue to grow; I’m not concerned that it’s not growing fast enough," he said in an interview.
An LVAD or a Heart Transplant?
Experts also note an important shift in attitude about the role for LVADs, a change that has brought them to the brink of replacing transplanted hearts as the default treatment for patients with end-stage heart failure.

"The current outcomes with HeartMate II have already changed the field. We published a paper last year that showed there was no difference between LVADs and transplant in costs and outcomes at 1 year (Ann. Thorac. Surg. 2011;91:1330-4)," said Dr. Mark S. Slaughter, professor of surgery and chief of thoracic and cardiovascular surgery at the University of Louisville (Ky.). Between that and the competitive mortality benefit from LVADs "we need to rethink the role of transplant. In appropriately selected patients, we can achieve outstanding long-term results with limited adverse events."
In their 2011 paper, Dr. Slaughter and his associates put the pending change more bluntly: "As outcomes for continuous-flow LVADs and heart transplantation converge, the therapy of heart transplantation could emerge as salvage therapy for major device-related complications or dysfunction or progressive right heart failure as opposed to the default option for all patients who are eligible for transplant."

Despite their huge promise, some experts have lingering concerns about the current generation of continuous-flow LVADs that make them reluctant to say that heart transplant has unquestionably stepped down from its pedestal as the gold standard treatment for patients with severe, end-stage heart failure.
"There is no question that [current continuous-flow LVADs] are very good technology. But whether they are great technology remains to be proven," said Dr. James K. Kirklin, professor of surgery and director of the division of cardiothoracic surgery at the University of Alabama at Birmingham.
"We still have important concerns about things like drive-line infections and thromboembolic complications. Neither rate is terribly high, but they are high enough to be a problem if you apply LVADs to patients who are doing okay. We still have important doubts about whether we can really mimic the quality of life" achieved by transplant, he said in an interview.
LVAD placement also poses an operative risk, which may be as high as 10%, said Dr. David Taylor, a cardiologist and advanced heart failure specialist at the Cleveland Clinic. "If the risk was 2% or 3%, I’d say do it, but a risk closer to 10% makes you hesitate.
"There are a lot of really sick, elderly patients with heart failure who could unquestionably benefit from an LVAD. But as soon as you push the envelope [by treating sicker patients] you increase mortality. A large group of heart failure patients are very ill with chronic disease and feel terrible, and these are the patients where you’d see the greatest benefit, but we can’t afford to do that. We can’t afford to allocate this resource to patients with a 30% mortality risk, because that would limit our ability to extend it to other patients," Dr. Kirklin said in an interview.
"Most patients appreciate an LVAD and like it, but they still look forward to getting a transplant," said Dr. Stephen H. Bailey, director of cardiothoracic surgery at Allegheny General Hospital in Pittsburgh. "In July 2012, while the margin between LVAD and transplant is narrowing, transplant is still the gold standard for definitive treatment. Most patients still favor transplant, but that might change. It’s not quite there yet that an LVAD is equal to a transplant for the long term. It also requires getting rid of the drive line. That would be a game changer. It would also help if we could reduce gastrointestinal bleeds, but that is more of a nuisance."
Part of the bleeding problem comes from anticoagulation treatment to prevent clots from forming in the LVAD, but another facet is an acquired Von Willebrand factor deficiency caused by absent pulsatile blood flow, Dr. Bailey said. Early clinical results suggest that allowing the heart to beat every few seconds, by adjusting the LVAD’s continuous flow rate, can minimize the acquired deficiency.
Which INTERMACS Level?
The most widely used gauge physicians and surgeons have for assessing LVAD candidates is their level in a seven-step sequence created by INTERMACS (the Interagency Registry for Mechanically Assisted Circulatory Support), which subdivides a patient’s descent from advanced New York Heart Association class III heart failure through the strata of class IV disease. (See table.) The most severe INTERMACS stage, level 1 patients (also known as "crash and burn") are those in cardiogenic shock. During the past year or two, about 16% of U.S. LVAD recipients have been level 1 patients, a percentage that should ideally drop much closer to zero, experts say.

But beyond the maxim that an advanced heart failure patient should get an LVAD before reaching level 1, opinions vary on the best target.
"Everyone agrees that inotrope-dependent patients [profile 2 and 3] should get an LVAD," said Dr. Lanfear. "But you definitely should not wait until INTERMACS 1 or 2. Everyone tries hard to treat level 3 and 4 patients. Level 5 is controversial. We need more data about these clearly less- sick patients."
"INTERMACS 2 and 3 is the sweet spot. Patients in INTERMACS 4, 5, or 6 are not as motivated to be connected to a device and undergo big open heart surgery," said Dr. Bailey.
"When patients are on the cusp of inotrope dependence [before they reach level 3], I start to think about implanting," said Dr. Jeffrey J. Teuteberg, a cardiologist and associate director of the cardiac transplant program at the University of Pittsburgh. "It’s nice to get patients who are pre–inotrope dependent – profile 4, 5, or 6 – but are symptom limited and quality of life limited. For destination therapy, you want patients with function and good quality of life. Mechanical support provides more than just improved survival; it also reduces adverse events and raises quality of life."
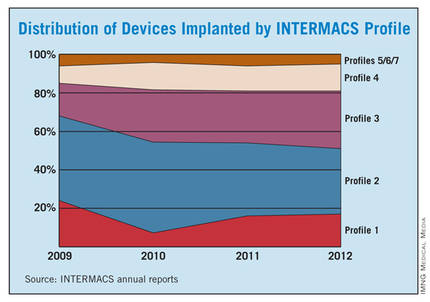
"For level 2 patients, there is no question that an LVAD as bridge to transplant is better than continued medical treatment," said Dr. Taylor. "For level 3 patients, it’s a little trickier, but I think the majority would say that if a patient is stable and inotropic dependent, use an LVAD unless you believe a transplant will occur quickly. An LVAD would reduce the risk for developing cardiogenic shock. The patient who does the best with an LVAD is the one who is [relatively] healthy when implanted, but the patient who needs it most is the one who is literally dying."
"Everyone knows that if you take patients who are sicker [for LVADs], you’ll have trouble reaching 80% survival after 2 years. But there is not yet enough confidence in the treatment to extend it to INTERMACS level 4, 5, or 6 patients," said Dr. Kirklin. "That’s where the big potential is. The action currently is in INTERMACS 2 and 3. For patients who are INTERMACS 3, there is no question that if they can’t get a transplant they need a VAD. The sweet spot will be patients at INTERMACS 4."
The INTERMACS registry numbers show that the field is currently stuck when it comes to INTERMACS levels, with the greatest number of LVADs going into level 2 patients, who received 34% of U.S LVADS during January-March 2012. The 34% level in early 2012 was down from 38% of LVAD recipients in 2011 and 47% in 2010, but growth in less-severe levels has been slow. At the start of 2012, 30% of LVAD recipients were at level 3, a small increase from the 27% rate in 2011 and 2010. Level 4 patients constituted 14% of LVAD recipients in early 2012, essentially unchanged from the prior 2 years, and level 5, 6, or 7 patients have consistently been a small slice of the U.S. LVAD pie, roughly 5% of recipients each year.
Beyond the INTERMACS Level
Heart failure specialists now recognize that INTERMACS level tells just part of the story.
"INTERMACS profiles depend on heart failure symptoms but not comorbidities. Severe diabetes, obstructive pulmonary disease, cardiorenal syndrome, chronic malnourishment, morbid obesity, and other factors all fall outside the INTERMACS profile criteria," said Dr. Kirklin.
He and his associates are formulating a risk assessment equation that will take comorbidities into account for a more global patient assessment. The most recently published INTERMACS registry analysis, which he first authored using data through the end of June 2011 with a total of 4,366 patients who received left ventricular support since 2006 (J. Heart Lung Transplant 2012;31:117-26), identified several comorbidity markers that each significantly linked with increased mortality. For example, a 1-unit increase in bilirubin linked with a 10% boost in mortality, a 1-unit increase in creatinine raised mortality by 16%, and a 0.5-unit increase in body surface area linked with a 48% rise in deaths. One of the strongest risk factors was being at INTERMACS level 1, which linked with a more-than-threefold higher mortality rate.
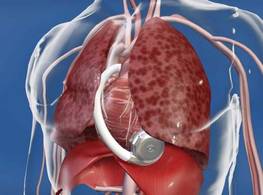
But more work must be done before the risk assessment formula is ready for clinical use. Right now, the formula "is not very reliable yet, because the maximum patient follow-up is 2 years. We need a little more follow-up," Dr. Kirklin said.
Growing the LVAD Numbers
Further growth in LVAD placements will happen on two fronts: broader use in patients at INTERMACS levels 2 and 3, and possibly level 4, where a strong consensus exists for LVAD support; and new evidence to document efficacy and safety in patients with less-severe disease at INTERMACS levels 4, 5, 6, and 7.
For the existing population, it’s a matter of physician and patient awareness. "We need to educate physicians that this is an option," said Dr. Lanfear. "Awareness is lacking. There is a lot of heart failure out there that is underrecognized and underreferred. Neither patients nor their physicians realize how sick they are, and that they are at a high enough risk to justify this."
Furthermore, there are parts of the country where the technology is underused. "In areas without a big center nearby, some physicians may not recognize patients who are sick enough to be LVAD candidates. We are trying to spread the word on who these patients are and when they should be referred."
According to Dr. Teuteberg, major warning signs that should flag patients who are potential LVAD candidates include advanced symptoms, increasing numbers of hospitalizations for heart failure, dwindling responses to ACE inhibitors and beta-blockers, increasing dosages of diuretics, a persistently high serum level of brain natriuretic peptide (BNP) despite good medical treatment, and lack of response to cardiac resynchronization therapy.
Expansion of the evidence base to show LVAD benefits in patients with INTERMACS level 5, 6, or 7 disease depends on a trial just starting, the REVIVE-IT (Randomized Evaluation of VAD Intervention Before Inotropic Therapy) study that’s set to enroll about 100 patients. But at press time, REVIVE-IT had not yet begun, posing doubts about LVAD use in the study’s targeted patients. "We haven’t really demonstrated reproducibly good survival [with LVADs] to compete with medical therapy in level 5 and 6 patients," said Dr. Kirklin. "The FDA put the study on hold while they reflected on that."
Making LVADs Better
With fast-paced technological advancement, continuous-flow LVADs will continue to evolve and improve. In June, results appeared on a new continuous-flow LVAD, the HeartWare device (Circulation 2012;125:3191-200), and last April an FDA advisory committee recommended that the agency approve the HeartWare LVAD for use as a bridge to (a trial testing the HeartWare LVAD for destination therapy is ongoing).
But no one interviewed for this article anticipates that the HeartWare LVAD will be a major advance. "Fundamentally, the major components and the implant technique are the same for the two devices," the HeartWare and the HeartMate II, said Dr. Slaughter. The HeartWare LVAD is smaller and designed to be placed completely in the pericardial space, but any clinical advantages based on these differences remain to be proved, he said in an interview.
A more meaningful improvement in LVAD design is in the works, and may reach initial clinical testing within a couple of years: a fully implantable LVAD with no transcutaneous drive line, a part that is subject to infection, prevents patients from submerging, and physically and psychologically limits patients by tethering them to equipment. "If there were one thing that could make a dramatic difference, it would be getting rid of the drive line. That is the Holy Grail for the field," said Dr. Bailey.
When a fully implantable LVAD becomes available for routine use, it will complete the LVAD revolution and help device therapy for advanced heart failure reach its full potential.
Dr. Lanfear has received research support and has received honoraria as a speaker for Thoratec, the company that markets the HeartMate II, and has received research support from HeartWare, the company developing the HeartWare LVAD. Dr. Slaughter has had contracts for services to Thoratec and HeartWare. Dr. Kirklin, Dr. Bailey, Dr. Teuteberg, and Dr. Taylor said that they had no disclosures.
*CORRECTION 8/10/12: The credit for the above photo was misstated and should have been ©2012 HeartWare International, Inc.
A sea change in management of severe heart failure began two and a half years ago, in January 2010, when the Food and Drug Administration approved U.S. marketing of the HeartMate II continuous-flow, left ventricular assist device for destination therapy. Patients, cardiologists, and cardiothoracic surgeons at last had a reasonably durable, effective, and relatively safe alternative to heart transplant to offer patients at the end stage of deteriorating heart function.
The Heart Mate II quickly supplanted the prior-generation, pulsatile-flow devices, and 2010 also saw a 10-fold spike in the number of left ventricular assist devices (LVADs) placed in U.S. patients as destination therapy.
During the first half of 2011, 6-month survival among U.S. patients with a continuous-flow LVAD (which means the HeartMate II, the only continuous-flow device on the U.S. market) was 89%, and 12-month survival during 2010 was 81% (J. Heart Lung Transplant 2012;31:117-26, putting the continuous-flow LVAD in at least the same ballpark as heart transplant, which has a 5-year survival of about 80% and a median survival of about 10 years in U.S. patients.
But despite estimates that many thousands, if not tens of thousands, of Americans meet the clinical criteria to qualify for placement of an LVAD, the reality is that in the 30 months since the HeartMate II became available for routine destination therapy through mid-2012, only about 4,600 were placed in U.S. patients. In addition, just slightly more than a third of those patients, roughly 1,700, received their LVAD explicitly for destination therapy.
Even in 2012, a majority of patients who received an LVAD got their device with the understanding that it was as a bridge to transplant, an LVAD placed with temporary intent to help patients survive and improve clinically until their LVAD could be swapped out and replaced by a transplanted heart.
LVAD Numbers Up, but Still Low
If the availability of the HeartMate II for destination therapy in routine practice was a revolution for the management of many patients with New York Heart Association class IV heart failure – which is how many experts in this field view the device – it has so far been a revolution played out in slow motion. In large part that’s because the field started small, and stayed small until recently. At the end of 2008, fewer than 1,000 patients had ever received a ventricular assist device or a total artificial heart. Even though the number nearly doubled in 2009, by decade’s end the cumulative total still remained under 2,000.
Despite the low numbers compared with the estimated need, many physicians and surgeons who specialize in advanced heart failure say they are satisfied with the pace at which continuous-flow LVADs have entered patients over the past 2 years. Though they acknowledge that the number of potential U.S. candidates for an LVAD undoubtedly is far larger than the nearly 4,600 who received one since early 2010, they note that the implantation rate grew in a steady and robust way during the past 30 months, as has the number of centers performing the surgery. With a better-than-50% year-over-year growth spurt in both 2010 and 2011, and with that pace continuing into the first months of 2012, the 140 U.S. centers that now place LVADs are on track to do nearly 3,000 this year (although only 42% were for destination therapy during January-March 2012).
"You probably don’t want it to increase too rapidly, because it is still high risk and expensive," said Dr. David E. Lanfear, a cardiologist specializing in advanced heart failure and transplantation at Henry Ford Hospital in Detroit. LVAD placements "have not reached the maximum number of patients who they could help, but you need to be cautious because it requires specialized expertise to do it properly. I wouldn’t expect that [during 2 years] it would immediately reach maximum use. It’s still in the growth phase. There are a lot of patients out there, so it will continue to grow; I’m not concerned that it’s not growing fast enough," he said in an interview.
An LVAD or a Heart Transplant?
Experts also note an important shift in attitude about the role for LVADs, a change that has brought them to the brink of replacing transplanted hearts as the default treatment for patients with end-stage heart failure.
"The current outcomes with HeartMate II have already changed the field. We published a paper last year that showed there was no difference between LVADs and transplant in costs and outcomes at 1 year (Ann. Thorac. Surg. 2011;91:1330-4)," said Dr. Mark S. Slaughter, professor of surgery and chief of thoracic and cardiovascular surgery at the University of Louisville (Ky.). Between that and the competitive mortality benefit from LVADs "we need to rethink the role of transplant. In appropriately selected patients, we can achieve outstanding long-term results with limited adverse events."
In their 2011 paper, Dr. Slaughter and his associates put the pending change more bluntly: "As outcomes for continuous-flow LVADs and heart transplantation converge, the therapy of heart transplantation could emerge as salvage therapy for major device-related complications or dysfunction or progressive right heart failure as opposed to the default option for all patients who are eligible for transplant."
Despite their huge promise, some experts have lingering concerns about the current generation of continuous-flow LVADs that make them reluctant to say that heart transplant has unquestionably stepped down from its pedestal as the gold standard treatment for patients with severe, end-stage heart failure.
"There is no question that [current continuous-flow LVADs] are very good technology. But whether they are great technology remains to be proven," said Dr. James K. Kirklin, professor of surgery and director of the division of cardiothoracic surgery at the University of Alabama at Birmingham.
"We still have important concerns about things like drive-line infections and thromboembolic complications. Neither rate is terribly high, but they are high enough to be a problem if you apply LVADs to patients who are doing okay. We still have important doubts about whether we can really mimic the quality of life" achieved by transplant, he said in an interview.
LVAD placement also poses an operative risk, which may be as high as 10%, said Dr. David Taylor, a cardiologist and advanced heart failure specialist at the Cleveland Clinic. "If the risk was 2% or 3%, I’d say do it, but a risk closer to 10% makes you hesitate.
"There are a lot of really sick, elderly patients with heart failure who could unquestionably benefit from an LVAD. But as soon as you push the envelope [by treating sicker patients] you increase mortality. A large group of heart failure patients are very ill with chronic disease and feel terrible, and these are the patients where you’d see the greatest benefit, but we can’t afford to do that. We can’t afford to allocate this resource to patients with a 30% mortality risk, because that would limit our ability to extend it to other patients," Dr. Kirklin said in an interview.
"Most patients appreciate an LVAD and like it, but they still look forward to getting a transplant," said Dr. Stephen H. Bailey, director of cardiothoracic surgery at Allegheny General Hospital in Pittsburgh. "In July 2012, while the margin between LVAD and transplant is narrowing, transplant is still the gold standard for definitive treatment. Most patients still favor transplant, but that might change. It’s not quite there yet that an LVAD is equal to a transplant for the long term. It also requires getting rid of the drive line. That would be a game changer. It would also help if we could reduce gastrointestinal bleeds, but that is more of a nuisance."
Part of the bleeding problem comes from anticoagulation treatment to prevent clots from forming in the LVAD, but another facet is an acquired Von Willebrand factor deficiency caused by absent pulsatile blood flow, Dr. Bailey said. Early clinical results suggest that allowing the heart to beat every few seconds, by adjusting the LVAD’s continuous flow rate, can minimize the acquired deficiency.
Which INTERMACS Level?
The most widely used gauge physicians and surgeons have for assessing LVAD candidates is their level in a seven-step sequence created by INTERMACS (the Interagency Registry for Mechanically Assisted Circulatory Support), which subdivides a patient’s descent from advanced New York Heart Association class III heart failure through the strata of class IV disease. (See table.) The most severe INTERMACS stage, level 1 patients (also known as "crash and burn") are those in cardiogenic shock. During the past year or two, about 16% of U.S. LVAD recipients have been level 1 patients, a percentage that should ideally drop much closer to zero, experts say.
But beyond the maxim that an advanced heart failure patient should get an LVAD before reaching level 1, opinions vary on the best target.
"Everyone agrees that inotrope-dependent patients [profile 2 and 3] should get an LVAD," said Dr. Lanfear. "But you definitely should not wait until INTERMACS 1 or 2. Everyone tries hard to treat level 3 and 4 patients. Level 5 is controversial. We need more data about these clearly less- sick patients."
"INTERMACS 2 and 3 is the sweet spot. Patients in INTERMACS 4, 5, or 6 are not as motivated to be connected to a device and undergo big open heart surgery," said Dr. Bailey.
"When patients are on the cusp of inotrope dependence [before they reach level 3], I start to think about implanting," said Dr. Jeffrey J. Teuteberg, a cardiologist and associate director of the cardiac transplant program at the University of Pittsburgh. "It’s nice to get patients who are pre–inotrope dependent – profile 4, 5, or 6 – but are symptom limited and quality of life limited. For destination therapy, you want patients with function and good quality of life. Mechanical support provides more than just improved survival; it also reduces adverse events and raises quality of life."
"For level 2 patients, there is no question that an LVAD as bridge to transplant is better than continued medical treatment," said Dr. Taylor. "For level 3 patients, it’s a little trickier, but I think the majority would say that if a patient is stable and inotropic dependent, use an LVAD unless you believe a transplant will occur quickly. An LVAD would reduce the risk for developing cardiogenic shock. The patient who does the best with an LVAD is the one who is [relatively] healthy when implanted, but the patient who needs it most is the one who is literally dying."
"Everyone knows that if you take patients who are sicker [for LVADs], you’ll have trouble reaching 80% survival after 2 years. But there is not yet enough confidence in the treatment to extend it to INTERMACS level 4, 5, or 6 patients," said Dr. Kirklin. "That’s where the big potential is. The action currently is in INTERMACS 2 and 3. For patients who are INTERMACS 3, there is no question that if they can’t get a transplant they need a VAD. The sweet spot will be patients at INTERMACS 4."
The INTERMACS registry numbers show that the field is currently stuck when it comes to INTERMACS levels, with the greatest number of LVADs going into level 2 patients, who received 34% of U.S LVADS during January-March 2012. The 34% level in early 2012 was down from 38% of LVAD recipients in 2011 and 47% in 2010, but growth in less-severe levels has been slow. At the start of 2012, 30% of LVAD recipients were at level 3, a small increase from the 27% rate in 2011 and 2010. Level 4 patients constituted 14% of LVAD recipients in early 2012, essentially unchanged from the prior 2 years, and level 5, 6, or 7 patients have consistently been a small slice of the U.S. LVAD pie, roughly 5% of recipients each year.
Beyond the INTERMACS Level
Heart failure specialists now recognize that INTERMACS level tells just part of the story.
"INTERMACS profiles depend on heart failure symptoms but not comorbidities. Severe diabetes, obstructive pulmonary disease, cardiorenal syndrome, chronic malnourishment, morbid obesity, and other factors all fall outside the INTERMACS profile criteria," said Dr. Kirklin.
He and his associates are formulating a risk assessment equation that will take comorbidities into account for a more global patient assessment. The most recently published INTERMACS registry analysis, which he first authored using data through the end of June 2011 with a total of 4,366 patients who received left ventricular support since 2006 (J. Heart Lung Transplant 2012;31:117-26), identified several comorbidity markers that each significantly linked with increased mortality. For example, a 1-unit increase in bilirubin linked with a 10% boost in mortality, a 1-unit increase in creatinine raised mortality by 16%, and a 0.5-unit increase in body surface area linked with a 48% rise in deaths. One of the strongest risk factors was being at INTERMACS level 1, which linked with a more-than-threefold higher mortality rate.
But more work must be done before the risk assessment formula is ready for clinical use. Right now, the formula "is not very reliable yet, because the maximum patient follow-up is 2 years. We need a little more follow-up," Dr. Kirklin said.
Growing the LVAD Numbers
Further growth in LVAD placements will happen on two fronts: broader use in patients at INTERMACS levels 2 and 3, and possibly level 4, where a strong consensus exists for LVAD support; and new evidence to document efficacy and safety in patients with less-severe disease at INTERMACS levels 4, 5, 6, and 7.
For the existing population, it’s a matter of physician and patient awareness. "We need to educate physicians that this is an option," said Dr. Lanfear. "Awareness is lacking. There is a lot of heart failure out there that is underrecognized and underreferred. Neither patients nor their physicians realize how sick they are, and that they are at a high enough risk to justify this."
Furthermore, there are parts of the country where the technology is underused. "In areas without a big center nearby, some physicians may not recognize patients who are sick enough to be LVAD candidates. We are trying to spread the word on who these patients are and when they should be referred."
According to Dr. Teuteberg, major warning signs that should flag patients who are potential LVAD candidates include advanced symptoms, increasing numbers of hospitalizations for heart failure, dwindling responses to ACE inhibitors and beta-blockers, increasing dosages of diuretics, a persistently high serum level of brain natriuretic peptide (BNP) despite good medical treatment, and lack of response to cardiac resynchronization therapy.
Expansion of the evidence base to show LVAD benefits in patients with INTERMACS level 5, 6, or 7 disease depends on a trial just starting, the REVIVE-IT (Randomized Evaluation of VAD Intervention Before Inotropic Therapy) study that’s set to enroll about 100 patients. But at press time, REVIVE-IT had not yet begun, posing doubts about LVAD use in the study’s targeted patients. "We haven’t really demonstrated reproducibly good survival [with LVADs] to compete with medical therapy in level 5 and 6 patients," said Dr. Kirklin. "The FDA put the study on hold while they reflected on that."
Making LVADs Better
With fast-paced technological advancement, continuous-flow LVADs will continue to evolve and improve. In June, results appeared on a new continuous-flow LVAD, the HeartWare device (Circulation 2012;125:3191-200), and last April an FDA advisory committee recommended that the agency approve the HeartWare LVAD for use as a bridge to (a trial testing the HeartWare LVAD for destination therapy is ongoing).
But no one interviewed for this article anticipates that the HeartWare LVAD will be a major advance. "Fundamentally, the major components and the implant technique are the same for the two devices," the HeartWare and the HeartMate II, said Dr. Slaughter. The HeartWare LVAD is smaller and designed to be placed completely in the pericardial space, but any clinical advantages based on these differences remain to be proved, he said in an interview.
A more meaningful improvement in LVAD design is in the works, and may reach initial clinical testing within a couple of years: a fully implantable LVAD with no transcutaneous drive line, a part that is subject to infection, prevents patients from submerging, and physically and psychologically limits patients by tethering them to equipment. "If there were one thing that could make a dramatic difference, it would be getting rid of the drive line. That is the Holy Grail for the field," said Dr. Bailey.
When a fully implantable LVAD becomes available for routine use, it will complete the LVAD revolution and help device therapy for advanced heart failure reach its full potential.
Dr. Lanfear has received research support and has received honoraria as a speaker for Thoratec, the company that markets the HeartMate II, and has received research support from HeartWare, the company developing the HeartWare LVAD. Dr. Slaughter has had contracts for services to Thoratec and HeartWare. Dr. Kirklin, Dr. Bailey, Dr. Teuteberg, and Dr. Taylor said that they had no disclosures.
*CORRECTION 8/10/12: The credit for the above photo was misstated and should have been ©2012 HeartWare International, Inc.
A sea change in management of severe heart failure began two and a half years ago, in January 2010, when the Food and Drug Administration approved U.S. marketing of the HeartMate II continuous-flow, left ventricular assist device for destination therapy. Patients, cardiologists, and cardiothoracic surgeons at last had a reasonably durable, effective, and relatively safe alternative to heart transplant to offer patients at the end stage of deteriorating heart function.
The Heart Mate II quickly supplanted the prior-generation, pulsatile-flow devices, and 2010 also saw a 10-fold spike in the number of left ventricular assist devices (LVADs) placed in U.S. patients as destination therapy.
During the first half of 2011, 6-month survival among U.S. patients with a continuous-flow LVAD (which means the HeartMate II, the only continuous-flow device on the U.S. market) was 89%, and 12-month survival during 2010 was 81% (J. Heart Lung Transplant 2012;31:117-26, putting the continuous-flow LVAD in at least the same ballpark as heart transplant, which has a 5-year survival of about 80% and a median survival of about 10 years in U.S. patients.
But despite estimates that many thousands, if not tens of thousands, of Americans meet the clinical criteria to qualify for placement of an LVAD, the reality is that in the 30 months since the HeartMate II became available for routine destination therapy through mid-2012, only about 4,600 were placed in U.S. patients. In addition, just slightly more than a third of those patients, roughly 1,700, received their LVAD explicitly for destination therapy.
Even in 2012, a majority of patients who received an LVAD got their device with the understanding that it was as a bridge to transplant, an LVAD placed with temporary intent to help patients survive and improve clinically until their LVAD could be swapped out and replaced by a transplanted heart.
LVAD Numbers Up, but Still Low
If the availability of the HeartMate II for destination therapy in routine practice was a revolution for the management of many patients with New York Heart Association class IV heart failure – which is how many experts in this field view the device – it has so far been a revolution played out in slow motion. In large part that’s because the field started small, and stayed small until recently. At the end of 2008, fewer than 1,000 patients had ever received a ventricular assist device or a total artificial heart. Even though the number nearly doubled in 2009, by decade’s end the cumulative total still remained under 2,000.
Despite the low numbers compared with the estimated need, many physicians and surgeons who specialize in advanced heart failure say they are satisfied with the pace at which continuous-flow LVADs have entered patients over the past 2 years. Though they acknowledge that the number of potential U.S. candidates for an LVAD undoubtedly is far larger than the nearly 4,600 who received one since early 2010, they note that the implantation rate grew in a steady and robust way during the past 30 months, as has the number of centers performing the surgery. With a better-than-50% year-over-year growth spurt in both 2010 and 2011, and with that pace continuing into the first months of 2012, the 140 U.S. centers that now place LVADs are on track to do nearly 3,000 this year (although only 42% were for destination therapy during January-March 2012).
"You probably don’t want it to increase too rapidly, because it is still high risk and expensive," said Dr. David E. Lanfear, a cardiologist specializing in advanced heart failure and transplantation at Henry Ford Hospital in Detroit. LVAD placements "have not reached the maximum number of patients who they could help, but you need to be cautious because it requires specialized expertise to do it properly. I wouldn’t expect that [during 2 years] it would immediately reach maximum use. It’s still in the growth phase. There are a lot of patients out there, so it will continue to grow; I’m not concerned that it’s not growing fast enough," he said in an interview.
An LVAD or a Heart Transplant?
Experts also note an important shift in attitude about the role for LVADs, a change that has brought them to the brink of replacing transplanted hearts as the default treatment for patients with end-stage heart failure.
"The current outcomes with HeartMate II have already changed the field. We published a paper last year that showed there was no difference between LVADs and transplant in costs and outcomes at 1 year (Ann. Thorac. Surg. 2011;91:1330-4)," said Dr. Mark S. Slaughter, professor of surgery and chief of thoracic and cardiovascular surgery at the University of Louisville (Ky.). Between that and the competitive mortality benefit from LVADs "we need to rethink the role of transplant. In appropriately selected patients, we can achieve outstanding long-term results with limited adverse events."
In their 2011 paper, Dr. Slaughter and his associates put the pending change more bluntly: "As outcomes for continuous-flow LVADs and heart transplantation converge, the therapy of heart transplantation could emerge as salvage therapy for major device-related complications or dysfunction or progressive right heart failure as opposed to the default option for all patients who are eligible for transplant."
Despite their huge promise, some experts have lingering concerns about the current generation of continuous-flow LVADs that make them reluctant to say that heart transplant has unquestionably stepped down from its pedestal as the gold standard treatment for patients with severe, end-stage heart failure.
"There is no question that [current continuous-flow LVADs] are very good technology. But whether they are great technology remains to be proven," said Dr. James K. Kirklin, professor of surgery and director of the division of cardiothoracic surgery at the University of Alabama at Birmingham.
"We still have important concerns about things like drive-line infections and thromboembolic complications. Neither rate is terribly high, but they are high enough to be a problem if you apply LVADs to patients who are doing okay. We still have important doubts about whether we can really mimic the quality of life" achieved by transplant, he said in an interview.
LVAD placement also poses an operative risk, which may be as high as 10%, said Dr. David Taylor, a cardiologist and advanced heart failure specialist at the Cleveland Clinic. "If the risk was 2% or 3%, I’d say do it, but a risk closer to 10% makes you hesitate.
"There are a lot of really sick, elderly patients with heart failure who could unquestionably benefit from an LVAD. But as soon as you push the envelope [by treating sicker patients] you increase mortality. A large group of heart failure patients are very ill with chronic disease and feel terrible, and these are the patients where you’d see the greatest benefit, but we can’t afford to do that. We can’t afford to allocate this resource to patients with a 30% mortality risk, because that would limit our ability to extend it to other patients," Dr. Kirklin said in an interview.
"Most patients appreciate an LVAD and like it, but they still look forward to getting a transplant," said Dr. Stephen H. Bailey, director of cardiothoracic surgery at Allegheny General Hospital in Pittsburgh. "In July 2012, while the margin between LVAD and transplant is narrowing, transplant is still the gold standard for definitive treatment. Most patients still favor transplant, but that might change. It’s not quite there yet that an LVAD is equal to a transplant for the long term. It also requires getting rid of the drive line. That would be a game changer. It would also help if we could reduce gastrointestinal bleeds, but that is more of a nuisance."
Part of the bleeding problem comes from anticoagulation treatment to prevent clots from forming in the LVAD, but another facet is an acquired Von Willebrand factor deficiency caused by absent pulsatile blood flow, Dr. Bailey said. Early clinical results suggest that allowing the heart to beat every few seconds, by adjusting the LVAD’s continuous flow rate, can minimize the acquired deficiency.
Which INTERMACS Level?
The most widely used gauge physicians and surgeons have for assessing LVAD candidates is their level in a seven-step sequence created by INTERMACS (the Interagency Registry for Mechanically Assisted Circulatory Support), which subdivides a patient’s descent from advanced New York Heart Association class III heart failure through the strata of class IV disease. (See table.) The most severe INTERMACS stage, level 1 patients (also known as "crash and burn") are those in cardiogenic shock. During the past year or two, about 16% of U.S. LVAD recipients have been level 1 patients, a percentage that should ideally drop much closer to zero, experts say.
But beyond the maxim that an advanced heart failure patient should get an LVAD before reaching level 1, opinions vary on the best target.
"Everyone agrees that inotrope-dependent patients [profile 2 and 3] should get an LVAD," said Dr. Lanfear. "But you definitely should not wait until INTERMACS 1 or 2. Everyone tries hard to treat level 3 and 4 patients. Level 5 is controversial. We need more data about these clearly less- sick patients."
"INTERMACS 2 and 3 is the sweet spot. Patients in INTERMACS 4, 5, or 6 are not as motivated to be connected to a device and undergo big open heart surgery," said Dr. Bailey.
"When patients are on the cusp of inotrope dependence [before they reach level 3], I start to think about implanting," said Dr. Jeffrey J. Teuteberg, a cardiologist and associate director of the cardiac transplant program at the University of Pittsburgh. "It’s nice to get patients who are pre–inotrope dependent – profile 4, 5, or 6 – but are symptom limited and quality of life limited. For destination therapy, you want patients with function and good quality of life. Mechanical support provides more than just improved survival; it also reduces adverse events and raises quality of life."
"For level 2 patients, there is no question that an LVAD as bridge to transplant is better than continued medical treatment," said Dr. Taylor. "For level 3 patients, it’s a little trickier, but I think the majority would say that if a patient is stable and inotropic dependent, use an LVAD unless you believe a transplant will occur quickly. An LVAD would reduce the risk for developing cardiogenic shock. The patient who does the best with an LVAD is the one who is [relatively] healthy when implanted, but the patient who needs it most is the one who is literally dying."
"Everyone knows that if you take patients who are sicker [for LVADs], you’ll have trouble reaching 80% survival after 2 years. But there is not yet enough confidence in the treatment to extend it to INTERMACS level 4, 5, or 6 patients," said Dr. Kirklin. "That’s where the big potential is. The action currently is in INTERMACS 2 and 3. For patients who are INTERMACS 3, there is no question that if they can’t get a transplant they need a VAD. The sweet spot will be patients at INTERMACS 4."
The INTERMACS registry numbers show that the field is currently stuck when it comes to INTERMACS levels, with the greatest number of LVADs going into level 2 patients, who received 34% of U.S LVADS during January-March 2012. The 34% level in early 2012 was down from 38% of LVAD recipients in 2011 and 47% in 2010, but growth in less-severe levels has been slow. At the start of 2012, 30% of LVAD recipients were at level 3, a small increase from the 27% rate in 2011 and 2010. Level 4 patients constituted 14% of LVAD recipients in early 2012, essentially unchanged from the prior 2 years, and level 5, 6, or 7 patients have consistently been a small slice of the U.S. LVAD pie, roughly 5% of recipients each year.
Beyond the INTERMACS Level
Heart failure specialists now recognize that INTERMACS level tells just part of the story.
"INTERMACS profiles depend on heart failure symptoms but not comorbidities. Severe diabetes, obstructive pulmonary disease, cardiorenal syndrome, chronic malnourishment, morbid obesity, and other factors all fall outside the INTERMACS profile criteria," said Dr. Kirklin.
He and his associates are formulating a risk assessment equation that will take comorbidities into account for a more global patient assessment. The most recently published INTERMACS registry analysis, which he first authored using data through the end of June 2011 with a total of 4,366 patients who received left ventricular support since 2006 (J. Heart Lung Transplant 2012;31:117-26), identified several comorbidity markers that each significantly linked with increased mortality. For example, a 1-unit increase in bilirubin linked with a 10% boost in mortality, a 1-unit increase in creatinine raised mortality by 16%, and a 0.5-unit increase in body surface area linked with a 48% rise in deaths. One of the strongest risk factors was being at INTERMACS level 1, which linked with a more-than-threefold higher mortality rate.
But more work must be done before the risk assessment formula is ready for clinical use. Right now, the formula "is not very reliable yet, because the maximum patient follow-up is 2 years. We need a little more follow-up," Dr. Kirklin said.
Growing the LVAD Numbers
Further growth in LVAD placements will happen on two fronts: broader use in patients at INTERMACS levels 2 and 3, and possibly level 4, where a strong consensus exists for LVAD support; and new evidence to document efficacy and safety in patients with less-severe disease at INTERMACS levels 4, 5, 6, and 7.
For the existing population, it’s a matter of physician and patient awareness. "We need to educate physicians that this is an option," said Dr. Lanfear. "Awareness is lacking. There is a lot of heart failure out there that is underrecognized and underreferred. Neither patients nor their physicians realize how sick they are, and that they are at a high enough risk to justify this."
Furthermore, there are parts of the country where the technology is underused. "In areas without a big center nearby, some physicians may not recognize patients who are sick enough to be LVAD candidates. We are trying to spread the word on who these patients are and when they should be referred."
According to Dr. Teuteberg, major warning signs that should flag patients who are potential LVAD candidates include advanced symptoms, increasing numbers of hospitalizations for heart failure, dwindling responses to ACE inhibitors and beta-blockers, increasing dosages of diuretics, a persistently high serum level of brain natriuretic peptide (BNP) despite good medical treatment, and lack of response to cardiac resynchronization therapy.
Expansion of the evidence base to show LVAD benefits in patients with INTERMACS level 5, 6, or 7 disease depends on a trial just starting, the REVIVE-IT (Randomized Evaluation of VAD Intervention Before Inotropic Therapy) study that’s set to enroll about 100 patients. But at press time, REVIVE-IT had not yet begun, posing doubts about LVAD use in the study’s targeted patients. "We haven’t really demonstrated reproducibly good survival [with LVADs] to compete with medical therapy in level 5 and 6 patients," said Dr. Kirklin. "The FDA put the study on hold while they reflected on that."
Making LVADs Better
With fast-paced technological advancement, continuous-flow LVADs will continue to evolve and improve. In June, results appeared on a new continuous-flow LVAD, the HeartWare device (Circulation 2012;125:3191-200), and last April an FDA advisory committee recommended that the agency approve the HeartWare LVAD for use as a bridge to (a trial testing the HeartWare LVAD for destination therapy is ongoing).
But no one interviewed for this article anticipates that the HeartWare LVAD will be a major advance. "Fundamentally, the major components and the implant technique are the same for the two devices," the HeartWare and the HeartMate II, said Dr. Slaughter. The HeartWare LVAD is smaller and designed to be placed completely in the pericardial space, but any clinical advantages based on these differences remain to be proved, he said in an interview.
A more meaningful improvement in LVAD design is in the works, and may reach initial clinical testing within a couple of years: a fully implantable LVAD with no transcutaneous drive line, a part that is subject to infection, prevents patients from submerging, and physically and psychologically limits patients by tethering them to equipment. "If there were one thing that could make a dramatic difference, it would be getting rid of the drive line. That is the Holy Grail for the field," said Dr. Bailey.
When a fully implantable LVAD becomes available for routine use, it will complete the LVAD revolution and help device therapy for advanced heart failure reach its full potential.
Dr. Lanfear has received research support and has received honoraria as a speaker for Thoratec, the company that markets the HeartMate II, and has received research support from HeartWare, the company developing the HeartWare LVAD. Dr. Slaughter has had contracts for services to Thoratec and HeartWare. Dr. Kirklin, Dr. Bailey, Dr. Teuteberg, and Dr. Taylor said that they had no disclosures.
*CORRECTION 8/10/12: The credit for the above photo was misstated and should have been ©2012 HeartWare International, Inc.